Область изобретения
Настоящее изобретение принадлежит к области медицины и относится к производному пиперидина, к способу его получения и к его фармацевтическому применению. В частности, настоящее изобретение относится к производному пиперидина, представленному формулой (I), к способу его получения, к содержащей его фармацевтической композиции и к его применению в качестве модулятора рецептора эстрогена в лечении опосредованного рецептором экстрогена или зависимого от него заболевания или состояния, особенно предпочтительно рака молочной железы.
Предшествующий уровень техники
После длительного периода базовых исследований и клинического мониторинга обнаружено, что заболевания, такие как рак молочной железы, рак яичника, остеопороз, шизофрения и болезнь Альцгеймера, тесно связаны с отклонениями пути передачи сигнала эстрогена. Эстроген представляет собой секретируемый эндокринной системой стероидный гормон и играет важную роль в репродуктивной системе, костной ткани, сердечно-сосудистой системе, иммунной системе и центральной нервной системе. Система преобразования сигнала эстрогена играет важную роль в регуляции роста, дифференцировки и апоптоза клеток. Возникновение и развитие эстрогензависимых опухолей, таких как рак молочной железы, рак яичника и рак эндометрия, тесно связано с эстрогеном. В настоящее время основная химиотерапия рака молочной железы состоит в применении антиэстрогенных средств, таких как тамоксифен. Тем не менее тамоксифен проявляет свойства агониста эстрогена в матке, стимулируя, таким образом, раковые клетки в матке. В связи с этими серьезными побочными эффектами первостепенная задача состоит в поиске нового безопасного и эффективного вида лечения.
Одним из важных белков пути передачи сигнала эстрогена является рецептор эстрогена (ER). ER представляет собой рецептор стероидного гормона и принадлежит к семейству активируемых лигандом факторов транскрипции надсемейства ядерных рецепторов, которое содержит два подтипа: ERα (открытый в 1950 г.) и ERβ (открытый в 1996 г.), соответственно, кодируемых различными генами. ERα и ERβ характеризуются высокой степенью подобия на аминокислотном уровне, где подобие их ДНК-связывающего домена составляет вплоть до 97%, а подобие лиганд-связывающего домена составляет 56%, но в Ν-конце их гомология низка и составляет 24%. ER содержит 6 доменов от А до F, которые состоят из четырех основных функциональных областей. Функциональная область А/В Ν-концевого домена обладает функцией лиганд-независимой активации транскрипции, a AF-1 характеризуется конститутивной активностью. Транскрипция генов-мишеней активируется в результате взаимодействия с основными факторами транскрипции, факторами реактивации и другими факторами транскрипции. В этом функциональном участке имеются множественные сайты фосфорилирования, и сообщают, что роль AF-1 зависит от фосфорилирования белка. ДНК-связывающий домен (DBD), состоящий из домена С, высоко консервативен и содержит 2 домена типа «цинковых пальцев», которые могут специфично связываться с ДНК-мишенью, в то же время, этот домен играет важную роль в димеризации рецептора. Домен D представляет собой шарнирную область, соединяющую домен DBD и лиганд-связывающий домен (LBD), обладающую низкой консервативностью (только 30% гомологии между двумя подтипами). Лиганд-связывающий домен (LBD), состоящий из С-концевого домена Е, определяет специфичное связывание ER с лигандами, такими как эстроген, селективный модулятор эстрогеновых рецепторов (СМЭР) и селективный супрессор эстрогеновых рецепторов (ССЭР). LBD обладает функцией лиганд-зависимой активации транскрипции AF-2, которая характеризуется синергетической реакцией с AF-1 при выполнении роли рецептора ER в активации транскрипции гена-мишени. В то же время, LBD имеет выраженную границу димеризации и может еще функционировать в отсутствие лигандов. Следовательно, LBD является ключевым центром димеризации рецептора.
ERα распространен, в основном, в матке, яичнике, семеннике, гипофизе, почках, придатке семенника и надпочечнике, тогда как ERβ распространен, в основном, в предстательной железе, яичнике, легком, мочевом пузыре, головном мозге и кровеносных сосудах. Исследование СМЭР начато в связи с серьезными побочными эффектами полных агонистов или полных антагонистов. Селективность означает, что СМЭР действует как агонист в некоторых тканях, таких как костная ткань, печень и сердечно-сосудистая система, обогащенных ERβ, где в некоторых других тканях, таких как молочная железа, он действует как антагонист. В матке, являющейся значительной областью действия ERα, он может быть либо агонистом, либо антагонистом. СМЭР, к настоящему времени имеющиеся в продаже, включают тамоксифен, ралоксифен, базедоксифен, торемифен и т.п. Тем не менее в исследованиях обнаружено, что имеющиеся в продаже СМЭР все же обладают серьезными побочными эффектами, например, длительное применение тамоксифена и торемифена может вызывать гиперплазию эндометрия, полипы и рак эндометрия, а частые побочные эффекты ралоксифена включают приливы жара, боль в икрах ног, болезненность в молочной железе и тромбоз вен и т.п. Поэтому исследование и разработка новых соединений все же являются неотложными проблемами, которые необходимо решить.
Тамоксифен принадлежит к классу соединений, известных как селективные модуляторы эстрогеновых рецепторов (СМЭР), и обладает способностью к стабилизации ERα и к некоторой повышающей регуляции уровня рецепторов ERα. В отличие от него, фулвестрант индуцирует быстрый распад ERα и усиливает блокирование биохимического пути передачи сигнала ER, и такие соединения называют селективными супрессорами эстрогеновых рецепторов (ССЭР). Различия механизмов действия этих СМЭР и ССЭР, по-видимому, являются механизмами, ответственными за резистентность к этим соединениям. Значительное число резистентных к тамоксифену и ER-положительных опухолей все же чувствительны к фулвестранту. На основании клинических данных обнаружено, что ССЭР, такими как фулвестрант, можно эффективно лечить некоторые виды рака молочной железы, которые являются ERα-положительными и резистентными к тамоксифену. Таким образом, соединения, ответственные за распад ERα, можно применять для продления периода эффективности у пациентов с раком молочной железы, у которых успешно проходит лечение антиэстрогеновой терапией, где можно успешно применять СМЭР, ингибиторы ароматазы и ССЭР.
Заявки на патенты, в которых раскрыты селективные модуляторы, опосредованные эстрогеновыми рецепторами, включают WO 2014165723, WO 2014151899, WO 2014141292, WO 2014135834 и WO20 14106848.
Для достижения лучшего терапевтического эффекта и для лучшего удовлетворения потребностей рынка авторы изобретения надеются разработать новое поколение высокоэффективных и низкотоксичных ССЭР, нацеленных на путь передачи сигнала эстрогена. В настоящем изобретении предложена новая структура ССЭР, и обнаружено, что соединения с такой структурой обладают высокой активностью и проявляют превосходный антагонизм в отношении рецептора ER.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединению формулы (I) или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси или к их фармацевтически приемлемой соли, где структура соединения формулы (I) является такой, как показано ниже:
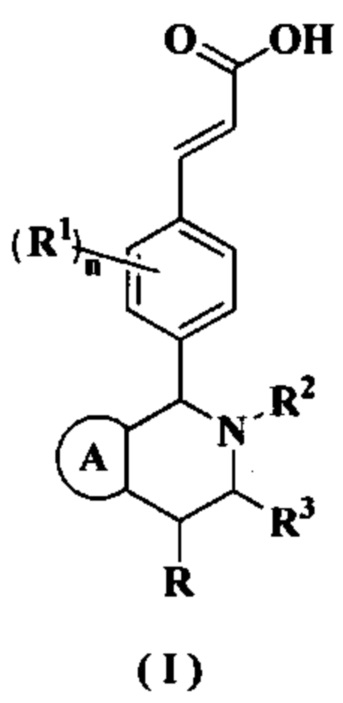
или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или их фармацевтически приемлемой соли,
где
кольцо А выбрано из группы, состоящей из:
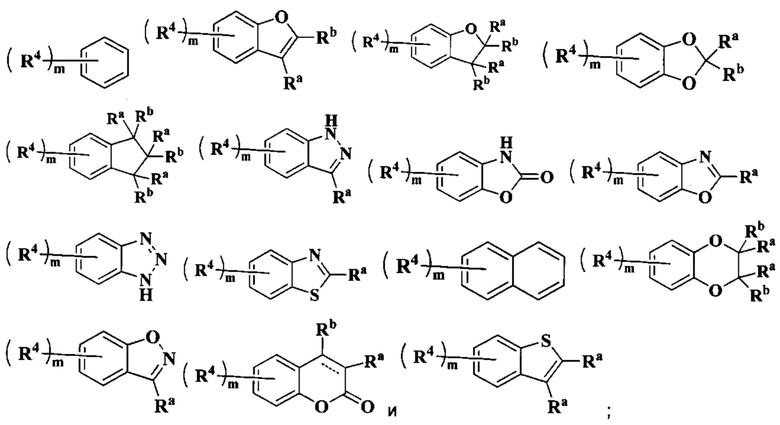
R выбран из группы, состоящей из атома водорода, алкила и циклоалкила, где каждый указанный алкил и циклоалкил необязательно замещен одной или более групп, выбранных из группы, состоящей из атома галогена, амино, циано, гидрокси, алкокси, карбокси, циклоалкила, арила и гетероарила;
каждый из R1 идентичен другому или отличен от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, атома галогена, циано и алкокси, где каждый указанный алкил и алкокси необязательно замещен одной или более групп, выбранных из группы, состоящей из атома галогена, амино, циано и гидрокси;
R2 выбран из группы, состоящей из алкила, галогеналкила и циклоалкила, где каждый указанный алкил и циклоалкил необязательно замещен одной или более групп, выбранных из группы, состоящей из атома галогена, амино, циано, гидрокси, алкокси, карбокси, циклоалкила, арила и гетероарила;
R3 выбран из группы, состоящей из атома водорода, алкила и циклоалкила, где каждый указанный алкил и циклоалкил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, амино, циано, гидрокси, алкокси, карбокси и циклоалкила;
каждый из R4 идентичен другому или отличен от него, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, циклоалкила, алкокси, амино, атома галогена, циано, карбокси, алкенила, алкинила, гетероциклила, арила, гетероарила, -OR5, -NHC(O)OR5 и -NHC(O)NR6R7, где каждый указанный алкил, алкенил, алкинил, циклоалкил, алкокси, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из Rc, алкила, галогеналкила, гидроксиалкила, атома галогена, амино, нитро, циано, гидрокси, оксо, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
Rc выбран из группы, состоящей из алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый указанный алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R5 выбран из группы, состоящей из атома водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый указанный алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила и -C(O)NR6R7;
R6 и R7 идентичны или отличны друг от друга, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, гидрокси, атома галогена, циано, амино, нитро, алкокси, циклоалкила, гетероциклила, арила и гетероарила, где каждый указанный алкил, алкокси, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
Ra и Rb идентичны или отличны друг от друга, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, гидрокси, атома галогена, циано, амино, нитро, алкокси, циклоалкила, гетероциклила, арила и гетероарила, где каждый указанный алкил, алкокси, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, -OR5, арила и гетероарила;
m равно 0, 1, 2, 3 или 4; и
n равно 0, 1, 2, 3 или 4.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли кольцо А выбрано из группы, состоящей из:
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли n равно 2.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R1 представляет собой атом галогена.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R2 представляет собой алкил, где указанный алкил необязательно замещен одной или более групп, выбранных из группы, состоящей из атома галогена, амино, циано, гидрокси, алкокси, карбокси и циклоалкила; предпочтительно алкила или галогеналкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R3 представляет собой алкил.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R представляет собой атом водорода или алкил.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли необязательно выбран из группы, состоящей из соединения формулы (ΙΑ), формулы (IB) и формулы (IC):
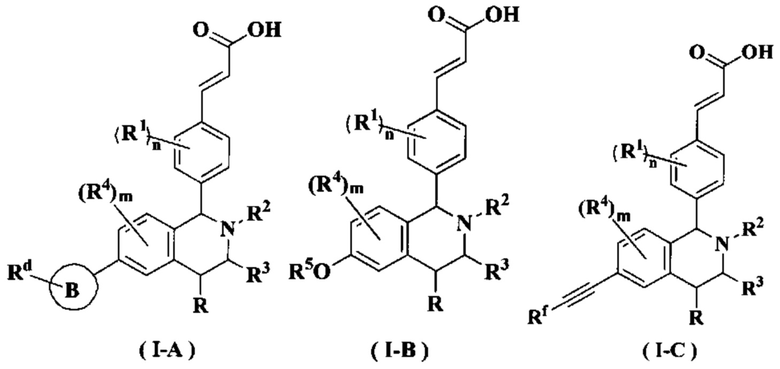
или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или их фармацевтически приемлемой соли,
где
кольцо В выбрано из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила;
Rd выбран из группы, состоящей из атома водорода, алкила, атома галогена, галогеналкила, гидроксиалкила, оксо, амино, циано, циклоалкила, гетероциклила, арила и гетероарила, где каждый указанный алкил, алкокси, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкил, циклоалкила, гетероциклила, арила и гетероарила;
Rf выбран из группы, состоящей из атома водорода, алкила, атома галогена, галогеналкила, гидроксиалкила, гидроксиалкила, амино, циано, циклоалкила, гетероциклила, арила и гетероарила, где каждый указанный алкил, алкокси, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
радикалы с R по R5, m и n являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль необязательно выбраны из группы, состоящей из соединения формулы (II), формулы (III) и формулы (IV):
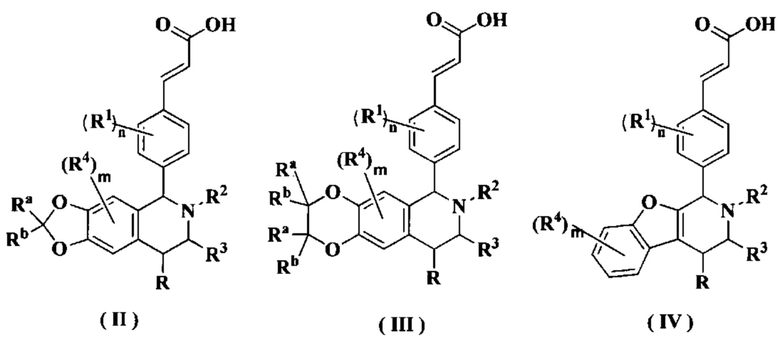
или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или их фармацевтически приемлемой соли,
где
радикалы с R по R3, Ra, Rb, m и n являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль необязательно представляет собой соединение формулы (I-D):
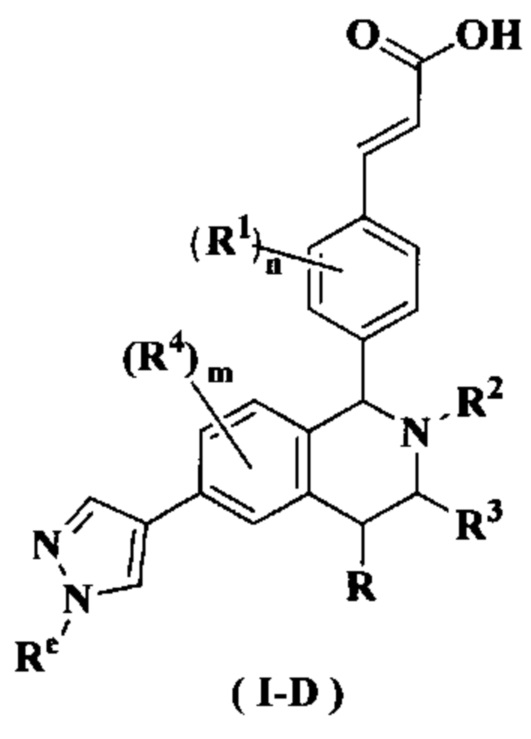
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь или их фармацевтически приемлемую соль,
где
Re выбран из группы, состоящей из алкила, галогеналкила, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила, где каждый указанный алкил, алкокси, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
радикалы с R по R5 и n являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль необязательно представляет собой соединение формулы (I-I):
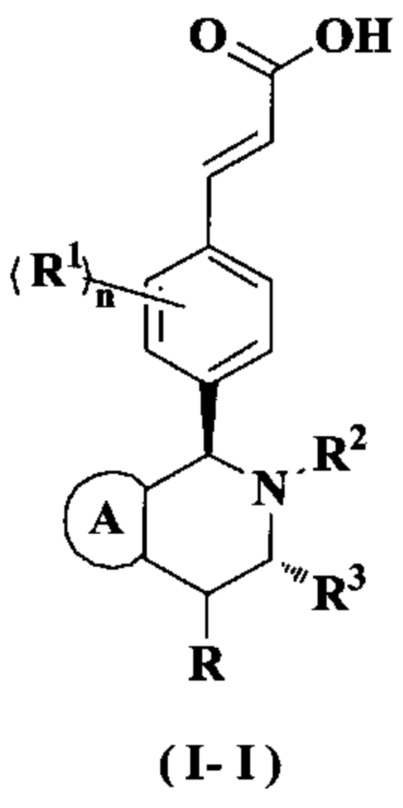
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь или их фармацевтически приемлемая соль,
где
кольцо А, радикалы с R по R3 и n являются такими, как определено в формуле (I).
В настоящем изобретении дополнительно предложено соединение формулы (V) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль,
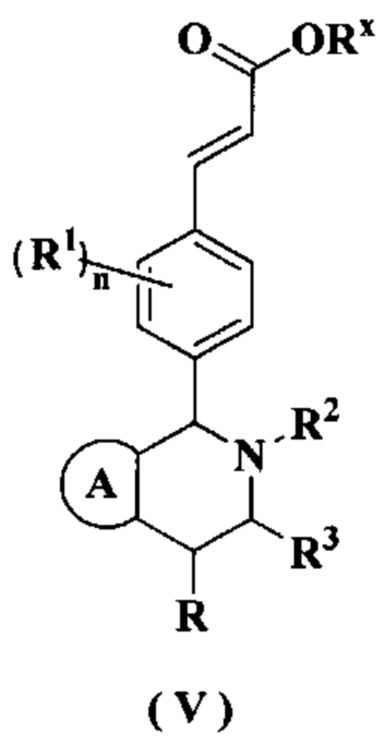
где
Rx представляет собой алкил или циклоалкил, где каждый указанный алкил и циклоалкил необязательно замещен одной или более групп, выбранных из группы, состоящей из алкила, атома галогена, амино, циано, гидрокси, алкокси, карбокси и циклоалкила;
кольцо A, R, радикалы с R1 по R3 и n являются такими, как определено в формуле (I)·
В настоящем изобретении дополнительно предложен способ получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий стадию:
гидролиз соединения формулы (V) в щелочных условиях с получением соединения формулы (I);
где
кольцо А, радикалы с R1 по R3 и n являются такими, как определено в формуле (I).
Другой аспект данного изобретения относится к фармацевтической композиции, содержащей соединение каждой из указанных выше формул или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Далее настоящее изобретение относится к способу получения фармацевтической композиции, включающий стадию смешивания соединения каждой из указанных выше формул или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли с одним или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
Далее настоящее изобретение относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции в получении модулятора рецептора эстрогена.
Далее настоящее изобретение относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции для лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, выбрано из группы, состоящей из рака, нарушений центральной нервной системы (ЦНС), нарушений сердечно-сосудистой системы, нарушений гематологической системы, иммунных и воспалительных заболеваний, подверженности инфекции, нарушений метаболизма, неврологических нарушений, нарушений психики и репродуктивных нарушений, где рак может представлять собой рак молочной железы, рак эндометрия, рак шейки матки, рак кожи, рак предстательной железы, рак яичника, рак фаллопиевой трубы, оофорому, гемофилию или лейкоз; предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки; более предпочтительно рак молочной железы; где нарушения центральной нервной системы (ЦНС) могут представлять собой алкоголизм или мигрень; где нарушения сердечно-сосудистой системы могут представлять собой аневризму аорты, подверженность инфаркту миокарда, склероз клапанов аорты, сердечно-сосудистые заболевания, ишемическую болезнь сердца или гипертензию; где иммунные и воспалительные заболевания могут представлять собой болезнь Грейвса, артрит, рассеянный склероз или цирроз; где подверженность инфекции может представлять собой гепатит В или хроническое заболевание печени; где нарушения метаболизма могут представлять собой холестаз, гипоспадии, ожирение, остеоартрит, остеопению или остеопороз; где неврологические нарушения могут представлять собой болезнь Альцгеймера, болезнь Паркинсона, мигрень или головокружение; где нарушения психики могут представлять собой нервную анорексию, синдром дефицита внимания и гиперактивности (СДВГ), деменцию, тяжелое депрессивное расстройство или психоз; и репродуктивные нарушения могут представлять собой возраст менархе, эндометриоз и бесплодие и т.п.
Далее настоящее изобретение относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо их фармацевтически приемлемой соли для применения в качестве лекарственного средства для лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, выбрано из группы, состоящей из рака, нарушений центральной нервной системы (ЦНС), нарушений сердечно-сосудистой системы, нарушений гематологической системы, иммунных и воспалительных заболеваний, подверженности инфекции, нарушений метаболизма, неврологических нарушений, нарушений психики и репродуктивных нарушений, где рак может представлять собой рак молочной железы, рак эндометрия, рак шейки матки, рак кожи, рак предстательной железы, рак яичника, рак фаллопиевой трубы, оофорому, гемофилию или лейкоз; предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки; более предпочтительно рак молочной железы; где нарушения центральной нервной системы (ЦНС) могут представлять собой алкоголизм или мигрень; где нарушения сердечно-сосудистой системы могут представлять собой аневризму аорты, подверженность инфаркту миокарда, склероз клапанов аорты, сердечно-сосудистые заболевания, ишемическую болезнь сердца или гипертензию; где иммунные и воспалительные заболевания могут представлять собой болезнь Грейвса, артрит, рассеянный склероз или цирроз; где подверженность инфекции может представлять собой гепатит В или хроническое заболевание печени; где нарушения метаболизма могут представлять собой холестаз, гипоспадии, ожирение, остеоартрит, остеопению или остеопороз; где неврологические нарушения могут представлять собой болезнь Альцгеймера, болезнь Паркинсона, мигрень или головокружение; где нарушения психики могут представлять собой нервную анорексию, синдром дефицита внимания и гиперактивности (СДВГ), деменцию, тяжелое депрессивное расстройство или психоз; и репродуктивные нарушения могут представлять собой возраст менархе, эндометриоз и бесплодие и т.п.
Настоящее изобретение дополнительно относится к способу лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или их фармацевтически приемлемой соли. Этот способ показал исключительную эффективность и меньшие побочные эффекты, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, выбрано из группы, состоящей из рака, нарушений центральной нервной системы (ЦНС), нарушений сердечно-сосудистой системы, нарушений гематологической системы, иммунных и воспалительных заболеваний, подверженности инфекции, нарушений метаболизма, неврологических нарушений, нарушений психики и репродуктивных нарушений, где рак может представлять собой рак молочной железы, рак эндометрия, рак шейки матки, рак кожи, рак предстательной железы, рак яичника, рак фаллопиевой трубы, оофорому, гемофилию или лейкоз; предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки; более предпочтительно рак молочной железы; где нарушения центральной нервной системы (ЦНС) могут представлять собой алкоголизм или мигрень; где нарушения сердечно-сосудистой системы могут представлять собой аневризму аорты, подверженность инфаркту миокарда, склероз клапанов аорты, сердечно-сосудистые заболевания, ишемическую болезнь сердца или гипертензию; где иммунные и воспалительные заболевания могут представлять собой болезнь Грейвса, артрит, рассеянный склероз или цирроз; где подверженность инфекции может представлять собой гепатит В или хроническое заболевание печени; где нарушения метаболизма могут представлять собой холестаз, гипоспадии, ожирение, остеоартрит, остеопению или остеопороз; где неврологические нарушения могут представлять собой болезнь Альцгеймера, болезнь Паркинсона, мигрень или головокружение; где нарушения психики могут представлять собой нервную анорексию, синдром дефицита внимания и гиперактивности (СДВГ), деменцию, тяжелое депрессивное расстройство или психоз; и репродуктивные нарушения могут представлять собой возраст менархе, эндометриоз и бесплодие и т.п.
В другом аспекте настоящее изобретение относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо их фармацевтически приемлемой соли для применения в качестве лекарственного средства для лечения рака. Этот способ показал исключительную эффективность и меньшие побочные эффекты в лечении рака, где рак может быть выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, рака фаллопиевой трубы, оофоромы, гемофилии и лейкоза; предпочтительно рака молочной железы, рака яичника, рака эндометрия, рака предстательной железы или рака матки; более предпочтительно рака молочной железы.
В другом аспекте настоящее изобретение относится к способу лечения рака, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли. Этот способ показал исключительную эффективность и меньшие побочные эффекты, где рак может быть выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, рака фаллопиевой трубы, оофоромы, гемофилии и лейкоза; предпочтительно рака молочной железы, рака яичника, рака эндометрия, рака предстательной железы или рака матки; более предпочтительно рака молочной железы.
В другом аспекте настоящее изобретение относится к соединению формулы (I) или к его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо к их фармацевтически приемлемой соли для применения в качестве лекарственного средства для лечения рака кости, рака молочной железы, колоректального рака, рака эндометрия, рака предстательной железы, рака яичника, рака матки, рака шейки матки, рака легкого, лейомиоматоза, лейомиом матки, алкоголизма, мигрени, аневризмы аорты, подверженности инфаркту миокарда, склероза клапанов аорты, сердечно-сосудистого заболевания, ишемической болезни сердца, гипертензии, тромбоза глубоких вен, болезни Грейвса, артрита, рассеянного склероза, цирроза, гепатита В, хронического заболевания печени, холестаза, гипоспадий, ожирения, остеоартрита, остеопороза, болезни Альцгеймера, болезни Паркинсона, мигрени, головокружения, нервной анорексии, синдрома дефицита внимания и гиперактивности (СДВГ), деменции, тяжелого депрессивного расстройства, психоза, возраста менархе, эндометриоза или бесплодия у млекопитающих.
В другом аспекте настоящее изобретение относится к способу лечения рака кости, рака молочной железы, колоректального рака, рака эндометрия, рака предстательной железы, рака яичника, рака матки, рака шейки матки, рака легкого, лейомиоматоза, лейомиом матки, алкоголизма, мигрени, аневризмы аорты, подверженности инфаркту миокарда, склероза клапанов аорты, сердечно-сосудистого заболевания, ишемической болезни сердца, гипертензии, тромбоза глубоких вен, болезни Грейвса, артрита, рассеянного склероза, цирроза, гепатита В, хронического заболевания печени, холестаза, гипоспадий, ожирения, остеоартрита, остеопороза, болезни Альцгеймера, болезни Паркинсона, мигрени, головокружения, нервной анорексии, синдрома дефицита внимания и гиперактивности (СДВГ), деменции, тяжелого депрессивного расстройства, психоза, возраста менархе, эндометриоза или бесплодия у млекопитающих, включающему стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли.
Содержащие активный ингредиент фармацевтические композиции могут иметь форму, приемлемую для перорального применения, например, таблетки, пастилки, лепешки, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, твердой или мягкой капсулы, либо сиропа или эликсира. Композиции для перорального применения можно готовить в соответствии с любым известным в данной области техники способом приготовления фармацевтических композиций. Такие композиции могут содержать одну или более добавок, выбранных из группы, состоящей из подсластителей, корригентов, красителей и консервантов, с целью получения фармацевтического препарата привлекательного внешнего вида и вкуса. Таблетка содержит активный ингредиент и нетоксичные фармацевтически приемлемые эксципиенты, пригодные для производства таблеток. Эти эксципиенты могут представлять собой инертные эксципиенты, гранулирующие агенты, разрыхлители и смазывающие вещества. Таблетка может быть непокрытой или покрытой известным методом, чтобы замаскировать вкус лекарственного средства или замедлить распад и абсорбцию лекарственного средства в желудочно-кишечном тракте, обеспечивая, таким образом, пролонгированное высвобождение в течение продолжительного периода времени.
Пероральная лекарственная форма может быть также представлена в виде мягких желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, либо активный ингредиент смешан с водорастворимым носителем или масляной средой, либо с оливковым маслом.
Водная суспензия содержит активный ингредиент в смеси с эксципиентами, приемлемыми для производства водной суспензии. Такие эксципиенты представляют собой суспендирующие агенты, диспергирующие или увлажняющие агенты. Водная суспензия может также содержать один или более консервантов, один или более красителей, один или более корригентов и один или более подсластителей.
Масляную суспензию можно готовить в виде лекарственной формы путем суспендирования активного ингредиента в растительном масле или в минеральном масле. Масляная суспензия может содержать загуститель. Для обеспечения приемлемого вкуса препарата можно добавлять указанные выше подсластители и корригенты. Эти композиции можно сохранять путем добавления антиоксиданта.
Активный ингредиент и диспергирующие агенты или смачивающие вещества, суспендирующий агент или один или более консервантов можно готовить в виде диспергируемого порошка или гранулы, приемлемых для приготовления водной суспензии путем добавления воды. Примерами приемлемых диспергирующих или смачивающих агентов и суспендирующих агентов являются уже приведенные выше. Могут быть также добавлены дополнительные эксципиенты, такие как подсластители, корригенты и красители.
Настоящая фармацевтическая композиция может также принимать форму эмульсии масло-в-воде. Масляная фаза может представлять собой растительное масло, либо минеральное масло, либо их смесь. Приемлемые эмульгирующие агенты могут представлять собой встречающиеся в природе фосфатиды или частичные сложные эфиры. Эмульсия может также содержать подсластители, корригенты, консерванты и антиоксиданты. Фармацевтическая композиция может иметь форму стерильного инъекционного водного раствора. Приемлемыми носителями и растворителями, которые можно применять, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Стерильный инъекционный препарат может также представлять собой стерильную инъекционную микроэмульсию масло-в-воде, в которой активный ингредиент растворен в масляной фазе. Инъекционный раствор или микроэмульсию можно вводить в кровоток индивида путем локальной болюсной инъекции.
Фармацевтическая композиция может иметь форму стерильной инъекционной водной или масляной суспензии для внутримышечного и подкожного введения. Такую суспензию можно готовить в лекарственной форме с приемлемыми диспергирующими агентами или смачивающими агентами и суспендирующими агентами, как описано выше, в соответствии с известными методами. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор или суспензию, приготовленные в нетоксичном разбавителе или растворителе, приемлемом для парентерального применения. Кроме того, в качестве растворителя или суспензионной среды можно легко использовать стерильные нелетучие масла.
Настоящее соединение можно вводить в форме суппозитория для ректального введения. Эти фармацевтические композиции можно готовить путем смешивания лекарственного средства с приемлемым нераздражающим эксципиентом, который является твердым при обычных температурах, но жидким в прямой кишке, таким образом, он плавится в прямой кишке с высвобождением лекарственного средства.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от ряда факторов, включающих без ограничений следующие факторы: активность конкретного соединения, возраст, пол, общее состояние здоровья, поведение, рацион питания пациента, время введения, путь введения, скорость экскреции, комбинацию лекарственных средств и т.п. Кроме того, лучший вариант лечения, такой как метод лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, может быть подтвержден на основании традиционных схем терапии.
Подробное описание изобретения
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют описанные ниже значения.
«Алкил» относится к насыщенной алифатической углеводородной группе, включающей С1-С20 прямоцепочечные и разветвленные группы, предпочтительно алкил, имеющий от 1 до 12 атомов углерода, и более предпочтительно алкил, имеющий от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1 -этил-2-метил пропил, 1,1,2-триметилпропил, 1,1 -диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексили их разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, n-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-д и метил бутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. В случае ее замещения группа (-ы) заместителей может (-гут) быть замещена (-ы) в любой доступной точке соединения. Группа (-ы) заместителя (-ей) предпочтительно представляет (-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Циклоалкил» относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода и более предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
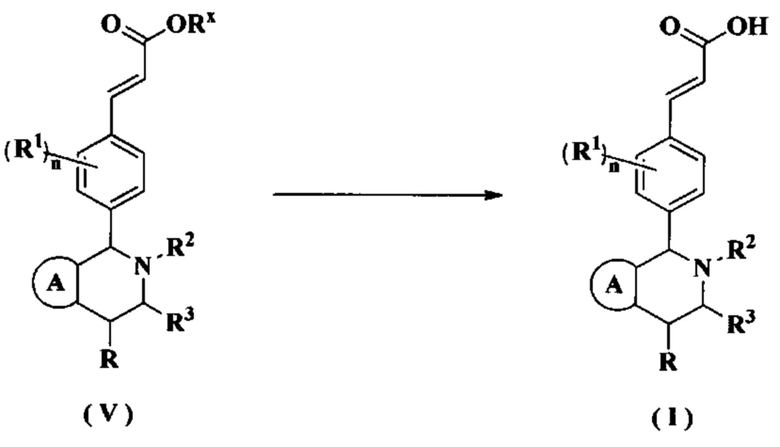
«Спиро-циклоалкил» относится к 5-20-членной полициклической группе, в которой кольца соединены посредством одного общего атома углерода (называемого спиро-атомом), где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному спиро-циклоалкилу и более предпочтительно к 7-10-членному спиро-циклоалкилу. В зависимости от числа спиро-атомов, общих для колец, спиро-циклоалкил можно разделить на моно-спиро-циклоалкил, ди-спиро-циклоалкил или поли-спиро-циклоалкил и предпочтительно моно-спиро-циклоалкил или ди-спиро-циклоалкил, более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-циклоалкил. Неограничивающие примеры спиро-циклоалкилов включают:
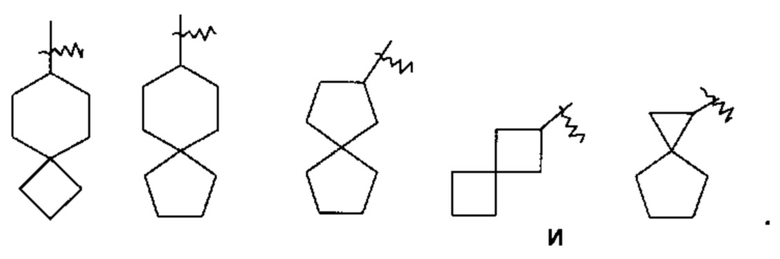
«Конденсированный циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, в которой каждое кольцо в системе имеет общую с другим кольцом пару примыкающих атомов углерода, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному конденсированному циклоалкилу, более предпочтительно к 7-10-членному конденсированному циклоалкилу. В зависимости от числа колец, содержащих члены, конденсированный циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, предпочтительно бициклический или трициклический конденсированный циклоалкил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
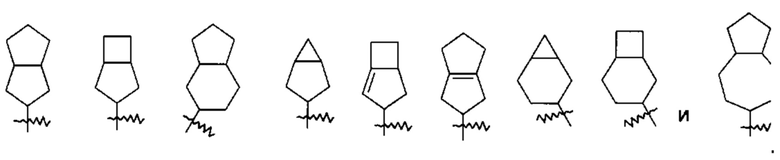
«Мостиковый циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, в которой каждые два кольца в системе имеют общую пару не соединенных атомов углерода, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному мостиковому циклоалкилу и более предпочтительно к 7-10-членному мостиковому циклоалкилу. В зависимости от числа колец, содержащих члены, мостиковый циклоалкил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и предпочтительно бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостиковых циклоалкилов включают:
Циклоалкильное кольцо может быть конденсировано с кольцом арила, гетероарила или гетероциклила, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и т.п. Циклоалкил может быть необязательно замещенным или незамещенным. В случае замещения группа (-ы) заместителя (-ей) предпочтительно представляет (-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Гетероциклил» относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатомов, выбранных из группы, состоящей из Ν, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве кольцевых атомов, но за исключением -O-O-, -O-S- или -S-S- в кольце, где остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 атомов, где от 1 до 4 атомов представляют собой гетероатомы, более предпочтительно от 3 до 6 атомов и наиболее предпочтительно от 5 до 6 атомов. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидротиенил, дигидроимидазолил, дигидрофурил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.п., предпочтительно пиранил, пиперазинил, морфолинил, тетрагидрофуранил, пиперидинил или пирролидинил. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
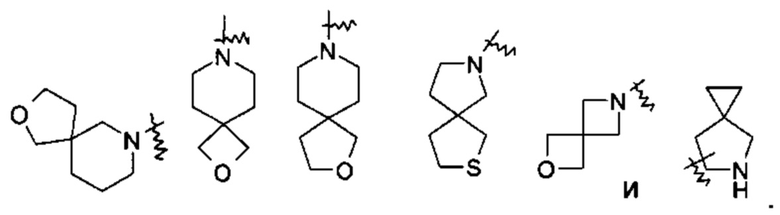
«Спиро-гетероциклил» относится к 5-20-членному полициклическому гетероциклилу, кольца которого соединены через один общий атом (называемый спиро-атомом), где указанные кольца имеют один или более гетероатомов, выбранных из группы, состоящей из Ν, О и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, где остальные кольцевые атомы представляют собой атомы углерода, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно к 6-14-членному спиро-гетероциклилу и более предпочтительно к 7-10-членному спиро-гетероциклилу. В зависимости от числа спиро-атомов, общих для колец, спиро-гетероциклил можно разделить на моно-спиро-гетероциклил, ди-спиро-гетероциклил или поли-спиро-гетероциклил и предпочтительно моно-спиро-гетероциклил или ди-спиро-гетероциклил, более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-гетероциклил. Неограничивающие примеры спиро-гетероциклилов включают:
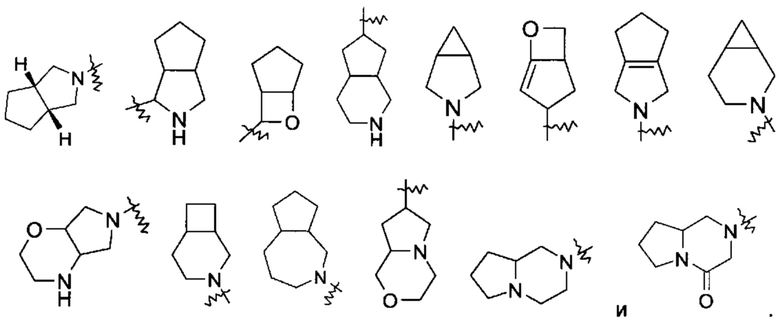
«Конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую пару смежных атомов с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и где указанные кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, где остальные кольцевые атомы представляют собой атомы углерода, предпочтительно к 6-14-членному конденсированному гетероциклилу и более предпочтительно к 7-10-членному конденсированному гетероциклилу. В зависимости от числа колец, содержащих члены, конденсированный гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
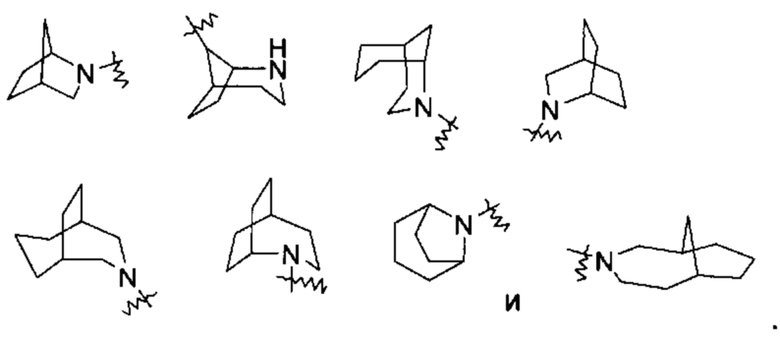
«Мостиковый гетероциклил» относится к 5-14-членной полициклической гетероциклильной группе, где каждые два кольца в системе имеет общую пару не соединенных друг с другом атомов с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и кольца имеют один или более гетероатомов, выбранных из группы, состоящей из Ν, О и S(O)m (где m представляет собой целое число от 0 до 2) в качестве кольцевых атомов, где остальные кольцевые атомы представляют собой атомы углерода, предпочтительно к 6-14-членному мостиковому гетероциклилу и более предпочтительно к 7-10-членному мостиковому гетероциклилу. В зависимости от числа колец, содержащих члены, мостиковый гетероциклил можно разделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и предпочтительно бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостиковых гетероциклилов включают:
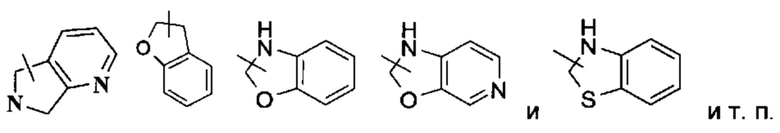
Гетероциклильное кольцо может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры включают:
Гетероциклил может быть необязательно замещенным или незамещенным. В случае замещения группа (-ы) заместителя (-ей) предпочтительно представляет (-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
"Арил" относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в системе имеет общую пару смежных атомов углерода с другим кольцом в системе), имеющему полностью конъюгированную пи-электронную систему, предпочтительно к 6-10-членному арилу, например фенилу и нафтилу. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Неограничивающие примеры включают:
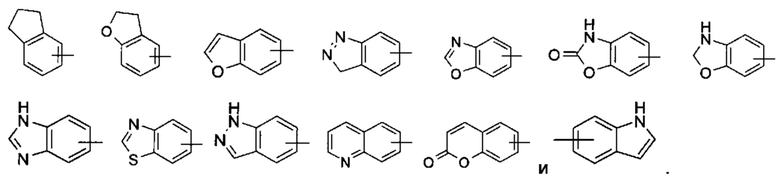
Арил может быть необязательно замещенным или незамещенным. В случае замещения группа (-ы) заместителя (-ей) предпочтительно представляет (-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N в качестве кольцевых атомов, предпочтительно к 5-10-членному гетероарилу, более предпочтительно к 5- или 6-членному гетероарилу, например, к имидазолилу, фурилу, тиенилу, тиазолилу, пиразолилу, оксазолилу, пирролилу, тетразолилу, пиридинилу, пиримидинилу, тиадиазолу, пиразинилу и т.п., предпочтительно к имидазолилу, пиразолилу, пиримидинилу, пиридинилу, тиазолилу или тетразолилу; более предпочтительно к пиразолилу. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Неограничивающие примеры включают:
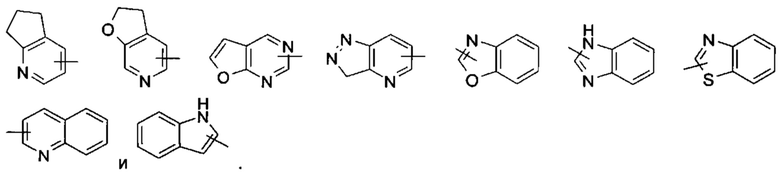
Гетероарил может быть необязательно замещенным или незамещенным. В случае замещения группа (-ы) заместителя (-ей) предпочтительно представляет (-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Алкокси» относится к группе -О-(алкил) или -O-(незамещенный циклоалкил), где алкил является таким, как определено выше. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т.п. Алкоксигруппа может быть необязательно замещенной или незамещенной. В случае замещения группа (-ы) заместителя (-ей) предпочтительно представляет (-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Гидроксиалкил» относится к алкильной группе, замещенной гидроксигруппой, где алкильная группа является такой, как определено выше.
«Гидрокси» относится к группе -ОН.
«Атом галогена» относится к атому фтора, хлора, брома или йода.
«Амино» относится к группе -ΝΗ2.
«Циано» относится к группе -CN.
«Нитро» относится к группе -ΝO2.
«Карбокси» относится к группе -С(O)ОН.
«Алкоксикарбонил» относится к группе -С(O)O(алкил) или (циклоалкил), где алкил и циклоалкил являются такими, как определено выше.
«Ацилгалогенид» относится к соединению, содержащему группу -С(O)-атом галогена.
«Необязательный» или «необязательно» означает, что описанное впоследствии событие или обстоятельство может произойти, но необязательно произойдет, и такое описание включает ситуацию, где это событие или обстоятельство происходит или не происходит. Например, «гетероциклическая группа, необязательно замещенная алкилом» означает, что алкильная группа может присутствовать, но необязательно присутствует, и такое описание включает ситуацию замещения гетероциклической группы алкилом и отсутствия замещения гетероциклической группы алкилом.
«Замещенный» относится к одному или более атомов водорода в группе, предпочтительно вплоть до 5, более предпочтительно 1-3 атомам водорода, независимо замещенным соответствующим количеством заместителей. Безусловно, заместители могут существовать в их возможном химическом положении. Специалист в данной области техники способен определить, является ли замещение возможным или невозможным, с помощью экспериментов или теории, не прилагая слишком больших усилий. Например, комбинация амино- или гидроксигруппы, имеющих свободные атомы водорода, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые связи), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси одного или более соединений в соответствии с настоящим изобретением или их физиологически/фармацевтически приемлемых солей или пролекарств и других химических соединений, таких как физиологически/фармацевтически приемлемые носители и эксципиенты. Цель фармацевтической композиции состоит в том, чтобы способствовать введению соединения в организм, обеспечивающему возможность абсорбции и, следовательно, проявления биологической активности активного ингредиента.
Фармацевтически приемлемая соль относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной и обладает желаемой биологической активностью в организме млекопитающего.
В настоящем изобретении различные термины, такие как «X выбран из группы, состоящей из А, В или С», «X выбран из группы, состоящей из А, В и С», «X представляет собой А, В или С» и «X представляет собой А, В и С», имеют одно и то же значение. Это означает, что X может представлять собой один или более из А, В и С.
Способ синтеза соединения по настоящему изобретению
Для достижения цели настоящего изобретения в настоящем изобретении применяют следующие технические решения синтеза.
Способ получения соединения формулы (I) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли включает следующие стадии:
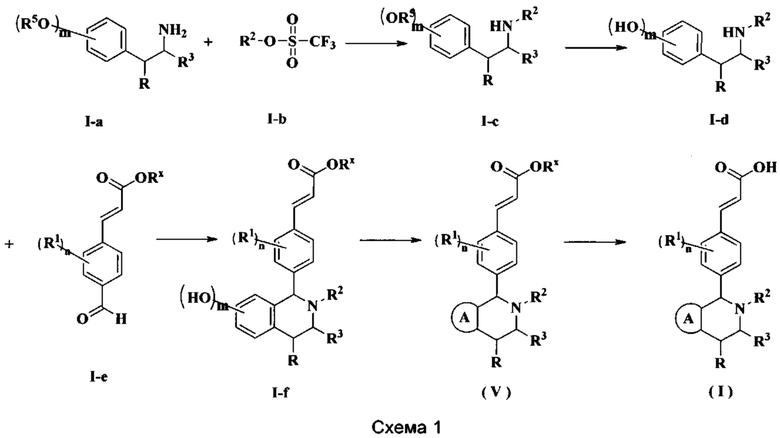
Соединение формулы (I-а) подвергают взаимодействию с соединением формулы (I-b) при высокой температуре в щелочных условиях с получением соединения формулы (I-c), где щелочной реагент, обеспечивающий щелочные условия для данного взаимодействия, предпочтительно представляет собой Ν,Ν-диизопропилэтиламин. Полученное в результате соединение формулы (I-c) восстанавливают при комнатной температуре с получением соединения формулы (I-d), где катализатор в этих условиях предпочтительно представляет собой палладий на углероде, а восстанавливающий агент предпочтительно представляет собой водород. Полученное в результате соединение формулы (I-d) подвергают реакции замыкания кольца с соединением формулы (I-е) при нагревании в кислых условиях с получением соединения формулы (I-f), где кислый реагент, обеспечивающий кислые условия для данного взаимодействия, предпочтительно представляет собой уксусную кислоту. Полученное в результате соединение формулы (I-f) необязательно подвергают реакции замыкания кольца с дигалогенидом при высокой температуре в щелочных условиях с получением соединения формулы (V), где щелочной реагент, обеспечивающий щелочные условия для данного взаимодействия, предпочтительно представляет собой карбонат цезия. Полученное в результате соединение формулы (V) подвергают гидролизу в щелочных условиях с получением соединения формулы (I), где щелочной реагент, обеспечивающий щелочные условия для данного взаимодействия, предпочтительно представляет собой гидроксид лития и гидроксид натрия.
