Область изобретения
Настоящее изобретение относится к синергической комбинации альфа-2-дельта лиганда и ингибитора обратного захвата серотонина-норадреналина двойного действия (DSNRI) или одного или обоих из селективного ингибитора обратного захвата серотонина (SSRI) и селективного ингибитора обратного захвата норадреналина (SNRI) для лечения боли. Оно относится также к способу лечения боли посредством применения эффективных количеств синергических комбинаций альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI.
Уровень техники
Лиганд рецептора альфа-2-дельта представляет собой любую молекулу, которая связывается с любым подтипом субъединицы альфа-2-дельта кальциевого канала человека. Субъединица альфа-2-дельта кальциевого канала включает в себя ряд подтипов рецептора, которые описаны в литературе: например, N.S.Gee, J.P.Brown, V.U.Dissanayake, J.Offord, R.Thurlow, and G.N.Woodruff, J-Biol-Chem 271 (10):5768-76, 1996, (тип 1); Gong, J.Hang, W.Kohler, Z.Li, and T-Z.Su, J. Membr. Biol. 184 (1):35-43, 2001 (типы 2 и 3); E.Marais, N.Klugbauer and F.Hofmann, Mol. Pharmacol. 59 (5):1243-1248, 2001 (типы 2 и 3); N.Qin, S.Yagel, M.L.Momplaisir, E.E.Codd and M.R.D'Andrea, Mol. Pharmacol. 62 (3):485-496, 2002 (тип 4). Они также могут быть известны как аналоги GABA.
Лиганды альфа-2-дельта описаны для ряда показаний. Наиболее известный лиганд альфа-2-дельта, габапентин (Neurontin®), 1-(аминометил)циклогексилуксусная кислота, впервые был описан в патентной литературе в патентном семействе, включающем US 4024175. Соединение разрешено для лечения эпилепсии и невропатической боли.
Второй лиганд альфа-2-дельта, прегабалин, (S)-(+)-4-амино-3-(2-метилпропил)бутаноевая кислота описан в публикации европейской патентной заявки номер ЕР 641330 в качестве противосудорожного лекарственного средства, пригодного для лечения эпилепсии, и ЕР 0934061 для лечения боли.
Кроме того, международная публикация патентной заявки WO 0128978 описывает ряд новых бициклических аминокислот, их фармацевтически приемлемые соли и их пролекарства формулы:

где n представляет собой целое число от 1 до 4, в которых существуют стереоцентры, каждый центр независимо может представлять собой R или S; предпочтительными соединениями являются соединения формул I-IV, выше, в которых n представляет собой целое число от 2 до 4.
Позднее международная публикация патентной заявки WO 02/85839 описывает лиганды альфа-2-дельта следующих формул:






где R1 и R2 каждый независимо выбран из Н, линейного или разветвленного алкила из 1-6 атомов углерода, циклоалкила из 3-6 атомов углерода, фенила и бензила, при условии, что, за исключением случая трициклооктанового соединения формулы (XVII), R1 и R2 не являются одновременно водородом; для применения в лечении по ряду показаний, включая боль, вместе с комбинациями с: селективными ингибиторами обратного захвата серотонина, например, флуоксетином, пароксетином, циталопрамом и сертралином; смешанными ингибиторами обратного захвата серотонина-норадреналина, например, милнаципраном, венлафаксином и дулоксетином, и селективными ингибиторами обратного захвата норадреналина, например, ребоксетином.
Международная патентная заявка №PCT/IB03/00976, не опубликованная на дату подачи настоящего изобретения, описывает соединения формулы I, ниже:
где R1 представляет собой водород или (С1-С6)алкил, необязательно замещенный от одного до пяти атомами фтора;
R2 представляет собой водород или (С1-С6)алкил, необязательно замещенный от одного до пяти атомами фтора; или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют трех-шестичленное циклоалкильное кольцо;
R3 представляет собой (С1-С6)алкил, (С3-С6)циклоалкил, (С3-С6)циклоалкил-(С1-С3)алкил, фенил, фенил-(С1-С3)алкил, пиридил, пиридил-(С1-С3)алкил, фенил-N(H)- или пиридил-N(H)-, где каждая из перечисленных алкильных частей может быть, необязательно, замещена от одного до пяти атомами фтора, предпочтительно, от нуля до трех атомами фтора, и где указанный фенил и указанный пиридил и фенильная и пиридильная части указанного фенил-(С1-С3)алкила и указанного пиридил-(С1-С3)алкила, соответственно, могут быть, необязательно, замещены заместителями от одного до трех, предпочтительно, заместителями от нуля до двух, независимо выбранными из хлора, фтора, аминогруппы, нитрогруппы, цианогруппы, (С1-С3)алкиламино, (С1-С3)алкила, необязательно замещенного от одного до трех атомами фтора, и (С1-С3)алкокси, необязательно замещенного от одного до трех атомами фтора;
R4 представляет собой водород или (С1-С6)алкил, необязательно замещенный от одного до пяти атомами фтора;
R5 представляет собой водород или (С1-С6)алкил, необязательно замещенный от одного до пяти атомами фтора;
R6 представляет собой водород или (С1-С6)алкил;
или его фармацевтически приемлемые соли.
Многие типы неврологических расстройств происходят в силу нарушений в цепочках головного мозга, которые передают сигналы с использованием определенных моноаминовых нейротрансмиттеров. Моноаминовые нейротрансмиттеры включают, например, серотонин (5-НТ), норэпинефрин (норадреналин) и дофамин. Указанные нейротрансмиттеры перемещаются от терминальной части нейрона через малый зазор (синаптическую щель) и связываются с рецепторными молекулами на поверхности второго нейрона. Указанное связывание вызывает внутриклеточные изменения, которые инициируют или активируют ответ или изменение в постсинаптическом нейроне. Инактивация возникает, прежде всего, вследствие транспорта (т.е. обратного захвата) нейротрансмиттера обратно в пресинаптический нейрон.
Селективные ингибиторы обратного захвата серотонина (SSRI) действуют посредством ингибирования обратного захвата серотонина афферентными нейронами. SSRI, хорошо известные специалистам, включают, без ограничения, сертралин (Zoloft®), метаболит сертралина деметилсертралин, флуоксетин (Prozac®), норфлуоксетин (дезметиловый метаболит флуоксетина), флувоксамин (Luvox®), пароксетин (Seroxat®, Paxil®) и его альтернативная композиция, Paxil-CR®, циталопрам (Celexa®), метаболит циталопрама дезметилциталопрам, эсциталопрам (Lexapro®), d,l-фенфлурамин (Pondimin®), фемоксетин, ифоксетин, цианодотиепин, литоксетин, дапоксетин, нефазодон (Serxone®), церикламин и тразодон (Desyrel®).
Селективные ингибиторы обратного захвата норадреналина (или норэпинефрина) (SNRI) действуют посредством повышения уровней норадреналина. SNRI, хорошо известные специалистам, включают, без ограничения, ребоксетин (Edronax®) и все энантиомеры ребоксетина, т.е. (R/R, S/S, R/S, S/R), дезипрамин (Norpramin®), мапротилин (Ludiomil®), лофепрамин (Gamanil®), миртазепин (Remeron®), оксапротилин, фезоламин, томоксетин, миансерин (Bolvidon®), бупроприон (Wellbutrin®), метаболит бупроприона гидроксибупроприон, номифензин (Merital®) и вилоксазин (Vivalan®).
Ингибиторы обратного захвата серотонина-норадреналина двойного действия (DSNRI), которые ингибируют обратный захват серотонина и норэпинефрина, включают венлафаксин (Effexor®), метаболит венлафаксина О-дезметилвенлафаксин, кломипрамин (Anafranil®), метаболит кломипрамина дезметилкломипрамин, дулоксетин (Cymbalta®), милнаципран и имипрамин (Tofranil® или Janimine®).
Содержание всех патентов и публикаций, процитированных в настоящей заявке, включено в настоящий документ в качестве ссылок.
Сущность изобретения
В настоящее время установлено, что комбинированная терапия альфа-2-дельта лигандом и ингибитором обратного захвата серотонина-норадреналина двойного действия (DSNRI) или одним или обоими из селективного ингибитора обратного захвата серотонина (SSRI) и селективного ингибитора обратного захвата норадреналина (SNRI) приводит к улучшению при лечении боли. Кроме того, при одновременном введении, последовательно или раздельно, альфа-2-дельта лиганд и DSNRI или один или оба из SSRI и SNRI могут действовать синергически для борьбы с болью. Указанный синергизм позволяет уменьшить требующуюся дозу каждого соединения, что приводит к уменьшению побочных эффектов и повышению клинической пригодности соединений.
Соответственно, настоящее изобретение относится, в первом аспекте, к комбинированному продукту, содержащему альфа-2-дельта лиганд и ингибитор обратного захвата серотонина-норадреналина двойного действия (DSNRI) или один или оба из селективного ингибитора обратного захвата серотонина (SSRI) и селективного ингибитора обратного захвата норадреналина (SNRI) или их фармацевтически приемлемые соли, при условии, что исключаются соединения (i)-(xxv) WO 02/85839 в комбинации с ингибитором обратного захвата серотонина, особенно, флуоксетином, пароксетином, циталопрамом и сертралином, смешанным ингибитором обратного захвата серотонина-норадреналина, особенно, милнаципраном, венлафаксином и дулоксетином, и ингибитором обратного захвата норадреналина, особенно, ребоксетином.
В качестве альтернативы или еще в одном аспекте, настоящее изобретение относится к синергическому комбинированному продукту, содержащему альфа-2-дельта лиганд и DSNRI или один или оба из SSRI и SNRI или их фармацевтически приемлемые соли.
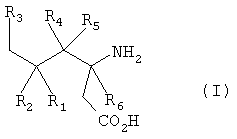
Пригодные циклические альфа-2-дельта лиганды по настоящему изобретению иллюстрируются следующей формулой (I):

где Х представляет собой карбоновую кислоту или биоизостер карбоновой кислоты;
n равно 0, 1 или 2; и
R1, R1а, R2, R2а, R3, R3а, R4 и R4а независимо выбраны из Н и С1-С6 алкила, или
R1 и R2 или R2 и R3, взятые вместе, образуют С3-С7 циклоалкильное кольцо, которое необязательно замещено одним или двумя заместителями, выбранными из С1-С6 алкила, или его фармацевтически приемлемой солью.
В формуле (I), соответственно, R1, R1а, R2а, R3а, R4 и R4а представляют собой Н, а R2 и R3 независимо выбраны из Н и метила, или R1а, R2а, R3а и R4а представляют собой Н, а R1 и R2 или R2 и R3, взятые вместе, образуют С3-С7 циклоалкильное кольцо, которое необязательно замещено одним или двумя метильными заместителями. Подходящий биоизостер карбоновой кислоты выбран из тетразолила и оксадиазолонила. Х предпочтительно представляет собой карбоновую кислоту.
В формуле (I), предпочтительно, R1, R1а, R2а, R3а, R4 и R4а представляют собой Н, а R2 и R3 независимо выбраны из Н и метила, или R1а, R2а, R3а и R4а представляют собой Н, а R1 и R2 или R2 и R3, взятые вместе, образуют С4-С5 циклоалкильное кольцо, или, когда n равно 0, R1, R1а, R2а, R3а, R4 и R4а представляют собой Н, а R2 и R3 образуют циклопентильное кольцо, или, когда n равно 1, R1, R1а, R2а, R3а, R4 и R4а представляют собой Н, а R2 и R3 оба представляют собой метил, или R1, R1а, R2а, R3а, R4 и R4а представляют собой Н, а R2 и R3 образуют циклобутильное кольцо, или, когда n равно 2, R1, R1а, R2, R2а, R3, R3а, R4 и R4а представляют собой Н, или n равно 0, R1, R1а, R2а, R3а, R4 и R4а представляют собой Н, а R2 и R3 образуют циклопентильное кольцо.
Пригодные ациклические альфа-2-дельта лиганды по настоящему изобретению иллюстрируются следующей формулой (II):

где n равно 0 или 1, R1 представляет собой водород или (С1-С6)алкил; R2 представляет собой водород или (С1-С6)алкил; R3 представляет собой водород или (С1-С6)алкил; R4 представляет собой водород или (С1-С6)алкил; R5 представляет собой водород или (С1-С6)алкил и R6 представляет собой водород или (С1-С6)алкил, или его фармацевтически приемлемой солью.
Согласно формуле (II), соответственно, R1 представляет собой С1-С6 алкил, R2 представляет собой метил, R3-R6 представляют собой водород, а n равно 0 или 1. Более подходяще, если R1 представляет собой метил, этил, н-пропил или н-бутил, R2 представляет собой метил, R3-R6 представляют собой водород, а n равно 0 или 1. Когда R2 представляет собой метил, R3-R6 представляют собой водород и n равно 0, R1 соответственно представляет собой этил, н-пропил или н-бутил. Когда R2 представляет собой метил, R3-R6 представляют собой водород и n равно 1, R1 соответственно представляет собой метил или н-пропил. Соединения формулы (II) имеют, соответственно, 3S,5R-конфигурацию.
