ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к области медицинского лечения и диагностики заболевания. Более конкретно, настоящее изобретение относится к новой сульфонилмочевине и родственным соединениям и их применению в лечении или идентификации заболевания или состояния, ответственного за модуляцию NLRP3 или ингибирование активации NLRP3 или родственных компонентов воспалительного процесса.
УРОВЕНЬ ТЕХНИКИ
[0002] Любая ссылка на уровень техники в настоящем документе не должна истолковываться как допущение того, что такой уровень техники составляет общеизвестные знания в Австралии или где-либо еще.
[0003] Инфламмасома с содержащим пириновый домен протеином 3 (NLRP3) семейства NOD-подобных рецепторов (NLR) является компонентом воспалительного процесса, и ее аномальная активация является патогенной в унаследованных нарушениях, таких как криопирин-ассоциированные периодические синдромы (КАПС (CAPS)) и комплексные заболевания, такие как рассеянный склероз, диабет 2-го типа, болезнь Альцгеймера и атеросклероз.
[0004] NLRP3 представляет собой внутриклеточную сигнальную молекулу, которая воспринимает многие факторы патогенного происхождения, окружающей среды и происходящие от хозяина. При активации NLRP3 связывается с крапчатоподобным ассоциированным с апоптозом белком, содержащим домен активации и рекрутирования каспазы (ASC). Затем ASC полимеризуется с образованием большого агрегата, известного как комплекс ASC. Полимеризованный ASC в свою очередь взаимодействует с цистеиновой протеазой каспазой-1 с образованием комплекса, называемого инфламмасомой. Это приводит к активации каспазы-1, которая расщепляет провоспалительные цитокины ИЛ-1β и ИЛ-18 до их активных форм и участвует в типе гибели воспалительных клеток, известном как пироптоз. Комплекс ASC может также рекрутировать и активировать каспазу-8, которая может процессировать про-ИЛ-1β и про-ИЛ-18 и запускать апоптотическую гибель клеток.
[0005] Каспаза-1 расщепляет про-ИЛ-1β и про-ИЛ-18 до их активных форм, которые выделяются из клетки. Активная каспаза-1 также расщепляет гасдермин-D с запуском пироптоза. Посредством ее управления пироптотическим путем гибели клеток каспаза-1 также участвует в высвобождении молекул алармина, таких как ИЛ-33 и белок группы высокой мобильности бокс 1 (HMGB1). Каспаза-1 также расщепляет внутриклеточный ИЛ-Ш2, приводя к его деградации и обеспечивая высвобождение ИЛ-1α. В клетках человека каспаза-1 может также управлять процессингом и секрецией ИЛ-37. Ряд других субстратов каспазы-1, таких как компоненты цитоскелета и гликолизного пути, могут вносить вклад в зависимое от каспазы-1 воспаление.
[0006] Зависимые от NLRP3 комплексы ASC высвобождаются во внеклеточное окружение, где они могут активировать каспазу-1, вызывать процессинг субстратов каспазы-1 и распространять воспаление.
[0007] Активные цитокины, происходящие из активации инфламмасомы NLRP3, являются важными движущими факторами воспаления и взаимодействуют с другими путями цитокинов для формирования иммунного ответа на заражение и повреждение. Например, передача сигнала ИЛ-1β вызывает секрецию провоспалительных цитокинов ИЛ-6 и TNF. ИЛ-1β и ИЛ-18 действуют синергически с ИЛ-23, вызывая образование ИЛ-17 с помощью клеток памяти Th17 CD4 и Т-клеток γδ в отсутствие действия рецептора Т-клеток. ИЛ-18 и ИЛ-12 также действуют синергически, вызывая образование IFN-γ из Т-клеток памяти и натуральной клетки-киллера, управляя реакцией Th1.
[0008] Другие внутриклеточные образраспознающие рецепторы (ОРР) также способны образовывать инфламмасомы. Они включают другие члены семейства NLR, такие как NLRP1 и NLRC4, а также ОРР не NLR-типа, такие как сенсоры двухцепочечной ДНК (dsDNA), белок 2, отсутствующий при меланоме (AIM2) и интерферон гамма-индуцируемый белок 16 (IFI16). Зависящий от NLRP3 процессинг ИЛ-1β также может активироваться непрямым, неканоническим путем после каспазы-11.
[0009] Унаследованные КАПС (CAPS) заболевания синдром Макла-Уэльса (СМУ (MWS)), семейный холодовой аутовоспалительный синдром и мультисистемное воспалительное заболевание неонатального возраста вызваны мутациями с приобретением функции в NLRP3, что определяет NLRP3 как критический компонент воспалительного процесса. NLRP3 также вовлечен в патогенез ряда комплексных заболеваний, в частности, включающих метаболические нарушения, таких как диабет 2-го типа, атеросклероз, ожирение и подагра.
[0010] Выявляется роль NLRP3 в заболеваниях центральной нервной системы, и показано, что на заболевания легких также оказывает влияние NLRP3. Кроме того, NLRP3 играет роль в развитии заболевания печени, заболевания почек и старении. Многие из этих связей были установлены с использованием мышей Nlrp3-/-, но появилось также понимание о специфичной активации NLRP3 в этих заболеваниях. При диабете 2-го типа отложение островкового амилоидного полипептида в поджелудочной железе активирует передачу сигнала NLRP3 и ИЛ-1β, приводя к гибели клеток и воспалению.
[0011] Было показано, что несколько небольших молекул ингибируют инфламмасому NLRP3. Глибурид ингибирует образование ИЛ-1β в микромолярных концентрациях в ответ на активацию NLRP3, но не NLRC4 или NLRP1. Другие ранее охарактеризованные ингибиторы NLRP3 включают партенолид, 3,4-метилендиокси-β-нитростирол и диметилсульфоксид (ДМСО), хотя эти средства имеют ограниченную активность и являются неспецифичными.
[0012] Современные способы лечения связанных с NLRP3 заболеваний включают биологические агенты, воздействующие на ИЛ-1. Таковыми являются рекомбинантный антагонист рецептора ИЛ-1 анакинра, нейтрализующее ИЛ-1β антитело канакинумаб и растворимый рецептор-ловушка ИЛ-1 рилонацепт. Эти подходы доказали свою эффективность в лечении КАПС (CAPS), и эти биологические агенты применяются в клинических испытаниях для других связанных с ИЛ-1β заболеваниях.
[0013] Было показано, что несколько небольших молекул ингибируют инфламмасому NLRP3. Глибурид ингибирует образование ИЛ-1β в микромолярных концентрациях в ответ на активацию NLRP3, но не NLRC4 или NLRP1. Другие ранее охарактеризованные ингибиторы NLRP3 включают партенолид, 3,4-метилендиокси-β-нитростирол и диметилсульфоксид (ДМСО), хотя эти средства имеют ограниченную активность и являются неспецифичными.
[0014] Некоторые содержащие диарилсульфонилмочевину соединения были идентифицированы как ингибирующие высвобождение цитокинов препараты (ИВЦП (CRID)) (Perregaux et al.; J. Pharmacol. Exp.Ther. 299, 187-197, 2001). ИВЦП (CRID) представляют собой класс содержащих диарилсульфонилмочевину соединений, которые ингибируют посттрансляционный процессинг ИЛ-1β. Посттрансляционный процессинг ИЛ-1β сопровождается активацией каспазы-1 и гибелью клеток. ИВЦП (CRID) блокируют активированные моноциты так, что каспаза-1 остается неактивной и задержка, связанная с клеточной мембраной, сохраняется.
[0015] Существует потребность в обеспечении соединений с улучшенными фармакологическими и/или физиологическими и/или физико-химическими свойствами и/или таких, которые обеспечивают полезную альтернативу известным соединениям.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0016] Согласно первому аспекту изобретения, предусматривается соединение формулы (I), или его фармацевтически приемлемая соль, сольват или пролекарство:
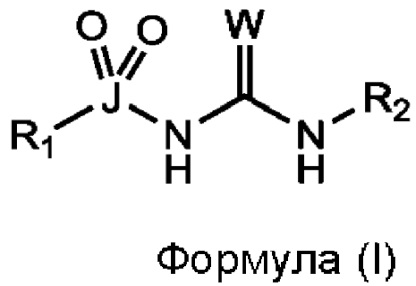
где W выбран из О, S и Se;
J выбран из S и Se;
R1 выбран из группы, состоящей из циклоалкила, арила, гетероарила и гетероциклила, все из которых могут быть необязательно замещенными;
R2 выбран из группы, состоящей из циклоалкила, арила, гетероарила и гетероциклила, все из которых могут быть необязательно замещенными; и
как R1 непосредственно связан с J, так и R2 непосредственно связан с примыкающим атомом азота, через атом углерода.
[0017] Согласно второму аспекту изобретения предусматривается фармацевтическая композиция, содержащая соединение из первого аспекта или его фармацевтически приемлемую соль, сольват или пролекарство, и фармацевтически приемлемый носитель, разбавитель и/или вспомогательное вещество.
[0018] Третий аспект изобретения заключается в способе лечения или профилактики заболевания, нарушения или состояния, включающем стадию введения эффективного количества соединения из первого аспекта или его фармацевтически эффективной соли, сольвата или пролекарства, или фармацевтической композиции из второго аспекта для лечения или профилактики этим заболевания, нарушения или состояния.
[0019] Четвертый аспект изобретения предусматривает соединение из первого аспекта или его фармацевтически эффективную соль, сольват или пролекарство, или фармацевтическую композицию из второго аспекта для применения в лечении или профилактике заболевания, нарушения или состояния.
[0020] Пятый аспект изобретения предусматривает применение соединения из первого аспекта или его фармацевтически эффективной соли, сольвата или пролекарства, в производстве лекарства для лечения или профилактики заболевания, нарушения или состояния.
[0021] В одном варианте реализации заболевания нарушение или состояние реагирует на ингибирование активации инфламмасомы NLRP3.
[0022] В конкретных неограничивающих вариантах реализации вышеупомянутых аспектов заболевание, нарушение или состояние представляет собой заболевание, нарушение или состояние иммунной системы, сердечно-сосудистой системы, эндокринной системы, желудочно-кишечного тракта, почечной системы, органов дыхания, центральной нервной системы, представляет собой рак или другую злокачественную опухоль и/или вызвано патогенным фактором или связано с ним.
[0023] В шестом аспекте изобретения предусматривается способ диагностики заболевания, нарушения или состояния у млекопитающего, включающий стадию введения меченого соединения формулы (I), (Ia), (Ib), (Ic) или (II) или его фармацевтически эффективной соли, сольвата или пролекарства, млекопитающему или в биологический образец, полученный из млекопитающего, для содействия диагностике заболевания, нарушения или состояния у млекопитающего.
[0024] Седьмой аспект изобретения заключается в способе модуляции активности биологической мишени, включающем стадию воздействия на биологическую мишень соединения из первого аспекта или его фармацевтически приемлемой соли.
[0025] Биологическая мишень может быть выбрана из группы, состоящей из инфламмасомы NLRP3, ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17.
[0026] Различные признаки и варианты реализации настоящего изобретения, указанные в отдельных разделах выше, относятся, в соответствующих случаях, к другим разделам, с соответствующими изменениями. Следовательно, признаки, указанные в одном разделе, могут сочетаться с признаками, указанными в других разделах, если это возможно.
[0027] Дополнительные признаки и преимущества настоящего изобретения будут очевидны из приведенного ниже подробного описания.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0028] Чтобы изобретение могло быть легко понято и осуществлено на практике, предпочтительные варианты реализации будут описаны с помощью примеров со ссылкой на приложенные графические материалы, в которых:
[0029] ФИГ. 1А-1С представляют собой ряд графических представлений концентраций в плазме известной сульфонилмочевины (МСС950), отображающих различные уровни дозировки в мышах; и
[0030] ФИГ. 2А-2С представляют собой ряд графических представлений концентраций в плазме сульфонилмочевины по настоящему изобретению (МСС7840) отображающих различные уровни дозировки в мышах.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0031] Настоящее изобретение основано, по крайней мере частично, на обнаружении, что определенные сульфонилмочевины и родственные соединения имеют выгодные свойства и проявляют полезную активность в ингибировании активации инфламмасомы NLRP3 и/или ингибировани ИЛ-1β и/или ИЛ-17 и/или ИЛ-18, и/или ИЛ-1α, и/или ИЛ-37, и/или ИЛ-33, а также препятствуют активности клеток Т-хелперов, таких как Th17, или модулируют ее. В частности, соединения по настоящему изобретению полезны в лечении широкого ряда нарушений, в которых играет роль воспалительный процесс, или инфламмасома NLRP3 и/или ИЛ-1β и/или ИЛ-17 и/или ИЛ-18, и/или ИЛ-1α, и/или ИЛ-37, и/или ИЛ-33 и/или клетки Th17.
[0032] Данные, полученные из пациентов КАПС (CAPS) человека и мышиных моделей КАПС (CAPS), позволили авторам настоящего изобретения считать, что ингибирование NLRP3 будет лучшим лечением по сравнению с биопрепаратами, воздействующими на ИЛ-1, поскольку ингибирование всех зависящих от NLRP3 процессов будет более эффективным, чем ингибирование одного зависящего от NLRP3 процесса, такого как передача сигнала с участием ИЛ-1.
[0033] Индивидуумы с КАПС (CAPS) проявляют разрегулированное выделение как ИЛ-1β, так и ИЛ-18, и пациенты с КАПС (CAPS), получающие лечение биопрепаратами, противодействующими ИЛ-1, имеют остаточные явления заболевания. Такие симптомы, как гиперостоз и суставная деформация, не предотвращаются биопрепаратами, воздействующими на ИЛ-1. Кроме того, симптомы с участием центральной нервной системы, такие как потеря слуха, сложно контролировать с помощью биопрепаратов, воздействующих на ИЛ-1, по-видимому, недостаточно проникающих в центральную нервную систему. Исследования в мышиных моделях КАПС (CAPS) показывают, что нарушения в передаче сигнала с участием либо ИЛ-1, либо только ИЛ-18, недостаточно для блокирования системного воспаления, в особенности у более старых животных. В тяжелой модели КАПС (CAPS) только полная потеря передачи сигнала каспазой-1 полностью избавила от заболевания.
[0034] Специфичное ингибирование NLRP3 содержащими сульфонилмочевину соединениями, такими как соединения из первого аспекта, может блокировать все процессы после NLRP3, включая образование комплекса ASC и активацию каспазы-8 и каспазы-1. Следовательно, ингибирование NLRP3 будет блокировать все зависимые от каспазы-1 процессы, такие как процессинг и секрецию ИЛ-1β, ИЛ-18 и ИЛ-37, расщепление гасдермина D, пироптоз и высвобождение ИЛ-1α, ИЛ-33 и HMGB. Помимо этого, будет блокироваться зависящее от NLRP3 внеклеточное высвобождение комплекс ASC, и будут предотвращаться зависимое от каспазы-8 расщепление про-ИЛ-1β и про-ИЛ-18 и апоптотическая гибель клеток. Таким образом, специфичное ингибирование NLRP3 соединениями из первого аспекта будет предотвращать ряд последующих воспалительных сигналов и поэтому должно показать более эффективную противовоспалительную терапию, чем только блокада ИЛ-1.
[0035] Биопрепараты, противодействующие ИЛ-1, блокируют ИЛ-1, происходящие из независимых от NLRP3 источников, такие ИЛ-1, произведенные другими инфламмасомами (например, NLRC4, NLRP1, NLRP6, AIM2) и ИЛ-1, образованные последующими путями, могут быть важны для иммунной защиты от патогенных факторов. Например, пациенты, принимающие антагонисты ИЛ-1/ИЛ-1 R, проявляют повышенную заболеваемость инфекциями верхних дыхательных путей. Специфичное ингибирование NLRP3 представленными соединениями, таким образом, может проявлять менее генерализованную иммуносупрессию по сравнению с биопрепаратами, противодействующими ИЛ-1.
[0036] ИЛ-1β и ИЛ-18, образованные по линии Nlrp3/каспаза-1, играют критические роли в управлении выработкой ИЛ-17 клетками Th17 CD4 и γδ Т-клетками. ИЛ-1β и ИЛ-18 действуют синергетически с ИЛ-23, вызывая выработку ИЛ-17 клетками памяти Th17 CD4 и γδ Т-клетками в отсутствие задействования рецепторов Т-клеток. ИЛ-17, управляемый ИЛ-1, также задействован в псориазе, диабете I типа, ревматоидном артрите, сахарном диабете 2-го типа, атеросклерозе, ожирении, подагре, и как недавно установлено, в астме.
[0037] По существу, показано, что каждое из этих заболеваний включает активацию тканевых макрофагов, дендритных клеток или микроглии мозга, управляемую либо растворимыми аларминами, либо незавершенным фагоцитозом метаболитов, которые накапливаются внеклеточно. NLRP3 реагирует на эти события, приводя к высвобождению ИЛ-1, запуская воспаление для очистки от вредного материала. Результатом будет заболевание, если этот процесс станет хроническим или сверхактивированным, что объясняет, почему так много заболеваний проявили включение NLRP3. Следовательно, ингибиторы, действующие с предотвращением активации NLRP3, могут иметь пользу в управляемых ИЛ-17, как и в управляемых ИЛ-1 заболеваниях.
[0038] В настоящем описании патента термины «содержит», «содержащий», «включает», «включающий», или подобные термины подразумевают значение неисключительного включения, так, что способ или композиция, содержащие ряд элементов, включает не только эти элементы, но вполне может включать другие не перечисленные элементы.
[0039] Если не определено иное, все технические и научные термины, использованные в настоящем документе, имеют то же значение, которое будет обычно подразумеваться специалистами в области техники, к которой относится данное изобретение.
[0040] Термин «фармацевтически приемлемая соль» в данном контексте относится к солям, которые являются токсикологически безопасными для системного или локализованного введения, такие как соли, полученные из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Фармацевтически приемлемые соли могут быть выбраны из группы, включающей соли щелочных и щелочноземельных металлов, аммония, алюминия, железа, амин, глюкозамин, хлорид, сульфат, сульфонат, бисульфат, нитрат, цитрат, тартрат, битартрат, фосфат, карбонат, бикарбонат, малат, малеат, напсилат, фумарат, сукцинат, ацетат, бензоат, терефталат, пальмоат, пиперазин, пектинат и S-метилметионин и подобные.
[0041] Термин «алкил» относится к прямоцепочечному или разветвленному алкильному заместителю, содержащему, например, от 1 до около 12 атомов углерода, предпочтительно от 1 до около 9 атомов углерода, более предпочтительно от 1 до около 6 атомов углерода, еще более предпочтительно от 1 до около 4 атомов углерода, еще более предпочтительно от 1 до 2 атомов углерода. Примеры таких заместителей могут быть выбраны из группы, состоящей из метила, этила, пропила, изопропила, н-бутила, втор-бутила, изобутила, трет-бутила, пентила, изоамила, 2-метилбутила, 3-метилбутила, гексила, гептила, 2-метилпентила, 3-метилпентила, 4-метилпентила, 2-этилбутила, 3-этилбутила, октила, нонила, децила, ундецила, додецила и подобных. Указанное количество углеродов относится к углеродному скелету и углеродным ответвлениям, но не включает атомы углерода, принадлежащие каким-либо заместителям, например, атомы углерода алкокси-заместителя, ответвленного от основной углеродной цепи. Замещенный алкил включает алкил, замещенный одним или более фрагментами, выбранными из группы, состоящей из гало (например, Cl, F, Br, и I); галогенированного алкила (например, CF3, 2-Br-этил, CH2F, CH2Cl, CH2CF3 или CF2CF3); гидроксила; амино; карбоксилата; карбоксамидо; алкиламино; ариламино; алкокси; арилокси; нитро; азидо; циано; тио; сульфоновой кислоты; сульфата; фосфоновой кислоты; фосфата и фосфоната, а также описываемые определением «необязательно замещенные».
[0042] Термин «алкенил» относится к необязательно замещенным ненасыщенным линейным или разветвленным углеводородным группам, имеющим от 2 до 12 атомов углерода, предпочтительно от 2 до 9 атомов углерода, более предпочтительно от 2 до 6 атомов углерода, и имеющим по меньшей мере одну углерод-углеродную двойную связь. В требуемых случаях алкенильная группа может иметь определенное количество атомов углерода, например, С2-С6 алкенил, включающий алкенильные группы, имеющие 2, 3, 4, 5 или 6 атомов углерода в линейных или разветвленных расположениях. Указанное количество углеродов относится к углеродному скелету и углеродным ответвлениям, но не включает атомы углерода, принадлежащие каким-либо заместителям. Примеры таких заместителей могут быть выбраны из группы, состоящей из этенила, пропенила, изопропенила, бутенила, втор- и трет-бутенила, пентенила, гексенила, гепт-l,3-диена, гекс-l,3-диена, нон-l,3,5-триена и подобных. Замещенный алкенил включает алкенил, замещенный одним или более фрагментами, выбранными из группы, состоящей из гало (например, Cl, F, Br, и I); галогенированного алкила (например, CF3, 2-Br-этил, CH2F, CH2Cl, CH2CF3 или CF2CF3); гидроксила; амино; карбоксилата; карбоксамидо; алкиламино; ариламино; алкокси; арилокси; нитро; азидо; циано; тио; сульфоновой кислоты; сульфата; фосфоновой кислоты; фосфата и фосфоната, а также описываемые определением «необязательно замещенные».
[0043] Термин «алкокси» в данном контексте означает алкильные группы с прямой или разветвленной цепью, соединенный атомом кислорода (т.е. -О-алкил), где алкил представляет собой описанный выше. В конкретных вариантах реализации алкокси относится к соединенным кислородом группам, содержащим от 1 до 10 атомов углерода («С1-10 алкокси»). В дополнительных вариантах реализации алкокси относится к соединенным кислородом группам, содержащим от 1 до 8 атомов углерода («С1-8 алкокси»), от 1 до 6 атомов углерода («С1-6-алкокси»), от 1 до 4 атомов углерода («С1-4 алкокси») или от 1 до 3 атомов углерода («С1-3 алкокси»).
[0044] Термины «циклоалкил» и «циклоалкенил» относятся к необязательно замещенным насыщенным и ненасыщенным моноциклическим, бициклическим или три циклическим углеродным группам. В требуемых случаях циклоалкильная или циклоалкенильная группа может иметь определенное количество атомов углерода, например, С3-С6 циклоалкил или циклоалкенил включает в свои рамки карбоциклическую группу, имеющую 3, 4, 5 или 6 атомов углерода. Примеры таких заместителей могут быть выбраны из группы, состоящей из циклопропила, циклобутила, циклопентила, циклопентенила, циклогексила, циклогексенила, циклогексадиенила и подобных. Замещенный циклоалкил или циклоалкенил включает замещения одним или более фрагментами, выбранными из группы, состоящей из гало (например, Cl, F, Br, и I); галогенированного алкила (например, CF3, 2-Br-этил, CH2F, CH2Cl, CH2CF3 или CF2CF3); гидроксила; амино; карбоксилата; карбоксамидо; алкиламино; ариламино; алкокси; арилокси; нитро; азидо; циано; тио; сульфоновой кислоты; сульфата; фосфоновой кислоты; фосфата и фосфоната, а также описываемые определением «необязательно замещенные».
[0045] Термин «алкилтио» в данном контексте означает тиогруппу с одним или более алкильными заместителями, где определение алкила приведено выше.
[0046] Термин «амино» в данном контексте означает фрагмент, который представлен структурой NR23 и включает первичные амины и вторичные и третичные амины, замещенные алкилом (т.е. алкиламино). Так, R23 может представлять, например, два атома водорода, два алкильных фрагмента или один атом водорода и один алкильный фрагмент.
[0047] Термин «арил» относится к стабильному моноциклическому, бициклическому или трициклическому углеродному кольцу, состоящему из до 8 членов в каждом кольце, где по меньшей мере одно кольцо является ароматическим согласно определению правила Хюккеля 4n+2. Термин включает полициклические системы, содержащие насыщенные углеродные кольца или гетероарильные или гетероциклические группы в случае, если по меньшей мере одно кольцо является арилом, как описано.
[0048] Термины «аралкил» и «арилалкил» в данном контексте означают арильную группу, определенную выше, соединенную с молекулой через алкильную группу, определенную выше.
[0049] Термин «гетероарил» относится к арильной группе, содержащей от одного или более (в частности, от одного до четырех) неуглеродного(ых) атома(ов) (в частности, N, О или S) или их комбинацию, при этом гетероарильная группа необязательно замещена по одному или более атому(ам) углерода или азота. Гетероарильные кольца могут также быть конденсированы с одним или более циклическим углеводородом, гетероциклическим, арильным или гетероарильным кольцами. Гетероарил включает, но без ограничения таковыми, 5-членные гетероарилы, имеющие один гетероатом (например, тиофены, пирролы, фураны); 5 членные гетероарилы, имеющие два гетероатома в 1,2- или 1,3- положениях (например, оксазолы, пиразолы, имидазолы, тиазолы, пурины); 5-членные гетероарилы, имеющие три гетероатома (например, триазолы, тиадиазолы); 5-членные гетероарилы, имеющие четыре гетероатома (например, тетразолы); 6-членные гетероарилы с одним гетероатомом (например, пиридин, хинолин, изохинолин, фенантрин, 5,6-циклогептенопиридин); 6-членные гетероарилы с двумя гетероатомами (например, пиридазины, циннолины, фталазины, пиразины, пиримидины, хиназолины); 6-членные гетероарилы с тремя гетероатомами (например, 1,3,5-триазин) и 6-членные гетероарилы с четырьмя гетероатомами. «Замещенный гетероарил» означает гетероарил, имеющий одну или более не взаимосвязанных групп в качестве заместителей и включающий определяемые как «необязательно замещенные».
[0050] «Гетероциклил» в данном контексте относится к неароматическому кольцу, имеющему от 5 до 8 атомов в кольце, и из этих атомов от 1 до 4 являются гетероатомами. Гетероциклические кольца могут также быть конденсированы с одним или более циклическим углеводородным, гетероциклическим, арильным или гетероарильным кольцами. Гетероциклические включают частично и полностью насыщенные гетероциклические группы. Гетероциклические системы могут быть соединены с другим фрагментом через любое количество атомов углерода или гетероатомы радикала и могут быть и насыщенными, и ненасыщенными. Неограничивающие примеры гетероциклов включают С4-С6 селеноциклы, пирролидинил, пирролинил, пиранил, пиперидинил, пиперазинил, морфолинил, тетрагидрофуранил, тетрагидротиофенил, пиразолинил, дитиолил, оксатиолил, диоксанил, диоксинил, оксазинил, азепинил, диазепинил, триазепинил, оксепинил и тиапинил, имидазолинил, тиоморфолинил и подобные.
[0051] «Необязательно замещенный» в отношении замещающей группы относится к замещающим группам, необязательно замещенным одним или более фрагментами, например, выбранным из группы, состоящей из необязательно замещенного С1-10 алкила (например, необязательно замещенного С1-6 алкила); необязательно замещенного С3-6 циклоалкила (например, необязательно замещенного циклопропила); необязательно замещенного гидроксиалкила; необязательно замещенного С1-10 алкокси (например, необязательно замещенного С1-6 алкокси); необязательно замещенного С2-10 алкенила; необязательно замещенного С2-10 алкинила; необязательно замещенного С6-С12 арила; арилокси; необязательно замещенного гетероарила; необязательно замещенного гетероциклила; гало (например, Cl, F, Br, и I); гидроксила; галогенированного алкила (например, CF3, 2-Br-этила, CH2F, CH2CF3, и CF2CF3); амино (например, NH2, NR12H, и NR12R13); алкиламино; ариламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфата; сульфоновой кислоты; эфиров сульфокислот, таких как алкил- или аралкилсульфонил, включая метансульфонил; фосфоновой кислоты; фосфата; фосфоната; моно-, ди-, или трифосфатных эфиров; тритила или монометокситритила; R24SO; R24SO2; CF3S; и CF3SO2; триалкилсилила, такого как диметил-трет-бутилсилил или дифенилметилсилил; и R24 и R25 каждый независимо выбран из Н или необязательно замещенного С1-10 алкила, С1-6 алкила или С1-4 алкила.
[0052] Во всех случаях, когда указан диапазон количества атомов в структуре (например, С1-С12, C1-C10, С1-С9, C1-С6, C1-C4, или С2-С20, C2-C12, С2-С10, С2-С9, С2-С8, С2-С6, С2-С4 алкил, алкенил и т.д.), конкретно предполагается, что может также использоваться любой поддиапазон или отдельное количество атомов углерода, попадающие в указанный диапазон. Так, например, приведение диапазона 1-12 атомов углерода (например, С1-С12), 1-9 атомов углерода (например, С1-С9), 1-6 атомов углерода (например, C1-C6), 1-4 атома углерода (например, С1-С4), 1-3 атома углерода (например, С1-С3), или 2-8 атомов углерода (например, С2-С8), используемое в отношении любой химической группы (например, алкила и т.д.), упомянутое в данном контексте, охватывает и в частности описывает 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 и/или 12 атомов углерода, если это уместно, а также любой его поддиапазон (например, 1-2 атома углерода, 1-3 атома углерода, 1-4 атома углерода, 1-5 атомов углерода, 1-6 атомов углерода, 1-7 атомов углерода, 1-8 атомов углерода, 1-9 атомов углерода, 1-10 атомов углерода, 1-11 атомов углерода, 1-12 атомов углерода, 2-3 атома углерода, 2-4 атома углерода, 2-5 атомов углерода, 2-6 атомов углерода, 2-7 атомов углерода, 2-8 атомов углерода, 2-9 атомов углерода, 2-10 атомов углерода, 2-11 атомов углерода, 2-12 атомов углерода, 3-4 атома углерода, 3-5 атомов углерода, 3-6 атомов углерода, 3-7 атомов углерода, 3-8 атомов углерода, 3-9 атомов углерода, 3-10 атомов углерода, 3-11 атомов углерода, 3-12 атомов углерода, 4-5 атомов углерода, 4-6 атомов углерода, 4-7 атомов углерода, 4-8 атомов углерода, 4-9 атомов углерода, 4-10 атомов углерода, 4-11 атомов углерода и/или 4-12 атомов углерода, и т.д., если это уместно).
[0053] Согласно первому аспекту изобретения, предусматривается соединение формулы (I) или его фармацевтически приемлемая соль, сольват или пролекарство:
где W выбран из О, S и Se;
J выбран из S и Se;
R1 выбран из группы, состоящей из циклоалкила, арила, гетероарила и гетеро цикл ил а, все из которых могут быть необязательно замещенными;
R2 выбран из группы, состоящей из циклоалкила, арила, гетероарила и гетеро цикл ил а, все из которых могут быть необязательно замещенными; и
как R1 непосредственно связан с J, так и R2 непосредственно связан с примыкающим атомом азота, через атом углерода.
[0054] В одном предпочтительном варианте реализации W представляет собой О.
[0055] В одном предпочтительном варианте реализации J представляет собой S.
[0056] В особенно предпочтительном варианте реализации W представляет собой О, a J представляет собой S.
[0057] В одном варианте реализации R1 выбран из группы, состоящей из С5 или С6 циклоалкила, 5-членного или 6-членного гетероарила, бициклического гетероарила, в котором по меньшей мере одно кольцо представляет собой гетероарил, фенил, бифенил, фенилгетероциклил, 5-членный или 6-членный гетероциклил, и гетероциклилциклоалкил, все из которых могут быть необязательно замещенными.
[0058] В определенных вариантах реализации W представляет собой О, J представляет собой S, a R1 выбран из группы, состоящей из пиразола, фурана, тетрагидрофурана, тетрагидропирана, пирана, пирролидина, пиррола, триазола, тетразола, имидазола, пиридина, морфолина, пиперазина, пиперидина, замещенного фенила, фенилгетероарила, фенилгетероциклила, бифенила, хинолина, изохинолина, нафтила, пиразина и пиримидина, все из которых могут быть необязательно замещенными по необходимости.
[0059] В одном варианте реализации, где W представляет собой О, J представляет собой S, a R1 представляет собой 2-фуран или 2-тиофен, он выбран из незамещенного 2-фурана или 2,5-замещенного фурана и незамещенного 2-тиофена или 2,5-замещенного тиофена.
[0060] В одном варианте реализации, где W представляет собой О, J представляет собой S, a R1 представляет собой 2,5-замещенный фуран или 2,5-замещенный тиофен, то 2,5-замещенный фуран или 2,5-замещенный тиофен не замещен группой третичного спирта.
[0061] В определенных вариантах реализации было обнаружено, что если Ri представляет собой незамещенный фуран, то он обладает способностью пересекать гематоэнцефалический барьер в концентрациях примерно в 10 раз больше, чем CRID3, сульфонилмочевина предшествующего уровня техники.
[0062] В вышеупомянутых вариантах реализации указание на 2,5-замещение не исключает присутствия дополнительных замещений по кольцу, а лишь показывает, что должны присутствовать перечисленные замещения. Например, 2,4,5-замещения считаются находящимися в рамках таких терминов.
[0063] Указание на 2-фуран и 2-тиофен означает, что кольцо соединено с серой сульфонила по положению 2 кольца, как показано ниже:
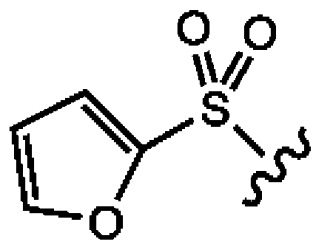
[0064] В одном варианте реализации R1 представляет собой 5-членный гетероциклил или гетероарил, каждый из которых может быть необязательно замещенным, содержащий по меньшей мере один, предпочтительно по меньшей мере два кольцевых гетероатома, выбранных из N, О и S.
[0065] В определенных вариантах реализации R1 представляет собой азотсодержащий гетероциклил или азотсодержащий гетероарил, каждый из которых может быть необязательно замещенным.
[0066] В одном варианте реализации R1 представляет собой 5-членный азотсодержащий гетероциклил или 5-членный азотсодержащий гетероарил, каждый из которых может быть необязательно замещенным.
[0067] В варианте реализации R1 представляет собой 5-членный гетероциклил или 5-членный гетероарил, каждый из которых содержит по меньшей мере два кольцевых атома азота и каждое из колец которых может быть необязательно замещено.
[0068] В одном варианте реализации W представляет собой О, J представляет собой S, a R1 выбран из группы, состоящей из хинолина, изохинолина, нафтила, пиразина, тетразола, имидазола, пирролидина, пиррола, тетрагидропирана, пирана, пиперидина, пиперазина, пиразола, пиридина, пиримидина и триазола, каждый из которых может быть необязательно замещенным.
[0069] В одном варианте реализации R1 и/или R2 могут содержать селеноцикл.
[0070] В одном варианте реализации R2 может быть выбран из бициклических и трициклических углеводородов, 5-, 6- и 7-членного гетероцикла или гетероарила, каждое из колец которых может быть необязательно замещено, и замещенного фенила.
[0071] Подходящим образом, трициклический углеводород может представлять собой индацен.
[0072] В одном варианте реализации R2 может быть выбран из 5-, 6- или 7-членных азотсодержащих гетероциклов, 6-членного азотсодержащего гетероарила и арила с конденсированным циклоалкильным кольцом.
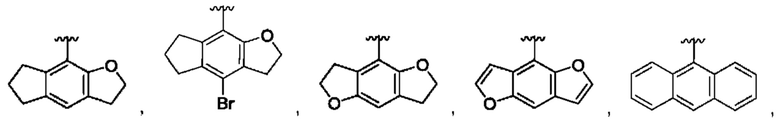
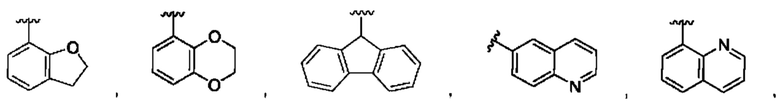
[0073] В одном варианте реализации соединения формулы (I) W представляет собой О, J представляет собой S, a R1 может быть выбран из группы, состоящей из:
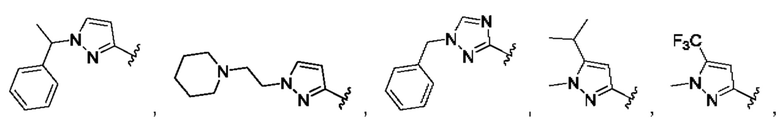
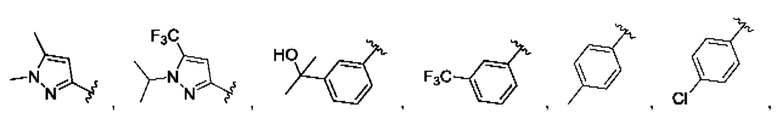
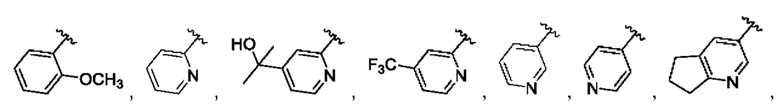

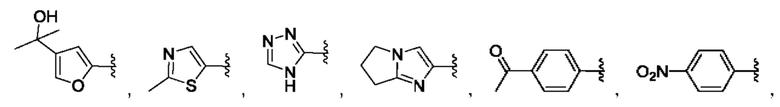
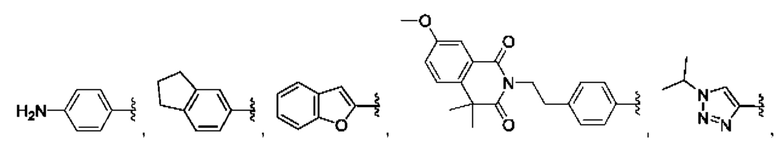
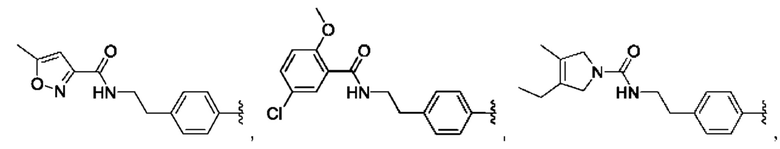
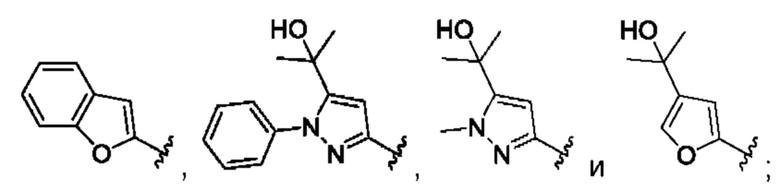
и для каждой из этих групп R1, R2 может быть независимо выбран из группы, состоящей из:
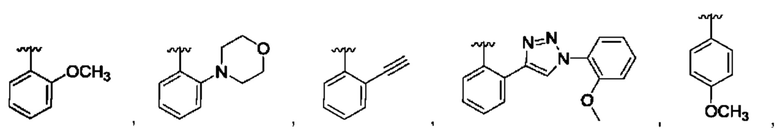
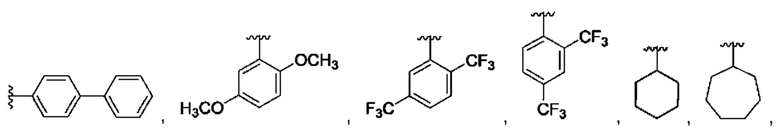
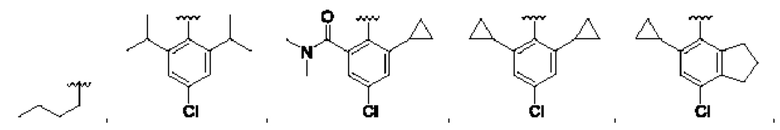
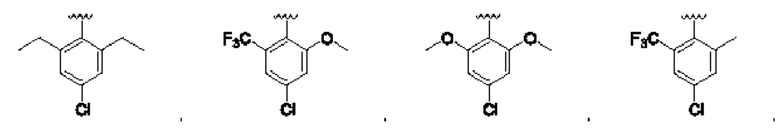
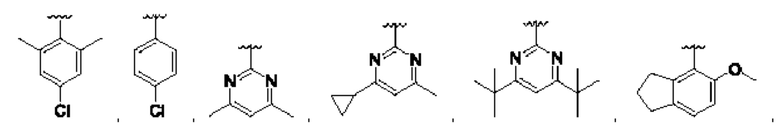
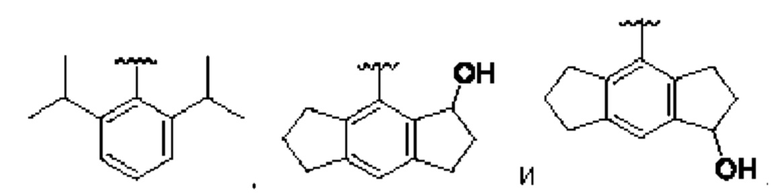
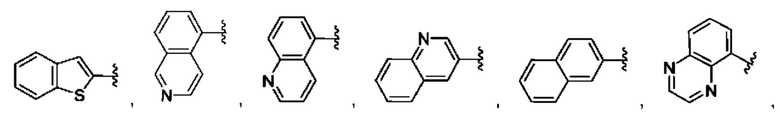
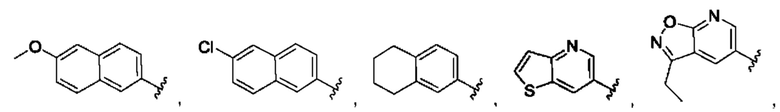
[0074] В любом варианте реализации первого аспекта, если J представляет собой S и W представляет собой О, и в комбинации с любой из перечисленных выше групп R1, R2 может быть выбран из:
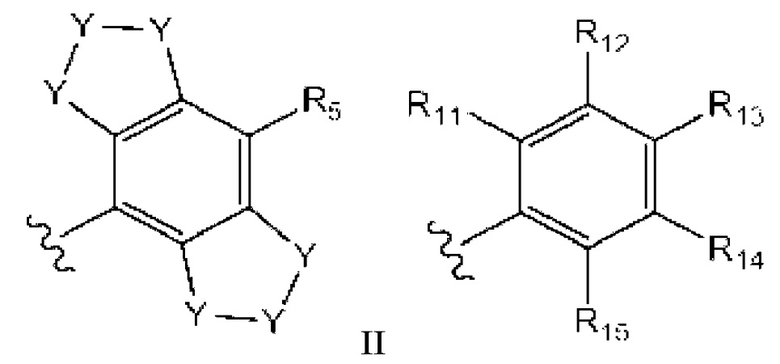
где каждый случай Y независимо выбран из С, N, S и О, и который может быть необязательно замещенным, по необходимости;
R5, R11, R12, R13, R14 и R15 независимо выбраны из группы, состоящей из водорода, гало, циано, амида, сульфонамида, ацила, гидроксила, C1-C6алкила, C1-C6галоалкила, С3-С5циклоалкила, С1-С6алкокси, все из которых могут быть необязательно замещены, по необходимости, на гало, циано или алкокси C1-C6; и
где R11 и R12 могут быть объединены с образованием фенила, 5- или 6-членного кислородсодержащего гетероцикла или 5- или 6-членного азотсодержащего гетероарила, каждый из которых может быть необязательно замещенным;
R12 и R13 могут быть объединены с образованием 5- или 6-членного азотсодержащего гетероарила, который может быть необязательно замещенным; и
R14 и R15 могут быть объединены с образованием 5- или 6-членного циклоалкильного кольца, фенила, 5- или 6-членного кислородсодержащего гетероцикла или 5- или 6-членного азотсодержащего гетероарила, каждый из которых может быть необязательно замещенным.
[0075] Подходящим образом, каждый случай Y представляет собой углерод, a R5 представляет собой водород или гало.
[0076] В одном варианте реализации R12 и R14 представляют собой водород, R11 и R15 представляют собой С1С6 алкил, a R13 представляет собой водород или гало.
[0077] Предпочтительно R2 выбран из замещенного или гидрированного индацена, 2,6-диалкилфенила, 2,6-диалкил-4-галофенила, 2,6-дициклоалкилфенила и 2,6-дициклоалкил-4-галофенила.
[0078] В определенных предпочтительных вариантах реализации, и в комбинации с любой группой R1, описанной для любой из формул первого аспекта, R2 выбран из гексагидроиндацена, 2,6-диизопропилфенила, 2,6-диизопропил-4-хлорфенила, 2,6-дициклопропилфенила и 2,6-дициклопропил-4-хлорфенила.
[0079] В одном варианте реализации W представляет собой О, a J представляет собой S, R1 представляет собой гетероарил и R2 представляет собой

где каждый Y представляет собой СН, a R5 представляет собой Н или галоген, предпочтительно R5 представляет собой Н.
[0080] В одном варианте реализации W представляет собой О, a J представляет собой S, R1 представляет собой гетероарил и R2 представляет собой
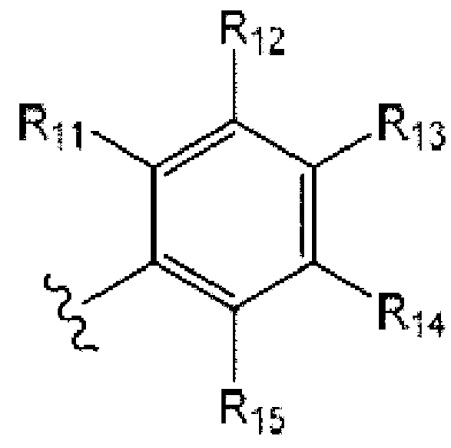
где
R11 и R15 представляют собой C1-6 алкил, предпочтительно изопропил;
R12 и R14 представляют собой Н,
R13 представляет собой Н или галоген, предпочтительно Н или Cl.
[0081] В одном варианте реализации W представляет собой О, a J представляет собой S, R1 представляет собой гетероарил и R2 представляет собой
где R11 и R15 представляют собой изопропил, R12 и R14 представляют собой Н, и R13 представляет собой Н или Cl.
[0082] В конкретных вариантах реализации соединение формулы (I) может быть выбрано из соединения формулы (Ia), (Ib) и (Ic), или его фармацевтически приемлемой соли, сольвата или пролекарства:
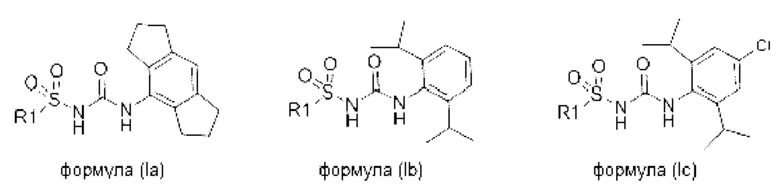
где R1 соответствует описанному ранее для любого варианта реализации формулы (I).
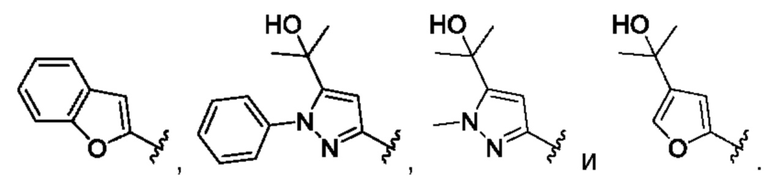
[0083] В одном варианте реализации соединения формулы (Ia), (Ib) и (Ic) R1 выбран из группы, состоящей из пиразола, фурана, тетрагидрофурана, тетрагидропирана, пирана, пирролидина, пиррола, триазола, тетразола, имидазола, пиридина, морфолина, пиперазина, пиперидина, замещенного фенила, фенилгетероарила, фенилгетероциклила, бифенила, хинолина, изохинолина, нафтила, пиразина и пиримидина, все из которых могут быть необязательно замещенными по необходимости.
[0084] В одном варианте реализации соединения формулы (Ia), (Ib) и (Ic) R1 выбран из группы, состоящей из:
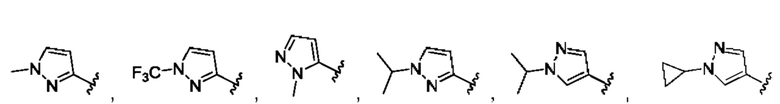
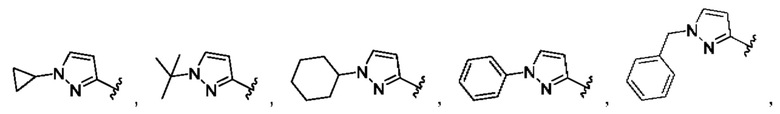
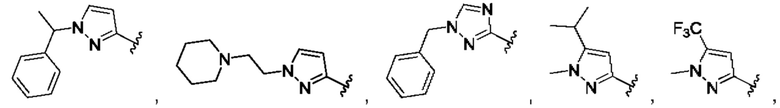
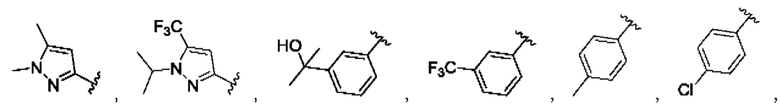
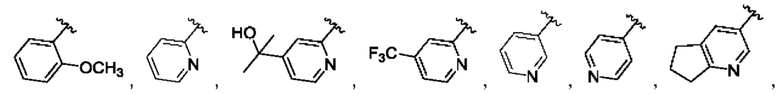
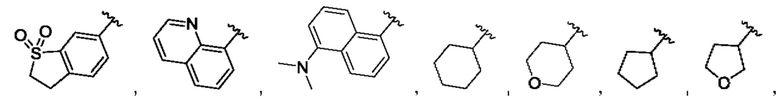
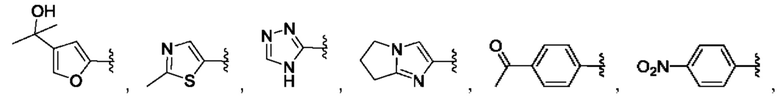
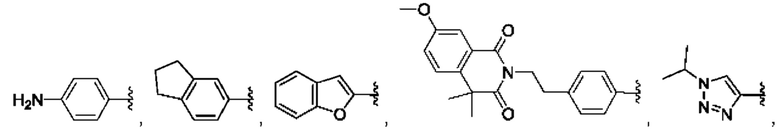
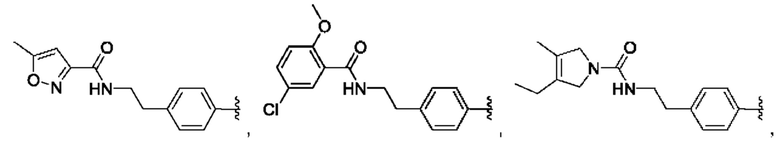
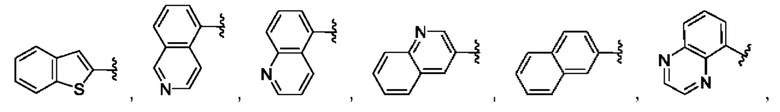
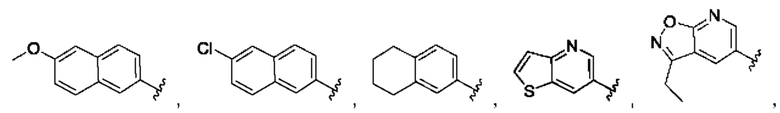
[0085] В одном варианте реализации соединение формулы (I) может быть выбрано из соединения формулы (II) или его фармацевтически приемлемой соли, сольвата или пролекарства:
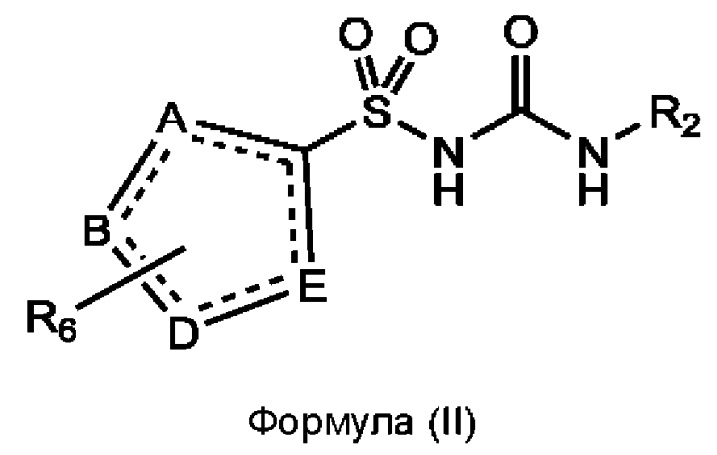
где А, В, D и Е независимо выбраны из С, N, О, S и Se, но по меньшей мере один из них представляет собой С;
каждая пунктирная линия может обозначать связь;
R2 соответствует определенному ранее для любого варианта реализации формулы (I), (Ia), (Ib) или (Ic), или может представлять собой флуоресцентную группу;
каждый случай R6 независимо выбран из группы, состоящей из водорода, гало, циано, C1-C6 алкила, C1-C6 алкиламино, C1-C6 алкилгидрокси, С3-С6 циклоалкила, алкилфенила, фенила, бензила, С1-С6 сложного эфира, С2-С6 алкенила, C1-C6 трифторалкила и С1-С6 алкокси, каждый из которых может быть необязательно замещенным, или R6 может представлять собой флуоресцентную группу.
[0086] В одном предпочтительном варианте реализации соединения формулы (II) по меньшей мере один из А, В, D и Е представляет собой N (т.е. азот).
[0087] В дополнительном предпочтительном варианте реализации соединения формулы (II) по меньшей мере два из А, В, D и Е представляют собой N.
[0088] В одном варианте реализации соединения формулы (II) А, В, D и Е выбраны из N и С.
[0089] В дополнительном варианте реализации соединения формулы (II) А представляет собой С и по меньшей мере два из В, D и Е представляют собой N.
[0090] В одном варианте реализации А, В, D и Е образуют кольцо, выбранное из пиразола, имидазола, триазола, и тетразола.
[0091] Предпочтительно А, В, D, и Е образуют кольцо, выбранное из пиразольного или имидазольного кольца, наиболее предпочтительно пиразольного кольца.
[0092] В одном варианте реализации А, В, D и Е и/или R2 могут составлять селеноцикл.
[0093] В одном варианте реализации соединение формулы (I) может быть выбрано из соединения формулы (IIa) или его фармацевтически приемлемой соли, сольвата или пролекарства:
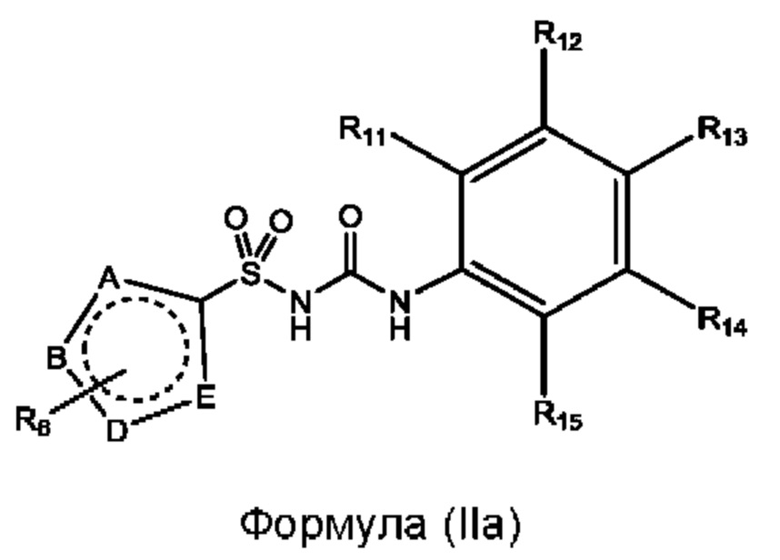
где R11 R12 R13 R14 и R15 соответствуют определенным выше;
А, В, D и Е выбраны из N и С и по меньшей мере два из А, В, D, и Е представляют собой N;
каждый случай R6 независимо выбран из группы, состоящей из водорода, галогенида, циано, C1-C6 алкила, C1-C6 алкиламино, C1-C6 алкилгидрокси, С3-С6 циклоалкила, алкилфенила, фенила, бензила, C1-C6 сложного эфира, С2-С6
алкенила, C1-C6 трифторалкила и C1-C6 алкокси, каждый из которых может быть необязательно замещенным.
[0094] В одном варианте реализации соединение формулы (I) может быть выбрано из соединения формулы (IIb) или его фармацевтически приемлемой соли, сольвата или пролекарства:
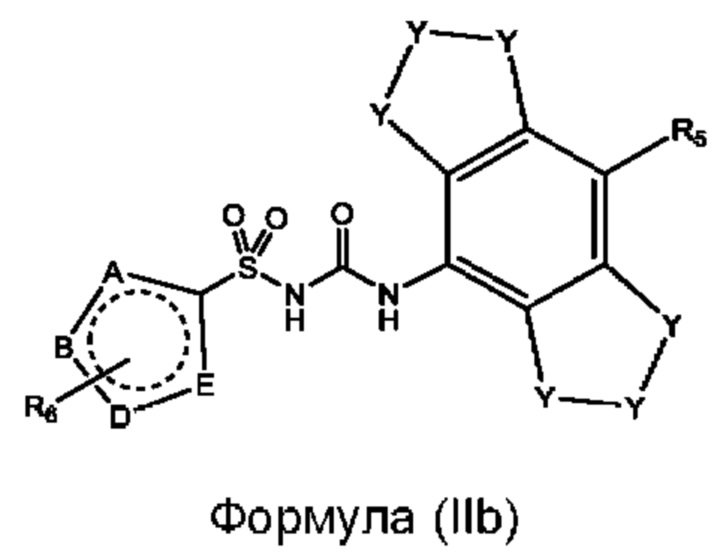
где Y и R5 соответствуют определенным выше;
А, В, D и Е выбраны из N и С и по меньшей мере два из А, В, D, и Е представляют собой N;
каждый случай R6 независимо выбран из группы, состоящей из водорода, галогенида, циано, C1-C6 алкила, C1-C6 алкиламино, С1-С6 алкилгидрокси, С3-С6 циклоалкила, алкилфенила, фенила, бензила, С1-С6 сложного эфира, С2-С6 алкенила, C1-C6 трифторалкила и С1С6 алкокси, каждый из которых может быть необязательно замещенным.
[0095] В одном варианте реализации соединение формулы (II) выбрано из:
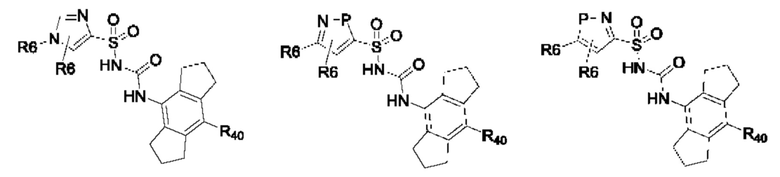
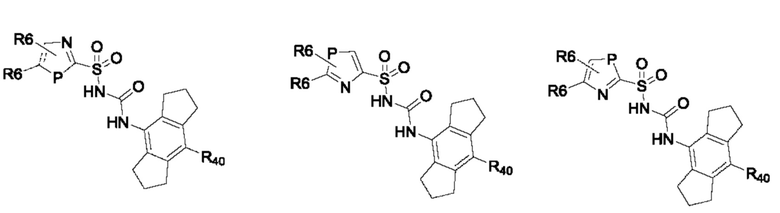
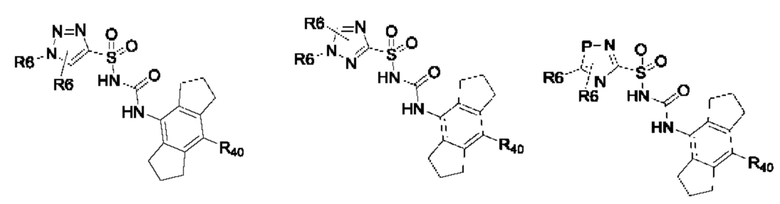
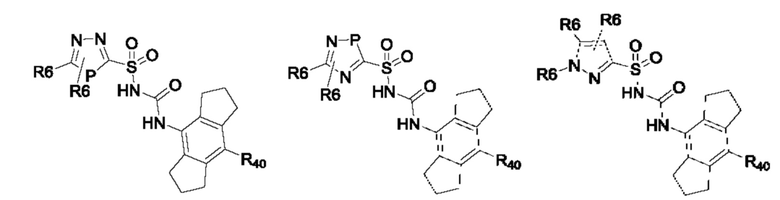
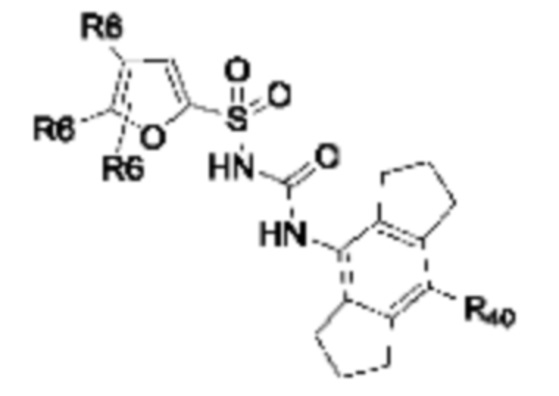

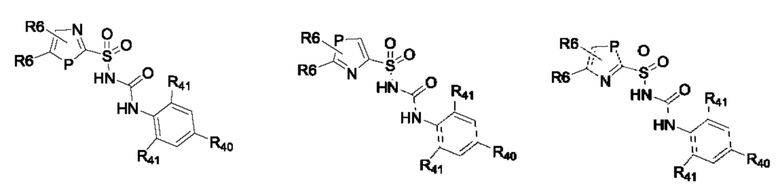
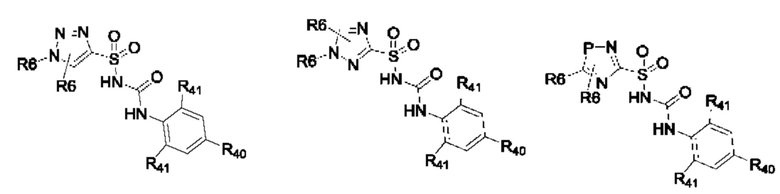
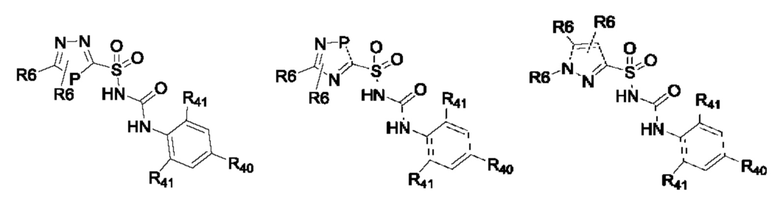
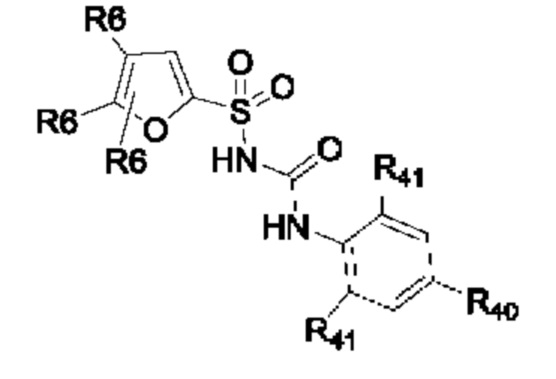
где R40 выбран из Н, алкила и гало;
R41 выбран из Н и алкила;
каждый случай Р независимо выбран из С, О или S; и
где каждый случай R6, если присутствует, независимо выбран из групп, определенных для формулы (II).
[0096] Будет понятно, что фрагмент R6, выходящий из центра каждого кольца, может обозначать группу, соединенную с углеродами кольца или гетероатомами кольца, по необходимости, с учетом валентности, или может не присутствовать.
[0097] В одном варианте реализации формулы (II) R6 представляет собой C1-C6 алкил или C1-C6 алкилгидрокси.
[0098] В определенных вариантах реализации соединения формулы (II), например, если R2 представляет собой гексагидроиндацен, a R1 представляет собой фуран, R6 не может быть третичным спиртовым заместителем.
[0099] В одном варианте реализации соединение из первого аспекта может быть выбрано из соединения формулы (IIIa), (IIIb) или (IIIc), или его фармацевтически приемлемой соли, сольвата или пролекарства:
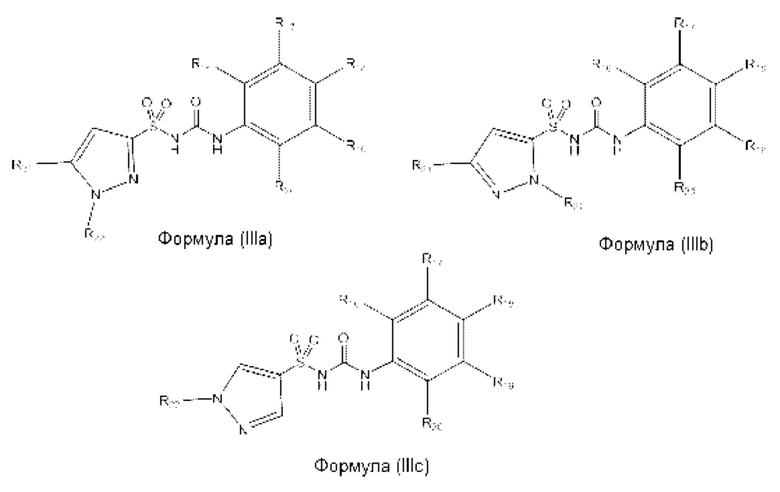
где R21 выбран из Н, алкила, пергалоалкила или гидроксилалкила;
R22 выбран из Н, алкила, пергалоалкила, С3-С6 циклоалкила, фенила или бензила;
R18 представляет собой Н или галоген;
R16 и R17 представляют собой Н или алкил; или R16 и R17, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
R19 и R20 представляют собой Н или алкил; или R19 и R20, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
при условии, что R21 и R22 не представляют собой одновременно Н; и при условии, что R16, R17, R18, R19 и R20 не представляют собой одновременно Н.
[00100] В предпочтительном варианте реализации соединений формул (IIIa), (IIIb) и (IIIc):
R21 выбран из Н, алкила, пергалоалкила или гидроксилалкила; предпочтительно С1-6-пергалоалкила или гидроксилалкила;
R22 выбран из Н, алкила, пергалоалкила, С3-С6 циклоалкила, фенила или бензила;
R16 и R17, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R19 и R20, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R18 представляет собой Н или галоген, предпочтительно R18 представляет собой Н; и
при условии, что R21 и R22 не представляют собой одновременно Н.
[00101] В еще одном предпочтительном варианте реализации соединений формул (IIIa), (IIIb) и (IIIc):
R21 выбран из Н, алкила, пергалоалкила или гидроксилалкила; предпочтительно С1-6-пергалоалкила или гидроксилалкила;
R22 выбран из Н, алкила, пергалоалкила, С3-С6 циклоалкила, фенила и бензила;
R16 и R20 представляют собой С1-6 алкил, предпочтительно изопропил;
R17 и R19 представляют собой Н,
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н или Cl; и
при условии, что R21 и R22 не представляют собой одновременно Н.
[00102] В одном варианте реализации соединение из первого аспекта может быть выбрано из соединения формулы (IVa), (IVb) или (IVc) или его фармацевтически приемлемой соли, сольвата или пролекарства:
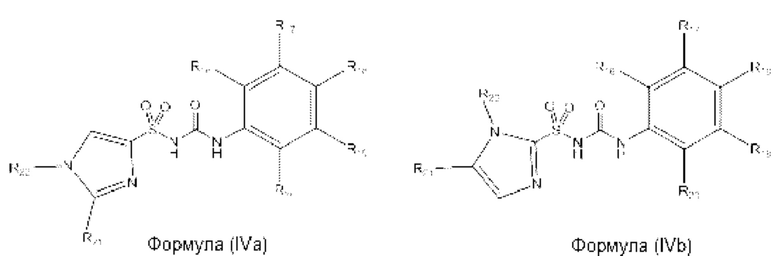
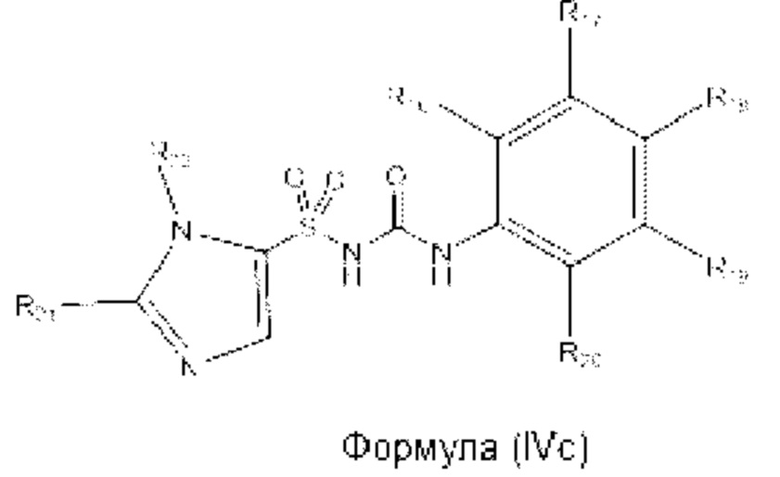
где R21 и R22 выбраны из Н, алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила, фенила и бензила или R21 и R22, вместе с атомами углерода, к которым они присоединены, могут образовывать циклопентильное или циклогексильное кольцо;
R18 представляет собой Н или галоген;
R16 и R17 представляют собой Н или алкил; или R16 и R17, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
R19 и R20 представляют собой Н или алкил; или R19 и R20, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
при условии, что R21 и R22 не представляют собой одновременно Н; и
при условии, что R16, R17, R18, R19 и R20 не представляют собой одновременно Н.
[00103] В предпочтительном варианте реализации соединений формул (IVa), (IVb) и (IVc):
R21 и R22 выбраны из Н, алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила, фенила и бензила; предпочтительно пергалоалкил и гидроксилалкил представляют собой С1-6 пергалоалкил и гидроксилалкил;
R16 и R17, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R19 и R20, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н; и
при условии, что R21 и R22 не представляют собой одновременно Н.
[00104] В еще одном предпочтительном варианте реализации соединений формул (IVa), (IVb) и (IVc):
R21 и R22 выбраны из Н, алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила, фенила и бензила; предпочтительно пергалоалкил и гидроксилалкил представляют собой C1-6 пергалоалкил и гидроксилалкил;
R16 и R20 представляют собой С1-6 алкил, предпочтительно изопропил;
R17 и R19 представляют собой Н,
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н или Cl;
при условии, что R21 и R22 не представляют собой одновременно Н.
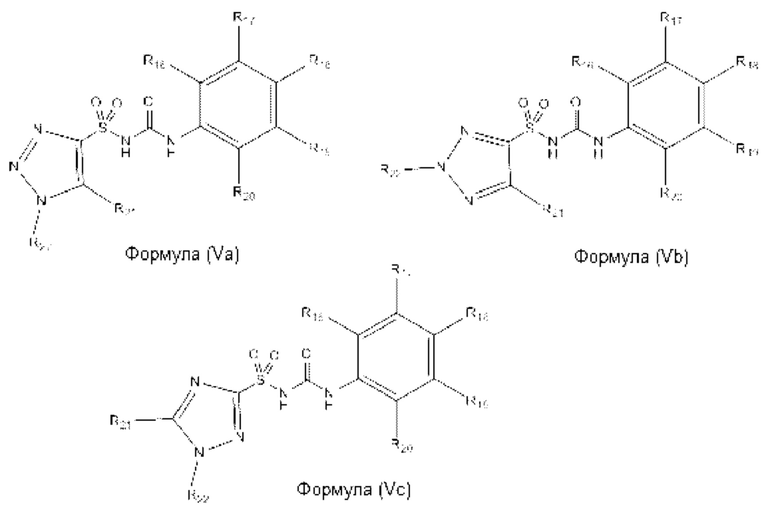
[00105] В одном варианте реализации соединение из первого аспекта может быть выбрано из соединения формулы (Va), (Vb) или (Vc) или его фармацевтически приемлемой соли, сольвата или пролекарства:
где R21 и R22 выбраны из Н, алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила, фенила и бензила;
R18 представляет собой Н или галоген;
R16 и R17 представляют собой Н или алкил; или R16 и R17, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
R19 и R20 представляют собой Н или алкил; или R19 и R20, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
при условии, что R21 и R22 не представляют собой одновременно Н; и
при условии, что R16, R17, R18, R19 и R20 не представляют собой одновременно Н.
[00106] В предпочтительном варианте реализации соединений формул (Va), (Vb) и (Vc):
R21 и R22 выбраны из Н, алкила, пергалоалкила, гидроксилалкила, С3-С6-циклоалкила, фенила и бензила; предпочтительно пергалоалкил и гидроксилалкил представляют собой C1-6 пергалоалкил и гидроксилалкил;
R16 и R17, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R19 и R20, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н; и
при условии, что R21 и R22 не представляют собой одновременно Н.
[00107] В еще одном предпочтительном варианте реализации соединений формул (Va), (Vb) и (Vc):
R21 и R22 выбраны из Н, алкила, пергалоалкила, гидроксилалкила, С3-С6-циклоалкила, фенила и бензила; предпочтительно пергалоалкил и гидроксилалкил представляют собой Q.6 пергалоалкил и гидроксилалкил;
R16 и R20 представляют собой C1-6-алкил, предпочтительно изопропил;
R17 и R19 представляют собой Н;
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н или Cl; и
при условии, что R21 и R22 не представляют собой одновременно Н.
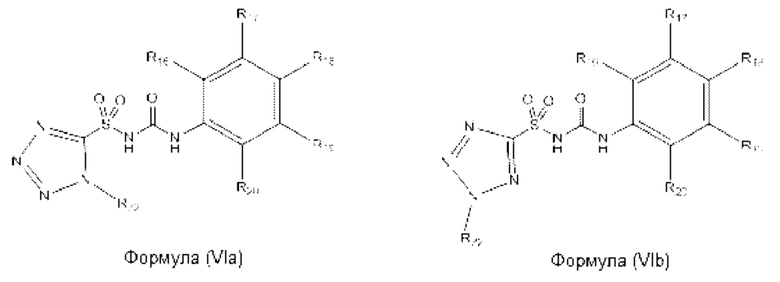
[00108] В одном варианте реализации соединение из первого аспекта может быть выбрано из соединения формулы (VIa) или (VIb) или его фармацевтически приемлемой соли, сольвата или пролекарства:
где R22 выбран из алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила, фенила и бензила;
R18 представляет собой Н или галоген;
R16 и R17 представляют собой Н или алкил; или R16 и R17, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
R19 и R20 представляют собой Н или алкил; или R19 и R20, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S; и
при условии, что R16, R17, R18, R19 и R20 не представляют собой одновременно Н.
[00109] В предпочтительном варианте реализации соединений формул (VIa) и (VIb):
R22 выбран из алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила, фенила и бензила; предпочтительно пергалоалкил и гидроксилалкил представляют собой С1-6 пергалоалкил и гидроксилалкил;
R16 и R17, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R19 и R20, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо; и
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н.
[00110] В еще одном предпочтительном варианте реализации соединений формул (VIa) и (VIb):
R22 выбран из алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила, фенила и бензила; предпочтительно пергалоалкил и гидроксилалкил представляют собой С1-6-пергалоалкил и гидроксилалкил;
R16 и R20 представляют собой C1-6 алкил, предпочтительно изопропил;
R17 и R19 представляют собой Н; и
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н или Cl.
[00111] В одном варианте реализации соединение из первого аспекта может быть выбрано из соединения формулы (VII) или его фармацевтически приемлемой соли, сольвата или пролекарства:

где Q представляет собой О или S;
каждый случай R30 независимо выбран из алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила и алкиламино;
R18 представляет собой Н или галоген;
R16 и R17 представляют собой Н или алкил; или R16 и R17 вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
R19 и R20 представляют собой Н или алкил; или R19 и R20, вместе с атомами углерода, к которым они присоединены, образуют 5- или 6-членное кольцо, при этом указанное кольцо является насыщенным, частично ненасыщенным или ненасыщенным, указанное кольцо необязательно содержит один или два гетероатома, выбранных из N, О и S;
при условии, что R16, R17, R18, R19 и R20 не представляют собой одновременно Н; и
при условии, что если Q представляет собой О, a R16 и R17, и отдельно R19 и R20, вместе с соответствующими атомами углерода, к которым они присоединены, образуют циклопентильное кольцо, то R30 не является С-3 гидроксилалкилом.
[00112] В предпочтительном варианте реализации соединений формул (VII):
Q представляет собой О или S;
каждый случай R30 независимо выбран из алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила и алкиламино; предпочтительно С1-6 алкила, пергалоалкила, гидроксилалкила и алкиламино;
R16 и R17, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо;
R19 и R20, вместе с атомами, к которым они присоединены, образуют циклопентильное кольцо; и
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н и
при условии, что если Q представляет собой О, то R30 не является С-3-гидроксилалкилом.
[00113] В еще одном предпочтительном варианте реализации соединений формул (VII):
Q представляет собой О или S;
каждый случай R30 независимо выбран из алкила, пергалоалкила, гидроксилалкила, С3-С6 циклоалкила и алкиламино; предпочтительно С1-6 алкила, пергалоалкила, гидроксилалкила и алкиламино;
R16 и R20 представляют собой С1-6-алкил, предпочтительно изопропил;
R17 и R19 представляют собой Н; и
R18 представляет собой Н или галоген; предпочтительно R18 представляет собой Н или Cl.
[00114] Соединения из первого аспекта, и, в частности, формул от (II) до (VI), обеспечивают ряд неожиданных преимуществ по сравнению с сульфонилмочевинами предшествующего уровня техники, эти преимущества могут быть выбраны из: улучшенной микросомальной стабильности; улучшенной всасываемости; пониженной подверженности Pgp (Р-гликопротеину); пониженного связывания с белками плазмы; увеличенного времени полувыведения; улучшенной биодоступности при пероральном введении; улучшенной ППК; улучшенной Cmax; пониженного ингибирования Сур; улучшенного ингибирования активации инфламмасомы NLRP3 и улучшенной растворимости. Растворимость и определенные другие улучшения могут быть особенно заметны в водной среде.
[00115] В одном варианте реализации соединения из первого аспекта обеспечивают улучшенные фармакокинетические характеристики. CRID3, известная сульфонилмочевина, имеет время полувыведения 3,2 часа (мыши), что может приводить к существенным остаточным уровням из дозирования один или два раза в сутки, если время полувыведения экстраполировать для человека. Соединения из первого аспекта могут различаться, например, в их связывании белка, метаболизме и доступности при пероральном введении.
[00116] В частности, было обнаружено, что соединения из первого аспекта, в особенности те, в которых А, В, D и Е образуют 5-членный азотсодержащий гетероарил, например, пиразольное кольцо, менее метаболически лабильны и/или имеют улучшенные фармакокинетические свойства по сравнению со структурно подобными в остальном фуранами и тиофенами, известными из предшествующего уровня техники.
[00117] В одном варианте реализации соединения из первого аспекта имеют tPSA менее чем 90 А2.
[00118] Одним преимуществом представленных соединений из первого аспекта является то, что они могут проявлять значительно сниженную площадь полярной поверхности по сравнению с сульфонилмочевинами предшествующего уровня техники, такими как CRID3.
[00119] В одном дополнительном варианте реализации соединения из первого аспекта имеют tPSA менее чем 90 А2 и молекулярную массу менее чем 405.
[00120] Отсутствие третичной спиртовой группы в некоторых вариантах реализации приводит к увеличению концентрации в плазме и способствует снижению как MW, так и площади полярной поверхности, давая тем самым общее улучшение в проникновении через гематоэнцефалический барьер.
[00121] В любом из вариантов реализации, описанных для соединений из первого аспекта, включая соединения формулы (I) - (VII), один или более водородов заместителей или необязательного замещения по ним может быть дейтерирован.
[00122] Дейтерированные аналоги соединений по настоящему изобретению могут проявлять увеличенную метаболическую стабильность благодаря кинетическому изотопному эффекту.
[00123] В одном варианте реализации соединение из первого аспекта выбрано из группы, состоящей из:
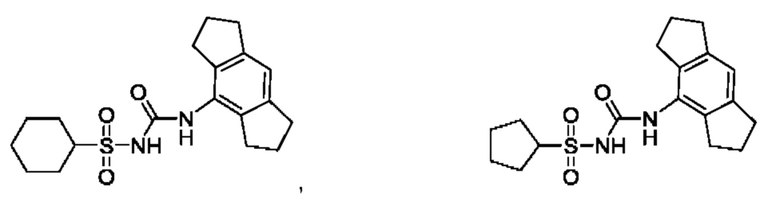
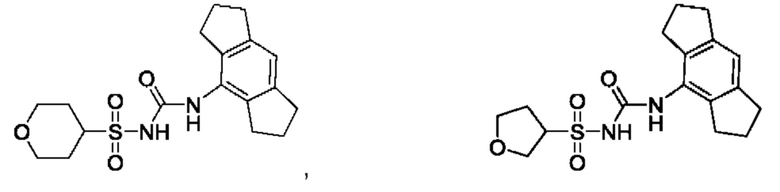
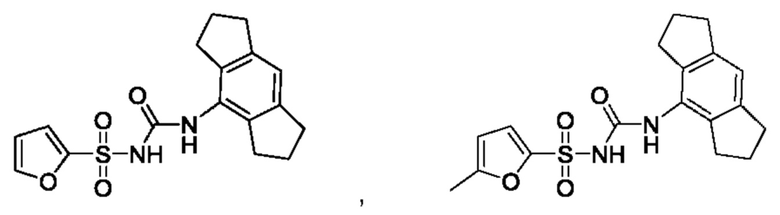
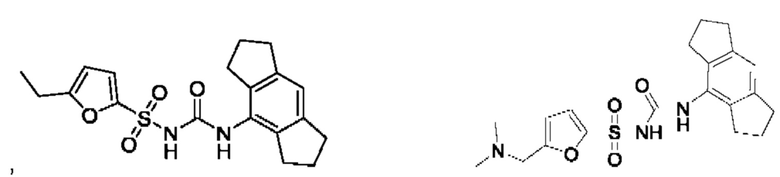
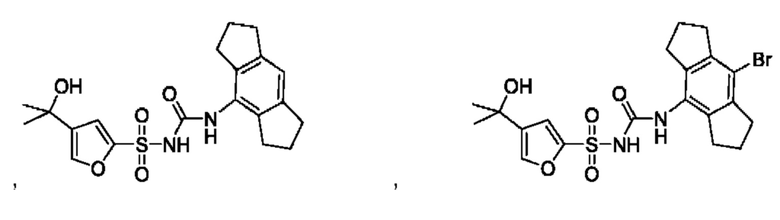

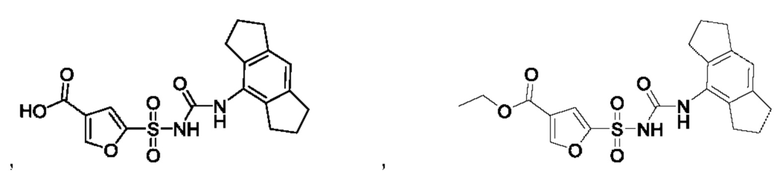
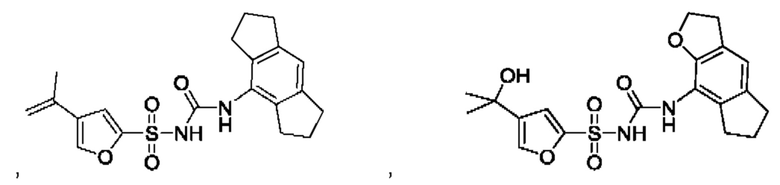

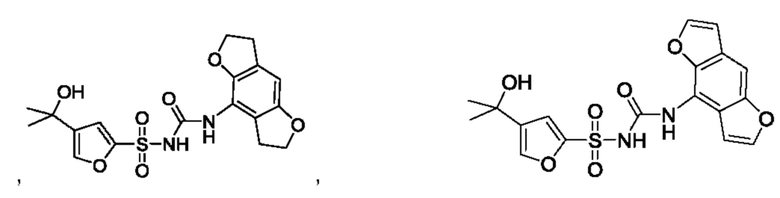
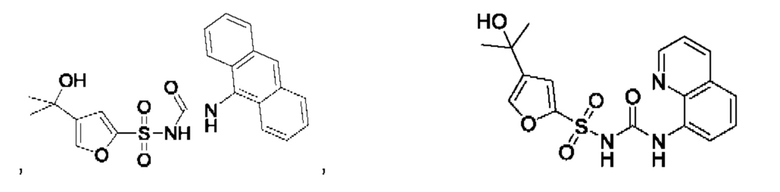
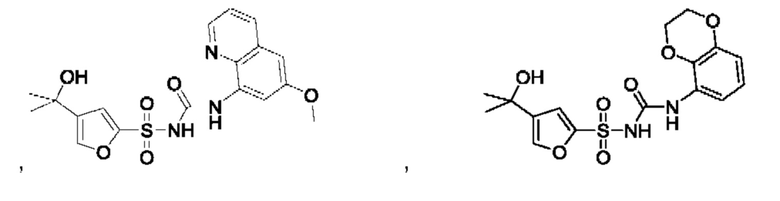
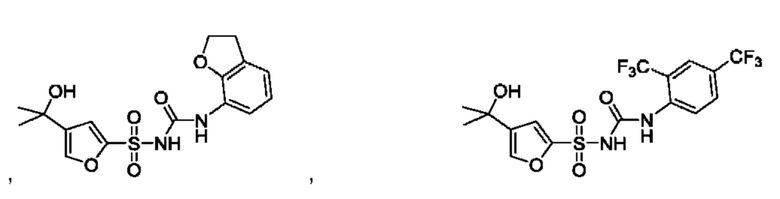
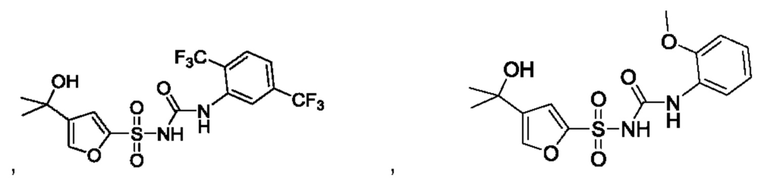
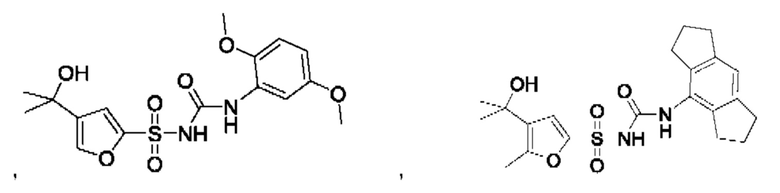
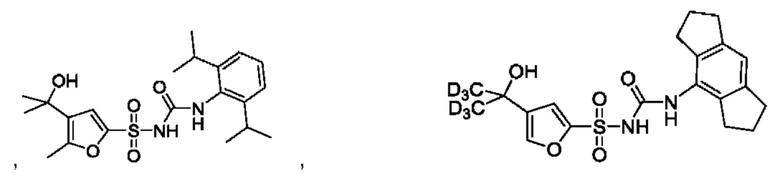
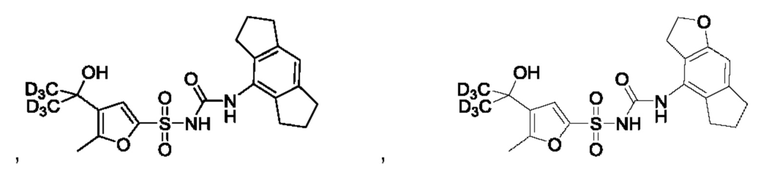
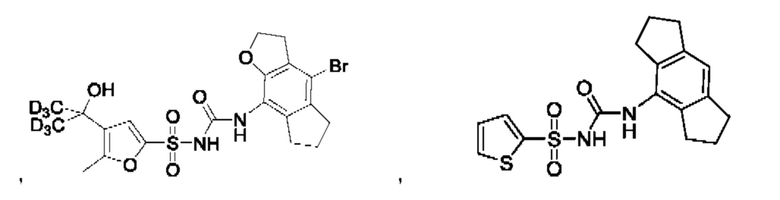
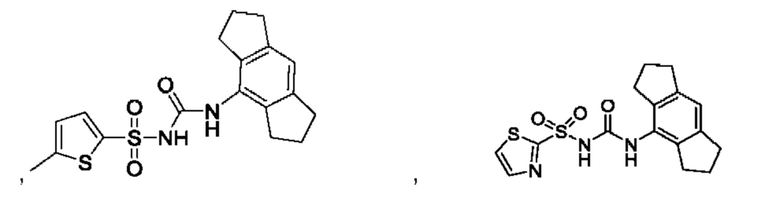
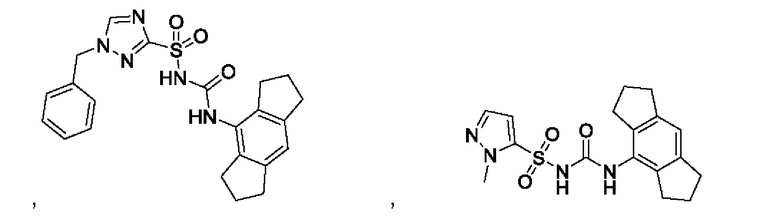
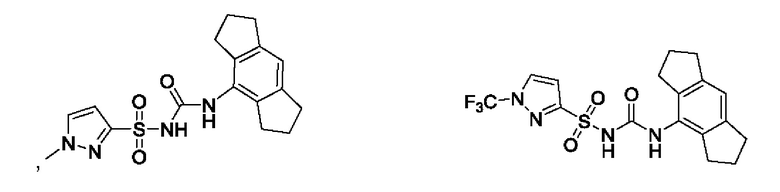
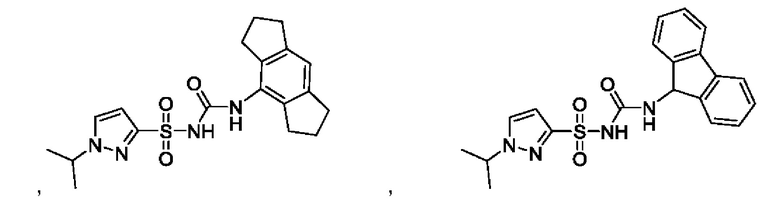
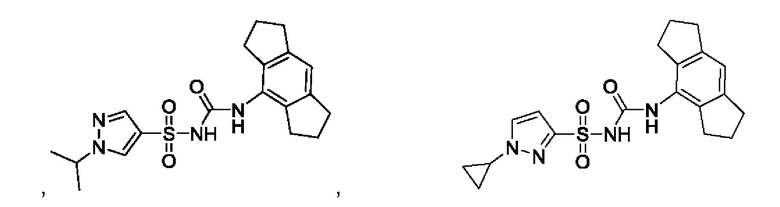

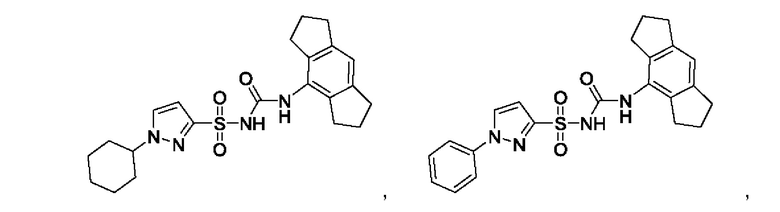
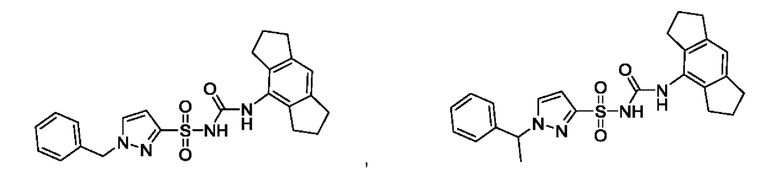
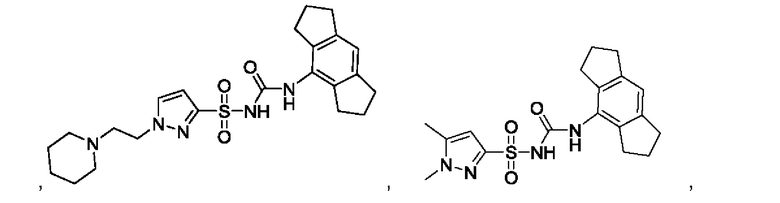


