Область техники, к которой относится изобретение
Данное изобретение относится к новым соединениям, которые представляют собой лиганды тиреоидных рецепторов, а также к способам получения указанных соединений и к способам их применения, таким как регулирование метаболизма.
Предшествующий уровень техники
Хотя важная роль тиреоидных гормонов в регулировании метаболизма у людей хорошо известна, открытие и разработка новых специфических лекарственных средств для лучшего лечения гипертиреоза и гипотиреоза осуществляется медленно. Такое положение ограничивает также разработку тиреоидных агонистов и антагонистов для лечения других важных клинических симптомов, таких как гиперхолестеринемия, ожирение и сердечная аритмия.
Тиреоидные гормоны фактически воздействуют на метаболизм каждой клетки организма. При нормальных уровнях содержания данные гормоны поддерживают массу тела, показатели обмена веществ, температуру тела и настроение, а также влияют на уровни содержания в крови сывороточного липопротеина низкой плотности (low density lipoprotein - LDL). Поэтому при гипотиреозе имеет место повышение массы тела, высокие уровни содержания холестерина LDL и депрессия. При гипертиреозе данные гормоны приводят к потере массы тела, гиперметаболизму, снижению уровней содержания сывороточного LDL, сердечной аритмии, сердечной недостаточности, мышечной слабости, остеопорозу у женщин в постклемактерический период и состоянию тревоги.
В настоящее время тиреоидные гормоны используются главным образом в заместительной терапии при лечении пациентов, страдающих гипотиреозом. Терапия с использованием L-тироксина возвращает метаболические функции к нормальному состоянию и может легко контролироваться стандартными измерениями уровней содержания тиреостимулирующего гормона (ТСГ), тироксина (3,4,3',5'-тетрайод-L-тиронина или Т4) и трийодтиронина (3,5,3'-трийод-L-тиронина или Т3). Однако заместительная терапия, особенно для более пожилых пациентов, может ограничиваться некоторыми вредными эффектами тиреоидных гормонов.
Кроме того, некоторые эффекты гормонов щитовидной железы могут быть терапевтически полезными для расстройств, не связанных со щитовидной железой, если снизить до минимума или исключить их побочные эффекты. Такие потенциально полезные эффекты включают снижение массы, снижение уровней содержания сывороточного LDL, облегчение депрессии и стимулирование образования костной массы. Ранее попытки фармацевтического применения тиреоидных гормонов для лечения таких расстройств ограничивались проявлениями гипертиреоза и особенно токсичностью для сердечно-сосудистой системы.
Более того, полезные в качестве лекарственных средств тиреоидные агонисты должны сводить к минимуму возможность нежелательных последствий вследствие локально индуцированного гипотиреоза, то есть суб-нормальных уровней тиреоидной гормональной активности в некоторых тканях и органах. Такое состояние может возникать вследствие того, что повышенные концентрации циркулирующих агонистов тиреоидных гормонов могут вызывать подавление секреции гипофизом тиреостимулирующего гормона (ТСГ), в результате чего снижается синтез тиреостимулирующего гормона щитовидной железой (отрицательная регуляция с обратной связью). Поскольку уровни содержания эндогенных тиреоидных гормонов снижаются, в любом месте, где введенное лекарственное средство, представляющее собой тиреоидный агонист, недостаточно компенсирует снижение содержания эндогенных гормонов в специфических тканях, может возникать локализованный гипотиреоз. Например, если лекарственное средство, представляющее собой тиреоидный агонист, не преодолевает гематоэнцефалический барьер, влияния подавления секреции ТСГ могут приводить к гипотиреозу ЦНС и ассоциированным с ним рискам, таким как депрессия.
Разработка специфических селективных лигандов рецепторов тиреоидных гормонов, особенно агонистов рецепторов тиреоидных гормонов, может привести к получению специфических терапевтических средств для лечения этих общих расстройств, не проявляющих токсичности в отношении сердечно-сосудистой системы и другой токсичности, характерной для нативных гормонов щитовидной железы. Агонисты тиреоидных гормонов, обладающие тканевой селективностью, могут быть получены посредством селективного тканевого поглощения или проникновения, местной или локальной доставкой, направленной на клетки через другие лиганды, присоединенные к агонисту, и направленной на рецепторные подтипы. Тканевая селективность может также достигаться селективной регуляцией в ткани генов, чувствительных к тиреоидным гормонам, специфическим образом.
Соответственно, открытие соединений, которые являются лигандами рецепторов тиреоидных гормонов, в частности селективными агонистами рецепторов тиреоидных гормонов, может продемонстрировать их применимость для лечения или профилактики заболеваний или расстройств, связанных с активностью тиреоидных гормонов, например, (1) в заместительной терапии для пожилых субъектов с гипотиреозом и риском сердечно-сосудистых осложнений; (2) в заместительной терапии пожилых субъектов с субклиническим гипотиреозом и риском сердечно-сосудистых осложнений; (3) при ожирении; (4) при гиперхолестеринемии, обусловленной повышенными уровнями содержания в плазме LDL; (5) при депрессии; и (6) при остеопорозе в сочетании с ингибитором резорбции кости.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
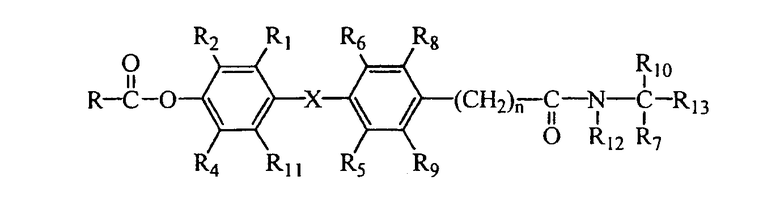
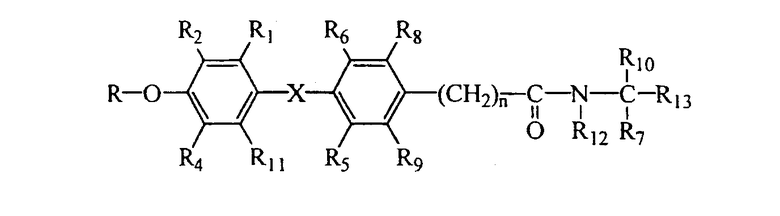
В соответствии с типичными вариантами осуществления изобретения и продемонстрированными признаками данного изобретения предлагаются соединения, которые являются лигандами тироидных рецепторов и представлены общей формулой I:
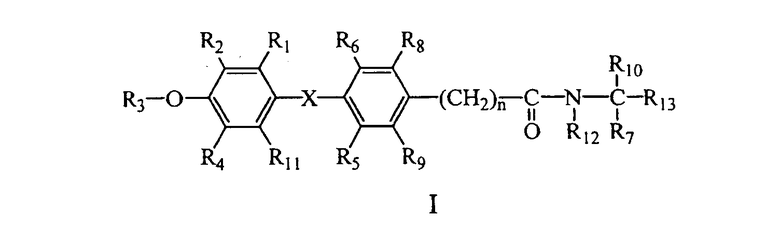
или их фармацевтически приемлемая соль, где:
R1 выбран из группы, включающей водород, галоген и С1 - С6;
R2 выбран из группы, включающей галоген, С1-С6 алкил, С2-С6 алкенил, С2-С6 алкинил, С4-С7 циклоалкенил, С3-С7 циклоалкокси, SO2(NR14R15),N(R16)SO2R17, SR17, SOR17, SO2R17, COR16 и CR18(OR16)R19; или R2 представляет собой водород, когда R4 представляет собой алкил и R1 представляет собой галоген;
R3 выбран из группы, включающей водород, алкил, бензил, ароил и алканоил;
R4 представляет собой галоген, циано или алкил;
R5 и R6 независимо выбраны из водорода, галогена, циано, С1-4 алкила, С3-С6 циклоалкила; где, по меньшей мере, один из R5 и R6 не является водородом;
R7 и R10 независимо выбраны из водорода, галогена, арила и алкила, и R7 и R10 могут соединяться, составляя цепь, содержащую от 2 до 6 метиленовых групп, с образованием 3-7-членного кольца;
R8 и R9 каждый независимо выбран из группы, включающей водород, галоген, алкокси, гидрокси (ОН), циано и алкил;
при условии, что не более чем один из R2, R4, R8 и R9 представляет собой водород;
R11 представляет собой водород, галоген или алкил;
R12 представляет собой водород или алкил;
R13 представляет собой карбоновую кислоту (СООН) или ее сложные эфиры, фосфоновую или фосфиновую кислоту или их сложные эфиры, сульфоновую кислоту, тетразол, гидроксамовую кислоту, тиазолидиндион, ацилсульфонамид или другие производные карбоновых кислот, известные в данной области техники;
R14 и R15 в каждом случае независимо выбраны из группы, включающей водород, алкил, циклоалкил, арил, гетероарил, арилалкил и гетероарилалкил, и R14 и R15 могут соединяться, составляя цепь из 3-6 метиленовых групп, с образованием 4-7-членного кольца;
R16 выбран из группы, включающей водород, алкил, циклоалкил, арил, гетероарил, арилалкил и гетероарилалкил;
R17 выбран из группы, включающей алкил, циклоалкил, арил, гетероарил, арилалкил и гетероарилалкил;
R18 и R19 в каждом случае независимо выбраны из группы, включающей водород, алкил, циклоалкил, арил, гетероарил, арилалкил или гетероарилалкил;
n представляет собой целое число 0, 1 или 2;
Х выбран из -О-, -СН2-, -CF2-, -Se-, -NH-, -S-, -SO-, -SO2- и -СО-.
Определение формулы I, представленной выше, включает все пролекарства, стереоизомеры и фармацевтически приемлемые соли формулы I.
Соединения формулы I представляют собой лиганды рецепторов тиреоидных гормонов и включают соединения, которые являются, например, селективными агонистами, частичными агонистами, антагонистами или частичными антагонистами тиреоидного рецептора. Предпочтительно соединения формулы I обладают активностью в качестве агонистов тиреоидного рецептора и могут использоваться для лечения заболеваний или расстройств, связанных с активностью тиреоидного рецептора. В частности, соединения формулы I могут использоваться для лечения заболеваний или расстройств, которые связаны с нарушением метаболизма или которые зависят от экспрессии Т3 регулируемого гена, таких как ожирение, гиперхолестеринемия, атеросклероз, сердечная аритмия, депрессия, остеопороз, гипотиреоз, зоб, рак щитовидной железы, глаукома, кожные расстройства или заболевания и застойная сердечная недостаточность.
Данное изобретение представляет соединения формулы I, фармацевтические композиции, содержащие данные соединения, и способы применения указанных соединений. В частности, данное изобретение представляет фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы I, одного или в сочетании с фармацевтически приемлемым носителем.
Кроме того, в соответствии с данным изобретением предложен способ профилактики, ингибирования или лечения прогрессирования или начала заболеваний или расстройств, связанных с тиреоидным рецептором, таких как заболевания или расстройства, определенные выше и далее, где терапевтически эффективное количество соединения формулы I вводится млекопитающему, например человеку, нуждающемуся в таком лечении.
Соединения данного изобретения могут использоваться сами по себе, в комбинации с другими соединениями данного изобретения или в комбинации с одним или несколькими другими лекарственными средствами, активными в терапевтических областях, определенных выше.
Кроме того, представлен способ профилактики, ингибирования или лечения заболеваний, определенных выше и далее, где терапевтически эффективное количество смеси соединения формулы I и другого соединения данного изобретения и/или терапевтического средства другого типа вводится млекопитающему, нуждающемуся в таком лечении.
Соединения данного изобретения включают, но без ограничения, следующие соединения:
N-[3,5-дихлор-4-(4-гидрокси-3-изопропил-5-метилфенокси)бензоил]глицин (Е1);
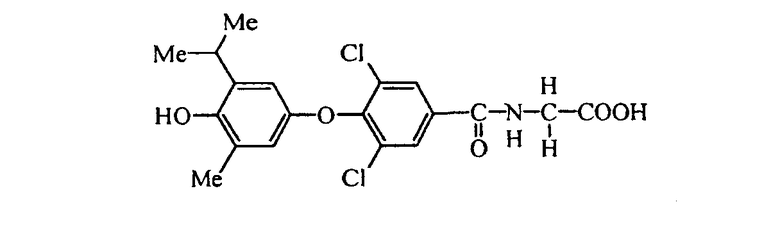
N-[3,5-дихлор-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е2);
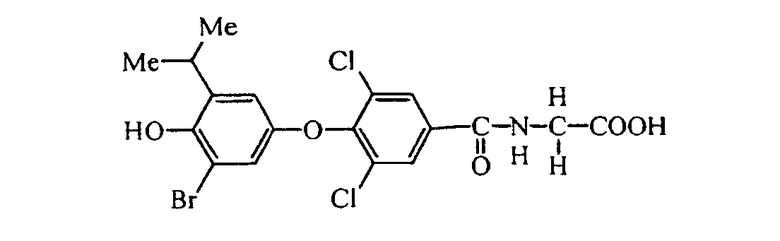
N-[3,5-дихлор-4-(2-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е3);
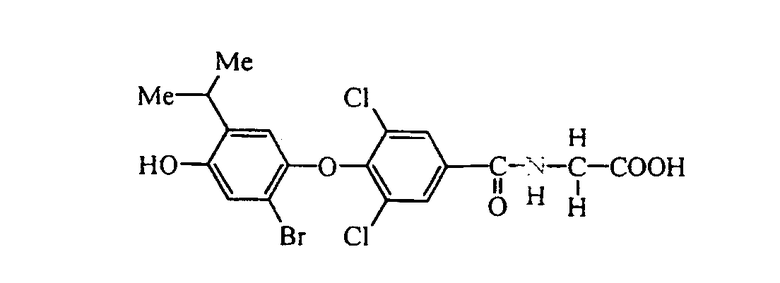
N-[3,5-дихлор-4-(3-хлор-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е4);
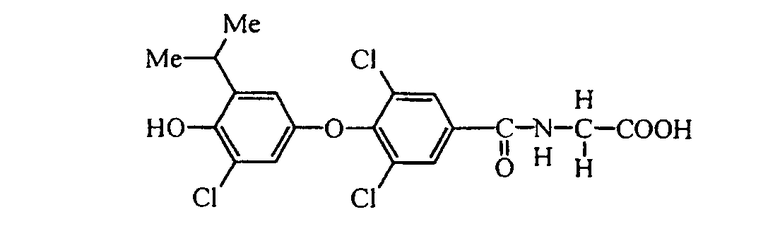
N-[3,5-дихлор-4-(3-циано-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е5);
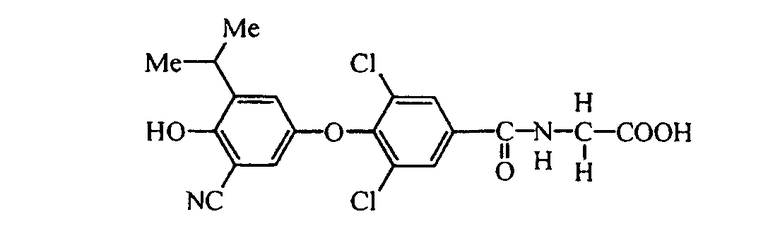
N-[3,5-дихлор-4-(3-фтор-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е6);
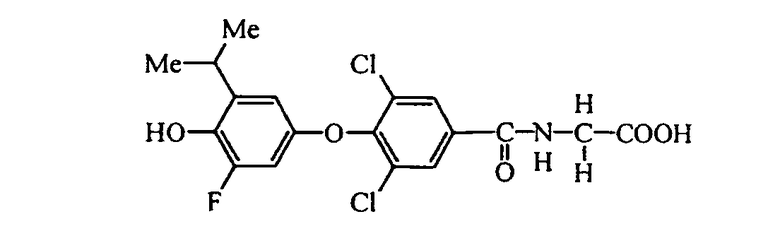
N-[3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е7);
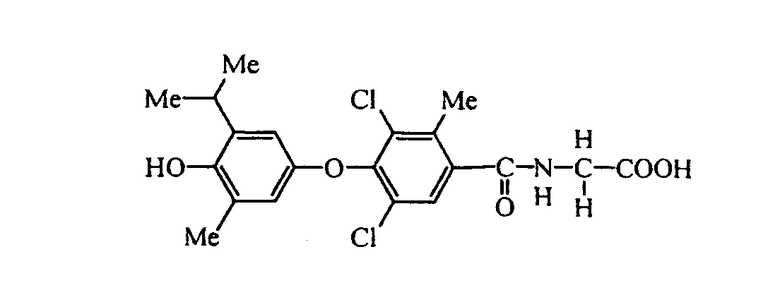
N-[3,5-дибром-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е8);
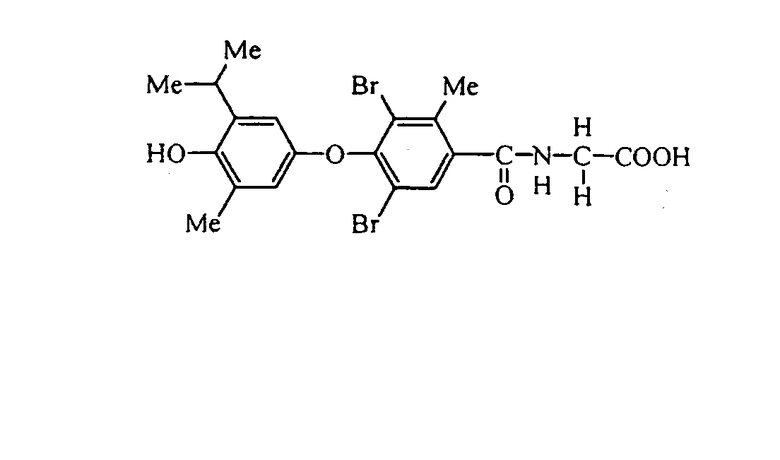
N-[3,5-диметил-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е9);
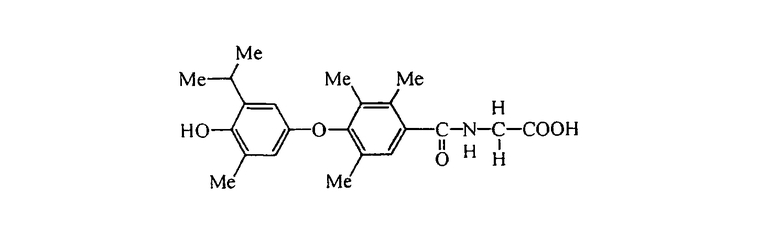
L-N-[3,5-дибром-4-(3-фтор-4-гидрокси-5-изопропилфенокси)фенилацетил]валин (Е10);
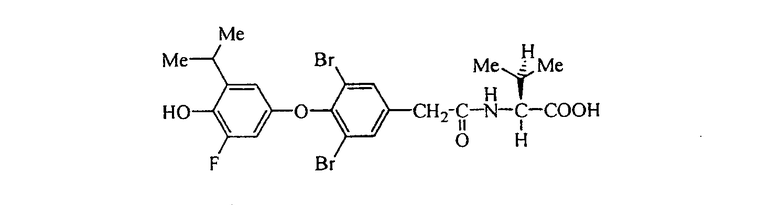
D-N-[3,5-дибром-4-(3-хлор-4-гидрокси-5-изопропилфенокси)фенилацетил]фенилглицин (Е11);
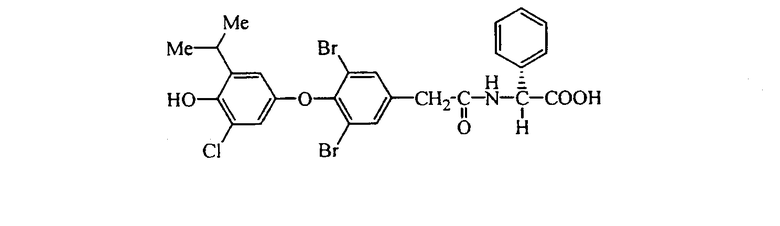
L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]валин (Е12);
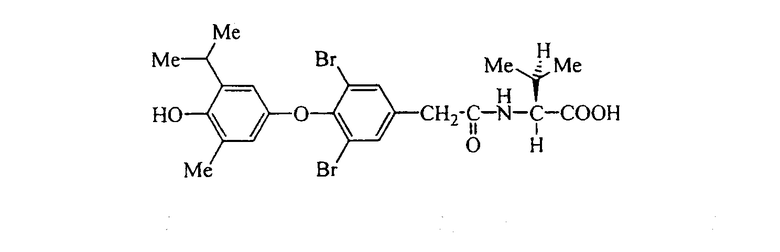
L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]фенилглицин (Е13);
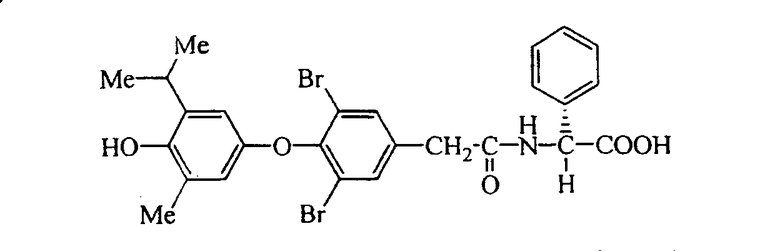
L-N-[3,5-дибром-4-(3,5-диметил-4-гидроксифенокси)фенилацетил]фенилглицин (Е14)
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее приводятся определения, которые относятся к терминам, используемым в данном описании, если конкретные примеры не ограничены другими определениями.
