Область изобретения
Данное изобретение относится к новым соединениям замещенных эфиров фенилуксусной кислоты, применимым в качестве седативных снотворных агентов кратковременного действия для анестезии и седативного эффекта. Кроме того, данное изобретение относится к фармацевтическим композициям, содержащим подобные соединения, способам использования подобных соединений для создания или поддержания анестезии или седативного эффекта и промежуточным соединениям для получения подобных соединений.
Состояние области техники
Пропофол, 2,6-диизопропилфенол (Diprivan® Injectable Emulsion, AstraZeneca), представляет собой анестезирующее средство для инъекций, обладающее снотворными свойствами. Оно может быть использовано для создания или поддержания общей анестезии и для седативного эффекта. Несмотря на то, что пропофол является широко используемым анестетиком, его применимость до некоторой степени ограничена по причине длительной и непредсказуемой продолжительности его действия после инфузии. Подобная непредсказуемая продолжительность действия приводит к нерегулярным и часто длительным периодам восстановления пациента, которые нежелательны.
Пропанидид (пропиловый эфир [4-[(N,N-диэтилкарбамоил)метокси]-3-метоксифенил]уксусной кислоты является другим инъецируемым анестезирующим средством, которое было одобрено для использования в нескольких странах за пределами Соединенных Штатов. Хотя пропанидид обеспечивает намного более короткий и предсказуемый период восстановления, чем пропофол, он не является эффективным анестезирующим средством. Кроме того, Epontol®, инъецируемый эмульсионный лекарственный препарат пропанидида, предлагаемый фирмой Байер, был изъят с рынка в Великобритании в 1983 из-за вопроса относительно анафилактоидных реакций. Таким образом, несмотря на то, что пропанидид обеспечивает более короткие и более предсказуемые периоды восстановления, чем пропофол, он не был широко принят в качестве инъецируемого анестезирующего средства.
В настоящее время есть необходимость в новых инъецируемых анестезирующих агентах для инъекций. Предпочтительные агенты будут обладать более короткой и более предсказуемой продолжительностью действия, чем пропофол. Кроме того, предпочтительные агенты будут эффективнее пропанидида.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Заявителями найдены новые соединения замещенных эфиров фенилуксусных кислот, применимые в качестве седативных снотворных агентов кратковременного действия. Данные агенты обладают более короткой и более предсказуемой продолжительностью действия, чем пропофол, а также более эффективны, чем пропанидид.
Соответственно, в изобретении предлагается соединение формулы (I):
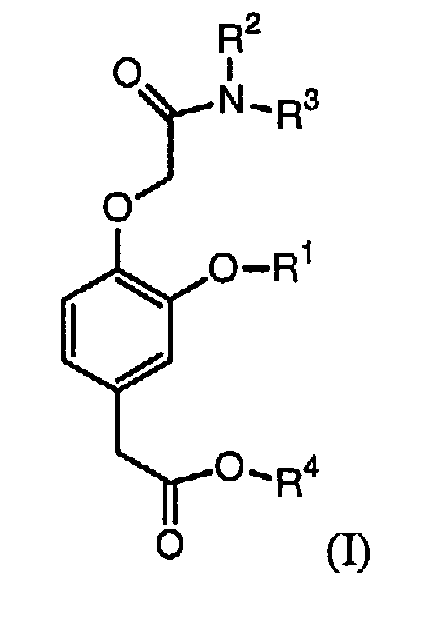
в которой:
R1 выбирают из группы, включающей (С2-С6)алкил, (С2-С6)алкенил, (С2-С6)алкинил, (С3-С6)циклоалкил(С1-С6)алкил, фенил и бензил,
каждый из R2 и R3 независимо выбирают из группы, включающей (С1-С6)алкил, (С2-С6)алкенил и (С2-С6)алкинил, или R2 и R3 вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, содержащее от 5 до 7 атомов, а
R4 выбирают из группы, включающей (С1-С6)алкил, (С2-С6)алкенил и (С2-С6)алкинил,
при условии, что сумма числа атомов углерода в R1, R2, R3 и R4 больше 7.
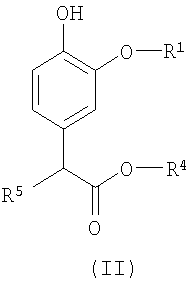
Изобретение относится также к промежуточным соединениям для получения соединений формулы (I). Соответственно, в изобретении предлагается соединение формулы (II):
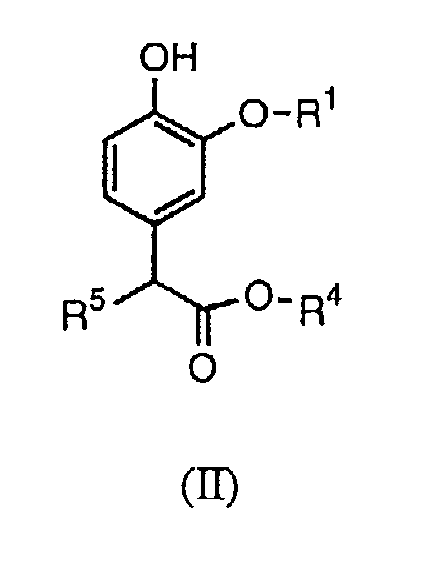
в которой R1 и R4 определены выше, а R5 представляет собой водород или гидроксил.
Кроме того, в изобретении предлагается фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы (I).
Соединения по изобретению являются высокоэффективными седативными снотворными агентами кратковременного действия для использования при создании или поддержании анестезии и седативного эффекта. Соответственно, в изобретении также предлагается способ создания или поддержания анестезии или седативного эффекта у млекопитающего, включающий введение данному млекопитающему эффективного количества соединения по изобретению. В изобретении также предлагается способ создания или поддержания анестезии или седативного эффекта у млекопитающего, включающий введение данному млекопитающему эффективного количества фармацевтической композиции изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
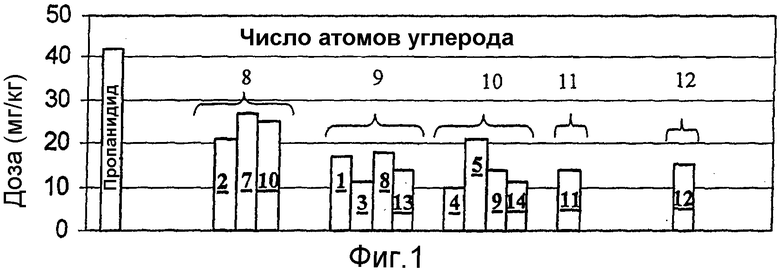
На фиг.1 приведено сопоставление дозы соединений изобретения в мг/кг, приводящей к средней потере рефлекса выпрямления за две минуты у крыс, с требуемой дозой соединения предшествующего уровня - пропанидида.
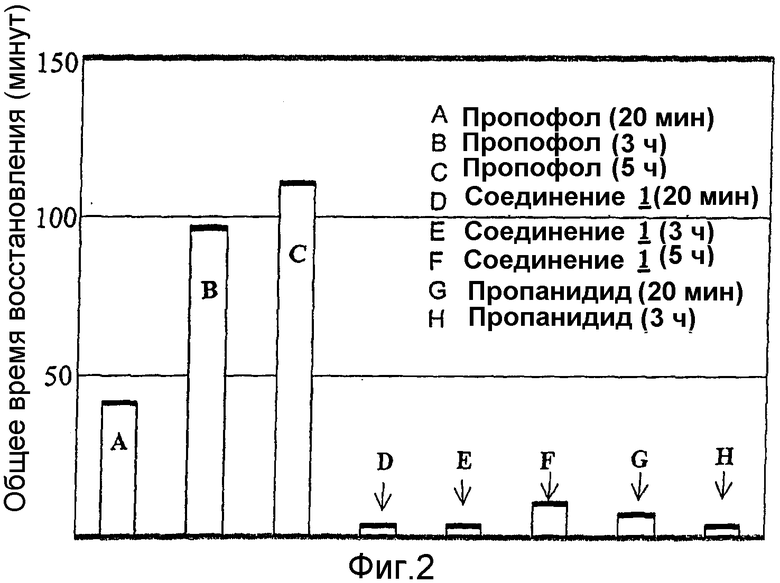
На фиг.2 приведено сопоставление общего периода восстановления в минутах после окончания инфузии соединения 1 настоящего изобретения в течение 20 минут, 3 часов и 5 часов у крыс с периодом восстановления после окончания инфузии соединений пропанидида и пропофола предшествующего уровня.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
При описании соединений, композиций и способов изобретения следующие термины имеют следующие значения, если не указано иначе.
Термин «(С1-С6)алкил» относится к монорадикальной разветвленной или неразветвленной насыщенной углеводородной цепи, содержащей от 1 до 6 атомов углерода. Примерами данного термина являются такие группы, как метильная, этильная, н-пропильная, изопропильная, н-бутильная, изобутильная, н-гексильная и так далее. Как использовано здесь, «Ме» представляет собой метил, «Et» представляет собой этил, «пропил» и «Pr» представляют собой н-пропил, а «iPr» представляет собой изопропил.
Термин «(С2-С6)алкенил» относится к монорадикалу разветвленной или неразветвленной ненасыщенной углеводородной группы, содержащему от 2 до 6 атомов углерода, и содержащему, по меньшей мере, 1 участок винильной ненасыщенности. Предпочтительные алкенильные группы включают этенильную (-СН=СН2), н-пропенильную (-СН2СН=СН2), изопропенильную (-С(СН3)=СН2) и так далее.
Термин «(С2-С6)алкинил» относится к монорадикалу ненасыщенного углеводорода, содержащему от 2 до 6 атомов углерода, и содержащему, по меньшей мере, 1 тройную связь. Предпочтительные алкинильные группы включают этинильную (-С≡СН), пропаргильную (-СН2С≡СН) и так далее.
Термин «(С3-С6)циклоалкил» относится к циклическим алкильным группам, содержащим от 3 до 6 атомов углерода, состоящим из одного циклического кольца. Подобные циклоалкильные группы включают, в качестве примера, моноциклические структуры, такие, как циклопропил, циклобутил, циклопентил, циклогексил и так далее.
Термин «(С3-С6)циклоалкил(С1-С6)алкил» относится к группе формулы (С3-С6)циклоалкил(С1-С6)алкил-, в которой (С3-С6)циклоалкил и (С1-С6)алкил определены выше.
Соединения данного изобретения могут содержать один или более хиральных центров. Соответственно подразумевается, что настоящее изобретение включает в себя рацемические смеси, диастереомеры, энантиомеры и смеси, обогащенные одним или более стереоизомерами. В описанную и заявленную область изобретения входят рацемические формы соединений, а также индивидуальные энантиомеры и их нерацемические смеси.
Термин «снотворный агент», обычно относится к соединению, способствующему засыпанию. Как используется в фармакологии, термином «снотворные агенты» описывают агенты, используемые для создания или поддержания анестезии, седативного эффекта, или сна.
Используемый здесь термин «анестезия» относится к потере чувствительности или сознания, происходящей по причине фармакологического подавления нервной деятельности.
Термин «седативный эффект» определен здесь как успокаивание психического возбуждения или снижение физиологической деятельности путем введения лекарственного средства.
Термин «эффективное количество» относится к такому количеству, которое эффективно для создания или поддержания анестезии или седативного эффекта при введении млекопитающему. Эффективное количество будет изменяться в зависимости от субъекта и способа введения и может быть определено обычным образом специалистом в данной области.
Термин «болеутоляющее средство» относится к соединению, которое облегчает боль за счет изменения восприятия болевых стимулов, не вызывая при этом значительной анестезии или потери сознания.
Термин «опиоид» относится к синтетическим наркотическим веществам, обладающим опиатоподобной активностью (например, обезболивающим действием), но полученными не из опиума.
Используемый здесь термин «кратковременное действие» относится к агентам, которые являются фармакокинетически восприимчивыми. При введении агентов кратковременного действия путем инфузии действие данных агентов заканчивается сразу же по окончании инфузии.
Хотя в кратком описании изобретения приведено широкое определение, некоторые агенты или композиции могут быть предпочтительными. Конкретные и предпочтительные значения, приведенные здесь для радикалов, заместителей и интервалов являются лишь иллюстративными; они не исключают других определенных значений внутри определенных интервалов для радикалов и заместителей.
Предпочтительный агент, который может быть включен в композиции изобретения, и который может быть введен в соответствии со способами данного изобретения, представляет собой соединение формулы (I), описанное выше, в котором сумма числа атомов углерода в R1, R2, R3 и R4 изменяется в пределах от 8 до 15.
Более предпочтительно, сумма числа атомов углерода в R1, R2, R3 и R4 изменяется в пределах от 8 до 12.
Предпочтительно, R1 выбирают из группы, включающей (С2-С6)алкил, (С2-С6)алкенил и (С2-С6)алкинил.
В другом предпочтительном способе воплощения R1 выбирают из группы, включающей (С3-С6)циклоалкил, фенил и бензил.
В следующем более предпочтительном способе воплощения R1 выбирают из группы, включающей (С2-С4)алкил, (С2-С4)алкенил и (С2-С4)алкинил.
В следующем более предпочтительном способе воплощения R1 выбирают из группы, включающей (С2-С4)алкил, циклопропил и циклобутил.
Даже более предпочтительно, R1 представляет собой (С2-С4)алкил.
Наиболее предпочтительно, R1 представляет собой этил или пропил.