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания, где органические основания включают без ограничений триэтиламин, Ν,Ν-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия, и где неорганические основания включают без ограничений гидрид натрия, фосфат калия, карбонат натрия, карбонат калия и карбонат цезия, гидроксид натрия и гидроксид лития.
Реагент, обеспечивающий кислые условия, включает без ограничений муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту и метансульфоновую кислоту.
Используемый в данном случае восстанавливающий агент включает без ограничений порошок Fe, порошок Zn, Н2, боргидрид натрия, триацетоксиборгидрид натрия, нитрил натрия - боргидрид натрия и алюмогидрид лития.
Используемый в данном случае растворитель включает без ограничений уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и Ν,Ν-диметилформамид,
где
кольцо А, радикалы с R1 по R3, R5, m и n являются такими, как определено в формуле (I).
Соединения формулы (I) по настоящему изобретению также могут быть получены следующим образом:
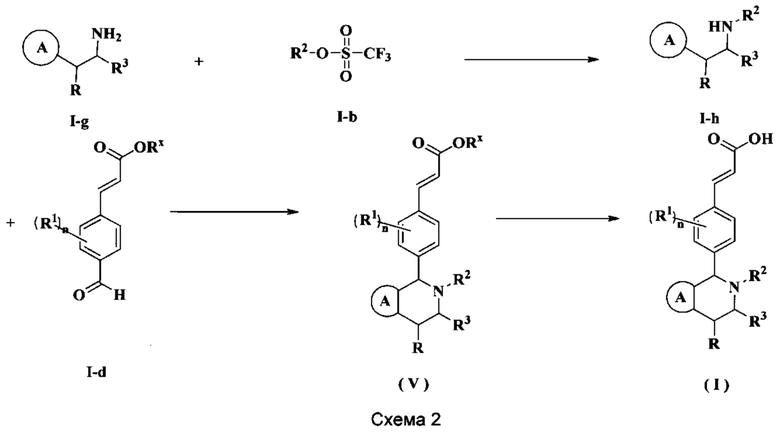
Соединение формулы (I-g) подвергают взаимодействию с соединением формулы (I-b) при нагревании и в щелочных условиях с получением соединения формулы (I-h), где щелочной реагент, обеспечивающий щелочные условия для данного взаимодействия, предпочтительно представляет собой Ν,Ν-диизопропилэтиламин. Полученное в результате соединение формулы (I-h) подвергают реакции замыкания кольца с триизопропилсилилхлоридом и соединением формулы (I-d) при нагревании с получением соединения формулы (V). Полученное в результате соединение формулы (V) подвергают гидролизу в щелочных условиях с получением соединения формулы (I), где щелочной реагент, обеспечивающий щелочные условия для данного взаимодействия, предпочтительно представляет собой гидроксид лития и гидроксид натрия.
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания, где органические основания включают без ограничений триэтиламин, Ν,Ν-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия, и где неорганические основания включают без ограничений гидрид натрия, фосфат калия, карбонат натрия, карбонат калия и карбонат цезия, гидроксид натрия и гидроксид лития.
Используемый в данном случае растворитель включает без ограничений уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и Ν,Ν-диметилформамид,
где
кольцо А, радикалы с R1 по R3 и n являются такими, как определено в формуле (I).
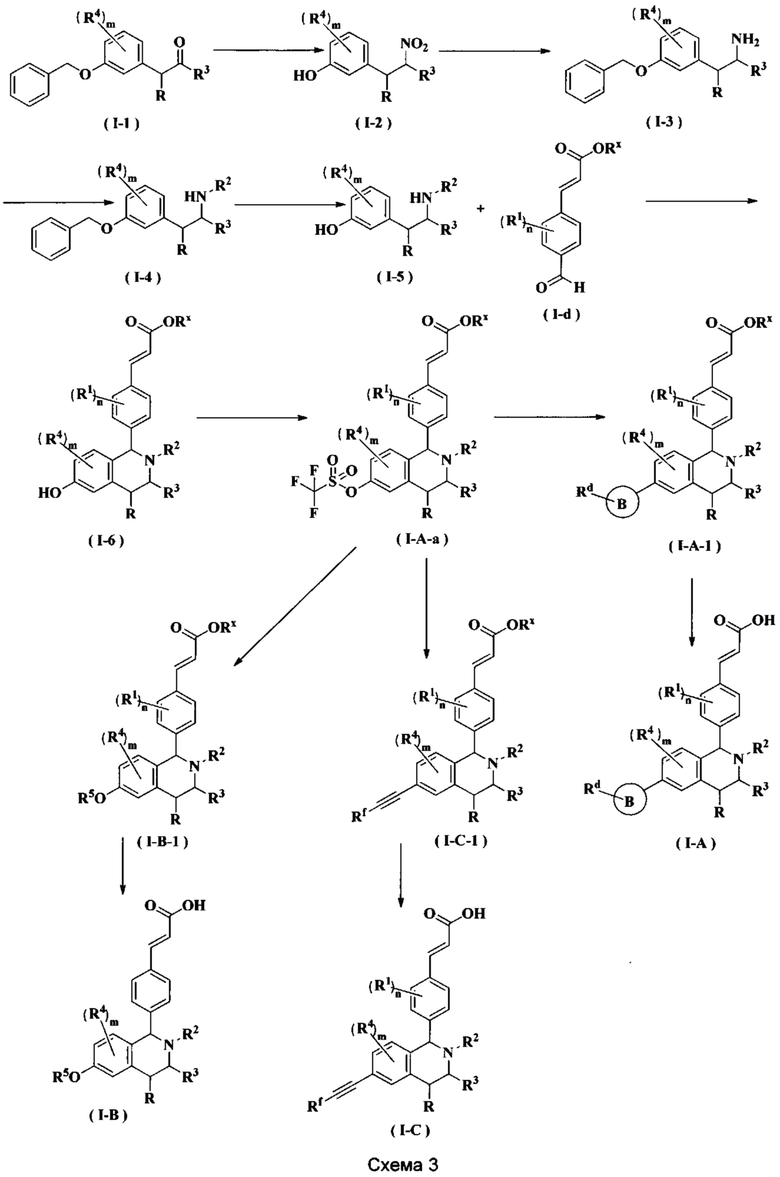
Соединение формулы (I-1) подвергают взаимодействию с ацетатом аммония и нитроэтаном при высокой температуре с получением соединения формулы (Ι-2). Полученное в результате соединение формулы (Ι-2) восстанавливают восстанавливающим агентом в этих условиях с получением соединения формулы (Ι-3), где восстанавливающий агент предпочтительно представляет собой алюмогидрид лития. Полученное в результате соединение формулы (Ι-3) подвергают взаимодействию с галогеналкилтрифторметансульфонатом при высокой температуре в щелочных условиях с получением соединения формулы (Ι-4), где щелочной реагент, обеспечивающий щелочные условия в этих условиях, предпочтительно представляет собой Ν,Ν-диизопропилэтиламин. Полученное в результате соединение формулы (Ι-4) подвергают воздействию высокой температуры и кислых условий с получением соединения формулы (Ι-5), где кислый реагент, обеспечивающий кислые условия в данных условиях, предпочтительно представляет собой трифторуксусную кислоту. Полученное в результате соединение формулы (Ι-5) подвергают взаимодействию с соединением формулы (I-d) при высокой температуре в кислых условиях с получением соединения формулы (I-6), где кислый реагент, обеспечивающий кислые условия в данных условиях, предпочтительно представляет собой уксусную кислоту. Полученное в результате соединение формулы (I-6) подвергают взаимодействию с трифторметансульфоновым ангидридом в щелочных условиях при низкой температуре с получением соединения формулы (I-A-a), где щелочной реагент, обеспечивающий щелочные условия в этих условиях, предпочтительно представляет собой 2,6-лутидин. Полученное в результате соединение формулы (I-A-a) подвергают взаимодействию в различных условиях с получением соединения соответствующей формулы.
(1) Соединение формулы (I-A-a) подвергают взаимодействию с боратом при высокой температуре в присутствии щелочного катализатора с получением соединения формулы (I-A-1), где щелочной реагент, обеспечивающий щелочные условия в данных условиях, предпочтительно представляет собой карбонат натрия, а катализатор предпочтительно представляет собой тетракис(трифенилфосфин)палладий. Полученное в результате соединение формулы (I-A-1) подвергают гидролизу в щелочных условиях с получением соединения формулы (I-A), где щелочной реагент, обеспечивающий щелочные условия в данных условиях, предпочтительно представляет собой гидроксид лития.
(2) Соединение формулы (I-A-a) подвергают взаимодействию с R5-содержащим галогенидом с получением соединения формулы (I-B-1). Полученное в результате соединение формулы (I-B-1) подвергают гидролизу в щелочных условиях с получением соединения формулы (I-В), где щелочной реагент, обеспечивающий щелочные условия в данных условиях, предпочтительно представляет собой гидроксид натрия.
(3) Соединение формулы (I-А-а) подвергают взаимодействию с йодидом меди и алкинил-содержащим соединением при высокой температуре в присутствии щелочного катализатора с получением соединения формулы (I-C-1), где щелочной реагент, обеспечивающий щелочные условия в данных условиях, предпочтительно представляет собой карбонат натрия, а катализатор предпочтительно представляет собой Ν,Ν-диизопропилэтиламин, и катализатор предпочтительно представляет собой бис(трифенилфосфин)палладия (II) дихлорид. Полученное в результате соединение формулы (I-C-1) подвергают гидролизу в щелочных условиях с получением соединения формулы (I-C), где щелочной реагент, обеспечивающий щелочные условия в данных условиях, предпочтительно представляет собой гидроксид натрия.
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания, где органические основания включают без ограничений триэтиламин, Ν,Ν-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия, и где неорганические основания включают без ограничений гидрид натрия, фосфат калия, карбонат натрия, карбонат калия и карбонат цезия, гидроксид натрия и гидроксид лития.
Реагент, обеспечивающий кислые условия, включает без ограничений муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту и метансульфоновую кислоту.
Используемый в данном случае восстанавливающий агент включает без ограничений порошок Fe, порошок Zn, Н2, боргидрид натрия, триацетоксиборгидрид натрия, нитрил натрия - боргидрид натрия и алюмогидрид лития.
Используемый в данном случае растворитель включает без ограничений уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и Ν,Ν-диметилформамид.
Катализатор включает без ограничений тетракис(трифенилфосфин)палладий, бис(трифенилфосфин)палладия (II) дихлорид, дихлорид палладия, ацетат палладия, [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) дихлорид или трис(дибензилиденацетон)дипалладий,
где
кольцо В, радикалы с R1 по R5, Rx, m и n являются такими, как определено в формуле (I) и в формуле (V).
Предпочтительные воплощения изобретения
Далее настоящее изобретение описано со ссылкой на следующие примеры, но эти примеры не следует истолковывать как ограничивающие объем изобретения.
Примеры
Структуры соединений идентифицированы по данным ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Химические сдвиги ЯМР (δ) приведены в 10-6 (млн-1). ЯМР определяют с помощью прибора Bruker AVANCE-400. Растворы для определения представляют собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), и внутренний стандарт представляет собой триметилсилан (ТМС).
МС определяют с помощью спектрометра FINNIGAN LCQAd (ионизация электрораспылением (ИЭР)) (производитель: компания Thermo, тип: Finnigan LCQ advantage MAX).
Данные высокоэффективной жидкостной хроматографии (ВЭЖХ) определяют на спектрометре для жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и на спектрометре для жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Данные хиральной ВЭЖХ определяют на приборе LC-10A vp (Shimadzu) или SFC-analytical (Berger Instruments Inc.).
Значения IC50 средней скорости ингибирования киназы определяют с помощью набора реагентов для твердофазного иммуносорбентного ферментного анализа (ИФА) NovoStar (BMG Co., Германия).
Для тонкослойной хроматографии на силикагеле (ТСХ) используют пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размеры пластины силикагеля, используемой для ТСХ, составляют от 0,15 мм до 0,2 мм, а размеры пластины силикагеля, используемой для очистки продукта, составляют от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии используют силикагель Yantai Huanghai от 200 до 300 меш.
Для хиральной препаративной колоночной хроматографии используют прибор Prep Star SD-1 (Varian Instruments Inc.) или SFC-multigram (Berger Instruments Inc.).
Прибор для быстрой препаративной хроматографии CombiFlash представляет собой Teledyne Isco CombiFlash®Rf200 от компании America.
Известные исходные вещества по настоящему изобретению могут быть получены традиционными способами синтеза в данной области техники или приобретены у компаний ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari Chemical Company и т.д.
Если не указано иное, реакции проводят в атмосфере азота или в атмосфере аргона.
Термин «атмосфера азота» или «атмосфера аргона» означает, что реакционная колба оборудована баллоном азота или аргона емкостью 1 л.
Термин «атмосфера водорода» означает, что реакционная колба оборудована баллоном водорода емкостью 1 л.
Реакции гидрогенизации под давлением проводят с помощью аппарата Парра для гидрогенизации Parr 3916ЕКХ и генератора водорода QL-500 или аппарата для гидрогенизации HC2-SS.
В реакциях гидрогенизации в реакционной системе обычно создают вакуум и заполняют ее водородом, и описанную выше операцию повторяют три раза.
В микроволновой реакции используют микроволновой реактор типа СЕМ Discover-S 908860.
Если не указано иное, раствор, используемый в реакциях, относится к водному раствору.
Если не указано иное, температура реакции в реакциях относится к комнатной температуре от 20°С до 30°С.
Мониторинг хода реакции проводят с помощью тонкослойной хроматографии (ТСХ), и система растворителей для проявления включает: А: дихлорметан и метанол, В: н-гексан и этилацетат, С: петролейный эфир и этилацетат, D: ацетон. Объемное отношение растворителей можно регулировать в соответствии с полярностью соединений. Система элюции соединений колоночной хроматографией, тонкослойной хроматографией и быстрой препаративной флэш-хроматографией с помощью аппарата CombiFlash включает: А: дихлорметан и метанол, В: н-гексан и этилацетат, С: дихлорметан и ацетон. Объемное отношение растворителей можно регулировать в соответствии с полярностью соединений, и иногда можно добавлять слабощелочной реагент, такой как триэтиламин, или кислый реагент, такой как уксусная кислота.
Пример 1
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловая кислота
Стадия 1
N-(1-(3,4-бис(бензилокси)фенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин
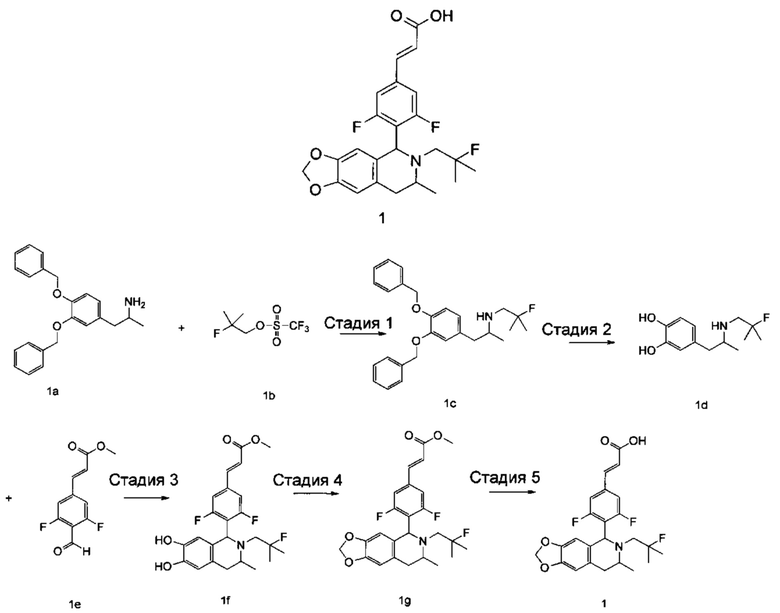
1-(3,4-Бис(бензилокси)фенил)пропан-2-амин 1а (0,8 г, 2,3 ммоль, полученный хорошо известным способом, раскрытым в публикации Bioorganic & Medicinal Chemistry, 2002, 10 (4), 1085-1092), 2-фтор-2-метилпропилтрифторметансульфонат 1b (671 мг, 3 ммоль, полученный хорошо известным способом, раскрытым в публикации Journal of Organic Chemistry, 2005, 70 (6), 2372-2375), и Ν,Ν-диизопропилэтиламин (595 мг, 4,6 ммоль) растворяли в 8 мл 1,4-диоксана. Полученную в результате смесь перемешивали в течение 12 часов при 90°С. Реакцию останавливали, и реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, N-(1-(3,4-бис(бензилокси)фенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина 1с (527 мг, выход 54,3%) в виде желтого масла.
Стадия 2
4-(2-((2-Фтор-2-метилпропил)амино)пропил)бензол-1,2-диол
N-(1-(3,4-бис(бензилокси)фенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 1с (100 мг, 0,237 ммоль) растворяли в 10 мл метанола, добавляли Pd/C (20 мг) в атмосфере аргона, затем реакционную систему продували водородом три раза. Реакционную смесь перемешивали в течение 12 часов при нормальных значениях давления и температуры. Реакцию останавливали, и смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, 4-(2-((2-фтор-2-метилпропил)амино)пропил)бензол-1,2-диола 1d (57 мг) в виде светло-желтого масла, которое использовали непосредственно в следующей стадии.
Стадия 3
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6,7-дигидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
Неочищенный 4-(2-((2-фтор-2-метилпропил)амино)пропил)бензол-1,2-диол 1d (57 мг, 0,236 ммоль) растворяли в 1,5 мл метанола, затем добавляли (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е (53,4 мг, 0,236 ммоль, полученный способом, раскрытым в заявке на патент WO 2014191726) и уксусную кислоту (28,4 мг, 0,472 ммоль). Полученную в результате смесь нагревали до 55°С и перемешивали в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении для удаления метанола и уксусной кислоты, добавляли дихлорметан, и остаток очищали хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6,7-дигидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 1f (51 мг, выход 48%) в виде коричневого масла.
Стадия 4
(Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6,7-дигидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 1f (51 мг, 0,113 ммоль) растворяли в 15 мл Ν,Ν-диметилформамида, затем добавляли дибромметан (29,6 мг, 0,17 ммоль) и карбонат цезия (55,5 мг, 0,17 ммоль). Полученную в результате смесь нагревали до 110°С и перемешивали в течение 3 часов. Реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении. Добавляли 10 мл этилацетата, затем смесь промывали водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акрилата 1g (50 мг) в виде коричневого твердого вещества, которое использовали непосредственно в следующей стадии.
Стадия 5
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловая кислота
Неочищенный (Е)-метил-3-(3,5-дифтор-4-(6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акрилат 1g (50 мг, 0,11 ммоль) растворяли в 2 мл смеси тетрагидрофурана и метанола (об./об. составляет 3:1). Реакционную смесь охлаждали до 0°С, затем добавляли 0,54 мл 1 Μ водного раствора гидроксида лития. Реакционную смесь естественным путем подогревали до комнатной температуры и перемешивали в течение 0,5 часа. Реакцию останавливали, и реакционный раствор концентрировали при пониженном давлении для удаления метанола и тетрагидрофурана. Добавляли по каплям 0,5 н. разбавленную соляную кислоту для доведения рН до 5, и смесь экстрагировали этилацетатом (5 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали высокоэффективной жидкостной хроматографией с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловой кислоты 1 (10 мг, выход 20,6%), в виде желтого твердого вещества.
МС m/z (ИЭР): 448,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.52 (d, 1Н), 7.04 (d, 2Н), 6.65 (s, 1Н), 6.37 (d, 1Н), 6.30 (s, 1H), 5.94 (d, 2H), 5.78 (s, 1H), 4.15-4.17 (m, 1H), 3.22-3.41 (m, 2H), 2.98 (t, 1H), 2.71 (dd, 1H), 1.58 (d, 3H), 1.26-1.43 (m, 7H).
Пример 2
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2,3,6,7,8,9-гексагидро-[1,4]диоксино[2,3-г]изохинолин-6-ил)фенил)акриловая кислота
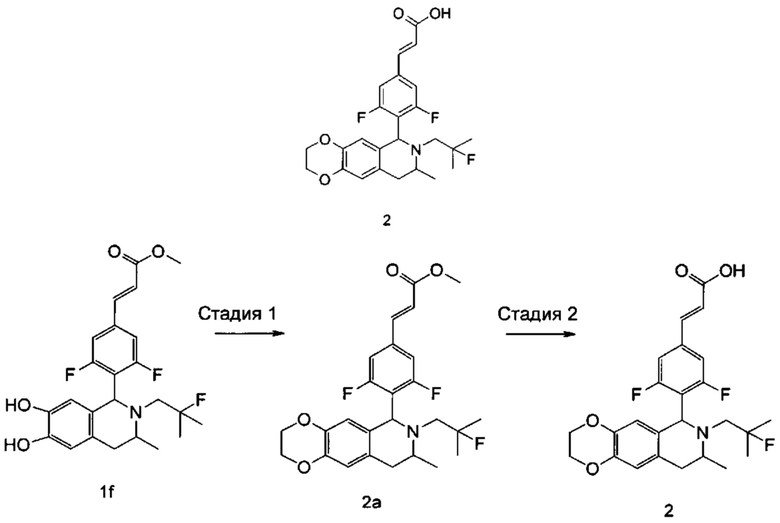
Стадия 1
(Е)-метил 3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2,3,6,7,8,9-гексагидро-[1,4]диоксино[2,3-г]изохинолин-6-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6,7-дигидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 1f (58 мг, 0,129 ммоль) растворяли в 1,5 мл Ν,Ν-диметилформамида, затем добавляли 1,2-дибромметан (36,4 мг, 0,194 ммоль) и карбонат цезия (63 мг, 0,194 ммоль). Полученную в результате смесь нагревали до 70°С и перемешивали в течение 16 часов. Реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении. Добавляли 10 мл этилацетата, и смесь промывали водой (10 мл) и насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке,
(Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2,3,6,7,8,9-гексагидро-[1,4]диоксино[2,3-г]изохинолин-6-ил)фенил)акрилата 2а (61 мг), в виде коричневого твердого вещества, которое использовали непосредственно в следующей стадии.
Стадия 2
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2,3,6,7,8,9-гексагидро-[1,4]диоксино[2,3-г]изохинолин-6-ил)фенил)акриловая кислота
Неочищенный (Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2,3,6,7,8,9-гексагидро-[1,4]диоксино[2,3-г]изохинолин-6-ил)фенил)акрилат 2а (61 мг, 0,128 ммоль) растворяли в 2 мл смеси тетрагидрофурана и метанола (об./об. составляет 3:1). Реакционную смесь охлаждали до 0°С и добавляли 0,65 мл 1 Μ водного раствора гидроксида лития. Реакционную смесь естественным путем подогревали до комнатной температуры и перемешивали в течение 0,5 часа. Реакцию останавливали, и реакционный раствор концентрировали при пониженном давлении для удаления метанола и тетрагидрофурана. Добавляли по каплям 0,5 н. разбавленную соляную кислоту до доведения рН до 5, и смесь экстрагировали этилацетатом (5 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали высокоэффективной жидкостной хроматографией с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2,3,6,7,8,9-гексагидро-[1,4]диоксино[2,3-г]изохинолин-6-ил)фенил)акриловой кислоты 2 (45 мг, выход 76%), в виде белого твердого вещества.
МС m/z (ИЭР): 462,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) 57.52 (d, 1Н), 7.03 (d, 2Н), 6.69 (s, 1Н), 6.41-6.37 (m, 2Н), 5.69 (s, 1Н), 4.20-4.25 (m, 4Н), 3.17-3.40 (m, 3Н), 2.90 (t, 1Н), 2.66 (dd, 1Н), 1.52 (d, 3Н), 1.26-1.40 (m, 6Н).
Пример 3
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1 -ил)фенил)акриловая кислота
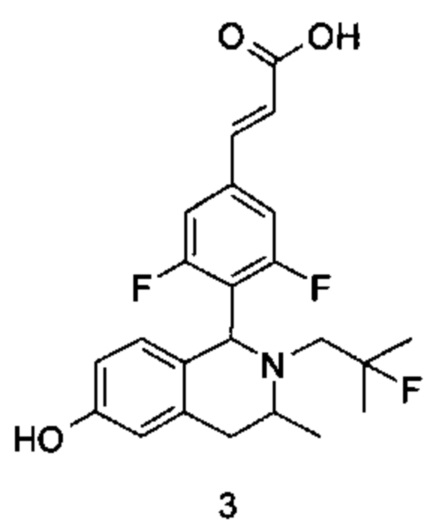
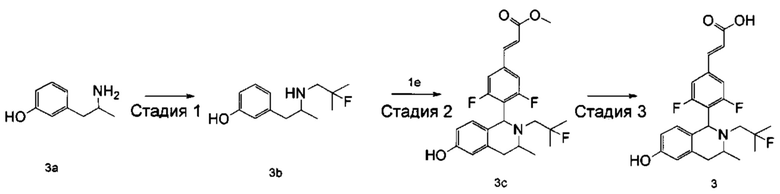
Стадия 1
3-(2-((2-фтор-2-метилпропил)амино)пропил)фенол
3-(2-Аминопропил)фенол 3а (220 мг, 2,3 ммоль, получен способом, раскрытым в заявке на патент WO 2009068177), 2-фтор-2-метилпропилтрифторметансульфонат 1b (651 мг, 2,9 ммоль) и Ν,Ν-диизопропилэтиламин (559 мг, 4,365 ммоль) растворяли в 5 мл 1,4-диоксана. Полученную в результате смесь нагревали до 90°С и перемешивали в течение 12 часов в атмосфере аргона. Реакцию останавливали, и реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, 3-(2-((2-фтор-2-метилпропил)амино)пропил)фенола 3b (113 мг, выход 35%) в виде коричневого твердого вещества.
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
3-(2-((2-фтор-2-метилпропил)амино)пропил)фенол 3b (113 мг, 0,5 ммоль) растворяли в 3 мл метанола, затем добавляли (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е (113 мг, 0,5 ммоль) и уксусную кислоту (60 мг, 1 ммоль). Полученную в результате смесь нагревали до 55°С и перемешивали в течение 12 часов. После остановки реакции реакционный раствор концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 3с (15 мг, выход 7%), в виде желтого твердого вещества.
Стадия 3
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1 -ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 3с (15 мг, 0,0346 ммоль) растворяли в 5 мл метанола, затем добавляли 2 мл водного раствора гидроксида натрия (14 мг, 0,346 ммоль). Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Реакцию останавливали. Добавляли по каплям 0,5 н. разбавленную соляную кислоту до доведения рН до 2, и смесь экстрагировали дихлорметаном (5 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 3 (5 мг, выход 34,5%), в виде желтого твердого вещества.
МС m/z (ИЭР): 420,4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.18-7.60 (m, 4Н), 6.51-6.56 (m, 3Н), 5.17 (s, 1Н), 3.69 (s, 1Н), 3.01 (s, 1Н), 2.07-2.55 (m, 3Н), 0.92-1.19 (m, 9Н).
Пример 4
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
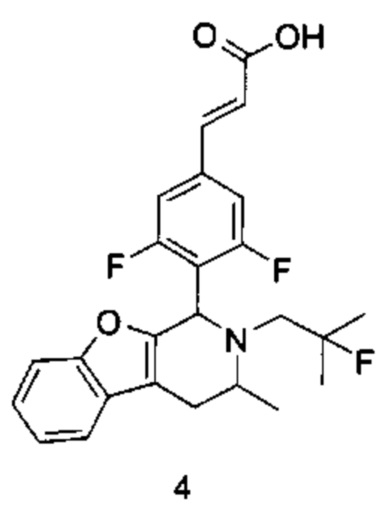
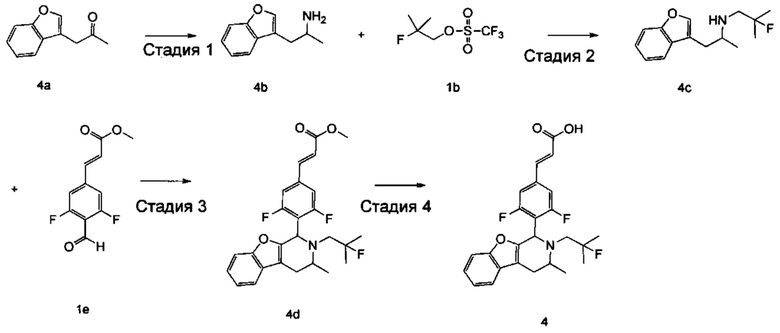
Стадия 1
1-(Бензофуран-3-ил)пропан-2-амин
1-(Бензофуран-3-ил)пропан-2-он 4а (1 г, 5,74 ммоль, получен хорошо известным способом, раскрытым в публикации Khimiya Geterotsiklicheskikh Soedinenii, 1987, (7), 889-93), ацетат аммония (4,42 г, 57,4 ммоль) и ацетат натрия (470 мг, 5,74 ммоль) растворяли в 30 мл метанола. Добавляли цианоборгидрид натрия (540 мг, 8,6 ммоль) и 10 капель уксусной кислоты по каплям. После этого полученную в результате смесь перемешивали в течение 12 часов при 25°С, реакцию останавливали. Добавляли 1 н. раствор NaOH до доведения рН до 7-8. Смесь высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, 1-(бензофуран-3-ил)пропан-2-амина 4b (600 мг, выход 60%) в виде коричневой жидкости.
Стадия 2
N-(1-(Бензофуран-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин
1-(Бензофуран-3-ил)пропан-2-амин 4b (50 мг, 0,285 ммоль), 2-фтор-2-метилпропилтрифторметансульфонат 1b (128 мг, 0,57 ммоль, получен хорошо известным способом, описанном в публикации Journal of Organic Chemistry, 2005, 70 (6), 2372-2375) и Ν,Ν-диизопропилэтиламин (109 мг, 0,855 ммоль) растворяли в 3 мл 1,4-диоксана. Полученную в результате смесь нагревали до 90°С и перемешивали в течение 12 часов в атмосфере аргона. Реакцию останавливали, и реакционный раствор концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, N-(1-(бензофуран-3-ил)-пропан-2-ил)-2-фтор-2-метилпропан-1-амина 4с (50 мг, выход 70%), в виде коричневой жидкости.
Стадия 3
(Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акрилат
N-(1-(бензофуран-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 4с (50 мг, 0.2 ммоль), (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е (90 мг, 0,4 ммоль, получен способом, раскрытым в заявке на патент WO 2014191726) и триизопропилсилилхлорид (193 мг, 1 ммоль) растворяли в 2 мл N,N-диметилформамиде и помещали в пробирку, которую герметично закрывали. Реакционную смесь нагревали до 130°С. После перемешивания в течение 3 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акрилата 4d (15 мг, выход 16%), в виде белого твердого вещества.
Стадия 4
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акрилат 4d (15 мг, 0.033 ммоль) растворяли в 5 мл метанола. Добавляли 2 мл водного раствора гидроксида натрия (13 мг, 0,33 ммоль). После перемешивания в течение 12 часов при комнатной температуре реакцию останавливали. Добавляли по каплям лимонную кислоту до доведения рН до 2-3, и смесь экстрагировали дихлорметаном (5 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-5,6,7,8-тетрагидро-[2,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловой кислоты 4 (10 мг, выход 67%), в виде желтого твердого вещества.
МС m/z (ИЭР): 444 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.60-7.64 (m, 2Н), 7.27-7.40 (m, 5Н), 6.61 (d, 1Н), 5.60 (s, 1Н), 3.86-3.88 (m, 1Н), 3.10-3.25 (m, 2Н), 2.76-2.78 (m, 2Н), 1.28-1.37 (m, 9Н).
Пример 5
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[2,3-с]пиридин-1-ил)фенил)акриловая кислота
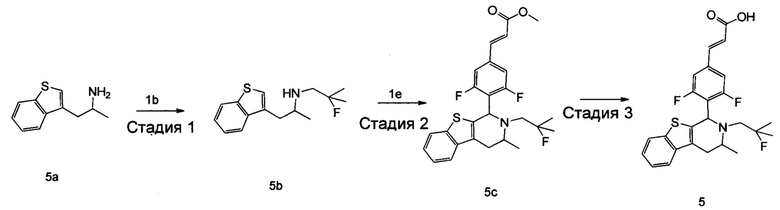
Стадия 1
N-(1-(Бензо[b]тиофен-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин-1-(Бензо[b]тиофен-3-ил)пропан-2-амин 5а (100 мг, 0,523 ммоль, получен хорошо известным способом, раскрытым в публикации Bioorganic & Medicinal Chemistry, 2005, 13(14), 4450-4457), 2-фтор-2-метилпропил трифторметансульфонат 1b (152 мг, 0,680 ммоль) и N,N-диизопропилэтиламин (135 мг, 1,046 ммоль) растворяли в 4 мл 1,4-диоксана. После перемешивания в течение 3 часов при 90°С реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, N-(1-(бензо[b]тиофен-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина 5b (100 мг, выход 72%), в виде коричневой жидкости.
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[2,3-с]пиридин-1-ил)фенил)акрилат
N-(1-(бензо[b]тиофен-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 5b (30 мг, 0,113 ммоль), (Е)-метил 3-(3,5-дифтор-4-формилфенил)акрилат 1е (31 мг, 0,136 ммоль) и триизопропилсилилхлорид (109 мг, 0,565 ммоль) растворяли в 1,5 мл N,N-диметилформамида и помещали в пробирку, которую герметично закрывали. Реакционную смесь нагревали до 140°С. После перемешивания в течение 6 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении и экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[2,3-с]пиридин-1-ил)фенил)акрилата 5с (10 мг, выход 20%), в виде желтого масла.
Стадия 3
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[2,3-с]пиридин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[2,3-с]пиридин-1-ил)фенил)акрилат 5с (10 мг, 0,021 ммоль) и гидроксид натрия (4 мг, 0,105 ммоль) растворяли в 3 мл метанола. После перемешивания в течение 3 часов при 50°С реакцию останавливали. Добавляли по каплям 1 н. соляную кислоту до доведения рН до 2-3, и смесь экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке,
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[2,3-с]пиридин-1-ил)фенил)акриловой кислоты 5 (2,5 мг, выход 26%), в виде желтого твердого вещества.
МС m/z (ИЭР): 460,2 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (d, 1Н), 7.69 (d, 1Н), 7.58 (d, 1Н), 7.37 (t, 1Н), 7.32 (t, 1Н), 7.24 (d, 1Н), 6.97 (d, 1Н), 6.57 (d, 1Н), 5.39 (s, 1Н), 3.27 (s, 1Н), 3.10-3.14 (m, 1Н), 2.99-3.04 (m, 1Н), 2.75-2.79 (m, 1Н), 2.35-2.46 (m, 1Н), 1.25 (d, 3Н), 1.19 (d, 6Н).
Пример 6
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[3,2-с]пиридин-1-ил)фенил)акриловая кислота
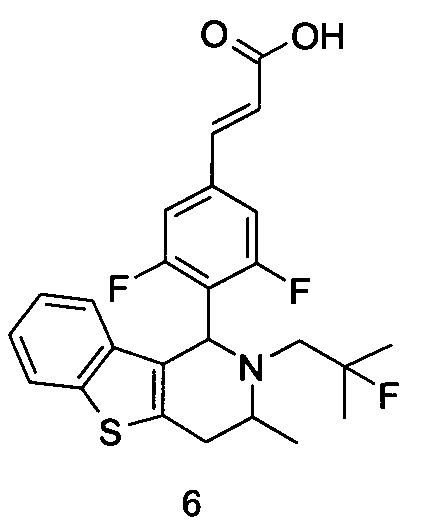
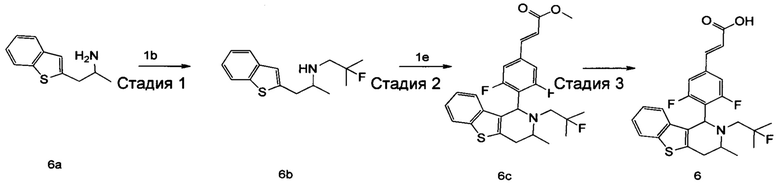
Стадия 1
N-(1-(бензо[b]тиофен-2-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин
1-(Бензо[b]тиофен-2-ил)пропан-2-амин 6а (155 мг, 0,811 ммоль, получен способом, раскрытым в заявке на патент WO 2009117097) и 2-фтор-2-метилпропилтрифторметансульфонат 1b (545 мг, 2,43 ммоль) растворяли в 3 мл 1,4-диоксана, затем добавляли N,N-диизопропилэтиламин (524 мг, 4,055 ммоль). После перемешивания в течение 5 часов при 90°С реакцию останавливали. Реакционную смесь концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, N-(1-(бензо[b]тиофен-2-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина 6b (75 мг, выход 35%), в виде желтого масла.
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[3,2-с]пиридин-1-ил)фенил)акрилат
N-(1-(бензо[b]тиофен-2-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 6b (82 мг, 0,309 ммоль) и (Е)-метил 3-(3,5-дифтор-4-формилфенил)акрилат 1е (84 мг, 0,371 ммоль) растворяли в 2 мл толуола, затем добавляли уксусную кислоту (37 мг, 0,618 ммоль). Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 24 часов, затем реакцию останавливали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[3,2-с]пиридин-1-ил)фенил)акрилата 6с (32 мг, выход 22%), в виде желтого твердого вещества.
МС m/z (ИЭР): 373,9 [М+1]
Стадия 3
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[3,2-с]пиридин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[3,2-с]пиридин-1-ил)фенил)акрилат 6с (32 мг, 0,068 ммоль) растворяли в 2 мл смеси тетрагидрофурана и метанола (об./об. составляет 3:1), затем добавляли 0,12 мл 6 н. водного раствора гидроксида натрия (27,2 мг, 0,68 ммоль). После перемешивания в течение 2 часов при комнатной температуре реакцию останавливали. Добавляли по каплям 10% лимонную кислоту до доведения рН до 3-4, и смесь экстрагировали дихлорметаном (5 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензо[4,5]тиено[3,2-с]пиридин-1-ил)фенил)акриловой кислоты 6 (8 мг, выход 26%), в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 460,3 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.66-7.80 (m, 1Н), 7.42-7.56 (m, 1Н), 6.43-7.23 (m, 5Н), 6.27-6.42 (m, 1Н), 5.32 (s, 1Н), 3.63-3.81 (m, 1Н), 3.22-3.37 (m, 1Н), 2.82-3.01 (m, 1Н), 2.61-2.80 (m, 1Н), 2.27-2.49 (m, 1Н), 1.26 (s, 3Н), 1.12-1.22 (m, 3Н), 1.07 (s, 3Н).
Пример 7
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 

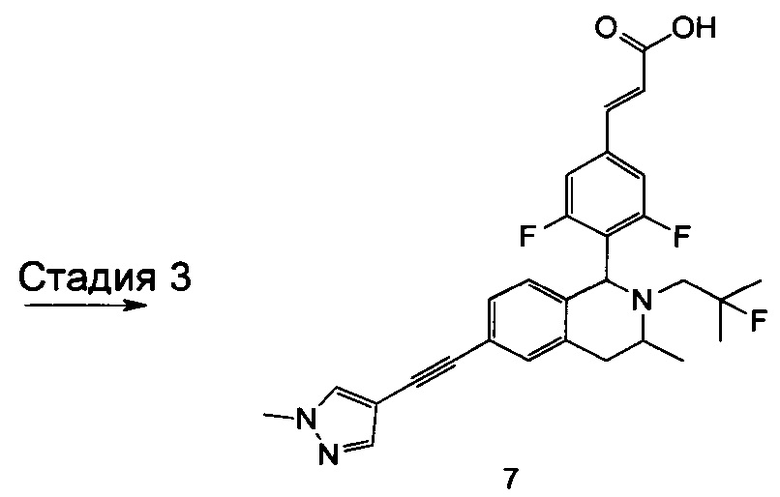
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 3с (43 мг, 0,1 ммоль) растворяли в 1 мл дихлорметана, затем добавляли 2,6-лутидин (21 мг, 0,2 ммоль). Трифторметансульфоновый ангидрид (42 мг, 0,15 ммоль) добавляли по каплям при 0°С. Реакционную смесь подогревали до комнатной температуры. После перемешивания в течение 2 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 7а (38 мг, выход 68%) в виде желтого масла.
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-(2-((1S,3R/1R,3S)-2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (38 мг, 0,067 ммоль) и 4-этинил-1-метил-1Н-пиразол 7b (17 мг, 0,16 ммоль, получен хорошо известным способом, раскрытым в публикации Journal of Medicinal Chemistry, 56(24), 10045-10065; 2013) растворяли в 1 мл N,N-диметилформамида, затем добавляли бис(трифенилфосфин)палладия (II) дихлорид (5 мг, 0,0067 ммоль), йодид меди (1,3 мг, 0,0067 ммоль) и N,N-диизопропилэтиламин (28 мг, 0,214 ммоль). Реакционную смесь нагревали до 120°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли воду, и смесь экстрагировали этилацетатом три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 7с (15 мг, выход 43%), в виде желтого твердого вещества.
Стадия 3
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7с (15 мг, 0,029 ммоль) растворяли в 1 мл смеси тетрагидрофурана и метанола (об./об. составляет 3:1), затем добавляли 0,05 мл 6 М раствор гидроксида натрия. После перемешивания в течение 2 часов реакцию останавливали. Добавляли по каплям 10% лимонную кислоту до доведения рН до 3-4, и смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 7 (5 мг, выход 33%), в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 508,1 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.82 (s, 1Н), 7.61 (s, 1Н), 7.45 (dd, 1H), 7.24 (s, 1Н), 7.12-7.17 (m, 3Н), 6.71 (dd, 1Н), 6.54 (dd, 1Н), 5.22 (s, 1Н), 3.92(s, 3Н) 3.72 (s, 1Н), 3.49 (dd, 1Н), 3.00 (t, 1Н), 2.61 (dd, 1Н), 2.35-2.24 (t, 1Н), 1.18-1.10 (t, 6Н), 1.02 (t, 3Н).
Примеры 8, 9
(Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1 -ил)фенил)акриловая кислота
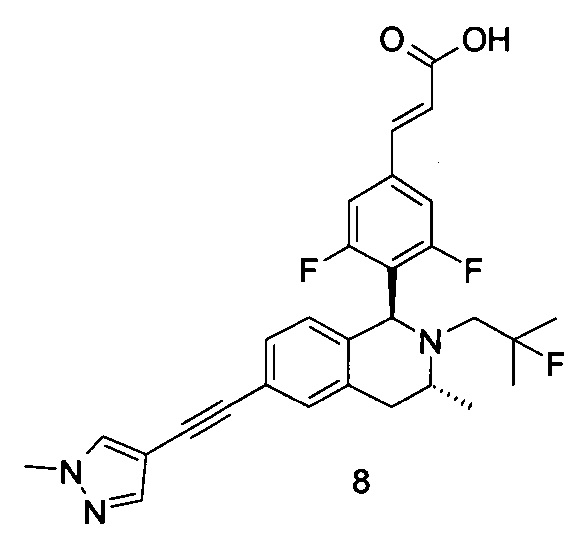
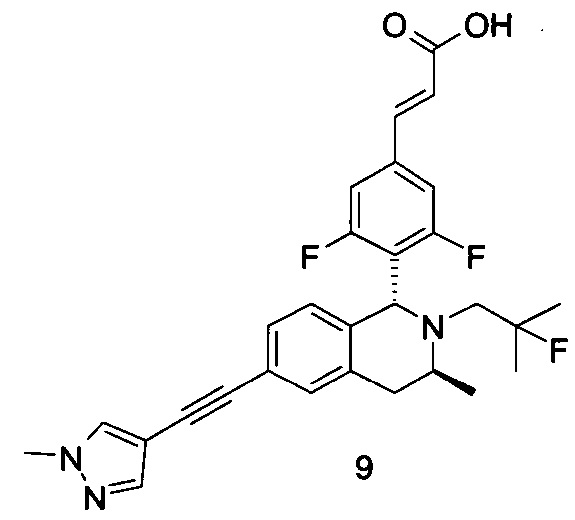
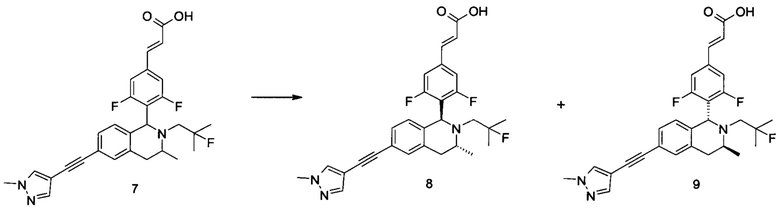
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловую кислоту 7 (242 мг, 0,48 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak AD, внутренний диаметр 2,5 см × длина 25 см; подвижная фаза: н-гексан: этанол: трифторуксусная кислота составляет 70:30:0,1, скорость потока: 30 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 8 (65 мг, желтое твердое вещество) и (Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((1-метил-1Н-пиразол-4-ил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 9 (70 мг, желтое твердое вещество).
Пример 8
МС m/z (ИЭР): 508,1 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 7,340 минут, хиральная чистота: 99,240% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 70/30/0,1(об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 7.82 (s, 1Н), 7.61 (s, 1Н), 7.45 (dd,1H), 7.24 (s, 1Н), 7.12-7.17 (m, 3Н), 6.71 (dd, 1Н), 6.54 (dd, 1Н), 5.22 (s, 1Н), 3.92(s, 3Н) 3.72 (s, 1Н), 3.49 (dd, 1Н), 3.00 (t, 1Н), 2.61 (dd, 1Н), 2.35-2.24 (t, 1Н), 1.18-1.10 (t, 6Н), 1.02 (t,3H).
Пример 9
МС m/z (ИЭР): 508,1 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 3,948 минут, хиральная чистота: 98,052% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 70/30/0,1(об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 7.82 (s, 1Н), 7.61 (s, 1Н), 7.45 (dd,1H), 7.24 (s, 1Н),7.12-7.17 (m, 3Н), 6.71 (dd, 1Н), 6.54 (dd, 1Н), 5.22 (s, 1Н), 3.92(s, 3Н) 3.72 (s, 1Н), 3.49 (dd, 1Н), 3.00 (t, 1Н), 2.61 (dd, 1Н), 2.35-2.24 (t, 1Н), 1.18-1.10 (t, 6Н), 1.02 (t, 3Н).
Пример 10
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
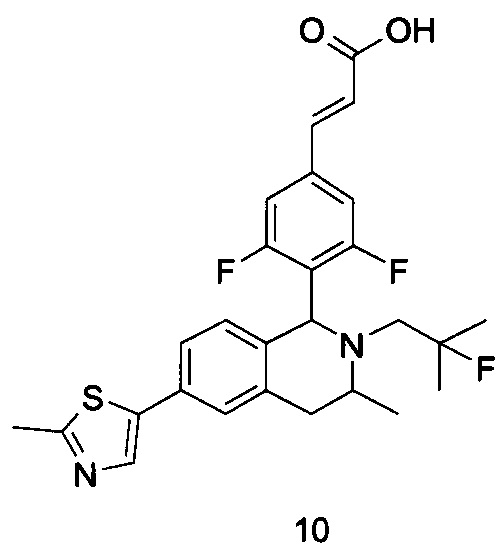
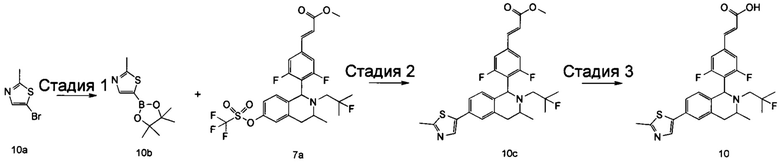
Стадия 1
2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол
5-Бром-2-метилтиазол 10а (2 г, 11,23 ммоль) растворяли в 70 мл тетрагидрофурана и добавляли 5,6 мл 2,4 М раствора н-бутиллития при -78°С. После перемешивания смеси в течение 30 минут добавляли 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (2,5 г, 13,44 ммоль). Реакционную смесь перемешивали в течение 1,5 часов, затем реакцию останавливали. Реакционный раствор подогревали до комнатной температуры, затем добавляли 10 мл смеси насыщенного раствора хлорида аммония и воды (об./об. составляет 1:1) для гашения реакции. Затем добавляли 50 мл этилацетата, и две фазы разделяли. Водную фазу экстрагировали 30 мл этилацетата. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазола 10b (1,6 г, выход 64%) в виде светло-желтого масла.