Термин «лиганд тиреоидного рецептора» в данном изобретении относится к любому фрагменту, который связывается с тиреоидным рецептором. Лиганд может выступать в качестве агониста, антагониста и частичного агониста или частичного антагониста. Синонимом термина «лиганд тиреоидного рецептора» является «тиреомиметик».
Если не указано другого определения, термин «алкил», используемый в данном описании сам по себе или как часть другой группы, включает углеводороды с прямой и разветвленной цепью, содержащие в нормальной цепи от 1 до 12 атомов углерода (в случае алкила или алк), предпочтительно от 1 до 4 атомов углерода, такие как метил, этил, пропил, изопропил, бутил, трет-бутил, или изобутил, пентил, гексил, изогексил, гептил, 4,4-диметилпентил, окстил, 2,2,4-триметилфенил, нонил, децил, ундецил, додецил. Как определено и заявлено в данном изобретении, термин «алкил» включает алкильные группы, определенные выше, необязательно замещенные 1-4 заместителями, которые могут представлять собой галоген, например F, Br, Cl или I, или CF3, алкил, алкокси, арил, арилокси, арил(арил) или диарил, арилалкил, арилалкилокси, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкилалкилокси, необязательно замещенный амино, гидрокси, гидроксиалкил, ацил, оксо, алканоил, гетероарил, гетероарилокси, циклогетероалкил, арилгетероарил, арилалкоксикарбонил, гетероарилалкил, гетероарилалкокси, арилоксиалкил, арилоксиарил, алкиламидо, алканоиламино, арилкарбониламино, алкоксикарбонил, алкиаминокарбонил, нитро, циано, тиол, галогеналкил, тригалогеналкил, алкилтио или карбоксил (или его алкиловый эфир).
Если не указано другого определения, термин «циклоалкил», используемый в данном описании сам по себе или как часть другой группы, включает насыщенные циклические углеводородные группы или частично ненасыщенные (содержащие 1 или 2 двойные связи) циклические углеводородные группы, содержащие одно кольцо и включающие от 3 до 8 атомов углерода, предпочтительно от 3 до 6 атомов углерода, образующих кольцо. Как определено и заявлено в данном изобретении, термин «циклоаклил» включает циклоалкильные группы, как определено выше, необязательно замещенные 1 или несколькими заместителями, такими как заместители, определенные для алкила.
Термин «арил» или «Ar», используемый в данном описании сам по себе или как часть другой группы, относится к моноциклическим или бициклическим ароматическим группам, содержащим от 6 до 10 атомов углерода в циклической части (таким как фенил или нафтил, включая 1-нафтил или 2-нафтил). Как определено и заявлено в данном изобретении, термин «арил» включает арильные группы, как определено выше, необязательно замещенные на любом(ых) доступном(ых) атоме(ах) углерода 1 или несколькими заместителями, такими как галоген, алкил, галогеналкил, алкокси, галогеналкокси, алкенил, трифтометил, трифторметокси, алкинил, гидрокси, амино, нитро, циано, карбоксил (или его алкиловый эфир), или любыми другими заместителями, описанными для алкила.
Если не указано другого определения, термин «гетероарил» или «гетероароматический», используемый в данном описании сам по себе или как часть другой группы, относится к 5- или 6-членному ароматическому кольцу, которое включает 1, 2, 3 или 4 гетероарома, таких как атом азота, атом кислорода или атом серы, и таким кольцам, которые конденсированы с арилом, циклоалкилом, гетероарилом или циклогетероалкилом (например, бензотиофенил, индол), и включает их возможные N-оксиды. Термин «замещенная гетероарильная» группа включает гетероарил, необязательно замещенный одним или несколькими заместителями, такими как любой заместитель алкила или арила, которые описаны выше. Как определено и заявлено в данном изобретении, термин «гетероарил» включает гетероарильные группы, как определено выше, необязательно замещенные на любом(ых) доступном(ых) атоме(ах) углерода 1 или несколькими заместителями, такими как любой из заместителей, определенных для алкила или арила.
Если не указано другого определения, термин «алкенил», используемый сам по себе или как часть другой группы, относится к радикалам с прямой или разветвленной цепью, содержащим в нормальной цепи от 2 до 20 атомов углерода, предпочтительно от 2 до 12 атомов углерода, более предпочтительно от 2 до 8 атомом углерода, которые включают в нормальной цепи одну или несколько двойных связей, таким как винил, 2-пропенил, 3-бутенил, 2-бутенил, 4-пентенил, 3-пентенил, 2-гексенил, 3-гексенил, 2-гептенил, 3-гептенил, 4-гептенил, 3-октенил, 3-ноненил, 4-деценил, 3-ундеценил, 4-додеценил, 4,8,12-тетрадекатриенил и т.п. Как определено и заявлено в данном изобретении, термин «алкенил» включает алкенильные группы, определенные выше, необязательно замещенные на любом(ых) доступном(ых) атоме(ах) углерода 1 или несколькими заместителями, такими как любой из заместителей, определенных для алкила или арила.
Если не указано другого определения, термин «алкинил», используемый в данном описании сам по себе или как часть другой группы, относится к радикалам с прямой или разветвленной цепью, содержащим в нормальной цепи от 2 до 20 атомов углерода, предпочтительно от 2 до 12 атомов углерода, более предпочтительно от 2 до 8 атомов углерода, которые включают в нормальной цепи одну или несколько тройных связей, таким как 2-пропинил, 3-бутинил, 2-бутинил, 4-пентинил, 3-пентинил, 2-гексинил, 3-гексинил, 2-гептинил, 3-гептинил, 4-гептинил, 3-октинил, 3-нонинил, 4-децинил, 3-ундецинил, 4-додецинил и т.п. Как определено и заявлено в данном изобретении, термин «алкинил» включает алкинильные группы, определенные выше, необязательно замещенные на любом(ых) доступном(ых) атоме(ах) углерода 1 или несколькими заместителями, такими как любой из заместителей, определенных для алкила или арила.
Термин «циклоалкенил», используемый в данном описании сам по себе или как часть другой группы, относится к циклическим углеводородам, содержащим от 3 до 12 атомов углерода, предпочтительно от 5 до 10 атомов углерода, и 1 или 2 двойные связи. Типичные примеры циклоалкенильных групп включают циклопентенил, циклогексенил, циклогексадиенил или циклогептадиенил, которые могут быть необязательно замещенными, как определено для циклоалкила. Как определено и заявлено в данном определении, термин «циклоалкенил» включает циклоалкенильные группы, определенные выше, необязательно замещенные на любом(ых) доступном(ых) атоме(ах) углерода 1 или несколькими заместителями, такими, как любой из заместителей, определенных для алкила или арила.
Термин «галоген» или «гало», используемый в данном описании сам по себе или как часть другой группы, относится к хлору, брому, фтору или йоду, а также к группе CF3, где предпочтительны хлор или бром.
Термин «алканоил», используемый в данном описании сам по себе или как часть другой группы, относится к алкилу или циклоалкилу, соединенному с карбонильной группой.
Термин «ароил», используемый в данном описании сам по себе или как часть другой группы, означает арил или гетероарил, соединенный с карбонильной группой.
Если не указано другого определения, термины «алкокси», «арилокси» или «гетероарилокси», применяемые сами по себе или как часть другой группы, включают любую из описанных выше алкильных, арильных или гетероарильных групп, присоединенных через атом кислорода.
Термин «циано» в данном описании относится к -CN группе.
Термин «арилалкил» и термин «гетероарилалкил», используемые в данном описании сами по себе или как часть другой группы, относятся к алкильным группам, описанным выше, содержащим арильный или гетероарильный заместитель. Типичные примеры арилалкила включают, но без ограничения, бензил, 2-фенилэтил, 3-фенилпропил.
Если не указано другого определения, термины «арилалкокси» и «циклоалкокси», используемые в данном описании по себе или как часть другой группы, включают арильные и циклоалкильные группы, присоединенные через атом кислорода.
Термин «карбоновая кислота» или «карбокси», используемый в данном описании, относится к -СООН группе.
Термин «бензил», используемый в данном описании, относится к группе -СН2С6Н5, которая может быть необязательно замещенной, как определено выше для алкила.
Соединения формулы I могут быть представлены в виде солей, которые также включены в объем данного изобретения. Фармацевтически приемлемые соли (то есть нетоксичные, физиологически приемлемые) являются предпочтительными. Если соединения формулы I имеют, например, по меньшей мере, один основный центр, они могут образовывать кислотно-аддитивные соли. Такие соли образуются, например, с сильными неорганическими кислотами, такими как минеральные кислоты, например серная кислота, фосфорная кислота или галогенводородная кислота, с сильными органическими карбоновыми кислотами, такими как алканкарбоновые кислоты, содержащие от 1 до 4 атомов углерода, которые являются незамещенными или замещенными, например галогеном, например уксусная кислота, такими как насыщенные или ненасыщенные дикарбоновые кислоты, например щавелевая, малоновая, янтарная, малеиновая, фумаровая, фталевая или терефталевая кислота, такими как оксикарбоновые кислоты, например аскорбиновая, гликолевая, молочная, яблочная, винная или лимонная кислота, такими как аминокислоты (например, аспаргиновая, глутаминовая кислота, лизин или аргинин), или бензойная кислота, или с органическими сульфоновыми кислотами, такими как (С1-С4)алкил- или арилсульфоновые кислоты, которые являются незамещенными или замещенными, например, галогеном, например метил- или п-толуолсульфоновая кислота. Могут быть получены соответствующие кислотно-аддитивные соли, содержащие, если необходимо, дополнительно присутствующий основный центр. Соединения формулы I, содержащие, по меньшей мере, одну кислотную группу (например, СООН), могут также образовывать соли с основаниями. Подходящими солями с основаниями являются, например, соли металлов, такие как соли щелочных металлов или соли щелочно-земельных металлов, например натриевые, калиевые или магниевые соли, или аммониевые соли или соли с органическим амином, таким как морфолин, тиоморфолин, пиперидин, пирролидин, низший моно-, ди- или три- алкиламин, например этил-, трет-бутил-, диэтил-, диизопропил-, триэтил-, трибутил- или диметилпропиламин, или моно-, ди- или тригидрокси-низший алкиламин, например моно-, ди- или триэтаноламин. Могут быть получены соответствующие внутренние соли. В объем данного изобретения включены также соли, которые не подходят для фармацевтического применения, но которые могут применяться, например, для выделения или очистки свободных соединений формулы I или их фармацевтически приемлемых солей. Предпочтительные соли соединений формулы I, которые содержат основную группу, включают моногидрохлорид, гидросульфат, метансульфонат, фосфат или нитрат. Предпочтительные соли соединений формулы I, которые содержат кислотную группу, включают натриевые, калиевые и магниевые соли и соли фармацевтически приемлемых органических аминов.
Соединения формулы I могут также иметь пролекарственные формы. Любое соединение, которое будет превращаться in vivo в биоактивное средство (то есть соединение формулы I), является пролекарством и относится к данному изобретению.
Различные формы пролекарств хорошо известны в данной области техники. Подробное описание пролекарств и производных пролекарств можно найти в следующих публикациях:
(i) The Practice of Medicinal Chemistry, Camille G. Wermuth et al, Ch 31, (Academic Press, 1996), (ii) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985); and (iii) A Textbook of Drug Design and Development, P.Krogsgaard-Larson and H.Bundgaard, eds. Ch 5, pgs 113-191 (Harwood Academic Publishers, 1991).
Указанные публикации введены в данное описание посредством ссылок.
Примеры пролекарств, подходящих для применения в данном изобретении, включают сложные низшие алкиловые эфиры, такие как сложный этиловый эфир, или сложные ацилоксиалкиловые эфиры, такие как пивалоилоксиметил (POM), группы карбоновой кислоты для R13.
Варианты воплощений пролекарств, подходящих для применения в данном изобретении, включают пролекарства, которые защищают свободную фенольную гидроксильную группу, присутствующую в формуле I, как показано на структуре, представленной ниже, где пролекарственная ароильная или алканоильная группа представляет собой фрагмент R-CO-, в котором R представляет собой алкил, гетероарил или арил:
Кроме того, воплощения пролекарств, подходящих для защиты фенольной гидроксильной группы, как указано выше, включают простые фенольные алкиловые эфиры, такие как представленные на структуре ниже, где R=алкил. Метаболическое гидроксилирование углерода алкильной группы R, которая присоединена к фенольному кислороду, приводит к получению промежуточного продукта, способного подвергаться дальнейшему разложению с высвобождением свободной фенольной формы соединений формулы I:
Подразумевается, что все стереомеры соединений данного изобретения в смеси или в чистом форме, или по существу, в чистой форме относятся к данному изобретению. Соединения данного изобретения могут иметь асимметричные центры на любом из атомов углерода, включая любой один или R заместители. Следовательно, соединения формулы I могут существовать в энантиомерных или диастереомерных формах или в их смесях. В способах получения в качестве исходных веществ могут использоваться рацематы, энантиомеры или дистереомеры. При получении диастереомерных или энантиомерных продуктов они могут разделяться стандартными способами, например хроматографией или фракционированной кристаллизацией.