Предпочтительно, R2 выбирают из группы, включающей (С1-С4)алкил, (С2-С4)алкенил и (С2-С4)алкинил.
В альтернативном, предпочтительном способе воплощения R2 и R3 вместе с атомом азота, с которым они связаны, образуют пиперидинильный цикл.
Более предпочтительно, R2 представляет собой (С1-С4)алкил.
Предпочтительно, R3 выбирают из группы, включающей (С1-С4)алкил, (С2-С4)алкенил и (С2-С4)алкинил.
Более предпочтительно, R3 представляет собой (С1-С4)алкил.
Предпочтительно, R4 выбирают из группы, включающей (С1-С4)алкил, (С2-С4)алкенил и (С2-С4)алкинил.
Более предпочтительно, R4 представляет собой (С1-С4)алкил.
В предпочтительном способе воплощения R1 представляет собой (С2-С4)алкил, каждый из R2 и R3 независимо представляет собой (С1-С4)алкил, а R4 представляет собой (С1-С4)алкил.
Предпочтительной подгруппой соединений является подгруппа, в которой R1 представляет собой (С2-С4)алкил, каждый из R2 и R3 независимо представляет собой (С1-С4)алкил, R4 представляет собой (С1-С4)алкил, а сумма числа атомов углерода в R1, R2, R3 и R4 изменяется в пределах от 8 до 12.
В пределах данной подгруппы R1 предпочтительно представляет собой этил или пропил, каждый из R2, R3 и R4 независимо выбирают из группы, включающей метил, этил и пропил, а сумма числа атомов углерода в R1, R2, R3 и R4 изменяется в пределах от 8 до 11. Конкретными предпочтительными значениями для суммы числа атомов углерода являются 9, 10 и 11.
Предпочтительными соединениями изобретения являются соединения формулы (I), в которых R1, R2, R3 и R4 представляют собой значения, приведенные ниже в таблице 1.
Особенно предпочтительными являются соединения, в которых R1 представляет собой этил или пропил, каждый из R2 и R3 представляет собой этил, а R4 представляет собой пропил. В частности, наиболее предпочтительным является соединение 1.
Общие методики синтеза
Промежуточные соединения и соединения настоящего изобретения можно получить из легко доступных исходных веществ по известным методикам синтеза. Например, эти соединения можно получить, как в общем виде описано ниже, и, кроме того, описано в примерах. Понятно, что в случаях, в которых приведены типичные или предпочтительные условия способа (то есть температура, продолжительность реакции, молярные соотношения реагентов, давление и так далее), можно также использовать и другие условия способа, если не указано иначе. Оптимальные условия реакции можно варьировать в зависимости от конкретных используемых реагентов или растворителя, но подобные условия может определить специалист в данной области при помощи обычных методик оптимизации.
Кроме того, как очевидно для специалиста в данной области, для предотвращения протекания нежелательных реакций по некоторым функциональным группам, могут потребоваться обычные защитные группы. Выбор подходящей защитной группы для конкретной функциональной группы, а также подходящие условия для защиты и снятия защиты хорошо известны в данной области. Например, многочисленные защитные группы и их введение и удаление описаны у T.W. Greene and G.M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999 и в приведенных там ссылках.
В настоящих способах синтеза используются новые промежуточные соединения формулы (II), в частности (IIa) или (IIb):
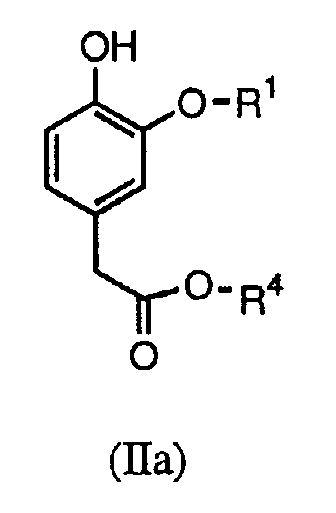
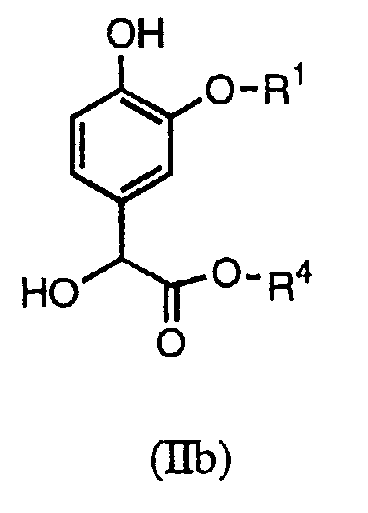
В первом способе синтеза соединения формулы (I) получают алкилированием соединения формулы (IIa) требуемым соединением формулы X-CH2C(=O)NR2R3, в котором Х является подходящей уходящей группой (например, хлором, бромом, тозильной или мезильной).
Во втором способе синтеза соединения формулы (IIb) алкилируют требуемыми ацетамидными соединениями формулы X-CH2C(=O)NR2R3, получая соединение формулы (III):
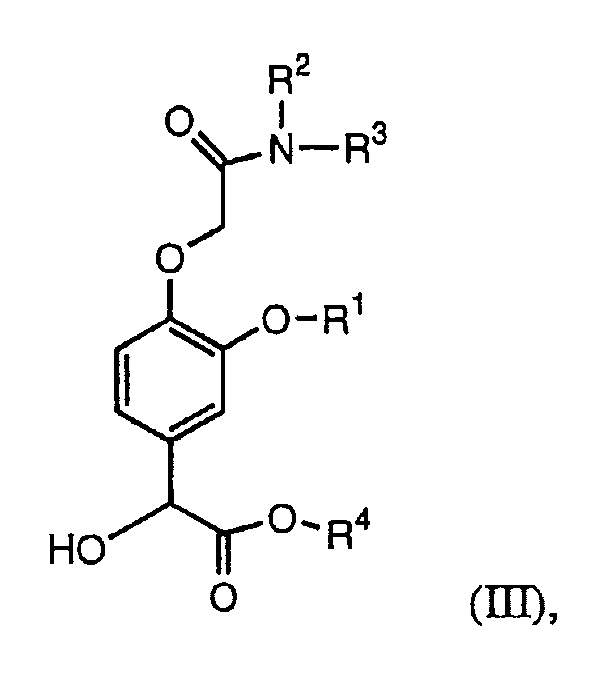
которое восстанавливают, получая соединение формулы (I). Как описано в примерах 4А, 4В и 10-13, удобный способ восстановления основан на двухстадийной реакции, в которой гидроксильную группу формулы (III) сначала ацетилируют перед взаимодействием с водородом.
Промежуточное соединение формулы (IIb) в описанной выше методике получают из коммерчески доступных исходных веществ и реагентов с использованием обычных методик. Например, данное промежуточное соединение можно получить, как показано на схеме А:
Схема А

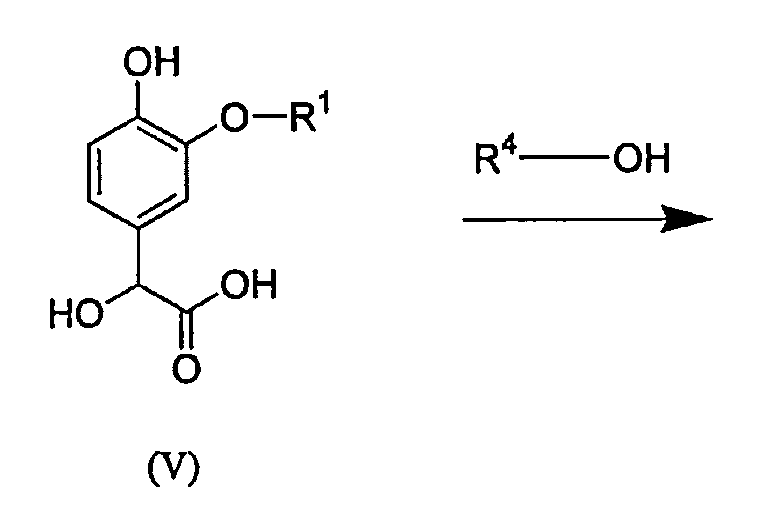
Как показано выше, катехин сочетают с соединением формулы R1X, в котором Х представляет собой уходящую группу, с образованием простого эфира (IV), который вводят во взаимодействие с глиоксиловой кислотой, получая соединение (V). Последующая реакция (V) с избытком спирта R4ОН приводит к получению промежуточного соединения формулы (IIb). Данное промежуточное соединение формулы (IIb) можно алкилировать, как описано выше, получая соединение формулы (III).
Промежуточное соединение формулы (Ia) можно получить, например, как описано в примере 1 подраздела (1), а также как показано на схеме В в примере 1 подраздела (2) ниже.
Фармацевтические композиции
Из соединений формулы I можно составить фармацевтические композиции и вводить их хозяину-млекопитающему, такому как пациент, являющийся животным или человеком, в виде разнообразных форм, адаптированных для выбранного способа введения, то есть перорального или парентерального, внутривенного, внутримышечного, местного или подкожного способа.
Таким образом, соединения согласно настоящему изобретению можно вводить систематически, например, в сочетании с фармацевтически приемлемым носителем, таким как инертный разбавитель или пищевой носитель. Их можно заключить в твердые или мягкие оболочки желатиновых капсул, можно спрессовать в таблетки, или можно вносить непосредственно в пищу рациона пациента. Для перорального терапевтического введения активное соединение можно соединить с одним или несколькими эксципиентами и использовать в форме проглатываемых таблеток, трансбуккальных таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, облаток и так далее. Подобные композиции и препараты могут содержать, по меньшей мере, 0,1% активного соединения. Процентный состав композиций и препаратов, конечно, можно менять и обычно можно установить в интервале от около 2 до около 60% от массы данной разовой дозированной формы. Количество активного соединения в подобных терапевтически применимых композициях такое, что будет достигнут уровень эффективной дозы.
Таблетки, облатки, пилюли, капсулы и так далее могут также содержать следующее: можно добавить связывающие вещества, такие как смола трагаканта, аравийская камедь, кукурузный крахмал или желатин, эксципиенты, такие как дикальций фосфат, разрыхлитель, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота и так далее, смазывающее вещество, такое как стеарат магния, и подсластитель, такой как сахароза, фруктоза, лактоза, или аспартам, или ароматизирующий агент, такой как перечная мята, масло грушанки или вишневый ароматизатор. В случае, когда разовая дозированная форма представляет собой капсулу, в дополнение к веществам перечисленного выше типа она может содержать жидкий носитель, такой как растительное масло или полиэтиленгликоль. Могут присутствовать другие различные вещества, такие как покрытия, или модифицирующие физическую форму твердой дозированной лекарственной формы иным образом. Например, таблетки, пилюли или капсулы могут быть покрыты желатином, воском, шеллаком или сахаром и так далее. Сироп или эликсир может содержать активное соединение, сахарозу или фруктозу в качестве подсластителя, метил и пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневый или апельсиновый ароматизатор. Конечно, любое используемое вещество, использованное при приготовлении любой разовой дозированной формы должно быть фармацевтически приемлемым и по существу нетоксичным в применяемых количествах. Кроме того, активное соединение может содержаться в препаратах и устройствах длительного высвобождения.
Описанные здесь активные агенты обычно используют для составления фармацевтических композиций, пригодных для внутривенного введения. Данные активные агенты относительно нерастворимы в воде. Таким образом, для внутривенного введения из данных агентов обычно составляют препараты в водных средах с использованием одного или более несмешивающихся с водой растворителей и одного или более эмульгаторов. Некоторые эмульгаторы в литературе называют различными поверхностно-активными веществами. Отдельные препараты могут содержать один или несколько дополнительных компонентов, таких как стабилизаторы, модификаторы тоничности, основания или кислоты для регулирования рН и солюбилизаторы. Данные препараты могут также необязательно содержать консервант, такой как, если упоминать лишь несколько, этилендиаминтетрауксусная кислота (ЭДТУ), или метабисульфит натрия.
В композициях настоящего изобретения можно использовать широкий ряд несмешивающихся с водой растворителей. Несмешивающийся с водой растворитель может представлять собой растительное масло, например соевое, сафлоровое, масло семян хлопчатника, кукурузное, подсолнечное, арахисовое, касторовое или оливковое масло. Альтернативно, несмешивающийся с водой растворитель представляет собой эфир средне- или длинноцепочечной жирной кислоты, например моно-, ди- или триглицерид, эфир комбинации средне- и длинноцепочечной кислоты, или представляет собой химически модифицированное или полученное вещество, такое как этилолеат, изопропилмиристат, изопропилпальмитат, сложный эфир глицерина, полиоксил, или гидрированное касторовое масло. Несмешивающийся с водой растворитель может также представлять собой жир морских животных, например жир печени трески, или другое полученное из рыбы масло. Подходящие растворители также включают в себя фракционированные масла, например фракционированное кокосовое масло или модифицированное соевое масло.
Композиции могут также содержать эмульгатор. Подходящие эмульгаторы включают в себя синтетические неионогенные эмульгаторы, например этоксилированные простые эфиры и сложные эфиры и полиоксипропилен-полиоксиэтиленовые блок-сополимеры, и фосфолипиды. Можно использовать как природные фосфолипиды, такие как фосфолипиды яйца и сои, и модифицированные или полученные искусственным путем, например, полученные в результате физической перегонки и/или хроматографии, или их смеси. Фосфолипиды иначе называют фосфатидами. Предпочтительными эмульгаторами являются фосфолипиды яичного желтка и сои. Фосфолипиды яичного желтка состоят, главным образом, из фосфатидилхолина и фосфатидилэтаноламина. Лецитин, который классифицируют как фосфатидилхолин и который можно получить из яичного желтка или сои, представляет собой другой обычно используемый эмульгатор.