МС m/z (ИЭР): 226,1 [М+1]
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-(2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-(2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (3,3 г, 5,835 ммоль) растворяли в 36 мл смеси 1,4-диоксана и воды (об./об. составляет 35:1). Затем добавляли 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b (1,6 г, 7,107 ммоль), тетракис(трифенилфосфин)палладий (0,674 г, 0,583 ммоль) и карбонат натрия (1,86 г, 17,55 ммоль). Полученную в результате смесь подогревали до 90°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-(2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 10с (650 мг, выход 22%) в виде желтого масла.
МС m/z (ИЭР): 515,0 [М+1]
Стадия 3
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-(2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 10с (650 мг, 1,263 ммоль) растворяли в 12 мл смеси тетрагидрофурана и метанола (об./об. составляет 5:1), затем добавляли 6,5 мл 1 М раствора гидроксида лития. После перемешивания в течение 2 часов реакцию останавливали. Добавляли по каплям 10% лимонную кислоту до доведения рН до 3-4 и добавляли 50 мл этилацетата. Две фазы разделяли, и водную фазу экстрагировали этилацетатом (30 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке,
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 10 (250 мг, выход 40%) в виде желтого твердого вещества.
МС m/z (ИЭР): 501,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.95 (s, 1Н), 7.64 (d, 1Н), 7.35 (s, 1Н), 7.25 (d, 1Н), 7.07 (d, 2Н), 6.86 (d, 1Н), 6.45 (d, 1Н), 5.43 (s, 1Н), 3.88 (s, 1Н), 3.54-3.47 (m, 1Н), 3.08 (t, 1Н), 2.88 (s, 3Н), 2.73 (dd, 1Н), 2.52-2.41 (m, 1Н), 1.35-1.14 (m, 9Н).
Примеры 11, 12
(Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
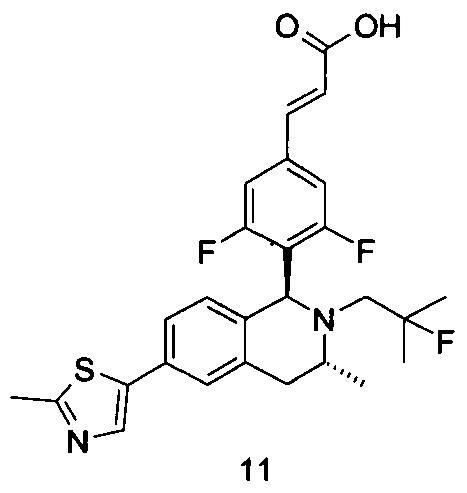
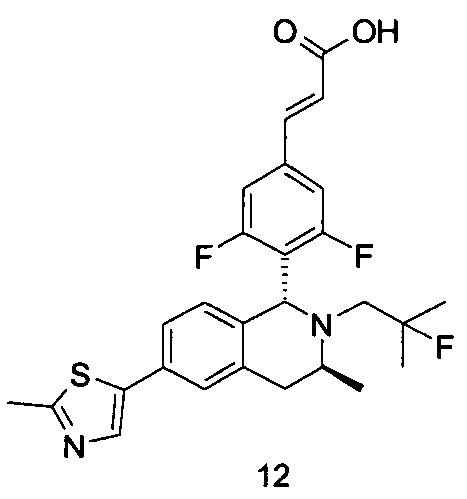
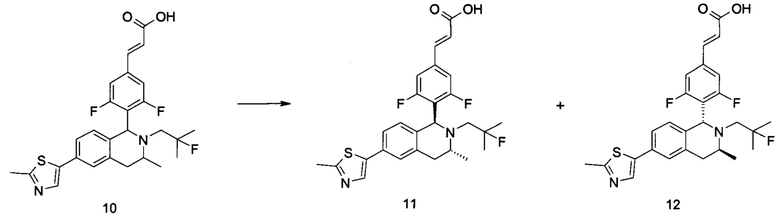
(Е)-3-(3,5)-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловую кислоту 10 (250 мг, 0,5 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak AD, внутренний диаметр 5,0 см × длина 25 см; подвижная фаза: н-гексан: этанол: трифторуксусная кислота составляет 60: 40: 0,1, скорость потока: 60 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 11 (117,9 мг, желтое твердое вещество) и (Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 12 (116,7 мг, желтое твердое вещество).
Пример 11
МС m/z (ИЭР): 501,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 8,585 минут, хиральная чистота: 99,989% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 60/40/0,1(об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 7.85 (s, 1Н), 7.60-7.56 (m, 1Н), 7.38 (s, 1Н), 7.30-7.21 (m, 3Н), 6.80-6.78 (d, 1Н), 6.57-6.53 (d, 1Н), 5.25 (s, 1Н), 3.74 (s, 1Н), 3.42-3.39 (m, 1Н), 3.05-2.98 (m, 1Н), 2.72 (s, 3Н), 2.36-2.19 (m, 2Н), 1.32-0.91 (m, 9Н).
Пример 12
МС m/z (ИЭР): 501,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 5,254 минут, хиральная чистота: 99,804% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 25 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 60/40/0,1 (об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 7.85 (s, 1Н), 7.60-7.56 (m, 1Н), 7.38 (s, 1Н), 7.30-7.21 (m, 3Н), 6.80-6.78 (d, 1Н), 6.57-6.53 (d, 1Н), 5.25 (s, 1Н), 3.74 (s, 1Н), 3.42-3.39 (m, 1Н), 3.05-2.98 (m, 1Н), 2.72 (s, 3Н), 2.36-2.19 (m, 2Н), 1.32-0.91 (m, 9Н).
Пример 13
(Е)-3-(4-((1S,3R/1R,3S)-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
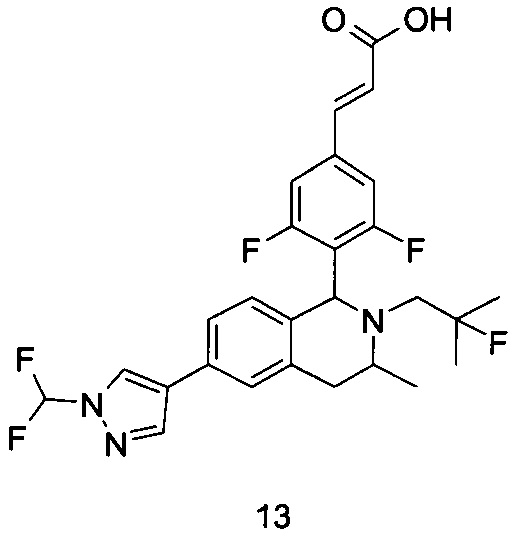
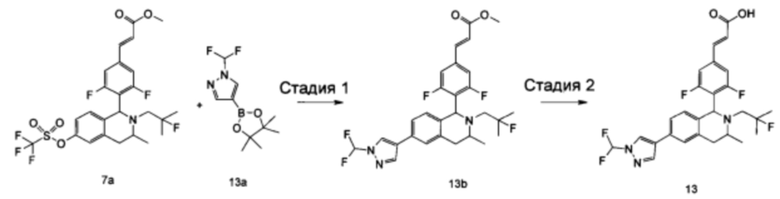
Стадия 1
(Е)-метил-3-(4-((1S,3R/1R,3S)-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (200 мг, 0,353 ммоль), 1-(дифторметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 13а (129 мг, 0,529 ммоль, получен способом, раскрытым в заявке на патент WO 2014159224) и карбонат калия (146 мг, 1,06 ммоль) растворяли в 5,5 мл смеси 1,4-диоксана и воды (об./об. составляет 10:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (12,9 мг, 0,0176 ммоль). Реакционную смесь нагревали до 80°С. После перемешивания в течение 18 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры. Добавляли этилацетат, и смесь промывали водой. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 13b (52 мг, выход 27,6%) в виде желтого твердого вещества.
Стадия 2
(Е)-3-(4-((1S,3R/1R,3S)-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-метил-3-(4-((1S,3R/1R,3S)-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил) акрилат 13b (50 мг, 0,093 ммоль) растворяли в 3,5 мл смеси тетрагидрофурана и метанола (об./об. составляет 6:1), затем добавляли 0,5 мл 1 М раствора гидроксида лития. После перемешивания в течение 3 часов реакцию останавливали. Добавляли по каплям 10% лимонную кислоту до доведения рН до 3-4, и смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 13 (18 мг, выход 37%) в виде желтого твердого вещества.
МС m/z (ИЭР): 520,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.66 (s, 1Н), 8.23 (s, 1Н), 7.97-7.68 (m, 1Н), 7.55-7.45 (m, 4Н), 7.36 (d, 1Н), 6.73 (d, 1Н), 6.65 (d, 1Н), 5.15 (s, 1Н), 3.63-3.54 (m, 1Н), 3.29-3.21 (m, 1Н), 3.00-2.89 (m, 1Н), 2.70-2.62 (s, 1Н), 2.29-2.23 (m, 1Н), 1.25-0.94 (m, 9Н).
Примеры 14, 15
(Е)-3-(4-((1R,3S)-6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота (E)-3-(4-((1R,3S)-6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
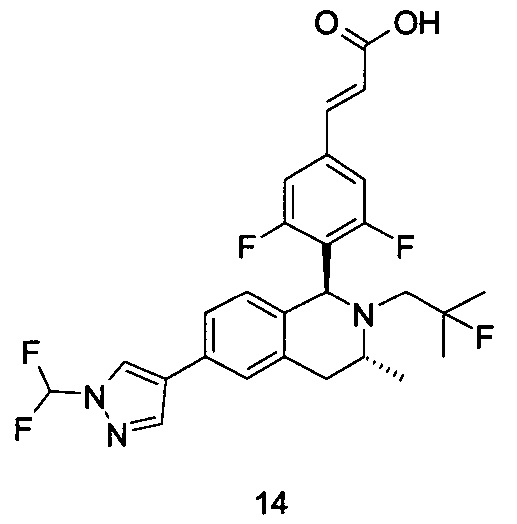
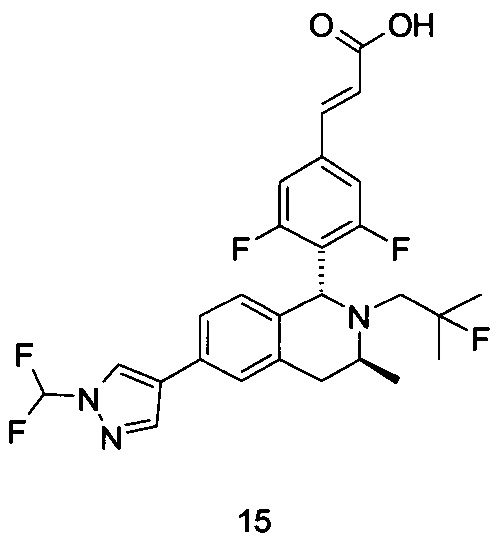
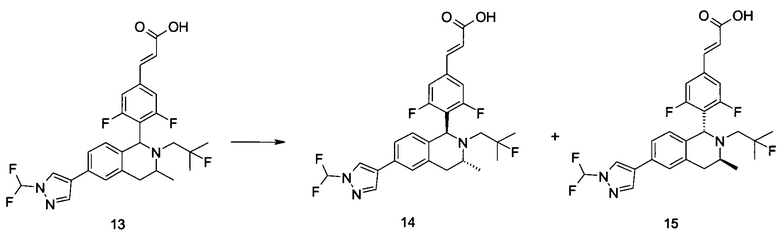
(Е)-3-(4-((1S,3R/1R,3S)-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловую кислоту 13 (1,4 г, 2,7 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak AD, внутренний диаметр 5,0 см × длина 25 см; подвижная фаза: н-гексан: этанол: трифторуксусная кислота составляет 85: 15: 0,1, скорость потока: 60 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(4-((1S,3R)-6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 14 (210 мг, желтое твердое вещество) и (Е)-3-(4-((1R,3S)-6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 15 (200 мг, желтое твердое вещество).
Пример 14:
МС m/z (ИЭР): 520,2 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 15,403 минут, хиральная чистота: 99,90% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 85/15/0,1 (об./об./об.));
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.66 (s, 1Н), 8.23 (s, 1Н), 7.97-7.68 (m, 1Н), 7.55-7.45 (m, 4Н), 7.36 (d, 1Н), 6.73 (d, 1Н), 6.65 (d, 1Н), 5.15 (s, 1Н), 3.63-3.54 (m, 1Н), 3.29-3.21 (m, 1Н), 3.00-2.89 (m, 1Н), 2.70-2.62 (s, 1Н), 2.29-2.23 (m, 1Н), 1.25-0.94 (m, 9Н).
Пример 15:
МС m/z (ИЭР): 520,2 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 10,902 мин, хиральная чистота: 99,90% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 85/15/0,1(об./об./об.));
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.66 (s, 1Н), 8.23 (s, 1Н), 7.97-7.68 (m, 1Н), 7.55-7.45 (m, 4Н), 7.36 (d, 1Н), 6.73 (d, 1Н), 6.65 (d, 1Н), 5.15 (s, 1Н), 3.63-3.54 (m, 1Н), 3.29-3.21 (m, 1Н), 3.00-2.89 (m, 1Н), 2.70-2.62 (s, 1Н), 2.29-2.23 (m, 1Н), 1.25-0.94 (m, 9Н).
Пример 16
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
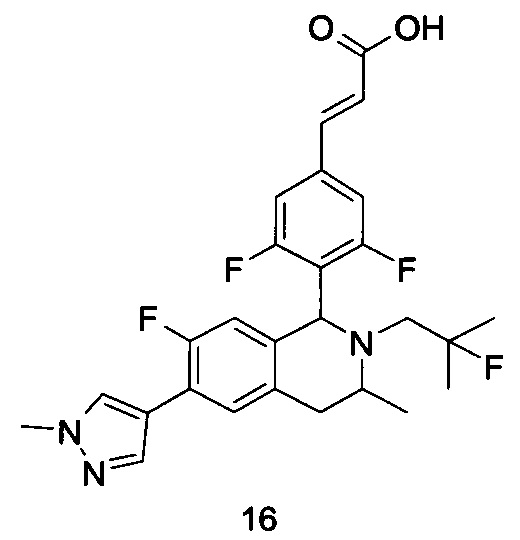
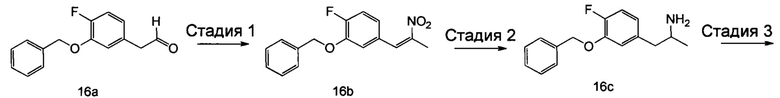
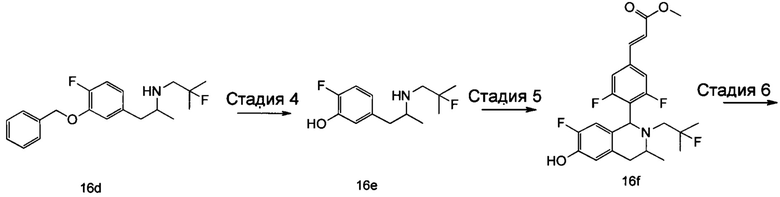
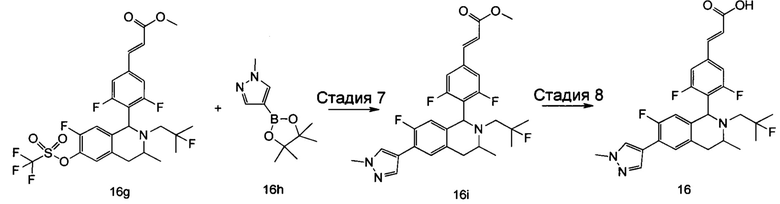
Стадия 1
2-(Бензилокси)-1-фтор-4-(2-нитропроп-1-ен-1-ил)бензол
2-(3-(Бензилокси)-4-фторфенил)ацетальдегид 16а (4 г, 0,017 ммоль, получен хорошо известным способом, раскрытым в публикации Bioorganic & Medicinal Chemistry, 9(3), 2001, 677-694) и ацетат аммония (1,6 г, 0,021 моль) смешивали с 30 мл нитроэтана. Полученную в результате смесь подогревали до 90°С и перемешивали в течение 12 часов. Реакцию останавливали, и смесь охлаждали до комнатной температуры. Смесь концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, 2-(бензилокси)-1-фтор-4-(2-нитропроп-1-ен-1-ил)бензола 16b (3,5 г, выход 70%) в виде желтого масла.
МС m/z(ИЭР): 288 [М+1]
Стадия 2
1-(3-(Бензилокси)-4-фторфенил)пропан-2-амин
2-(Бензилокси)-1-фтор-4-(2-нитропроп-1-ен-1-ил)бензол 16b (3,5 г, 0,012 ммоль) растворяли в 50 мл тетрагидрофурана, затем добавляли алюмогидрид лития (926 мг, 0,024 моль) при 0°С. Реакционный раствор подогревали до комнатной температуры.
После перемешивания в течение 12 часов реакцию останавливали. Добавляли 3 мл 15% раствора гидроксида натрия для гашения реакции, и реакционный раствор фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 1-(3-(бензилокси)-4-фторфенил)пропан-2-амина 16с (3,1 г), в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
N-(1-(3-(бензилокси)-4-фторфенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин
Неочищенный 1-(3-(бензилокси)-4-фторфенил)пропан-2-амин 16с (3,1 г, 0,012 ммоль), 2-фтор-2-метилпропил трифторметансульфонат 1b (4,02 г, 0,018 ммоль) и N,N-диизопропилэтиламин (3,1 г, 0,024 ммоль) растворяли в 30 мл 1,4-диоксана. Полученную в результате смесь подогревали до 90°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, N-(1-(3-(бензилокси)-4-фторфенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина 16d (1,3 г, выход 30%), в виде желтой жидкости.
МС m/z (ИЭР): 334,0 [М+1]
Стадия 4
2-Фтор-5-(2-((2-фтор-2-метилпропил)амино)пропил)фенол
N-(1-(3-(бензилокси)-4-фторфенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 16d (1 г, 3 ммоль) растворяли в 8 мл трифторуксусной кислоты. Полученную в результате смесь подогревали до 50°С. После перемешивания в течение 48 часов реакцию останавливали. Раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. К остатку добавляли насыщенный раствор бикарбоната натрия до доведения рН до 8-9. Смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, 2-фтор-5-(2-((2-фтор-2-метилпропил)амино)пропил)фенола 16е (300 мг, выход 41%) в виде желтого масла.
МС m/z (ИЭР): 244,0 [М+1]
Стадия 5
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
2-Фтор-5-(2-((2-фтор-2-метилпропил)амино)пропил)фенол 16е (300 мг, 1,233 ммоль) растворяли в 5 мл метанола, затем добавляли (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е (418 мг, 1,896 ммоль) и уксусную кислоту (740 мг, 12,33 ммоль). Полученную в результате смесь нагревали до 80°С и перемешивали в течение 48 часов. После остановки реакции реакционный раствор подогревали до комнатной температуры. К реакционному раствору добавляли 1 М раствор бикарбоната натрия до доведения рН до 7-8. Смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 16f (300 мг, выход 54%), в виде желтого масла.
МС m/z (ИЭР): 451,9 [М+1]
Стадия 6
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил) фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 16f (300 мг, 0,664 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 2,6-лутидин (142 мг, 1,329 ммоль) и трифторметансульфоновый ангидрид (281 мг, 0,996 ммоль) при 0°С. Реакционную смесь перемешивали в течение 2 часов при 0°С. После остановки реакции реакционный раствор подогревали до комнатной температуры. Добавляли воду, и смесь экстрагировали дихлорметаном. Органическую фазу концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 16g (300 мг, выход 77%), в виде желтого масла.
МС m/z (ИЭР): 583,8 [М+1]
Стадия 7
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил) -3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акрилат 16g (350 мг, 0,6 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 16h (250 мг, 1.2 ммоль, получен хорошо известным способом, раскрытым в публикации Tetrahedron Letters, 50 (49), 2009, 6783-6786) и карбонат калия (248 мг, 1,8 ммоль) растворяли в 10 мл смеси 1,4-диоксана и воды (об./об. составляет 8:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (44 мг, 0,06 ммоль). Реакционную смесь нагревали до 90°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 16i (300 мг) в виде коричневого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 515,9 [М+1]
Стадия 8
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
Неочищенный
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 16i (300 мг, 0,582 ммоль) растворяли в 10 мл метанола, затем добавляли 2 мл 3 М раствор гидроксида натрия. После перемешивания в течение 12 часов реакцию останавливали. Добавляли по каплям 1 М раствор соляной кислоты до доведения рН до 5-6. Смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали высокоэффективной жидкостной хроматографией с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 16 (120 мг, выход 41%), в виде желтого твердого вещества.
МС m/z (ИЭР): 502,5 [М+1]
1Н ЯМР (400 МГц, CD3OD): δ 8.01 (s, 1Н), 7.89 (s, 1Н), 7.66 (d, 1Н), 7.50 (d, 1Н), 7.31 (d, 2Н), 6.59 (d, 2Н), 5.52 (s, 1Н), 3.95 (s, 3Н), 3.83-3.78 (m, 1Н), 3.37-3.34 (m, 1Н), 3.32-3.30 (m, 1Н), 2.82 (s, 2Н), 1.38-1.11 (m, 9Н).
Пример 17
(Е)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
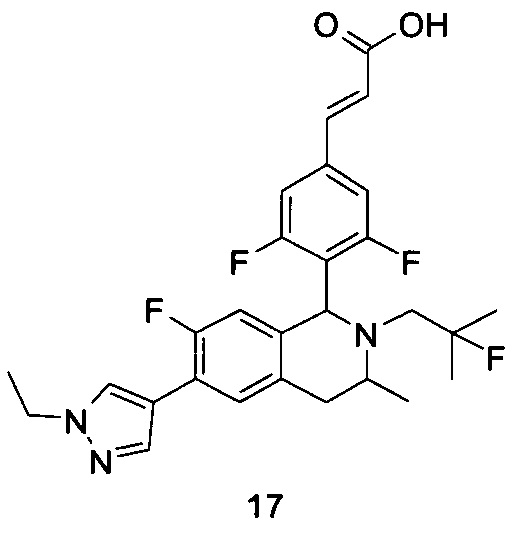
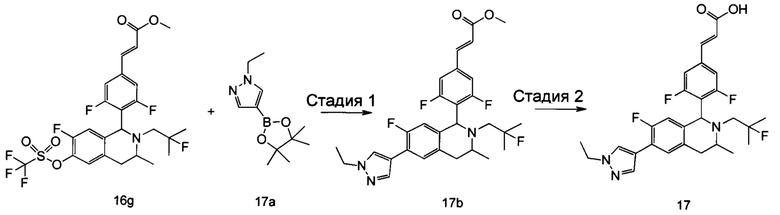
Стадия 1
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 16g (300 мг, 0,514 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 17а (228 мг, 1,028 ммоль, получен хорошо известным способом, раскрытым в публикации Bioorganic & Medicinal Chemistry Letters, 2008, 18 (19), 5299-5302) и карбонат калия (213 мг, 1,542 ммоль) растворяли в 12 мл смеси 1,4-диоксана и воды (об./об. составляет 5:1), добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (37 мг, 0,051 ммоль). Реакционную смесь нагревали до 85°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 17b (250 мг) в виде коричневого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 530,0 [М+1]
Стадия 2
(Е)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
Неочищенный (Е)-метил-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил) акрилат 17b (250 мг, 0,472 ммоль) растворяли в 10 мл метанола, затем добавляли 2 мл 4 М раствор гидроксида натрия. Реакционную смесь охлаждали до комнатной температуры и перемешивали 1 час, затем реакцию останавливали. После этого реакционную смесь охлаждали до комнатной температуры, добавляли по каплям 1 М раствор лимонной кислоты до доведения рН до 5-6. Смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали высокоэффективной жидкостной хроматографией с получением соединения, указанного в заголовке,
(E)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 17 (30 мг, выход 13%), в виде желтого твердого вещества.
МС m/z (ИЭР): 515,9 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.06 (s, 1Н), 7.91 (s, 1Н), 7.61 (d, 1Н), 7.53 (d, 1Н), 7.32 (d, 2Н), 6.68-6.58 (m, 2Н), 5.58 (s, 1Н), 4.22-4.24 (m, 2Н), 3.86 (s, 1Н), 3.37-3.34 (m, 1Н), 3.32-3.30 (m, 1Н), 2.86 (s, 2Н), 1.51 (t, 3Н), 1.34-1.22 (m, 9Н).
Пример 18
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-пропил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
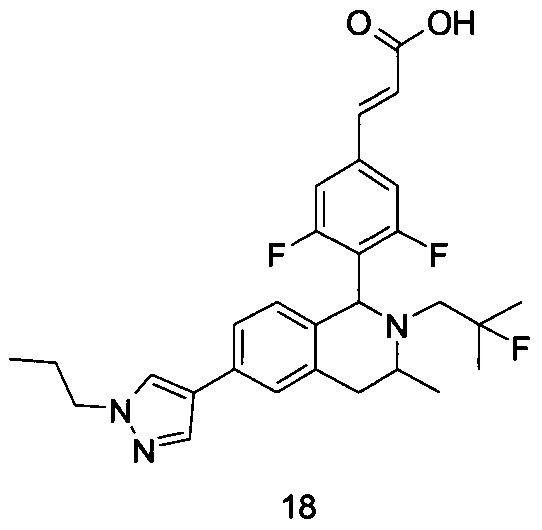
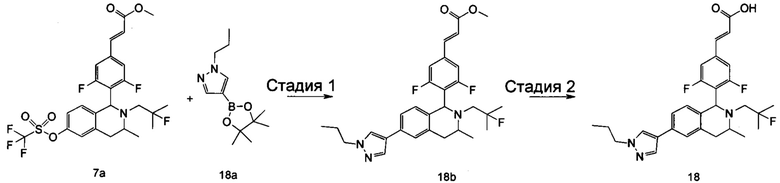
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-пропил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (200 мг, 0,35 ммоль), 1-пропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 18а (167 мг, 0,70 ммоль, получен хорошо известным способом, раскрытым в публикации Journal of Heterocyclic Chemistry, 41 (6), 2004, 931-939") и карбонат калия (145 мг, 1,05 ммоль) растворяли в 5 мл смеси 1,4-диоксана и воды (об./об. составляет 4:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (12,8 мг, 0,0175 ммоль). Реакционную смесь нагревали до 90°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-пропил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 18b (128 мг, выход 69,9%) в виде маслянистой жидкости.
Стадия 2
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-пропил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-6-(1-пропил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 18b (58 мг, 0,11 ммоль) растворяли в 10 мл метанола, затем добавляли 1 мл 1 М раствора гидроксида натрия. После перемешивания в течение 4 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-пропил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 18 (29 мг, выход 51,8%), в виде желтого твердого вещества.
МС m/z (ИЭР): 510,5 [М-1]
1Н ЯМР (400 МГц, CD3OD) δ 7.94 (s, 1Н), 7.79 (s, 1Н), 7.56 (d, 1Н) 7.33 (s, 1Н), 7.21 (t, 3Н), 6.71 (d, 1Н), 6.53 (d, 1Н), 5.23 (s, 1Н), 4.13 (t, 2Н), 3.70-3.74 (m, 1Н), 3.38-3.42 (dd, 1Н), 3.01 (t, 1Н), 2.69-2.64 (dd, 1Н), 2.36-2.25 (m, 1Н), 1.93-1.88 (m, 2Н), 1.19-1.10 (dd, 6Н), 1.03 (d, 3Н), 0.94 (t, 3Н).
Пример 19
(Е)-3-(4-((1S,3R/1R,3S)-6-(3-этил-1,2,4-оксадиазол-5-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
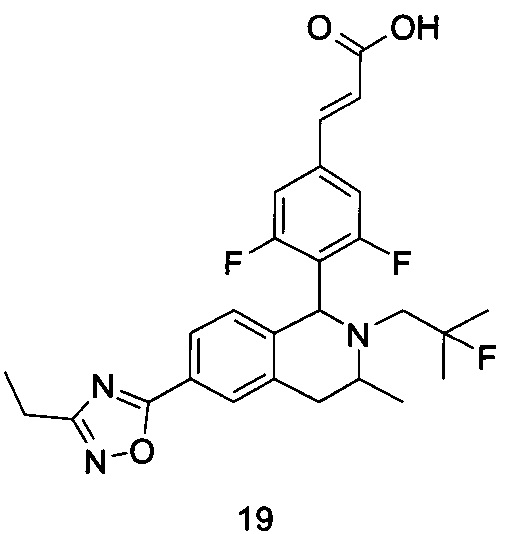
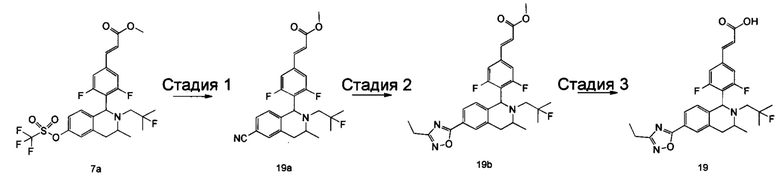
Стадия 1
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-циано-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (2 г, 3,54 ммоль), цианид цинка (0,41 г, 3,54 ммоль), порошок цинка (368 мг, 5,66 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (155 мг, 0,21 ммоль) растворяли в 65 мл смеси N,N-диэтилацетамида и воды (об./об. составляет 12:1). Реакционную смесь нагревали до 120°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Добавляли воду, и смесь экстрагировали этилацетатом (50 мл). Органические фазы объединяли, промывали водой, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-циано-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 19а (1,17 г, выход 75%) в виде белого твердого вещества.
Стадия 2
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(3-этил-1,2,4-оксадиазол-5-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-циано-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 19а (200 мг, 0,45 ммоль), N-гидроксипропионамидин (40 мг, 0,45 ммоль), 0,15 мл 1 М раствора хлорида цинка в диэтиловом эфире и гидрат пара-толуолсульфоновой кислоты (25,7 мг, 0,135 ммоль) последовательно добавляли к 5 мл N,N-диметилформамида. Реакционную смесь нагревали до 80°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-(3-этил-1,2,4-оксадиазол-5-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил) акрилата 19b (210 мг, выход 90,5%) в виде бесцветного масла.
МС m/z (ИЭР): 514,0 [М+1]
Стадия 3
(Е)-3-(4-((1S,3R/1R,3S)-6-(3-этил-1,2,4-оксадиазол-5-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(3-этил-1,2,4-оксадиазол-5-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 19b (210 мг, 0,41 ммоль) растворяли в 5 мл метанола, затем добавляли 1 мл 2 М раствор гидроксида натрия. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении и добавляли по каплям соляную кислоту до доведения рН до 3. Смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали высокоэффективной жидкостной хроматографией с получением соединения, указанного в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(3-этил-1,2,4-оксадиазол-5-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 19 (20 мг, выход 10%), в виде желтого твердого вещества.
МС m/z (ИЭР): 500,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): δ 12.04 (s, 1Н), 7.91 (s, 1Н), 7.78 (d, 1Н), 7.53-7.46 (m, 3Н), 6.99 (d, 1Н), 6.68 (d, 1Н), 5.23 (s, 1Н), 3.61-3.56 (m, 1Н), 2.93 (t, 1Н), 2.76 (d, 1Н), 2.55 (q, 2Н), 2.27-2.17 (m, 2Н), 1.11 (dd, 6Н), 1.02 (t, 3Н), 0.97 (d, 3Н).
Пример 20
(Е)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
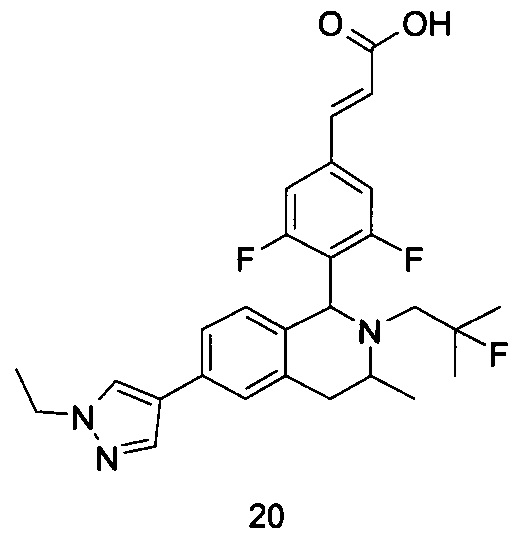
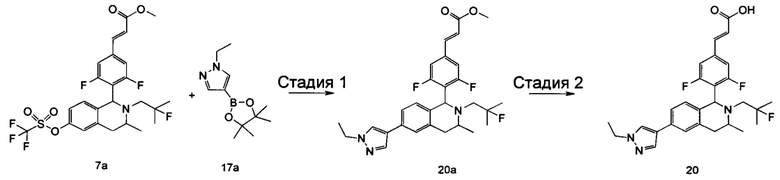
Стадия 1
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акрилат 7а (3,9 г, 6,896 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 17а (3 г, 13,792 ммоль) и карбонат калия (2,86 г, 20,689 ммоль) растворяли в 60 мл смеси 1,4-диоксана и воды (об./об. составляет 5:1), затем добавляли [1,1'-бис (дифенилфосфино)ферроцен]дихлорпалладий (II) (503 мг, 0,689 ммоль). Реакционную смесь нагревали до 80°С. После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат экстрагировали этилацетатом, и органическую фазу концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 20а (3,2 г, выход 91%), в виде желтого твердого вещества.
МС m/z (ИЭР): 512,0 [М+1]
Стадия 2
(Е)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 20а (3,2 г, 6,255 ммоль) растворяли в 30 мл метанола, затем добавляли 3 мл 6 М раствора гидроксида натрия. После перемешивания в течение 12 часов реакцию останавливали. Добавляли по каплям 1 М раствор соляной кислоты до достижения кислого рН реакционного раствора. Смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором бикарбоната натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 20 (1,5 г, выход 48%), в виде желтого твердого вещества.
МС m/z (ИЭР): 498,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.96 (s, 1Н), 7.80 (s, 1Н), 7.56 (d, 1Н), 7.33 (s, 1Н), 7.25-7.17 (m, 3Н), 6.71 (d, 1Н), 6.58-6.50 (m, 1Н), 5.23 (s, 1Н), 4.25-4.18 (m, 2Н), 3.76-3.69 (m, 1Н), 3.44-3.37 (m, 1Н), 3.06-2.96 (m, 1Н), 2.70-2.63 (m, 1Н), 2.37-2.23 (m, 1Н), 1.51-1.48 (m, 3Н), 1.22-1.03 (m, 9Н).
Примеры 21, 22
(E)-3-(4-((1S,3R)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-3-(4-((1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
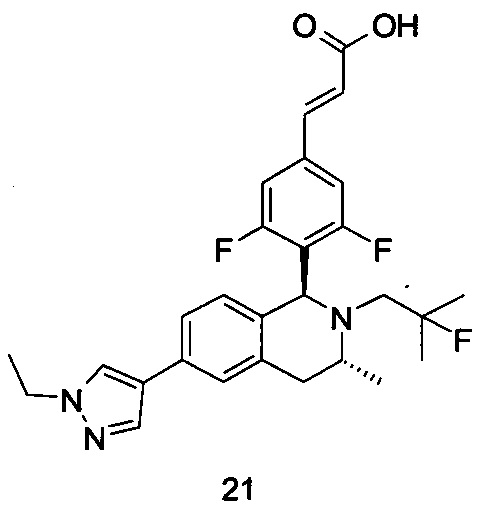
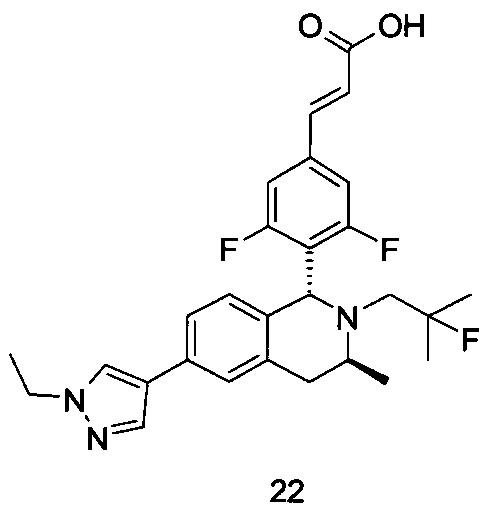
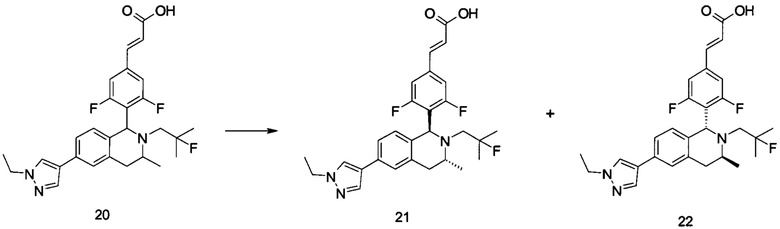
(E)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловую кислоту 20 (1,5 г, 3,01 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Superchiral S-AD (Chiralway), внутренний диаметр 2 см * длина 25 см, 5 мкм; подвижная фаза: диоксид углерода:этанол:диэтиламин составляет 65:35:0,05, скорость потока: 50 г/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке (Е)-3-(4-((1S,3R)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил) акриловой кислоты 21 (650 мг, желтое твердое вещество) и (E)-3-(4-((1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 22 (600 мг, желтое твердое вещество).
Пример 21
МС m/z(ИЭР): 498,0 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 5,292 минут, хиральная чистота: 99,512% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см × длина 25 см; подвижная фаза: диоксид углерода/этанол/диэтиламин составляет 65/35/0,05 (об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 8.05 (s, 1Н), 7.86 (s, 1Н), 7.62 (d, 1Н), 7.50 (s, 1Н), 7.42-7.37 (m, 3Н), 7.00 (s, 1Н), 6.63 (d, 1Н), 5.96 (s, 1Н), 4.27-4.20 (m, 2Н), 4.02 (s, 1Н), 3.57 (s, 1Н), 3.44-3.38 (dd, 1Н), 3.06 (s, 2Н), 1.52-1.32 (m, 12Н).
Пример 22
МС m/z (ИЭР): 498,6 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 4,568 минут, хиральная чистота: 100% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см × длина 25 см; подвижная фаза: диоксид углерода/этанол/диэтиламин составляет 65/35/0,05 (об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 7.95 (s, 1Н), 7.79 (s, 1Н), 7.58 (d, 1Н), 7.33 (s, 1Н), 7.24-7.19 (m, 3Н), 6.70 (d, 1Н), 6.53 (d, 1Н), 5.23 (s, 1Н), 4.21 (q, 2Н), 3.71 (s, 1Н), 3.40 (dd, 1Н), 3.01 (t, 1Н), 2.65 (d, 1Н), 2.30 (q, 1Н), 1.50 (t, 3Н), 1.02-1.19 (m, 9Н).
Пример 23
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-(2-гидроксиэтил)-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
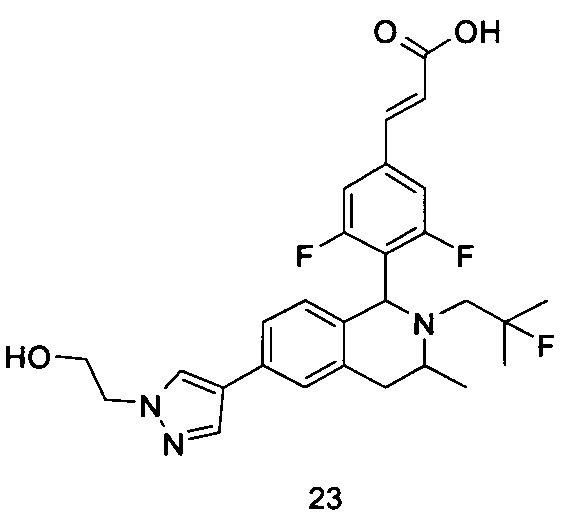
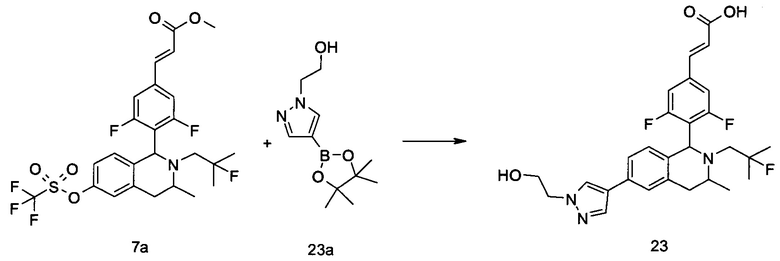
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акрилат 7а (60 мг, 0,106 ммоль), 2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)этанол 23а (38 мг, 0,16 ммоль, получен хорошо известным способом, раскрытым в публикации Bioorganic & Medicinal Chemistry, 2013, 21(21), 6804-6820) и карбонат калия (36,6 мг, 0,265 ммоль) растворяли в 3,5 мл смеси 1,4-диоксана и воды (об./об. составляет 6:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (7,8 мг, 0,0106 ммоль). Реакционную смесь нагревали до 80°С. После перемешивания в течение 18 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры. Добавляли этилацетат, и смесь промывали водой. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-(2-гидроксиэтил)-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 23 (9 мг, выход 20%), в виде желтого твердого вещества.
МС m/z (ИЭР): 514,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.96 (s, 1Н), 7.80 (s, 1Н), 7.56 (d, 1Н), 7.33 (s, 1Н), 7.25-7.17 (m, 3Н), 6.71 (d, 1Н), 6.50-6.58 (m, 1Н), 5.23 (s, 1H), 4.02 (t, 2H), 3.72-3.62 (m, 2H), 3.76-3.69 (m, 1H), 3.44-3.37 (m, 1H), 3.06-2.96 (m, 1H), 2.70-2.63 (m, 1H), 2.37-2.23 (m, 1H), 1.22-1.03 (m, 9H).
Пример 24
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
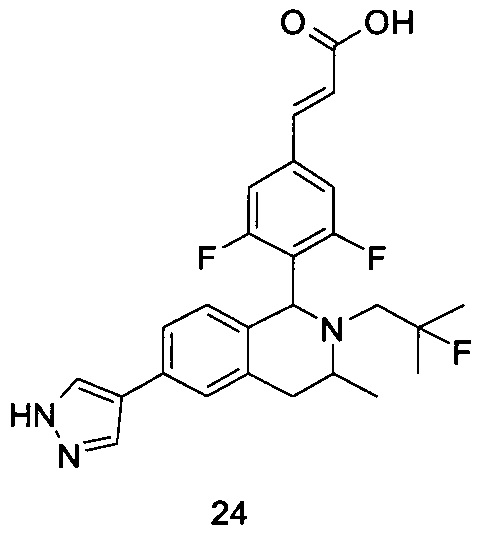
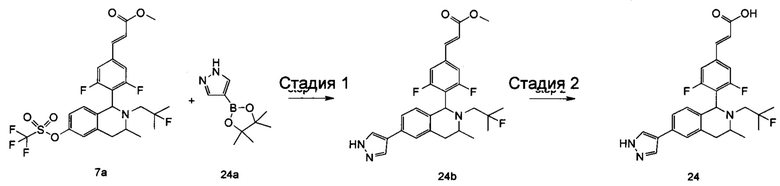
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (100 мг, 0,177 ммоль), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 24а (75 мг, 0,265 ммоль, получен хорошо известным способом, раскрытым в публикации Journal of the American Chemical Society, 2014, 136 (11), 4287-4299) и карбонат калия (73 мг, 0,531 ммоль) растворяли в 20 мл смеси 1,4-диоксана и воды (об./об. составляет 9:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (12,9 мг, 0,0177 ммоль). Реакционную смесь нагревали до 95°С. После перемешивания в течение 22 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры. Добавляли воду, и смесь экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 24b (31 мг, выход 36%), в виде желтого твердого вещества.
Стадия 2
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 24b (30 мг, 0,062 ммоль) растворяли в 3 мл этанола, затем добавляли 2 мл 1 М раствора гидроксида натрия. После перемешивания в течение 2 часов реакцию останавливали. К остатку добавляли 10% раствор лимонной кислоты до доведения рН до 6-7. Смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором бикарбоната натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 24 (5 мг, выход 16,6%), в виде желтого твердого вещества.
МС m/z (ИЭР): 470,4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.04 (s, 2Н), 7.63 (d, 1Н), 7.58 (br, 1Н), 7.51-7.41 (m, 3Н), 7.08 (d, 1Н), 6.65 (d, 1Н), 6.14-6.09 (m, 1Н), 4.12-4.06 (m, 1Н), 3.77-3.68 (m, 1Н), 3.46-3.40 (m, 1Н), 3.20-3.13 (m, 2Н), 1.61-1.49 (m, 9Н).
Примеры 25, 26
(Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
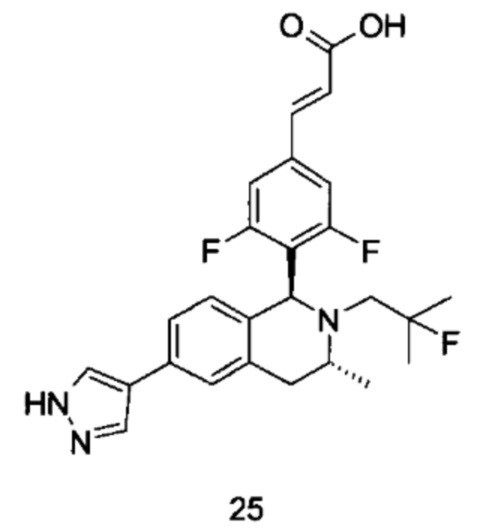
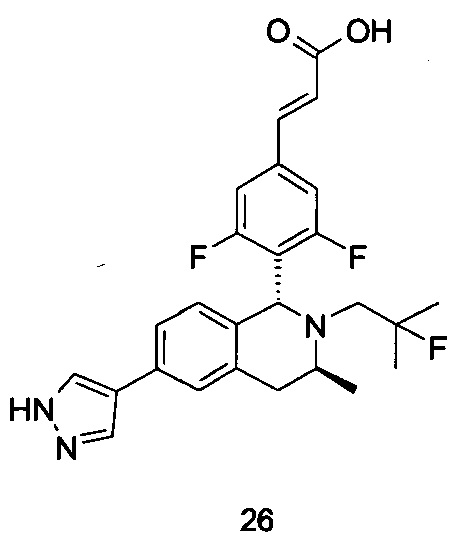
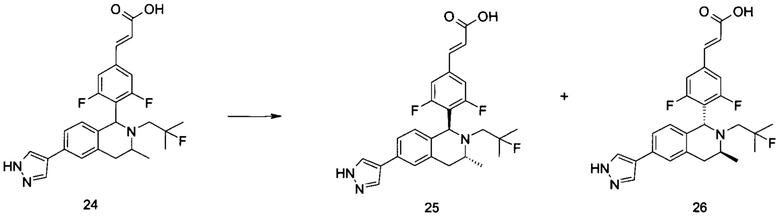
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловую кислоту 24 (860 мг, 1,83 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak IE, внутренний диаметр 5,0 см × длина 25 см; подвижная фаза: н-гексан: этанол: трифторуксусная кислота составляет 70: 30: 0,1, скорость потока: 60 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 25 (250 мг, желтое твердое вещество) и (Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 26 (240 мг, желтое твердое вещество).
Пример 25
МС m/z (ИЭР): 470,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 4,266 минут, хиральная чистота: 99,767% (хроматографическая колонка: Chiralpak IE, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 70/30/0,1 (об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 8.04 (s, 2Н), 7.63 (d, 1Н), 7.58 (br, 1Н), 7.51-7.41 (m, 3Н), 7.08 (d, 1Н), 6.65 (d, 1Н), 6.14-6.09 (m, 1Н), 4.12-4.06 (m, 1Н), 3.77-3.68 (m, 1Н), 3.46-3.40 (m, 1Н), 3.20-3.13 (m, 2Н), 1.61-1.49 (m, 9Н).