Введение терапевтического средства данного изобретения включает введение терапевтически эффективного количества средства данного изобретения. Термин «терапевтически эффективное количество», используемый в данном изобретении, относится к количеству терапевтического средства для лечения или профилактики состояния, которое может лечиться введением композиции данного изобретения. Указанное количество является количеством, достаточным для проявления заметного терапевтического, профилактического или облегчающего эффекта. Эффект может включать, например, лечение или профилактику состояний, перечисленных в данном описании. Точное эффективное количество для субъекта будет зависеть от массы тела субъекта и его состояния здоровья, природы и тяжести состояния, подлежащего лечению, рекомендаций лечащего врача и терапевтических средств или комбинации терапевтических средств, выбранных для введения. Следовательно, не следует заранее указывать точное эффективное количество.
Соединения формулы I могут быть получены типичными способами, представленными в приведенных далее схемах, а также в соответствии с описанными в литературе методиками, которые используются квалифицированными специалистами в данной области. Типичные реагенты и последовательности этих реакций описаны далее и в рабочих примерах. Защита и удаление защитных групп, описанные на схемах ниже, могут проводиться с использованием методик, хорошо известных в данной области (см., например, T.W. Greene&P.G.M.Wuts, "Pretecting Groups in Organic Synthesis", 3rd Edition, Wiley, 1999).
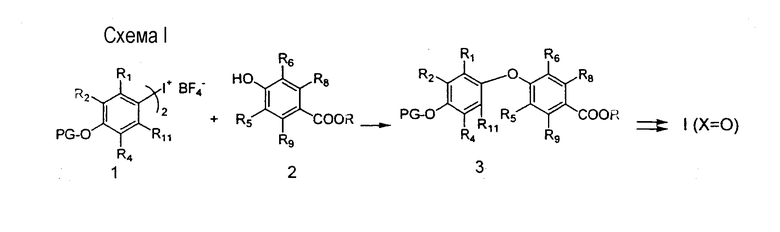
Методика получения соли йодония, представленная на схеме I, описана в литературе для синтеза аналогов тиреоидных гормонов («Novel Thyroid Receptor Ligands and Methods, Y.-L.Li, Y.Liu, A.Hedfors, J.Malm, C.Mellin, M.Zhang, PCT Int. App. WO 9900353 Al 990107; D.M.B. Hickey et al., J. Chem. Soc. Perkin Trans. I, 3103-3111, 1988; N.Yokoyama et al., J.Med.Chem, 38, 695-707, 1995)
и в целом для простых диариловых эфиров (E.A. Couladouros, V.I. Moutsos. Tetrahedron Lett., 40, 7023-7026). Взаимодействие соли йодония I с подходящей промежуточной гидроксибензойной кислотой 2 приводит к получению простого диарилового эфира 3, который способом, известным квалифицированному специалисту, может легко подвергаться превращению в соединения формулы I, где Х=О.
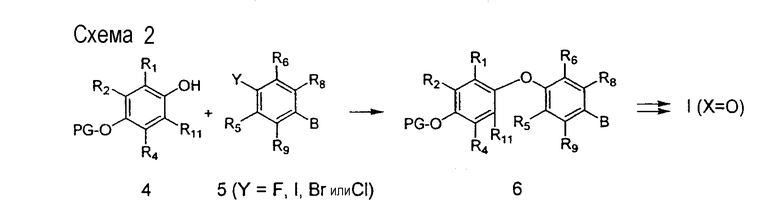
На схеме 2 представлен другой общий подход к синтезу соединений формулы I, в которых Х=О. Соответствующий замещенный фенол 4 подвергается алкилированию соответствующим промежуточным соединением 5, где Y представляет собой способную к замещению группу, такую как фтор, хлор, йод или бром, и активирующая группа В представляет собой группу, которая активирует группу Y для нуклеофильного замещения и способна впоследствии трансформироваться в группу карбоновой кислоты (такую как формил (СНО) или нитро (NO2)) с получением промежуточного продукта 6. Активирующая группа В в промежуточном продукте 6 затем последовательно трансформируется в группу карбоновой кислоты или ее производного, которое затем может дополнительно превращаться в соединения формулы I, где Х=О. Такие активирующие функциональные группы и их способности к превращению хорошо известны специалистам в данной области. Например, когда В представляет собой функциональную нитрогруппу в промежуточном продукте 6, нитрогруппа может восстанавливаться до аминогруппы хорошо известными способами, такими как применение каталитического гидрирования в присутствии, например, катализатора - никеля Ренея или палладия на углероде, в полярном растворителе, таком как ледяная уксусная кислота или этанол. Альтернативно, восстановление может осуществляться с использованием железного порошка в водной ледяной уксусной кислоте при температурах окружающей среды. Полученный ариламин может подвергаться превращению в соответствующую соль диазония применением, например, смеси нитрата натрия и серной кислоты в подходящих растворителях. Образующаяся диазониевая группа может затем подвергаться превращению в формильную группу (СНО) взаимодействием с моноксидом углерода в присутствии подходящего паладиевого катализатора, такого как ацетат палладия. После связывания с промежуточным продуктом 5 последовательные превращения защитной и функциональной группы приводят к получению целевых соединений формулы I, где Х=О. Другой активирующей группой является формил (С=-СНО). После связывания со вторым ароматическим кольцом альдегидная группа может подвергаться окислению до группы карбоновой кислоты и подвергаться дальнейшему превращению с получением соединений формулы I, в которых n=0. Кроме того, та же самая образующаяся группа карбоновой кислоты может подвергаться гомологизации до группы уксусной кислоты хорошо известными способами, такими как гомологизация Арндта-Эйстерта (Arndt-Eistert) для получения, в конечном счете, примеров соединений I, в которых n=1. Подход к общему синтезу простых диариловых эфиров для тиреомиметиков, представленный на схеме 2, подробно описан в литературе (P.D. Leeson, J.C. Emmett, J. Chem. Perkin Trans. I, 3085-3096, 1988; N. Yokoyama et al., J. Med, Chem., 38, 695-7078, 1995).
Дополнительные способы синтеза соединений формулы I, в которых Х=О, NH, S, CO или СН2 в целом описаны в литературе (для Х=О: D. M. B. Hickey et al., J. Chem. Soc. Perkin Trans. I, 3097-3102, 1988; Z-W. Guo et al., J. Org. Chem., 62, 6700-6701, 1997; D.M.T. Chan et al., Tetrahedron Lett., 39, 2933-2936, 1998; D. A. Evans et al., Tetrahedron Lett., 39, 2937-2940, 1998; G.M. Salamonczyk et al., Tetrahedron Lett., 38, 6965-6968, 1997; J.-F.Marcoux, J. Am. Chem. Soc., 119, 10539-10540, 1997; A. V. Kalinin et al., J. Org. Chem., 64, 2986-2987, 1999; для X =N:D.M.T. Chan et al., Tetrahedron Lett., 39, 2933-2936, 1998; J.P. Wolfe et al., J. Am. Chem. Soc., 118, 7215, 1996; M.S. Driver, J.F. Hartwig, J. Am. Chem. Soc., 118, 7217,1996, см. ссылки в обзоре C.G.Frost, P. Mendonca, J. Chem. Soc. Perkin I, 2615-2623, 1998; для X=S: C.R. Harrington, Biochem. J., 43, 434-437, 1948; A. Dibbo et al., J. Chem. Soc., 2890-2902, 1961; N. Yokoyama et al., United States Patent 5401772, 1995; для X=CO или CH2: L. Horner, H.H.G. Medem, Chem. Ber., 85, 520-530, 1952; G. Chiellini et al., Chemistry @ Biology, 5, 299-306, 1998; и для Х=CF2, см. G.S. Lal et al., J. Org. Chem., 65, 4830-4832, 2000).
Способы, применимые для синтеза соединений формулы I, в которых Х=О и R2 и R3 независимо выбраны из водорода, галогена и алкила, описаны в "Novel Thyroid Receptor Ligands and Methods", Y.-L. Li, Liu, A. Hedfors, J. Malm, C. Mellin, M. Zhang, PCT Int. App. WO 9900353 A1 990107.
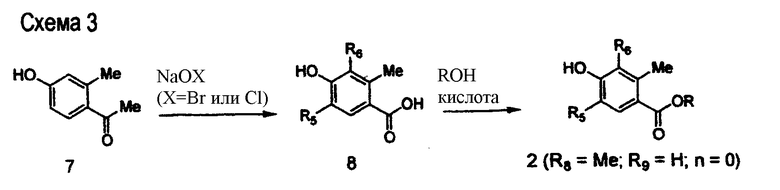
На схеме 3 представлен синтез полупродукта 2, в котором R5=R6=Br или Cl, R8=Me, R9=H, R=H и n=0. Ацетофенон 7 (коммерчески доступен) подвергается взаимодействию с гипохлоритом натрия или гипобромитом натрия в условиях реакции образования галогенпроизводных для одновременного превращения ацетильной функции в карбоксильную и введения галогена в два орто-положения относительно фенольной гидроксильной группы. Этерификация приводит к получению целевого полупродукта 2, который применяется в последовательности химических превращений, описанных на схеме 4.
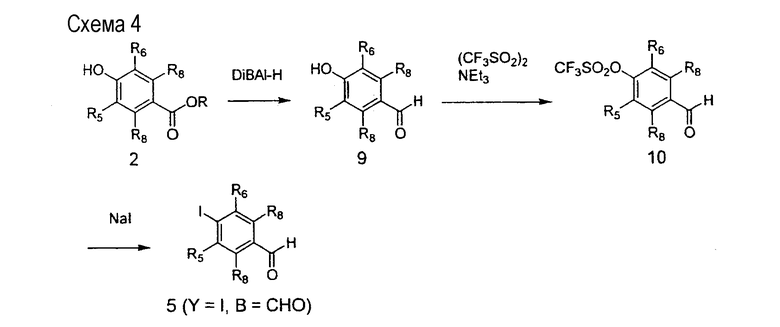
На схеме 4 описано применение полупродукта 2 для синтеза полупродукта 5, в котором Y=I и В=СНО. Промежуточный сложный эфир 2 подвергается восстановлению до альдегида 9 с помощью восстановителей, таких как гидрид диизобутилалюминия (DiBAl-H). Соединение 9 подвергается превращению в трифлат 10 применением ангидрида трифторметансульфоновой кислоты и органического аминного основания, такого как триэтиламин. Взаимодействия трифлата 10 с йодидом натрия приводит к получению целевого арилйодид-альдегида 5, который может использоваться в последовательности химических превращений, описанных на схеме 2 выше.
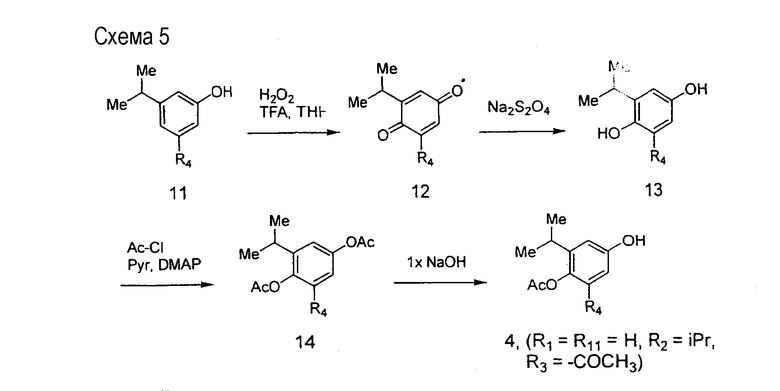
На схеме 5 описан синтез полупродукта 4, в котором R1 и R11 =H, R2 = изопропил и R3 представляет собой защитную ацетильную группу (-СОСН3). Фенол 11 подвергается окислению до хинона 12, который после восстановления гидросульфитом натрия (Na2S2O4) превращается в гидрохинон 13. Ацетилирование соединения 13 в стандартных условиях приводит к получению бис-ацетата 14, который после обработки 1 эквивалентом гидроксида натрия с удалением наименее заторможенной ацетатной группы приводит к получению целевого соединения 4, которое может использоваться в химических превращениях, описанных на схеме 2 выше.
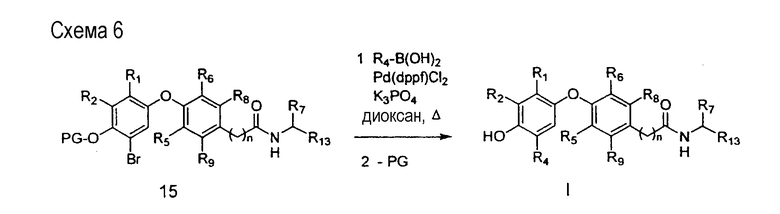
На схеме 6 описан подход к синтезу соединений формулы I, в которых группа R4 представляет собой низшую алкильную группу, такую как метил или этил. Промежуточный арилбромид 15, полученный бромированием соответствующего свободного фенола (R3=OH; получен в соответствии с методиками, описанными на схеме 1 и схеме 2) с последующим введением защитной группы (PG) на фенольный гидроксил. Группа R4 вводится сочетанием промежуточного арилбромида 15 с алкилбороновой кислотой в присутствии соответствующего палладиевого катализатора, такого как [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (Harada et al., Synlett, 1995, 283). Промежуточный арилбромид может заменяться на соответствующий арилйодид или арилтрифлат и подвергаться такому же превращению.
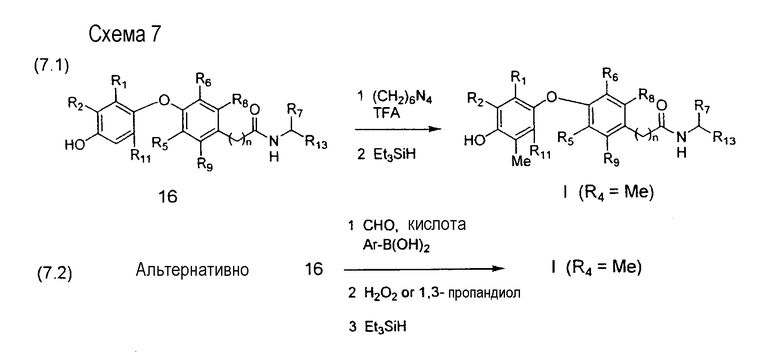
На схеме 7 описаны две методики специфического введения метильной группы в положение R4. Оба способа осуществляются (1) гидроксиметилированием в орто-положение относительно фенольной гидроксильной группы (R3) в полупродукте 16 (полученном в соответствии с методиками, описанными на схеме 1 и схеме 2), с последующим (2) восстановлением гидроксиметильной группы до метильной группы с использованием такого реагента, как триэтилсилан. В реакции 7.1 гидроксиметилирование достигается применением гексаметилентетрамина в присутствии трифторуксусной кислоты. В реакции 7.2 стадия гидроксиметилирования ускоряется применением бензолбороновой кислоты, которая образует комплекс с первичным продуктом присоединения в форме диоксаборонового полупродукта. Целевое гидроксиметил-производное выделяется либо реакцией обмена с этиленгликолем, либо реакцией с пероксидом водорода (Nagata et al., Synthesis, 1979, 365).
Другие способы введения метильной группы в ароматическое кольцо известны специалистам в данной области. Например: (1) арилбромиды и арилйодиды могут метилироваться обработкой с Me4Sn (тетраметилоловом) в присутствии палладиевого катализатора (Tet. Lett., 1999, 40, 2719-2722); (2) арилтрифлаты метилируются обработкой с Me3In (триметилиндием) в присутствии палладиевого катализатора (Org. Lett., 1999, 1, 1267-1269); (3) арилтрифлаты превращаются в арилметил-производные обработкой с Me3Al (триметилалюминием) в присутствии палладиевого катализатора; (4) арилтрифлаты также метилируются обработкой метилбороновой кислотой в присутствии палладиевого катализатора (Synlett, 1995, 283-284).
ПРИМЕНЕНИЯ И КОМБИНАЦИИ
ПРИМЕНЕНИЯ: Соединения данного изобретения являются лигандами тиреоидных рецепторов и включают соединения, которые представляют собой, например, селективные агонисты, частичные агонисты, антагонисты или частичные антагонисты тиреоидного рецептора. Предпочтительные соединения данного изобретения обладают активностью в качестве агонистов тиреоидного рецептора, предпочтительно селективных агонистов тиреоидного рецептора-бэта, и могут использоваться для лечения заболеваний или расстройств, связанных с активностью тиреоидного рецептора. В частности, соединения данного изобретения могут использоваться для лечения заболеваний или расстройств, которые связаны с нарушением метаболизма или зависят от экспрессии Т3 регулируемого гена.