Кроме того, фармацевтические композиции могут содержать стабилизирующие агенты, которые, альтернативно, можно считать со-эмульгаторами. Анионогенные стабилизаторы включают фосфатидилэтаноламины, связанные с полиэтиленгликолем, (PEG-PE) и фосфатидилглицерины, конкретным примером которых является димиристилфосфатидилглицерин (DMPG). Дополнительные примеры применимых стабилизаторов включают олеиновую кислоту и ее натриевую соль, холевую кислоту и дезоксихолевую кислоту и их соответствующие соли, катионогенные липиды, такие как стеариламин и олеиламин и 3β-[N-(N',N'-диметиламиноэтан)карбамоил]холестерол (DC-Chol).
Фармацевтические композиции данного изобретения можно сделать изотоничными крови за счет введения подходящего модификатора тоничности. Наиболее часто в качестве модификатора тоничности используют глицерин. Альтернативные модифицирующие агенты включают в себя ксилит, маннит и сорбит. Фармацевтические композиции обычно составляют таким образом, чтобы у них был физиологически нейтральный рН, обычно в интервале 6,0-8,5. рН можно регулировать добавлением основания, например NaOH или NaHCO3, или, в некоторых случаях, кислоты, такой как HCl.
Фармацевтически безопасные эмульсии масла в воде, содержащие растительное масло, фосфатидный эмульгатор, обычно яичный лецитин или соевый лецитин, и модификатор тоничности, предлагаются промышленностью для парентерального употребления, например, под торговыми марками Lyposyn® II и Lyposyn® III (Abbott Laboratories, North Chicago, IL) и Intralipid® (Fresenius Kabi AB, Упсала, Швеция). Из описанных здесь агентов можно составить препараты с данными или другими сходными эмульсиями масла в воде, как показано, например, в инъекциях с 5 по 9 в приведенном ниже примере 16.
Из соединений данного изобретения можно также составить препараты с триглицерид-содержащими эфирами, по меньшей мере, одной жирной кислоты со средней длиной цепи (С6-С12). Предпочтительно данный глицерид включает в себя эфир С8-С10 жирной кислоты. Триглицериды, пригодные для составления препарата из соединения данного изобретения, предлагаются под торговой маркой Miglyol® Condea Chemie GmbH (Виттен, Германия). Например, Miglyol® 810 или 812 (каприловый (С10)/каприновый (С8) глицерид) подходят для составления препаратов из настоящих агентов. В приведенной ниже инъекции 11 примера 16 представлен препарат, содержащий фосфатиды яичного желтка в качестве эмульгатора, DMPG в качестве анионного стабилизатора и глицерин в качестве модификатора тоничности, в котором в качестве масляной фазы используют Miglyol® 810.
Кроме того, из описанных здесь агентов можно составить препарат по аналогии с фармацевтическими композициями пропанидида, известными в данной области. Например, соединения данного изобретения можно внести в смеси, содержащие эфир жирной кислоты со средней длиной цепи, как описано в патенте США № 4711902. Кроме того, из соединений данного изобретения можно составить препараты по аналогии с композициями пропофола, известными в данной области, как описано, например, в патентах США № 4056635, 4452817 и 4798846.
В еще одном варианте из соединений данного изобретения можно составить препараты с использованием солюбилизатора, например гидроксипропил-β-циклодекстрина, с образованием комплекса включения.
Другие подходящие препараты для использования в настоящем изобретении можно найти в Remington's Pharmaceutical Sciences, Mace Publishing Company, Philadelphia, PA. 17th ed. (1985).
Соединения в соответствии с настоящим изобретением представляют собой эффективные снотворныеагенты, которые in vivo быстро подвергаются метаболизму в неактивные и хорошо переносимые метаболиты карбоновых кислот (формула I, где R4 представляет собой водород). По сравнению с предшествующими агентами настоящие соединения проявляют одно или более следующих полезных свойств: повышенная эффективность, более короткие периоды восстановления, сниженные воздействия на сердечно-сосудистую систему, более низкая токсичность и более высокий терапевтический индекс, где терапевтический индекс определяют как соотношение максимальной переносимой дозы и эффективной дозы.
Таким образом, соединения настоящего изобретения можно использовать для создания и/или поддержания общей анестезии, для создания и/или поддержания седативного эффекта у самостоятельно дышащих пациентов в сознании, или для создания и/или поддержания седативного эффекта у интубированных пациентов в сознании с искусственной вентиляцией.
Количество активного агента, необходимое для использования в способах данного изобретения, может изменяться в зависимости от способа введения, возраста и состояния пациента и степени необходимой анестезии или седативного эффекта, и оно будет, в конечном итоге, представлено на усмотрение лечащего врача или клинициста.
В целом, для получения анестезии или седативного эффекта агенты можно вводить в виде начального болюса с последующей непрерывной инфузией агента со скоростью, которая достаточна для достижения и поддержания желательного уровня анестезии или седативного эффекта. Альтернативным образом, для поддержания анестезии или седативного эффекта можно использовать непрерывную инфузию агента настоящего изобретения после создания или создания и поддержания при помощи другого седативного гипнотического агента (например, пропофола, барбитурата, такого как nembutal® (пентобарбитал натрия), или brevital® натрия (метогекситал натрия), или бензодиазепина, такого как valium®).
Например, подходящая болюсная доза настоящего агента для пациента-человека обычно будет находиться в интервале от около 0,1 до около 50 миллиграмм/килограмм (мг/кг), предпочтительно, от около 0,5 до около 20 мг/кг. Скорость инфузии обычно будет находиться в интервале от около 5 до около 5000 микрограмм/килограмм/минуту (мкг/кг/мин), предпочтительно, от около 10 до около 2000 мкг/кг/мин.
Соединения настоящего изобретения можно также вводить в сочетании с другими терапевтическими агентами, например, такими как другие седативные снотворные агенты, болеутоляющие средства (например, опиоид, такой как μ-опиоидный агонист ремифентанил, фентанил, сульфентанил или альфентанил) или парализующие агенты, такие как безилат атракурия или бромид панкурония. Соответственно, композиции настоящего изобретения могут необязательно содержать, кроме того, другой терапевтический агент, например седативный снотворный агент, болеутоляющее средство или парализующий агент. Аналогичным образом, терапевтические способы данного изобретения могут необязательно включать введение млекопитающему другого терапевтического агента (например, седативного снотворного агента, болеутоляющего средства или парализующего агента).
Способность какого-либо агента действовать в качестве анестезирующего или седативного средства можно определить при помощи анализов, известных в данной области (смотри, например, патент США номер 5908869, или R. James and J. Glen, J. Med. Chem., 23, 1350 (1980)), или при помощи анализа, описанного ниже в тесте А.
ТЕСТ А
Способы
Препарат
Из тестируемых соединений, например, представительных соединений изобретения, а также соединения сравнения, пропанидида, были составлены препараты в (1) 10% кремофоре EL/90% D5W (5% декстрозы в дистиллированной воде), (2) 10% Liposyn® III (эмульсия жира для внутривенного введения, содержащая (на 100 мл) 10 г соевого масла, 1,2 г фосфатидов яичного желтка и 25 г глицерина), поставляемых Abbott Laboratories, North Chicago, IL и (3) инъекций (10) или (11) (описанных в примере 16) с Miglyol® 810 (каприловый/каприновый глицерид). Обычно описанный выше препарат (1) использовали для болюсных доз и препаратов (2) или (3) для введения путем инфузии. Соединения по изобретению и пропанидид синтезировали, как описано в примерах 1-15, приведенных далее. Препарат с пропофолом в соевом масле, продаваемый как эмульсия для инъекций Diprivan®, получали от AstraZeneca (Wilmington, DE).
Введение болюса (крысы)
Крыс (взрослых самцов Sprague-Dawley) помещали в приспособление для фиксации конечностей из Perspex и делали им инъекцию (1 или 2 мл/кг приблизительно в течение 3 секунд) интересующего соединения через хвостовую вену. Регистрировали время до начала анестезии (определяемой как потеря рефлекса выпрямления), продолжительности анестезии (то есть продолжительности потери рефлекса выпрямления) и восстановления поведения (то есть продолжительность атаксии, седативного эффекта и/или летаргии после восстановления рефлекса выпрямления). Продолжительность анестезии определяли, помещая крыс на спину после начала действия анестезии и регистрировали время до восстановления рефлекса выпрямления при помощи секундомера. Глубину анестезии оценивали с перерывами, наблюдая за величиной рефлекса отдергивания при болезненном щипке задней лапки. Восстановление поведения оценивали путем визуального наблюдения.
Введение болюса (морские свинки)
Взрослые самцы морских свинок получали дозу при введении болюса (объем 0,1-0,25 мл) через ушную вену. Продолжительность потери рефлекса выпрямления определяли, как описано выше для крыс.
Введение при помощи инфузии (крысы)
Крыс (взрослых Sprague-Dawley) помещали в приспособление для фиксации конечностей из Perspex и вызывали у них анестезию путем инъекции болюса через хвостовую вену (0,15-1 мл/кг приблизительно в течение 3 секунд при дозе, определенной в более ранних опытах с болюсами, получая анестезию продолжительности около 2 минут). Сразу после введение болюса начинали инфузию (обычно с продолжительностью 20, 180, или 300 минут) через хвостовую вену (0,075-0,5 мл/кг/мин при половине болюсной дозы/мин). В некоторых опытах первоначальную скорость инфузии поддерживали на протяжении всей инфузии, тогда как в других скорость изменяли по необходимости для поддержания постоянной глубины анестезии (определяемой по среднему отдергиванию лапки в ответ на болезненный щипок). После окончания инфузии регистрировали продолжительность анестезии (то есть продолжительность потери рефлекса выпрямления) и восстановления поведения (то есть продолжительность атаксии, седативного эффекта, или летаргии после восстановления рефлекса выпрямления).
Результаты
Введение болюса (крысы): Определили кривую отклика, в зависимости от дозы, продолжительности потери рефлекса выпрямления у крыс, происходящей при инъекции болюса тестируемых соединений, полученных в препарате (1). Для количественной оценки эффективности анестезии вычисляли дозы тестируемого соединения, приводившие к средней потере рефлекса выпрямления в течение 2 минут. На фиг. 1 приведено сопоставление болюсной дозы соединений изобретения в мг/кг, приводящей к 2-минутной потере рефлекса выпрямления, с требуемой дозой соединения сравнения, пропанидидом.
Введение болюса (морские свинки): По аналогичной методике эффективность соединения 1 исследовали также на морских свинках. Рассчитанная доза соединения 1, необходимая для получения 2-минутной потери рефлекса выпрямления у морских свинок, составила 8 мг/кг по сравнению с дозой 13 мг/кг для пропанидида.
Введение при помощи инфузии (крысы): Периоды восстановления после окончания введения путем инфузии у крыс определяли для соединения 1 и для соединений сравнения пропофола и пропанидида. Продолжительность потери рефлекса выпрямления (в минутах) после окончания инфузии приведена в виде функции продолжительности инфузии в таблице 2 далее.
На фиг.2 общие периоды восстановления после окончания инфузии определенной продолжительности у крыс приведены в виде суммы продолжительности потери рефлекса выпрямления, указанного в таблице 2, и продолжительности восстановления поведения после восстановления рефлекса выпрямления.
Как показывают приведенные выше данные для животных моделей на крысах и морских свиньях, протестированные соединения данного изобретения представляют собой более эффективные общие анестезирующие средства, чем пропанидид, и обеспечивают значительно более быструю скорость общего восстановления, чем пропофол, даже после длительных (5-часовых) инфузий. Кроме того, продолжительность потери рефлекса выпрямления после окончания инфузии тестируемого соединения по изобретению не зависела от продолжительности инфузии в пределах ошибки результатов эксперимента.
Устойчивость представительных соединений изобретения in vitro можно определить, как описано в тесте В.
ТЕСТ В
Источник проб цельной крови
Пробы цельной крови крыс и свиней, полученные путем прокола сердца, собирали в пробирки vacutainer, содержащие гепарин натрия. Пробы хранили на льду и использовали в день отбора. Цельную кровь собаки, обезьяны и человека, полученную от поставщиков, хранили на льду (водном) и использовали на следующий день после отбора.
Анализ на метаболизм
Тестируемые соединения, пропанидид и представительное соединение изобретения, добавляли в 300 мкл пробы цельной крови до конечной концентрации 100 мкМ. Белки мгновенно осаждались при добавлении двойного объема охлажденного льдом этанола и вихревом перемешивании. Это представляет собой нулевую временную точку. В идентичных 300 мкл инкубационных образцах пробы цельной крови с добавкой затем инкубировали при 37°С в течение от 30 секунд до 60 минут. В заранее определенное время к смеси добавляли 600 мкл охлажденного льдом этанола для прекращения инкубации. После окончания инкубации пробы центрифугировали, а надосадочную жидкость сушили в токе азота при комнатной температуре. Остаток вновь растворяли в 150 мкл стерильной воды, а затем центрифугировали. Аликвоту (50 мкл) надосадочной жидкости вводили в ВЭЖХ-УФ для анализа.