Пример 26
МС m/z (ИЭР): 470,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 3,138 минут, хиральная чистота: 96,048% (хроматографическая колонка: Chiralpak IE, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/трифторуксусная кислота составляет 70/30/0,1 (об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 8.04 (s, 2Н), 7.63 (d, 1Н), 7.58 (br, 1Н), 7.51-7.41 (m, 3Н), 7.08 (d, 1Н), 6.65 (d, 1Н), 6.14-6.09 (m, 1Н), 4.12-4.06 (m, 1Н), 3.77-3.68 (m, 1Н), 3.46-3.40 (m, 1Н), 3.20-3.13 (m, 2Н), 1.61-1.49 (m, 9Н).
Пример 27 (Е)-3-(4-((1S,3R/1R,3S)-6-(6-аминопиридин-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
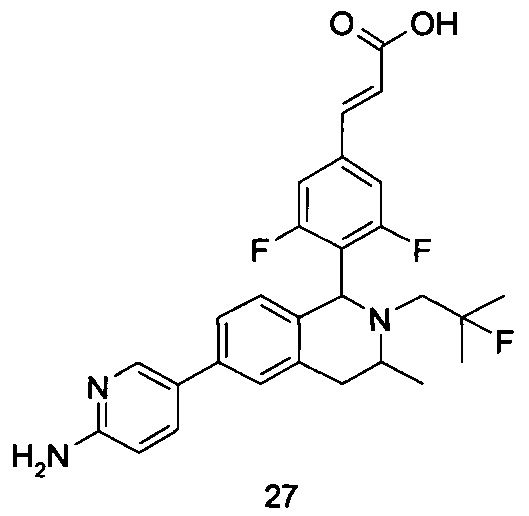
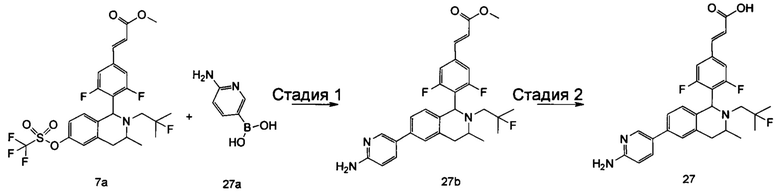
Стадия 1
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(6-аминопиридин-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (100 мг, 0,177 ммоль), (6-аминопиридин-3-ил)бороновую кислоту 27а (36,6 мг, 0,265 ммоль, получена способом, раскрытым в заявке на патент 2014180735) и карбонат калия (73,4 мг, 0,531 ммоль) растворяли в 3,5 мл смеси 1,4-диоксана и воды (об./об. составляет 6:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (6,5 мг, 0,00885 ммоль). Реакционную смесь нагревали до 80°С. После перемешивания в течение 18 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры. Добавляли этилацетат, и смесь промывали водой. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-(6-аминопиридин-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 27b (20 мг, выход 22%), в виде желтого твердого вещества.
Стадия 2
(Е)-3-(4-((1S,3R/1R,3S)-6-(6-аминопиридин-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(6-аминопиридин-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 27b (15 мг, 0,029 ммоль) растворяли в 3 мл этанола, затем добавляли 0,15 мл 1 М раствора гидроксида лития. После перемешивания в течение 3 часов реакцию останавливали. Добавляли по каплям 10% раствор лимонной кислоты до достижения нейтрального рН реакционного раствора. Смесь экстрагировали этилацетатом. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(6-аминопиридин-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 27 (9 мг, выход 62%), в виде желтого твердого вещества.
МС m/z (ИЭР): 496,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.44 (s, 1Н), 8.20-8.17 (m, 1Н), 7.55 (d, 1Н), 7.44 (s, 1Н), 7.33 (d, 1Н), 7.21 (d, 2Н), 7.16-7.14 (m, 1Н), 6.87 (d, 1Н), 6.58-6.54 (m, 1Н), 5.29 (s, 1Н), 3.78-3.75 (m, 1Н), 3.47-3.43 (m, 1Н), 3.07-2.99 (m, 1Н), 2.77-2.73 (m, 1Н), 2.38-2.27 (s, 1Н), 1.20-1.20 (m, 9Н).
Пример 28
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
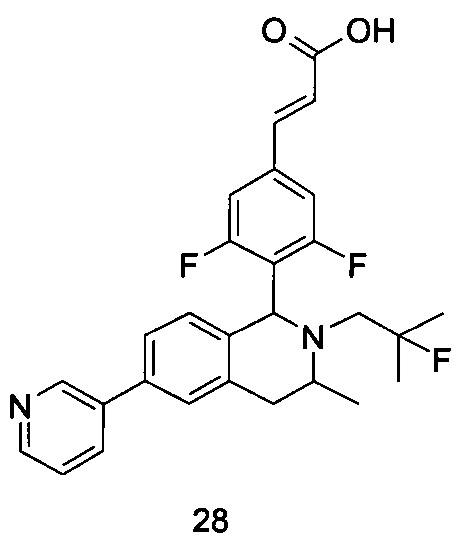
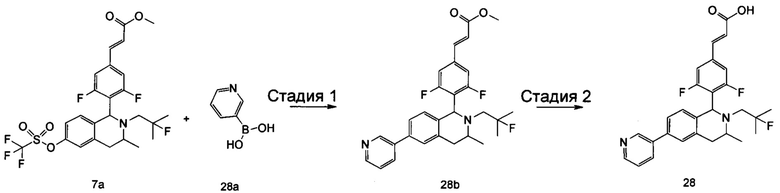
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил) фенил)акрилат 7а (100 мг, 0,177 ммоль), пиридин-3-илбороновую кислоту 28а (28 мг, 0,23 ммоль, получена хорошо известным способом, раскрытым в публикации Tetrahedron Letters, 2002, 43 (23), 4285-4287), и карбонат калия (74 мг, 0,531 ммоль) растворяли в 3,5 мл смеси 1,4-диоксана и воды (об./об. составляет 6:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (6,5 мг, 0,00885 ммоль). Реакционную смесь нагревали до 80°С. После перемешивания в течение 18 часов реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры. Добавляли этилацетат, и смесь промывали водой. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 28b (25 мг, выход 28,5%), в виде желтого твердого вещества.
Стадия 2
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 28b (50 мг, 0,1 ммоль) растворяли в 2,5 мл этанола, затем добавляли 0,5 мл 1 М раствора гидроксида лития. После перемешивания в течение 2 часов реакцию останавливали. Добавляли по каплям 10% раствор лимонной кислоты до достижения нейтрального рН реакционного раствора. Смесь экстрагировали этилацетатом. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 28 (16,9 мг, выход 35%), в виде желтого твердого вещества.
МС m/z (ИЭР): 481,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.80 (s, 1Н), 8.51 (d, 1Н), 8.10 (d, 1Н), 7.60-7.48 (m, 3Н), 7.37 (m, 1Н), 7.23 (d, 2Н), 6.68 (d, 1Н), 6.55 (d, 1Н), 5.30 (s, 1Н), 3.79-3.75 (m, 1Н), 3.49-3.44 (m, 1Н), 3.07-3.00 (m, 1Н), 2.77-2.74 (m, 1Н), 2.38-2.27 (m, 1Н), 1.20-1.05 (m, 9Н).
Пример 29
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(3-метилуреидо)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
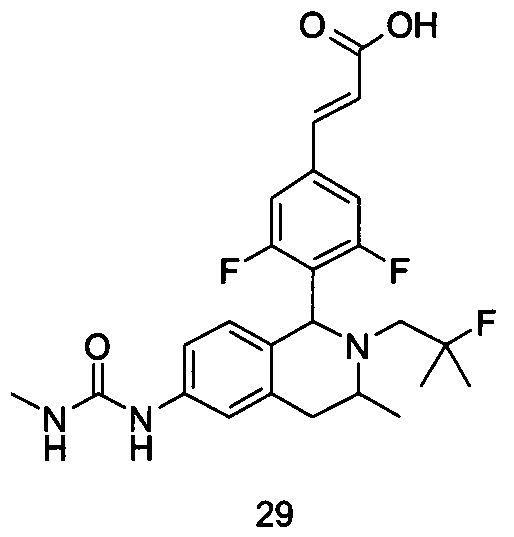
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 1-метилмочевиной, соответственно было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(3-метилуреидо)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 29.
МС m/z (ИЭР): 476,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.58-7.54 (d, 1Н), 7.39-7.35 (m, 1Н), 7.23-7.17 (m, 3Н), 6.61-6.51 (m, 2Н), 5.18 (s, 1Н), 3.69 (s, 1Н), 3.50-3.33 (m, 1Н), 3.03-2.95 (m, 1Н), 2.77 (s, 3Н), 2.59-2.56 (m, 1Н), 2.34-2.23 (m, 1Н), 1.18-0.92 (m, 9Н).
Пример 30
(Е)-3-(4-((1S,3R/1R,3S)-6-(2-(циклопропиламино)-2-оксоэтокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
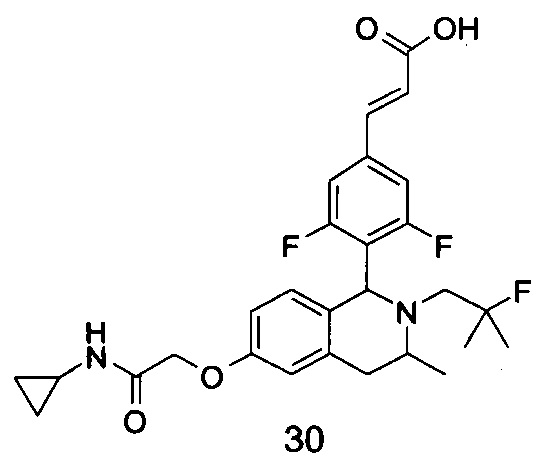
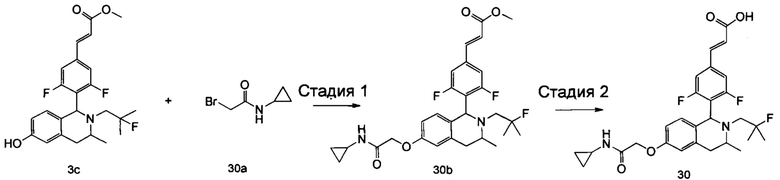
Стадия 1
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(2-(циклопропиламино)-2-оксоэтокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 3с (173 мг, 0,4 ммоль) и 2-бром-М-циклопропилацетамид 30а (107 мг, 0,6 ммоль, получен хорошо известным способом, раскрытым в публикации Organic & Biomolecular Chemistry, 2014, 12 (44), 8952-8965") растворяли в 3 мл N,N-диметилформамида, затем добавляли карбонат калия (110 мг, 0,8 ммоль). Реакционную смесь подогревали до 90°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры реакционный раствор концентрировали при пониженном давлении. Полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-(2-(циклопропиламино)-2-оксоэтокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 30b (12 мг, выход 5,6%), в виде светло-желтого твердого вещества.
Стадия 2
(Е)-3-(4-((1S,3R/1R,3S)-6-(2-(циклопропиламино)-2-оксоэтокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(2-(циклопропиламино)-2-оксоэтокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил) акрилат 30b (12 мг, 0,023 ммоль) растворяли в 2,5 мл смеси метанола и тетрагидрофурана (об./об. составляет 1:4), затем добавляли 0,04 мл 6 М раствора гидроксида натрия. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Добавляли по каплям 1 М раствор соляной кислоты до доведения рН до 5-6. Смесь экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(2-(циклопропиламино)-2-оксоэтокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 30 (8 мг, выход 67,2%), в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 517,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.35 (d, 1Н), 7.10 (d, 2Н), 6.74 (s, 1Н), 6.67-6.64 (m, 2Н), 6.52 (d, 1Н), 5.51 (s, 1Н), 5.16 (s, 1Н), 4.45 (s, 2Н), 3.69 (s, 1Н), 3.37 (s, 1Н), 2.98 (t, 1Н), 2.73 (s, 1Н), 2.59 (d, 1Н), 2.33-2.21 (m, 1Н), 1.17-1.12 (m, 6Н), 1.06 (d, 3Н), 0.75 (d, 2Н), 0.56 (d, 2Н).
Пример 31
(Е)-3-(4-((1S,3R/1R,3S)-6-((трет-бутоксикарбонил)амино)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
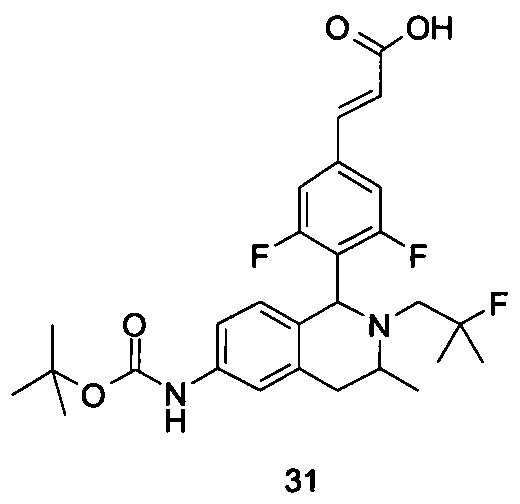
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено трет-бутил-карбаматом, соответственно было получено соединение, указанное в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-((трет-бутоксикарбонил)амино)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 31.
МС m/z (ИЭР): 519,6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.60-7.56 (d, 1Н), 7.27-7.18 (m, 3Н), 7.07-7.05 (m, 1Н), 6.62-6.51 (m, 2Н), 5.18 (s, 1Н), 3.70 (s, 1Н), 3.37-3.36 (m, 1Н), 3.03-2.95 (m, 1Н), 2.60-2.55 (m, 1Н), 2.33-2.21 (m, 1Н), 1.38 (s, 9Н), 1.18-0.92 (m, 9Н).
Пример 32
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-(метиламино)-2-оксоэтокси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
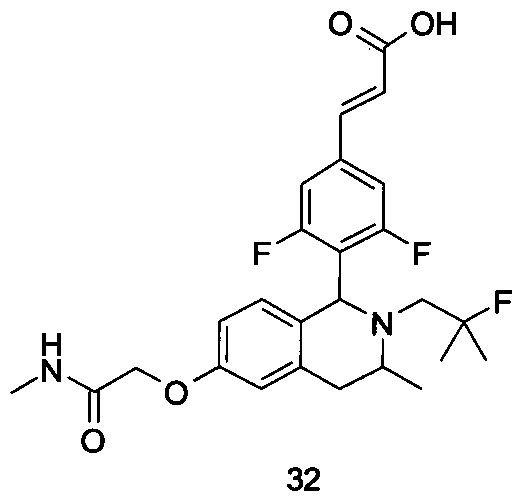
В соответствии с путем синтеза примера 30 исходное вещество 2-бром-N-циклопропилацетамид 30а, используемое на стадии 1, было заменено 2-бром-N-метилацетамидом, который был получен хорошо известным способом, раскрытым в публикации Bioorganic & Medicinal Chemistry, 2011, 19(3), 1106-1114, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-(метиламино)-2-оксоэтокси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 32.
МС m/z (ИЭР): 491,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.54 (d, 1Н), 7.17 (d, 2Н), 6.77 (s, 1Н), 6.68-6.63 (m, 2Н), 6.53 (d, 1Н), 5.18 (s, 1Н), 4.47 (s, 2Н), 3.69 (s, 1Н), 3.38 (s, 1Н), 2.98 (t, 1Н), 2.80 (s, 3Н), 2.60 (s, 1Н), 2.40-2.20 (m, 1Н), 1.18-1.09 (m, 6Н), 1.00 (d, 3Н).
Пример 33
(Е)-3-(4-((1S,3R/1R,3S)-6-(цианометокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
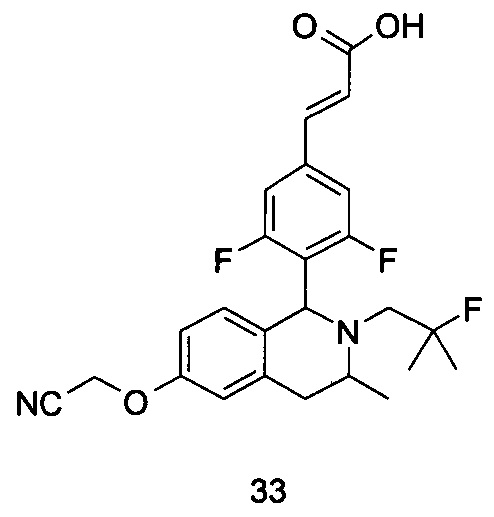
В соответствии с путем синтеза примера 30 исходное вещество 2-бром-N-циклопропилацетамид 30а, используемое на стадии 1, было заменено 2-бромацетонитрил, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(цианометокси)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 33.
МС m/z (ИЭР): 459,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.64-7.60 (d, 1Н), 7.35 (s, 2Н), 6.99-6.93 (m, 3Н), 6.64-6.60 (d, 1Н), 6.00-5.89 (m, 1Н), 5.02 (s, 2Н), 4.11 (s, 1Н), 3.41-3.37 (m, 2Н), 3.18-2.88 (m, 2Н), 1.49-1.31 (m, 9Н).
Пример 34
(Е)-3-(4-((1S,3R/1R,3S)-6-циклопропил-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
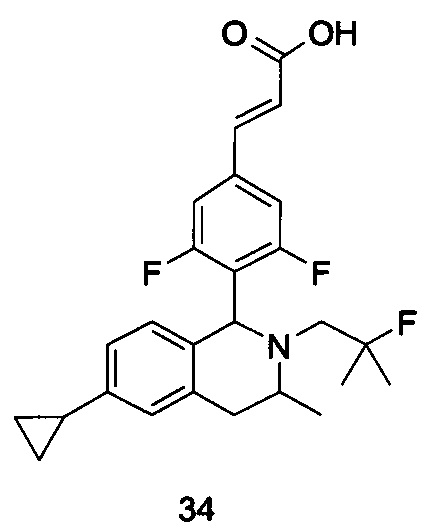
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено циклопропилборатом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-циклопропил-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 34.
МС m/z (ИЭР): 444,4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.60-7.55 (m, 2Н), 7.40 (d, 1Н), 7.23 (d, 2Н), 6.93 (d, 1Н), 6.56 (d, 1Н), 5.30 (s, 1Н), 3.78-3.70 (m, 1Н), 3.67-3.41 (m, 1Н), 3.04-2.96 (m, 1Н), 2.75-2.70 (m, 1Н), 2.36-2.25 (m, 1Н), 1.52-1.48 (m, 1Н), 1.33-1.01 (m, 13Н).
Пример 35
(Е)-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-диметокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
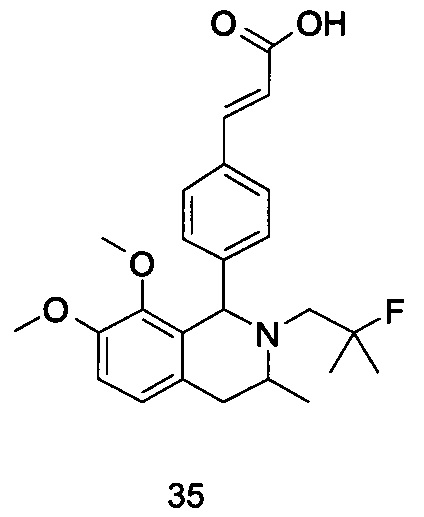
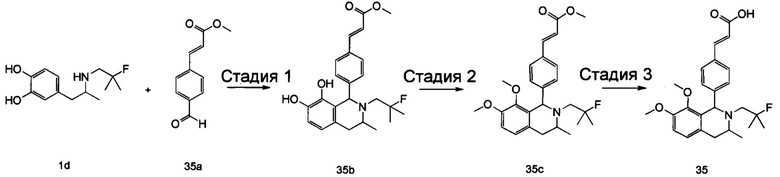
Стадия 1
(Е)-метил-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-дигидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
Неочищенный 4-(2-((2-фтор-2-метилпропил)амино)пропил)бензол-1,2-диол 1d (200 мг, 0,83 ммоль) растворяли в 8 мл метанола, затем добавляли (Е)-метил-3-(4-формилфенил)акрилат 35а (165 мг, 0,87 ммоль, полученный хорошо известным способом, раскрытым в публикации Applied Organometallic Chemistry, 2014, 28(7), 529-536, и уксусную кислоту (100 мг, 1,66 ммоль). Полученную в результате смесь нагревали до 55° и перемешивали в течение 5 часов. Реакционный раствор концентрировали при пониженном давлении для удаления метанола и уксусной кислоты. Добавляли дихлорметан, и смесь очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-дигидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 35b (100 мг, выход 29%) в виде коричневого масла.
Стадия 2
(Е)-метил-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-диметокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-дигидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 35b (100 мг, 0,24 ммоль) растворяли в 3 мл ацетона, затем добавляли диметилсульфат (92 мг, 0,72 ммоль) и карбонат калия (133 мг, 0,96 ммоль). Полученную в результате смесь перемешивали при кипячении с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 100 мл этилацетата, затем смесь промывали насыщенным раствором хлорида натрия (100 мл × 2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-диметокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 35с (18 мг, выход 17%) в виде коричневого твердого вещества.
Стадия 3
(Е)-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-диметокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-диметокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 35с (18 мг, 0,04 ммоль) растворяли в 4 мл смеси тетрагидрофурана и метанола (об./об. составляет 1:1), затем добавляли 0,5 мл 1 М раствора гидроксида натрия. Реакционную смесь перемешивали в течение 10 часов при комнатной температуре. Добавляли по каплям 1 М раствор соляной кислоты до доведения рН до 5-6. Смесь экстрагировали этилацетатом (50 мл). Органическую фазу промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-7,8-диметокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 35 (5 мг, выход 28%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 428,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.76-7.72 (d, 1Н), 7.44-7.42 (d, 2Н), 7.29-7.26 (m 2Н), 6.87 (s 2Н), 6.41-6.37 (d 1Н), 5.37 (s, 1Н), 3.85 (s 3Н), 3.52 (s 3Н), 3.12-3.05 (m 1Н), 2.64-2.44 (m, 4Н), 1.35-1.25 (m, 6Н), 0.99-0.97 (m, 3Н).
Пример 36
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((тетрагидро-2Н-пиран-4-ил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акриловая кислота
В соответствии с путем синтеза примера 30 исходное вещество 2-бром-N-циклопропилацетамид 30а, используемое на стадии 1, было заменено тетрагидро-4-пиранолом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((тетрагидро-2Н-пиран-4-ил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 36.
МС m/z (ИЭР): 504,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.57 (d, 1Н), 7.19 (d, 2Н), 6.73 (s, 1Н), 6.64-6.60 (m, 2Н), 6.53 (d, 1Н), 5.17 (s, 1Н), 4.50 (s, 1Н), 3.98-3.91 (m, 2Н), 3.68 (s, 1Н), 3.59 (t, 1Н), 3.36 (s, 1Н), 2.99 (t, 1Н), 2.58 (d, 1Н), 2.34-2.22 (m, 2Н), 2.12-2.01 (m, 2Н), 1.70-1.66 (m, 2Н), 1.17-1.09 (m, 6Н), 1.01 (d, 3Н).
Пример 37
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((S)-тетрагидрофуран-3-ил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
В соответствии с путем синтеза примера 30 исходное вещество 2-бром-N-циклопропилацетамид 30а, используемое на стадии 1, было заменено (R)-3-гидрокситетрагидрофураном, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((S)-тетрагидрофуран-3-ил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 37.
MC m/z (ИЭР): 490,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.51 (d, 1Н), 7.03 (d, 2Н), 6.76 (d, 1Н), 6.69-6.66 (m, 2Н), 6.35 (d, 1Н), 5.79 (s, 1Н), 4.91 (s, 1Н), 4.17 (d, 1Н), 4.03-3.92 (m, 4Н), 3.27-3.20 (m, 2Н), 2.99-2.95 (m, 1Н), 2.75 (dd, 1Н), 2.21-2.16 (m, 2Н), 1.54 (d, 3Н), 1.41-1.26 (m, 6Н).
Пример 38
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилморфолино)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота

В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 2-метилморфолином, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилморфолино)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 38.
МС m/z (ИЭР): 503,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.43- 7.39 (m, 1Н), 7.15-7.01 (m, 3Н), 6.74-6.48 (m, 3Н), 5.22-5.14 (m, 1Н), 4.05-3.95 (m, 1Н), 3.82-3.37 (m, 4Н), 3.25-3.19 (m, 1Н), 2.81-2.21 (m, 6Н), 1.23-0.99 (m, 12Н).
Пример 39
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(4-метил-3-оксопиперазин-1-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 1-метилпиперазин-2-оном, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(4-метил-3-оксопиперазин-1-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 39.
МС m/z (ИЭР): 516,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.68-7.55 (m, 2Н), 7.44 (d, 1Н), 7.19-7.08 (m, 2Н), 6.82-6.60 (m, 2Н), 6.56-6.50 (m, 1Н), 5.24-5.15 (m, 1Н), 4.01-3.94 (m, 1Н), 3.78-3.37 (m, 6Н), 3.16-2.91 (m, 5Н), 2.68-2.56 (m, 1Н), 2.33-2.22 (m, 1Н), 1.19-0.99 (m, 9Н).
Пример 40
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(3-метилморфолино)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
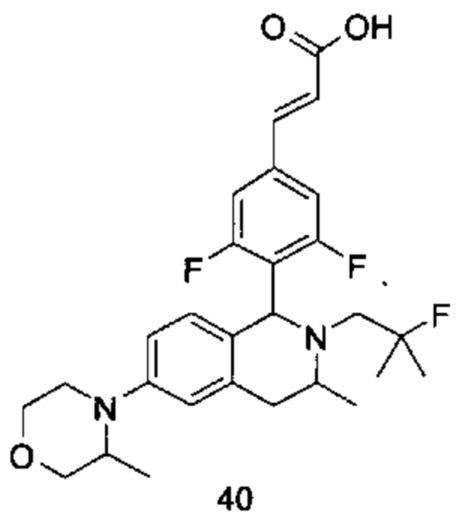
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 3-метилморфолином, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(3-метилморфолино)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 40.
МС m/z (ИЭР): 503,0 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.59 (d, 1Н), 7.22 (d, 2Н), 6.83-6.77 (m, 2Н), 6.75-6.71 (m, 1Н), 6.54 (d, 1Н), 5.29 (s, 1Н), 3.95 (d, 1Н), 3.87 (d, 1Н), 3.77-3.66 (m, 4Н), 3.38-3.37 (m, 1Н), 3.26-3.18 (m, 2Н), 3.18-3.08 (m, 1Н), 2.73-2.69 (m, 1Н), 2.41-2.21 (m, 1Н), 1.18-1.02 (m, 12Н).
Пример 41
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-морфолино-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
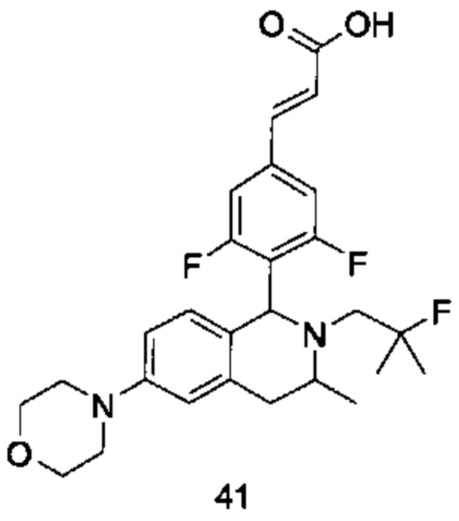
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено морфолином, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-морфолино-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 41.
МС m/z (ИЭР): 489,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.60-7.55 (m, 2Н), 7.40 (d, 1Н), 7.23 (d, 2Н), 6.93 (d, 1Н), 6.56 (d, 1Н), 5.30 (s, 1Н), 3.78-3.70 (m, 1Н), 3.67-3.41 (m, 5Н), 3.04-2.80 (m, 5Н), 2.75-2.70 (m, 1Н), 2.36-2.25 (m, 1Н), 1.33-1.01 (m, 9Н).
Пример 42
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пирроло[3,4-с]пиридин-2(3Н)-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
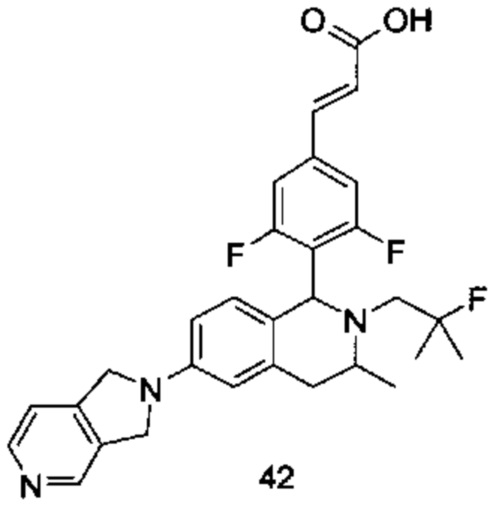
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 2,3-дигидро-1Н-пирроло[3,4-с]пиридина гидрохлоридом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пирроло[3,4-с]пиридин-2(3Н)-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 42.
МС m/z (ИЭР): 522,6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.60-8.48 (m, 2Н), 7.90-7.83 (m, 1Н), 7.65-7.57 (m, 2Н), 7.23-7.16 (m, 2Н), 6.65-6.52 (m 3Н), 5.19 (s, 1Н), 4.69 (s, 4Н), 3.71 (s 1Н), 3.05-3.01 (m, 1Н), 2.64-2.61 (m, 1Н), 2.35-2.22 (m, 2Н), 1.22-0.93 (m, 9Н).
Пример 43
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(5-метилтиофен-2-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
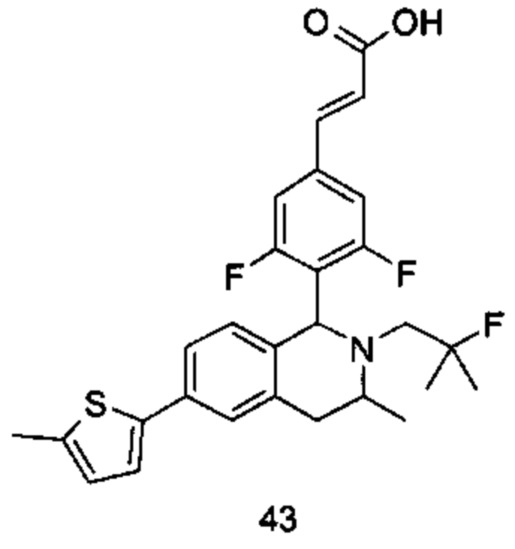
В соответствии с путем синтеза примера 30 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 4,4,5,5-тетраметил-2-(5-метилтиофен-2-ил)-1,3,2-диоксабороланом, который был получен хорошо известным способом, раскрытым в Organometallics, 2015, 34(19), 4732-4740, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(5-метилтиофен-2-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 43.
МС m/z (ИЭР): 500,4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.60-7.56 (d, 1Н), 7.33 (s, 1Н), 7.26-7.20 (m, 3Н), 7.13 (s, 1Н), 6.74-6.70 (m 2Н), 6.56-6.52 (d 1Н), 5.23 (s, 1Н), 3.73 (s 1Н), 3.41-3.37 (m 1Н), 3.05-2.97 (m, 1Н), 2.68-2.64 (m, 1Н), 2.49 (s, 3Н), 2.36-2.25 (m, 1Н), 1.19-0.92 (m, 9Н).
Пример 44
(Е)-3-(4-((1S,3R/1R,3S)-6-(3,3-диметилуреидо)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 1-изопропилмочевиной, соответственно, было получено соединение, указанное в заголовке, (E)-3-(4-((1S,3R/1R,3S)-6-(3,3-диметилуреидо)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 44.
МС m/z (ИЭР): 490,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.58 (d, 1Н), 7.00 (m, 3Н), 6.64 (d, 1Н), 6.40 (d, 1Н), 6.24 (s, 1Н), 5.17 (s, 1Н), 3.36 (dd, 1Н), 3.02 (s, 6Н), 2.94 (t, 1Н), 2.55 (dd, 1Н), 2.24 (m, 1Н), 1.93 (s, 1Н), 1.32-1.09 (m, 6Н), 0.99 (d, 3Н).
Пример 45
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
В соответствии с путем синтеза примера 16 исходное вещество 2-(3-(бензилокси)-4-фторфенил)ацетальдегид 16а, используемое на стадии 1, было заменено 2-(3-(бензилокси)-2-фторфенил)ацетальдегидом, который был получен хорошо известным способом, раскрытым в публикации Environmental Science and Pollution Research, 2014, 21(7), 4861-4870), соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 45. МС m/z (ИЭР): 502,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.05 (s, 1Н), 7.90 (s, 1Н), 7.61 (d, 1Н), 7.49-7.47 (m, 1Н), 7.36-7.33 (m, 2Н), 6.80 (s, 1Н), 6.61 (d, 1Н), 5.79 (s, 1H), 3.97-3.94 (m, 4H), 3.30-3.27 (m, 2H), 3.03-2.68 (m, 2H), 1.42-1.33 (m, 9H).
Пример 46
(E)-3-(4-(((1S,3R/1R,3S)-6-(5-этил-1,2,4-оксадиазол-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(N-гидроксикарбамимидоил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-циано-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 19а (300 мг, 0,68 ммоль), гидроксиламина гидрохлорид (56 мг, 0,81 ммоль) и триэтиламин (151 мг, 1,49 ммоль) растворяли в 10 мл этанола. Полученную в результате смесь перемешивали при кипячении с обратным холодильником в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(N-гидроксикарбамимидоил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 46а (350 мг) в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-((2)-N'-гидрокси-N-пропионилкарбамимидоил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(N-гидроксикарбамимидоил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 46а (350 мг, 0,68 ммоль) и карбонат калия (187 мг, 1,36 ммоль) растворяли в ацетоне и добавляли по каплям пропионилхлорид (94 мг, 1,02 ммоль). Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Добавляли воду, и смесь экстрагировали этилацетатом (50 мл ×2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-((2)-N'-гидрокси-N-пропионилкарбамимидоил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 46b (350 мг) в виде желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(5-этил-1,2,4-оксадиазол-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
Неочищенный (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-((Z)-N'-гидрокси-N-пропионилкарбамимидоил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 46b (350 мг, 0,68 ммоль) растворяли в 15 мл толуола. Реакционную смесь подогревали до 90°С и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-(5-этил-1,2,4-оксадиазол-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 46с (50 мг, выход 14,3%), в виде желтого масла.
Стадия 4
(Е)-3-(4-((1S,3R/1R,3S)-6-(5-этил-1,2,4-оксадиазол-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-(5-этил-1,2,4-оксадиазол-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 46с (50 мг, 0,1 ммоль) растворяли в 2 мл метанола, затем добавляли 0,3 мл 1 М раствор гидроксида лития. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении для удаления метанола. Добавляли по каплям 1 М раствор соляной кислоты до доведения рН до 3. Смесь экстрагировали этилацетатом (30 мл ×2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(5-этил-1,2,4-оксадиазол-3-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 46 (10 мг, выход 20%), в виде желтого твердого вещества.
МС m/z (ИЭР): 500,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 12.57 (s, 1Н), 7.80 (d, 1Н), 7.68 (d, 1Н), 7.55-7.46 (m, 3Н), 6.90 (d, 1Н), 6.66 (d, 1Н), 5.21 (s, 1Н), 3.60-3.53 (m, 1Н), 3.30-3.24 (m, 1Н), 3.04-2.91 (m, 3Н), 2.75 (dd, 1Н), 2.29-2.18 (m, 1Н), 1.33 (t, 3Н), 1.18-1.04 (m, 6H), 0.97 (d, 3Н).
Пример 47
(Е)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
В соответствии с путем синтеза примера 16 исходное вещество 2-(3-(бензилокси)-4-фторфенил)ацетальдегид 16а, используемое на стадии 1, было заменено 2-(3-(бензилокси)-2-фторфенил)ацетальдегидом, который был получен хорошо известным способом, раскрытым в публикации Environmental Science and Pollution Research, 2014, 21(7), 4861-4870, и исходное вещество 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 16h, используемое на стадии 7, было заменено 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолане-2-ил)-1Н-пиразолом с получением соединения, указанного в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(1-этил-1Н-пиразол-4-ил)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловой кислоты 47.
МС m/z (ИЭР): 516,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.03 (s, 1Н), 7.87 (s, 1Н), 7.59 (d, 1Н), 7.36-7.32 (m, 1Н), 7.23 (d, 2Н), 6.59-6.53 (m, 2Н), 5.27 (s, 1Н), 4.27-4.22 (m, 2Н), 3.76 (s, 1Н), 3.15-2.84 (m, 3Н), 2.37-2.26 (m, 1H), 1.52-1.50 (m, 3Н), 1.21-1.05 (m, 9H).
Пример 48
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-(2-фторэтил)-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
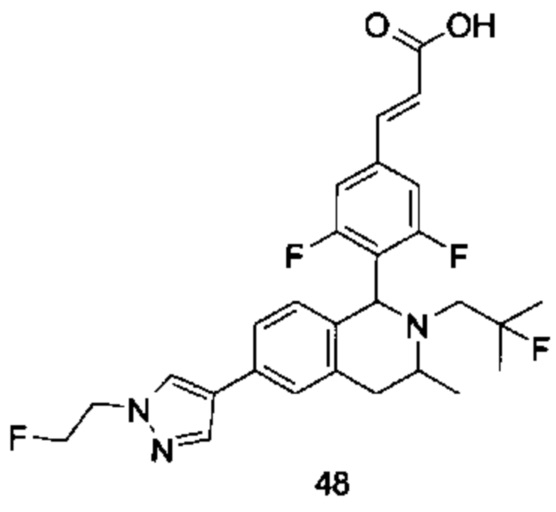
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 1-(2-фторэтил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, который был получен способом, раскрытым в заявке на патент WO 2013162061), соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-(2-фторэтил)-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 48.
МС m/z (ИЭР): 516,0 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.99 (s, 1Н), 7.85 (s, 1Н), 7.56 (d, 1Н), 7.34 (s, 1Н), 7.25-7.19 (m, 3Н), 6.73-6.71 (d, 1Н), 6.56-6.52 (d, 1Н), 5.23 (s, 1Н), 4.84-4.82 (m, 1Н), 4.73-4.70 (m, 1Н), 4.52-4.49 (m, 1Н), 4.45-4.43 (m, 1Н), 4.75-4.71 (m, 1Н), 3.43-3.38 (m, 1Н), 3.05-2.97 (s, 1Н), 2.69-2.64 (m, 1Н), 2.36-2.25 (m, 1Н), 1.19-1.03 (m, 9Н).
Пример 49
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилоксазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
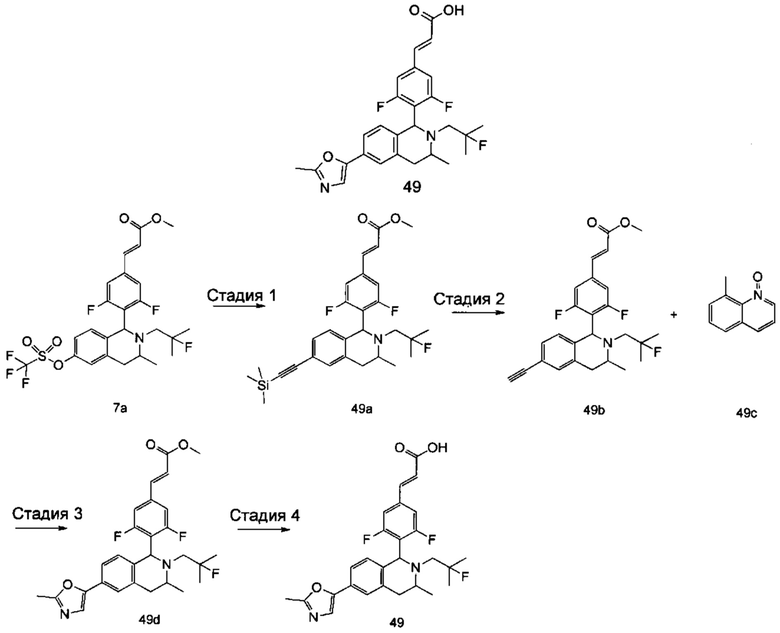
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((триметилсилил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (800 мг, 1,42 ммоль), триметилсилилацетилен (418 мг, 4,26 ммоль), диметиламин (44 мг, 0,98 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (52,6 мг, 0,072 ммоль) и йодид меди (27,1 мг, 0,142 ммоль) растворяли в N,N-диметилформамиде. Полученную в результате смесь подогревали до 80°С и перемешивали в течение 18 часов. Реакционный раствор охлаждали до комнатной температуры и добавляли этилацетат. Смесь промывали водой, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил 3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((триметилсилил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 49а (590 мг, выход 81%), в виде светло-желтого твердого вещества.
Стадия 2
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-этинил-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-((триметилсилил)этинил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 49а (590 мг, 1,15 ммоль) растворяли в 5 мл 1 М тетрабутиламмония фторида. Реакционную смесь перемешивали в течение 5 часов при комнатной температуре. Добавляли воду, и смесь экстрагировали этилацетатом три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-этинил-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 49b (60 мг, выход 11,8%), в виде белого твердого вещества.
Стадия 3
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилоксазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-этинил-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 49b (20 мг, 0,045 ммоль) растворяли в 4 мл ацетонитрила, затем добавляли 8-метилхинолин-1-оксид 49с (9,4 мг, 0,058 ммоль, получен хорошо известным способом, раскрытым в публикации ChemMedChem, 2009, 4(2), 249-260) и [бис(трифторметансульфонил)имидат](трифенилфосфин)золото (I) (1,7 мг, 0,00225 ммоль), и реакционную смесь подогревали до 60°С и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилоксазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 49d (13 мг, выход 58%), в виде светло-желтого твердого вещества.
Стадия 4
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилоксазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилоксазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 49d (13 мг, 0,026 ммоль) растворяли в 1 мл метанола, затем добавляли 1 мл 1 М раствора гидроксида лития, и смесь перемешивали в течение 1 часа при комнатной температуре. Добавляли по каплям 10% раствор лимонной кислоты до доведения рН до 6-7. Полученную в результате смесь экстрагировали этилацетатом, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали тонкослойной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилоксазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 49 (6 мг, выход 48%) в виде желтого твердого вещества.
МС m/z (ИЭР): 485,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.63 (d, 1Н), 7.47 (s, 1Н), 7.36 (d, 1Н), 7.31 (s, 1Н), 7.22 (d, 2Н), 6.80 (d, 1Н), 6.55 (d, 1Н), 5.26 (s, 1Н), 3.76-3.72 (m, 1Н), 3.43-3.39 (m, 1Н), 3.05-2.98 (m, 1Н), 2.72-2.68 (m, 1Н), 2.52 (s, 3Н), 2.37-2.25 (m, 1Н), 1.20-1.03 (m, 9Н).
Пример 50
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-изопропил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
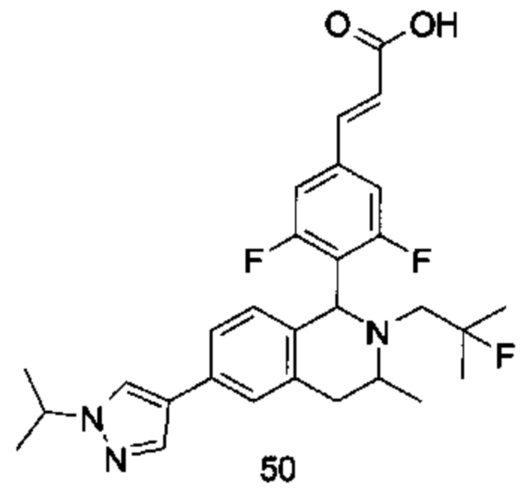
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 1-изопропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, который был получен хорошо известным способом, раскрытым в публикации Journal of Medicinal Chemistry, 2011, 54(18), 6342-6363, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-изопропил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 50.
МС m/z (ИЭР): 510,5 [М-1]
1Н ЯМР (400 МГц, CD3OD) δ 7.99 (s, 1Н), 7.79 (s, 1Н), 7.59 (d, 1Н), 7.34 (s, 1Н), 7.22 (t, 3Н), 6.71 (d, 1Н), 6.54 (d, 1Н), 5.24 (s, 1Н), 3.70-3.74 (m, 1Н), 3.38-3.42 (dd, 1Н), 3.01 (t, 1Н), 2.69-2.64 (dd, 1Н), 2.36-2.25 (m, 2Н), 1.53 (d, 6Н), 1.19-1.10 (dd, 6H), 1.04 (s, 3Н).
Пример 51
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метил-2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота

Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(E)-метил-3-(4-((1S,3R/1R,3S)-6-циано-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 19а (20 мг, 0,045 ммоль), азид натрия (17 мг, 0,271 ммоль) и хлорид аммония (15 мг, 0,271 ммоль) добавляли к N,N-диметилформамиду. Смесь подогревали до 150°С и перемешивали в микроволновом реакторе в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры и добавляли этилацетат. Смесь экстрагировали этилацетатом (20 мл ×2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 51а (20 мг) в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительно очистки.
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метил-2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 51а (20 мг, 0,041 ммоль), метилйодид (9 мг, 0,061 ммоль) и карбонат калия (11 мг, 0,082 ммоль) растворяли в 1 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Добавляли воду, и реакционный раствор экстрагировали этилацетатом (20 мл ×2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метил-2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 51 b (20 мг), в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метил-2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
Неочищенный (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метил-2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 51b (20 мг, 0,04 ммоль) растворяли в 1,5 мл смеси метанола и тетрагидрофурана (об./об. составляет 2:1), затем добавляли 0,2 мл 1 М раствор гидроксида лития. После перемешивания при комнатной температуре в течение 12 часов реакционный раствор концентрировали при пониженном давлении, и к остатку добавляли 1 М HCl до рН 4. Смесь экстрагировали этилацетатом (20 мл ×2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали тонкослойной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метил-2Н-тетразол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 51 (10 мг, выход 50%), в виде белого твердого вещества.
МС m/z (ИЭР): 486,2 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 12.53 (s, 1Н), 7.82 (d, 1Н), 7.75 (d, 1Н), 7.55-7.46 (m, 3Н), 6.87 (d, 1Н), 6.66 (d, 1Н), 5.19 (s, 1H), 4.52 (s, 3Н), 3.59-3.51 (m, 1H), 3.33-3.28 (m, 1H), 3.04-2.91 (m, 1H), 2.75 (dd, 1H), 2.29-2.18 (m, 1H), 1.18-1.04 (m, 6H), 0.97 (d, 3H).
Пример 52
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-изобутил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
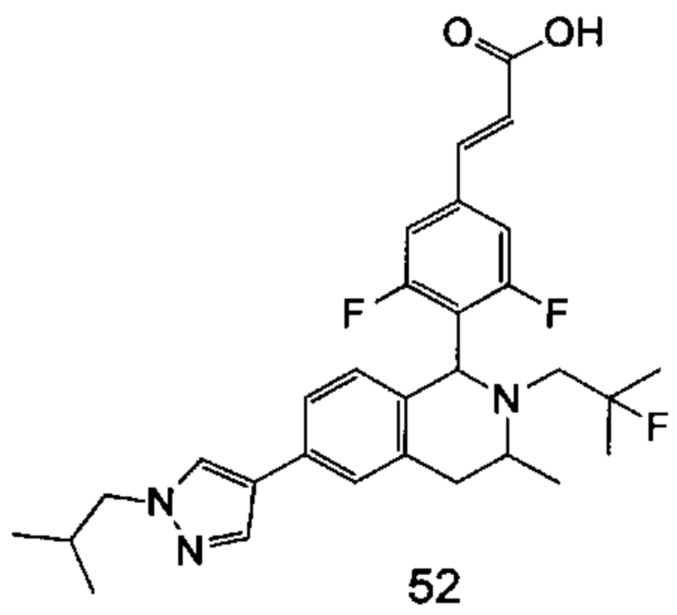
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 1-изобутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, который был получен способом, раскрытым в заявке на патент WO 2014153214, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1-изобутил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 52.