Соответственно, соединения данного изобретения могут вводиться млекопитающим, предпочтительно людям, для лечения различных состояний или расстройств, таких как, но без ограничения, гипотиреоз; субклинический гипертиреоз; нетоксический зоб; атеросклероз; тиреогормональная заместительная терапия (например, у пожилых пациентов); клетки злокачественной опухоли, содержащие тиреоидный рецептор; папиллярный или фолликулярный рак; поддержание силы и функции мышц (например, у пожилых пациентов); обращение или профилактика ухудшения состояния здоровья или возрастного функционального ухудшения здоровья (age-related functional decline - ARFD) в пожилом возрасте (например, саркопения); лечение катаболических побочных эффектов глюкокортикоидов; профилактика и/или лечение снижения костной массы, плотности или роста костной массы (например, остеопороза и остеопении); лечение синдрома хронической усталости (chronic fatigue syndrom - CFS); ускорение заживления осложненных переломов, например остеогенез с дистракцией; восполнение сустава; расстройства аппетита (например, анорексия); лечение ожирение и замедление роста, связанного с ожирением; лечение депрессии, повышенной возбудимости, слезливости и состояния стресса; лечение сниженной умственной активности и самооценки (например, мотивации/уверенности в себе); улучшение познавательной функции (например, лечение слабоумия, включая болезнь Альцгеймера и кратковременную потерю памяти); лечение катаболизма в связи с легочной дисфункцией и зависимости от аппарата искусственной вентиляции легких; лечение сердечной дисфункции (например, связанной с пороком клапана сердца, инфарктом миокарда, сердечной гипертрофией или застойной сердечной недостаточностью); снижение кровяного давления; защита от вентрикулярной дисфункции или профилактика случаев реперфузии; лечение гиперинсулинемии; стимулирование остеобластов, восстановления кости и роста хряща; регулирование поглощения пищи; лечение инсулинорезистентности, включая NIDDM, у млекопитающих (например, у людей); лечение инсулинорезистентности в сердце; лечение застойной сердечной недостаточности; лечение скелетно-мышечного ослабления (например, у пожилых пациентов); улучшение общей легочной функции; кожные расстройства или заболевания, такие как дермальная атрофия, вызванная глюкокортикоидами, включая реконструкцию дермальной атрофии, вызванной местными глюкокортикоидами, и профилактику дермальной атрофии, вызванной местными глюкокортикоидами (например, одновременным лечением местным глюкокортикоидом или фармакологическим средством, включающим глюкокортикоид и соединение данного изобретения), реконструкция/профилактика дермальной атрофии, вызванной системным лечением глюкокортикоидами, реконструкция/профилактика атрофии дыхательной системы, вызванной местным лечением глюкокортикоидами, дермальной атрофии, вызванной УФ-облучением, дермальной атрофии, вызванной старением (морщины и т.п.), заживления мелких ран, келоидов, атрофических полосок кожи, целюлита, бугристости кожи, актинического повреждения кожи, плоского лишая, ихтиоза, акнэ, псориаза, болезни Дернера, экземы, атопическиго дерматита, хлоракне, питириаза и кожных рубцов.
Подразумевается, что термин «лечение» включает также профилактическое лечение. Кроме того, с помощью соединений данного изобретения могут лечиться состояния, заболевания и болезни, объединяемые общим названием «синдром Х» или метаболический синдром, как описано в публикации Johannsson, J. Clin. Endocrinol. Metab., 82, 727-34 (1997).
КОМБИНАЦИИ: Данное изобретение включает фармацевтические композиции, содержащие в качестве активного ингредиента терапевтически эффективное количество, по меньшей мере, одного из соединений формулы I, отдельно или в комбинации с фармацевтическим носителем или разбавителями. Необязательно, соединения данного изобретения могут использоваться сами по себе, в комбинации с другими соединениями данного изобретения или в комбинации с одним или несколькими другими терапевтическими средствами, например противодиабетическим средством или другим фармацевтически активным веществом.
Соединения данного изобретения могут применяться в комбинации с другими модуляторами и/или лигандами тиреоидного рецептора или другими подходящими терапевтическими средствами, полезными для лечения указанных выше расстройств, включая противодиабетические средства, средства против остеопороза, средства против ожирения, средства, промотирующие рост (включая усилители секреции гормона роста), противовоспалительные средства, средства против тревоги, антидепрессанты, противогипертензивные средства, сердечные гликозиды, средства для снижения содержания холестерина/липидов, средства для подавления аппетита, ингибиторы резорбции кости, тиреомиметики (включая другие агонисты тиреоидного рецептора), анаболические средства и противоопухолевые средства.
Примеры противодиабетических средств, подходящих для применения в комбинации с соединениями данного изобретения, включают бигуаниды (например, метформин или фенформин), ингибиторы глюкозидазы (например, акарбозу или миглитол), инсулины (включая усилители секреции инсулина и сенсибилизаторы инсулина), меглитиниды (например, репаглинид), сульфонилмочевины (например, глимепирид, глибурид, гликлазид, клорпропамид и глипизид), комбинации бигуанид/глибурид (например, Glucovance®), тиазолидиндионы (например, троглитазон, росиглитазон и пиоглитазон), PPAR-альфа агонисты, PPAR-гамма агонисты, двойные PPAR альфа/гамма агонисты, ингибиторы SGLT2, ингибиторы гликогенфосфорилазы, ингибитор белка, связывающего жирные кислоты (аР2), глюкагон-подобный пептид-1 (GLP-1) и ингибиторы дипептидилпептидазы IV (DP4).
Примеры средств против остеопороза, подходящих для применения в комбинации с соединениями данного изобретения, включают алендронат, ризедронат, РТН, РТН фрагмент, ралоксифен, кальцитонин, антагонисты RANK лигандов, антагонисты рецепторов, чувствительных к кальцию, TRAP ингибиторы, селективные модуляторы эстрогенового рецептора (SERM) и АР-1 ингибиторы.
Примеры средств против ожирения, подходящих для применения в комбинации с соединениями данного изобретения, включают аР2 ингибиторы, PPAR-гамма антагонисты, PPAR-дельта агонисты, бэта 3 адренергические агонисты, такие как AJ9677 (Takeda/Dainippon), L750355 (Merk), CP331648 (Pfizer) или другие известные бэта-3 агонисты, описанные в патентах США №№ 5541204, 5770615, 5491134, 5776983 и 5488064, ингибитор липазы, такой как орлистат или ATL-962 (Alizymer), ингибитор повторного поглощения серотонина (и дофамина), такой как сибутрамин, топирамат (Jonson&Johnson) или аксокин (Regeneron), другие лекарственные средства тиреоидного бэта рецептора, такие как лиганд тиреоидного рецептора, описанный в WO 97/21993 (U. Cal SF), WO 99/003353 (KaroBio) и GB98/284425 (KaroBio), CB-1 (каннабиноидный рецептор) антагонисты (см. G. Colombo et al., "Appetite Suppression and Weight Loss After the Cannabionid Antagonist SR 141716", Life Sciences, Vol. 63, PL 113-117 (1998)), а также средство, снижающее аппетит, такое как дексамфетамин, фентермин, фенилпропаноламин или мазиндол.
Соединения данного изобретения могут объединяться со средствами, промотирующими рост, такими как, но без ограничения, TRH, диэтилстилбестерол, теофиллин, энкефалины, простагландины серии Е, соединения, описанные в патенте США № 3239345, например зеранол, соединения, описанные в патенте США № 4036979, например сулбенокс, или пептиды, описанные в патенте США № 4411890.
Соединения данного изобретения могут применяться в комбинации со средствами, усиливающими секрецию гормона роста, такими как GYRP-6, GHRP-2 (описанные в патенте США № 4411890 и публикациях WO 89/07110 и WO 89/07111), GHRP-2 (описанный в WO 93/04081), NN703 (Novo Nordisk), LY444711 (Lilly), MK-677 (Merk), CP424391 (Pfizer) и В-НТ920, или другим фактором высвобождения гормона роста или его аналогами, или гормоном роста и его аналогами или соматомединами, включая IGF-1 и IGF-2, или альфа-адренергическими агонистами, такими как агонисты клонидина или серотонина 5-НТD, такими как суматриптан, или средствами, которые ингибируют соматостатин или его высвобождение, такими как физостигмин и пиридостигмин. Соединения данного изобретения могут также применяться в комбинации с паратиреоидным гормоном, РНТ (1-32) или бисфосфонатами, такими как МК-217 (алендронат).
Соединения данного изобретения могут применяться также в комбинации с эстрогеном, тестостероном, селективным модулятором эстрогенового рецептора, таким как тамоксифен или ралоксифен, или другими модуляторами андрогенового рецептора, такими, которые описаны, например, в публикациях Edwards, J.O. et al., Bio. Med Chem. Let., 9, 1003-1008 (1999); Hamann, L.G. et al., J. Med. Chem., 42, 210-212 (1999).
Соединения данного изобретения могут применяться в комбинации со стероидными или нестероидными агонистами прогестеронового рецептора (progesterone receptor agonista - "PRA"), такими как левоноргестрел, медроксипрогестерон ацетат (МРА).
Примеры противовоспалительных средств, подходящих для применения в комбинации с соединениями данного изобретения, включают преднизон, дексаметазон, Enbrel®, ингибиторы циклооксигеназы (то есть СОХ-1 и/или СОХ-2 ингибиторы, такие как NSAID, аспирин, индометацин, ибупрофен, пироксикам, Naproxen®, Celebrex®, Vioxx®), CTLA4-Ig агонисты/антагонисты, антагонисты лиганда CD40, ингибиторы IMPDH, такие как микофенолят (CellCept®), интегриновые антагонисты, антагонисты альфа-4 бэта-7 интегрина, ингибиторы клеточной адгезии, антагонисты гамма-интерферона, ICAM-1, антагонисты фактора некроза опухоли (TNF) (например, инфликсимаб, OR1384), ингибиторы синтеза простагландина, будезонид, клофазимин, CNI-1493, CD4 антагонисты (например, приликсимаб), ингибиторы р38 митоген-активируемой протеинкиназы, ингибиторы протеинтирозинкиназы (РТК), IKK ингибиторы, и терапевтические средства для лечения синдрома раздраженного кишечника (например, слабительные Zelmac® и Maxi-K® например, описанные в патенте США № 6184231 В1).
Примеры средств для лечения состояния тревоги, подходящих для применения в комбинации с соединениями данного изобретения, включают диазепам, лоразепам, буспирон, оксазепам и гидроксизин памоат.
Примеры антидепрессантов, подходящих для применения в комбинации с соединениями данного изобретения, включают циталопрам, флуоксетин, нефазодон, сертралин и пароксетин.
Для лечения кожных расстройств или заболеваний, описанных выше, соединения данного изобретения могут использоваться сами по себе или, необязательно, в комбинации с ретиноидом, таким как третиноин, или аналогом витамина D.
Примеры противогипертензивных средств, подходящих для применения в комбинации с соединениями данного изобретения, включают бэта-адренергические блокаторы, блокаторы кальциевых каналов (L-типа и Т-типа; например, дилтиазем, верапамил, нифедипин, амлодипин и мибефрадил), диуретические средства (например, хлоротиазид, гидрохлоротиазид, флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлоротиазид, трихлорометиазид, политиазид, бензтиазид, трикринафен этакриновой кислоты, хлорталидон, фуросемид, музолимин, буметанид, триамтренен, амилорид, спиронолактон) ингибиторы ренина, ингибиторы АСЕ (например, каптоприл, зофеноприл, фозиноприл, эналаприл, сераноприл, силазоприл, делаприл, пентоприл, квинаприл, рамиприл, лисиноприл), антагонисты АТ-1 рецептора (например, лосартан, ирбесартан, валсартан), антагонисты ЕТ рецептора (например, ситакссентан, атрсентат и соединения, описанные в патента США №№ 5612359 и 6043265), двойные ET/AII антагонисты (например, соединения, описанные в WO 00/01389), ингибиторы нейтральной эндопептидазы (NEP), ингибиторы вазопепсидазы (двойные NEP-ACE ингибиторы) (например, омапатрилат и гемопатрилат) и нитраты.
Примеры сердечных гликозидов, подходящих для применения в комбинации с соединениями данного изобретения, включают дигиталис и оуабаин.
Примеры средств, снижающих содержание холестерина/липидов и подходящих для применения в комбинации с соединениями данного изобретения, включают ингибиторы HMG-CoA редуктазы, ингибиторы скваленсинтетазы, фибраты, средства, усиливающие секрецию желчной кислоты, ингибиторы АСАТ, ингибиторы МТР, ингибиторы липоксигеназы, ингибитор совместного транспорта Na+/желчной кислоты в подвздошной кишке, ингибиторы абсорбции холестерина и ингибиторы белка, переносящего сложные эфиры холестерина (например, CP-529414).
МТР ингибиторы, которые могут применяться в комбинации с одним или несколькими соединениями формулы I, включают МТР ингибиторы, описанные в патенте США № 5595872, в патенте США № 5739135, в патенте США № 5712279, в патенте США № 5760246, в патенте США № 5827875, в патенте США № 5885983 и в патенте США № 5962440, которые введены в данное описание посредством ссылок.
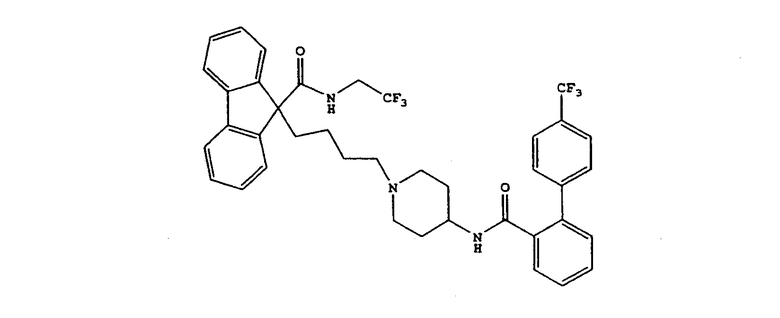
Предпочтительным МТР ингибитором является
9-[4-[4-[[2-(2,2,2-трифторэтокси)бензоил]амино]-1-пиперидинил]бутил]-N-(2,2,2-трифторэтил)-9Н-флуорен-9-карбоксамид
Ингибиторы HMG CoA редуктазы, которые могут применяться в комбинации с одним или несколькими соединениями формулы I, включают мевастатин и родственные соединения, описанные в патенте США № 3983140, ловастатин (мевинолин) и родственные соединения, описанные в патенте США № 4231938, правастатин и родственные соединения, описанные в патенте США № 4346227, симвастатин и родственные соединения, описанные в патентах США №№ 4448784 и 4450171. Ингибиторы HMG CoA редуктазы, которые также могут использоваться в данном изобретении, включают флувастати, описанный в Патенте США № 535 772, церивастатин, описанный в патентах США №№ 4681893, 5273995, 5385929 и 5686104, пиразольные аналоги производных мевалонолактона, описанные в патенте США № 4613610, инденовые аналоги производных мевалонолактона, описанные в PCT заявке WO 86/03488, 6-[2-(замещенный пиррол-1-ил)алкил]пиран-2-оны и его производные, описанные в патенте США № 4647576, SC-45355 (Searle) (производное 3-замещенной пентадиовой кислоты), дихлорацетат, имидазоловые аналоги мевалонолактона, описанные в РСТ заявке WO 86/07054, производные 3-карбокси-2-гидроксипропанфосфоновой кислоты, описанные в патенте Франции № 2596393, производные 2,3-дизамещенных пиррола, фурана и тиофена, описанные в Европейской патентной заявке на патент № 0221025, нафтиловые аналоги мевалонолактона, описанные в патенте США №4686237, октагидронафталины, описанные в патенте США №4499289, кето-аналоги мевинолина (ловастатин), описанные в Европейской патентной заявке № 142146, а также другие известные ингибиторы HMG CoA редуктазы.
Ингибиторы скваленсинтетазы, которые могут использоваться в комбинации с соединениями данного изобретения, включают, но без ограничения, α-фосфоносульфонаты, описанные в патенте США № 5712396 и в публикации Biller et al., J. Med. Chem., 1988, Vol. 31, No. 10, pp. 1869-1871, включая изопреноидные (фосфинилметил)фосфонаты, терпеноидные пирофосфаты, описанные в публикации P. Ortiz de Montellano et al., J. Med. Chem., 1977, 20, 243-249, фарнезил-дифосфатный А аналог и пресквален-пирофосфатные (PSQ-PP) аналоги, описанные в публикации Corey and Volante, J. Am. Chem. Soc., 1976, 98, 1291-1293, фосфинилфосфонаты, описанные в публикации McClard, R.W. et al., J.A.C.S., 1987, 109, 5544, и циклопропаны, описанные в публикации Capson, T.L., PhD Dissertation, June, 1987, Dept. Med. Chem. U of Utah, Abstract, Table of Contents, pp. 16, 17, 40-43, 48-51, а также другие ингибиторы скваленсинтетазы, описанные в патентах США №№ 4871721 и 4924024 и в публикации Biller, S.A., Neuenschwander, K., Ponpipom M.M., and Poulter C.D., Current Pharmaceutical Design, 2, 1-40 (1996).