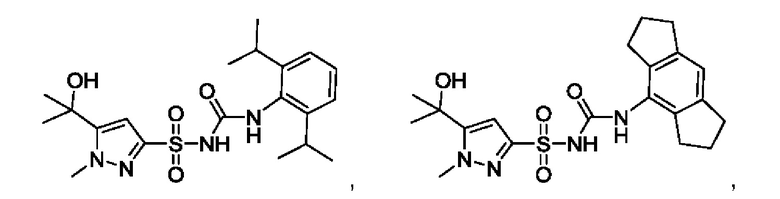
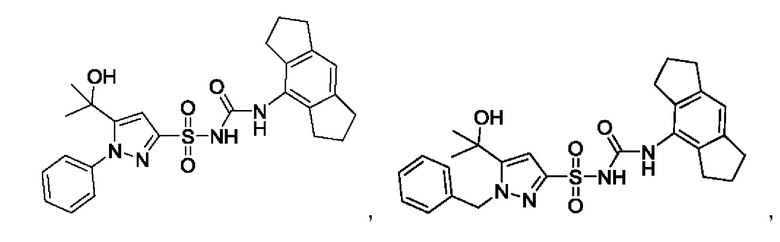
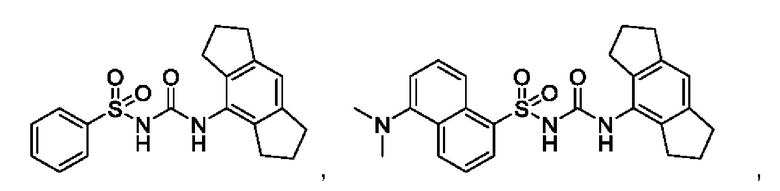

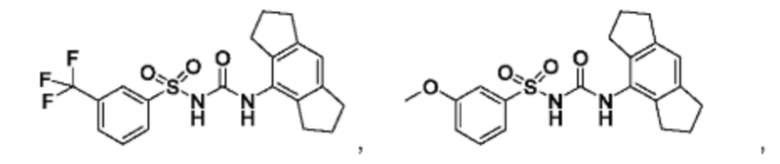

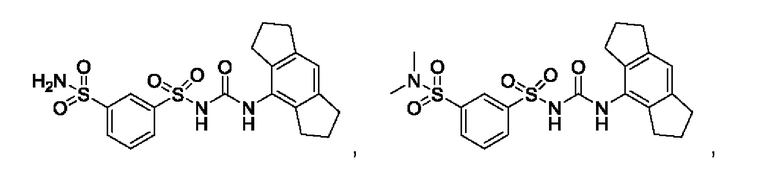
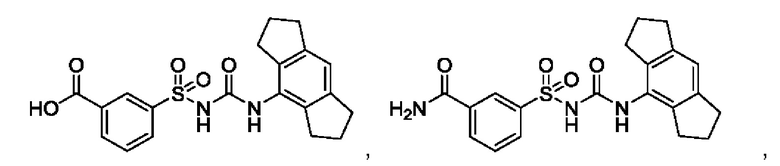
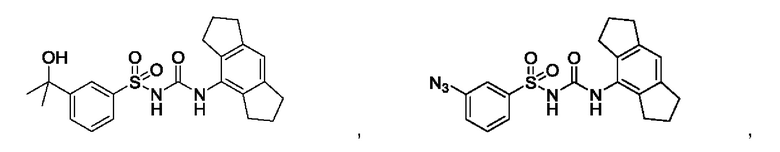
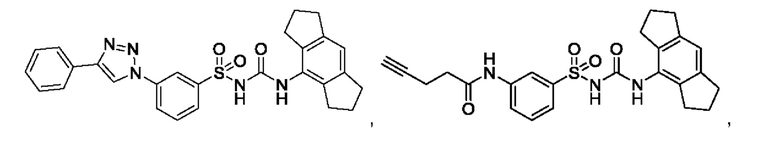
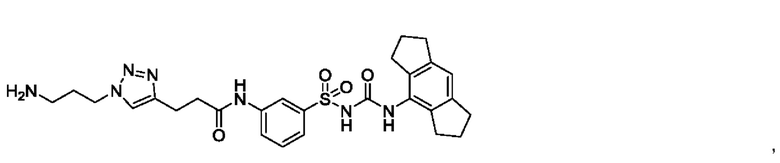
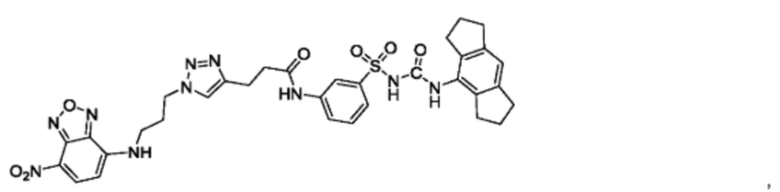
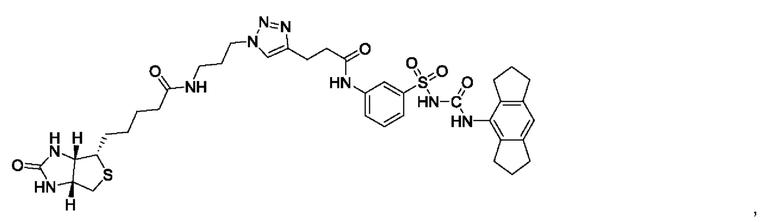
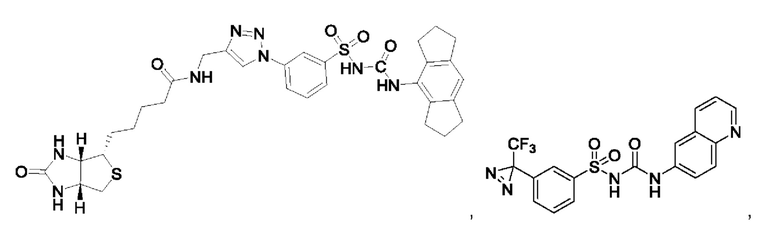
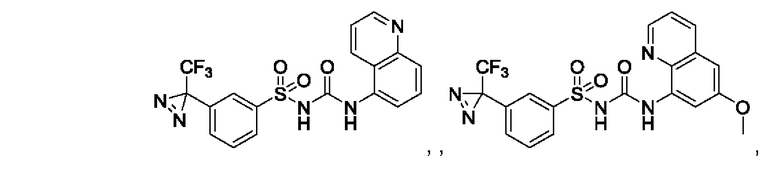
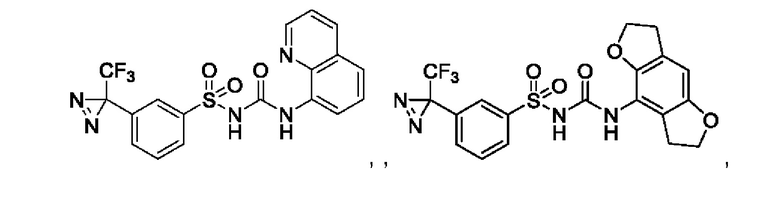
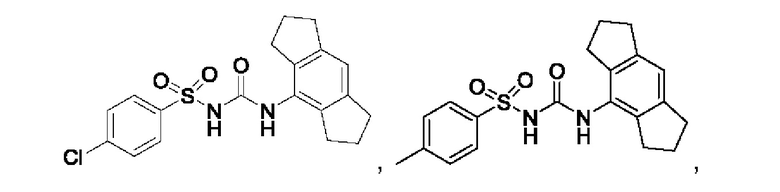
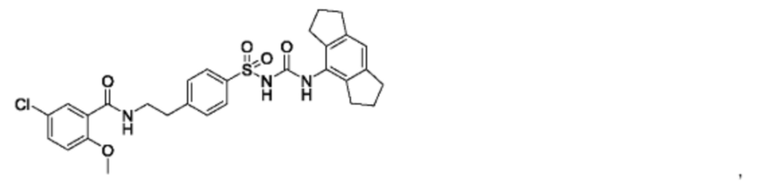
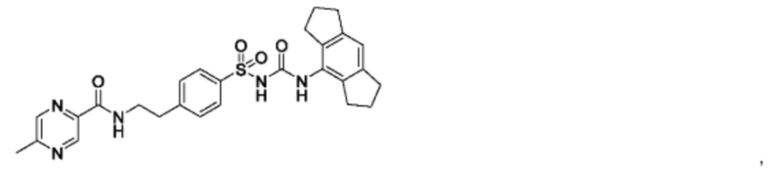
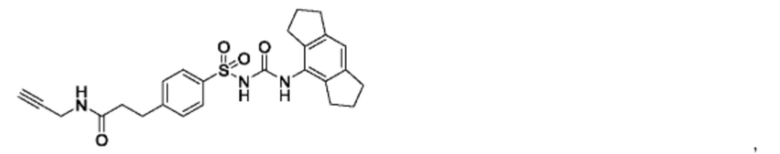
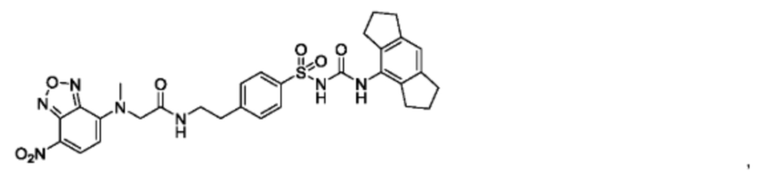
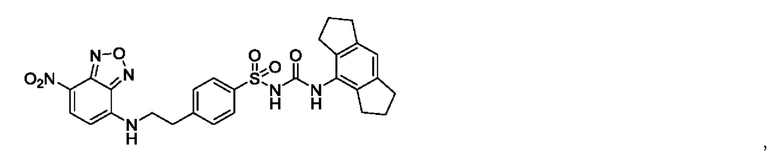

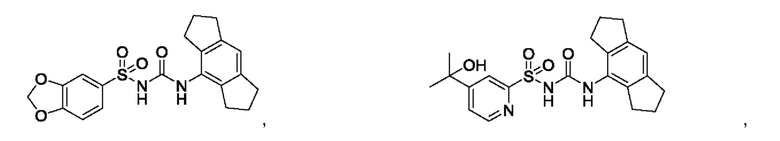
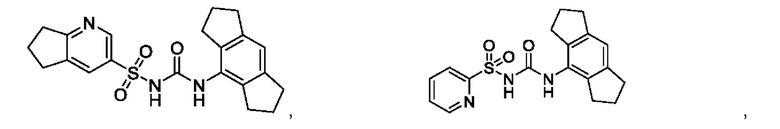
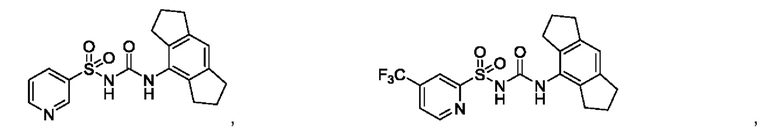
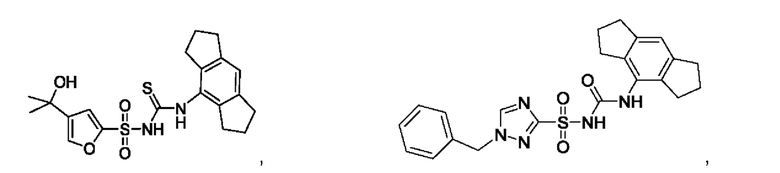
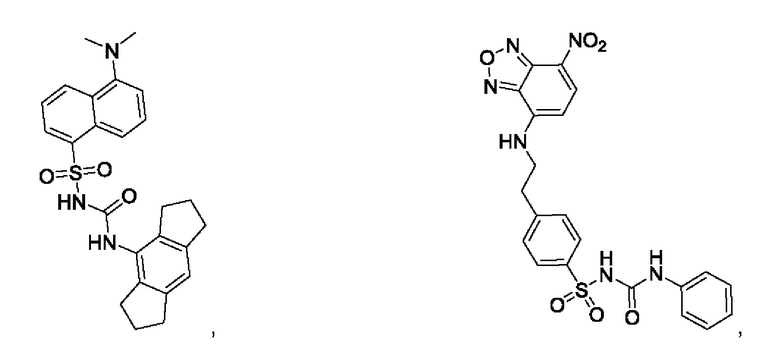
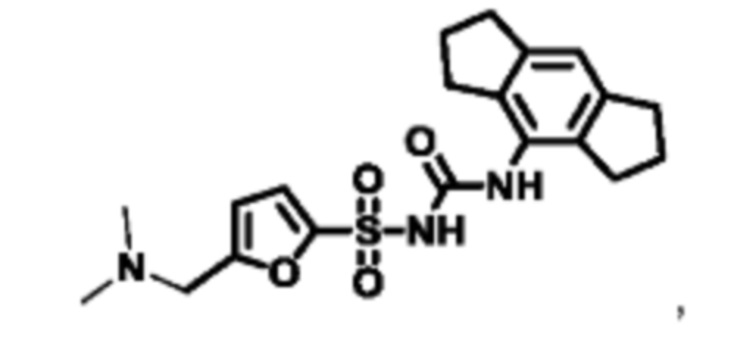
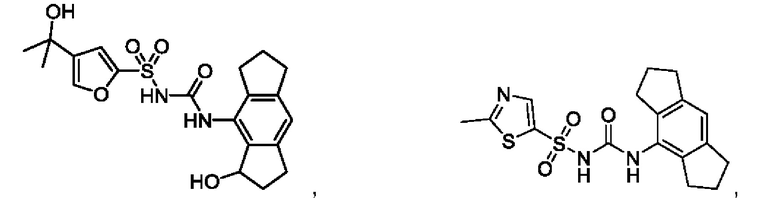
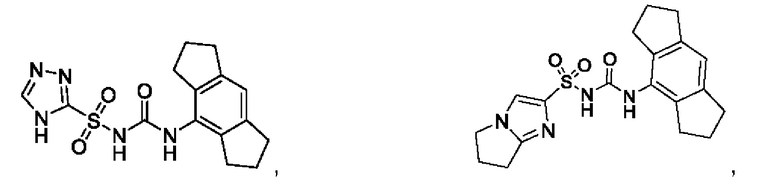
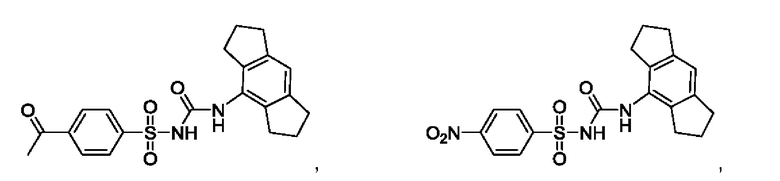
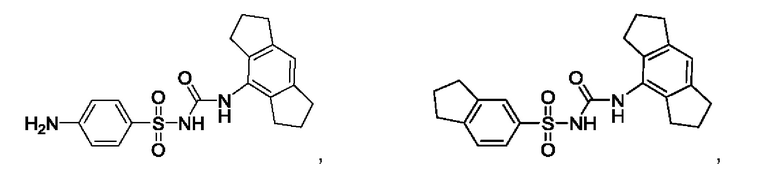
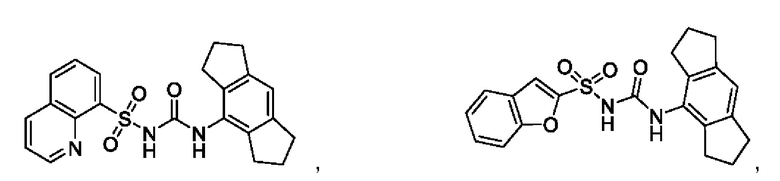
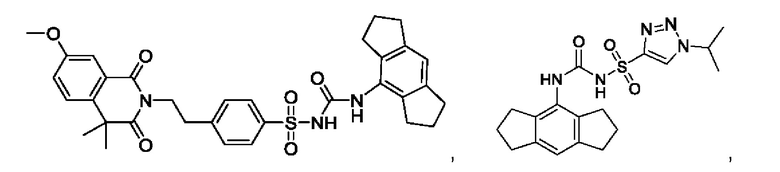
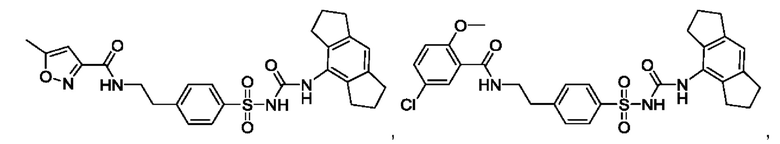
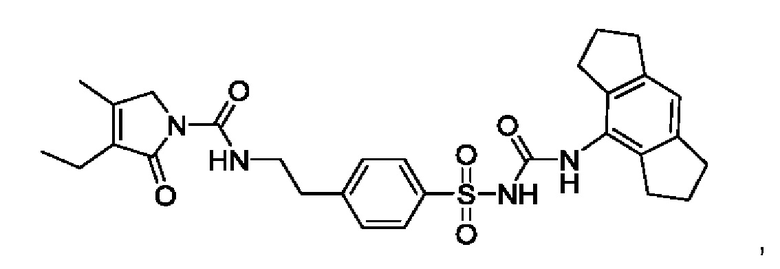
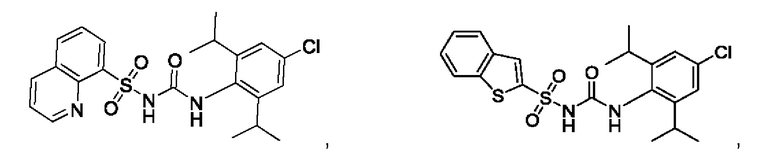
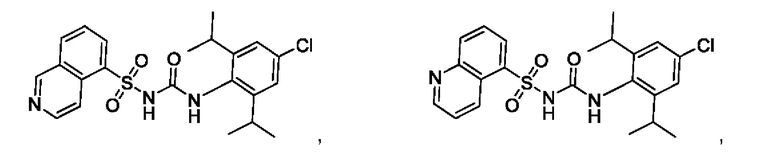
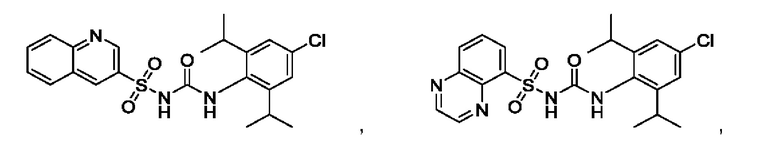
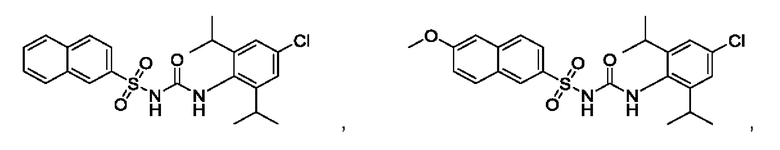
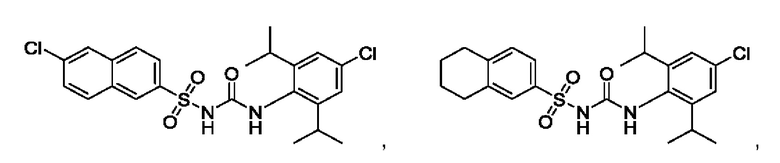
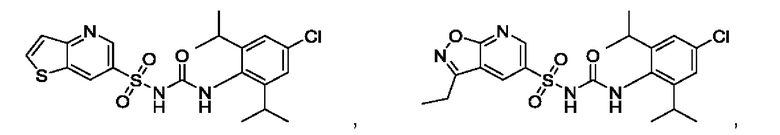
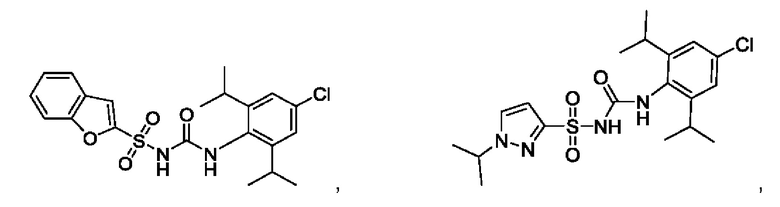
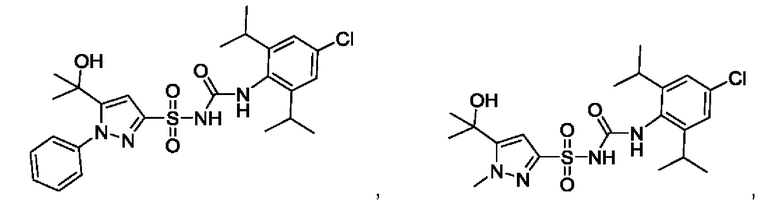
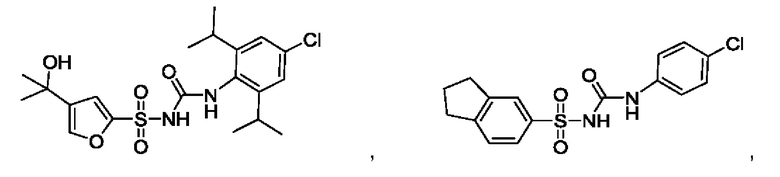
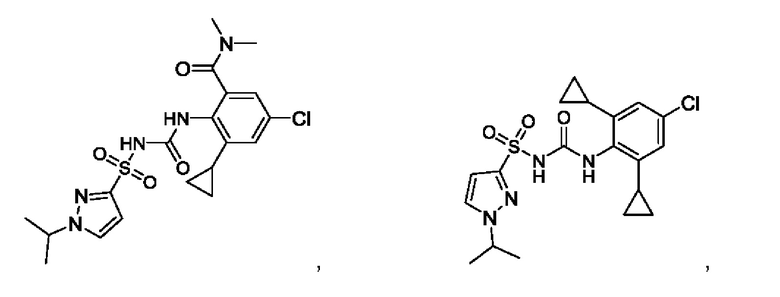

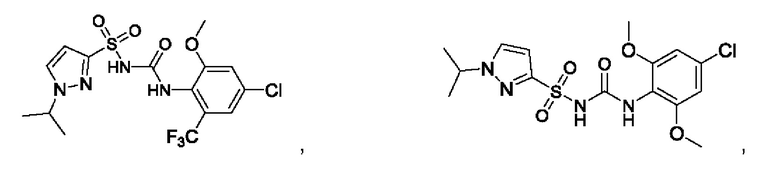
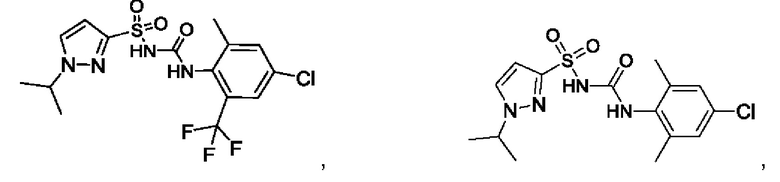
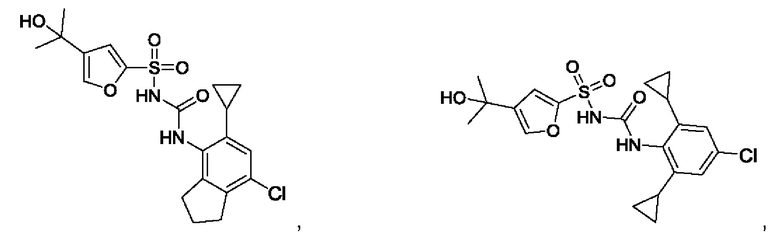


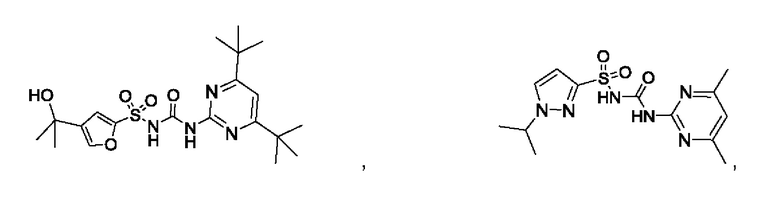
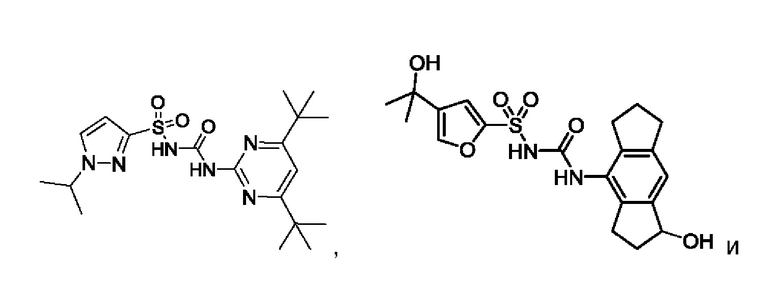

[00124] В определенных вариантах реализации соединения из первого аспекта могут проявлять улучшенные свойства по сравнению с известными антидиабетическими препаратами. Примеры таких соединений могут включать приведенные ниже:
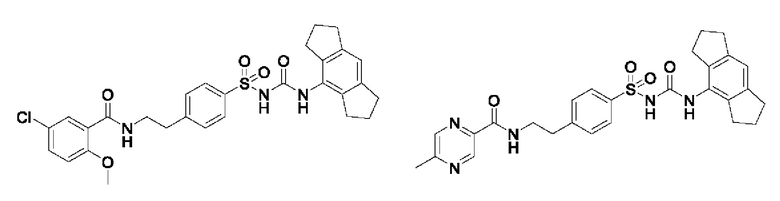
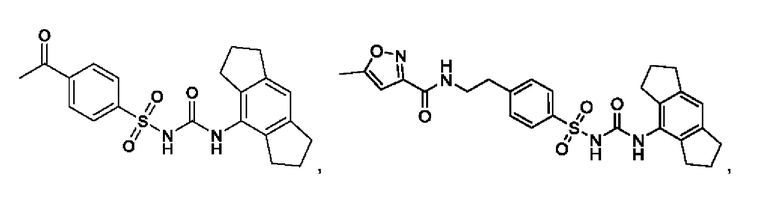
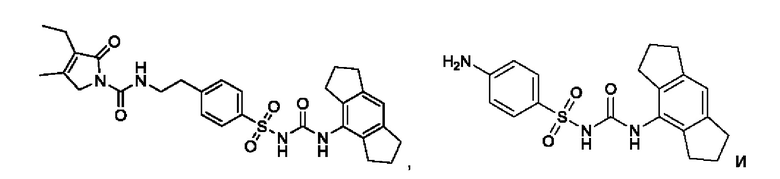
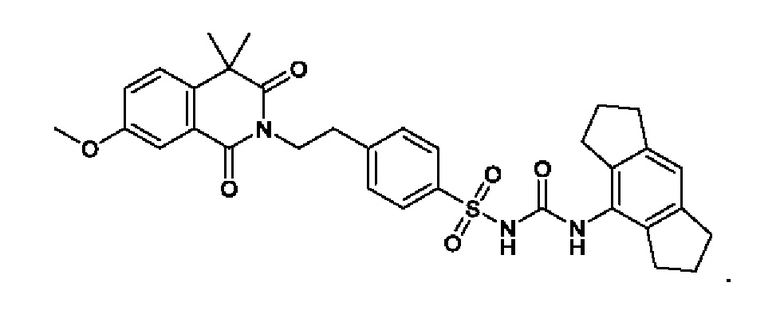
[00125] Эти четыре соединения формулы (I) могут рассматриваться как очень сильнодействующие версии существующих на данный момент антидиабетических препаратов на основе сульфонилмочевины. Данные IC50, представленные в экспериментальной части, отражают эту точку зрения. Считается, что известные препараты не воздействуют на NLRP3 в какой-либо терапевтически существенной степени, и поэтому необходимо использовать очень высокие дозы для получения какого-либо значительного действия на инфламмасому NLRP3. Четыре соединения, представленные выше, и другие из первого аспекта, проявляют выгодно улучшенные свойства в значительном снижении IC50 по сравнению с инфламмасомой NLRP3 и дополнительно имеют преимущества, не реализованные существующими антидиабетическими и другими препаратами, связанные с ингибированием NLRP3, такие как улучшенное заживление ран и другие преимущества, описанные в настоящем документе.
[00126] В любом одном или более вариантах реализации первого аспекта и в отношении любого одного или более из соединений формулы (I)-(VII), соединение представляет собой ингибитор активации инфламмасомы NLRP3.
[00127] Поэтому следует понимать, что настоящее изобретение предусматривает сульфонилмочевину и родственные препараты, проявляющие значительно более низкие значения IC50 NLRP3 в исследовании на клетках с использованием ММПЧ (HMDM, макрофаг моноцитарного происхождения человека) (протоколы см. в экспериментальной части), чем вышеупомянутые соединения сравнения. Известные на настоящее время антидиабетические препараты не являются сильнодействующими ингибиторами инфламмасомы NLRP3 в терапевтических дозах и для достижения какого-либо подобного ингибирования будут требовать дозировки за пределами рекомендуемых уровней. Представленные соединения позволяют использовать более низкие дозы и поэтому ограничивают опасность токсических действий.
[00128] В дополнительном варианте реализации одно или более из соединений из первого аспекта могут быть полезны в качестве фотопереключаемых соединений, которые можно использовать в ряде применений, включая, но без ограничения ею, секрецию инсулина. Такие соединения в одном варианте реализации могут быть выбраны из группы, состоящей из:

где R2 соответствует определенному в любом одном или более вариантах реализации соединений формулы (I)-(VII), описанных ранее.
[00129] В определенных вариантах реализации изобретения одно или более соединений из первого аспекта могут быть подходящими для применения в качестве зондов, таких как фотоаффинные зонды, или в качестве реакционных промежуточных продуктов, которые можно модифицировать либо непосредственно, либо через связующий фрагмент с получением биотинилированных, флуоресцентных или фотоаффинных зондов, включая, но без ограничения ими, показанные ниже:
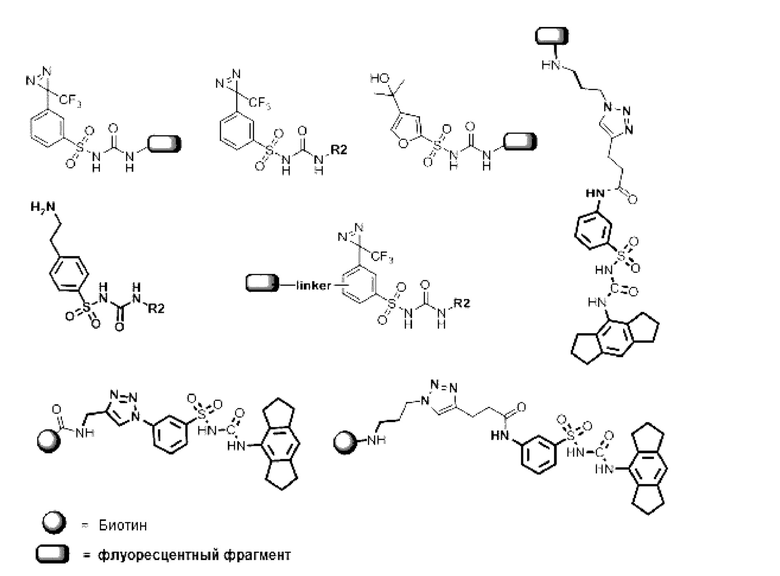
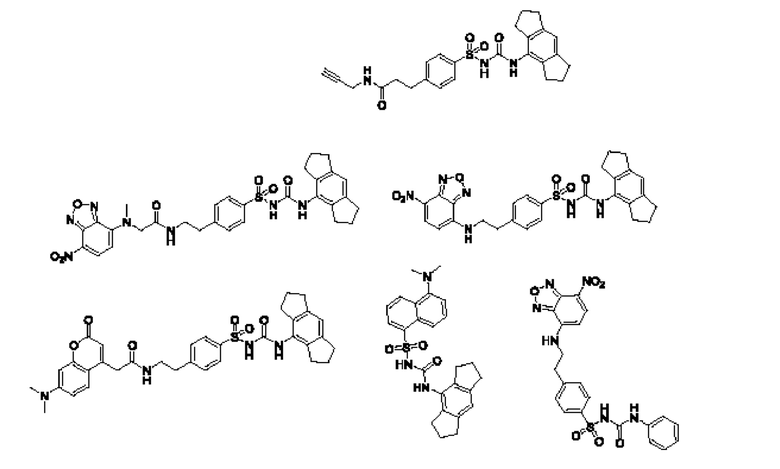
где R2 соответствует определенному в любом одном или более вариантах реализации, описанных для формулы (I)-(VII).
[00130] В частности, такие соединения, как зонды или реакционные промежуточные продукты, могут быть выбраны из приведенных ниже:

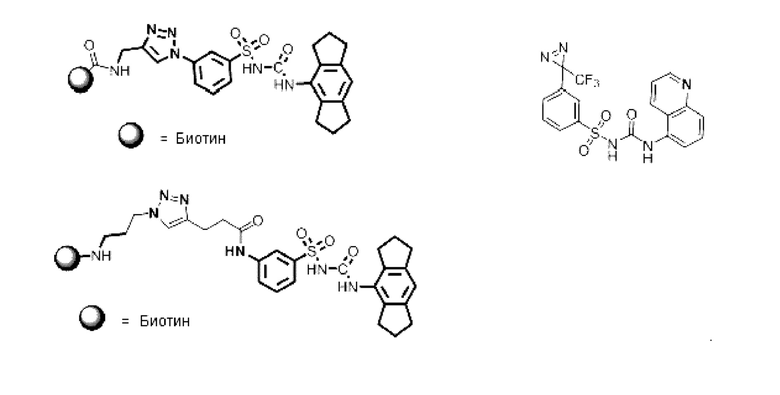
[00131] Следует понимать, что соединения из первого аспекта могут быть модифицированы или дериватизированы средствами, хорошо известными в технике, для обеспечения связывания с молекулой, такой как биотин, или флуоресцентной группой или фотоаффинной меткой, как показано с некоторыми из соединений выше.
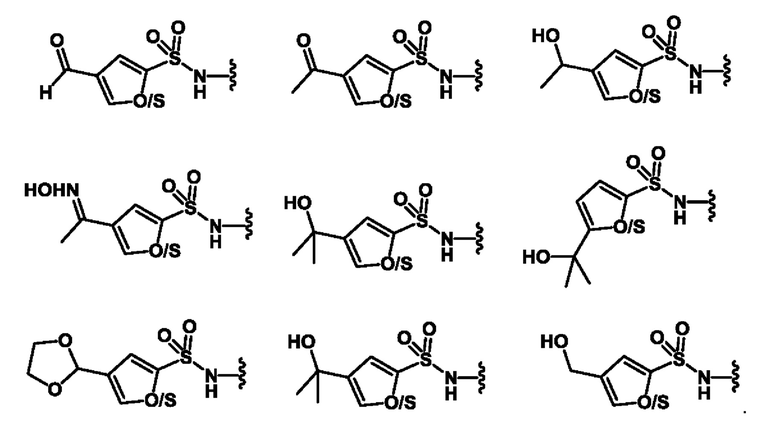
[00132] В одном варианте реализации соединение формулы (I) или (II) не содержит структуру, выбранную из групп, показанных ниже, соединенную с сульфонильным фрагментом (т.е. в качестве группы R1):
[00133] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S, W в виде О, a R2 выбран из гексагидроиндацена, 2,6-диизопропилфенила и 2,6-диизопропил-4-хлорфенила, то R1 не является одним из 2,4-дизамещеного фурана, 2,4-дизамещенного тиофена, 2,5-дизамещенного фурана и 2,5-дизамещенного тиофена.
[00134] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S, W в виде О, a R1 выбран из замещенного триазола, тиадиазола, 4-замещенного пиридина и 1,2-дизамещенного имидазола, то R2 не является незамещенным фенилом, 2-или 4-хлорфенилом или 3,4- замещенным фенилом, замещенным одним или более из гало, трифторметила, нитро или тиометила.
[00135] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S, W в виде О, a R1 выбран из замещенного триазола, тиадиазола, бензотиазола и замещенного пиримидина, то R2 не является тиофеном, 3-хлорфенилом, 4-этоксифенилом, замещенным бензимидазолом или замещенным бензотиазолом.
[00136] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S, W в виде О, a R1 представляет собой этоксизамещенный бензотиазол, то R2 не является 2,6-диизопропилфенилом.
[00137] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S, W в виде О, a R1 выбран из бензофурана, бензотиофена и индола, то R2 не является 3- или 3,4-гало-, метил-, этил- или трифторметилзамещенным фенилом.
[00138] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S, W в виде О, a R2 представляет собой замещенный пиримидин, то R-i не является пиразолом, замещенным сложным эфиром или карбокси.
[00139] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S и W в виде О, то атом углерода из R2, который непосредственно связан с азотом мочевины, не является карбонильным углеродом.
[00140] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S и W в виде О, то атом углерода из R2, который непосредственно связан с азотом мочевины, представляет собой углерод арила, гетероарила или гетероциклического кольца.
[00141] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S и W в виде О, R2 представляет собой замещенный фенил, a R-i представляет собой пиразол, то пиразол R1 не замещен арильной или гетероарильной группой.
[00142] В одном варианте реализации, если соединение из первого аспекта, включая любое соединение формулы (I)-(VII), имеет J в виде S, W в виде О, a R1 представляет собой пиразол и линкер сульфонилмочевины разветвлен в положении 4, пиразол не конденсирован в положениях 1 и 5 6-членным гетероциклом с образованием пиразолопиримидинового производного.
[00143] В одном варианте реализации соединение из первого аспекта, включая любое соединение формулы (I)-(VII), не является соединением, выбранным из группы, состоящей из:
1. 1-(4-Хлор-2,6-диизопропил-фенил)-3-[3-(1-гидрокси-1-метил-этил)-бензол сульфонил]-мочевины;
2. 1-(1,2,3,5,6,7-Гексагидро-s-индацен-4-ил)-3-[4-(1-гидрокси-1-метил-этил)-фуран-2-сульфонил]-мочевины;
3. 1-(1,2,3,5,6,7-Гексагидро-4-аза-э-индацен-8-ил)-3-[4-(1-гидрокси-1-метил-этил)-фуран-2-сульфонил]-мочевины;
4. 1-(1,2,3,5,6,7-Гексагидро-s-индацен-4-ил)-3-[4-(1-гидрокси-1-метил-этил)-тиофен-2-сульфонил]-мочевины;
5. 1-(4-[1,3]Диоксолан-2-ил-фуран-2-сульфонил)-3-(1,2,3,5,6,7-гексагидро-э-индацен-4-ил)-мочевины;
6. 1-(2,6-Диизопропил-фенил)-3-[4-(1-гидрокси-1-метил-этил)-фуран-2-сульфонил]-мочевины;
7. 1-(2,6-Диизопропил-фенил)-3-[4-(1-гидрокси-1-метил-этил)-тиофен-2-сульфонил]-мочевины;
8. 1-(4-Ацетил-тиофен-2-сульфонил)-3-(1,2,3,5,6,7-гексагидро-s-индацен-4-ил)-мочевины;
9. 1-(1Н-Бензимидазол-5-сульфонил)-3-(1,2,3,5,6,7-гексагидро-s-индацен-4-ил)-мочевины;
10. 1-(1,2,3,5,6,7-Гексагидро-s-индацен-4-ил)-3-[4-(1-гидрокси-1-метил-этил)-тиофен-2-сульфонил]-мочевины;
11. 1-(8-Хлор-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)-3-[4-(1-гидрокси-1-метил-этил)-фуран-2-сульфонил]-мочевины;
12. 1-(4-Ацетил-фуран-2-сульфонил)-3-(1,2,3,5,6,7-гексагидро-s-индацен-4-ил)-мочевины;
13. 1-(8-Фтор-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)-3-[4-(1-гидрокси-1-метил-этил)-фуран-2-сульфонил-мочевины;
14. 1-(4-Фтор-2,6-диизопропил-фенил)-3-[3-(1-гидрокси-1-метил-этил)-бензолсульфонил]-мочевины; и
15. 1-(6-Фтор-1Н-бензимидазол-5-сульфонил)-3-(1,2,3,5,6,7-гексагидро-s-индцен-4-ил)-мочевины;
16. 1-(4-Хлор-2,6-диизопропил-фенил)-3-(1Н-индол-6-сульфонил)-мочевины;
17. 1-(4-Хлор-2,6-диизопропил-фенил)-3-(5-фтор-1Н-индол-6-сульфонил)-мочевины;
18. 1-[1,2,3,5,6,7-Гексагидро-s-индацен-4и-ил)-3-(1Н-индол-6-сульфонил)-мочевины;
19. 1-(5-Фтор-1Н-индол-6-сульфонил)-3-(1,2,3,5,6,7-гексагидро-5-индацен-4-ил)-мочевины;
20. 1-[4-Хлор-2,6-диизопропил-фенил]-3-[2-фтор-5-(2-метил-(1,3)диоксолан-2-ил)-бензолсульфонил]-мочевины;
21. 3-[3-[4-Хлор-2,6-диизопропил-фенил]-уреидосульфонил]-N-метил-бензолсульфонамида;
22. 1-[2-Фтор-5-(2-метил-(1,3)диоксолан-2-ил)бензолсульфонил]-3-1,2,3,5,6,7-гексагидро-индацен-4-ил)-мочевины;
23. 3-[3-(1,2,3,5,6,7-Гексагидро-5-индацен-4-ил)-уреидосульфонил]-N-метил-бензолсульфонамида;
24. 4-(1-гидрокси-1-метил-этил)-фуран-2-сульфонамида.
[00144] В некоторых вариантах реализации настоящего изобретения обеспечиваются терапевтически неактивные пролекарства соединений из первого аспекта. Пролекарства представляют собой соединения, которые при введении млекопитающему преобразуются полностью или частично в соединение по настоящему изобретению. В большинстве вариантов реализации пролекарства представляют собой фармакологически инертные химические производные, которые могут быть преобразованы in vivo в активные молекулы препарата для проявления терапевтического эффекта. Любые из соединений, описанных в данном контексте, могут вводиться в виде пролекарства для увеличения активности, биодоступности или устойчивости соединения или для изменения иным образом свойств соединения. Типичные примеры пролекарств включают соединения, имеющие биологически лабильные защитные группы на функциональном фрагменте активного соединения. Пролекарства включают, но без ограничения ими, соединения, которые могут быть окислены, восстановлены, аминированы, деаминированы, гидроксилированы, дегидроксилированы, гидролизованы, дегидролизованы, алкилированы, деалкилированы, ацилированы, деацилированы, фосфорилированы и/или дефосфорилированы с получением активного соединения.
[00145] Известней ряд лигандов пролекарств. В целом, алкилирование, ацилирование или другая липофильная модификация одного или более гетероатомов соединения, таких как свободный остаток амина или карбоновой кислоты, могут снизить полярность и позволить соединениям проходить внутрь клеток. Примеры замещающих групп, которые могут заменять один или более атомов водорода по фрагменту свободного амина и/или карбоновой кислоты включают, но без ограничения ими, следующие: арил; стероиды; углеводы (включая сахара); 1,2-диацилглицерин; спирты; ацил (включая низший ацил); алкил (включая низший алкил); эфир сульфокислоты (включая алкил- или арилалкилсульфонил, такие как метансульфонил и бензил, где фенильная группа необязательно замещена одним или более заместителей, предусмотренными в определении арила, приведенном в настоящем документе); необязательно замещенный арилсульфонил; липиды (включая фосфолипиды); фосфатидилхолин; фосфохолин; остатки или производные аминокислот; ацильные остатки или производные аминокислот; пептиды; холестерины или другие фармацевтически приемлемые уходящие группы, которые при введении in vivo обеспечивают свободный амин. Любой из этих фрагментов может быть применен в комбинации с представленными активными агентами для достижения желаемого эффекта.
[00146] В некоторых вариантах реализации предусматриваются соединения с одним или более хиральными центрами. Хотя рацемические смеси соединений по настоящему изобретению могут быть активными, селективными и биодоступными, выделенные изомеры также могут представлять интерес.
[00147] Соединения из первого аспекта могут содержать хиральные центры, которые могут иметь либо (R), либо (S)-конфигурацию, или которые могут содержать их смесь. Соответственно, настоящее изобретение также включает стереоизомеры соединений, описанных в настоящем документе, где это применимо, либо индивидуально, либо смешанными в любых соотношениях.
Стереоизомеры могут включать, но без ограничения ими, энантиомеры, диастереомеры, рацемические смеси и их комбинации. Такие стереоизомеры могут быть получены и разделены с использованием традиционных методик, либо путем введения в реакцию энантиомерных исходных материалов, либо путем разделения изомеров соединений и пролекарств по настоящему изобретению. Изомеры могут включать геометрические изомеры. Примеры геометрических изомеров включают, но без ограничения ими, цис-изомеры или транс-изомеры по двойной связи. Среди соединений по настоящему изобретению предусматриваются другие изомеры. Изомеры могут применяться либо в чистом виде, либо в смеси с другими изомерами соединений, описанных в настоящем документе.
[00148] Для получения оптически активных форм и определения активности в технике известны различные способы. Такие способы включают стандартные испытания, описанные в настоящем документе, и другие аналогичные испытания, хорошо известные в технике. Примеры способов, которые можно применять для получения оптических изомеров соединений по настоящему изобретению, включают следующие:
i) физическое разделение кристаллов, при котором макроскопические кристаллы индивидуальных энантиомеров разделяют вручную. Эта методика, в частности, может применяться, если кристаллы отдельных энантиомеров существуют (т.е. материал представляет собой конгломерат), и кристаллы визуально различимы;
ii) одновременную кристаллизацию, при которой индивидуальные энантиомеры кристаллизуются отдельно из раствора рацемата, что возможно только, если последний представляет собой конгломерат в твердом состоянии;
iii) ферментативные разделения, при которых рацемат разделяется частично или полностью благодаря различным скоростям реакции энантиомеров с ферментом;
iv) ферментативный асимметричный синтез, синтетическая методология, при которой по меньшей мере одна стадия синтеза использует ферментативную реакцию для получения энантиомерно чистого или обогащенного синтетического предшественника желаемого энантиомера;
v) химический асимметричный синтез, при котором желаемый энантиомер синтезируют из ахирального предшественника в условиях, создающих асимметрию (т.е. хиральность) в продукте, что может достигаться применением хиральных катализаторов или хиральных вспомогательных добавок;
vi) диастереомерные разделения, при которых рацемическое соединение вводят в реакцию с энантиомерно чистым реагентом (хиральным вспомогательным веществом), который преобразует индивидуальные энантиомеры в диастереомеры. Полученные диастереомеры затем разделяют хроматографией или кристаллизацией благодаря их теперь более выраженным структурным различиям, а хиральное вспомогательное вещество после этого удаляют для получения желаемого энантиомера;
vii) асимметричные преобразования первого и второго порядка, при которых диастереомеры из рацемата приводят в равновесие с получением преобладания диастереомера из желаемого энантиомера в растворе, или при которых предпочтительная кристаллизация диастереомера из желаемого энантиомера смещает равновесие таким образом, что в итоге по существу весь материал преобразуется в кристаллический диастереомер из желаемого энантиомера. Затем желаемый энантиомер выделяют из диастереомеров;
viii) кинетические разделения, включающие частичное или полное разделение рацемата (или дополнительное разделение частично разделенного соединения) на основании неравных скоростей реакции энантиомеров с хиральным, нерацемическим реагентом или катализатором в кинетических условиях;
ix) энантиоспецифичный синтез из нерацемических предшественников, при котором желаемый энантиомер получают из нехиральных исходных материалов и в котором стереохимическая чистота не нарушается или лишь минимально нарушается в ходе синтеза;
x) хиральную жидкостную хроматографию, при которой энантиомеры рацемата разделяются в жидкой подвижной фазе благодаря их различающимся взаимодействиям с неподвижной фазой. Неподвижная фаза может быть изготовлена из хирального материала, или подвижная фаза может содержать дополнительный хиральный материал с целью вызвать различающиеся взаимодействия;
xi) хиральную газовую хроматографию, при которой рацемат испаряют и энантиомеры разделяются благодаря их различающимся взаимодействиям в газообразной подвижной фазе с колонной, содержащей неподвижную фазу из нерацемического хирального адсорбента;
xii) экстракцию хиральными растворителями, при которой энантиомеры разделяются благодаря предпочтительному растворению одного энантиомера в конкретном хиральном растворителе; и
xiii) перенос сквозь хиральные мембраны, при котором рацемат приводят в контакт с тонким мембранным барьером. Барьер, как правило, разделяет две смешивающиеся текучие среды, одна из которых содержит рацемат, и движущая сила, такая как разность концентрации или давления, вызывает предпочтительный перенос сквозь мембранный барьер. Разделение происходит в результате нерацемической хиральной природы мембраны, позволяющей проходить только одному энантиомеру рацемата.
[00149] Соединение необязательно может быть предусмотрено в композиции, являющейся энантиомерно обогащенной, такой как смесь энантиомеров, в которой один энантиомер присутствует в избытке, в частности, до величины 95% или более, 96% или более, 97% или более, 98% или более, или 99% или более, включая 100%.
[00150] Термины (R), (S), (R,R), (S,S), (R,S) и (S,R) в данном контексте означают, что композиция содержит большую долю указанного изомера соединения по сравнению с другими изомерами. В предпочтительном варианте реализации эти термины показывают, что композиция содержит по меньшей мере 90% по массе указанного изомера и 10% по массе или менее одного или более других изомеров; или более предпочтительно около 95% по массе указанного изомера и 5% или менее одного или более других изомеров. В некоторых вариантах реализации композиция может содержать по меньшей мере 99% по массе указанного изомера и 1% или менее по массе одного или более других изомеров, или может содержать 100% по массе указанного изомера и 0% по массе одного или более других изомеров. Эти процентные содержания приведены в пересчете на общее количество соединения по настоящему изобретению, присутствующего в композиции.
[00151] Соединения из первого аспекта можно применять сами по себе или в виде фармацевтически приемлемого сложного эфира, амида, соли, сольвата, пролекарства или изомера по необходимости. Например, соединение может быть предусмотрено в виде фармацевтически приемлемой соли. В случае применения соль лекарственного соединения должна быть как фармакологически, так и фармацевтически приемлемой, но фармацевтически неприемлемые соли могут удобным образом применяться для получения свободного активного соединения или его фармацевтически приемлемых солей и не исключаются из объема настоящего изобретения. Такие фармакологически и фармацевтически приемлемые соли могут быть получены путем реакции препарата с органической или неорганической кислотой с использованием стандартных способов, подробно изложенных в литературе.
[00152] Примеры фармацевтически приемлемых солей соединений, применимых в соответствии с изобретением, включают соли присоединения кислоты. Однако соли фармацевтически неприемлемых кислот могут быть полезны, например, в получении и очистке соединений. Подходящие соли присоединения кислоты согласно настоящему изобретению включают органические и неорганические кислоты. Предпочтительные соли включают образованные из соляной, бромистоводородной, серной, фосфорной, лимонной, винной, молочной, пировиноградной, уксусной, янтарной, фумаровой, малеиновой, щавелевоуксусной, метансульфоновой, этансульфоновой, л-толуолсульфоновой, бензолсульфоновой и изэтионовой кислот. Другие пригодные соли присоединения кислоты включают пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, яблочную кислоту, малоновую кислоту, бензойную кислоту, коричную кислоту, миндальную кислоту, салициловую кислоту и подобные. Конкретные примеры фармацевтически приемлемых солей включают, но без ограничения ими, сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиоляты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, малеаты, бутин-1,4-диоляты, гексин-1,6-диоляты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, γ-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты и манделаты.
[00153] Соль присоединения кислоты может быть снова преобразована в свободное основание посредством воздействия подходящего основания. Получение основных солей кислотных фрагментов, которые могут присутствовать в соединении или пролекарстве, пригодных согласно настоящему изобретению, может быть осуществлено аналогичным образом с использованием фармацевтически приемлемого основания, такого как гидроксид натрия, гидроксид калия, гидроксид аммония, гидроксид кальция, триэтиламин или подобные.
[00154] Сложные эфиры соединений действующих веществ по настоящему изобретению могут быть получены через функционализацию гидроксильных и/или карбоксильных групп, которые могут присутствовать в молекулярной структуре соединения. Амиды и пролекарства также могут быть получены с использованием методологий, известных специалистам в данной области. Например, амиды могут быть получены из сложных эфиров с использованием подходящих аминных реагентов, или они могут быть получены из ангидрида или хлорида кислоты при реакции с аммиаком или низшим алкиламином. Кроме того, сложные эфиры и амиды соединений по настоящему изобретению могут быть получены реакцией с карбонилирующим агентом (например, этилформиатом, уксусным ангидридом, метоксиацетилхлоридом, бензоилхлоридом, метилизоцианатом, этилхлорформиатом, метансульфонилхлоридом) и подходящим основанием (например, 4-диметиламинопиридином, пиридином, триэтиламином, карбонатом калия) в подходящем органическом растворителе (например, тетрагидрофуране, ацетоне, метаноле, пиридине, N,N-диметилформамиде) при температуре от 0°С до 60°С. Пролекарства, как правило, получают ковалентным присоединением фрагмента, что приводит к соединению, являющемуся терапевтически неактивным до модификации метаболической системой индивидуума. Примеры фармацевтически приемлемых сольватов включают, но без ограничения ими, соединения по настоящему изобретению в комбинации с водой, изопропанолом, этанолом, метанолом, ДМСО, этилацетатом, уксусной кислотой или этаноламином.
[00155] В случае твердых композиций понятно, что соединения, применяемые в способах по настоящему изобретению, могут существовать в различных формах. Например, соединения могут существовать в стабильных и метастабильных кристаллических формах и изотропных и аморфных формах, все из которых подразумеваются находящимися в рамках настоящего изобретения.
[00156] Если соединение, пригодное в качестве действующего вещества согласно изобретению, представляет собой основание, желаемая соль может быть получена любым подходящим способом, известным в технике, включая действие на свободное основание неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные, или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидильные кислоты, такие как глюкуроновая кислота и галактуроновая кислота, альфа-гидроксикислоты, такие как лимонная кислота и винная кислота, аминокислоты, такие как аспарагиновая кислота и глутаминовая кислота, ароматические кислоты, такие как бензойная кислота и коричная кислота, сульфокислоты, такие как л-толуолсульфокислота или этансульфокислота, или подобные.
[00157] Если соединение, описанное в настоящем документе в качестве действующего вещества, представляет собой кислоту, желаемая соль может быть получена любым подходящим способом, известным в технике, включая действие на свободную кислоту неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или щелочноземельного металла или подобными. Иллюстративные примеры подходящих солей включают органические соли, происходящие из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов, и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, происходящие из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
[00158] Согласно второму аспекту изобретения предусмотрена фармацевтическая композиция, содержащая соединение формулы от (I) до (VII), или его фармацевтически приемлемую соль, сольват или пролекарство, и фармацевтически приемлемый носитель, разбавитель и/или вспомогательное вещество.
[00159] Подходящим образом, фармацевтически приемлемый носитель, разбавитель и/или вспомогательное вещество могут представлять собой или включать один или более из разбавителей, растворителей, буферов рН, связующих, наполнителей, эмульгаторов, разрыхлителей, полимеров, смазывающих веществ, масел, жиров, восков, покрытий, модификаторов вязкости, глидантов и подобных.
[00160] Солевые формы соединений по настоящему изобретению могут быть особенно пригодными благодаря их улучшенной растворимости.
[00161] В одном варианте реализации фармацевтическая композиция включает циклодекстрин.
[00162] Циклодекстрин может быть выбран из альфа-, бета- или гамма-циклодекстринов.
[00163] В одном варианте реализации циклодекстрин выбран из метилциклодекстрина, гидроксипропилциклодекстрина и сульфобутилового эфира циклодекстрина.
[00164] Было обнаружено, что циклодекстрины обеспечивают значительные преимущества в технологии изготовления и доставке соединений по настоящему изобретению.
[00165] Циклодекстриновые составы, такие как, например, одно или более соединений по настоящему изобретению с гидроксипропил-бета-циклодекстрином или метил-бета-циклодекстрином, могут иметь применения в секвестрации холестерина/снижении уровня холестерина или через ингибирование NLRP3 для неалкогольного стеатогепатита (НАСГ), алкогольной болезни печени, атеросклероза, а также при болезни Альцгеймера (БА).
[00166] Разбавители могут включать один или более из микрокристаллической целлюлозы, лактозы, маннита, фосфата кальция, сульфата кальция, каолина, сухого крахмала, порошкообразного сахара и подобных. Связующие могут включать одно или более из повидона, крахмала, стеариновой кислоты, камедей, гидроксипропилметилцеллюлозы и подобных. Разрыхлители могут включать один или более из крахмала, кроскармеллозы натрия, кросповидона, крахмалгликолята натрия и подобных. Растворители могут включать один или более из этанола, метанола, изопропанола, хлороформа, ацетона, метилэтилкетона, метиленхлорида, воды и подобных. Смазывающие вещества могут включать одно или более из стеарата магния, стеарата цинка, стеарата кальция, стеариновой кислоты, стеарилфумарата натрия, гидрированного растительного масла, глицерилбегената и подобных. Глидант может быть одним или более из коллоидного диоксида кремния, талька или кукурузного крахмала и подобных. Буферы могут включать фосфатные буферы, боратные буферы и карбонатные буферы, хотя и без ограничения ими. Наполнители могут включать один или более гелей, включая желатин, крахмал и синтетические полимерные гели, хотя и без ограничения ими. Покрытия могут включать одно или более из пленкообразователей, растворителей, пластификаторов и подобных. Подходящие пленкообразователи могут быть одним или более из гидроксипропилметилцеллюлозы, метилгидроксиэтилцеллюлозы, этилцеллюлозы, гидроксипропилцеллюлозы, повидона, натрий-карбоксиметилцеллюлозы, полиэтиленгликоля, акрилатов и подобных. Подходящие растворители могут быть одним или более из воды, этанола, метанола, изопропанола, хлороформа, ацетона, метилэтилкетона, метиленхлорида и подобных. Пластификаторы могут быть одним или более из пропиленгликоля, касторового масла, глицерина, полиэтиленгликоля, полисорбатов и подобных.
[00167] Делается ссылка на Handbook of Excipients 6th Edition, Eds. Rowe, Sheskey & Quinn (Pharmaceutical Press), где предусматриваются неограничивающие примеры вспомогательных веществ, которые могут быть пригодны согласно изобретению.
[00168] Следует понимать, что выбор фармацевтически приемлемых носителей, разбавителей и/или вспомогательных веществ будет, по меньшей мере частично, зависеть от режима введения состава. Только в качестве примера, композиция может иметь форму таблетки, капсулы, каплета, порошка, жидкости для инъекций, суппозитория, состава медленного высвобождения, состава для осмотического насоса или любую другую форму, являющуюся эффективной и безопасной для введения.
[00169] Подходящим образом, фармацевтическая композиция предназначена для лечения или профилактики заболевания, нарушения или состояния у млекопитающего.
[00170] Третий аспект изобретения заключается в способе лечения или профилактики заболевания, нарушения или состояния, включающем стадию введения эффективного количества соединения формулы (I)-(VII), или его фармацевтически эффективной соли, сольвата или пролекарства, или фармацевтической композиции из второго аспекта для лечения или профилактики этим заболевания, нарушения или состояния.
[00171] Четвертый аспект изобретения предусматривает соединение формулы (I)-(VII), или его фармацевтически эффективную соль, сольват или пролекарство, или фармацевтическую композицию из второго аспекта для применения в лечении или профилактике заболевания, нарушения или состояния.
[00172] Пятый аспект изобретения предусматривает применение соединения формулы (I)-(VII), или его фармацевтически эффективной соли, сольвата или пролекарства, в производстве лекарства для лечения или профилактики заболевания, нарушения или состояния.
[00173] При обычном использовании в данном контексте термины «введение» или «ввод» и подобные описывают введение соединения или композиции млекопитающему, например, через конкретный путь или среду. Пути введения могут включать местный, парентеральный и энтеральный, которые включают пероральный, буккальный, подъязычный, назальный, анальный, желудочно-кишечный, подкожный, внутримышечный и внутрикожный пути введения, хотя и без ограничения ими.
[00174] Под «лечить» или «лечением» подразумевается введение соединения или композиции субъекту с целью по меньшей мере улучшения, снижения или подавления существующих признаков или симптомов заболевания, нарушения или состояния, испытываемых субъектом.
[00175] Под «предотвращением», «профилактикой» или «профилактическим» подразумевается профилактическое введение состава субъекту, который не проявляет признаков или симптомов заболевания, нарушения или состояния, но для которого ожидается или предполагается вероятное проявление таких признаков или симптомов при отсутствии профилактики. Профилактическое лечение может по меньшей мере уменьшить или частично исправить ожидаемые симптомы или признаки.
[00176] В данном контексте «эффективное количество» относится к введению количества соответствующего соединения или композиции, достаточного для предотвращения возникновения симптомов состояния, подлежащего лечению, или для вызывания прекращения ухудшения симптомов, или для лечения и исправления или по меньшей мере снижения тяжести симптомов. Эффективное количество будет варьироваться способом, понятным специалисту в данной области, с возрастом, полом, весом пациента и т.д. Подходящая дозировка или режим дозирования могут быть установлены при стандартном обследовании.
[00177] В данном контексте термины «субъект» или «индивидуум» или «пациент» могут относиться к любому субъекту, в частности, позвоночному субъекту, и, еще более конкретно, млекопитающему субъекту, для которого требуется лечение. Подходящие позвоночные животные включают, но не ограничены ими, приматов, птиц, сельскохозяйственных животных (например, овец, коров, лошадей, ослов, свиней), животных для лабораторных исследований (например, кролей, мышей, крыс, морских свинок, хомяков), домашних животных (например, кошек, собак) и пойманных диких животных (например, лис, оленей, динго). Предпочтительным субъектом является человек, нуждающийся в лечении заболевания, нарушения или состояния, описанных в настоящем документе. Однако будет понятно, что вышеупомянутые термины не означают, что симптомы обязательно присутствуют.
[00178] В одном конкретном варианте реализации заболевание, нарушение или состояние является таким, которое реагирует на ингибирование активации инфламмасомы NLRP3.
[00179] Согласно этому варианту реализации, соединение из первого аспекта или его фармацевтически эффективная соль, сольват или пролекарство представляет собой специфичный ингибитор NLRP3.
[00180] В дополнительном варианте реализации заболевание, нарушение или состояние реагирует на модуляцию одного или более из ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17.
[00181] В одном варианте реализации модуляция представляет собой ингибирование одного или более из ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, и ИЛ-33.
[00130] В одном варианте реализации модуляция клеток Th17 осуществляется ингибированием выработки и/или секреции ИЛ-17.
[00182] В общих вариантах реализации заболевание, нарушение или состояние представляет собой заболевание, нарушение или состояние иммунной системы, сердечно-сосудистой системы, эндокринной системы, желудочно-кишечного тракта, почечной системы, органов дыхания, центральной нервной системы, представляет собой рак или другую злокачественную опухоль и/или вызвано патогенным фактором или связано с ним.
[00183] Следует понимать, что эти общие варианты реализации, определенные согласно широким категориям заболеваний, нарушений и состояний, не являются взаимоисключающими. В этом отношении любое конкретное заболевание, нарушение или состояние может быть классифицировано в соответствии более чем одному из вышеупомянутых общих вариантов реализации. Неограничивающим примером является диабет I типа, который является аутоиммунным заболеванием и заболеванием эндокринной системы.
[00184] В одном варианте реализации заболевание, нарушение или состояние относится к иммунной системе. В конкретных вариантах реализации заболевание, нарушение или состояние представляет собой воспалительное заболевание, нарушение или состояние или аутоиммунное заболевание, нарушение или состояние.
[00185] В одном варианте реализации заболевание, нарушение или состояние относится к коже.
[00186] В одном варианте реализации заболевание, нарушение или состояние относится к сердечно-сосудистой системе.
[00187] В одном варианте реализации заболевание, нарушение или состояние представляет собой рак, опухоль или другое злокачественное новообразование. В данном контексте раки, опухоли и злокачественные новообразования относятся к заболеваниям, нарушениям или состояниям, или к клеткам или тканям, связанным с заболеваниями, нарушениями или состояниями, характеризующимися нарушенной или аномальной пролиферацией клеток, дифференциацией и/или миграцией, часто сопровождаемой нарушенным или аномальным молекулярным фенотипом, который включает одну или более генетических мутаций или других генетических изменений, связанных с онкогенезом, экспрессией маркеров опухоли, потерей экспрессии или активности опухолевого супрессора экспрессии и/или нарушенной или аномальной экспрессией маркера клеточной поверхности. В общих вариантах реализации раки, опухоли и злокачественные новообразования могут включать саркомы, лимфомы, лейкемии, плотные опухоли, бластомы, глиомы, карциномы, меланомы и метастатические раки, хотя и без ограничения ими. Более исчерпывающее перечисление раковых опухолей и злокачественных новообразований можно найти на веб-сайте Национального института рака http://www.cancer.gov/cancertopics/types/alphalist.
[00188] В одном варианте реализации заболевание, нарушение или состояние относится к почечной системе.
[00189] В одном варианте реализации заболевание, нарушение или состояние относится к желудочно-кишечному тракту.
[00190] В одном варианте реализации заболевание, нарушение или состояние относится к органам дыхания.
[00191] В дополнительном варианте реализации заболевание, нарушение или состояние относится к эндокринной системе.
[00192] В одном варианте реализации заболевание, нарушение или состояние относится к центральной нервной системе (ЦНС).
[00193] В одном варианте реализации заболевание, нарушение или состояние вызвано патогенным фактором или связано с ним. Патогенный фактор может представлять собой вирус, бактерию, простейшее, червя или грибок или любой другой организм, способный заражать млекопитающее, хотя и без ограничения ими.
[00194] Неограничивающие примеры вирусов включают вирус гриппа, цитомегаловирус, вирус Эпштейна-Барр, вирус иммунодефицита человека (ВИЧ), альфавирус, такой как чикунгунья и вирус реки Росс, флавивирусы, такие как вирус Денге, вирус Зика и папилломавирус, хотя и без ограничения ими.
[00195] Неограничивающие примеры патогенных бактерий включают Staphylococcus aureus, Helicobacter pylori, Bacillus anthracis, Bordatella pertussis, Corynebactehum dipthehae, Clostridium tetani, Clostridium botulinum, Streptococcus pneumoniae, Streptococcus pyogenes, Listeria monocytogenes, Hemophilus influenzae, Pasteurella multicida, Shigella dysenteriae, Mycobacterium tuberculosis, Mycobacterium leprae, Mycoplasma pneumoniae, Mycoplasma hominis, Neisseria meningitidis, Neisseria gonorrhoeae, Rickettsia rickettsii, Legionella pneumophila, Klebsiella pneumoniae, Pseudomonas aeruginosa, Propionibacterium acnes, Treponema pallidum, Chlamydia trachomatis, Vibrio cholerae, Salmonella typhimurium, Salmonella typhi, Borrelia burgdorferi и Yersinia pestis, хотя и без ограничения ими.
[00196] Неограничивающие примеры простейших включают плазмодий, бабезиоз, лямблию, энтамебу, лейшманию и трипаносомы, хотя и без ограничения ими.
[00197] Неограничивающие примеры червей включают гельминтов, включая шистосомы, круглых червей, ленточных червей и трематод, хотя и без ограничения ими.
[00198] Неограничивающие примеры грибков включают виды кандида и аспергиллы, хотя и без ограничения ими.
[00199] Дополнительные связанные заболевания, нарушения или состояния могут быть выбраны из группы, состоящей из упомянутых в статье журнала, находящейся по адресу:
http://onlinelibrary.wiley.com/store/10.1111/j.1365-2249.2011.04440.x/asset/j.1365-2-249.2011.04440.x.pdf?v=1&t=i60c1phf&s=d26f50a2622926cc6b4bc855bd911 ae9dc 9750cf.
[00200] В конкретных вариантах реализации заболевание, нарушение или состояние выбрано из группы, состоящей из конститутивного воспаления, включая криопирин-ассоциированные периодические синдромы (КАПС (CAPS)): синдром Макла-Уэльса (СМУ (MWS)), семейный холодовой аутовоспалительный синдром (СХАС (FCAS)) и мультисистемное воспалительное заболевание неонатального возраста (МВЗНВ (NOMID)); включая аутовоспалительные заболевания: периодическую болезнь (ПБ (FMF)), периодический синдром, связанный с рецепторами TNF (ПССРТ (TRAPS)), дефицит мевалонаткиназы (ДМК (MKD)), гипериммуноглобулинемия D и синдром периодической лихорадки (ГИДС (HIDS)), дефицит антагониста рецептора интерлейкина 1 (ДАРИ (DIRA)), синдром Маджида, гнойный артрит, гангренозную пиодермию и акне (ГАПА (PAPA)), гаплонедостаточность А20 (ГА20 (НА20)), педиатрический гранулематозный артрит (ПГА (PGA)), связанный с PLCG2 дефицит антител и иммунную дисрегуляцию (СПЛДАИД(PLAID)), связанное с PLCG2 аутовоспалительное заболевание, дефицит антител и иммунную дисрегуляцию (СПЛАДАИД (APLAID)), сидеробластическую анемию с В-клеточным иммунодефицитом, периодические лихорадки и задержку в развитии (СИЛЗ (SIFD)); синдром Свита, хронический небактериальный остеомиелит (ХНО (CNO)), хронический рецидивирующий мультифокальный остеомиелит (ХРМО (CRMO)) и синовит, акне, пустулез, гиперостоз, синдром остита (САПГО (SAPHO)); аутоиммунные заболевания, включая рассеянный склероз (PC(MS)), диабет типа 1, псориаз, ревматоидный артрит, болезнь Бехчета, синдром Шегрена и синдром Шницлера; респираторные заболевания, включая хроническую обструктивную болезнь легких (ХОБЛ (COPD)), стероидорезистентную астму, асбестоз, силикоз и кистозный фиброз; заболевания центральной нервной системы, включая болезнь Паркинсона, болезнь Альцгеймера, болезнь моторных нейронов, болезнь Хантингтона, церебральную малярию и повреждение мозга от пневмококкового менингита; метаболические заболевания, включая диабет типа 2, атеросклероз, ожирение, подагру, псевдо-подагру; болезни глаз, в том числе глазного эпителия, возрастную макулярную дегенерацию (ВМД (AMD)), инфекцию роговицы, увеит и сухой глаз; заболевания почек, включая хроническую болезнь почек, оксалатную нефропатию и диабетическую нефропатию; заболевания печени, включая безалкогольный стеатогепатит и алкогольную болезнь печени; воспалительные реакции на коже, включая контактную гиперчувствительность и солнечные ожоги; воспалительные реакции в суставах, включая остеоартрит, системный ювенильный идиопатический артрит, приобретенную болезнь Стилла, рецидивирующий полихондрит; вирусные инфекции, включая альфа-вирус (чикунгунья, вирус реки Росса) и флавивирус (вирус Денге и Зика), грипп, ВИЧ; гнойный гидраденит (ГГ (HS)) и другие вызывающие кисту кожные заболевания; раки, включая метастазы рака легкого, рак поджелудочной железы, рак желудка, миелодиспластический синдром, лейкемию; полимиозит; апоплексию; инфаркт миокарда; реакцию трансплантата против хозяина; повышенное давление; колит; гельминтовую инфекцию; бактериальную инфекцию; аневризму брюшной аорты; заживление ран; депрессию, психологический стресс; перикардит, включая синдром Дресслера, ишемически-реперфузионное повреждение и любое заболевание, при котором определено, что индивидуум несет генеративную или соматическую мутацию в NLRP3, не являющуюся молчащей.
[00201] В одном неограничивающем примере из описанных, заболевание, нарушение или состояние, подлежащее лечению, представляет собой НАСГ. Активация инфламмасомы NLRP3 является главным в воспалительном накоплении в НАСГ, и ингибирование NLRP3 может как предотвращать, так и обращать вспять фиброз печени. Соединения по настоящему изобретению, путем нарушения функции инфламмасом NLRP3 в ткани печени, могут вызывать восстановление ткани при воспалении печени, сниженное накопление макрофагов и нейтрофилов и подавление активации NF-κВ. Ингибирование NLRP3 может снижать экспрессию в печени про-ИЛ-1β и нормализовывать уровни ИЛ-1β, ИЛ-6 и МСР-1 в печени и в циркулирующей крови, тем самым способствуя лечению заболевания.
[00202] В дополнительном неограничивающем примере из описанных, заболевание, нарушение или состояние, подлежащее лечению, представляет собой тяжелую астму со стероидной резистентностью (ТАСР). Респираторные инфекции индуцируют в легких сигнальный путь инфламмасома NLRP3/каспаза-1/ИЛ-1β, который провоцирует астму ТАСР. Инфламмасома NLRP3 рекрутирует и активирует прокаспазу-1 для индуцирования реакций ИЛ-1(3. Поэтому индуцированные инфламмасомой NLRP3 реакции ИЛ-1(3 важны в сдерживании инфекций, однако чрезмерная активация приводит к аномальному воспалению и связывалась с патогенезом астмы ТАСР и ХОБЛ. Введение соединений из первого аспекта, которые воздействуют на конкретные процессы заболевания, является более терапевтически привлекательными, чем неспецифичное ингибирование воспалительных реакций с помощью стероидов или ИЛ-1β. Поэтому воздействие на сигнальный путь инфламмасома NLRP3/каспаза-1/ИЛ-1β соединениями из первого аспекта может быть полезным в лечении астмы ТАСР и других стероид-резистентных воспалительных состояний.
[00203] В одном дополнительном неограничивающем примере из описанных, заболевание, нарушение или состояние, подлежащее лечению, представляет собой болезнь Паркинсона. Болезнь Паркинсона представляет собой наиболее распространенное нейродегенеративное двигательное расстройство и характеризуется выборочной потерей дофаминергических нейронов, сопровождаемой накоплением неправильно свернутого α-синуклеина (Syn) в тельца Леви, которые являются патологическими отличительными признаками заболевания. На ранней стадии заболевания проявляется хроническое микроглиальное нейровоспаление, и было предположено, что оно вызывает патологию.
[00204] В развитии болезни Паркинсона центральная роль отводится микроглиальной NLRP3. Инфламмасома NLRP3 активируется фибриллярным Syn через зависимый от Syk-киназы механизм, а также происходит в отсутствие патологии Syn на ранних стадиях дофаминергической дегенерации и влечет за собой потерю нейронов. Соединения из первого аспекта могут блокировать активацию инфламмасомы NLRP3 фибриллярным Syn или митохондриальной дисфункцией и тем самым обеспечивать эффективную нейропротекцию нигростриарной дофаминергической системы и способствовать лечению болезни Паркинсона.
[00205] В шестом аспекте изобретения предусматривается способ диагностики заболевания, нарушения или состояния у млекопитающего, включающий стадию введения меченого соединения формулы (I)-(VII) или его фармацевтически эффективной соли, сольвата или пролекарства, млекопитающему или в биологический образец, полученный из млекопитающего, для содействия диагностике заболевания, нарушения или состояния у млекопитающего.
[00206] Известно, что активация инфламмасомы, в частности, инфламмасомы NLRP3, приводит к запуску, прогрессированию и хроническому развитию огромного количества воспалительных заболеваний. Сульфонилмочевины и родственные соединения из первого аспекта являются сильнодействующими и специфичными прямыми ингибиторами NLRP3. Соответственно, химический зонд, специфичный для NLRP3, которая присутствует в иммунных клетках при воспалении, имеет потенциальную ценность для диагностики воспалительных и других родственных заболеваний. Зонд активации NRLP3, содержащий соединение из первого аспекта, может действовать как эффективный суррогатный биомаркер воспалительного заболевания для диагностики ex vivo (в крови) или in vivo (MPT, ПЭТ и т.д.).
[00207] Применение соединений из первого аспекта в диагностике воспалительных и других родственных заболеваниях, таких как перечисленные выше, может быть достигнуто флуоресцентной визуализацией в ближнем инфракрасном диапазоне и характеризацией ex vivo иммунных клеток по степени ингибирования ИЛ-1бета, расщеплению прокаспазы 1 и уровней ИЛ-18. В частности, имеют значение моноциты периферической крови (МПК), макрофаги, дендритные клетки, Т-клетки CD4+, клетки Th17, клетки Th1 и клетки Th2. В диагностике in vivo применяют магнитно-резонансную томографию (МРТ). Меченые атомами Н2 (дейтерия), 13С, 19F, 15N варианты [классов соединений] вводят пациенту внутривенно, внутримышечно, подкожно, перорально, местно, интратекально и т.д.
[00208] Подходящей также является диагностика in vivo с применением позитронно-эмиссионной томографии (ПЭТ). ПЭТ представляет собой технологию молекулярной визуализации, требующую специфичных зондов, меченых короткоживущими излучающими позитроны радионуклидами. Типичные изотопы включают 11С, 13N, 15O, 18F, 64Cu, 62Cu, 124I, 76Br, 82Rb и 68Ga, при этом наиболее клинически используемым является 18F. В частности, возможно простым способом получать устойчивую соль 64Cu или 62Cu одного или более соединений формулы (I) путем простого ионного обмена с натриевой (или другого одновалентного катиона) солью указанных соединений. Это позволяет быстро получать диагностические зонды для радиовизуализации, ПЭТ и подобных, при которых интенсивность, расположение и накопление во времени диагностического зонда позволяет идентифицировать степень и/или расположение иммунных клеток с активированным NLRP3 в качестве суррогатного биомаркера воспалительного заболевания пациента, и место воспаления внутри тела. Они также будут полезны для применения к биологическим образцам, извлеченным из тела, т.е. для диагностики in vitro.
[00209] Седьмой аспект изобретения заключается в способе модуляции активности биологической мишени, включающем стадию воздействия на биологическую мишень соединения формулы (I)-(VII) или его фармацевтически эффективной соли, сольвата или пролекарства.
[00210] Биологическая мишень может быть выбрана из группы, состоящей из инфламмасомы NLRP3, ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1αх, ИЛ-37, ИЛ-33 и клеток Th17.
[00211] Модуляция может быть такой, как описана ранее для аспектов от третьего до пятого.
[00212] При обычном использовании в данном контексте биологический образец может включать клетки, ткани, текучие среды, молекулы или другие биологические материалы, полученные, или которые можно получить, из млекопитающего. Неограничивающие примеры включают мочу, кровь и ее фракции, такие как сыворотка, плазма, лимфоциты и эритроциты, спинномозговую жидкость, мазок Папаниколау, носовые и глазные выделения, околоплодную жидкость, кал, сперму, биопсии тканей и/или органов и образцы нуклеиновых кислот (например, ДНК, РНК) или белка, хотя и без ограничения ими.
[00213] В следующей экспериментальной части более подробно описаны характеристики определенных соединений по настоящему изобретению и их эффективность. Целью является проиллюстрировать определенные конкретные варианты реализации соединений по настоящему изобретению и их эффективность без ограничения изобретения каким-либо образом.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Общие способы синтеза
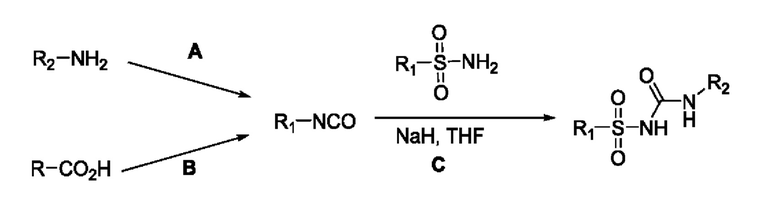
Способ А:
А1: К раствору полупродукта амина R2 (1 экв.) с основанием или без него, таким как, но не исключительно, триэтиламин (1,2 экв.) в безводном апротонном растворителе, таком как, но не исключительно, тетрагидрофуран или дихлорметан, добавили трифосген (от 0,4 до 1,1 экв.). Реакционную смесь перемешивали при температуре окружающей среды или, если необходимо, нагревали с обратным холодильником до завершения, как правило, от 2 до 18 ч.
А2: К ди-трет-бутилдикарбонату (1,2-1,4 экв.) в безводном ацетонитриле или ТГФ добавили ДМАП (15-100 мол. %), через 5 минут добавили раствор полупродукта амина R2 (1,0 экв.) в ацетонитриле. Реакционную смесь перемешивали в течение 30-60 мин при комнатной температуре.
Способ В:
В1: Полупродукт карбоновую кислоту R2 (1 экв.) растворили в апротонном растворителе, таком как толуол, с 2 каплями ДМФА или без него, и добавили хлорирующий агент, такой как тионилхлорид (2 экв.). Реакционную смесь нагревали с обратным холодильником до завершения, затем концентрировали при пониженном давлении. Получили соответствующий полупродукт хлорангидрид кислоты R2.
В данном случае в равной степени также пригодны альтернативные способы образования хлорангидрида кислоты, например, вышеупомянутую процедуру можно проводить без толуола и ДМФА, используя при этом тионилхлорид в качестве и растворителя, и хлорирующего агента.
Полупродукт хлорангидрид кислоты R2 растворили в ацетоне и добавили по каплям к раствору азида натрия (1,5 экв.) в растворе вода:ацетон (50:50) при 0°С. Добавили ледяную воду для осаждения полученного полупродукта ацилазида R2, который растворили в толуоле и высушили (MgSO4) с последующим добавлением раствора по каплям к безводному толуолу при кипячении с обратным холодильником, поддерживая при этом постоянный ток инертного газа. Реакционную смесь нагревали до завершения, как правило, 2 ч, с получением изоцианата R2.
В2: Хлорангидрид кислоты R2 (образованный, как указано в способе В1) в сухом CH2Cl2 добавили к NaN3 (2,0 экв.) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и экстрагировали в EtOAc. Органический слой промыли H2O (15 мл), высушили (MgSO4), и осторожно упарили с получением ацилазида. Ацилазид растворили в сухом толуоле и нагревали до 100°С в течение 2 ч. Растворитель удалили с получением технического изоцианата R2.
Способ С:
С1: Полупродукт сульфонамид R1 (1 экв.) растворили в безводном ТГФ и подействовали NaH (1 экв.) при пониженном давлении. Смесь нагревали с обратным холодильником в течение 2 ч, затем охладили до комнатной температуры и добавили полупродукт изоцианат R2 в ТГФ в атмосфере азота. Реакционную смесь перемешивали при кипячении с обратным холодильником до завершения.
С2: Полупродукт сульфонамид R1 (1 экв.) растворили в безводном ТГФ или безводном метаноле и подействовали NaH (1 экв.) при пониженном давлении. После прекращения выделения газа добавили полупродукт изоцианат R2 и реакционную смесь перемешивали при температуре окружающей среды на протяжении ночи.
С3: К полупродукт сульфонамиду R1 (1 экв.) в безводном ТГФ (5 мл/ммоль) добавили NaH (1 экв.) при 0°С и перемешивали в течение от 30 мин до 2 ч, или до завершения, при температуре окружающей среды в атмосфере азота. Снова охладили до 0°С, добавили изоцианат R2 (1,0 экв.) в ТГФ и перемешивали при температуре окружающей среды до завершения, как правило, от 2 до 16 ч.
С4: К техническому изоцианату R2 (1,0 экв.) в безводном ТГФ или ДХМ (5-11 мл/ммоль) добавили сульфонамид R1 (1,0 экв.), затем основание, такое как триэтиламин, DIPEA или DBU (1-2 экв.), и реакционную смесь перемешивали при температуре окружающей среды на протяжении ночи.
С5: К полупродукту сульфонамиду R1 (1 экв.) в безводном МеОН (5 мл/ммоль) добавили NaOMe (1 экв.) [альтернативно: раствор свежеприготовленного метилата натрия 1,0 ммоль/л (1 экв.) добавили к раствору сульфонамида R1 1,0 ммоль/л (1 экв.) в безводном метаноле]. Затем растворитель удалили при пониженном давлении. Соль суспендировали в безводном апротонном растворителе, таком как ацетонитрил или ТГФ, добавили изоцианат R2 (1,0 экв.) в безводном апротонном растворителе, таком как ацетонитрил или ТГФ и смесь перемешивали при температуре окружающей среды на протяжении ночи. Затем раствор нагревали с обратным холодильником до завершения, как правило, 90 мин.
С6: Сульфонамид R1 (1,0 экв.) растворили в безводном ТГФ в атмосфере азота. Добавили твердый метилат натрия (1,0 экв ммоль) одной порцией. Эту смесь перемешивали при температуре окружающей среды в течение 3 ч. Добавили по каплям раствор изоцианата R2 (1,17 экв.) в ТГФ. Реакционную смесь перемешивали при комнатной температуре на протяжении ночи.
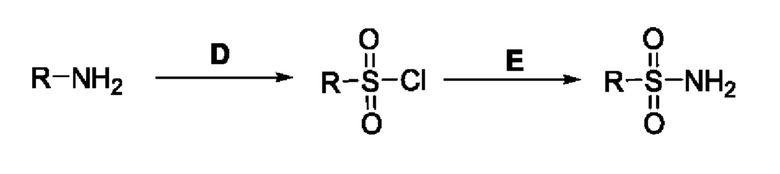
Способ D:
На раствор амина (1,0 экв.) в ацетонитриле (7-12 мл/ммоль) при 0°С подействовали конц. HCl (1,25-2,25 мл/ммоль) в H2O (0,5-1,2 мл/ммоль), а затем водным раствором NaNO2 (1,2 экв.), растворенным в H2O (0,3-0,5 мл/ммоль NaNO2). Полученный раствор перемешивали при 0°С в течение 45 мин. К вышеуказанной смеси последовательно добавили АсОН (0,5-1,2 мл/ммоль), CuCl2.2H2O (0,5 экв.) и CuCl (0,05 экв.) и продули газообразным SO2 в течение 20 мин при 0°С. Полученную реакционную смесь перемешивали при 0°С - 10°С до завершения.
Способ Е:
Е1: Раствор сульфонилхлорида (1 экв.) в ТГФ (10-20 мл/ммоль) охладили до -78°С и через раствор пропускали газообразный аммиак в течение 15 мин, перемешивание продолжали еще 30 мин, затем оставили прогреться до температуры окружающей среды и перемешивали в течение 2 ч или до завершения.
Е2: На раствор сульфонилхлорида (1 экв.) в ацетоне (20 мл/ммоль) подействовали раствором NH4HCO3 (4 экв.), растворенного в воде 1,5 мл/ммоль NH4HCO3) при температуре окружающей среды и перемешивали в течение 4 ч или до завершения.
Е3: На раствор сульфонилхлорида (1 экв.) в ацетоне (2,5 мл/ммоль) подействовали NH3 (3,5 мл/ммоль, NH4OH в H2O, на основе 28% NH3) при 0°С и перемешивали в течение 2 ч или до завершения.
Способ F
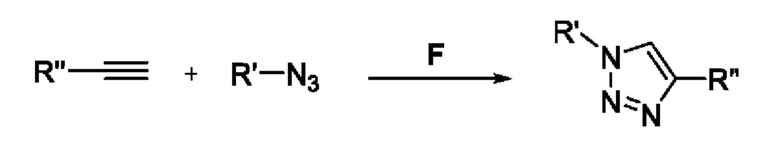
Общая методика для синтеза триазолов
Алкин (1 экв.) и азид (1,2 экв.), 5 мол .% CuSO4, 10 мол. % раствор NaAsc в ДМСО (500 мкл) перемешивали при комнатной температуре до завершения, как правило, 12 ч.
Синтез полупродуктов сульфонамидов R1:
Циклогексансульфонамид
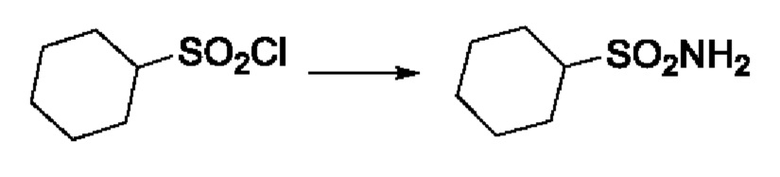
К раствору циклогексансульфонилхлорида (0,1 г, 0,54 ммоль) в ацетоне (1 мл) добавили водный NH3 (2 мл, 28% NH4OH в H2O) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение ~2 ч. Растворитель удалили при пониженном давлении и добавили МеОН/дихлорметан (1:9) (5 мл), побочный продукт NH4Cl удалили фильтрованием и оставшийся раствор концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 0,2% MeOH-CH2Cl2 с получением циклогексансульфонамида в виде белого с желтоватым оттенком твердого вещества (30 мг, 34%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=6,61 (ш с, 2Н), 2,76-2,70 (м, 1 Н), 2,09-2,04 (м, 2Н), 1,80-1,76 (м, 2Н), 1,65-1,60 (м, 1Н), 1,31-1,19 (м, 4Н), 1,16-1,06 (м, 1 Н).
Циклопентансульфонамид
К раствору циклопентансульфонилхлорида (0,1 г, 0,59 ммоль) в ацетоне (1 мл) добавили водный NH3 (1 мл, 28% NH4OH в H2O) при 0°С и реакционную смесь перемешивали при комнатной температуре в течение ~2 ч. Растворитель удалили при пониженном давлении и добавили МеОН/дихлорметан(1:9) (5 мл), побочный продукт NH4Cl удалили фильтрованием и оставшийся раствор концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 35% EtOAc-гексаны с получением циклопентансульфонамида в виде белого с желтоватым оттенком твердого вещества (72 мг, 81%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=6,69 (ш с, 2Н), 3,42-3,32 (м, 1Н), 1,89-1,84 (м, 4Н), 1,68-1,64 (м, 2Н), 1,61-1,52 (м, 2Н).
5-((диметиламино)метил)фуран-2-сульфонамид
Фуран-2-карбоновую кислоту (5 г, 44,6 ммоль) растворили в этаноле (100 мл), добавили конц. H2SO4 (1,0 мл) и раствор нагревали с обратным холодильником на протяжении ночи. Реакционную смесь концентрировали при пониженном давлении, затем распределили между этилацетатом (100 мл) и насыщенным NaHCO3 (100 мл). Органическую фазу промыли водой, затем солевым раствором, высушили (MgSO4) и концентрировали при пониженном давлении с получением этилфуран-2-карбоксилата (4,5 г, 80%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,57 (д, J=1,2 Гц, 1Н), 7,18 (д, J=3,5 Гц, 1Н), 6,51 (дд, J=3,5, 1,2 Гц, 1Н), 4,37 (кв, J=7,1 Гц, 2Н), 1,38 (т, J=7,1 Гц, 3Н).
Этилфуран-2-карбоксилат (9,0 г, 64,3 ммоль) растворили в дихлорметане (200 мл) и добавили хлорсульфоновую кислоту (7,5 г, 64,3 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 6 часов, или до завершения, затем добавили порциями пиридин (5,6 г, 70,7 ммоль) и PCl5 (14,7 г, 70,7 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов, затем разбавили ледяной водой и перемешивали в течение 30 мин. Смесь экстрагировали с помощью ДХМ и объединенные органические фазы промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт, этил-5-(хлорсульфонил)фуран-2-карбоксилат (7 г, 46%), использовали сразу без дополнительной очистки. 1Н-ЯМР (300 МГц, CDCl3) δ=7,33 (д, J=3,9 Гц, 1Н), 7,27 (д, J=3,9 Гц, 1 Н), 4,44 (кв, J=7,1 Гц, 1 Н), 1,42 (т, J=7,1 Гц, 2Н).
Технический этил-5-(хлорсульфонил)фуран-2-карбоксилат (7 г) преобразовали с использованием общего способа Е1 с получением этил-5-сульфамоилфуран-2-карбоксилата (5 г, 78%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ=8,05 (с, 2Н), 7,38 (д, J=3,7 Гц, 1 Н), 7,09 (д, J=3,7 Гц, 1Н), 4,32 (кв, J=7,1 Гц, 2Н), 1,29 (т, J=7,1 Гц, 3Н).
Этил 5-сульфамоилфуран-2-карбоксилат (2 г, 9,13 ммоль) в сухом ТГФ (40 мл) охладили до 0°С и добавили порциями алюмогидрид лития (1,05 г, 27,3 ммоль) на протяжении 30 мин. Реакционную смесь нагревали до 70°С в течение 4 часов. Реакционную смесь охладили до 0°С и с большой осторожностью добавили по каплям насыщенный NH4Cl (20 мл) на протяжении 30 мин. Реакционную смесь разбавили этилацетатом (100 мл) и профильтровали через слой целита. Органическую фазу промыли водой (100 мл), солевым раствором (100 мл), высушили (MgSO4) и концентрировали при пониженном давлении с получением 5-(гидроксиметил)фуран-2-сульфонамида (1,25 г, 78%) в виде светло-коричневой жидкости. 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,71 (с, 2Н), 6,88 (д, J=3,5 Гц, 1Н), 6,44 (д, J=3,4 Гц, 1Н), 5,47 (с, 1Н), 4,44 (д, J=5,7 Гц, 2Н), 3,36 (с, 7Н), 2,51 (кв, J=1,8 Гц, 5Н), 1,36 (с, 1Н).
5-(гидроксиметил)фуран-2-сульфонамид (0,3 г, 1,7 ммоль) в ТГФ (5 мл) охладили до 0°С и медленно добавили POCl3 (0,4 г, 2,54 ммоль). Реакционную смесь перемешивали при 75°С в течение 2 часов, затем охладили до температуры окружающей среды. Техническую смесь разделили между этилацетатом (50 мл) и насыщенн. водн. NaHCO3 (50 мл) и органическую фазу промыли водой (50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 30% EtOAc-гексаны с получением 5-(хлорметил)фуран-2-сульфонамида в виде светло-коричневого полутвердого вещества (0,25 г, 76%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ=7,85 (с, 2Н), 6,93 (дд, J=3,5, 1,3 Гц, 1Н), 6,69 (дд, J=3,5, 1,3 Гц, 1Н), 4,89 (д, J=1,3 Гц, 2Н).
5-(хлорметил)фуран-2-сульфонамид (0,4 г, 2,05 ммоль) в ТГФ (20 мл) охладили до 0°С, добавили конц. HCl (7,5 мг, 2,05 ммоль) и раствор перемешивали в течение 20 мин при этой же температуре. Добавили N,N-диметиламин в этаноле 5,6 моль/л (0,28 г, 6,15 ммоль, 3 экв.) при 0°С и герметизировали реакционный сосуд с последующим перемешиванием при комнатной температуре на протяжении ночи. Растворители удалили при пониженном давлении и азеотропной отгонкой с толуолом (×2) с получением 5-((диметиламино)метил)фуран-2-сульфонамида в виде смолы (0,25 г, 60%). Технический продукт использовали сразу без дополнительной очистки.
Фуран-2-сульфонамид
Фуран-2-сульфонилхлорид (0,30 г, 1,8 ммоль) добавили к водному аммиаку (1,0 мл) при 0°С и смесь перемешивали при температуре окружающей среды в течение 1 ч. По завершении реакции избыток водного аммиака удалили при пониженном давлении. Остаток подвергли азеотропной отгонке с изопропанолом и растерли с пентаном с получением указанного в подзаголовке соединения в виде светло-коричневого твердого вещества (0,21 г, 79%), 1Н-ЯМР (400 МГц, ДМСО-d6): 5=7,91 (с, 1 Н), 7,45 (ш.с, 2Н), 6,95 (д, J=3,6 Гц, 1Н), 6,63 (дд, J=2,8, 1,6 Гц, Н). ЖХ-МС 97,4% (ELSD (испарительный детектор светорассеяния)); m/z 146,11 [М-Н]+.
5-метил фуран-2-сульфонамид
К раствору 2-метилфурана (2,0 г, 24,3 ммоль) в безводном ацетонитриле (4 мл) добавили комплекс SO3⋅Py (5,0 г, 31,6 ммоль) и реакционную смесь нагревали при 40°С под азотом на протяжении ночи. Реакционную смесь разбавили EtOAc (5 мл) и перемешивали в течение 2 ч при 0°С, полученные осадки удалили фильтрованием и высушили с получением пиридиний 5-метилфуран-2-сульфоната в виде белого с желтоватым оттенком твердого вещества (2,93 г, 50%). 1Н-ЯМР (400 МГц, ДМСО): δ=8,90 (дд, J1=4 Гц, J2=8 Гц, 2Н), 8,57 (тт, J,=1,5 Гц, J2=8,1 Гц, 1Н), 8,04 (дд, J,=4 Гц, J2=8 Гц, 2Н), 6,27 (д, J=3,0 Гц, 1Н), 5,98-5,94 (м, 1Н), 2,19 (с, 3Н).
На суспензию пиридиний 5-метилфуран-2-сульфоната (1,0 г, 4,41 ммоль) в безводном ДМЭ) подействовали оксалилхлоридом (0,53 мл, 6,21 ммоль), затем ДМФА (0,32 мл, 4,41 ммоль) при 0°С под аргоном и реакционную смесь перемешивали при комнатной температуре до завершения. Реакционную смесь разбавили ледяной водой и экстрагировали толуолом (2×50 мл), объединенные органические фазы промыли водным насыщенным NaHCO3 (20 мл), солевым раствором (20 мл), высушили (MgSO4) и концентрировали при пониженном давлении с получением 5-метилфуран-2-сульфонилхлорида в виде бледно-желтого масла (350 мг, 47%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,23-7,21 (м, 1Н), 6,27-6,25 (м, 1Н), 2,47 (с, 3Н).
К раствору 5-метилфуран-2-сульфонилхлорида (0,2 г, 1,10 ммоль) в ацетоне (1 мл) добавили водн. NH3 (1 мл, 28% NH4OH в H2O) при 0 °С.Реакционную смесь перемешивали при температуре окружающей среды в течение ~2 ч, затем концентрировали при пониженном давлении. Остаток суспендировали в дихлорметане (5 мл), побочный продукт NH4Cl удалили фильтрованием и оставшийся раствор концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 40% EtOAc-гексаны с получением 5-метилфуран-2-сульфонамида в виде белого с желтоватым оттенком твердого вещества (130 мг, 73%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,60 (с, 2Н), 6,83-6,82 (д, J=4,0 Гц, 1Н), 6,26-6,25 (д, J=4,0 Гц, 1Н), 2,34 (с, 3Н).
5-этил-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульф онамид
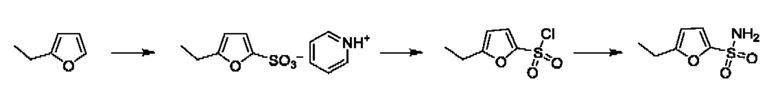
К раствору 2-этилфурана (2,0 г, 20,8 ммоль) в безводном ацетонитриле (3 мл) добавили комплекс SO3⋅Py (4,30 г, 27,0 ммоль). Полученную реакционную смесь нагревали при 40°С в атмосфере азота в течение 23 ч или до завершения. Добавили EtOAc (5 мл) и раствор перемешивали в течение 2 ч при 0°С. Полученный осадок удалили фильтрованием и высушили с получением пиридин-1-ий 5-этилфуран-2-сульфоната в виде коричневого гигроскопичного твердого вещества (3,2 г, 60%), который использовали сразу на следующей стадии без очистки.
К суспензии пиридиний 5-этилфуран-2-сульфоната (3,2 г, 12,5 ммоль) в ДМЭ (15 мл) добавили оксалилхлорид (1,62 мл, 27,0 ммоль) и затем ДМФА (0,97 мл, 12,5 ммоль) при 0°С в атмосфере аргона, полученную реакционную смесь перемешивали при комнатной температуре до завершения. Реакционную смесь разбавили ледяной водой и затем экстрагировали толуолом (2×50 мл), органический слой промыли водным насыщенным NaHCO3(20 мл) и солевым раствором (20 мл), высушили (MgSO4) и концентрировали при пониженном давлении с получением 5-этилфуран-2-сульфонилхлорида в виде светло-коричневого масла (510 мг, 21%). 1Н-ЯМР (400 МГц, CDCl3): δ 7,23 (д, J=4 Гц, 1Н), 6,26 (д, J=8 Гц, 1Н), 2,80 (кв, J=8 Гц, 2Н), 1,33 (т, J=8 Гц, 3Н).
К раствору 5-этилфуран-2-сульфонилхлорида в ацетоне (1 мл) добавили водный NH3 (1,5 мл, NH4OH в H2O, основа 28% NH3) при 0°С, полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч или до завершения. Растворитель удалили при пониженном давлении и азеотропной отгонкой с толуолом (×2). Остаток очистили колоночной хроматографией на силикагеле с использованием элюента 1% МеОН/ДХМ с получением 5-этилфуран-2-сульфонамида в виде смолы коричневого цвета (0,36 г, 78%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ 7,63 (шс, 2Н), 6,85 (д, J=4 Гц, 1Н), 6,28 (д, J=4 Гц, 1Н), 2,70 (кв, J=8 Гц, 2Н), 1,21 (т, J=6 Гц, 3Н).
4-(проп-1-ен-2-ил)фуран-2-сульфонамид
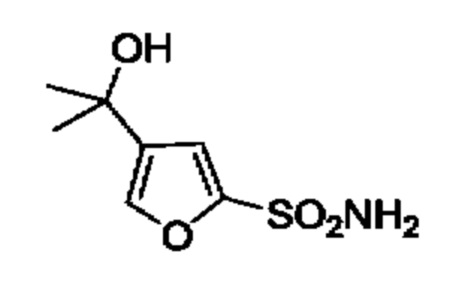
Синтез 4-(проп-1-ен-2-ил)фуран-2-сульфонамида осуществили из этилфуран-3-карбоксилата с использованием методик, подробно описанных Urban et.al. Synth.Commun. 2003, 33(12), 2029-2043, с получением указанного в подзаголовке соединения в виде белого твердого вещества со всеми спектральными данными, соответствующими указанной литературной ссылке.
d6-4-(проп-1-ен-2-ил)фуран-2-сульфонамид
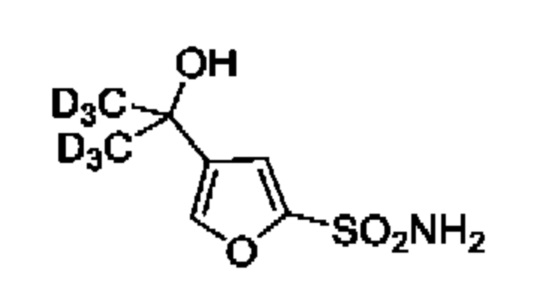
Модификация методик, приведенных в Urban et.al, Synth.Commun. 2003, 33(12), 2029-2043, с применением метил-d3-магниййодида вместо метилмагнийхлорида приводит к соответствующему d6-4-(проп-1-ен-2-ил)фуран-2-сульфонамиду.
4-(проп-1-ен-2-ил)фуран-2-сульфонамид
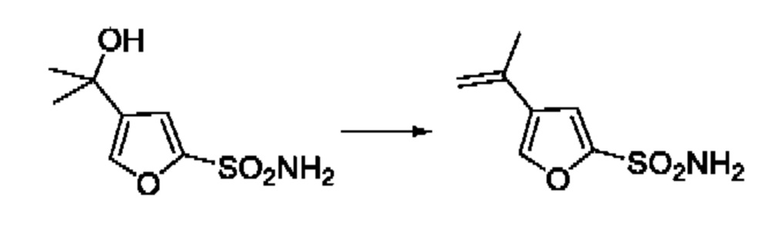
К раствору трифенилфосфина (0,3 г, 1,16 ммоль) в безводном ТГФ (5,0 мл) добавили йод (1,0 экв.) и смесь перемешивали при комнатной температуре в течение 10 мин. Медленно добавили раствор 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида в ТГФ (3,0 мл) и перемешивание продолжали в течение 2 ч или до завершения. Раствор разбавили EtOAc (20 мл), промыли 10% водн. бисульфитом натрия (20 мл), водой (20 мл). Органический слой отделили, высушили (MgSO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 20% EtOAc: гексаны с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,1 г, 58%). 1Н-ЯМР (400 МГц, CDCl3) δ=7,51 (с, 1Н), 7,16 (с, 1Н), 5,27 (с, 2Н), 2,02 (с, 3Н). 13С-ЯМР (101 МГц, CDCl3) δ 149,3, 140,7, 140,7, 132,3, 127,8, 112,1, 111,9, 76,0, 7,7, 28,7, 19,8.
4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид

К раствору этилового эфира 2-метил-3-фуранкарбоновой кислоты (30 г, 0,195 моль) в ДХМ (300 мл) при -10°С добавили хлорсульфоновую кислоту (23,8 г, 0,204 моль) по каплям в течение ~15 мин. Реакционную смесь оставили прогреться до температуры окружающей среды и перемешивали в течение 72 часов. Раствор охладили до -10°С и добавили по каплям безводный пиридин (16,9 г, 0,214 моль), затем добавили пентахлорид фосфора (44,6 г, 0,214 моль) порциями по ~10 г в течение 10 мин. Перемешивали при <0°С в течение 30 мин, затем перемешивали при температуре окружающей среды на протяжении ночи. Реакционную смесь добавили по каплям к воде (550 мл) при перемешивании и перемешивание продолжали в течение 2 часов. Органическую фазу отделили и водную фазу экстрагировали с помощью ДХМ (150 мл). Объединенные органические фазы промыли водой (300 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением 44 г темно-красного масла. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексан с получением этил-5-(хлорсульфонил)-2-метилфуран-3-карбоксилата в виде оранжевого масла (36 г, 73%). 1Н-ЯМР (400 МГц, CDCl3) δ=7,55 (с, 1Н), 4,63 (кв, J=7,2 Гц, 2Н), 2,75 (с, 3Н), 1,38 (т, J=7,2 Гц, 3Н).
Этил-5-(хлорсульфонил)-2-метилфуран-3-карбоксилат (30 г, 0,12 моль) в ацетоне (200 мл) добавили по каплям в течение 15 мин к раствору бикарбоната аммония (37,6 г, 0,475 моль) в воде (630 мл). Реакционную смесь перемешивали при температуре окружающей среды до завершения (~3 ч). Добавили EtOAc (250 мл) и довели рН с помощью добавления по каплям конц. HCl до рН~2. Органическую фазу отделили, а оставшуюся водную фазу насытили хлоридом натрия и повторно экстрагировали с помощью EtOAc (250 мл). Объединенные органические фазы промыли солевым раствором (300 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением коричневого маслянистого твердого вещества, которое перекристаллизовали с использованием EtOAc-гексана с получением этил-2-метил-5-сульфамоилфуран-3-карбоксилата в виде бежевого твердого вещества (11,4 г, 41%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=7,8 (с, 2Н), 7,02 (с, 1Н), 4,26 (кв, J=7,2 Гц, 2Н), 2,62 (с, 3Н), 1,3 (т, J=7,2 Гц, 3Н).
На этил-2-метил-5-сульфамоилфуран-3-карбоксилат (10 г, 0,043 моль) в безводном ТГФ (400 мл) при -10°С подействовали раствором метилмагнийхлорида (3,0 моль/л в ТГФ, 64,3 мл) по каплям в течение 5 минут при интенсивном перемешивании. Затем раствор перемешивали при температуре окружающей среды в течение 6 часов, затем охладили до -5°С и подействовали по каплям раствором хлорида аммония (51,8 г в 265 мл воды). Водный раствор экстрагировали с использованием EtOAc (2×250 мл), объединенные органические фазы промыли солевым раствором (250 мл), высушили (Na2SO4) и концентрировали при пониженном давлении до оранжевого масла (10 г). Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 40% EtOAc-гексан с получением указанного в подзаголовке соединения в виде белого твердого вещества (6,1 г, 42%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=7,54 (ш.с, 2Н), 6,78 (с, 1Н), 4,95 (с, 1Н), 2,42 (с, 3Н), 1,4 (с, 6Н).
(d6-4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид
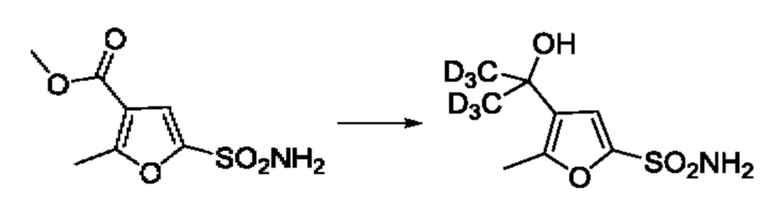
Метил-2-метил-5-сульфамоилфуран-3-карбоксилат можно получить модификацией методик, применяемых для синтеза этил-2-метил-5-сульфамоилфуран-3-карбоксилата, но с применением в качестве исходного материала метил-2-метилфуран-3-карбоксилата вместо этил-2-метилфуран-3-карбоксилата.
Метил-2-метил-5-сульфамоилфуран-3-карбоксилат получили в виде белого твердого вещества (3 г, 29%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=7,89 (с, 2Н), 7,03 (с, 1Н), 3,79 (с, 3Н), 2,61 (с, 3Н).
На метил-2-метил-5-сульфамоилфуран-3-карбоксилат (0,7 г, 3,2 ммоль) в безводном ТГФ (20 мл) при -10°С подействовали раствором с/з-метилмагниййодида (1,0 моль/л в Et2O, 26 мл) по каплям в течение 10 минут при интенсивном перемешивании. Затем раствор перемешивали при температуре окружающей среды в течение 12 ч, затем охладили до 0°С и подействовали по каплям насыщенным раствором хлорида аммония. Водный раствор экстрагировали с использованием EtOAc (2×25 мл), объединенные органические фазы промыли солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента градиента 40-70% EtOAc-гексан с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,37 г, 51%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=7,57 (с, 2Н), 6,79 (с, 1Н), 4,99 (с, 1Н), 2,4 (с, 3Н). 13С-ЯМР (100 МГц, CD3OD) δ=150,3, 147,3, 128,4, 113,4, 67,3, 28,5 (мультиплет), 12,2.
1-бензил-1Н-1,2,4-триазол-3-сульфонамид
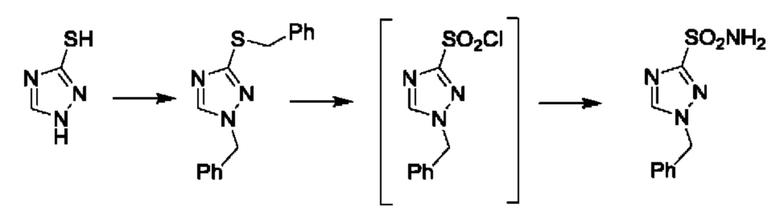
На раствор 1Н-1,2,4-триазол-3-тиола (1 г, 9,90 ммоль) в ДМФА (20 мл) подействовали K2CO3 (4,8 г, 34,7 ммоль), охладили до 0°С, затем добавили по каплям бензилбромид (4,2 г, 24,8 ммоль) в течение 5 мин. Полученную реакционную смесь прогрели до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь разбавили водой (25 мл) и экстрагировали этилацетатом (2×25 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 20% EtOAc-гексаны с получением 1-бензил-3-(бензилтио)-1Н-1,2,4-триазола в виде белого твердого вещества (1,5 г, 54%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,67 (с, 1Н), 7,39-7,32 (м, 5Н), 7,27-7,21 (м, 5Н), 5,36 (с, 2Н), 4,29 (с, 2Н).
К раствору 1-бензил-3-(бензилтио)-1Н-1,2,4-триазола, 2 (0,5 г, 1,77 ммоль) в ацетонитриле (5 мл) при 0°С добавили АсОН (3 мл) и затем H2O (2 мл) и через раствор пропускали газообразный Cl2 в течение 45 мин. Перемешивание продолжали при 0°С в течение 30 мин, затем при 20°С в течение 1,5 ч. Реакционную смесь разбавили водой (20 мл) и экстрагировали EtOAc (2×20 мл).
Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением бесцветной жидкости. Остаток разбавили ТГФ и охладили до -78°С. Через раствор пропускали газообразный аммиак в течение 20 мин и перемешивание продолжали в течение еще 30 мин с последующим прогреванием до температуры окружающей среды и перемешиванием в течение 1 ч. Реакционную смесь разбавили водой (25 мл) и экстрагировали этилацетатом (2×25 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Остаток растерли с диэтиловым эфиром с получением 1-бензил-1Н-1,2,4-триазол-3-сульфонамида в виде белого с желтоватым оттенком твердого вещества (0,25 г, 60%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,88 (с, 1 Н), 7,77 (с, 2Н), 7,39-7,33 (м, 5Н), 5,45 (с, 2Н).
1-изопропил-1Н-1,2,3-триазол-4-сульфонамид
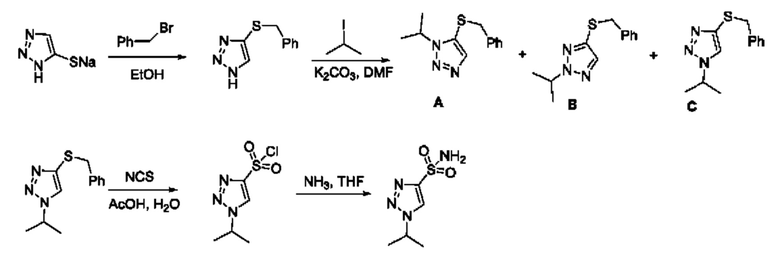
1Н-1,2,3-триазол-5-тиолят натрия (500 мг, 4,06 ммоль) растворили в EtOH (5 мл) и охладили до 0°С. Добавили по каплям бензилбромид (0,69 г, 4,06 ммоль) на протяжении 5 мин. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 1 ч. По завершении реакционную смесь концентрировали при пониженном давлении и полученный остаток разбавили насыщенным раствором NaHCO3 и экстрагировали EtOAc (2×20 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток перемешали с н-пентаном (30 мл), отфильтровали и высушили в вакууме с получением 4-(бензилтио)-1H-1,2,3-триазола в виде белого твердого вещества (0,7 г, 90%), который использовали без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3): δ=7,40-7,38 (м, 1Н), 7,35-7,21 (м, 5Н), 4,12 (с, 2Н). ЖХМС (m/z): 192,0 [М+Н]+
Раствор 4-(бензилтио)-1H-1,2,3-триазола (5 г, 26,1 ммоль) в ДМФА (50 мл) охладили до 0°С и подействовали на него K2CO3 (9,03 г, 65,4 ммоль). Реакционную смесь перемешивали в течение 5 минут при той же температуре. К описанной выше смеси добавили по каплям изопропилйодид (8,89 г, 52,3 ммоль) в течение 5 мин. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 2 ч. По завершении реакционную смесь разбавили водой (30 мл) и экстрагировали этилацетатом (50 мл). Органический экстракт промыли водой, солевым раствором и высушили над безводным Na2SO4. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 8% EtOAc-гексан с получением 5-(бензилтио)-1-изопропил-1Н-1,2,3-триазола А (0,9 г),
4-(бензилтио)-2-изопропил-2Н-1,2,3-триазола В (1 г) и желаемого продукта 4-(бензилтио)-1-изопропил-1Н-1,2,3-триазола С (1,4 г, 23%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,29-7,18 (м, 5Н), 4,78-4,71 (м, 1Н), 4,09 (с, 2Н), 1,4 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 234,30 [М+Н]+.
Раствор 4-(бензилтио)-1-изопропил-1Н-1,2,3-триазола (75 мг, 0,32 ммоль) в уксусной кислоте (2,25 мл) и H2O (1,12 мл) охладили до 0°С.Добавили N-хлорсукцинамид (170 мг, 1,28 ммоль) при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 1 ч. По завершении реакционную смесь разбавили водой и экстрагировали этилацетатом (2×10 мл). Объединенные органические экстракты промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 8% EtOAc-гексаны с получением 1-изопропил-1Н-1,2,3-триазол-4-сульфонилхлорида (0,1 г, 100%) в виде светло-коричневой жидкости, использованной без дополнительной очистки. ЖХМС (m/z): 210,10 [М+Н]+.
Раствор 1-изопропил-1Н-1,2,3-триазол-4-сульфонилхлорида (100 мг) в ТГФ (5 мл) охладили до -40°С. Через вышеупомянутый раствор пропускали газообразный аммиак в течение 15 мин. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 2 ч. По завершении реакционную смесь концентрировали при пониженном давлении и полученный остаток разбавили этилацетатом (25 мл) и водой (10 мл). Органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1-изопропил-1H-1,2,3-триазол-4-сульфонамида (0,07 г, 78%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,71 (с, 1Н), 7,66 (с, 2Н), 4,91-4,87 (м, 1Н), 1,5 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 191,30 [М+Н]+.
1-метил-1Н-пиразол-3-сульфонамид
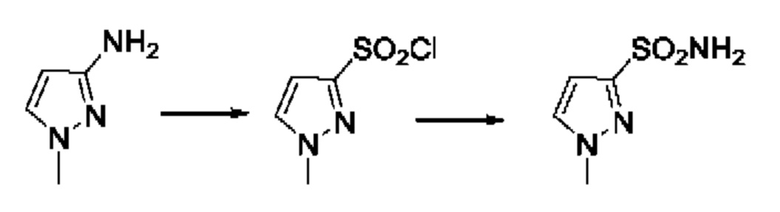
Гидрохлорид 1-метил-1Н-пиразол-3-амина преобразовали в 1-метил-1Н-пиразол-3-сульфонилхлорид, бледно-желтую жидкость, с использованием общего способа D (0,7 г, 38%). 1Н-ЯМР (300 МГц, CDCl3): δ=7,51-7,50 (д, J=2,1 Гц, 1Н), 6,89-6,88 (д, J=2,4 Гц, 1Н), 4,06 (с, 3Н). ЖХМС (m/z): 160,9 (М-1)-. Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,4 г, 69%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,80 (д, J=2,1 Гц, 1Н), 7,36 (с, 2Н), 6,53 (д, J=2,1 Гц, 1Н), 3,88 (с, 3Н). ЖХМС (m/z): 162,05 (М+1)+.
1-(трифторметил)-1Н-пиразол-3-сульфонамид
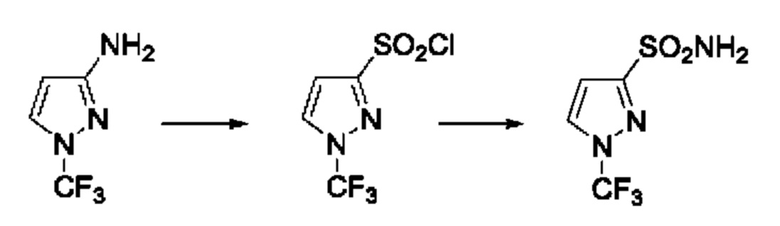
1-(трифторметил)-1Н-пиразол-3-амин преобразовали в 1-(трифторметил)-1Н-пиразол-3-сульфонилхлорид, коричневую жидкость, с использованием общего способа D (0,4 г, 43%). 1Н-ЯМР (300 МГц, CDCl3) δ=8,02 (д, J=2,8 Гц, 1Н), 7,06 (д, J=2,8 Гц, 1Н). 19F-ЯМР (282 МГц, CDCl3) δ=-60,46.
Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения (0,22 г, 46%). 1Н-ЯМР (300 МГц, CDCl3) 5=7,92 (дд, J=2,8, 0,3 Гц, 1Н), 6,91 (дд, J=2,8, 0,7 Гц, 1Н), 5,28 (с, 2Н). 19F-ЯМР (282 МГц, CDCl3) δ=-60,41.
1-изопропил-1Н-пиразол-3-сульфонамид
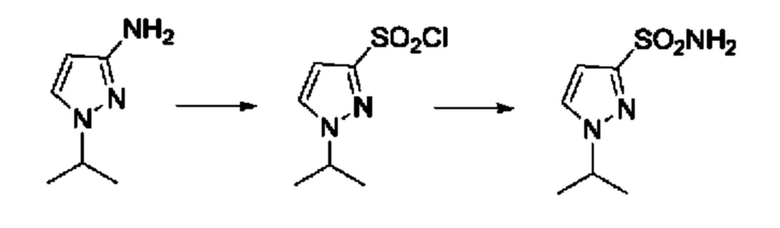
1-Изопропил-1Н-пиразол-3-амин преобразовали в 1-изопропил-1H-пиразол-3-сульфонилхлорид, коричневую жидкость, с использованием общего способа D (0,5 г, 43%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,55 (с, 1Н), 6,88 (с, 1Н), 4,66-4,63 (м, 1Н), 3,6 (ш.с, 2Н), 1,59 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 209,0 (М+1)+. Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде желтого твердого вещества (0,45 г, 82%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=7,9 (д, J=2,4 Гц, 1 Н), 7,36 (с, 2Н), 6,55 (д, J=2,1 Гц, 1Н), 4,57-4,53 (м, 1Н), 1,42 (д, J=6,9 Гц, 6Н). ЖХМС (m/z): 190,0 (М+1)+.
1-изопропил-1Н-пиразол-4-сульфонамид
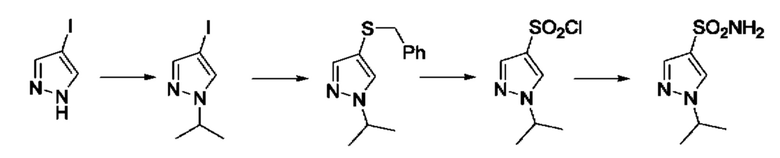
На раствор 4-йод-1H-пиразола (1 г, 5,15 ммоль) в ДМФА (20 мл) подействовали K2CO3 (1,42 г, 10,30 ммоль) и изопропилйодидом (1,05 г, 6,19 ммоль) при температуре окружающей среды в атмосфере азота. Полученную реакционную смесь нагрели до 90°С и перемешивали в течение 12 ч. Смесь охладили, разбавили водой (50 мл) и экстрагировали диэтиловым эфиром (2×50 мл). Объединенные органические фазы промыли водой (2×50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением 4-йод-1-изопропил-1Н-пиразола в виде бесцветной жидкости (1,1 г, 92%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,50-7,46 (м, 2Н), 4,53-4,47 (м, 1Н), 1,50 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 237,2 (М+1)+.
На раствор 4-йод-1-изопропил-1Н-пиразола (1 г, 4,24 ммоль) в диоксане (20 мл) подействовали последовательно бензилмеркаптаном (0,8 г, 6,35 ммоль) и DIPEA (1,1 г, 8,47 ммоль) в атмосфере азота. Раствор дегазировали посредством продувки газообразным аргоном в течение 15 мин. Добавили Pd2(dba)3 (40 мг, 0,0423 ммоль) и Ксантфос (50 мг, 0,0847 ммоль) в атмосфере аргона, затем полученную смесь загерметизировали в реакционном сосуде и нагревали при 75°С в течение 6 ч. Реакционную смесь охладили, концентрировали при пониженном давлении, разбавили водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические фазы промыли водой (2×50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением 4-(бензилтио)-1-изопропил-1Н-пиразола в виде желтой жидкости (650 мг, 66%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,36 (с, 1Н), 7,26-7,22 (м, 4Н), 7,11-7,09 (м, 2Н), 4,41-4,36 (м, 1Н), 3,76 (с, 2Н), 1,42 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 233,3 (М+1)+.
К раствору 4-(бензилтио)-1-изопропил-1H-пиразола, 3 (0,35 г, 1,508 ммоль) в ацетонитриле (10 мл) при 0°С добавили АсОН (0,7 мл) и H2O (0,35 мл), затем добавили порциями DCDMH (0,6 г, 3,017 ммоль) в течение 5 мин. Раствор перемешивали в течение 30 мин, затем прогрели до температуры окружающей среды и перемешивали в течение еще 2 ч. Реакционную смесь разбавили водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1-изопропил-1Н-пиразол-4-сульфонилхлорида в виде бесцветной жидкости. Сульфонилхлорид разбавили ТГФ и охладили до -78°С, затем через раствор пропускали газообразный NH3 в течение 15 минут. Реакционную смесь перемешивали при -78°С в течение 1 ч и при температуре окружающей среды в течение 2 ч. Реакционную смесь разбавили водой и соединение экстрагировали этилацетатом (2×25 мл). Объединенные органические экстракты промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток растерли с диэтиловым эфиром и высушили при пониженном давлении с получением 1-изопропил-1Н-пиразол-4-сульфонамида в виде светло-коричневого твердого вещества (0,2 г, 71%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,21 (с, 1Н), 7,71 (с, 1Н), 7,22 (с, 2Н), 4,59-4,53 (м, 1Н), 1,4 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 190,2 (М+1)+.
1-циклопропил-1Н-пиразол-3-сульфонамид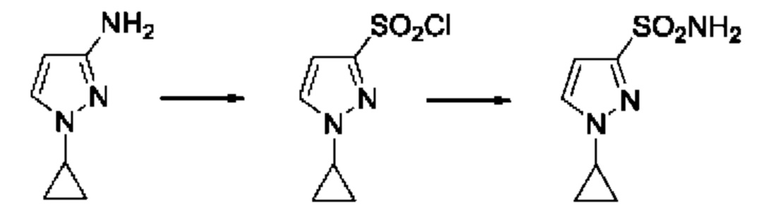
1-Циклопропил-1Н-пиразол-3-амин преобразовали в 1-циклопропил-1Н-пиразол-3-сульфонилхлорид с использованием общего способа D, затем преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде светло-коричневого твердого вещества (0,2 г, 33%). 1Н-ЯМР (400 МГц, CDCl3) δ=7,51 (д, J=2,4 Гц, 1Н), 6,69 (д, J=2,4 Гц, 1Н), 5,04 (с, 2Н), 3,67 (м, 1Н), 1,28-1,05 (м, 4Н).
1-(трет-бутил)-1Н-пиразол-3-сульфонамид
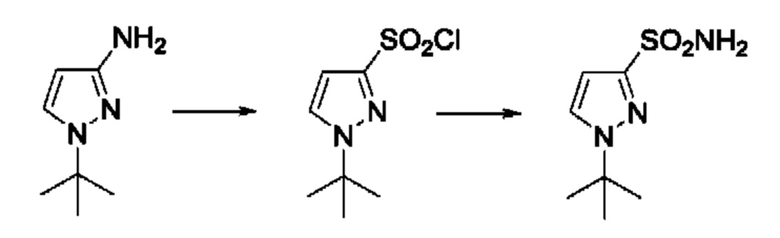
1-(трет-бутил)-1Н-пиразол-3-амин преобразовали в 1-(трет-бутил)-1Н-пиразол-3-сульфонилхлорид с использованием общего способа D, затем преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде светло-коричневого твердого вещества (150 мг, 26%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,56 (д, J=3,6 Гц, 1Н), 6,7 (д, J=3,2 Гц, 1Н), 4,75 (ш.с, 1Н), 1,60 (с, 9Н). ЖХМС (m/z): 204,15 (М+1)+.
1-циклогексил-1Н-пиразол-3-сульфонамид
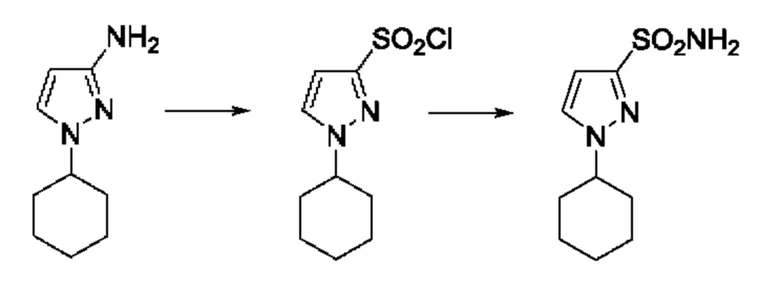
1-Циклогексил-1Н-пиразол-3-амин преобразовали в 1-циклогексил-1Н-пиразол-3-сульфонилхлорид с использованием общего способа D, затем преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,35 мг, 50%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ=7,89 (д, J=2,3 Гц, 1Н), 7,36 (с, 2Н), 6,55 (д, J=2,3 Гц, 1Н), 4,28-4,08 (м, 1 Н), 2,0-1,1 (м, 6Н).
1-фенил-1Н-пиразол-3-сульфонамид
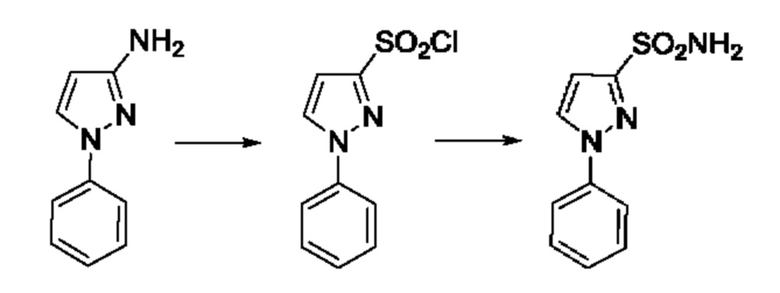
1-фенил-1 Н-пиразол-3-амин преобразовали в 1-фенил-1Н-пиразол-3-сульфонилхлорид, желтую жидкость, с использованием общего способа D (0,5 г, 47%). 1Н-ЯМР (400 МГц, CDCl3): δ=8,04 (д, J=2,4 Гц, 1Н), 7,73 (д, J=9,2 Гц, 2Н), 7,58 (т, J=7,6 Гц, 2Н), 7,47 (т, J=7,2 Гц, 1Н), 7,08 (д, J=2,8 Гц, 1Н). Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде желтого твердого вещества (0,4 г, 87%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,62 (д, J=2,7 Гц, 1Н), 7,86 (д, J=8,7 Гц, 2Н), 7,61 (ш.с, 2Н), 7,57 (т, J=7,8 Гц, 2Н), 7,41 (т, J=7,2 Гц, 1Н), 6,85 (д, J=2,4 Гц, 1Н). ЖХМС (m/z): 224,1 (М+1)+.
1-бензил-1Н-пиразол -3-сульфонилхлорид
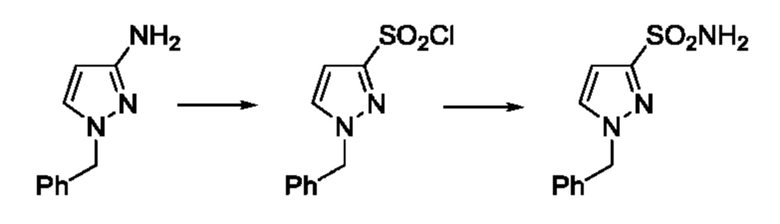
1-бензил-1Н-пиразол-3-амин преобразовали в 1-бензил-1Н-пиразол-3-сульфонилхлорид, светло-коричневую жидкость, с использованием общего способа D (0,2 г, 45%). 1Н-ЯМР (300 МГц, CDCl3): δ 7,42-7,38 (м, 3Н), 7,33-7,28 (м, 3Н), 6,8 (д, J=2,4 Гц, 1Н), 5,42 (с, 2Н). Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде светло-коричневой жидкости (0,15 г, 81%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,42-7,36 (м, 4Н), 7,24 (д, J=1,6 Гц, 2Н), 6,7 (д, J=2,4 Гц, 1Н), 5,35 (с, 2Н), 5,10 (с, 2Н). ЖХМС (m/z): 238,10 (М+1)+.
1-(1-фенилэтил)-1Н-пиразол-3-сульфонамид
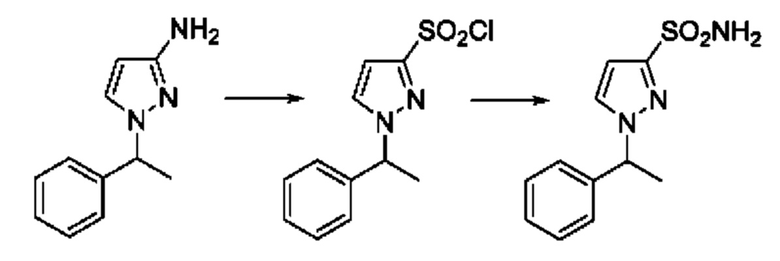
1-(1-фенилэтил)-1Н-пиразол--амин преобразовали в 1-(1-фенилэтил)-1Н-пиразол-3-сульфонилхлорид с использованием общего способа D, затем преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,25 мг, 68%). 1Н-ЯМР (300 МГц, CDCl3) δ=7,43-7,18 (м, 6Н), 6,72 (д, J=2,4 Гц, 1Н), 5,57 (кв, J=7,1 Гц, 1Н), 5,02 (с, 2Н), 1,92 (д, J=7,1 Гц, 3Н).
1-(2-(пиперидин-1-ил)этил)-1Н-пиразол-3-сульфонамид
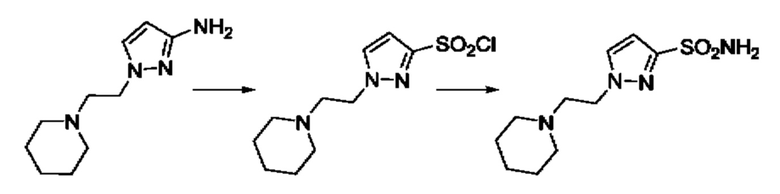
1-(2-(пиперидин-1-ил)этил)-1Н-пиразол-3-амин преобразовали в 1-(2-(пиперидин-1-ил)этил)-1Н-пиразол-3-сульфонилхлорид, светло-коричневую жидкость, с использованием общего способа D, затем преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,3 г, 46%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=7,84 (д, J=2,1 Гц, 1Н), 7,36 (с, 2Н), 6,54 (с, J=2,4 Гц, 1Н), 4,26 (т, J=6,9 Гц, 2Н), 2,66 (т, J=6,6 Гц, 2Н), 2,36 (с, 4Н), 1,46-1,34 (м, 6Н). ЖХМС (m/z): 259,10 (М+1)+.
1,5-диметил-1Н-пиразол-3-сул ьфонамид

1,5-диметил-1Н-пиразол-3-амин преобразовали в 1,5-диметил-1Н-пиразол-3-сульфонилхлорид, желтую жидкость, с использованием общего способа D (0,45 г, 26%). 1Н-ЯМР (300 МГц, CDCl3): δ=5,92 (с, 1Н), 3,71 (с, 3Н), 2,23 (с, 3Н). ЖХМС (m/z): 217 (М+Na)+. Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,25 г, 55%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,30 (с, 2Н), 6,36 (с, 1Н), 3,76 (с, 3Н), 2,27 (с, 3Н). ЖХМС (m/z): 175,9 (М+1)+.
1-метил-5-(трифторметил)-1Н-пиразол-3-сульфонамид
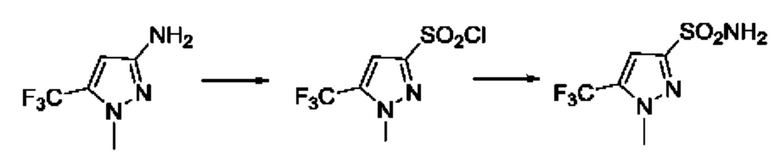
1-метил-5-(трифторметил)-1Н-пиразол-3-амин преобразовали в 1-метил-5-(трифторметил)-1Н-пиразол-3-сульфонилхлорид, светло-коричневую жидкость, с использованием общего способа D (1,1 г, 37%). 1Н-ЯМР (300 МГц, CDCl3): 5=7,21 (с, 1Н), 4,16 (с, 3Н). Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде желтого твердого вещества (0,45 мг, 82%). 1Н-ЯМР (300 МГц, CDCl3): δ=7,06 (с, 1Н), 5,02 (ш.с, 2Н), 4,03 (с, 3Н).
1-изопропил-5-(трифторметил)-1Н-пиразол-3-сульфонамид
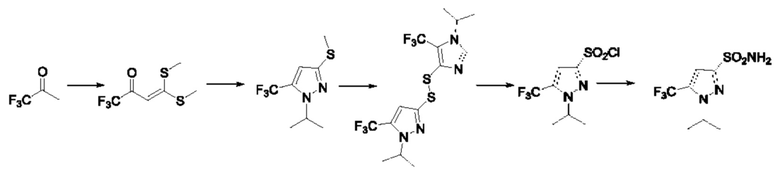
Смесь NaH (2,14 г, 89,3 ммоль) в ДМФА (20 мл) охладили до -10°С. К указанной смеси очень осторожно добавили раствор 1,1,1-трифторпропан-2-она (5 г, 44,6 ммоль) в ДМФА (80 мл) и перемешивали при -10°С в течение 5 мин. К описанной выше смеси добавили по каплям CS2 (10,2 г, 133,9 ммоль) в течение 30 мин, затем реакционную смесь прогрели до температуры окружающей среды и перемешивали в течение 1 ч. Реакционную смесь охладили до 0°С и подействовали CH3 (7,5 мл) в течение 10 мин. Полученную реакционную смесь прогрели до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь разбавили холодной водой (50 мл) и экстрагировали диэтиловым эфиром (2×100 мл). Объединенные органические экстракты промыли водой (50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% EtOAc-гексаны с получением 1,1,1-трифтор-4,4-бис(метилтио)бут-3-ен-2-она в виде светло-коричневого твердого вещества (3,5 г, 36%). 1Н-ЯМР (300 МГц, CDCl3): δ=6,24 (с, 1Н), 2,57 (м, 6Н). ЖХМС (m/z): 217,20 (М+1)+.
На раствор 1,1,1-трифтор-4,4-бис(метилтио)бут-3-ен-2-она (2,5 г, 11,6 ммоль) в EtOH (25 мл) подействовали гидрохлоридом изопропилгидразина (2 г, 13,9 ммоль) при 0°С, добавили Et3N (2,4 г, 40,98 ммоль) и смесь нагревали при 80°С в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении, разбавили насыщенн. водн. раствором NaHCO3 и экстрагировали EtOAc (2×250 мл). Объединенные органические фазы промыли водой (200 мл), солевым раствором (200 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 100% EtOAc с получением 1-изопропил-3-(метилтио)-5-(трифторметил)-1Н-пиразола в виде светло-коричневой жидкости (1,5 г, 58%). 1Н-ЯМР (400 МГц, CDCl3): δ=6,47 (с, 1Н), 4,58-4,53 (м, 1Н), 2,49 (с, 3Н), 1,50 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 225,20 (М+1)+.
На раствор 1-изопропил-3-(метилтио)-5-(трифторметил)-1H-пиразола (0,5 г, 2,23 ммоль) в хлороформе (10 мл) при 0°С подействовали mCPBA (0,38 г, 2,23 ммоль) и перемешивали при 10°С в течение 1 ч. Реакционную смесь разбавили насыщенным раствором NaHCO3 (10 мл) и экстрагировали CHCl3 (2×30 мл). Объединенные органические фазы промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток растворили в CHCl3 (10 мл) и подействовали трифторуксусным ангидридом (1,4 г, 6,7 ммоль), реакционную смесь нагревали при 50°С в течение 3 ч, охладили до температуры окружающей среды и концентрировали при пониженном давлении. Полученный остаток разбавили МеОН (5 мл)-ТГФ (5 мл)-H2O (5 мл), охладили до 0°С, подействовали Na2CO3 (0,7 г, 6,7 ммоль) и перемешивали в течение 3 ч. Раствор разбавили водой (30 мл) и экстрагировали CHCl3 (2×50 мл). Объединенные органические фазы промыли водой (50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический остаток (0,2 г), содержащий 1-изопропил-3-((1-изопропил-5-(трифторметил)-1Н-имидазол-4-ил)дисульфанил)-5-(трифторметил)-1Н-пиразол, использовали на следующей стадии без дополнительной очистки. 1Н-ЯМР (400 МГц, CDCl3): δ=6,85 (с, 2Н), 6,70 (с, 1Н), 6,60 (с, 1Н), 4,6 (м, 2Н), 1,53 (м, 6Н). ЖХМС (m/z): 416,75 (М-1)-.
Раствор технического 1-изопропил-3-((1-изопропил-5-(трифторметил)-1Н-имидазол-4-ил)дисульфанил)-5-(трифторметил)-1Н-пиразола (0,2 г технического, 0,478 ммоль) в ацетонитриле (10 мл) охладили до 0°С и подействовали на него АсОН (1 мл) и H2O (1,5 мл). Добавили порциями DCDMH (0,19 г, 0,956 ммоль) в течение 5 минут и перемешивали в течение 2 ч. Смесь разбавили водой (20 мл) и экстрагировали ДХМ (2×20 мл). Объединенные органические фазы промыли водой (50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1-изопропил-5-(трифторметил)-1H-пиразол-3-сульфонилхлорида в виде бесцветной жидкости.
(1-изопропил-5-(трифторметил)-1Н-пиразол-3-сульфонилхлорид) разбавили ТГФ, охладили до -78°С и через раствор пропускали газообразный NH3 в течение 10 мин, затем перемешивали в течение 1 ч с последующим прогреванием до температуры окружающей среды и перемешиванием в течение еще 1 ч. Реакционную смесь разбавили водой и экстрагировали этилацетатом (2×25 мл). Объединенные органические фазы промыли водой (50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток растерли с диэтиловым эфиром и н-пентаном с получением 1-изопропил-5-(трифторметил)-1Н-пиразол-3-сульфонамида в виде белого твердого вещества (75 мг, 61%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,01 (с, 1Н), 5,06 (с, 2Н), 4,73-4,70 (м, 1Н), 1,5 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 256,0 (М-1)-.
5-изопропил -1-метил -1Н-пиразол -3-сульфонамид
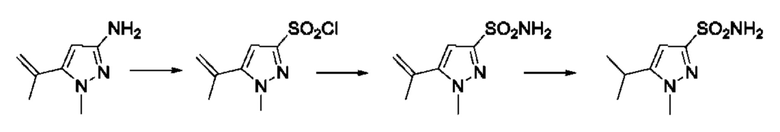
На раствор 1-метил-5-(проп-1-ен-2-ил)-1Н-пиразол-3-амина (0,25 г, 1,824 ммоль) в ацетонитриле (10 мл) при 0°С подействовали конц. HCl (1,2 мл) в H2O (0,5 мл), а затем водным раствором NaNO2 (0,15 г, 2,19 ммоль), растворенным в H2O (2 мл). Полученный раствор перемешивали при 0°С в течение 45 мин. К описанной выше смеси последовательно добавили АсОН (0,25 мл), CuCl2⋅2H2O (0,15 г, 0,91 ммоль) и CuCl (10 мг, 0,091 ммоль) и продували газообразным SO2 в течение 20 мин при 0°С. Полученную реакционную смесь перемешивали при 0°С - 10°С в течение 60 мин. По завершении реакционную смесь разбавили водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 20% EtOAc-гексаны с получением 1-метил-5-(проп-1-ен-2-ил)-1Н-пиразол-3-сульфонилхлорида в виде бесцветной жидкости (0,15 г, 38%). 1Н-ЯМР (300 МГц, CDCl3): δ=6,77 (с, 1Н), 5,51 (с, 1Н), 5,28 (с, 1Н), 4,02 (с, 3Н), 2,11 (с, 3Н).
Раствор 1-метил-5-(проп-1-ен-2-ил)-1Н-пиразол-3-сульфонилхлорида (0,075 г, 0,34 ммоль) в ТГФ (7 мл) охладили до -78°С и через раствор пропускали газообразный аммиак в течение 15 мин, перемешивание продолжали в течение еще 30 мин, затем оставили прогреться до температуры окружающей среды и перемешивали в течение 2 ч или до завершения. Реакционную смесь разбавили этилацетатом (25 мл) и профильтровали через слой целита. Фильтрат высушили (Na2SO4) и концентрировали при пониженном давлении с получением 0,04 г (технического) 1-метил-5-(проп-1-ен-2-ил)-1Н-пиразол-3-сульфонамида в виде белого с желтоватым оттенком твердого вещества, использованного без очистки.
На раствор технического 1-метил-5-(проп-1-ен-2-ил)-1Н-пиразол-3-сульфонамида (0,12 г, 0,6 ммоль) в МеОН (10 мл) - EtOAc (4 мл) подействовали 10% палладия на угле (30 мг) в атмосфере азота. Реакционную колбу вакуумировали, заполнили водородом (надувной шар) и перемешивали в течение 4 ч. Реакционную смесь разбавили этилацетатом (25 мл), профильтровали через слой целита, высушили (Na2SO4) и концентрировали при пониженном давлении. Полученное твердое вещество дополнительно промыли диэтиловым эфиром с получением 5-изопропил-1-метил-1Н-пиразол-3-сульфонамида в виде белого с желтоватым оттенком твердого вещества (0,11 г, 91%). 1Н-ЯМР (400 МГц, CDCl3): δ=6,50 (с, 1 Н), 5,00 (ш.с., 2Н), 3,87 (с, 3Н), 2,97-2,93 (м, 1Н), 1,28 (д, J=7,2 Гц, 6Н).
5-(2-гидроксипропан-2-ил)-1-метил-1Н-пиразол-3-сульфонамид
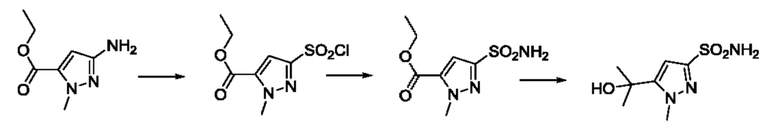
Этил-3-амино-1-метил-1Н-пиразол-5-карбоксилат преобразовали в этил 3-(хлорсульфонил)-1-метил-1Н-пиразол-5-карбоксилат, светло-желтую жидкость, с использованием общего способа D (0,35 г, 47%). 1Н-ЯМР (300 МГц, хлороформ-d) δ 7,39 (с, 1 Н), 4,40 (кв, J=7,1 Гц, 2Н), 4,32 (с, 3Н), 1,40 (т, J=7,1 Гц, 3Н). Сульфонилхлорид преобразовали с использованием общего способа Е2 с получением этил-1-метил-3-сульфамоил-1Н-пиразол-5-карбоксилата в виде белого с желтоватым оттенком твердого вещества (0,3 г, 94%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ=7,59 (с, 2Н), 7,09 (с, 1Н), 4,33 (кв, J=7,1 Гц, 2Н), 4,14 (с, 3H), 1,31 (т, J=7,1 Гц, 3Н).
К раствору этил-1-метил-3-сульфамоил-1Н-пиразол-5-карбоксилата (0,25 г, 1,07 ммоль) в безводном ТГФ (10 мл) при 0°С добавили по каплям метилмагнийхлорид (3 моль/л в ТГФ, 5 эквивалентов). Полученную реакционную смесь постепенно прогрели до температуры окружающей среды и перемешивали в течение 6 ч или до завершения. Раствор охладили до 0°С, разбавили насыщенн. водн. NH4Cl (2,0 мл), затем разбавили холодной водой (20 мл) и экстрагировали EtOAc (2×25 мл). Объединенные органические фазы промыли солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента градиента 50% EtOAc в гексанах с получением указанного в подзаголовке соединения в виде белого твердого вещества. (0,2 г, 87%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=7,34 (с, 2Н), 6,40 (с, 1Н), 5,48 (с, 1Н), 4,0 (с, 3Н), 1,50 (с, 6Н).
1-бензил-5-(2-гидроксипропан-2-ил)-1Н-пиразол-3-сульфонамид
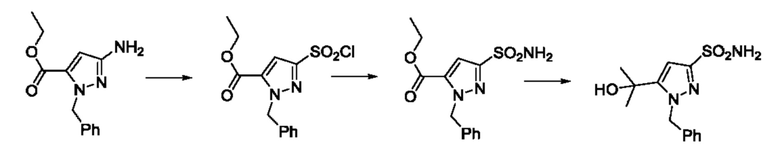
Этил-3-амино-1-бензил-1Н-пиразол-5-карбоксилат преобразовали в этил-1-бензил-3-(хлорсульфонил)-1Н-пиразол-5-карбоксилат, светло-коричневую жидкость, с применением способа D (0,35 г, 47%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,41 (с, 1Н), 7,34-7,26 (м, 5Н), 5,87 (с, 2Н), 4,37 (кв, J=7,2 Гц, 2Н), 1,38 (т, J=7,2 Гц, 3Н). Сульфонилхлорид преобразовали с использованием общего способа Е2 с получением этил-1-бензил-3-сульфамоил-1Н-пиразол-5-карбоксилата в виде белого твердого вещества (0,7 г, 88%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,66 (с, 2Н), 7,39-7,27 (м, Н), 7,2-7,18 (м, 3Н), 5,77 (с, 2Н), 4,33 (кв, J=7,2 Гц, 2Н), 1,29 (т, J=7,2 Гц, 3Н). ЖХМС (m/z): 310,05 (М+1)+.
К раствору этил-1-бензил-3-сульфамоил-1Н-пиразол-5-карбоксилата (0,5 г, 1,62 ммоль) в безводном ТГФ (10 мл) при 0°С добавили по каплям метилмагнийхлорид (3 моль/л в ТГФ, 2,77 мл, 8,1 ммоль). Полученную реакционную смесь постепенно прогрели до температуры окружающей среды и перемешивали в течение 4 ч или до завершения. Раствор охладили до 0°С, разбавили насыщенн. водн. NH4Cl (2,0 мл), затем разбавили холодной водой (20 мл) и экстрагировали EtOAc (2×25 мл). Объединенные органические фазы промыли солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента градиента 70-100% EtOAc в гексанах с получением указанного в подзаголовке соединения в виде белого твердого вещества. (0,27 г, 57%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=7,37 (с, 2Н), 7,39-7,27 (м, 3Н), 7,2-7,18 (м, 2Н), 6,45 (с, 1Н), 5,66 (с, 2Н), 5,60 (с, 1Н), 1,44 (с, 6Н). ЖХМС (m/z): 296,1 (М+1)+.
5-(2-гидроксипропан-2-ил)-1-фенил-1Н-пиразол-3-сульфонамид 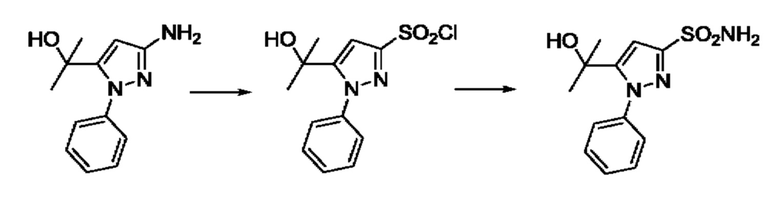
2-(3-амино-1-фенил-1Н-пиразол-5-ил)пропан-2-ол преобразовали в 5-(2-гидроксипропан-2-ил)-1-фенил-1Н-пиразол-3-сульфонилхлорид, желтую жидкость, с применением способа D (0,4 г, 36%). 1Н-ЯМР (300 МГц, CDCl3): δ=7,55-7,45 (м, 5Н), 6,91 (с, 1Н), 1,51 (с, 6Н). Сульфонилхлорид преобразовали с использованием общего способа Е2 с получением указанного в подзаголовке соединения в виде желтого твердого вещества (0,32 г, 87%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=7,5 (с, 5Н), 7,47 (с, 2Н), 6,65 (с, 1Н), 5,41 (с, 1Н), 1,30 (с, 6Н).
5-(диметил амино)нафталин -1-сульфонамид
5-(диметиламино)нафталин-1-сульфонамид 3-азидобензолсульфонамид синтезировали согласно методикам, приведенным в Satish K. Nair, Daniel Elbaum and David W. Christianson. J. Biol. Chem. 1996, 2711003-100 и Lixuan Mu, Wensheng Shi, Guangwei She, Jack C. Chang, and Shuit-Tong Lee. Angew. Chem. Int. Ed. 2009, 48, 3469-3472.
Раствор 5-(диметиламино)нафталин-1-сульфонилхлорида (0,12 г, 0,44 ммоль) в ацетоне (5 мл) добавили по каплям к раствору бикарбоната аммония (0,17 г, 1,76 ммоль) в воде (1,0 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч или до завершения. рН с помощью конц. HCl довели до рН 2,0. Органическую фазу отделили, а водную фазу насытили NaCl и экстрагировали этилацетатом. Объединенные органические фазы промыли солевым раствором, высушили (MgSO4) и концентрировали при пониженном давлении с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,075 г, выход 67%). 1Н-ЯМР (600 МГц, CD3OD) δ=8,54 (д, J=8,5 Гц, ОН), 8,36 (д, J=8,7 Гц, 1 Н), 8,23 (д, J=7,3 Гц, 1Н), 7,58 (ддд, J=17,0, 8,6, 7,4 Гц, 2Н), 7,28 (д, J=7,6 Гц, 1Н), 2,89 (с, 6Н). 13С-ЯМР (151 МГц, CD3OD) δ=151,6, 138,9, 129,7, 129,4, 129,2, 127,4, 126,6, 122,9, 119,5, 114,8, 44,4.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-2,3-дигидробензо[b]тиофен-6-сульфонамид-1,1-диоксид
м-Хлорнадбензойную кислоту (77%, 6,35 г, 27,9 ммоль) добавили порциями к раствору бензо[b]тиофена (1,50 г, 11,1 ммоль) в безводном дихлорметане (100 мл) при комнатной температуре при интенсивном перемешивании, полученную реакционную смесь перемешивали в течение 16 ч при той же температуре. Добавили насыщенный водный раствор NaHCO3 (250 мл) и водный слой экстрагировали дихлорметаном (2×100 мл), органический слой отделили, объединенные органические слои высушили (MgSO4) и концентрировали при пониженном давлении. При кристаллизации из этанола получили бензо[b]тиофен-1,1-диоксид (1,56 г, 84%) в виде белого с желтоватым оттенком твердого вещества. 1Н-ЯМР (600 МГц, CDCl3): δ=7,73 (д, J=6 Гц, 1Н), 7,58-7,53 (м, 2Н), 7,38 (д, J=12 Гц, 1Н), 7,23 (д, J=6 Гц, 1 Н), 6,73 (д, J=6 Гц, 1Н). ЖХМС (m/z): 167 [М+Н]+
Раствор бензо[b]тиофен-1,1-диоксида (0,75 г, 4,51 ммоль) в этаноле (55 мл) дегазировали с помощь азота в течение 10 минут, затем добавили 10% Pd/C (10 мг) и смесь перемешивали в атмосфере водорода (1 атм) в течение 24 ч. Реакционную смесь профильтровали через слой целита, фильтрат концентрировали с получением 2,3-дигидробензо[b]тиофен-1,1-диоксида (0,74 г, 97%) в виде белого с желтоватым оттенком твердого вещества. 1Н-ЯМР (600 МГц, CDCl3): δ=7,75 (д, J=6 Гц, 1Н), 7,59 (т, J=9 Гц, 1Н), 7,49 (т, J=6 Гц, 1Н), 7,40 (д, J=6 Гц, 1Н), 3,51 (т, J=6 Гц, 2Н), 3,41 (т, J=6 Гц, 2Н). ЖХМС (m/z): 169 [М+Н]+
2,3-дигидробензо[b]тиофен-1,1-диоксид (0,75 г, 4,45 ммоль) нагревали в хлорсульфоновой кислоте (1,5 мл, 22,2 ммоль) при 80°С в течение 4 ч. Реакционную смесь вылили на измельченный лед и перемешивали в течение 5 минут. Водный раствор экстрагировали дихлорметаном (2×50 мл) и объединенные органические фазы высушили (MgSO4) и концентрировали при пониженном давлении с получением
2,3-дигидробензо[b]тиофен-6-сульфонилхлорид-1,1-диоксида (0,45 г, 38%) в виде светло-коричневого масла. Технический продукт использовали сразу на следующей стадии без очистки. 1Н-ЯМР (600 МГц, CDCl3): δ=8,42 (с, 1Н), 8,25 (д, J=12 Гц, 1Н), 7,69 (д, J=6 Гц, 1Н), 3,64 (т, J=9 Гц, 2Н), 3,55 (т, J=6 Гц, 2Н).
К раствору 2,3-дигидробензо[b]тиофен-6-сульфонилхлорид-1,1-диоксида (0,45 г, 1,68 ммоль) в ацетоне (1 мл) добавили водный NH3 (2 мл, 28% NH4OH в H2O) при 0°С, полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч или до завершения. Растворитель удалили при пониженном давлении и азеотропной отгонкой с толуолом (×2). Технический остаток очистили колоночной хроматографией на силикагеле с использованием элюента 4% МеОН/CH2Cl2 с получением 2,3-дигидробензо[b]тиофен-6-сульфонамид-1,1-диоксида (0,16 мг, 39%) в виде белого с желтоватым оттенком твердого вещества. 1Н-ЯМР (ДМСО-d6): δ=8,09 (с, 1Н), 8,06 (д, J=12 Гц, 1Н), 7,75 (д, J=6 Гц, 1Н), 7,60 (шс, 2Н) 3,70 (т, J=6 Гц, 2Н), 3,44 (т, J=9 Гц, 2Н).
3-азидобензолсульфонамид
Синтезировали в соответствии с методикой, приведенной в Pawan Kumar, Navneet Chandak, Poul Nielsen, Pawan K. Sharma. Bioorg. Med. Chem. 2012, 20, 3843-3849. Раствор 3-аминобензолсульфонамида (0,3 г, 1,7 ммоль) в CH3CN (8 мл) охладили до 0°С. К этой перемешиваемой смеси добавили трет-BuONO (250 мкл, 2,1 ммоль), а затем TMSN3 (276 мкл, 2,1 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 100% гексаны с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (0,31 г, 91%). 1Н-ЯМР (600 МГц, CD3OD) δ 7,68-7,62 (м, 1Н), 7,56 (д, J=1,8 Гц, 1Н), 7,47 (д, J=7,8 Гц, 1Н), 7,19-7,15 (м, 1Н). 13С-ЯМР (151 МГц, ДМСО-d6) δ 146,2, 140,8, 131,2, 122,9, 122,4, 116,5.
N-(3-Сульфамоилфенил)пент-4-инамид
К раствору пент-4-иновой кислоты (0,1 г, 1,02 ммоль) и 3-аминобензолсульфонамида (0,21 г, 1,22 ммоль) в сухом ДМФА (5,0 мл) добавили HBTU (0,46 г, 1,22 ммоль), а затем DIPEA (212 мкл, 1,22 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч, или до завершения. Смесь разбавили EtOAc (30 мл), промыли H2O (20 мл), солевым раствором (20 мл), затем органическую фазу высушили (MgSO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 100% гексаны с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (0,2 г, 79%). 1Н-ЯМР (400 МГц, CD3OD) δ=8,22 (дд, J=2,2, 1,7 Гц, 1 Н), 7,75-7,68 (м, 1Н), 7,65-7,58 (м, 1Н), 7,51-7,42 (м, 2Н), 2,64-2,59 (м, 2Н), 2,58-2,54 (м, 2Н), 2,32-2,25 (м, 1Н). 13С-ЯМР (101 МГц, CD3OD) δ=171,3, 143,8, 138,9, 129,2, 122,9, 121,0, 117,1, 82,1, 69,1, 35,4, 14,0.
Бензол-1,3-дисульфонамид

Бензол-1,3-дисульфонилдихлорид (0,50 г, 0,726 ммоль) растворили в тетрагидрофуране (4 мл) и раствор охладили до 0°С. Добавили водный аммиак (0,4 мл) при 0°С и смесь перемешивали при температуре окружающей среды в течение 1 ч. По завершении реакции смесь вылили в охлажденную воду и экстрагировали этилацетатом. Объединенные органические экстракты промыли солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Полученное твердое вещество растерли с пентаном с получением указанного в подзаголовке соединения в виде светло-коричневого твердого вещества (0,16 г, 87%), 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,27 (т, J=2,0 Гц, 1Н), 8,06 (дд, J=2,0, 8,0 Гц, 2Н), 7,81 (т, J=8,0 Гц, 1Н), 7,64 (с, 4Н).
N1,N1-диметил бензол-1,3-дисульфонамид
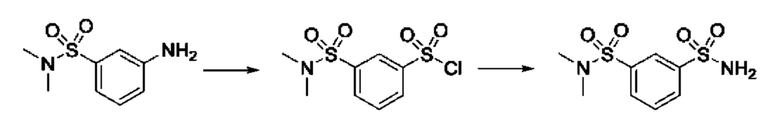
3-амино-N,N-диметилбензолсульфонамид преобразовали в 3-(N,N-диметилсульфамоил)бензолсульфонилхлорид (0,45 г, 80%) с применением способа D. 1Н-ЯМР (300 МГц, CDCl3) δ=8,42 (т, J=2,0 Гц, 1Н), 8,27 (д, J=7,9 Гц, 1Н), 8,14 (д, J=7,9 Гц, 1Н), 7,85 (т, J=7,9 Гц, 1Н), 2,79 (с, 6Н). Сульфонилхлорид преобразовали с использованием общего способа Е1 с получением указанного в подзаголовке соединения в виде желтого твердого вещества (0,45 г, 93%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=8,13 (м, 2Н), 7,98 (д, J=7,9 Гц, 1Н), 7,87 (т, J=7,9 Гц, 1Н), 7,65 (с, 2Н), 2,65 (с, 6Н).
Метил 3-сульфамоилбензоат
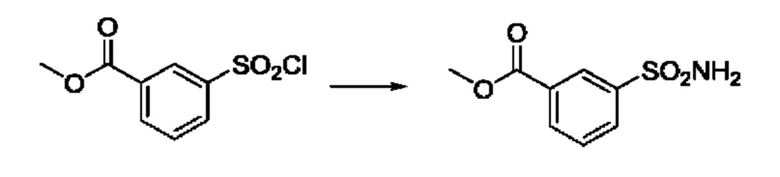
Метил-3-(хлорсульфонил)бензоат (1,00 г, 4,26 ммоль) растворили в безводном тетрагидрофуране (15 мл) и раствор охладили до 0°С. Добавили по каплям водный аммиак (5,0 мл) и смесь перемешивали при температуре окружающей среды в течение 2 ч. По завершении реакционную смесь вылили в охлажденную воду и экстрагировали этилацетатом. Объединенные органические экстракты промыли солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Полученное твердое вещество растерли с пентаном с получением указанного в подзаголовке соединения в виде светло-коричневого твердого вещества (0,75 г, 82%), 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,40 (с, 1Н), 8,19 (д, J=8 Гц, 1Н), 8,1 (д, J=8 Гц, 1Н), 7,77 (т, J=8 Гц, 1Н), 7,6 (с, 2Н), 3,92 (с, 3Н); m/z 214,0 [М-Н+]-
3-(4-фенил-1Н-1,2,3-триазол-1-ил)бензолсульфонамид
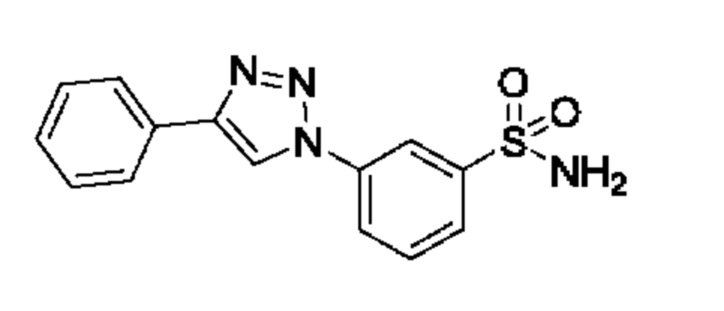
Этинилбензол (1 экв.) и 3-азидобензолсульфонамид (1,2 экв.), 5 мол. % CuSO4, 10 мол. % раствор NaAsc в ДМСО (500 мкл) перемешивали при комнатной температуре в течение 12 ч. Технический продукт очистили непосредственно из реакционной смеси с помощью хроматографии на колонне с обращенной фазой (система для колоночной флэш-хроматографии Reveleris, 4 г, 18 мл/мин.) и высушили сублимацией с получением продукта в виде белого твердого вещества (32 мг, 70%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=9,57 - 9,36 (м, 1Н), 8,46 (д, J=5,7 Гц, 1Н), 8,20 (с, 1Н), 7,98 (д, J=8,1 Гц, 3Н), 7,88 (д, J=7,6 Гц, 1Н), 7,62 (с, 2Н), 7,53 (д, J=7,4 Гц, 2Н), 7,42 (д, J=7,5 Гц, 1Н).
N-(проп-2-ин-1-ил)-3-(4-сульфамоилфенил)пропанамид
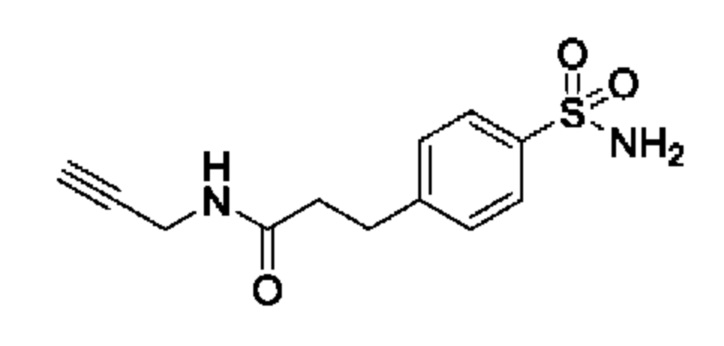
К раствору 3-(4-сульфамоилфенил)пропионовой кислоты (0,3 г, 1,5 ммоль) и пропаргиламина (0,11 г, 1,5 ммоль) в сухом ДМФА (5,0 мл) добавили HBTU (0,74 г, 1,5 ммоль), а затем DIPEA (342 мкл, 1,22 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию контролировали по ЖХМС и после завершения реакции, смесь разбавили EtOAc (30 мл), промыли H2O (20 мл), солевым раствором (20 мл). Органический слой отделили; высушили (MgSO4) и упарили с получением технического продукта. Технический продукт очистили колоночной хроматографией на силикагеле (1:1, EtOAc:Гексан) с выделением указанного в подзаголовке соединения в виде белого твердого вещества (0,22 г, 63%). 1Н-ЯМР (400 МГц, CD3OD) δ=7,85 (д, J=7,9 Гц, 2Н), 7,38 (д, J=7,9 Гц, 2Н), 3,97 (т, J=2,4 Гц, 2Н), 3,04 (т, J=7,6 Гц, 2Н), 2,55 (т, J=7,6 Гц, 2Н), 2,33 (д, J=2,8 Гц, 1Н).
бензо[d][1,3]диоксол-5-сульфонамид
К безводному ДМФА (2,10 мл, 26,7 ммоль) добавили сульфурилхлорид (2,18 мл, 26,7 ммоль) при 0°С в атмосфере азота, затем ледяную баню удалили и раствор перемешивали в течение 15 минут. Раствор охладили еще раз до 0°С и добавили бензо[d][1,3]диоксол. Реакционную смесь оставили до достижения комнатной температуры, затем нагревали при 100°С в течение 2 ч. Реакционную смесь вылили на измельченный лед, перемешивали в течение 5 минут, затем экстрагировали дихлорметаном (100 мл, затем 2×50 мл). Объединенные органические фазы высушили (MgSO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 15% ДХМ-гексаны с получением бензо[d][1,3]диоксол-5-сульфонилхлорида в виде белого с желтоватым оттенком твердого вещества (1,78 г, 33%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,64 (д, J=8,0 Гц, 1 Н), 7,43 (с, 1 Н), 6,95 (д, J=8,0 Гц, 1Н), 6,16 (с, 2Н).
К раствору бензо[d][1,3]диоксол-5-сульфонилхлорида (0,30 г, 1,35 ммоль) в ацетоне (1 мл) добавили водн. NH3 (1,5 мл, 28% NH4OH в H2O) при 0°С, реакционную смесь перемешивали при комнатной температуре до завершения, как правило, 2 ч, затем концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 2% МеОН-ДХМ с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (210 мг, 77%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,32 (д, J=8,0 Гц, 1Н), 7,25 (с, 1Н), 7,21 (ш.с, 2Н), 7,02 (д, J=8,0 Гц, 1Н), 6,11 (с, 2Н).
Пиридин-4-сульфонамид
Пиридин-4-сульфонилхлорид преобразовали с использованием общего способа Е3 с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (50 мг, 56%), 1Н-ЯМР (300 МГц, ДМСО-d6): δ=8,56 (д, J=4,5 Гц, 1Н), 7,49 (д, J=4,5 Гц, 1Н), 7,24 (ш.с, 2Н).
Пиридин-3-сульфонамид
Пиридин-3-сульфонилхлорид преобразовали с использованием общего способа Е3 с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (0,7 г, 79%), 1Н-ЯМР (300 МГц, ДМСО-d6) δ=8,96 (дд, J=2,5, 0,9 Гц, 1Н), 8,77 (дд, J=4,8, 1,6 Гц, 1Н), 8,17 (ддд, J=8,0, 2,4, 1,6 Гц, 1Н), 7,67 -7,56 (м, 3Н).
Пиридин-2-сульфонамид
Раствор HCl 1,0 моль/л (45 мл) и ДХМ (45 мл) охладили до -10°С и добавили пиридин-2-тиол (1,0 г, 9,0 ммоль). Через 10 мин добавили по каплям NaOCl (6% раствор, 47 мл, 3,3 экв.) в течение 5 мин и перемешивание продолжали при -10°С в течение 10 мин. Органическую фазу отделили, высушили с помощью Na2SO4 и профильтровали. Полученный раствор добавили по каплям к предварительно охлажденному насыщенному раствору аммиака в метаноле и ДХМ (1:1, 40 мл) при 0°С, затем оставили прогреться до температуры окружающей среды и перемешивали до завершения, как правило, 2 ч. Растворитель удалили при пониженном давлении с получением белого твердого вещества, которое растворили в горячем EtOAc и отфильтровали для удаления твердых примесей. Растворитель удалили при пониженном давлении и перекристаллизовали из смеси EtOAc-гексаны с получением указанного в подзаголовке соединения в виде желтого твердого вещества (0,5 г, 35%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ=8,70 (ддд, J=4,7, 1,7, 0,9 Гц, 1Н), 8,05 (тд, J=7,7, 1,7 Гц, 1Н), 7,91 (дт, J=7,9, 1,1 Гц, 1Н), 7,62 (ддд, J=7,6, 4,7, 1,2 Гц, 1Н), 7,45 (с, 2Н).
4-(трифторметил)пиридин-2-сульфонамид
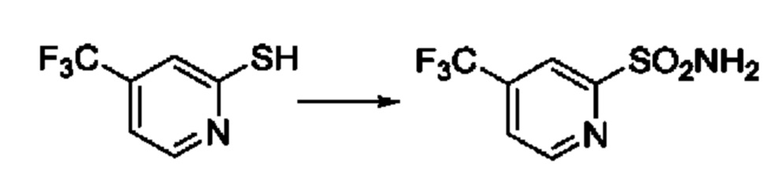
4-(трифторметил)пиридин-2-сульфонамид синтезировали согласно методикам, использованным для синтеза пиридин-2-сульфонамида, но с применением 4-(трифторметил)пиридин-2-тиола вместо пиридин-2-тиола. Продукт 4-(трифторметил)пиридин-2-сульфонамид получили в виде твердого вещества (0,7 г, 56%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ=9,02 (д, J=5,0 Гц, 1Н), 8,16 (с, 1Н), 8,07 (д, J=5,0 Гц, 1Н), 7,68 (с, 2Н).
3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид 
3-(3-(трифторметил)-3Н-диазирин-3-ил)анилин преобразовали с использованием общего способа D в 3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонилхлорид, желтую жидкость (1,1 г, 52%). 1Н-ЯМР (300 МГц, CDCl3) δ=8,15-8,08 (м, 1Н), 7,82-7,77 (м, 1 Н), 7,76-7,68 (м, 1 Н), 7,68-7,61 (м, 1Н). 19F-ЯМР (282 МГц, CDCl3) δ -65,06.
3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонилхлорид преобразовали с использованием общего способа Е2 в указанное в подзаголовке соединение в виде белого твердого вещества (0,6 г, 60%). 1Н-ЯМР (300 МГц, CDCl3) δ=7,99 (дт, J=7,9,1,5 Гц, 1Н), 7,71 (т, J=2,0 Гц, 1Н), 7,60 (т, J=7,9 Гц, 1Н), 7,49 (д, J=7,9 Гц, 1Н), 4,87 (с, 2Н). 19F-ЯМР (282 МГц, CDCl3) δ-65,13.
2-(метил(7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)-N-(4-сульфамоилфенэтил)ацетамид
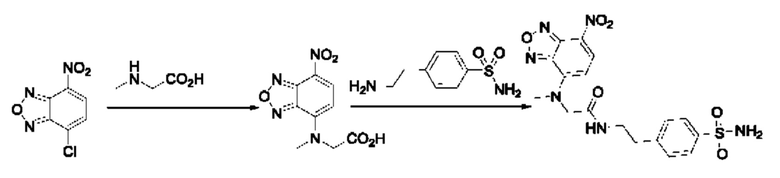
2-(Метиламино)уксусную кислоту (0,24 г, 2,75 ммоль) и гидрокарбонат натрия (0,694 г, 8,26 ммоль) растворили в смеси воды (10 мл) и МеОН (20 мл). Затем добавили 4-хлор-7-нитробензо[с][1,2,5]оксадиазол (0,50 г, 2,50 ммоль) и смесь перемешивали при 60°С в течение 2 ч. По завершении реакции летучие компоненты удалили при пониженном давлении и технический полученный остаток очистили колоночной хроматографией на силикагеле с использованием градиента 0-5% метанола в дихлорметане с получением 2-(метил(7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)уксусной кислоты в виде кирпично-красного твердого вещества (1,10 г, 87%).
2-(Метил(7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)уксусную кислоту (1,00 г, 3,96 ммоль) растворили в безводном тетрагидрофуране (25 мл) в атмосфере азота и раствор охладили до 0°С. Добавили диизопропилэтиламин (0,76 г, 5,55 ммоль) и 1,1'-карбонилдиимидазол (0,90 г, 4,75 ммоль) и смесь перемешивали при 50°С до тех пор, пока не прореагирует вся 2-(метил(7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)уксусная кислота. Затем реакционную смесь охладили до 0°С, добавили 4-(2-аминоэтил)бензолсульфонамид (0,95 г, 4,75 ммоль) и перемешивали при температуре окружающей среды до завершения, как правило, 6 ч. Растворители удалили при пониженном давлении и остаток очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде кирпично-красного твердого вещества (1,20 г, 70%). ЖХМС (m/z): 435,4 (М+1)+.
4-(2-(7-Нитробензо[с][1,2,5]оксадиазол4-иламино)этил)бензолсульфонамид
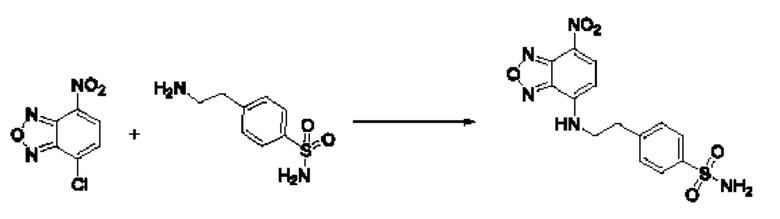
4-(2-аминоэтил)бензолсульфонамид (0,55 г, 2,75 ммоль) и диизопропилэтиламин (0,64 г, 2,75 ммоль) растворили в этаноле (20 мл) и раствор охладили до 0°С. Добавили 4-хлор-7-нитробензо[с][1,2,5]оксадиазол (0,50 г, 2,50 ммоль) при 0°С и смесь перемешивали при температуре окружающей среды в течение 16 ч. По завершении реакции реакционную массы вылили в солевой раствор и экстрагировали этилацетатом. Растворители упарили из объединенного органического экстракта при пониженном давлении и полученный технический продукт очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке продукта в виде темно-желтого твердого вещества (0,250 г, 7%). 1Н-ЯМР (400 МГц, CD3OD) δ=8,5 (д, J=8,8 Гц, 1Н), 7,82 (д, J=8,4 Гц. 2Н). 7,47 (д, J=8,4 Гц, 2Н),6,35 (д, J=8,8 Гц, 1Н), 3,83 (м, 2Н), 3,15 (т, J=7,6 Гц, 2Н).
2-(7-(Диметилам ино)-2-оксо-2Н-хромен-4-ил)-N-(4-сульфамоилфенэтил)ацетамид (171):
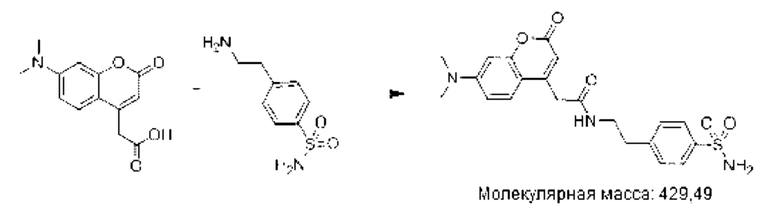
2-(7-(диметиламино)-2-оксо-2Н-хромен-4-ил)уксусную кислоту (0,50 г, 2,02 ммоль), EDCHCl (0,47 г, 3,03 ммоль), HOBt (0,464 г, 3,03 ммоль) и N-метилморфолин (0,409 г, 4,04 ммоль) смешали в безводном тетрагидрофуране (5 мл) и перемешивали при 0°С в течение 30 мин. Добавили 4-(2-аминоэтил)бензолсульфонамид (0,445 г, 2,224 ммоль) и перемешивание продолжали при температуре окружающей среды в течение 18 ч. По завершении реакционную смесь вылили в охлажденную воду и экстрагировали этилацетатом. Растворитель удалили при пониженном давлении и остаток очистили колоночной хроматографией на силикагеле с использованием градиента 0-5% метанола в дихлорметане с получением 2-(7-(диметиламино)-2-оксо-2Н-хромен-4-ил)-N-(4-сульфамоилфенэтил)ацетамида в виде зеленовато-желтого твердого вещества (0,25 г, 29%). ЖХМС (m/z): 430,2 (М+1)+.
6,7-Дигидро-5Н-пирроло[1,2-а]имидазол-2-сульфонамид
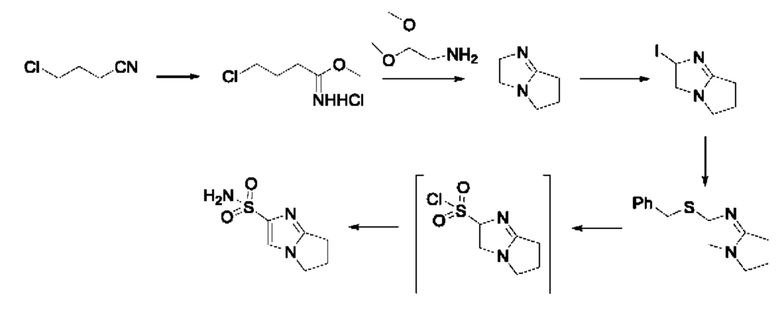
На раствор 3-хлорбутаннитрила (20 г, 193,1 ммоль) в диэтиловом эфире (100 мл) подействовали МеОН (7,41 г, 231,7 ммоль) и охладили до 0°С. В реакционную смесь пропускали газообразный HCl в течение 4 ч при 0°С. Реакционную смесь перемешивали при -20°С в течение 24 ч и реакционную смесь концентрировали при пониженном давлении. Полученный твердый остаток промыли диэтиловым эфиром (3×100 мл), н-пентаном (2×100 мл) и высушили в вакууме при 45°С с получением гидрохлорида метил-4-хлорбутанимидата в виде белого твердого вещества. Гидрохлорид метил-4-хлорбутанимидата (25 г, 146,1 ммоль) растворили в ДХМ (250 мл) и подействовали на него Et3N (44,3 г, 4,38 ммоль) и полученный раствор охладили до 0°С. К описанной выше смеси добавили по каплям 2,2-диметоксиэтан-1-амин (12,2 г, 116,9 ммоль) на протяжении 5 мин. Полученную реакционную смесь прогрели до 60°С и перемешивали в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении и на полученный остаток подействовали муравьиной кислотой (150 мл) и нагревали при 80°С в течение 24 ч. По завершении реакционную смесь концентрировали при пониженном давлении и полученный остаток подвергли азеотропной отгонке с толуолом (2×100 мл). Повысили основность технической смеси насыщенным раствором NaHCO3 и экстрагировали ДХМ (3×200 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 6,7-дигидро-5Н-пирроло[1,2-а]имидазола (8 г, 39% по 3 стадиям) в виде низкоплавкого темного твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,0 (с, 1Н), 6,83 (с, 1Н), 3,95 (т, J=7,2 Гц, 2Н), 2,84 (т, J=7,2 Гц, 2Н), 2,61-2,51 (м, 2Н).
Раствор 6,7-дигидро-5Н-пирроло[1,2-а]имидазола (4 г, 37,0 ммоль) в ацетонитриле (120 мл) охладили до 0°С. Добавили порциями N-йодсукцинимид (9,16 г, 40,7 ммоль) при 0°С.Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили насыщенным раствором Na2S2O3 и экстрагировали этилацетатом (2×50 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 4-40% EtOAc-гексаны с получением 2-йод-6,7-дигидро-5Н-пирроло[1,2-а]имидазола (1,0 г, 19%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,03 (с, 1Н), 3,89 (т, J=7,2 Гц, 2Н), 3,02 (т, J=7,2 Гц, 2Н), 2,65-2,55 (м, 2Н). ЖХМС (m/z): 235 [М+Н]+
В герметизируемом реакционном сосуде емкостью 50 мл, на раствор 2-йод-6,7-дигидро-5Н-пирроло[1,2-а]имидазола (0,3 г, 1,28 ммоль) и фенилметантиола (0,24 г, 1,92 ммоль) в 1,4-диоксане (10 мл) подействовали DIPEA (0,41 г, 3,20 ммоль) при комнатной температуре в атмосфере азота. Через раствор пропускали газообразный азот в течение 5 минут. К вышеупомянутому раствору последовательно добавили Ксантфос (74 мг, 0,128 ммоль) и Pd2(dba)3 (60 мг, 0,064 ммоль) и сосуд продували газообразным азотом в течение 5 минут. Полученную смесь перемешивали при 110°С в течение 12 ч. По завершении смесь охладили до комнатной температуры, разбавили EtOAc (25 мл) и профильтровали через целит. Фильтрат высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 50-70% EtOAc-гексаны с получением 2-(бензилтио)-6,7-дигидро-5Н-пирроло[1,2-а]имидазола, (0,15 г, 51%) в виде коричневой жидкости. 1Н-ЯМР (300 МГц, CDCl3): δ=7,26-7,22 (м, 3Н), 7,12 (с, 1Н), 7,05-7,02 (м, 2Н), 3,73 (с, 2Н), 3,14 (т, J=6,9 Гц, 2Н), 2,81 (т, J=7,2 Гц, 2Н), 2,32-2,27 (м, 2Н). ЖХМС (m/z): 231,3 [М+Н]+.
Раствор 2-(бензилтио)-6,7-дигидро-5Н-пирроло[1,2-а]имидазола (250 мг, 1,08 ммоль) в ацетонитриле (2,5 мл), уксусной кислоте (0,5 мл) и H2O (1,2 мл) охладили до 0°С. Добавили DCDMH (170 мг, 0,869 ммоль) при 0°С и полученную реакционную смесь перемешивали при 0°С в течение 2 ч. Реакционную смесь разбавили водой и экстрагировали этилацетатом (2×20 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 6,7-дигидро-5Н-пирроло[1,2-а]имидазол-2-сульфонилхлорида в виде светло-коричневой жидкости, использованной сразу на следующей стадии.
Раствор 6,7-дигидро-5Н-пирроло[1,2-а]имидазол-2-сульфонилхлорида (300 мг) в ТГФ (15 мл) охладили до -40°С. Через вышеупомянутый раствор пропускали газообразный аммиак в течение 15 мин и раствор перемешивали при -40°С в течение 1 ч. Реакционную смесь прогрели до комнатной температуры, перемешивали в течение 1 ч, затем, по завершении, концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 10% MeOH-CHCl3 с получением 6,7-дигидро-5Н-пирроло[1,2-а]имидазол-2-сульфонамида (117 мг, 88%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, ДМСО-d6): δ=7,54 (шс, 2Н), 7,26 (с, 1Н), 4,06 (с, 2Н), 2,80 (с, 2Н), 2,32 (с, 2Н). ЖХМС (m/z): 187,95 [М+Н]+.
4-Нитробензолсульфонамид