МС m/z (ИЭР): 526,6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.93 (s, 1Н), 7.80 (s, 1Н), 7.58 (d, 1Н), 7.33 (s, 1Н), 7.21 (t, 3Н), 6.70 (d, 1Н), 6.53 (d, 1Н), 5.23 (s, 1Н), 3.96 (d, 2Н), 3.70-3.74 (m, 1Н), 3.38-3.42 (dd, 1Н), 3.01 (t, 1Н), 2.63-2.68 (dd, 1Н), 2.36-2.17 (m, 2Н), 1.19-1.10 (dd, 6Н), 1.03 (s, 3Н), 0.93 (d, 6Н).
Пример 53
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(3-метил-1,2,4-оксадиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
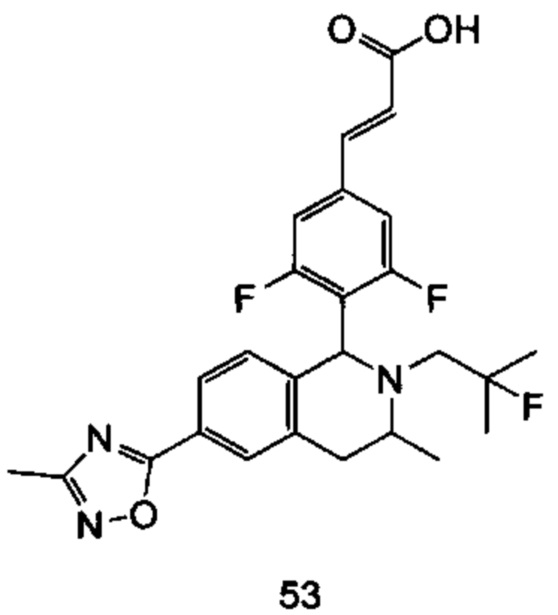
В соответствии с путем синтеза примера 19 исходное вещество N-гидроксипропионамидин, используемое на стадии 2, было заменено N-гидроксиацетамидином, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(3-метил-1,2,4-оксадиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 53.
МС m/z (ИЭР): 486,0 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6): 5 12.05 (s, 1Н), 7.90 (s, 1Н), 7.77 (d, 1Н), 7.52 -7.46 (m, 3Н), 6.98 (d, 1Н), 6.66 (d, 1Н), 5.23 (s, 1Н), 3.62-3.57 (m, 1Н), 2.95 (t, 1Н), 2.79 (d, 1Н), 2.41 (s, 3Н), 2.29-2.19 (m, 2Н), 1.11 (dd, 6Н), 0.97 (d, 3Н).
Пример 54
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(оксазол-2-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
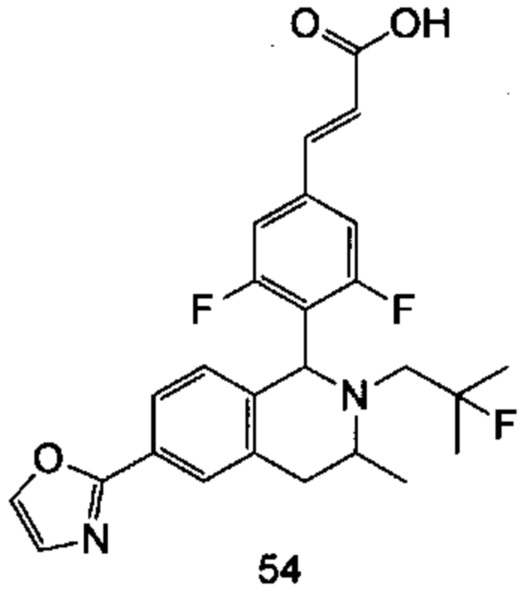
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено оксазолом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(оксазол-2-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 54.
МС m/z (ИЭР): 471,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.98 (s, 1Н), 7.83 (s, 1Н), 7.71 (d, 1Н), 7.57 (d, 1Н), 7.30 (s, 1Н), 7.23 (d, 2Н), 6.89 (d, 1Н), 6.55 (d, 1Н), 5.30 (s, 1Н), 3.78-3.74 (m, 1H), 3.47-3.42 (m, 1H), 3.06-2.99 (m, 1H), 2.77-2.72 (m, 1H), 2.38-2.27 (m, 1H), 1.20-1.04 (m, 9H).
Пример 55
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
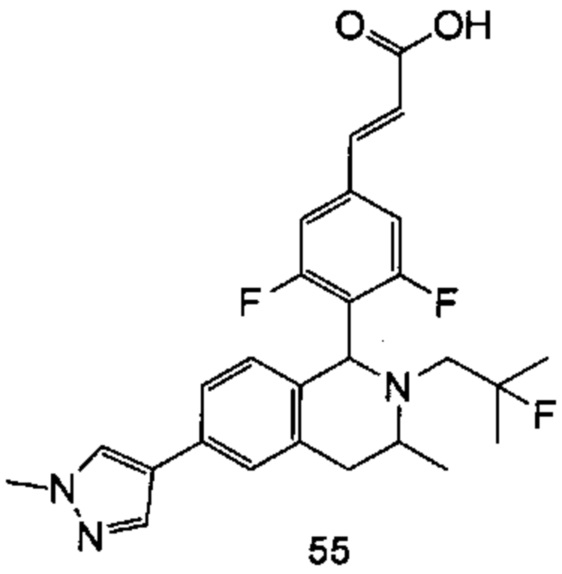
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 55.
МС m/z (ИЭР): 484,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.99 (s, 1Н), 7.84 (s, 1Н), 7.63-7.40 (m, 5Н), 7.06-7.00 (m, 1Н), 6.63 (d, 1Н), 6.10-6.02 (m, 1Н), 4.09-4.00 (m, 1Н), 3.93 (s, 3Н), 3.43-3.37 (m, 2Н), 3.17-3.10 (m, 2Н), 1.56-1.44 (m, 9Н).
Пример 56 и пример 57
(Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
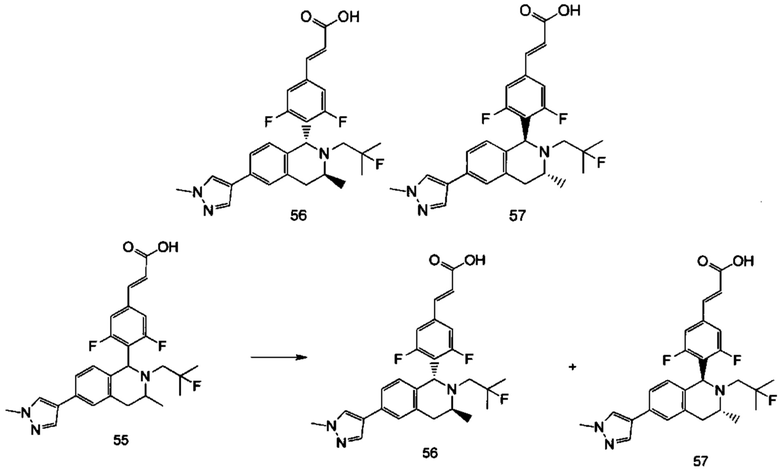
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловую кислоту 55 (605 мг, 1,25 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak AD-H, внутренний диаметр 0,46 см × длина 25 см; подвижная фаза: этанол: ТФУ составляет 100:0,1, скорость потока: 1,0 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 56 (419,7 мг) в виде желтого твердого вещества и (Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 57 (325.8 мг) в виде желтого твердого вещества.
Пример 56:
МС m/z (ИЭР): 484,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания: 6,654 минуты, хиральная чистота: 99,20% (хроматографическая колонка: Chiralpak IE, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: этанол/ТФУ составляет 100/0,1(об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 8.00 (s, 1Н), 7.85 (s, 1Н), 7.62 (d, 1Н), 7.51 (s, 1Н), 7.43-7.40 (m, 3Н), 7.02 (m, 1Н), 6.63 (d, 1Н), 5.98 (s, 1Н), 4.04 (s, 1Н), 3.95 (s, 3Н), 3.62-3.58 (m, 1Н), 3.44-3.38 (dd, 1Н), 3.15-3.08 (m, 2Н), 1.52-1.32 (m, 9Н).
Пример 57:
МС m/z (ИЭР): 484,5 [М+1]
Анализ хиральной ВЭЖХ: время удерживания 11,021 минут, хиральная чистота: 99,9% (колонка: Chiralpak IE, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: этанол/ТФУ составляет 100/0,1(об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 8.00 (s, 1Н), 7.85 (s, 1Н), 7.62 (d, 1Н), 7.51 (s, 1Н), 7.43-7.40 (m, 3Н), 7.02 (m, 1Н), 6.63 (d, 1Н), 5.98 (s, 1Н), 4.04 (s, 1Н), 3.95 (s, 3Н), 3.62-3.58 (m, 1Н), 3.44-3.38 (dd, 1Н), 3.15-3.08 (m, 2Н), 1.52-1.32 (m, 9Н).
Пример 58
(E)-3-(4-((1S,3R/1R,3S)-6-(1-циклопропил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота

В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 1-циклопропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-((1S,3R/1R,3S)-6-(1-циклопропил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 58.
МС m/z (ИЭР): 510,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.04 (s, 1Н), 7.82 (s, 1Н), 7.57 (d, 1Н), 7.48 (s, 1Н), 7.37-7.33 (m, 3Н), 6.96 (s, 1Н), 6.61 (d, 1Н), 5.96 (s, 1Н), 3.99 (s, 1Н), 3.71-3.67 (m, 1Н), 3.44-3.38 (dd, 1Н), 3.25-3.23 (m, 1Н), 3.15-2.88 (m, 2Н), 1.64-1.21 (m, 9Н), 1.14-1.08 (m, 4Н).
Пример 59
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-фенил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
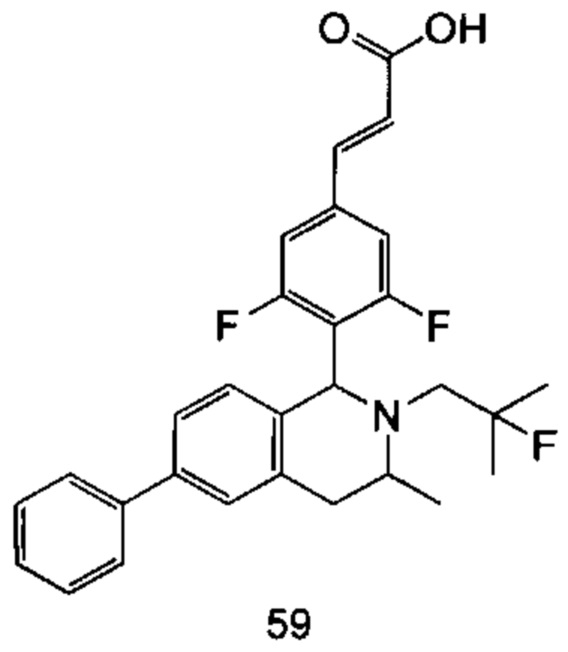
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено фенилбороновой кислотой, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-фенил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 59.
МС m/z (ИЭР): 480,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.12 (s, 1Н), 7.78-7.76 (s, 1Н), 7.66-7.62 (m, 1Н), 7.46-7.44 (m, 2Н), 7.30 (s, 1Н), 7.24-7.14 (m, 4Н), 6.77-6.67 (m, 2Н), 5.23 (s, 1Н), 3.74-3.72 (m, 1Н), 3.43-3.38 (m, 1Н), 3.03-2.96 (m, 1Н), 2.70-2.68 (m, 1Н), 2.35-2.20 (m, 1Н), 1.77-0.99 (m, 9Н).
Пример 60
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(4-морфолинофенил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
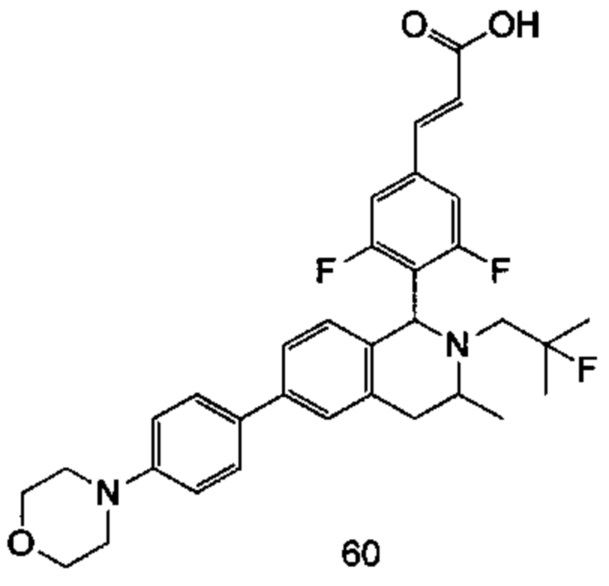
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)морфолином, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(4-морфолинофенил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 60.
МС m/z (ИЭР): 565,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.56-7.55 (m, 2Н), 7.54-7.36 (m, 3Н), 7.30 (m, 1Н), 7.00-6.98 (m, 2Н), 6.87-6.81 (m, 2Н), 6.43 (d, 1Н), 5.27 (s, 1Н), 3.92 (s, 4Н), 3.48 (d, 1Н), 3.25 (d, 4Н), 3.00 (t, 1Н), 2.68 (d, 1Н), 2.36-2.25 (m, 1Н), 1.48 (s, 1Н), 1.30-1.15 (m, 6Н), 1.07-1.06 (m, 3Н).
Пример 61
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(6-(1-метил-1Н-пиразол-4-ил)пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
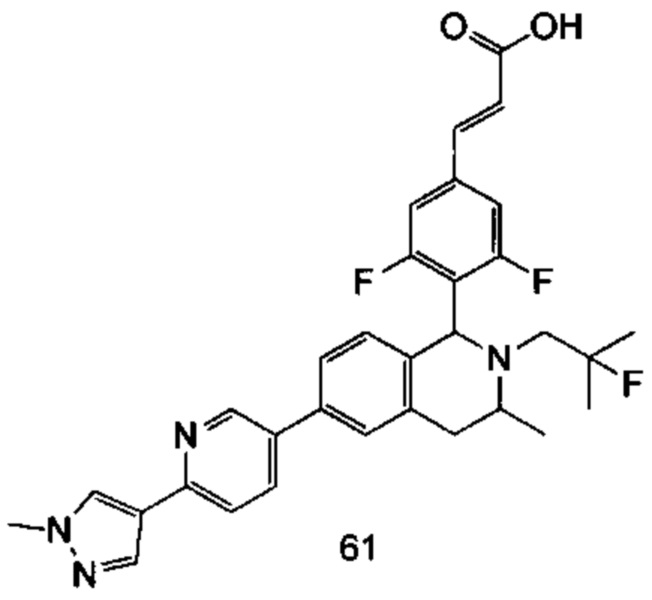
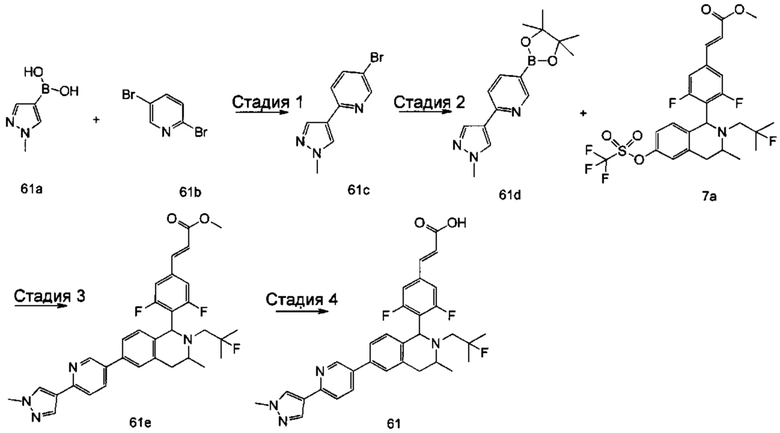
Стадия 1
5-Бром-2-(1-метил-1Н-пиразол-4-ил)пиридин
(1-Метил-1Н-пиразол-4-ил)бороновую кислоту 61а (0,1 г, 0,794 ммоль) растворяли в 5,5 мл смеси 1,4-диоксана и воды (об./об. составляет 10:1), затем добавляли 2,5-дибромпиридин 61b (0,188 г, 0,784 ммоль), тетракис(трифенилфосфин)палладий (92 мг, 0,796 ммоль) и карбонат натрия (0,168 г, 1,585 ммоль). Смесь подогревали до 85°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры добавляли 20 мл этилацетата. Реакционную смесь промывали водой, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, 5-бром-2-(1-метил-1Н-пиразол-4-ил)пиридина 61с (0,13 г, выход 68%) в виде белого твердого вещества.
Стадия 2
2-(1-метил-1Н-пиразол-4-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
5-Бром-2-(1-метил-1Н-пиразол-4-ил)пиридин 61с (0,12 г, 0,504 ммоль) растворяли в 3 мл 1,4-диоксана, затем добавляли бис(пинаколато)дибор (0,192 г, 0,756 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалалдий (II) (22,13 мг, 0,0302 ммоль) и ацетат калия (0,15 г, 1,528 ммоль). Реакционную смесь подогревали до 85°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, 2-(1-метил-1Н-пиразол-4-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина 61d (35 мг, выход 24%) в виде желтого масла.
Стадия 3
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(6-(1-метил-1Н-пиразол-4-ил)пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7а (69 мг, 0,122 ммоль) растворяли в 3,3 мл смеси 1,4-диоксана и воды (об./об. составляет 10:1), затем добавляли 2-(1-метил-1Н-пиразол-4-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин 61d (35 мг, 0,123 ммоль), тетракис(трифенилфосфин)палладий (14,1 мг, 0,122 ммоль) и карбонат натрия (25,9 г, 0,244 ммоль). Реакционную смесь подогревали до 85°С и перемешивали в течение 12 часов. После охлаждения до комнатной температуры добавляли 20 мл этилацетата. Реакционную смесь промывали водой, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(6-(1-метил-1Н-пиразол-4-ил)пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 61е (25 мг, выход 36%) в виде желтого масла.
Стадия 4
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(6-(1-метил-1Н-пиразол-4-ил)пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(6-(1-метил-1Н-пиразол-4-ил)пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 61е (25 мг, 0,0435 ммоль) растворяли в 4 мл смеси тетрагидрофурана и метанола (об./об. составляет 3:1), затем добавляли 0,22 мл 1 М раствора гидроксида лития. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Добавляли 10% раствор лимонной кислоты до доведения рН до 3-4. Добавляли 20 мл этилацетата, затем реакционную смесь высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(6-(1-метил-1Н-пиразол-4-ил)пиридин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 61 (10 мг, выход 41%) в виде желтого твердого вещества.
МС m/z (ИЭР): 561,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.73 (s, 1Н), 8.27 (s, 1Н), 8.19 (s, 2Н), 7.74-7.72 (d, 1Н), 7.59-7.55 (d 1Н), 7.48 (s 1Н), 7.39-7.37 (d 1Н), 7.24-7.22 (d 2Н), 6.88-6.86 (d, 1Н), 6.57-6.53 (d 1Н), 5.30 (s, 1Н), 3.99 (s, 3Н), 3.77 (s 1Н), 3.49-3.45 (m 1Н), 3.07-3.00 (m, 1Н), 2.77-2.74 (m, 1Н), 2.38-2.28 (m, 1Н), 1.16-0.92 (m, 9Н).
Пример 62
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
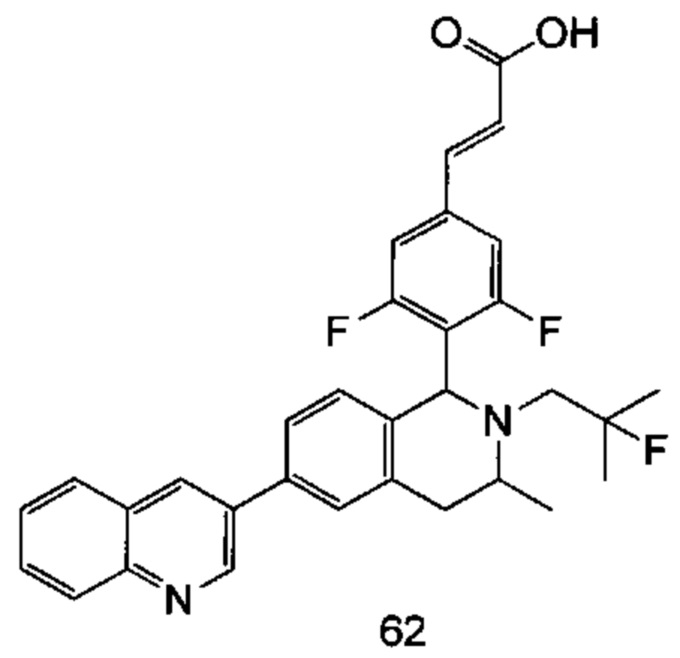
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хинолином, который был получен хорошо известным способом, раскрытым в публикации Organic & Biomolecular Chemistry, 2014, 12(37), 7318-7327, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-3-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 62.
МС m/z (ИЭР): 531,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 9.16 (s, 1Н), 8.59 (s, 1Н), 8.09-8.03 (m, 2Н), 7.80-7.78 (m 1Н), 7.68-7.53 (m 4Н), 7.26-7.23 (d, 2Н), 6.94-6.92 (d 1Н), 6.58-6.54 (d 1Н), 5.33 (s, 1Н), 3.79 (s 1Н), 3.52-3.49 (m 1Н), 3.09-3.01 (m, 1H), 2.82-2.79 (m, 1H), 2.40-2.29 (m, 1H), 1.18-0.90 (m, 9H).
Пример 63
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
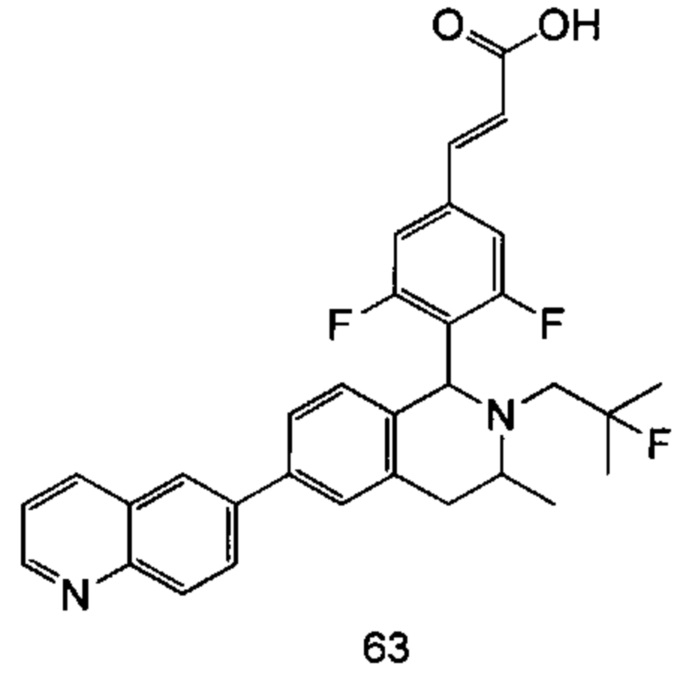
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хинолином, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 63.
МС m/z (ИЭР): 531,6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.85 (dd, 1Н), 8.45 (dd, 1Н), 8.19 (t, 1Н), 8.10 (d, 2Н), 7.62-7.55 (m, 3Н), 7.50 (dd, 1Н), 7.23 (d, 2Н), 6.88 (d, 1Н), 6.55 (d, 1Н), 5.33 (s, 1Н), 3.78 (s, 1Н), 3.49 (dd, 1Н), 3.05 (t, 1Н), 2.78 (dd, 1Н), 2.40-2.29 (t, 1Н), 1.21-1.12 (t, 6Н), 1.08 (t, 3Н).
Пример 64 и пример 65
(Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
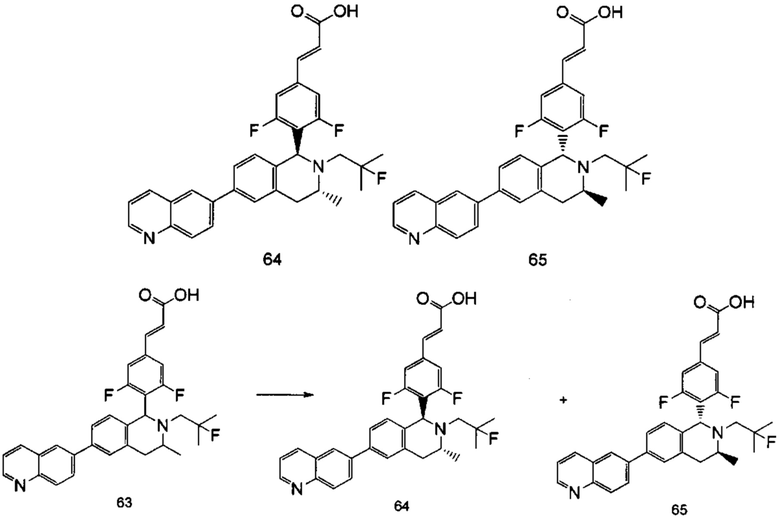
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловую кислоту 63 (290 мг, 0,547 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak AD-H, внутренний диаметр 0,46 см × длина 25 см; подвижная фаза: н-гексан: этанол: ТФУ составляет 70:30:0,1, скорость потока: 1,0 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 64 (210 мг) в виде белого твердого вещества и (Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(хинолин-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловой кислоты 65 (248 мг) в виде белого твердого вещества.
Пример 64:
МС m/z (ИЭР): 531,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 6,604 минут, хиральная чистота: 100% (хроматографическая колонка: Chiralpak IE, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/ТФУ составляет 70/30/0,1 (об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 9.16 (s, 1Н), 8.59 (s, 1Н), 8.09-8.03 (m, 2Н), 7.80-7.78 (m 1Н), 7.68-7.53 (m 4Н), 7.26-7.23 (d, 2Н), 6.94-6.92 (d 1Н), 6.58-6.54 (d 1Н), 5.33 (s, 1Н), 3.79 (s 1Н), 3.52-3.49 (m 1H), 3.09-3.01 (m, 1H), 2.82-2.79 (m, 1H), 2.40-2.29 (m, 1H), 1.18-0.90 (m, 9H).
Пример 65:
МС m/z (ИЭР): 531,5 [M+1];
Анализ хиральной ВЭЖХ: время удерживания 10,592 минут, хиральная чистота: 99,2% (хроматографическая колонка: Chiralpak IE, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан/этанол/ТФУ составляет 70/30/0,1(об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 9.16 (s, 1Н), 8.59 (s, 1Н), 8.09-8.03 (m, 2Н), 7.80-7.78 (m 1Н), 7.68-7.53 (m 4Н), 7.26-7.23 (d, 2Н), 6.94-6.92 (d 1Н), 6.58-6.54 (d 1Н), 5.33 (s, 1Н), 3.79 (s 1Н), 3.52-3.49 (m 1Н), 3.09-3.01 (m, 1Н), 2.82-2.79 (m, 1Н), 2.40-2.29 (m, 1Н), 1.18-0.90 (m, 9Н).
Пример 66
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилбензо[d]тиазол-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
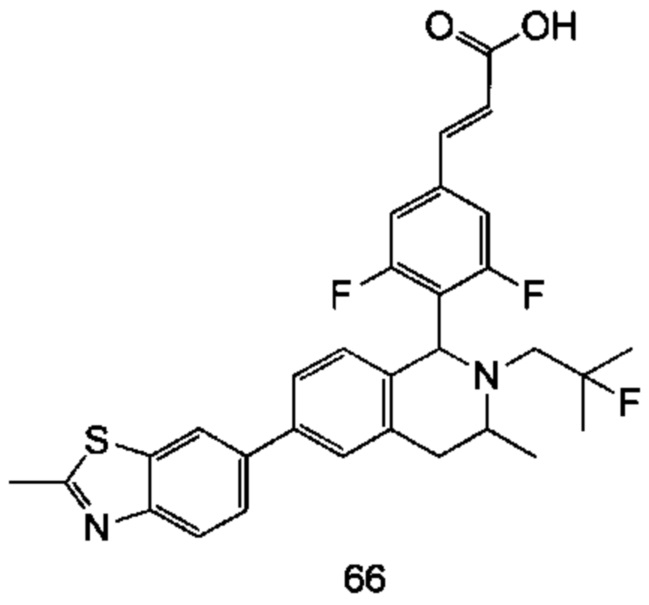
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 2-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил) бензо[d]тиазолом, который был получен хорошо известным способом, раскрытым в публикации Journal of the American Chemical Society, 2014, 136(11), 4287-4299, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(2-метилбензо[d]тиазол-6-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 66.
МС m/z (ИЭР): 551,0 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.13 (s, 1Н), 7.91 (d, 1Н), 7.73 (d, 1Н), 7.58 (d, 1Н), 7.46 (s, 1H), 7.36 (d, 1H), 7.20 (d, 2H), 6.82 (d, 1H), 6.53 (d, 1H), 5.29 (s, 1H), 3.75 (s, 1H), 3.45 (d, 1H), 3.03 (t, 1H), 2.86 (s, 3H), 2.73 (d, 1H), 2.38-2.27 (m, 1H), 1.20-1.11 (m, 6H), 1.05 (d, 3H).
Пример 67
(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1Н-индазол-5-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
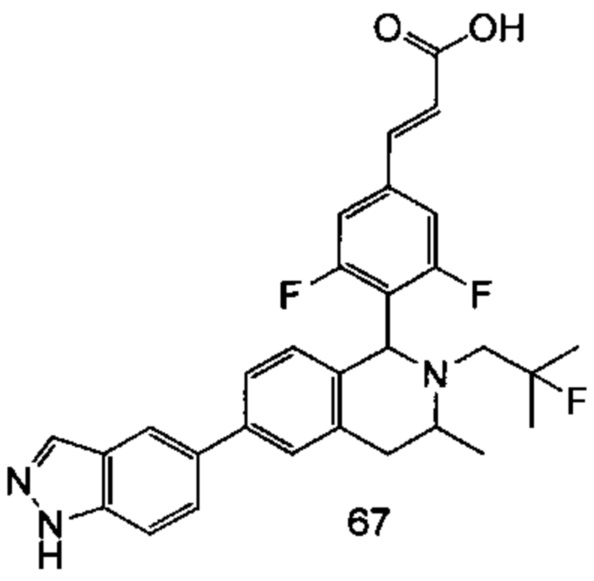
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индолом, который получен хорошо известным способом, раскрытым в публикации Organic Letters, 2012, 14(2), 600-603), соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-(1Н-индазол-5-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 67.
МС m/z (ИЭР): 520,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.10 (s, 1Н), 7.99 (s, 1Н), 7.71-7.09 (m, 1Н), 7.68-7.59 (m 2Н), 7.56 (s 1Н), 7.37-7.35 (d 2Н), 7.22-7.20 (d, 2Н), 6.82-6.80 (d 1Н), 6.57-6.53 (d 1Н), 5.29 (s, 1Н), 3.77 (s 1Н), 3.50-3.45 (m 1Н), 3.08-3.00 (m, 1Н), 2.76-2.72 (m, 1Н), 2.39-2.28 (m, 1Н), 1.20-1.06 (m, 9Н).
Пример 68 и пример 69
(Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота

(Е)-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловую кислоту 4 (1,1 г, 2,48 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak IF, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: н-гексан: этанол: ДЭА составляет 95:5:0,1, скорость потока: 1,0 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловой кислоты 68 (414 мг) в виде желтого твердого вещества и (Е)-3-(3,5-дифтор-4-((1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловой кислоты 69 (562,9 мг) в виде желтого твердого вещества.
Пример 68:
МС m/z (ИЭР): 444,2 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 6,878 минут, хиральная чистота: 99,52% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 25 см; подвижная фаза: н-гексан/метанол/этанол/ТФУ составляет 90/5/5/0,01 (об./об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 7.61 (d, 1Н), 7.53-7.51 (m, 1Н), 7.34-7.21 (m, 5Н), 6.57 (d, 1Н), 5.28 (s, 1Н), 3.73-3.71 (m, 1Н), 3.03-2.94 (m, 2Н), 2.64-2.59 (m, 1Н), 2.49-2.39 (m, 1Н), 1.31-1.14 (m, 9Н).
Пример 69:
МС m/z (ИЭР): 444,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 5,320 минут, хиральная чистота: 98,99% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 25 см, подвижная фаза: н-гексан/метанол/этанол/ТФУ составляет 90/5/5/0,01 (об./об./об./об.));
1Н ЯМР (400 МГц, CD3OD) δ 7.61 (d, 1Н), 7.53-7.51 (m, 1Н), 7.34-7.21 (m, 5Н), 6.57 (d, 1Н), 5.28 (s, 1Н), 3.73-3.71 (m, 1Н), 3.03-2.94 (m, 2Н), 2.64-2.59 (m, 1Н), 2.49-2.39 (m, 1Н), 1.31-1.14 (m, 9Н).
Пример 70
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3Н-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота
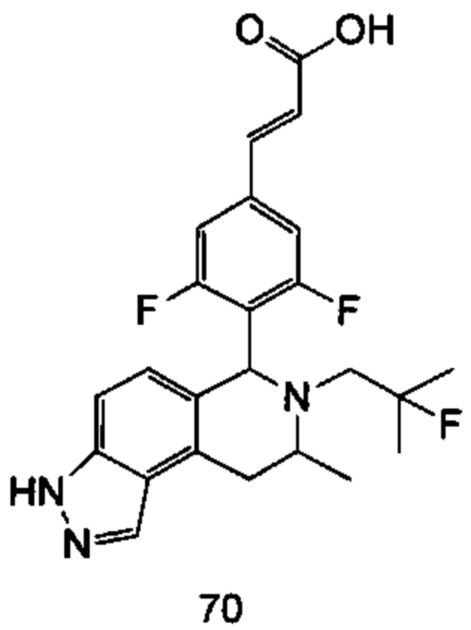
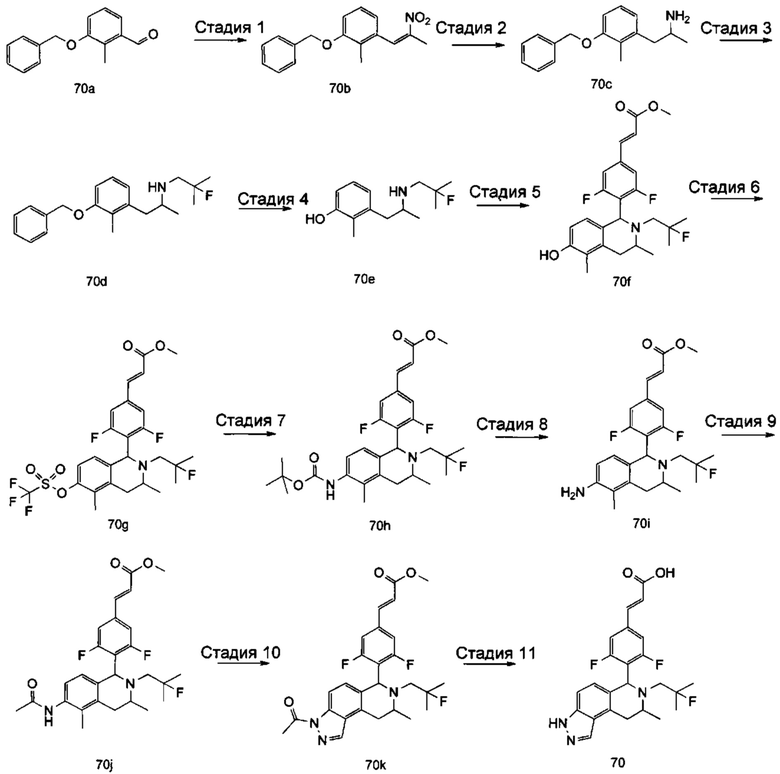
Стадия 1
1-(Бензилокси)-2-метил-3-(2-нитропроп-1-ен-1-ил)бензол
3-(Бензилокси)-2-метилбензальдегид 70а (5 г, 22 ммоль, полученный способом, раскрытым в заявке на патент WO 2008080015), и ацетат аммония (2 г, 26,5 ммоль) смешивали в 100 мл нитроэтана. Реакционную смесь подогревали до 80°С и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры сконцентрировали при пониженном давлении. К остатку добавляли воду, и реакционный раствор экстрагировали дихлорметаном три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, 1-(бензилокси)-2-метил-3-(2-нитропроп-1-ен-1-ил)бензола 70b (5,35 г, выход 85%) в виде желтого масла.
Стадия 2
1-(3-(Бензилокси)-2-метилфенил)пропан-2-амин
1-(Бензилокси)-2-метил-3-(2-нитропроп-1-ен-1-ил)бензол 70b (5,35 г, 18,9 ммоль) растворяли в 100 мл тетрагидрофурана, затем добавляли алюмогидрид лития (1,51 г, 37,8 ммоль) при 0°С. Реакционную смесь подогревали до 50°С и перемешивали в течение 12 часов, затем реакцию останавливали. После охлаждения до комнатной температуры добавляли сульфат натрия декагидрат для гашения реакционной смеси. Реакционный раствор фильтровали, и фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, 1-(3-(бензилокси)-2-метилфенил)пропан-2-амина 70с (4,78 г), в виде светло-желтого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
N-(1-(3-(бензилокси)-2-метилфенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин
Неочищенный 1-(3-(бензилокси)-2-метилфенил)пропан-2-амин 70с (4,78 г, 18,7 ммоль), 2-фтор-2-метилпропилтрифторметансульфонат 1b (8,4 г, 37,4 ммоль) и N,N-диизопропилэтиламин (7,24 г, 56,1 ммоль) растворяли в 80 мл 1,4-диоксана. Реакционную смесь нагревали до 100°С и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, N-(1-(3-(бензилокси)-2-метилфенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина 70d (1,25 г, выход 20%) в виде желто-коричневого масла.
Стадия 4
3-(2-((2-Фтор-2-метилпропил)амино)пропил)-2-метилфенол
N-(1-(3-(бензилокси)-2-метилфенил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 70d (1,2 г, 3,64 ммоль) растворяли в 50 мл метанола, затем добавляли формиат аммония (4,5 г, 72,8 ммоль) и Pd/C (300 мг, 10%). Реакционную смесь подогревали до образования флегмы и перемешивали в течение 5 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции А с получением соединения, указанного в заголовке, 3-(2-((2-фтор-2-метилпропил)амино)пропил)-2-метилфенола 70е (740 мг, выход 91%) в виде желтого масла.
Стадия 5
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
3-(2-((2-фтор-2-метилпропил)амино)пропил)-2-метилфенол 70е (740 мг, 3,1 ммоль) растворяли в 30 мл метанола, затем добавляли (Е)-метил 3-(3,5-дифтор-4-формилфенил)акрилат 1е (1,05 г, 4,6 ммоль) и уксусную кислоту (560 мг, 9,3 ммоль). Реакционную смесь подогревали до 90°С и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-6-гидрокси-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 70f (900 мг, выход 65%) в виде желтого твердого вещества.
Стадия 6
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3,5-диметил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-6-гидрокси-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 70f (900 мг, 2 ммоль) растворяли в 50 мл дихлорметана, затем добавляли 2,6-диметилпиридин (428 мг, 4 ммоль) и трифторметансульфоновый ангидрид (850 мг, 3 ммоль) при 0°С. Реакционную смесь перемешивали в течение 1,5 часов при 0°С, затем реакцию останавливали. Реакционный раствор подогревали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3,5-диметил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 70g (955 мг, выход 82%) в виде бесцветного масла.
Стадия 7
(Е)-метил-3-(4-((1S,3R/1R,3)-6-((трет-бутоксикарбонил)амино)-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3,5-диметил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 70g (250 мг, 0,43 ммоль), трет-бутилкарбамат (76 мг, 0,65 ммоль), трис(дибензилиденацетон)дипалладий (79 мг, 0,086 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (100 мг, 0,172 ммоль), карбонат цезия (280 мг, 0,86 ммоль) и 3 мл 1,4-диоксана помещали в пробирку для микроволнового реактора. Смесь подогревали до 125°С и перемешивали в микроволновом реакторе в течение 55 минут, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-((трет-бутоксикарбонил)амино)-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 70h (117 мг, выход 50%) в виде желтого масла.
Стадия 8
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-амино-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-((трет-бутоксикарбонил)амино)-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 70h (50 мг, 0,0915 ммоль) растворяли в 10 мл дихлорметана, затем добавляли 5 мл ТФУ. После перемешивания в течение 2 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении. Остаток растворяли в дихлорметане. Добавляли насыщенный раствор карбоната натрия, пока рН этой системы не стал щелочным. Реакционный раствор экстрагировали дихлорметаном три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-амино-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 70i (35 мг), в виде коричневого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 9
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-ацетамидо-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-амино-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 70i (70 мг, 0,16 ммоль) растворяли в 10 мл дихлорметана, затем добавляли уксусный ангидрид (80 мг, 0,78 ммоль) и N,N-диизопропилэтиламин (100 мг, 0,78 ммоль). После перемешивания в течение 12 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-ацетамидо-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 70j (50 мг, выход 64%) в виде желтого твердого вещества.
Стадия 10
(Е)-метил-3-(4-((6S,8R/6R,8S)-3-ацетил-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3Н-пиразоло[4,3-f]изохинолин-6-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-ацетамидо-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 70j (10 мг, 0,02 ммоль) растворяли в 1,5 мл этилацетата, затем добавляли уксусный ангидрид (6 мг, 0,06 ммоль), тетрабутиламмония бромид (0,6 мг, 0,002 ммоль), ацетат калия (5 мг, 0,05 ммоль) и 3-метил-1-нитробутан (5 мг, 0,04 ммоль). После перемешивания в течение 48 часов реакцию останавливали. Реакционный раствор концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((6S,8R/6R,8S)-3-ацетил-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3Н-пиразоло[4,3-f]изохинолин-6-ил)-3,5-дифторфенил)акрилата 70k (7 мг, выход 70%), в виде желтого твердого вещества.
Стадия 11
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3Н-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота
(Е)-метил-3-(4-((6S,8R/6R,8S)-3-ацетил-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3Н-пиразоло[4,3-f]изохинолин-6-ил)-3)5-дифторфенил)акрилат 70k (5 мг, 0,01 ммоль) растворяли в 10 мл метанола, затем добавляли 5 мл 0,2 М раствора гидроксида натрия. После перемешивания в течение 2 часов реакцию останавливали. Добавляли 10% раствор лимонной кислоты, пока рН реакционного раствора не становился кислым. Смесь экстрагировали дихлорметаном три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3Н-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловой кислоты 70 (3 мг, выход 68%), в виде желтого твердого вещества.
МС m/z (ИЭР): 444,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 8.10 (s, 1Н), 7.58 (d, 1Н), 7.25-7.19 (m, 2Н), 6.76 (d, 1Н), 6.54 (d, 1Н), 5.35 (s, 1Н), 3.82 (s, 1Н), 3.50-3.45 (m, 1Н), 3.08-2.99 (m, 2Н), 2.42-2.35 (m, 1Н), 2.06 (s, 1Н), 1.22-1.07 (m, 9Н).
Пример 71 и пример 72
(Е)-3-(3,5-дифтор-4-((5S,7R)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-g]изохинолин-5-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловая кислота
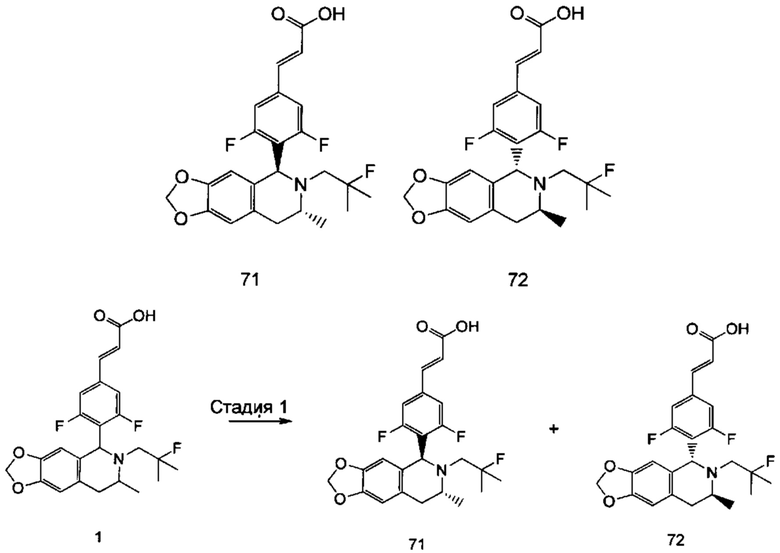
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловую кислоту 1 (595 мг, 1.33 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak AD, внутренний диаметр 5,0 см × длина 25 см; подвижная фаза: метанол/CO2/ТФУ составляет 20:80:0,1, скорость потока: 60 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((5S,7R)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловой кислоты 71 (278 мг) в виде желтого твердого вещества и (Е)-3-(3,5-дифтор-4-((5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-г]изохинолин-5-ил)фенил)акриловой кислоты 72 (249 мг) в виде желтого твердого вещества.
Пример 71:
МС m/z (ИЭР): 448,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 3,307 минут, хиральная чистота: 96,79% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 15 см, подвижная фаза:  составляет 20:80:0,1 (об./об./об.));
составляет 20:80:0,1 (об./об./об.));
1Н ЯМР (400 МГц, CDCl3) δ 7.55 (d, 1Н), 7.07 (s, 2Н), 6.69 (s, 1Н), 6.42-6.36 (m, 2Н), 6.09 (s, 1Н), 5.97 (s, 2Н), 4.35 (s, 1Н), 3.37-3.25 (m, 3Н), 2.93-2.83 (m, 1Н), 1.73 (d, 3Н), 1.51 (m, 6Н).
Пример 72:
МС m/z (ИЭР): 448,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 2,173 минут, хиральная чистота: 99,37% (хроматографическая колонка: Chiralpak AD-H, внутренний диаметр 0,46 см × длина 15 см, подвижная фаза: составляет 20:80:0,1 (об./об./об.));
1Н ЯМР (400 МГц, CDCl3) δ 7.54 (d, 1Н), 7.06 (d, 2Н), 6.67 (s, 1Н), 6.39 (d, 1Н), 6.33 (s, 1Н), 5.97-5.95 (m, 3Н), 4.25 (s, 1Н), 3.39-3.27 (m, 2Н), 3.11 (t, 1Н), 2.79-2.72 (m, 1Н), 1.66 (d, 3Н), 1.48-1.43 (m, 6Н).
Пример 73
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
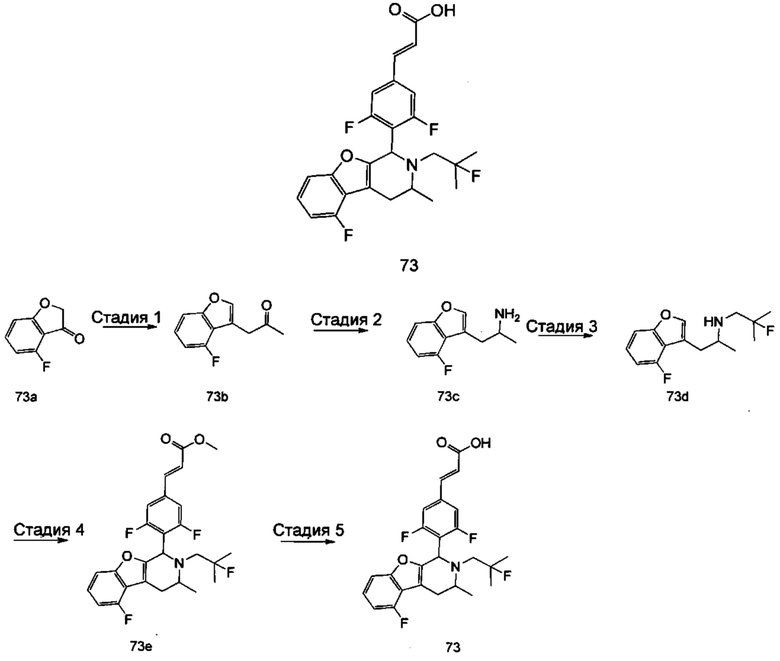
Стадия 1
1-(4-Фторбензофуран-3-ил)пропан-2-он
4-Фторбензофуран-3(2Н)-он 73а (500 мг, 3,29 ммоль, получен хорошо известным способом, раскрытым в публикации Journal of Medicinal Chemistry, 2011, 54(15), 5395-5402), ацетонилтрифенилфосфония хлорид (1,754 г, 4,93 ммоль) и N,N-диизопропилэтиламин (1,27 г, 9,87 ммоль) растворяли в 15 мл диметилбензола. Реакционную смесь подогревали до 140°С и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции В с получением соединения, указанного в заголовке, 1-(4-фторбензофуран-3-ил)пропан-2-она 73b (350 мг, выход 55%) в виде желтого твердого вещества.