Вещества, усиливающие секрецию желчной кислоты, которые могут использоваться в комбинации с соединениями данного изобретения, включают холестирамин, колестипол и DEAE-Sephadex (Secholex®, Policexide®), а также липостабил (Rhone-Poulenc), Eisai E-5050 (N-замещенное этаноламин-производное), иманиксил (НОЕ-402), тетрагидролипстатин (THL), истигмастанилфосфорилхолин (SPC, Roche), аминоциклодекстрин (Tanabe Seiyoku), Ajinomoto AJ-814 (азулен-производное), мелинамид (Sumitomo), Sandoz 58-035, American Cyamamid CL-277082 и CL-283546 (дизамещенные производные мочевины), никотиновую кислоту, аципимокс, асифран, неомицин, п-аминосалициловую кислоту, аспирин, поли(диаллилметиламин)-производные, например, описанные в патенте США № 4759923, четвертичный амин поли(диаллилдиметиламмоний хлорид) и ионены, описанные в патенте США № 4027008, а также другие известные средства, снижающие содержание холестерина в сыворотке крови.
АСАТ ингибиторы, подходящие для применения в комбинации с соединениями данного изобретения, включают АСАТ ингибиторы, описанные в следующих публикациях: Drugs of the Future 24, 9-15 (1999), (Avasimibe); "The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters", Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137(1), 77-85; "The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoBl00-containing lipoprotein", Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16(1), 16-30; "RP 73163: a bioavailable alkylsulfinyl-diphenylimidazole ACAT inhibitor", Smith, C., et al, Bioorg. Med. Chem. Lett. (1996), 6(1), 47-50; "ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals", Krause et al, Editor(s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation: Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton, Fla.; "ACAT inhibitors: potential anti-atherosclerotic agents", Sliskovic et al, Curr. Med. Chem. (1994), 1(3), 204-25; "Inhibitors of acyl-CoA:cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid-regulating activity. Inhibitors of acyl-CoA:cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(1-phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity". Stout et al, Chemtracts: Org. Chem. (1995), 8(6), 359-62.
Примеры ингибитора абсорбиции холестерина, подходящего для применения в комбинации с соединениями данного изобретения, включают SCH48461 (Schering-Plough), а также соединения, описанные в публикациях Atherosclerosis 115, 45-63 (1995) и J. Med. Chem. 41, 973 (1998).
Примеры ингибиторов совместного переноса подвздошных Na+/желчной кислоты, подходящих для применения в комбинации с соединениями данного изобретения, включают соединения, описанные в публикации Drugs of the Future, 24, 425-430 (1999).
Примеры тиреомиметиков, подходящих для применения в комбинации с соединениями данного изобретения, включают тиреотропин, политиреоид, КВ-130015 и дронедарон.
Примеры анаболических средств, подходящих для применения в комбинации с соединениями данного изобретения, включают тестостерон, ТСГ диэтилстилбестерол, эстрогены, β-агонисты, теофиллин, анаболические стероиды, дегидроэпиандростерон, энкефалины, простагландины Е-серии, ретиноевую кислоту и соединения, описанные в патенте США № 3239345, например Zeranol®; в патенте США № 4036979, например Sulbenox®, или пептиды, описанные в патенте США № 4411890.
Все указанные выше патенты и заявки на патенты введены в данное описание посредством ссылок.
Перечисленные выше терапевтические средства при применении в комбинации с соединениями данного изобретения могут использоваться, например, в количествах, указанных в Physicians' Desk Reference (PDR) или иначе определенных квалифицированным специалистом.
Когда соединения данного изобретения используются в комбинации с одним или несколькими другими терапевтическими средствам, одновременно или последовательно, предпочтительны следующие соотношения и интервалы дозировок.
При применении в комбинации с гиполипидемическим средством, антидепрессантом, ингибитором резорбции кости и/или средством, подавляющим аппетит, соединения формулы I могут использоваться при массовом отношении к дополнительному средству в интервале от примерно 500:1 до примерно 0,005:1, предпочтительно в интервале от примерно 300:1 до примерно 0,01:1.
В том случае, когда противодиабетическое средство представляет собой бигуанид, соединения формулы I могут применяться при массовом отношении к бигуаниду в интервале от примерно 0,01:1 до примерно 100:1, предпочтительно от примерно 0,5:1 до примерно 2:1.
Соединения формулы I могут применяться при массовом отношении к ингибитору глюкозидазы в интервале от примерно 0,01:1 до примерно 100:1, предпочтительно от примерно 0,5:1 до примерно 50:1.
Соединения формулы I могут применяться при массовом отношении к сульфонилмочевине в интервале от примерно 0,01:1 до примерно 100:1, предпочтительно от примерно 0,2:1 до примерно 10:1.
Соединения формулы I могут применяться при массовом отношении к тиазолидиндиону в интервале от примерно 0,01 до примерно 100:1, предпочтительно от примерно 0,5:1 до примерно 5:1. Тиазолидиндион может применяться в количестве в интервале от примерно 0,01 до примерно 2000 мг/день, которое может необязательно вводиться в одной дозе или в разделенных дозах от одного до четырех раз в день. Кроме того, когда сульфонилмочевина и тиазолидиндион должны вводиться перорально в количестве менее примерно 150 мг, эти дополнительные средства могут помещаться в одну таблетку с терапевтически эффективным количеством соединений формулы I.
Метформин или его соль может применяться с соединениями формулы I в количествах в интервале от примерно 500 до примерно 2000 мг в день, которые могут вводиться в одной дозе или в разделенных дозах от одного до четырех раз в день.
Соединения формулы I могут применяться при массовом отношении к PPAR-альфа агонисту, PPAR-гамма агонисту, двойному PPAR-альфа/гамма агонисту, ингибитору SGLT2 и/или аР2 ингибитору в интервале от примерно 0,01:1 до примерно 100:1, предпочтительно от примерно 0,5:1 до примерно 5:1.
МТР ингибитор может вводиться перорально с соединениями формулы I в количестве в интервале от примерно 0,01 мг/кг до примерно 100 мг/кг, предпочтительно от примерно 0,1 мг/кг до примерно 75 мг/кг, от одного до четырех раз в день. Предпочтительная дозированная форма для перорального введения, такая как таблетки или капсулы, может содержать МТР ингибитор в количестве от примерно 1 до примерно 500 мг, предпочтительно от примерно 2 до примерно 400 мг, более предпочтительно от примерно 5 до примерно 250 мг, и вводиться по схеме от одного до четырех раз в день. Для парентерального введения МТР ингибитор может применяться в количестве в интервале от примерно 0,005 мг/кг до примерно 10 мг/кг, предпочтительно от примерно 0,005 мг/кг до примерно 8 мг/кг, и вводиться по схеме от одного до четырех раз в день.
Ингибитор HMG CoA редуктазы может вводиться перорально с соединениями формулы I в количестве в интервале от примерно 1 до 2000 мг, предпочтительно от примерно 4 до примерно 200 мг. Предпочтительная дозированная форма для перорального введения, такая как таблетки или капсулы, будет содержать ингибитор HMG CoA редуктазы в количестве от примерно 0,1 до примерно 100 мг, предпочтительно от примерно 5 до примерно 80 кг, более предпочтительно от примерно 10 до примерно 40 мг.
Ингибитор скваленсинтетазы может вводиться с соединениями формулы I в количестве в интервале от примерно 10 мг до примерно 2000 мг, предпочтительно от примерно 25 мг до примерно 200 мг. Предпочтительная дозированная форма для перорального введения, такая как таблетки или капсулы, будет содержат ингибитор скваленсинтетазы в количестве от примерно 10 до примерно 500 мг, предпочтительно от примерно 25 до примерно 200 мг
Соединения формулы I данного изобретения могут вводиться перорально или парентерально, например, подкожно или внутривенно, а также назально, ректально или подъязычным введением различным млекопитающим, которые, как известно, подвержены таким заболеваниям, например людям, с дозировкой эффективного количества в интервале от примерно 0,01 мкг/кг до примерно 1000 мкг/кг, предпочтительно от примерно 0,1 мкг/кг до 100 мкг/кг, более предпочтительно от примерно 0,2 мг/кг до примерно 50 мкг/кг (или от примерно 0,5 до 2500 мг, предпочтительно от примерно 1 до 2000 мг), по схеме в одной дозе или в разделенных дозах от двух до четырех раз в день.
Соединения формулы I могут вводиться для любого из применений, описанных в данном изобретении, любым подходящим способом, например перорально - в форме таблеток, капсул, гранул или порошков; подъязычно; буккально; парентерально, например, методами подкожной, внутривенной, внутримышечной или интрастернальной инъекций или вливаний (в виде стерильных водных или неводных растворов или суспензий для инъекций); назально, включая способы введения в назальные мембраны, такие как распылительная ингаляция; местно, например, в форме крема или мази; или ректально, например, в форме суппозиториев; композиции единичной дозы содержат нетоксичные фармацевтически приемлемые носители или разбавители. Соединения данного изобретения могут вводиться, например, в форме, подходящей для быстрого высвобождения или длительного высвобождения действующего вещества. Быстрое высвобождение или длительное высвобождение могут достигаться применением для соединений данного изобретения подходящих фармацевтических композиций или особенно в случае длительного высвобождения, применением устройств, таких как подкожные имплантаты или осмотические насосы. Данные соединения могут также вводиться липосомально.
Типичные композиции для перорального введения включают суспензии, которые могут содержать, например, микрокристаллическую целлюлозу для придания объема, альгиновую кислоту или альгинат натрия в качестве суспендирующего средства, метилцеллюлозу в качестве добавки, повышающей вязкость, и подсластители или вкусовые агенты, которые известны в данной области; таблетки немедленного высвобождения действующего вещества, которые могут содержать, например, микрокристаллическую целлюлозу, дикальцийфосфат, крахмал, стеарат магния, и/или лактозу, и/или другие эксципиенты, связующие вещества, наполнители, дезинтегранты, разбавители и лубриканты, которые известны специалистам в данной области. Соединения формулы I также могут доставляться через ротовую полость подъязычным и/или буккальным введением. Литые таблетки, прессованные таблетки или таблетки, полученные сушкой сублимацией, представляют собой типичные примеры форм, которые могут использоваться. Типичные композиции включают композиции, в которых присутствует(ют) соединение(я) данного изобретения с быстро растворяющимися разбавителями, такими как маннит, лактоза, сахароза и/или циклодекстрины. В такие композиции могут также включаться высокомолекулярные эксципиенты, такие как целлюлозы (avicel) или полиэтиленгликоли (PEG). Такие композиции могут также включать эксципиент, способствующий адгезии в слизистой оболочке, такой как гидроксипропилцеллюлоза (НРС), гидроксипропилметилцеллюлоза (НРМС), натрийкарбоксиметилцеллюлоза (SCMC), сополимер малеинового ангидрида (например, Gantrez) и средства для контроля высвобождения, такие как полиакриловый сополимер (например, Carbopol 934). Лубриканты, добавки, усиливающие скольжение, вкусовые добавки, красители и стабилизаторы также могут добавляться для облегчения производства и применения.
Типичные композиции для назального аэрозольного или ингаляционного введения включают растворы в физиологическом растворе, которые могут содержать, например, бензиловый спирт или другие подходящие консерванты, промоторы абсорбции для повышения биодоступности и/или другие добавки, способствующие растворению или диспергированию, которые хорошо известны в данной области.
Типичные композиции для парентерального введения включают растворы или суспензии для инъекций, которые могут содержать, например, подходящие нетоксичные приемлемые для парентерального введения разбавители или растворители, такие как маннит, 1,3-бутандиол, вода, раствор Рингера, изотонический раствор хлорида натрия или другие подходящие диспергирующие, смачивающие или суспендирующие средства, включая синтетические моно- или диглицериды, жирные кислоты, в том числе олеиновую кислоту, или Cemaphor.
Типичные композиции для ректального введения включают суппозитории, которые могут содержать, например, подходящий, не оказывающий раздражающего воздействия эксципиент, такой как какао-масло, синтетические сложные эфиры глицеридов или полиэтиленгликоли, которые являются твердыми при обычных температурах, но становятся жидкими и/или растворяются в ректальной полости для высвобождения лекарственного средства.
Типичные композиции для местного применения включают носитель для местного применения, такой как Plastibase (минеральное масло, желатинированное с полиэтиленом).
Следует представлять, что конкретная дозировка и частота приема для любого конкретного субъекта может изменяться и будет зависеть от различных факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия данного соединения, разновидность субъекта, его возраст, массу тела, общее состояния здоровья, пол и режим питания, способа и времени введения, скорости выделения, сочетания с другими лекарственными средствами и тяжести конкретного состояния.
ПРИМЕРЫ
В приведенных далее примерах представлены предпочтительные варианты осуществления данного изобретения. Однако данные примеры никоим образом не следует рассматривать как ограничивающие данное изобретение. 1Н ЯМР спектры соответствуют предполагаемым структурам. Масс-спектры записаны на спектрометре Perkin-Elmer API 150Ex с ионным турбораспылением с отрицательно заряженными ионами (ES-1) или положительно заряженными ионами (ES+1) с использованием колонки Zorbax SB-C8 (ЖХ-МС). Подходящие методики получения метил[3,5-хлор-4-(4-метокси-3-изопропилфенокси)]бензоата и метил-N-[3,5-дихлор-4-(4-метокси-3-изопропилфенокси)бензоил]глицина можно найти соответственно в следующих публикациях: «Novel Thyroid Receptor Ligands and Methods» Li, Y.-L.; Liu, Y.; Hedfors, A.; Malm, J.; Mellin, C.; Zhang, M. WO99/00353, PCT/EP98/04039 и "Novel diphenyl ether derivatives which are thyroid hormone beta-receptor ligands useful for treating metabolic disorders." Hangeland. J.; Zhang, M.; Caringal, Y.; Ryono, D.; Li.Y-L.; Malm, J.; Liu, Y.; Garg, N.; Litten, C.; Garcia Collazo, A.M.; Koehler, K.WO00/039077, PCT/IB99/02084 соответственно.
Методики получения 3,5-дихлор-2-метил-4-(4-гидрокси-3-изопропилфенокси)бензойной кислоты можно найти в "Benzamide ligands for the thyroid receptor", Ryono, Denis E., WO 0194293, патент США № 6395784.
Пример 1: N-[3,5-дихлор-4-(4-гидрокси-3-изопропил-5-метилфенокси)бензоил]глицин (Е1)
(а) Азотную кислоту (2,84 мл, 65%) при перемешивании добавляют к раствору метил[3,5-хлор-4-(3-изопропил-4-метоксифенокси)]бензоата (2,84 г, 7,64 ммоль) в бензоле (200 мл). Полученную реакционную смесь желтого цвета перемешивают при комнатной температуре в течение трех часов и затем выливают в насыщенный раствор гидрокарбоната натрия. Образующиеся органическую и водную фазы разделяют и водную фазу экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и концентрируют с получением 2,40 г твердого вещества светло-желтого цвета. Твердый продукт растворяют в этаноле (150 мл, 95,5%), добавляют дитионит натрия (Na2S2O4, чистота 85%, 7,12 г, 35,0 ммоль) и реакционную смесь кипятят с обратным холодильником в течение 16 часов. После этого к реакционной смеси добавляют вторую порцию дитионита натрия (3,00 г, 14,7 ммоль) и реакционную смесь кипятят в течение трех часов. Реакционную смесь охлаждают до комнатной температуры, нейтрализуют гидрокарбонатом натрия (насыщенный раствор) и концентрируют. Остаток разбавляют этилацетатом (75 мл) и промывают водой (50 мл). Органическую фазу сушат над безводным сульфатом натрия и концентрируют, получая твердое вещество светло-желтого цвета. Твердое вещество желтого цвета фильтруют через тонкий слой диоксида кремния, получая 2,2 г (75%) метил[3,5-дихлор-4-(3-амино-5-изопропил-4-метоксифенокси)]бензоата.
(b) Раствор нитрита натрия (0,30 г, 13 ммоль) в воде (5 мл) при энергичном перемешивании добавляют к смеси метил[3,5-дихлор-4-(3-амино-5-изопропил-4-метоксифенокси)]бензоата (1,1 г, 2,86 ммоль), метанола (100 мл) и соляной кислоты (75 мл, 37%). Реакционную смесь перемешивают в течение часа, затем добавляют раствор йодида калия (1,43 г, 8,6 ммоль) в воде (5 мл) и реакционную смесь перемешивают в течение 30 минут. Температуру внутри колбы в течение всей реакции поддерживают на уровне 0°С. По достижении комнатной температуры реакционную смесь коричневатого цвета экстрагируют хлороформом (3×50 мл), объединенные органические фазы промывают насыщенным раствором гидросульфата натрия, затем насыщенным раствором тиосульфата натрия. Органическую фазу концентрируют, получая маслянистый остаток темно-красного цвета, который очищают колоночной хроматографией (силикагель, н-гептан/этилацетат 95:5), получая 0,78 г (71%) метил[3,5-дихлор-4-(3-йод-5-изопропил-4-метоксифенокси)]бензоата в виде массы бледно-желтого цвета.