4-Нитробензолсульфонилхлорид (1,0 экв.), растворенный в ацетоне (0,8 мл/ммоль), добавили по каплям к бикарбонату аммония (4,0 экв.), растворенному в воде (0,8 мл/ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч с последующим подкислением 1 М HCl (рН ~2). Смесь экстрагировали этилацетатом (3×10 мл), высушили (MgSO4) и концентрировали при пониженном давлении с получением указанного в подзаголовке соединения в виде бледно-оранжевого твердого вещества (157 мг, 57%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=8,42 (д, J=8,8 Гц, 2Н), 8,06 (д, J=8,8 Гц, 2Н), 7,74 (с, 2Н). МСВР вычислено для C6H5N2O4S1 [М-Н]- 200,9976, найдено 200,9984.
5-Метил-N-(4-сульфамоилфенэтил)изоксазол-3-карбоксамид

5-Метилизоксазол-3-карбонилхлорид (1,0 экв.) (полученный с использованием общего способа В1) растворили в безводном ТГФ (4 мл/ммоль) и подействовали триэтиламином (1,0 экв.). После перемешивания в течение 5 минут к раствору хлорангидрида кислоты добавили 4-(2-аминоэтил)бензолсульфонамид (1,0 экв.). Реакционную смесь перемешивали при комнатной температуре, в атмосфере аргона на протяжении ночи. Растворитель удалили при пониженном давлении и остаток очистили колоночной хроматографией на обращенной фазе с использованием ацетонитрила/10 ммоль/л бикарбоната аммония (водного) в качестве подвижной фазы с получением указанного в подзаголовке соединения в виде белого твердого вещества (205 мг, 48%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=8,79 (т, J=5,8 Гц, 1Н), 7,74 (д, J=8,3 Гц, 2Н), 7,41 (д, J=8,3 Гц, 2Н), 7,30 (с, 2Н), 6,50 (кв, J=0,6 Гц, 1Н), 3,52-3,46 (м, 2Н), 2,91 (т, J=7,2 Гц, 2Н), 2,45 (д, J=0,6 Гц, 3Н). МСВР вычислено для C13H14N3O4S1 [М-Н]- 308,0711, найдено 308,0708.
4-(2-(7-Метокси-4,4-диметил-1,3-диоксо-3,4-дигидроизохинолин-2(1Н)-ил)этил)бензолсульфонамид
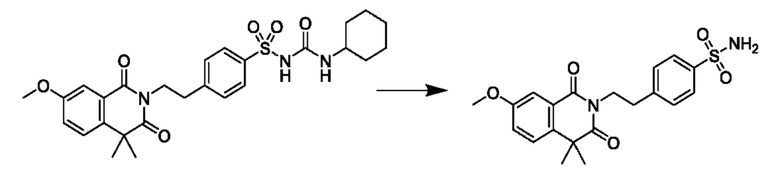
N-(циклогексилкарбамоил)-4-(2-(7-метокси-4,4-диметил-1,3-диоксо-3,4-дигидроизохинолин-2(1Н)-ил)этил)бензолсульфонамид (1,0 экв.) растворили в безводном пиридине (8 мл/ммоль), подействовали фталевым ангидридом (1 экв.) и ДМАП (0,1 экв.) и нагревали с обратным холодильником в инертной атмосфере в течение 4 часов. Растворитель удалили при пониженном давлении и остаток очистили колоночной хроматографией на обращенной фазе с использованием ацетонитрила/10 ммоль/л бикарбоната аммония (водного) в качестве подвижной фазы с получением указанного в подзаголовке соединения в виде белого твердого вещества (291 мг, 75%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=7,72 (д, J=8,4 Гц, 2Н), 7,61 (д, J f=8,7 Гц, 1Н), 7,53 (д, J=2,8 Гц, 1Н), 7,40 (д, J=8,2 Гц, 2Н), 7,33-7,25 (м, 3Н), 4,13 (д, J=7,5 Гц, 2Н), 3,83 (с, 3Н), 2,93 (т, J=7,3 Гц, 2Н), 1,45 (с, 6Н). МСВР вычислено для C20H21N2O5S1 [М-Н]- 401,1177, найдено 401,1174.
Синтез полупродуктов аминов R1 и R2:
1-метил-1Н-пиразол-3-амин HCl
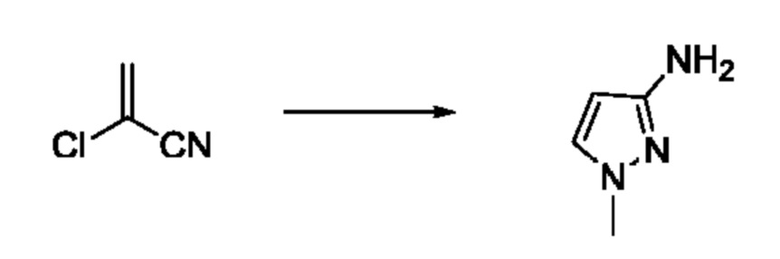
В микроволновой виале емкостью 20 мл на раствор 2-хлоракрилонитрила (2 г, 22,85 ммоль) в EtOH (10 мл) подействовали метилгидразином (1,93 г, 41,13 ммоль). Полученную реакционную смесь нагревали при 100°С в течение 10 минут в микроволновом синтезаторе Biotage. Реакционную смесь оставили при <5°С в течение 12 ч, за это время выпало в осадок твердое вещество. Осадок удалили фильтрованием и высушили в вакууме с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,12 г, 55%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ 7,76 (с, 1Н), 6,13 (с, 1Н), 3,80 (с, 3Н), 2,58 (с, 2Н). ЖХМС (m/z): 98,3 (М+1)+.
1-(трифторметил)-1Н-пиразол-3-амин
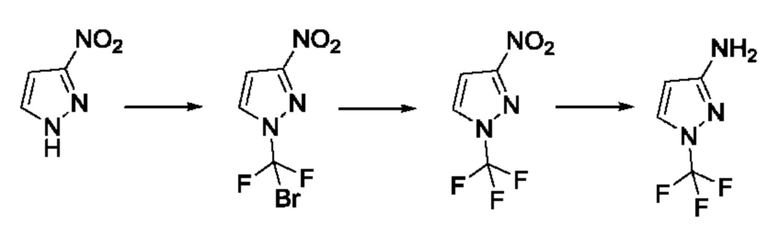
3-Нитро-1Н-пиразол (5 г, 44 ммоль) растворили в N,N-диметилформамиде (100 мл), охладили до -5°С и добавили порциями NaH (3,8 г, 93,6 ммоль). Реакционную смесь перемешивали в течение 15 мин с последующим добавлением дибромдифторметана (8,6 г, 44 ммоль) и прогреванием до температуры окружающей среды на протяжении ночи. Реакционную смесь разбавили ледяной водой и экстрагировали с помощью этилацетата. Органическую фазу промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 3-нитро-1-(трифторметил)-1Н-пиразола (2,1 г, 22%), который использовали без дополнительной очистки. 19F-ЯМР (282 МГц, CDCl3) δ=-34,20.
1-(бромдифторметил)-3-нитро-1Н-пиразол (2,1 г, 9,76 ммоль) растворили в ДХМ (50 мл) и охладили до -78°С с последующим добавлением AgBF4 (5,7 г, 28,3, 3 эквивалента). Реакционную смесь оставили прогреться до температуры окружающей среды на протяжении ночи, затем охладили до 0°С и остановили реакцию добавлением насыщенн. водн. NaHCO3 (50 мл). Водную фазу экстрагировали с помощью ДХМ и объединенные органические фазы промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 3-нитро-1-(трифторметил)-1Н-пиразола (0,9 г, 51%). 19F-ЯМР (282 МГц, CDCl3) δ=-60,96.
3-нитро-1-(трифторметил)-1 Н-пиразол (1,0 г) растворили в ТГФ:EtOAc (1:1, 50 мл), добавили Pd/C (200 мг) и смесь перемешивали в атмосфере водорода (надувной шар) на протяжении ночи. Смесь профильтровали через целит и промыли с помощью этилацетата. Растворитель удалили при пониженном давлении и остаток очистили колоночной хроматографией на силикагеле с использованием элюента 40% EtOAc в гексанах с получением указанного в подзаголовке продукта (0,75 г, 87%). 1Н-ЯМР (300 МГц, CDCl3) δ=7,54 (д, J=2,7 Гц, 1Н), 5,84 (д, J=2,7 Гц, 1Н). 19F-ЯМР (282 МГц, CDCl3) δ=-61,13.
1-изопропил-1 Н-пиразол-3-амин
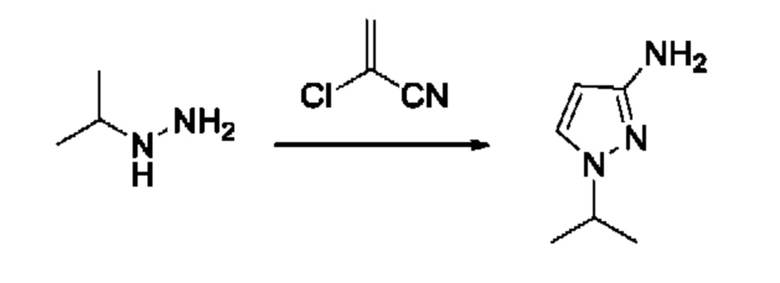
На гидрохлорид изопропилгидразина (5 г, 45,45 ммоль) в воде (40 мл) подействовали последовательно K2CO3 (12,5 г, 91 ммоль) и 2-хлоракрилонитрилом (4 г, 45,45 ммоль). Полученную реакционную смесь перемешивали при 50°С в течение 1 ч, охладили до комнатной температуры и экстрагировали этилацетатом (50 мл). Органический экстракт промыли водой (40 мл), солевым раствором(40 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением указанного в подзаголовке соединения в виде желтого твердого вещества (3,5 г, 62%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,15 (с, 1Н), 5,56 (с, 1Н), 4,27-4,23 (м, 1Н), 3,6 (ш.с, 2Н), 1,43 (д, J=6,4 Гц, 6Н). ЖХМС (m/z): 126,0 (М+1)+.
1-циклопропил-1Н-пиразол-3-амин
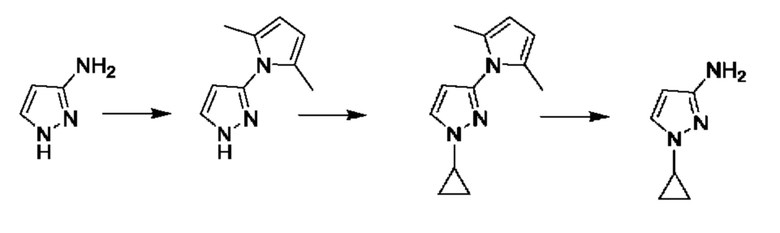
На раствор 1H-пиразол-3-амина (2 г, 24,1 ммоль) в АсОН (20 мл) подействовали 2,5-гександионом (5,7 г, 50,6 ммоль) при температуре окружающей среды в атмосфере азота. Полученную реакционную смесь грели до 100°С в течение 6 h. Реакционную смесь концентрировали при пониженном давлении и подвергли азеотропной отгонке с толуолом. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента градиента 50-100% EtOAc-гексаны с получением 3-(2,5-диметил-1Н-пиррол-1-ил)-1Н-пиразола в виде красного твердого вещества (2,25 г, 59%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 12,92 (с, 1Н), 7,85 (т, J=1,8 Гц, 1Н), 6,28 (т, J=2,1 Гц, 1Н), 5,75 (с, 2Н), 2,00 (с, 6Н). Ацетат меди (II) (0,56 г, 3,1 ммоль), 2,2'-бипиридин (0,48 г, 3,1 ммоль) и дихлорэтан (10 мл) нагревали до 75°С в течение 20 мин. 5 мл этого заранее приготовленного раствора добавили к смеси 3-(2,5-диметил-1Н-пиррол-1-ил)-1Н-пиразола (0,5 г, 3,1 ммоль), циклопропилтрифторбората калия (2 экв.) и карбоната натрия (2 экв.) в дихлорэтане (5 мл), затем реакционную смесь перемешивали при 75°С в течение 6 ч. Реакционную смесь разбавили ДХМ, промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением 1-циклопропил-3-(2,5-диметил-1Н-пиррол-1-ил)-1Н-пиразола в виде желтой жидкости (0,2 г, 32%). 1Н-ЯМР (300 МГц, Хлороформ-d) δ 7,48 (дд, J=2,3, 0,5 Гц, 1Н), 6,12 (д, J=2,3 Гц, 1Н), 5,84 (с, 2Н), 3,61 (тт, J=7,3, 3,6 Гц, 1Н), 2,09 (с, 6Н), 1,22-0,95 (м, 4Н).
К раствору гидроксида гидрохлорида аммония (1,64 г, 11,8 ммоль) в этаноле ((10 мл) добавили раствор гидроксида калия (0,66 г) в воде (10 мл) при 0°С. Через 10 мин перемешивания добавили раствор 1-циклопропил-3-(2,5-диметил-1Н-пиррол-1-ил)-1Н-пиразола (0,95 г) в этаноле (10 мл) и реакционную смесь нагревали при 100°С в течение 20 ч. Растворитель удалили при пониженном давлении и остаток разделили между этилацетатом и водой. Органическую фазу промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента градиента 70-100% EtOAc-гексаны с получением указанного в подзаголовке продукта в виде коричневого твердого вещества (0,4 г, 69%). 1Н-ЯМР (400 МГц, CDCl3) δ=7,51 (д, J=2,4 Гц, 1Н), 6,69 (д, J=2,4 Гц, 1Н), 5,04 (с, 2Н), 3,67 (м, 1Н), 1,19 (м, 2Н), 1,12 (м, 2Н).
1-(трет-бутил)-1Н-пиразол-3-амин
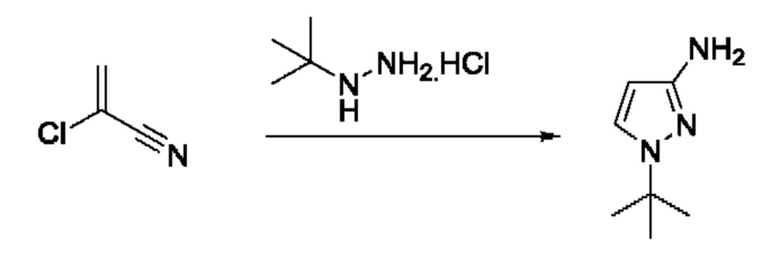
На гидрохлорид изопропилгидразина (1,42 г, 11,4 ммоль) в воде (25 мл) при 0°С подействовали последовательно K2CO3 (1,57 г, 11,4 ммоль), NaHCO3 (1,91 г, 22,9 ммоль) и 2-хлоракрилонитрилом (1 г, 11,4 ммоль), затем прогрели до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь разбавили водой (20 мл) и экстрагировали этилацетатом (2×25 мл). Объединенные органические фазы промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением указанного в подзаголовке соединения в виде коричневой жидкости (0,9 г, 60%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,34 (с, 1Н), 5,35 (с, 1Н), 4,51 (ш.с, 2Н), 1,40 (с, 9Н). ЖХМС (m/z): 140,10 (М+1)+.
1-циклогексил-1Н-пиразол-3-амин
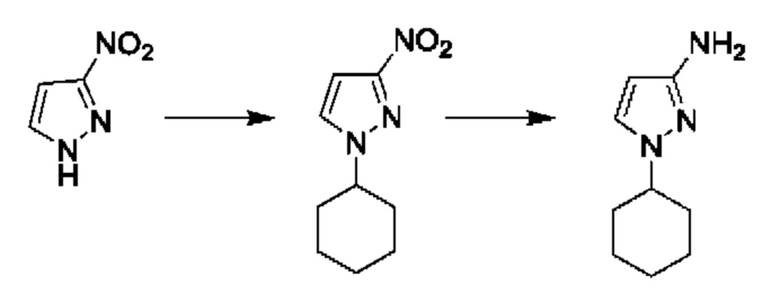
3-нитро-1Н-пиразол (1 г, 8,85 ммоль) растворили в N,N-диметилформамиде (20 мл) и подействовали карбонатом калия (1,47 г, 10,62 ммоль) и бромциклогексаном (1,8 г, 10,62 ммоль). Смесь нагревали при 100°С в течение 16 часов (или до завершения), затем охладили до температуры окружающей среды, разбавили водой (100 мл) и экстрагировали с помощью этилацетата (2×75 мл). Объединенные органические фазы промыли водой (100 мл), солевым раствором (100 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента градиента 10% EtOAc-гексаны с получением 1-циклогексил--нитро-1Н-пиразола в виде бесцветной жидкости (1,3 г, 76%). 1Н-ЯМР (300 МГц, CDCl3) δ=7,47 (д, J=2,5 Гц, 1Н), 6,88 (д, J=2,5 Гц, 1Н), 4,26-4,11 (м, 1Н), 2,18 (м, 2Н), 1,93 (м, 2Н), 1,82-1,20 (м, 6Н).
В реакционном сосуде к аппарату Парра емкостью 100 мл на раствор 1,5-диметил-3-нитро-1Н-пиразола (0,65 г, 3,3 ммоль) в МеОН (4 мл) и EtOAc (20 мл) подействовали 10% палладия на угле (200 мг) в атмосфере азота. Колбу вакуумировали, затем заполнили газообразным водородом (60 фунт/кв.дюйм (414 кПа)) и перемешивали при температуре окружающей среды в течение 12 ч.
Реакционную смесь разбавили этилацетатом (50 мл) и фильтровали через слой целита. Фильтрат высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1-циклогексил-1Н-пиразол-3-амина в виде светло-коричневого твердого вещества (0,3 г, 55%). 1Н-ЯМР (300 МГц, CDCl3) δ=7,15 (д, J=2,3 Гц, 1Н), 5,56 (д, J=2,3 Гц, 1Н), 3,85 (м, 1Н), 3,62 (с, 1Н), 2,1 (м, 2Н), 1,8 (м, 2Н), 1,77-1,10 (м, 6Н).
1-фенил-1Н-пиразол-3-амин

Трет-бутилат калия (11,9 г, 106,3 ммоль) растворили в трет-BuOH (100 мл) и раствор нагрели до 100°С. Последовательно добавили фенилгидразин (5 г, 46,2 ммоль) и 3-этоксиакрилонитрил (4,5 г, 46,2 ммоль) и нагрев продолжили в течение 16 ч. Смесь концентрировали при пониженном давлении. Полученный остаток разделили между водой (500 мл) и этилацетатом (500 мл). Органический экстракт промыли водой (250 мл), солевым раствором (250 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 25% EtOAc-гексаны с получением 1-фенил-1Н-пиразол-3-амина в виде светло-коричневого твердого вещества (3,5 г, 48%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,69 (с, 1H), 7,57 (д, J=8,0 Гц, 2Н), 7,41 (д, J=8,0 Гц, 2Н), 7,2 (т, J=7,6 Гц, 1H), 5,85 (с, 1Н), 3,83 (ш.с, 2Н). ЖХМС (m/z): 160,3 (М+1)+.
1-бензил-1Н-пиразол-3-амин
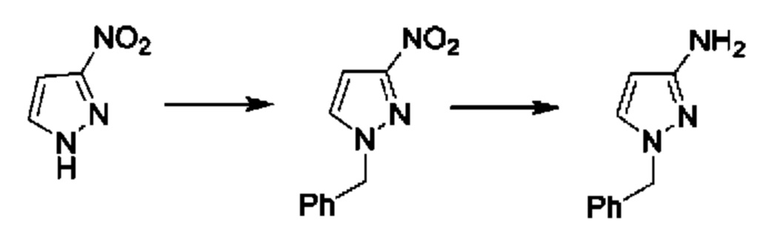
Раствор 3-нитро-1H-пиразола (1 г, 8,85 ммоль) в ТГФ (20 мл) охладили до 0°С и добавили NaH (0,53 г, 13,27 ммоль). Суспензию перемешивали в течение 20 мин, затем добавили по каплям бензилбромид (1,5 г, 8,85 ммоль). Реакционную смесь перемешивали до завершения ~6 ч, разбавили насыщенным раствором NaHCO3 (20 мл) и экстрагировали EtOAc (2 × 50 мл). Органические фазы промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1-бензил-3-нитро-1H-пиразола в виде белого твердого вещества (1,5 г, 84%). 1Н-ЯМР (300 МГц, CDCl3): δ=7,40-7,36 (м, 4Н), 7,31-7,27 (м, 2Н), 6,90 (д, J=2,7 Гц, 1Н), 5,37 (с, 2Н). ЖХМС (m/z): 204,20 (М+1)+
Раствор 1-бензил-3-нитро-1H-пиразола (1,5 г, 7,39 ммоль) в ТГФ (20 мл) и МеОН (5 мл) охладили до 0°С. Добавили порошок цинка (2,4 г, 36,9 ммоль) и раствор NH4Cl (1,97 г, 36,94 ммоль; в 5 мл воды). Полученную реакционную смесь нагревали при 70°С в течение 12 ч. Реакционную смесь охладили до температуры окружающей среды, разбавили EtOAc (50 мл) и фильтровали через слой целита. Фильтрат высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 50% EtOAc-гексаны с получением 1-бензил-1H-пиразол-3-амина в виде светло-коричневой жидкости (0,85 г, 67%). 1Н-ЯМР (300 МГц, CDCl3): δ=7,34-7,26 (м, 3H), 7,14-7,11 (м, 2Н), 7,05 (д, J=2,4 Гц, 1H), 5,59 (д, J=2,4 Гц, 1H), 5,14 (с, 2Н). ЖХМС (m/z): 174,10 (М+1)+
1-(1-фенилэтил)-1Н-пиразол-3-амин
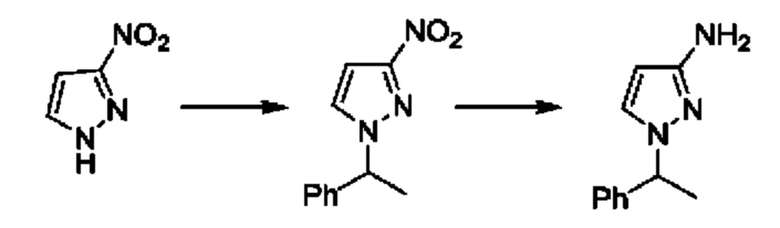
Раствор 3-нитро-1H-пиразола (1 г, 8,85 ммоль) в ТГФ (20 мл) охладили до 0°С и добавили NaH (0,7 г, 17,7 ммоль). Суспензию перемешивали в течение 30 мин, затем добавили по каплям (1-бромэтил)бензол (1,96 г, 10,6 ммоль). Реакционную смесь нагревали при 80°С на протяжении ночи или до завершения, охладили до температуры окружающей среды, разбавили водой (40 мл) и экстрагировали EtOAc (2 × 50 мл). Органические фазы промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 20% EtOAc-гексаны с получением 3-нитро-1-(1-фенилэтил)-1Н-пиразола в виде желтой жидкости (1,2 г, 63%).
1Н-ЯМР (300 МГц, CDCl3) δ 7,46-7,18 (м, 6Н), 6,88 (д, J=2,5 Гц, 1H), 5,59 (кв, J=7,1 Гц, 1H), 1,96 (д, J=7,1 Гц, 3Н).
Раствор 3-нитро-1-(1-фенилэтил)-1Н-пиразола (1 г, 4,6 ммоль) в ТГФ (20 мл) и МеОН (5 мл) охладили до 0°С. Добавили порошок цинка (1,49 г, 23,04 ммоль) и раствор NH4Cl (1,23 г, 23,04 ммоль; в 5 мл воды). Полученную реакционную смесь перемешивали в течение 30 мин, затем нагревали при 80°С в течение 6 ч. Реакционную смесь охладили до температуры окружающей среды, разбавили EtOAc (50 мл) и фильтровали через слой целита. Органическую фазу промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 50% EtOAc-гексаны с получением 1-(1-фенилэтил)-1H-пиразол-3-амина в виде желтой жидкости (0,85 г, 67%). 1Н-ЯМР (300 МГц, CDCl3) δ=7,39-7,04 (м, 6Н), 5,59 (д, J=2,4 Гц, 1H), 5,35 (кв, J=7,1 Гц, 1Н), 3,83 (с, 2Н), 1,78 (д, J=7,1 Гц, 3Н).
1-(2-(пиперидин-1-ил)этил)-1Н-пиразол-3-амин
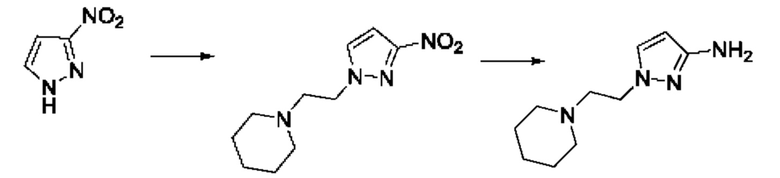
На раствор 3-нитро-1Н-пиразола (2 г, 17,7 ммоль) в ДМФА (20 мл) подействовали гидрохлоридом 1-(2-хлорэтил)пиперидина (4,8 г, 26,5 ммоль) при температуре окружающей среды. Раствор охладили до 0°С и подействовали K2CO3 (6,1 г, 44,27 ммоль) порциями на протяжении 5 мин. Полученную реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Реакционную смесь разбавили водой и экстрагировали этилацетатом (2 × 40 мл). Объединенные органические фазы промыли водой (40 мл), солевым раствором (40 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 25% EtOAc-гексаны с получением 1-(2-(3-нитро-1Н-пиразол-1-ил)этил)пиперидина в виде бледно-желтого твердого вещества (2,5 г, 64%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,60 (д, J=2,4 Гц, 1Н), 6,81 (д, J=2,4 Гц, 1H), 4,29 (т, J=6,4 Гц, 2Н), 2,78 (т, J=6,4 Гц, 2Н), 2,41 (с, 4Н), 1,57-1,53 (м, 4Н), 1,45 (т, J=6 Гц, 2Н). ЖХМС (m/z): 225,10 (М+1)+.
Раствор 1-(2-(3-нитро-1Н-пиразол-1-ил)этил)пиперидина 3 (2,5 г, 11,16 ммоль) в ТГФ (20 мл) и МеОН (5 мл) охладили до 0°С. На раствор последовательно подействовали порошком цинка (3,6 г, 55,8 ммоль) и водного NH4Cl (3 г, 55,8 ммоль), затем раствор прогрели до температуры окружающей среды и перемешивали в течение 5 ч. Реакционную смесь разбавили этилацетатом (50 мл), профильтровали через слой целита и концентрировали при пониженном давлении. Остаток разбавили этилацетатом (60 мл) и промыли водой (40 мл), солевым раствором (40 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1-(2-(пиперидин-1-ил)этил)-1H-пиразол-3-амина в виде светло-желтой жидкости (1,75 г, 81%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,28 (д, J=2 Гц, 1Н), 5,30 (с, J=2 Гц, 1H), 4,50 (с, 2Н), 3,90 (т, J=6,8 Гц, 2Н), 2,5-2,53 (м, 4Н), 2,39-2,33 (м, 6Н), 1,2 (с, 2Н). ЖХМС (m/z): 195,10 (М+1)+.
1,5-диметил-1Н-пиразол-3-амин
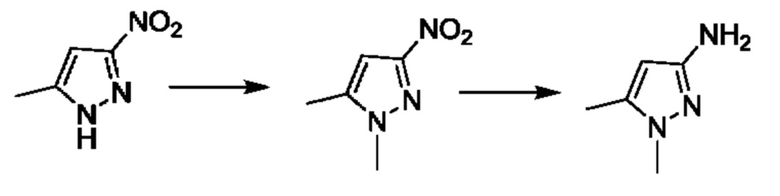
Раствор 5-метил-3-нитро-1H-пиразола (2 г, 15,7 ммоль) в ТГФ (20 мл) охладили до 0°С. Добавили порциями NaH (0,7 г, 17,32 ммоль) в течение 10 мин в атмосфере азота. Полученную суспензию перемешивали в течение 10 мин, затем подействовали Mel (2,2 г, 15,7 ммоль), прогрели до температуры окружающей среды и перемешивали в течение 4 ч. Реакционную смесь разбавили насыщенным раствором NH4Cl (20 мл) и экстрагировали EtOAc (2 × 30 мл). Органические фазы промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1,5-диметил-3-нитро-1Н-пиразола в виде белого твердого вещества (2 г, 91%). 1Н-ЯМР (300 МГц, CDCl3): δ=6,71 (с, 1Н), 3,87 (с, 3H), 2,34 (с, 3H).
В реакционном сосуде к аппарату Парра емкостью 100 мл на раствор 1,5-диметил-3-нитро-1H-пиразола (2 г, 14,18 ммоль) в МеОН (4 мл) и EtOAc (20 мл) подействовали 10% палладия на угле (400 мг) в атмосфере азота. Колбу вакуумировали, затем заполнили газообразным водородом (60 фунт/кв. дюйм (414 кПа)) и перемешивали при температуре окружающей среды в течение 12 ч. Реакционную смесь разбавили этилацетатом (50 мл) и фильтровали через слой целита. Фильтрат высушили (Na2SO4) и концентрировали при пониженном давлении с получением 1,5-диметил-1Н-пиразол-3-амина в виде светло-коричневого твердого вещества (1,36 г, 87%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=5,19 (с, 1H), 4,33 (ш.с, 2Н), 3,43 (с, 3H), 2,07 (с, 3H). ЖХМС (m/z): 112,3 (М+1)+
1-метил-5-(трифторметил)-1Н-пиразол-3-амин
Раствор 1,1,1-триэтоксиэтана (20 г, 123 ммоль) в ДХМ (250 мл) и пиридине (20,5 г, 259 ммоль) охладили до 0°С. Добавили по каплям раствор трифторуксусного ангидрида (52 г, 246 ммоль) в ДХМ (50 мл) на протяжении 30 мин. Реакционную смесь прогрели до температуры окружающей среды, перемешивали в течение 12 ч, затем разбавили насыщенн. водн. раствором NaHCO3 и экстрагировали ДХМ (2 × 250 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением 4,4-диэтокси-1,1,1-трифторбут-3-ен-2-она в виде светло-коричневой жидкости (20 г, 76%). 1Н-ЯМР (400 МГц, CDCl3): δ=4,93 (с, 1Н), 4,39 (кв, J=7,2 Гц, 2Н), 4,18 (кв, J=7,2 Гц, 4Н), 1,46-1,40 (м, 6Н).
На раствор 4,4-диэтокси-1,1,1-трифторбут-3-ен-2-она (10 г, 47,16 ммоль) в ацетонитриле (100 мл) подействовали водным раствором NH3 (15 мл) при 0°С, затем перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении, затем на остаток подействовали водой (250 мл) и экстрагировали ДХМ (2 × 250 мл). Объединенные органические фазы промыли водой (250 мл), солевым раствором (250 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением (E)-4-амино-4-этокси-1,1,1-трифторбут-3-ен-2-она в виде белого с желтоватым оттенком твердого вещества (7,5 г, 87%). 1Н-ЯМР (300 МГц, CDCl3): δ=5,6 (ш.с, 1H), 4,17 (кв, J=7,2 Гц, 2Н), 1,42 (т, J=7,2 Гц, 3H).
На раствор (Е)-4-амино-4-этокси-1,1,1-трифторбут-3-ен-2-она (5 г, 27,3 ммоль) в EtOH (30 мл) подействовали сульфатом метилгидразина (4,72 г, 32,8 ммоль) и Et3N (4,1 г, 41,0 ммоль) при температуре окружающей среды. Полученную реакционную смесь нагревали при 85°С в течение 12 ч, затем охладили до температуры окружающей среды и концентрировали при пониженном давлении. Полученный остаток разбавили насыщенн. водн. раствором NaHCO3 (250 мл) и экстрагировали EtOAc (2 × 250 мл). Объединенные органические фазы промыли водой (250 мл), солевым раствором (250 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 20% EtOAc-гексаны с получением 1-метил-5-(трифторметил)-1Н-пиразол-3-амина в виде светло-коричневой жидкости (0,17 г, 38%). 1Н-ЯМР (400 МГц, CDCl3): δ=5,93 (с, 1Н), 3,79 (с, 3H), 3,68 (ш.с, 2Н).
1-метил-5-(проп-1-ен-2-ил)-1Н-пиразол-3-амин
На раствор 1-метил-1Н-пиразол-3-амина (2 г, 20,6 ммоль) в АсОН (50 мл) подействовали 2,5-гександионом (4,9 г, 43,29 ммоль) при температуре окружающей среды в атмосфере азота. Полученную реакционную смесь грели до 100°С в течение 1 ч, затем перемешивали при температуре окружающей среды в течение 5 ч. Реакционную смесь концентрировали при пониженном давлении и подвергли азеотропной отгонке с толуолом. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением 3-(2,5-диметил-1Н-пиррол-1-ил)-1-метил-1Н-пиразола в виде жидкости (2,5 г, 69%). 1Н-ЯМР (300 МГц, CDCl3): δ=7,39 (д, J=2,1 Гц, 1Н), 6,15 (д, J=2,4 Гц, 1H), 5,84 (с, 2Н), 3,92 (с, 3H), 2,10 (с, 6Н).
Раствор 3-(2,5-диметил-1Н-пиррол-1-ил)-1-метил-1Н-пиразола (1 г, 5,71 ммоль) в сухом ТГФ (10 мл) охладили до -78°С в атмосфере азота, к указанному раствору добавили по каплям H-BuLi (1,6 моль/л в гексанах, 4,4 мл, 6,86 ммоль) на протяжении 10 минут, затем перемешивали при -78°С в течение 1 ч с последующим воздействием раствором I2 (1,54 г, 5,71 ммоль) в ТГФ (5 мл) при -78°С, перемешивание продолжали при этой температуре до завершения (2 ч). Реакционную смесь разбавили насыщенн. водн. раствором NH4Cl и экстрагировали этилацетатом (2 × 25 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 50% EtOAc-гексаны с получением 3-(2,5-диметил-1H-пиррол-1-ил)-5-йод-1-метил-1H-пиразола в виде белого с желтоватым оттенком твердого вещества (0,75д, 43,6%). 1Н-ЯМР (400 МГц, CDCl3): δ=6,33 (с, 1H), 5,84 (с, 2Н), 3,95 (с, 3H), 2,09 (с, 6Н).
На раствор 3-(2,5-диметил-1H-пиррол-1-ил)-5-йод-1-метил-1H-пиразола (1 г, 3,32 ммоль) в смеси ДМЭ:вода (8:2, 10 мл) подействовали 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2-диоксабороланом (0,67 г, 3,98 ммоль) и Na2CO3 (0,52 г, 4,98 ммоль) при температуре окружающей среды в атмосфере азота. Полученный раствор дегазировали посредством продувки аргоном в течение 15 мин, затем подействовали Pd(PPh3)4 (190 мг, 0,166 ммоль) в атмосфере аргона. Полученную смесь нагревали при 90°С в течение 24 ч, затем охладили до температуры окружающей среды и концентрировали при пониженном давлении. Полученный остаток разбавили холодной водой (20 мл) и экстрагировали EtOAc (2 × 20 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% EtOAc-гексаны с получением 3-(2,5-диметил-1H-пиррол-1-ил)-1-метил-5-(проп-1-ен-2-ил)-1H-пиразола в виде бледно-желтой жидкости (0,765 г, 92%). 1Н-ЯМР (300 МГц, CDCl3): δ=6,08 (с, 1Н), 5,84 (с, 2Н), 5,39 (с, 1H), 5,23 (с, 1H), 3,92 (с, 3H), 2,12 (с, 9Н).
На раствор 3-(2,5-диметил-1H-пиррол-1-ил)-1-метил-5-(проп-1-ен-2-ил)-1H-пиразола (0,7 г, 3,25 ммоль) в EtOH-H2O (8:2, 12 мл) подействовали NH2OH.HCl (2,26 г, 32,55 ммоль) и KOH (1,8 г, 32,55 ммоль). Полученную реакционную смесь нагревали при 100°С в течение 48 ч. Реакционную смесь охладили и концентрировали при пониженном давлении. На остаток подействовали насыщенным NaHCO3 с получением раствора с рН~8, затем экстрагировали EtOAc (2 × 50 мл). Объединенные органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 100% EtOAc с получением 1-метил-5-(проп-1-ен-2-ил)-1H-пиразол-3-амина в виде светло-коричневой жидкости (0,4 г, 91%). 1Н-ЯМР (400 МГц, CDCl3): δ=5,54 (с, 1H), 5,27 (с, 1H), 5,08 (с, 1H), 3,71 (с, 3H), 2,41 (с, 2Н), 1,92 (с, 3H).
Этил-1-бензил-3-нитро-1Н-пиразол-5-карбоксилат
На раствор 3-нитро-1H-пиразол-5-карбоновой кислоты (5 г, 31,8 ммоль) в этаноле (50 мл) подействовали тионилхлоридом (4,5 г, 38,2 ммоль) по каплям на протяжении 10 мин при 0°С в атмосфере азота. Полученную смесь перемешивали при 80°С в течение 6 ч, затем охладили до температуры окружающей среды и концентрировали при пониженном давлении. Повысили основность полученного остатка до рН 8 насыщенным раствором NaHCO3 с последующим экстрагированием этилацетатом (2 × 100 мл). Объединенные органические фазы промыли водой (100 мл), солевым раствором (100 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт растерли с диэтиловым эфиром, отфильтровали и высушили при пониженном давлении с получением этил-3-нитро-1Н-пиразол-5-карбоксилата в виде белого твердого вещества (5 г, 85%). 1Н-ЯМР (400 МГц, -d6): δ=7,44 (с, 1H), 4,36 (кв, J=6,8 Гц, 2Н), 1,33 (т, J=7,2 Гц, 3H). ЖХМС (m/z): 184 (М-1)-.
Этил-3-нитро-1H-пиразол-5-карбоксилат (1 г, 5,4 ммоль) растворили в ДМФА (10 мл) при температуре окружающей среды и подействовали на него (1,34 г, 9,7 ммоль). Полученную смесь охладили до 0°С и добавили по каплям метил йодид (1,15 г, 8,1 ммоль), реакционную смесь герметизировали, оставили прогреться до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь разбавили водой (20 мл) и экстрагировали EtOAc (2 × 30 мл).
Объединенные органические фазы промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% EtOAc-гексаны с получением этил-1-метил-3-нитро-1H-пиразол-5-карбоксилата в виде белого твердого вещества (0,65 г, 61%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=7,54 (с, J=1,1 Гц, 1Н), 4,35 (кв, J=7,1 Гц, 2Н), 4,19 (д, J=1,2 Гц, 3H), 1,33 (т, J=7,1 Гц, 3H).
Этил-1-метил-3-нитро-1H-пиразол-5-карбоксилат (0,65 г, 3,3 ммоль) растворили в ТГФ (20 мл) и МеОН (5 мл) при 0°С. Добавили последовательно порошок цинка (1,0 г, 16,3 ммоль) и водный NH4Cl (0,87 г, 16,3 ммоль). Полученную реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч, затем нагрели до 70°С в течение 1 часа. Растворители удалили при пониженном давлении. Полученный остаток растворили в EtOAc (30 мл) и профильтровали через слой целита. Фильтрат промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением этил-3-амино-1-метил-1H-пиразол-5-карбоксилата в виде белого твердого вещества (0,5 г, 91%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 5,95 (с, 1H), 4,24 (кв, J=7,1 Гц, 2Н), 3,83 (с, 3H), 3,41 (с, 2Н), 1,27 (т, J=7,1 Гц, 4Н).
Этил-1-бензил-3-нитро-1Н-пиразол-5-карбоксилат
На раствор 3-нитро-1H-пиразол-5-карбоновой кислоты (5 г, 31,8 ммоль) в этаноле (50 мл) подействовали тионилхлоридом (4,5 г, 38,2 ммоль) по каплям на протяжении 10 мин при 0°С в атмосфере азота. Полученную смесь перемешивали при 80°С в течение 6 ч, затем охладили до температуры окружающей среды и концентрировали при пониженном давлении. Повысили основность полученного остатка до рН 8 насыщенным раствором NaHCO3 с последующим экстрагированием этилацетатом (2 × 100 мл). Объединенные органические фазы промыли водой (100 мл), солевым раствором (100 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт растерли с диэтиловым эфиром, отфильтровали и высушили при пониженном давлении с получением этил-3-нитро-1Н-пиразол-5-карбоксилата в виде белого твердого вещества (5 г, 85%). 1Н-ЯМР (400 МГц, -d6): δ=7,44 (с, 1H), 4,36 (кв, J=6,8 Гц, 2Н), 1,33 (т, J=7,2 Гц, 3H). ЖХМС (m/z): 184 (М-1)-.
Этил-3-нитро-1H-пиразол-5-карбоксилат (2 г, 10,8 ммоль) растворили в ДМФА (10 мл) при температуре окружающей среды и подействовали (3 г, 21,6 ммоль). Полученную смесь охладили до 0°С и добавили по каплям бензилбромид (2,7 г, 16,2 ммоль), реакционную смесь оставили прогреться до температуры окружающей среды и перемешивали в течение 2 ч. Реакционную смесь разбавили водой (20 мл) и экстрагировали EtOAc (2 × 30 мл). Объединенные органические фазы промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% EtOAc-гексаны с получением этил-1-бензил-3-нитро-1Н-пиразол-5-карбоксилата в виде белого твердого вещества (1,2 г, 40%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,44 (с, 1Н), 7,34-7,31 (м, 5Н), 5,83 (с, 2Н), 4,39 (кв, J=7,2 Гц, 2Н), 1,38 (т, J=7,2 Гц, 3H). ЖХМС (m/z): 276,15 (М
Этил-1 -бензил-3-нитро-1H-пиразол-5-карбоксилат (1,2 г, 4,36 ммоль) растворили в ТГФ (20 мл) и МеОН (5 мл) при 0°С. Добавили последовательно порошок цинка (1,4 г, 21,8 ммоль) и водный NH4Cl (1,16 г, 21,8 ммоль). Полученную реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч, затем концентрировали при пониженном давлении. Полученный остаток растворили в EtOAc (30 мл) и фильтровали через слой целита. Фильтрат промыли водой (30 мл), солевым раствором (30 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением этил-3-амино-1-бензил-1H-пиразол-5-карбоксилата в виде белого твердого вещества (1 г, 94%). 1Н-ЯМР (300 МГц, ДМСО-d6): δ=7,30-7,19 (м, 3H), 7,11-7,08 (м, 2Н), 6,00 (с, 1H), 5,43 (с, 2Н), 4,91 (с, 2Н), 4,23 (кв, J=7,2 Гц, 2Н), 1,24 (т, J=7,2 Гц, 3H). ЖХМС (m/z): 245,9 (М+1)+.
3-(2,5-диметил-1Н-пиррол-1-ил)-1-фенил-1Н-пиразол

На раствор 1-фенил-1H-пиразол-3-амина (3,5 г, 21,9 ммоль) в уксусной кислоте (20 мл) подействовали 2,5-гександионом (5,2 г, 45,9 ммоль) и нагревали при 100°С в течение 4 ч. Смесь охладили и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% EtOAc-гексаны с получением 3-(2,5-диметил-1H-пиррол-1-ил)-1-фенил-1H-пиразола в виде бесцветной жидкости (2,8 г, 54%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,98 (с, 1H), 7,74 (д, J=8,4 Гц, 2Н), 7,49 (д, J=8,4 Гц, 2Н), 7,32 (т, J=7,6 Гц, 1H), 6,39 (с, 1H), 5,9 (с, 2Н), 2,19 (с, 6Н).
На раствор 3-(2,5-диметил-1H-пиррол-1-ил)-1-фенил-1H-пиразола (2,7 г, 11,4 ммоль) в ТГФ (70 мл) при -78°С подействовали по каплям н-BuLi (1,6 моль/л в ТГФ, 10 мл, 23,91 ммоль) в течение 10 мин. Реакционную смесь перемешивали при -78°С в течение 1,5 ч, затем подействовали свежевысушенным ацетоном (1 г, 17,0 ммоль) и перемешивание продолжали при -78°С в течение 1,5 ч. Реакционную смесь разбавили насыщенным хлоридом аммония (2 мл), концентрировали при пониженном давлении, затем распределили между водой (100 мл) и этилацетатом (100 мл). Органический экстракт промыли водой (100 мл), солевым раствором (100 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 20% EtOAc-гексаны с получением 2-(3-(2,5-диметил-1H-пиррол-1-ил)-1-фенил-1H-пиразол-5-ил)пропан-2-ола в виде белого с желтоватым оттенком твердого вещества (1,4 г, 42%). 1Н-ЯМР (400 МГц, CDCl3): δ=7,57-7,56 (м, 2Н), 7,47-7,46 (м, 3H), 6,23 (с, 1Н), 5,85 (с, 2Н), 2,19 (с, 6Н), 1,52 (с, 6Н). ЖХМС (m/z): 296,1 (М+1)+.
В герметизируемом реакционном сосуде емкостью 100 мл 2-(3-(2,5-диметил-1H-пиррол-1-ил)-1-фенил-1H-пиразол-5-ил)пропан-2-ол(1,4 г, 4,74 ммоль) растворили в - (1:1, 50 мл) при температуре окружающей среды. Добавили последовательно гидрохлорид гидроксиламина (3,3 г, 47,45 ммоль) и KOH (2,6 г, 47,45 ммоль) и полученную реакционную смесь нагревали при 120°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении, разбавили водой (50 мл) и экстрагировали этилацетатом (2 × 50 мл). Органические экстракты промыли водой (50 мл), солевым раствором (50 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 50% EtOAc-гексаны с получением 2-(3-амино-1-фенил-1Н-пиразол-5-ил)пропан-2-ола в виде бесцветной жидкости (0,8 г, 78%). ЖХМС (m/z): 218,1 (М+1)+.
8-бром-1,2,3,5,6,7-гексагидро-5-индацен-4-амин

N-Бромсукцинимид (1,02 г, 5,78 ммоль) добавили порциями к раствору 1,2,3,5,6,7-гексагидро-s-индацен-4-амина (1 г, 5,78 ммоль) в ДХМ (20 мл) при 0°С. Раствор постепенно прогрели до температуры окружающей среды и перемешивали в течение 12 ч. Реакционную смесь разбавили насыщенным водным Na2S2O3 (50 мл) и экстрагировали ДХМ (2 × 25 мл). Объединенные органические экстракты промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% EtOAc-гексан с получением 8-бром-1,2,3,5,6,7-гексагидро-s-индацен-4-амина в виде коричневого твердого вещества (1,2 г, 83%). 1Н-ЯМР (300 МГц, CDCl3): δ=3,45 (ш.с, 2Н), 2,92-2,88 (м, 4Н), 2,81-2,77 (м, 4Н), 2,16-2,09 (м, 4Н); ЖХ-МС 94% (210 нм); m/z 252,15 [М+Н]+.
8-хлор-1,2,3,5,6,7-гексагидро-5-индацен-4-амин
N-Хлорсукцинимид (0,46 г, 3,46 ммоль) добавили порциями к раствору 1,2,3,5,6,7-гексагидро-s-индацен-4-амина, 1 (0,6 г, 3,46 ммоль) в CHCl3 (10 мл) при 0°С. Раствор постепенно прогрели до температуры окружающей среды и перемешивали в течение 10 ч. Реакционную смесь разбавили насыщенным водным Na2S2O3 (50 мл) и экстрагировали ДХМ (2 × 25 мл). Объединенные органические экстракты промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% -гексан с получением 8-хлор-1,2,3,5,6,7-гексагидро-s-индацен-4-амина в виде коричневого твердого вещества (0,45 г, 63%). 1Н-ЯМР (300 МГц, CDCl3): δ=2,94 (т, J=7,2 Гц, 4Н), 2,77 (т, J=8,1 Гц, 4Н), 2,18 (м, 4Н); m/z 207,8 [М+Н]+.
8-метил 1,2,3,5,6 7-гексагидро-s-индацен-4-амин
8-Бром-1,2,3,5,6,7-гексагидро-s-индацен-4-амин (400 мг, 1,59 ммоль) растворили в смеси 1,4-диоксан-вода (8:2, 10 мл) и реакционную колбу продули аргоном в течение 15 мин. Последовательно добавили (650 мг, 4,78 ммоль), метилбороновую кислоту (100 мг, 1,75 ммоль) и Pd(PPh3)4 (100 мг, 0,079 ммоль) в атмосфере аргона. Полученную смесь загерметизировали и нагревали при 100°С в течение 2 ч. Реакционную смесь охладили, разбавили водой и экстрагировали с помощью EtOAc (2 × 20 мл). Объединенные органические экстракты промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% - гексан с получением 8-метил-1,2,3,5,6,7-гексагидро-s-индацен-4-амина в виде бесцветной жидкости (0,220 г, 76%). 1Н-ЯМР (400 МГц, CDCl3): 5=3,41 (ш.с, 2Н), 2,88-2,8 (м, J=7,5 Гц, 4Н), 2,75-2,67 (м, 4Н), 2,18-2,09 (м, 7Н); m/z 188,2 [М+Н]+.
3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-амин
Раствор 2,3-дигидробензофуран-5-карбальдегида, (10 г, 67,6 ммоль), малоновой кислоты (10,5 г, 101,35 ммоль) и пиперидина (0,47 мл, 4,73 ммоль, 0,07 экв.) нагревали в пиридине (60 мл) при 100°С в течение 5 ч. Реакционную смесь подкислили до ~рН 3 с помощью 1N HCl и продукт экстрагировали с помощью смеси 10% ИПС/хлороформ (2 × 250 мл). Объединенные органические экстракты промыли водой (250 мл), солевым раствором (250 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт растерли с диэтиловым эфиром с получением
(Е)-3-(2,3-дигидробензофуран-5-ил)акриловой кислоты в виде желтого твердого вещества (10 г, 78%). 1Н-ЯМР (300 МГц, Хлороформ-d) δ=7,73 (д, J=15,9 Гц, 1H), 7,43 (с, 1H), 7,33 (дд, J=8,1, 1,8 Гц, 1H), 6,80 (д, J=8,1 Гц, 1H), 6,29 (д, J=15,9 Гц, 1H), 4,64 (т, J=8,7 Гц, 2Н), 3,24 (т, J=8,7 Гц, 2Н).
На раствор (Е)-3-(2,3-дигидробензофуран-5-ил)акриловой кислоты (8,0 г, 42,1 ммоль) в уксусной кислоте (80 мл) и воде (1,0 мл) подействовали 10% палладия на угле (1,0 г) двумя порциями. Реакционную смесь перемешивали в атмосфере газообразного водорода (надувной шар) до завершения, как правило, 4 ч. Смесь разбавили этилацетатом (100 мл) и фильтровали через слой целита с промывкой дополнительным количеством этилацетата. Растворители удалили при пониженном давлении и технический остаток подвергли азеотропной отгонке с толуолом (2 × 50 мл) с получением белого с желтоватым оттенком твердого вещества, которое растерли с диэтиловым эфиром (50 мл) с получением 3-(2,3-дигидробензофуран-5-ил)пропионовой кислоты в виде белого твердого вещества (6,5 г, 80%). 1Н-ЯМР (400 МГц, CDCl3) δ=7,04 (с, 1H), 6,93 (д, J=8,4, 1H), 6,7 (д, J=8,4 Гц, 1H), 4,55 (т, J=8,4 Гц, 2Н), 3,18 (т, J=8,4 Гц, 2Н), 2,89 (т, J=7,6 Гц, 2Н), 2,64 (т, J=7,6 Гц, 2Н).
Раствор 3-(2,3-дигидробензофуран-5-ил)пропионовой кислоты (6,0 г, 31 ммоль) в тионилхлориде (8 мл) нагревали при 80°С в течение 1 ч. По завершении реакции тионилхлорид удалили при пониженном давлении и технический 3-(2,3-дигидробензофуран-5-ил)пропаноилхлорид растворили в безводном 1,2-дихлорэтане (30 мл). В отдельной колбе к безводному 1,2-дихлорэтану (40 мл) добавили трихлорид алюминия (2 г, 15 ммоль) при 0°С, а затем раствор хлорангидрида кислоты (10 мл) по каплям в течение 5 мин и полученный раствор перемешивали в течение 30 мин при 0°С. Добавили дополнительную порцию трихлорида алюминия (3 г, 22,5 ммоль), с последующим добавлением по каплям оставшегося раствора хлорангидрида кислоты (20 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч или до завершения, разбавили водой и экстрагировали с помощью (2 × 50 мл). Объединенные органические экстракты промыли 1N HCl (50 мл), 1N NaOH (50 мл), водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением 2,3,5,6-тетрагидро-7Н-индено[5,6-b]фуран-7-она в виде белого твердого вещества (3,8 г, 70%). 1Н-ЯМР (300 МГц, CD3OD) δ=7,36 (с, 1Н), 6,91 (с, 1H), 4,61 (т, J=8,6 Гц, 3H), 3,26 (т, J=8,6 Гц, 2Н), 3,05 (т, J=5,5 Гц, 3H), 2,68 (т, J=5,5 Гц, 2Н).
2,3,5,6-тетрагидро-7Н-индено[5,6-b]фуран-7-он (1,5 г, 8,61 ммоль) растворили в конц. H2SO4 (6,0 мл) при 0°С с последующим добавленим по каплям дым. HNO3:конц. H2SO4, 1:1 (1,2 мл), перемешивание продолжали при 0°С в течение 1 ч. Реакционную смесь добавили к ледяной воде (60 мл) и перемешивали в течение 10 мин, полученный светло-коричневый осадок удалили фильтрованием, промыли ледяной водой (20 мл) и высушили в вакууме с получением 8-нитро-2,3,5,6-тетрагидро-7Н-индено[5,6-b]фуран-7-она (1,2 г, 64%). 1Н-ЯМР (300 МГц, CD3OD) δ=7,54 (с, 1H), 4,80 (т, J=8,6 Гц, 2Н), 3,42 (т, J=8,6 Гц, 2Н), 3,09 (т, J=5,6 Гц, 2Н), 2,74 (т, J=5,6 Гц, 2Н).
На раствор 8-нитро-2,3,5,6-тетрагидро-7Н-индено[5,6-b]фуран-7-она (1,0 г, 4,57 ммоль) в метаноле (20 мл) 0°С подействовали метансульфоновой кислотой (0,2 мл), а затем 20% гидроксидом палладия (0,5 г). Реакционную смесь перемешивали в атмосфере водорода при давлении 60 фунт/кв. дюйм (414 кПа) до завершения. Реакционную смесь профильтровали через слой целита с промывкой метанолом (50 мл) и концентрировали при пониженном давлении. Остаток разбавили этилацетатом (50 мл) и промыли насыщенн. водн. NaHCO3 (50 мл), водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% -гексаны с получением 3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-амина в виде белого твердого вещества (0,5 г, 63%). 1Н-ЯМР (300 МГц, CDCl3) δ=6,54 (с, 1H), 5,30 (с, 2Н), 4,61 (т, J=8,7 Гц, 2Н), 3,21 (т, J=8,7 Гц, 2Н), 2,95 (т, J=5,5 Гц, 2Н), 2,66 (т, J=5,5 Гц, 2Н).
4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-амин
На 3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-амин (0,5 г, 2,86 ммоль) и триэтиламин (0,51 мл, 3,71 ммоль) в дихлорметане (6,0 мл) при 0°С подействовали по каплям раствором хлорангидрида пивалиновой кислоты (0,41 г, 3,43 ммоль) в ДХМ (4,0 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 6 ч. Реакционную смесь добавили к насыщенн. водн. NaHCO3 (30 мл), и экстрагировали с помощью ДХМ (2 × 25 мл). Объединенные органические фазы промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении с получением N-(3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)пиваламида в виде белого твердого вещества (0,55 г, 74%). 1Н-ЯМР (300 МГц, Хлороформ-d) δ=6,91 (с, 1H), 4,56 (т, J=8,6 Гц, 2Н), 3,17 (т, J=8,6 Гц, 2Н), 2,83 (т, J=7,4 Гц, 2Н), 2,75 (т, J=7,4 Гц, 2Н), 2,04 (п, J=7,4 Гц, 2Н), 1,32 (с, 9Н).
На N-(3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)пиваламид (0,55 г, 2,12 ммоль) в уксусной кислоте (10 мл) подействовали по каплям раствором брома (0,4 г, 2,55 ммоль) в уксусной кислоте (2,0 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 3 ч. К реакционной смеси добавили ледяную воду и перемешивали в течение 10 мин. Полученный осадок удалили фильтрованием, промыли водой (20 мл) и высушили в вакууме с получением
N-(4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)пиваламида в виде светло-коричневого твердого вещества (0,65 г, 91%). 1Н-ЯМР (300 МГц, Хлороформ-d) δ=6,94 (с, 1Н), 4,61 (т, J=8,7 Гц, 2Н), 3,18 (т, J=8,7 Гц, 2Н), 2,92-2,80 (м, 4Н), 2,06 (п, J=7,4 Гц, 2Н), 1,31 (с, 9Н).
N-(4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)пиваламид (0,6 г, 1,78 ммоль) в EtOH (10 мл) и конц. HCI (15 мл) нагревали при 90°С в течение 36 ч. Раствор концентрировали при пониженном давлении, затем повысили основность с помощью водного раствора NH4OH. Водную фазу экстрагировали с помощью этилацетата (2 × 20 мл) и объединенные органические фазы высушили (Na2SO4) и концентрировали при пониженном давлении с получением 4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-амина в виде коричневого твердого вещества (0,3 г, 67%). 1Н-ЯМР (400 МГц, Хлороформ-d) δ=4,61 (т, J=8,6 Гц, 2Н), 3,49 (с, 2Н), 3,17 (т, J=8,6 Гц, 2Н), 2,84 (т, J=7,4 Гц, 2Н), 2,78 (т, J=7,4 Гц, 2Н),2,12(п, J=7,4 Гц, 2Н).
3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-4-амин
На 3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-амин (0,5 г, 1,98 ммоль) в этаноле (10 мл) и уксусной кислоте (1,5 мл) подействовали раствором нитрата натрия (1,3 г, 19,8 ммоль) в воде (3,0 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 4 ч. Этанол удалили при пониженном давлении, затем остаток разбавили водой (30 мл), экстрагировали с помощью смеси 10% ИПС/хлороформ (2 × 25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% EtOAc-гексаны с получением 4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фурана в виде желтого твердого вещества (0,28 г, 60%).
На 4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран (0,28 г, 1,18 ммоль) в ДМСО (10 мл) подействовали йодидом меди (0,22 г, 1,18 ммоль), L-пролином (0,21 г, 1,88 ммоль) и азидом натрия (0,19 г, 2,94 ммоль). Реакционную смесь нагревали в герметизированном сосуде при 135°С в течение 36 ч. Реакционную смесь охладили, разбавили водой и экстрагировали с помощью EtOAc (2 × 25 мл). Объединенные органические экстракты промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением 3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-4-амина в виде серого твердого вещества (0,17 г, 85%), 1Н-ЯМР (300 МГц, Хлороформ-d) δ=6,21 (с, 1Н), 4,59 (т, J=8,5 Гц, 2Н), 3,51 (с, 1Н), 2,98 (т, J=8,5 Гц, 2Н), 2,83 (т, J=7,5 Гц, 2Н), 2,64 (т, J=7,5 Гц, 2Н), 2,10 (п, J=7,5 Гц, 2Н).
бензо[1,2-b:4,5-b']дифуран-4-амин
2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-карбоновую кислоту (0,8 г, 3,88 ммоль), 2, 3-дихлор-5,6-дицианобензохинон (2,64 г, 11,65 ммоль) в безводном диоксане (20 мл) нагревали в герметичном сосуде при 120°С в течение 18 ч. Реакционную смесь охладили до комнатной температуры и добавили насыщенн. водн. Na2S2O3 (30 мл) с последующей экстракцией этилацетатом (2 × 25 мл). Объединенные органические фазы высушили (Na2SO4) и концентрировали при пониженном давлении с получением технической бензо[1,2-b:4,5-b']дифуран-4-карбоновой кислоты (1,5 г). Техническую кислоту (1,5 г), триэтиламин (2,05 мл) и дифенилфосфорилазид (4,08 г, 14,85 ммоль) в третичном бутаноле (20 мл) нагревали в герметичном сосуде при 90°С в течение 12 ч. Раствор охладили до комнатной температуры, разбавили водой (50 мл) и экстрагировали с помощью (2 × 50 мл). Объединенные органические экстракты промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением трет-бутилбензо[1,2-b:4,5-b']дифуран-4-илкарбамата (0,75 г) с незначительными количествами примесей из фосфинового реагента, продукт растворили в ДХМ (10 мл) и добавили по каплям ТФУК (3,0 мл) в течение 5 мин при 0°С. Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч, затем осторожно прибавили к насыщенн. водн. NaHCO3 (50 мл). Водную фазу экстрагировали с помощью ДХМ (2 × 30 мл) и объединенные органические экстракты промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% EtOAc-гексаны с получением бензо[1,2-b:4,5-b']дифуран-4-амина в виде белого с желтоватым оттенком твердого вещества (0,2 г, 30% по трем стадиям). 1Н-ЯМР (400 МГц, CDCl3): δ=7,6 (д, J=2,2 Гц, 1H), 7,53 (д, J=2,2 Гц, 1Н), 7,12 (с, 1Н), 6,78 (м, 2Н), 4,17 (ш.с, 1Н).
3-(3-(трифторметил)-3H-диазирин-3-ил)анилин
К раствору 2,2,2-трифтор-1-фенилэтан-1-она (5 г, 28,7 ммоль) в конц. H2SO4 (10 мл) при -5°С добавили раствор конц. H2SO4 и дым. HNO3 (1:1, 16 мл) и реакционную смесь перемешивали в течение 3 часов. Полученный раствор вылили на лед с водой (100 мл) и экстрагировали с помощью этилацетата (2 × 100 мл). Объединенные органические фазы промыли водой (100 мл), солевым раствором (100 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 20% EtOAc-гексаны с получением 2,2,2-трифтор-1-(3-нитрофенил)этан-1-она в виде желтой жидкости (4,2 г, 67%).
1Н-ЯМР (400 МГц, CDCl3) δ=8,92 (с, 1H), 8,59 (дд, J=8,1, 1,4 Гц, 1H), 8,41 (д, J=7,8 Гц, 1H), 7,82 (т, J=8,1 Гц, 1Н). 19F-ЯМР (233,33 МГц, CDCl3): -71,82 (с, 3F).
Раствор 2,2,2-трифтор-1-(3-нитрофенил)этан-1-она (4,2 г, 19,2 ммоль), гидрохлорида гидроксиламина (4,0 г, 57,5 ммоль) и пиридина (25 мл) в этаноле (25 мл) нагревали с обратным холодильником в течение 3 ч. или до завершения. Растворитель удалили при пониженном давлении и технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 40% - гексаны с получением оксима 2,2,2-трифтор-1-(3-нитрофенил)этан-1-она в виде бесцветной жидкости (4,0 г, 89%). 19F-ЯМР (233,33 МГц, CDCl3): -66,42 и 62,28 (Е- и Z-оксим).
Раствор оксима 2,2,2-трифтор-1-(3-нитрофенил)этан-1-она (4,0 г, 17,1 ммоль) в дихлорметане (20 мл) охладили до 0°С, подействовали на него триэтиламином (1,5 экв.), N,N-диметиламинпиридином (0,5 экв.), тозилхлоридом (1,1 экв.) и перемешивали при температуре окружающей среды до завершения, как правило, 16 ч. Реакционную смесь разбавили дихлорметаном (50 мл), промыли насыщенн. водн. NH4Cl (100 мл), водой (100 мл), солевым раствором (100 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 5% -гексаны с получением 2,2,2-трифтор-1-(3-нитрофенил)этан-1-он-O-тозилоксима в виде белого твердого вещества (4,0 г, 60%). 1Н-ЯМР (300 МГц, CDCl3) δ=8,41 (ддд, J=5,6, 3,5, 2,2 Гц, 1Н), 8,21 (т, J=1,5 Гц, 1Н), 7,89 (д, J=8,3 Гц, 2Н), 7,81 - 7,65 (м, 2Н), 7,42 (д, J=7,8 Гц, 2Н), 2,50 (с, 3H). 19F-ЯМР (282 МГц, CDCl3) δ -61,55, -66,90.
Раствор 2,2,2-трифтор-1-(3-нитрофенил)этан-1-он-O-тозилоксима (4,0 г, 10,3 ммоль) в диэтиловом эфире охладили до -78°С и пропускали через него раствор аммиака в течение 30 мин. Реакционную смесь герметизировали, оставили прогреться до температуры окружающей среды, затем перемешивали в течение 16 ч. Смесь профильтровали через слой целита и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 7% -гексаны с получением 3-(3-нитрофенил)-3-(трифторметил)диазирина в виде бесцветной жидкости (2,4 г, 100%). 1Н-ЯМР (300 МГц, CDCl3) δ=8,52 (т, J=2,0 Гц, 1Н), 8,33 (ддд, J=8,3, 2,3, 1,1 Гц, 1H), 7,99 (д, J=8,0 Гц, 1Н), 7,65 (тт, J=7,8, 0,4 Гц, 1Н), 2,95 (д, J=8,8 Гц, 1Н), 2,31 (д, J=8,9 Гц, 1H). 19F-ЯМР (282 МГц, CDCl3) δ=-75,10.
На раствор 3-(3-нитрофенил)-3-(трифторметил)диазирина (2,4 г, 10,3 ммоль) в метаноле (30 мл) подействовали триэтиламином (2 экв.) и йодом (1 экв.) и реакционную смесь перемешивали до завершения, как правило, 2 ч. Раствор разбавили диэтиловым эфиром, промыли 10% водной лимонной кислотой, водой, водным тиосульфатом натрия, солевым раствором затем высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% -гексаны с получением 3-(3-нитрофенил)-3-(трифторметил)-3H-диазирина в виде бесцветной жидкости (2,1 г, 88%). 1Н-ЯМР (300 МГц, CDCl3) δ 8,30 (ддд, J=7,9, 2,2, 1,4 Гц, 1Н), 8,09-8,01 (м, 1H), 7,70-7,54 (м, 2Н). 19F-ЯМР (282 МГц, CDCl3) δ=-65,14.
На 3-(3-нитрофенил)-3-(трифторметил)-3H-диазирин (3,0 г, 13 ммоль) в ТГФ (70 мл) подействовали раствором дитионата натрия (10 экв.) в воде (30 мл) и смесь перемешивали при температуре окружающей среды до завершения, как правило, на протяжении ночи. Раствор разбавили водой, экстрагировали с помощью этилацетата (х2), промыли водой, солевым раствором, затем высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 40% -гексаны с получением указанного в подзаголовке соединения, 3-(3-(трифторметил)-3H-диазирин-3-ил)анилина, в виде желтого твердого вещества (1,5 г, 58%). 1Н-ЯМР (300 МГц, CDCl3) δ=7,16 (т, J=7,9 Гц, 1H), 6,70 (ддд, J=8,1, 2,3, 0,9 Гц, 1H), 6,52 (ддт, J=7,9, 1,9, 0,9 Гц, 1Н), 6,45 (шс, 1H), 3,77 (с, 2Н). 19Р-ЯМР (282 МГц, CDCl3) 5 -65,07.
4,6-ди-трет-бутилпиримидин-2-амин
2,4,6-Трихлорпиримидин (2,7 г, 14,7 ммоль) растворили в безводном ТГФ (30 мл) при 0°С в атмосфере азота. К вышеупомянутому раствору добавили Cul (280 мг, 1,47 ммоль) и затем подействовали трет-бутилмагнийхлоридом 2 моль/л в ТГФ (3,78 г, 16,15 мл, 32,3 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 3 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl и экстрагировали EtOAc (2 × 50 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 100% гексаны с получением 4,6-ди-трет-бутил-2-хлорпиримидина (1,3 г, 39%) в виде светло-коричневой жидкости. 1Н-ЯМР (300 МГц, CDCl3): δ=7,20 (с, 1Н), 1,33 (с, 18Н). ЖХМС (m/z): 227,3 [М+Н]+
В герметизируемом реакционном сосуде емкостью 100 мл раствор 4,6-ди-трет-бутил-2-хлорпиримидина (1,3 г) в EtOH (15 мл) охладили до -50°С. Через вышеупомянутый раствор пропускали газообразный аммиак в течение 15 мин. Реакционную смесь нагрели до 70°С и перемешивали в течение 12 ч. По завершении реакционную смесь концентрировали при пониженном давлении и полученный остаток разбавили водой и экстрагировали этилацетатом (50 мл). Органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 4,6-ди-трет-бутилпиримидин-2-амина (0,7 г, 59%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=6,64 (с, 1H), 4,83 (с, 2Н), 1,26 (с, 18Н). ЖХМС (m/z): 208,4 [М+Н]+
4-хлор-2,6-диизопропиланилин
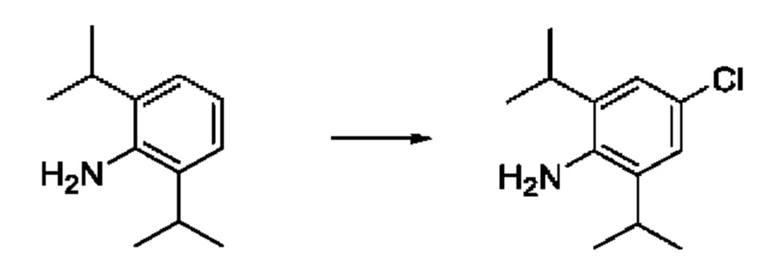
На 2,6-диизопропиланилин (5,0 г, 28,2 ммоль) в ДМФА (100 мл) подействовали N-хлорсукцинимидом (3,97 г, 29,7 ммоль) и реакционную смесь перемешивали при комнатной температуре на протяжении ночи. Раствор вылили в воду (500 мл) и экстрагировали с помощью диэтилового эфира (2 × 150 мл). Объединенные органические фазы промыли водой (2 × 200 мл), солевым раствором (200 мл), высушили (MgSO4) и концентрировали при пониженном давлении. Продукт очистили посредством перегонки на коротком пути с получением указанного в подзаголовке соединения в виде красного масла (3,0 г, 50%). 1Н-ЯМР (600 МГц, ДМСО-d6): δ=6,84 (с, 2Н), 4,75 (с, 2Н), 3,01 (гепт, J=6,8 Гц, 2Н), 1,13 (д, J=6,8 Гц, 12Н). 13С-ЯМР (151 МГц, ДМСО-d6): δ=141,1, 133,8, 122,5, 120,5, 27,2, 22,8.
4-хлор-2,6-дициклопропиланилин
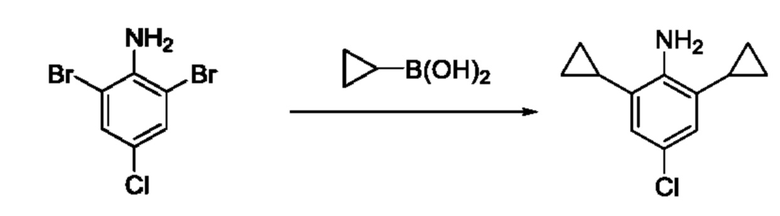
В герметизируемом реакционном сосуде емкостью 50 мл раствор 2,6-дибром-4-хлоранилина (0,25 г, 0,88 ммоль) и циклопропилбороновую кислоту (0,22 г, 2,62 ммоль) вместе с K3PO4 (0,74 г, 3,50 ммоль) растворили в смеси толуол:вода (10 мл:1 мл). Полученный раствор дегазировали посредством продувки азотом в течение 5 минут. Добавили Pd(OAc)2 (20 мг, 0,087 ммоль) и трициклогексилфосфин (25 мг, 0,087 ммоль) и раствор продували газообразным азотом в течение еще 5 минут. Полученную смесь перемешивали при 100°С в течение 12 ч. По завершении реакции смесь разбавили водой (25 мл), экстрагировали EtOAc (2 × 25 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 5% EtOAc-гексаны с получением 4-хлор-2,6-дициклопропиланилина (150 мг, 83%) в виде коричневой жидкости. 1Н-ЯМР (300 МГц, ДМСО-d6): δ=6,69 (с, 2Н), 4,98 (с, 2Н), 1,74-1,64 (м, 2Н), 0,90-0,84 (м, 4Н), 0,52 -0,47 (м, 4Н). ЖХМС (m/z): 208,30 [М+Н]+.
Синтез 4-хлор-2-метил-6-(трифторметил)анилина
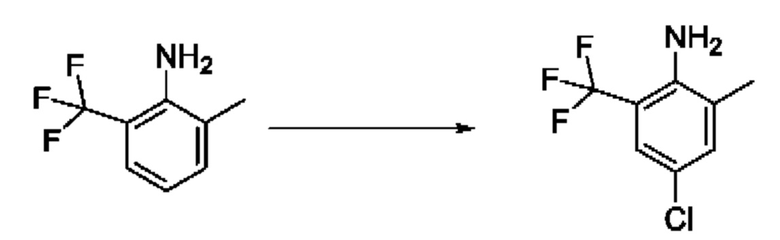
Раствор 2-метил-6-(трифторметил)анилина (0,4 г, 2,20 ммоль) в ацетонитриле (4 мл) и АсОН (0,3 мл) охладили до 0°С. Добавили N-хлорсукцинимид (0,36 г, 2,70 ммоль) при 0°С и раствор затем оставили прогреться до комнатной температуры и перемешивали в течение 12 ч. По завершении реакции реакционную смесь разбавили ледяной водой и полученный осадок удалили фильтрованием и промыли последовательно насыщенным NaHCO3, раствором Na2S2O3, н-пентаном и высушили в вакууме с получением 4-хлор-2-метил-6-(трифторметил)анилина (0,25 г, 52%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ=7,30 (д, J=2,4 Гц, 1H), 7,18 (д, J=2,0 Гц, 1H), 2,17 (с, 3H). 19F-ЯМР (400 МГц, CDCl3): δ=-63,03
4-хлор-2,6-диэтил анилин
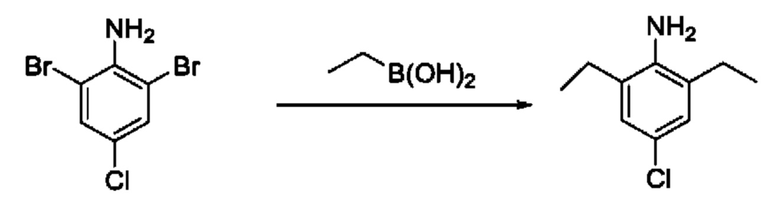
На раствор 2,6-дибром-4-хлоранилина (0,5 г, 1,75 ммоль) и этилбороновой кислоты (0,4 г, 5,25 ммоль) в толуоле (15 мл) и воде (4 мл) подействовали (1,5 г, 7,0 ммоль) при комнатной температуре в атмосфере аргона. Продували раствор аргоном в течение 5 минут с последующим воздействием Pd(OAc)2 (40 мг, 0,175 ммоль) и трициклогексилфосфином (50 мг, 0,175 ммоль). Реакционную смесь снова продували аргоном в течение 5 минут. Полученную смесь перемешивали при 100°С в течение 12 ч. По завершении реакционную смесь разбавили водой, экстрагировали EtOAc (2 × 25 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 8% EtOAc-гексаны с получением 4-хлор-2,6-дициклопропиланилина (100 мг, 31%) в виде желтой жидкости. 1Н-ЯМР (300 МГц, ): δ=6,94 (с, 2Н), 3,61 (с, 2Н), 2,53 (кв, J=7,5 Гц, 4Н), 1,27 (т, J=7,5 Гц, 6Н). ЖХМС (m/z): 184,00 [М+Н]+.
4-хлор-2,6-диметоксианилин
2,6-дибром-4-хлоранилин (4 г, 14,0 ммоль) растворили в 25% растворе NaOMe в МеОН (48 мл) и подействовали Cul (2,9g, 15,4 ммоль) при комнатной температуре в атмосфере азота. Полученную смесь перемешивали при 70°С в течение 12 ч в атмосфере азота. По завершении реакционную смесь охладили до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 × 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием 1% EtOAc-гексаны с получением 4-хлор-2,6-диметоксианилина (1,0 г, 38%) в виде светло-коричневой жидкости. 1Н-ЯМР (300 МГц, CDCl3): δ=6,52 (с, 2Н), 3,83 (с, 6Н). ЖХМС (m/z): 187,9 [М+Н]+
2-амино-5-хлор-3-циклопропил-N,N-диметилбензамид

В герметизируемом реакционном сосуде емкостью 50 мл 2-амино-3-бромбензойную кислоту (2,0 г, 9,25 ммоль) растворили в ДМФА (20 мл) и охладили до 0°С. Последовательно добавили EDC-HCl (2,1 г, 11,0 ммоль), HOBt (1,49 г, 11,0 ммоль), DIPEA (2,8 мл, 27,7 ммоль) и гидрохлорид диметиламина (1,13 г, 13,8 ммоль) при 0°С. Реакционную смесь нагрели до 70°С и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили водой, экстрагировали (2 × 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 2-амино-3-бром-N,N-диметилбензамида (2,0 г, 89%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,44 (дд, J=7,8, 1,5 Гц, 1Н), 7,06 (дд, J=7,8, 1,5 Гц, 1H), 6,61 (т, J=7,8 Гц, 1H), 4,82 (ш.с, 2Н), 3,05 (с, 6Н). ЖХМС (m/z): 243,10, 245,10 [М+Н]+.
В герметизируемом реакционном сосуде емкостью 50 мл раствор 2-амино-3-бром-N,N-диметилбензамида (2 г, 8,23 ммоль), циклопропилбороновую кислоту (850 мг, 9,87 ммоль) и (5,23 г, 24,06 ммоль) растворили в толуоле (30 мл) и воде (3 мл). Раствор дегазировали посредством продувки азотом в течение 5 минут, затем добавили Pd(OAc)2 (184 мг, 0,823 ммоль) и трициклогексилфосфин (230 мг, 0,823 ммоль) и раствор снова продували азотом в течение 5 минут. Полученную смесь перемешивали при 100°С в течение 12 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали (2 × 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием 20% -гексаны с получением 2-амино-3-циклопропил-N,N-диметилбензамида (1,0 г, 60%) в виде твердого вещества светло-коричневого цвета. 1Н-ЯМР (300 МГц, CDCl3): δ=7,06 (д, J=7,5 Гц, 1Н), 6,99 (дд, J=7,8, 1,5 Гц, 1Н), 6,66 (т, J=7,8 Гц, 1H), 4,70 (шс, 2Н), 3,05 (с, 6Н), 1,68-1,61 (м, 1Н), 0,94-0,88 (м, 2Н), 0,62-0,57 (м, 2Н). ЖХМС (m/z): 205,3 [М+Н]+.
Раствор 2-амино-3-циклопропил-N,N-диметилбензамида (0,5 г, 2,44 ммоль) в ацетонитриле (10 мл) и АсОН (0,3 мл) охладили до 0°С. Добавили N-хлорсукцинимид (0,5 г, 3,67 ммоль) при 0°С и полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили насыщенным раствором Na2S2O3 и экстрагировали этилацетатом (2 × 50 мл). Объединенные органические экстракты промыли раствором NaHCO3, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 25% -гексаны с получением 2-амино-5-хлор-3-циклопропил-N,N-диметилбензамида (0,2 г, 34%) в виде коричневого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,02 (д, J=1,5 Гц, 1H), 6,96 (д, J=2,7 Гц, 1H), 4,66 (шс, 2Н), 3,05 (с, 6Н), 1,68-1,61 (м, 1H), 0,94-0,88 (м, 2Н), 0,62-0,57 (м, 2Н). ЖХМС (m/z): 239,0 [М+Н]+.
4-хлор-2-метокси-6-(трифторметил)анилин
В герметизируемом реакционном сосуде емкостью 50 мл раствор 2-бром-4-хлор-6-(трифторметил)анилина (0,5 г, 1,82 ммоль), бис(пинаколато)дибора (0,92 г, 3,64 ммоль) и KOAc (0,44 г, 4,55 ммоль) в 1,4-диоксане (10 мл) дегазировали посредством продувки азотом в течение 5 минут. Добавили Pd(dppf)Cl2 (015 г, 0,182 ммоль) и раствор снова продували газообразным азотом в течение 5 минут. Полученную смесь перемешивали при 110°С в течение 12 ч. По завершении реакционную смесь разбавили водой, экстрагировали (2 × 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 2% EtOAc-гексаны с получением 4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-(трифторметил)анилина (0,5 г, 85%). ЖХМС (m/z): 324,10 [М+Н]+.
4-Хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-6-(трифторметил)анилин (500 мг, 1,55 ммоль) растворили в ТГФ (5 мл) и H2O (2 мл) при комнатной температуре. Добавили порциями NaBO3.H2O (0,62 г, 6,23 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. По завершении реакционную смесь разбавили водой, экстрагировали (2 × 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 15% EtOAc-гексаны с получением 2-амино-5-хлор-3-(трифторметил)фенола (0,5 г, 100%) в виде желтой жидкости. 1Н-ЯМР (300 МГц, -d6): δ=10,50 (с, 1Н), 6,85 (с, 2Н), 5,12 (ш.с, 2Н). 19F-ЯМР (300 МГц, ДМСО-d6): δ=-61,46. ЖХМС (m/z): 211,6 [М+Н]+.
2-амино-5-хлор-3-(трифторметил)фенол (250 мг, 1,18 ммоль) растворили в безводном ДМФА (5 мл) и подействовали (240 мг, 1,77 ммоль).
Полученную смесь перемешивали при комнатной температуре в течение 30 минут. Добавили по каплям метилйодид (185 мг, 1,303 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По завершении реакционную смесь разбавили водой, экстрагировали (2 × 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 10% EtOAc-гексаны с получением 4-хлор-2-метокси-6-(трифторметил)анилина (0,2 г, 75%) в виде светло-коричневого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,10 (д, J=2,0 Гц, 1Н), 6,98 (д, J=2,0 Гц, 1Н), 5,34 (ш.с, 2Н) 3,85 (с, 3H). 19F-(400 МГц, ДМСО-d6): δ=-61,45.
7-хлор-5-циклопропил-2,3-дигидро-1Н-инден-4-амин

Раствор 2,3-дигидро-1H-инден-4-амина, 1 (500 мг, 3,75 ммоль) в EtOH (5 мл) охладили до 0°С и подействовали по каплям уксусным ангидридом (0,95 г, 9,37 ммоль) в атмосфере азота. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 3 ч. По завершении реакции (ТСХ, 30% этилацетат-гексаны, Rf, 0,2), реакционную смесь концентрировали при пониженном давлении. Полученный остаток разбавили диэтиловым эфиром, отфильтровали и высушили в вакууме с получением N-(2,3-дигидро-1H-инден-4-ил)ацетамида (0,3 г, 45%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ=9,29 (с, 1H), 7,41 (д, J=8,0 Гц, 1H), 7,08 (т, J=7,6 Гц, 1Н), 6,99 (д, J=6,8 Гц, 1H), 2,87 (т, J=7,2 Гц, 2Н), 2,80 (т, J=7,2 Гц, 2Н), 2,04 (с, 3H), 1,99-1,95 (м, 2Н). ЖХМС (m/z): 176,40 [М+Н]+
N-(2,3-дигидро-1Н-инден-4-ил)ацетамид (200 мг, 1,11 ммоль) растворили в АсОН (5 мл) и охладили до 0°С. Добавили N-хлорсукцинимид (220 мг, 1,69 ммоль), затем реакционную смесь прогрели до комнатной температуры и перемешивали на протяжении ночи. По завершении реакционную смесь разбавили ледяной водой и образовавшееся твердое вещество удалили фильтрованием, промыли насыщенным NaHCO3, раствором Na2S2O3 и высушили в вакууме с получением N-(7-хлор-2,3-дигидро-1H-инден-4-ил)ацетамида (0,12 г, 50%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,73 (д, J=8,4 Гц, 1Н), 7,14 (д, J=8,4 Гц, 1Н), 6,86 (с, 1Н), 3,02-2,85 (м, 4Н), 2,19 (с, 3H), 1,99 (м, 2Н). ЖХМС (m/z): 209,80 [М+Н]+
N-(7-хлор-2,3-дигидро-1H-инден-4-ил)ацетамид (120 мг, 0,57 ммоль) растворили в 3 М HCl (5 мл) и нагревали при 90°С в течение 4 ч. По завершении реакционную смесь охладили до комнатной температуры и повысили основность (рН ~8) насыщенным раствором NaHCO3 с последующим экстрагированием EtOAc (2 × 20 мл). Объединенные органические экстракты промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением 7-хлор-2,3-дигидро-1H-инден-4-амина (70 мг, 74%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, ДМСО-d6): δ=6,85 (д, J=8,4 Гц, 1Н), 6,40 (д, J=8,4 Гц, 1H), 4,97 (с, 2Н), 2,82 (т, J=8,1 Гц, 2Н), 2,71 (т, J=7,5 Гц, 2Н), 2,01-1,96 (м, 2Н). ЖХМС (m/z): 168,20 [М+Н]+.
Раствор 7-хлор-2,3-дигидро-1Н-инден-4-амина (0,8 г, 4,79 ммоль) в ацетонитриле (10 мл) охладили до 0°С и подействовали N-йодсукцинимидом (1,61 г, 7,18 ммоль) при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили насыщенным раствором Na2S2O3 и экстрагировали этилацетатом (2 × 50 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 4-5% -гексаны с получением 7-хлор-5-йод-2,3-дигидро-1Н-инден-4-амина (0,45 г, 32%) в виде светло-коричневого твердого вещества. 1Н-ЯМР (300 МГц, ДМСО-d6): δ=7,36 (с, 1H), 5,04 (с, 2Н), 2,82-2,72 (м, 4Н), 2,03-1,98 (м, 2Н). ЖХМС (m/z): 293,7 [М+Н]+.
В герметизируемом реакционном сосуде емкостью 50 мл на раствор 7-хлор-5-йод-2,3-дигидро-1Н-инден-4-амина (0,35 г, 1,19 ммоль) и циклопропилбороновую кислоту (0,41 г, 4,77 ммоль) в 1,4-диоксане (14 мл) и воде (4 мл) подействовали Cs2CO3 (1,16 г, 3,57 ммоль) при комнатной температуре в атмосфере азота. Через раствор пропускали азот в течение 5 минут и подействовали Pd(OAc)2 (26 мг, 0,119 ммоль) и Catacxium-A (42 мг, 0,119 ммоль) в атмосфере азота. Полученную смесь снова дегазировали азотом в течение еще 5 минут. Полученную смесь перемешивали при 100°С в течение 24 ч. По завершении реакционную смесь разбавили водой и экстрагировали EtOAc (2 × 25 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 5% EtOAc-гексаны с получением 7-хлор-5-циклопропил-2,3-дигидро-1H-инден-4-амина (70 мг, 28%) в виде светло-коричневого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=6,86 (с, 1H), 2,94 (т, J=7,5 Гц, 2Н), 2,80 (т, J=7,5 Гц, 2Н), 2,15-2,10 (м, 2Н), 1,44-1,43 (м, 1H), 0,91-0,88 (м, 2Н), 0,58-0,55 (м, 2Н). ЖХМС (m/z): 208,3 [М+Н]+.
Синтез полупродуктов кислот R2:
2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-карбоновая кислота
Синтез 2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-карбоновой кислоты осуществили из гидрохинона с использованием методик, подробно описанных Monte et.al. J.Med.Chem. 1996, 39, 2953-2961, с получением 2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-карбальдегида в виде светло-желтого твердого вещества; 1Н-ЯМР (400 МГц, CDCl3): δ=10,27 (с, 1Н), 6,87 (с, 1H), 4,67 (т, J=8,8 Гц, 2Н), 4,59 (т, J=8,8 Гц, 2Н), 4,59 (т, J=8,8 Гц, 2Н), 3,46 (т, J=8,8 Гц, 2Н), 3,18 (т, J=8,8 Гц, 2Н).
Альдегид (0,68 г, 3,58 ммоль) окислили с помощью оксида серебра (I) (1,5 экв.) в 5% водном гидроксиде натрия при комнатной температуре в течение 20 дней. Техническую реакционную смесь профильтровали через целит, экстрагировали с помощью диэтилового эфира (2 × 50 мл) для удаления непрореагировавшего альдегида, затем водную фазу подкислили до рН 1 с помощью 3,0 моль/л водной HCl по каплям при 0°С. Продукт экстрагировали с помощью дихлорметана (2 × 50 мл) и объединенные органические фазы промыли солевым раствором (50 мл), высушили (MgSO4) и концентрировали при пониженном давлении с получением 2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-карбоновой кислоты в виде белого твердого вещества (0,44 г; 60%).
Альтернативно, на альдегид (0,5 г, 2,77 ммоль) в ацетоне (5,0 мл) подействовали аминосульфоновой кислотой (0,4 г, 4,17 ммоль) двумя порциями при 0°С. Через 2 мин добавили раствор хлорита натрия (0,32 г, 3,6 ммоль) в воде (1,0 мл) по каплям и перемешивание продолжали при 0°С в течение 4 ч. Реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью смеси 10% ИПС/хлороформ (2 × 20 мл). Объединенные органические фазы промыли водой (25 мл), солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Техническое твердое вещество растерли с диэтиловым эфиром с получением 2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-карбоновой кислоты (0,4 г, 70%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=6,86 (с, 1H), 4,52 (т, J=8,8 Гц, 2Н), 4,47 (т, J=8,8 Гц, 2Н), 3,30 (т, J=8,8 Гц, 2Н), 3,10 (т, J=8,8 Гц, 2Н). 13С (100 МГц, ДМСО-d6): δ=166,4, 154,2, 153,9, 128,9, 127,2, 111,4, 110,43, 71,9, 71,6, 31,5, 29,5.
Бензо[d][1,3]диоксол-4-карбоновая кислота
Синтезирована с использованием модифицированных методик из Pie et.al. J.Med.Chem. 2004, 47, 871-887 следующим образом:
На 2,3-дигидроксибензойную кислоту (5,0 г, 32,4 ммоль) в безводном метаноле (50 мл) подействовали концентрированной серной кислотой (10 капель) и нагревали с обратным холодильником на протяжении ночи. Реакционную смесь концентрировали при пониженном давлении, разбавили EtOAc (100 мл), промыли насыщ. водн NaHCO3 (2 × 50 мл), солевым раствором (50 мл), затем высушили (MgSO4) и концентрировали при пониженном давлении с получением метил 2,3-дигидроксибензоата (2,92 г, 54%). 1Н-ЯМР (400 МГц, CDCl3): δ=10,9 (с, 1H), 7,32 (дд, J=8,0, 1,2 Гц, 1Н), 7,09 (м, 1Н), 6,78 (т, J=8,0 Гц, 1H), 5,65 (с, 3H). 13С-ЯМР (100 Гц, CDCl3) 170,7, 148,8, 145,0, 120,5, 119,8, 119,2, 112,4, 52,4.
На метил 2,3-дигидроксибензоат (1,0 г, 5,95 ммоль) в ДМФА (16 мл) подействовали KF (1,79 г, 30,9 ммоль) и перемешивали при температуре окружающей среды в течение 30 минут. Добавили дийодметан (1,79 г, 6,7 ммоль) и реакционную смесь нагревали при 100°С в течение 5 часов. Реакционную смесь охладили до комнатной температуры, вылили в воду (100 мл) и экстрагировали с помощью диэтилового эфира (2 × 50 мл). Объединенные органические фазы промыли водой (50 мл), солевым раствором (50 мл), высушили (MgSO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 10% - петролейный эфир с получением метилбензо[d][1,3]диоксол-4-карбоксилата в виде белого кристаллического твердого вещества (0,56 г, 52%); 1Н-ЯМР (400 МГц, CDCl3): δ=7,41 (дд, J=8,0, 1,2 Гц, 1H), 6,97 (дд, J=8,0, 1,2 Гц, 1Н), 6,86 (т, J=8,0 Гц, 1H), 6,1 (с, 2Н), 3,93 (с, 3H).
На раствор метил бензо[d][1,3]диоксол-4-карбоксилата (0,4 г, 2,22 ммоль) в метаноле (8,0 мл) подействовали 2,0 М водн KOH (2,2 мл) и раствор перемешивали при комнатной температуре в течение 3 часов. Смесь концентрировали до объема ~3 мл, разбавили водой (5 мл) и подкислили до рН ~3 с помощью 2,0 моль/л HCl. Полученный осадок удалили фильтрованием, промыли водой, затем диэтиловым эфиром, и высушили в вакууме с получением бензо[d][1,3]диоксол-4-карбоновой кислоты в виде бежевого твердого вещества (0,38 г, 97%). 1Н-ЯМР (400 МГц, -d6): δ=7,28 (дд, J=8,0, 1,2 Гц, 1H), 6,97 (дд, J=8,0, 1,2 Гц, 1Н), 6,89 (т, J=8,0 Гц, 1Н), 6,12 (с, 2Н); 13С-ЯМР (100 Гц, ДМСО-d6) 165,5, 148,9, 148,5, 122,9, 121,6, 113,8, 112,5, 102,1.
Синтезированные соединения по классу заместителя
Алифатические заместители
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)циклогексансульфонамид