Стадия 2
1-(4-Фторбензофуран-3-ил)пропан-2-амин
1-(4-Фторбензофуран-3-ил)пропан-2-он 73b (300 мг, 1,56 ммоль), ацетат аммония (1,2 г, 15,6 ммоль) и ацетат натрия (128 мг, 1,56 ммоль) растворяли в 10 мл метанола, затем добавляли цианоборгидрид натрия (147 мг, 2,34 ммоль) и добавляли по каплям 0,5 мл уксусной кислоты. После перемешивания в течение 12 часов реакцию останавливали. Добавляли воду, и реакционный раствор экстрагировали дихлорметаном три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией на препаративном хроматографе CombiFlash с системой элюции А с получением соединения, указанного в заголовке, 1-(4-фторбензофуран-3-ил)пропан-2-амина 73с (300 мг, выход 66,7%) в виде желтого масла.
Стадия 3
2-Фтор-N-(1-(4-фторбензофуран-3-ил)пропан-2-ил)-2-метилпропан-1-амин
1-(4-Фторбензофуран-3-ил)пропан-2-амин 73с (200 мг, 1 ммоль), 2-фтор-2-метилпропилтрифторметансульфонат 1b (448 мг, 2 ммоль) и N,N-диизопропилэтиламин (384 мг, 3 ммоль) растворяли в 10 мл 1,4-диоксана. Реакционный раствор подогревали до 90°С и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, 2-фтор-N-(1-(4-фторбензофуран-3-ил)пропан-2-ил)-2-метилпропан-1-амина 73d (200 мг, выход 74%) в виде желтого масла.
Стадия 4
(Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акрилат
2-Фтор-N-(1-(4-фторбензофуран-3-ил)пропан-2-ил)-2-метилпропан-1-амин 73d (200 мг, 0,75 ммоль), (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е (254 мг, 1,12 ммоль) и триизопропилсилилхлорид (723 мг, 3,75 ммоль) растворяли в 5 мл N,N-диметилформамида и помещали в герметичную пробирку. Реакционную смесь нагревали до 130°С и подвергали взаимодействию в течение 3 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил) акрилата 73е (300 мг, выход 84%), в виде желтого масла.
Стадия 5
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1R,3R/1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акрилат 73е (300 мг, 0,63 ммоль) растворяли в 20 мл метанола, затем добавляли 10 мл 0,6 М раствора гидроксида натрия. После перемешивания в течение 12 часов реакцию останавливали. Добавляли 10% раствор лимонной кислоты, пока рН реакционного раствора не становился кислым. Смесь экстрагировали дихлорметаном три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали тонкослойной хроматографией с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловой кислоты 73 (200 мг, выход 69%), в виде желтого твердого вещества.
МС m/z (ИЭР): 462,4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.62 (d, 1Н), 7.33-7.22 (m, 4Н), 7.00-6.95 (m, 1Н), 6.60 (d, 1Н), 5.50 (s, 1Н), 3.79 (d, 1Н), 3.23-3.12 (m, 2Н), 2.88-2.83 (m, 1Н), 2.68 (s, 1Н), 1.40-1.24 (m, 9Н).
Пример 74 и пример 75
(Е)-3-(3,5-дифтор-4-((1R,3R)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловую кислоту 73 (410 мг, 0,89 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak IF, внутренний диаметр 5,0 см × длина × 25 см; подвижная фаза: н-гексан:этанол:ТФУ составляет 95:5:0,1, скорость потока: 60 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3R)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловой кислоты 74 (110 мг) в виде желтого твердого вещества и (Е)-3-(3,5-дифтор-4-((1S,3S)-5-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловой кислоты 75 (200 мг) в виде желтого твердого вещества.
Пример 74:
МС m/z (ИЭР): 462,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 8,381 минут, хиральная чистота: 99,54% (хроматографическая колонка: Chiralpak IF, внутренний диаметр 0,46 см × длина 15 см, подвижная фаза: н-гексан:этанол:ТФУ составляет 95:5:0,1 (об./об./об.));
1H ЯМР (400 МГц, CD3OD) δ 7.62 (d, 1H), 7.33-7.22 (m, 4H), 7.00-6.95 (m, 1H), 6.60 (d, 1H), 5.50 (s, 1H), 3.79 (d, 1H), 3.23-3.12 (m, 2H), 2.88-2.83 (m, 1H), 2.68 (s, 1H), 1.40-1.24 (m, 9H).
Пример 75
MC m/z (ИЭР): 462,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 5,321 минут, хиральная чистота: 99,56% (хроматографическая колонка: Chiralpak IF, внутренний диаметр 0,46 см × длина 15 см, подвижная фаза: н-гексан : этанол : ТФУ составляет 95:5:0,1 (об./об./об.));
1H ЯМР (400 МГц, CD3OD) δ 7.62 (d, 1H), 7.33-7.22 (m, 4H), 7.00-6.95 (m, 1H), 6.60 (d, 1H), 5.50 (s, 1H), 3.79 (d, 1H), 3.23-3.12 (m, 2H), 2.88-2.83 (m, 1H), 2.68 (s, 1H), 1.40-1.24 (m, 9H).
Пример 76
(E)-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота
Стадия 1
1-(5-Бромбензофуран-3-ил)пропан-2-он
5-Бромбензофуран-3(2Н)-он 76а (5 г, 23,471 ммоль, получен способом, раскрытым в заявке на патент 2008068974), ацетонилтрифенилфосфония хлорид (12,5 г, 35,206 ммоль) и N,N-диизопропилэтиламин (9,1 г, 70,413 ммоль) растворяли в 60 мл диметилбензола. Реакционную смесь нагревали до 120°C и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, 1-(5-бромбензофуран-3-ил)пропан-2-она 76b (3,1 г, выход 53%) в виде желтого масла.
Стадия 2
1-(5-Бромбензофуран-3-ил)пропан-2-амин
1-(5-Бромбензофуран-3-ил)пропан-2-он 76b (3,1 г, 12,249 ммоль), ацетат аммония (9,4 г, 122,486 ммоль) и ацетат натрия (1 г, 12,249 ммоль) растворяли в 10 мл метанола, затем добавляли цианоборгидрид натрия (1,15 г, 18,374 ммоль) и добавляли по каплям 30 капель уксусной кислоты. После перемешивания в течение 3 часов реакцию останавливали. Добавляли воду, и реакционный раствор экстрагировали этилацетатом три раза. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, 1-(5-бромбензофуран-3-ил)пропан-2-амина 76с (1,2 г, выход 39%) в виде желтого масла.
Стадия 3
N-(1-(5-Бромбензофуран-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин
1-(5-Бромбензофуран-3-ил)пропан-2-амин 76с (1,2 г, 4,722 ммоль), 2-фтор-2-метилпропилтрифторметансульфонат 1b (1,59 г, 7,083 ммоль) и N,N-диизопропилэтиламин (1,83 г, 14,166 ммоль) растворяли в 25 мл 1,4-диоксана. Реакционную смесь нагревали до 90°C и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, N-(1-(5-бромбензофуран-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина 76d (1,3 г, выход 84%) в виде желтого масла.
Стадия 4
(Е)-метил-3-(4-((1R,3R/1S,3S)-6-бром-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акрилат
N-(1-(5-бромбензофуран-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 76d (1,3 г, 3,961 ммоль), (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е (1,34 г, 5,941 ммоль) и триизопропилсилилхлорид (3,8 г, 19,805 ммоль) растворяли в 6 мл N,N-диметилформамида и помещали в герметичную пробирку. Реакционную смесь нагревали до 120°C и подвергали взаимодействию в течение 3 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и добавляли этилацетат. Реакционный раствор экстрагировали этилацетатом, и органическую фазу концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1R,3R/1S,3S)-6-бром-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акрилата 76е (460 мг, выход 22%), в виде желтого твердого вещества.
Стадия 5
(Е)-метил-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(4-((1R,3R/1S,3S)-6-бром-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акрилат 76е (200 мг, 0,373 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 17а (166 мг, 0,746 ммоль) и карбонат калия (154 мг, 1,119 ммоль) растворяли в 6 мл смеси 1,4-диоксана и воды (об./об. составляет 5:1), затем добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (27 мг, 0,037 ммоль). Смесь подогревали до 85°C и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акрилата 76f (110 мг, выход 53%), в виде желтого масла.
МС m/z (ИЭР): 551,9 [М+1].
Стадия 6
(E)-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-метил-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акрилат 76f (110 мг, 0,199 ммоль) растворяли в 5 мл метанола, затем добавляли 1 мл 2 М гидроксида натрия. Реакционную смесь подогревали до 40°C и перемешивали в течение 1 часа при 40°C, затем реакцию останавливали. Реакционный раствор охлаждали до комнатной температуры, затем добавляли 1 М раствор лимонной кислоты до доведения рН до 5-6. Реакционный раствор экстрагировали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с получением соединения, указанного в заголовке, (E)-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловой кислоты 76 (100 мг, выход 93%), в виде желтого твердого вещества.
МС m/z (ИЭР): 535,8 [М-1]
1H ЯМР (400 МГц, CD3OD) δ 8.04 (s, 1H), 7.87 (s, 1H), 7.69 (s, 1H), 7.62 (d, 1H), 7.49 (d, 1H), 7.39-7.32 (m, 3Н), 6.61 (d, 1H), 5.62 (s, 1H), 4.30-4.22 (m, 2Н), 3.88 (s, 1H), 3.27-3.13 (m, 2Н), 2.82-2.79 (m, 2Н), 1.52 (t, 3Н), 1.39-1.31 (m, 9Н).
Пример 77
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2-оксо-6,7,8,9-тетрагидро-2Н-пирано[2,3-г]изохинолин-6-ил)фенил)акриловая кислота
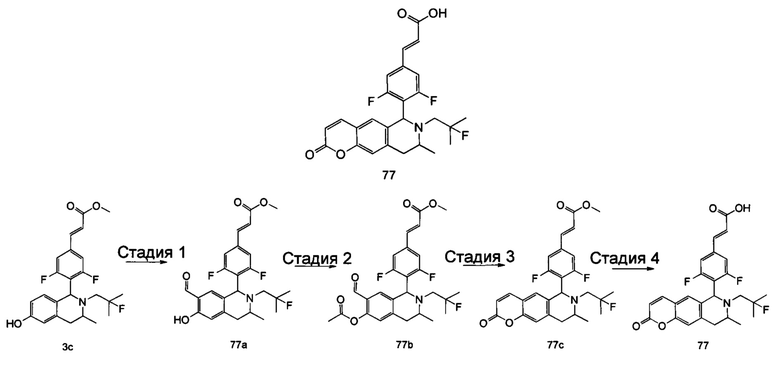
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-7-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 3с (1 г, 2,3 ммоль), параформальдегид (692,5 мг, 2,3 ммоль), хлорид магния (658 мг, 6,93 ммоль) и триэтиламин (1,168 г, 11,5 ммоль) растворяли в 33 мл тетрагидрофурана. Реакционную смесь подогревали до образования флегмы в течение 4 часов. После охлаждения до комнатной температуры добавляли ледяную воду для гашения реакционной смеси. Реакционную смесь экстрагировали дихлорметаном (50 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-7-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 77а (1 г) в виде светло-желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-ацетокси-2-(2-фтор-2-метилпропил)-7-формил-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-7-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 77а (300 мг, 0.65 ммоль) и ацетат натрия (160 мг, 1,95 ммоль) растворяли в 9 мл уксусного ангидрида. Реакционную смесь подогревали до 150°C и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-ацетокси-2-(2-фтор-2-метилпропил)-7-формил-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 77b (150 мг, выход 47,5%), в виде бесцветного масла.
Стадия 3
(Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2-оксо-6,7,8,9-тетрагидро-2Н-пирано[2,3-г]изохинолин-6-ил)фенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-ацетокси-2-(2-фтор-2-метилпропил)-7-формил-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 77b (150 мг, 0,3 ммоль) и ацетат натрия (150 мг, 1,8 ммоль) смешивали, подогревали до 180°C и перемешивали в течение 8 часов. Реакционный раствор охлаждали до комнатной температуры. Остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2-оксо-6,7,8,9-тетрагидро-2Н-пирано[2,3-г]изохинолин-6-ил)фенил)акрилата 77с (70 мг, выход 48,1%) в виде бесцветного масла.
Стадия 4
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2-оксо-6,7,8,9-тетрагидро-2Н-пирано[2,3-г]изохинолин-6-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2-оксо-6,7,8,9-тетрагидро-2Н-пирано[2,3-г]изохинолин-6-ил)фенил)акрилат 77с (60 мг, 0,124 ммоль) растворяли в 2,4 мл смеси тетрагидрофурана и метанола (об./об. составляет 3:1). Реакционную смесь охлаждали до 0°C и добавляли 0,62 мл 1 М раствора гидроксида лития. Реакционную смесь естественным путем подогревали до комнатной температуры и перемешивали в течение 0,5 часа. Реакционный раствор концентрировали при пониженном давлении для удаления метанола и тетрагидрофурана. Добавляли 0,5 М HCl до доведения рН до 5, и смесь экстрагировали этилацетатом (5 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали высокоэффективной жидкостной хроматографией с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-2-оксо-6,7,8,9-тетрагидро-2Н-пирано[2,3-г]изохинолин-6-ил)фенил)акриловой кислоты 77 (30 мг, выход 51,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 472,4 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.79 (d, 1H), 7.61 (dd, 1H), 7.29 (d, 2H), 7.23 (s, 1H), 7.16 (s, 1H), 6.58 (d, 1H), 6.35 (d, 1H), 5.54 (s, 1H), 3.84 (s, 1H), 3.47 (dd, 1H), 3.29-3.15 (m, 1H), 2.91 (dd, 2H), 1.30-1.16 (m, 9Н).
Пример 78 и пример 79
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидроизоксазоло[4,5-f]изохинолин-6-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидроизоксазоло[5,4-g]изохинолин-5-ил)фенил)акриловая кислота
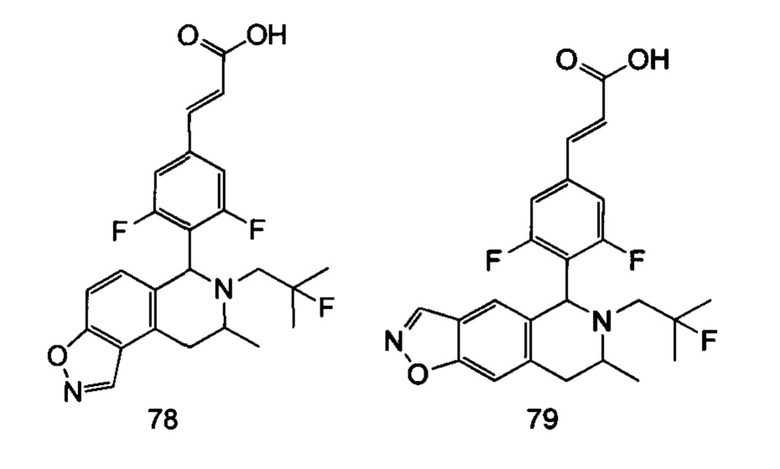
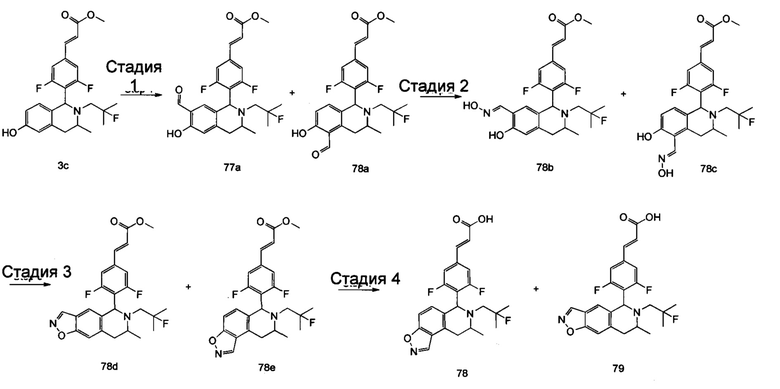
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-7-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-5-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 3с (150 мг, 0,346 ммоль), параформальдегид (104 мг, 3,464 ммоль) и хлорид магния (99 мг, 1,03 ммоль) растворяли в 10 мл тетрагидрофурана. Реакционную смесь подогревали до 65°C и перемешивали в течение 12 часов. После охлаждения до комнатной температуры добавляли ледяную воду для гашения реакционной смеси. Реакционную смесь экстрагировали дихлорметаном (50 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенных соединений, указанных в заголовке: смесь (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-7-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 77а и (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-5-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 78а (150 мг) в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-7-((гидроксиимино)метил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-6-гидрокси-5-((гидроксиимино)метил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
Неочищенную смесь (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-7-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акрилата 77а и (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-5-формил-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акрилата 78а (150 мг, 0,325 ммоль) и гидроксиламина гидрохлорида (27 мг, 0,390 ммоль) растворяли в 4 мл пиридина, затем смесь перемешивали в течение 2,5 часов при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении, добавляли этилацетат, промывали насыщенным раствором карбоната натрия и водой, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенных соединений, указанных в заголовке: смесь (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-7-((гидроксиимино)метил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 78b и (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-6-гидрокси-5-((гидроксиимино)метил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 78с (120 мг) в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
(Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидроизоксазоло[5,4-g]изохинолин-5-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидроизоксазоло[4,5-f]изохинолин-6-ил)фенил)акрилат
Смесь (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-7-((гидроксиимино)метил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 78b и (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-6-гидрокси-5-((гидроксиимино) метил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 78с (120 мг, 0,252 ммоль) и трифенилфосфина (142 мг, 0,504 ммоль) растворяли в 10 мл тетрагидрофурана, затем медленно добавляли диэтилазодикарбоксилат (88 мг, 0,504 ммоль). Реакционный раствор подогревали до 40°C и перемешивали в течение 1 часа, затем концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединений, указанных в заголовке: смесь (Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидроизоксазоло[5,4-g]изохинолин-5-ил)фенил)акрилата 78d и (Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидроизоксазоло[4,5-f]изохинолин-6-ил)фенил)акрилата 78е (40 мг, выход 35%) в виде прозрачного масла.
Стадия 4
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидроизоксазоло[4,5-f]изохинолин-6-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидроизоксазоло[5,4-g]изохинолин-5-ил)фенил)акриловая кислота
Смесь (Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидроизоксазоло[5,4-g]изохинолин-5-ил)фенил) акрилата 78d и (Е)-метил-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидроизоксазоло[4,5-f]изохинолин-6-ил)фенил) акрилата 78е (40 мг, 0,087 ммоль) растворяли в 5 мл метанола, затем добавляли гидроксид натрия (35 мг, 0,873 ммоль). Реакционную смесь подогревали до 30°C и перемешивали в течение 1 часа. Добавляли 1 М HCl до доведения рН до 5, и смесь экстрагировали этилацетатом. Органические фазы высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографии на силикагеле с системой элюции А с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидроизоксазоло[4,5-f]изохинолин-6-ил)фенил)акриловой кислоты 78 (7,9 мг) в виде белого твержого вещества и (Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидроизоксазоло[5,4-g]изохинолин-5-ил)фенил)акриловой кислоты 79 (7,8 мг) в виде белого твердого вещества, и общий выход составляет 40,3%.
Пример 78
МС m/z (ИЭР): 445,2 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.60 (d, 1H), 7.31 (d, 2H), 7.00 (s, 1H), 6.77 (d, 1H), 6.59 (d, 1H), 5.58 (s, 1H), 3.87 (s, 1H), 3.50-3.36 (m, 1H), 3.00-2.96 (m, 1H), 2.85-2.42 (m, 2H), 1.36-1.25 (m, 9H).
Пример 79
МС m/z (ИЭР): 445,2 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.67 (s, 1H), 7.62 (d, 1H), 7.31 (d, 2H), 7.11 (s, 1H), 6.82 (d, 1H), 6.60 (d, 1H), 5.62 (s, 1H), 3.85 (s, 1H), 3.36-3.38 (m, 1H), 2.69-3.88 (m, 3H), 1.37-1.23 (m, 9H).
Пример 80
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-2,7-диметил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акриловая кислота
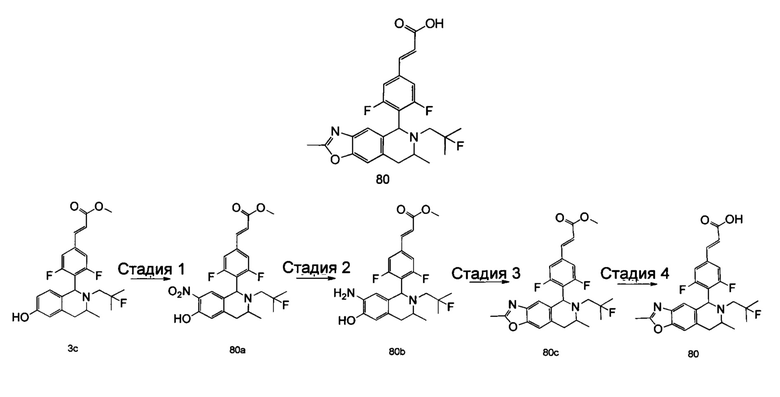
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-7-нитро-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 3с (1,3 г, 3 ммоль) растворяли в 13 мл уксусной кислоты, затем добавляли азотную кислоту (189 мг, 3,6 ммоль). Реакционную смесь перемешивали в течение 30 часов при комнатной температуре. В реакционный раствор добавляли ледяную воду и добавляли аммиак, пока рН не становился щелочным. Реакционный раствор экстрагировали дихлорметаном. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-7-нитро-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 80а (460 мг, выход 32%) в виде желтого масла.
Стадия 2
(Е)-метил-3-(4-((1S,3R/1R,3S)-7-амино-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-7-нитро-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 80а (460 мг, 0,961 ммоль) растворяли в 15 мл дихлорметана, затем добавляли тетрахлорид олова (365 мг, 1,923 ммоль). Реакционный раствор подогревали до 50°C и перемешивали в течение 2 часов при 50°C, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Добавляли 10 мл этанола, а затем одну каплю концентрированной HCl и порошок Fe (161 мг, 2,883 ммоль). Реакционный раствор подогревали до 80°C и перемешивали в течение 2 часов при 50°C, затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-7-амино-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 80b (400 мг), в виде черного твердого вещества, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 3
(Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-2,7-диметил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-7-амино-2-(2-фтор-2-метилпропил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 80b (100 мг, 0,223 ммоль), триметилортоацетат (80 мг, 0,669 ммоль) и пиридиния пара-толуолсульфонат (6 мг, 0,022 ммоль) растворяли в 3 мл N,N-диметилформамида. Реакционный раствор подогревали до 80°C и перемешивали в течение 1 часа при 80°C, затем охлаждали до комнатной температуры, добавляли воду и экстрагировали этилацетатом. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали тонкослойной хроматографией с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-2,7-диметил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акрилата 80с (20 мг, выход 19%) в виде желтого твердого вещества.
Стадия 4
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-2,7-диметил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-2,7-диметил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акрилат 80с (20 мг, 0,042 ммоль) растворяли в 5 мл метанола, затем добавляли гидроксид натрия (17 мг, 0,423 ммоль). Реакционный раствор подогревали до 30°C, и реакционную смесь перемешивали в течение 1 часа при 30°C. Добавляли 1 М HCl до доведения рН до 5. Реакционный раствор экстрагировали этилацетатом, и органическую фазу высушивали над безводным сульфатом натрия и концентрировали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-2,7-диметил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акриловой кислоты 80 (4 мг, выход 20%), в виде белого твердого вещества.
МС m/z (ИЭР): 459,2 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.34 (s, 1H), 7.27 (d, 1H), 7.10 (d, 2H), 6.96 (s, 1H), 6.52 (d, 1H), 5.34 (s, 1H), 3.73 (s, 1H), 3.49-3.47 (m, 1H), 3.01 (t, 1H), 2.77 (d, 1H), 2.60 (s, 3Н), 2.34-2.27 (m, 1H), 1.15 (t, 6H), 1.04 (d, 3H).
Пример 81
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акриловая кислота
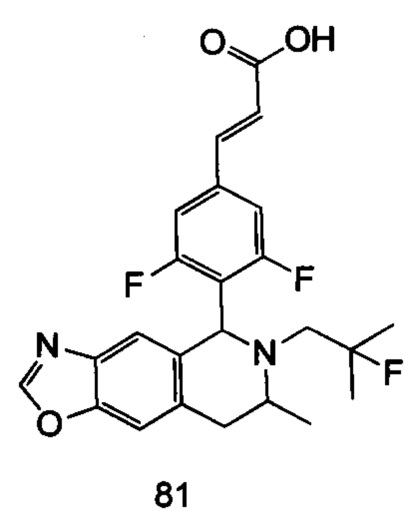
В соответствии с путем синтеза примера 80 исходное вещество триметилортоацетат на стадии 3 было заменено триметилортоформиатом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидрооксазоло[5,4-g]изохинолин-5-ил)фенил)акриловая кислота 81.
МС m/z (ИЭР): 445,2 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 8.35 (s, 1H), 7.46 (s, 1H), 7.32 (d, 1H), 7.15-7.12 (m, 3Н), 6.53 (d, 1H), 5.39 (s, 1H), 3.75 (s, 1H), 3.53-3.51 (m, 1H), 3.23-3.21 (m, 1H), 3.02 (t, 1H), 2.83 (d, 1H), 1.21 (t, 6H), 1.15 (d, 3H).
Пример 82
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-[1,3]диоксоло[4,5-f]изохинолин-6-ил)фенил)акриловая кислота
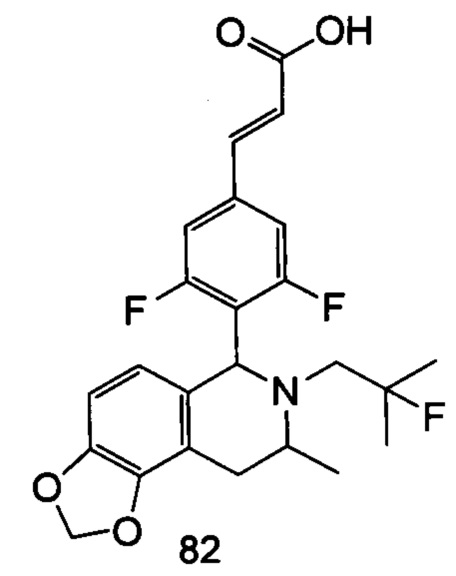
В соответствии с путем синтеза примера 35 исходное вещество (Е)-метил-3-(4-формилфенил)акрилат 35а, используемое на стадии 1, было заменено (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилатом 1е, а исходное вещество диметилсульфат, используемое на стадии 2, было заменено дибромметаном, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-[1,3]диоксоло[4,5-f]изохинолин-6-ил)фенил)акриловая кислота 82.
МС m/z (ИЭР): 448,4 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.59-7.55 (d, 1H), 7.20-7.18 (d, 2H), 6.71-6.51 (m 2Н), 6.24-6.22 (d 1H), 5.97-5.96 (m 2H), 5.21 (s, 1H), 3.68 (s 1H), 3.04-2.88 (m 1H), 2.68-2.63 (m, 1H), 2.34-2.23 (m, 2H), 1.19-0.90 (m, 9Н).
Пример 83
(Е)-3-(4-(6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-g]изохинолин-5-ил)фенил)акриловая кислота
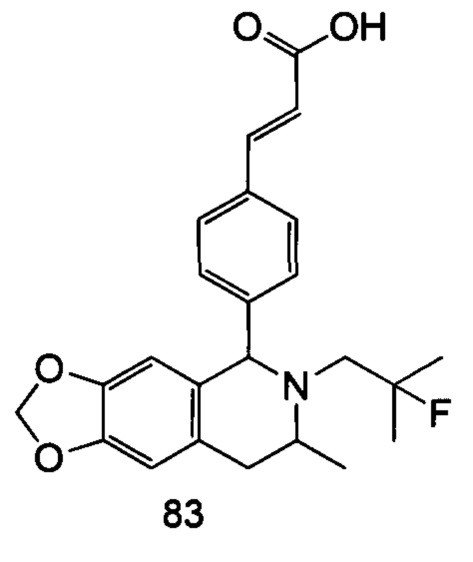
В соответствии с путем синтеза примера 1 исходное вещество (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е, используемое на стадии 3, было заменено (Е)-метил 3-(4-формилфенил)акрилатом 35а, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-(6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-[1,3]диоксоло[4,5-g]изохинолин-5-ил)фенил)акриловая кислота 83.
MC m/z (ИЭР): 412,3 [M+1]
1H ЯМР (400 МГц, CD3OD) δ 7.45-7.42 (m, 2H), 7.28-7.25 (m, 2H), 6.88 (d, 1H), 6.75 (s, 1H), 6.63 (s, 1H), 6.55 (d, 1H), 6.30 (s, 2H), 5.33 (s, 1H), 3.75 (s, 1H), 3.50 (dd, 1H), 3.03 (t, 1H), 2.75 (dd, 1H), 2.40-2.24 (t 1H), 1.21-1.15 (t, 6H), 1.06 (t, 3H).
Пример 84
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-8-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1 -ил)фенил)акриловая кислота

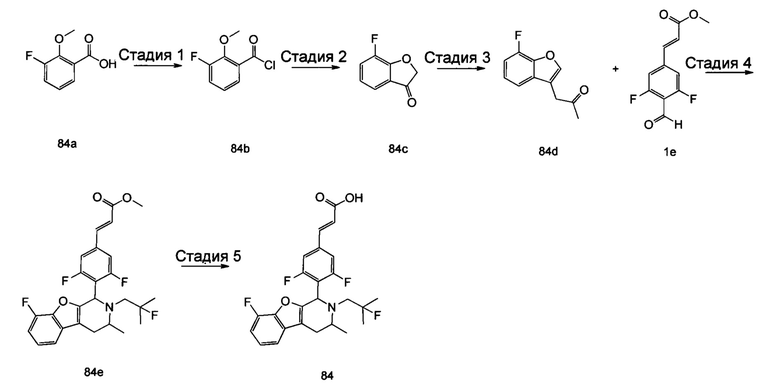
Стадия 1
3-Фтор-2-метоксибензоилхлорид
3-Фтор-2-метоксибензойную кислоту 84а (1,5 г, 8,8 ммоль, получена способом, раскрытым в заявке на патент WO 2014031937) растворяли в 2,0 мл дихлорметана, затем добавляли 9 капель N,N-диметилформамида с последующим добавлением 1,4 мл тионилхлорида в ледяной бане. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре, затем концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, 3-фтор-2-метоксибензоилхлорида 84b (1,86 г), в виде желтой жидкости, которую использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
7-Фторбензофуран-3(2Н)-он
Неочищенный 3-Фтор-2-метоксибензоилхлорид 84b (1,86 г, 8,8 ммоль) растворяли в 20 мл эфира, затем добавляли по каплям 10 мл 2 М раствора (триметилсилил)диазометана в н-гексане в ледяной бане. Затем реакционный раствор перемешивали в течение 3 часов при комнатной температуре, затем концентрировали при пониженном давлении для удаления растворителя. В ледяной бане добавляли 20 мл уксусной кислоты, и реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении, затем добавляли воду. Реакционный раствор экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором карбоната натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, 7-фторбензофуран-3(2Н)-она 84 с (340 мг, выход 27%), в виде желтого твердого вещества.
Стадия 3
1-(7-Фторбензофуран-3-ил)пропан-2-он
7-Фторбензофуран-3(2Н)-он 84с (340 мг, 2,2 ммоль) и ацетонилтрифенилфосфония хлорид (1,2 г, 3,3 ммоль) растворяли в 10 мл ксилола, затем добавляли 1,1 мл N,N-диизопропилэтиламина. Реакционный раствор подогревали до 140°C и перемешивали в течение 12 часов при 50°C, затем охлаждали до комнатной температуры и добавляли воду. Реакционный раствор экстрагировали этилацетатом. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, 1-(7-фторбензофуран-3-ил)пропан-2-она 84d (171 мг, выход 40%), в виде красно-коричневой жидкости.
В соответствии с путем синтеза примера 4 исходное вещество N-(1-(бензофуран-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин 4с, используемое на стадии 3, было заменено 1-(7-фторбензофуран-3-ил)пропан-2-оном 84d, соответственно, было получено соединение, указанное в заголовке, 1-(7-фторбензофуран-3-ил)пропан-2-он 84.
MC m/z (ИЭР): 462,2 [М+1]
1H ЯМР (400 МГц, ДМСО-d6) δ 12.05 (s, 1H), 7.42 (d, 1H), 7.29-7.23 (m, 3H), 7.17 (t, 1H), 7.08 (d, 1H), 6.53 (d, 1H), 5.23 (s, 1H), 3.54-3.48 (m, 1H), 2.95-2.86 (m, 2H), 2.45-2.34 (m, 2H), 1.24 (d, 3H), 1.18 (d,3H), 1.11 (d, 3H).
Пример 85
(E)-3-(4-((1R,3R/1S,3S)-7-хлор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота
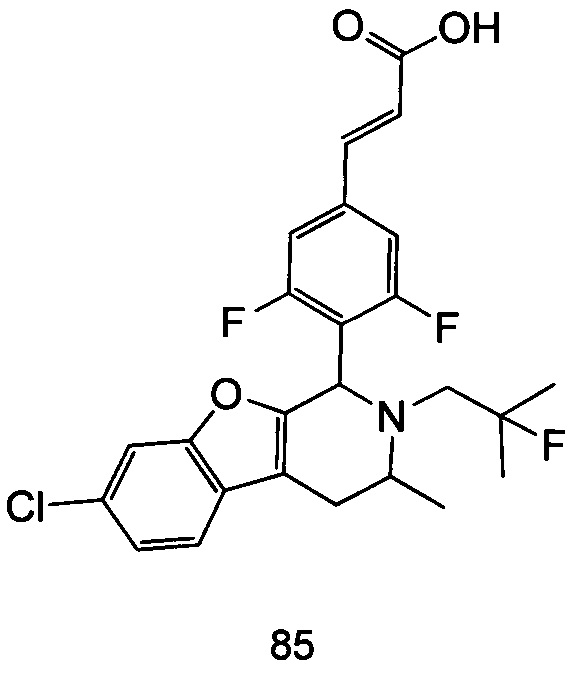
В соответствии с путем синтеза примера 84 исходное вещество 1-(7-фторбензофуран-3-ил)пропан-2-он 84d было заменено 1-(фторбензофуран-3-ил)пропан-2-оном, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-((1R,3R/1S,3S)-7-хлор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота 85.
МС m/z (ИЭР): 478,4 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.50-7.42 (m, 3Н), 7.27-7.20 (m, 3Н), 6.57 (d, 1H), 5.27 (s, 1Н), 3.70 (d, 1H), 3.27-2.92 (m, 2Н), 2.63-2.38 (m, 2Н), 1.43-1.13 (m, 9Н).
Пример 86
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
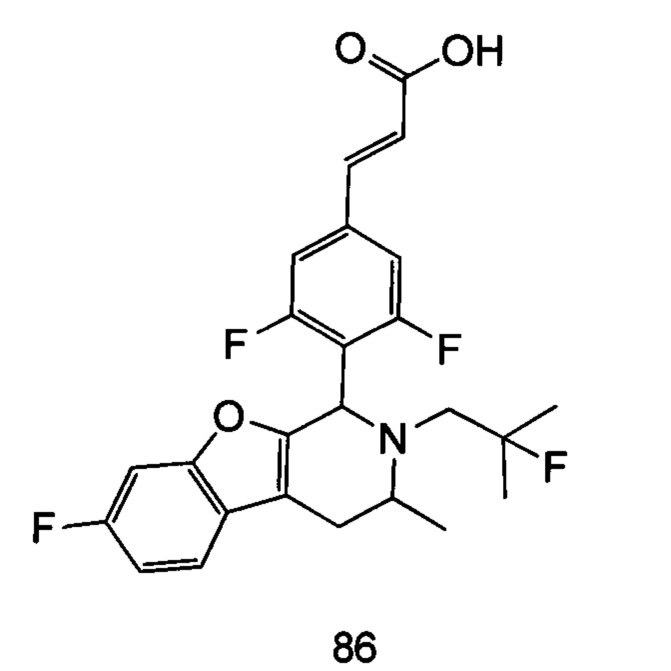
В соответствии с путем синтеза примера 80 исходное вещество 3-фтор-2-метоксибензойную кислоту 84а, используемое на стадии 1, было заменено 4-фтор-2-метоксибензойной кислотой, которая была получена способом, раскрытым в заявке на патент WO 201153359, соответственно, было получено соединение, указанное в заголовке,
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-7-фтор-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота 86.
МС m/z (ИЭР): 462,4 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.55 (d, 1Н), 7.45 (dd, 1Н), 7.20 (d, 2H), 7.11 (dd, 1H), 7.01 (m, 1Н), 6.53 (d, 1Н), 5.24 (s, 1Н), 3.66 (m, 1Н), 2.94 (m, 2H), 2.56 (dd, 1Н), 2.41 (dd, 1Н), 1.30-1.10 (m, 9Н).
Пример 87
(Е)-3-(4-((1R,3R/1S,3S)-6-бром-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота
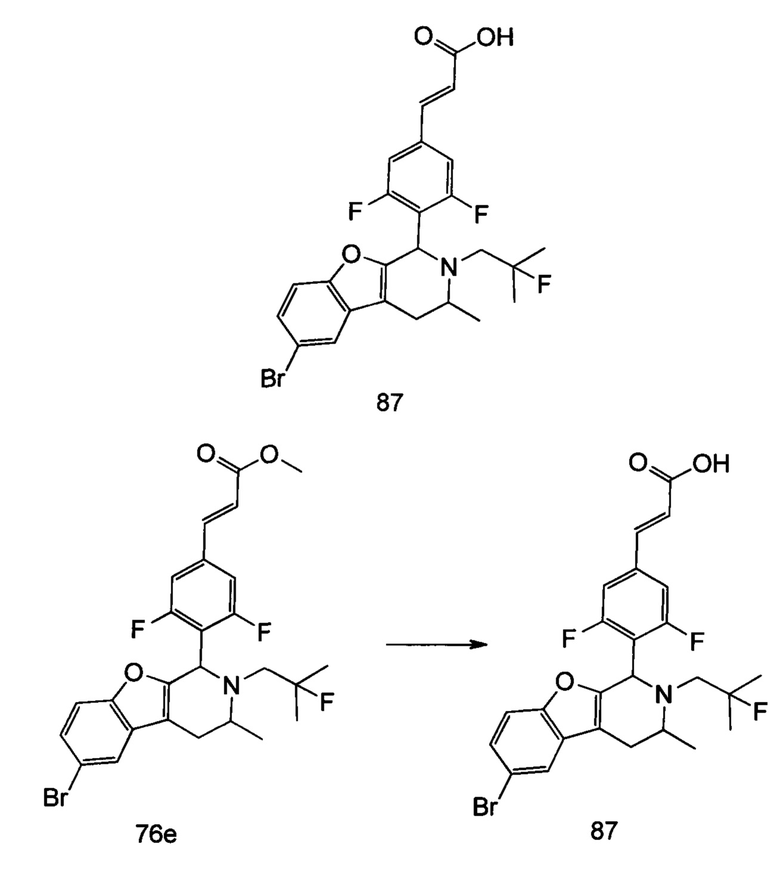
(Е)-метил-3-(4-(6-бром-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акрилат 76е (10 мг, 0,042 ммоль) растворяли в 5 мл метанола, затем добавляли гидроксид натрия (7 мг, 0,187 ммоль) и добавляли 1 мл воды. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Добавляли по каплям 1 М HCl до доведения рН до 5. Реакционный раствор экстрагировали дихлорметаном. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(4-((1R,3R/1S,3S)-6-бром-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловой кислоты 87 (5 мг, выход 50%), в виде желтого твердого вещества.
МС m/z (ИЭР): 523,3 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.67 (s, 1Н), 7.42 (d, 1H), 7.35-7.33 (dd, 1H), 7.28 (d, 1H), 7.18 (d, 2H), 6.57 (d, 1H), 5.26 (s, 1H), 3.69 (s, 1H), 3.02-2.92 (m, 2Н), 2.60-2.56 (m, 1H), 2.48-2.38 (m, 1 Н), 1.20-1.11 (m, 9Н).
Пример 88
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
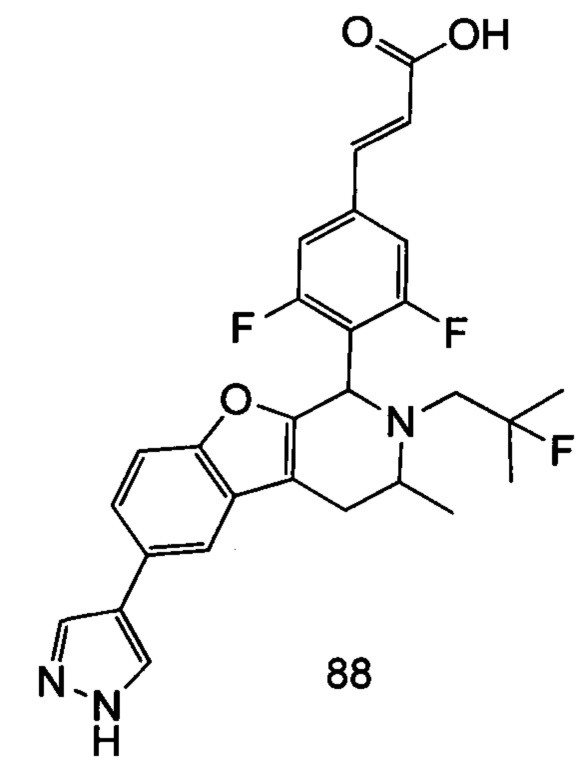
В соответствии с путем синтеза примера 76 исходное вещество 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 17а используемое на стадии 5, было заменено трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-карбоксилатом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота 88.
МС m/z (ИЭР): 510,5 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 8.06 (s, 2H), 7.77 (s, 1H), 7.62 (d, 1H), 7.51 (d, 1H), 7.39-7.31 (m, 3H), 6.60 (d, 1H), 5.56 (s, 1H), 3.85 (s, 1H), 3.16-3.11 (m, 2H), 2.79-2.75 (m, 2H), 1.36-1.27 (m, 9H).
Пример 89
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
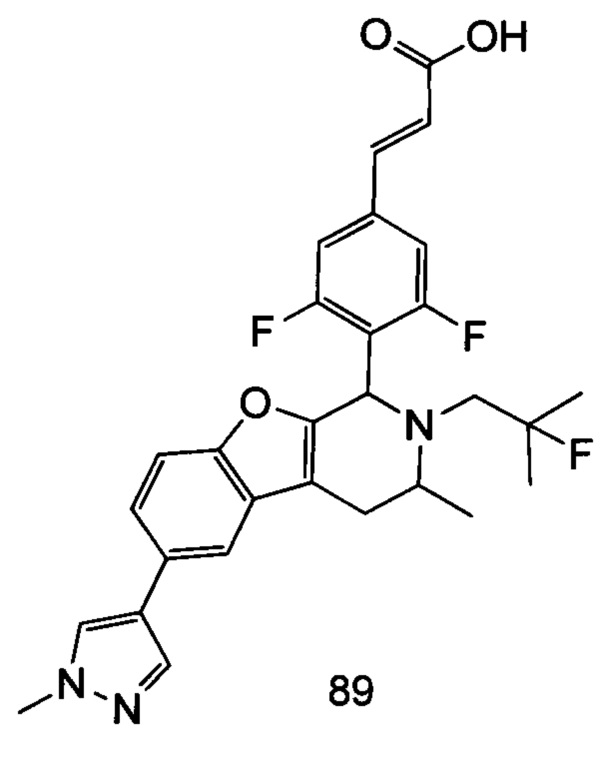
В соответствии с путем синтеза примера 76 исходное вещество 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 17а, используемое на стадии 5, было заменено 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом 16h, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота 89.
МС m/z (ИЭР): 524,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.98 (s, 1H), 7.86 (s, 1H), 7.72 (s, 1H), 7.62 (d, 1H), 7.37-7.48 (m, 1H), 7.35-7.32 (m, 3H), 6.60 (d, 1H), 5.55 (s, 1H), 3.98 (s, 3H), 3.86 (s, 1H), 3.22-3.11 (m, 2H), 2.79-2.75 (m, 2H), 1.36-1.27 (m, 9H).
Пример 90
(Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота
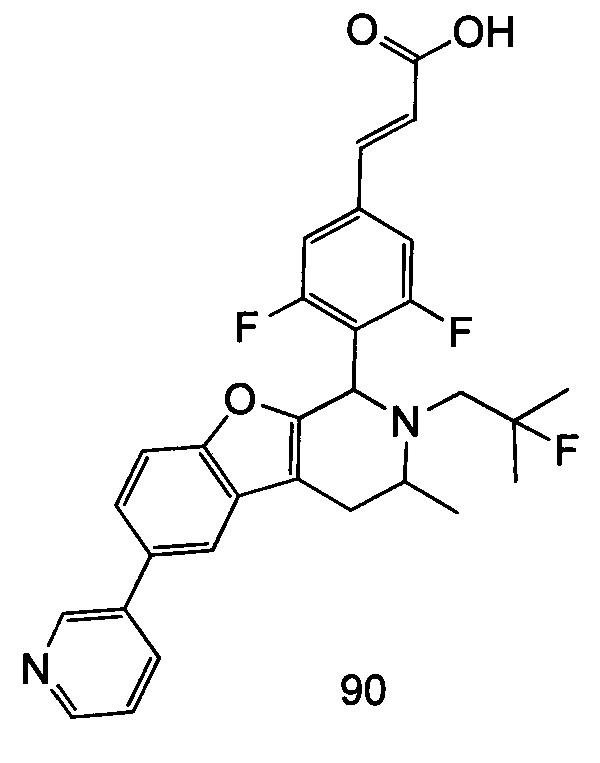
В соответствии с путем синтеза примера 76 исходное вещество 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 17а, используемое на стадии 5, было заменено пиридин-3-илбороновой кислотой 28а, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((1R,3R/1S,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(пиридин-3-ил)-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)фенил)акриловая кислота 90.
MC m/z (ИЭР): 521,5 [M+1]
1H ЯМР (400 МГц, CD3OD) δ 9.19 (s, 1Н), 8.89-8.80 (m, 2H), 8.10 (t, 1H), 8.01 (s, 1H), 7.70-7.57 (m, 3Н), 7.28 (d, 2H), 6.58 (d, 1H), 5.36 (s, 1H), 3.78 (s, 1H), 3.13-2.99 (m, 2H), 2.50 (d, 1H), 2.71-2.44 (m, 1H), 1.29-1.16 (m, 9H).