(с) Метил[3,5-дихлор-4-(3-йод-5-изопропил-4-метоксифенокси)]бензоат (0,500 мг, 1,01 ммоль), фосфат калия (1,07 г, 5,05 ммоль), метилбороновую кислоту (0,303 мг, 5,05 ммоль) и диоксан смешивают в трубке Шленка (Schlenk) в атмосфере газообразного азота. В трубку под атмосферой азота добавляют PdCl2 (dppf) и реакционную смесь выдерживают при 100°Св течение 16 часов. Реакционную смесь фильтруют через небольшой слой диоксида кремния и концентрируют. Темный остаток очищают колоночной хроматографией (силикагель, н-гептан/этилацетат 95:5), получая смесь (85:15) метил[3,5-дихлор-4-(3-изопропил-4-метокси-5-метилфенокси)]бензоата и метил[3,5-дихлор-4-(3-изопропил-4-метоксифенокси)]бензоата. Смесь получают в виде твердого белого вещества (0,250 мг, 55%) и используют в следующей стадии без дополнительной очистки.
(d) Смесь метил[3,5-дихлор-4-(3-изопропил-4-метокси-5-метилфенокси)]бензоата и метил[3,5-дихлор-4-(3-изопропил-4-метоксифенокси)]бензоата, полученную на предыдущей стадии (250 мг), растворяют в тетрагидрофуране (15 мл) и добавляют гидроксид лития (1Н, 15 мл). Смесь перемешивают при комнатной температуре в течение 90 минут, затем подкисляют соляной кислотой (1Н) и водную и органическую фазы разделяют. Органическую фазу сушат над сульфатом натрия и концентрируют, получая 0,250 г твердого белого вещества, которое смешивают с метиловым эфиром глицина (164 мг, 1,30 ммоль), гидрохлоридом 3-этил-1-[3-(диметиламино)пропил]карбодиимида (EDCl) (118 мг, 0,978 ммоль) и N,N-диметилформамидом (20 мл). Реакционную смесь перемешивают в течение 10 минут при комнатной температуре, после чего добавляют 1-гидроксибензотриазолгидрат (HOBt) (150 мл, 0,978 ммоль) и триэтиламин (0,272 мл, 1,96 ммоль). Смесь выдерживают при комнатной температуре в течение 48 часов, затем выливают в воду (150 мл) и нейтрализуют гидрокарбонатом натрия. Водную фазу экстрагируют этилацетатом (4×100 мл), объединенные органические фазы сушат над сульфатом магния и концентрируют. Остаток очищают колоночной хроматографией (силикагель, элюирование с градиентом: н-гептан/этилацетат от 7:3 до 5:5). Получают 165 мг (60%) метил-N-[3,5-дихлор-4-(5-изопропил-4-метокси-3-метилфенокси)бензоил]глицина в виде твердого белого вещества.
(е) Диметилсульфид трифторида бора (1,48 мг, 13,7 ммоль) при 4°С при перемешивании добавляют к раствору метил-N-[3,5-дихлор-4-(5-изопропил-4-метокси-3-метилфенокси)бензоил]глицина (155 мг, 0,352 ммоль) в дихлорметане (15 мл). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов, затем обрабатывают ледяной водой (30 мл), экстрагируют этилацетатом и концентрируют. Остаток очищают колоночной хроматографией (ЖХСД, силикагель, хлороформ/метанол/уксусная кислота, элюирование с градиентом: от 1/0/0 до 95/5/0,5), получая 110 мг (76%) N-[3,5-дихлор-4-(4-гидрокси-3-изопропил-5-метилфенокси)бензоил]глицина. ЖХ-МС (ES-1): m/z 410.
Пример 2: N-[3,5-дихлор-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е2)
(а) Трибромид бора (1Н, в дихлорметане) добавляют к раствору метил-N-[3,5-дихлор-4-(4-метокси-3-изопропилфенокси)бензоил]глицина (114 мг, 0,267 ммоль) в дихлорметане (5 мл) при -78°С. Полученную реакционную смесь коричневого цвета выдерживают при -25°С в течение 16 часов и при 4°С в течение двух часов. Добавляют смесь метанола (5 мл) и воды (5 мл) при -70°С, реакционную смесь концентрируют и разбавляют этилацетатом. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют. В результате получают 107 мг метил-N-[3,5-дихлор-4-(4-гидрокси-3-изопропилфенокси)бензоил]глицина в виде твердого вещества бежевого цвета.
(b) Бром (33 мкл) при перемешивании по каплям добавляют к смеси метил-N-[3,5-дихлор-4-(4-гидрокси-3-изопропилфенокси)бензоил]глицина (240 мг, 0,582 ммоль), уксусной кислоты (4 мл), ацетата натрия (88 мл, 0,64 ммоль) и нескольких капель воды. Реакционную смесь перемешивают при комнатной температуре в течение 16 часов, добавляют тиосульфат натрия (насыщенный раствор) и полученную смесь желтого цвета концентрируют. Остаток разбавляют этилацетатом и промывают водой. Водную фазу экстрагируют дихлорметаном, объединенные органические фазы промывают насыщенным раствором соли и сушат над сульфатом натрия. После концентрирования получают 330 мг твердого вещества желтого цвета, которое очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 100% н-гептана до смеси 20% н-гептана и 80% этилацетата), получая 200 мг (70%) метил-N-[3,5-дихлор-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицина в виде твердого белого вещества.
(С) Гидроксид лития (1Н, 9 мл) при комнатной температуре добавляют к смеси метил-N-[3,5-дихлор-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицина (150 мл, 0,305 ммоль) и тетрагидрофурана (9 мл). По истечении 16 часов реакционную смесь подкисляют соляной кислотой (1Н) и экстрагируют этилацетатом. Фильтрация через рыхлый слой диоксида кремния приводит к получению N-[3,5-дихлор-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицина с количественным выходом в виде твердого вещества светло-желтого цвета. ЖХ-МС (ES-1); m/z 476.
Пример 3: N-[3,5-дихлор-4-(2-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е3)
(а) Бром (13 мкл, 0,26 ммоль) по каплям при тщательном перемешивании добавляют к смеси метил-N-[3,5-дихлор-4-(4-метокси-3-изопропилфенокси)бензоил]глицина (50 мг, 0,12 ммоль), уксусной кислоты (1,0 мл), ацетата натрия (35 мг, 0,26 ммоль) и нескольких капель воды. Реакционную смесь перемешивают при комнатной температуре в течение трех часов, в течение 90 минут при 40°С и наконец при комнатной температуре в течение 16 часов. Добавляют ацетат натрия (17 мг) и бром (6 мкл) и реакционную смесь выдерживают при 40°С в течение двух часов. После этого реакционную смесь выдерживают в течение трех дней при 4°С. Добавляют тиосульфат натрия (насыщенный раствор) и полученную реакционную смесь желтого цвета концентрируют. Остаток разбавляют этилацетатом и промывают водой. Водную фазу экстрагируют дихлорметаном, объединенные органические фазы промывают насыщенным раствором соли и сушат над сульфатом натрия. После концентрирования остаток очищают колоночной хроматографией (ВЭЖХ, С8, ацетонитрил/вода/муравьиная кислота, элюирование с градиентом: от 20/80/0,5 до 100/0/0), получая 1,0 мг (2%) метил-N-[3,5-дихлор-4-(2-бром-4-метокси-5-изопропилфенокси)бензоил]глицина.
(b) Диметилсульфид трифторида бора (10 мкл, 80 мкмоль) при комнатной температуре с перемешиванием добавляют к раствору метил-N-[3,5-дихлор-4-(2-бром-4-метокси-5-изопропилфенокси)бензоил]глицина (1,0 мг, 2,0 мкмоль) и дихлорметана (0,50 мл). Реакционную смесь перемешивают при комнатной температуре в течение 8 часов и затем обрабатывают ледяной водой, экстрагируют этилацетатом и промывают водой. Органическую фазу сушат над сульфатом натрия и концентрируют, получая твердое вещество светло-желтого цвета. Твердый продукт очищают колоночной хроматографией (ЖХСД, силикагель, хлороформ/метано/уксусная кислота, элюирование с градиентом: от 1/0/0 до 95/5/0,5), получая 0,90 мг (94%) N-[3,5-дихлор-4-(2-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицина. ЖХ-МС (ES-1): m/z 476.
Пример 4: N-[3,5-дихлор-4-(3-хлор-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е4)
Метил[3,5-дихлор-4-(3-изопропил-4-метоксифенокси)]бензоат (664 мг, 1,8 ммоль), растворенный в ацетоне (40 мл), в течение 5 минут при 0°С добавляют при перемешивании к смеси гипохлорита кальция (515 мг, 3,6 ммоль), воды (10 мл) и уксусной кислоты (4 мл). Реакционную смесь перемешивают при 0°С в течение 30 минут и затем в течение 30 минут при комнатной температуре. Реакционную смесь выливают в воду, экстрагируют этилацетатом (3×50 мл), объединенные органические фазы промывают водой (4×30 мл) и концентрируют. Остаток очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 98/2 до 90/10), получая 240 мг (60%) метил[3,5-дихлор-4-(3-хлор-5-изопропил-4-метоксифенокси)]бензоата. Гипохлорит кальция, используемый в данной стадии, может быть замещен трет-бутилгипохлоритом.
(b) Метил[3,5-дихлор-4-(3-хлор-5-изопропил-4-метоксифенокси)]бензоат, метанол (25 мл) и гидроксид натрия (6Н, 4 мл) перемешивают при комнатной температуре в течение 8 часов. Реакционную смесь нейтрализуют водной соляной кислотой и экстрагируют этилацетатом (3×30 мл). Объединенные органические фазы промывают водой, концентрируют и остаток растирают с н-гептаном. В результате получают 133 мг (58%) 3,5-дихлор-4-(3-хлор-5-изопропил-4-метоксифенокси)бензойной кислоты в виде твердого белого вещества.
(с) Гидрохлорид метилового эфира глицина (83 мг, 0,66 ммоль) и триэтиламин (100 мг, 0,99 ммоль) при перемешивании добавляют к смеси 3,5-дихлор-4-(3-хлор-5-изопропил-4-метоксифенокси)бензойной кислоты (130 мг, 0,33 ммоль), EDCI (88 мг, 0,46 ммоль), HOBt (86 мг, 0,56 ммоль) и N,N-диметилформамида (15 мл). Реакционную смесь перемешивают в течение 48 часов при комнатной температуре, выливают в смесь соляной кислоты (1Н, 5,0 мл) и воды (50 мл) и экстрагируют этилацетатом (4×20 мл). Объединенные органические фазы сушат, промывают водой (4×15 мл), органическую фазу концентрируют и остаток очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 90/10 до 75/25). В результате получают 90 мг (59%) метил-N-[3,5-дихлор-4-(3-хлор-5-изопропил-4-метоксифенокси)бензоил]глицина.
(d) Диметилсульфид трифторида бора (1,0 мл) при комнатной температуре при перемешивании добавляют к раствору метил-N-[3,5-дихлор-4-(3-хлор-5-изопропил-4-метоксифенокси)бензоил]глицина (90 мг) в дихлорметане (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов, выливают в воду и экстрагируют этилацетатом (3×30 мл). Объединенные органические фазы промывают водой (4×20 мл), концентрируют и полученный остаток очищают колоночной хроматографией (силикагель, хлороформ/метанол/уксусная кислота 94:6:0,65). В результате получают 53 мг (63%) N-[3,5-дихлор-4-(3-хлор-4-гидрокси-5-изопропилфенокси)бензоил]глицина. ЖХ-МС (ES+1): m/z 434.
Пример 5: N-[3,5-дихлор-4-(3-циано-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е5)
(а) Метил[3,5-дихлор-4-(3-йод-5-изопропил-4-метоксифенокси)]бензоат (методика получения приведена в прим. 1 (a-b)) (200 мг, 0,40 ммоль), цианид меди (50 мг, 0,56 ммоль) и ДМФА (4 мл) нагревают до 120°С и выдерживают при этой температуре в течение 16 часов. После охлаждения до комнатной температуры темную реакционную смесь гасят насыщенным раствором гидрокарбоната натрия и экстрагируют этилацетатом. Органическую фазу сушат над сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией (ЖХСД, силикагель, н-гептан/этилацетат, элюирование с градиентом: от 1/0 до 9/1), получая 65 мг (41%) метил[3,5-дихлор-4-(3-циано-5-изопропил-4-метоксифенокси)]бензоата.
(b) Трибромид бора (1Н в дихлорметане, 0,93 мл) добавляют к раствору метил[3,5-дихлор-4-(3-циано-5-изопропил-4-метоксифенокси)]бензоата (32 мг, 81 мкмоль) в дихлорметане (0,75 мл) при -78°С. Смесь выдерживают в течение 20 часов при -25°С и в течение 16 часов при комнатной температуре, после чего гасят ледяной водой при 0°С и органический растворитель удаляют выпариванием. Остаток подкисляют соляной кислотой (1Н) и экстрагируют этилацетатом. Органическую фазу сушат над сульфатом натрия и фильтруют через рыхлый слой диоксида кремния. Полученное твердое вещество желтого цвета смешивают с метиловым эфиром глицина (23 мг, 0,18 ммоль), EDCI (35 мг, 0,18 ммоль) и N,N-диметилформамидом (1 мл). Реакционную смесь перемешивают в течение 10 минут при комнатной температуре, затем добавляют HOBt (28 мг, 0,18 ммоль) и триэтиламин (38 мкл, 0,27 ммоль). Реакционную смесь выдерживают в течение 16 часов при комнатной температуре, затем выливают в воду (5 мл) и нейтрализуют гидрокарбонатом натрия. Водную фазу экстрагируют этилацетатом (2×10 мл), объединенные органические фазы сушат над сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией (ЖХСД, силикагель, н-гептан/этилацетат, элюирование с градиентом: от 9/1 до 7/3). В результате получают 15 мг (42%) метил-N-[3,5-дихлор-4-(3-циано-4-гидрокси-5-изопропилфенокси)бензоил]глицина в виде твердого белого вещества.
(с) Трибромид бора (1Н в дихлорметане, 0,11 мл) добавляют к раствору метил-N-[3,5-дихлор-4-(3-циано-4-гидрокси-5-изопропилфенокси)бензоил]глицина (7 мл, 16 мкмоль) в дихлорметане (0,5 мл) при -78°С. Полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов и затем гасят ледяной водой. Органический растворитель удаляют выпариванием, остаток подкисляют соляной кислотой (1Н) и экстрагируют этилацетатом. Сушка над сульфатом натрия, концентрирование и очистка колоночной хроматографией (SPE, диоксид кремния (0,5 г, 3 мл), хлороформ/метанол/уксусная кислота, элюирование с градиентом: от 1/0/0 до 98/2/0,5) приводят к получению 3 мг (44%) N-[3,5-дихлор-4-(3-циано-4-гидрокси-5-изопропилфенокси)бензоил]глицина. ЖХ-МС (ES-1): m/z 421.
Пример 6: N-[3,5-дихлор-4-(3-фтор-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е6)
(а) К раствору метил[3,5-дихлор-4-(3-изопропил-4-метоксифенокси)]бензоата (2,0 г, 5,4 ммоль) в бензоле (200 мл) по каплям добавляют азотную кислоту (2,07 мл, 65%). Смесь перемешивают в течение 1 часа при комнатной температуре. Ход реакции контролируют ТСХ (65:35 н-гептан/этилацетат) и реакцию гасят насыщенным водным раствором гидрокарбоната натрия. Образующиеся органическую и водную фазы разделяют и водную фазу экстрагируют этилацетатом. Объединенную органическую фазу промывают водой, насыщенным раствором соли, органическую фазу сушат над сульфатом магния и концентрируют. Сырой продукт кристаллизуют из метанола, получая 1,8 г (80%) метил[3,5-дихлор-4-(5-изопропил-4-метокси-3-нитрофенокси)]бензоата.
(b) К раствору метил[3,5-дихлор-4-(5-изопропил-4-метокси-3-нитрофенокси)]бензоата (500 мг, 1,21 ммоль) в тетрагидрофуране (10 мл) добавляют гидроксид лития (5 мл, 1М). Смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь подкисляют соляной кислотой (1Н) и водную и органическую фазы разделяют. Водную фазу экстрагируют этилацетатом. Объединенную органическую фазу промывают водой, сушат над сульфатом магния и концентрируют. Сырой продукт очищают колоночной хроматографией (силикагель, хлороформ/метанол/уксусная кислота 98/2/0,3), получая 418 мг (86%) 3,5-дихлор-4-(5-изопропил-4-метокси-3-нитрофенокси)бензойной кислоты.