4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и циклогексансульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (12 мг, 41%). 1Н-ЯМР (400 МГц, CD3OD): δ=6,97 (с, 1H), 3,50-3,43 (м, 1H), 2,87 (т, 4Н, J=8,0 Гц), 2,78 (т, 4Н, J=8,0 Гц), 2,22-2,18 (м, 2Н), 2,10-2,02 (м, 4Н), 1,94-1,71 (м, 2Н), 1,63-1,59 (м, 1Н), 1,64-1,53 (м, 2Н); 1,41-1,21 (м, 3H); 13С-ЯМР (100 МГц, CD3OD): δ=143,7, 137,8, 126,4, 118,4, 110,2, 59,9, 35,5, 30,0, 28,5, 25,8, 25,1, 24,8; Чистота по ЖХМС: >95%; ЖХМС (m/z): 363 [М+Н]+; МСВР вычислено для C19H26N2O3S [М+Н]+:363,1737, найдено 363,1729.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)циклопентансульфонамид
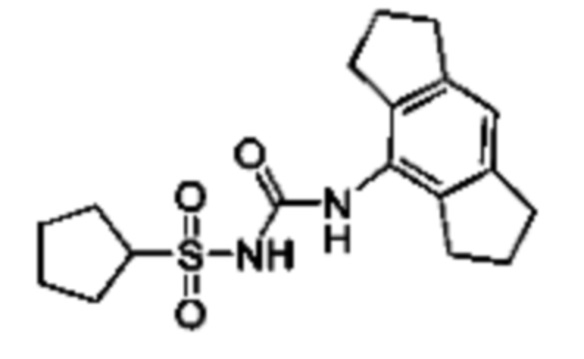
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и циклопентансульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (26 мг, 42%) 1Н-ЯМР (400 МГц, CD3OD): δ=6,97 (с, 1Н), 4,08-4,02 (м, 1H), 2,83 (т, J=8,0 Гц, 4Н), 2,80 (т, J=8,0 Гц, 4Н), 2,13-2,01 (м, 8Н), 1,84-1,77 (м, 2Н), 1,71-1,65 (м, 2Н); 13С-ЯМР (100 МГц, CD3OD): δ=145,1, 139,2, 127,8, 119,8, 111,7, 62,2, 33,9, 31,4, 29,9, 28,6, 26,9; Чистота по ЖХМС: >95%; ЖХМС (m/z): 349 [М+Н]+; МСВР вычислено для C18H24N2O3S: 349,1580, найдено 349,1588.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)тетрагидро-2Н-пиран-4-сульфонамид.
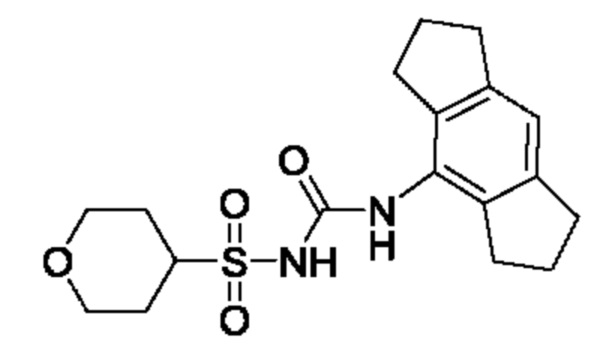
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и тетрагидро-2Н-пиран-4-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (12 мг, 57%). 1Н-ЯМР (400 МГц, CD3OD): δ=7,00 (с, 1H), 4,09 (дд, J1=4 Гц, J2=12 Гц, 2Н), 3,82-3,76 (м, 1H), 3,49-3,43, (м, 2Н), 2,89 (т, J=8 Гц, 4Н), 2,81 (т, J=8 Гц, 4Н), 2,12-2,05 (м, 6Н), 1,98-1,87 (м, 2Н); 13С-ЯМР (100 МГц, CD3OD): δ=154,0, 143,1, 137,7, 126,5, 110,4, 66,0, 57,0, 32,5, 28,5, 25,9, 25,1; Чистота по ЖХМС: >95%; ЖХМС (m/z): 365 [М+Н]+; МСВР вычислено для C18H24N2O4S, 365,1530, найдено 365,1541.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)тетрагидрофуран-3-сульфонамид
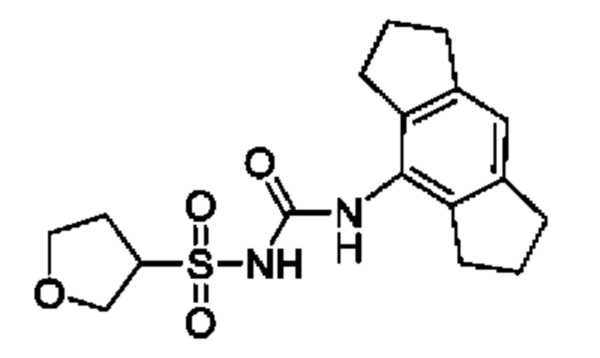
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и тетрагидрофуран-3-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (12 мг, 60%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,04 (с, 1Н), 6,93 (с, 1H), 4,33-4,27 (м, 1H), 4,04-4,00 (м, 1H),3,91-3,89 (м, 1Н), 3,85-3,79 (м, 1H), 3,72-3,66 (м, 1H), 2,80 (т, J=16,0 Гц, 4Н), 2,70 (т, J=16,0 Гц, 4Н), 2,24-2,17 (м, 2Н), 1,99 -1,95 (м, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=142,4, 139,6, 136,6, 124,7, 108,2, 68,7, 61,7, 32,5, 30,3, 28,8, 28,1, 24,9; Чистота по ЖХМС: >95%; ЖХМС (m/z): 351 [М+Н]+; МСВР вычислено для C17H22N2O4S 351,1373, найдено 351,1389.
ФУРАНЫ
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамид
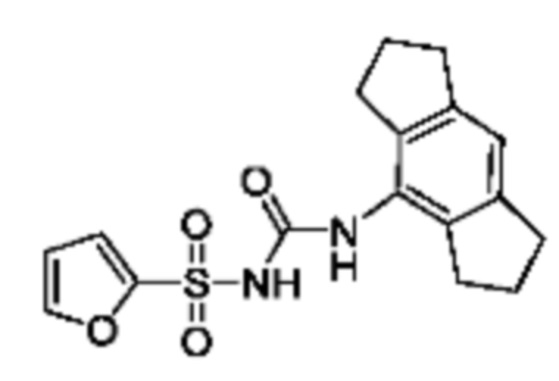
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и фуран-2-сульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого твердого вещества (75 мг, 16%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=11,08 (ш.с, 1H), 8,08 (с, 1H), 8,02 (с, 1H), 7,22 (кв, J=2,0 Гц, 1Н), 6,94 (с, 1Н), 6,71 (кв, J=2,0 Гц, 1H), 2,78 (т, J=7,2 Гц, 4Н), 2,59 (т, J=7,2 Гц, 4Н), 1,94 (квинт, J=7,2 Гц, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=148,9, 147,9, 147,3, 143,1, 137,3, 128,7, 118,0, 117,5, 111,7, 54,9, 32,5, 30,1, 25,1. ЖХМС, Чистота: 96,26%; m/z 345,1 (М-Н+). МСВР (FAB-(бомбардировка быстрыми атомами)) вычислено для C17H18N2O4S [М-Н]-: 345,0987, найдено: 345,0866.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-5-метилфуран-2-сульфонамид
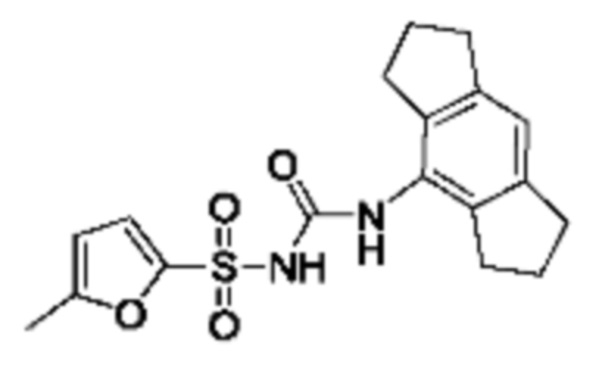
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 5-метилфуран-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (28 мг, 53%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,96 (с, 1H), 7,00-6,99 (д, J=4,0 Гц, 1Н), 6,91 (с, 1H), 6,29-6,28 (д, J=4,0 Гц, 1H), 1Н), 2,78 (т, J=8,0 Гц, 4Н), 2,61 (т, J=8,0 Гц, 4Н), 2,34 (с, 3H), 2 (т, J=8,0 Гц, 4Н), 1,98-1,90 (м, 4Н); 13С-ЯМР (100 МГц, ДМСО-d6): δ=143,3, 137,6, 129,9, 125,2, 118,0, 114,6, 108,7, 108,2, 107,8, 32,9, 30,6, 25,4, 13,8; Чистота по ЖХМС: >95%; ЖХМС (m/z): 361 [М+Н]+; МСВР вычислено для C18H20N2O4S [М+Н]+ 361,1216, найдено 361,1217.
5-этил-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамид
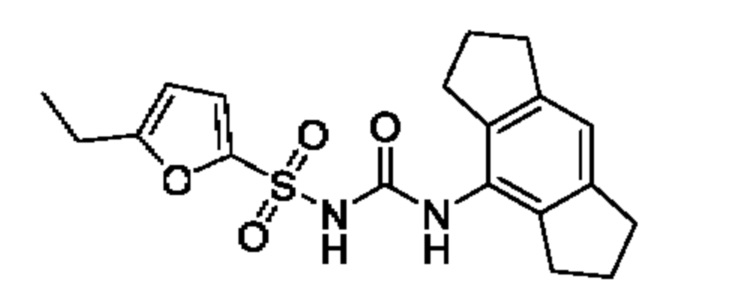
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 5-этилфуран-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (51 мг, 47%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,97 (шс, 1H), 7,02 (с, 1H), 6,91 (д, J=4,0 Гц, 1Н), 6,31 (д, J=4,0 Гц, 1Н), 2,78 (т, J=8,0 Гц, 4Н), 2,68 (кв, J=8,0 Гц, 2Н), 2,59 (т, J=6,0 Гц, 4Н), 1,97-1,90 (м, 4Н), 1,19 (т, J=8,0 Гц, 3H). 13С-ЯМР (150 МГц, ДМСО-d6): δ=143,5, 143,3, 142,9, 137,6, 129,8, 118,0, 108,7, 106,8, 106,3, 32,9, 30,5, 25,4, 21,3, 12,1; Чистота по ЖХМС: >95%; ЖХМС (m/z): 375 [М+Н]+; МСВР вычислено для C19H22N2O4S [М+Н]+ 375,13730, найдено 375,13910.
5-((диметиламино)метил)-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамид

4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 5-((диметиламино)метил)фуран-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (25 мг, 6%), 1Н-ЯМР (400 МГц, CD3OD) δ=7,17 (д, J=3,5 Гц, 1Н), 6,96 (с, 1Н), 6,86 (д, J=3,5 Гц, 1H), 4,43 (с, 2Н), 2,86 (с, 3H),2,86 (т, J=7,4 Гц, 4Н), 2,73 (т, J=7,4 Гц, 4Н), 2,04 (п, J=7,4 Гц, 4Н).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
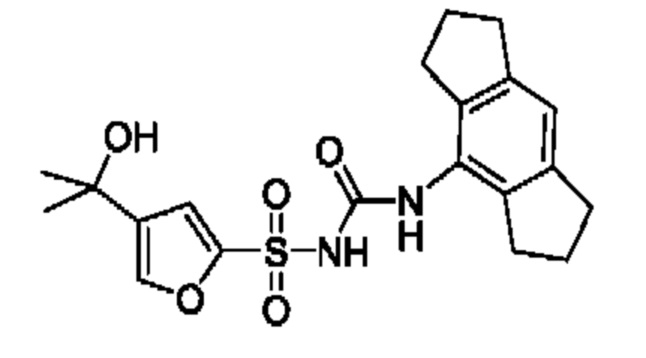
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С5 с получением указанного в подзаголовке соединения в виде белого твердого вещества (2,5 г, 63%). 1Н-ЯМР (600 МГц, ДМСО-d6): δ=7,61 (ш.с, 1Н), 7,37 (д, J=0,9 Гц, 1Н), 6,77 (с, 1Н), 6,58 (д, J=0,9 Гц, 1H), 2,74 (т, J=7,3 Гц, 4Н), 2,65 (т, J=7,3 Гц, 4Н), 1,89 (тт, J=7,3, 7,3 Гц, 4Н), 1,34 (с, 6Н). 13С-ЯМР (101 Гц, ДМСО-d6): δ=157,4, 155,7, 142,2, 137,3, 136,7, 135,7, 132,4, 115,7, 109,3, 66,6, 32,6, 31,1, 30,6, 25,1.
N-((8-бром-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
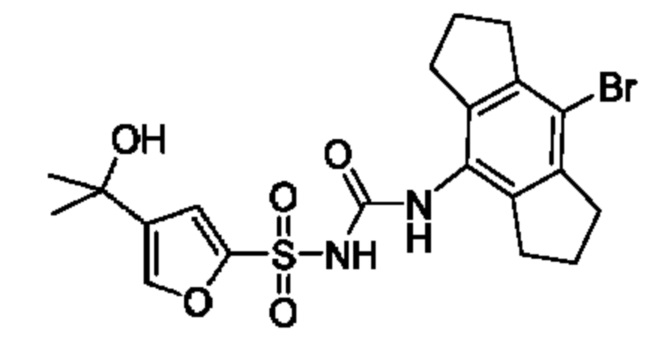
4-Бром-8-изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (40 мг, 7%). 1Н-ЯМР (400 МГц, CD3OD): δ=7,68 (с, 1Н), 7,23 (с, 1Н), 2,91 (т, J=7,6 Гц, 4Н), 2,85 (т, J=7,6 Гц, 4Н), 2,11 (м, 4Н), 1,51 (с, 6Н). ЖХМС (m/z): 482,9 [М-Н]-; 97,64% (210 нм), 99% (254 нм). ВЭЖХ: 96,70% (210 нм), 97,22% (254 нм).
N-((8-хлор-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
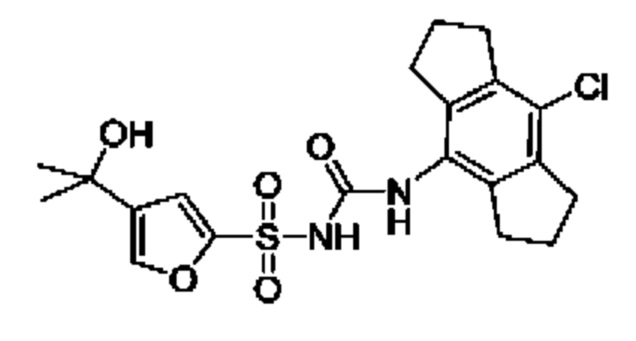
4-Хлор-8-изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (50 мг, 16%). 1Н-ЯМР (400 МГц, CD3OD) δ=7,55 (с, 1Н), 7,02 (с, 1H), 2,91 (т, J=7,2 Гц, 4Н), 2,85 (т, J=7,2 Гц, 4Н), 2,09 (м, 4Н), 1,5 (с, 6Н). ЖХМС (m/z): 460,9 (М+Na)-; 95,16% (210 нм), 95,07% (254 нм). ВЭЖХ: 97,91% (210 нм), 98,04% (254 нм).
4-(2-гидроксипропан-2-ил)-N-((8-метил-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамид
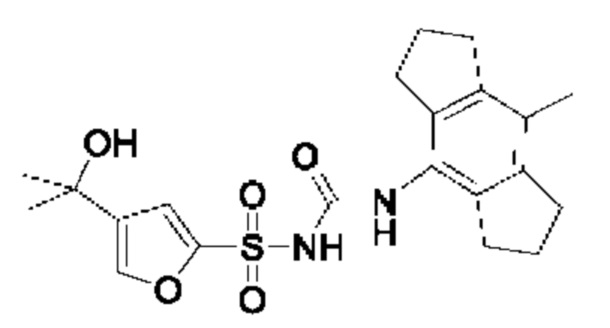
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (15 мг, 3%). 1Н-ЯМР (400 МГц, CD3OD): δ=7,58 (с, 1Н), 7,07 (с, 1Н), 6,46 (с, 1H), 2,82-2,73 (м, J=7,5 Гц, 8Н), 2,12 (с, 3H), 2,05-2,02 (м, 4Н), 1,508 (с, 6Н). ЖХМС(m/z): 417,10(М-1)-; 99,59% (210 нм), 99,33% (254 нм). ВЭЖХ: 97,92% (210 нм), 97,53% (254 нм).
5-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фуран-3-карбоновая кислота
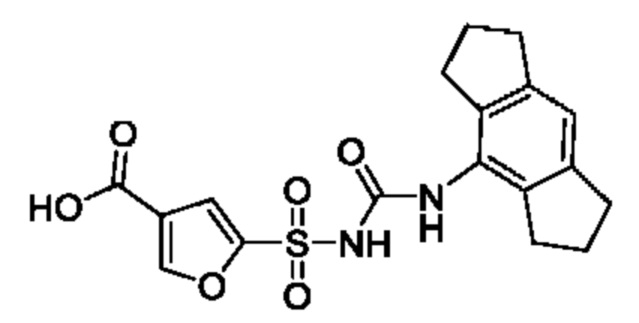
На этил-5-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фуран-3-карбоксилат (0,1 г, 0,24 ммоль) в ТГФ (8 мл) при 0°С подействовали раствором LiOH (0,1 г, 2,4 ммоль) в воде (2 мл). Охлаждающую баню удалили и реакционную смесь перемешивали в течение 3 ч. Раствор подкислили 10% лимонной кислотой и сразу экстрагировали с помощью этилацетата (2 × 25 мл). Органические фазы промыли водой (20 мл), солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде белого твердого вещества (5,0 мг, 5%). 1Н-ЯМР (400 МГц, CD3OD): δ=8,14 (с, 1H), 7,28 (с, 1H), 6,93 (с, 1H), 2,85 (т, J=7,6 Гц, 4Н), 2,74 (т, J=7,6 Гц, 4Н), 2,04 (квинт, J=7,6 Гц, 4Н).
Этил-5-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фуран-3-карбоксилат
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и этил-5-сульфамоилфуран-3-карбоксилат использовали в общем способе С3. Реакционную смесь разбавили водой (50 мл), экстрагировали с помощью этилацетата (2 × 25 мл) и органические фазы промыли солевым раствором (25 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 50% -гексаны с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,45 г, 63%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ=8,31 (с, 1Н), 7,59 (с, 1Н), 6,77 (с, 1H), 4,22 (кв, J=7,2 Гц, 2Н), 2,75 (т, J=7,3 Гц, 4Н), 2,65 (т, J=7,3 Гц, 4Н) 1,90 (пент, J=7,6 Гц, 4Н), 1,26 (т, J=7,2 Гц, 3H).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(проп-1-ен-2-ил)фуран-2-сульфонамид
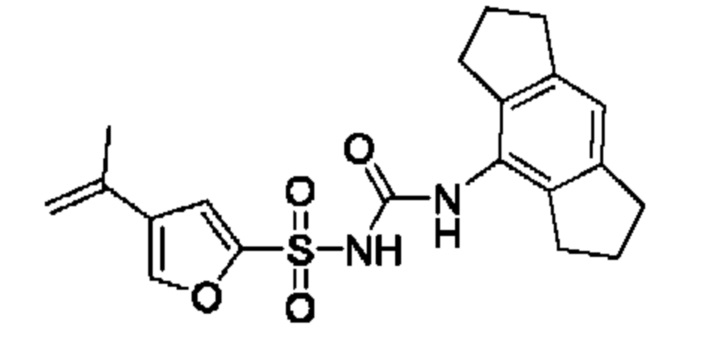
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4-(проп-1-ен-2-ил)фуран-2-сульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (85 мг, 51%). 1Н-ЯМР (400 МГц, CDCl3) δ 7,54 (с, 1H), 7,28 (с, 1H), 7,00 (с, 1H), 5,26 (с, 1H), 5,05 (с, 1H), 2,86 (т, J=7,4 Гц, 4Н), 2,69 (т, J=7,5 Гц, 4Н), 2,09- 1,98 (м, 7Н). 13С-ЯМР (101 МГц, CDCl3) δ 144,4, 142,8, 137,8, 132,8, 129,2, 127,2, 119,4, 115,4, 113,6, 32,9, 30,5, 25,5, 20,9. МСВР (ESI (ионизация распылением в электрическом поле)) вычислено для C20H23N2O4S [М+Н] 387,1373, найдено 387,1379.
4-(2-гидроксипропан-2-ил)-N-((3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)карбамоил)фуран-2-сульфонамид
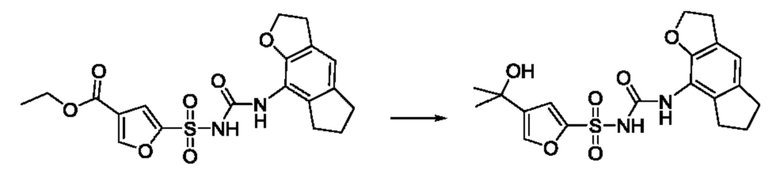
8-изоцианато-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран (полученный с использованием общего способа А1) и этил-5-сульфамоилфуран-3-карбоксилат использовали в общем способе С3 с получением этил-5-(N-((3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)карбамоил)сульфамоил)фуран-3-карбоксилата в виде светло-коричневого твердого вещества (0,25 г, 50%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ 8,32 (с, 1H), 7,17 (с, 1H), 6,77 (д, J=5,2 Гц, 1H), 4,43 (т, J=8,6 Гц, 2Н), 4,23 (кв, J=7,1 Гц, 2Н), 3,07 (т, J=8,6 Гц, 2Н), 2,71 (т, J=7,3 Гц, 2Н), 2,63 (т, J=7,3 Гц, 2Н), 1,89 (п, J=7,4 Гц, 2Н), 1,26 (т, J=7,1 Гц, 3H).
На этил-5-(N-((3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)карбамоил)сульфамоил)фуран-3-карбоксилат (0,25 г, 0,6 ммоль) в безводном ТГФ (10 мл) при 0°С подействовали раствором метилмагнийхлорида (3,0 моль/л в Et2O, 6 экв.) по каплям в течение 5 минут при интенсивном перемешивании. Затем раствор перемешивали при 0°С в течение 30 мин, затем при температуре окружающей среды в течение 4 ч с последующей остановкой реакции добавлением по каплям насыщенного раствора хлорида аммония. Водный раствор экстрагировали с использованием (2 × 25 мл), объединенные органические фазы промыли солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт растерли с диэтиловым эфиром, затем очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде белого твердого вещества (32 мг, 13%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,58 (с, 1Н), 7,08 (с, 1H), 6,84 (с, 1H), 5,02 (с, 1H), 4,47 (т, J=8,6 Гц, 2Н), 3,10 (т, J=8,6 Гц, 2Н), 2,73 (т, J=7,3 Гц, 2Н), 2,59 (т, J=7,5 Гц, 2Н), 1,90 (д, J=7,4 Гц, 2Н), 1,36 (с, 6Н).
N-((4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
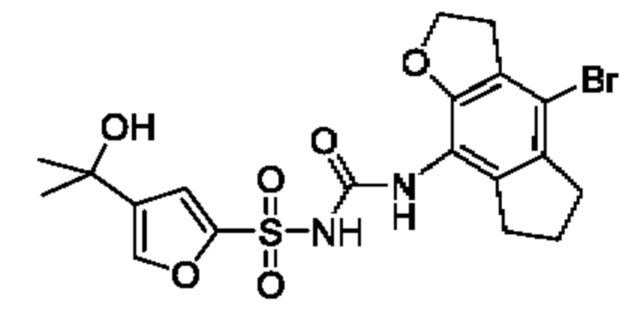
4-бром-8-изоцианато-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран(полученный с использованием общего способа А1) и 4-(проп-1-ен-2-ил)фуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (20 мг, 9%). 1Н-ЯМР (400 МГц, CD3OD) δ=7,48 (д, J=1,2 Гц, 1H), 6,93 (д, J=1,2 Гц, 1Н), 4,60 (т, J=8,7 Гц, 2Н), 3,16 (т, J=8,6 Гц, 2Н), 2,85 (м, 4Н), 2,03 (п, J=7,5 Гц, 2Н), 1,50 (с, 6Н).
4-(2-гидроксипропан-2-ил)-N-((3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-4-ил)карбамоил)фуран-2-сульфонамид
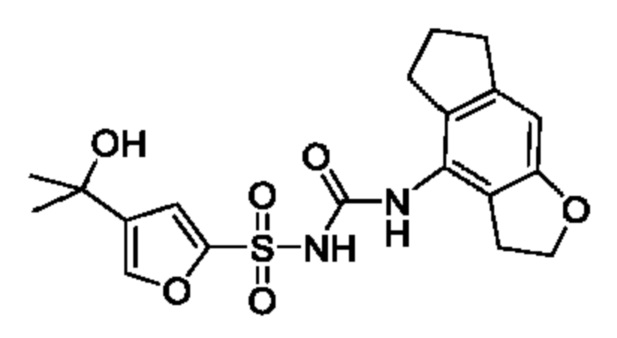
4-изоцианато-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран (полученный с использованием общего способа А1) и 4-(проп-1-ен-2-ил)фуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (20 мг, 9%). 1Н-ЯМР (400 МГц, CD3OD) δ=7,58 (с, 1H), 7,07 (с, 1Н), 6,46 (с, 1H), 4,49 (д, J=8,9 Гц, 2Н), 3,05 (т, J=8,7 Гц, 2Н), 2,82 (т, J=7,4 Гц, 2Н), 2,70 (т, J=7,4 Гц, 2Н), 2,04 (п, J=7,4 Гц, 2Н), 1,51 (д, J=1,9 Гц, 6Н).
4-(2-гидроксипропан-2-ил)-N-((2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-ил)карбамоил)фуран-2-сульфонамид
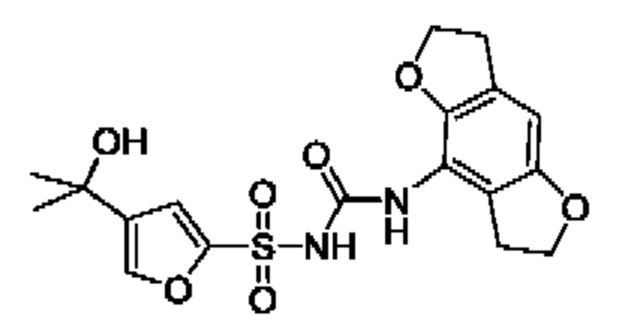
4-изоцианато-2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран(полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (285 мг, 96%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ 7,76 (с, 1Н), 7,66 (с, 1H), 7,01 (с, 1Н), 6,45 (с, 1Н), 5,04 (с, 1H), 4,46 (т, J=8,6 Гц, 2Н), 4,39 (т, J=8,6 Гц, 2Н), 3,08 (т, J=8,6 Гц, 2Н), 2,94 (т, J=8,6 Гц, 2Н), 1,37 (с, 6Н).
N-(бензо[1,2-b:4,5-b']дифуран-4-илкарбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
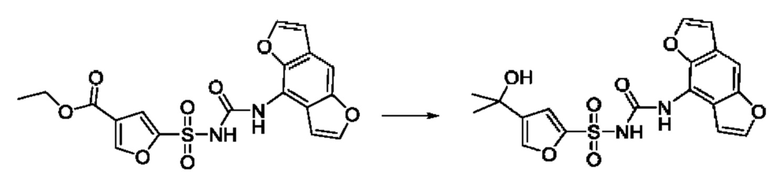
4-изоцианатобензо[1,2-b:4,5-b']дифуран (полученный с использованием общего способа А1) и этил-5-сульфамоилфуран-3-карбоксилат использовали в общем способе C3 с получением этил-5-(N-(бензо[1,2-b:4,5-b']дифуран-4-илкарбамоил)сульфамоил)фуран-3-карбоксилата в виде белого твердого вещества (0,05 г, 53%). 1Н-ЯМР (300 МГц, CD3OD) δ=8,25 (с, 1Н), 7,72 (д, J 2,1 Гц, 1Н), 7,63 (д, J 2,1 Гц, 1Н), 7,46 (с, 1Н), 7,27 (с, 1Н), 6,93 (с, 1H), 6,89 (д, J 2,1 Гц, 1Н), 6,86 (д, J 2,1 Гц, 1Н), 4,30 (кв, J 6,9 Гц, 2Н), 1,4 (т, J 6,9 Гц, 3H).
На этил-5-(N-(бензо[1,2-b:4,5-b']дифуран-4-илкарбамоил)сульфамоил)фуран-3-карбоксилат (0,25 г, 0,6 ммоль) в безводном ТГФ (10 мл) при 0°С подействовали раствором метилмагнийхлорида (3,0 моль/л в Et2O, 10 экв.) по каплям в течение 10 минут при интенсивном перемешивании. Затем раствор перемешивали при 0-10°С в течение 3 ч, затем остановили реакцию добавлением по каплям насыщенного раствора хлорида аммония. Водный раствор экстрагировали с использованием EtOAc (2 × 20 мл), объединенные органические фазы промыли солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт растерли с диэтиловым эфиром, затем очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде белого твердого вещества (15 мг, 6%), 1H-MP (400 МГц, CD3OD) δ = 7,76 (д, J=2,0 Гц, 1Н), 7,65 (д, J=2,4 Гц, 1Н), 7,55 (с, 1Н), 7,47 (с, 1Н), 7,05 (с, 1Н), 6,93 (д, J=2,0 Гц, 1Н), 6,89 (д, J=2,4 Гц, 1Н), 1,5 (с, 6Н).
N-(антрацен-9-илкарбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
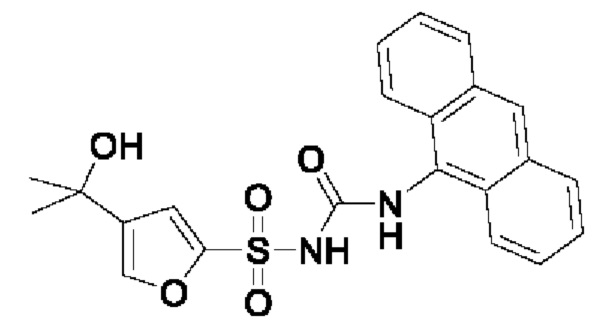
9-изоцианатоантрацен (полученный с использованием общего способа В2) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (24 мг, 23%). 1Н-ЯМР (400 МГц, CD3OD) δ = 8,49 (с, 1Н), 8,07-7,98 (м, 4Н), 7,75 (с, 1Н), 7,55-7,44 (м, 4Н), 7,27-7,22 (м, 1Н), 1,49 (с, 6Н). 13С-ЯМР (101 МГц, CD3OD) δ = 153,8, 149,4, 141,4, 136,6, 131,7, 128,8, 128,2, 127,4, 126,4, 125,9, 124,9, 122,8, 115,2, 111,1, 67,2, 29,6.
4-(2-гидроксипропан-2-ил)-N-(хинолин-8-илкарбамоил)фуран-2-сульфонамид
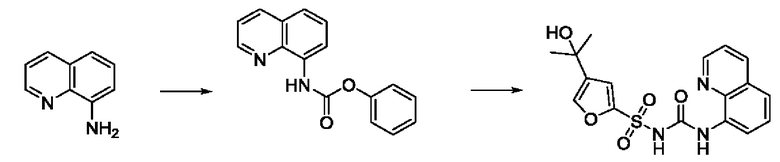
К раствору хинолин-8-амина (1 г, 6,9 ммоль) в ТГФ (10 мл) и триэтиламине (2 экв.) медленно добавили фенилхлорформиат (1,5 экв.) при 0°С. Раствор перемешивали при комнатной температуре в течение 2 ч или до завершения. Раствор разбавили насыщенн.. водн. раствором NaHCO3, экстрагировали с помощью этилацетата (2 х 50 мл), промыли водой, солевым раствором, затем высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием смеси 10% EtOAc-гексаны с получением фенилхинолин-8-илкарбамата (1,5 г, 83%) в виде белого твердого вещества, которое использовали сразу на следующей стадии реакции.
На 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (0,2 г, 0,98 ммоль) в ТГФ (5 мл) при 0°С подействовали гидридом натрия (3 экв.) порциями и суспензию перемешивали при температуре окружающей среды в течение 45 минут (до прекращения выделения газа). Технический фенилхинолин-8-илкарбамат растворили в ТГФ (5 мл), затем медленно добавили к реакционной смеси и раствор перемешивали при температуре окружающей среды до завершения, как правило, 4 ч. Реакционную смесь разбавили насыщенным водным NH4Cl, экстрагировали этилацетатом (х2), промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили посредством ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения, 4-(2-гидроксипропан-2-ил)-N-(хинолин-8-илкарбамоил)фуран-2-сульфонамида в виде белого твердого вещества (40 мг, 11%). 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,63 (с, 1Н), 8,89 (д, J=4,3 Гц, 1Н), 8,37 (м, 2Н), 7,80-6,76 (м, 5Н), 5,09 (с, 1Н), 1,38 (с, 6Н).
4-(2-гидроксипропан-2-ил)-N-((6-метоксихинолин-8-ил)карбамоил)фуран-2-сульфонамид
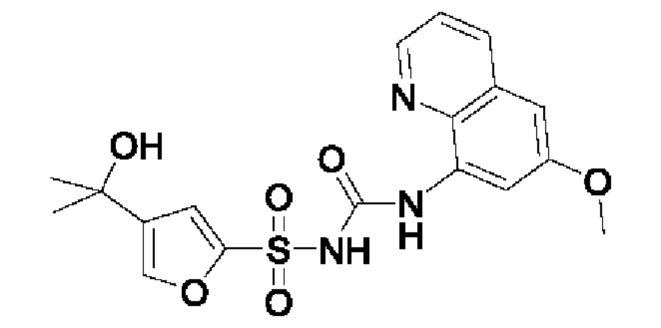
4-(2-гидроксипропан-2-ил)-N-((6-метоксихинолин-8-ил)карбамоил)фуран-2-сульфонамид синтезировали с использованием модификации методик, использованных для получения 4-(2-гидроксипропан-2-ил)-N-(хинолин-8-илкарбамоил)фуран-2-сульфонамида, но с применением 6-метоксихинолин-8-амина вместо хинолин-8-амина. Указанное в подзаголовке соединение получили в виде белого с желтоватым оттенком твердого вещества (75 мг, 38%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,79 (с, 1Н), 8,63 (м, 1Н), 8,17 (м, 1Н), 8,09 (д, J=2,7 Гц, 1Н), 7,49 (дд, J=8,3, 4,2 Гц, 1Н), 7,40 (с, 1Н), 6,79 (д, J=2,8 Гц, 1Н), 6,69 (с, 1Н), 4,96 (с, 1Н), 3,84 (с, 3Н), 1,36 (с, 6Н).
N-((2,3-дигидробензо[b][1,4]диоксин-5-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
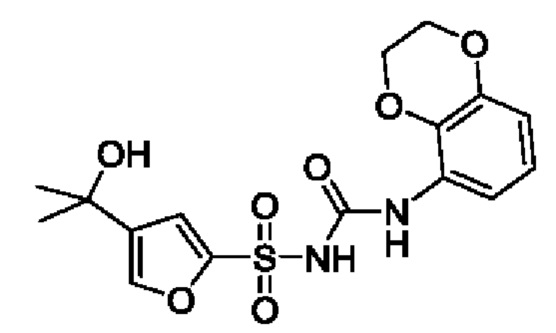
5-Изоцианато-2,3-дигидробензо[b][1,4]диоксин (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (49 мг, 39%). 1H-ЯМР (600 МГц, Ацетонитрил-d3) δ = 7,56 (дд, J=8,4, 1,5 Гц, 1Н), 7,45 (д, J=1,0 Гц, 1Н), 6,98 (д, J=1,0 Гц, 1Н), 6,7 (т, J=8,4 Гц, 1Н), 6,48 (дд, J=8,4, 1,5 Гц, 1Н), 4,22 (м, 4Н), 1,43 (с, 6Н).
N-((2,3-дигидробензофуран-7-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
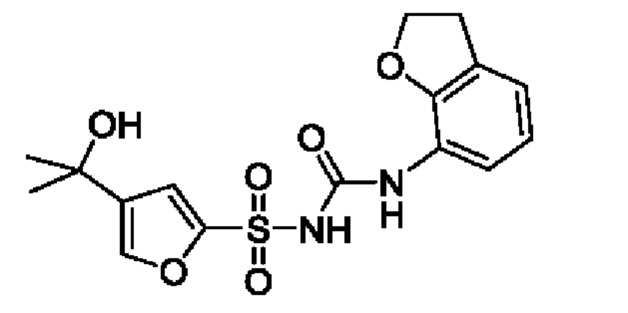
7-Изоцианато-2,3-дигидробензофуран (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (32 мг, 39%). 1H-ЯМР (600 МГц, Ацетонитрил-d3) δ 7,64 (д, J=7,7 Гц, 1Н), 7,48 (д, J=1,1 Гц, 1Н), 7,00 (д, J=1,1 Гц, 1Н), 6,89 (м, 1Н), 6,74 (т, J=7,7 Гц, 1Н), 4,56 (т, J=8,7 Гц, 1Н), 3,2 (т, J=8,7 Гц, 1Н), 1,43 (с, 6Н).
N-((2,4-бис(трифторметил)фенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
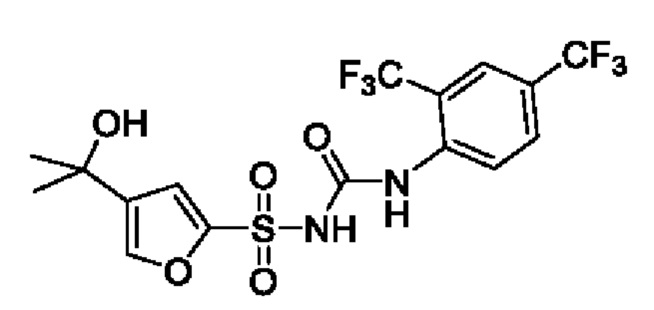
1-Изоцианато-2,4-бис(трифторметил)бензол (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,12 г, 33%). 1H-ЯМР (400 МГц, ДМСО-d6): δ = 8,59 (д, J=8,8 Гц, 1Н), 7,87 (д, J=9,2 Гц, 1Н), 7,81 (с, 1Н), 7,67 (с, 1Н), 7,43 (с, 1Н), 6,68 (с, 1Н), 4,94 (с, 1Н), 1,36 (с, 6Н). 13С-ЯМР (100 МГц, ДМСО-d6: δ = 156,0, 154,4, 142,5, 138,1, 135,8, 129,9, 125,2, 124,9, 123,0, 122,5, 121,3, 120,7, 120,4, 115,5, 115,2, 110,2, 66,5, 31,0. ЖХМС, Чистота: 90,47%, tr=3,84 мин, m/z 459,25 (M-H+). MCBP (FAB-) вычислено для C16H14F6N2O5S [M-H]-: 459,0528, найдено: 459,0512.
N-((2,5-бис(трифторметил)фенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
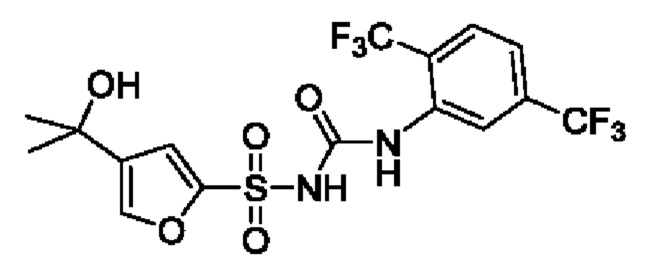
2-Изоцианато-1,4-бис(трифторметил)бензол (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (55 мг, 12%). 1H-ЯМР (400 МГц, CD3OD): δ = 8,61 (с, 1Н), 7,75 (д, J=7,6 Гц, 1Н), 7,48 (с, 1Н), 7,37 (д, J=8,4 Гц, 1Н), 6,95 (с, 1Н), 1,41 (с, 6Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ 156,4, 154,5, 139,7, 138,1, 132,9, 127,2, 124,9, 122,3, 118,9, 117,6, 117,0, 110,0, 66,5, 31,0. ЖХМС, Чистота: 95,02%, tr=2,09 мин, m/z 558,94 (M-H+). MCBP (FAB-) вычислено для C16H14F6N2O5S [M-H]-: 459,0528, найдено: 459,0224.
4-(2-гидроксипропан-2-ил)-N-((2-метоксифенил)карбамоил)фуран-2-сульфонамид
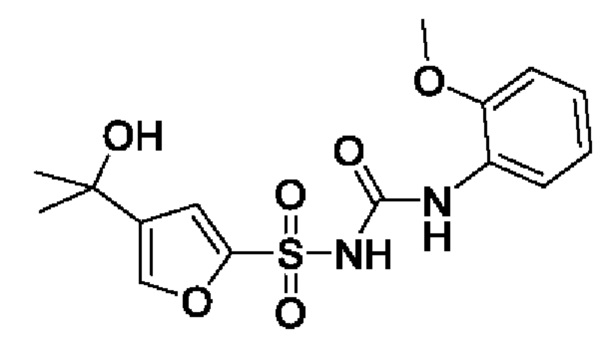
1-изоцианато-2-метоксибензол (полученный с использованием общего способа А2) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (30 мг, 38%).
N-((2,5-диметоксифенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
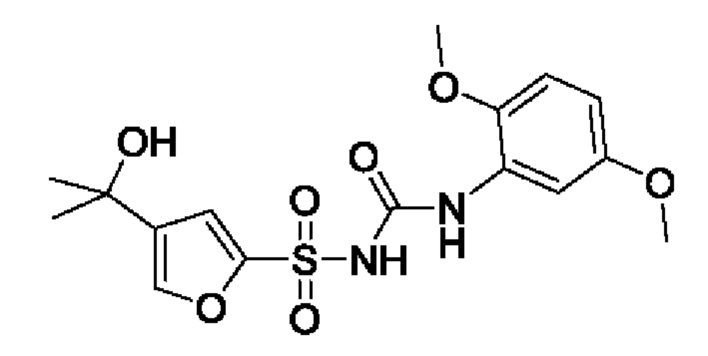
2-изоцианато-1,4-диметоксибензол и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (52 мг, 55%).
N-((4-хлор-2,6-диметилфенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
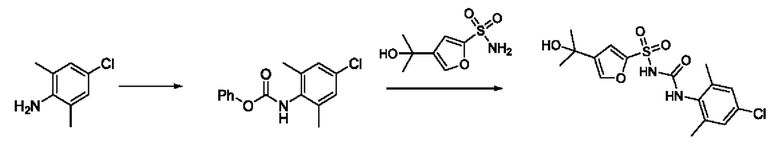
4-хлор-2,6-диметиланилин, 1 (300 мг, 1,92 ммоль) растворили в ТГФ (50 мл) и охладили до 0°С. К вышеупомянутому раствору добавили порциями NaH (100 мг, 2,49 ммоль) в атмосфере азота и перемешивали смесь в течение 15 мин. К вышеупомянутому раствору добавили по каплям фенилхлорформиат (0,33 мл, 0,72 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 30% EtOAc-гексаны с получением фенил (4-хлор-2,6-диметилфенил)карбамата (0,2 г, 85%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ = 7,41-7,36 (м, 2Н), 7,21-7,19 (м, 2Н), 7,11-7,10 (м, 3Н), 6,30 (шс, 1Н), 2,33 (с, 6Н).
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (133 мг, 0,64 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (65 мг, 1,63 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 45 мин, затем подействовали раствором фенил (4-хлор-2,6-диметилфенил)карбамата (200 мг, 0,73 ммоль) в ТГФ (3 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 2 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны с получением
N-((4-хлор-2,6-диметилфенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (20 мг, 31%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CDCl3): δ = 7,83 (с, 1Н), 7,56 (с, 1Н), 7,19 (с, 1Н), 7,10-7,05 (м, 2Н), 2,16 (с, 6Н), 1,55 (с, 6Н). ЖХМС (m/z): 385,05 [M-H]-, 94,12% (210 нм). ВЭЖХ: 92,60% (210 нм). МСВР вычислено для C16H18Cl1N2O5S1 [M-H]- 385,0630, найдено 365,0621.
N-((2,4-диметил-6-(трифторметил)фенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
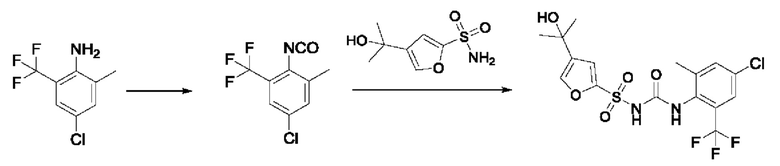
4-Хлор-2-метил-6-(трифторметил)анилин (230 мг, 1,1 ммоль) растворили в безводном ТГФ (20 мл) и подействовали Et3N (0,17 мл, 1,32 ммоль) при комнатной температуре. На раствор подействовали трифосгеном (130 мг, 0,44 ммоль) и полученную смесь перемешивали при 60°С в течение 4 ч, затем концентрировали при пониженном давлении. Полученный остаток перемешали с н-пентаном (20 мл) в течение 10 мин, профильтровали через слой целита и концентрировали при пониженном давлении с получением 5-хлор-2-изоцианато-1-метил-3-(трифторметил)бензола (0,2 г) в виде белого твердого вещества. Продукт использовали на следующей стадии без дополнительной очистки.
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид, 3 (150 мг, 0,731 ммоль) растворили в безводном ТГФ (50 мл) и осторожно подействовали NaH (44 мг, 1,096 ммоль) при 0°С в атмосфере азота. Полученную реакционную смесь перемешивали при комнатной температуре в течение 30 минут и подействовали раствором 5-хлор-2-изоцианато-1-метил-3-(трифторметил)бензола (0,2 г) в ТГФ (30 мл) в атмосфере азота. Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны. Затем продукт растерли с диэтиловым эфиром и н-пентаном с получением N-((2,4-диметил-6-(трифторметил)фенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (15 мг, 5%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD): δ = 7,52-7,48 (м, 3Н), 6,99 (с, 1Н), 2,19 (с, 3Н), 1,47 (с, 6Н). 19F-ЯМР (400 МГц, CD3OD): δ = -63,09. ЖХМС (m/z): 439,05 [М-Н]-; 94,86% (210 нм), 96,92% (254 нм). ВЭЖХ: 98,90% (210 нм). МСВР вычислено для C16H15Cl1F3N2O5S1 [M-H]- 439,0348, найдено 439,0339. N-((4-хлор-2,6-диизопропилфенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
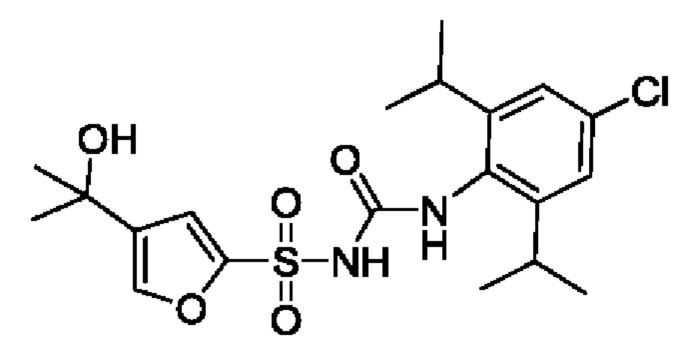
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (161 мг, 34%). 1H-ЯМР (600 МГц, ДМСО-d6) δ = 7,82 (с, 1Н), 7,61 (с, 1Н), 7,09 (с, 2Н), 6,93 (с, 1Н), 5,04 (с, 1Н), 3,05 -2,99 (м, 2Н), 1,35 (с, 6Н), 1,05 (д, J=6,9 Гц, 12Н). МСВР вычислено для C20H26Cl1N2O5S1 [M-H]- 441,1256, найдено 441,1264.
N-((4-хлор-2,6-дициклопропилфенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
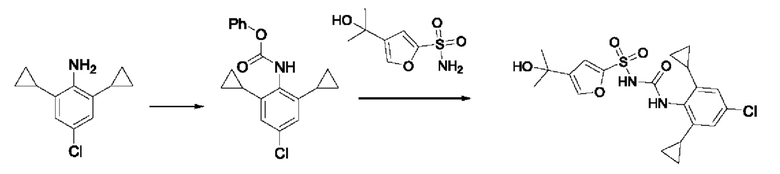
4-Хлор-2,6-дициклопропиланилин, 1 (250 мг, 1,20 ммоль) растворили в ТГФ (50 мл) и охладили до 0°С. Добавили порциями NaH (72 мг, 1,80 ммоль) и полученную смесь перемешивали в течение 20 мин в атмосфере азота. Добавили по каплям фенилхлорформиат (370 мг, 2,40 ммоль) при 0°С, затем реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 8% EtOAc-гексаны с получением фенил (4-хлор-2,6-дициклопропилфенил)карбамата (0,2 г, 51%) в виде белого твердого вещества. 1H-ЯМР (300 МГц, CDCl3): δ = 7,38-7,35 (м, 2Н), 7,21-7,19 (м, 3Н), 6,84-6,83 (м, 2Н), 2,06-2,04 (м, 2Н), 1,04-1,02 (м, 4Н), 0,69-0,68 (м, 4Н).
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (75 мг, 0,365 ммоль) растворили в безводном ТГФ (50 мл) и осторожно подействовали NaH (36 мг, 0,914 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем подействовали раствором фенил (4-хлор-2,6-дициклопропилфенил)карбамата (135 мг, 0,402 ммоль) в ТГФ (3 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 4 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием 50-100% EtOAc-гексаны с получением
N-((4-хлор-2,6-дициклопропилфенил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (10 мг, 6%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD): δ = 7,59 (с, 1Н), 7,13 (с, 1Н), 6,76 (с, 2Н), 1,88-1,86 (м, 2Н), 1,47 (с, 6Н), 0,89-0,84 (м, 4Н), 0,55-0,54 (м, 4Н). ЖХМС (m/z): 436,95 [М-Н]-; 96,29% (210 нм). ВЭЖХ: 98,29% (210 нм). МСВР вычислено для C20H22Cl1N2O5S1 [М-Н]- 437,0943, найдено 437,0945. МСВР вычислено для C20H22Cl1N2O5S1 [М-Н]- 437,0943, найдено 437,0945.
4-(2-гидроксипропан-2-ил)-N-((5-метокси-2,3-дигидро-1Н-инден-4-ил)карбамоил)фуран-2-сульфонамид
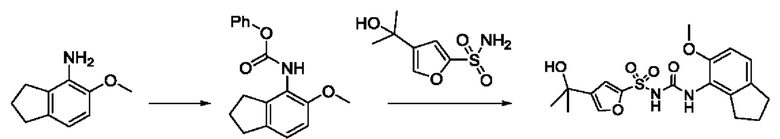
5-метокси-2,3-дигидро-1Н-инден-4-амин (150 мг, 0,59 ммоль) растворили в ТГФ (15 мл) и охладили до 0°С. К вышеупомянутому раствору добавили NaH (35 мг, 0,89 ммоль) и перемешивали в течение 20 мин. Добавили по каплям фенилхлорформиат (150 мг, 0,932 ммоль) при 0°С, затем раствор оставили прогреться до комнатной температуры на протяжении ночи. По завершении реакционную смесь разбавили насыщенным NaHCO3 и экстрагировали EtOAc (30 мл), органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении с получением фенил (5-метокси-2,3-дигидро-1H-инден-4-ил)карбамата (100 мг, 59%) в виде белого твердого вещества. 1H-ЯМР (300 МГц, CDCl3): δ = 7,40-7,35 (м, 2Н), 7,22-7,18 (м, 3Н), 7,06 (д, J=8,1 Гц, 1Н), 6,73 (д, J=8,4 Гц, 1Н), 3,86 (с, 3Н), 2,98-2,84 (м, 4Н), 2,08 (т, J=7,5 Гц, 2Н). ЖХМС (m/z): 284,3 [M+H]+
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (87 мг, 0,424 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (44 мг, 1,097 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 1 ч, затем подействовали раствором фенил (5-метокси-2,3-дигидро-1H-инден-4-ил)карбамата (120 мг, 0,424 ммоль) в ТГФ (5 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 6 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl и экстрагировали EtOAc (2 х 30 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Х bridge (150 мм х 19 мм, размер частиц 5 мкм); расход: 15 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: 17% В=0/10, 2/10, 9/70]. Фракции высушили сублимацией с получением 4-(2-гидроксипропан-2-ил)-N-((5-метокси-2,3-дигидро-1Н-инден-4-ил)карбамоил)фуран-2-сульфонамида (45 мг, 17%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD): δ = 7,67 (с, 1Н), 7,21 (с, 1Н), 7,04 (д, J=8 Гц, 1Н), 6,77 (д, J=8 Гц, 1H), 3,79 (с, 3Н), 2,84 (т, J=7,2 Гц, 2Н), 2,69 (т, J=7,2 Гц, 2Н), 2,01-1,97 (м, 2Н), 1,49 (с, 6Н). ЖХМС (m/z): 393,10 [М-Н]-; 98,97% (210 нм), 99,47% (254 нм). ВЭЖХ: 92,07% (210 нм), 93,87% (254 нм). МСВР вычислено для C18H21N2O6S1 [М-H]+ 393,1126, найдено 392,1113.
N-((7-хлор-5-циклопропил-2,3-дигидро-1Н-инден-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
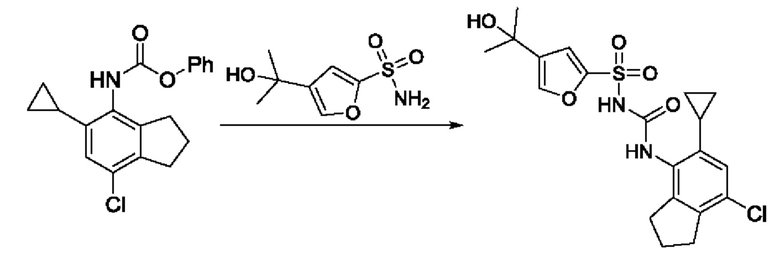
7-хлор-5-циклопропил-2,3-дигидро-1Н-инден-4-амин, 6 (70 мг, 0,33 ммоль) растворили в ТГФ (5 мл) и охладили до 0°С. К вышеупомянутому раствору добавили NaH (20 мг, 0,505 ммоль) в атмосфере азота и перемешивали в течение 15 мин с последующим добавлением фенилхлорформиата (100 мг, 0,674 ммоль) по каплям при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 10% EtOAc-гексаны с получением фенил(7-хлор-5-циклопропил-2,3-дигидро-1H-инден-4-ил)карбамата (80 мг, 73%) в виде коричневого твердого вещества. 1H-ЯМР (300 МГц, CDCl3): δ = 7,39-7,37 (м, 3Н), 7,25-7,24 (м, 2Н), 6,85 (с, 1Н), 3,0-2,94 (м, 4Н), 2,12-2,10 (м, 2Н), 1,34 (м, 1Н), 0,96-0,95 (м, 2Н), 0,59-0,57 (м, 2Н). ЖХМС (m/z): 328,30 [M+H]+.
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (56 мг, 0,274 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (27 мг, 0,685 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 15 мин и подействовали раствором фенил (7-хлор-5-циклопропил-2,3-дигидро-1H-инден-4-ил)карбамата (100 мг, 0,244 ммоль) в ТГФ (2 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 3 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 30 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Gemini NX C18 (21,2 мм х 150 мм, размер частиц 5 мкм); расход: 18 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: Т/% В=0/20, 2/30, 10/50]. Фракции высушили сублимацией с получением
N-((7-хлор-5-циклопропил-2,3-дигидро-1H-инден-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (45 мг, 38%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6): δ = 7,95 (д, J=2,4 Гц, 1Н), 6,74 (с, 1Н), 6,69(д, J=2,4 Гц, 1Н), 4,49 (м, 1Н), 2,84 (т, J=7,6 Гц, 2Н), 2,66 (т, J=7,6 Гц, 2Н), 1,95 (м, 2Н), 1,78 (м, 1Н), 1,41 (д, J=6,4 Гц, 6Н), 0,82 (м, 2Н), 0,54 (м, 2Н). ЖХМС (m/z): 437,0 [М-Н]-, 97,99% (210 нм). ВЭЖХ: 98,26% (210 нм). МСВР вычислено для C20H22Cl1N2O5S1 [M-H]+ 437,0943, найдено 437,0927.
4-(2-гидроксипропан-2-ил)-N-((3-оксо-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамид
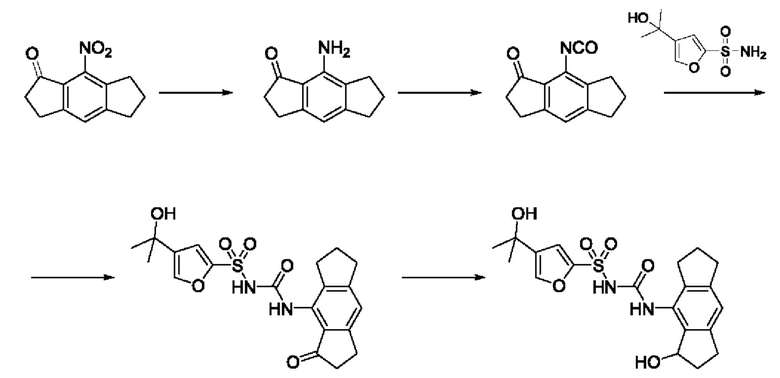
Раствор 8-нитро-3,5,6,7-тетрагидро-з-индацен-1(2Н)-она (200 мг, 0,92 ммоль) в МеОН (5 мл) дегазировали с помощью азота в течение 5 минут, добавили 10% Pd/C (20 мг, 10% масс/масс) и смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 ч. Реакционную смесь профильтровали через целит и фильтрат концентрировали при пониженном давлении с получением 8-амино-3,5,6,7-тетрагидро-s-индацен-1(2Н)-она в виде белого с желтоватым оттенком твердого вещества (160 мг, 93%). 1H-ЯМР (600 МГц, ДМСО-d6) δ 6,49 (с, 1Н), 6,34 (с, 2Н), 2,90-2,84 (м, 2Н), 2,80 (т, J=7,5 Гц, 2Н), 2,62 (т, J=7,4 Гц, 2Н), 2,56-2,51 (м, 2Н), 2,04-1,99 (м, 2Н). 13С-ЯМР (150 МГц, ДМСО-d6): 206,6, 155,4, 153,7, 144,1, 125,3, 118,6, 109,4, 36,8, 33,7, 28,6, 25,0, 24,9. ЖХМС (m/z): 188 [M+H]+. MCBP вычислено для C2H14N1O1 [M+H]+ 188,1070, найдено 188,1077.
К ди-трет-бутилдикарбонату (163 мг, 0,74 ммоль) в безводном ацетонитриле (1 мл) добавили ДМАП (26,1 мг, 0,21 ммоль) при комнатной температуре, перемешивали в течение 5 минут, добавили раствор 8-амино-3,5,6,7-тетрагидро-s-индацен-1(2Н)-она (100 мг, 0,53 ммоль) в ацетонитриле. Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Реакционную смесь использовали непосредственно на следующей стадии без выделения.
К полупродукту 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамиду (100 мг, 0,48 ммоль) в безводном ТГФ (1 мл) добавили NaH (18,3 мг, 0,48 ммоль) при 0°С и перемешивали в течение 30 минут при температуре окружающей среды в атмосфере азота. Снова охладили до 0°С, добавили 8-изоцианато-3,5,6,7-тетрагидро-s-индацен-1(2Н)-он (реакционная смесь из предыдущей стадии) и перемешивали при температуре окружающей среды в течение 16 ч. К реакционной смеси добавили 0,5 мл H2O, загрузили непосредственно в колонну С18 для очистки с использованием водного раствора (NH4)НСО3 10 ммоль/л и ацетонитрила в качестве подвижной фазы, с получением
4-(2-гидроксипропан-2-ил)-N-((3-оксо-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамида в виде белого твердого вещества (150 мг, 67%), 1Н-ЯМР (600 МГц, ДМСО-d6): δ 8,79 (с, 1Н), 7,37 (с, 1Н), 6,94 (с, 1Н), 6,61 (с, 1Н), 4,92 (с, 1Н), 2,92 (т, J=5,6 Гц, 2Н), 2,82 (т, J=7,5 Гц, 2Н), 2,75 (т, J=7,5 Гц, 2Н), 2,63-2,57 (м, 2Н), 1,97-1,80 (м, 2Н), 1,34 (с, 6Н); ЖХМС (m/z): 417 [М-Н]-. МСВР вычислено для C20H23N2O6S1 [M+H]+ 419,1271, найдено 419,1291
К раствору 4-(2-гидроксипропан-2-ил)-N-((3-оксо-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамида (70 мг, 0,16 ммоль) в МеОН (2 мл) добавили NaBH4 (63 мг, 1,67 ммоль) при 0°С в атмосфере азота, полученную реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавили H2O (2 мл, отогнали МеОН, водный слой загрузили непосредственно в колонну С18 для очистки с использованием водного раствора (NH4)НСО3 10 ммоль/л и ацетонитрила в качестве подвижной фазы, с получением
N-((3-гидрокси-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида в виде белого с желтоватым оттенком твердого вещества (60 мг, 86%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ 7,73 (ш.с, 1Н), 7,38 (с, 1Н), 6,81 (с, 1Н), 6,60 (с, 1Н), 5,63 (ш.с, 1Н), 4,92 (ш.с, 1Н), 4,87 (д, J=6,0 Гц, 1Н), 3,00-2,84 (м, 2Н), 2,77 (т, J=7,4 Гц, 2Н), 2,64-2,53 (м, 2Н), 2,07-2,00 (м, 1Н), 1,97-1,92 (м, 1Н), 1,91-1,81 (м, 2Н), 1,35 (с, 6Н); 13С-ЯМР (150 МГц, ДМСО-d6): 159,5, 156,0, 144,9, 143,3, 138,3, 137,7, 136,8, 136,0, 133,0, 72,6, 67,0, 35,4, 33,1, 31,5, 31,4, 31,0, 30,4, 25,5; ЖХМС (m/z): 419 [М-Н]-; МСВР вычислено для C20H23N2O6S1 [М-Н]-419,1282, найдено 419,1263.
N-((1-гидрокси-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
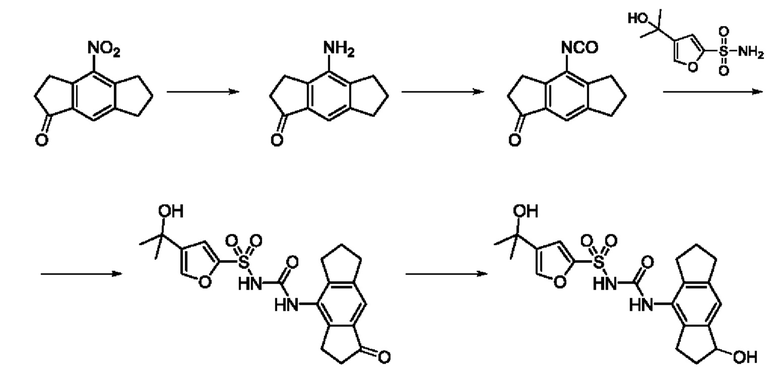
Раствор 4-нитро-3,5,6,7-тетрагидро-s-индацен-1(2Н)-она (110 мг, 0,50 ммоль) в МеОН (5 мл) дегазировали с помощь азота в течение 5 минут, добавили 10% Pd/C (11 мг, 10% масс/масс), перемешивали в атмосфере водорода при комнатной температуре на протяжении около 2 ч. Реакционную смесь профильтровали через слой целита, фильтрат концентрировали с получением 4-амино-3,5,6,7-тетрагидро-s-индацен-1(2Н)-она в виде белого с желтоватым оттенком твердого вещества (75 мг, 80%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ 6,72 (с, 1Н), 5,11 (с, 2Н), 2,87-2,74 (м, 4Н), 2,70 (т, J=7,4 Гц, 2Н), 2,62-2,54 (м, 2Н), 2,06-1,99 (м, 2Н); 13С-ЯМР (150 МГц, ДМСО-d6) δ 206,7, 144,6, 141,7, 139,9, 136,9, 136,8, 104,2, 39,9, 36,7, 30,6, 30,5, 24,5, 23,7. ЖХМС (m/z): 188 [М+Н]-; МСВР вычислено для C12H14N1O1 [M+H]+ 188,1070, найдено 188,1074.
К ди-трет-бутилдикарбонату (81,6 мг, 0,37 ммоль) в безводном ацетонитриле (1 мл) добавили ДМАП (13,0 мг, 0,04 ммоль) при комнатной температуре, перемешивали в течение 5 минут, добавили раствор 4-амино-3,5,6,7-тетрагидро-s-индацен-1(2Н)-она (50 мг, 0,26 ммоль) в ацетонитриле (1 мл). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Реакционную смесь использовали непосредственно на следующей стадии без выделения.
К полупродукту 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамиду (50 мг, 0,24 ммоль) в безводном ТГФ (1 мл) добавили NaH (9,3 мг, 0,24 ммоль) при 0°С и перемешивали в течение 30 минут при температуре окружающей среды в атмосфере азота. Снова охладили до 0°С, добавили 4-изоцианато-3,5,6,7-тетрагидро-s-индацен-1(2Н)-он (реакционная смесь из предыдущей стадии) и перемешивали при температуре окружающей среды в течение 16 ч. К реакционной смеси добавили 0,5 мл H2O, загрузили непосредственно в колонну С18 для очистки с использованием водного раствора (NH4)НСО3 10 ммоль/л и ацетонитрила в качестве подвижной фазы, с получением
4-(2-гидроксипропан-2-ил)-N-((1-оксо-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамида (70 мг, 63%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ 7,94 (с, 1Н), 7,38 (с, 1Н), 7,17 (с, 1Н), 6,60 (с, 1Н), 4,92 (с, 1Н), 2,94-2,89 (м, 2Н), 2,85 (т, J=7,4 Гц, 2Н), 2,80 (т, J=7,4 Гц, 2Н), 2,56-2,52 (м, 2Н), 2,00-1,94 (м, 2Н), 1,35 (с, 6Н); ЖХМС (m/z): 417 [М-Н]-; МСВР вычислено для C20H21N2O6S1 [М-Н]- 417,1126, найдено 417,1113.
К раствору 4-(2-гидроксипропан-2-ил)-N-((1-оксо-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)фуран-2-сульфонамида (50 мг, 0,11 ммоль) в МеОН (2 мл) добавили NaBH4 (45 мг, 1,19 ммоль) при 0°С в атмосфере азота, полученную реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавили Н2О (1 мл), отогнали МеОН, водный слой загрузили непосредственно в колонну С18 для очистки с использованием водного раствора (NH4)НСО3 10 ммоль/л и ацетонитрила в качестве подвижной фазы, с получением
N-((1-гидрокси-1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (20 мг, 40%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ 7,77 (с, 1Н), 7,51 (с, 1Н), 6,93 (с, 1Н), 6,79 (с, 1Н), 6,55 (с, 1Н), 5,05 (д, J=5,8 Гц, 1Н), 4,99 (с, 1Н), 4,94 (кв, J=6,4 Гц, 1Н), 2,79 (т, J=7,5 Гц, 2Н), 2,70-2,61 (м, 3Н), 2,52-2,49 (м, 1Н), 2,2-2,21 (м, 1Н), 1,95-1,90 (м, 2Н), 1,74-1,59 (м, 1Н), 1,36 (с, 6Н); 13С-ЯМР (150 МГц, ДМСО-d6): 163,5, 146,0, 143,1, 140,6, 138,5, 136,5, 136,2, 122,0, 116,5, 112,5, 108,5, 74,9, 67,0, 36,0, 33,0, 31,5, 30,9, 27,8, 25,6; ЖХМС (m/z): 419 [М-Н]-. МСВР вычислено для C20H23N2O6S1 [М-Н]- 419,1282, найдено 419,1265.
N-((4,6-диметилпиримидин-2-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид

Раствор 4,6-диметилпиримидин-2-амина (200 мг, 1,62 ммоль) в ТГФ (5 мл) охладили до 0°С и подействовали NaH (130 мг, 3,24 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 15 мин и подействовали фенилхлорформиатом (380 мг, 2,43 ммоль) при 0°С в атмосфере азота. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны с получением фенил (4,6-диметилпиримидин-2-ил)карбамата (200 мг, 51%) в виде белого твердого вещества. 1H-ЯМР (300 МГц, CDCl3): δ = 7,91 (с, 1Н), 7,40-7,35 (м, 2Н), 7,24-7,19 (м, 3Н), 6,78 (с, 1Н), 2,41 (с, 6Н). ЖХМС (m/z): 244,20 [M+H]+.
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (150 мг, 0,731 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (75 мг, 1,83 ммоль) при 0°С в атмосфере азота. Полученную смесь нагрели до 60°С и перемешивали в течение 2 ч. Раствор охладили до 0°С и подействовали раствором фенил (4,6-диметилпиримидин-2-ил)карбамата (195 мг, 0,804 ммоль) в ТГФ (5 мл) в атмосфере азота при 0°С. Реакционную смесь нагрели до 50°С в течение 4 ч и затем перемешивали при комнатной температуре в течение 4 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 30 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: X-bridge (150 мм х 19 мм, размер частиц 5 мкм);
расход: 15 мл/мин; элюент: 10 ммоль/л ацетат аммония в 0,1% АсОН в воде (А) & MeCN (В); градиент: Т/% В=0/15, 2/25, 8/40]. Фракции высушили сублимацией с получением
N-((4,6-диметилпиримидин-2-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (25 мг, 7%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CDCl3): δ = 8,18 (с, 1Н), 7,51 (д, J=0,8 Гц, 1Н), 7,40 (д, J=1,2 Гц, 1Н), 6,78 (с, 1Н), 2,48 (с, 6Н), 1,57 (с, 6Н). ЖХМС (m/z): 355,0 [M+H]+, 100% (210 нм), 100% (254 нм). ВЭЖХ: 96,49% (210 нм), 98,76% (254 нм). МСВР вычислено для C14H17N4O5S1 [M-H]- 353,0925, найдено 353,0921.
N-((4-циклопропил-6-метилпиримидин-2-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
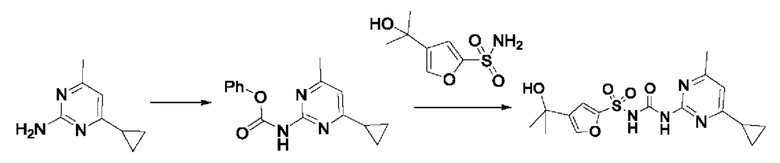
4-циклопропил-6-метилпиримидин-2-амин (50 мг, 0,33 ммоль) растворили в ТГФ (2 мл) и охладили до 0°С. К вышеупомянутому раствору осторожно добавили NaH (16 мг, 0,40 ммоль) и перемешивали в течение 20 мин. Добавили по каплям фенилхлорформиат (80 мг, 0,503 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали при комнатной температуре в течение 12 ч. По завершении реакции (ТСХ, 50% этилацетат-гексаны, Rf, 0,4), реакционную смесь разбавили EtOAc и профильтровали через слой целита. Фильтрат концентрировали при пониженном давлении и технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 30% EtOAc-гексаны с получением фенил (4-циклопропил-6-метилпиримидин-2-ил)карбамата (40 мг, 44%) в виде белого с желтоватым оттенком твердого вещества. 1H-ЯМР (300 МГц, CDCl3): δ = 8,06 (с, 1Н), 7,40-7,37 (м, 2Н), 7,24-7,15 (м, 3Н), 6,73 (с, 1Н), 2,4 (с, 3Н), 1,94-1,86 (м, 1Н), 1,15-1,1 (м, 2Н), 1,0-0,99 (м, 2Н). ЖХМС (m/z): 270,3 [M+H]+
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (150 мг, 0,731 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (73 мг, 1,829 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 1 ч и подействовали раствором фенил (4-циклопропил-6-метилпиримидин-2-ил)карбамата (190 мг, 0,731 ммоль) в ТГФ (5 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали при комнатной температуре в течение 6 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl и экстрагировали EtOAc (2 х 30 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Х bridge (150 мм х 19 мм, размер частиц 5 мкм);
расход: 15 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: 17% В=0/15, 2/25, 8/40]. Фракции высушили сублимацией с получением
N-((4-циклопропил-6-метилпиримидин-2-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (30 мг, 11%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CD3OD): δ = 7,53 (с, 1Н), 7,07 (с, 1Н), 6,80 (с, 1Н), 2,35 (с, 3Н), 1,97-1,93 (м, 1Н), 1,42 (с, 6Н), 1,08-1,02 (м, 4Н). ЖХМС (m/z): 381,00 [M+H]+; 98,60% (210 нм), 99,49% (254 нм). ВЭЖХ: 98,05% (210 нм), 99,01% (254 нм). МСВР вычислено для C16H19N4O5S1 [M-H]+ 379,1082, найдено 379,1082.
N-((4,6-ди-трет-бутилпиримидин-2-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
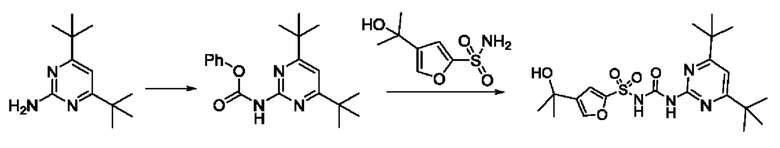
4,6-ди-трет-бутилпиримидин-2-амин (0,15 г, 0,72 ммоль) растворили в ТГФ (5 мл) и охладили до 0°С. К вышеупомянутому раствору добавили NaH (37 мг, 0,93 ммоль) и полученную смесь перемешивали при 15 мин в атмосфере азота. К вышеупомянутому раствору добавили по каплям фенилхлорформиат (0,16 г, 1,08 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакции (ТСХ, 10% этилацетат-гексаны, Rf, 0,5), реакционную смесь концентрировали при пониженном давлении. Полученный остаток разбавили смесью 10% ИПС/CHCl3, профильтровали через слой целита и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 4% EtOAc-гексаны с получением фенил(4,6-ди-трет-бутилпиримидин-2-ил)карбамата (0,1 г, 43%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ = 7,65 (с, 1Н), 7,38 (м, 2Н), 7,22 (м, 3Н), 7,01 (с, 1Н), 1,32 (с, 18Н). ЖХМС (m/z): 327,80 [M+H]+
4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (65 мг, 0,305 ммоль) растворили в безводном ТГФ (8 мл) и осторожно подействовали NaH (30 мг, 0,764 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 45 минут и подействовали раствором фенил (4,6-ди-трет-бутилпиримидин-2-ил)карбамата (100 мг, 0,305 ммоль) в ТГФ (5 мл) по каплям в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 3 ч. По завершении реакции (ТСХ, 50% этилацетат-гексаны, Rf, 0,5) реакционную смесь разбавили насыщенным раствором NH4CI и экстрагировали EtOAc (2 х 20 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны с получением N-((4,6-ди-трет-бутилпиримидин-2-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамида (0,07 г, 52%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6): δ = 13,75 (с, 1Н), 10,71 (с, 1Н), 7,83 (с, 1Н),7,37(с, 1Н),7,20 (с, 1Н), 5,17 (с, 1Н), 1,39 (с, 6Н), 1,31 (с, 18Н). ЖХМС (m/z): 439,55 [M+H]+; 94,58% (210 нм), 97,94% (254 нм). ВЭЖХ: 98,51% (210 нм), 99,27% (254 нм). МСВР вычислено для C20H29N4O5S1 [M-H]+ 437,1864, найдено 437,1846.
МЕТИЛФУРАНЫ
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид
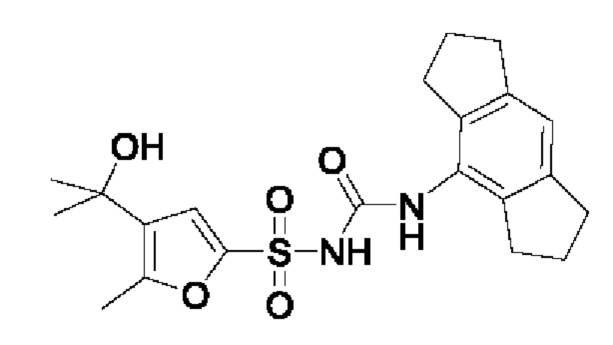
4-изоцианато-1,2,3,5,6,7-гексагидро-8-индацен (полученный с использованием общего способа А2) и 4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (52 мг, 51%).
N-((2,6-диизопропилфенил)карбамоил)-4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид
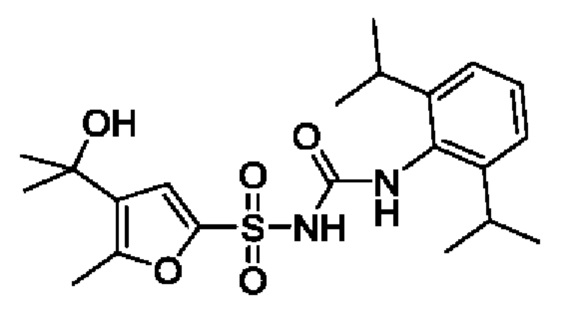
2-Изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А1) и 4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (14 мг, 4%). 1H-ЯМР (400 МГц, CD3OD): δ = 7,24 (т, J=7,6 Гц, 1Н), 7,15 (д, J=7,6 Гц, 2Н), 7,04 (с, 1Н), 3,10 (септ, J=6,8 Гц, 2Н), 2,50 (с, 3Н), 1,50 (с, 6Н), 1,17 (д, J=6,8 Гц, 12Н).
ДЕЙТЕРИРОВАННЫЕ ФУРАНЫ
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил-1,1,1,3,3,3-d6)фуран-2-сульфонамид
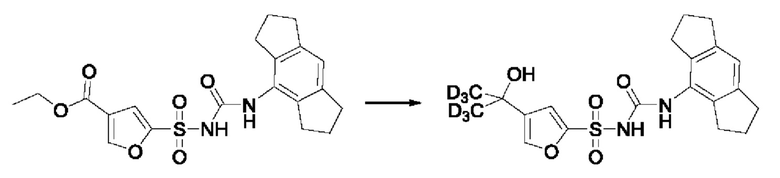
N-((1,2,3,5,6,7-гексагидро-з-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид можно синтезировать с использованием 4-изоцианато-1,2,3,5,6,7-гексагидро-5-индацена (полученного с использованием общего способа А1) и d6-4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамида в общем способе С1.
Альтернативно, на этил-2-метил-5-сульфамоилфуран-3-карбоксилат (0,4 г, 0,96 ммоль) в безводном ТГФ (30 мл) при -10°С подействовали раствором d3-метилмагниййодида (1,0 моль/л в Et2O, 10 экв.) по каплям в течение 10 минут при интенсивном перемешивании. Затем раствор перемешивали при температуре окружающей среды в течение 12 ч, затем охладили до 0°С и остановили реакцию добавлением по каплям насыщенного раствора хлорида аммония. Водный раствор экстрагировали с использованием EtOAc (2 х 20 мл), объединенные органические фазы промыли солевым раствором (20 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде белого твердого вещества (5 мг, 1%). 1H-ЯМР (300 МГц, CD3OD): δ = 7,50 (д, J=1,2 Гц, 1Н), 6,95 (д, J=1,2 Гц, 1Н), 6,89 (с, 1Н), 2,83 (т, J=7,2 Гц, 4Н), 2,75 (т, J=7,2 Гц, 4Н), 2,02 (квинт, J=7,2 Гц, 4Н).
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил-1,1,1,3,3,3-d6)-5-метилфуран-2-сульфонамид
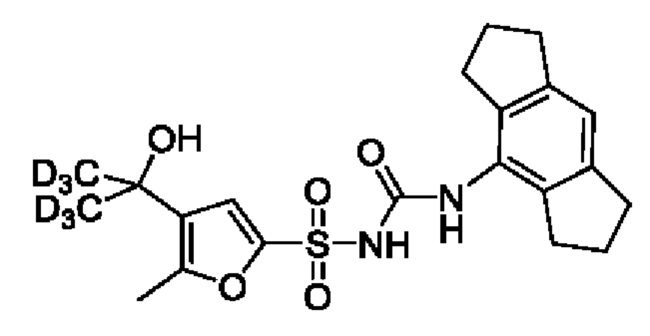
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и d6-4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (10 мг, 3%), 1Н-ЯМР (400 МГц, CD3OD) δ = 7,03 (с, 1Н), 6,95 (с, 1Н), 2,86 (т, J=7,4 Гц, 4Н), 2,73 (т, J=7,4 Гц, 4Н), 2,48 (с, 3Н), 2,04 (п, J=7,4 Гц, 4Н).
4-(2-гидроксипропан-2-ил-1,1,1,3,3,3-d6)-5-метил-N-((3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-4-ил)карбамоил)фуран-2-сульфонамид
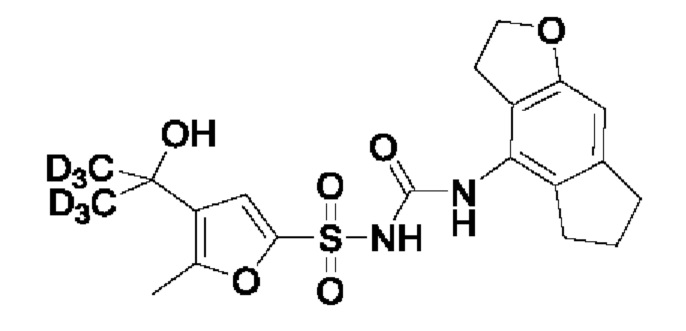
4-изоцианато-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран (полученный с использованием общего способа А1) и d6-4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (20 мг, 5%). 1Н-ЯМР (400 МГц, CD3OD) δ = 7,13 (с, 1Н), 6,52 (с, 1Н), 4,51 (т, J=8,6 Гц, 2Н), 3,03 (т, J=8,6 Гц, 2Н), 2,84 (т, J=7,4 Гц, 2Н), 2,68 (т, J=7,4 Гц, 2Н), 2,50 (с, 3Н), 2,05 (п, J=7,4 Гц, 2Н).
N-((4-бром-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран-8-ил)карбамоил)-4-(2-гидроксипропан-2-ил-1,1,1,3,3,3-d6)-5-метилфуран-2-сульфонамид
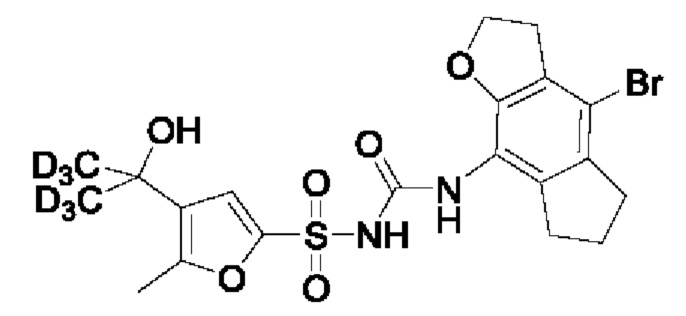
4-бром-8-изоцианато-3,5,6,7-тетрагидро-2Н-индено[5,6-b]фуран (полученный с использованием общего способа А1) и d6-4-(2-гидроксипропан-2-ил)-5-метилфуран-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (32 мг, 39%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ = 8,01 (с, 1Н), 7,07 (с, 1Н), 5,04 (с, 1Н), 4,59 (т, J=8,7 Гц, 2Н), 3,14 (т, J=8,7 Гц, 2Н), 2,77 (т, J=7,4 Гц, 2Н), 2,69 (т, J=7,4 Гц, 2Н), 2,43 (с, 3Н), 1,98 (кв, J=7,4 Гц, 2Н).
ТИОФЕНЫ
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)тиофен-2-сульфонамид
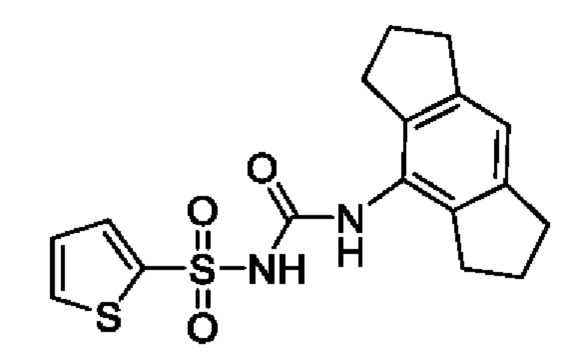
4-Изоцианато-1,2,3,5,6,7-гексагидро-5-индацен (полученный с использованием общего способа А2) и тиофен-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (11 мг, 11%). 1Н-ЯМР (400 МГц, CD3OD): δ = 7,79 (д, J=4,0 Гц, 1Н), 7,76 (д, J=4,0 Гц, 1Н), 7,73 (т, J=4,0 Гц, 1Н), 6,93 (с, 1Н), 2,83 (т, J=12 Гц, 4Н), 2,66 (т, J=12 Гц, 4Н), 2,04-1,96 (м, 4Н). 13С-ЯМР (100 МГц, CD3OD): δ 143,5, 143,2, 137,8, 132,7, 132,2, 126,6, 126,4, 118,2, 110,3, 32,5, 29,9, 25,1; Чистота по ЖХМ С: >95%; ЖХМС (m/z): 363 [М+H]+; МСВР вычислено для C17H18N2O3S2 (M+H)+, 363,0832, найдено 363,0819.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-5-метилтиофен-2-сульфонамид
4-Изоцианато-1,2,3,5,6,7-гексагидро-5-индацен (полученный с использованием общего способа А2) и 5-метилтиофен-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (12 мг, 18%). 1H-ЯМР (400 МГц, ДМСО-d6): δ = 7,88 (с, 1Н), 7,43 (д, J=4,0 Гц, 1Н), 6,89 (с, 1Н), 6,82 (д, J=4,0 Гц, 1Н), 2,78 (т, J=12 Гц, 4Н), 2,61 (т, J=12 Гц, 4Н), 2,47 (с, 3Н), 1,97-1,89 (м, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ = 143,2, 142,9, 137,3, 130,6, 126,0, 125,6, 125,2, 117,5, 108,7, 32,69, 30,7, 25,5, 15,4; Чистота по ЖХМС: >95%; ЖХМС (m/z): 377 [М+Н]+; МСВР вычислено для C18H20N2O3S2, (M+H)+ 377,0988, найдено 377,0994.
ТИАЗОЛЫ
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)тиазол-2-сульфонамид
4-Изоцианато-1,2,3,5,6,7-гексагидро-8-индацен (полученный с использованием общего способа А2) и тиазол-2-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (8 мг, 20%). 1H-ЯМР (400 МГц, CD3OD): δ = 7,02 (д, 1Н, J=4,0 Гц), 6,99 (с, 1Н), 6,60 (д, 1Н, J=4,0 Гц), 2,88 (т, 4Н, J=8,0 Гц), 2,76 (т, 4Н, J=8,0 Гц), 2,08-2,02 (м, 4Н); 13С-ЯМР (100 МГц, CD3OD): δ = 169,6, 144,2, 144,1, 137,7, 137,5, 132,4, 118,1, 106,9, 32,4, 29,9, 25,2.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-2-метилтиазол-5-сульфонамид
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 2-метилтиазол-5-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (35 мг, 65%); 1H-ЯМР (600 МГц, ДМСО-d6) δ = 7,73 (с, 1Н), 7,53 (с, 1Н), 6,78 (с, 1Н), 2,75 (т, J=7,4 Гц, 4Н), 2,66 (т, J=7,4 Гц, 4Н), 2,59 (с, 3Н), 1,93-1,88 (м, 4Н); 13С-ЯМР (150 МГц, ДМСО-d6): 166,8, 158,3, 143,9, 142,0, 141,4, 136,6, 132,3, 115,5, 32,5, 30,4, 25,0, 18,6; Чистота по ЖХМС: >95%; ЖХМС (m/z): 378 [M+H]+; MCBP вычислено для C17H18N3O3S2 [М-Н]+ 376,0795, найдено 376,0791.
ТРИАЗОЛЫ
1-бензил-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1Н-1,2,4-триазол-3-сульфонамид
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-бензил-1H-1,2,4-триазол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (40 мг, 15%) 1H-ЯМР (400 МГц, ДМСО-d6): δ = 8,9 (с, 1Н), 8,0 (с, 1Н), 7,35-7,28 (м, 5Н), 6,90 (с, 1Н), 5,48 (с, 2Н), 2,77 (т, J=7,2 Гц, 4Н), 2,59 (т, J=7,2 Гц, 4Н), 1,95-1,90 (м, 4Н). ЖХМС (m/z): 438,10 (М+1)+ 95,84% (210 нм), 97,84% (254 нм). ВЭЖХ: 95,99% (210 нм), 95,31% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4Н-1,2,4-триазол-3-сульфонамид
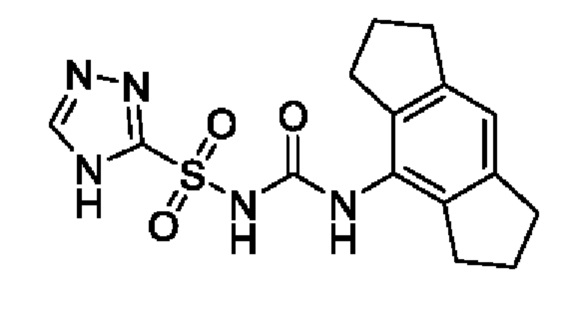
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4Н-1,2,4-триазол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (31 мг, 62%); 1H-ЯМР (600 МГц, ДМСО-d6) δ = 9,83 (шс, 1Н), 7,99 (с, 1Н), 7,62 (с, 1Н), 6,77 (с, 1Н), 2,73 (т, J=7,4 Гц, 4Н), 2,64 (т, J=7,4 Гц, 4Н), 1,91-1,86 (м, 4Н); 13С-ЯМР (150 МГц, ДМСО-d6): 159,2, 149,8, 148,7, 142,5, 137,2, 132,8, 116,1, 33,0, 30,9, 25,6; Чистота по ЖХМС: >95%; ЖХМС (m/z): 348 [M+H]+;
MCBP вычислено для C15H16N5O3S1 [M-H]- 346,0979, найдено 346,0983.
N((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-изопропил-1Н-1,2,3-триазол-4-сульфонамид
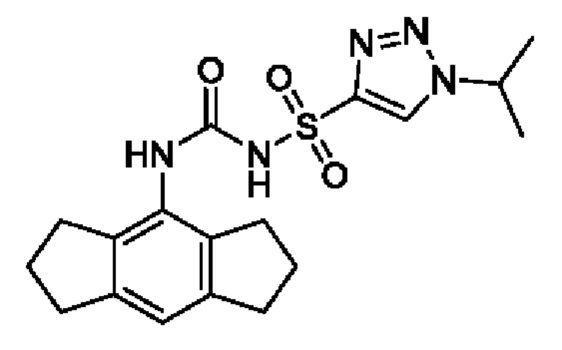
1,2,3,5,6,7-гексагидро-s-индацен-4-амин, 7 (100 мг, 0,578 ммоль) растворили в безводном ТГФ (5 мл) и подействовали Et3N (70 мг, 0,693 ммоль) при комнатной температуре. На раствор подействовали трифосгеном (70 мг, 0,231 ммоль) и полученную смесь перемешивали при 70°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток перемешали с н-пентаном (20 мл) в течение 10 мин и профильтровали через целит. Фильтрат концентрировали при пониженном давлении с получением изоцианата в виде белого твердого вещества. В другой круглодонной колбе емкостью 50 мл 1-изопропил-1Н-1,2,3-триазол-4-сульфонамид (95 мг, 0,50 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (42 мг, 1,05 ммоль) при 0°С под азотом. Перемешивали при комнатной температуре в течение 45 минут и подействовали вышеупомянутым раствором изоцианата в ТГФ под азотом.
Полученную реакционную смесь перемешивали при комнатной температуре в течение 5 ч. По завершении (ТСХ, 70% этилацетат-гексаны, Rf, 0,3) реакционную смесь разбавили насыщенным раствором NH4Cl и экстрагировали EtOAc (2 х 25 мл). Объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Gemini NX С18 (21,5 мм х 150 мм, размер частиц 5 мкм); расход: 15 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: Т/% В=0/10, 2/20, 8/65]. Фракции высушили сублимацией с получением N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-изопропил-1Н-1,2,3-триазол-4-сульфонамида (25 мг, 12%), в виде белого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6): δ = 8,75 (с, 1Н), 7,91 (с, 1Н), 6,89 (с, 1Н), 4,9 (м, 1Н), 2,79 (т, J=7,2 Гц, 4Н), 2,60 (т, J=7,2 Гц, 4Н), 1,96-1,89 (м, 4Н), 1,5 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 390,10 [M+H]+; 100% (210 нм),100% (254 нм). ВЭЖХ: 96,05% (210 нм), 96,13% (254 нм). МСВР вычислено для C18H22N5O3S1 [М-Н]- 388,1449, найдено 388,1457.
ПИРАЗОЛЫ
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-метил-1Н-пиразол-5-сульфонамид
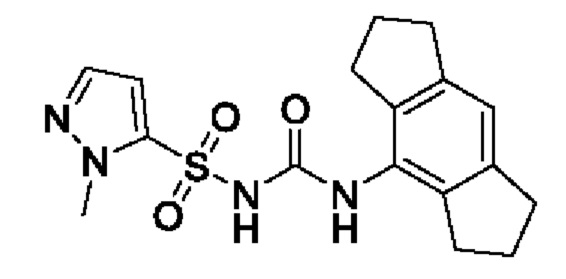
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 1-метил-1Н-пиразол-5-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (8 мг) 20%. 1H-ЯМР (400 МГц, ДМСО-d6): δ = 7,95 (шс, 1Н), 7,45 (с, 1Н), 6,88 (с, 1Н), 6,66 (с, 1Н), 4,02 (с, 3Н), 2,77 (т, J=16 Гц, 4Н), 2,60 (т, J=16 Гц, 4Н), 1,96-1,88 (м, 4Н). 13С-ЯМР (150 МГц, ДМСО-d6): δ = 143,1, 142,9, 137,2, 125,2, 117,4, 110,0, 109,0, 108,7, 38,6, 33,0, 30,7, 25,5; Чистота по ЖХМС: >95%; ЖХМС (m/z): 361 [М+H]+; МСВР вычислено для C17H20N4O3S (M+H)+, 361,13289, найдено 361,13213.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-метил-1Н-пиразол-3-сульфонамид
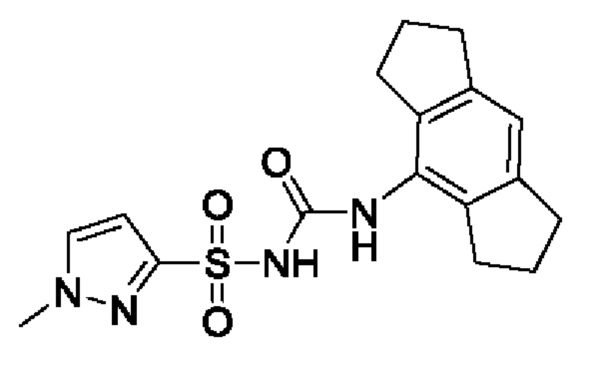
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-метил-1H-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (40 мг, 8%). 1H-ЯМР (400 МГц, ДМСО-d6): δ = 10,8 (шс, 1Н), 8,02 (с, 1Н), 7,86 (с, 1Н), 6,92 (с, 1Н), 6,69 (с, 1Н), 3,91 (с, 3Н), 2,80 (т, J=7,2 Гц, 4Н), 2,62 (т, J=7,2 Гц, 4Н), 1,96 (т, J=7,2 Гц, 4Н). ЖХМС (m/z): 383,10 (М+Na)+; 96,00% (210 нм), 93,44% (254 нм). ВЭЖХ: 97,86% (210 нм), 97,44% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-(трифторметил)-1Н-пиразол-3-сульфонамид
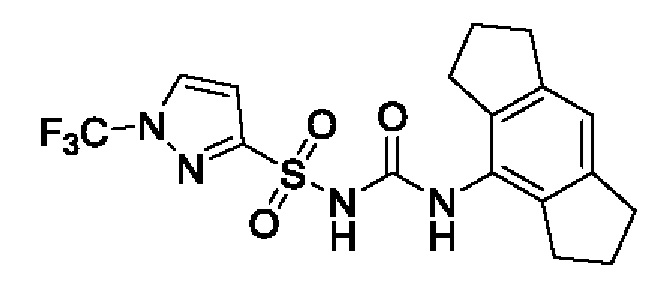
4-Изоцианато-1,2,3,5,6,7-гексагидро-5-индацен (полученный с использованием общего способа А1) и 1-(трифторметил)-1Н-пиразол-3-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (5 мг, 1%). 1H-ЯМР (400 МГц, CD3OD) δ = 8,28 (д, J=2,8 Гц, 1Н), 6,96 (д, J=2,8, 1Н), 6,91 (с, 1Н), 2,84 (т, J=7,4 Гц, 4Н), 2,75 (т, J=7,4 Гц, 4Н), 2,03 (м, J=7,4 Гц, 4Н).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид 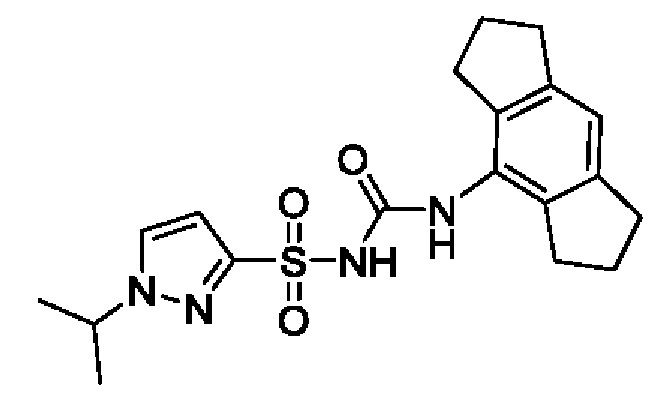
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-изопропил-1Н-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (40 мг, 9%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ = 10,92 (с, 1Н), 8,02 (с, 1Н), 8,0 (с, 1Н), 6,94 (с, 1Н), 6,74 (с, 1Н), 4,67-4,59 (м, 1Н), 2,78 (т, J=7,2 Гц, 4Н), 2,58 (т, J=7,2 Гц, 4Н), 1,95-1,91 (м, 4Н), 1,44 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 387,1 (М -1)-; 97,14% (210 нм), 95,11% (254 нм). ВЭЖХ: 95,57% (210 нм), 93,53% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-изопропил-1Н-пиразол-4-сульфонамид
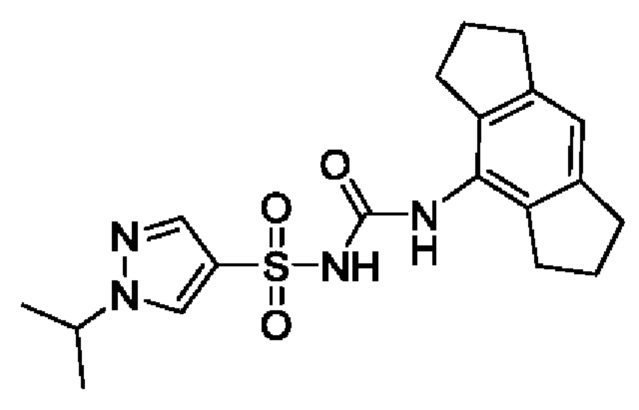
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-5-индацен (полученный с использованием общего способа А1) и 1-изопропил-1H-пиразол-4-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (40 мг, 10%) 1Н-ЯМР (400 МГц, ДМСО-d6): δ = 10,6 (с, 1Н), 8,44 (с, 1Н), 8,05 (с, 1Н), 7,86 (с, 1Н), 6,94 (с, 1Н), 4,63-4,57 (м, 1Н), 2,80 (т, J=7,2 Гц, 4Н), 2,57 (т, J=7,6 Гц, 4Н), 1,94-1,89 (м, 4Н), 1,42 (д, J=6,8 Гц 6Н). ЖХМС (m/z): 389,20 (М+1)+; 97,25% (210 нм), 94,22% (254 нм). ВЭЖХ: 97,13% (210 нм), 95,06% (254 нм).
1-циклопропил-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1Н-пиразол-3-сульфонамид
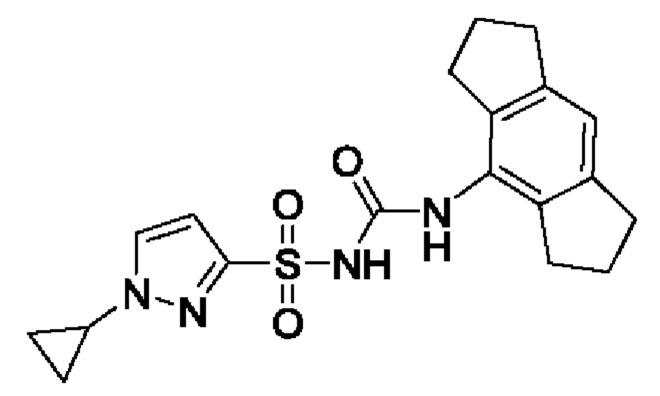
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и N-циклопропил-1Н-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (20 мг, 6%). 1H-ЯМР (400 МГц, ДМСО-d6) δ = 7,83 (с, 1Н), 7,8 (с, 1Н), 6,84 (с, 1Н), 6,48 (с, 1Н), 3,81-3,71 (м, 1Н), 2,77 (т, J=7,4 Гц, 4Н), 2,64 (т, J=7,4 Гц, 4Н), 2,02-1,86 (м, 4Н), 1,09-0,93 (м, 4Н).
1-(трет-бутил)-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1Н-пиразол-3-сульфонамид
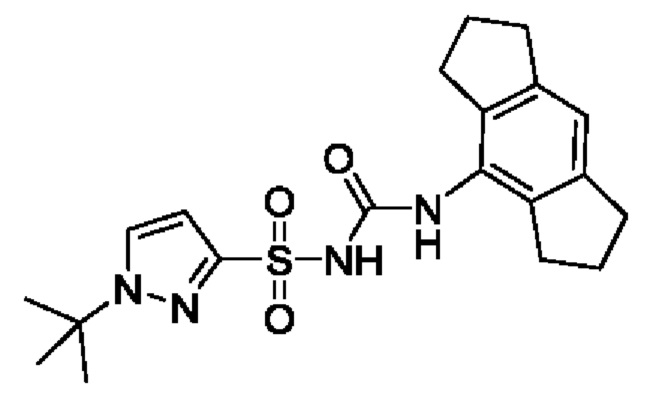
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-5-индацен (полученный с использованием общего способа А1) и 1-(трет-бутил)-1H-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (120 мг, 51%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ = 10,85 (ш.с., 1Н), 7,95 (с, 1Н), 7,88 (ш.с., 1Н), 6,88 (с, 1Н), 6,63 (с, 1Н), 2,79 (т, J=7,2 Гц, 4Н), 2,61 (т, J=7,2 Гц, 4Н), 1,96 (м, 4Н), 1,55 (с, 9Н). ЖХМС (m/z): 403,15 (М +1)+; 97,86% (210 нм), 96,50% (254 нм). ВЭЖХ: 96,45% (210 нм), 95,89% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-(1-метилпиперидин-4-ил)-1Н-пиразол-3-сульфонамид
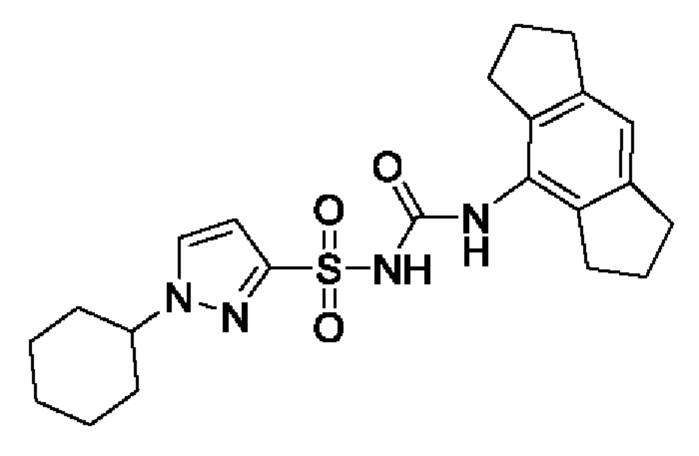
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-циклогексил-1Н-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (20 мг, 6%). 1H-ЯМР (400 МГц, ДМСО-d6) δ = 10,8 (с, 1Н), 8,03 (с, 1Н), 7,99 (д, J=2,4, 1Н), 6,95 (с, 1Н), 6,75 (д, J=2,4 Гц, 1Н), 4,33-4,20 (м, 1Н), 2,79 (т, J=7,4 Гц, 4Н), 2,58 (т, J=7,4 Гц, 4Н), 2,05-1,88 (м, 6Н), 1,86-1,63 (м, 6Н), 1,48-1,33 (м, 2Н).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-фенил-1Н-пиразол-3-сульфонамид
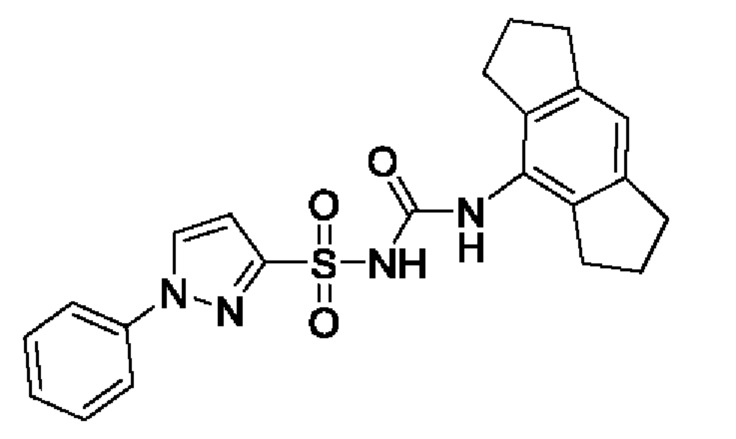
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-фенил-1H-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (110 мг, 27%). 1H-ЯМР (400 МГц, ДМСО-d6): δ = 10,92 (c, 1Н),8,61 (д, J=2,4 Гц, 1Н), 7,95 (ш.с, 1Н), 7,86 (д, J=8,4 Гц, 2Н), 7,56 (т, J=7,6 Гц, 2Н), 7,41 (т, J=7,2 Гц, 1Н), 6,9 (д, J=2,0 Гц, 1Н), 6,86 (с, 1Н), 2,77 (т, J=7,2 Гц, 4Н), 2,62 (т, J=7,2 Гц, 4Н), 1,91-1,83 (м, 4Н). ЖХМС (m/z): 421,05 (М -1)-; 96,62% (210 нм), 95,12% (254 нм). ВЭЖХ: 95,2% (210 нм), 95,77% (254 нм).
1-бензил-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1Н-пиразол-3-сульфонамид
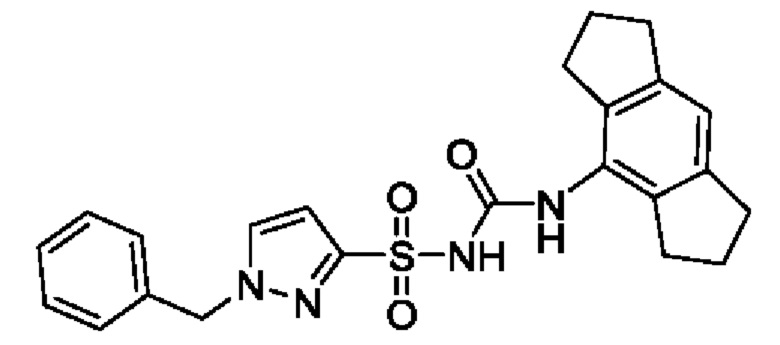
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-5-индацен (полученный с использованием общего способа А1) и 1-бензил-1H-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (85 мг, 34%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ = 10,85 (с, 1Н), 8,05 (д, J=2,4 Гц, 1Н), 7,99 (с, 1Н), 7,32-7,31 (м, 3Н), 7,24-7,22 (м, 2Н), 6,93 (с, 1Н), 6,78 (д, J=2,4 Гц, 1Н), 5,44 (с, 2Н), 2,80 (т, J=7,6 Гц, 4Н), 2,57 (т, J=7,2 Гц, 4Н), 1,96 (м, 4Н). ЖХМС (m/z): 437,15 (М +1)+; 97,70% (210 нм), 96,86% (254 нм). ВЭЖХ: 98,05% (210 нм), 97,56% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-(1-фенилэтил)-1Н-пиразол-3-сульфонамид
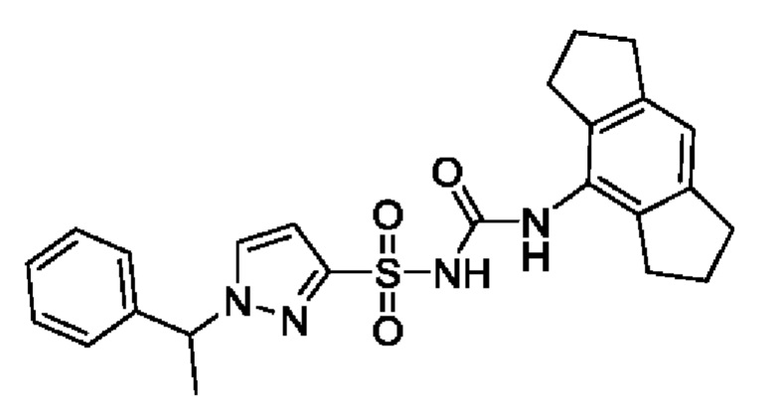
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-(1-фенилэтил)-1Н-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,13 г, 38%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ = 7,94 (д, J=2,4 Гц, 1Н), 7,80 (с, 1Н), 7,34-7,18 (м, 5Н), 6,85 (с, 1Н), 6,62 (д, J=2,3 Гц, 1Н), 5,68 (кв, J=7,0 Гц, 1Н), 2,76 (т, J=7,4 Гц, 4Н), 2,58 (т, J=7,4 Гц, 4Н), 1,90 (п, J=7,4 Гц, 4Н), 1,8 (д, J=7,1 Гц, 3Н).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-(2-(пиперидин-1-ил)этил)-1Н-пиразол-3-сульфонамид
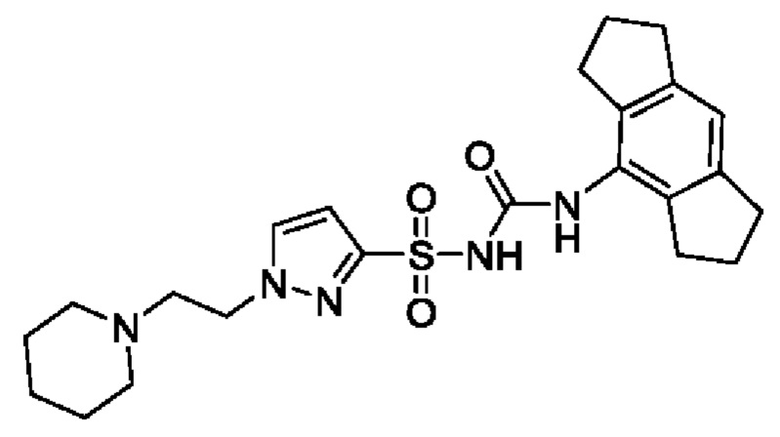
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-(2-(пиперидин-1-ил)этил)-1H-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (110 мг, 25%). 1H-ЯМР (400 МГц, CD3OD): δ = 7,76 (д, J=2,4 Гц, 1Н), 6,91 (с, 1Н), 6,73 (д, J=2,4 Гц 1Н), 4,55 (т, J=6,4 Гц, 2Н), 3,41 (т, J=6,0 Гц, 2Н), 3,02 (с, 4Н), 2,86 (т, J=7,2 Гц, 4Н), 2,78 (т, J=7,2 Гц, 4Н), 2,06-1,99 (м, 4Н), 1,74-1,70 (м, 4Н), 1,51 (д, J=5,2 Гц, 2Н). ЖХМС (m/z): 458,20 (М +1)+; 100% (210 нм), 100% (254 нм). ВЭЖХ: 98,70% (210 нм), 98,31% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1,5-диметил-1Н-пиразол-3-сульфонамид
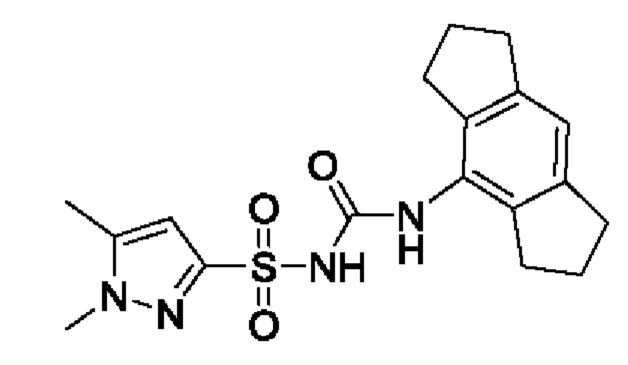
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1,5-диметил-1H-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (15 мг, 4%). 1H-ЯМР (400 МГц, ДМСО-d6): δ = 10,7 (ш.с., 1Н), 7,98 (с, 1Н), 6,93 (с, 1Н), 6,52 (с, 1Н), 3,79 (с, 3Н), 2,80 (т, J=7,2 Гц, 4Н), 2,62 (т, J=7,6 Гц, 4Н), 2,28 (с, 3Н), 1,98-1,93 (м, 4Н). ЖХМС (m/z): 397,10 (M+Na)+; 97,75% (210 нм), 88,23% (254 нм). ВЭЖХ: 94,42% (210 нм), 95,19% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-метил-5-(трифторметил)-1Н-пиразол-3-сульфонамид
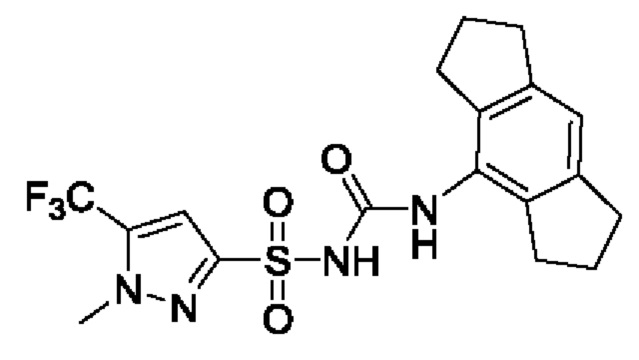
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-метил-5-(трифторметил)-1H-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (200 мг, 48%). 1H-ЯМР (400 МГц, CD3OD): δ = 7,10 (с, 1Н), 6,87 (с, 1Н), 4,03 (с, 3Н), 2,83 (т, J=7,2 Гц, 4Н), 2,74 (т, J=7,2 Гц, 4Н), 2,03-1,99 (м, 4Н). ЖХМС(m/z): 429,10 (М +1)+; 97,73% (210 нм), 95,71% (254 нм). ВЭЖХ: 94,95% (210 нм), 93,52% (254 нм).
N-((2,6-диизопропилфенил)карбамоил)-1-метил-5-(трифторметил)-1Н-пиразол-3-сульфонамид
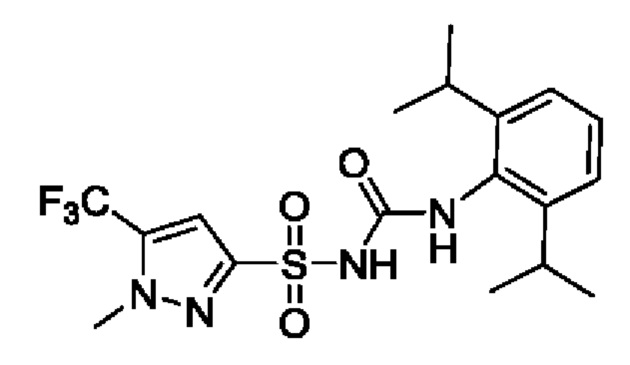
2-Изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А1) и 1-метил-5-(трифторметил)-1H-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (70 мг, 39%). 1H-ЯМР (400 МГц, CD3OD): δ = 7,18-7,16 (м, 1Н), 7,10-7,08 (м, 3Н), 4,03 (с, 3Н), 3,17-3,13 (м, 2Н), 1,03 (д, J=6,0 Гц, 12Н). ЖХМС (m/z): 433,15 (М +1)+; 99,73% (210 нм), 98,16% (254 нм). ВЭЖХ: 97,51% (210 нм), 95,47% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-1-изопропил-5-(трифторметил)-1Н-пиразол-3-сульфонамид
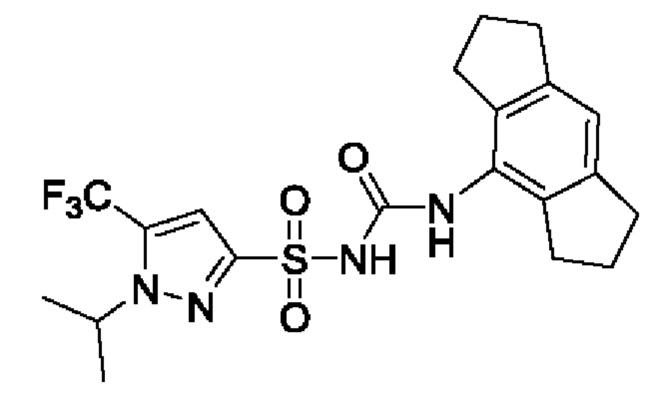
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 1-изопропил-5-(трифторметил)-1Н-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (15 мг, 12%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ = 8,54 (с, 1Н), 6,90 (с, 1Н), 6,77 (с, 1Н), 4,62-4,56 (м, 1Н), 2,76 (т, J=7,2 Гц, 4Н), 2,67 (т, J=7,6 Гц, 4Н), 1,92-1,84 (м, 4Н), 1,44 (д, J=6,4 Гц, 6Н). ЖХМС (m/z): 455,05 (М -1)-; 96,13% (210 нм), 95,41% (254 нм). ВЭЖХ: 95,71% (210 нм), 95,12% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-5-изопропил-1-метил-1H-пиразол-3-сульфонамид
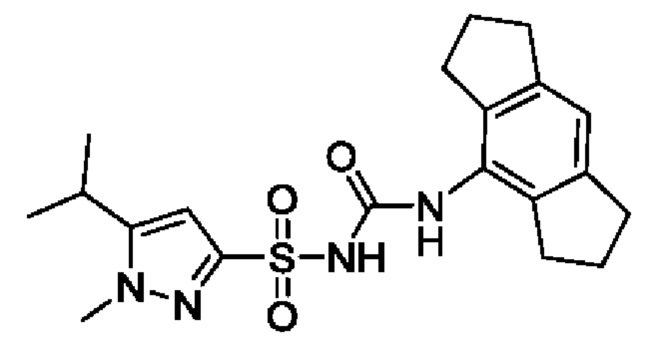
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и
5-изопропил-1-метил-1Н-пиразол-3-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (10 мг, 17%). 1H-ЯМР (400 МГц, CD3OD): δ = 6,88 (с, 1Н), 6,50 (с, 1Н), 3,82 (с, 3Н), 3,08-3,03 (м, 1Н), 2,83 (т, J=7,2 Гц, 4Н), 2,71 (т, J=7,2 Гц, 4Н), 2,04-1,96 (м, 4Н), 1,21 (д, J=6,8 Гц, 6Н). ЖХМС(m/z): 403,20 (М +1)+; 98,39% (210 нм), 94,19% (254 нм). ВЭЖХ: 95,62% (210 нм), 93,00% (254 нм).
N-((2,6-диизопропилфенил)карбамоил)-5-(2-гидроксипропан-2-ил)-1-метил-1H-пиразол-3-сульфонамид

2-Изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А1) и 5-(2-гидроксипропан-2-ил)-1-метил-1H-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (90 мг, 26%). 1Н-ЯМР (400 МГц, CD3OD): δ = 7,25-7,24 (м, 1Н), 7,16-7,14 (м, 2Н), 6,67 (с, 1Н), 4,13 (с, 3Н), 3,11-3,08 (м, 2Н), 1,61 (с, 6Н), 1,16 (д, J=6,8 Гц, 12Н). ЖХМС (m/z): 423,20 (М +1)+; 99,16% (210 нм), 97,19% (254 нм). ВЭЖХ: 98,16% (210 нм), 97,09% (254 нм).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-5-(2-гидроксипропан-2-ил)-1-метил-1Н-пиразол-3-сульфонамид.
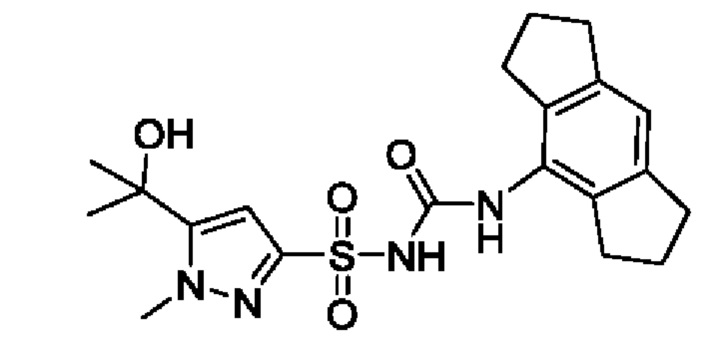
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и
5-(2-гидроксипропан-2-ил)-1-метил-1Н-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (70 мг, 15%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 8,01 (с, 1Н), 6,92 (с, 1Н), 6,53 (с, 1Н), 5,51 (с, 1Н), 4,02 (с, 3Н), 2,79 (т, J=7,4 Гц, 4Н), 2,62 (т, J=7,4 Гц, 4Н), 1,95 (п, J=7,4 Гц, 4Н), 1,50 (с, 6Н).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-5-(2-гидроксипропан-2-ил)-1-фенил-1Н-пиразол-3-сульфонамид

4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и
5-(2-гидроксипропан-2-ил)-1-фенил-1H-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (10 мг, 2%). 1Н-ЯМР (400 МГц, CD3OD): δ = 7,54 (с, 5Н), 6,59 (с, 1Н), 6,91 (с, 1Н), 2,86 (т, J=7,2 Гц, 4Н), 2,69 (т, J=7,2 Гц, 4Н), 2,05-1,96 (м, 4Н), 1,44 (с, 6Н). ЖХМС (m/z): 481,20 (М -1)-; 93,76% (210 нм), 93,24% (254 нм). ВЭЖХ: 95,86% (210 нм), 93,93% (254 нм).
1-бензил-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-5-(2-гидроксипропан-2-ил)-1Н-пиразол-3-сульфонамид
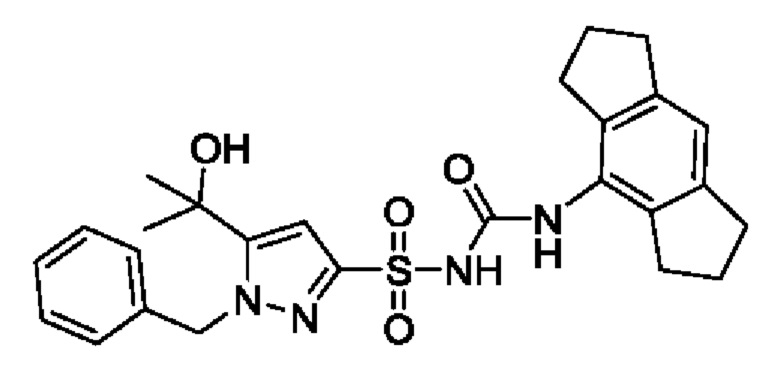
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-5-индацен (полученный с использованием общего способа А1) и 1-бензил-5-(2-гидроксипропан-2-ил)-1H-пиразол-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (40 мг, 7%). 1Н-ЯМР (400 МГц, CD3OD): δ = 7,20-7,14 (м, 5Н), 6,95 (с, 2Н), 6,73 (с, 1Н), 5,77 (с, 2Н), 2,86 (т, J=7,2 Гц, 4Н), 2,68 (т, J=7,6 Гц, 4Н), 2,01-1,94 (м, 4Н), 1,51 (с, 6Н). ЖХМС (m/z): 494,7 (М +1)+; 98,74% (210 нм), 96,05% (254 нм). ВЭЖХ: 95,11% (210 нм), 95,08% (254 нм).
N-((4-хлор-2,6-диизопропилфенил)карбамоил)-5-(2-гидроксипропан-2-ил)-1-метил-1H-пиразол-3-сульфонамид
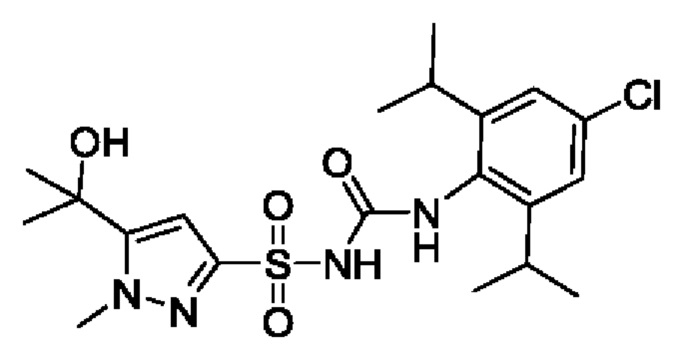
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и 5-(2-гидроксипропан-2-ил)-1-метил-1H-пиразол-3-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (83 мг, 17%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ = 7,81 (с, 1Н), 7,10 (с, 2Н), 6,42 (с, 1Н), 5,45 (с, 1Н), 3,99 (с, 3Н), 3,03 (гепт, J=7,0 Гц, 2Н), 1,47 (с, 6Н), 1,05 (д, J=1,8 Гц, 12Н). МСВР вычислено для C20H28Cl1N4O4S1 [M-H]- 455,1525, найдено 455,1515.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)-5-(2-гидроксипропан-2-ил)-1-фенил-1H-пиразол-3-сульфонамид
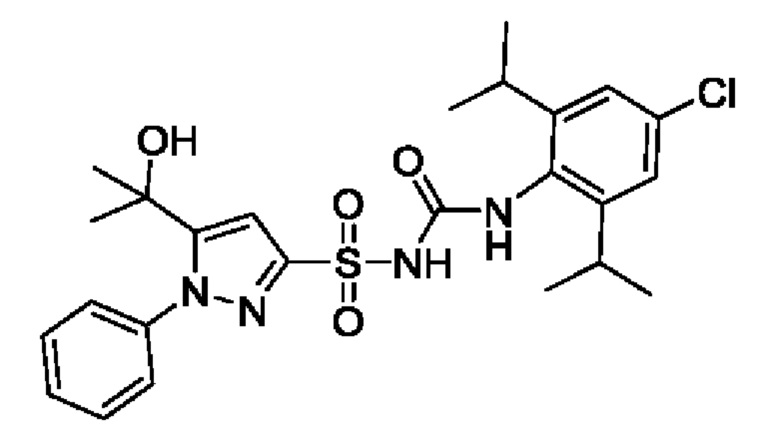
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и 5-(2-гидроксипропан-2-ил)-1-фенил-1H-пиразол-3-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (168 мг, 31%). 1H-ЯМР (600 МГц, ДМСО-d6) δ = 7,87 (с, 1Н), 7,52 (с, 5Н), 7,10 (с, 2Н), 6,71 (с, 1Н), 5,42 (с, 1Н), 3,10-2,92 (м, 2Н), 1,31 (с, 6Н), 1,02 (д, J=7,0 Гц, 12Н). МСВР вычислено для C25H30Cl1N4O4S1 [М-Н]- 517,1682, найдено 517,1671.
N-((4-хлор-2,6-диметилфенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
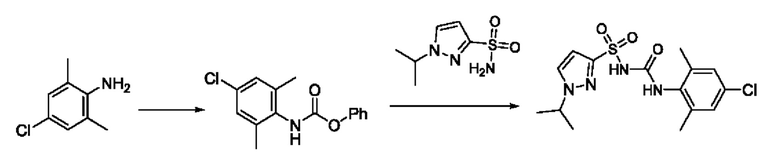
На раствор 4-хлор-2,6-диметиланилина (50 мг, 0,321 ммоль) в ДХМ (5 мл) подействовали Et3N (50 мг, 0,48 ммоль) и охладили до 0°С. Добавили фенилхлорформиат (60 мг, 0,39 ммоль) по каплям при 0°С.Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили насыщенным раствором NaHCO3, экстрагировали ДХМ (2 х 20 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток промыли н-пентаном и высушили в вакууме с получением фенил(4-хлор-2,6-диметилфенил)карбамата (75 мг, 85%) в виде коричневого твердого вещества. 1H-ЯМР (300 МГц, CDCl3): δ = 7,46-7,37 (м, 4Н), 7,26-7,20 (м, 2Н), 7,12-7,11 (м, 2Н), 2,33 (с, 6Н). ЖХМС (m/z): 275,9 [M+H]+.
1-изопропил-1Н-пиразол-3-сульфонамид (150 мг, 0,79 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (80 мг, 1,98 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем подействовали раствором фенил (4-хлор-2,6-диметилфенил)карбамата (240 мг, 0,87 ммоль) в ТГФ (3 мл) в атмосфере азота при 0°С.Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 3 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 30 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны с получением
N-((4-хлор-2,6-диметилфенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (90 мг, 31%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6): δ = 11,05 (с, 1Н), 7,99 (д, J=2,8 Гц, 1Н), 7,93 (с, 1Н), 7,13 (с, 2Н), 6,73 (д, J=2,4 Гц, 1Н), 4,64-4,57 (м, 1Н), 2,03 (с, 6Н), 1,43 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 370,95 [М+Н]+.; 97,62% (210 нм), 97,48% (254 нм). ВЭЖХ: 97,20% (210 нм). МСВР вычислено для C15H18Cl1N4O3S1 [М-Н]- 369,0794, найдено 369,0785.
N-((4-хлор-2,6-диметоксифенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
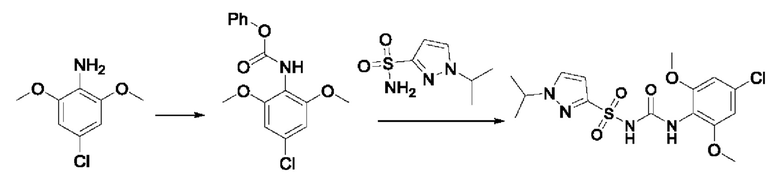
Раствор 4-хлор-2,6-диметоксианилина (200 мг, 1,06 ммоль) в ТГФ (8 мл) охладили до 0°С и подействовали NaH (62 мг, 1,59 ммоль) в атмосфере азота. Полученную смесь перемешивали в течение 15 мин. с последующим добавлением фенилхлорформиата (330 мг, 2,13 ммоль) по каплям при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc и профильтровали через слой целита и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 10% EtOAc-гексаны с получением фенил (4-хлор-2,6-диметоксифенил)карбамата (0,2 г, 61%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,35-7,33 (м, 2Н), 7,20-7,19 (м, 3Н), 6,61 (с, 2Н), 3,83 (с, 6Н).
1-изопропил-1Н-пиразол-3-сульфонамид (100 мг, 0,53 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (52 мг, 1,32 ммоль) при 0°С в атмосфере азота.). Полученную смесь перемешивали при комнатной температуре в течение 40 мин, затем подействовали раствором фенил (4-хлор-2,6-диметоксифенил)карбамата (180 мг, 0,58 ммоль) в ТГФ (3 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Объединенный органический экстракт промыли водой, солевым раствором, высушили над безводным Na2SO4 и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Gemini NX C18 (21,2 мм х 150 мм, размер частиц 5 мкм); расход: 20 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: Т/% В=0/20, 2/20, 8/70]. Фракции высушили сублимацией с получением
N-((4-хлор-2,6-диметоксифенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (20 мг, 9%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ = 7,90 (д, J=2,0 Гц, 1Н), 7,28 (с, 1Н), 6,72 (с, 2Н), 6,63 (д, J=2,0 Гц, 1Н), 4,60-4,54 (м, 1Н), 3,70 (с, 6Н), 1,43(д, J=6,8 Гц, 6Н). ЖХМС (m/z): 403,0 [M+H]+.; 90,61% (210 нм). ВЭЖХ: 91,63% (210 нм). МСВР вычислено для C15H18Cl1N4O5S1 [M-H]- 401,0692, найдено 401,0684.
N-((4-хлор-2-метил-6-(трифторметил)фенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
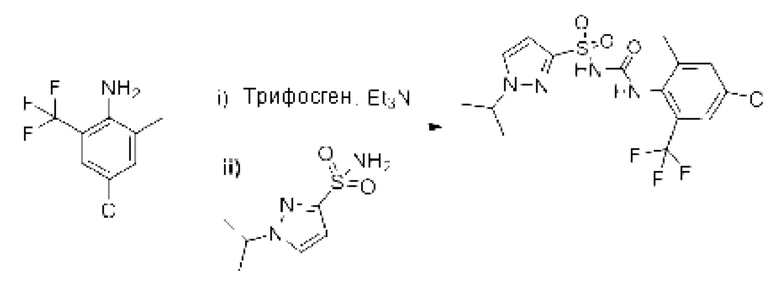
4-Хлор-2-метил-6-(трифторметил)анилин (50 мг, 0,24 ммоль) растворили в безводном ТГФ (5 мл) и подействовали Et3N (30 мг, 0,29 ммоль) при комнатной температуре. На раствор подействовали трифосгеном (30 мг, 0,095 ммоль) и полученную смесь перемешивали при 60°С в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток перемешал и с н-пентаном (20 мл) в течение 10 мин, профильтровали через слой целита и концентрировали при пониженном давлении с получением соответствующего изоцианата в виде белого твердого вещества. В другой кругодонной колбе емкостью 50 мл, 1-изопропил-1Н-пиразол-3-сульфонамид (40 мг, 0,212 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (22 мг, 0,529 ммоль) при 0°С в атмосфере азота. Перемешивали при комнатной температуре в течение 30 минут. Вышеупомянутый раствор изоцианата добавили к ТГФ в атмосфере азота. Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По завершении реакции реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 25 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны с получением N-((4-хлор-2-метил-6-(трифторметил)фенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида. Его растерли с диэтиловым эфиром и н-пентаном с получением
N-((4-хлор-2-метил-6-(трифторметил)фенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (35 мг, 35%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6): δ = 11,05 (с, 1Н), 8,09 (с, 1Н), 7,6 (с, 1Н), 7,70 (с, 1Н), 7,61 (с, 1Н), 6,67 (д, J=0,4 Гц, 1Н), 4,62 (м, 1Н), 2,05 (с, 3Н), 1,43 (д, J=6,8 Гц, 6Н). 19F-ЯМР (400 МГц, ДМСО-d6): δ = -60,82. ЖХМС (m/z): 425,00 [М+H]+; 94,05% (210 нм).
ВЭЖХ 98,03% (210 нм). МСВР вычислено для C1H15Cl1F3N4O3S1 [M-H]- 423,0511, найдено 423,0513.
N-((4-хлор-2-метокси-6-(трифторметил)фенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
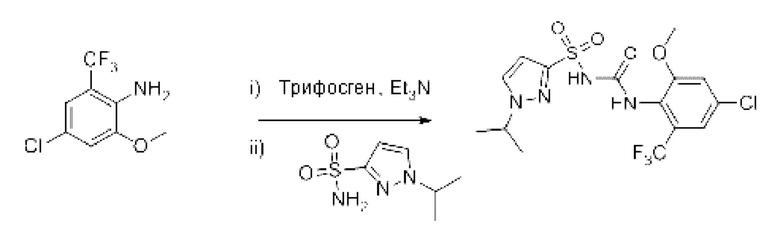
4-Хлор-2-метокси-6-(трифторметил) анилин (50 мг, 0,22 ммоль) растворили в безводном ТГФ (2 мл) и подействовали Et3N (27 мг, 0,27 ммоль) при комнатной температуре. На раствор подействовали трифосгеном (32 мг, 0,11 ммоль) и полученную смесь перемешивали при 70°С в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении и полученный остаток перемешивали с 5% EtOAc-гексаны (20 мл) в течение 10 мин, профильтровали через целит и концентрировали при пониженном давлении с получением целевого изоцианата в виде белого твердого вещества. В другой круглодонной колбе емкостью 50 мл 1-изопропил-1Н-пиразол-3-сульфонамид (42 мг, 0,22 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (18 мг, 0,44 ммоль) при 0°С в атмосфере азота. Смесь перемешивали при 0°С в течение 20 минут и подействовали вышеупомянутым раствором изоцианата в ТГФ в атмосфере азота. Полученную реакционную смесь перемешивали при 0-10°С в течение 2 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 50% EtOAc-гексаны с получением N-((4-хлор-2-метокси-6-(трифторметил)фенил)карбамоил)-1-изопропил-1Н-пира зол-3-сульфонамида (10 мг, 10%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, CDCl3): δ = 7,81 (с, 1Н), 7,53 (д, J=2,4 Гц, 1Н), 7,22 (д, J=1,6 Гц, 1Н), 7,10 (с, 1Н), 6,82 (д, J=2,0 Гц, 1Н), 4,64-4,57 (м, 1Н), 3,84 (с, 3Н), 1,54 (д, J=6,8 Гц, 6Н). 19F-ЯМР (400 МГц, CDCl3): δ = -61,55. ЖХМС (m/z): 441,05 [М+Н]+.; 94,58% (210 нм). ВЭЖХ: 92,16% (210 нм). МСВР вычислено для C15H15Cl1F3N4O4S1 [M-H]- 439,0460, найдено 439,0478.
N-((4-хлор-2,6-диэтилфенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
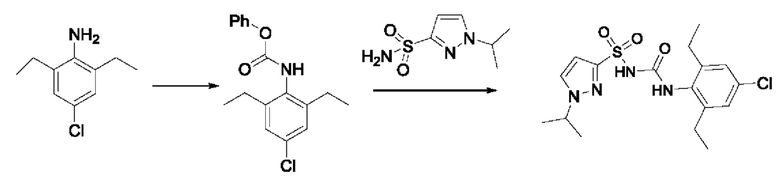
Раствор 4-хлор-2,6-дициклопропиланилина (100 мг, 0,546 ммоль) в ТГФ (5 мл) охладили до 0°С и подействовали NaH (30 мг, 0,66 ммоль) в атмосфере азота и перемешивали в течение 15 мин. К вышеупомянутому раствору добавили по каплям фенилхлорформиат (130 мг, 0,819 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении смесь разбавили EtOAc, профильтровали через слой целита и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием смеси 10% EtOAc-гексаны с получением фенил(4-хлор-2,6-диэтилфенил)карбамата (0,15 г, 91%) в виде белого твердого вещества. 1H-ЯМР (300 МГц, CDCl3): δ = 7,38-7,33 (м, 2Н), 7,23-7,18 (м, 3Н), 7,13 (м, 2Н), 6,27 (с, 1Н), 2,75-2,64 (м, 4Н), 1,28-1,22 (м, 6Н).
1-изопропил-1Н-пиразол-3-сульфонамид (75 мг, 0,40 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (40 мг, 0,99 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем подействовали раствором фенил(4-хлор-2,6-диэтилфенил)карбамата (130 мг, 0,44 ммоль) в ТГФ (3 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 4 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2 х 30 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 30-40% EtOAc-гексаны с последующим растиранием с диэтиловым эфиром и н-пентаном с получением N-((4-хлор-2,6-диэтилфенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (40 мг, 24%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ=11,05 (с, 1Н), 7,99 (д, J=2,4 Гц, 1Н), 7,89 (с, 1Н), 7,12 (с, 2Н), 6,72 (д, J=2,4 Гц, 1Н), 4,62-4,59 (м, 1Н), 2,42 (кв, J=7,6 Гц, 4Н), 1,43 (д, J=6,8 Гц, 6Н), 1,02 (т, J=7,6 Гц, 6Н). ЖХМС (m/z): 399,0 [М+Н]+, 96,72% (210 нм). ВЭЖХ: 97,13% (210 нм). МСВР вычислено для C17H22Cl1N4O3S1 [М-Н]- 397,1107, найдено 397,1090.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
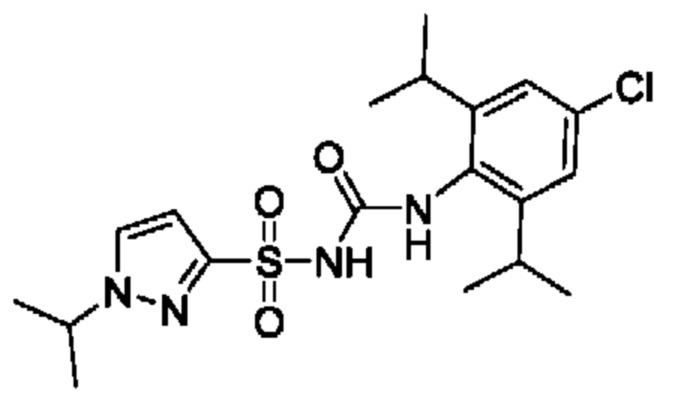
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и 1-изопропил-1Н-пиразол-3-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (221 мг, 49%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=7,70 (д, J=2,3 Гц, 1Н), 7,49 (с, 1Н), 7,00 (с, 2Н), 6,36 (с, 1Н), 4,62-4,29 (м, 1Н), 3,11 (д, J=6,4 Гц, 2Н), 1,38 (д, J=6,8 Гц, 6Н), 1,01 (д, J=6,8 Гц, 12Н). 13С-ЯМР (151 МГц, ДМСО) δ=160,96, 156,43, 150,19, 135,25, 131,49, 128,53, 123,27, 105,21, 54,23, 28,80, 24,15, 23,55. МСВР вычислено для C19H26Cl1N4O3S1 [М-Н]- 425,1420, найдено 425,1409.
N-((4-хлор-2,6-дициклопропилфенил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
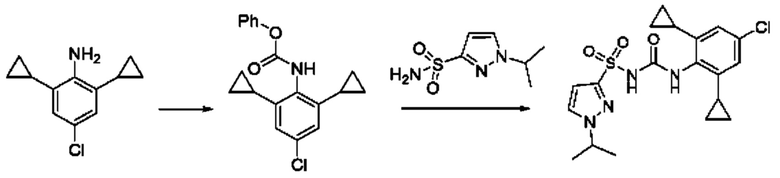
Раствор 4-хлор-2,6-дициклопропиланилина (150 мг, 0,724 ммоль) в ТГФ (5 мл) охладили до 0°С. К вышеупомянутому раствору добавили порциями NaH (35 мг, 0,87 ммоль) и перемешивали в течение 20 мин. К вышеупомянутому раствору добавили по каплям фенилхлорформиат (170 мг, 1,08 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении смесь разбавили EtOAc, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 15% EtOAc-гексаны с получением фенил(4-хлор-2,6-дициклопропилфенил)карбамата (195 мг, 83%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,37-7,35 (м, 2Н), 7,21-7,19 (м, 3Н), 6,84-6,83 (м, 2Н), 2,08-2,04 (м, 2Н), 1,04-1,02 (м, 4Н), 0,69-0,68 (м, 4Н). ЖХМС (m/z): 328,2 [М+Н]+.
1-изопропил-1H-пиразол-3-сульфонамид (100 мг, 0,53 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (53 мг, 1,32 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем подействовали раствором фенил(4-хлор-2,6-дициклопропилфенил)карбамата (190 мг, 0,582 ммоль) в ТГФ (3 мл) в атмосфере азота при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 4 ч. По завершении реакции реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2×30 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны с получением N-((4-хлор-2,6-дициклопропилфенил)карбамоил)-1-изопропил-1H-пиразол-3-сульфонамида. Его растерли с диэтиловым эфиром и н-пентаном с получением белого твердого вещества (25 мг, 11%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=11,05 (с, 1Н), 8,01-7,98 (м, 2Н), 6,74 (с, 3Н), 4,59-4,56 (м, 1Н), 1,77-1,76 (м, 2Н), 1,41 (д, J=6,8 Гц, 6Н), 0,77-0,75 (м, 4Н), 0,56-0,55 (м, 4Н). ЖХМС (m/z): 423,00 [М+Н]+.; 93,58% (210 нм). ВЭЖХ: 92,87% (210 нм). МСВР вычислено для C19H22Cl1N4O3S1 [М-Н]- 421,1107, найдено 421,1107.
N-((7-хлор-5-циклопропил-2,3-дигидро-1Н-инден-4-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
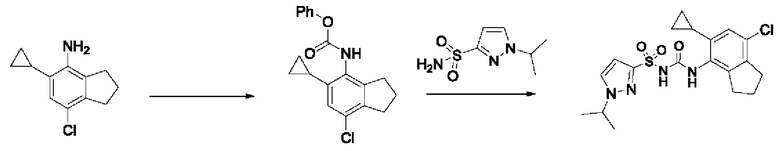
7-хлор-5-циклопропил-2,3-дигидро-1Н-инден-4-амин, 6 (70 мг, 0,33 ммоль) растворили в ТГФ (5 мл) и охладили до 0°С. К вышеупомянутому раствору добавили NaH (20 мг, 0,505 ммоль) в атмосфере азота и перемешивали в течение 15 мин с последующим добавлением фенилхлорформиата (100 мг, 0,674 ммоль) по каплям при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 10% EtOAc-гексаны с получением фенил(7-хлор-5-циклопропил-2,3-дигидро-1H-инден-4-ил)карбамата (80 мг, 73%) в виде коричневого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=7,39-7,37 (м, 3Н), 7,25-7,24 (м, 2Н), 6,85 (с, 1Н), 3,0-2,94 (м, 4Н), 2,12-2,10 (м, 2Н), 1,34 (м, 1Н), 0,96-0,95 (м, 2Н), 0,59-0,57 (м, 2Н). ЖХМС (m/z): 328,30 [М+Н]+.
1-изопропил-1H-пиразол-3-сульфонамид (41 мг, 0,219 ммоль) растворили в безводном ТГФ (3 мл) и осторожно подействовали NaH (21 мг, 0,549 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 30 мин и подействовали раствором фенил (7-хлор-5-циклопропил-2,3-дигидро-1H-инден-4-ил)карбамата (80 мг, 0,244 ммоль) в ТГФ (2 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 4 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2×30 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Gemini NX С18 (21,2 мм×150 мм, размер частиц 5 мкм); расход: 20 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: Т/% В=0/20, 2/30, 8/70]. Фракции высушили сублимацией с получением N-((7-хлор-5-циклопропил-2,3-дигидро-1Н-инден-4-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (20 мг, 20%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ=7,95 (д, J=2,4 Гц, 1Н), 6,74 (с, 1Н), 6,69 (д, J=2,4 Гц, 1Н), 4,60-4,55 (м, 1Н), 2,84 (т, J=7,6 Гц, 2Н), 2,66 (т, J=7,6 Гц, 2Н), 1,95-1,92 (м, 2Н), 1,79-1,76 (м, 1Н), 1,41 (д, J=6,4 Гц, 6Н), 0,82-0,78 (м, 2Н), 0,54-0,50 (м, 2Н). ЖХМС (m/z): 421,15 [М-Н]-; 94,19% (210 нм). ВЭЖХ: 95,46% (210 нм). МСВР вычислено для C19H22Cl1N4O3S1 [М-Н]- 421,1107, найдено 421,1110.
5-хлор-3-циклопропил-2-(3-((1-изопропил-1Н-пиразол-3-ил)сульфонил)уреидо)-N,N-диметилбензамид
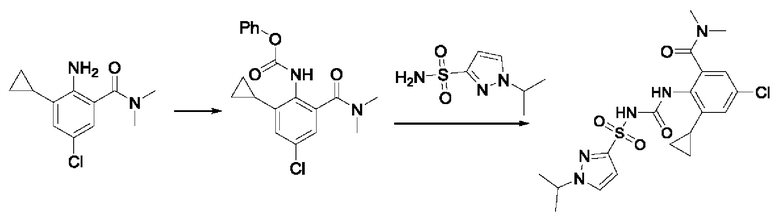
2-амино-5-хлор-3-циклопропил-N,N-диметилбензамид (200 мг, 0,84 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (50 мг, 1,26 ммоль) при 0°С в атмосфере азота. Полученную реакционную смесь перемешивали в течение 15 мин., затем добавили по каплям фенилхлорформиат (262 мг, 1,68 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 15% EtOAc-гексаны с получением фенил(4-хлор-2-циклопропил-6-(диметилкарбамоил)фенил)карбамата (0,14, 47%) в виде темно-коричневой жидкости. 1Н-ЯМР (300 МГц, CDCl3): δ=7,39-7,34 (м, 2Н), 7,23-7,15 (м, 3Н), 7,08-7,02 (м, 2Н), 3,09 (с, 3Н), 2,96 (с, 3Н), 2,05-2,0 (м, 1Н), 1,05-1,02 (м, 2Н), 0,71-0,69 (м, 2Н). ЖХМС (m/z): 358,60 [М+Н]+.
1-изопропил-1H-пиразол-3-сульфонамид (57 мг, 0,30 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (18 мг, 0,451 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 45 мин, затем подействовали раствором фенил (4-хлор-2-циклопропил-6-(диметилкарбамоил)фенил)карбамата (120 мг, 0,335 ммоль) в ТГФ (3 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 2 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2×30 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Gemini NX С18 110A AXIA (21,2 мм×150 мм, размер частиц 5 мкм); расход: 18 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: Т/% В=0/20, 2/20, 10/60]. Фракции высушили сублимацией с получением 5-хлор-3-циклопропил-2-(3-((1-изопропил-1H-пиразол-3-ил)сульфонил)уреидо)-N,N-диметилбензамида (10 мг, 7%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): δ=7,77 (с, 1Н), 7,11 (д, J=2,4 Гц, 1Н), 7,05 (д, J=2,0 Гц, 1Н), 6,76 (с, 1Н), 4,62-4,53 (м, 1Н), 2,96 (с, 3Н), 2,84 (с, 3Н), 1,88-1,87(м, 1Н), 1,50 (д, J=6,8 Гц, 6Н), 0,90-0,88 (м, 2Н), 0,63-0,61 (м, 2Н). ЖХМС (m/z): 454,0 [М+Н]+.; 97,52% (210 нм). ВЭЖХ: 92,05% (210 нм). МСВР вычислено для C19H23Cl1N5O4S1 [М-Н]- 452,1165, найдено 452,1180.
N-((4,6-диметилпиримидин-2-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
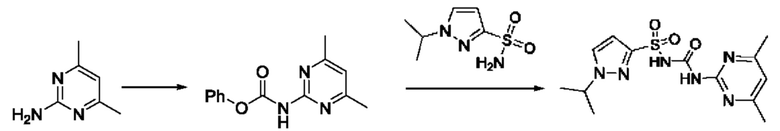
Раствор 4,6-диметилпиримидин-2-амина (300 мг, 2,43 ммоль) в ТГФ (10 мл) охладили до 0°С и подействовали NaH (140 мг, 3,64 ммоль) в атмосфере азота. Полученную смесь перемешивали в течение 15 мин, затем подействовали фенилхлорформиатом (0,6 мл, 4,87 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь разбавили EtOAc (30 мл), профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 30% EtOAc-гексаны с получением фенил(4,6-диметилпиримидин-2-ил)карбамата (250 мг, 42%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ=8,14 (с, 1Н), 7,45-7,34 (м, 2Н), 7,25-7,18 (м, 3Н), 6,78 (с, 1Н), 2,46 (с, 6Н). ЖХМС (m/z): 244,30 [М+Н]+.
1-изопропил-1H-пиразол-3-сульфонамид (75 мг, 0,396 ммоль) растворили в безводном ТГФ (50 мл) и осторожно подействовали NaH (40 мг, 0,99 ммоль) при 0°С в атмосфере азота. Полученную смесь нагрели до комнатной температуры и перемешивали в течение 30 мин. Реакционную смесь охладили до 0°С, затем подействовали раствором фенил(4,6-диметилпиримидин-2-ил)карбамата (100 мг, 0,436 ммоль) в ТГФ (5 мл) в атмосфере азота при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 3 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2×50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили препаративной ВЭЖХ на обращенной фазе [колонна: Gemini NX-bridge (150 мм×21,2 мм, размер частиц 5 мкм); расход: 15 мл/мин; элюент: 10 ммоль/л бикарбонат аммония в воде (А) & MeCN (В); градиент: Т/% В=0/10, 2/20, 10/60]. Фракции высушили сублимацией с получением N-((4,6-диметилпиримидин-2-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (10 мг, 13%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ=13,0 (с, 1Н), 7,49-7,46 (м, 2Н), 7,02 (д, J=2,4 Гц, 1Н), 6,74 (с, 1Н), 4,62-4,57 (м, 1Н), 2,45 (с, 6Н), 1,53 (д, J=6,8 Гц, 6Н). ЖХМС (m/z): 339,10 [М+Н]+. 99,70% (210 нм), 100% (254 нм). ВЭЖХ: 97,22% (210 нм). МСВР вычислено для C13H17N6O3S1 [М-Н]- 337,1088, найдено 337,1099.
N-((4,6-ди-трет-бутилпиримидин-2-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамид
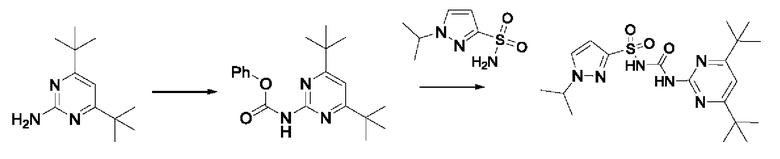
Раствор 4,6-ди-трет-бутилпиримидин-2-амина (0,15 г, 0,72 ммоль) в ТГФ (5 мл) охладили до 0°С и подействовали NaH (35 мг, 0,86 ммоль) в атмосфере азота. Полученную смесь перемешивали в течение 15 мин и к вышеупомянутому раствору добавили по каплям фенилхлорформиат (0,17 г, 1,08 ммоль) при 0°С. Реакционную смесь прогрели до комнатной температуры и перемешивали в течение 12 ч. По завершении реакционную смесь концентрировали при пониженном давлении и полученный остаток разбавили этилацетатом, профильтровали через целит и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 15% EtOAc-гексаны с получением фенил (4,6-ди-трет-бутилпиримидин-2-ил)карбамата (140 мг, 59%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ=7,95 (с, 1Н), 7,65-7,61 (м, 2Н), 7,50-7,45 (м, 4Н), 1,32 (с, 18Н). ЖХМС (m/z): 328,40 [М+Н]+;
1-Изопропил-1Н-пиразол-3-сульфонамид (50 мг, 0,264 ммоль) растворили в безводном ТГФ (40 мл) и осторожно подействовали NaH (27 мг, 0,661 ммоль) при 0°С в атмосфере азота. Полученную смесь перемешивали в течение 30 минут, затем подействовали раствором фенил(4,6-ди-трет-бутилпиримидин-2-ил)карбамата (95 мг, 0,29 ммоль) в ТГФ (5 мл) в атмосфере азота при 0°С. Полученную реакционную смесь прогрели до комнатной температуры и перемешивали в течение 4 ч. По завершении реакционную смесь разбавили насыщенным раствором NH4Cl, экстрагировали EtOAc (2×50 мл) и объединенный органический экстракт промыли водой, солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле (60-120 меш) с использованием элюента 40% EtOAc-гексаны с получением N-((4,6-ди-трет-бутилпиримидин-2-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (38 мг, 25%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6): δ=13,75 (с, 1Н), 10,71 (с, 1Н), 8,01 (д, J=2,4 Гц, 1Н), 7,18 (с, 1Н), 6,81 (д, J=2,4 Гц, 1Н), 4,61-4,54 (м, 1Н), 1,38 (д, J=6,8 Гц, 6Н), 1,31 (с, 18Н). ЖХМС (m/z): 423,50 [М+Н]+; 99,88% (210 нм). ВЭЖХ: 98,49% (210 нм). МСВР вычислено для C19H29N6O3S1 [М-Н]- 421,2027, найдено 421,2008.
ФЕНИЛ/БИЦИКЛЫ
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)бензолсульфонамид
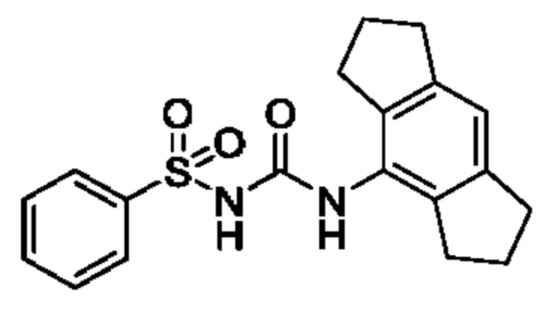
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и фенилсульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (50 мг, 13%). 1Н-ЯМР (400 МГц, CD3OD): δ=8,05 (д, J=7,6 Гц, 2Н), 7,72 (т, J=7,6 Гц, 1Н), 7,63 (т, J=7,6 Гц, 2Н), 6,96 (с, 1Н), 2,84 (т, J=7,2 Гц, 4Н), 2,59 (т, J=7,2 Гц, 4Н), 2,00 (квинт, J=7,2 Гц, 4Н).
5-(Диметиламино)-N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)нафталин-1-сульфонамид

На раствор 5-(диметиламино)нафталин-1-сульфонамида (20 мг, 0,08 ммоль) в ТГФ (5,0 мл) подействовали DIPEA (17 мкл, 0,09 ммоль), перемешивали при температуре окружающей среды в течение 15 мин, затем добавили по каплям раствор 4-изоцианато-1,2,3,5,6,7-гексагидро-s-индацена (полученного с использованием общего способа А2) (19 мг, 0,09 ммоль) в ТГФ (1,0 мл). Реакционную смесь перемешивали при температуре окружающей среды на протяжении ночи, затем концентрировали при пониженном давлении. Технический продукт очистили посредством ВЭЖХ с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (24 мг, 66%). 1Н-ЯМР (400 МГц, CD3OD) δ=8,62 (д, J=8,4 Гц, 1Н), 8,36 (дд, J=9,5, 8,1 Гц, 2Н), 7,67-7,56 (м, 2Н), 7,26 (д, J=7,8 Гц, 1Н), 6,91 (с, 1Н), 2,91 (с, 6Н), 2,79 (т, J=7,4 Гц, 4Н), 2,40 (т, J=7,4 Гц, 4Н), 1,92 (квинт, J=7,4 Гц, 4Н). 13С-ЯМР (101 МГц, CD3OD) δ=152,5, 150,3, 144,4, 138,3, 131,9, 131,7, 131,1, 130,3, 130,1, 129,9, 128,9, 127,2, 123,6, 119,4, 119,1, 118,9, 115,7, 78,4, 78,0, 77,7, 49,6, 48,3, 45,6, 33,4, 33,2, 30,5, 29,3, 25,9, 25,8; МСВР (ESI) вычислено для C25H27N3O3S [М+Н] 450,1846, найдено 450,1859.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-2,3-дигидро бензо[b]тиофен-6-сульфонамид-1,1-диоксид
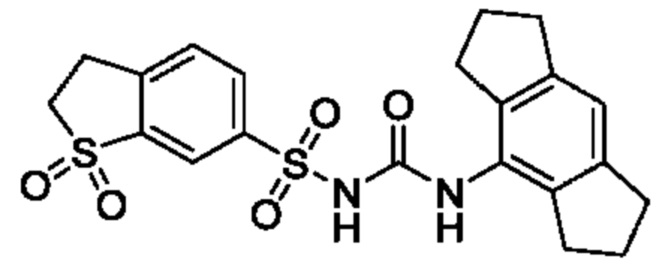
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 2,3-дигидробензо[b]тиофен-6-сульфонамид-1,1-диоксид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (33 мг, 28%). 1Н-ЯМР (600 МГц, ДМСО-d6): δ=8,17 (шс, 1Н), 8,15 (с, 1Н), 8,13 (д, J=9 Гц, 1Н), 7,73 (д, J=12 Гц, 1Н), 6,89 (с, 1Н), 3,68 (т, J=9 Гц, 2Н), 3,43 (т, J=6 Гц, 2Н), 2,75 (т, J=6 Гц, 4Н), 2,55 (т, J=6 Гц, 4Н), 1,93-1,88 (м, 4Н). 13С-ЯМР (150 МГц, ДМСО-d6): δ=151,6, 143,3, 143,0, 142,7, 139,6, 137,6, 137,5, 132,2, 128,9, 120,1, 117,9, 50,9, 32,9, 30,6, 25,6, 25,4 ЖХМС (m/z): 447 [М+Н]+
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-2-метоксибензолсульфонамид
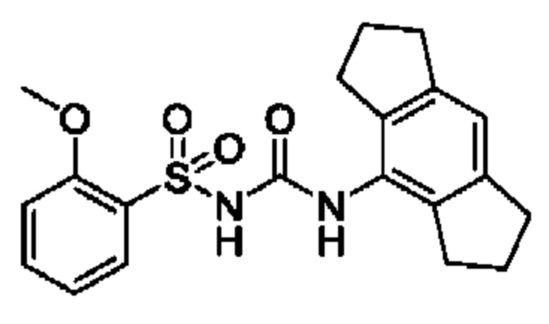
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 2-метоксибензолсульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (30 мг, 48%). 1Н-ЯМР (400 МГц, CD3OD) δ 7,96 (д, J=7,9 Гц, 1Н), 7,62 (т, J=8,3 Гц, 1Н), 7,14-7,05 (м, 2Н), 6,97 (с, 1Н), 4,00 (с, 3Н), 2,83 (т, J=7,3 Гц, 4Н), 2,56 (т, J=7,3 Гц, 4Н), 2,09-1,90 (м, 4Н). 13С-ЯМР (101 МГц, CD3OD) δ 155,9, 143,2, 136,6, 134,7, 129,5, 127,2, 126,6, 125,9, 119,5, 118,0, 111,6, 111,2, 55,3, 31,9, 29,3, 24,5. МСВР (ESI) вычислено для C20H23N2O4S [М+Н] 387,1373, найдено 387,1378.
N-(1,2,3,5,6,7-Гексагидро-s-индацен-4-илкарбамоил)-3-(трифторметил)бензолсульфонамид
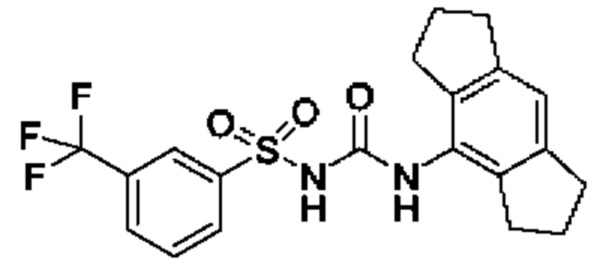
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 3-(трифторметил)бензолсульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,015 г, 12%); белого цвета с желтоватым оттенком, вязкого. 1Н-ЯМР (400 МГц, ДМСО-d6): δ=11,01 (ш.с, 1Н), 8,34 (с, 1Н), 8,25-8,23 (м, 2Н), 8,11 (д, J=7,6 Гц, 1Н), 7,90 (т, J=8,0 Гц, 1Н), 6,93 (с, 1Н), 2,77 (т, J=7,2 Гц, 4Н), 2,50 (м, 4Н), 1,90 (квинт, J=7,6 Гц, 4Н). ЖХМС, Чистота: 96,69%, m/z 425,1 (М+Н+). МСВР (FAB+) вычислено для C20H19F3N2O3S [М+Н]+: 425,1068, найдено: 425,1009.
N-(1,2,3,5,6,7-гексагидро-5-индацен-4-илкарбамоил)-3-метоксибензолсульфонамид
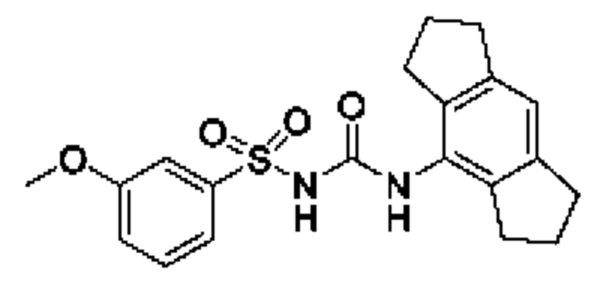
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 3-метоксибензолсульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,025 г, 23%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=10,77 (ш.с, 1Н), 8,15 (с, 1Н), 7,56-7,45 (м, 3Н), 7,27 (д, J=8,0 Гц, 1Н), 6,93 (с, 1Н), 3,82 (с, 3Н), 2,77 (т, J=7,2 Гц, 4Н), 2,53 (т, J=7,6 Гц, 4Н), 1,92 (квинт, J=7,2 Гц, 4Н). ЖХМС, Чистота: 95,02%, tr=3,77 мин, m/z 387,28 (М+Н+). МСВР (FAB+) вычислено для C20H22N2O4S [М+Н]+: 387,1300, найдено: 387,1301.
N-(1,2,3,5,6,7-Гексагидро-s-индацен-4-илкарбамоил)-3-(трифторметокси)бензолсульфонамид
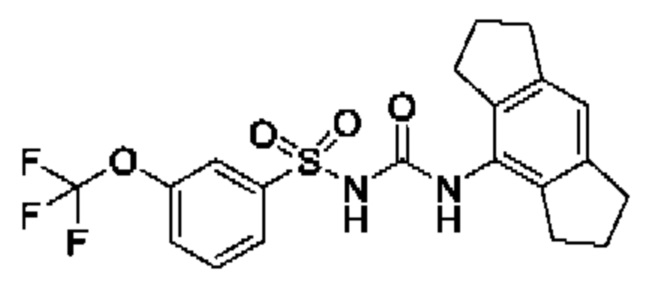
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 3-(трифторметокси)бензолсульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,045 г, 43%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,04 (д, J=8,0 Гц, 1Н), 7,95 (с, 1Н), 7,72 (т, J=8,4 HZ, 1Н), 7,62 (д, J=8,4 Гц, 1Н), 6,95 (с, 1Н), 2,82 (т, J=7,6 Гц, 4Н), 2,59 (т, J=7,6 Гц, 4Н), 1,99 (квинт, J=7,6 Гц, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=149,2, 147,9, 143,1, 142,2, 137,3, 131,5, 128,5, 126,4, 125,9, 121,2, 119,7, 118,6, 118,1, 32,5, 29,4, 25,0. 19F-ЯМР (233,33 МГц, ДМСО-d6): -57,10 (с, 3F). ЖХМС, Чистота: 95,56%, m/z 441,20 (М+Н+). МСВР (FAB+) вычислено для C20H19F3N2O4S [М+Н]+: 441,1018, найдено: 441,1015.
3-(дифторметокси)-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)бензолсульфонамид
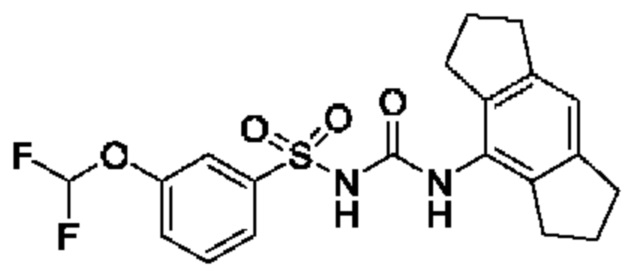
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 3-(дифторметокси)бензолсульфонамид использовали в общем способе С5 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,056 г, 50%). 1Н-ЯМР (600 МГц, Ацетонитрил-d3) δ=7,85 (д, J=8,0 Гц, 1Н), 7,75 (т, J=2,1 Гц, 1Н), 7,60 (т, J=8,0 Гц, 1Н), 7,53 (с, 1Н), 7,43 (дд, J=8,0, 2,1 Гц, 1Н), 6,95 (с, 1Н), 2,81 (т, J=7,5 Гц, 4Н), 2,55 (т, J=7,5 Гц, 4Н), 1,95 (квинт, J=7,5 Гц, 4Н).
N1-(1,2,3,5,6,7-Гексагидро-s-индацен-4-илкарбамоил)бензол-1,3-дисульфонамид
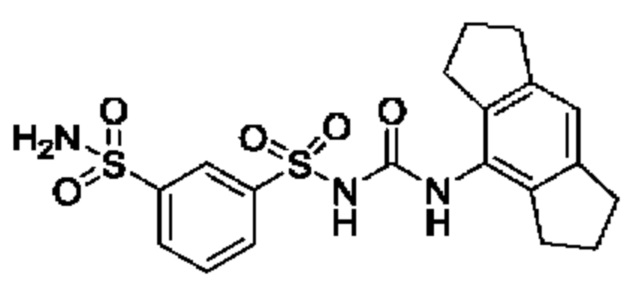
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и бензол-1,3-дисульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,080 г, 12%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=11,02 (ш.с, 1Н), 8,36 (с, 1Н), 8,24 (с, 1Н), 8,15 (д, J=7,8 Гц, 1Н), 8,11 (д, J=8,0 Гц, 1Н), 7,84 (т, J=8,0 Гц, 1Н), 7,63 (с, 2Н), 6,93 (с, 1Н), 2,77 (т, J=7,2 Гц, 4Н), 2,54 (т, J=7,6 Гц, 4Н), 1,92 (квинт, J=7,2 Гц, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=149,0, 144,9, 143,1, 140,9, 137,3, 130,4, 130,2, 128,5, 124,4, 118,1, 32,4, 30,0, 25,0. ЖХМС, Чистота: 98,63%, m/z 436,03 (М+Н+). МСВР (FAB+) вычислено для C19H21N3O5S2 [М+Н]+: 436,0923, найдено: 436,0919.
N1-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)-N3,N3-диметилбензол-1,3-дисульфонамид
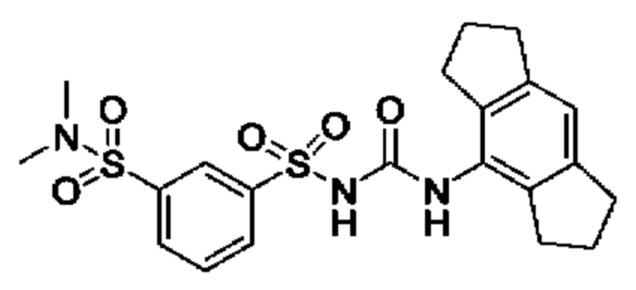
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и N1,N1-диметилбензол-1,3-дисульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,019 г, 5%). 1Н-ЯМР (400 МГц, CD3OD) δ 8,41 (т, J=1,4 Гц, 1Н), 8,32 (дт, J=7,9, 1,4 Гц, 1Н), 8,08 (дт, J=7,9, 1,4 Гц, 1Н), 7,87 (т, J=7,9 Гц, 1Н), 6,95 (с, 1Н), 2,84 (т, J=7,4 Гц, 4Н), 2,73 (с, 6Н), 2,61 (т, J=7,4 Гц, 4Н), 2,00 (п, J=7,4 Гц, 4Н).
3-(N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)сульфамоил)бензойная кислота
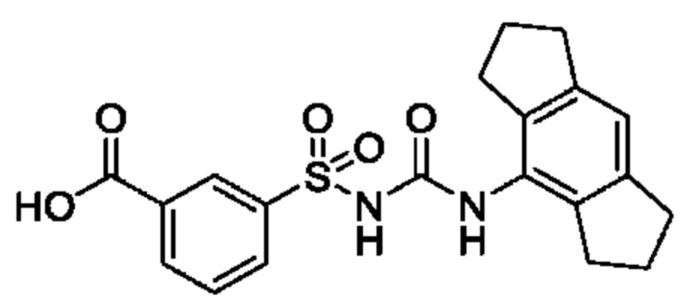
Метил-3-(N-(1,2,3,5,6,7-гексагидро-s-индацен-4-илкарбамоил)сульфамоил)бензоат (0,25 г, 0,603 ммоль) растворили в смеси тетрагидрофуран : метанол : вода (9 мл, 1:1:1) и смесь охладили до 0°С. Добавили моногидрат гидроксида лития (0,75 г, 1,81 ммоль, 3 экв.) и смесь перемешивали при температуре окружающей среды в течение 3 ч. По завершении реакционную смесь вылили в охлажденную воду и экстрагировали этилацетатом. Объединенные органические экстракты промыли солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Продукт очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,017 г, 3%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=13,26 (ш.с, 1Н), 8,43 (с, 1Н), 8,13-8,08 (м, 2Н), 7,99 (ш.с, 1Н), 7,67 (т, J=8,0 Гц, 1Н), 6,87 (с, 1Н), 6,52 (с, 1Н), 2,75 (т, J=7,2 Гц, 4Н), 2,55 (т, J=7,6 Гц, 4Н), 1,89 (квинт, J=7,6 Гц, 4Н). ЖХМС, Чистота: 96%, m/z 400,98 (М+Н+). МСВР (FAB+) вычислено для C20H20N2O5S [М+Н]+: 401,1093, найдено: 401,4514.
3-(N-(1,2,3,5,6,7-Гексагидро-s-индацен-4-илкарбамоил)сульфамоил)бензамид
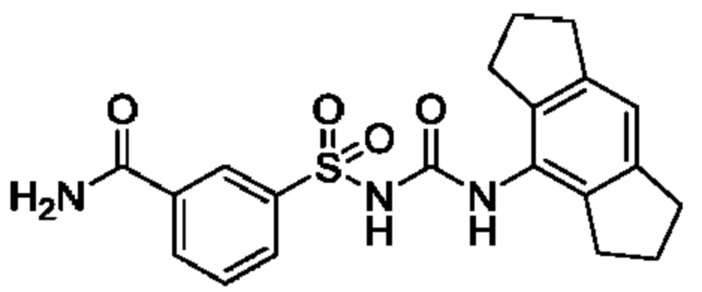
3-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)бензойную кислоту (0,06 г, 0,074 ммоль) растворили в безводном N,N-диметилформамиде (4 мл) и раствор охладили до 0°С. Добавили диизопропилэтиламин (3,0 экв.) и HATU (2,0 экв.) и смесь перемешивали при 0°С в течение 15 мин. Добавили хлорид аммония (3,0 экв.) и смесь перемешивали при температуре окружающей среды в течение 5 ч. По завершении реакционную смесь вылили в солевой раствор (20 мл) и экстрагировали этилацетатом (2×10 мл). Объединенные органические экстракты промыли солевым раствором (10 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический остаток очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,011 г, 37%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,23 (д, J=9,2 Гц, 2Н), 8,02 (с, 1Н), 7,89 (д, J=7,6 Гц, 1Н), 7,84 (д, J=7,6 Гц, 1Н), 7,42 (т, J=8,0 Гц, 1Н), 7,38 (с, 1Н), 7,33 (с, 1Н). 6,74 (с, 1Н), 2,73 (т, J=6,8 Гц, 4Н), 2,62 (т, J=6,8 Гц, 4Н), 1,87 (квинт, J=7,6 Гц, 4Н). ЖХМС, Чистота: 93%, m/z 400,05 (М+Н+). МСВР (FAB+) вычислено для C20H21N3O4S [М+Н]+: 400,1253, найдено: 400,1378.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-3-(2-гидроксипропан-2-ил)бензолсульфонамид
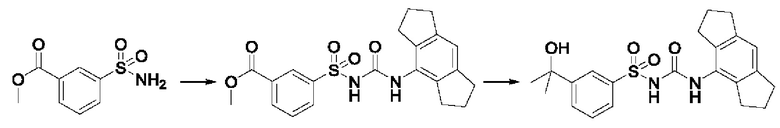
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) добавили непосредственно к метил-3-сульфамоилбензоату (0,447 г, 2,07 ммоль, 1,20 экв) при температуре окружающей среды и смесь перемешивали на протяжении ночи. Реакционную смесь вылили в охлажденную воду и экстрагировали этилацетатом. Объединенные органические экстракты промыли солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Полученный остаток очистили колоночной хроматографией на силикагеле с использованием градиента метанола в дихлорметане 0-10% с получением метил-3-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)бензоата в виде светло-коричневого твердого вещества (0,36 г, 50%).
Метил-3-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)бензоат (0,06 г, 0,144 ммоль) растворили в безводном ТГФ и раствор охладили до 0°С. Добавили метилмагнийбромид (раствор в диэтиловом эфире 3 моль/л, 0,14 мл, 0,42 ммоль, 3,0 экв.) и смесь перемешивали при температуре окружающей среды в течение 4 ч. По завершении к реакционной смеси добавили насыщенный водный хлорид аммония и экстрагировали этилацетатом. Объединенные органические экстракты промыли солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Очисткой технического остатка препаративной ВЭЖХ на обращенной фазе получили указанное в подзаголовке соединение в виде белого с желтоватым оттенком твердого вещества (0,015 г, 25%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,16 (с, 1Н), 7,91 (с, 1Н), 7,62 (д, J=7,2 Гц, 1Н), 7,59-7,48 (м, 2Н), 7,32 (т, J=7,6 Гц, 1Н), 6,78 (с, 1Н), 5,10 (с, 1Н), 2,74 (т, J=7,2 Гц, 4Н), 2,60 (т, J=6,8 Гц, 4Н), 1,88 (квинт, J=7,6 Гц, 4Н), 1,42 (с, 6Н). ЖХМС, Чистота: 91%, m/z 415,05 (М+Н+).
3-Азидо-N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)бензолсульфонамид
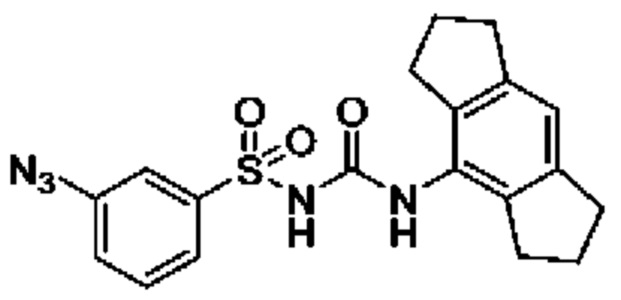
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 3-азидобензолсульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (70 мг, 50%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=8,22 (с, 1Н), 7,72 (м, J=5,2 Гц, Н), 7,65 (т, J=8,0 Гц, 1Н), 7,59 (с, 1Н), 7,46-7,42 (м, 1Н), 6,93 (с, 1Н), 2,77 (т, J=7,4 Гц, 4Н), 2,53 (т, J=7,4 Гц, 4Н), 1,92 (м, 4Н). 13С-ЯМР (101 МГц, CD3OD) δ 151,2, 144,9, 144,6, 142,9, 142,5, 138,9, 131,5, 131,4, 128,6, 124,9, 124,6, 124,6, 119,8, 119,2, 118,9, 111,9, 33,7, 33,6, 31,1, 29,7, 26,3. МСВР (ESI) вычислено для C19H20N5O3S [М+Н] 398,1281, найдено 398,1272.
N-((1,2,3,5,6,7-Гексагидро-5-индацен-4-ил)карбамоил)-3-(4-фенил-1Н-1,2,3-тр иазол-1-ил)бензолсульфонамид