Метод ВЭЖХ
Использовали колонку для ВЭЖХ с обращенной фазой А С18 5 мкм, 2 × 150 мм в.д. (LUNA, Phenomenex) и градиент от 10% до 68% ацетонитрила в течение 15 минут с последующим изократическим проходом при 10% ацетонитрила. Компоненты подвижной фазы содержали 0,1% ТФУ. Мониторинг аналитических образцов проводили при помощи УФ детектирования при 214 нм.
Анализ данных
Концентрацию субстрата в инкубационных образцах определяли в виде соотношений площадей пиков с использованием метода внутреннего стандарта, а процент разложения определяли относительно значений нулевого времени.
Результаты
Тестируемые соединения формулы (I) быстро подвергались метаболизму в соответствующие карбоновые кислоты (формула (I), в которой R4 =водород). Было найдено, что кислотные метаболиты неактивны в качестве анестезирующих средств в тесте А. Быстрое превращение соединений формулы (I) в их кислотные метаболиты и неактивность данных кислотных метаболитов в качестве анестезирующих средств может быть, по крайней мере, отчасти, причиной более коротких и более предсказуемых периодов восстановления, наблюдаемых для соединений формулы (I).
Далее изобретение будет проиллюстрировано следующими неограничивающими примерами.
ПРИМЕРЫ
В приведенных далее примерах следующие сокращения имеют следующие значения. Любые нерасшифрованные сокращения имеют свои общепринятые значения. Если не указано иначе, все температуры приведены в градусах Цельсия.
ДМСО = диметилсульфоксид
EtOAc = этилацетат
DCM = хлористый метилен
PPTS = паратолуолсульфонат пиридиния
ДМФА = диметилформамид
Общее: если не указано иначе, реагенты, исходное вещество и растворители приобретали у поставщиков, например у Sigma-Aldrich (St. Louis, MO) и Trans World Chemicals, Inc. (TCI) (Rockville, MD), и использовали без дополнительной очистки; реакции проводили в атмосфере азота; контроль реакционных смесей осуществляли при помощи тонкослойной хроматографии (ТСХ на силикагеле), аналитической высокоэффективной жидкостной хроматографии (анал. ВЭЖХ), или масс-спектрометрии; реакционные смеси обычно очищали колоночной флэш-хроматографией на силикагеле, или перегонкой в вакууме; образцы для ЯМР растворяли в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6), а спектры регистрировали на приборе Varian Gemini 2000 (300 МГц) с использованием растворителя в качестве внутреннего стандарта, если не указано иначе; а масс-спектрометрическое определение проводили методом электроспреевой ионизации (ESMS) на приборе Perkin Elmer (PE SCIEX API 150 EX).
Пример 1. Соединение 1: пропиловый эфир [4-[(N,N-диэтилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты
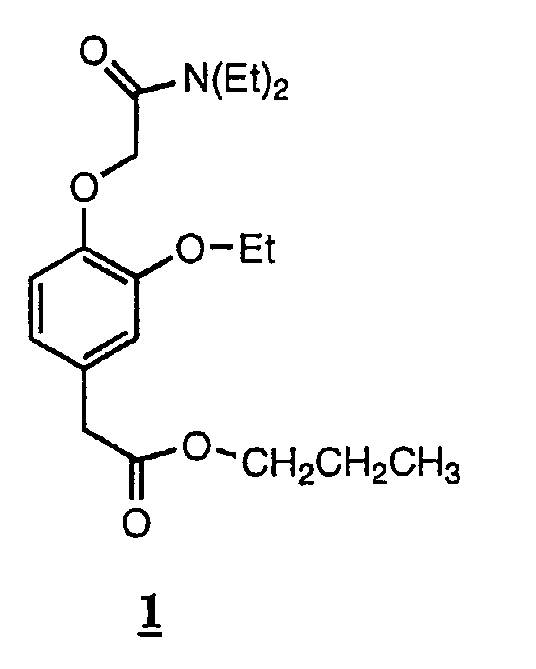
В круглодонной колбе на 50 мл, снабженной магнитной мешалкой, пропиловый эфир 3-этокси-4-гидроксифенилуксусной кислоты (800 мг, 3,4 ммоль, 1,0 эквив.) растворяли в сухом ацетоне (20 мл). К раствору добавляли К2СО3 (705 мг, 5,1 ммоль, 1,5 эквив.), затем 2-хлор-N,N-диэтилацетамид (0,55 мл, 4,0 ммоль, 1,2 эквив., поставляемый Aldrich). При энергичном перемешивании данную суспензию нагревали до кипения с обратным холодильником и выдерживали в этих условиях в течение 15 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали через складчатый бумажный фильтр, а из оставшегося раствора удаляли растворитель при пониженном давлении. Маслообразный продукт очищали колоночной хроматографией (SiO2, 50% EtOAc/гексан), получая 630 мг (53% от теории) бесцветного масла, чистота которого составляла 99,6% по ВЭЖХ.
ТСХ (силикагель, 50% EtOAc/гексан) Rf 0,25; ЯМР 1Н (CDCl3, 300 МГц), д 0,90 (3H, т, пропилат СН3), 1,13 и 1,20 (каждый 3Н, т, N-этил СН3), 1,43 (3Н, т, этокси СН3), 1,60-1,67 (2Н, м, пропилат СН2), 3,35-3,46 (4Н, м, N-этил СН2), 3,53 (2Н, с, ОСН2СО), 4,01-4,11 (4Н, м, 2хОСН2), 4,70 (2Н, с, ArCH2CO), 6,75-6,91 (3H, м, ArH). m/z: [M+H+] вычисл. для С19Н29NO5 352,22; найдено 352.
(1) Получение промежуточного соединения формулы (IIa), R1 =этил и R4 =пропил (пропиловый эфир 3-этокси-4-гидроксифенилуксусной кислоты)
В стеклянную емкость (пробирку) с тефлоновой завинчивающейся крышкой помещали магнитную мешалку и заполняли 3-этокси-4-гидроксифенилуксусной кислотой (2,5 г, 12,7 ммоль, 1,0 эквив., поставляемой Trans World Chemicals). Добавляли 1-пропанол (20 мл, 270 ммоль, ˜20 эквив.) и перемешивали смесь до растворения. Добавляли концентрированную серную кислоту (2 капли). Крышку емкости плотно завинчивали вручную и погружали пробирку в масляную баню. Оставляли реакционную смесь перемешиваться при 90°С в течение 15 часов. Давали емкости остыть до комнатной температуры, после чего содержимое переносили в круглодонную колбу и отгоняли избыток спирта в вакууме. Оставшееся масло растворяли в этилацетате (50 мл) и промывали насыщенным раствором гидрокарбоната натрия. После сушки над сульфатом магния и фильтрования растворитель отгоняли при пониженном давлении, получая в остатке 2,6 г (выход 85%) сложного эфира в виде светло-желтого масла.
(2) Получение промежуточного соединения формулы (IIa), R1 =этил и R4 =пропил (пропиловый эфир 3-этокси-4-гидроксифенилуксусной кислоты)
Указанное в заголовке промежуточное соединение также получали по приведенной ниже схеме В
Схема В
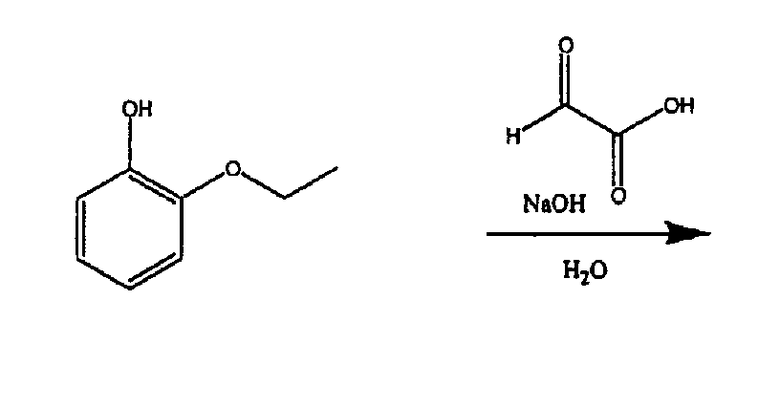

(а) Получение соединения В1
Смешивали 2-этоксифенол (56,6, 0,401 моль, 1 экв.), глиоксиловую кислоту (50%-ный водный раствор) (41,0 мл, 0,396 моль, 0,99 экв.) и дистиллированную воду (110 мл). Смесь охлаждали на бане со льдом и медленно добавляли через капельную воронку раствор 10%-ного NaOH (32,2 г NaOH в 300 мл дистиллированной воды, 0,805 моль, 2 экв.). Реакционной смеси давали медленно нагреться до комнатной температуры и через ˜18 часов раствор промывали этилацетатом (4×250 мл), потом подкисляли 6н. HCl до рН˜3. Добавляли NaCl, а затем продукт экстрагировали этилацетатом (4×200 мл). Органическую фазу промывали насыщенным раствором соли, сушили над сульфатом магния и удаляли растворитель в вакууме, получая 51,8 г В1 в виде светло-розового твердого вещества.
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 1,24 (т, 3Н), 3,90 (кв, 2Н), 4,79 (с, 1Н), 5,59 (ушир.с, 1Н), 6,67 (кв, 2Н), 6,86 (с, 1Н), 8,81 (с, 1Н), 12,35(ушир.с, 1Н).
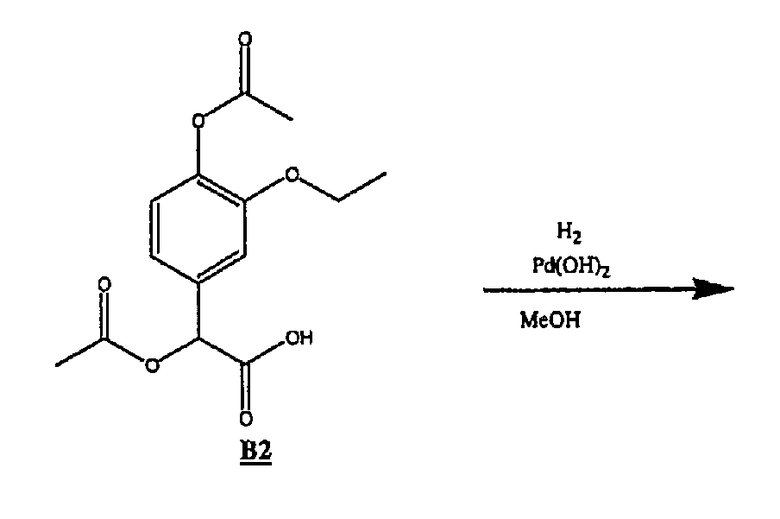
(b) Получение соединения В2
Соединение В1 (45,0 г, 0,212 моль, 1 экв.) растворяли в DCM (225 мл), добавляли пиридин (80 мл, 0,989 моль, 6 экв.) и охлаждали смесь на бане со льдом в атмосфере азота. Через капельную воронку медленно добавляли уксусный ангидрид (100 мл, 1,06 моль, 4 экв.). Смесь перемешивали (˜3 ч) до завершения реакции, а затем разбавляли диэтиловым эфиром (500 мл) и промывали 1н. HCl (4×250 мл). Смесь экстрагировали 8%-ным раствором гидрокарбоната натрия (4 × 80 мл), подкисляли до ˜рН 4 6н HCl и экстрагировали продукт диэтиловым эфиром, получая 41,1 г В2 в виде белого кристаллического вещества.
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 1,12 (т, 3Н), 2,05 (с, 3Н), 2,17 (с, 3Н), 3,95 (кв, 2Н), 5,72 (с, 1Н), 6,69 (д, 1Н), 7,04 (д, 1Н), 7,12 (с, 1Н).
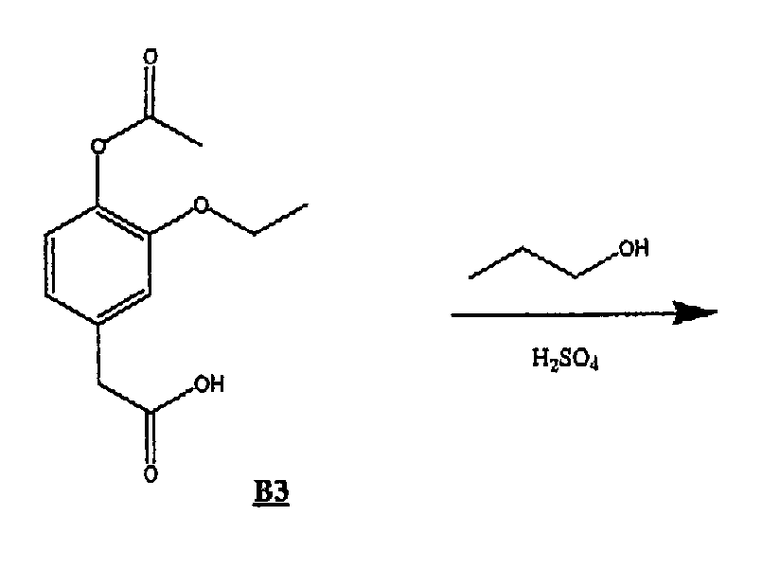
(с) Получение соединения В3
Соединение В2 (30,9 г, 0,104 моль) растворяли в метаноле (500 мл), добавляли Pd(OH)2 (5,0 г), смоченный дистиллированной водой, и помещали данную смесь в атмосферу водорода при давлении 30 фунтов/кв.дюйм при встряхивании. Спустя 48 ч Pd(OH)2 удаляли фильтрованием, а растворитель удаляли в вакууме, получая 22 г В3 в виде желтого масла.