(с) Гидрохлорид метилового эфира глицина (262 мг, 2,08 ммоль) и триэтиламин (317 мг, 3,13 ммоль) при перемешивании добавляют к смеси 3,5-дихлор-4-(5-изопропил-4-метокси-3-нитрофенокси)]бензойной кислоты (418 мг, 1,04 ммоль), EDCI (401 мг, 2,08 ммоль), HOBt (320 мг, 2,08 ммоль) в N,N-диметилформамиде (50 мл). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре, выливают в смесь соляной кислоты (1Н, 10 мл) и воды (50 мл) и экстрагируют этилацетатом. Объединенные органические фазы промывают водой, сушат, органическую фазу концентрируют и остаток очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 98/2 до 60/40), получая 380 мг (78%) метил-N-[3,5-дихлор-4-(5-изопропил-4-метокси-3-нитрофенокси)бензоил]глицина.
(d) Смесь метил-N-[3,5-дихлор-4-(5-изопропил-4-метокси-3-нитрофенокси)бензоил]глицина (380 мг, 0,81 ммоль) и PtO2 (40 мг) в этилацетате (10 мл) перемешивают при комнатной температуре в атмосфере водорода (атмосферное давление) в течение 24 часов. Pt катализатор удаляют фильтрованием через целит, фильтрат концентрируют и очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 98/2 до 60/40), получая 320 мг (89%) метил-N-[3,5-дихлор-4-(3-амино-5-изопропил-4-метоксифенокси)бензоил]глицина.
(е) Раствор тетрафторбората нитрозония (55 мг, 0,47 ммоль) в дихлорметане (5 мл) охлаждают до 0°С и к раствору добавляют метил-N-[3,5-дихлор-4-(3-амино-5-изопропил-4-метоксифенокси)бензоил]глицин (189 мг, 0,43 ммоль). Реакционную смесь дополнительно перемешивают при 0°С в течение 1 часа. Дихлорметан удаляют продувкой азотом и к реакционной смеси добавляют о-ксилол (10 мл). Реакционную смесь кипятят с обратным холодильником в течение 1 часа и растворитель удаляют. Реакционную смесь очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 98/2 до 60/40), получая 50 мг (26%) метил-N-[3,5-дихлор-4-(3-фтор-5-изопропил-4-метоксифенокси)бензоил]глицина.
(f) Диметилсульфид трифторида бора (2,0 мл) при комнатной температуре при перемешивании добавляют к раствору метил-N-[3,5-дихлор-4-(3-фтор-5-изопропил-4-метоксифенокси)бензоил]глицина (50 мг, 0,11 ммоль) в дихлорметане (8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов, выливают в воду и экстрагируют этилацетатом. Объединенные органические фазы промывают водой, концентрируют и полученный остаток очищают колоночной хроматографией (ВЭЖХ, ACE-C8, ацетонитрил/0,05% муравьиная кислота в воде, элюирование с градиентом: от 5/95 до 60/40), получая 5 мг (20%) N-[3,5-дихлор-4-(3-фтор-4-гидрокси-5-изопропилфенокси)бензоил]глицина. ЖХ-МС (ES-1): m/z 414.
Пример 7: N-[3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е7)
(а) Суспензию трибромида бензилтриметиламмония (2,2 г, 5,63 ммоль) и СаСО3 (0,28 г, 2,82 ммоль) перемешивают в течение 2 часов при 20°С в растворе MeOH/CH2Cl2 (1:1) (10 мл), содержащем 3,5-дихлор-2-метил-4-(4-гидрокси-3-изопропилфенокси)бензойную кислоту (1,0 г, 2,82 ммоль). Когда ВЭЖХ показывает, что конверсия в желательный монобромированный фенол оптимальна, реакцию гасят добавлением 15 мл 1Н HCl. После удаления органических растворителей в вакууме роторного испарителя водную суспензию дважды экстрагируют EtOAc. Объединенные слои EtOAc сушат над MgSO4, фильтруют и EtOAc удаляют в вакууме. Полученный остаток очищают колоночной (С18) хроматографией с обращенной фазой (элюирование: смесь МеОН/Н2О, содержащая 0,1% ТФУК), получая 3,5-дихлор-2-метил-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензойную кислоту (400 мг).
(b) Газообразый HCl барботируют через раствор 3,5-дихлор-2-метил-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензойной кислоты (375 мл, 0,86 ммоль) в МеОН (20 мл) в течение 5 минут. Смесь перемешивают при 20°С в течение 22 часов, после чего ВЭЖХ показывает, что этерификация прошла полностью. Растворитель удаляют в вакууме, остаток растворяют в EtOAc, слой EtOAc промывают (2×) Н2О, затем насыщенным раствором соли и сушат над MgSO4. Летучие растворители удаляют в вакууме, получая сырой метил-3,5-дихлор-2-метил-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоат в виде твердого вещества коричневого цвета, которое используют в следующих стадиях без дополнительной очистки.
(с) Через суспензию порошка безводного К3РО4 (473 мг, 2,23 ммоль) и метилбороновой кислоты (133 мг, 2,23 ммоль) в диоксане (5 мл), содержащем метил-3,5-дихлор-2-метил-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоат (263 мг, 0,59 ммоль) в течение 15 минут пропускают Ar, затем добавляют Pd(dppf)Cl2 (168 мг, 0,21 ммоль). Смесь кипятят с обратным холодильником в течение 3 часов, затем охлаждают, разбавляют EtOAc и фильтруют. Остаток, образующийся после удаления летучих растворителей, хроматографируют на силикагеле. Прогрессивное элюирование смесью CH2Cl2/гексан с соотношением сначала 1:1, затем 3:1 приводит к получению 125 мг метил-3,5-дихлор-2-метил-4-(3-метил-4-гидркоси-5-изопропилфенокси)бензоата в виде пены оранжевого цвета.
(d) Раствор метил-3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоата (125 мг, 0,33 ммоль) и LiOH H2O (68 мг, 1,63 ммоль) в смеси ТГФ/Н2О (5:1) (6 мл) перемешивают в течение ночи при 20°С в атмосфере N2. Раствор концентрируют в вакууме и подкисляют до рН 1 с помощью 1Н HCl. Полученное твердое вещество желтовато-коричневого цвета собирают фильтрованием, промывают Н2О и сушат на воздухе, получая 114 мг 3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензойной кислоты.
(е) К раствору неочищенной 3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензойной кислоты (114 мг, 0,31 ммоль) в ДМФА (5 мл) при перемешивании в атмосфере N2 последовательно добавляют EDCI, (83 мг, 0,43 ммоль), HOBt (80 мг, 0,53 ммоль), метилглицинат гидрохлорид (78 мг, 0,62 ммоль) и Et3N (0,13 мл, 0,93 ммоль). Смесь перемешивают в течение 21 часа при 20°С, после чего ЖХМС показывает наличие исходной бензойной кислоты, поэтому добавляют 8 мг метилглицината гидрохлорида и 13 мл Et3N. Реакцию завершают через 3 часа разбавлением 1Н HCl и 25 мл Н2О. Водную фазу экстрагируют (1×) EtOAc, объединенные слои EtOAc промывают (2×) Н2О и (1×) насыщенным раствором соли, после чего сушат над MgSO4. После концентрирования получают 163 мг неочищенного глицинамида, который хроматографируют на силикагеле (элюирование: EtOAc/гексан 1:4), получая 96 мг целевого метил-N-(3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил)глицината в виде твердого вещества бежевого цвета.
(f) Раствор метил-N-(3,5-дихлор-2-метил-4-(3-метил-4-гидроксид-5-изопропилфенокси)бензоил)глицината (96 мг, 0,22 ммоль) и LiOH Н2О (46 мг, 1,09 ммоль) в смеси ТГФ/Н2O (5:1) (6 мл) перемешивают в течение ночи при 20°С в атмосфере N2. Раствор концентрируют в вакууме и подкисляют до рН 1 добавлением 1Н HCl. После экстракции водной фазы (3×) EtOAc объединенные слои EtOAc сушат над MgSO4. Концентрирование приводит к получению 92 мг неочищенного глицинамида, который очищают колоночной (С18) хроматографией с обращенной фазой (элюирование с градиентом: смесь MeCN/Н2О, содержащая 0,1 ТФУК), получая 72 мг целевого N-(3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил)глицина: m/z 426 (М+Н) и 424 (М-Н).
Пример 8: N-[3,5-дибром-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е8)
Соединение Е8 получают в соответствии с методикой, описанной для получения соединения Е7 в примере 7.
Пример 9: N-[3,5-диметил-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е9).
Соединение Е9 получают в соответствии с методикой, описанной в примере 7 для получения соединения Е7.
Пример 10: L-N-[3,5-дибром-4-(3-фтор-4-гидрокси-5-изопропилфенокси)фенилацетил]валин (Е10)
(а) К раствору метил[3,5-дибром-4-(3-изопропил-4-метоксифенокси)фенил]ацетата (1,0 г, 2,12 ммоль) в бензоле (150 мл) по каплям добавляют азотную кислоту (0,81 мл, 65%). Смесь дополнительно перемешивают в течение 1 часа при комнатной температуре. Ход реакции контролируют ТСХ (65:35 н-гептан/этилацетат) и реакцию гасят насыщенным водным раствором гидрокарбоната натрия. Полученные органическую и водную фазы разделяют и водную фазу экстрагируют этилацетатом. Объединенные органические фазы промывают водой, насыщенным раствором соли, органическую фазу сушат над сульфатом магния и концентрируют. Сырой продукт кристаллизуют из метанола, получая 1,1 г (99%) метил[3,5-дибром-4-(5-изопропил-4-метокси-3-нитрофенокси)фенил]ацетата.
(b) Метил[3,5-дибром-4-(5-изопропил-4-метокси-3-нитрофенокси)фенил]ацетат (1,1 г, 2,12 ммоль) растворяют в этаноле (50 мл) и добавляют гидросульфит натрия (1,85 г, 10,76 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 48 часов. Ход реакции контролируют ТСХ (65:35 н-гептан:EtOAc) и после завершения реакции реакционную смесь разбавляют этилацетатом и промывают водой и насыщенным раствором соли. Органический слой суша над MgSO4 и концентрируют. Остаток очищают колоночной хроматографией (силикагель, н-гептан/EtOAc 1:1), получая метил[3,5-дибром-4-(3-амино-5-изопропил-4-метоксифенокси)фенил]ацетат (457 мг, 44%).
(с) Раствор тетрафторбората нитрозония (121 мг, 1,03 ммоль) в дихлорметане (10 мл) охлаждают до 0°С и добавляют метил[3,5-дибром-4-(3-амино-5-изопропил-4-метоксифенокси)фенил]ацетат (457 мг, 0,94 ммоль). Реакционную смесь перемешивают при 0°С в течение часа. Дихлорметан удаляют, пропуская через раствор струю газообразного азота, и к полученному остатку добавляют о-ксилол (10 мл). Реакционную смесь кипятят с обратным холодильником в течение часа и органическую фазу концентрируют в вакууме. Остаток очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 98/2 до 60/40), получая 205 мг (44%) метил[3,5-дибром-4-(3-фтор-5-изопропил-4-метоксифенокси)фенил]ацетата.
(d) К раствору метил[3,5-дибром-4-(3-фтор-5-изопропил-4-метоксифенокси)фенил]ацетата (205 мг, 0,42 ммоль) в тетрагидрофуране (30 мл) добавляют гидроксид лития (30 мл, 1Н). Реакционную смесь перемешивают в течение ночи при комнатной температуре, подкисляют соляной кислотой (1Н) и водную и органическую фазы разделяют. Водную фазу экстрагируют этилацетатом. Объединенную органическую фазу промывают водой, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают колоночной хроматографией (силикагель, хлороформ/метанол/уксусная кислота 98/2/0,3), получая 210 мг (99%) 3,5-дибром-4-(3-фтор-5-изопропил-4-метоксифенокси)фенилуксусной кислоты.
(е) Гидрохлорид метилового эфира L-валина (148 мг, 0,88 ммоль) и триэтиламин (134 мг, 1,32 ммоль) при перемешивании добавляют к смеси 3,5-дибром-4-(3-фтор-5-изопропил-4-метоксифенокси)фенилуксусной кислоты (210 мг, 0,44 ммоль), EDCI (169 мг 0,88 ммоль), HOBt (135 мг, 0,88 ммоль) в N,N-диметилформамиде (25 мл). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре, выливают в смесь соляной кислоты (1Н, 5 мл) и воды (30 мл) и экстрагируют этилацетатом. Объединенные органические фазы сушат, органическую фазу концентрируют и остаток очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 98/2 до 60/40), получая 224 мг (86%) L-метил-N-[3,5-дибром-4-(3-фтор-5-изопропил-4-метоксифенокси)фенилацетил]валината.
(f) Диметилсульфид трифторида бора (4,0 мл) при комнатной температуре при перемешивании добавляют к раствору L-метил-N-[3,5-дибром-4-(3-фтор-5-изопропил-4-метоксифенокси)фенилацетил]валина (224 мг, 0,38 ммоль) в дихлорметане (25 мл). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов, выливают в воду и экстрагируют этилацетатом. Объединенные органические фазы промывают водой, концентрируют и полученный остаток очищают колоночной хроматографией (силикагель, хлороформ/метанол/уксусная кислота 98/2/0,3), получая 93 мг (44%) L-N-[3,5-дибром-4-(3-фтор-4-гидрокси-5-изопропилфенокси)-фенилацетил]валина: m/z 562/2 (М+Н); 560,0 (М-Н)
Пример 11: D-N-[3,5-дибром-4-(3-хлор-4-гидрокси-5-изопропилфенокси)фенилацетил]фенилглицин (Е11)
(а) К раствору 3,5-дибром-4-(3-изопропил-4-метоксифенокси)фенилуксусной кислоты (1,5 г, 3,4 ммоль) в уксусной кислоте (40 мл) при комнатной температуре при перемешивании добавляют тетрахлорйодат бензилтриметиламмония (1,42 г, 3,4 ммоль). Реакционную смесь перемешивают в течение часа и фильтруют. Фильтрат концентрируют, продукт экстрагируют этилацетатом, промывают насыщенным водным раствором NaHCO3, водным раствором NaCl, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают колоночной хроматографией (силикагель, хлороформ/метанол/уксусная кислота 98/2/0,3), получая 802 мг (50%) 3,5-дибром-4-(3-хлор-4-гидрокси-5-изопропилфенокси)фенилуксусной кислоты.
(b) Гидрохлорид метилового эфира D-фенилглицина (674 мг, 3,34 ммоль) и триэтиламин (507 мг, 5,02 ммоль) при перемешивании добавляют к смеси 3,5-дибром-4-(3-хлор-4-гидрокси-5-изопропилфенокси)фенилуксусной кислоты (800 мг, 1,7 ммоль), EDCI (641 мг, 3,34 ммоль) и HOBt (512 мг, 3,34 ммоль) в N,N-диметилформамиде (100 мл). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре, выливают в смесь соляной кислоты (1Н, 5 мл) и воды (30 мл) и экстрагируют этилацетатом. Объединенные органические фазы сушат, органическую фазу концентрируют и остаток очищают колоночной хроматографией (силикагель, н-гептан/этилацетат, элюирование с градиентом: от 98/2 до 60/40), получая 800 мг (75%) D-метил-N-[3,5-дибром-4-(3-хлор-5-изопропил-4-метоксифенокси)фенилацетил]фенилглицината.
(с) Диметилсульфид трифторида бора (10 мл) при комнатной температуре при перемешивании добавляют к раствору D-метил-N-[3,5-дибром-4-(3-хлор-5-изопропил-4-метоксифенокси)фенилацетил]фенилглицината (800 мг, 1,3 ммоль) и дихлорметана (80 мл). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов, выливают в воду и экстрагируют этилацетатом. Объединенные органические фазы промывают водой, концентрируют и остаток очищают колоночной хроматографией (силикагель, хлороформ/метанол/уксусная кислота 98/2/0,3), получая 350 мг (44%) D-N-[3,5-дибром-4-(3-хлор-4-гидрокси-5-изопропилфенокси)фенилацетил]фенилглицина: m/z 512,0 (М+Н); 510,0 (М-Н).
Пример 12: L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]валин (Е12)
(a) Метил[3,5-дибром-4-(4-гидрокси-3-изопропилфенокси)-фенил] ацетат (2,0 г, 4,4 ммоль), SnCl4 (25 мкл, 0,2 ммоль), триоктиламин (0,77 мл, 1,76 ммоль) и толуол (15 мл) смешивают в реакционной емкости. Смесь перемешивают при комнатной температуре в течение 20 минут, затем к реакционному раствору добавляют параформальдегид (0,264 г, 8,8 ммоль). Реакционную емкость герметично закрывают, и температура возрастает до 105°С. Смесь перемешивают в течение 20 часов, после чего реакцию гасят добавлением ледяной воды и подкислением соляной кислотой (1Н). Экстракция этилацетатом, промывка насыщенным раствором соли, сушка над Na2SO4 и концентрирование приводят к получению сырого продукта. Остаток очищают колоночной хроматографией (диоксид кремния, элюирование с градиентом: н-гептан/этилацетат от 1/0 до 4/1), получая 1,08 г (51%) метил[3,5-дибром-4-(3-формил-4-гидрокси-5-изопропилфенокси)фенил]ацетата.