4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 3-(4-фенил-1Н-1,2,3-триазол-1-ил)бензолсульфонамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (10 мг, 49%). 1Н-ЯМР (400 МГц, CD3OD) δ=8,89 (с, 1Н), 8,55 (с, 1Н), 8,21 (д, J=8,2 Гц, 1Н), 8,13 (д, J=7,8 Гц, 1Н), 7,92 (д, J=7,6 Гц, 2Н), 7,79 (т, J=8,0 Гц, 1Н), 7,68 (с, 1Н), 7,48 (т, J=7,6 Гц, 2Н), 7,39 (т, J=7,4 Гц, 1Н), 6,92 (с, 1Н), 2,82 (т, J=7,4 Гц, 4Н), 2,70-2,63 (м, 4Н), 1,98 (м, 4Н). 13С-ЯМР (151 МГц, CD3OD) δ=148,8, 143,9, 143,6, 137,7, 137,2, 137,0, 130,6, 130,3, 129,7, 129,6, 128,8, 128,5, 127,6, 127,4, 126,7, 125,6, 124,5, 124,0, 119,3, 118,7, 110,8, 32,7, 32,6, 30,2, 28,7, 25,3. МСВР (ESI) вычислено для C27H26N5O3S [М+Н] 500,1751, найдено 500,1735.
N-(3-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенил)пент-4-инамид
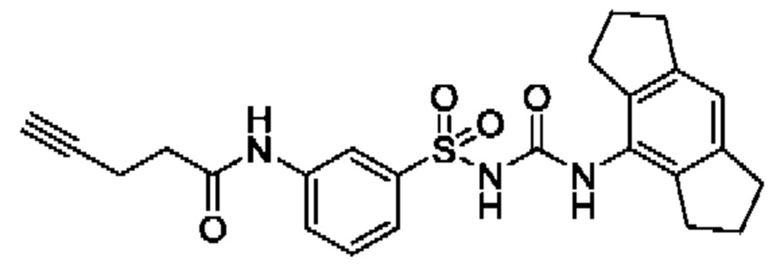
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и N-(3-сульфамоилфенил)пент-4-инамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (116 мг, 61%). 1Н-ЯМР (400 МГц, CD3OD) δ=8,18 (с, 1Н), 7,81 (д, J=8,3 Гц, 1Н), 7,68 (д, J=8,3 Гц, 1Н), 7,43 (дд, J=8,3, 7,8 Гц, 1Н), 6,87 (с, 1Н), 2,79 (т, J=7,2 Гц, 4Н), 2,67-2,60 (м, 4Н), 2,60-2,48 (м, 4Н), 2,28-2,22 (м, 1Н), 2,04-1,89 (м, 4Н). 13С-ЯМР (101 МГц, CD3OD) δ=170,9, 143,3, 143,0, 138,8, 137,7, 128,7, 128,3, 126,4, 122,8, 122,0, 117,9, 117,7, 82,0, 68,9, 35,4, 32,4, 29,9, 25,1, 13,9. МСВР (ESI) вычислено для C24H26N3O4S [М+Н] 452,1639, найдено 452,1658.
3-(1-(3-аминопропил)-1Н-1,2,3-триазол-4-ил)-N-(3-(N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)сульфамоил)фенил)пропанамид
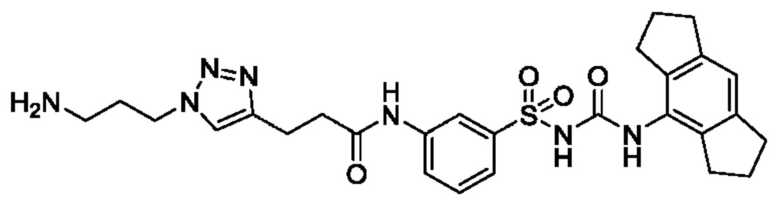
N-(3-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенил)пент-4-инамид и 3-азидопропан-1-амин использовали в общем способе F с получением указанного в подзаголовке соединения в виде белого твердого вещества (6 мг, 43%). 1Н-ЯМР (600 МГц, CD3OD) δ=7,85 (с, 1Н), 7,55 (т, J=3,8 Гц, 2Н), 7,50 (д, J=8,0 Гц, 2Н), 7,29 (т, J=7,9 Гц, 1Н), 6,78 (с, 1Н), 4,26 (т, J=6,4 Гц, 2Н), 3,00 (т, J=6,6 Гц, 2Н), 2,71 (т, J=7,3 Гц, 4Н), 2,64-2,50 (м, 8Н), 1,94-2,02 (м, 2Н), 1,92-1,83 (м, 4Н). 13С-ЯМР (151 МГц, CD3OD) δ=173,0, 147,4, 146,8, 144,7, 144,6, 139,5, 139,2, 131,6, 130,0, 129,8, 124,2, 123,9, 123,2, 119,5, 118,6, 48,3, 37,7, 34,0, 31,6, 26,7, 26,6, 22,9. МСВР (ESI) вычислено для C27H34N7O4S [М+Н] 552,2387, найдено 552,2368.
N-(3-(N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)сульфамоил)фенил)-3-(1-(3-((7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)пропил)-1Н-1,2,3-триазол-4-ил)пропанамид
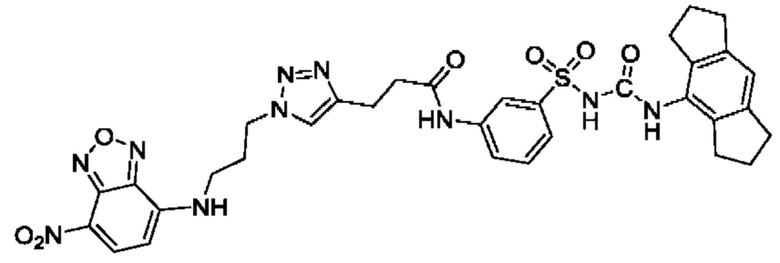
N-(2-Азидопропил)-7-нитробензо[с][1,2,5]оксадиазол-4-амин синтезировали способами, приведенными в Chun Li, Etienne Henry, Naresh Kumar Mani, Jie Tang, Jean-Claude Brochon, Eric Deprez, и Juan Xie Eur. J. Org. Chem. 2010, 2395-2405. К раствору 4-хлор-7-нитробензо[с][1,2,5]оксадиазола (300 мг, 1,5 ммоль) в ТГФ (10 мл) добавили 3-азидопропиламин (160 мг, 1,65 ммоль) и Cs2CO3 (480 мг, 1,5 ммоль). Реакционную смесь перемешивали при 50°С в течение 4 ч. Реакционную смесь распределили между EtOAc (50 мл) концентрировали при пониженном давлении. Остаток очистили колоночной хроматографией на силикагеле с использованием элюента 30% EtOAc-петролейный эфир с получением N-(2-азидопропил)-7-нитробензо[с][1,2,5]оксадиазол-4-амина (240 мг, 76%). 1Н-ЯМР (400 МГц, CDCl3): δ=8,50 (д, J=8,8 Гц, 1Н), 6,57 (с, 1Н, NH), 6,23 (д, J=8,8 Гц, 1Н), 3,66 (кв, J=6,8 Гц, 2Н), 3,59 (J=6,0 Гц, 2Н), 2,00-2,16 (м, 2Н). 13С-ЯМР (101 МГц, CDCl3) δ 144,2, 144,0, 143,8, 136,7, 123,7, 98,8, 49,1, 41,6, 27,6. МСВР (ESI): вычислено для C9H10N7O3 264,0840; найдено 264,0711.
N-(3-(N-((1,2,3,5,6,7-Гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенил)пент-4-инамид (10 мг, 0,022 ммоль) и N-(2-азидопропил)-7-нитробензо[с][1,2,5]оксадиазол-4-амин (7,0 мг, 0,026 ммоль), 10 мол. % ТНРТА, 5 мол. % CuSO4, 10 мол. % аскорбата натрия в ДМСО (500 мкл) перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь подвергли очистке с помощью обращенной фазы (система для колоночной флэш-хроматографии Reveleris, 4 г, 18 мл/мин., подвижная фаза; 10 ммоль водн. NH4CO3, MeCN) и высушили сублимацией с получением продукта в виде белого твердого вещества (7,0 мг, 44%). 1Н-ЯМР (600 МГц, CD3OD) δ=8,46 (д, J=8,7 Гц, 1Н), 8,18 (с, 1Н), 7,79 (д, J=8,4 Гц, 1Н), 7,67 (д, J=7,9 Гц, 1Н), 7,61 (с, 1Н), 6,94 (с, 1Н), 6,15 (д, J=9,0 Гц, 1Н), 4,46 (т, J=6,7 Гц, 2Н), 3,09 (т, J=7,0 Гц, 2Н), 2,82 (т, J=7,4 Гц, 4Н), 2,77 (т, J=7,0 Гц, 2Н), 2,70-2,56 (м, 6Н), 2,37-2,26 (м, 2Н), 1,99 (кв, J=7,3 Гц, 4Н). 13С-ЯМР (151 МГц, CD3OD) δ=172,9, 147,9, 145,4, 140,5, 139,0, 138,4, 130,6, 129,1, 128,0, 125,6, 124,2, 123,6, 120,3, 119,6, 112,4, 70,6, 48,9, 37,2, 34,3, 34,2, 31,7, 30,2, 26,8, 22,3; МСВР (ESI) вычислено для C33H34N10O7S [М-Н] 713,2260, найдено 713,2290.
N-(3-(4-(3-((3-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенил)амино)-3-оксопропил)-1Н-1,2,3-триазол-1-ил)пропил)-5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)пентанамид
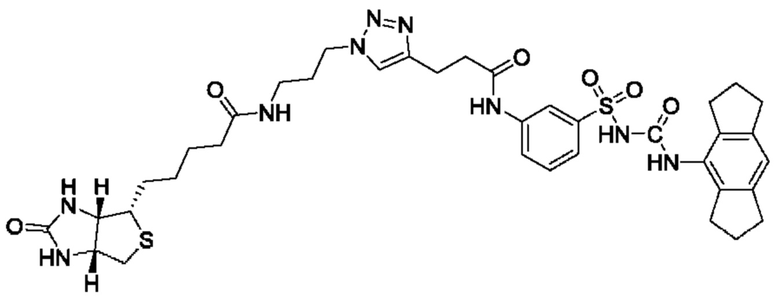
К раствору биотина (0,4 г, 1,63 ммоль) и 3-азидопропиламина (0,2 г, 1,96 ммоль) в сухом ДМФА (10,0 мл) добавили HBTU (0,93 г, 2,45 ммоль), а затем DIPEA (428 мкл, 2,45 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Реакцию контролировали по ЖХМС и после завершения реакции разбавили EtOAc (50 мл), промыли H2O (25 мл), солевым раствором (25 мл). Органический слой отделили; высушили (MgSO4) и упарили с получением технического продукта. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента 50% EtOAc-гексан с выделением N-(3-азидопропил)-5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)пентанамида в виде белого твердого вещества (0,13 г, 24%). Н-ЯМР (400 МГц, CD3OD) δ=4,52 (дд, J=7,9, 5,0 Гц, 1Н), 4,32 (дд, J=7,9, 4,5 Гц, 1Н), 3,36 (т, J=6,7 Гц, 2Н), 3,28 (д, J=6,8 Гц, 2Н), 3,21-3,14 (м, 1Н), 2,93 (дд, J=12,8, 5,0 Гц, 1Н), 2,75 (д, J=12,8 Гц, 1Н), 2,20 (т, J=7,3 Гц, 2Н), 1,78 (кв, J=6,8 Гц, 2Н), 1,74-1,57 (м, 4Н), 1,45 (кв, J=7,5 Гц, 2Н). 13С-ЯМР (101 МГц, CD3OD) δ=173,5, 163,4, 61,0, 59,3, 54,7, 48,2, 39,4, 35,8, 34,8, 27,7, 27,5, 27,2, 24,6.
N-(3-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенил)пент-4-инамид (1,0 ммоль) и N-(3-азидопропил)-5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)пентанамид (2,0 ммоль), 10 мол. % ТНРТА, 5 мол. % CuSO4, 10 мол. % аскорбата натрия в ДМСО перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь очистили с помощью хроматографии на колонне с обращенной фазой с получением указанного в подзаголовке соединения в виде белого твердого вещества (8,0 мг, 31%); 1Н-ЯМР (600 МГц, CD3OD) δ=8,26 (с, 1Н), 7,83-7,68 (м, 3Н), 7,50-7,43 (м, 1Н), 6,92 (с, 1Н), 4,48 (дд, J=8,0, 4,8 Гц, 1Н), 4,41-4,22 (м, 3Н), 3,18 (дд, J=6,9, 3,5 Гц, 1Н), 3,14 (тд, J=6,7, 1,7 Гц, 2Н), 3,12-3,06 (м, 2Н), 2,90 (дд, J=12,8, 4,9 Гц, 1Н), 2,81 (т, J=7,7 Гц, 4Н), 2,77 (д, J=7,1 Гц, 1Н), 2,71 (с, 1Н), 2,62 (т, J=7,3 Гц, 4Н), 2,19 (тд, J=7,4, 1,7 Гц, 2Н), 2,05-2,01 (м, 2Н), 2,00-1,95 (м, 4Н), 1,76-1,57 (м, 4Н), 1,43 (кв, J=7,6, 7,1 Гц, 2Н). 13С-ЯМР (151 МГц, CD3OD) δ=174,8, 174,8, 171,6, 171,5, 164,5, 146,2, 143,6, 139,1, 137,7, 129,1, 129,1, 128,7, 128,1, 126,5, 123,7, 122,9, 122,4, 122,2, 120,9, 118,4, 118,4, 118,3, 118,2, 118,2, 117,2, 110,5, 69,0, 61,9, 60,2, 55,6, 39,8, 36,1, 36,0, 35,8, 35,4, 35,4, 32,6, 32,6, 30,0, 29,7, 29,7, 28,6, 28,3, 28,0, 25,3, 25,2, 25,2, 20,9. МСВР (ESI) вычислено для C37H48N9O6S2 [М+Н] 778,3163, найдено 778,3145.
N-((1-(3-(N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)сульфамоил)фенил)-1Н-1,2,3-триазол-4-ил)метил)-5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)пентанамид
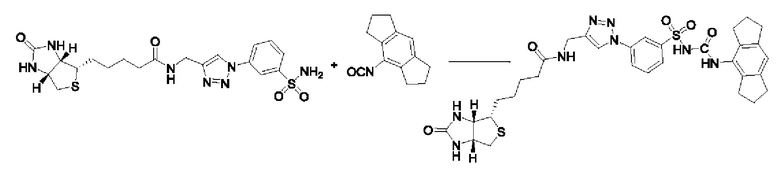
5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)-N-((1-(3-сульфамоилфенил)-1Н-1,2,3-триазол-4-ил)метил)пентанамид синтезировали, используя 5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)-N-(проп-2-ин-1-ил)пентанамид (1,0 ммоль) и 3-азидобензолсульфонамид (2,0 ммоль), 10 мол. % ТНРТА, 5 мол. % CuSO4, 10 мол. % NaAsc в ДМСО, перемешивали при комнатной температуре в течение 12 ч. Образование продукта наблюдали по ЖХМС. После завершения реакции реакционную смесь подвергли очистке с помощью ВЭЖХ (система для колоночной флэш-хроматографии Reveleris, 4 г, 18 мл/мин., подвижная фаз; 10 ммоль водн. NH4CO3, MeCN) с выделением 5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)-N-((1-(3-сульфамоилфенил)-1Н-1,2,3-триазол-4-ил)метил)пентанамида в виде белого твердого вещества (24 мг, 47%) который использовали сразу.
К раствору 5-((3aS,4S,6aR)-2-оксогексагидро-1Н-тиено[3,4-d]имидазол-4-ил)-N-((1-(3-сульфамоилфенил)-1Н-1,2,3-триазол-4-ил)метил)пентанамида (15 мг, 0,031 ммоль) в ТГФ (5,0 мл) в атмосфере азота добавили DIPEA (605 мкл, 0,037 ммоль). Эту смесь перемешивали при комнатной температуре в течение 15 мин. Добавили по каплям раствор 4-изоцианато-1,2,3,5,6,7-гексагидро-s-индацена (полученного с использованием общего способа А2) (705 мг, 0,037 ммоль) в ТГФ. Реакционную смесь перемешивали при комнатной температуре на протяжении ночи, затем растворитель удалили при пониженном давлении с получением технического соединения, которое очистили колоночной хроматографией на обращенной фазе с использованием подвижной фазы водн. (NH4)2CO3 10 ммоль/л и MeCN с выделением указанного в подзаголовке соединения в виде белого твердого вещества (5,2 мг, 24%). 1Н-ЯМР (600 МГц, CD3OD) δ=8,53 (д, J=2,4 Гц, 1Н), 8,50 (д, J=7,5 Гц, 1Н), 8,22-8,11 (м, 2Н), 7,86-7,78 (м, 1Н), 6,99 (с, 1Н), 4,62 (с, 2Н), 4,57-4,49 (м, 1Н), 4,38-4,31 (м, 1Н), 3,27-3,20 (м, 1Н), 3,00-2,84 (м, 4Н), 2,78-2,70 (м, 4Н), 2,36 (т, J=7,2 Гц, 2Н), 2,06 (кв, J=7,4 Гц, 4Н), 1,83-1,73 (м, 3Н), 1,71-1,63 (м, 1Н), 1,54-1,43 (м, 3Н). 13С-ЯМР (151 МГц, CD3OD) δ=174,6, 164,5, 146,1, 145,4, 143,6, 137,7, 137,2, 130,5, 125,9, 123,4, 121,1, 117,8, 110,6, 61,8, 60,2, 55,5, 47,7, 47,6, 39,8, 3,2, 34,3, 32,6, 30,14, 28,2, 27,9, 25,3, 25,2. МСВР (ESI) вычислено для C32H39N8O5S2 [М+Н] 679,2479, найдено 679,2456.
N-(хинолин-6-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид
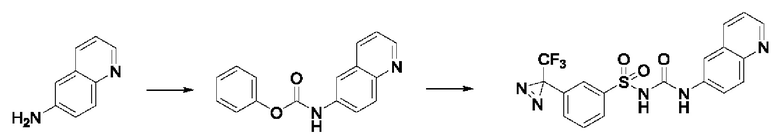
К раствору хинолин-6-амина (0,1 г, 0,69 ммоль) в ТГФ (50 мл) и триэтиламине (1,5 экв.) добавили фенилхлорформиат (1 экв.) при 0°С. Раствор разбавили водой, экстрагировали с помощью этилацетата (×2), промыли водой, солевым раствором, затем высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт растерли с пентаном с получением фенилхинолин-6-илкарбамата в виде белого с желтоватым оттенком твердого вещества, которое использовали сразу на следующей стадии реакции.
На 3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид (0,185 г, 0,69 ммоль) в ТГФ (30 мл) при 0°С подействовали гидридом натрия (3 экв.) порциями и суспензию перемешивали в течение 30 минут (до прекращения выделения газа). Технический фенилхинолин-6-илкарбамат растворили в ТГФ (20 мл), затем медленно добавили к реакционной смеси и перемешивание продолжали при температуре окружающей среды до завершения, как правило, 2 ч. Реакционную смесь разбавили насыщенн.. водн. NH4Cl, экстрагировали этилацетатом (×2), промыли водой (100 мл), солевым раствором (100 мл), высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт растерли с диэтиловым эфиром, затем с пентаном с получением указанного в подзаголовке соединения, N-(хинолин-6-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамида, в виде белого твердого вещества (10 мг, 3%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=8,88 (с, 1Н), 8,59 (д, J=3,5 Гц, 1Н), 8,11 (с, 1Н), 8,04 (д, J=8,3 Гц, 1Н), 7,96 (д, J=7,9 Гц, 1Н), 7,74 (д, J=9,1 Гц, 1Н), 7,70-7,60 (м, 2Н), 7,56 (т, J=7,8 Гц, 1Н), 7,34 (дд, J=8,3, 4,2 Гц, 1Н), 7,30 (д, J=8,0 Гц, 1Н). 19F-ЯМР (376 МГц, ДМСО-d6) δ -64,49.
N-(хинолин-5-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид
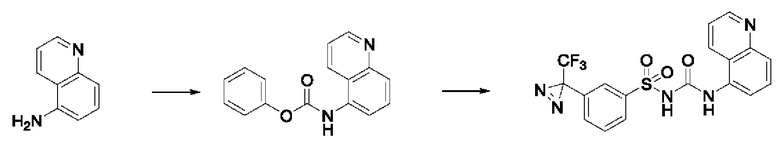
N-(хинолин-5-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид синтезировали с использованием модификации методик, использованных для получения N-(хинолин-6-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамида, но с применением хинолин-5-амина вместо хинолин-6-амина. Указанное в подзаголовке соединение получили в виде белого с желтоватым оттенком твердого вещества (10 мг, 3%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ=8,79 (д, J=4,1 Гц, 1Н), 8,61 (с, 1Н), 8,57 (д, J=8,7 Гц, 1Н), 7,95 (д, J=7,8 Гц, 1Н), 7,88 (д, J=7,2 Гц, 1Н), 7,70 (с, 1Н), 7,59-7,50 (м, 2Н), 7,40 (дд, J=8,7, 4,1 Гц, 1Н), 7,29 (д, J=7,9 Гц, 1Н). 19F-ЯМР (376 МГц, ДМСО-d6) δ -64,51.
N-((6-метоксихинолин-8-ил)карбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид
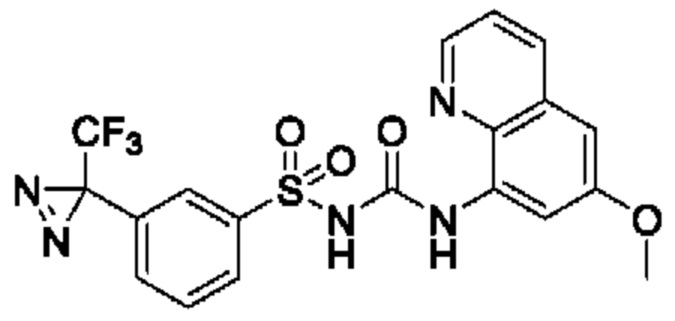
N-((6-метоксихинолин-8-ил)карбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид синтезировали с использованием модификации методик, использованных для получения N-(хинолин-6-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамида, но с применением 6-метоксихинолин-8-амина вместо хинолин-6-амина. Указанное в подзаголовке соединение получили в виде белого с желтоватым оттенком твердого вещества (35 мг, 20%). 1Н-ЯМР (400 МГц, CD3OD) δ 8,64 (дд, J=4,2, 1,6 Гц, 1Н), 8,18-8,02 (м, 3Н), 7,88 (с, 1Н), 7,60 (т, J=7,9 Гц, 1Н), 7,50-7,36 (м, 2Н), 6,79 (д, J=2,6 Гц, 1Н), 3,88 (с, 3Н). 19F-ЯМР (376 МГц, CD3OD) δ -67,04.
N-(хинолин-8-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид
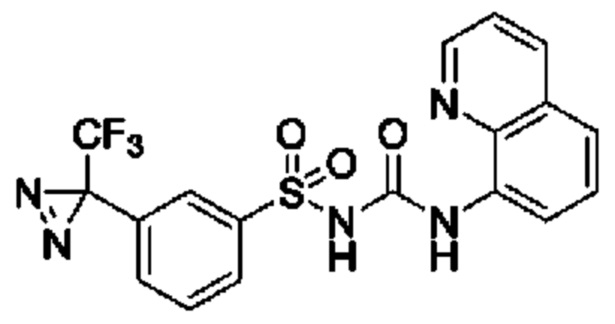
N-(Хинолин-8-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид синтезировали с использованием модификации методик, использованных для получения N-(хинолин-6-илкарбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамида, но с применением хинолин-8-амина вместо хинолин-6-амина. Указанное в подзаголовке соединение получили в виде белого твердого вещества (20 мг, 16%). 1Н-ЯМР (400 МГц, CD3OD) δ=8,82 (дд, J=4,3, 1,6 Гц, 1Н), 8,35 (дд, J=7,4, 1,8 Гц, 1Н), 8,21 (дд, J=8,3, 1,7 Гц, 1Н), 8,12 (д, J=7,9 Гц, 1Н), 7,88 (с, 1Н), 7,59 (т, J=7,9 Гц, 1Н), 7,52-7,38 (м, 4Н).
N-((2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран-4-ил)карбамоил)-3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид
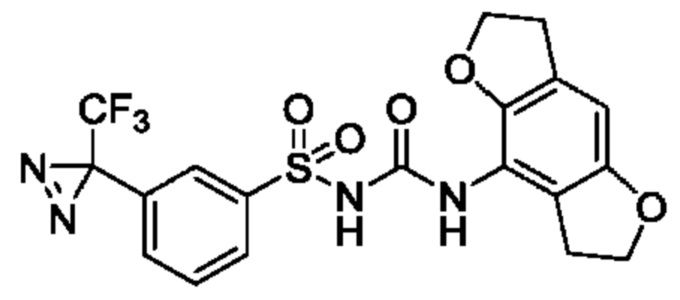
4-изоцианато-2,3,6,7-тетрагидробензо[1,2-b:4,5-b']дифуран (полученный с использованием общего способа А1) и 3-(3-(трифторметил)-3Н-диазирин-3-ил)бензолсульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,01 г, 2%). 1Н-ЯМР (400 МГц, CD3OD) δ=8,06 (дт, J=7,9, 1,3 Гц, 1Н), 7,80 (с, 1Н), 7,57 (т, J=7,9 Гц, 1Н), 7,44 (д, J=7,9 Гц, 1Н), 6,39 (с, 1Н), 4,49 (т, J=8,6 Гц, 2Н), 4,42 (т, J=8,6 Гц, 2Н), 3,09 (т, J=8,4 Гц, 2Н), 3,02 (т, J=8,6 Гц, 2Н). 19F-ЯМР (376 МГц, CD3OD) δ -67,06.
4-хлор-N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)бензолсульфонамид
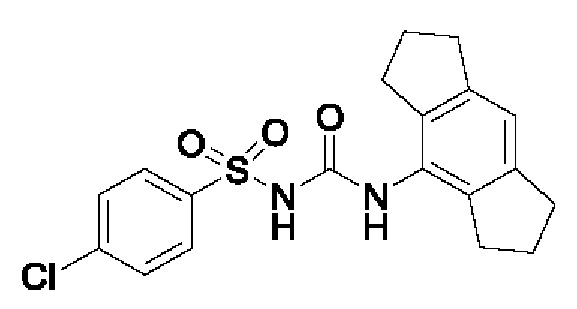
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4-хлорбензолсульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (48 мг, 43%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,13 (с, 1Н), 7,93 (д, J=8,0 Гц, 2Н), 7,68 (д, J=12 Гц, 2Н), 6,92 (с, 1Н), 2,77 (т, J=8,0 Гц, 4Н), 2,54 (т, J=8,0 Гц, 4Н), 1,95-1,88 (м, 4Н); 13С-ЯМР (100 МГц, ДМСО-d6): δ=150,1, 143,4, 139,9, 138,1, 137,6, 129,6, 129,4, 129,2, 32,8, 30,5, 25,9; Чистота по ЖХМС: >95%; ЖХМС (m/z): 391 [М+Н]+; МСВР вычислено для C19H19ClN2O3S [М+Н]+: 391,0878, найдено: 391,0895.
N-(1,2,3,5,6,7-гексагидро-5-индацен-4-илкарбамоил)-4-метилбензолсульфонамид
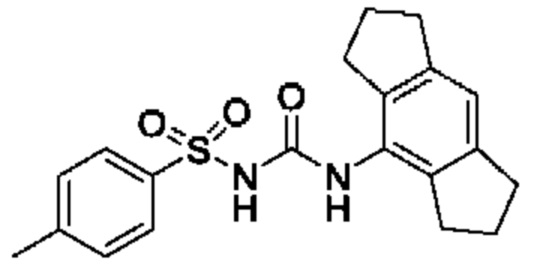
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 4-метилбензолсульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,045 г, 27%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=10,70 (ш.с, 1Н), 8,08 (с, 1Н), 7,82 (д, J=8 Гц, 2Н), 7,41 (д, J=8,0 Гц, 2Н), 6,92 (с, 1Н), 2,79-2,68 (м, 4Н), 2,58-2,50 (м, 4Н), 2,39 (с, 3Н), 1,97-1,87 (м, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=149,0, 143,6, 143,0, 137,1, 129,4, 128,6, 127,3, 117,9, 32,4, 30,0, 25,0, 21,0. ЖХМС, Чистота: 95,08%, m/z 371,07 (М+Н+). МСВР (FAB+) вычислено для C20H22N2O3S [М+Н]+: 371,1351, найдено: 371,1419.
5-Хлор-N-(4-(N-(1,2,3,5,6,7-гексагидро-5-индацен-4-илкарбамоил)сульфамоил)фенэтил)-2-метоксибензамид
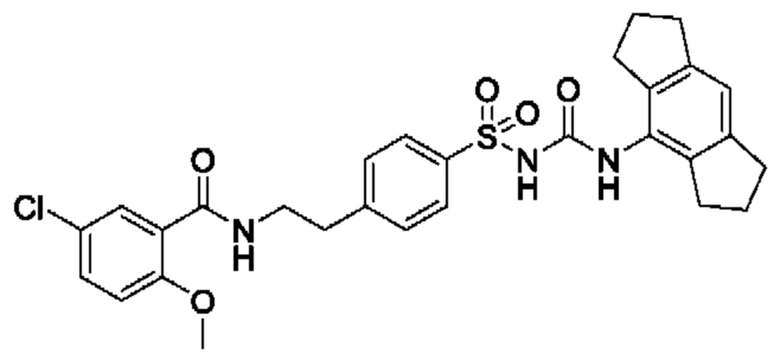
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 5-хлор-2-метокси-N-(4-сульфамоилфенэтил)бензамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого твердого вещества (45 мг, 10%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=10,73 (с, 1Н), 8,27 (т, J=5,2 Гц, 1Н), 8,09 (с, 1Н), 7,89 (д, J=8,4 Гц, 2Н), 7,65 (д, J=2,4 Гц, 1Н), 7,50 (д, J=8,4 Гц, 2Н), 7,49 (д, J=2,4 Гц, 1Н), 7,13 (д, J=9,2 Гц, 1Н), 6,92 (с, 1Н), 3,78 (с, 3Н), 3,54 (кв, J=6,4 Гц, 2Н), 2,94 (т, J=6,8 Гц, 2Н), 2,75 (т, J=7,2 Гц, 4Н), 2,50 (м, 4Н), 1,89 (квинт, J=7,6 Гц, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=163,6, 155,7, 145,3, 143,6, 143,0, 142,4, 142,1, 139,6, 137,1, 131,5, 129,5, 129,2, 127,4, 125,7, 124,8, 124,3, 117,9, 114,1, 108,3, 56,2, 34,7, 32,6, 32,4, 30,0, 28,9, 24,9. ЖХМС, Чистота: 90,06%, tr=3,38 мин, m/z 566,37 (М-Н+). МСВР (FAB+) вычислено для C29H30ClN3O5S [М+Н]+: 568,1595, найдено: 568,1589.
N-(4-(N-(1,2,3,5,6,7-Гексагидро-s-индацен-4-илкарбамоил)сульфамоил)фенэтил)-5-метилпиразин-2-карбоксамид
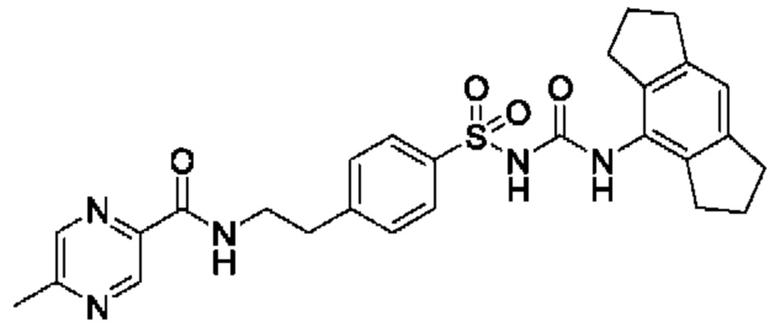
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 5-метил-N-(4-сульфамоилфенэтил)пиразин-2-карбоксамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (0,02 г, 4%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=10,71 (с, 1Н), 9,02 (с, 1Н), 8,96 (т, J=6 Гц, 1Н), 8,59 (с, 1Н), 8,07 (с, 1Н), 7,85 (д, J=8,4 Гц, 2Н), 7,47 (д, J=8,0 Гц, 2Н), 6,92 (с, 1Н), 3,57 (кв, J=6,8 Гц, 2Н), 2,97 (т, J=7,4 Гц, 2Н), 2,82-2,73 (м, 4Н), 2,53 (с, 3Н), 2,57-2,50 (м, 4Н), 1,97-1,84 (м, 4Н). ЖХМС, Чистота: 88,15%, m/z 520,28 (М+Н+). МСВР (FAB+) вычислено для C27H29N5O4S [М+Н]+: 520,1940, найдено: 520,1977.
3-(4-(N-((1,2,3,5,6,7-Гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенил)-N-(проп-2-ин-1-ил)пропанамид
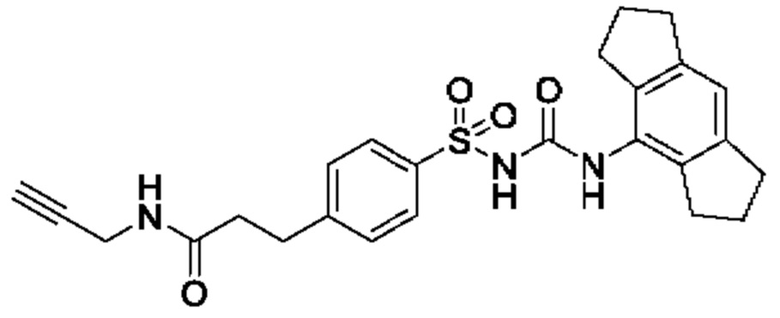
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и N-(проп-2-ин-1-ил)-3-(4-сульфамоилфенил)пропанамид использовали в общем способе С6 с получением указанного в подзаголовке соединения в виде белого твердого вещества (120 мг, 68%). 1Н-ЯМР (400 МГц, CD3OD) δ=7,91 (д, J=7,8 Гц, 2Н), 7,39 (д, J=7,8 Гц, 2Н), 6,98 (с, 1Н), 3,95 (д, J=2,9 Гц, 2Н), 3,03 (т, J=7,7 Гц, 2Н), 2,85 (т, J=7,4 Гц, 4Н), 2,62 (т, J=6,9 Гц, 4Н), 2,55-2,46 (м, 2Н), 2,25 (т, J=2,6 Гц, 1Н), 2,02 (м, 4Н). 13С-ЯМР (101 МГц, CD3OD) δ=172,0, 147,2, 144,1, 143,8, 137,5, 129,0, 128,8, 128,1, 127,4, 126,5, 118,9, 79,2, 71,0, 36,8, 32,8, 32,8, 31,2, 30,7, 28,8, 28,7, 25,4, 25,3. МСВР (ESI) вычислено для C25H28N3O4S [М+Н] 466,1795, найдено 466,1794.
N-(4-(N-(1,2,3,5,6,7-Гексагидро-s-индацен-4-илкарбамоил)сульфамоил)фенэтил)-2-(метил(7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)ацетамид
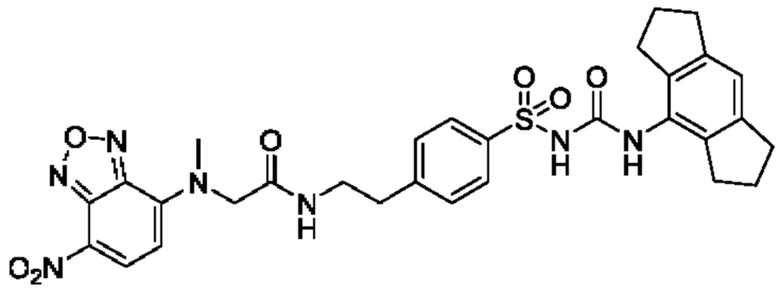
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 2-(метил(7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)-N-(4-сульфамоилфенэтил)ацетамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде оранжевого твердого вещества (0,003 г, 1%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=10,74 (с, 1Н), 8,51 (д, J=8,8 Гц, 1Н), 8,31 (т, J=7,6 Гц, 1Н), 8,09-7,96 (м, 1Н), 7,82 (д, J=8,4 Гц, 2Н), 7,41 (д, J=7,6 Гц, 2Н), 6,89 (с, 1Н), 6,42-6,32 (м, 1Н), 4,74 (шс, 2Н), 3,44-3,30 (м, 5Н), 2,80 (т, J=7,6 Гц, 2Н), 2,73-2,69 (м, 4Н), 2,61-2,50 (м, 4Н), 1,92-1,88 (м, 4Н). ЖХМС, Чистота: 92,20%, m/z 632,35 (М-Н+).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-((7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)этил)бензолсульфонамид
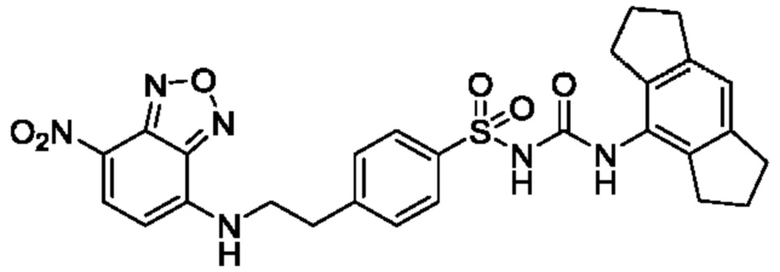
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 4-(2-((7-нитробензо[с][1,2,5]оксадиазол-4-ил)амино)этил)бензолсульфонамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде желтого твердого вещества (0,047 г, 15%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=10,69 (ш.с, 1Н), 9,55 (с, 1Н), 8,50 (д, J=8,8 Гц, 1Н), 8,09 (с, 1Н), 7,87 (д, J=8,0 Гц, 2Н), 7,56 (д, J=8,0 Гц, 2Н), 6,92 (с, 1Н), 6,50 (д, J=8,8 Гц, 1Н), 3,76 (ш с, 2Н), 3,11 (т, J=6,8 Гц, 2Н), 2,76 (т, J=7,6 Гц, 4Н), 2,53 (т, J=6,8 Гц, 4Н), 1,90 (квинт, J=7,6 Гц, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=149,1, 144,8, 144,3, 142,4, 138,3, 137,8, 137,1, 129,3, 128,6, 127,3, 125,7, 121,0, 117,9, 108,3, 99,5, 44,1, 33,2, 32,5, 30,1, 28,9, 25,0. ЖХМС, Чистота: 96,50%, tr=2,29 мин, m/z 563,20 (М+Н+). МСВР (FAB+) вычислено для C27H26N6O6S [М+Н]+: 563,1635, найдено: 563,1641.
2-(7-(Диметиламино)-2-оксо-2Н-хромен-4-ил)-N-(4-(N-(1,2,3,5,6,7-гексагидро-5-индацен-4-илкарбамоил)сульфамоил)фенэтил)ацетамид
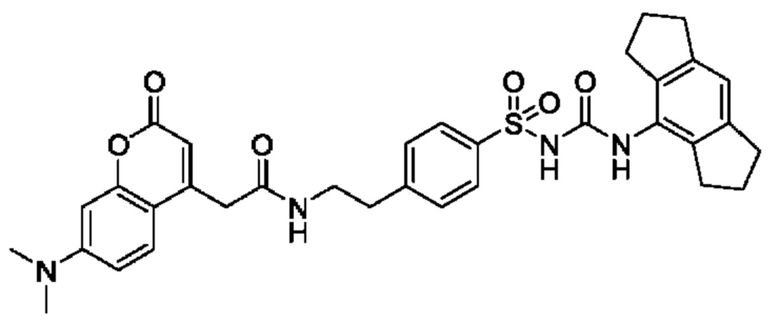
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 2-(7-(диметиламино)-2-оксо-2Н-хромен-4-ил)-N-(4-сульфамоилфенэтил)ацетамид использовали в общем способе С4 с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (0,008 г, 0,44%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=9,53 (с, 1Н), 8,29 (т, J=4,8 Гц, 1Н), 8,22 (с, 1Н), 7,68 (д, J=8,0 Гц, 2Н), 7,46 (д, J=8,8 Гц, 1Н), 8,42 (с, 1Н), 7,16 (д, J=8,0 Гц, 2Н), 6,74-6,70 (м, 2Н), 6,54 (д, J=2,4 Гц, 1Н), 5,99 (с, 1Н), 3,56-3,52 (м, 2Н), 3,48 (т, J=6,0 Гц, 2Н), 3,31-3,24 (м, 2Н), 2,76-2,70 (м, 4Н), 3,02 (с, 6Н), 2,63 (т, J=7,2 Гц, 4Н), 1,88 (квинт, J=7,6 Гц, 4Н). ЖХМС, Чистота: 92,26%, m/z 629,40 (МН+).
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)бензо[d][1,3]диоксол-5-сульфонамид
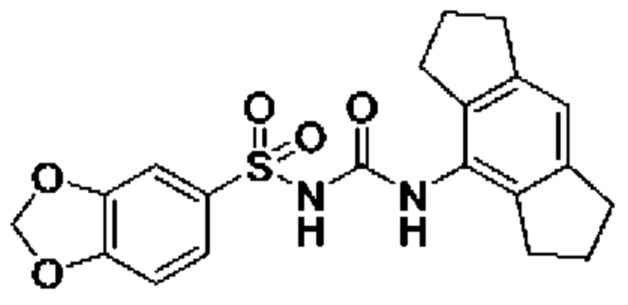
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и бензо[d][1,3]диоксол-5-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (28 мг, 27%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,04 (ш.с., 1Н), 7,47 (д, J=8,0 Гц, 1Н), 7,38 (с, 1Н), 7,09 (д, J=4,0 Гц, 1Н), 6,91 (с, 1Н), 6,16 (с, 2Н) 2,77 (т, J=8,0 Гц, 4Н), 2,56 (т, J=8,0 Гц, 4Н), 1,96-1,89 (м, 4Н); Чистота по ЖХМС: >95%; ЖХМС (m/z): 401 [М+Н]+; МСВР вычислено для C20H20N2O5S [М+Н]+ 401,1166, найдено 401,1182.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-6,7-дигидро-5Н-пирроло[1,2-а]имидазол-2-сульфонамид
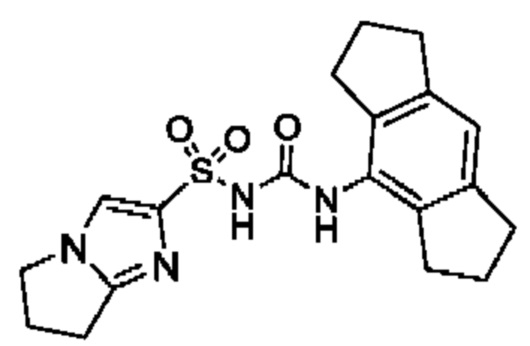
1,2,3,5,6,7-Гексагидро-s-индацен-4-амин (70 мг, 0,40 ммоль) растворили в безводном ТГФ (5 мл) и подействовали Et3N (49 мг, 0,49 ммоль) при комнатной температуре. На раствор подействовали трифосгеном (48 мг, 0,161 ммоль) и полученную смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток перемешивали с 5% EtOAc-гексаны (20 мл) в течение 10 мин, профильтровали через слой целита и концентрировали при пониженном давлении с получением соответствующего изоцианата в виде белого твердого вещества. В отдельной колбе 6,7-дигидро-5Н-пирроло[1,2-а]имидазол-2-сульфонамид (115 мг, 0,61 ммоль) растворили в безводном ТГФ (5 мл) и осторожно подействовали NaH (25 мг, 0,61 ммоль) при 0°С в атмосфере азота и перемешивали в течение 20 минут. К реакционной смеси добавили вышеупомянутый изоцианат в ТГФ в атмосфере азота. Реакционную смесь прогрели до комнатной температуры, перемешивали в течение 4 ч, затем концентрировали при пониженном давлении. Полученный остаток разбавили бикарбонатом аммония в воде 10 ммоль/л (20 мл), ацетонитрилом (20 мл), этилацетатом (10 мл) и образовавшееся твердое вещество удалили фильтрованием и промыли диэтиловым эфиром с получением N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-6,7-дигидро-5H-пирроло[1,2-а]имидазол-2-сульфонамида (50 мг, 32%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): δ=7,29 (с, 1Н), 6,85 (с, 1Н), 4,23 (т, J=7,2 Гц, 1Н), 2,86-2,79 (м, 6Н), 2,72 (т, J=7,2 Гц, 4Н), 2,65-2,60 (м, 2Н), 2,02-1,95 (м, 4Н). ЖХМС (m/z): 387,10 [M+H]+; 95,53% (210 нм). ВЭЖХ: 94,43% (210 нм). МСВР вычислено для C19H21N4O3S1 [М-Н]- 385,1340, найдено 385,1331.
4-Ацетил-N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)бензолсульфонамид
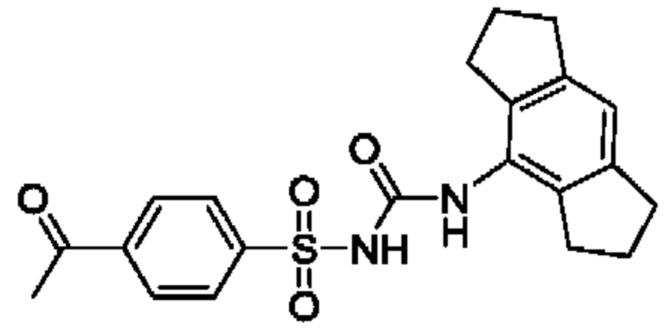
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4-ацетилбензолсульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (31 мг, 16%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=11,03 (шс, 1Н) 8,08 (д, J=8,5 Гц, 2Н), 7,99 (д, J=8,5 Гц, 2Н), 7,03 (ш.с, 1Н), 6,87 (с, 1Н), 2,75 (т, J=7,4 Гц, 4Н), 2,62 (с, 3Н), 2,56 (т, J=7,4 Гц, 4Н), 1,90 (п, J=7,4 Гц, 4Н). МСВР вычислено для C21H21N2O4S1 [М-Н]- 397,1128, найдено 397,1225.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-нитробензолсульфонамид

4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4-нитробензолсульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде бледно-желтого твердого вещества (148 мг, 60%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=10,00 (ш.с, 1Н), 8,21 (д, J=9,0 Гц, 2Н), 7,97 (д, J=9,0 Гц, 2Н), 7,45 (с, 1Н), 6,75 (с, 1Н), 2,73 (т, J=7,4 Гц, 4Н), 2,61 (т, J=7,4 Гц, 4Н), 1,87 (п, J=7,4 Гц, 4Н). МСВР вычислено для C19H18N3O5S1 [М-Н]- 400,0973, найдено 400,0979.
4-Амино-N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)бензолсульфонамид
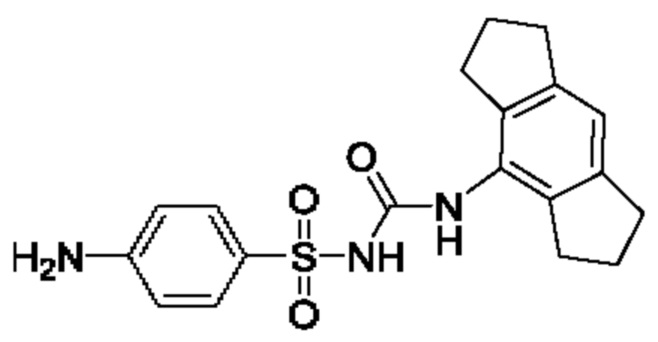
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-нитробензолсульфонамид, растворенный в растворе этилацетат/ДМФА (4:1, 25 мл/ммоль), перемешивали при комнатной температуре в течение 1 ч в атмосфере водорода с каталитическим количеством Pd/C (0,1 мол. %) с получением указанного в подзаголовке соединения в виде белого твердого вещества (16 мг, 43%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=7,95 (с, 1Н), 7,54 (д, J=8,8 Гц, 2Н), 6,91 (с, 1Н), 6,59 (д, J=8,8 Гц, 2Н), 6,05 (с, 2Н), 2,77 (т, J=7,4 Гц, 4Н), 2,55 (т, J=7,4 Гц, 4Н), 1,93 (кв, J=7,4 Гц, 4Н). МСВР вычислено для C19H20N3O3S1 [М-Н]- 370,1231, найдено 370,1225.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-2,3-дигидро-1Н-инден-5-сульфонамид
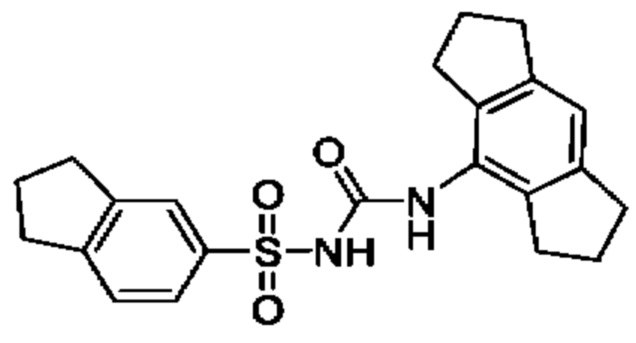
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 2,3-дигидро-1Н-инден-5-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (48 мг, 12%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=10,68 (ш.с, 1Н), 8,02 (с, 1Н), 7,75 (д, J=1,7 Гц, 1Н), 7,68 (дд, J=7,9, 1,7 Гц, 1Н), 7,41 (д, J=7,9 Гц, 1Н), 6,90 (с, 1Н), 2,91 (т, J=7,5 Гц, 4Н), 2,76 (т, J=7,4 Гц, 4Н), 2,53 (т, J=7,4 Гц, 4Н), 2,05 (п, J=7,5 Гц, 2Н), 1,91 (п, J=7,4 Гц, 4Н). МСВР вычислено для C22H23N2O3S1 [М-Н]- 395,1435, найдено 395,1430.
N-((4-хлорфенил)карбамоил)-2,3-дигидро-1Н-инден-5-сульфонамид

1-Хлор-4-изоцианатобензол (полученный с использованием общего способа В1) и 2,3-дигидро-1Н-инден-5-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (60 мг, 32%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ 10,90 (ш.с, 1Н), 8,90 (с, 1Н), 7,74 (д, J=1,8 Гц, 1Н) 7,68 (дд, J=7,9, 1,7 Гц, 1Н), 7,41-7,35 (м, 3Н), 7,26 (дт, 2Н), 2,91 (м, 4Н), 2,05 (п, J=7,5 Гц, 2Н). МСВР вычислено для C16H14Cl1N2O3S1 [М-Н]- 349,0419, найдено МСВР 349,0418.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)хинолин-8-сульфонамид
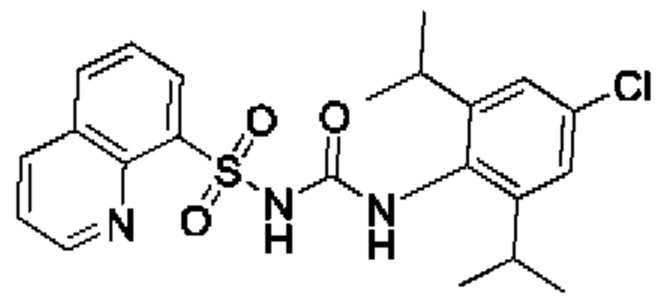
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и хинолин-8-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (75 мг, 71%). 1Н-ЯМР (600 МГц, CD3OD): δ=9,13 (дд, J=4,2, 1,6 Гц, 1Н), 8,57-8,49 (м, 2Н), 8,26 (д, J=8,2 Гц, 1Н), 7,77-7,67 (м, 2Н), 6,99 (с, 2Н), 2,65-2,60, (м, 2Н), 0,85 (д, 12Н); 13С-ЯМР (150 МГц, CD3OD) δ=151,2, 149,0, 143,3, 136,8, 136,7, 133,8, 133,5, 132,3, 129,4, 129,1, 125,3, 123,0, 122,1, 109,1, 28,3, 22,5; Чистота по ЖХМС: >95%; ЖХМС (m/z): 446 [М+Н]+; МСВР вычислено для C22H25Cl1N3O3S1 [М+Н]+ 446,1300, найдено 446,1314.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)изохинолин-5-сульфонамид
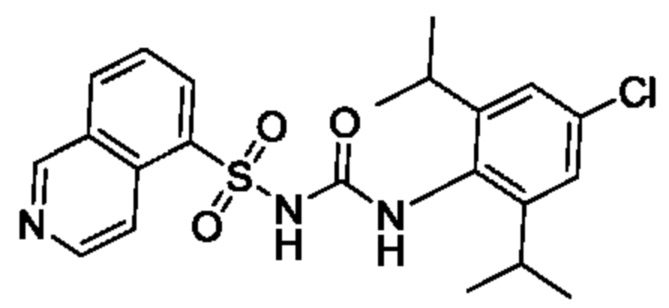
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и изохинолин-5-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (70 мг, 67%). 1Н-ЯМР (600 МГц, CD3OD): δ=9,41 (с, 1Н), 8,82 (с, 1Н), 8,59 (д, J=7,3 Гц, 2Н), 8,35 (д, J=8,2 Гц, 1Н), 7,79 (т, J=7,6 Гц, 1Н), 6,96 (с, 2Н), 2,74-2,70 (м, 2Н), 0,96 (с, 6Н), 0,85 (д, 12Н); 13С-ЯМР (150 МГц, CD3OD) δ=156,3, 152,5, 149,1, 143,8, 137,2, 133,9, 133,1, 132,6, 131,5, 130,4, 126,3, 124,8, 122,8, 122,1, 28,3, 22,4; Чистота по ЖХМС: >95%; ЖХМС (m/z): 446 [М+Н]+; МСВР вычислено для C22H25Cl1N3O3S1 [М+Н]+ 446,1300, найдено 446,1319.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)хинолин-5-сульфонамид
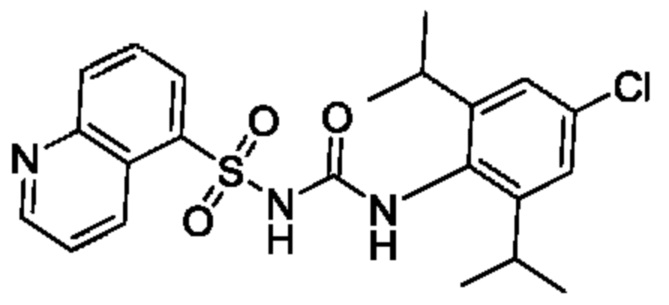
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и хинолин-5-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (31 мг, 60%). 1Н-ЯМР (600 МГц, CD3OD): δ=9,53 (д, J=8,9 Гц, 1Н), 8,94 (д, J=3,8 Гц, 1Н), 8,35 (дд, J=7,3, 1,2 Гц, 1Н), 8,15 (д, J=8,5 Гц, 1Н), 7,79 (дд, J=8,5, 7,3 Гц, 1Н), 7,69 (дд, J=8,7, 4,3 Гц, 1Н), 2,81-2,76 (м, 2Н), 0,85 (д, 12Н); 13С-ЯМР (150 МГц, CD3OD) δ=161,4, 151,3, 150,7, 149,1, 142,2, 137,4, 134,0, 133,0, 132,8, 129,9, 129,4, 126,0, 124,1, 122,9, 29,6, 24,0; Чистота по ЖХМС: >95%; ЖХМС (m/z): 446 [М+Н]+; МСВР вычислено для C22H25Cl1N3O3S1 [М+Н]+ 446,1300, найдено 446,1317.
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)хинолин-8-сульфонамид
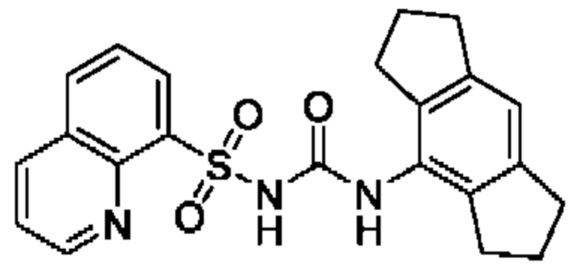
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4Н-хинолин-8-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (60 мг, 51%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ=9,11 (д, J=2,7 Гц, 1Н), 8,56 (д, J=8,3 Гц, 1Н), 8,40 (д, J=7,4 Гц, 1Н), 8,32 (д, J=8,2 Гц, 1Н), 8,17 (с, 1Н), 7,76 (т, J=7,7 Гц, 1Н), 7,73 (дд, J=8,4, 4,2 Гц, 1Н), 6,82 (с, 1Н), 2,67 (т, J=7,4 Гц, 4Н), 2,26 (т, J=7,4 Гц, 4Н), 1,79 (п, J=7,5 Гц, 4Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ=151,8, 151,7, 143,3, 143,2, 137,5, 137,1, 134,5, 133,4, 132,8, 129,9, 126,0, 122,8, 118,0, 108,7, 32,7, 30,2, 25,3. ЖХМС (m/z): 408 [М+Н]+. МСВР вычислено для C22H22N3O3S1 [М+Н]+ 408,1376, найдено 408,1371.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)хинолин-3-сульфонамид
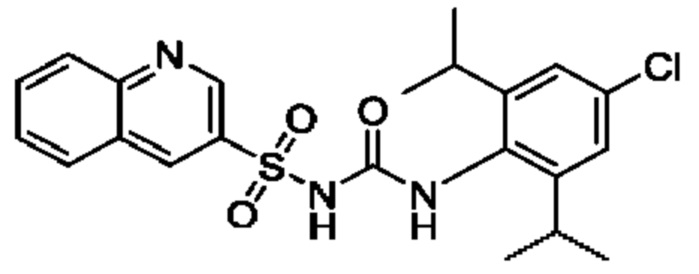
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и хинолин-3-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (30 мг, 57%). 1Н-ЯМР (600 МГц, ДМСО-d6): δ=9,24 (с, 1Н), 8,92 (с, 1Н), 8,20 (д, J=8,3 Гц, 1Н), 8,12 (д, J=8,5 Гц, 1Н), 7,98 (с, 1Н), 7,93 (т, J=7,7 Гц, 1Н), 7,74 (т, J=7,6 Гц, 1Н), 7,02 (с, 2Н), 2,81-2,78 (м, 2Н), 0,84 (д, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ=153,7, 149,3, 148,7, 147,6, 141,5, 136,8, 132,6, 132,4, 131,5, 129,9, 129,2, 128,4, 126,4, 123,3, 28,5, 23,5; Чистота по ЖХМС: >95%; ЖХМС (m/z): 446 [М+Н]+; МСВР вычислено для C22H25Cl1N3O3S1 [М+Н]+ 446,1300, найдено 446,1315.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)хиноксалин-5-сульфонамид
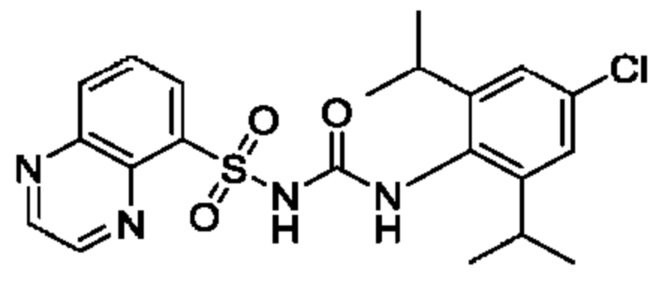
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и хиноксалин-5-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (39 мг, 75%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ=9,18 (д, J=3,7 Гц, 2Н), 8,46 (д, J=7,3 Гц, 1Н), 8,38 (дд, J=8,1, 2,7 Гц, 1Н), 8,04-7,95 (м, 1Н), 7,83 (с, 1Н), 6,99 (с, 2Н), 2,55-2,49 (м, 2Н), 0,74 (д, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): 149,2, 147,1, 146,8, 146,2, 142,7, 140,5, 138,5, 138,2, 134,2, 133,4, 132,6, 129,6, 123,4, 28,4, 22,7; Чистота по ЖХМС: >95%; ЖХМС (m/z): 447 [М+Н]+; МСВР вычислено для C21H24Cl1N4O3S1 [М+Н]+ 447,1252, найдено 447,1266.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)нафталин-2-сульфонамид
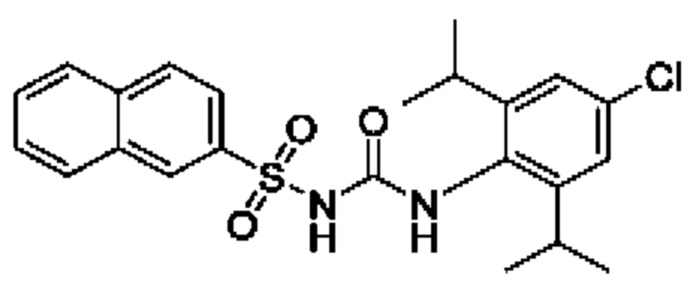
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и нафталин-2-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (35 мг, 67%). 1Н-ЯМР (600 МГц, CD3OD): δ=8,55 (с, 1Н), 8,05-7,92 (м, 4Н), 7,64-7,58 (м, 2Н), 6,99 (с, 2Н), 2,94-2,89 (м, 2Н), 0,94 (шс, 12Н); 13С-ЯМР (150 МГц, CD3OD) δ 159,9, 150,6, 142,2, 135,9, 134,0, 133,7, 132,6, 130,2, 129,5, 129,1, 128,8, 128,6, 128,1, 124,3, 124,2, 29,7, 24,0; Чистота по ЖХМС: >95%; ЖХМС (m/z): 445 [М+Н]+; МСВР вычислено для C23H26Cl1N2O3S1 [М+Н]+ 445,1347, найдено 445,1349.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)-6-метоксинафталин-2-сул фонамид
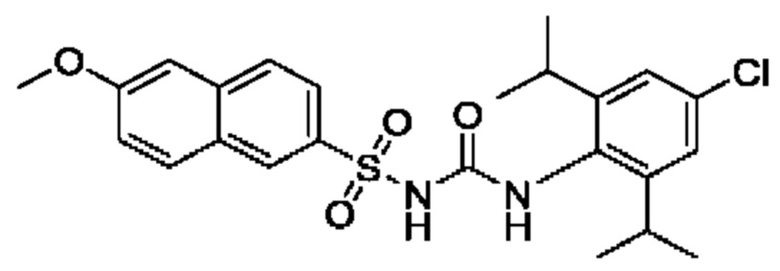
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и 6-метоксинафталин-2-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (39 мг, 70%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ=8,23 (с, 1Н), 7,86 (д, J=9,0 Гц, 1Н), 7,78 (дд, J=9,1, 6,4 Гц, 2Н), 7,49 (с, 1Н), 7,35 (д, J=2,6 Гц, 1Н), 7,20 (дд, J=8,9, 2,6 Гц, 1Н), 6,96 (с, 2Н), 3,08-2,98 (м, 2Н), 0,93 (ш.с, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ=158,0, 149,6, 135,2, 134,4, 131,0, 130,5, 128,1, 127,5, 127,3, 126,5, 124,7, 124,6, 122,7, 119,4, 106,2, 55,7, 28,3, 23,4; Чистота по ЖХМС: >95%; ЖХМС (m/z): 475 [М+Н]+; МСВР вычислено для C24H28Cl1N2O4S1 [М+Н]+ 475,1453, найдено 475,1474.
6-хлор-N-((4-хлор-2,6-диизопропилфенил)карбамоил)нафталин-2-сульфонамид
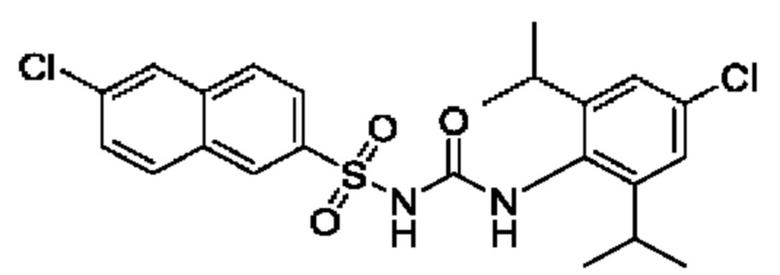
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и 6-хлорнафталин-2-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (34 мг, 61%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ=8,30 (с, 1Н), 8,06 (д, J=2,2 Гц, 1Н), 8,01 (д, J=8,8 Гц, 1Н), 7,93-7,85 (м, 2Н), 7,55 (дд, J=8,7, 2,2 Гц, 1Н), 7,40 (с, 1Н), 6,95 (с, 2Н), 3,09-2,97 (м, 2Н), 0,92 (ш.с, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ=160,2, 149,6, 145,8, 134,8, 134,2, 131,7, 131,1, 130,8, 130,7, 127,2, 126,8, 126,6, 125,8, 125,7, 122,7, 28,3, 23,6; Чистота по ЖХМС: >95%; ЖХМС (m/z): 479 [М+Н]+; МСВР вычислено для C23H25Cl2N2O3S1 [М+Н]+ 479,0957, найдено 479,0937.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)-5,6,7,8-тетрагидронафталин-2-сульфонамид
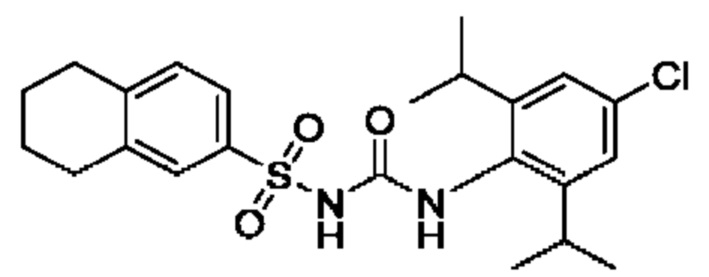
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и 5,6,7,8-тетрагидронафталин-2-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (8 мг, 38%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ=7,85 (с, 1Н), 7,58 (с, 1Н), 7,57 (д, J=8,0 Гц, 1Н), 7,22 (д, J=7,2 Гц, 1Н), 7,08 (с, 2Н), 2,85-2,81 (м, 2Н), 2,78-2,74 (м, 4Н), 1,74 (т, J=3,3 Гц, 4Н), 0,98 (ш.с, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ=149,3, 137,7, 137,5, 132,4, 129,8, 129,5, 129,7, 126,3, 124,3, 123,4, 122,9, 29,3, 29,2, 28,5, 23,4, 22,8, 22,7; Чистота по ЖХМС: >95%; ЖХМС (m/z): 449 [М+Н]+; МСВР вычислено для C23H30Cl1N2O3S1 [М+Н]+ 449,1660, найдено 449,1664.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)тиено[3,2-b]пиридин-6-сульфонамид
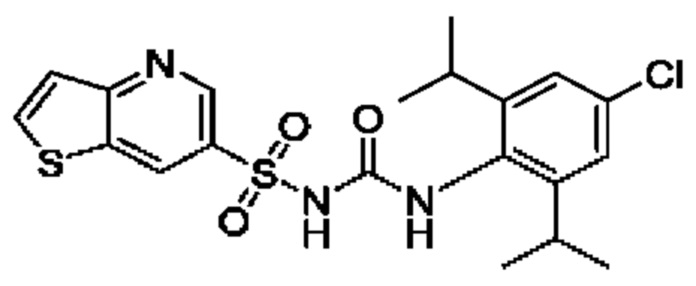
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и тиено[3,2-b]пиридин-6-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (35 мг, 66%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ=9,07 (д, J=2,0 Гц, 1Н), 9,05 (с, 1Н), 8,45 (д, J=5,5 Гц, 1Н), 8,01 (с, 1Н), 7,70 (д, J=5,5 Гц, 1Н), 7,05 (с, 2Н), 2,79-2,75 (м, 2Н), 0,87 (д, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ=158,2, 152,7, 149,3, 145,8, 138,2, 132,7, 132,3, 132,2, 131,6, 131,1, 124,7, 123,4, 28,5, 22,9; Чистота по ЖХМС: >95%; ЖХМС (m/z): 452 [М+Н]+; МСВР вычислено для C20H23Cl1N3O3S2 [М+Н]+ 452,0864, найдено 452,0884.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)-3-этилизоксазоло[5,4-b]пиридин-5-сульфонамид
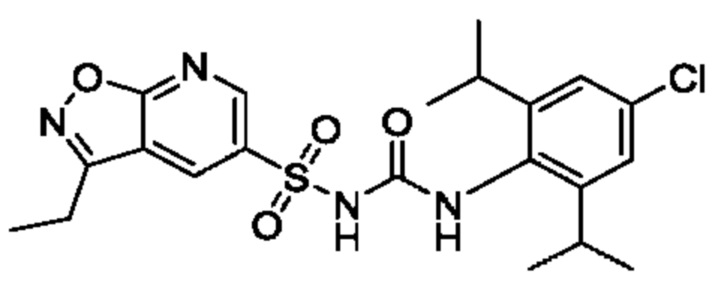
5-Хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и тиено[3,2-b]пиридин-6-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (38 мг, 64%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ=9,11 (с, 1Н), 8,98 (с, 1Н), 8,14 (с, 1Н), 7,08 (с, 2Н), 3,09 (кв, J=7,5 Гц, 2Н), 2,82-2,77 (м, 2Н), 1,34 (т, J=7,5 Гц, 3Н), 1,02-0,90 (д, 12Н). 13С-ЯМР (150 МГц, ДМСО-d6): δ=170,2, 161,9, 150,5, 149,3, 134,3, 134,2, 132,8, 131,0, 123,5, 112,7, 109,9, 28,5, 23,0, 19,2, 11,9; Чистота по ЖХМС: >95%; ЖХМС (m/z): 465 [М+Н]+; МСВР вычислено для C21H26Cl1N4O4S1 [М+Н]+ 465,1358, найдено 465,1354.
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)бензофуран-2-сульфонамид
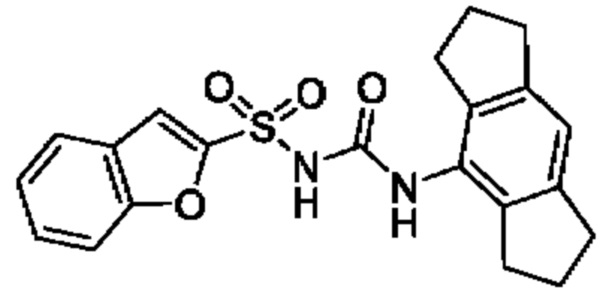
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и бензофуран-2-сульфонамид использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (60 мг, 52%); 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,00 (ш.с, 1Н), 7,77 (д, J=8 Гц, 1Н), 7,69 (д, J=8 Гц, 1Н), 7,51 (с, 1Н), 7,49 (д, J=8 Гц, 1Н), 7,36 (т, J=8 Гц, 1Н), 7,08 (ш.с, 1Н), 6,87 (с, 1Н), 2,75 (т, J=8 Гц, 4Н), 2,59 (т, J=8 Гц, 4Н), 1,92-1,85 (м, 4Н). 13С-ЯМР (150 МГц, ДМСО-d6): δ=154,9, 143,2, 137,6, 130,3, 127,5, 126,7, 124,4, 123,3, 117,7, 112,3, 110,0, 109,4, 107,4, 32,9, 30,6, 25,5. ЖХМС (m/z): 397 [М+Н]+. МСВР вычислено для C21H21N2O4S1 [М+Н]+ 397,1217, найдено 397,1215.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)бензофуран-2-сульфонамид
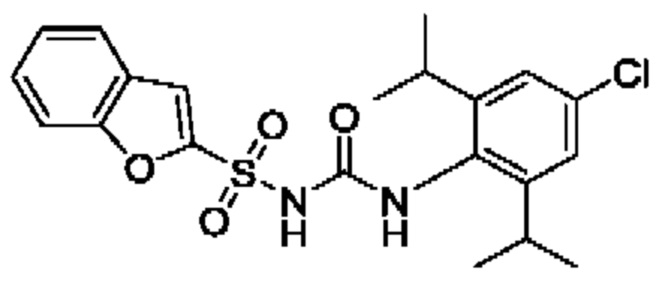
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и бензофуран-2-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (25 мг, 49%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=7,85 (с, 1Н), 7,73 (д, J=7,8 Гц, 1Н), 7,66 (д, J=8,4 Гц, 1Н), 7,47 (с, 1Н), 7,34 (т, J=7,5 Гц, 1Н), 7,17 (с, 1Н), 7,04 (с, 2Н), 2,99-2,95 (м, 2Н), 0,94 (шс, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ=154,9, 149,5, 132,6, 132,3, 132,0, 127,2, 126,9, 126,8, 123,3, 123,2, 112,1, 112,0, 109,8, 28,5, 23,3; Чистота по ЖХМС: >95%; ЖХМС (m/z): 435 [М+Н]+; МСВР вычислено для C21H24Cl1N2O4S1 [М+Н]+ 435,1140, найдено 435,1140.
N-((4-хлор-2,6-диизопропилфенил)карбамоил)бензо[b]тиофен-2-сульфонамид
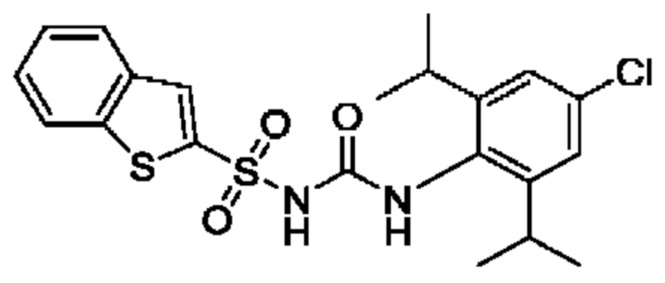
5-хлор-2-изоцианато-1,3-диизопропилбензол (полученный с использованием общего способа А2) и бензо[b]тиофен-2-сульфонамид (полученный с использованием общего способа Е3) использовали в общем способе С3 с получением указанного в подзаголовке соединения в виде белого твердого вещества (38 мг, 72%); 1Н-ЯМР (600 МГц, ДМСО-d6) δ 8,03 (д, J=7,9 Гц, 1Н), 7,93 (д, J=8,0 Гц, 2Н), 7,80 (с, 1Н), 7,46 (дт, J=15,4, 7,0 Гц, 2Н), 7,04 (с, 2Н), 3,05-2,83 (м, 2Н), 0,94 (шс, 12Н); 13С-ЯМР (150 МГц, ДМСО-d6): δ 155,3, 149,5, 141,1, 138,0, 132,4, 132,0, 126,8, 125,7, 125,4, 123,1, 123,0, 122,9, 109,7, 28,5, 23,3; Чистота по ЖХМС: >95%; ЖХМС (m/z): 451 [М+Н]+; МСВР вычислено для C21H24Cl1N2O3S2 [М+Н]+ 451,0911, найдено 451,0900.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(2-(7-метокси-4,4-диметил-1,3-диоксо-3,4-дигидроизохинолин-2(1Н)-ил)этил)бензолсульфонамид
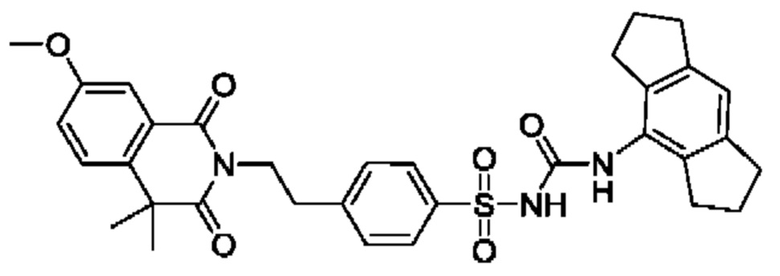
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 4-(2-(7-метокси-4,4-диметил-1,3-диоксо-3,4-дигидроизохинолин-2(1Н)-ил)этил)бензолсульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (85 мг, 52%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=10,72 (шс, 1Н), 7,91 (с, 1Н), 7,80 (д, J=8,1 Гц, 2Н), 7,58 (д, J=8,7 Гц, 1Н), 7,53 (д, J=2,9 Гц, 1Н), 7,39 (д, J=7,9 Гц, 2Н), 7,29 (дд, J=8,7, 2,9 Гц, 1Н), 6,88 (с, 1Н), 4,13 (т, J=7,5 Гц, 2Н), 3,83 (с, 3Н), 2,93 (т, J=7,5 Гц, 2Н), 2,76 (т, J=7,4 Гц, 4Н), 2,55 (т, J=7,4 Гц, 4Н), 1,90 (п, J=7,4 Гц, 4Н), 1,42 (с, 6Н). МСВР вычислено для C33H34N3O6S1 [М-Н]- 600,2174, найдено 600,2183.
N-(4-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенэтил)-5-метилизоксазол-3-карбоксамид

4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 5-метил-N-(4-сульфамоилфенэтил)изоксазол-3-карбоксамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (14 мг, 62%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ 8,78 (т, J=5,8 Гц, 1Н), 7,80 (с, 1Н), 7,74 (д, J=8,3 Гц, 2Н), 7,41 (д, J=8,3 Гц, 2Н), 6,85 (с, 1Н), 6,50 (кв, J=1,0 Гц, 1Н), 3,49 (м, 2Н), 2,91 (т, J=7,0 Гц, 2Н), 2,75 (т, J=7,4 Гц, 4Н), 2,56 (т, J=7,4 Гц, 4Н), 2,45 (д, J=0,9 Гц, 3Н), 1,89 (п, J=7,4 Гц, 4Н). МСВР вычислено для C26H27N4O5S1 [М-Н]- 507,1708, найдено 507,1709.
3-Этил-N-(4-(N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)сульфамоил)фенэтил)-4-метил-2-оксо-2,5-дигидро-1Н-пиррол-1-карбоксамид
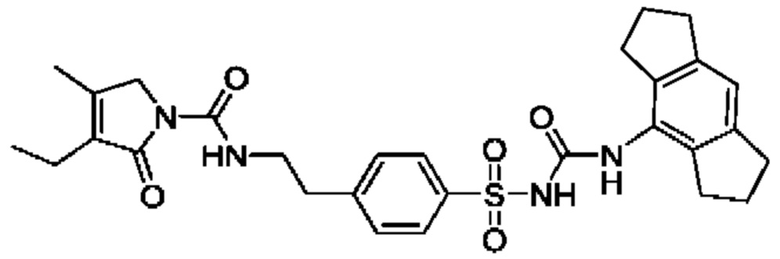
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 3-этил-4-метил-2-оксо-N-(4-сульфамоилфенэтил)-2,5-дигидро-1H-пиррол-1-карбоксамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (78 мг, 50%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=10,78 (шс, 1Н), 8,38 (т, J=7,6 Гц, 1Н), 7,97 (с, 1Н), 7,82 (д, J=8,2 Гц, 2Н), 7,42 (д, J=8,2 Гц, 2Н), 6,88 (с, 1Н), 4,16 (с, 2Н), 3,48 (кв, J=6,7 Гц, 2Н), 2,88 (т, J=7,2 Гц, 2Н), 2,75 (т, J=7,4 Гц, 4Н), 2,53 (т, J=7,4 Гц, 4Н), 2,18 (кв, J=7,5 Гц, 2Н), 2,01 (с, 3Н), 1,90 (п, J=7,4 Гц, 4Н), 0,97 (т, J=7,5 Гц, 3Н). МСВР вычислено для C29H33N4O5S1 [М-Н]- 549,2177, найдено 549,2169.
5-Хлор-N-(4-(N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)сульфамоил)фенэтил)-2-метоксибензамид
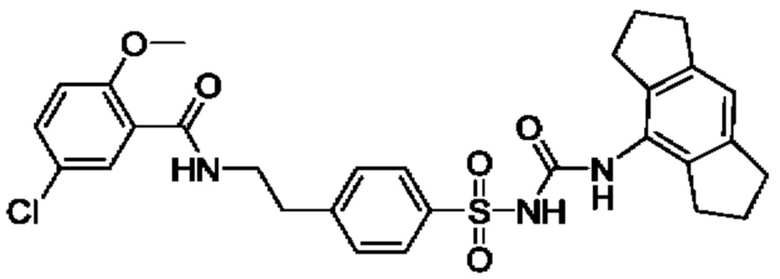
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 5-хлор-2-метокси-N-(4-сульфамоилфенэтил)бензамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (325 мг, 70%). 1Н-ЯМР (600 МГц, ДМСО-d6) δ=10,83 (шс, 1Н), 8,27 (т, J=6,0 Гц, 1Н), 7,96 (с, 1Н), 7,84 (д, J=8,0 Гц, 2Н), 7,66 (д, J=2,8 Гц, 1Н), 7,50 (дд, J=8,9 Гц, 2,8 Гц, 1Н, 7,44 (т, J=8,0 Гц, 2Н), 7,13 (д, J=8,9 Гц, 1Н), 6,87 (с, 1Н), 3,78 (с, 3Н), 3,53 (кв, J=6,6 Гц, 2Н), 2,91 (т, J=7,2 Гц, 2Н), 2,74 (т, J=7,4 Гц, 4Н), 2,53 (т, J=7,4 Гц, 4Н), 1,88 (п, J=7,3 Гц, 4Н). МСВР вычислено для C29H29Cl1N3O5S1 [М-Н]- 566,1522, найдено 566,1543. 13С-ЯМР (100 МГц, ДМСО-d6): δ=163,6, 155,7, 145,3, 143,6, 143,0, 142,4, 142,1, 139,6, 137,1, 131,5, 129,5, 129,2, 127,4, 125,7, 124,8, 124,3, 117,9, 114,1, 108,3, 56,2, 34,7, 32,6, 32,4, 30,0, 28,9, 24,9. ЖХМС, Чистота: 90,06%, tr=3,38 мин, m/z 566,37 (М-Н+). МСВР (FAB+) вычислено для C29H30ClN3O5S [М+Н]+: 568,1595, найдено: 568,1589.
ПИРИДИНЫ
N-(1,2,3,5,6,7-гексагидро-s-индацен-4-илкарбамоил)-4-(2-гидроксипропан-2-ил)пиридин-2-сульфонамид
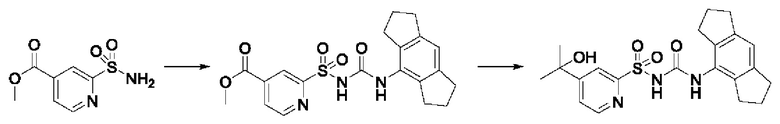
К раствору 1,2,3,5,6,7-гексагидро-s-индацен-4-амина (0,20 г, 1,15 ммоль) в безводном ТГФ (5 мл) добавили триэтиламин (0,35 г, 3,47 ммоль, 3,0 экв.), а затем трифосген (0,265 г, 0,86 ммоль, 0,5 экв.) при 0°С и смесь перемешивали при температуре окружающей среды в течение 3 ч. Смесь охладили до 0°С, добавили метил-2-сульфамоилизоникотинат (0,27 г, 1,27 ммоль, 1,1 экв.) и перемешивание продолжали при температуре окружающей среды на протяжении ночи. По завершении реакционную смесь вылили в солевой раствор и экстрагировали этилацетатом. Объединенные органические экстракты промыли солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический продукт очистили колоночной хроматографией на силикагеле с использованием элюента градиента 20-50% EtOAc-гексаны с получением метил-2-(N-(1,2,3,5,6,7-гексагидро-s-индацен-4-илкарбамоил)сульфамоил)изоникотината в виде светло-коричневого твердого вещества (0,31 г, 65%).
Метил-2-(N-(1,2,3,5,6,7-гексагидро-s-индацен-4-илкарбамоил)сульфамоил)изоникотинат (0,30 г, 0,72 ммоль) растворили в безводном ТГФ (8 мл) и раствор охладили до 0°С. Добавили метилмагнийбромид (раствор в диэтиловом эфире 3 моль/л, 0,96 мл, 2,88 ммоль, 4,0 экв.) при 0°С в атмосфере азота и перемешивание продолжали при температуре окружающей среды в течение 3 ч. По завершении реакционную смесь вылили в насыщенный водный хлорид аммония и экстрагировали этилацетатом. Объединенные органические экстракты промыли солевым раствором, высушили (Na2SO4) и концентрировали при пониженном давлении. Технический остаток очистили препаративной ВЭЖХ на обращенной фазе с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,016 г, 5%). 1Н-ЯМР (400 МГц, CD3OD): δ=8,45 (ш.с, 1Н), 8,16 (с, 1Н), 7,55 (ш.с, 1Н), 6,87 (с, 1Н), 2,80 (т, J=7,2 Гц, 4Н), 2,66 (т, J=7,2 Гц, 4Н), 1,96 (квинт, J=7,6 Гц, 4Н), 1,53 (с, 6Н). ЖХМС, Чистота: 98%, m/z 416,09 (М+Н+). МСВР (FAB+) вычислено для C21H25N3O4S [М+Н]+: 416,1566, найдено: 416,1556.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-6,7-дигидро-5Н-циклопента[b]пиридин-3-сульфонамид
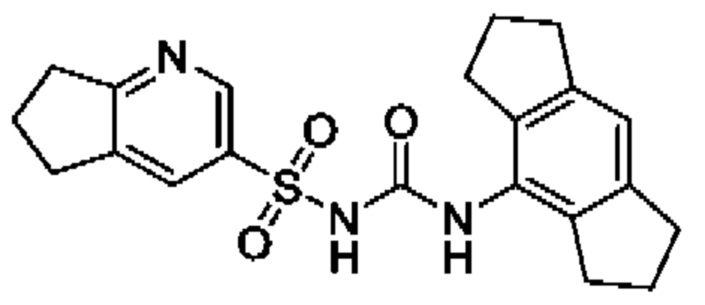
4-Изоцианато-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А2) и 6,7-дигидро-5Н-циклопента[b]пиридин-3-сульфонамид использовали в общем способе С2 с получением указанного в подзаголовке соединения в виде белого твердого вещества (12 мг, 27%). 1Н-ЯМР (600 МГц, ДМСО-d6): δ=8,71 (с, 1Н), 8,01 (с, 1Н), 7,96 (шс, 1Н), 6,87 (с, 1Н), 2,97-2,93 (м, 4Н), 2,75 (т, J=6 Гц, 4Н), 2,55 (т, J=6 Гц, 4Н), 2,11-2,07 (м, 2Н), 1,93-1,88 (м, 4Н). 13С-ЯМР (150 МГц, ДМСО-d6): δ 169,6, 146,4, 144,9, 143,2, 137,4, 137,2, 131,0, 129,6, 117,7, 108,7, 34,0, 32,9, 30,6, 30,3, 25,4, 23,2. ЖХМС (m/z): 398 [М+Н]+.
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)пиридин-2-сульфонамид
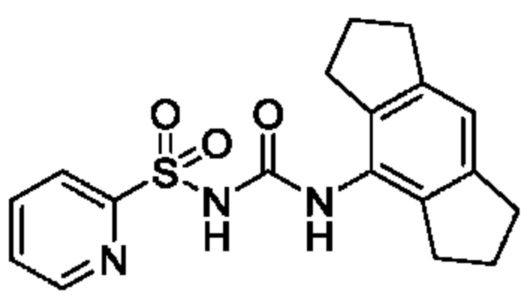
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и пиридин-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (40 мг, 10%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=8,5 (д, J=4,0 Гц, 1Н), 7,88 (т, J=7,6 Гц, 1Н), 7,81 (т, J=7,6 Гц, 1Н), 7,59 (с, 1Н), 7,4 (т, J=5,8 Гц, 1Н), 6,76 (с, 1Н), 2,73 (т, J=7,2 Гц, 4Н), 2,61 (т, J=7,2 Гц, 4Н), 1,88 (квинт, J=7,2 Гц, 4Н).
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)пиридин-3-сульфонамид
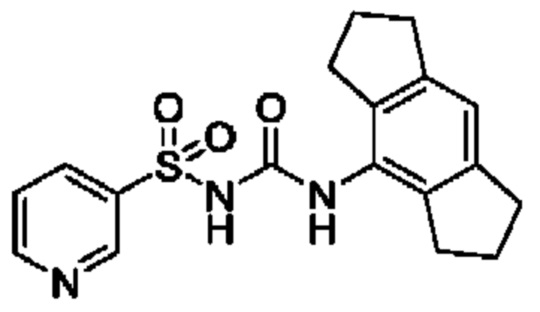
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и пиридин-3-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (12 мг, 3%). 1Н-ЯМР (400 МГц, CD3OD): δ=9,08 (с, 1Н), 8,65 (д, J=4,4 Гц, 1Н), 8,36 (д, J=8,0 Гц, 1Н), 7,56 (дд, J=8,0, 4,8 Гц, 1Н), 6,88 (с, 1Н), 2,82 (т, J=7,2 Гц, 4Н), 2,69 (т, J=7,2 Гц, 4Н), 2,0 (квинт, J=7,2 Гц, 4Н).
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамоил)-4-(трифторметил)пиридин-2-сульфонамид 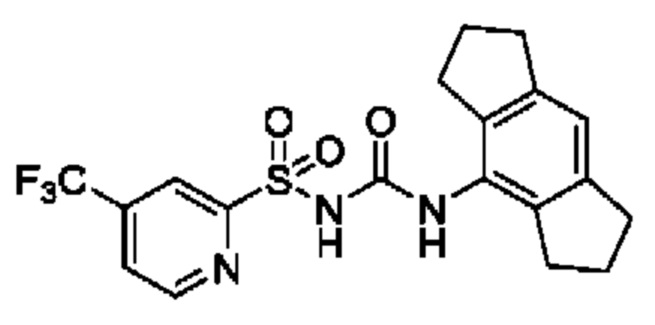
4-Изоцианато-8-метил-1,2,3,5,6,7-гексагидро-s-индацен (полученный с использованием общего способа А1) и 4-(трифторметил)пиридин-2-сульфонамид использовали в общем способе С1 с получением указанного в подзаголовке соединения в виде белого твердого вещества (16 мг, 3%). 1Н-ЯМР (400 МГц, CD3OD): δ=8,47 (с, 1Н), 8,22 (с, 1Н), 7,42 (с, 1 Н), 6,98 (с, 1 Н), 2,82 (т, J=7,2 Гц, 4Н), 2,66 (т, J=7,2 Гц, 4Н), 1,95 (квинт, J=7,2 Гц, 4Н); 19Р-ЯМР (233,33 МГц, ДМСО-d6): -63,48 (с, 3F).
ЛИНКЕР
N-((1,2,3,5,6,7-гексагидро-s-индацен-4-ил)карбамотиоил)-4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид
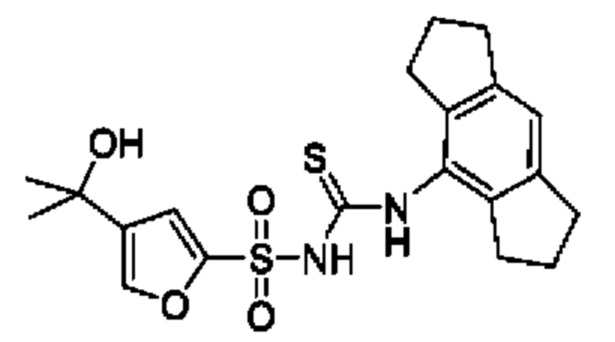
К раствору 1,2,3,5,6,7-гексагидро-з-индацен-4-амина (0,10 г, 0,58 ммоль) в безводном ДХМ (2,0 мл) добавили 1,1'-тиокарбонилдиимидазол (1,1 экв.) и реакционную смесь перемешивали в течение 4 ч при температуре окружающей среды. Растворитель удалили при пониженном давлении, затем остаток перенесли в ацетон (2,0 мл) и добавили карбонат калия (2,5 экв.), а затем 4-(2-гидроксипропан-2-ил)фуран-2-сульфонамид (1,2 экв.). Реакционную смесь нагревали с обратным холодильником на протяжении ночи, концентрировали при пониженном давлении, затем нейтрализовали с помощью 10% лимонной кислоты (10 мл) и сразу экстрагировали с помощью этилацетата (2×10 мл), высушили (MgSO4) и концентрировали при пониженном давлении. Технический продукт очистили посредством колоночной хроматографии на силикагеле с элюентом МеОН/ДХМ, а затем с помощью ВЭЖХ с получением указанного в подзаголовке соединения в виде белого с желтоватым оттенком твердого вещества (13 мг, 4%). 1Н-ЯМР (400 МГц, ДМСО-d6): δ=12,0 (шс, 1Н), 9,72 (с, 1 Н), 7,88 (с, 1 Н), 7,43 (с, 1Н), 7,01 (с, 1 Н), 5,15 (ш.с., 1Н), 2,81 (т, J=6,8 Гц, 4Н), 2,59 (т, J=6,8 Гц, 4Н), 1,95 (квинт, J=7,6 Гц, 4Н), 1,39 (с, 6Н). 13С-ЯМР (100 МГц, ДМСО-d6): δ=176,9, 143,1, 142,7, 138,7, 137,1, 130,4, 119,3, 117,7, 66,6, 32,4, 30,9, 29,9, 24,9. ЖХМС: Чистота = 95,08%, tr=3,45 мин, m/z 421,30 (М+Н+).
Методология биологических испытаний
Испытания ингибирования NLRP3
Для определения ингибирующей активности испытуемых соединений на инфламмасому NLRP3 можно применять следующие испытания с использованием обычных стимулов, таких как аденозинтрифосфат, нигерицин, LeuLeu-OMe или кристаллы моноурата натрия (MSU).
Культура клеток
Для выработки ММПЧ моноциты человека выделили из крови с лейкоцитарной пленкой с помощью Ficoll-Plaque Plus (GE Healthcare) и центрифугирования по плотности. Отбор клеток CD14+ осуществили с помощью магнитных частиц MACS (Miltenyl Biotec). Выделенные моноциты CD14+ дифференцировали в культуре в течение 7 дней с КСФ-1 (колониестимулирующим фактором) человека 10 нг/мл (Miltenyl Biotec) в среде Дульбекко, модифицированной по способу Исков (IMDM), содержащей L-глутамин, с добавкой 10% FBS (фетальной бычьей сыворотки) и 1% пенициллина/стрептомицина (Life Technologies), как описано в Croker et al 2013 Immunol Cell Biol 91:625.
Макрофаги из костного мозга мышей (BMDM) получили из клеток-предшественников костного мозга, выделенных из бедер и большеберцовых костей мышей C57BL/6. Кости промыли средой, и клетки костного мозга культивировали 7 дней в среде RPMI 1640 с добавкой 10% термоинактивированной FCS (фетальной бычьей сыворотки), 2 ммоль/л GlutaMAX (Life Technologies), 50 мкмоль/мл пенициллина-стрептомицина (Life Technologies) и 150 нг/мл рекомбинантного M-CSF (макрофагального колониестимулирующего фактора) человека (без эндотоксинов, экспрессированного и очищенного на технических средствах Квинслендского университета по экспрессии белков).
Испытания активации инфламмасом NLRP3
Произвели посев ММПЧ при 1×105/мл. На следующий день среду после ночи заменили и клетки стимулировали с помощью серотипа Escherichia coli 0111:В4 (Sigma Aldrich) в течение 3 ч. Среду удалили и заменили на среду без сыворотки (SFM), содержащую испытуемое соединение, за 30 мин до стимуляции NLRP3. Затем клетки стимулировали с помощью: гидрата двунатриевой соли аденозин-5'-трифосфата (5 ммоль/л 1 ч), нигерицина (10 мкмоль/л 1 ч), LeuLeu-OMe (1 ммоль/л 2 ч) или MSU (200 мкг/мл 15 ч). АТФ можно приобрести у Sigma Aldrich, нигерицин и MSU у Invivogen, a LeuLeu-Ome у Chem-Impex International.
Произвели посев BMDM при 1×105/мл. На следующий день среду после ночи заменили и клетки стимулировали с помощью липополисахарида Ultrapure из штамма K12 Escherichia coli (InvivoGen) в течение 3 ч. Среду удалили и заменили на среду без сыворотки (SFM), содержащую испытуемое соединение, за 30 мин до стимуляции NLRP3. Затем клетки стимулировали с помощью: гидрата двунатриевой соли аденозин-5'-трифосфата (1,25-5 ммоль/л 1 ч), нигерицина (5 мкмоль/л 1 h), LeuLeu-OMe (1 ммоль/л 2 ч) или MSU (200 мкг/мл 15 ч). АТФ можно приобрести у Sigma Aldrich, нигерицин и MSU у Invivogen, a LeuLeu-Ome у Chem-lmpex International.
Измерение ИЛ-1β, ИЛ-18, TNFα и гибели клеток
Для испытаний ИФА (иммуноферментного анализа, ELISA) и гибели клеток произвели посев клеток в 96-луночных планшетах. Надосадочные жидкости удалили и анализировали с помощью комплектов ИФА согласно указаниям производителя (DuoSet® R&D Systems, ReadySetGo!® eBioscience, BD OptEIA™ или Perkin Elmer AlphaLISA®). Гибель клеток оценивали измерением высвобождения ЛДГ в сравнении с контрольным образцом 100% лизиса клеток с помощью нерадиоактивного анализа цитотоксичности CytoTox96® (Promega).
Исследования уровней соединений в плазме крови и мозге на мышах
Общая экспериментальная часть: Карбутамид приобрели у Sigma Aldrich (Номер по каталогу 381578). Ацетонитрил представлял собой Chromasolv® квалификации для ВЭЖХ (Sigma Aldrich, Сидней, Австралия), муравьиная кислота была квалификации AR (чда) 99%-100% Normapur (VWR International Pty Ltd, Брисбен, Австралия), ДМСО был квалификации ReagentPlus® (D5879, Sigma Aldrich, Сидней, Австралия), а H2O Milli-Q была профильтрована. Виалы для ВЭЖХ и полипропиленовые вставки от Agilent Technologies (Мельбурн, Австралия), а пробирки Эппендорф 1,5 мл Protein LoBind Tubes были от VWR International Pty Ltd (Брисбен, Австралия).
Подготовка раствора для осаждения: 100 мл ацетонитрила и 5 мкл карбутамида в ДМСО 10 ммоль/л (внутренний стандарт МС ацетонитрил с 135 нг/мл карбутамида).
Подготовка калибровочной кривой в плазме: Приготовили 1 мг/мл испытуемого соединения в NH4HCO3 10 ммоль/л и 10-кратно разбавили с получением базового раствора 100000 нг/мл. Последовательностью 10-кратных разбавлений базового раствора 100000 нг/мл с помощью NH4HCO3 10 ммоль/л получили концентрации 10000, 1000, 100 и 10 нг/мл. Базовый раствор 100000 нг/мл разбавили до 3:7 с помощью NH4HCO3 10 ммоль/л с получением концентрации 30000 нг/мл и последовательностью 10-кратных разбавлений получили концентрации 3000, 300, 30 и 3 нг/мл.
20 мкл раствора, содержащего испытуемое соединение, и 160 мкл раствора для осаждения добавили к 20 мкл плазмы мыши в пробирке Эппендорф низкого удержания. Пробы перемешали вихревым способом, оставили стоять при 4°С в течение 10 мин и центрифугировали при 14000 × g в течение 8 мин. 150 мкл надосадочной жидкости перенесли во вставку для виалы ВЭЖХ. Пробы хранили при 4°С до анализа.
Подготовка калибровочной кривой в гомогенате мозга: исследуемые растворы, приготовленные для калибровочной кривой плазмы, использовали для калибровочной кривой гомогената мозга.
Гомогенат мозга мышей из контрольной группы, получавшей физиологический раствор, оттаивали и перемешивали вихревым способом в течение 3 мин или до однородности, обрабатывали ультразвуком в течение 1 мин. После оседания пены 50 мкл гомогената мозга мышей переносили в пробирку Эппендорф, а затем 50 мкл испытуемого соединения в NH4HCO3 10 ммоль/л, 150 мкл H2O и 500 мкл ледяного раствора для осаждения с перемешиванием вихревым способом после каждого добавления. Стандартные растворы оставили стоять при 4°С в течение 10 мин и затем центрифугировали при 14000 × g в течение 8 мин. 200 мкл надосадочной жидкости перенесли во вставку для виалы ВЭЖХ, обеспечивая, чтобы не присутствовали пузырьки воздуха, и пробы хранили при 4°С до анализа.
Дозирование мышам и транскардиальная перфузия
Дозирование: Пероральное принудительное питание в дозе 20 мг/кг
Момент времени: 2 часа
Приготовили основные соединения для дозирования при 4 мг/мл в стерильном PBS (фосфатно-буферном физиологическом растворе). Мышей взвесили и дозировали путем принудительного перорального питания в дозе 20 мг/кг для каждого соединения. Через 2 часа мышей анестезировали с помощью комбинации Золетила (50 мг/кг) и Ксилазина (10 мг/кг) и собрали кровь пункцией сердца в пробирки, содержащие 20 мкл 100 ммоль/л ЭДТК. Кровь центрифугировали при 2000 × g в течение 15 минут при 4°С для сбора плазмы.
Подготовка проб плазмы для анализа: 20 мкл NH4HCO3 и 160 мкл раствора для осаждения добавили к 20 мкл плазмы мыши в пробирке Эппендорф низкого удержания. Пробы перемешали вихревым способом, оставили стоять при 4°С в течение 10 мин и центрифугировали при 14,000 × g в течение 8 мин. 150 мкл надосадочной жидкости перенесли во вставку для виалы ВЭЖХ, обеспечивая, чтобы не присутствовали пузырьки воздуха. Пробы хранили при 4°С до анализа.
Приготовление гомогената мозга: мозги мышей перфузировали PBS в течение 5 минут, затем разрезали и взвесили. Гомогенат мозга готовили путем гомогенизации всего мозга (0,5 г) с 4 объемами (2 мл) деионизированной воды и хранили при -20°С до анализа. Гомогенат оттаивали, перемешивали вихревым способом в течение 3 мин или до однородности и обрабатывали ультразвуком в течение 1 мин. После оседания пены 50 мкл гомогената мозга мышей переносили в пробирку Эппендорф, а затем 50 мкл NH4HCO3 10 ммоль/л, 150 мкл H2O и 500 мкл ледяного раствора для осаждения с перемешиванием вихревым способом после каждого добавления. 200 мкл надосадочной жидкости переносили во вставку для виалы ВЭЖХ, обеспечивая, чтобы не присутствовали пузырьки воздуха, и пробы хранили при 4°С до анализа.
Приготовление проб мозга для анализа: 50 мкл мозга мыши переносили в пробирку Эппендорф, а затем 50 мкл NH4HCO3 10 ммоль/л, 150 мкл H2O и 500 мкл ледяного раствора для осаждения с перемешиванием вихревым способом после каждого добавления. Растворы оставили стоять при 4°С в течение 10 мин и затем центрифугировали при 14000 × g в течение 8 мин. 200 мкл надосадочной жидкости переносили во вставку для виалы ВЭЖХ, обеспечивая, чтобы не присутствовали пузырьки воздуха, и пробы хранили при 4°С до анализа.
ЖХ-МС/МС: пробы анализировали на АВ Sciex 4000QTrap MS с 2 устройствами подачи растворителя Shimadzu Nexera LC-30AD, автодозатором Shimadzu Nexera SIL-30AC, дегазатором Shimadzu Prominence DGU-20A5, контроллером системы Shimadzu Prominence CBM-20A и колоночным термостатом Shimadzu Prominence CTO-20A. Колоночный термостат установили на 40°С, тогда как автодозатор установили на 15°С. Производили вводы 2 мкл и проводили МС-анализы в режиме селективного мониторинга реакций (SRM) с использованием Turbo Spray (-)-ESI (ионизации распылением в электрическом поле) с Q1 низкого разрешения и Q3 низкого разрешения. Параметры МС: CUR: 30,00, IS: -4300,00, ТЕМ: 500,00, GS1: 50,00, GS2: 50,00, ihe: ON, CAD: High, DP -60,00, ЕР -10,00, СХР -15,00. MCC950 SRM: Q1 403,2 до Q3 204,3 Da, время измерения в точке 150 мс, SRM СЕ -27 и карбутамида (IS): Q1 270,0 до Q3 171,0 Da, время измерения в точке 100 мс, СЕ -25. Колонна ВЭЖХ: Waters Atlantis® Т3 5 мкм 2,1 × 50 мм с предколонкой Atlantis® Т3 5 мкм 2,1 × 10 мм. Расходы и растворители: 0,35 мл/мин, растворитель А: 0,1% муравьиной кислоты в H2O, растворитель В: 0,1% муравьиной кислоты в ацетонитриле; изократичный 2% В от 0→2 мин, градиент 2%→100% В от 2→5 мин, изократичный 100% от 5→9 мин, градиент 100%→2% В от 9→9,1 мин и изократичный 2% В от 9,1→13 мин. Площади пиков из данных SRM для карбутамида и испытуемого соединения анализировали с помощью программного обеспечения АВ Sciex's Analyst с использованием Quantitation Wizard. Площадь пика откладывали в зависимости от концентрации в нг/мл в растворах испытуемого соединения 20 мкл от 3 до 30000 нг/мл и определяли нижний и верхний предел линейной реакции. Эти данные затем представляли в виде графика в Microsoft Excel и использовали уравнение линейной реакции для определения концентрации испытуемого соединения в растворах плазмы 20 мкл. Аналогично, для проб гомогената мозга, использовали площади пиков 50 мкл от 3 до 3000 нг/мл растворов испытуемых соединений для определения концентрации испытуемого соединения в растворах гомогената мозга 50 мкл.
РЕЗУЛЬТАТЫ
Полные ряды tPSA и биологические результаты приведены в таблицах ниже, однако отдельные данные для определенных соединений по настоящему изобретению представлены ниже.