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 1,19 (т, 3Н), 2,16 (с, 3Н), 3,47 (с, 2Н), 3,92 (кв, 2Н), 6,74 (д, 1Н), 6,91 (м, 2Н).
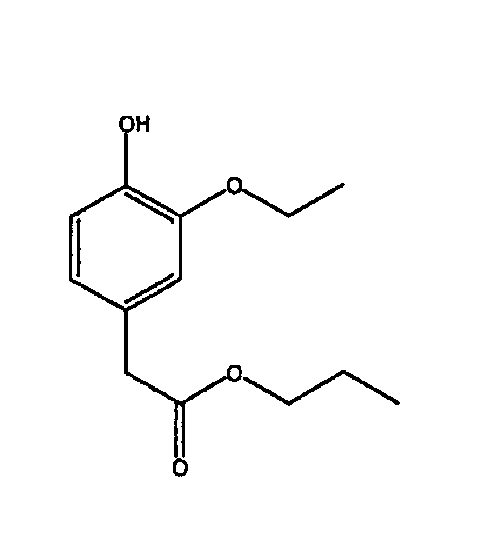
(d) Получение пропилового эфира 4-гидроксифенилуксусной кислоты
Соединение В3 (1,40 г, 5,87 ммоль) растворяли в избытке 1-пропанола (50 мл), добавляли концентрированную H2SO4 (3 капли) и нагревали смесь при 90°С в течение ˜18 часов. Объем 1-пропанола упаривали в вакууме, затем смесь разбавляли диэтиловым эфиром, промывали насыщенным раствором гидрокарбоната натрия (2х), дистиллированной водой (1х), насыщенным раствором соли (1х), сушили над сульфатом магния и удаляли растворитель в вакууме, получая пропиловый эфир 4-гидроксифенилуксусной кислоты в виде желтого масла.
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,78 (т, 3Н), 1,25 (т, 3Н), 1,48 (кв, 2Н), 3,44 (с, 2Н), 3,92 (м, 4Н), 6,58 (д, 1Н), 6,64 (д, 1Н), 6,74 (с, 1Н), 8,73 (с, 1Н).
Пример 2. Соединение 2: этиловый эфир [4-[(N,N-диэтилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты.
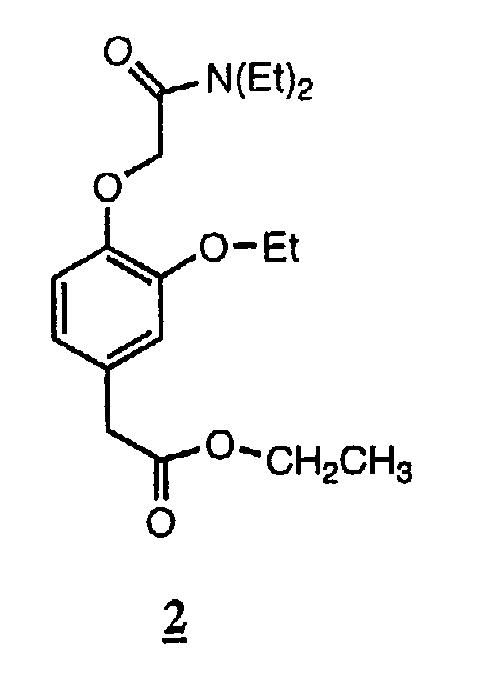
По методике, аналогичной описанной в примере 1, за исключением того, что 1-пропанол заменяли этанолом в синтезе промежуточного продукта для получения промежуточного соединения формулы (IIa) с R1 =этил и R4 =пропил, указанное в заголовке соединение получали с выходом 81% в виде бесцветного масла, чистота которого по данным ВЭЖХ составляла 96%.
ТСХ (силикагель, 50% EtOAc/гексан) Rf 0,25; ЯМР 1Н (CDCl3, 300 МГц): d 1,13-1,22 (6Н, м, N-этил СН3), 1,25 (3Н, т, этиловый эфир СН3), 1,43 (3Н, т, этокси СН3), 3,38-3,45 (4Н, м, N-этил СН2), 3,52 (2Н, с, ОСН2СО), 4,05-4,17 (4Н, м, 2хОСН2), 4,71 (2Н, с, ArCH2CO), 6,78-6,91 (3H, м, ArH). m/z: [M+H+] вычисл. для С18Н27NO5 338,20; найдено 338.
Пример 3. Соединение 3: изопропиловый эфир [4-[(N,N-диэтилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты.
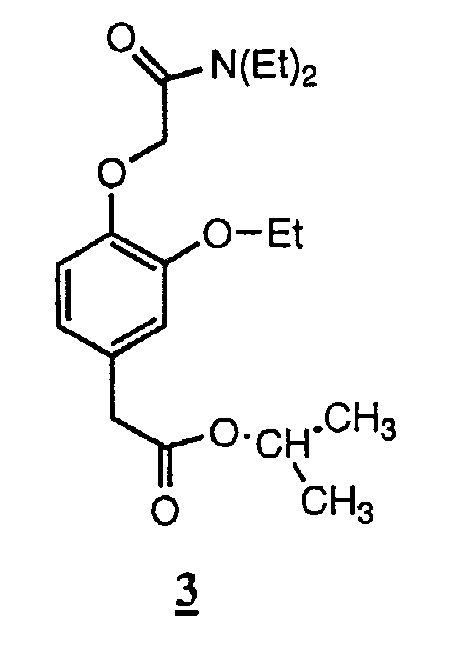
По методике, аналогичной описанной в примере 1, за исключением того, что 1-пропанол заменяли изопропанолом в синтезе промежуточного продукта для получения промежуточного соединения формулы (IIa) с R1=этил и R4=изопропил, указанное в заголовке соединение получали с выходом 63% в виде бесцветного масла, чистота которого по данным ВЭЖХ составляла 99%.
ТСХ (силикагель, 50% EtOAc/гексан) Rf 0,25; ЯМР 1Н (CDCl, 300 МГц) d 1,06-1,19 (6Н, м, N-этил СН3), 1,14 и 1,16 (2х3Н, 2с, изопропиловый эфир СН3), 1,36 (3Н, т, этокси СН3), 3,30-3,36 (4Н, м, N-этил СН2), 3,42 (2Н, с, ОСН2СО), 3,98-4,03 (2Н, м, ОСН2), 4,64 (2Н, с, ArCH2CO), 4,90-4,98 (1H, м, CH), 6,71-6,84 (3H, м, ArH). m/z: [M+H+] вычисл. для С19Н29NO5 352,22; найдено 352.
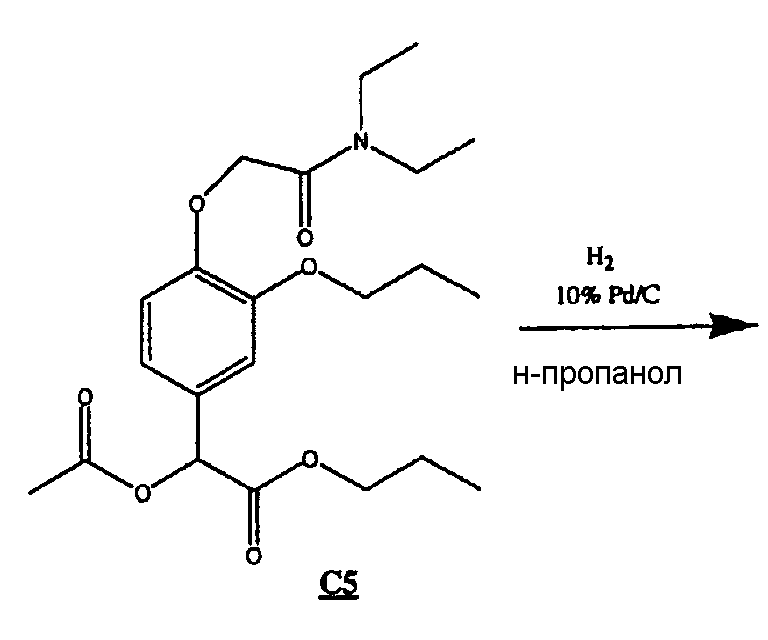
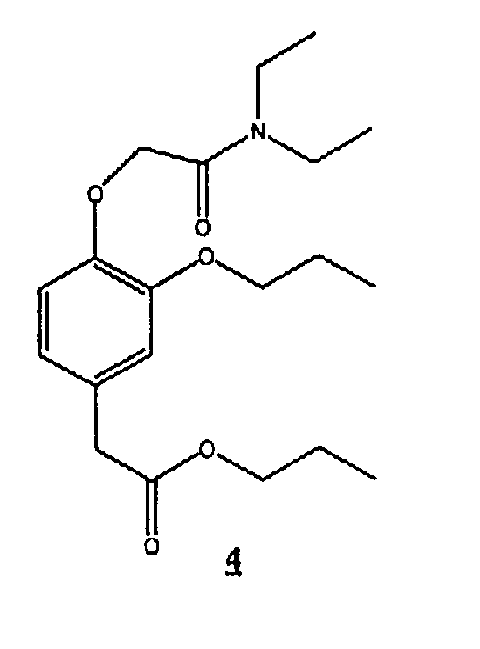
Пример 4А. Соединение 4: пропиловый эфир [4-[(N,N-диэтилкарбамоил)метокси]-3-пропоксифенил]уксусной кислоты.
Соединение 4 получали по приведенной далее схеме С
Схема С
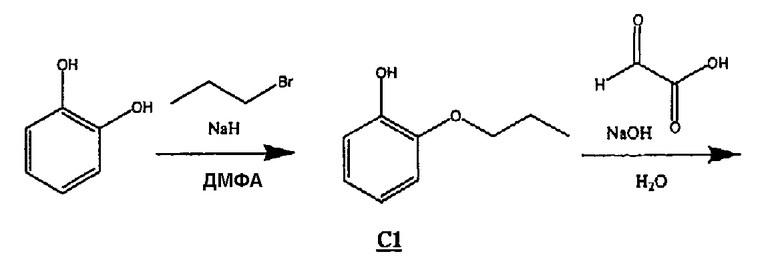

(1) Получение соединения С1 (формула (IV) R1 =пропил)
Готовили раствор катехина (81,0 г, 0,74 моль) в ДМФА (1,5 л) в 3 л колбе, снабженной верхней мешалкой, и охлаждали на бане со льдом. К раствору медленно добавляли NaH (60% в масле) (29 г, 0,73 моль), как только он полностью прореагировал (около 1 ч после последнего прибавления), добавляли 1-бромпропан (72 мл, 0,74 моль). Реакционную смесь перемешивали в течение ночи и оставляли медленно нагреваться до комнатной температуры.
Реакционную смесь выливали в делительную воронку, содержащую диэтиловый эфир, и промывали водой (3х), затем экстрагировали 1н. NaOH (3х), водную часть подкисляли 6н. HCl до рН ˜1 и экстрагировали продукт DCM (3х). DCM промывали насыщенным раствором соли (1х), сушили над сульфатом магния и удаляли растворитель в вакууме, получая красное масло. Данное масло очищали пропусканием через 6-дюймовый слой силикагеля, промывая смесью 10% этилацетата/гексан, затем растворитель удаляли в вакууме, получая 26,8 г бесцветного масла С1.
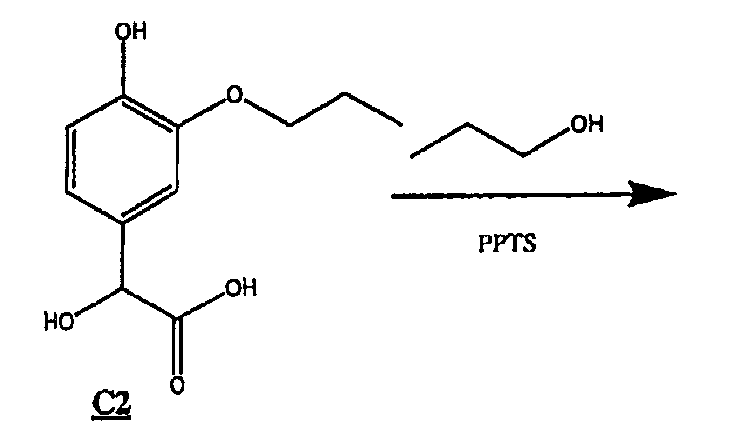
(2) Получение соединения С2 (формула (V) R1=пропил)
К смеси С1 (26,8 г, 0,176 моль) и глиоксиловой кислоты (50%-ный водный раствор) (17,6 мл, 0,160 моль), охлажденной на бане со льдом, добавляли раствор 10%-ного NaOH (128 мл, 0,320 моль). Смесь перемешивали в течение ночи и оставляли медленно нагреваться до комнатной температуры. Через ˜15 часов для растворения смеси добавляли 150 мл дистиллированной воды и реакционную смесь снова перемешивали в течение ночи при комнатной температуре.
Реакционную смесь промывали этилацетатом (4х), водную часть подкисляли ледяной уксусной кислотой до рН ˜3 и экстрагировали продукт этилацетатом (3х). Этилацетат промывали насыщенным раствором соли, сушили над сульфатом магния и удаляли растворитель в вакууме, получая 12 г белого твердого вещества С2.