Примеры альфа-2-дельта лигандов для применения по настоящему изобретению представляют собой соединения, в общих чертах или конкретно описанные в US 4024175, особенно, габапентин, ЕР 641330, особенно прегабалин, US 5563175, WO 9733858, WO 9733859, WO 9931057, WO 9931074, WO 9729101, WO 02085839, особенно, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусную кислоту, WO 9931075, особенно, 3-(1-аминометилциклогексилметил)-4Н-[1,2,4]оксадиазол-5-он и С-[1-(1H-тетразол-5-илметил)циклогептил]метиламин, WO 9921824, особенно, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусную кислоту, WO 0190052, WO 0128978, особенно, (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусную кислоту, ЕР 0641330, WO 9817627, WO 0076958, особенно, (3S,5R)-3-аминометил-5-метилоктановую кислоту, PCT/IB03/00976, особенно, (3S,5R)-3-амино-5-метилгептановую кислоту, (3S,5R)-3-амино-5-метилнонановую кислоту и (3S,5R)-3-амино-5-метилоктановую кислоту, ЕР 1178034, ЕР 1201240, WO 9931074, WO 03000642, WO 0222568, WO 0230871, WO 0230881, WO 02100392, WO 02100347, WO 0242414, WO 0232736 и WO 0228881 или их фармацевтически приемлемые соли, которые включены в настоящий документ в качестве ссылок.
Предпочтительные альфа-2-дельта лиганды по настоящему изобретению включают: габапентин, прегабалин, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусную кислоту, 3-(1-аминометилциклогексилметил)-4Н-[1,2,4]оксадиазол-5он, С-[1-(1H-тетразол-5-илметил)циклогептил]метиламин, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусную кислоту, (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусную кислоту, (3S,5R)-3-аминометил-5-метилоктановую кислоту, (3S,5R)-3-амино-5-метилгептановую кислоту, (3S,5R)-3-амино-5-метилнонановую кислоту и (3S,5R)-3-амино-5-метилоктановую кислоту или их фармацевтически приемлемые соли. Особенно предпочтительные альфа-2-дельта лиганды по настоящему изобретению выбраны из габапентина, прегабалина, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты или их фармацевтически приемлемых солей.
SSRI, подходящие для настоящего изобретения, включают SSRI, описанные в US 4536518, т.е. цис-изомерные соединения формулы (III):

где R1 выбран из группы, состоящей из водорода и нормального алкила из 1-3 атомов углерода, R2 представляет собой нормальный алкил из 1-3 атомов углерода, Z представляет собой

Х и Y каждый выбран из группы, состоящей из водорода, фтора, хлора, брома, трифторметила, алкокси из 1-3 атомов углерода и циано, по меньшей мере один из Х и Y не является водородом, а W выбран из группы, состоящей из водорода, фтора, хлора, брома, трифторметила и алкокси из 1-3 атомов углерода, и где термин «цис-изомерный» относится к относительной ориентации NR1R2 и Z частей на циклогексеновом кольце, указанное соединение представляет собой (1S)-энантиомер или рацемическую смесь (1S)-энантиомера и соответствующего (1R)-энантиомера, или их пролекарство или фармацевтически приемлемую соль указанного пролекарства. Особенно предпочтительным соединением формулы (III) является сертралин.
Примерами SSRI для применения по настоящему изобретению являются соединения, в общем или конкретно описанные в U.S. 4536518, особенно, сертралин, U.S. 4943590 [RE 34712], U.S. 4650884, особенно, циталопрам, U.S. 3198834, особенно, d,l-фенфлурамин, U.S. 3912743, 4571424, особенно, фемоксетин, U.S. 4314081, 4626549, особенно, флуоксетин, U.S. 4085225, особенно, флувоксетин, U.S. 3912743, 4007196, особенно, пароксетин, ифоксетин, цианодотиепин и литоксетин, или их фармацевтически приемлемые соли, которые включены в настоящий документ в качестве ссылок.
Подходящие SSRI для применения по настоящему изобретению включают сертралин, метаболит сертралина деметилсертралин, флуоксетин, норфлуоксетин (дезметиловый метаболит флуоксетина), флувоксамин, пароксетин и его альтернативная композиция, Paxil-CR®, циталопрам, метаболит циталопрама дезметилциталопрам, эсциталопрам, d,l-фенфлурамин, фемоксетин, ифоксетин, цианодотиепин, литоксетин, дапоксетин, нефазаодон, церикламин и тразодон или их фармацевтически приемлемые соли. Предпочтительно, SSRI представляет собой сертралин или его фармацевтически приемлемую соль.
SNRI, пригодные для настоящего изобретения, включают соединения, описанные в US 4229449, т.е. рацематы и оптические изомеры, соответствующие соединению формулы (IV)

предпочтительно, замещенные пропаноламиновые и морфолиновые производные, соответствующие формуле (IV), где
n и n1 равны, независимо, 1, 2 или 3;
каждая из групп R и R1, которые могут быть одинаковыми или разными, представляет собой водород; галоген; галоген-С1-С6алкил; гидрокси; С1-С6алкокси; С1-С6алкил, необязательно, замещенный; арил-С1-С6алкил, необязательно, замещенный; арил-С1-С6алкокси, необязательно, замещенный; -NO2;

где R5 и R6 представляют собой, независимо, водород или С1-С6алкил, или две соседние группы R или две соседние группы R1, взятые вместе, образуют радикал -О-СН2-О-;
R2 представляет собой водород; С1-С12алкил, необязательно, замещенный, или арил-С1-С6алкил;
каждая из групп R3 и R4, которые могут быть одинаковыми или разными, представляет собой водород, С1-С6алкил, необязательно, замещенный, С2-С4алкенил, С2-С4алкинил, арил-С2-С4алкил, необязательно, замещенный, С3-С7циклоалкил, необязательно, замещенный, или R3 и R4, вместе с атомом азота, с которым они связаны, образуют пентатомический или гексатомический, насыщенный или ненасыщенный, необязательно, замещенный, гетеромоноциклический радикал, необязательно, содержащий другие гетероатомы, принадлежащие к классу O, S и N; или R2 и R4, взятые вместе, образуют радикал -СН2-СН2-. Предпочтительное соединение формулы (IV) представлено ребоксетином.
Примерами SNRI для применения по настоящему изобретению являются соединения, в общем или конкретно описанные в U.S. 4229449, 5068433, 5391735, особенно, ребоксетин, ВР 908788980231, U.S. 3454554, особенно, дезипрамин, U.S. 3399201, особенно, мапротилин, ВР 1177525, U.S. 3637660, особенно, лофепрамин, в патентной заявке Нидерландов 6603256, U.S. 3534041, особенно, миансерин, U.S. 4062843, особенно, миртазепин; U.S. 4314081, 4018895, 4194009, особенно, томоксетин, U.S. 4535186, 4611078, особенно, венлафаксин, и U.S. 3819706, 3885046, особенно, бупроприон, и оксапротилин и фезоламин, или их фармацевтически приемлемые соли, которые включены в настоящий документ в качестве ссылок.
Конкретные примеры SNRI по настоящему изобретению включают ребоксетин и все энантиомеры ребоксетина, т.е. (R/R, S/S, R/S, S/R), дезипрамин, мапротилин, лофепрамин, миртазепин, венлафаксин (описанный в патенте США №4761501), оксапротилин, фезоламин, томоксетин, миансерин и бупроприон, метаболит бупроприона гидроксибупроприон, номифензин или вилоксазин или их фармацевтически приемлемые соли. Предпочтительно, SNRI выбран из мапротилина, дезипрамина, бупроприона, ребоксетина и S,S-ребоксетина или их фармацевтически приемлемых солей.
DSNRI, пригодные для настоящего изобретения, можно проиллюстрировать соединениями формулы (V)
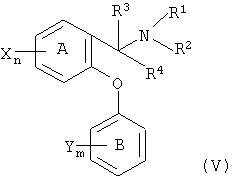
где фенильное кольцо А и фенильное кольцо В каждое может быть, независимо, заменено на нафтильную группу, и где, когда фенильное кольцо А заменено на нафтильную группу, эфирный кислород структуры I и атом углерода, к которому присоединены R3, R4 и NR1R2, присоединены к соседним кольцевым атомам углерода нафтильной группы, и ни один из указанных соседних кольцевых атомов углерода не является также смежным с атомом углерода слитого кольца указанной нафтильной группы;
n и m выбраны, независимо, из одного, двух и трех;
R1 и R2 выбраны, независимо, из водорода, (С1-С4)алкила, (С2-С4)алкенила и (С2-С4)алкинила, или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют четырех-восьмичленное насыщенное кольцо, содержащее один или два гетероатома, включая атом азота, к которому присоединены R1 и R2, где второй гетероатом, если он присутствует, выбран из кислорода, азота и серы, при условии, что указанное кольцо не может содержать два соседних атома кислорода или два соседних атома серы, и где указанное кольцо может быть, необязательно, замещено по доступным участкам связывания заместителями в количестве от одного до трех, выбранными, независимо, из гидрокси и (С1-С6)алкила;
R3 и R4 выбраны, независимо, из водорода, (С1-С4)алкила, необязательно замещенного от одного до трех атомами фтора, или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют четырех-восьмичленное насыщенное карбоциклическое кольцо, и где указанное кольцо может быть, необязательно, замещено по доступным участкам связывания заместителями в количестве от одного до трех, выбранными, независимо, из гидрокси и (С1-С6)алкила;
или R2 и R3 вместе с атомом азота, к которому присоединен R2, и атомом углерода, к которому присоединен R3, образуют четырех-восьмичленное насыщенное кольцо, содержащее один или два гетероатома, включая атом азота, к которому присоединен R2, где второй гетероатом, если он присутствует, выбран из кислорода, азота и серы, при условии, что указанное кольцо не может содержать два соседних атома кислорода или два соседних атома серы, и где указанное кольцо может быть, необязательно, замещено по доступным участкам связывания заместителями в количестве от одного до трех, выбранными, независимо, из гидрокси и (С1-С6)алкила;
каждый Х выбран, независимо, из водорода, галогена (т.е. хлора, фтора, брома или иода), (С1-С4)алкила, необязательно замещенного от одного до трех атомами фтора, (С1-С4)алкокси, необязательно замещенного от одного до трех атомами фтора, циано, нитро, амино, (С1-С4)алкиламино, ди-[(С1-С4)алкил]амино, NR5(C=O)(С1-С4)алкила, SO2NR5R6 и SOp(С1-С6)алкила, где R5 и R6 выбраны, независимо, из водорода и (С1-С6)алкила, а р равно нулю, одному или двум; и
каждый Y выбран, независимо, из водорода, (С1-С6)алкила и галогена;
при условии, что: (а) не более чем один из NR1R2, CR3R4 и R2NCR3 может образовывать кольцо; и (b) по меньшей мере один Х не должен представлять собой водород, когда (i) R3 и R4 оба представляют собой водород, (ii) R1 и R2 выбраны, независимо, из водорода и (С1-С4)алкила, и (iii) кольцо В является одно- или двузамещенным, соответственно, одной или двумя галогеновыми группами; и их фармацевтически приемлемыми солями. Соединения формулы V описаны в WO 00/50380.
Подходящие DSNRI по настоящему изобретению выбраны из венлафаксина, метаболита венлафаксина О-дезметилвенлафаксина, кломипрамина, метаболита кломипрамина дезметилкломипрамина, дулоксетина, милнаципрана и имипрамина, или их фармацевтически приемлемых солей. Предпочтительные DSNRI по настоящему изобретению выбраны из милнаципрана, дулоксетина и венлафаксина, или их фармацевтически приемлемых солей.
Пригодность любых конкретных DSNRI, SSRI или SNRI можно легко определить путем оценки его эффективности и селективности с использованием способов, описанных в литературе, с последующей оценкой его токсичности, всасывания, метаболизма, фармакокинетики и т.п., в соответствии с обычной фармацевтической практикой.
В качестве альтернативы или еще в одном аспекте, настоящее изобретение относится к комбинации, содержащей габапентин или его фармацевтически приемлемую соль и DSNRI, выбранный из венлафаксина, метаболита венлафаксина О-дезметилвенлафаксина, кломипрамина, метаболита кломипрамина дезметилкломипрамина, дулоксетина, милнаципрана и имипрамина, или один или оба из SSRI, выбранных из сертралина, флуоксетина, флувоксамина, пароксетина, циталопрама, d,l-фенфлурамина, фемоксетина, тразодона, церикламина, ифоксетина, цианодотиепина и литоксетина или их фармацевтически приемлемых солей, и SNRI, выбранных из ребоксетина, S,S-ребоксетина, дезипрамина, мапротилина, лофепрамина, миансерина, миртазепина, оксапротилина, фезоламина, томоксетина или бупроприона или их фармацевтически приемлемых солей, и их фармацевтически приемлемые соли. Особенно предпочтительная комбинация включает габапентин и один из сертралина, милнаципрана, дулоксетина, венлафаксина, мапротилина, дезипрамина, бупроприона, ребоксетина или S,S-ребоксетина, и их фармацевтически приемлемые соли.
В качестве альтернативы или еще в одном аспекте, настоящее изобретение относится к комбинации, содержащей прегабалин и DSNRI, выбранный из венлафаксина, метаболита венлафаксина О-дезметилвенлафаксина, кломипрамина, метаболита кломипрамина дезметилкломипрамина, дулоксетина, милнаципрана и имипрамина, или комбинацию с одними или обоими из SSRI, выбранных из сертралина, флуоксетина, флувоксамина, пароксетина, циталопрама, d,l-фенфлурамина, фемоксетина, тразодона, церикламина, ифоксетина, цианодотиепина и литоксетина или их фармацевтически приемлемых солей, и SNRI, выбранных из ребоксетина, S,S-ребоксетина, дезипрамина, мапротилина, лофепрамина, миансерина, миртазепина, оксапротилина, фезоламина, томоксетина или бупроприона или их фармацевтически приемлемых солей, и их фармацевтически приемлемые соли. Особенно предпочтительная комбинация включает прегабалин и один из сертралина, милнаципрана, дулоксетина, венлафаксина, мапротилина, дезипрамина, бупроприона, ребоксетина или S,S-ребоксетина, и их фармацевтически приемлемые соли.