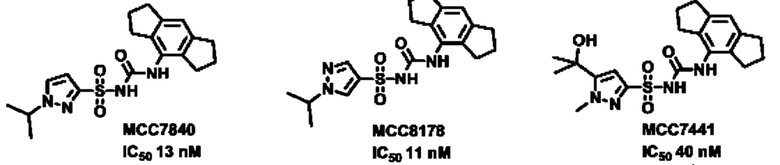
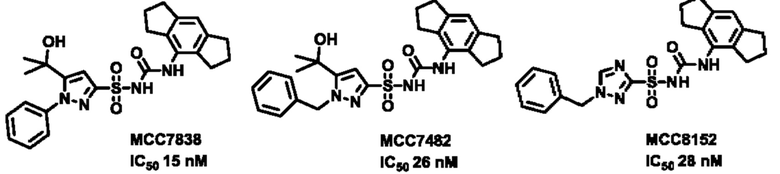
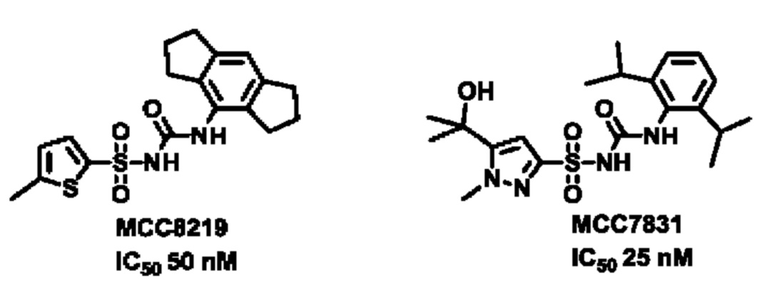
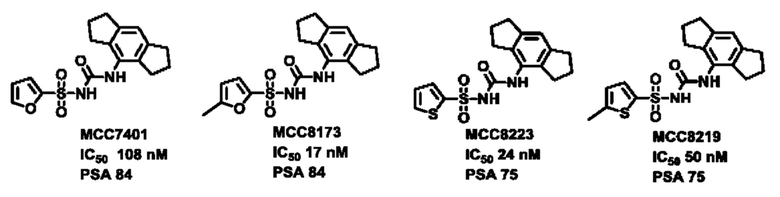
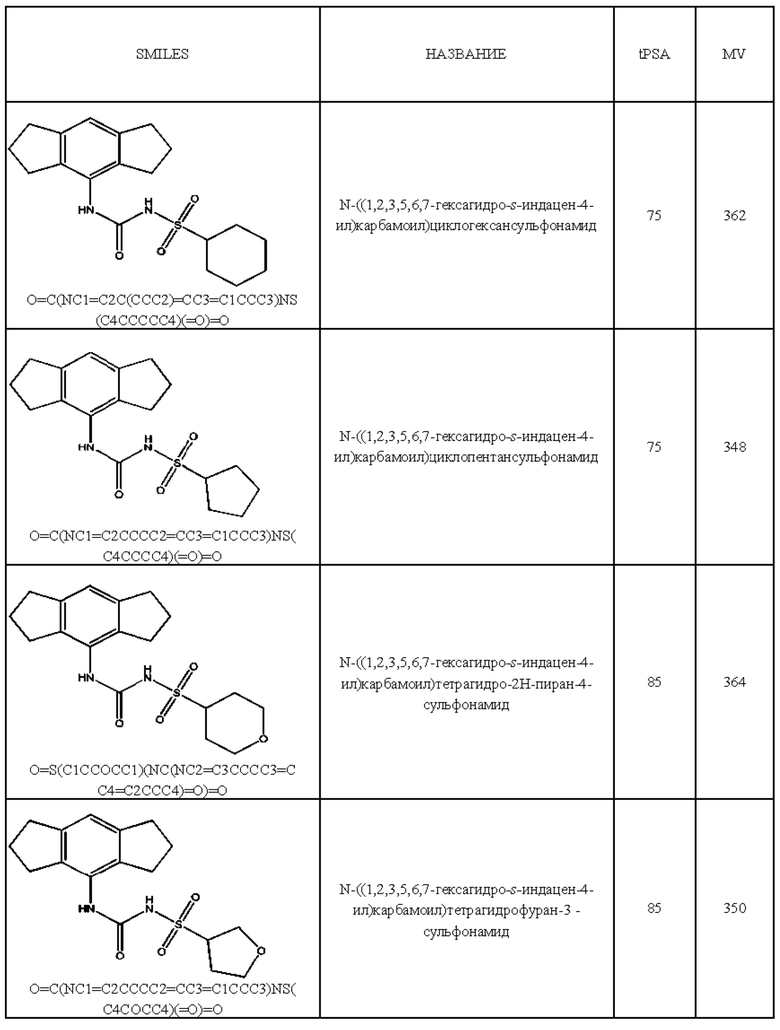
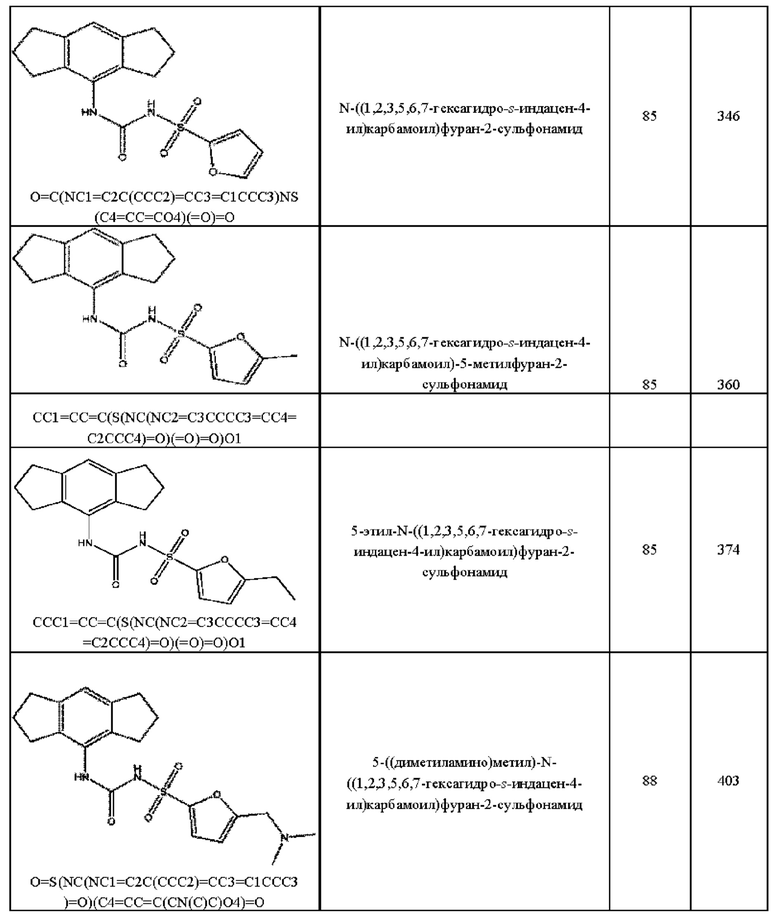

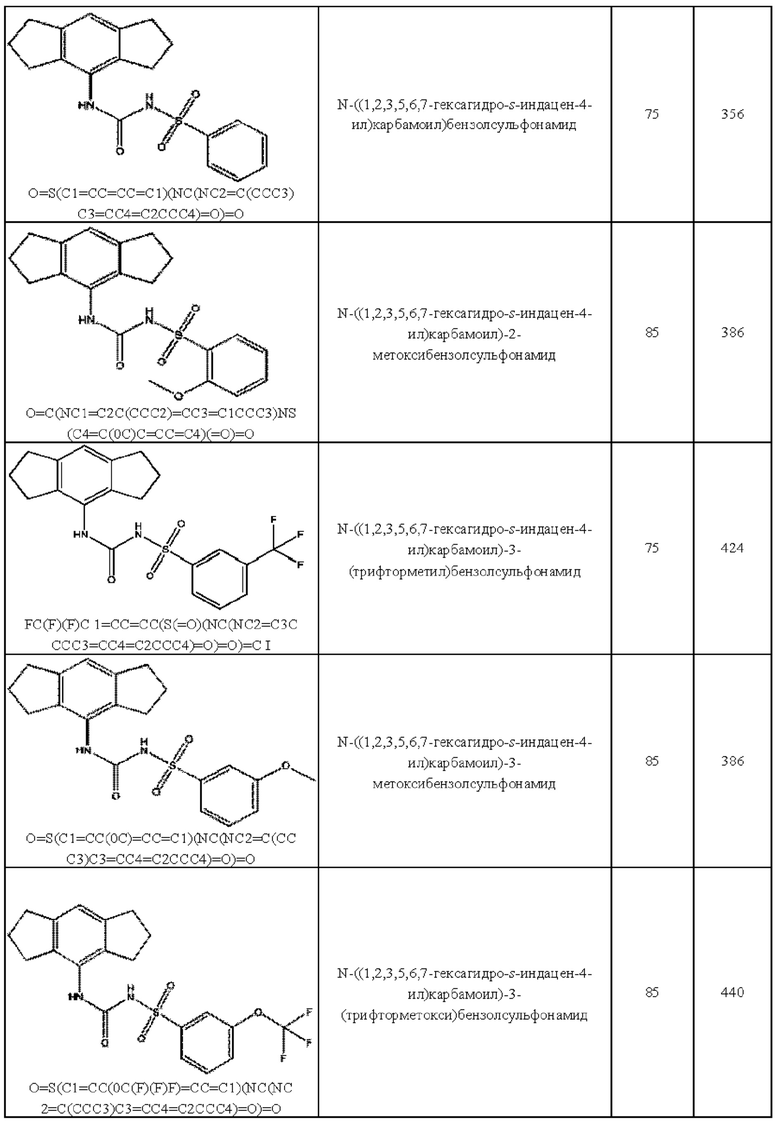
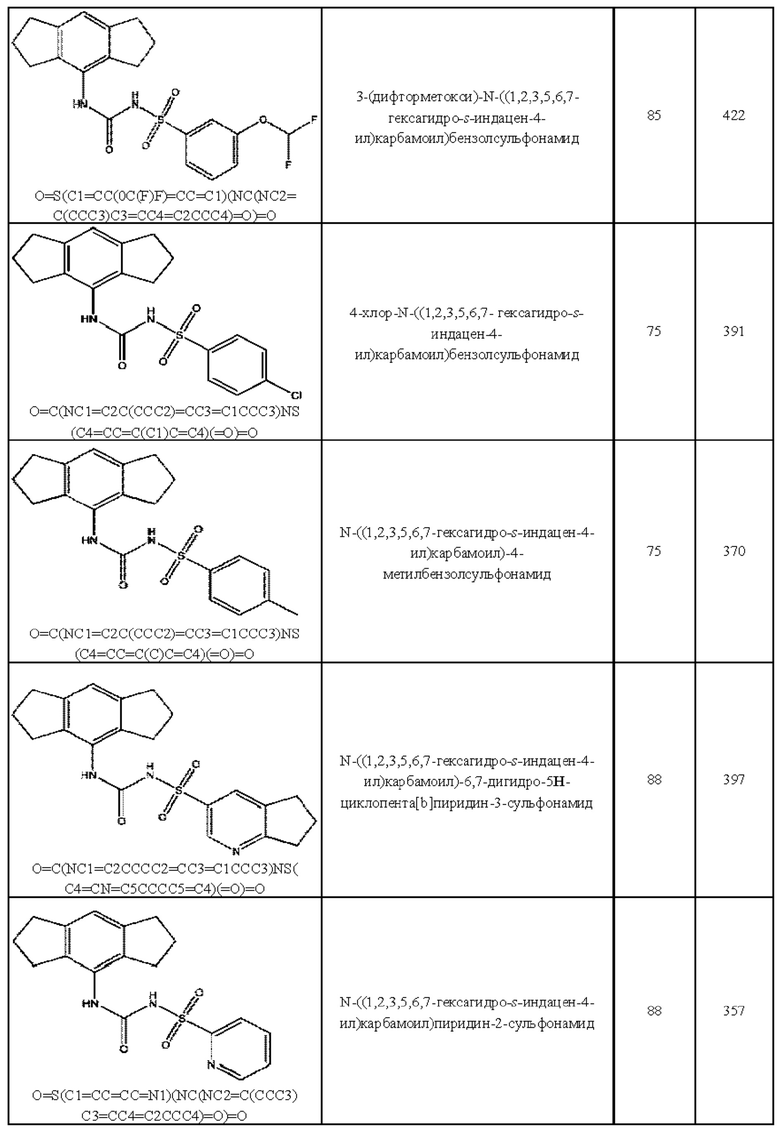

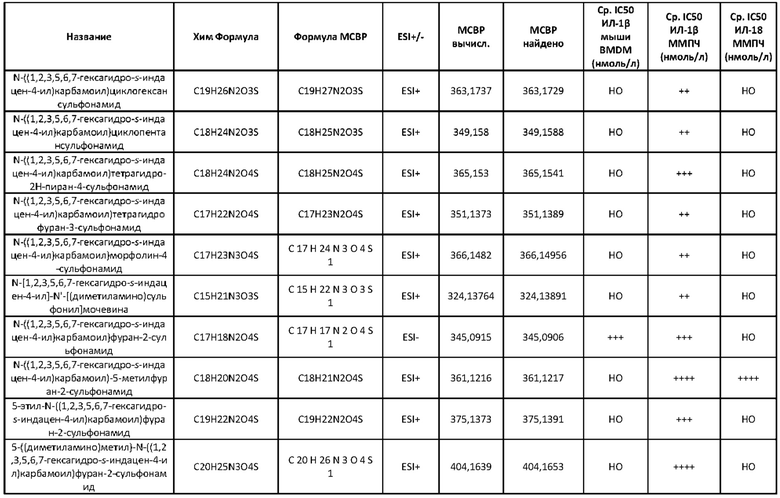
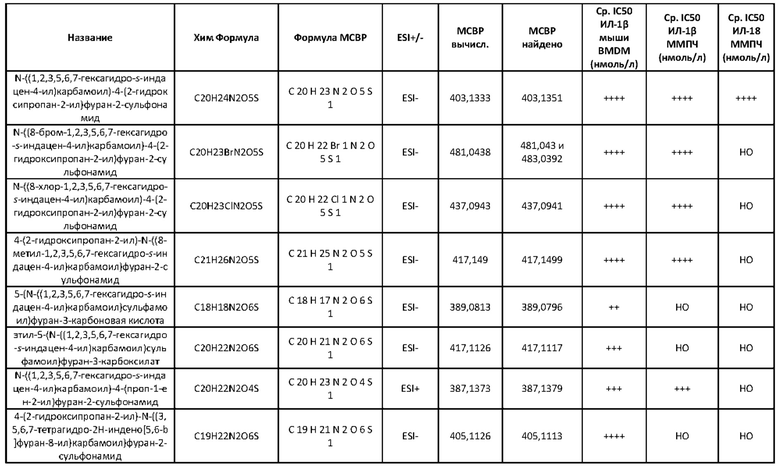
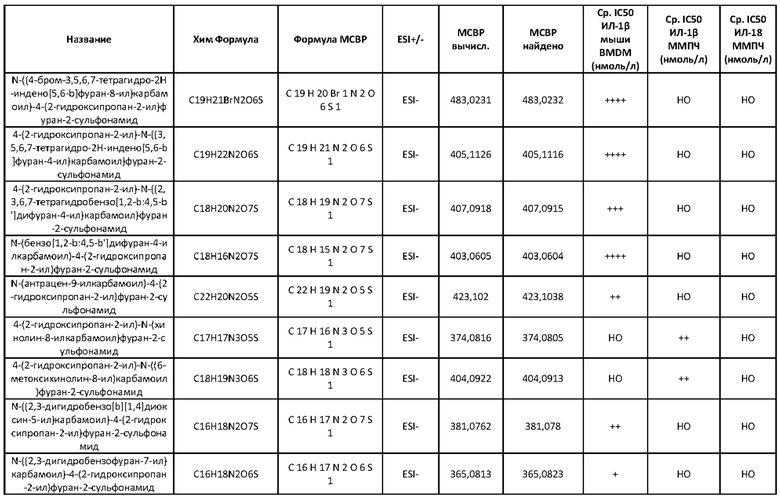
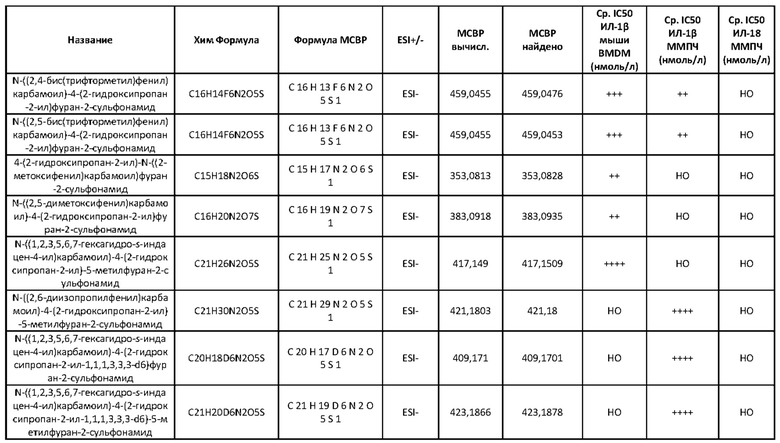
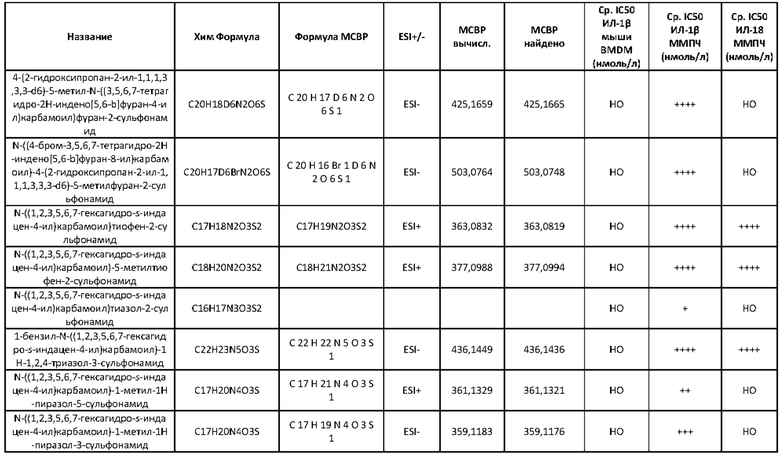
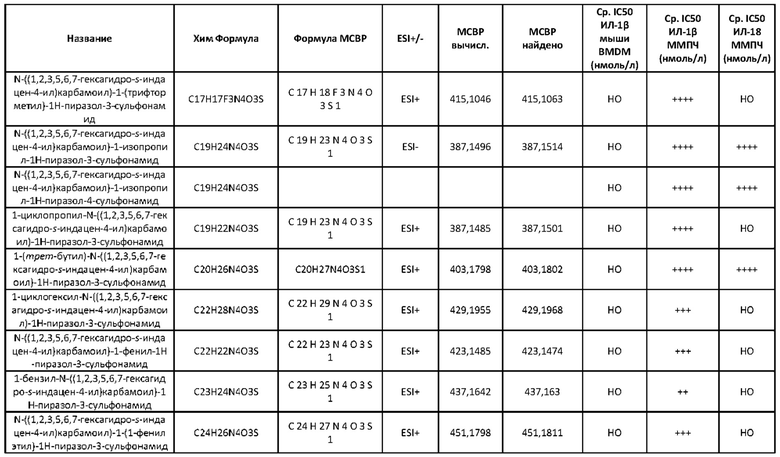
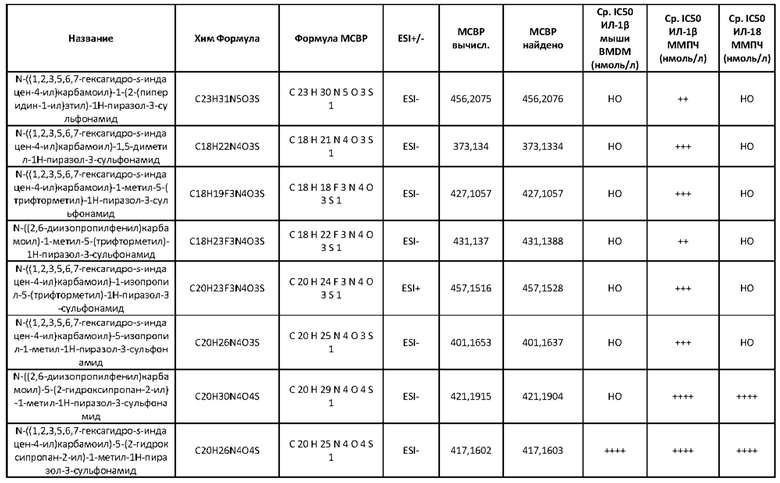
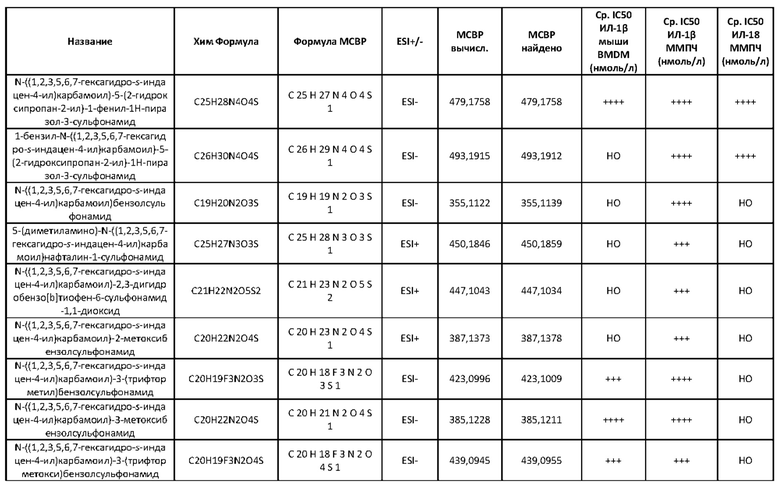
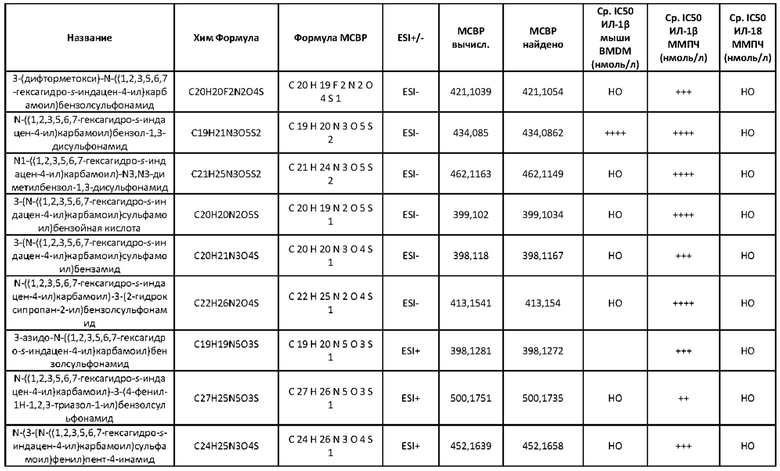
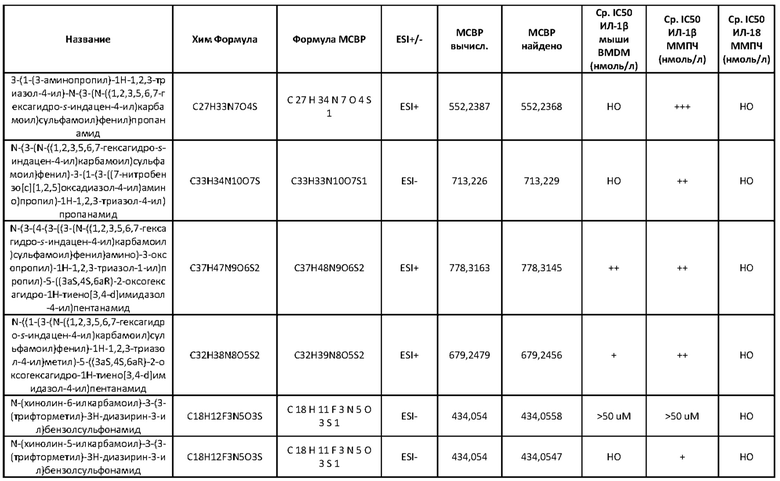
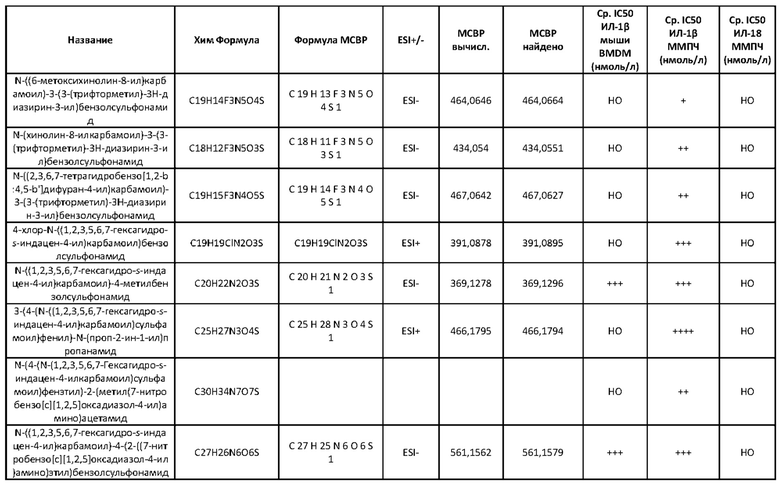
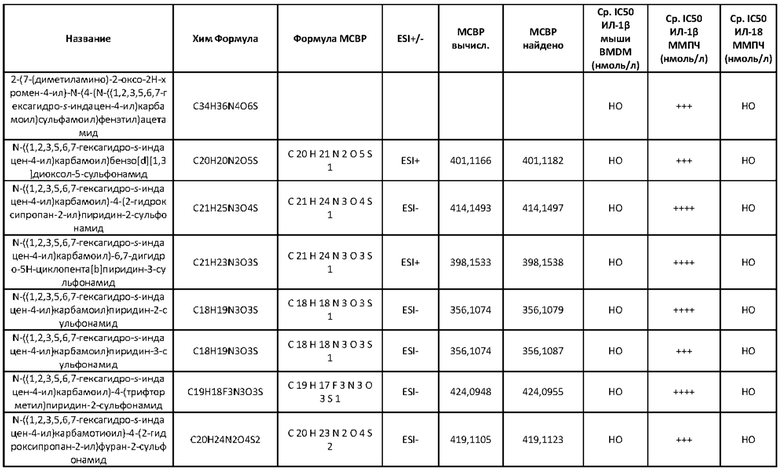
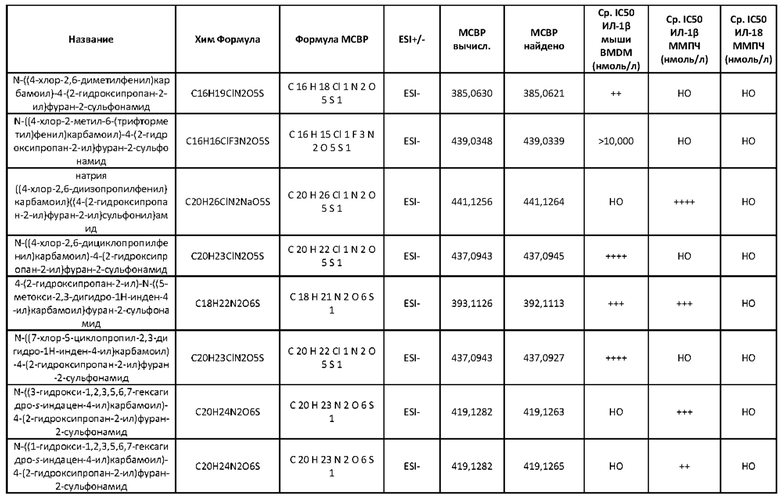
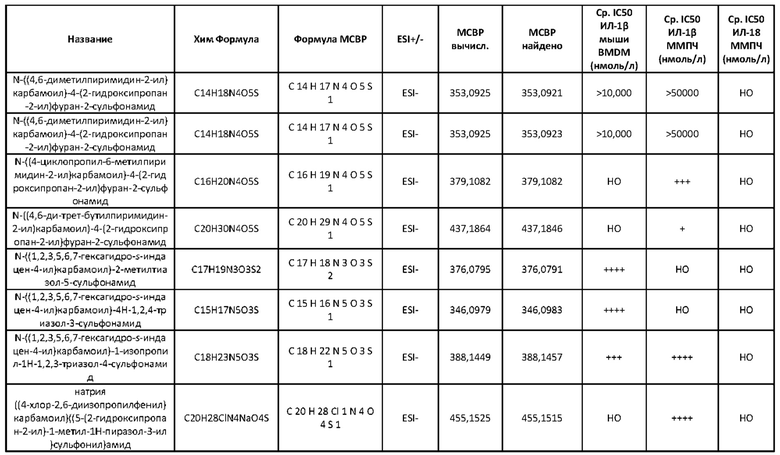
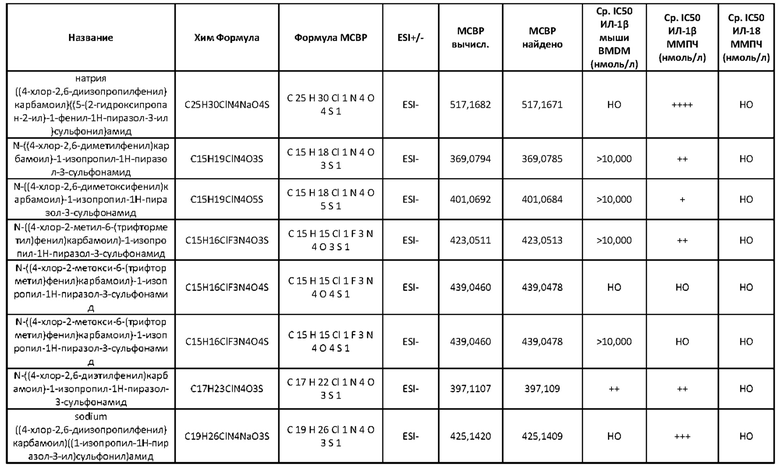
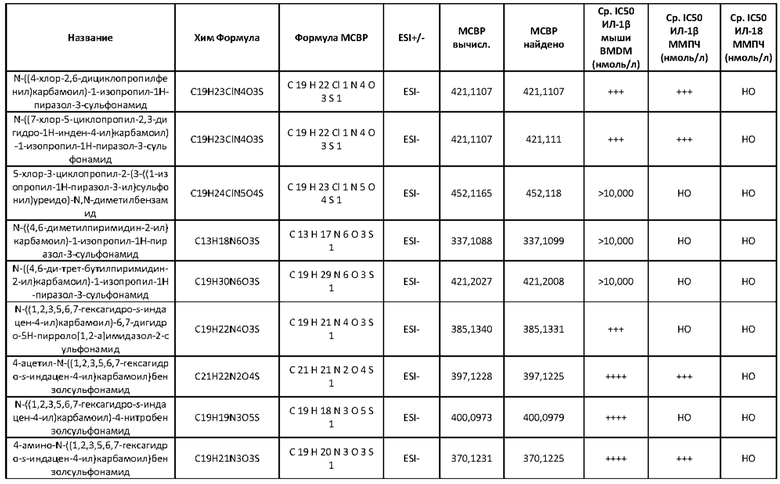

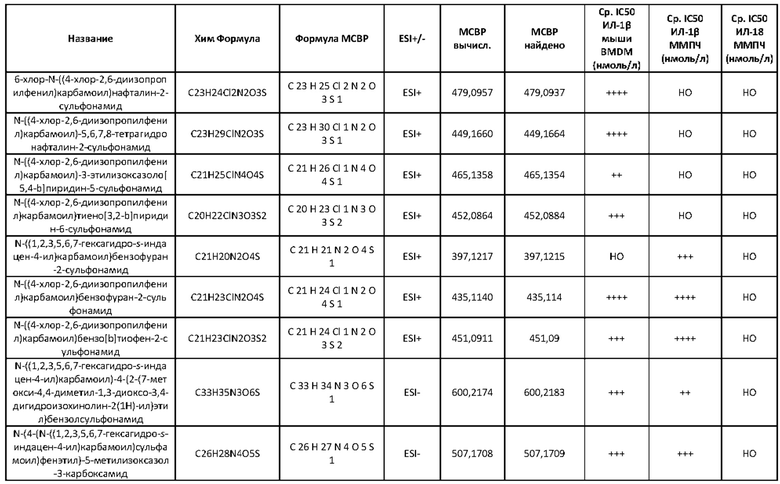

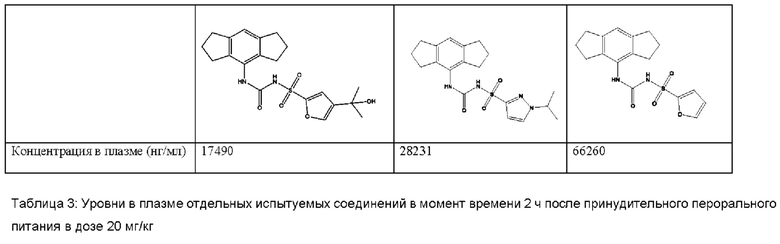
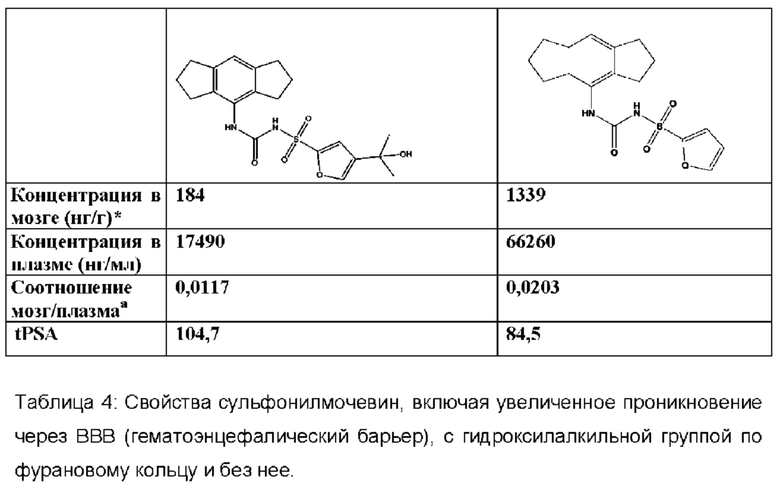
Концентрация в плазме после дозировки
Фармакокинетическое исследование разовой дозы
N-((1,2,3,5,6,7-гексагидро-з-индацен-4-ил)карбамоил)-1-изопропил-1Н-пиразол-3-сульфонамида (МСС7840 и являющееся соединением из первого аспекта) по сравнению с
N-((1,2,3,5,6,7-гексагидро-5-индацен-4-ил)карбамоил)-4-(2-гидроксипропан-2-ил) фуран-2-сульфонамидом (МСС950) с использованием внутривенной дозы 4 мг/кг и пероральной дозы 20 мг/кг однозначно показало более продолжительное время полувыведения, увеличенную максимальную концентрацию (Cmax) и площадь под кривой (ППК) для производного пиразола по сравнению с фураном. Это является преимуществом, приводящим к сравнительно меньшим дозам или менее частому введению.
Выполненная процедура была такой: использовали самцов мышей C57BL/6 возраста 7-9 недель с 3 животными на каждую группу. Мышам вводили дозу испытуемого соединения посредством одного внутривенного болюса или принудительного перорального питания. Пробы крови отбирали из подчелюстной или подкожной вены для анализа концентраций соединения в плазме с помощью ЖХ-МС/МС в следующие моменты времени: внутривенно (ВВ) (3 мыши): 0,083, 0,25, 0,5, 1, 2, 4, 8 и 24 часа после дозировки, перорально (ПО) (3 мыши): 0,25, 0,5,1, 2, 4, 8 и 24 часа после дозировки. Разработали способ ЖХ-МС/МС для количественного определения испытуемого соединения в соответствующей биологической матрице. Фармакокинетические (ФК) параметры рассчитали с помощью Phoenix WinNonlin 6.3. Результаты представлены графически на фиг. от 1А до 1С (МСС950) и фиг. от 2А до 2С (МСС7840).
Соответствующие структуры соединений приведены ниже, а таблицы 5-8 содержат соответствующие данные:
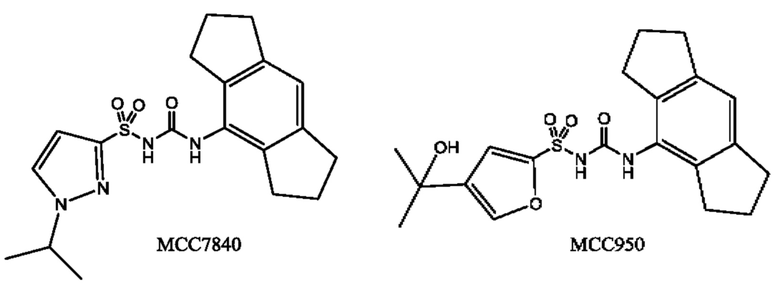
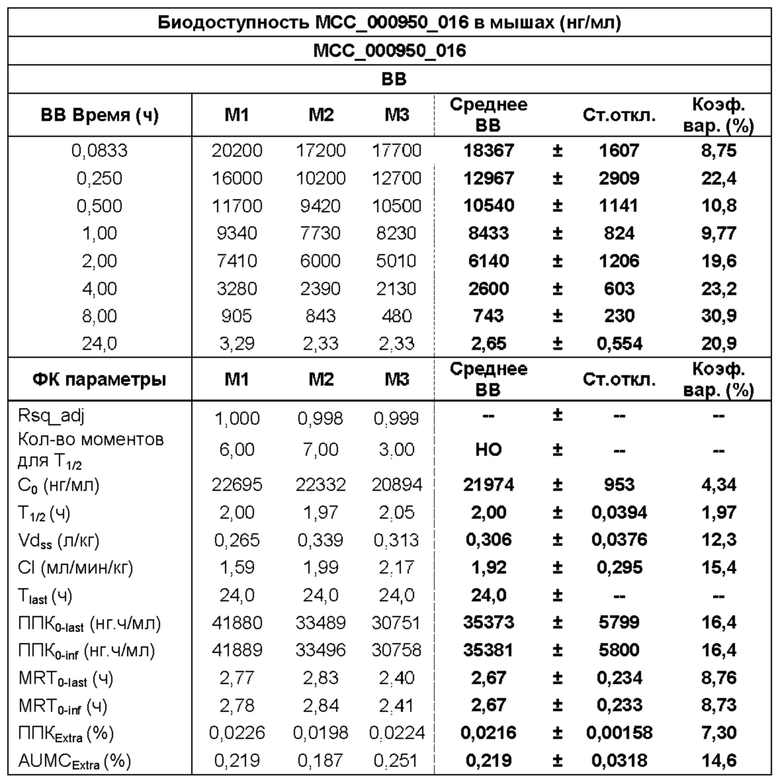
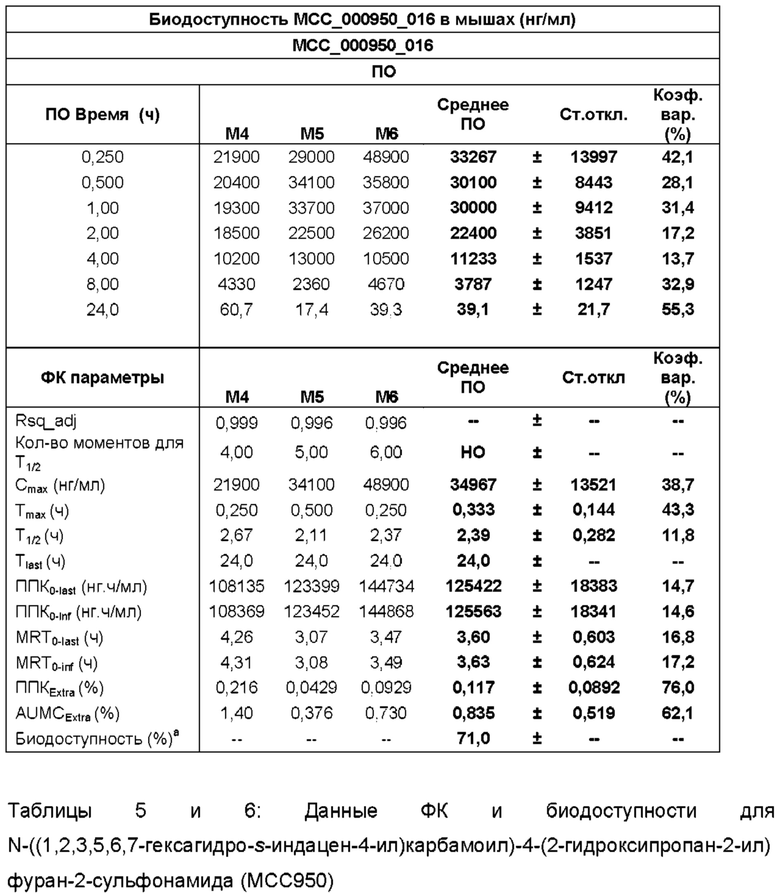
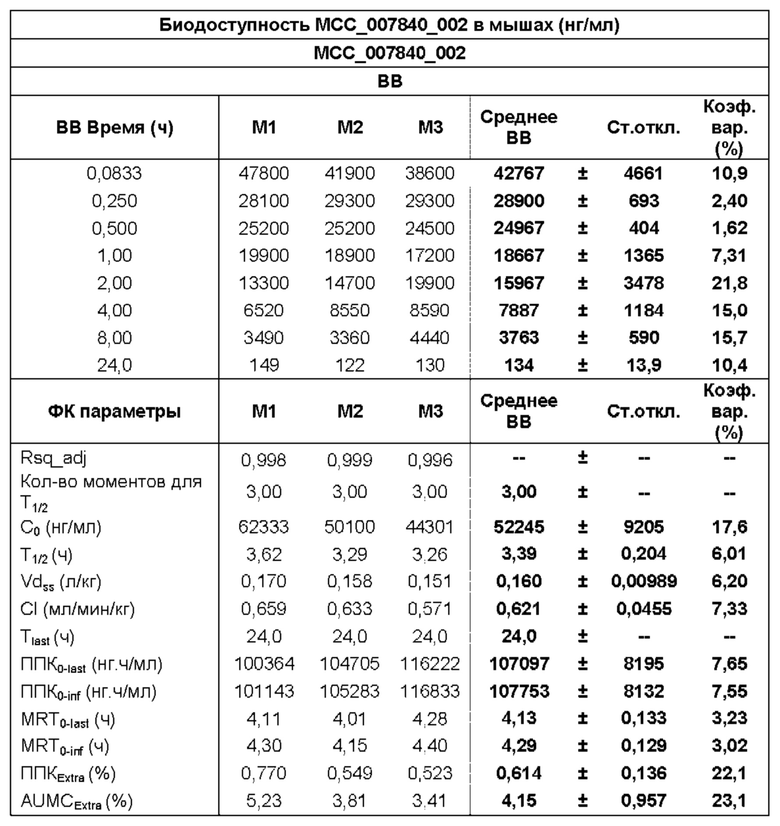
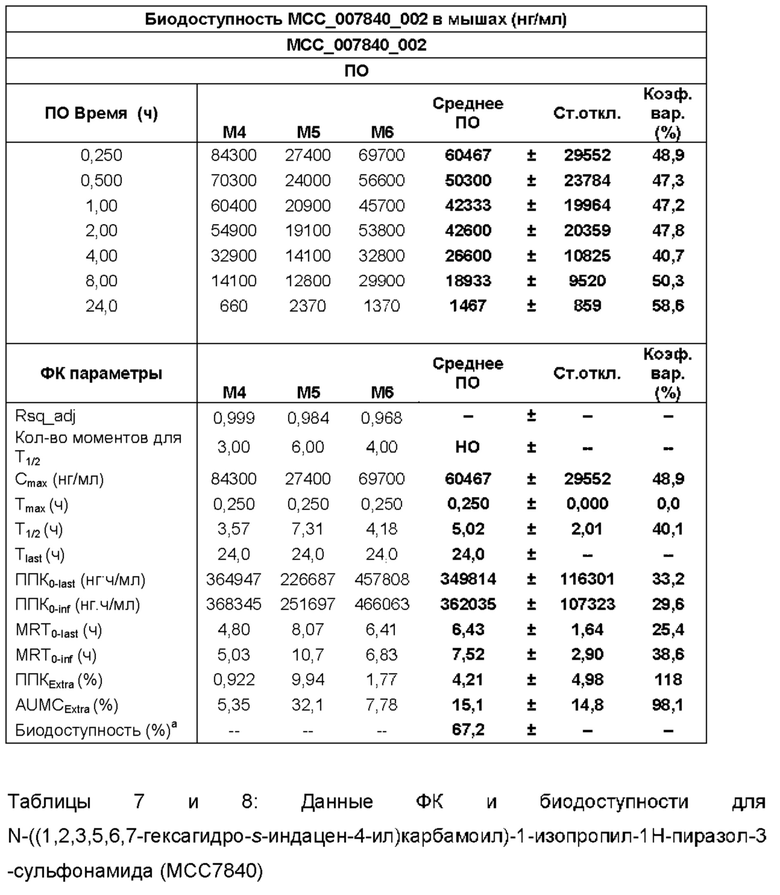

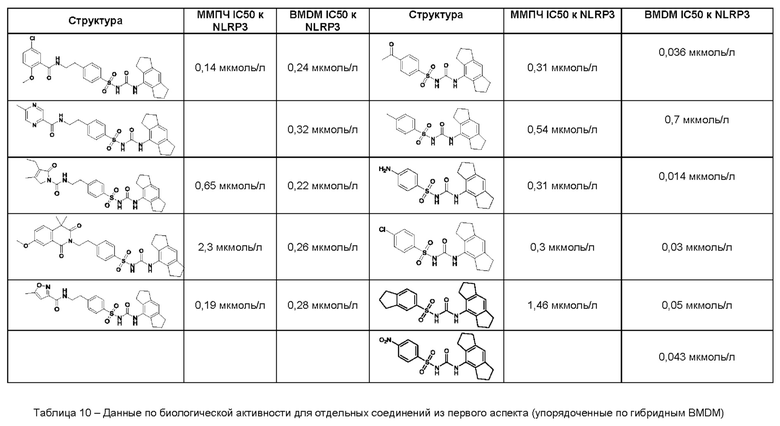
| название | год | авторы | номер документа |
|---|---|---|---|
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВНОСТИ ИНТЕРЛЕЙКИНА-1 | 2018 |
|
RU2792143C2 |
| СОЕДИНЕНИЯ СУЛЬФОНИМИДАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ АКТИВНОСТИ ИНТЕРЛЕЙКИНА-1 | 2019 |
|
RU2820289C2 |
| НОВЫЕ СУЛЬФОНАМИДКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2808572C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ТЕРАПИИ ПРОТИВ ВИЧ | 2019 |
|
RU2806030C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
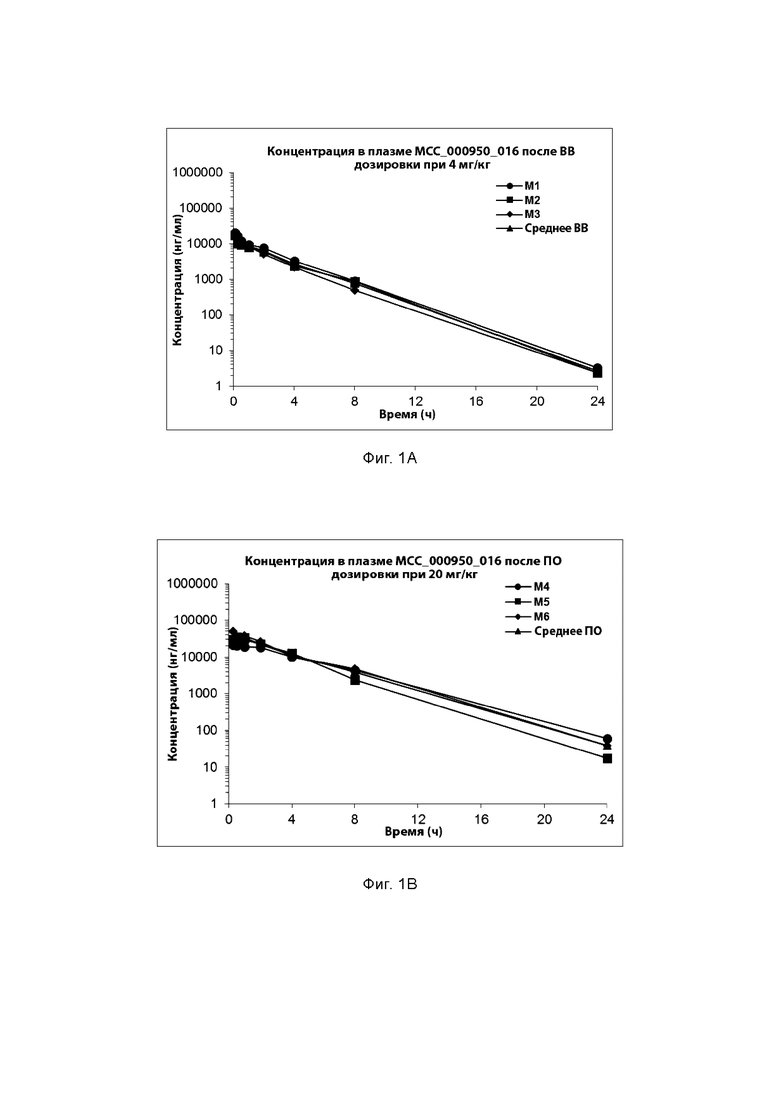
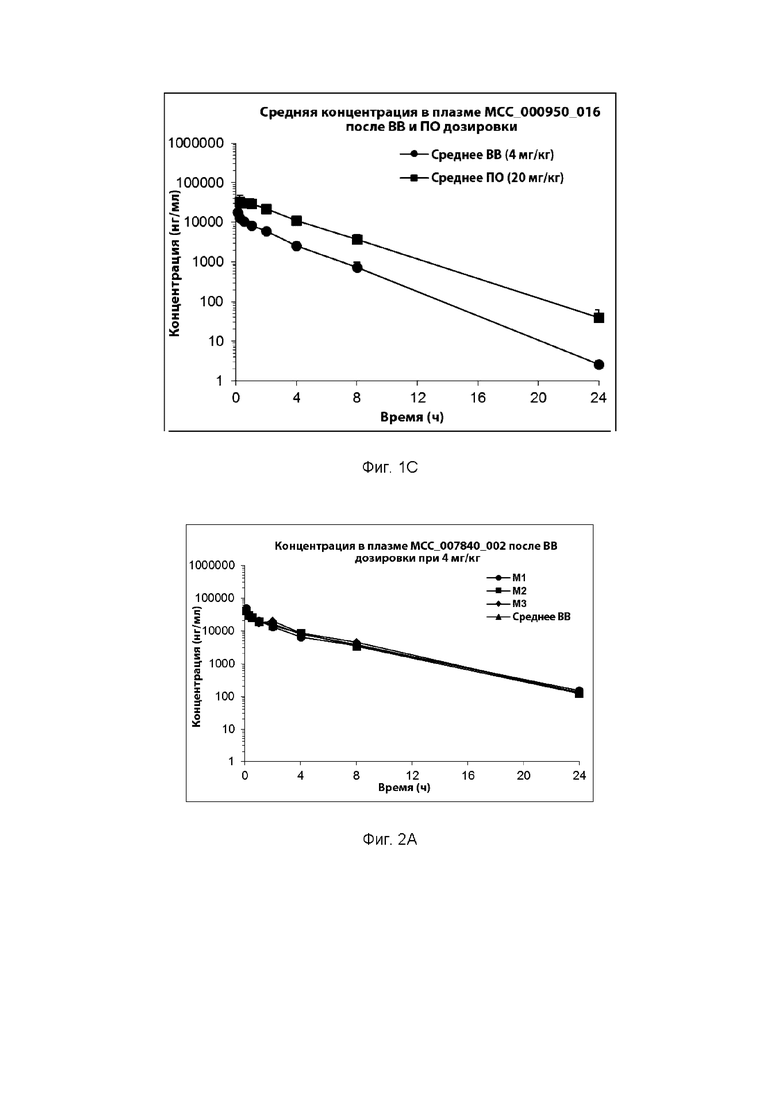
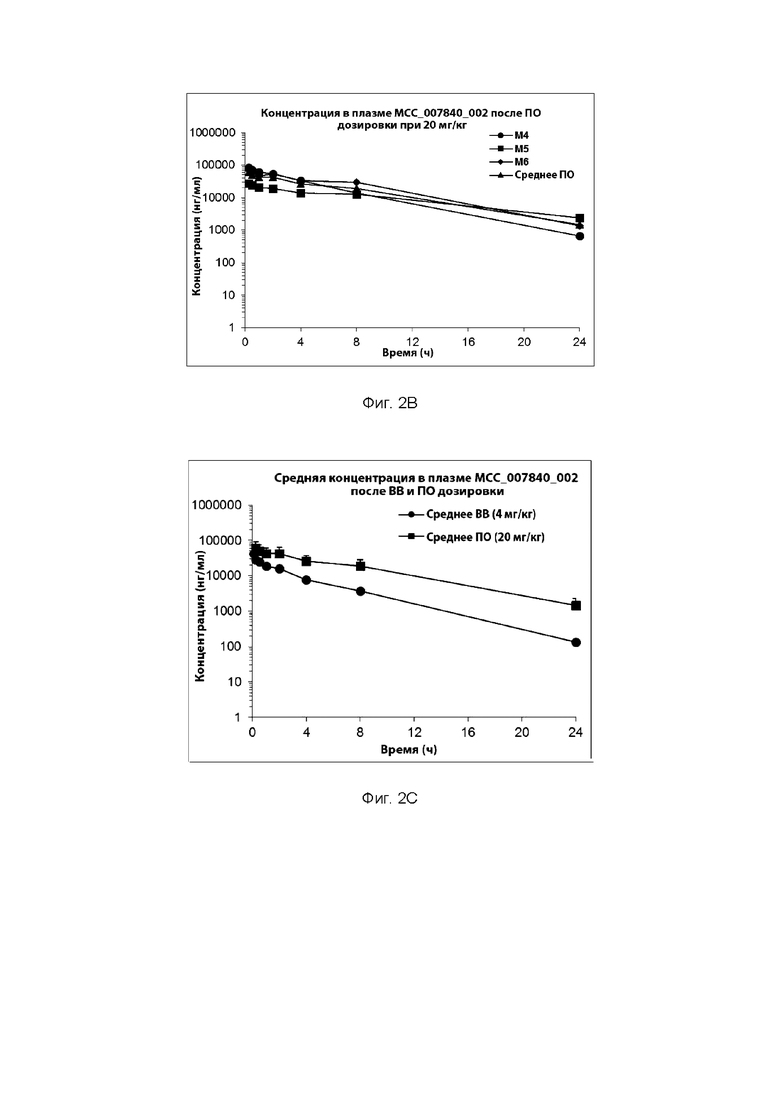
Группа изобретений относится к области фармацевтической химии и включает соединение формулы (I) или его фармацевтически приемлемую соль, фармацевтическую композицию, способ лечения, способ ингибирования активности биологической мишени и применение соединения формулы (I). В формуле (I): W выбран из О, S и Se; J выбран из S и Se; R1 выбран из группы, состоящей из 5-членного и 6-членного гетероциклила, каждый из которых содержит от 1 до 4 гетероатомов, выбранных из N, О и S, и каждый из которых может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; С1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; C6-C12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; С1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила; R2 выбран из 2,6-диалкилфенила, 2,6-диалкил-4-галофенила, 2,6-дициклоалкилфенила, 2,6-дициклоалкил-4-галофенила и 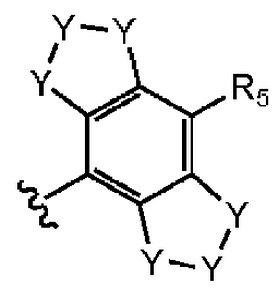 , где каждый случай Y независимо выбран из С, N, S и О, который может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; С1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; С1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила; R5 выбран из группы, состоящей из водорода, гало, циано, амида, сульфонамида, ацила, гидроксила, C1-C6 алкила, C1-C6 галоалкила, С3-С5 циклоалкила и С1-С6 алкокси, все из которых могут быть необязательно замещены гало, циано или C1-C6 алкокси; и как R1 непосредственно связан с J, так и R2 непосредственно связан с примыкающим атомом азота, через атом углерода; при условии, что, когда W представляет собой О, J представляет собой S и R1 представляет собой 5-членный гетероциклил, тогда указанный 5-членный гетероциклил не содержит никаких атомов азота в кольце. Технический результат: соединение формулы (I), обладающее свойствами ингибитора активации инфламмасомы NLRP3, используемое для лечения заболеваний, которые реагируют на ингибирование активации инфламмасомы NLRP3. 6 н. и 23 з.п. ф-лы, 6 ил., 10 табл.
, где каждый случай Y независимо выбран из С, N, S и О, который может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; С1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; С1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила; R5 выбран из группы, состоящей из водорода, гало, циано, амида, сульфонамида, ацила, гидроксила, C1-C6 алкила, C1-C6 галоалкила, С3-С5 циклоалкила и С1-С6 алкокси, все из которых могут быть необязательно замещены гало, циано или C1-C6 алкокси; и как R1 непосредственно связан с J, так и R2 непосредственно связан с примыкающим атомом азота, через атом углерода; при условии, что, когда W представляет собой О, J представляет собой S и R1 представляет собой 5-членный гетероциклил, тогда указанный 5-членный гетероциклил не содержит никаких атомов азота в кольце. Технический результат: соединение формулы (I), обладающее свойствами ингибитора активации инфламмасомы NLRP3, используемое для лечения заболеваний, которые реагируют на ингибирование активации инфламмасомы NLRP3. 6 н. и 23 з.п. ф-лы, 6 ил., 10 табл.
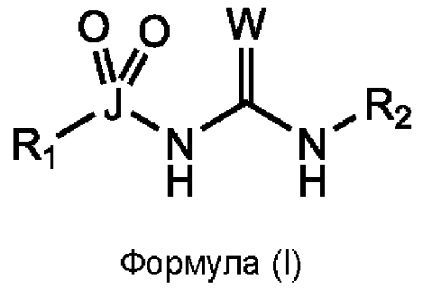
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
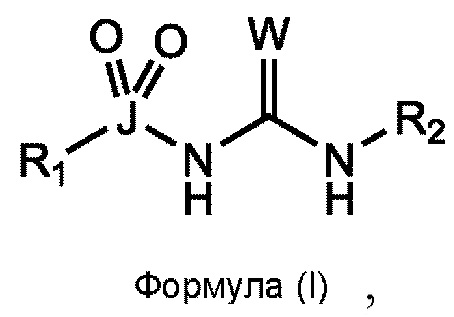
где W выбран из О, S и Se;
J выбран из S и Se;
R1 выбран из группы, состоящей из 5-членного и 6-членного гетероциклила, каждый из которых содержит от 1 до 4 гетероатомов, выбранных из N, О и S, и каждый из которых может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; С1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; C6-C12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; С1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила;
R2 выбран из 2,6-диалкилфенила, 2,6-диалкил-4-галофенила, 2,6-дициклоалкилфенила, 2,6-дициклоалкил-4-галофенила, и
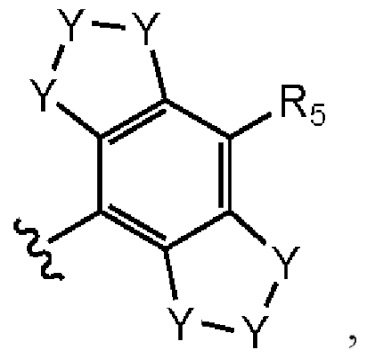
где каждый случай Y независимо выбран из С, N, S и О, который может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; С1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; С1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила;
R5 выбран из группы, состоящей из водорода, гало, циано, амида, сульфонамида, ацила, гидроксила, C1-C6 алкила, C1-C6 галоалкила, С3-С5 циклоалкила и С1-С6 алкокси, все из которых могут быть необязательно замещены гало, циано или C1-C6 алкокси; и
как R1 непосредственно связан с J, так и R2 непосредственно связан с примыкающим атомом азота, через атом углерода;
при условии, что, когда W представляет собой О, J представляет собой S и R1 представляет собой 5-членный гетероциклил, тогда указанный 5-членный гетероциклил не содержит никаких атомов азота в кольце.
2. Соединение по п. 1, в котором R1 представляет собой 6-членный гетероциклил, который содержит от 1 до 4 гетероатомов, выбранных из N, О и S, и который может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; C1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; C6-C12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; C1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или C1-10 алкила.
3. Соединение по любому из предшествующих пунктов, в котором R1 представляет собой полностью насыщенный гетероциклил, который содержит от 1 до 4 гетероатомов, выбранных из N, О и S, и который может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; C1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного C1-9 алкила; амино; C1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; С1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила.
4. Соединение по любому из предшествующих пунктов, в котором R1 представляет собой азотный гетероциклил, который содержит от 1 до 4 гетероатомов, выбранных из N, О и S, и который может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; C1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного C1-9 алкила; амино; C1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; С1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила.
5. Соединение по любому из предшествующих пунктов, в котором R1 выбран из группы, состоящей из тетрагидрофурана, тетрагидропирана, пирана, пирролидина, морфолина, пиперазина и пиперидина, все из которых могут быть необязательно замещенными по необходимости, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; C1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; С1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила.
6. Соединение по любому из предшествующих пунктов, в котором R1 выбран из группы, состоящей из тетрагидрофурана, тетрагидропирана, пирролидина, морфолина, пиперазина и пиперидина, все из которых могут быть необязательно замещенными, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; C1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; C1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила.
7. Соединение по любому из предшествующих пунктов, в котором R1 выбран из группы, состоящей из пирролидина, пиперазина и пиперидина, все из которых могут быть необязательно замещенными, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; C1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного С1-9 алкила; амино; С1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; C1-3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила.
8. Соединение по любому из предшествующих пунктов, в котором R1 представляет собой пиперидин, который может быть необязательно замещенным, где любой необязательный заместитель независимо выбран из группы, состоящей из С1-10 алкила; С3-6 циклоалкила; C1-9 гидроксиалкила; С1-10 алкокси; С2-10 алкенила; С2-10 алкинила; С6-С12 арила; гетероциклила, содержащего от 5 до 8 атомов в кольце и из этих атомов от 1 до 4 представляют собой гетероатомы, выбранные из N, О и S; гало; гидроксила; галогенированного C1-9 алкила; амино; C1-9 алкиламино; ацила; амидо; CN; NO2; N3; СН2ОН; CONH2; CONR24R25; CO2R24; CH2OR24; NHCOR24; NHCO2R24; d.3 алкилтио; сульфоновой кислоты; тритила; монометокситритила; R24SO; R24SO2; CF3S; CF3SO2; и триалкилсилила; где R24 и R25 каждый независимо выбран из Н или С1-10 алкила.
9. Соединение по любому из пп. 1, 2, 3, 5 или 6, в котором R1 выбран из группы, состоящей из
и в комбинации с каждой такой группой R1, R2 независимо выбран из группы, состоящей из:
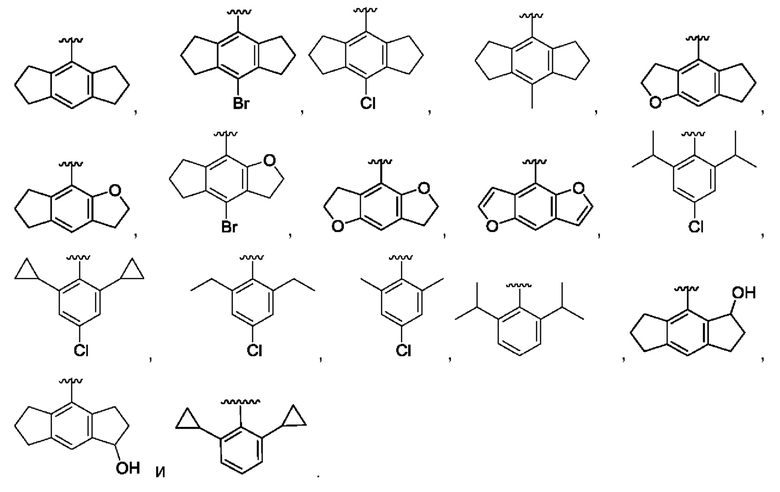
10. Соединение по любому из предшествующих пунктов, в котором R2 выбран из
где Y и R5 имеют значения, указанные в любом из предшествующих пунктов.
11. Соединение по любому из предшествующих пунктов, в котором R2 выбран из
где каждый случай Y представляет собой углерод и R5 представляет собой водород или гало.
12. Соединение по любому из предшествующих пунктов, в котором R2 выбран из замещенного или гидрированного индацена, 2,6-диалкилфенила, 2,6-диалкил-4-галофенила, 2,6-дициклоалкилфенила и 2,6-дициклоалкил-4-галофенила.
13. Соединение по любому из предшествующих пунктов, в котором R2 выбран из гексагидроиндацена, 2,6-диизопропилфенила, 2,6-диизопропил-4-хлорфенила, 2,6-дициклопропилфенила и 2,6-дициклопропил-4-хлорфенила.
14. Соединение по любому из предшествующих пунктов, в котором J представляет собой атом серы.
15. Соединение по любому из предшествующих пунктов, в котором W представляет собой атом кислорода.
16. Соединение по любому из предшествующих пунктов, которое представляет собой соединение формулы (Ia), (Ib) или (Ic) или его фармацевтически приемлемую соль:
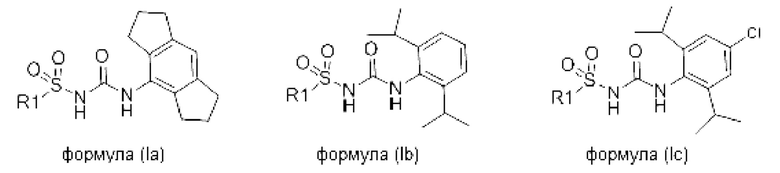
где R1 соответствует определенному в любом из предшествующих пунктов.
17. Соединение по любому из пп. 1-3, 5, 6 или 9-16, в котором соединение выбрано из группы, состоящей из:
и их фармацевтически приемлемых солей.
18. Соединение или фармацевтически приемлемая соль по любому из предшествующих пунктов, в котором соединение, или его фармацевтически приемлемая соль, представляет собой ингибитор инфламмасомы NLRP3.
19. Фармацевтическая композиция, обладающая активностью ингибирования активации инфламмасомы NLRP3 и содержащая эффективное количество соединения по любому из пп. 1-18, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель и/или вспомогательное вещество.
20. Способ лечения или профилактики заболевания, нарушения или состояния, включающий стадию введения эффективного количества соединения по любому из пп. 1-18 или его фармацевтически приемлемой соли или фармацевтической композиции по п. 19 для лечения или профилактики этим заболевания, нарушения или состояния, где заболевание, нарушение или состояние является таким, которое реагирует на ингибирование активации инфламмасомы NLRP3.
21. Способ по п. 20, где заболевание, нарушение или состояние реагирует на модуляцию одного или более из ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17.
22. Способ по п. 20 или 21, где заболевание, нарушение или состояние выбрано из группы, состоящей из конститутивного воспаления, включая криопирин-ассоциированные периодические синдромы (КАПС (CAPS)): синдром Макла-Уэльса (СМУ (MWS)), семейный холодовой аутовоспалительный синдром (СХАС (FCAS)) и мультисистемное воспалительное заболевание неонатального возраста (МВЗНВ (NOMID)); включая аутовоспалительные заболевания: периодическую болезнь (ПБ (FMF)), периодический синдром, связанный с рецепторами TNF (ПССРТ (TRAPS)), дефицит мевалонаткиназы (ДМК (MKD)), гипериммуноглобулинемию D и синдром периодической лихорадки (ГИДС (HIDS)), дефицит антагониста рецептора интерлейкина 1 (ДАРИ (DIRA)), синдром Маджида, гнойный артрит, гангренозную пиодермию и акне (ГАПА (PAPA)), гаплонедостаточность А20 (ГА20 (НА20)), педиатрический гранулематозный артрит (ПГА (PGA)), связанный с PLCG2 дефицит антител и иммунную дисрегуляцию (СПЛДАИД(PLAID)), связанное с PLCG2 аутовоспалительное заболевание, дефицит антител и иммунную дисрегуляцию (СПЛАДАИД (APLAID)), сидеробластическую анемию с В-клеточным иммунодефицитом, периодические лихорадки и задержку в развитии (СИЛЗ (SIFD)); синдром Свита, хронический небактериальный остеомиелит (ХНО (CNO)), хронический рецидивирующий мультифокальный остеомиелит (ХРМО (CRMO)) и синовит, акне, пустулез, гиперостоз, синдром остита (САПГО (SAPHO)); аутоиммунные заболевания, включая рассеянный склероз (PC(MS)), диабет типа 1, псориаз, ревматоидный артрит, болезнь Бехчета, синдром Шегрена и синдром Шницлера; респираторные заболевания, включая хроническую обструктивную болезнь легких (ХОБЛ (COPD)), стероидорезистентную астму, асбестоз, силикоз и кистозный фиброз; заболевания центральной нервной системы, включая болезнь Паркинсона, болезнь Альцгеймера, болезнь моторных нейронов, болезнь Хантингтона, церебральную малярию и повреждение мозга от пневмококкового менингита; метаболические заболевания, включая диабет типа 2, атеросклероз, ожирение, подагру, псевдоподагру; болезни глаз, в том числе глазного эпителия, возрастную макулярную дегенерацию (ВМД (AMD)), инфекцию роговицы, увеит и сухой глаз; заболевания почек, включая хроническую болезнь почек, оксалатную нефропатию и диабетическую нефропатию; заболевания печени, включая безалкогольный стеатогепатит и алкогольную болезнь печени; воспалительные реакции на коже, включая контактную гиперчувствительность и солнечные ожоги; воспалительные реакции в суставах, включая остеоартрит, системный ювенильный идиопатический артрит, приобретенную болезнь Стилла, рецидивирующий полихондрит; вирусные инфекции, включая альфа-вирус, включая чикунгунью и вирус реки Росса, и флавивирус, включая вирусы Денге и Зика, грипп, ВИЧ; гнойный гидраденит (ГГ (HS)) и другие вызывающие кисту кожные заболевания; раки, включая метастазы рака легкого, рак поджелудочной железы, рак желудка, миелодиспластический синдром, лейкемию; полимиозит; апоплексию; инфаркт миокарда; реакцию трансплантата против хозяина; повышенное давление; колит; гельминтовую инфекцию; бактериальную инфекцию; аневризму брюшной аорты; заживление ран; депрессию, психологический стресс; перикардит, включая синдром Дресслера, ишемически-реперфузионное повреждение и любое заболевание, при котором определено, что индивидуум несет генеративную или соматическую мутацию в NLRP3, не являющуюся молчащей.
23. Способ по любому из пп. 20-22, где лечение или профилактику заболевания, нарушения или состояния осуществляют на млекопитающем.
24. Способ по п. 23, где млекопитающее представляет собой субъект человека.
25. Способ ингибирования активности биологической мишени, включающий стадию воздействия на биологическую мишень соединением по любому из пп. 1-18 или его фармацевтически приемлемой солью, где биологическая мишень выбрана из группы, состоящей из инфламмасомы NLRP3, ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17.
26. Соединение или фармацевтически приемлемая соль по любому из пп. 1-18, где соединение или его фармацевтически приемлемая соль применяется в:
(a) лечении или профилактике заболевания, нарушения или состояния, реагирующего на ингибирование активации инфламмасомы NLRP3; и/или
(b) лечении или профилактике заболевания, нарушения или состояния, реагирующего на модуляцию одного или более из ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17; и/или
(c) лечении или профилактике заболевания, нарушения или состояния, выбранного из группы, состоящей из конститутивного воспаления, включая криопирин-ассоциированные периодические синдромы (КАПС (CAPS)): синдром Макла-Уэльса (СМУ (MWS)), семейный холодовой аутовоспалительный синдром (СХАС (FCAS)) и мультисистемное воспалительное заболевание неонатального возраста (МВЗНВ (NOMID)); включая аутовоспалительные заболевания: периодическую болезнь (ПБ (FMF)), периодический синдром, связанный с рецепторами TNF (ПССРТ (TRAPS)), дефицит мевалонаткиназы (ДМК (MKD)), гипериммуноглобулинемию D и синдром периодической лихорадки (ГИДС (HIDS)), дефицит антагониста рецептора интерлейкина 1 (ДАРИ (DIRA)), синдром Маджида, гнойный артрит, гангренозную пиодермию и акне (ГАПА (PAPA)), гаплонедостаточность А20 (ГА20 (НА20)), педиатрический гранулематозный артрит (ПГА (PGA)), связанный с PLCG2 дефицит антител и иммунную дисрегуляцию (СПЛДАИД(PLAID)), связанное с PLCG2 аутовоспалительное заболевание, дефицит антител и иммунную дисрегуляцию (СПЛАДАИД (APLAID)), сидеробластическую анемию с В-клеточным иммунодефицитом, периодические лихорадки и задержку в развитии (СИЛЗ (SIFD)); синдром Свита, хронический небактериальный остеомиелит (ХНО (CNO)), хронический рецидивирующий мультифокальный остеомиелит (ХРМО (CRMO)) и синовит, акне, пустулез, гиперостоз, синдром остита (САПГО (SAPHO)); аутоиммунные заболевания, включая рассеянный склероз (PC(MS)), диабет типа 1, псориаз, ревматоидный артрит, болезнь Бехчета, синдром Шегрена и синдром Шницлера; респираторные заболевания, включая хроническую обструктивную болезнь легких (ХОБЛ (COPD)), стероидорезистентную астму, асбестоз, силикоз и кистозный фиброз; заболевания центральной нервной системы, включая болезнь Паркинсона, болезнь Альцгеймера, болезнь моторных нейронов, болезнь Хантингтона, церебральную малярию и повреждение мозга от пневмококкового менингита; метаболические заболевания, включая диабет типа 2, атеросклероз, ожирение, подагру, псевдоподагру; болезни глаз, в том числе глазного эпителия, возрастную макулярную дегенерацию (ВМД (AMD)), инфекцию роговицы, увеит и сухой глаз; заболевания почек, включая хроническую болезнь почек, оксалатную нефропатию и диабетическую нефропатию; заболевания печени, включая безалкогольный стеатогепатит и алкогольную болезнь печени; воспалительные реакции на коже, включая контактную гиперчувствительность и солнечные ожоги; воспалительные реакции в суставах, включая остеоартрит, системный ювенильный идиопатический артрит, приобретенную болезнь Стилла, рецидивирующий полихондрит; вирусные инфекции, включая альфа-вирус, включая чикунгунью и вирус реки Росса, и флавивирус, включая вирусы Денге и Зика, грипп, ВИЧ; гнойный гидраденит (ГГ (HS)) и другие вызывающие кисту кожные заболевания; раки, включая метастазы рака легкого, рак поджелудочной железы, рак желудка, миелодиспластический синдром, лейкемию; полимиозит; апоплексию; инфаркт миокарда; реакцию трансплантата против хозяина; повышенное давление; колит; гельминтовую инфекцию; бактериальную инфекцию; аневризму брюшной аорты; заживление ран; депрессию, психологический стресс; перикардит, включая синдром Дресслера, ишемически-реперфузионное повреждение и любое заболевание, при котором определено, что индивидуум несет генеративную или соматическую мутацию в NLRP3, не являющуюся молчащей.
27. Соединение или фармацевтически приемлемая соль по любому из пп. 1-18, где соединение или его фармацевтически приемлемая соль применяется в ингибировании активности биологической мишени, выбранной из группы, состоящей из инфламмасомы NLRP3, ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17.
28. Применение соединения или фармацевтически приемлемой соли по любому из пп. 1 -18 для производства лекарственного средства для:
(a) лечения или профилактики заболевания, нарушения или состояния, реагирующего на ингибирование активации инфламмасомы NLRP3; и/или
(b) лечения или профилактики заболевания, нарушения или состояния, реагирующего на модуляцию одного или более из ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17; и/или
(c) лечения или профилактики заболевания, нарушения или состояния, выбранного из группы, состоящей из конститутивного воспаления, включая криопирин-ассоциированные периодические синдромы (КАПС (CAPS)): синдром Макла-Уэльса (СМУ (MWS)), семейный холодовой аутовоспалительный синдром (СХАС (FCAS)) и мультисистемное воспалительное заболевание неонатального возраста (МВЗНВ (NOMID)); включая аутовоспалительные заболевания: периодическую болезнь (ПБ (FMF)), периодический синдром, связанный с рецепторами TNF (ПССРТ (TRAPS)), дефицит мевалонаткиназы (ДМК (MKD)), гипериммуноглобулинемию D и синдром периодической лихорадки (ГИДС (HIDS)), дефицит антагониста рецептора интерлейкина 1 (ДАРИ (DIRA)), синдром Маджида, гнойный артрит, гангренозную пиодермию и акне (ГАПА (PAPA)), гаплонедостаточность А20 (ГА20 (НА20)), педиатрический гранулематозный артрит (ПГА (PGA)), связанный с PLCG2 дефицит антител и иммунную дисрегуляцию (СПЛДАИД(PLAID)), связанное с PLCG2 аутовоспалительное заболевание, дефицит антител и иммунную дисрегуляцию (СПЛАДАИД (APLAID)), сидеробластическую анемию с В-клеточным иммунодефицитом, периодические лихорадки и задержку в развитии (СИЛЗ (SIFD)); синдром Свита, хронический небактериальный остеомиелит (ХНО (CNO)), хронический рецидивирующий мультифокальный остеомиелит (ХРМО (CRMO)) и синовит, акне, пустулез, гиперостоз, синдром остита (САПГО (SAPHO)); аутоиммунные заболевания, включая рассеянный склероз (PC(MS)), диабет типа 1, псориаз, ревматоидный артрит, болезнь Бехчета, синдром Шегрена и синдром Шницлера; респираторные заболевания, включая хроническую обструктивную болезнь легких (ХОБЛ (COPD)), стероидорезистентную астму, асбестоз, силикоз и кистозный фиброз; заболевания центральной нервной системы, включая болезнь Паркинсона, болезнь Альцгеймера, болезнь моторных нейронов, болезнь Хантингтона, церебральную малярию и повреждение мозга от пневмококкового менингита; метаболические заболевания, включая диабет типа 2, атеросклероз, ожирение, подагру, псевдоподагру; болезни глаз, в том числе глазного эпителия, возрастную макулярную дегенерацию (ВМД (AMD)), инфекцию роговицы, увеит и сухой глаз; заболевания почек, включая хроническую болезнь почек, оксалатную нефропатию и диабетическую нефропатию; заболевания печени, включая безалкогольный стеатогепатит и алкогольную болезнь печени; воспалительные реакции на коже, включая контактную гиперчувствительность и солнечные ожоги; воспалительные реакции в суставах, включая остеоартрит, системный ювенильный идиопатический артрит, приобретенную болезнь Стилла, рецидивирующий полихондрит; вирусные инфекции, включая альфа-вирус, включая чикунгунью и вирус реки Росса, и флавивирус, включая вирусы Денге и Зика, грипп, ВИЧ; гнойный гидраденит (ГГ (HS)) и другие вызывающие кисту кожные заболевания; раки, включая метастазы рака легкого, рак поджелудочной железы, рак желудка, миелодиспластический синдром, лейкемию; полимиозит; апоплексию; инфаркт миокарда; реакцию трансплантата против хозяина; повышенное давление; колит; гельминтовую инфекцию; бактериальную инфекцию; аневризму брюшной аорты; заживление ран; депрессию, психологический стресс; перикардит, включая синдром Дресслера, ишемически-реперфузионное повреждение и любое заболевание, при котором определено, что индивидуум несет генеративную или соматическую мутацию в NLRP3, не являющуюся молчащей.
29. Применение соединения или фармацевтически приемлемой соли по любому из пп. 1-18 для производства фармацевтической композиции для ингибирования активности биологической мишени, выбранной из группы, состоящей из инфламмасомы NLRP3, ИЛ-1β, ИЛ-17, ИЛ-18, ИЛ-1α, ИЛ-37, ИЛ-33 и клеток Th17.
| WO 2001019390 A1, 22.03.2001 | |||
| WO 1998032733 A1, 30.07.1998 | |||
| 1,3-ЗАМЕЩЕННЫЕ СУЛЬФОНИЛМОЧЕВИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2022963C1 |
| Датчик скорости движения потока | 1978 |
|
SU885890A1 |
| Устройство для вычисления значения модуля вектора | 1984 |
|
SU1236468A1 |
| US 5169860 A1, 08.12.1992 | |||
| US 4018929 A1, 19.04.1977 | |||
| Howbert J | |||
| J | |||
| et al., Novel agents effective against solid tumors: the diarylsulfonylureas | |||
| Synthesis, activities, and analysis of quantitative | |||