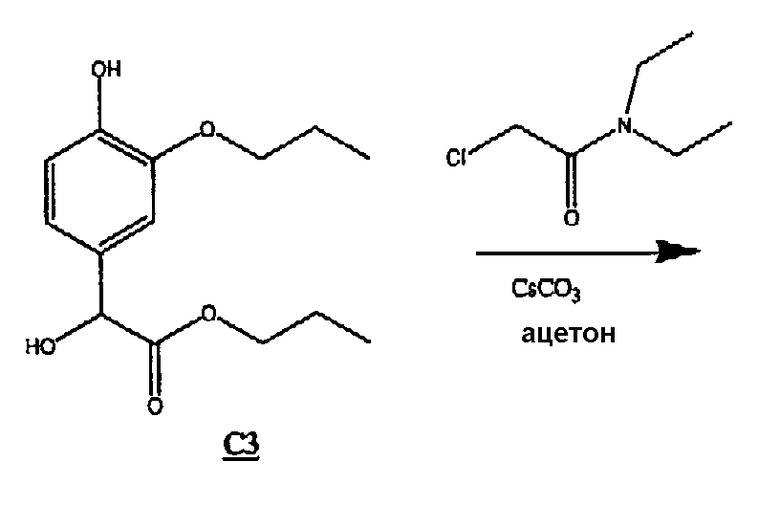
(3) Получение соединения С3 (формула (IIb) R1 и R4=пропил)
К раствору С2 (3,27 г, 1,44 ммоль), растворенному в избытке 1-пропанола (90 мл), добавляли PPTS (0,47 г, 1,87 ммоль). Раствор нагревали при 50оС в течение ночи.
Объем 1-пропанола упаривали в вакууме, разбавляли этилацетатом и промывали 1н. HCl (3х), насыщенным раствором гидрокарбоната натрия (3х) и насыщенным раствором соли (1х) и сушили над сульфатом магния. Растворитель удаляли в вакууме, а смесь очищали колоночной хроматографией, получая 1,7 г бесцветного масла С3.
(4) Получение соединения С4 (формула (III) R1 и R4 =пропил, R2 и R3 =этил)
К раствору С3 (1,70 г, 6,36 ммоль), растворенному в ацетоне (100 мл), добавляли карбонат цезия (10 г, 30,7 ммоль). После перемешивания в течение 10 минут добавляли 2-хлор-N,N-диэтилацетамид (0,95 мл, 6,91 ммоль) и нагревали реакционную смесь при 60°С в течение ночи.
По завершении реакции карбонат цезия отфильтровывали и удаляли растворитель в вакууме, смесь очищали колоночной хроматографией, получая 0,82 г бесцветного масла С4.
(5) Получение соединения С5
К раствору С4 (0,512 г, 1,40 ммоль), растворенному в DCM (50 мл) и пиридине (0,35 мл, 4,33 ммоль) и охлажденному на бане со льдом, добавляли бромистый ацетил (0,21 мл, 2,84 ммоль). Реакционную смесь перемешивали в течение ночи и оставляли медленно нагреваться до комнатной температуры.
Смесь выливали в диэтиловый эфир и промывали 1н. HCl (3х), насыщенным раствором гидрокарбоната натрия (3х), дистиллированной водой (1х) и насыщенным раствором соли (1х), затем сушили над сульфатом магния и удаляли растворитель в вакууме, получая 0,517 г розового масла С5.
(6) Синтез соединения 4
К раствору С5 (0,167 г, 0,394 ммоль) в 1-пропаноле (25 мл) добавляли 10% Pd/C (20 мг), смоченного 1-пропанолом, и обрабатывали водородом при давлении 28 фунтов/кв.дюйм. Через 1 час Pd/C удаляли и заменяли другой порцией 10% Pd/C (20 мг), смоченного 1-пропанолом, и снова обрабатывали водородом при давлении 28 фунтов/кв.дюйм в течение 3 часов. Pd/C удаляли фильтрованием и удаляли растворитель в вакууме, затем смесь очищали колоночной хроматографией, получая 90 мг бесцветного масла 4.
Альтернативным образом соединение 4 можно получить, как в следующем примере.
Пример 4В. Соединение 4: пропиловый эфир [4-[(N,N-диэтилкарбамоил)метокси]-3-пропоксифенил]уксусной кислоты
(1) Получение соединения С1 (формула (IV) R1 =пропил)
К раствору катехина (100,1 г, 0,91 моль), растворенному в ацетоне (1 л), медленно добавляли карбонат калия (125,1 г, 0,91 моль) при энергичном перемешивании; при нагревании добавляли 1-бромпропан (90,0 мл, 0,92 моль) и смесь кипятили с обратным холодильником в течение ночи.
Как только реакционная смесь остывала до комнатной температуры и карбонат калия удаляли фильтрованием, растворитель удаляли в вакууме. После этого продукт разбавляли диэтиловым эфиром, промывали дистиллированной водой (4х), затем экстрагировали 1н. NaOH. Водный слой собирали и подкисляли до рН˜1 6н. HCl и экстрагировали продукт диэтиловым эфиром, сушили над сульфатом магния и удаляли растворитель в вакууме. Продукт очищали пропусканием через 6-дюймовый слой силикагеля, промывая смесью 10% этилацетат/гексан и удаляли растворитель в вакууме, получая 45 г (0,30 моль, 32% выход) не совсем белого твердого вещества С1.
ТСХ (силикагель, 20% EtOAc/гексан) Rf 0,67; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,90 (т, 3Н), 1,64 (кв, 2Н), 3,80 (т, 2Н), 6,61-6,81 (м, 4Н), 8,70 (с, 1Н).
(2) Получение соединения С2 (формула (V) R1 =пропил)
К смеси С1 (100 г, 0,657 моль) и глиоксиловой кислоты (50%-ный водный раствор) (67 мл, 0,648 моль) в 1 л дистиллированной воды, охлажденной на бане со льдом, из капельной воронки медленно добавляли раствор 10%-ного NaOH (52 г NaOH в 500 мл деионизованной воды, 1,30 моль). Смесь перемешивали в течение ночи, в то время как она медленно нагревалась до комнатной температуры.
Реакционную смесь промывали этилацетатом (4х), водную часть собирали и подкисляли 6н. HCl до рН ˜3, а затем экстрагировали продукт этилацетатом (3х). Этилацетат промывали насыщенным раствором соли, сушили над сульфатом магния и удаляли растворитель в вакууме, получая 70 г (0,31 моль, выход 47%) светло-розового твердого вещества С2.
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,90 (т, 3Н), 1,64 (кв, 2Н), 3,79 (т, 2Н), 4,79 (с, 1Н), 5,58 (ушир.с, 1Н), 6,63-6,71 (м, 2Н), 6,85 (с, 1Н), 8,77 (с, 1Н), 12,3 (ушир.с, 1Н).
(3) Получение соединения С3 (формула (IIb) R1 и R4 =пропил)
К раствору С2 (70 г, 0,289 моль), растворенному в избытке 1-пропанола (550 мл), добавляли PPTS (7,5 г, 29,8 ммоль) и нагревали при 50°С в течение ночи.
Объем 1-пропанола упаривали в вакууме, затем разбавляли этилацетатом и промывали 1н. HCl (3х), насыщенным раствором гидрокарбоната натрия (3х) и насыщенным раствором соли (1х), после этого сушили над сульфатом магния. Растворитель удаляли в вакууме, а смесь затем очищали колоночной хроматографией, получая 55 г (0,20 моль, выход 71%) не совсем белого твердого вещества С3.
ТСХ (силикагель, 50% EtOAc/гексан) Rf 0,56; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,69 (т, 3Н), 0,89 (т, 3Н), 1,43 (кв, 2Н), 1,64 (кв, 2Н), 3,79 (т, 2Н), 3,89 (т, 2Н), 4,89 (д, 1Н), 5,76 (д, 1Н), 6,63-6,69 (м, 2Н), 6,84 (с, 1Н), 8,80 (с, 1Н).
(4) Получение соединения С4 (формула (III) R1 и R4 =пропил, R2 и R3 =этил)
К раствору С3 (85 г, 0,32 моль), растворенному в ацетоне (500 мл), медленно добавляли карбонат калия (95 г, 0,69 моль). Затем смесь нагревали при 60°С, после перемешивания в течение 1 часа добавляли 2-хлор-N,N-диэтилацетамид (43,5 мл, 0,32 моль) и нагревали реакционную смесь при 60°С в течение 48 часов.
По завершении реакции карбонат калия удаляли фильтрованием, а растворитель удаляли в вакууме, смесь очищали колоночной хроматографией, получая 50 г (0,13 моль, выход 46%) бесцветного масла С4.
ТСХ (силикагель, 50% EtOAc/гексан) Rf 0,18; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,70 (т, 3Н), 0,87-0,96 (м, 6Н), 1,03-1,09 (м, 3Н), 1,44 (кв, 2Н), 1,64 (кв, 2Н), 3,17-3,26 (м, 4Н), 3,82 (т, 2Н), 3,88 (т, 2Н), 4,66 (с, 2Н), 4,95 (д, 1Н), 5,86 (д, 1Н), 6,71 (д, 1Н), 6,78 (д, 1Н), 6,92 (с, 1Н).
(5) Получение соединения С5
К раствору С4 (50 г, 0,13 моль), растворенному в DCM (600 мл) и пиридине (30 мл, 0,37 моль), охлажденному на бане со льдом, добавляли бромистый ацетил (20 мл, 0,27 моль). Реакционную смесь перемешивали в течение ночи, пока она медленно нагревалась до комнатной температуры.
Объем растворителя уменьшали выпариванием в вакууме, затем разбавляли диэтиловым эфиром и промывали 1н. HCl (5х), насыщенным раствором гидрокарбоната натрия (4х) и насыщенным раствором соли (1х), затем сушили над сульфатом магния. Растворитель удаляли в вакууме, получая желтое масло, которое затем очищали колоночной хроматографией, получая 50 г (0,12 моль, выход 91%) желтого маслаС5.
ТСХ (силикагель, 50% EtOAc/гексан) Rf 0,31; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,70 (т, 3Н), 0,87-0,96 (м, 6Н), 1,03-1,09 (м, 3Н), 1,44 (кв, 2Н), 1,64 (кв, 2Н), 2,02 (с, 3Н), 3,17-3,26 (м, 4Н), 3,84 (м, 2Н), 3,95 (м, 2Н), 4,71 (с, 2Н), 5,73 (с, 1Н), 6,76 (д, 1Н), 6,90 (д, 1Н), 6,99 (с, 1Н).
(6) Синтез соединения 4
К раствору С5 (50 г, 0,12 моль) в 1-пропаноле (200 мл) добавляли 10% Pd/C (5 г), смоченного 1-пропанолом, и обрабатывали водородом при давлении 32 фунта/кв.дюйм в течение 48 часов при встряхивании. Pd/C удаляли и заменяли другой порцией 10% Pd/C (2 г), смоченного 1-пропанолом, и снова обрабатывали водородом при давлении 30 фунтов/кв.дюйм в течение 4 часов при встряхивании. Pd/C удаляли фильтрованием через миллипористый фильтр и удаляли растворитель в вакууме, затем продукт очищали колоночной хроматографией, получая 38 г (0,10 моль, выход 87%) бесцветного масла 4.
ТСХ (силикагель, 50% EtOAc/гексан) Rf 0,41; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,78 (т, 3Н), 0,86-0,96 (м, 6Н), 1,06 (т, 3Н), 1,49 (кв, 2Н), 1,64 (кв, 2Н), 3,17-3,26 (м, 4Н), 3,48 (с, 2Н), 3,82 (т, 2Н), 3,90 (т, 2Н), 4,64 (с, 2Н), 6,65-6,79 (м, 2Н), 6,80 (с, 1Н). ВЭЖХ (ОФ, 10-70% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания4,75 мин, чистота 100% по данным ВЭЖХ.
Пример 5. Соединение 5: этиловый эфир [4-[(N,N-дипропилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты
Соединение 5 получали по методике примера 2, заменяя 2-хлор-N,N-диэтилацетамид на 2-хлор-N,N-дипропилацетамид (выход 54%).
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,69-0,80 (м, 6Н), 1,09 (т, 3Н), 1,24 (т, 3Н), 1,37-1,47 (м, 4Н), 3,09-3,17 (м, 4Н), 3,46 (с, 2Н), 3,90-4,02 (м, 4Н), 4,66 (с, 2Н), 6,65 (м, 2Н), 6,78 (с, 1Н). ВЭЖХ (ОФ, 30-90% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания3,20 мин, чистота 97% по данным ВЭЖХ.
Пример 6. Соединение 6: пропиловый эфир [4-[(N,N-дипропилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты
Соединение 6 получали по методике примера 1, заменяя 2-хлор-N,N-диэтилацетамид на 2-хлор-N,N-дипропилацетамид (выход 51%).
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,81-0,91 (м, 9Н), 1,36 (т, 3Н), 1,46-1,66 (м, 6Н), 3,20-3,29 (м, 4Н), 3,60 (с, 2Н), 3,99-4,07 (м, 4Н), 4,78 (с, 2Н), 6,77 (м, 2Н), 6,91 (с, 1Н). ВЭЖХ (ОФ, 30-90% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания3,57 мин, чистота 100% по данным ВЭЖХ.