В качестве еще одной альтернативы или еще в одном аспекте, настоящее изобретение относится к комбинации, содержащей (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусную кислоту или ее фармацевтически приемлемую соль и DSNRI или один или оба из SSRI и SNRI. Соответственно, изобретение относится к комбинации, содержащей (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусную кислоту или ее фармацевтически приемлемую соль и DSNRI, выбранный из венлафаксина, метаболита венлафаксина О-дезметилвенлафаксина, кломипрамина, метаболита кломипрамина дезметилкломипрамина, дулоксетина, милнаципрана и имипрамина, или один или оба из SSRI, выбранных из сертралина, флуоксетина, флувоксамина, пароксетина, циталопрама, d,l-фенфлурамина, фемоксетина, тразодона, церикламина, ифоксетина, цианодотиепина и литоксетина или их фармацевтически приемлемых солей, и SNRI, выбранных из ребоксетина, S,S-ребоксетина, дезипрамина, мапротилина, лофепрамина, миансерина, миртазепина, оксапротилина, фезоламина, томоксетина или бупроприона или их фармацевтически приемлемых солей, и их фармацевтически приемлемые соли. Особенно предпочтительная комбинация включает (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусную кислоту и один из сертралина, милнаципрана, дулоксетина, веналфаксина, мапротилина, дезипрамина, бупроприона, ребоксетина или S,S-ребоксетина, и их фармацевтически приемлемые соли.
В качестве еще одного предпочтительноого аспекта настоящего изобретения комбинация выбрана из:
габапентина и сертралина;
габапентина и милнаципрана;
габапентина и дулоксетина;
габапентина и венлафаксина;
габапентина и мапротилина;
габапентина и дезипрамина;
габапентина и бупропиона;
габапентина и ребоксетина;
габапентина и S,S-ребоксетина;
прегабалина и сертралина;
прегабалина и милнаципрана;
прегабалина и дулоксетина;
прегабалина и венлафаксина;
прегабалина и мапротилина;
прегабалина и дезипрамина;
прегабалина и бупропиона;
прегабалина и ребоксетина;
прегабалина и S,S-ребоксетина;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и сертралина;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и милнаципрана;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и дулоксетина;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и венлафаксина;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и мапротилина;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и дезипрамина;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и бупропиона;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и ребоксетина;
[(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты и S,S-ребоксетина;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и сертралина;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и милнаципрана;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и дулоксетина;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и венлафаксина;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и мапротилина;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и дезипрамина;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и бупропиона;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и ребоксетина;
(1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты и S,S-ребоксетина;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и сертралина;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и милнаципрана;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и дулоксетина;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и венлафаксина;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и мапротилина;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и дезипрамина;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и бупропиона;
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и ребоксетина; и
(3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты и S,S-ребоксетина;
или их фармацевтически приемлемых солей.
Комбинация по настоящему изобретению в виде единой лекарственной формы является подходящей для введения любому млекопитающему, предпочтительно, человеку. Введение можно осуществлять один раз в день (o.d.), дважды в день (b.i.d.) или три раза в день (t.i.d.), подходяще b.i.d. или t.i.d., более подходяще, b.i.d., наиболее подходяще, o.d.
Таким образом, в качестве еще одного аспекта настоящее изобретение относится к применению комбинации, особенно, синергической, альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI для производства лекарственного средства, которое вводят один, два или три, подходяще, два или три, более подходяще, два, наиболее подходяще, один раз в день, для терапевтического, профилактического или паллиативного лечения боли.
Альтернативно, настоящее изобретение относится к способу терапевтического, профилактического или паллиативного лечения боли у млекопитающего, включающего введение один, два или три, подходяще, два или три, более подходяще, два, наиболее подходяще, один раз в день, эффективной, особенно, синергической, комбинации альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI.
Для определения синергического взаимодействия между одним или более компонентов, можно окончательно измерить оптимальные пределы для достижения эффекта и абсолютные пределы доз каждого компонента для достижения эффекта посредством введения компонентов в различных пределах массовых соотношений и дозах пациентам, которые нуждаются в лечении. Для людей сложность и дороговизна проведения клинических испытаний на пациентах делает непрактичным использование указанной формы тестирования в качестве первичной модели синергизма. Однако наблюдение синергизма на одном виде может иметь прогностическое значение в отношении эффекта для других видов, и существуют модели на животных, как описано в настоящем документе, для измерения синергического эффекта, и результаты подобных исследований также можно использовать для прогнозирования эффективных пределов соотношений доз и концентраций в плазме, а также абсолютных доз и концентраций в плазме, которые требуются другим видам, путем использования фармакокинетических/фармакодинамических методик. Установленные корреляции между моделями на животных и эффектами, наблюдающимися у человека, предполагают, что синергизм для животных наилучшим образом демонстрируется с использованием статических и динамических измерений аллодинии на грызунах, которым осуществили хирургические (например, повреждение хроническим сдавлением) или химические процедуры (например, стрептозоцин) для индуцирования аллодинии. Из-за эффектов плато на указанных моделях их ценность лучше оценивать с точки зрения синергических взаимодействий, которые у пациентов с невропатической болью будут трансформироваться в преимущества уменьшения доз. Другие модели, на которых существующие агенты, используемые для лечения невропатической боли, дают только частичный ответ, больше подходят для прогнозирования потенциала комбинаций, действующих синергически, для получения повышенной максимальной эффективности в максимально переносимых дозах двух компонентов.
Таким образом, в еще одном аспекте настоящее изобретение относится к синергической комбинации для введения человеку, включающей альфа-2-дельта лиганд и один из DSNRI, SSRI или SNRI, или их фармацевтически приемлемые соли, в пределах масс комбинации, которые соответствуют абсолютным пределам, наблюдающимся на модели, не являющейся человеком, предпочтительно, на крысиной модели, которую, преимущественно, используют для идентификации синергического взаимодействия. Соответственно, пределы соотношений для человека соответствуют пределам для животных, которые не являются человеком, выбранным из: от 1:50 до 50:1 массовых частей, от 1:50 до 20:1, от 1:50 до 10:1, от 1:50 до 1:1, от 1:20 до 50:1, от 1:20 до 20:1, от 1:20 до 10:1, от 1:20 до 1:1, от 1:10 до 50:1, от 1:10 до 20:1, от 1:10 до 10:1, от 1:10 до 1:1, от 1:1 до 50:1, от 1:1 до 20:1 и от 1:1 до 10:1. Более подходяще, если пределы для человека соответствуют пределам для модели, не являющейся человеком, от 1:10 до 20:1 массовых частей. Предпочтительно, пределы для человека соответствуют синергическим пределам для модели, не являющейся человеком, порядка от 1:1 до 10:1 массовых частей.
Для человека можно использовать несколько экспериментальных моделей боли у людей для демонстрации того, что агенты с подтвержденным синергизмом на животных также оказывают действие на человека, согласующееся с указанным синергизмом. Примеры модели на людях, которые могут подходить для указанной цели, включают модель жар/капсаицин (Petersen K.L. & Rowbotham M.C. (1999) NeuroReport 10, 1511-1516), модель капсаицин i.d. (Andersen O.L., Felsby S., Nicolaisen L., Bjerring P., Jsesn T.C. & Arendt-Nielsen L. (1996) Pain 66, 51-62), включающие использование повторных травм капсаицином (Witting N., Svesson P., Arendt-Nielsen L. & Jensen T.S. (2000) Somatosensory Motor Res. 17, 5-12) и суммирование реакций нервного возбуждения (Curatolo M. et al. (2000) Anesthesiology 93, 1517-1530). При использовании указанных моделей можно использовать в качестве конечных результатов субъективную оценку интенсивности боли или площадей гипералгезии или можно использовать более объективные конечные результаты, основанные на электрофизиологических или визуализирующих технологиях (таких как функциональная магнитно-резонансная визуализация) (Bornhovd K., Quante M., Glauche V., Bromm B., Weiller C. & Buchel C. (2002) Brain 125, 1326-1336). Все подобные модели требуют данных об объективной правильности до того, как можно сделать заключение о том, что они обеспечивают данные у человека, подтверждающие синергическое действие комбинации, которое наблюдалось в исследованиях на животных.
Для использования настоящего изобретения для человека подходящие пределы соотношений альфа-2-дельта лиганд:DSNRI, SSRI или SNRI выбирают из следующих соотношений: от 1:50 до 50:1 массовых частей, от 1:50 до 20:1, от 1:50 до 10:1, от 1:50 до 1:1, от 1:20 до 50:1, от 1:20 до 20:1, от 1:20 до 10:1, от 1:20 до 1:1, от 1:10 до 50:1, от 1:10 до 20:1, от 1:10 до 10:1, от 1:10 до 1:1, от 1:1 до 50:1, от 1:1 до 20:1 и от 1:1 до 10:1, более подходяще, от 1:10 до 20:1, предпочтительно, от 1:1 до 10:1.
Оптимальные дозы для каждого компонента для синергизма можно определить с использованием опубликованных процедур на экспериментальных животных. Однако для людей (даже на экспериментальных моделях боли) стоимость исследования для определения в полном объеме взаимоотношений воздействие-ответ при всех терапевтически релевантных дозах каждого компонента комбинации может быть очень высокой. Может оказаться необходимым, по крайне мере, сначала, оценить, можно ли наблюдать эффекты, которые соответствуют синергизму, в дозах, которые были экстраполированы, исходя из доз, которые обеспечивали оптимальный синергизм у животных. При пересчете доз для животных на дозы для человека необходимо принимать во внимание такие факторы как относительный показатель масса тела/площадь поверхности тела, относительное всасывание, распределение, метаболизм и экскреция каждого компонента и относительный показатель связывания белками плазмы, и, в силу указанных причин, оптимальное соотношение доз, прогнозируемое для человека (а также для пациентов), едва ли будет таким же, как соотношение доз, оптимальное для животных. Однако взаимоотношения между двумя показателями может понять и рассчитать специалист в области фармакокинетики животных и человека. Для установления связей между эффектами у животного и человека важным являются концентрации в плазме, полученные для каждого компонента, используемого в экспериментах на животных, поскольку указанные параметры связаны с концентрацией в плазме каждого компонента, которая, как следует ожидать, будет эффективной для человека. Фармакокинетическое/фармакодинамическое моделирование (включая такие методики, как изоболограммы, индекс взаимодействия и моделирование поверхности отклика) и имитации могут помочь в прогнозировании синергических соотношений доз у человека, особенно, в случае, когда один из указанных компонентов или оба уже были изучены на человеке.
Важно выяснить, происходит ли любой синергизм, наблюдающийся у животных или у человека, только вследствие фармакокинетических взаимодействий. Например, ингибирование метаболизма одного соединения другим может создать ложное впечатление о фармакодинамическом синергизме.
Таким образом, согласно еще одной особенности, настоящее изобретение относится к синергической комбинации для введения людям, содержащей альфа-2-дельта лиганд и DSNRI и один или оба из SSRI и SNRI или их фармацевтически приемлемые соли, в которой соотношение доз каждого компонента соответствует абсолютным пределам, которые наблюдаются на модели, не являющейся человеком, предпочтительно, на крысиной модели, которая преимущественно используется для идентификации синергического взаимодействия.
Соответственно, доза альфа-2-дельта лиганда для применения у человека находится в пределах, выбранных из 1-1200 мг, 1-500 мг, 1-100 мг, 1-50 мг, 1-25 мг, 500-1200 мг, 100-1200 мг, 100-500 мг, 50-1200 мг, 50-500 мг или 50-100 мг; подходяще, 50-100 мг, b.i.d. или t.i.d., подходяще, t.i.d., а доза SSRI и/или SNRI находится в пределах, выбранных из 1-200 мг, 1-100 мг, 1-50 мг, 1-25 мг, 10-100 мг, 10-50 мг или 10-25 мг, подходяще, 10-100 мг, b.i.d. или t.i.d., подходяще, t.i.d.
Специалисту будет понятно, что пределы концентраций в плазме комбинаций альфа-2-дельта лиганда и DSNRI и одного или обоих из SSRI и SNRI по настоящему изобретению, которые требуются для получения терапевтического эффекта, зависят от вида, который лечат, и используемых компонентов. Например, для габапентина у крыс величины Cmax варьируют от 0,520 мкг/мл до 10,5 мкг/мл.
Возможно, используя обычные ФК/ФД и аллометрические способы, экстраполировать величины концентрации в плазме, которые наблюдаются у экспериментальных животных, для прогнозирования величин у других видов, особенно, у человека.
Таким образом, согласно еще одного аспекта, настоящее изобретение относится к синергической комбинации для введения людям, содержащей альфа-2-дельта лиганд и DSNRI и один или оба из SSRI и SNRI, в которой пределы концентраций в плазме каждого компонента соответствует абсолютным пределам, которые наблюдаются на модели, не являющейся человеком, предпочтительно, на крысиной модели, которая преимущественно используется для идентификации синергического взаимодействия. Соответственно, пределы концентраций в плазме для человека соответствуют пределам от 0,05 мкг/мл до 10,5 мкг/мл для альфа-2-дельта лиганда для крысиной модели.
Особенно предпочтительные комбинации по настоящему изобретению включают такие комбинации, в которых каждая переменная комбинации выбрана из подходящих параметров для каждой переменной. Еще более предпочтительные комбинации по настоящему изобретению включают такие комбинации, в которых каждая переменная комбинации выбрана из более подходящих, наиболее подходящих, предпочтительных или более предпочтительных параметров для каждой переменной.