(b) Триэтилсилан при комнатной температуре добавляют к смеси метил[3,5-дибром-4-(3-формил-4-гидрокси-5-изопропилфенокси)фенил]ацетата (243 мг, 0,45 ммоль) и трифторуксусной кислоты (10 мл). Смесь перемешивают в течение 12 часов, затем концентрируют посредством совместного выпаривания с толуолом. Остаток очищают колоночной хроматографией (диоксид кремния, н-гептан/этилацетат, элюирование с градиентом: от 9/1 до 7/3), получая 197 мг (84%) метил[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенил]ацетата.
(с) Смесь метил[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенил]ацетата (194 мг, 0,41 ммоль) и гидроксида лития (20 мл, 1Н) в тетрагидрофуране (10 мл) перемешивают в течение 10 часов при комнатной температуре. Реакционную смесь нейтрализуют охлажденной водной соляной кислотой с последующей экстракцией этилацетатом (3×30 мл). Объединенные органические слои промывают водой (4×25 мл) и выпаривают совместно с ацетонитрилом. Остаток очищают колоночной хроматографией (диоксид кремния, элюирование с градиентом: хлороформ/метанол/уксусная кислота от 1/0/0 до 90/10/1), получая 169 мг (90%) 3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенил]уксусной кислоты.
(d) Смесь 3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]уксусной кислоты (0,44 г, 0,96 ммоль), метилового эфира L-валина (322 мг, 1,92 ммоль), 3-этил-1-[3-(диметиламино)пропил]карбодиимида гидрохлорида (EDCl) (174 мг, 1,44 ммоль) и N,N-диметилформамида (9 мл) перемешивают в течение 5 минут при комнатной температуре, затем добавляют 1-гидроксибензотриазолгидрат (HOBt) (220 мг, 1,44 ммоль) и триэтиламин (0,401 мл, 12,88 ммоль). Спустя 16 часов добавляют гексафторфосфат бромтрипирролидинфосфония (0,2 г, 0,43 ммол). Смесь перемешивают при комнатной температуре в течение 1,75 часа, затем разбавляют водой (10 мл) и соляной кислотой (10 мл, 1Н), водный слой экстрагируют этилацетатом (3×20 мл) и объединенные органические слои промывают насыщенным раствором соли (40 мл). После сушки над Na2SO4, фильтрации и концентрирования сырой продукт фильтруют через рыхлый слой диоксида кремния. Очистка на chromatotron (диоксид кремния, 4 мм, н-гептан/этилацетат 6/4) приводит к получению L-метил-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]валината.
(е) К раствору L-метил-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]валината (240 мг, 0,42 ммоль) в тетрагидрофуране (4 мл) при перемешивании добавляют гидроксид лития (4 мл, 1Н). Смесь перемешивают в течение часа, затем значение рН реакционной смеси доводят до 1 добавлением соляной кислоты (1Н). Органическую фазу концентрируют в вакууме. Остаток экстрагируют этилацетатом, объединенные органические фазы сушат над Na2SO4 и концентрируют. Остаток очищают колоночной хроматографией (ЖХСД, диоксид кремния, хлороформ/метанол/уксусная кислота, элюирование с градиентом: от 1/0/0 до 98/2/0,3), получая L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]валин в виде твердого белого вещества (0,14 г, 60%) ЖХ-МС (ES-1): m/z 554.
Пример 13: L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]фенилглицин (Е13)
(а) Раствор 3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилуксусной кислоты (18 мг, 0,04 ммоль), диизопропилэтиламина (33 мкл, 0,2 ммоль), гексафторфосфата бромтрипирролидинфосфония (22 мг, 0,05 ммоль), 1-гидроксибензотриазолгидрата (7 мг, 0,05 ммоль), метилового эфира D-фенилглицина (16 мг, 0,08 ммоль) и дихлорметана (1 мл) перемешивают в течение 30 минут при комнатной температуре. После этого реакционную смесь промывают соляной кислотой (2 мл, 1Н) и водный слой экстрагируют хлороформом (3×2 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют в вакууме. Очистка колоночной хроматографией (диоксид кремния, н-гептан/этилацетат, элюирование с градиентом: от 4/1 до 7/3) приводит к получению 16 мг (74%) L-метил-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]фенилглицината.
(b) L-метил-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]фенилглицинат (15 мл, 0,025 ммоль) растворяют в тетрагидрофуране (0,5 мл) и полученный раствор обрабатывают водным раствором гидроксидом лития (0,5 мл, 1Н) в течение 1 часа при комнатной температуре. Реакционную смесь подкисляют соляной кислотой (1Н) и органическую фазу удаляют в вакууме. Остаток экстрагируют этилацетатом (3×5 мл), объединенные органические фазы сушат над Na2SO4 и концентрируют в вакууме. Остаток очищают колоночной хроматографией (ВЭЖХ, С8, ацетонитрил/вода (0,5% муравьиной кислоты), элюирование с градиентом: от 1/4 до 1/0), получая L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]фенилглицин в виде твердого белого вещества (6 мг, 41%). ЖХ-МС (ES-1): m/z 588.
Пример 14: L-N-[3,5-дибром-4-(3,5-диметил-4-гидроксифенокси)фенилацетил]фенилглицин (Е14)
(а) Тетрафторборат бис-(4-метоксифенилйодония) (5,48 г, 12,8 ммол) и медный порошок (1,25 г, 19,6 ммоль) суспендируют в дихлорметане (25 мл) и полученную суспензию охлаждают до 0°С. К суспензии, накрытой алюминиевой фольгой, при перемешивании добавляют раствор метил(3,5-дибром-4-гидроксифенил)ацетата (3,19 г, 9,8 ммоль), триэтиламина (1,64 мл, 11,8 ммоль) и дихлорметана (35 мл). Смесь перемешивают в темноте при комнатной температуре в течение 20 часов, затем промывают соляной кислотой (1Н) и для разделения фаз используют сепаратор фаз (IST). Оставшуюся кислотную фазу экстрагируют хлороформом и объединенные органические фазы концентрируют в вакууме. Остаток очищают колоночной хроматографией (ЖХСД, силикагель, н-гептан/этилацетат, элюирование с градиентом: от 1/0 до 4/1), получая 2,5 г (45%) метил[3,5-дибром-4-(4-гидроксифенокси)фенил]ацетата.
(b) BF3-SMe2 (24 мл, 228 ммоль) при перемешивании добавляют к раствору метил[3,5-дибром-4-(4-метоксифенокси)фенил]ацетата (2,5 г, 5,8 ммоль) и дихлорметана (50 мл) при 0°С. Реакционную смесь перемешивают при комнатной температуре в течение 20 часов и затем гасят ледяной водой (50 мл). Два слоя разделяют в фазовом сепараторе и оставшуюся водную фазу экстрагируют этилацетатом. Фильтрование через слой диоксида кремния приводит к получению неочищенной 3,5-дибром-4-(4-гидроксифенокси)фенилуксусной кислоты. Остаток растворяют в метаноле (40 мл) и к реакционной смеси осторожно добавляют тионилхлорид (4 мл). Смесь кипятят с обратным холодильником в течение 1,5 часа, затем органическую фазу удаляют в вакууме и остаток растворяют в хлороформе. Органическую фазу промывают водой и концентрируют, получая метил[3,5-дибром-4-(4-гидроксифенокси)фенил]ацетат в виде твердого вещества бежевого цвета (1,8 г, 75%).
(с) Гексаметилентетрамин (262 мг, 1,87 ммоль) добавляют к раствору метил[3,5-дибром-4-(гидроксифенокси)фенил]ацетата (370 мг, 0,89 ммоль) и трифторуксусной кислоты (1,5 мл). Реакционную смесь перемешивают в течение 16 часов при 96°С, затем охлаждают до комнатной температуры. Добавляют соляную кислоту (3 мл, 2Н), полученную реакционную смесь перемешивают в течение часа и экстрагируют хлороформом. Органическую фазу концентрируют в вакууме и растворяют в этилацетате. Органическую фазу промывают соляной кислотой (2Н), водой и насыщенным раствором соли, сушат над Na2SO4 и концентрируют. Остаток очищают колоночной хроматографией (ЖХСД, диоксид кремния, н-гептан/этилацетат, элюирование с градиентом: от 1/0 до 4/1), получая метил[3,5-дибром-4-(3-формил-4-гидроксифенокси)фенил]ацетат в виде твердого белого вещества (150 мг, 38%).
(d) Смесь метил[3,5-дибром-4-(3-формил-4-гидроксифенокси)фенилацетата (250 мг, 0,56 ммоль) и трифторуксусной кислоты (5,5 мл) обрабатывают триэтилсиланом (0,34 мл, 2,13 ммоль) и реакционную смесь перемешивают в течение 16 часов при комнатной температуре. Реакционную смесь концентрируют и подвергают совместному выпариванию (толуол, дихлорметан), в результате получают остаток, который фильтруют через рыхлый слой диоксида кремния. Очистка колоночной хроматографией (ЖХСД, диоксид кремния, элюирование с градиентом: н-гептан/этилацетат от 1:0 до 4/1) приводит к получению 110 мг (46%) метил[3,5-дибром-4-(4-гидрокси-3-метилфенокси)фенил]ацетата.
(е) Раствор метил[3,5-дибром-4-гидрокси-3-метилфенокси)фенил]ацетата (110 мг, 0,26 ммоль), гексаметилентетрамина (77 мг, 0,55 ммоль) и трифторуксусной кислоты 92,5 мл) перемешивают при 100°С в течение 20 часов. Реакционный раствор разбавляют соляной кислотой (5 мл, 2Н), полученную смесь перемешивают при комнатной температуре в течение 30 минут и экстрагируют хлороформом. Органическую фазу концентрируют, разбавляют этилацетатом и промывают соляной кислотой (2×10 мл, 2Н), водой (10 мл) и насыщенным раствором соли (10 мл). Органический слой сушат над Na2SO4, концентрируют и фильтруют через рыхлый слой диоксида кремния, получая 60 мг (50%) метил[3,5-дибром-4-(3-формил-4-гидрокси-5-метилфенокси)фенил]ацетата.
(f) К смеси метил[3,5-дибром-4-(3-формил-4-гидрокси-5-метилфенокси)фенил]ацетата (60 мг, 0,13 ммоль) и трифторуксусной кислоты (1,5 мл) добавляют триэтилсилан (62 мкл, 0,39 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 20 часов, органическую фазу концентрируют и остаток подвергают совместному выпариванию (толуол), получая 60 мг неочищенного метил[3,5-дибром-4-(3,5-дибром-4-гидроксифенокси)фенил]ацетата. Неочищенный продукт используют на следующей стадии. Гидроксид лития (1,5 мл, 1Н) при перемешивании добавляют к раствору метил[3,5-дибром-4-(3,5-дибром-4-гидроксифенокси)фенил]ацетата в тетрагидрофуране (1,5 мл). Полученную смесь перемешивают в течение 2 часов при комнатной температуре, подкисляют соляной кислотой (1Н) и экстрагируют этилацетатом. Органическую фазу сушат над Na2SO4 и концентрируют в вакууме. Остаток очищают колоночной хроматографией (ВЭЖХ, С8, ацетонитрил/вода (0,5% муравьиная кислота), элюирование с градиентом: от 1/4 до 1/0), получая 16 мг (15% для двух стадий) 3,5-дибром-4-(3,5-дибром-4-гидроксифенокси)фенилуксусной кислоты.
(g) 3,5-Дибром-4-(3,5-дибром-4-гидроксифенокси)фенилуксусную кислоту (16 мг, 0,037 ммоль), диизопропилэтиламин (33 мкл, 0,2 ммоль), гексафторфосфат бромтрипирролидинофосфония (21 мг, 0,044 ммоль), 1-гидроксибензотриазолгидрат (7 мг, 0,05 ммоль), метиловый эфир D-фенилглицина (15 мг, 0,074 ммоль) и дихлорметан (0,5 мл) объединяют и перемешивают при комнатной температуре в течение 1 часа, после чего выдерживают при 4°С в течение 20 часов. Реакционную смесь промывают соляной кислотой (1Н) и полученный водный слой экстрагируют хлороформом. Объединенные органические слои концентрируют в вакууме и остаток очищают колоночной хроматографией (диоксид кремния, элюент: хлороформ), получая 10 мг (47%) L-метил-N-[3,5-дибром-4-(3,5-диметил-4-гидроксифенокси)фенилацетил]фенилглицината.
(h) L-Метил-N-[3,5-дибром-4-(3,5-диметил-4-гидроксифенокси)фенилацетил]фенилглицинат (10 мг, 0,017 ммоль) растворяют в тетрагидрофуране (0,25 мл) и добавляют гидроксид лития (0,25 мл, 1Н). Реакционную смесь перемешивают в течение 1 часа при комнатной температуре, затем добавляют 1М соляную кислоту (1Н) до достижения значения рН 1. Полученную смесь экстрагируют этилацетатом и объединенные органические слои сушат над Na2SO4. Смесь концентрируют и очищают препаративной ТСХ, получая 5 мг (52%) L-N-[3,5-дибром-4-(3,5-диметил-4-гидроксифенокси)фенилацетил]фенилглицина. ЖХ-МС (ES-1): m/z 560.
Соединения примеров 1-6 проявляют аффиности связывания с тиреоидным бэта-рецептором со значением IC50 в интервале от 1,0 до 100 нМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ПРИ ПОВЫШЕНИИ ЭНДОГЕННОГО ЭРИТРОПОЭТИНА | 2004 |
|
RU2379291C2 |
| ПРОЛЕКАРСТВА ПИРИДОНАМИДОВ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2014 |
|
RU2692766C1 |
| ПИРИДИН-2-ИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИММУНОМОДУЛИРУЮЩИХ АГЕНТОВ | 2009 |
|
RU2494099C2 |
| ПРОИЗВОДНЫЕ ПИРИДИН-3-ИЛА В КАЧЕСТВЕ ИММУНОМОДУЛИРУЮЩИХ АГЕНТОВ | 2007 |
|
RU2454413C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| ПРОЛЕКАРСТВА ПИРИДОНАМИДОВ, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ НАТРИЕВЫХ КАНАЛОВ | 2014 |
|
RU2811402C2 |
| АГЕНТ, ПРОМОТИРУЮЩИЙ ПРОДУЦИРОВАНИЕ/СЕКРЕЦИЮ НЕЙРОТРОФИНА, ВКЛЮЧАЮЩИЙ ПРОИЗВОДНОЕ АЗОЛА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2260003C2 |
| СОСТАВЫ И СМЕСИ ДЛЯ ДОСТАВКИ АКТИВНЫХ АГЕНТОВ | 2005 |
|
RU2403237C2 |
| ФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИММУНОМОДУЛЯТОРОВ | 2007 |
|
RU2442780C2 |
| ЗАМЕЩЕННЫЕ АМИНОИНДАНЫ И ИХ АНАЛОГИ, И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2009 |
|
RU2522586C2 |
Данное изобретение относится к производные бензамида или фенилацетамида, которые являются лигандами тиреоидных рецепторов, и к способам получения данных соединений. Кроме того, заявляется фармацевтическая композиция, обладающая активностью в отношении рецептора тиреоидного гормона, содержащая эффективное количество заявленного соединения или его фармацевтически эффективной соли в сочетании с фармацевтически приемлемым носителем, и способ ее получения. В настоящем изобретении также заявляется способ профилактики, ингибирования или лечения заболевания, которое зависит от экспрессии Т3 регулируемого гена или ассоциировано с нарушением метаболизма, который включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества заявленного соединения. Целью изобретения является применение соединения для изготовления лекарственного средства для ингибирования или лечения заболевания, которое зависит от экспрессии Т3 регулируемого гена или связанного с нарушением метаболизма. 8 н. и 4 з.п. ф-лы.
N-[3,5-дихлор-4-(3-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е2);
N-[3,5-дихлор-4-(2-бром-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е3);
N-[3,5-дихлор-4-(3-хлор-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е4);
N-[3,5-дихлор-4-(3-циано-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е5);
N-[3,5-дихлор-4-(3-фтор-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е6);
N-[3,5-дихлор-2-метил-4-(3-метил-4-гидрокси-5-изопропилфенокси)бензоил]глицин (Е7);
L-N-[3,5-дибром-4-(3-фтор-4-гидрокси-5-изопропилфенокси)фенилацетил]валин (Е10);
D-М-[3,5-дибром-4-(3-хлор-4-гидрокси-5-изопропилфенокси)фенилацетил]фенилглицин (Е11);
L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]валин (E12);
L-N-[3,5-дибром-4-(4-гидрокси-3-изопропил-5-метилфенокси)фенилацетил]фенилглицин (Е13);
L-N-[3,5-дибром-4-(3,5-диметил-4-гидроксифенокси)фенилацетил]фенилглицин (Е14).
| РЕТИНОИДОПОДОБНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2163590C2 |
| US 6395784 B1, 28.05.2002 | |||
| СПОСОБ ЭКСТРАКЦИОННОЙ ОБРАБОТКИ РАЗЛИЧНЫХ ВИДОВ ЕСТЕСТВЕННОГО ТВЕРДОГО СЫРЬЯ | 1933 |
|
SU39077A1 |
| Регулятор температуры | 1976 |
|
SU580550A1 |