Пример 7. Соединение 7: пропиловый эфир [4-[(N-этил-N-метилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты
Соединение 7 получали по методике примера 1, заменяя 2-хлор-N,N-диэтилацетамид на 2-хлор-N-этил-N-метилацетамид (выход 88%).
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,89 (т, 3Н), 1,28 (дт, 3Н), 1,36 (т, 3Н), 1,60 (кв, 2Н), 2,92 (д, 3Н), 3,35 (м, 2Н), 3,60 (с, 2Н), 3,99-4,07 (м, 4Н), 4,77 (с, 2Н), 6,79 (м, 2Н), 6,91 (с, 1Н). ВЭЖХ (ОФ, 30-90% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания2,45 мин, чистота 99% по данным ВЭЖХ.
Пример 8. Соединение 8: этиловый эфир [4-[(N-этил-N-пропилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты
Соединение 8 получали по методике примера 2, заменяя 2-хлор-N,N-диэтилацетамид на 2-хлор-N-этил-N-пропилацетамид (выход 64%).
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,88 (м, 3Н), 1,05 (т, 15Н), 1,21 (м, 4,5Н), 1,36 (т, 3Н), 1,47-1,65 (м, 2Н), 3,21-3,41 (м, 4Н), 3,59 (с, 2Н), 4,00-4,14 (м, 4Н), 4,77 (д, 2Н), 6,77 (м, 2Н), 6,90 (с, 1Н). ВЭЖХ (ОФ, 30-90% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания2,81 мин, чистота 95% по данным ВЭЖХ.
Пример 9. Соединение 9: пропиловый эфир [4-[(N-этил-N-пропилкарбамоил)метокси]-3-этоксифенил]уксусной кислоты
Соединение 9 получали по методике примера 1, заменяя 2-хлор-N,N-диэтилацетамид на 2-хлор-N-этил-N-пропилацетамид (выход 92%).
ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,89 (м, 6Н), 1,12 (дт, 3Н), 1,36 (т, 3Н), 1,47-1,67 (м, 4Н), 3,21-3,39 (м, 4Н), 3,60 (с, 2Н), 4,77 (д, 2Н), 6,79 (м, 2Н), 6,91 (с, 1Н). ВЭЖХ (ОФ, 30-90% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания2,95 мин, чистота 100% по данным ВЭЖХ.
Пример 10. Соединение 10: пропиловый эфир [4-[(N,N-диметилкарбамоил)метокси]-3-пропоксифенил]уксусной кислоты
(1) Получение пропилового эфира 2-[4-[(N,N-диметилкарбамоил)метокси]-3-пропоксифенил]-2-гидроксиуксусной кислоты (10-D)
Соединение 10-D (1,4 г) получали по методике примера 4В подраздела (4) с использованием в качестве реагентов соединения С (2,49 г, 9,28 ммоль), ацетона (60 мл), карбоната калия (2,55 г, 18,5 ммоль) и N,N-диметилацетамида (1,42 г, 11,5 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,11; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,71 (т, 3Н), 0,89 (т, 3Н), 1,44 (кв, 2Н), 1,65 (кв, 2Н), 2,75 (с, 3Н), 2,91 (с, 3Н), 3,80-3,91 (м, 4Н), 4,69 (с, 2Н), 4,95 (д, 1Н), 5,86 (д, 1Н), 6,72 (д, 1Н), 6,77 (д, 1Н), 6,91 (с, 1Н).
(2) Получение пропилового эфира 2-[4-[(N,N-диметилкарбамоил)метокси]-3-пропоксифенил]-2-ацетоксиуксусной кислоты (10-Е)
Соединение 10-Е (1,4 г) получали по методике примера 4В подраздела (5) с использованием в качестве реагентов соединения 10-D (1,4 г, 3,96 ммоль), DCM (100 мл), пиридина (1,0 мл, 12,4 ммоль) и бромистого ацетила (0,55 мл, 7,44 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,20; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,71 (т, 3Н), 0,89 (т, 3Н), 1,44 (кв, 2Н), 1,65 (кв, 2Н), 2,03 (с, 3Н), 2,75 (с, 3Н), 2,91 (с, 3Н), 3,84 (т, 2Н), 3,95 (м, 2Н), 4,74 (с, 2Н), 4,95 (д, 1Н), 5,68 (с, 1Н), 6,76 (д, 1Н), 6,83 (д, 1Н), 6,95 (с, 1Н).
(3) Синтез соединения 10
В результате обработки соединения 10-Е водородом в соответствии со способом примера 4В подраздела (6) получали соединение 10 в виде белого твердого вещества (0,80 г, 2,37 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,17; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,71 (т, 3Н), 0,89 (т, 3Н), 1,44 (кв, 2Н), 1,65 (кв, 2Н), 2,75 (с, 3Н), 2,91 (с, 3Н), 3,48 (с, 2Н), 3,84 (т, 2Н), 3,90 (т, 2Н), 4,67 (с, 2Н), 6,64 (д, 1Н), 6,70 (д, 1Н), 6,79 (с, 1Н). ВЭЖХ (ОФ, 10-70% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания4,23 мин, чистота 99,2% по данным ВЭЖХ.
Пример 11. Соединение 11: пропиловый эфир [4-[(N-этил-N-пропилкарбамоил)метокси]-3-пропоксифенил]уксусной кислоты
(1) Получение пропилового эфира 2-[4-[(N-этил-N-пропилкарбамоил)метокси]-3-пропоксифенил]-2-гидроксиуксусной кислоты (11-D)
Соединение 11-D (1,75 г) получали по методике примера 4В подраздела (4) с использованием в качестве реагентов соединения С (2,43 г, 9,06 ммоль), ацетона (60 мл), карбоната калия (2,50 г, 18,1 ммоль) и 2-хлор-N-этил-N-пропилацетамида (1,94 г, 11,9 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,28; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,67-0,79 (м, 6Н), 0,87-0,99 (м, 3Н), 1,00-1,07 (м, 3Н), 1,40-1,47 (м, 4Н), 1,65 (кв, 2Н), 3,11-3,31 (м, 4Н), 3,82 (т, 2Н), 3,88 (т, 2Н), 4,66 (д, 2Н), 4,94 (д, 1Н), 5,85 (д, 1Н), 6,74 (д, 1Н), 6,77 (д, 1Н), 6,92 (с, 1Н).
(2) Получение пропилового эфира 2-[4-[(N-этил-N-пропилкарбамоил)метокси]-3-пропоксифенил]-2-ацетоксиуксусной кислоты (11-Е)
Соединение 11-Е (2,0 г) получали по методике примера 4В подраздела (5) с использованием в качестве реагентов соединения 11-D (1,70 г, 4,29 ммоль), DCM (100 мл), пиридина (1,0 мл, 12,4 ммоль) и бромистого ацетила (0,60 мл, 4,77 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,49; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,67-0,79 (м, 6Н), 0,87-0,92 (м, 3Н), 1,00-1,07 (м, 3Н), 1,43-1,46 (м, 4Н), 1,65 (кв, 2Н), 2,03 (с, 3Н), 3,11-3,31 (м, 4Н), 3,83 (т, 2Н), 3,95 (т, 2Н), 4,72 (д, 2Н), 5,72 (д, 1Н), 6,74 (д, 1Н) 6,77 (д, 1Н), 6,92 (с, 1Н).
(3) Синтез соединения 11
В результате обработки соединения 11-Е водородом в соответствии со способом примера 4В подраздела (6) соединение 11 получали в виде бесцветного масла (0,95 г, 2,50 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,49; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,70-0,80 (м, 6Н), 0,87-0,95 (м, 4,5Н), 1,05 (т, 1,5Н), 1,45-1,52 (м, 4Н), 1,52-1,65 (м, 2Н), 3,11-3,27 (м, 4Н), 3,48 (с, 2Н), 3,82 (т, 2Н), 3,95 (т, 2Н), 4,64 (д, 2Н), 6,64-6,67 (кв, 2Н), 6,79 (с, 1Н). ВЭЖХ (ОФ, 10-70% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания5,26 мин, чистота 100% по данным ВЭЖХ.
Пример 12. Соединение 12: пропиловый эфир [4-[(N,N-дипропилкарбамоил)метокси]-3-пропоксифенил]уксусной кислоты
(1) Получение пропилового эфира [4-[(N,N-дипропилкарбамоил)метокси]-3-пропоксифенил]-2-гидроксиуксусной кислоты (12-D)
Соединение 12-D (1,0 г) получали по методике примера 4В подраздела (4) с использованием в качестве реагентов соединения С (2,27 г, 8,46 ммоль), ацетона (60 мл), карбоната калия (2,50 г, 18,1 ммоль) и 2-хлор-N,N-дипропилацетамида (1,65 г, 9,29 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,36; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,67-0,79 (м, 6Н), 0,87-0,92 (м, 3Н), 1,43-1,46 (м, 4Н), 1,65 (кв, 2Н), 2,03 (с, 3Н), 3,11-3,31 (м, 4Н), 3,81 (т, 2Н), 3,89 (т, 2Н), 4,67 (с, 2Н), 4,94 (д, 1Н), 5,86 (д, 1Н), 6,71 (д, 1Н), 6,78 (д, 1Н), 6,91 (с, 1Н).
(2) Получение пропилового эфира 2-[4-[(N,N-дипропилкарбамоил)метокси]-3-пропоксифенил]-2-ацетоксиуксусной кислоты (12-Е)
Соединение 12-Е (1,0 г) получали по методике примера 4В подраздела (5) с использованием в качестве реагентов соединения 12-D (1,70 г, 4,29 ммоль), DCM (100 мл), пиридина (1,0 мл, 12,4 ммоль) и бромистого ацетила (0,60 мл, 4,77 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,57; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,67-0,79 (м, 6Н), 0,90 (т, 3Н), 1,43-1,48 (м, 4Н), 1,65 (кв, 2Н), 2,03 (с, 3Н), 3,11-3,31 (м, 4Н), 3,83 (т, 2Н), 4,73 (с, 2Н), 5,72 (д, 1Н), 6,74 (д, 1Н), 6,85 (д, 1Н), 6,96 (с, 1Н).
(3) Синтез соединения 12
В результате обработки соединения 12-Е водородом в соответствии со способом примера 4В подраздела (6) соединение 12получали в виде бесцветного масла (0,80 г, 2,03 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,63; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,69-0,80 (м, 9Н), 0,89 (т, 3Н), 1,36-1,51 (м, 2Н), 1,64 (кв, 2Н), 3,08-3,17 (м, 4Н), 3,48 (с, 2Н), 3,81 (т, 2Н), 3,89 (т, 2Н), 4,65 (с, 2Н), 6,64-6,69 (м, 2Н), 6,79 (с, 1Н). ВЭЖХ (ОФ, 10-70% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания5,45 мин, чистота 100% по данным ВЭЖХ.
Пример 13. Соединение 13: пропиловый эфир [4-[(N-этил-N-метилкарбамоил)метокси]-3-пропоксифенил]уксусной кислоты
(1) Получение пропилового эфира 2-[4-[(N-этил-N-метилкарбамоил)метокси]-3-пропоксифенил]-2-гидроксиуксусной кислоты (13-D)
Соединение 13-D (1,6 г) получали по методике примера 4В подраздела (4) с использованием в качестве реагентов соединения С (2,26 г, 8,42 ммоль), ацетона (60 мл), карбоната калия (2,50 г, 18,1 ммоль) и 2-хлор-N-этил-N-метилацетамида (1,26 г, 9,29 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,16; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,71 (т, 3Н), 0,91 (кв, 4,5Н), 1,06 (т, 1,5Н), 1,45 (кв, 2Н), 1,65 (кв, 2Н), 2,80 (д, 3Н), 3,20-3,28 (м, 2Н), 3,84 (т, 2Н), 3,96 (м, 2Н), 4,73 (с, 2Н), 4,95 (д, 1Н), 5,73 (д, 1Н), 6,79 (д, 1Н), 6,85 (д, 1Н), 6,96 (с, 1Н).
(2) Получение пропилового эфира 2-[4-[(N-этил-N-метилкарбамоил)метокси]-3-пропоксифенил]-2-ацетоксиуксусной кислоты (13-Е)
Соединение 13-Е (1,9 г) получали по методике примера 4В подраздела (5) с использованием в качестве реагентов соединения 13-D (1,60 г, 4,35 ммоль), DCM (100 мл), пиридина (1,0 мл, 12,4 ммоль) и бромистого ацетила (0,60 мл, 4,77 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,25; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,71 (т, 3Н), 0,91 (кв, 4,5Н), 1,06 (т, 1,5Н), 1,45 (кв, 2Н), 1,65 (кв, 2Н), 2,04 (с, 3Н), 2,80 (д, 3Н), 3,20-3,28 (м, 2Н), 3,84 (т, 2Н), 3,96 (м, 2Н), 4,73 (с, 2Н), 5,73 (с, 1Н), 6,79 (д, 1Н), 6,85 (д, 1Н), 6,96 (с, 1Н).
(3) Синтез соединения 13
В результате обработки соединения 13-Е водородом в соответствии со способом примера 4В подраздела (6) соединение 13 получали в виде бесцветного масла (1,5 г, 4,27 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,28; ЯМР 1Н (ДМСО-d6, 300 МГц): δ 0,78 (т, 3Н), 0,90 (м, 4,5Н), 1,05 (т, 1,5Н), 1,48 (кв, 2Н), 1,64 (кв, 2Н), 2,80 (д, 3Н), 3,20-3,28 (м, 4Н), 3,48 (с, 2Н), 3,82 (т, 2Н), 3,89 (т, 2Н), 4,65 (с, 2Н), 6,65-6,69 (м, 2Н), 6,79 (с, 1Н). ВЭЖХ (ОФ, 10-70% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания4,47 мин, чистота 99% по данным ВЭЖХ.