Подробное описание изобретения
Соединения по настоящему изобретению получают способами, хорошо известными специалистам. Конкретно, патенты, патентные заявки и публикации, упомянутые выше в настоящем документе, каждая из которых включена в настоящий документ в качестве ссылки, приводят примеры соединений, которые можно использовать в комбинациях, фармацевтических композициях, способах и наборах в соответствии с настоящим изобретением, и отсылают к способам получения указанных соединений.
Соединения в комбинации по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы, включая гидратированные формы, которые могут содержать изотопные замещения (например, D2O, d6-ацетон, d6-DMSO), являются эквивалентными несольватированным формам и входят в объем настоящего изобретения.
Некоторые из соединений по настоящему изобретению имеют один или более хиральных центров, и каждый центр может существовать в R- или S-конфигурации. Настоящее изобретение включает в себя все энантиомерные и эпимерные формы, а также свойственные им смеси. Разделение диастереоизомеров или цис- и транс-изомеров можно осуществлять с использованием обычных методик, например, фракционной кристаллизацией, хроматографией или ВЭЖХ стереоизомерной смеси соединения по настоящему изобретению или его подходящей соли или производного.
Ряд альфа-2-дельта лигандов по настоящему изобретению представляют собой аминокислоты. Поскольку аминокислоты являются амфотерными, фармакологически совместимыми солями могут быть соли подходящих нетоксичных неорганических или органических кислот или оснований. Подходящими кислотно-аддитивными солями являются соли ацетат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат, камзилат, цитрат, эдизилат, эзилат, фумарат, глуцептат, глюконат, глюкуронат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидроиодид/иодид, гидрофосфат, изетионат, D- и L-лактат, малат, малеат, малонат, мезилат, метилсульфат, 2-напзилат, никотинат, нитрат, оротат, пальмоат, фосфат, сахарат, стеарат, сукцинат, сульфат, D- и L-тартрат и тозилат. Подходящие основные соли образуются из оснований, которые образуют нетоксичные соли, и примерами являются соли натрия, калия, алюминия, кальция, магния, цинка, холина, диоламина, оламина, аргинина, глицина, трометамина, бензатина, лизина, меглумина и диэтиламина. Соли с четвертичными аммониевыми ионами также можно получить, например, с ионом тетраметиламмония. Соединения по настоящему изобретению также могут быть образованы в виде цвиттериона.
Подходящей солью для аминокислотных соединений по настоящему изобретению является соль гидрохлорид. Обзор подходящих солей см. в Stahl and Wermuth, Handbook of Pharmaceutical Salts: Properties, Selection, and Use, Wiley-VCH, Weinheim, Germany (2000).
Также в объем настоящего изобретения входят клатраты, соединения включения лекарственное средство-реципиент, в которых, в отличие от упомянутых сольватов, лекарственное средство и реципиент присутствуют в нестехиометрических количествах. Обзор указанных соединений см. В J. Pharm. Sci., 64(8), 1269-1288, Haleblian (август 1975 г.).
Далее в настоящем документе все ссылки на соединения по настоящему изобретению включают ссылки на их соли и на сольваты и клатраты соединений по настоящему изобретению и их соли.
Также в объем настоящего изобретения включены полиморфы соединений по настоящему изобретению.
Пролекарства соединений по настоящему изобретению включены в объем настоящего изобретения. Химически модифицированное лекарственное средство или пролекарство должны обладать отличным от родительского соединения фармакокинетическим профилем, обеспечивающим более легкое всасывание через эпителий слизистых оболочек, лучшее образование солей и/или растворимость, улучшенную системную стабильность (например, для увеличения полупериода существования в плазме). Указанные химические модификации могут представлять собой:
(1) Сложноэфирные или амидные производные, которые могут расщепляться, например, эстеразами или липазами. В случае сложноэфирных производных, сложный эфир получают из части карбоновой кислоты молекулы лекарственного средства известными способами. В случае амидных производных, амид можно получить из части карбоновой кислоты или аминной части молекулы лекарственного средства известными способами.
(2) Пептиды, которые могут распознаваться специфичными или неспецифичными протеиназами. Пептид может быть присоединен к молекуле лекарственного средства посредством образования амидной связи с аминной частью или частью карбоновой кислоты молекулы лекарственного средства известными способами.
(3) Производные, которые накапливаются в месте действия через мембранную селекцию пролекарственной формы или модифицированной пролекарственной формы.
(4) Любую комбинацию от 1 до 3.
Аминоацил-гликолевые и молочные эфиры известны в качестве пролекарств аминокислот (Wermuth C.G., Chemistry and Industry, 1980:433-435). Карбонильная группа аминокислот может быть эстерифицирована известными способами. Пролекарства и мягкие лекарственные средства известны специалистам (Palomino E., Drugs of the Future, 1990; 15(4):361-368). Два последних источника включены в настоящий документ в качестве ссылки.
Комбинация по настоящему изобретению является пригодной для общего лечения боли, особенно, невропатической боли. Физиологическая боль является важным защитным механизмом, направленным на предупреждение об опасности потенциально повреждающих раздражителей, исходящих из внешнего окружения. Система действует через специфический ряд первичных чувствительных нейронов и активируется исключительно вредными раздражителями через периферические преобразующие механизмы (обзор Millan, 1999 Prog. Neuribio. 57:1-164). Указанные чувствительные волокна известны как ноцицепторы и отличаются наличием аксонов малого диаметра с низкими скоростями проведения. Ноцицепторы кодируют интенсивность, продолжительность и качество вредного раздражителя и, посредством их топографически организованной проекции на спинной мозг, локализацию раздражителя. Ноцицепторы находят на ноцицептивных нервных волокнах, которые существуют в двух главных типах, А-дельта волокна (миелинизированные) и С волокна (немиелинизированные). Активность, генерированная ноцицепторным входом, переносится после сложной обработки в дорсальном роге, непосредственно или через релейные ядра ствола головного мозга, в вентро-базальный таламус, а затем в кору, где генерируется ощущение боли.
Интенсивная острая боль и хроническая боль может вовлекать одни и те же пути, управляемые патофизиологическими процессами, и, в качестве таковых, прекращать обеспечение защитного механизма и, вместо этого, осуществлять свой вклад в обессиливающие симптомы, связанные с широким рядом болезненных состояний. Боль является признаком многих травматических и болезненных состояний. При значительном повреждении тканей тела через болезнь или травму характеристики ноцицепторной активации изменяются. Имеет место сенсибилизация на периферии, локально вокруг повреждения и центрально, где ноцицепторы заканчиваются. Это приводит к гиперчувствительности в месте повреждения и в расположенной поблизости нормальной ткани. При острой боли указанные механизмы могут быть полезными и позволяют происходить процессам восстановления, а гиперчувствительность возвращается к норме после заживления повреждения. Однако при многих состояниях хронической боли гиперчувствительность длится гораздо дольше, чем процесс заживления, и это обычно происходит в результате повреждения нервной системы. Указанное повреждение часто приводит к плохой адаптации афферентных волокон (Woolf & Salter 2000 Science 288:1765-1768). Клиническая боль имеет место, когда дискомфорт и патологическая чувствительность являются отличительным признаком симптомов у пациента. Состав пациентов бывает достаточно разнородным, и у них могут иметь место разнообразные симптомы боли. Существует ряд типичных подтипов боли: 1) спонтанная боль, которая может быть тупой, жгучей или колющей; 2) болевые реакции на вредные раздражители являются чрезмерными (гипералгезия); 3) боль вызывается раздражителями, которые в норме являются безвредными (аллодиния) (Meyer et al., 1994 Textbook of Pain 13-44). Хотя пациенты с болью в спине, артритной болью, травмой ЦНС или невропатической болью могут иметь сходные симптомы, лежащие в основе боли механизмы могут различаться и, таким образом, могут требовать различных стратегий лечения. Таким образом, боль можно разделить на ряд различных областей вследствие различающейся патфизиологии, что включает ноцицептивную, воспалительную, невропатическую боль и т.п. Следует отметить, что некоторые типы боли имеют множественную этиологию и, таким образом, могут быть классифицированы в более чем одной области, например, боль в спине, боль при злокачественной опухоли имеют как ноцицептивные, так и невропатические компоненты.
Ноцицептивную боль вызывает повреждение ткани или интенсивный раздражитель, который потенциально может вызвать повреждение. Афферентные пути боли активируются преобразованием раздражителей ноцицепторами в месте повреждения и сенсибилизируют спинной мозг на уровне своего окончания. Это затем передается вверх по спинальным путям в головной мозг, где боль воспринимается (Meyer et al., 1994 Textbook of Pain 13-44). Активация ноцицепторов активирует два типа афферентных нервных волокон. Миелинизированные волокна А-дельта осуществляют быстрое проведение и отвечают за ощущения острой и колющей боли, в то время как немиелинизированные волокна С осуществляют более медленное проведение и отвечают за ощущения тупой или ноющей боли. Острая ноцицептивная боль от умеренной до тяжелой является заметным признаком, но без ограничения боли от деформации/растяжения, послеоперационной боли (боли после хирургической процедуры любого типа), посттравматической боли, от ожогов, при инфаркте миокарда, остром панкреатите и почечной колике. Также связанные со злокачественными опухолями острые болевые синдромы обычно происходят в результате терапевтических взаимодействий, таких как химиотерапевтическая токсичность, иммунотерапия, гормональная терапия и лучевая терапия. Острая ноцицептивная боль от умеренной до тяжелой является заметным признаком, но без ограничения, боли при злокачественной опухоли, которая может представлять собой боль, связанную с опухолью (например, боль в костях, головная боль и лицевая боль, висцеральная боль), или боль, связанную с терапией злокачественной опухоли (например, постхимиотерапевтические синдромы, синдромы хронической послеоперационной боли, синдромы после облучения), боль в спине, которая может появляться в результате грыжи или повреждения межпозвоночных дисков или патологии суставов поясничного отдела, крестцово-подвздошных суставов, параспинальных мышц или задней продольной связки.
Невропатическую боль определяют как боль, инициированную или вызванную первичным повреждением или дисфункцией нервной системы (определение IASP). Повреждение нерва может быть вызвано травмой и заболеванием, и, таким образом, термин «невропатическая боль» охватывает многие расстройства с различной этиологией. Они включают, без ограничения, диабетическую невропатию, постгерпетическую невралгию, боль в спине, невропатию при злокачественной опухоли, невропатию при ВИЧ-инфекции, фантомную боль в конечности, кистевой туннельный синдром, хронический алкоголизм, гипотиреоз, невралгию тройничного нерва, уремию или дефициты витаминов. Невропатическая боль является патологической, поскольку она не играет защитной роли. Она часто имеет место после исчезновения причины ее возникновения, обычно длится годами, значительно ухудшая качество жизни пациентов (Woolf and Mannion 1999 Lancet 353:1959-1964). Симптомы невропатической боли плохо поддаются лечению, поскольку они зачастую являются разнородными даже у пациентов с одним и тем же заболеванием (Woolf & Decosterd 1999 Pain Supp.6:S141-S147; Woolf and Mannion 1999 Lancet 353:1959-1964). Они включают спонтанную боль, которая может быть непрерывной или приступообразной, и боль, вызываемую необычным образом, такую как гипералгезия (повышенная чувствительность к вредному раздражителю) и аллодиния (чувствительность к раздражителю, безвредному в норме).
Воспалительный процесс представляет собой сложный ряд биохимических и клеточных событий, которые активируются в ответ на повреждение ткани или присутствие чужеродных веществ, что приводит к отеку и боли (Levine and Taiwo 1994: Textbook of Pain 45-56). Артритная боль составляет основную часть популяции воспалительной боли. Ревматоидное заболевание представляет собой одно из наиболее распространенных хронических воспалительных состояний в развитых странах, а ревматоидный артрит является распространенной причиной инвалидности. Точная этиология RA неизвестная, но современные гипотезы предполагают, что важную роль могут играть как генетические, так и микробиологические факторы (Grennan & Jayson 1994 Textbook of Pain 397-407). Было подсчитано, что почти 16 миллионов американцев имеют клинически выраженный остеоартрит (ОА) или дегенеративное заболевание суставов, возраст большинства из которых более 60 лет, и, как ожидается, это количество возрастет до 40 миллионов по мере старения популяции, что делает это проблемой здравоохранения огромных масштабов (Houge & Mersfelder 2002 Ann. Pharmacother. 36:679-686; McCarthy et al., 1994 Textbook of Pain 387-395). Большинство пациентов с ОА обращаются за медицинской помощью в связи с болью. Артрит оказывает значительное влияние на физиологическую и физическую функцию и, как известно, является ведущей причиной инвалидности в дальнейшей жизни. Другие типы воспалительной боли включают, без ограничения, воспалительные заболевания кишечника (IBD).
Другие типы боли включают, без ограничения:
- мышечно-скелетные нарушения, включая, без ограничения, миалгию, фибромиалгию, спондилит, серонегативные (неревматоидные) артропатии, несуставной ревматизм, дистрофинопатию, гликогенолиз, полимиозит, пиомиозит;
- центральную боль или «таламическую боль», что определяет боль, вызванную повреждением или дисфункцией нервной системы, включая, без ограничения, центральную постинсультную боль, рассеянный склероз, повреждение спинного мозга, болезнь Паркинсона и эпилепсию;
- сердечную и сосудистую боль, включая, без ограничения, стенокардию, инфаркт миокарда, феномен Рейно, склередему, ишемию скелетных мышц;
- висцеральную боль и желудочно-кишечные расстройства. Внутренние органы включают органы брюшной полости. Указанные органы включают половые органы, селезенку и часть пищеварительной системы. Боль, связанная с внутренними органами, можно разделить на боль в органах пищеварения и боль в органах, не относящихся к системе пищеварения. Распространенные желудочно-кишечные (GI) нарушения включают функциональные расстройства кишечника (FBD) и воспалительные заболевания кишечника (IBD). Указанные GI нарушения включают обширный ряд болезненных состояний, которые в настоящее время контролируются лишь отчасти, включая - в случае FBD, желудочно-пищеводный рефлюкс, диспепсию, синдром раздраженной кишки (IBS) и синдром функциональной абдоминальной боли (FAPS), и, в случае IBD, болезнь Крона, илеит и язвенный колит, которые регулярно вызывают висцеральную боль. Другие типы висцеральной боли включают боль, связанную с дисменореей, тазовой болью, циститом и панкреатитом;
- головную боль, включая, без ограничения, мигрень, мигрень с аурой, мигрень без ауры, приступы головной боли, головную боль по типу напряжения;
- рото-лицевая боль, включая, без ограничения, зубную боль, височно-нижнечелюстную миофасциальную боль.