Пример 91
(Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-1Н-пиразоло[3,4-г]изохинолин-5-ил)фенил)акриловая кислота
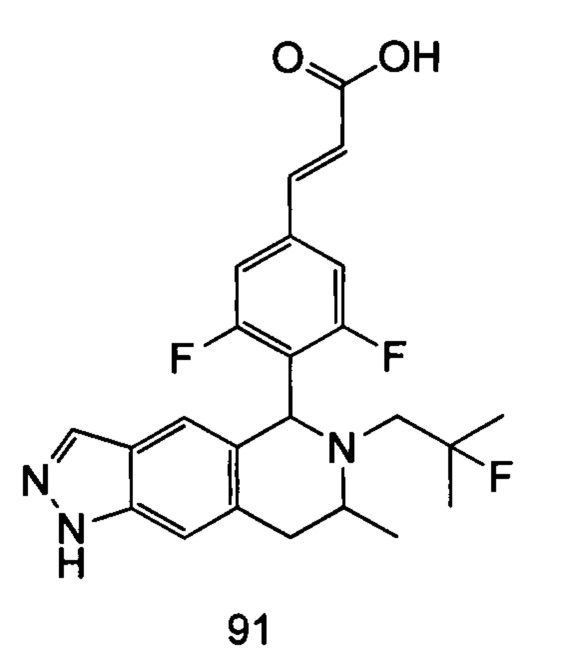
В соответствии с путем синтеза примера 70 исходное вещество 3-(бензилокси)-2-метилбензальдегид 70а, используемое на стадии 1, было заменено 3-(бензилокси)-4-метилбензальдегидом, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((5S,7R/5R,7S)-6-(2-фтор-2-метилпропил)-7-метил-5,6,7,8-тетрагидро-1Н-пиразоло[3,4-г]изохинолин-5-ил)фенил)акриловая кислота 91.
МС m/z (ИЭР): 444,5 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 8.10 (d, 1H), 7.58-7.18 (m, 3H), 6.78 (s, 1H), 6.55 (d, 1H), 5.36 (s, 1H), 3.81 (s, 1H), 3.25-3.30 (m, 2Н), 2.42-2.30 (m, 2Н), 2.07 (s, 1H), 1.22-0.92 (m, 9Н).
Пример 92
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-(7-изобутил-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота

Стадия 1
1-(3-(Бензилокси)-2-метилфенил)пропан-2-он
(Z)-1-(бензилокси)-2-метил-3-(2-нитроргор-1-en-1-ил)бензол 70b (37 г, 0,131 моль) растворяли в 600 мл уксусной кислоты, затем добавляли порошок железа (73 г, 1,306 моль). Реакционную смесь подогревали до 100°C и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. К остатку добавляли дихлорметан и фильтровали. Фильтрат промывали насыщенным раствором бикарбоната натрия. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке, 1-(3-(бензилокси)-2-метилфенил)пропан-2-она 92а (26 г), в виде коричневого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 2
N-(1-(3-(Бензилокси)-2-метилфенил)пропан-2-ил)-2-метилпропан-1-амин
1-(3-(Бензилокси)-2-метилфенил)пропан-2-он 92а (5 г, 0,019 моль) растворяли в 100 мл дихлорэтана, затем добавляли 2-метилпропан-1-амин (2,2 г, 0,029 моль) и триацетоксиборгидрид натрия (8 г, 0,038 ммоль). Реакционную смесь перемешивали в течение 48 часов при комнатной температуре, затем добавляли воду и экстрагировали дихлорметаном. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, N-(1-(3-(бензилокси)-2-метилфенил)пропан-2-ил)-2-метилпропан-1-амина 92b (5,2 г, выход 85%), в виде светло-желтого масла.
Стадия 3
3-(2-(Изобутиламино)пропил)-2-метилфенол
N-(1-(3-(бензилокси)-2-метилфенил)пропан-2-ил)-2-метилпропан-1-амин 92b (5,2 г, 16,696 ммоль) растворяли в 50 мл метанола, затем добавляли палладий на активированном углероде (1 г, 10%). Реакционную систему три раза продували водородом, чтобы удалить воздух. Реакционный раствор перемешивали в течение 12 часов при комнатной температуре, затем фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 3-(2-(изобутиламино)пропил)-2-метилфенола 92 с (4 г), в виде светло-коричневого масла, которое использовали непосредственно в следующей стадии без дополнительной очистки.
Стадия 4
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-6-гидрокси-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
3-(2-(Изобутиламино)пропил)-2-метилфенол 92 с (3,7 г, 0,017 ммоль) растворяли в 50 мл метанола, затем добавляли (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е (5,68 г, 0,025 моль) и уксусную кислоту (2,04 г, 0,03 ммоль). Реакционный раствор подогревали до 60°C и перемешивали в течение 12 часов, затем охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-6-гидрокси-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 92d (2,5 г, выход 34%), в виде желтого масла.
Стадия 5
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-изобутил-3,5-диметил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-6-гидрокси-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 92d (2,5 г, 5,821 ммоль) растворяли в 25 мл дихлорметана, затем добавляли 2,6-лутидин (1,25 г, 11,641 ммоль) и добавляли трифторметансульфоновый ангидрид (2,46 г, 8,732 ммоль) при 0°C. Реакционный раствор перемешивали в течение 2 часов при 0°C, затем концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-изобутил-3,5-диметил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилата 92е (2,5 г, выход 76%) в виде светло-желтого твердого вещества.
Стадия 6
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-((дифенилметилен)амино)-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-изобутил-3,5-диметил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 92е (2,5 г, 4,452 ммоль), бензофенонимин (1,05 г, 5,787 ммоль), ацетат палладия (200 мг, 0,89 ммоль), (R)-(+)-2,2'-бис(дифенилфосфино)-1,1'-динафтил (554 мг, 0,89 ммоль) и карбонат цезия (2,9 г, 8,89 ммоль) растворяли в 40 мл 1,4-диоксана. Реакционный раствор подогревали до 100°C и перемешивали в течение 12 часов, затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученные в результате остатки очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(4-((1S,3R/1R,3S)-6-((дифенилметилен)амино)-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 92f (1,8 г, выход 69%), в виде светло-желтого масла.
Стадия 7
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-амино-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат
(Е)-метил-3-(4-((1S,3R/1R,3S)-6-((дифенилметилен)амино)-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 92f (1,8 г, 3,037 ммоль) растворяли в 30 мл метанола. Реакционный раствор охлаждали до 0°C, добавляли 10 мл 1 М HCl и перемешивали в течение 2 часов. Добавляли насыщенный раствор карбоната натрия, пока рН этой системы не стал нейтральным. Реакционный раствор экстрагировали дихлорметаном, и органическую фазу высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил 3-(4-((1S,3R/1R,3S)-6-амино-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилата 92g (1,2 г, выход 92%), в виде светло-желтого масла.
В соответствии с путем синтеза примера 70 исходное вещество (Е)-метил-3-(4-((1S,3R/1R,3S)-6-амино-2-(2-фтор-2-метилпропил)-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 70i, используемое на стадии 9, было заменено (Е)-метил 3-(4-((1S,3R/1R,3S)-6-амино-2-изобутил-3,5-диметил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилатом 92g, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-изобутил-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота 92.
МС m/z (ИЭР): 426,2 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 8.07 (s, 1H), 7.22-7.30 (m, 2H), 7.08 (t, 2H), 6.77 (dd, 1H), 6.53 (dd, 1H), 5.23 (s, 1H), 3.54-3.58 (m, 1H), 3.36-3.42 (dd, 1H), 2.96-3.00 (dd, 1H), 2.51-3.56 (dd, 1H), 2.06-2.09 (m, 1H), 1.64-1.69 (m, 1H), 1.03 (d, 3H), 0.83 (d, 3H), 0.72 (d, 3H).
Пример 93
(Е)-3-(4-((1S,3R/1R,3S)-6-циано-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
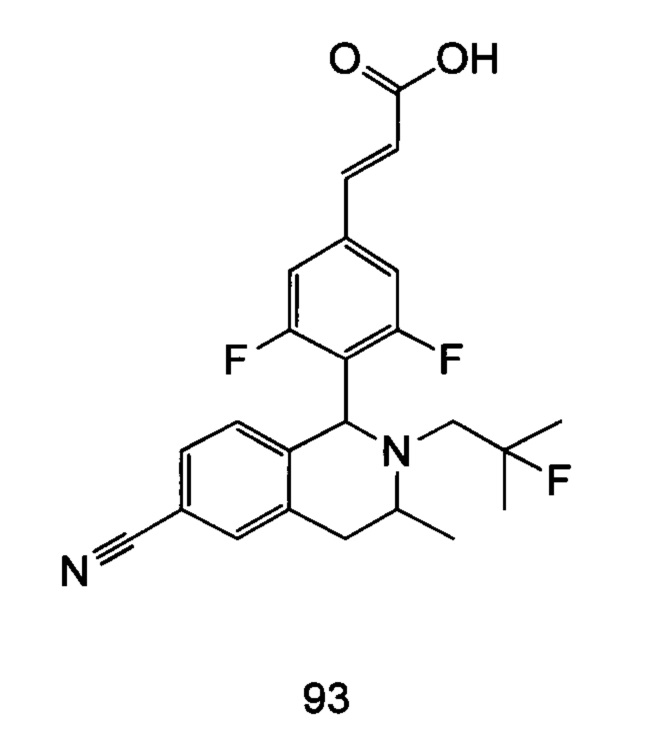
В соответствии с путем синтеза примера 10 исходное вещество 2-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол 10b, используемое на стадии 2, было заменено цианидом цинка, соответственно, было получено соединение, указанное в заголовке, (E)-3-(4-((1S,3R/1R,3S)-6-циано-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 93.
МС m/z (ИЭР): 429,5 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 7.60-7.55 (m, 2H), 7.40 (d, 1H), 7.23 (d, 2H), 6.93 (d, 1H), 6.56 (d, 1H), 5.30 (s, 1H), 3.78-3.70 (m, 1H), 3.67-3.41 (m, 1H), 3.04-2.96 (m, 1H), 2.75-2.70 (m, 1H), 2.36-2.25 (m, 1H), 1.33-1.01 (m, 9H).
Примеры 94, 95
(Е)-3-(3,5-дифтор-4-((6S,8R)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота
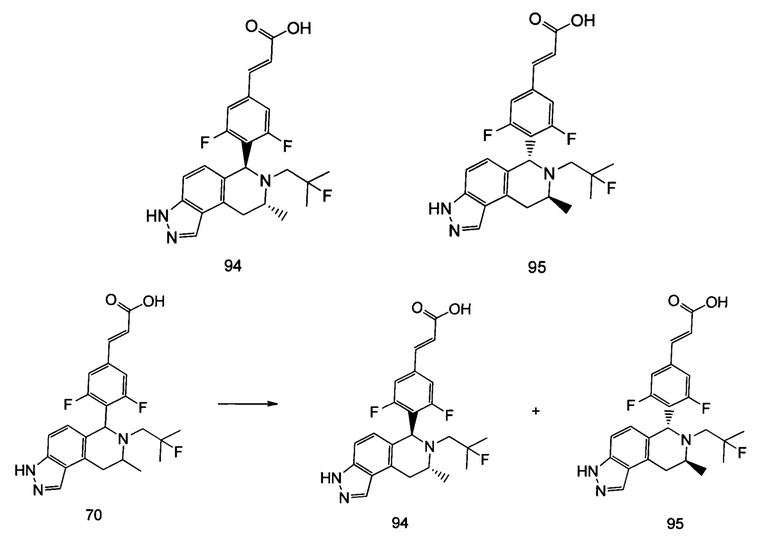
(Е)-3-(3,5-дифтор-4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловую кислоту 70 (250 мг, 0,56 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Superchiral S-AD (Chiralway), внутренний диаметр 2 см * длина 25 см, 5 мкм; подвижная фаза: CO2 : этанол составляет 60:40, скорость потока: 60 г/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((6S,8R)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловой кислоты 94 (100 мг) в виде желтого твердого вещества и (Е)-3-(3,5-дифтор-4-((6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловой кислоты 95 (100 мг) в виде желтого твердого вещества.
Пример 94
МС m/z (ИЭР): 444,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 5,299 минут, хиральная чистота: 99,286% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: CO2 : этанол составляет 60:40 (об./об.));
1H ЯМР (400 МГц, CD3OD) δ 8.10 (s, 1H), 7.58 (d, 1H), 7.25-7.19 (m, 2H), 6.76 (d, 1H), 6.54 (d, 1H), 5.35 (s, 1H), 3.82 (s, 1H), 3.50-3.45 (m, 1H), 3.08-2.99 (m, 2H), 2.42-2.35 (m, 1H), 2.06 (s, 1H), 1.22-1.07 (m, 9H).
Пример 95
MC m/z (ИЭР): 444,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 4,101 минут, хиральная чистота: 99,649% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см × длина 15 см; подвижная фаза: CO2 : этанол составляет 60:40 (об./об.));
1H ЯМР (400 МГц, CD3OD) δ 8.10 (s, 1H), 7.58 (d, 1H), 7.25-7.19 (m, 2H), 6.76 (d, 1H), 6.54 (d, 1H), 5.35 (s, 1H), 3.82 (s, 1H), 3.50-3.45 (m, 1H), 3.08-2.99 (m, 2H), 2.42-2.35 (m, 1H), 2.06 (s,1H), 1.22-1.07 (m, 9H).
Пример 96 и пример 97
(E)-3-(4-((1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота
(Е)-3-(4-((1R,3R)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота
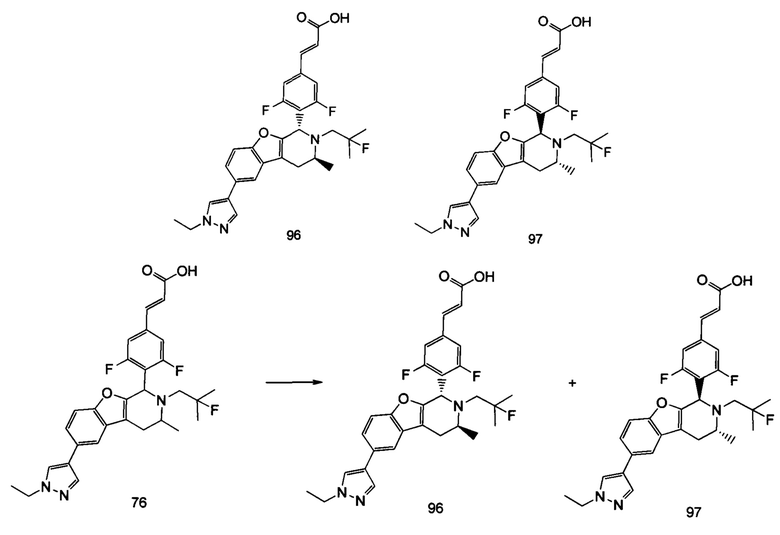
(Е)-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-мет ил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловую кислоту 76 (160 мг, 0,3 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Chiralpak AD, внутренний диаметр 2,5 см × длина × 25 см; подвижная фаза: н-гексан : этанол : ТФУ составляет 60:40:0,1, скорость потока: 60 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (E)-3-(4-((1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловой кислоты 96 (124,4 мг) в виде светло-желтого твердого вещества и (E)-3-(4-((1R,3R)-6-(1-этил-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловой кислоты 97 (170 мг) в виде светло-желтого твердого вещества.
Пример 96
МС m/z (ИЭР): 538,2 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 8,909 минут, хиральная чистота: 98,88% (хроматографическая колонка: Chiralpak IC, 4,6 мм × 250 мм, 5 мкм; подвижная фаза: н-гексан : этанол : ТФУ составляет 80:20:0,1 (об./об.));
1H ЯМР (400 МГц, CD3OD) δ 8.03 (s, 1H), 7.86 (s, 1H), 7.72 (s, 1H), 7.61 (d, 1H), 7.47 (d, 1H), 7.30-7.37 (dd, 3Н), 6.59 (d, 1H), 5.52 (s, 1H), 4.24 (q, 2H), 3.83 (s, 1H), 3.12 (d, 2H), 2.75 (d, 2H), 1.52 (t, 3Н), 1.25-1.39 (m, 9H).
Пример 97
MC m/z (ИЭР): 538,2 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 7,337 минут, хиральная чистота: 98,82% (хроматографическая колонка: Chiralpak IC, 4,6 мм × 250 мм, 5 мкм; подвижная фаза: н-гексан:этанол:ТФУ составляет 80:20:0,1 (об./об.));
1H ЯМР (400 МГц, CD3OD) δ 8.03 (s, 1H), 7.86 (s, 1H), 7.72 (s, 1H), 7.61 (d, 1H), 7.47 (d, 1H), 7.30-7.37 (dd, 3Н), 6.59 (d, 1H), 5.52 (s, 1H), 4.24 (q, 2H), 3.83 (s, 1H), 3.12 (d, 2H), 2.75 (d, 2H), 1.52 (t, 3Н), 1.25-1.35 (m, 9H).
Пример 98 и пример 99
(Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акриловая кислота
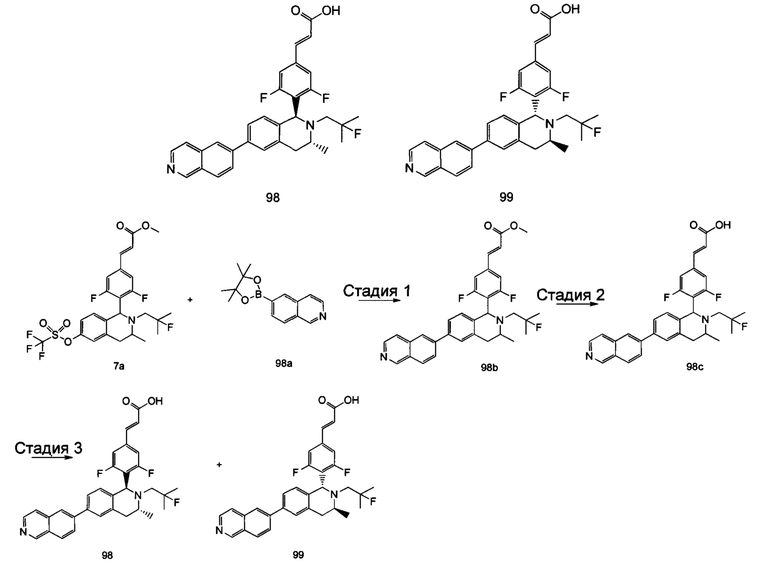
Стадия 1
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акрилат
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акрилат 7а (1.588 г, 2.8 ммоль) растворяли в 20 мл смеси 1,4-диоксана и воды (об./об. составляет 10:1), затем добавляли 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохинолин 98а (0,862 г, 3,36 ммоль), тетракис(трифенилфосфин)палладий (0,324 г, 0,28 ммоль) и карбонат натрия (0,89 г, 8,4 ммоль). Реакционную смесь подогревали до 90°C и перемешивали в течение 12 часов, затем реакцию останавливали. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением соединения, указанного в заголовке, (Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетра гидро-[6,6'-диизохинолин]-1-ил)фенил)акрилата 98b (550 мг, выход 36%) в виде белого твердого вещества.
Стадия 2
(Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акриловая кислота
(Е)-метил-3-(3,5-дифтор-4-((1S,3R/1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акрилат 98b (550 мг, 1,01 ммоль) растворяли в 12 мл смеси метанола и тетрагидрофурана (об./об. составляет 1:3), затем добавляли гидроксид натрия (404 мг, 10,1 ммоль) и 1,68 мл воды. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. В реакционную смесь добавляли 1 М раствор HCI до достижения рН 5-6, добавляли 10 мл воды. Реакционную смесь экстрагировали этилацетатом (15 млх4). Органическую фазу объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением соединения, указанного в заголовке, (Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил) акриловой кислоты 98 с (450 мг, выход 83%), в виде светло-желтого твердого вещества.
Стадия 3
(Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-биизохинолин]-1-ил)фенил)акриловая кислота
(Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акриловую кислоту 98 с (300 мг, 0,565 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Superchiral S-AD (Chiralway), внутренний диаметр 0,46 см * длина 25 см, 5 мкм; подвижная фаза: CO2/изопропанол/ДЭА составляет 60:40:0,05 (об./об./об.), скорость потока: 2,5 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением соединений, указанных в заголовке, (Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акриловой кислоты 98 (90 мг) в виде белого твердого вещества и (Е)-3-(3,5-дифтор-4-((1R,3S)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидро-[6,6'-диизохинолин]-1-ил)фенил)акриловой кислоты 99 (90 мг) в виде желтых кристаллов.
Пример 98:
МС m/z (ИЭР): 531,4[М+1];
Анализ хиральной ВЭЖХ: время удерживания 15,869 минут, хиральная чистота: 100% (хроматографическая колонка: Chiralpak IE, 4,6 мм × 150 мм, 5 мкм; подвижная фаза: н-гексан : этанол : ТФУ составляет 70:30:0,1 (об./об./об.));
1H ЯМР (400 МГц, CD3OD) δ 9.27 (s, 1H), 8.45 (d, 1H), 8.20 (d, 2H), 8.03 (d, 1H), 7.92 (d, 1H), 7.60 (d, 2H), 7.51 (d, 1H), 7.24 (d, 2H), 6.90 (d, 1H), 6.55 (d, 1H), 5.32 (s, 1H), 3.77 (s, 1H), 3.48 (d, 1H), 3.05 (t, 1H), 2.78 (s, 1H), 2.29-2.40 (m, 1H), 1.16-1.21 (m, 6H), 1.08 (d, 3H).
Пример 99:
МС m/z (ИЭР): 531,4 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 14,020 минут, хиральная чистота: 97,42% (хроматографическая колонка: Chiralpak IE, 4,6 мм × 150 мм, 5 мкм; подвижная фаза: н-гексан : этанол : ТФУ составляет 70:30:0,1 (об./об./об.));
1H ЯМР (400 МГц, CD3OD) δ 9.72 (s, 1H), 8.56 (t, 3H), 8.47 (d, 1H), 8.38 (d, 1H), 7.78 (s, 1H), 7.59-7.67 (m, 2H), 7.29 (d, 2H), 7.03 (d, 1 H), 6.58 (d, 1H), 5.50 (s, 1H), 3.84 (s, 1H), 3.51 (d, 1H), 3.17 (t, 1H), 2.89 (d, 1H), 2.18-2.38 (m, 1H), 1.19-1.30 (m, 6H), 1.16 (d, 3H).
Пример 100
(E)-3-(4-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота
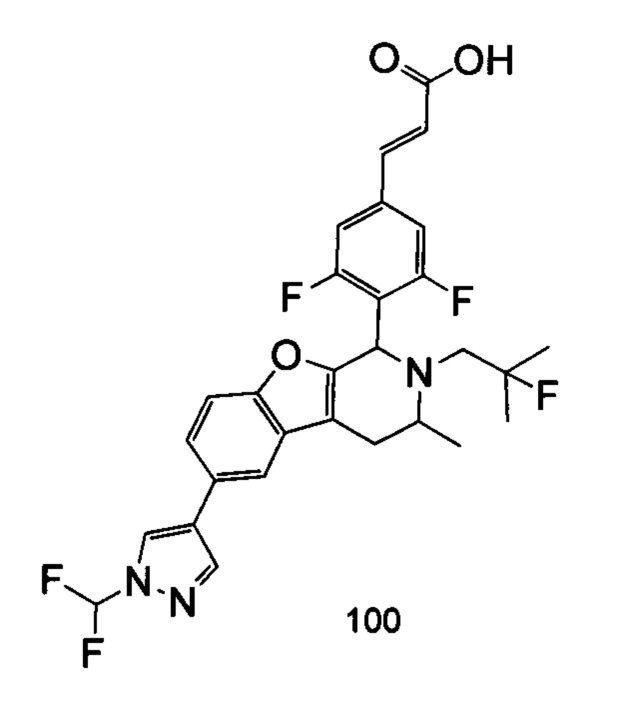
В соответствии с путем синтеза примера 76 исходное вещество 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 17а, используемое на стадии 5, было заменено 1-(дифторметил)-4п-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом 13а, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-(6-(1-(дифторметил)-1Н-пиразол-4-ил)-2-(2-фтор-2-метилпропил)-3-метил-1,2,3,4-тетрагидробензофуро[2,3-с]пиридин-1-ил)-3,5-дифторфенил)акриловая кислота 100.
МС m/z (ИЭР): 557,8 [М-1]
1H ЯМР (400 МГц, CD3OD) δ 8.44 (s, 1H), 8.14 (s, 1H), 7.82 (d, 1H), 7.53-7.68 (m, 3Н), 7.30-7.42 (m, 3Н), 6.60 (m, 1H), 5.49 (s, 1H), 3.82 (s, 1H), 3.13 (d, 2H), 2.77 (d, 2H), 1.24-1.34 (m, 9Н).
Пример 101
(Е)-3-(4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3Н-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота
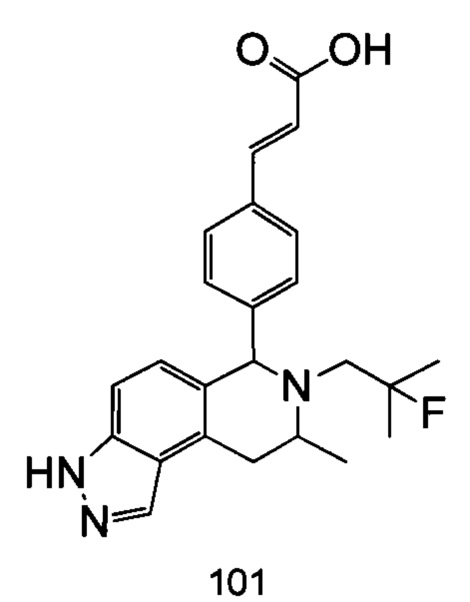
В соответствии с путем синтеза примера 92 исходные вещества 3-(2-(изобутиламино)пропил)-2-метилфенол 92 с и (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е, используемые на стадии 4, были заменены 3-(2-((2-фтор-2-метилпропил)амино)пропил)-2-метилфенолом 70е и (Е)-метил-3-(4-формилфенил)акрилатом 35а, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-((6S,8R/6R,8S)-7-(2-фтор-2-метилпропил)-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота 101.
МС m/z (ИЭР): 408,4 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 8.24 (s, 1H), 7.65-7.60 (m, 3Н), 7.47 (d, 1H) 7.39 (d, 2H), 6.96 (d, 1H), 6.55 (d, 1H), 6.07 (s, 1H), 4.24 (m, 1H), 3.69 (m, 1H), 3.66 (m, 1H), 3.35 (m, 1H), 3.26 (m, 1H), 1.66-1.29 (m, 9H).
Пример 102
(Е)-3-(4-((6R,8R/6S,8S)-7-изобутил-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота
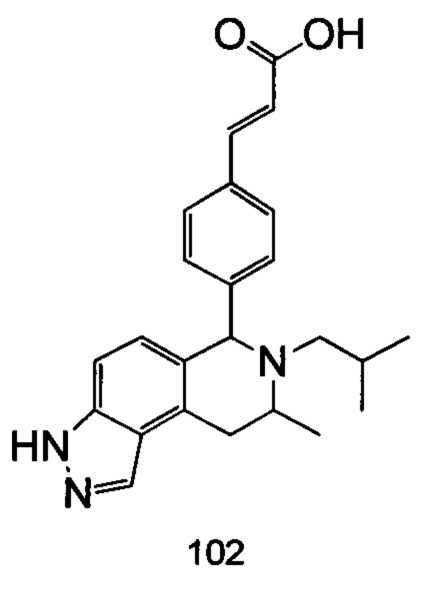
В соответствии с путем синтеза примера 92 исходное вещество (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат 1е, используемое на стадии 4, было заменено (Е)-метил 3-(4-формилфенил)акрилатом 35а, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-((6R,8R/6S,8S)-7-изобутил-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)фенил)акриловая кислота 102.
МС m/z (ИЭР): 390,4 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 8.10 (s, 1H), 7.52-7.47 (m, 3Н), 7.29-7.26 (m, 3Н), 6.87 (d, 1Н), 6.50 (d, 1H), 3.54-3.51 (m, 1H), 3.29-3.25 (m, 1H), 3.00-2.99 (m, 1H), 2.50-2.47 (m, 1H), 2.23-2.20 (m, 1H), 1.97 (s, 1H), 1.82-1.87 (m, 1H), 1.12 (d, 3Н), 0.95 (d, 3Н), 0.82 (d, 3Н).
Пример 103
(Е)-3-(4-((6S,8R/6R,8S)-7-циклопентил-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)-3,5-дифторфенил)акриловая кислота
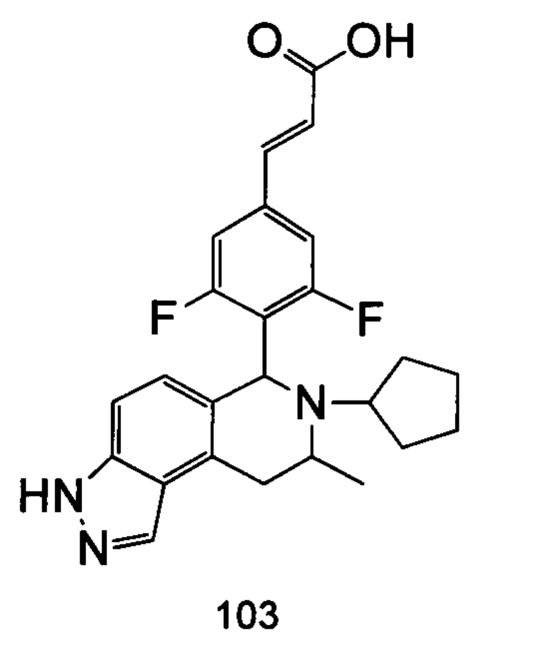
В соответствии с путем синтеза примера 92 исходное вещество 2-метилпропан-1-амин, используемое на стадии 2, было заменено циклопентиламином, соответственно, было получено соединение, указанное в заголовке, (Е)-3-(4-(7-циклопентил-8-метил-6,7,8,9-тетрагидро-3H-пиразоло[4,3-f]изохинолин-6-ил)-3,5-дифторфенил)акриловая кислота 103.
МС m/z (ИЭР): 438,4 [М+1]
1H ЯМР (400 МГц, CD3OD) δ 8.09 (s, 1H), 7.42 (d, 1H), 7.24 (d, 1H), 7.14 (d, 2H), 6.79 (d, 1H), 6.53 (d, 1H), 5.26 (s, 1H), 3.58 (s, 1H), 3.43-3.37 (m, 1H), 3.03-2.98 (m, 1H), 2.59-2.54 (m, 1H), 2.11-2.05 (m, 1H), 1.68-1.71 (m, 1H), 1.05 (d, 3Н), 0.84 (d, 3Н), 0.71 (d, 3Н).
БИОЛОГИЧЕСКИЙ АНАЛИЗ
Далее настоящее изобретение описано со ссылкой на следующие тестовые примеры, но эти примеры не следует истолковывать как ограничивающие объем изобретения.
Тестовый пример 1. Ингибирующее действие соединений по настоящему изобретению на связывание Е с ER
1. Цель эксперимента
Соединения по настоящему изобретению оказывают ингибиторное действие на связывание Е (эстрогена) с ER (эстрогеновым рецептором), блокируя, таким образом, образование комплекса Е и ER с ERE (эстрогенчувствительным элементом) и впоследствии блокируя экспрессию нижележащего белка люциферазы.
Ингибиторное действие соединений на связывание Е с ER in vitro исследовали описанным ниже способом.
Цель данного эксперимента состояла в определении ингибиторного действия соединений на связывание Е с ER, и оценку активности соединений in vitro проводили на основании значения IC50.
2. Методика эксперимента
ERE клонировали выше гена люциферазы, и моноклональные клетки MCF-7/ERE-люцифераза отбирали путем трансфекции MCF-7 (Банк клеток Комитета по сохранению типовых клеточных культур Китайской Академии Наук, TCHu74). Клетки MCF-7/ERE-люцифераза засевали в модифицированную питательную среду Игла (MEM) (Hyclone, SH30024.01B), содержащую 10% фетальную бычью сыворотку (ФБС), очищенную на активированном угле (Moregate, FBSF), 1% пируват натрия (Sigma, S8636), 1% неэссенциальные аминокислоты (Sigma, M7145) и 500 мкг/мл G418, в лунки 96-луночного планшета при плотности 30000 клеток/лунка, и клетки инкубировали в условиях 37°C и 5% CO2. Лекарственное средство готовили в виде 20 мМ исходного раствора, который затем разводили 100% ДМСО с получением градиента 10-кратной концентрации и впоследствии разводили 20-кратной питательной средой. После инкубации в течение 24 часов питательную среду удаляли, затем в каждую лунку добавляли 0,1 нМ эстрадиола (Sigma, E2758) и 10 мкл лекарственного средства, разведенного питательной средой, а в контрольную группу добавляли ДМСО. Планшет мягко встряхивали и инкубировали в инкубаторе при 37°C, 5% CO2. Через 24 часа питательную среду для культуры клеток сливали. В каждую лунку добавляли 50 мкл подготовленного субстрата люциферазы (Promega, E6110), и планшет помещали в темное место при комнатной температуре на 10-15 минут, затем определяли значение сигнала хемилюминесценции.
3. Результат теста
В описанном выше эксперименте было исследовано ингибиторное действие соединений на связывание Е с ER. Значения сигнала хемилюминесценции относительно логарифмических значений концентрации соединений наносили на график с помощью программы Graghpad Prism, и значения IC50 приведены в таблице 1.
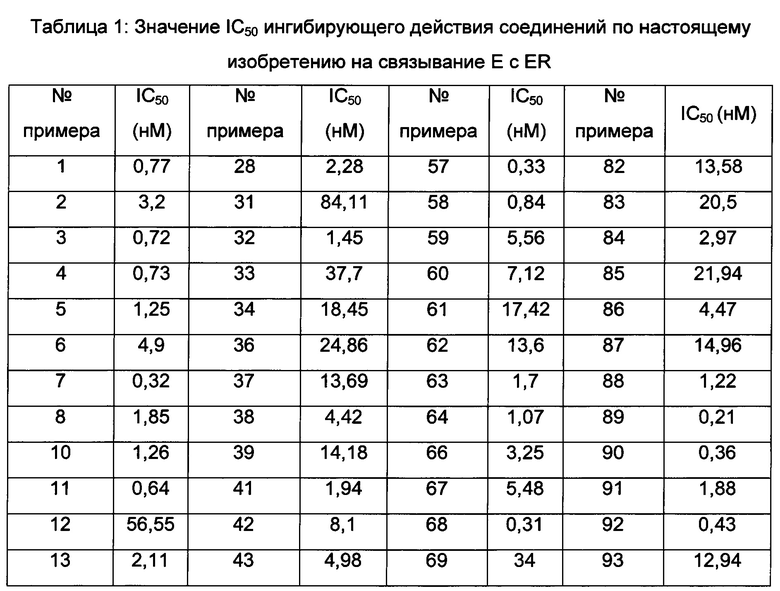
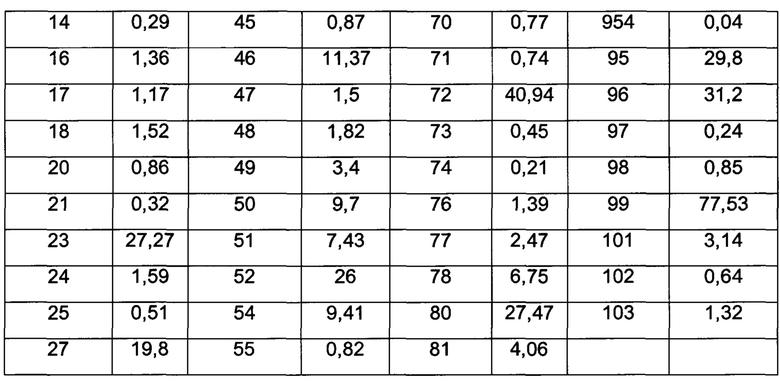
Вывод: Соединения по настоящему изобретению характеризуются значительным ингибирующим действием на связывание Е с ER.
Тестовый пример 2. Ингибирующее действие соединений по настоящему изобретению на пролиферацию клеток MCF7
1. Цель эксперимента
Цель данного эксперимента состояла в определении ингибиторного действия соединений на активность пролиферации клеток MCF7, и оценку активности соединений in vitro проводили на основании значения IC50.
2. Методика эксперимента
Клетки MCF-7 (Банк клеток Комитета по сохранению типовых клеточных культур Китайской Академии Наук, TChu 74) засевали в питательную среду MEM (Hyclone, SH30024.01B), содержащую 10% ФБС (Gibco, 10099-141), 1% пируват натрия (Sigma, S8636) и 1% неэссенциальные аминокислоты (Sigma, M7145), в лунки 96-луночного планшета при плотности 40000 клеток/лунка, и клетки инкубировали в условиях 37°C и 5% CO2. Соединение готовили в виде 20 мМ исходного раствора, который затем разводили до 1000-кратной конечной концентрации 100% ДМСО и впоследствии разводили 20-кратной питательной средой, содержащей 2% ФБС. После инкубации в течение 24 часов питательную среду удаляли, затем в каждую лунку добавляли 90 мкл питательной среды, содержащей 2% ФБС и 10 мкл лекарственного средства, в контрольную группу добавляли 10 мкл ДМСО, а холостая проба содержала только 100 мкл питательной среды, содержащей 2% ФБС. Планшет мягко встряхивали и инкубировали в инкубаторе при 37°C, 5% CO2. Через 72 часа в каждую лунку добавляли 50 мкл смешанного реагента Cell Titer-Glo (Promega, G7571). Планшет встряхивали, чтобы хорошо перемешать ингредиенты, и помещали с условия комнатной температуры на 10 минут, затем определяли сигнал хемилюминесценции.
3. Анализ данных
Значения сигнала хемилюминесценции относительно логарифмических значений концентрации соединений наносили на график с помощью программы Graghpad Prism, из которого были получены значения IC50. Результаты представлены в таблице 2.
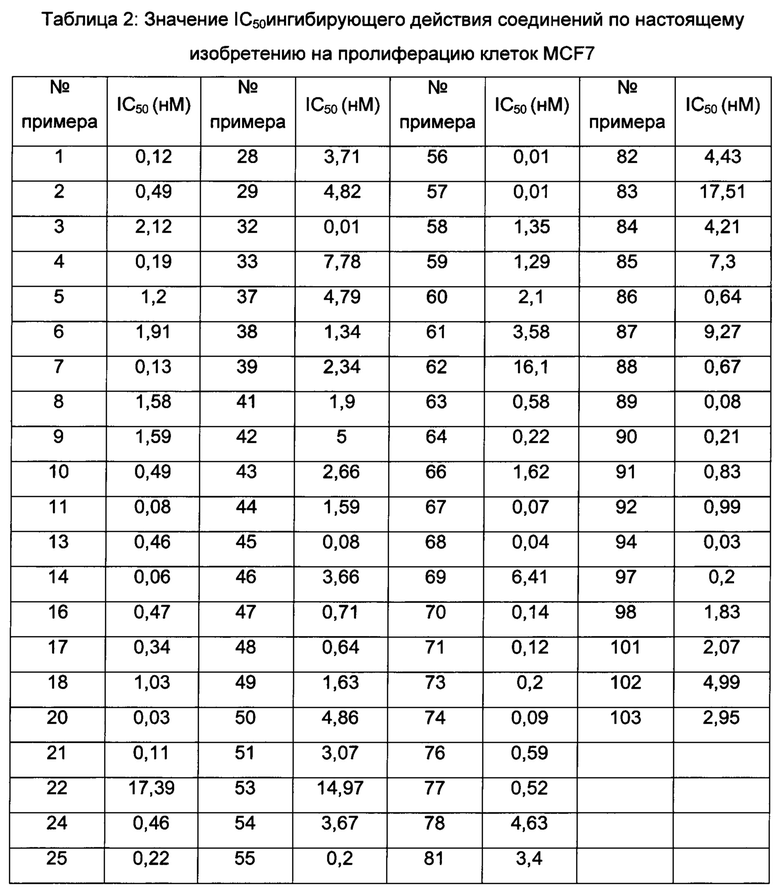
Тестовый пример 3. Действие соединений по настоящему изобретению, вызывающее деградацию ERα
1. Цель эксперимента
Для определения действия соединений по настоящему изобретению, вызывающего деградацию ER, использовали методику определения деградации ER под действием соединений по настоящему изобретению.
2. Материалы и оборудование
Считывающее устройство для планшетов с плоским дном BioTek Synergy HT
Линия клеток MCF-7 (TChu 74, Банк клеток Комитета по сохранению типовых клеточных культур Китайской Академии Наук)
Набор реагентов ERα Duoset (#DYC5715E, R&D System)
3. Методика эксперимента
Клетки MCF-7 инкубировали в питательной среде DMEM/F-12, содержащей 10% ФБС.
В первый день эксперимента клетки MCF-7 ресуспендировали в питательной среде DMEM/F-12, содержащей 10% ФБС, очищенной активированным углем, затем засевали в 48-луночный планшет при плотности 50000 клеток/лунка и инкубировали в течение 22-24 часов.
На второй день эксперимента тестируемое соединение разводили питательной средой и добавляли в лунки 48-луночного планшета. В лунки 96-луночного планшета вносили антитело захвата к ERα, разведенное до 1 мкг/мл фосфатно-солевым буферным раствором (ФСБ), в количестве 100 мкл/лунка. Планшет запечатывали и покрывали антителом в течение ночи при комнатной температуре.
На третий день эксперимента покрытый антителом 96-луночный планшет дважды промывали ФСБ, добавляли блокирующий раствор (1% бычий сывороточный альбумин в ФСБ) в количестве 110 мкл/лунка и блокировали в течение 1 часа при комнатной температуре. 48-луночный планшет промывали один раз ФСБ и удаляли остаточную жидкость. В каждую лунку добавляли 60 мкл буферного раствора для лизиса (6 М мочевина, 1 мМ ЭДТА, 0,5% Тритон Х-100, 1 мМ фенилметилсульфонилфторид (ФМСФ), смесь ингибиторов протеаз). После лизиса на льду в течение 15 минут добавляли разбавитель (1 мМ этилендиаминтетрауксусная кислота (ЭДТА), 0,5% Тритон Х-100, растворенный в ФСБ). Разведенный лизат клеток переносили в 96-луночный планшет в количестве 100 мкл/лунка, затем планшет инкубировали при комнатной температуре в течение 2 часов. Добавляли разведенное первичное антитело, после чего планшет промывали 4 раза промывочным раствором (фосфатно-солевой буферный раствор с Твином (ФСБТ)). После инкубации в течение 1 часа 96-луночный планшет промывали 4 раза и добавляли второе антитело, затем планшет инкубировали в течение 30 минут. После промывания планшета промывочным раствором добавляли хромогенный раствор тетраметилбензидина (ТМБ) и инкубировали в течение 15 минут. Реакцию останавливали добавлением 1 М H2SO4, затем регистрировали показания оптической плотности при 450 нм.
4. Результат теста
Значения IC50, определенные для действия соединений по настоящему изобретению, вызывающего деградацию ERα, приведены в таблице 3.
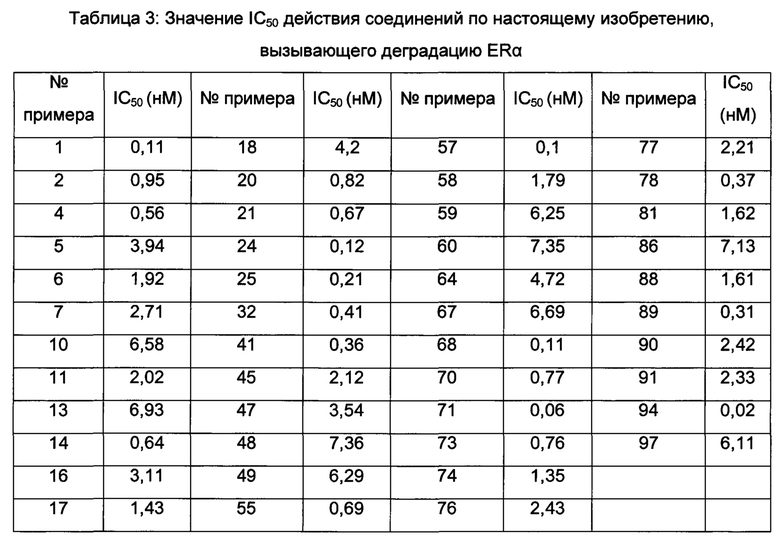
Вывод: Соединения по настоящему изобретению характеризуются значительным действием, вызывающим деградацию ERα.
Фармакокинетический анализ
Тестовый пример 4. Фармакокинетический анализ соединений примеров 10, 14, 20, 21, 57, 68, 71, 73, 74, 76 и 89 по настоящему изобретению
1. Краткий обзор
В качестве экспериментальных животных использовали мышей линии BALB/C. Концентрацию лекарственного средства в плазме крови в различные моменты времени определяли методом жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ/МС/МС после внутрижелудочного введения соединений примеров 10, 14, 20, 21, 57, 68, 71, 73, 74, 76 и 89 бестимусным мышам BALB/C. Фармакокинетические свойства соединений по настоящему изобретению изучали и оценивали на бестимусных мышах BALB/C.
2. Протокол
2.1 Исследуемые образцы
Соединения примеров 10, 14, 20, 21, 57, 68, 71, 73, 74, 76 и 89
2.2 Экспериментальные животные
99 самок бестимусных мышей BALB/C, приобретенных у компании SINO-BRITSH SIPPR/BK LAB, делили на 11 равных групп. ANIMAL LTD., CO, № сертификата: SCXK (Шанхай) 2008-0016.
2.3 Приготовление исследуемых соединений
Взвешивали соответствующее количество каждого исследуемого соединения и последовательно добавляли к навеске смесь 9% полиэтиленгликоля (ПЭГ) 400, 0,5% Твин 80, 0,5% поливинилпирролидона (ПВП) и 90% водного раствора 0,5% карбоксиметилцеллюлозы (КМЦ).
2.4 Введение
После нахождения в состоянии натощак в течение ночи 99 самок бестимусных мышей BALB/C делили на 11 равных групп и вводили исследуемые соединения внутрижелудочно в объеме введения 0,2 мл/10 г.
3. Способ
Брали 0,1 мл крови (3 мыши на каждый момент времени) через 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 11,0 и 24,0 часа после введения. Образцы хранили в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин, чтобы отделить плазму крови. Образцы плазмы крови хранили при -20°C. Концентрацию исследуемых соединений в плазме крови бестимусных мышей BALB/C после внутрижелудочного введения определяли методом ЖХ/МС/МС.
4. Результаты фармакокинетических параметров у бестимусных мышей BALB/C Фармакокинетические параметры соединений примеров 10, 14, 20, 21, 57, 68, 71, 73, 74, 76 и 89 по настоящему изобретению приведены ниже.