Пример 14. Соединение 14: этиловый эфир [4-[(N-этил-N-пропилкарбамоил)метокси]-3-пропоксифенил]уксусной кислоты
Соединение 11 (0,201 г, 0,510 ммоль) омыляли, растворяя в смеси (1:1) МеОН: деионизованная вода (10 мл). При погружении смеси в баню со льдом добавляли 0,1н. NaOH (5,1 мл, 0,51 ммоль) и перемешивали смесь в течение ночи, разбавляли деионизованной водой и промывали DCM. Водную часть подкисляли 1н HCl, а продукт экстрагировали DCM и сушили над сульфатом магния. Растворитель удаляли в вакууме.
Кислотный продукт вновь растворяли в этаноле (20 мл), добавляли серную кислоту (2 капли) и нагревали смесь при 110°С в течение ночи. Растворитель удаляли в вакууме, а продукт затем очищали колоночной хроматографией, получая соединение 14 в виде бесцветного масла (170 мг, 0,465 ммоль).
ТСХ (силикагель, 50% EtOAc/гексан), Rf 0,59; ЯМР 1Н (CDCl3, 300 МГц): δ 0,82 (кв, 3Н), 0,94-1,20 (м, 9Н), 1,52 (м, 2Н), 1,74 (м, 2Н), 3,22 (д, 2Н), 3,34 (кв, 2Н), 3,45 (с, 2Н), 3,89 (т, 2Н), 4,07 (кв, 2Н), 4,63 (с, 2Н), 6,68 (д, 1Н), 6,76 (с, 12Н), 6,81 (д, 1Н). ВЭЖХ (ОФ, 10-70% ацетонитрил/вода, проход 6 минут, 214 нм), время удерживания4,88 мин, чистота 95% по данным ВЭЖХ.
Пример 15. Соединение сравнения пропанидид: пропиловый эфир [4-[(N,N-диэтилкарбамоил)метокси]-3-метоксифенил]уксусной кислоты
(1) Получение пропилового эфира 3-метокси-4-гидроксифенилуксусной кислоты (15-А)
4-Гидрокси-3-метоксифенетиловый спирт (Sigma-Aldrich) растворяли в безводном 1-пропаноле. К данному раствору добавляли ˜5 капель концентрированной серной кислоты и нагревали раствор при 100°С в течение 3-5 часов в емкости под давлением. По завершении реакции 1-пропанол удаляли при пониженном давлении, полученное масло разбавляли этилацетатом и промывали насыщенным раствором гидрокарбоната натрия, дистиллированной водой, а затем насыщенным раствором соли. Раствор сушили над сульфатом магния и фильтровали и удаляли растворитель при пониженном давлении, получая 15-А в виде красного масла с практически количественным выходом.
ЯМР 1Н (ДМСО, 300 МГц): δ 0,77 (3Н, т, СН3), 1,47 (2Н, кв, СН2), 3,44 (2Н, с, ArCH2CO), 3,65 (3Н, с, OCH3), 3,89 (2Н, т, OCH2), 6,60 (2Н, м, ArH), 6,73 (1Н, с, ArH), 8,79 (1Н, с, ArOH).
(2) Получение пропилового эфира 4-[(N,N-диэтилкарбамоил)метокси]-3-метоксифенил]уксусной кислоты
Пропиловый эфир 3-метокси-4-гидроксифенилуксусной кислоты (15-А) растворяли в ацетоне. К данному раствору добавляли 2 эквивалента К2СО3, затем 1,2 эквивалента 2-хлор-N,N-диэтилацетамида. При энергичном перемешивании суспензию нагревали до кипения с обратным холодильником (60°С) в течение ˜15 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали и удаляли оставшийся растворитель при пониженном давлении, получая темное желтое масло с выходом 95%. Маслообразный продукт очищали колоночной хроматографией на силикагеле, получая указанное в заголовке соединение.
ЯМР 1Н (ДМСО, 300 МГц): δ 0,78 (3Н, т, СН3), 0,94 (3Н, т, СН3), 1,05 (3Н, т, СН3), 1,49 (2Н, кв, СН2), 3,20 (4Н, м, N-этил СН3), 3,49 (2Н, с, ArCH2CO), 3,66 (3Н, с, OCH3), 3,90 (2Н, т, OCH2), 4,63 (2Н, с, OCH2СО), 6,72 (2Н, м, ArH), 6,80 (1Н, с, ArH).
Пример 16. Далее приведена иллюстрация представительных фармацевтических дозированных форм, содержащих соединение изобретения «соединение Х»
«Соединение Х»
соевое масло
яичный фосфатид
глицерин
дигидрат эдетата динатрия
гидроксид натрия
вода для инъекций
2,0
10,0
1,2
2,25
0,0055
q.s.
до 100
«Соединение Х»
соевое масло
фракционированное кокосовое масло
яичный фосфатид
глицерин
дигидрат эдетата динатрия
гидроксид натрия
вода для инъекций
1,0
5,0
5,0
1,2
2,25
0,0055
q.s.
до 100
«Соединение Х»
N-метилпирролидидон
пропиленгликоль
вода для инъекций
1,0% мас./об.
30% мас./об.
40% мас./об.
«Соединение Х»
N-метилпирролидидон
пропиленгликоль
вода для инъекций
2,0% мас./об.
30% мас./об.
40% мас./об.
«Соединение Х»
соевое масло
лецитин
глицерин
гидроксид натрия
вода для инъекций
1,0
1,0-3,0
1,2
2,25
q.s.
до 100
«Соединение Х»
соевое масло
сафлоровое масло
яичные фосфатиды
глицерин
гидроксид натрия
вода для инъекций
1,0% мас./об.
10,0% мас./об.
10,0% мас./об.
1,2% мас./об.
2,5% мас./об.
q.s.
«Соединение Х»
соевое масло
яичные фосфатиды
глицерин
гидроксид натрия
вода для инъекций
1,0% мас./об.
10,0% мас./об.
1,2% мас./об.
2,5% мас./об.
q.s.
«Соединение Х»
соевое масло
фосфатидилхолин
из яичного желтка
глицерин
гидроксид натрия
вода для инъекций
1,0% мас./об.
30% мас./об.
1,2% мас./об.
1,67% мас./об.
q.s.
«Соединение Х»
соевое масло
лецитин
глицерин
олеиновая кислота
0,1н гидроксид натрия
вода для инъекций
4,0% мас./об.
20% мас./об.
2,4% мас./об.
2,5% мас./об.
0,03% мас./об.
q.s. до рН 8
«Соединение Х»
каприловый/каприновый триглицерид
яичные фосфатиды
глицерин
гидроксид натрия
вода для инъекций
10,0% мас./об.
10,0% мас./об.
1,2% мас./об.
2,5% мас./об.
q.s.
«Соединение Х»
каприловый/каприновый триглицерид
яичные фосфатиды
глицерин
гидроксид натрия
вода для инъекций
5,0% мас./об.
15,0% мас./об.
1,2% мас./об.
2,5% мас./об.
q.s.
«Соединение Х»
Migliyol®810
фосфатиды яичного желтка
DMPG
глицерин
гидроксид натрия
вода для инъекций
10,0% мас./об.
5,0-10,0% мас./об.
0,5-1,0% мас./об.
0,1% мас./об
2,25% мас./об.
q.s.
Описанные выше препараты можно получить по обычным методикам, хорошо известным в области фармации. Например, препарат соединения 1 в соответствии с инъекцией 9 получали по следующей методике.
Смесь L-α-фосфатадилхолина 60% (лецитин) (2,40 г), глицерина (98%) (2,50 г) (и то, и другое от Sigma-Aldrich), олеиновой кислоты (99%) (0,03 г) (Fluka-Sigma-Aldrich, Buchs, Switzerland) и деионизированной воды (71,1 г) нагревали до 60°С до полного растворения, получая непрозрачный раствор. Пока раствор был еще теплым, доводили рН до рН 8, добавляя 0,1н. NaOH. Смесь соединения 1 (4,0 г) и соевого масла (Sigma-Aldrich) (20,0 г) нагревали до 60°С до смешивания, а затем добавляли к первой смеси. Раствор недолго перемешивали при 60°С, а затем переносили в химический стакан и перемешивали при помощи гомогенизатора тканей Polytron в течение 5 мин на максимальной скорости, получая предварительно смешанный раствор.
Микрофлюидизатор (Microfluidics Corp., Newton, MA, модель № 110S) промывали изопропанолом, а затем деионизованной водой. Микрофлюидизатор заливали минимальным количеством предварительно смешанного раствора. Резервуар микрофлюидизатора заполняли предварительно смешанным раствором и данный раствор циркулировал через смесительную камеру в течение 30 сек при максимальном давлении (˜12000-15000 фунтов/кв.дюйм). Первые ˜10 капель микрофлюидизированного раствора собирали и отбрасывали, затем все последующие фракции собирали в стеклянный сосуд.
Все публикации, патенты и патентные документы включены здесь в виде ссылки. Изобретение описано со ссылкой на различные конкретные и предпочтительные способы воплощения и методы. Однако следует понимать, что возможно много изменений и модификаций, в пределах духа и рамок и данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АКТИВАТОРЫ РАСТВОРИМОЙ ГУАНИЛАТЦИКЛАЗЫ И ИНГИБИТОРЫ ФОСФОДИЭСТЕРАЗЫ С ДВОЙНЫМ МЕХАНИЗМОМ ДЕЙСТВИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2758373C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНА | 2011 |
|
RU2528386C2 |
| СОЕДИНЕНИЯ СУЛЬФОНИМИДАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВНОСТИ ИНТЕРЛЕЙКИНА-1 | 2019 |
|
RU2820289C2 |
| ЭФИРЫ 5-АМИНОЛЕВУЛИНОВОЙ КИСЛОТЫ КАК ФОТОЧУВСТВИТЕЛЬНЫЕ АГЕНТЫ В ФОТОХИМИОТЕРАПИИ | 2001 |
|
RU2246483C2 |
| АЛКАЛОИДНЫЕ ПРОИЗВОДНЫЕ СЛОЖНЫХ АМИНОЭФИРОВ И ВКЛЮЧАЮЩИЕ ИХ ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ | 2011 |
|
RU2569846C2 |
| БОРСОДЕРЖАЩИЕ МАЛЫЕ МОЛЕКУЛЫ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ АГЕНТОВ | 2009 |
|
RU2547441C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2013 |
|
RU2654327C2 |
Изобретение относится к соединению формулы (I), в которой R1 представляет (С2-С6)алкил, каждый из R2, R3 и R4 представляют (С1-С6)алкил, при условии, что сумма числа атомов углерода в R1, R2, R3 и R4 больше 7. Изобретение относится к соединению формулы (II), в которой R1 представляет собой этил, R4 представляет собой пропил, а R5 представляет собой водород. Изобретение относится к фармацевтической композиции, обладающей способностью создания или поддержания анестезии или седативного эффекта, содержащей эффективное количество соединения формулы I и фармацевтически приемлемый носитель. Также изобретение относится к применению соединения формулы I для получения лекарственного средства, применимого для создания или поддержания анестезии, или седативного эффекта у млекопитающего и к способу создания или поддержания анестезии или седативного эффекта у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы I. 5 н. и 7 з.п. ф-лы, 2 табл., 2 ил.
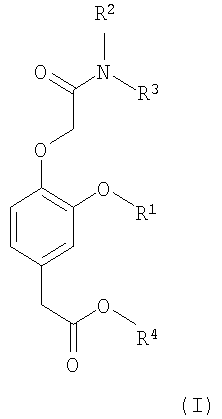
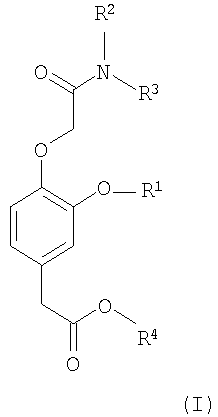
в которой R1 представляет (С2-С6)алкил,
каждый из R2, R3 и R4 представляют (С1-С6)алкил,
при условии, что сумма числа атомов углерода в R1, R2, R3 и R4 больше 7.
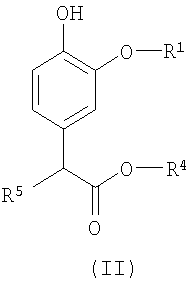
в которой R1 представляет собой этил, R4 представляет собой пропил, а R5 представляет собой водород.
| Щеточная траверса электрической машины | 1983 |
|
SU1134981A1 |
| АВТОМАТИЧЕСКИЙ КАРАНДАШ С ПЛОСКИМ ГРИФЕЛЕМ | 0 |
|
SU236280A1 |
| US 4918092 A, 17.04.1990 | |||
| BULLETIN OF THE KOREAN CHEMICAL SOCIETY, 1992, vol.13(1), P.87-91 | |||
| СПОСОБ ПОЛУЧЕНИЯ АМИДОВ АРИЛОКСИАЛКИЛКАРБОНОВЫХ КИСЛОТ | 0 |
|
SU259871A1 |