Комбинация по настоящему изобретению является также пригодной для лечения недержания мочи, такого как истинное стрессовое недержание (GSI), стрессовое недержание мочи (SUI) или недержание мочи у лиц старшего возраста; гиперактивного мочевого пузыря (ОАВ), включая идиопатическую нестабильность детрузора, гиперактивность детрузора, вторичную по отношению к неврологическим заболеваниям (например, болезни Паркинсона, рассеянному склерозу, повреждению спинного мозга и инсульту), и гиперактивность детрузора, вторичную по отношению к затруднению оттока из мочевого пузыря (например, к доброкачественной гиперплазии предстательной железы (ВРН), стриктуре или стенозу уретры); ночной энурез; недержание мочи вследствие комбинации перечисленных выше условий (например, истинное стрессовое недержание, связанное с гиперактивным мочевым пузырем) и уринарные симптомы, такие как частота и ургентность.
Комбинация также является пригодной для лечения недержания кала.
В еще одном аспекте настоящее изобретение относится к применению альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI, при условии, что исключаются соединения (i)-(xxv) WO 02/85839 в комбинации с ингибитором обратного захвата серотонина, особенно, флуоксетином, пароксетином, циталопрамом и сертралином, смешанным ингибитором обратного захвата серотонина-норадреналина, особенно, милнаципраном, венлафаксином и дулоксетином, и ингибитором обратного захвата норадреналина, особенно, ребоксетином, для производства лекарственного средства для терапевтического, профилактического или паллиативного лечения боли, особенно невропатической боли.
В качестве альтернативного признака, изобретение относится к применению синергического эффективного количества альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI для производства лекарственного средства для терапевтического, профилактического или паллиативного лечения боли, особенно невропатической боли.
В качестве альтернативной особенности изобретение относится к способу терапевтического, профилактического или паллиативного лечения боли, особенно невропатической боли, включающему одновременное, последовательное или раздельное введение терапевтически эффективного количества альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI млекопитающему, которое нуждается в указанном лечении, при условии, что исключаются комбинации, описанные в WO 02/85839, т.е. соединение формулы (i)-(xxv) в комбинации с ингибитором обратного захвата серотонина, особенно, флуоксетином, пароксетином, циталопрамом и сертралином, смешанным ингибитором обратного захвата серотонина-норадреналина, особенно, милнаципраном, венлафаксином и дулоксетином, и ингибитором обратного захвата норадреналина, особенно, ребоксетином.
В качестве альтернативной особенности изобретение относится к способу терапевтического, профилактического или паллиативного лечения боли, особенно невропатической боли, включающему одновременное, последовательное или раздельное введение терапевтически синергического количества альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI млекопитающему, которое нуждается в указанном лечении.
Биологическую активность альфа-2-дельта лигандов по настоящему изобретению можно измерить с помощью анализа связывания радиолиганда с использованием [3H]габапентина и субъединицы α2δ, полученной из ткани головного мозга свиньи (Gee N.S., Brown J.P., Dissanayake V.U.K., Offord J., Thurlow R., Woodruff G.N., J. Biol. Chem., 1996; 271:5879-5776). Результаты могут быть выражены как мкМ или нМ α2δ связывающей аффинности.
Способность соединений по настоящему изобретению действовать в качестве селективных ингибиторов обратного захвата серотонина можно измерить in vivo в соответствии с установленными процедурами, например, согласно примеру 68 US 4536518.
Способность соединений по настоящему изобретению действовать в качестве ингибиторов обратного захвата серотонина-норадреналина двойного действия или селективных ингибиторов обратного захвата норадреналина можно измерить в соответствии с установленными процедурами, особенно в соответствии с документами, упомянутыми выше в настоящем документе.
Элементы комбинации по настоящему изобретению можно вводить по отдельности, одновременно или последовательно, для лечения боли. Комбинацию можно также, необязательно, вводить с одним или более другими фармакологически активными агентами. Подходящие необязательные агенты включают:
(i) опиоидные анальгетики, например, морфин, героин, гидроморфон, оксиморфон, леворфанол, леваллорфан, метадон, меперидин, фентанил, кокаин, кодеин, дигидрокодеин, оксикодон, гидрокодон, пропоксифен, налмефен, налорфин, налоксон, налтрексон, бупренорфин, буторфанол, налбуфин и пентазоцин;
(ii) нестероидные противовоспалительные лекарственные средства (NSAID), например, аспирин, диклофенак, дифлузинал, этодолак, фенбуфен, фенопрофен, флуфенизал, флурбипрофен, ибупрофен, индометацин, кетопрофен, кеторолак, меклофенамовую кислоту, мефенамовую кислоту, набуметон, напроксен, оксапрозин, фенилбутазон, пироксикам, сулиндак, толметин, зомепирак и их фармацевтически приемлемые соли;
(iii) барбитуратные седативные средства, например, амобарбитал, апробарбитал, бутабарбитал, бутабитал, мефобарбитал, метарбитал, метогекситал, пентобарбитал, фенобарбитал, секобарбитал, талбутал, теамилал, тиопентал и их фармацевтически приемлемые соли;
(iv) бензодиазепины, обладающие седативным действием, например, хлордиазепоксид, клоразепат, диазепам, флуразепам, лоразепам, оксазепам, темазепам, триазолам и их фармацевтически приемлемые соли;
(v) Н1-антагонисты, обладающие седативным действием, например, дифенгидрамин, пириламин, прометазин, хлорфенирамин, хлорциклизин и их фармацевтически приемлемые соли;
(vi) смешанные седативные средства, такие как глутетимид, мепробамат, метаквалон, дихлоралфеназон и их фармацевтически приемлемые соли;
(vii) релаксанты скелетной мускулатуры, например, баклофен, каризопродол, хлорзоксазон, циклобензаприн, метокарбамол, орфренадин и их фармацевтически приемлемые соли;
(viii) антагонисты рецепторов NMDA, например, декстрометорфан ((+)-3-гидрокси-N-метилморфинан) и его метаболит декстрорфан ((+)-3-гидрокси-N-метилморфинан), кетамин, мемантин, пирролохинолинхинон и цис-4-(фосфонометил)-2-пиперидинкарбоновая кислота и их фармацевтически приемлемые соли;
(ix) альфа-адренергические активные соединения, например, доксазозин, тамзулозин, клонидин и 4-амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолин;
(х) трициклические антидепрессанты, например, дезипрамин, имипрамин, амитриптилин и нортриптилин;
(xi) противосудорожные средства, например, карбамазепин и вальпроат;
(xii) антагонисты тахикинина (NK), особенно, NK-3, NK-2 и NK-1, например, антагонисты, (αR,9R)-7-[3,5-бис(трифторметил)бензил]-8,9,10,11-тетрагидро-9-метил-5-(4-метилфенил)-7Н-[1,4]диазоцино[2,1-g][1,7]нифтридин-6-13-дион (ТАК-637), 5-[[(2R,3S)-2-[(1R)-1-[3,5-бис(трифторметил)фенил]этокси-3-(4-фторфенил)-4-морфолинил]метил]-1,2-дигидро-3Н-1,2,4-триазол-3-он (МК-869), ланепитант, дапитант и 3-[[2-метокси-5-(трифторметокси)фенил]метиламино]-2-фенилпиперидин (2S,3S);
(xiii) мускариновые антагонисты, например, оксибутин, толтеродин, пропиверин, тропсия хлорид и дарифенацин;
(xiv) ингибиторы СОХ-2, например, целекоксиб, рофекоксиб и вальдекоксиб;
(xv) неселективные ингибиторы СОХ (предпочтительно, с защитой GI), например, нитрофлурбипрофен (НСТ-1026);
(xvi) каменноугольные анальгетики, в частности, парацетамол;
(xvii) нейролептики, такие как дроперидол;
(xviii) агонисты ваниллоидных рецепторов, например, резинфератоксин;
(xix) бета-адренергические соединения, такие как пропранолол;
(хх) местные анестетики, такие как мексилетин;
(xxi) кортикостероиды, такие как дексаметазон;
(xxii) агонисты и антагонисты серотониновых рецепторов;
(xxiii) холинергические (никотиновые) анальгетики;
(xxiv) смешанные агенты, такие как Tramadol®;
(xxv) ингибиторы PDEV, такие как силденафил, варденафил или таладафил.
Настоящее изобретение относится к продукту, включающему альфа-2-дельта лиганд, DSNRI или один или оба из SSRI и SNRI и один или более других терапевтических агентов, таких, которые перечислены выше, для одновременного, отдельного или последовательного применения для терапевтического, профилактического лечения боли, особенно невропатической боли.
Комбинацию по настоящему изобретению можно вводить в отдельности, но один или оба элемента обычно будут вводить в смеси с подходящим фармацевтическим наполнителем (наполнителями), разбавителем (разбавителями) или носителем (носителями), выбранными с учетом планируемого пути введения и стандартной фармацевтической практики. Если необходимо, можно добавлять вспомогательные вещества. Вспомогательные вещества представляют собой консерванты, антиоксиданты, корригенты или красители. Соединения по настоящему изобретению могут высвобождаться по немедленному, отсроченному, модифицированному, замедленному, импульсному или контролируемому типу.
Элементы комбинации по настоящему изобретению можно вводить, например, без ограничения, следующим путем: перорально, трасбуккально или сублингвально, в форме таблеток, капсул, мульти- и наночастиц, гелей, пленок (включая мукоадгезивы), порошка, овул, эликсиров, лепешек (включая наполненные жидкостью), жевательных пастилок, растворов, суспензий и спреев. Соединения по настоящему изобретению также можно вводить в виде осмотической лекарственной формы, или в форме макроэнергической дисперсии или частиц с покрытием или в виде быстрорастворимой, быстро распадающейся лекарственной формы, как описано в Ashley Publications, 2001, Liang и Chen. Соединения по настоящему изобретению можно вводить в виде кристаллических или аморфных продуктов, лиофилизированных или высушенных с использованием распылительной сушки продуктов. Подходящие композиции соединений по настоящему изобретению могут находиться в гидрофильном или гидрофобном матриксе, комплексе ионообменной смолы, в форме с покрытием или без покрытия и в других типах, как описано в US 6106864, по желанию. Подобные фармацевтические композиции, например, таблетки, могут содержать наполнители, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция, глицин и крахмал (предпочтительно, кукурузный, картофельный крахмал или крахмал тапиоки), маннит, разрыхлители, такие как гликолят натрий-крахмала, натрий-кроскармеллоза и некоторые комплексные силикаты, и связующие агенты для гранулирования, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (НРМС), триглицериды, гидроксипропилцеллюлоза (НРС), бентонит, сахароза, сорбит, желатин и аравийская камедь. Кроме того, к твердым композициям можно добавлять смазывающие агенты, такие как стеарат магния, стеариновая кислота, глицерилбегенат, PEG и тальк, или увлажняющие агенты, такие как лаурилсульфат натрия. Кроме того, можно включать в состав полимеры, такие как углеводы, фосфолипиды и белки.
Лекарственные формы с быстрым диспергированием или быстрым растворением (FDDF) могут содержать следующие ингредиенты: аспартам, ацесульфам калия, лимонную кислоту, натрий-кроскармеллозу, кросповидон, диаскорбиновую кислоту, этилакрилат, этилцеллюлозу, желатин, гидроксипропилметилцеллюлозу, стеарат магния, маннит, метилметакрилат, мятный корригент, полиэтиленгликоль, мелкодисперсный диоксид кремния, диоксид кремния, гликолят натрий-крахмала, стеарилфумарат натрия, сорбит или ксилит. Термины «диспергирование» или «растворение», используемые в настоящем документе для описания FDDF, зависят от растворимости используемого лекарственного вещества, т.е. когда лекарственное вещество является нерастворимым, можно изготовить лекарственную форму с быстрым диспергированием, а когда лекарственное вещество является растворимым, можно изготовить лекарственную форму с быстрым растворением.
Твердую лекарственную форму, такую как таблетки, производят с использованием стандартного процесса, например, прямого прессования или влажного, сухого гранулирования или гранулирования расплава, замораживания расплава и экструдирования. На ядра таблеток, которые могут быть одно- или многослойными, можно наносить подходящие покрытия, известные специалистам.
Твердые композиции подобного типа могут также использоваться для наполнения капсул, таких как желатиновые, крахмальные или НРМС капсулы. Предпочтительные наполнители в данной связи включают лактозу, крахмал, целлюлозу, молочный сахар или высокомолекулярные полиэтиленгликоли. Жидкие композиции могут использоваться для наполнения мягких или твердых капсул, таких как желатиновые капсулы. Для водных или масляных суспензий, растворов, сиропов и/или эликсиров соединения по настоящему изобретению можно комбинировать с различными подсластителями или корригентами, окрашивающими веществами или красками, с эмульгирующими и/или суспендирующими агентами и с разбавителями, такими как вода, этанол, пропиленгликоль, метилцеллюлоза, альгиновая кислота или альгинат натрия, глицерин, масла, гидроколлоидные агенты и их комбинации. Кроме того, композиции, содержащие данные соединения и наполнители, могут быть представлены в виде сухого продукта для разведения водой или другими подходящими носителями перед использованием.