Вывод: Соединения по настоящему изобретению характеризуются высокой абсорбцией и обладают значительным фармакологическим эффектом абсорбции.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ БЕНЗОПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2727705C1 |
| ТРИЦИКЛИЧЕСКОЕ ТЕТРАГИДРОИЗОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2830109C1 |
| ПРОИЗВОДНЫЕ С-АРИЛГЛЮКОЗИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2011 |
|
RU2606501C2 |
| МОДУЛЯТОРЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2738646C2 |
| ПРОИЗВОДНОЕ ИНДАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2817737C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДО[3,4-b]ИНДОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2014 |
|
RU2664109C2 |
| ОКСАСПИРОПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЯ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 2016 |
|
RU2733373C2 |
| ПРОИЗВОДНОЕ ОКСОПИКОЛИНАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2742771C2 |
| ПРОИЗВОДНОЕ ИМИДАЗОИЗОИНДОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2717577C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I), к его энантиомеру или к его фармацевтически приемлемой соли, где кольцо А выбрано из группы, состоящей из структур, указанных в формуле изобретения; R представляет собой атом водорода; каждый R1 идентичен другому или отличен от него и каждый выбран из атома водорода и атома галогена; R2 выбран из С1-6-алкила и С3-6-циклоалкила, где каждый указанный С1-6-алкил необязательно замещен 1 группой, выбранной из атома галогена; R3 выбран из С1-6-алкила; каждый R4 идентичен другому или отличен от него и каждый независимо выбран из атома водорода, С3-6-циклоалкила, атома галогена, циано, С2-6-алкинила, гетероциклила, С6-арила, гетероарила, -OR5, -NHC(O)OR5 и -NHC(O)NR6R7, где каждый указанный С2-6-алкинил, гетероциклил, С6-арил и гетероарил необязательно замещен 1-2 группами, выбранными из Rc, С1-6-алкила, галоген-С1-6-алкила, гидрокси-С1-6-алкила, амино, оксо, С3-6-циклоалкила или гетероциклила; Rc представляет собой гетероарил, где указанный гетероарил необязательно замещен 1 группой, выбранной из С1-6-алкила; R5 выбран из атома водорода, С1-6-алкила и гетероциклила, где указанный С1-6-алкил необязательно замещен 1 группой, выбранной из циано и -C(O)NR6R7; R6 и R7 идентичны или отличны друг от друга и каждый выбран из атома водорода, С1-6-алкила и С3-6-циклоалкила; Ra и Rb идентичны или отличны друг от друга и каждый выбран из атома водорода и С1-6-алкила; m равно 0, 1 или 2; n равно 0, 1 или 2; где указанное С6-арильное кольцо может быть конденсировано с 5-6-членным гетероарилом; указанный гетероциклил представляет собой 5-6-членную насыщенную моноциклическую углеводородную группу, имеющую один или два гетероатома, выбранных из азота и кислорода, или указанное гетероциклильное кольцо может быть конденсировано с пиридином; и указанный гетероарил представляет собой 5-6-членную гетероароматическую систему, имеющую от 1 до 4 гетероатомов, выбранных из азота, кислорода и серы, или указанное гетероарильное кольцо может быть конденсировано с фенилом или пиридином. Также изобретение относится к промежуточному соединению формулы (V), способу получения целевого соединения из соединения формулы (V), фармацевтической композиции на основе соединения формулы (I), применению соединения формулы (I) или композиции на его основе, а также к способу лечения заболевания, опосредованного рецептором эстрогена. Технический результат: получены новые производные конденсированного пиперидина, полезные в качестве модулятора рецептора эстрогена в лечении опосредованных рецептором эстрогена или зависимых от рецептора эстрогена заболеваний или состояний. 8 н. и 13 з.п. ф-лы, 3 табл., 103 пр.
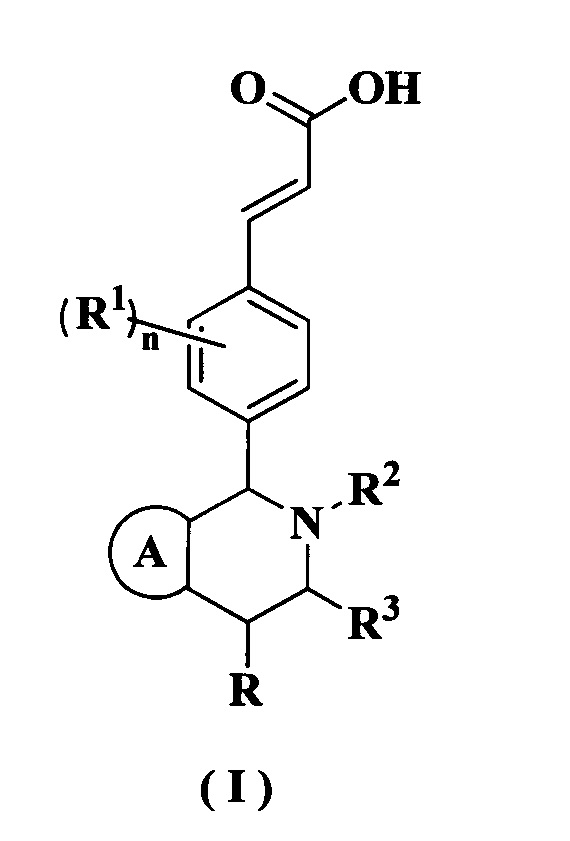
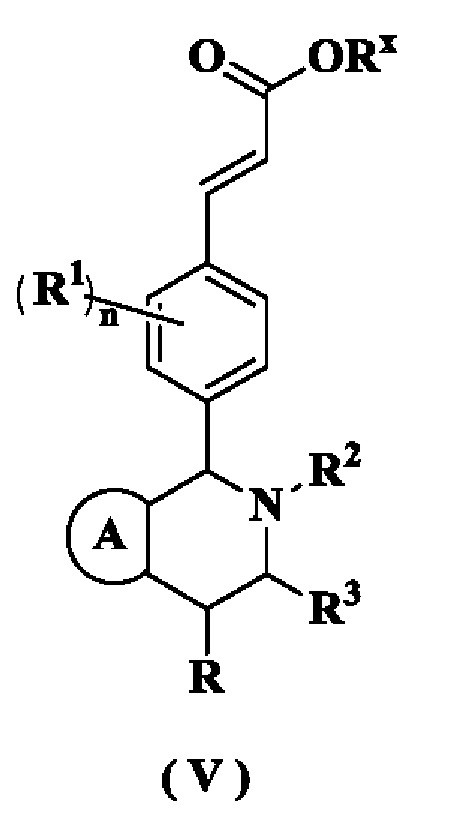
1. Соединение формулы (I),
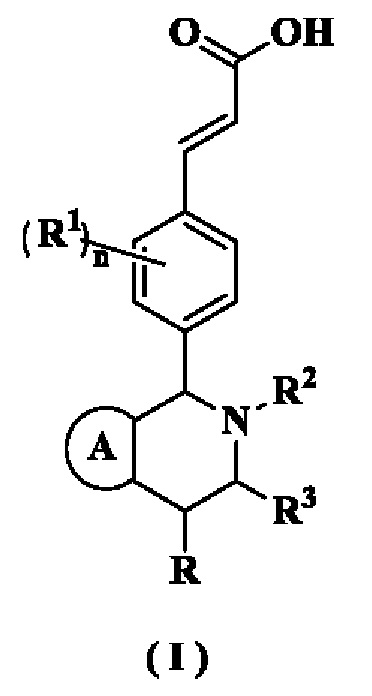
или его энантиомер, или их фармацевтически приемлемая соль,
где
кольцо А выбрано из группы, состоящей из:
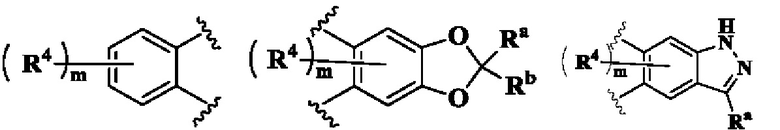
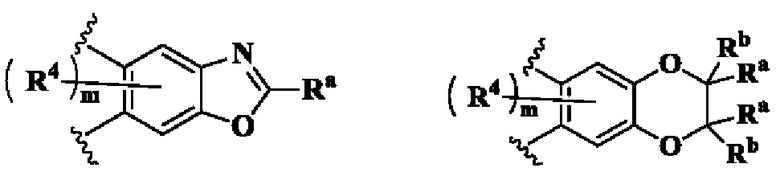
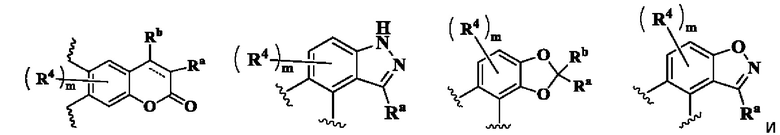
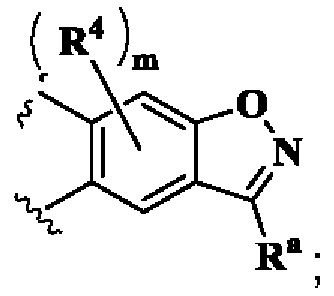
R представляет собой атом водорода;
каждый R1 идентичен другому или отличен от него и каждый независимо выбран из группы, состоящей из атома водорода и атома галогена;
R2 выбран из группы, состоящей из С1-6-алкила и С3-6-циклоалкила, где каждый указанный С1-6-алкил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена;
R3 выбран из группы, состоящей из С1-6-алкила;
каждый R4 идентичен другому или отличен от него и каждый независимо выбран из группы, состоящей из атома водорода, С3-6-циклоалкила, атома галогена, циано, С2-6-алкинила, гетероциклила, С6-арила, гетероарила, -OR5, -NHC(O)OR5 и -NHC(O)NR6R7, где каждый указанный С2-6-алкинил, гетероциклил, С6-арил и гетероарил необязательно замещен одной или двумя группами, выбранными из группы, состоящей из Rc, С1-6-алкила, галоген-С1-6-алкила, гидрокси-С1-6-алкила, амино, оксо, С3-6-циклоалкила или гетероциклила;
Rc представляет собой гетероарил, где указанный гетероарил необязательно замещен одной группой, выбранной из С1-6-алкила;
R5 выбран из группы, состоящей из атома водорода, С1-6-алкила и гетероциклила, где указанный С1-6-алкил необязательно замещен одной группой, выбранной из группы, состоящей из циано и -C(O)NR6R7;
R6 и R7 идентичны или отличны друг от друга и каждый независимо выбран из группы, состоящей из атома водорода, С1-6-алкила и С3-6-циклоалкила;
Ra и Rb идентичны или отличны друг от друга и каждый независимо выбран из группы, состоящей из атома водорода и С1-6-алкила;
m равно 0, 1 или 2;
n равно 0, 1 или 2;
где указанное С6-арильное кольцо может быть конденсировано с 5-6-членным гетероарилом;
указанный гетероциклил представляет собой 5-6-членную насыщенную моноциклическую углеводородную группу, имеющую один или два гетероатома, выбранных из азота и кислорода, или указанное гетероциклильное кольцо может быть конденсировано с пиридином; и
указанный гетероарил представляет собой 5-6-членную гетероароматическую систему, имеющую от 1 до 4 гетероатомов, выбранных из азота, кислорода и серы, или указанное гетероарильное кольцо может быть конденсировано с фенилом или пиридином.
2. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по п.1, где n равно 2.
3. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по любому из пп. 1, 2, где R1 представляет собой атом галогена.
4. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по любому из пп. 1-3, где R2 представляет собой С1-6-алкил, где указанный С1-6-алкил необязательно замещен одной группой, выбранной из группы, состоящей из атома галогена.
5. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по п. 1, необязательно выбранные из группы, состоящей из соединения формулы (I-A), формулы (I-B) и формулы (I-C):
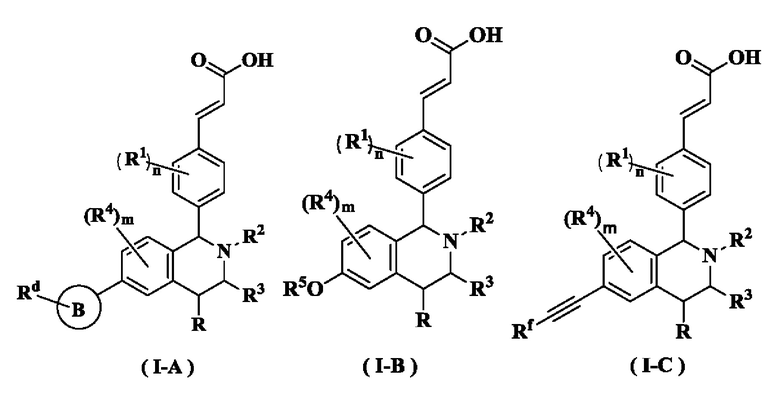
или его энантиомера, или их фармацевтически приемлемой соли,
где
кольцо В выбрано из группы, состоящей из С3-6-циклоалкила, гетероциклила, С6-арила и гетероарила;
Rd выбран из группы, состоящей из атома водорода, С1-6-алкила, галоген-С1-6-алкила, оксо, амино, С3-6-циклоалкила и гетероциклила, где указанный С1-6-алкил необязательно замещен одной группой, выбранной из группы, состоящей из гидрокси;
Rf выбран из группы, состоящей из гетероарила, где указанный гетероарил необязательно замещен С1-6-алкилом;
где указанное С6-арильное кольцо может быть конденсировано с 5-6-членным гетероарилом;
указанный гетероциклил представляет собой 5-6-членную насыщенную моноциклическую углеводородную группу, имеющую один или два гетероатома, выбранных из азота и кислорода, или указанное гетероциклильное кольцо может быть конденсировано с пиридином;
указанный гетероарил представляет собой 5-6-членную гетероароматическую систему, имеющую от 1 до 4 гетероатомов, выбранных из азота, кислорода и серы, или указанное гетероарильное кольцо может быть конденсировано с фенилом или пиридином; и
радикалы с R по R5, m и n являются такими, как определено в п. 1.
6. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по п. 1, необязательно выбранные из группы, состоящей из соединения формулы (II) и формулы (III):
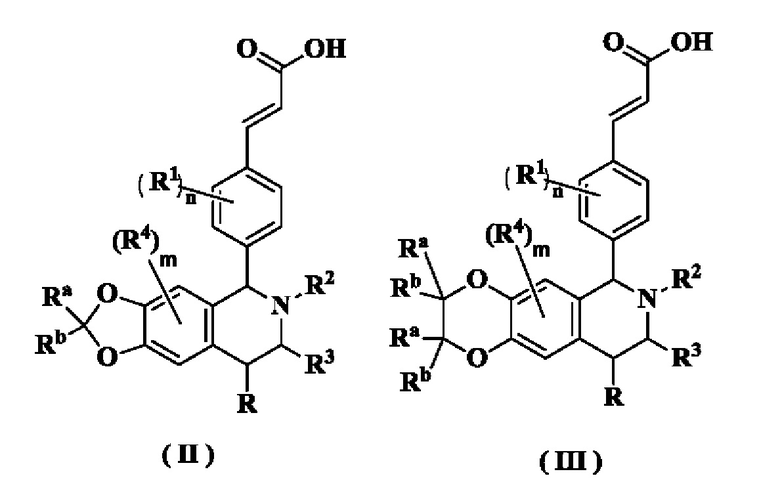
или его энантиомера, или их фармацевтически приемлемой соли,
где
радикалы с R по R4, Ra, Rb, m и n являются такими, как определено в п. 1.
7. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по п. 1, необязательно представляющие собой соединение формулы (I-D):
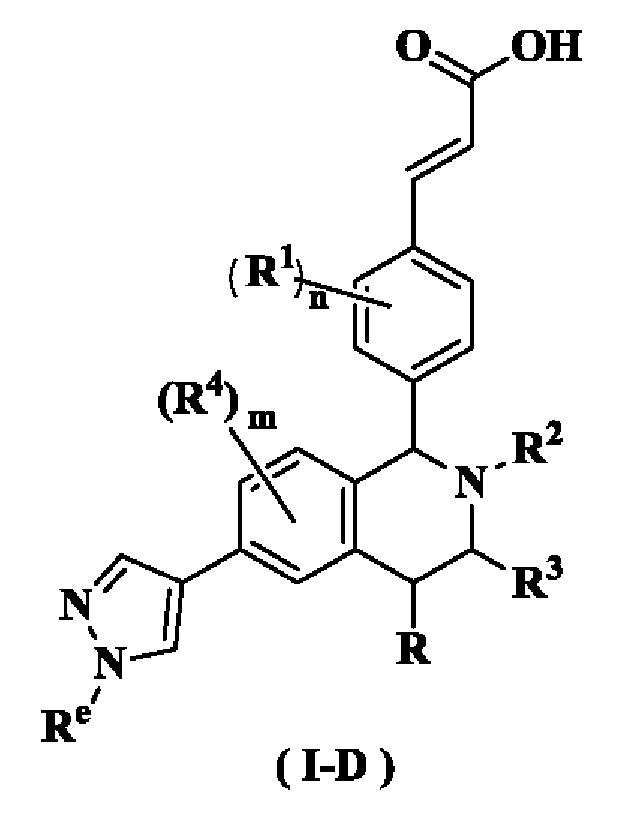
или его энантиомер, или их фармацевтически приемлемая соль,
где
Re выбран из группы, состоящей из С1-6-алкила, галоген-С1-6-алкила, гидрокси-С1-6-алкила, и С3-6-циклоалкила; и
радикалы с R по R5, m и n являются такими, как определено в п. 1.
8. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по п. 1, необязательно представляющие собой соединение формулы (I-I):
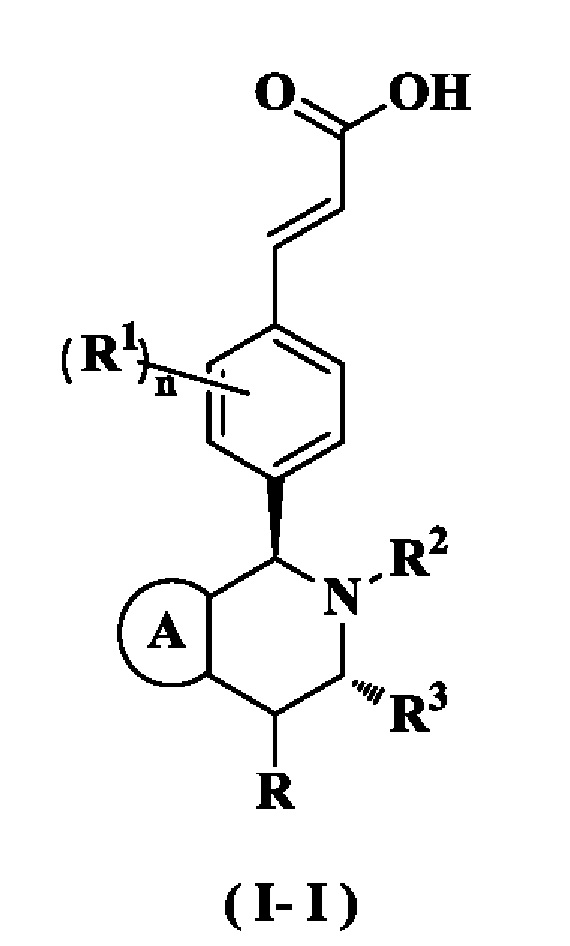
или его энантиомер, или их фармацевтически приемлемая соль,
где
кольцо А, радикалы с R по R3 и n являются такими, как определено в п. 1.
9. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по любому из пп. 1-8, где указанное соединение выбрано из группы, состоящей из:
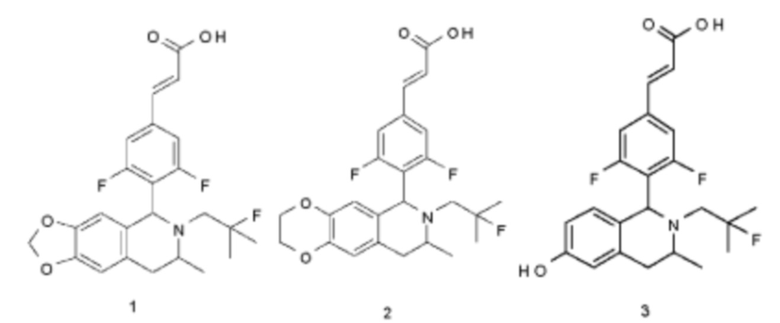
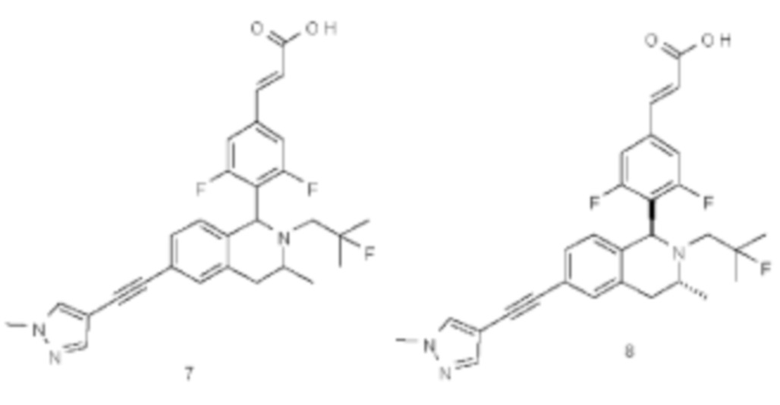
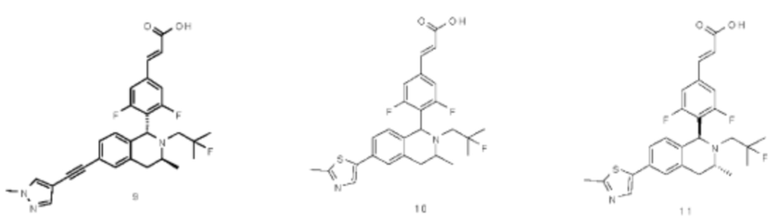
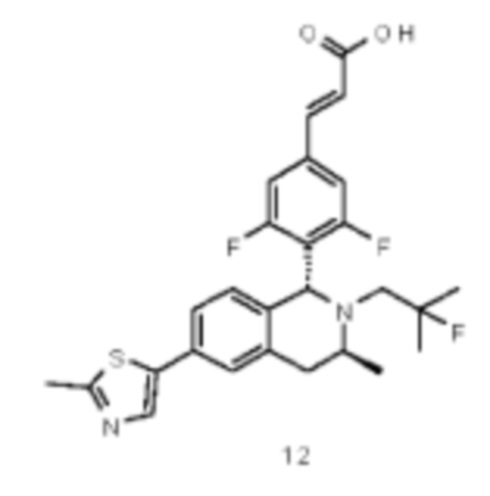
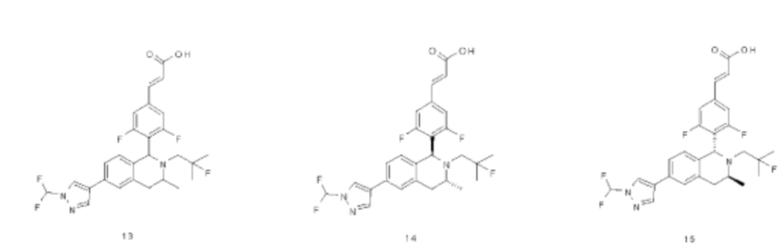
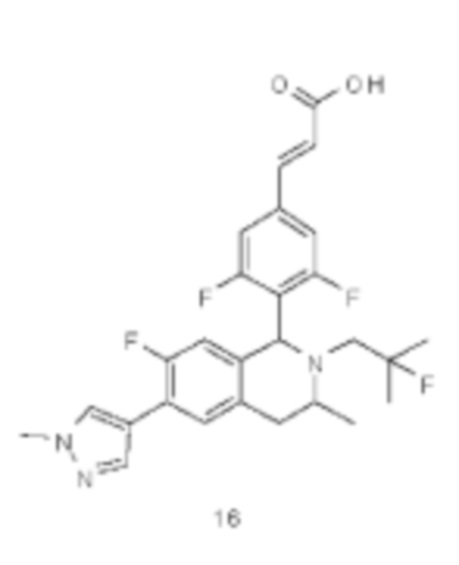
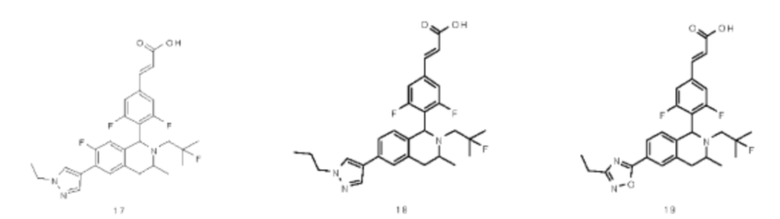
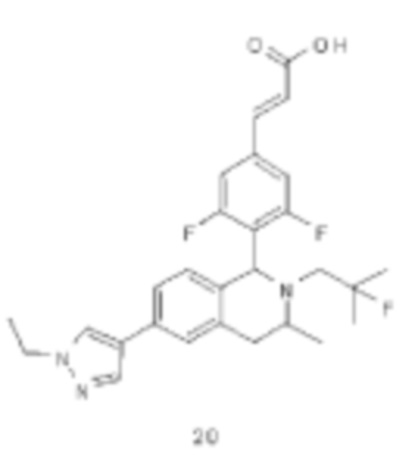
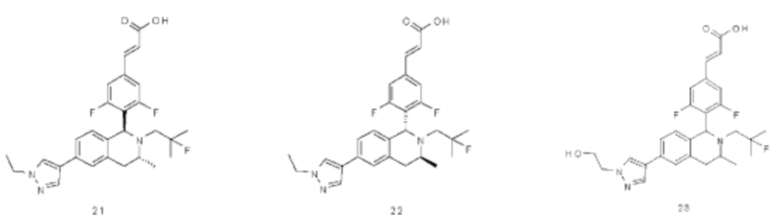
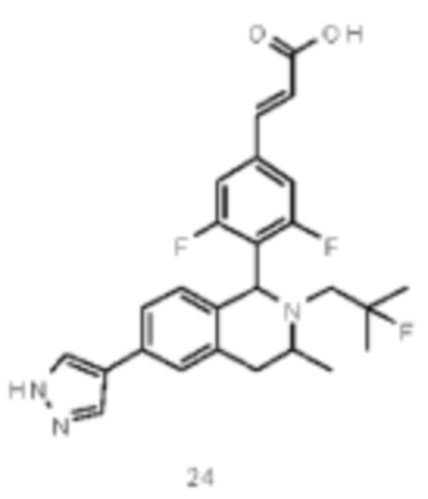
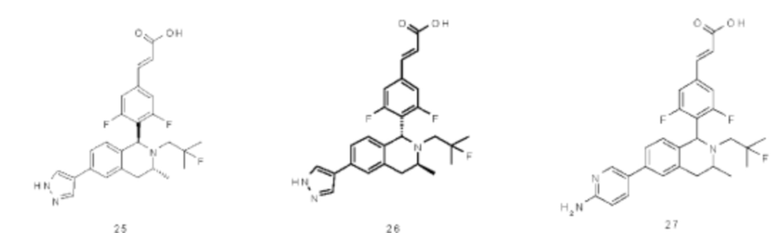
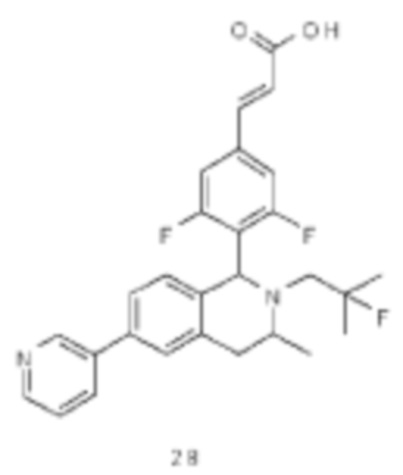
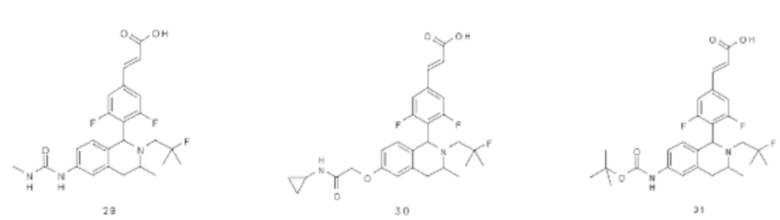
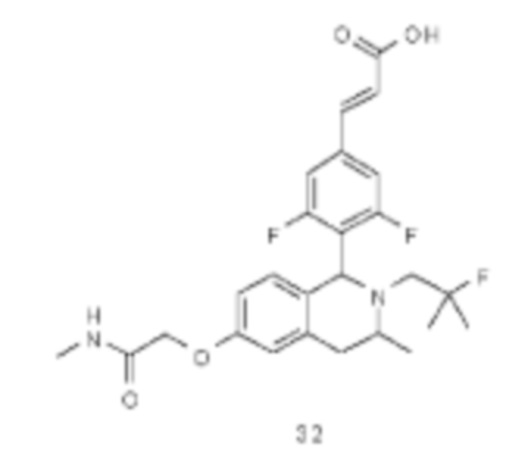
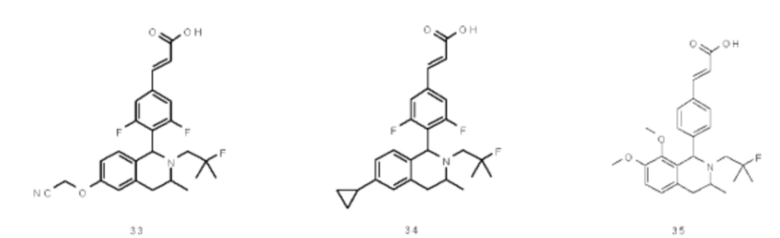
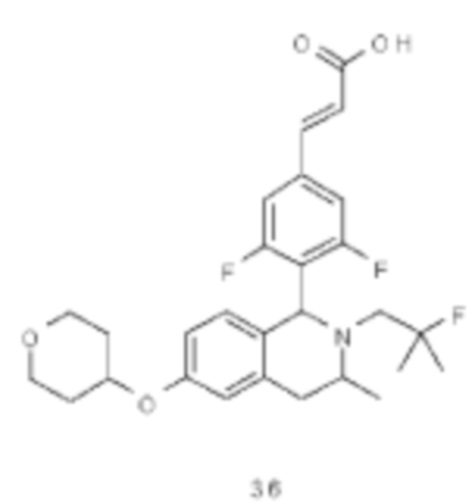
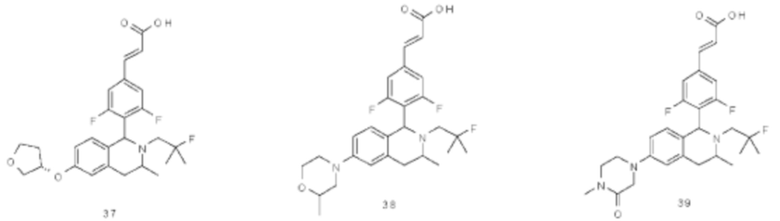
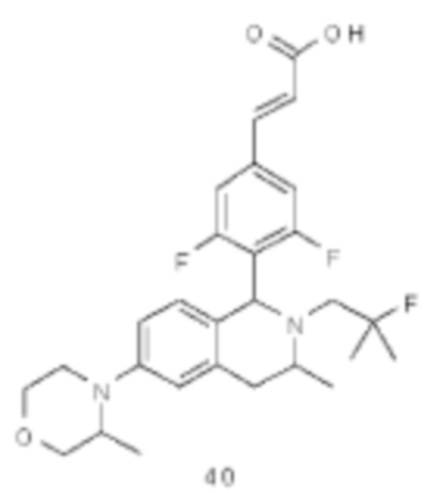
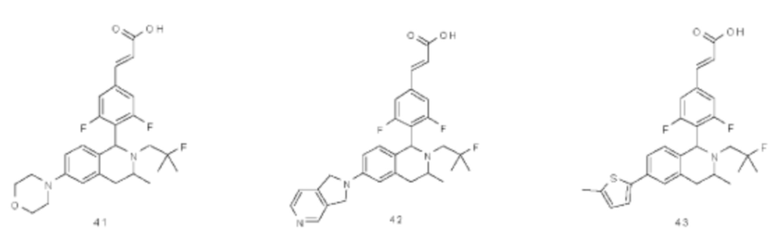

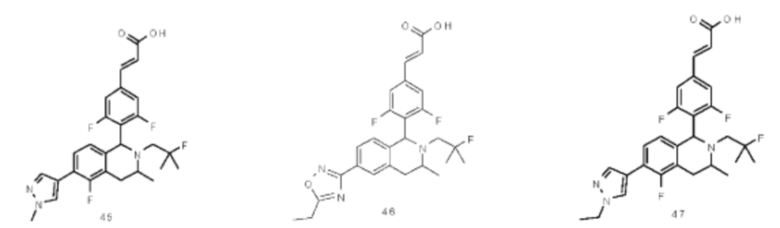
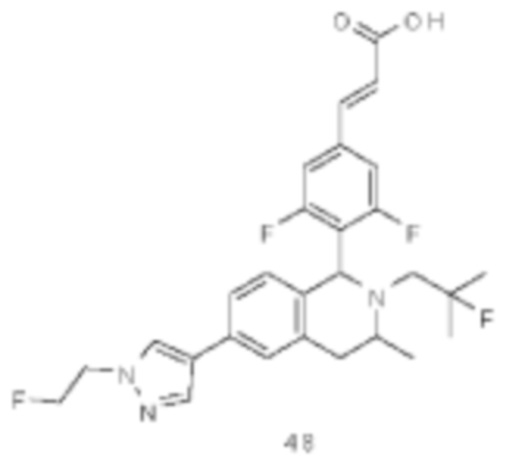
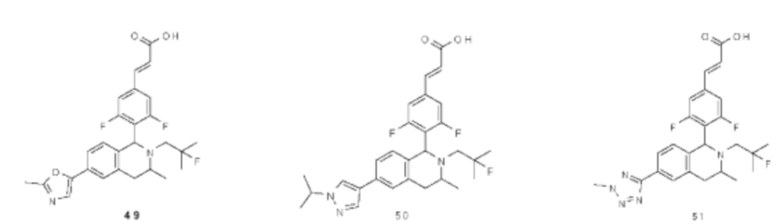
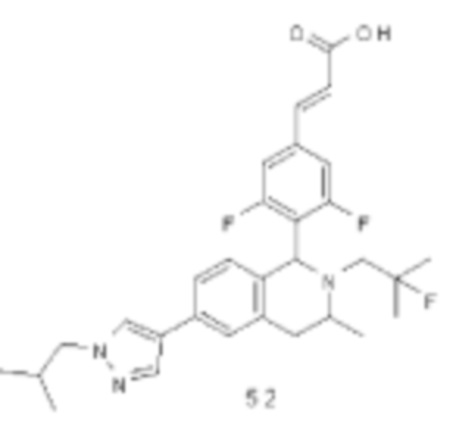
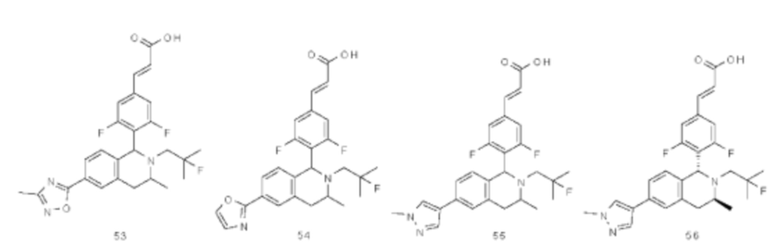
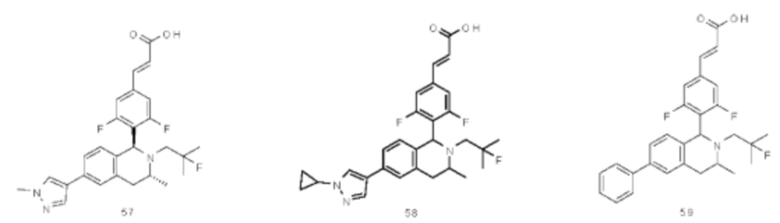
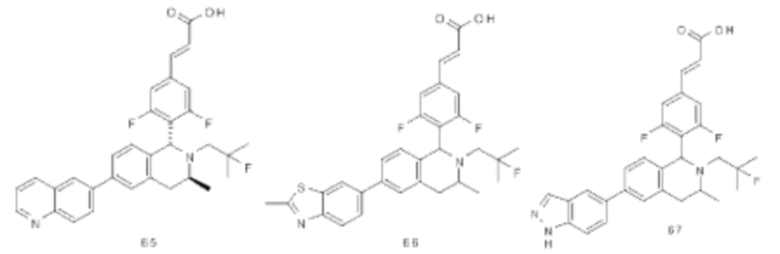
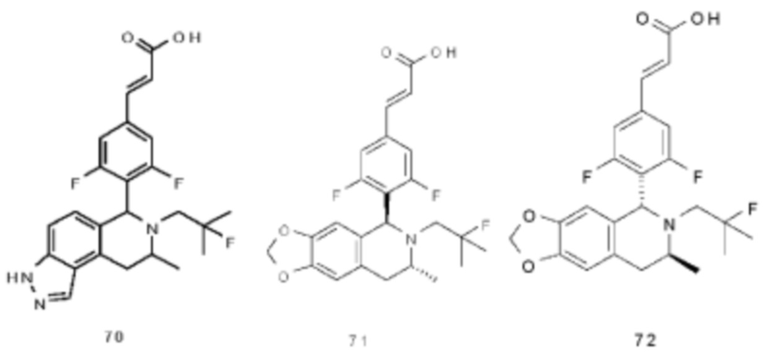
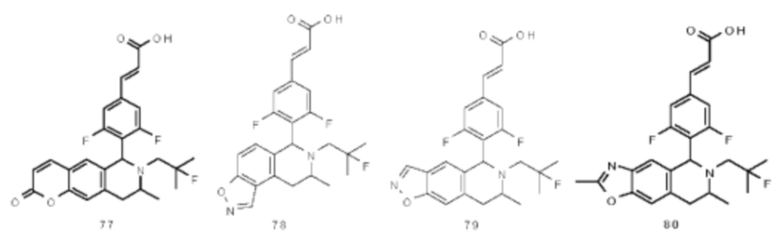
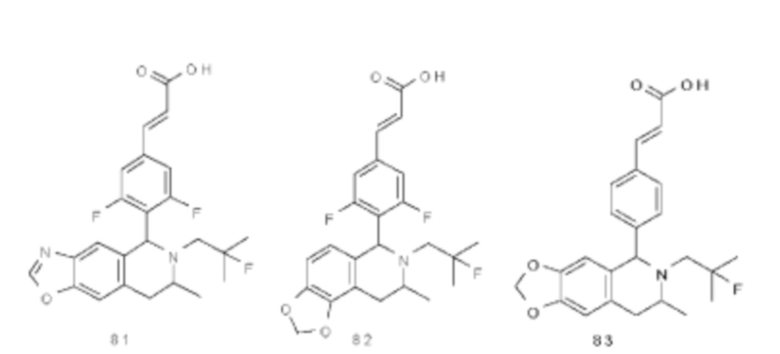
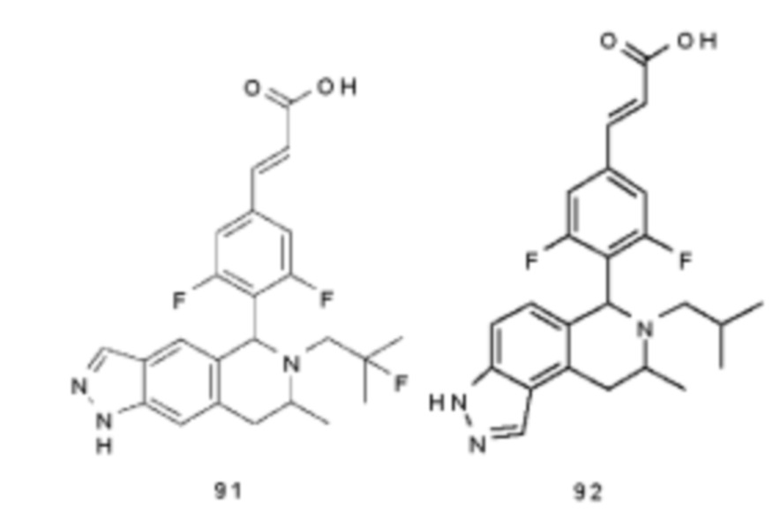
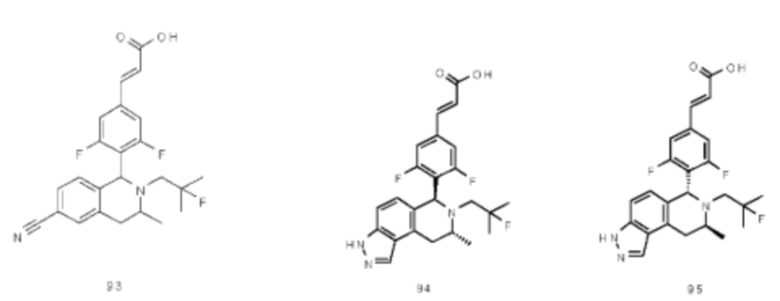
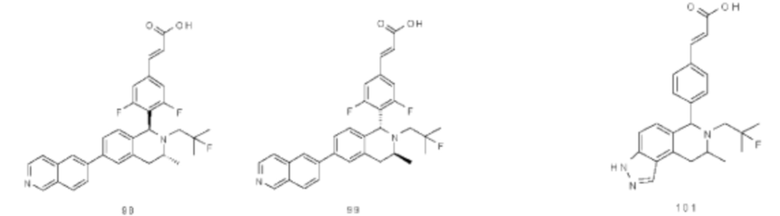
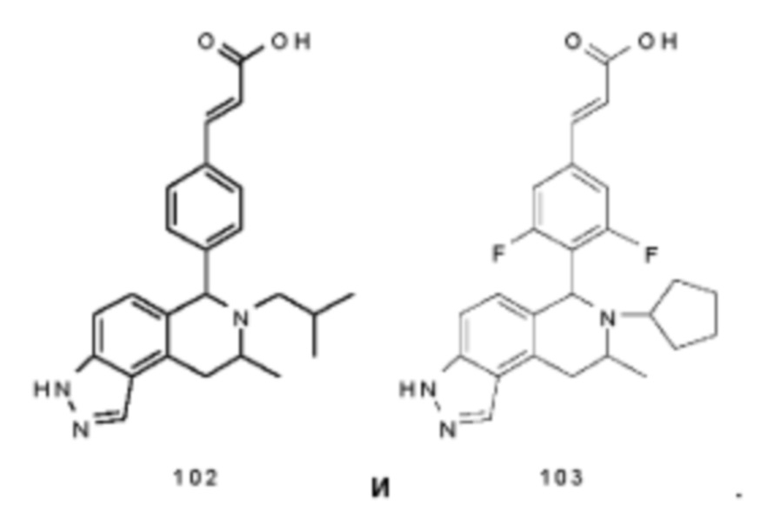
10. Соединение формулы (V):
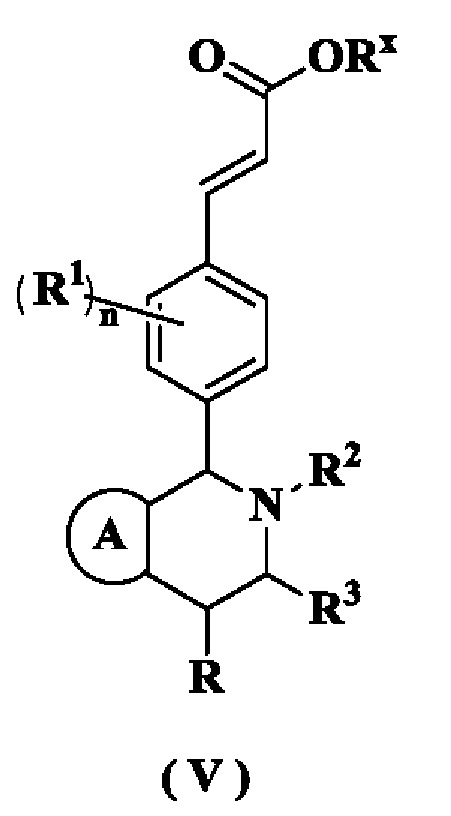
или его энантиомер, или их фармацевтически приемлемая соль,
где
Rx представляет собой С1-6-алкил;
кольцо A, R, радикалы с R1 по R3 и n являются такими, как определено в п. 1.
11. Способ получения соединения формулы (I) или его энантиомера, либо их фармацевтически приемлемой соли, включающий стадию:
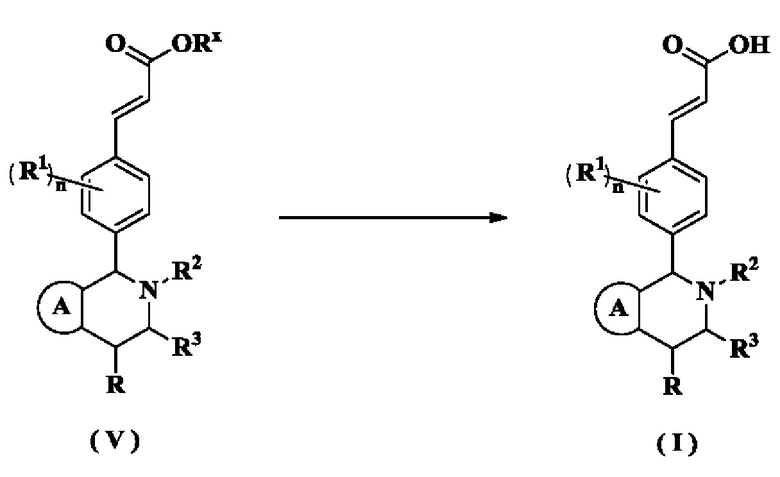
гидролиза соединения формулы (V) в щелочных условиях с получением соединения формулы (I);
где
кольцо A, R, радикалы с R1 по R3 и n являются такими, как определено в п. 1, Rx является таким, как определено в п. 10.
12. Фармацевтическая композиция, обладающая антагонистической активностью в отношении рецептора эстрогена, содержащая терапевтически эффективное количество соединения формулы (I) или его энантиомера, либо их фармацевтически приемлемой соли по любому из пп. 1-9 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
13. Применение соединения формулы (I) или его энантиомера, либо их фармацевтически приемлемой соли по любому из пп. 1-9 или фармацевтической композиции по п. 12 в получении модулятора рецептора эстрогена.
14. Применение соединения формулы (I) или его энантиомера, либо их фармацевтически приемлемой соли по любому из пп. 1-9 или фармацевтической композиции по п. 12 в получении лекарственного средства для лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена.
15. Применение по п. 14, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, выбрано из группы, состоящей из рака, нарушений центральной нервной системы, нарушений сердечно-сосудистой системы, иммунных и воспалительных заболеваний, подверженности инфекции, нарушений метаболизма, неврологических нарушений, нарушений психики и репродуктивных нарушений.
16. Применение по п. 15, где рак выбран из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, рака фаллопиевой трубы, оофоромы и лейкоза; предпочтительно рака молочной железы, рака яичника, рака эндометрия, рака предстательной железы или рака матки; более предпочтительно рака молочной железы.
17. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по любому из пп. 1-9 или фармацевтическая композиция по п. 12 для применения в качестве лекарственного средства для лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена.
18. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по любому из пп. 1-9 или фармацевтическая композиция по п. 12 для применения в качестве лекарственного средства для лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, выбрано из группы, состоящей из рака, нарушений центральной нервной системы, нарушений сердечно-сосудистой системы, иммунных и воспалительных заболеваний, подверженности инфекции, нарушений метаболизма, неврологических нарушений, нарушений психики и репродуктивных нарушений.
19. Соединение формулы (I) или его энантиомер, либо их фармацевтически приемлемая соль по любому из пп. 1-9 или фармацевтическая композиция по п. 12 для применения в качестве лекарственного средства для лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, представляет собой рак, выбранный из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, рака фаллопиевой трубы, оофоромы и лейкоза; предпочтительно рака молочной железы, рака яичника, рака эндометрия, рака предстательной железы или рака матки; более предпочтительно рака молочной железы.
20. Способ лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, включающий стадию введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его энантиомера, либо их фармацевтически приемлемой соли по любому из пп. 1-9 или фармацевтической композиции по п. 12, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, выбрано из группы, состоящей из рака, нарушений центральной нервной системы, нарушений сердечно-сосудистой системы, иммунных и воспалительных заболеваний, подверженности инфекции, нарушений метаболизма, неврологических нарушений, нарушений психики и репродуктивных нарушений.
21. Способ лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, включающий стадию введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его энантиомера, либо их фармацевтически приемлемой соли по любому из пп. 1-9 или фармацевтической композиции по п. 12, где заболевание или состояние, опосредованное рецептором эстрогена или зависимое от рецептора эстрогена, представляет собой рак, выбранный из группы, состоящей из рака молочной железы, рака эндометрия, рака шейки матки, рака кожи, рака предстательной железы, рака яичника, рака фаллопиевой трубы, оофоромы и лейкоза; предпочтительно рака молочной железы, рака яичника, рака эндометрия, рака предстательной железы или рака матки; более предпочтительно рака молочной железы.
| WO 2014191726 А1, 04.12.2014 | |||
| О.Н.Зефирова и др.: "Об истории возникновения и развития концепции биоизостеризма", Вестник Московского Университета, сер.2, Химия, 2002, т.43 | |||
| Каток для обработки почвы | 1983 |
|
SU1113007A1 |
| ПРОИЗВОДНЫЕ АКРИЛОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФУНГИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ БОРЬБЫ С ГРИБАМИ | 1993 |
|
RU2119483C1 |