Жидкие препараты включают растворы, суспензии и эмульсии, например, растворы в воде или в воде и пропиленгликоле. Для парентеральных инъекций жидкие препараты можно изготавливать в водном растворе полиэтиленгликоля. Водные растворы, подходящие для перорального введения, можно изготавливать растворением активного компонента в воде и добавлением подходящих красителей, корригентов, стабилизирующих и загущающих агентов, по желанию. Водные суспензии, подходящие для перорального введения, можно изготавливать диспергированием тонкоизмельченного активного компонента в воде с вязким материалом, таким как натуральные или синтетические камеди, смолы, метилцеллюлоза, натрий-карбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты.
Элементы комбинации по настоящему изобретению также можно вводить путем инъекции, т.е. внутривенно, внутримышечно, внутрикожно, интрадуоденально или интраперитонеально, интраартериально, интратекально, интравентрикулярно, интрауретрально, интрастернально, интракраниально, интраспинально или подкожно, или их можно вводить путем инфузии, безыгольными инъекторами или с использованием методик инъекции имплантата. Для указанного парентерального введения их лучше всего использовать в форме стерильного водного раствора, суспензии или эмульсии (или системы, которая может включать мицеллы), которые могут содержать другие вещества, известные специалистам, например, соли или углеводы, такие как глюкоза, в количестве, достаточном для придания раствору изотоничности по отношению к крови. Водные растворы должны содержать подходящее количество буфера (предпочтительно, до рН от 3 до 9), если это необходимо. Для некоторых форм парентерального введения их можно использовать в форме стерильной неводной системы, такой как нелетучие масла, включая моно- или диглицериды, и жирные кислоты, включая олеиновую кислоту. Изготовление подходящих композиций для парентерального введения в стерильных условиях, например, лиофилизацию, легко осуществить с использованием стандартных фармацевтических методик, хорошо известных специалистам. Альтернативно, активный ингредиент может быть в порошкообразной форме для разведения подходящим носителем (например, стерильной апирогенной водой) перед использованием.
Также элементы комбинации по настоящему изобретению можно вводить интраназально или путем ингаляции. Их удобно доставлять в форме сухого порошка (по отдельности или в виде смеси, например в виде сухой смеси с лактозой, или в виде смешанной многокомпонентной частицы, например, с фосфолипидами) из ингалятора для сухого порошка или в форме аэрозоля из контейнера под давлением, насоса, спрея, распылителя (предпочтительно, распылителя с использованием электрогидродинамики для получения мелкодисперсного тумана) или небулайзера, с использованием или без использования подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, гидрофторалкана, такого как 1,1,1,2-тетрафторэтан (HFA 134A [товарный знак] или 1,1,1,2,3,3,3-гептафторпропан (HFA 227ЕA [товарный знак], диоксида углерода, дополнительно перфторированного углеводорода, такого как Perflubron (товарный знак), или другого подходящего газа. В случае аэрозоля под давлением, стандартную дозу можно определять с помощью клапана для доставки отмеренного количества. Контейнер, насос, спрей, распылитель или небулайзер под давлением может содержать раствор или суспензию активного соединения, например, с использованием смеси этанола (необязательно, водного раствора этанола) или подходящего агента для диспергирования, солюбилизации или высвобождения с расширением и пропеллента в качестве растворителя, который дополнительно может содержать смазывающий агент, например, триолеат сорбитана. Капсулы, блистерные упаковки и картриджи (изготовленные, например, из желатина или НРМС) для использования в ингаляторе или инсуффляторе, могут содержать порошкообразную смесь соединения по настоящему изобретению, подходящей порошкообразной основы, такой как лактоза или крахмал, и модификатор действия, такой как 1-лейцин, маннит или стеарат магния.
Перед использованием в сухой порошкообразной композиции или суспензии для ингаляции элементы комбинации по настоящему изобретению должны быть микронизированы до размера, подходящего для доставки путем ингаляции (обычно менее 5 микрон). Микронизации можно добиться с использованием ряда способов, например размалывания на струйной мельнице, размалывания псевдоожиженного слоя на струйной мельнице, использования сверхкритической кристаллизации жидкости или распылительной сушки.
Подходящий раствор для использования в распылителе, использующем электрогидродинамику для получения мелкодисперсного тумана, может содержать от 1 мкг до 10 мг соединения по настоящему изобретению на одно срабатывание, а объем срабатывания может варьировать от 1 до 100 мкл. Типичная композиция может включать элементы комбинации по настоящему изобретению, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители можно использовать вместо пропиленгликоля, например, глицерин или полиэтиленгликоль.
Альтернативно, элементы комбинации по настоящему изобретению можно вводить местно на кожу, слизистые оболочки, дермально или трансдермально, например, в форме геля, гидрогеля, лосьона, раствора, крема, мази, распыляемого порошка, повязки, пенки, пленки, кожного пластыря, пластинок, имплантата, губок, волокон, бинта, микроэмульсий и их комбинаций. Для подобного применения соединения по настоящему изобретению можно суспендировать или растворять, например, в смеси с одним или более из следующих агентов: минеральным маслом, жидким вазелином, белым вазелином, пропиленгликолем, полиоксиэтиленполиоксипропиленовым соединением, эмульгирующим воском, нелетучими маслами, включая моно- или диглицериды, и жирными кислотами, включая олеиновую кислоту, водой, моностеаратом сорбитана, полиэтиленгликолем, жидким парафином, полисорбатом 60, воском цетиловых сложных эфиров, цетеариловым спиртом, 2-октилдодеканолом, бензиловым спиртом, спиртами, такими как этанол. Альтернативно, можно использовать усилители проникновения. Можно использовать также полимеры, углеводы, белки, фосфолипиды в форме наночастиц (таких как ниосомы или липосомы) или в суспендированной или растворенной форме. Кроме того, их можно доставлять с использованием ионофореза, электропорации, фонофореза или сонофореза.
Альтернативно, элементы комбинации по настоящему изобретению можно вводить ректально, например, в форме суппозитория или пессария. Их можно также вводить вагинальным путем. Например, указанные композиции можно изготавливать смешивания лекарственного средства с подходящими нераздражающими наполнителями, такими как масло какао, синтетические сложные эфиры глицерина или полиэтиленгликоли, которые являются твердыми при обычных температурах, но разжижаются и/или растворяются в полости и высвобождают лекарственное средство.
Элементы комбинации по настоящему изобретению можно также вводить окулярным путем. Для офтальмологического использования соединения можно изготавливать в виде микронизированных суспензий в изотоническом, с подобранной величиной рН, стерильном физиологическом растворе, или, предпочтительно, в виде растворов в изотоническом, с подобранной величиной рН, стерильном физиологическом растворе. Можно добавлять полимер, такой как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер (например, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза, метилцеллюлоза) или гетерополисахаридный полимер (например, гелановая камедь). Альтернативно, их можно изготавливать в виде мази, такой как мазь на основе вазелина или минерального масла, инкорпорированными в биоразлагаемые (например, всасываемые гелевые губки, коллаген) или в небиоразлагаемые (например, силиконовые) имплантаты, пластины, капли, линзы, или доставлять посредством частиц или пузырьковых систем, таких как ниосомы или липосомы. Композиции можно, необязательно, комбинировать с консервантом, таким как хлорид бензалкония. Кроме того, их можно доставлять с использованием ионофореза. Их можно также вводить в ухо, с использованием, например, без ограничения, капель.
Элементы комбинации по настоящему изобретению также можно использовать в комбинации с циклодекстрином. Циклодекстрины, как известно, образуют комплексы с включением и без включения с молекулами лекарственные средства. Образование комплекса лекарственное средство-циклодекстрин может модифицировать растворимость, скорость растворения, маскировку вкуса, биодоступность и/или стабильность молекулы лекарственного средства. Комплексы лекарственное средство-циклодекстрин обычно являются пригодными для большинства лекарственных форм и путей введения. В качестве альтернативы прямому комплексированию с лекарственным средством циклодекстрин можно использовать как вспомогательную добавку, например, в качестве носителя, разбавителя или солюбилизирующего агента. Чаще всего используются альфа-, бета- и гамма-циклодекстрины, и подходящие примеры описаны в WO-A-91/11172, WO-A-94/02518 и WO-A-98/55148.
Термин «вводить» включает доставку с использованием вирусных и невирусных методик. Вирусные механизмы доставки включают, без ограничения, аденовирусные векторы, адено-ассоциированные вирусные (AAV) векторы, герпетические векторы, ретровирусные векторы, лентивирусные векторы и бакуловирусные векторы. Невирусные механизмы доставки включают трансфекцию, опосредованную липидами, липосомы, иммунолипосомы, липофектин, катионоактивные лицевые амфифильные вещества (CFA) и их комбинации. Пути для указанных механизмов доставки включают без ограничения доставку через слизистые оболочки, назальный, пероральный, парентеральный, гастроинтестинальный, местный или сублингвальный пути.
Таким образом, в качестве еще одной особенности, настоящее изобретение относится к фармацевтической композиции, содержащей альфа-2-дельта лиганд, DSNRI или один или оба из SSRI и SNRI или их фармацевтически приемлемые соли, при условии, что исключаются соединения (i)-(xxv) WO 02/85839 в комбинации с ингибитором обратного захвата серотонина, особенно, флуоксетином, пароксетином, циталопрамом и сертралином, смешанным ингибитором обратного захвата серотонина-норадреналина, особенно, милнаципраном, венлафаксином и дулоксетином, и ингибитором обратного захвата норадреналина, особенно, ребоксетином, и подходящий наполнитель, разбавитель или носитель. Соответственно, композиция является подходящей для использования для лечения боли, особенно, невропатической боли.
В качестве альтернативной особенности, настоящее изобретение относится к фармацевтической композиции, содержащей синергическую комбинацию, включающую альфа-2-дельта лиганд, DSNRI или один или оба из SSRI и SNRI или их фармацевтически приемлемые соли и подходящий наполнитель, разбавитель или носитель. Соответственно, композиция является подходящей для использования для лечения боли, особенно, невропатической боли.
Для введения животному, которое не является человеком, термин «фармацевтический», используемый в настоящем документе, может быть заменен на «ветеринарный».
Фармацевтический препарат предпочтительно находится в виде стандартной лекарственной формы. В указанной форме препарат разделен на стандартные дозы, содержащие подходящие количества активного компонента. Стандартная лекарственная форма может представлять собой упакованный препарат; упаковка содержит отдельные количества препарата, такие как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Также стандартная лекарственная форма может представлять собой сами капсулы, таблетку, облатку или лепешку, или она может представлять собой подходящее количество, любую из указанных форм в упакованном виде. Количество активного компонента в препарате стандартной лекарственной формы может варьировать или доводиться до количества от 0,1 мг до 1 г, согласно конкретному применению и эффективности активных компонентов. При медицинском использовании лекарственное средство можно вводить три раза в день в виде, например, капсул, содержащих 100 или 300 мг. При терапевтическом применении соединения, которые используются в фармацевтическом способе по настоящему изобретению, вводят в первоначальной дозировке приблизительно от 0,01 мг до 100 мг/кг в день. Предпочтительны пределы суточных доз приблизительно от 0,01 мг до 100 мг/кг. Дозировки, однако, могут варьировать в зависимости от требований пациента, тяжести состояния, по поводу которого осуществляют лечение, и используемых соединений. Определение правильной дозировки для конкретной ситуации находится в компетенции специалиста. Обычно лечение начинают с более низких доз, которые меньше оптимальной дозы соединений. После этого дозировку понемногу увеличивают до достижения оптимального при данных обстоятельствах эффекта. Для удобства суммарную суточную дозу можно разделить и вводить частями в течение дня, если это желательно.
Для ветеринарного использования комбинацию по настоящему изобретению или ее ветеринарно-приемлемые соли или сольваты вводят в подходящей приемлемой композиции, в соответствии с обычной ветеринарной практикой, и ветеринарный врач определит режим дозирования и путь введения, которые являются наиболее подходящими для конкретного животного.
Биологические примеры
Методы
Животные
Самцов крыс Sprague Dawley (200-250 г), полученных из Charles River (Margate, Kent, U.K.), содержали группами по 6 животных в каждой. Всех животных содержали при 12-часовом цикле свет/темнота (свет включали в 07 час 00 мин), при свободном доступе к пище и воде. Все эксперименты осуществлял наблюдатель, не информированный о лекарственном лечении.
CCI оперативное вмешательство на крысах
Животных обезболивали изофлюраном. Седалищный нерв лигировали, как описано ранее Bennett и Xie, 1988. Животных помещали на гомеотермическое одеяло на всю продолжительность процедуры. После хирургической обработки обнажали общий седалищный нерв на середине бедра тупым рассечением через двуглавую мышцу бедра. Проксимальнее от разделения седалищного нерва на три ветви, приблизительно 7 мм нерва освобождали от прилегающей ткани и вокруг него неплотно завязывали 4 лигатуры (шелк 4-0), с расстоянием между ними около 1 мм. Разрез послойно ушивали, а рану обрабатывали местными антибиотиками.
Влияние комбинаций на сохранение CCI-индуцированной статической и динамической аллодинии
Определение взаимоотношений доза-ответы на габапентин, DSNRI, SSRI и SNRI сначала осуществляли по отдельности на модели CCI. Комбинации изучали в соответствии с фиксированной схемой соотношений. Осуществляли определение взаимоотношений доза-ответ для каждого фиксированного соотношения доз. В каждый экспериментальный день перед лекарственным воздействием определяли исходные пороги отдергивания лапы (PWT) от нитей фон Фрея и латентные периоды до отдергивания лапы (PWL) от раздражения ватным тампоном.
Оценка аллодинии
Статическую аллодинию измеряли с использованием нитей Semmes-Weinstein von Frey (Stoelting, Illinois, USA). Животных помещали в клетки с проволочным дном, которое обеспечивало доступ к нижней поверхности их лап. До начала эксперимента животные привыкали к указанному окружению. Статическую аллодинию изучали путем прикосновения к плантарной поверхности правой задней лапы животных нитей фон Фрея с увеличивающимся порядком усилия (0,7, 1,2, 1,5, 2, 3,6, 5,5, 8,5, 11,8, 15,1 и 29 г). После установления реакции отдергивания лапу повторно испытывали, начиная со следующей нисходящей серии нитей фон Фрея, до исчезновения реакции. Точку отсечения представляло самое большое усилие 29 г, которое приподнимало лапу, а также вызывало реакцию. Самое малое усилие, которое требовалось для получения реакции, отмечали как PWT в граммах.
Динамическую аллодинию оценивали легким ударом по плантарной поверхности задней лапы ватным тампоном. Были предприняты все меры для того, чтобы данную процедуру производили на полностью привыкших в данному окружению крысах, которые не проявляли активность для того, чтобы избежать записи общей двигательной активности. По меньшей мере три измерения проводили в каждый момент времени; среднее значение от указанных измерений представляло собой латентный период отдергивания лапы (PWL). Если в течение 15 сек реакции не наблюдалось, процедуру прекращали, а животным устанавливали указанное время отдергивания. Таким образом, период времени 15 сек эффективно представлял отсутствие отдергивания. Реакция одергивания часто сопровождалась повторным одергиванием или лизанием лапы. Считалось, что динамическая аллодиния имеет место, если животные отвечали на раздражение ватой до истечения 8 сек после удара.
Изучение комбинации
Определение взаимоотношений доза-ответы сначала осуществляли с использованием альфа-2-дельта лиганда (п/о) DSNRI или SSRI и SNRI (п/к или п/о) по отдельности. Затем можно было изучать ряд фиксированных соотношений доз в комбинации. Осуществляли определение взаимоотношений доза-ответ для каждого фиксированного соотношения доз с течением времени для каждого эксперимента, определенного продолжительностью антиаллодинического действия каждого отдельного соотношения. Можно исследовать различные фиксированные соотношения доз комбинаций по массе.
Подходящие DSNRI или SSRI и/или SNRI соединения по настоящему изобретению можно получить, как описано в ссылках, или как это очевидно для специалиста на основе указанных документов.
Подходящие альфа-2-дельта лиганды по настоящему изобретению можно получить, как описано ниже в настоящем документе, или в упомянутых выше ссылках на патентную литературу, которые иллюстрируются следующими неограничивающими примерами и промежуточными веществами.
Химические примеры
Пример 1
Гидрохлорид (3S,5R)-3-амино-5-метилоктановой кислоты
(R)-2,6-диметилнон-2-ен. К (S)цитронеллилбромиду (50 г, 0,228 моль) в THF (800 мл) при 0°С добавляли LiCl (4,3 г), а затем CuCl2 (6,8 г). Через 30 минут добавляли хлорид метилмагния (152 мл 3 М раствора в THF, Aldrich) и раствор нагревали до комнатной температуры. Через 10 часов раствор охлаждали до 0°С и осторожно добавляли насыщенный водный раствор хлорида аммония. Полученные два слоя разделяли и водную фазу экстрагировали эфиром. Объединенные органические фазы сушили (MgSO4) и концентрировали с получением (R)-2,6-диметилнон-2-ена. 32,6 г; 93%. Использовали без дополнительной очистки. 1H ЯМР (400 МГц; CDCl3) δ 5,1 (м, 1H), 1,95 (м, 2H), 1,62 (с, 3H), 1,6 (с, 3H), 1,3 (м, 4H), 1,2 (м, 2H), 0,8 (с, 6H).
(R)-4-метилгептановая кислота. К (R)-2,6-диметилнон-2-ену (20 г, 0,13 моль) в ацетоне (433 мл) добавляли раствор CrO3 (39 г, 0,39 моль) в H2SO4 (33 мл)/Н2О (146 мл) в течение 50 минут. Через 6 часов добавляли дополнительное количество CrO3 (26 г, 0,26 моль) в H2SO4 (22 мл)/Н2О (100 мл). Через 12 часов раствор разбавляли насыщенным солевым раствором и раствор экстрагировали эфиром. Объединенные органические фазы сушили (MgSO4) и концентрировали. Флэш-хроматография (градиент от 6:1 до 2:1 гексан/EtOAc) дала (R)-4-метилгептановую кислоту в виде масла, 12,1 г; 65%. МС, m/z (относительная интенсивность): 143 [M-H, 100%].
(4R,5S)-4-метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-он. К (R)-4-метилгептановой кислоте (19 г, 0,132 моль) и триэтиламину (49,9 г, 0,494 моль) в THF (500 мл) при 0°С добавляли триметилацетилхлорид (20 г, 0,17 моль). Через 1 час добавляли LiCl (7,1 г, 0,17 моль), а затем (4R,5S)-(+)-4-метил-5-фенил-2-оксазолидинон) 3 (30 г, 0,17 моль). Смесь затем нагревали до комнатной температуры и через 16 часов фильтрат удаляли фильтрованием и раствор концентрировали при пониженном давлении. Флэш-хроматография (7:1 гексан/EtOAc) дала (4R,5S)-4-метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-он в виде масла, 31,5 г; 79%. [α]D=+5,5 (с 1 в CHCl3). МС, m/z (относительная интенсивность): 304 [M+H, 100%].
Трет-бутиловый эфир (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты. К (4R,5S)-4-метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-ону (12,1 г, 0,04 моль) в THF (200 мл) при -50°С добавляли бис(триметилсилил)амид натрия (48 мл 1 М раствора в THF). Через 30 мин добавляли трет-бутилбромацетат (15,6 г, 0,08 моль). Раствор перемешивали в течение 4 часов при -50°С, а затем нагревали до комнатной температуры. Через 16 часов добавляли насыщенный водный раствор хлорида аммония, и два слоя разделялись. Водную фазу экстрагировали эфиром, и комбинированные органические фазы сушили (MgSO4) и концентрировали. Флэш-хроматография (9:1 гексан/EtOAc) дала трет-бутиловый эфир (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты в виде твердого вещества белого цвета, 12 г; 72%. [α]D=+30,2 (с 1 в CHCl3). 13С ЯМР (100 МГц; CDCl3) δ 176,47, 171,24, 152,72, 133,63, 128,87, 125,86, 80,85, 78,88, 55,34, 39,98, 38,77, 38,15, 37,58, 30,60, 28,23, 20,38, 20,13, 14,50, 14,28.
4-трет-бутиловый эфир (S)-2-((R)-2-метилпентил)янтарной кислоты. К трет-бутиловому эфиру (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты (10,8 г, 0,025 моль) в Н2О (73 мл) и THF (244 мл) при 0°С добавляли предварительно смешанный раствор LiOH (51,2 мл 0,8 М раствора) и Н2О2 (14,6 мл 30% раствора). Через 4 часа добавляли еще 12,8 мл LiOH (0,8 М раствор) и 3,65 мл Н2О2 (30% раствор). Через 30 минут добавляли бисульфит натрия (7 г), сульфит натрия (13 г) и воду (60 мл), а затем гексан (100 мл) и эфир (100 мл). Два слоя разделялись, и водный слой экстрагировали эфиром. Комбинированные органические фазы концентрировали до масла, которое растворяли в гептане (300 мл). Полученное твердое вещество отфильтровывали и фильтрат сушили (MgSO4) и концентрировали с получением 4-трет-бутилового эфира (S)-2-((R)-2-метилпентил)янтарной кислоты (6 г, 93%), который использовали немедленно без дополнительной очистки. МС, m/z (относительная интенсивность): 257 [M+H, 100%].
Трет-бутиловый эфир (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты. На раствор 4-трет-бутилового эфира (S)-2-((R)-2-метилпентил)янтарной кислоты (6,0 г, 23,22 ммоль) и триэтиламина (3,64 мл, 26,19 ммоль) в толуоле (200 мл) действовали дифенилфосфорилазидом (5,0 мл, 23,22 мл) и перемешивали при комнатной температуре в течение 0,5 часа. После того, как реакционную смесь нагревали с обратным холодильником в течение 3 часов и быстро охлаждали, добавляли бензиловый спирт (7,2 мл, 69,7 ммоль) и раствор нагревали еще 3 часа. После охлаждения реакционной смеси ее разбавляли этиловым эфиром (200 мл) и комбинированный органический слой промывали последовательно насыщенным NaHCO3 и насыщенным солевым раствором и сушили (Na2SO4). Концентрированный органический компонент очищали хроматографией (MPLC) с элюированием 8:1 гексаны: этилацетат, с получением трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты (6,4 г, 75,8%). МС: М+1: 364,2, 308,2.
Трет-бутиловый эфир (3S,5R)-3-амино-5-метилоктановой кислоты. На раствор трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты (2,14 г, 5,88 ммоль) в THF (50 мл) действовали Pd/C (0,2 г) и Н2 при 50 фунтах на кв.дюйм в течение 2 часов. Реакционную смесь затем фильтровали и концентрировали до масла в вакууме, с получением трет-бутилового эфира (3S,5R)-3-амино-5-метилоктановой кислоты с количественным выходом. МС: М+1: 230,2, 174,1.
Гидрохлорид (3S,5R)-3-амино-5-метилоктановой кислоты. Взвесь трет-бутилового эфира (3S,5R)-3-амино-5-метилоктановой кислоты (2,59 г, 11,3 ммоль) в 6 н. HCl (100 мл) нагревали с обратным холодильником в течение 18 часов, охлаждали и фильтровали через целит. Фильтрат концентрировали в вакууме до 25 мл и полученные кристаллы собирали и сушили, с получением гидрохлорида (3S,5R)-3-амино-5-метилоктановой кислоты, т.пл. 142,5-142,7°С (1,2 г, 50,56%). Вторую партию (0,91 г) получали из фильтрата. Аналитически рассчитано для C9H19NO2·HCl: C: 51,55, H: 9,61, N: 6,68, Cl: 16,91. Обнаружено: C: 51,69, H: 9,72, N: 6,56, Cl: 16,63.
Соль хлористоводородной кислоты (3S,5R)-3-амино-5-метилоктановой кислоты. 5,3 г 4-трет-бутилового эфира 2S-(2R-метилпентил)янтарной кислоты, содержащиеся в 30 мл метилтрет-бутилового эфира, взаимодействовали при комнатной температуре с 3,5 мл триэтиламина, а затем с 6,4 г дифенилфосфорилазида. После достижения реакционной смесью экзотерма до 45°С и перемешивания в течение по меньшей мере 4 часов, реакционную смесь оставляли охлаждаться до комнатной температуры и стоять до разделения фаз. Нижний слой сливали, а верхний слой промывали водой, а затем разбавленной водной HCl. Верхний слой затем объединяли с 10 мл 6 н. водной HCl и перемешивали при 45-65°С. Реакционную смесь концентрировали вакуумной дистилляцией приблизительно до 10-14 мл и позволяли кристаллизоваться при охлаждении приблизительно до 5°С. После сбора продукта фильтрованием продукт промывали толуолом и повторно суспендировали в толуоле. Продукт сушили нагреванием в условиях вакуума, с получением 2,9 г (67%) кристаллического продукта белого цвета. Продукт можно перекристаллизовать из водной HCl. Т.пл. 137°С.
Пример 2
(3S,5R)-амино-5-метилгептановая кислота
(S)-3,7-диметилокт-6-ениловый эфир метансульфоновой кислоты. К (S)-(-)цитронеллолу (42,8 г, 0,274 моль) и триэтиламину (91 мл, 0,657 моль) в CH2Cl2 (800 мл) при 0°С добавляли метансульфонилхлорид (26 мл, 0,329 моль) в CH2Cl2 (200 мл). После 2 часов при 0°С раствор промывали 1 н. HCl, а затем насыщенным солевым раствором. Органическую фазу сушили (MgSO4) и концентрировали, с получением указанного в заголовке соединения в виде масла (60,5 г, 94%), которое использовали без дополнительной очистки. МС, m/z (относительная интенсивность): 139 [100%], 143 [100%].
(R)-2,6-диметилокт-2-ен. К (S)-3,7-диметилокт-6-ениловый эфиру метансульфоновой кислоты (60 г, 0,256 моль) в THF (1 л) при 0°С добавляли гидрид литий-алюминия (3,8 г, 0,128 моль). Через 7 часов добавляли еще 3,8 г гидрида литий-алюминия и раствор нагревали до комнатной температуры. Через 18 часов добавляли еще 3,8 г гидрида литий-алюминия. Еще через 21 час реакцию осторожно гасили 1 н. лимонной кислотой и раствор дополнительно разбавляли насыщенным солевым раствором. Полученные две фазы разделялись, и органическую фазу сушили (MgSO4) и концентрировали, с получением указанного в заголовке соединения в виде масла, которое использовали без дополнительной очистки. МС, m/z (относительная интенсивность): 139 [100%].
(R)-4-метилгексановая кислота. Использовали процедуру, сходную с процедурой синтеза (R)-4-метилгептановой кислоты, с получением кислоты в виде масла (9,3 г, 56%). МС, m/z (относительная интенсивность): 129 [М-Н, 100%].
(4R,5S)-4-метил-3-((R)-4-метилгексаноил)-5-фенилоксазолидин-2-он. Использовали процедуру, сходную с процедурой синтеза (4R,5S)-4-метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-она, с получением указанного в заголовке соединения в виде масла (35,7 г, 95%). МС, m/z (относительная интенсивность): 290 [М+Н, 100%].
Трет-бутиловый эфир (3S,5R)-5-метил-3-[1-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-ил)метаноил]гептановой кислоты. Использовали процедуру, сходную с процедурой синтеза трет-бутилового эфира (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты с получением указанного в заголовке соединения в виде масла (7,48 г, 31%). МС, m/z (относительная интенсивность): 178 [100%], 169 [100%], [α]D=+21,6 (c 1 в CHCl3).
4-трет-бутиловый эфир (S)-2-((R)-2-метилбутил)янтарной кислоты. К трет-бутиловому эфиру (3S,5R)-5-метил-3-[1-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-ил)метаноил]гептановой кислоты (7,26 г, 0,018 моль) в Н2О (53 мл) и THF (176 мл) при 0°С добавляли предварительно смешанный раствор LiOH (37 мл 0,8 М раствора) и Н2О2 (10,57 мл 30% раствора) и раствор нагревали до комнатной температуры. Через 2 часа добавляли бисульфит натрия (7 г), сульфит натрия (13 г) и воду (60 мл), и два слоя разделялись, и водный слой экстрагировали эфиром. Объединенные органические фазы концентрировали до масла, которое растворяли в гептане (200 мл). Полученное твердое вещество отфильтровывали и фильтрат сушили (MgSO4) и концентрировали, с получением указанного в заголовке соединения в виде масла (4,4 г), которое использовали без дополнительной очистки. МС, m/z (относительная интенсивность): 243 [100%].
Трет-бутиловый эфир (3S,5R)-3-бензиоксикарбониламино-5-метилгептановой кислоты. Указанное соединение получали, как описано выше, начиная с 4-трет-бутилового эфира (S)-2-((R)-2-метилбутил)янтарной кислоты, с получением трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилгептановой кислоты в виде масла (выход 73,3%). 1H ЯМР (400 МГц; CDCl3) δ 0,84 (т, 3H, J=7,33 Гц), 0,89 (д, 3H, J=6,60 Гц), 1,12-1,38 (м, 4H), 1,41 (с, 9H), 1,43-1,59 (м, 2H), 2,42 (м, 2H), 4,05 (м, 1H), 5,07 (т, 2H, J=12,95 Гц) и 7,28-7,34 (м, 5H).
Трет-бутиловый эфир (3S,5R)-амино-5-метилгептановой кислоты. Указанное соединение получали, как описано выше, начиная с трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилгептановой кислоты вместо трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты, с получением указанного в заголовке соединения. 1H ЯМР (400 МГц; CDCl3) δ 0,84 (перекрывающиеся т и д, 6H), 1,08-1,16 (м, 2H), 1,27-1,30 (м, 2H), 1,42 (с, 9H), 1,62 (ушир.с, 2H), 2,15 (дд, 1H, J=8,54 и 15,62 Гц), 2,29 (дд, 1H, J=4,15 и 15,37 Гц) и 3,20 (ушир.с, 2H).
Гидрохлорид (3S,5R)-амино-5-метилгептановой кислоты. Взвесь трет-бутилового эфира (3S,5R)-амино-5-метилгептановой кислоты (1,44 г, 6,69 ммоль) в 3 н. HCl нагревали с обратным холодильником в течение 3 часов, фильтровали горячим через целит и концентрировали до сухости. Растирание полученного твердого вещества в этиловом эфире дало гидрохлорид (3S,5R)-3-амино-5-метилгептановой кислоты (0,95 г, 85%), т.пл. 126,3-128,3°С.
Пример 3
(3S,5R)-3-амино-5-метилнонановая кислота
(R)-4-метилоктановая кислота. Хлорид лития (0,39 г, 9,12 ммоль) и хлорид меди (I) (0,61 г, 4,56 ммоль) объединяли в 45 мл THF при комнатной температуре и перемешивали в течение 15 минут, затем охлаждали до 0°С, и в указанный момент добавляли бромид этилмагния (1 М раствор в THF, 45 мл, 45 ммоль). По каплям добавляли (S)-цитронеллилбромид (5,0 г, 22,8 ммоль) и раствору позволяли медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакцию гасили осторожным добавлением насыщенный NH4Cl (водный) и перемешивали с Et2O и насыщенный NH4Cl (водный) в течение 30 минут. Фазы разделялись, и органическую фазу сушили (MgSO4) и концентрировали. Сырой (R)-2,6-диметилдец-2-ен использовали без очистки. К раствору (R)-2,6-диметилдец-2-ена (3,8 г, 22,8 ммоль) в 50 мл ацетона при 0°С добавляли реагент Джонса (2,7 М в H2SO4 (водный), 40 мл, 108 ммоль) и раствору позволяли медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Смесь распределяли между Et2O и Н2О, фазы разделялись, и органическую фазу промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали. Остаток очищали флэш-хроматографией (8:1 гексаны: EtOAc), с получением 2,14 г (59%) указанного в заголовке соединения в виде бесцветного масла: LRMS: m/z 156,9 (M+). Реагент Джонса изготавливали как 2,7 М раствор объединением 26,7 г CrO3, 23 мл H2SO4 и разбавлением до 100 мл Н2О.
(4R,5S)-4-метил-3-((R)-4-метилоктаноил)-5-фенилоксазолидин-2-он. К (R)-4-метилоктановой кислоте (2,14 г, 13,5 ммоль) в 25 мл CH2Cl2 при 0°С добавляли 3 капли DMF, а затем оксалилхлорид (1,42 мл, 16,2 ммоль), после чего наблюдалось бурное газовыделение. Раствор затем непосредственно нагревали до комнатной температуры, перемешивали в течение 30 минут и концентрировали. Тем временем к раствору оксазолидинона (2,64 г, 14,9 ммоль) в 40 мл THF при -78°С добавляли н-бутиллитий (1,6 М раствор в гексанах, 9,3 мл, 14,9 ммоль) по каплям. Смесь перемешивали в течение 10 минут, после чего добавляли хлорангидрид кислоты в 10 мл THF по каплям. Реакционную смесь перемешивали в течение 30 минут при -78°С, затем непосредственно нагревали до комнатной температуры и гасили насыщенным NH4Cl. Смесь распределяли между Et2O и насыщенным NH4Cl (водный), фазы разделялись, и органическую фазу сушили (MgSO4) и концентрировали, с получением 3,2 г указанного в заголовке соединения в виде бесцветного масла. LRMS: m/z 318,2 (M+).
Трет-бутиловый эфир (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)нонановой кислоты. К раствору диизопропиламина (1,8 мл, 12,6 ммоль) в 30 мл THF при -78°С добавляли н-бутиллитий (1,6 М раствор в гексанах, 7,6 мл, 12,1 ммоль) и смесь перемешивали в течение 10 минут, после чего добавляли (4R,5S)-4-метил-3-((R)-4-метилоктаноил)-5-фенилоксазолидин-2-он (3,2 г, 10,1 ммоль) в 10 мл THF по каплям. Раствор перемешивали в течение 30 минут, быстро по каплям добавляли трет-бромацетат (1,8 мл, 12,1 ммоль) при -50°С, и смеси позволяли медленно нагреваться до 10°С в течение 3 часов. Смесь распределяли между Et2O и насыщенным NH4Cl (водный), фазы разделялись, и органическую фазу сушили (MgSO4) и концентрировали. Остаток очищали флэш-хроматографией (от 16:1 до 8:1 гексаны:EtOAc), с получением 2,65 г (61%) указанного в заголовке соединения в виде бесцветного твердого вещества, т.пл.=84-86°С. [δ]D 23+17,1 (c=1,00, CHCl3).
4-трет-бутиловый эфир (S)-2-((R)-2-метилгексил)янтарной кислоты. К раствору трет-бутилового эфира (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)нонановой кислоты (2,65 г, 6,14 ммоль) в 20 мл THF при 0°С добавляли предварительно охлажденный (0°С) раствор моногидрата LiOH (1,0 г, 23,8 ммоль) и пероксид водорода (30 мас.% водный раствор, 5,0 мл) в 10 мл Н2О. Смесь интенсивно перемешивали в течение 90 минут, затем нагревали до комнатной температуры и перемешивали в течение 90 минут. Реакцию гасили при 0°С добавлением 100 мл 10% NaHSO3 (водный), затем экстрагировали Et2O. Фазы разделялись, и органическую фазу промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали. Указанное в заголовке соединение использовали без очистки.
Трет-бутиловый эфир (3S,5R)-3-бензиоксикарбониламино-5-метилнонановой кислоты. Указанное соединение получали, как описано выше, начиная с 4-трет-бутилового эфира (S)-2-((R)-2-метилгексил)янтарной кислоты, вместо 4-трет-бутилового эфира (S)-2-((R)-2-метилпентил)янтарной кислоты с получением указанного в заголовке соединения в виде масла (выход 71,6%). 1H ЯМР (400 МГц; CDCl3) δ 0,81 (т, 3H, J=4,40 Гц), 0,85 (д, 3H, J=6,55 Гц), 1,06-1,20 (м, 7H), 1,36 (с, 9H), 1,38-1,50 (м, 2H), 2,36 (м, 2H), 3,99 (м, 1H), 5,02 (м+с, 3H) и 7,28-7,28 (м, 5H).
Трет-бутиловый эфир (3S,5R)-3-амино-5-метилнонановой кислоты. Указанное соединение получали, как описано выше, начиная с трет-бутилового эфира (3S,5R)-бензиоксикарбониламино-5-метилнонановой кислоты вместо трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты. Выход=97%. 1H ЯМР (400 МГц; CDCl3) δ 0,82 (перекрывающиеся д и т, 6H), 1,02-1,08 (м, 1H), 1,09-1,36 (м, 6H), 1,39 (с, 9H), 1,47 (ушир.с, 1H), 1,80 (с, 2H), 2,13 (дд, 1H, J=8,54 и 15,61 Гц) и 2,27 (дд, 1H, J=4,15 и 15,38 Гц).
Гидрохлорид (3S,5R)-3-амино-5-метилнонановой кислоты. Смесь трет-бутилового эфира (3S,5R)-3-амино-5-метилнонановой кислоты (1,50 г, 6,16 ммоль) в 3 н. HCl (100 мл) нагревали с обратным холодильником в течение 3 часов, фильтровали горячим через целит и концентрировали в вакууме до 30 мл. Полученные кристаллы собирали, промывали дополнительно 3 н. HCl и сушили, с получением указанного в заголовке соединения, т.пл. 142,5-143,3°С. Дополнительные партии получали из фильтрата, 1,03 г (70,4%). Анал. рассчитано для C10H21NO2·HCl: C: 53,68, H: 9,91, N: 6,26, Cl: 15,85. Обнаружено: C: 53,89, H: 10,11, N: 6,13. МС: М+1: 188,1.
Примеры фармацевтических композиций
В следующих примерах термин «активное соединение» или «активный ингредиент» относится к подходящей комбинации или отдельному элементу альфа-2-дельта лиганда и DSNRI или одного или обоих из SSRI и SNRI и/или их фармацевтически приемлемой соли, согласно настоящему изобретению.
(i) Композиции таблеток
Следующие композиции А и В можно изготовить влажным гранулированием ингредиентов от (а) до (с) и от (а) до (d) с использованием раствора повидона, с последующим добавлением стеарата магния и прессованием.
Следующие композиции D и Е можно изготовить прямым прессованием смешанных ингредиентов. Лактоза, используемая в композиции Е, представляет собой лактозу типа прямого прессования.
Композицию можно изготовить влажным гранулированием ингредиентов от (а) до (с) с использованием раствора повидона, с последующим добавлением стеарата магния и прессованием.
Композиция G (таблетка с энтеросолюбильным покрытием)
Таблетки с энтеросолюбильным покрытием композиции С можно изготовить нанесением на таблетки покрытия в количестве 25 мг на таблетку из энтеросолюбильного полимера, такого как ацетофталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы или из анионоактивных полимеров метакриловой кислоты и метилового эфира метакриловой кислоты (Eudragit L). За исключением Eudragit L, указанные полимеры также должны включать 10% (от массы используемого полимера) пластификатора, чтобы предотвратить растрескивание оболочки по время применения или хранения. Подходящие пластификаторы включают диэтилфталат, трибутилцитрат и триацетин.
Композиция Н (таблетка с энтеросолюбильным покрытием, с контролируемым высвобождением)
Таблетки с энтеросолюбильным покрытием композиции F можно изготовить нанесением на таблетки покрытия в количестве 50 мг на таблетку из энтеросолюбильного полимера, такого как ацетофталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы или из анионоактивных полимеров метакриловой кислоты и метилового эфира метакриловой кислоты (Eudragit L). За исключением Eudragit L, указанные полимеры также должны включать 10% (от массы используемого полимера) пластификатора, чтобы предотвратить растрескивание оболочки во время нанесения или хранения. Подходящие пластификаторы включают диэтилфталат, трибутилцитрат и триацетин.
(ii) Композиции для капсул
Композиция А
Капсулы можно изготовить смешиванием ингредиентов композиции D, выше, и наполнением состоящих из двух частей твердых желатиновых капсул полученной смесью. Композицию В (ниже) можно изготовить сходным образом.
Капсулы можно изготовить расплавлением Macrogol 4000 BP, диспергированием активного ингредиента в расплаве и наполнением им твердых желатиновых капсул.
Капсулы можно изготовить диспергированием активного ингредиента в лецитине и арахисовом масле и наполнением дисперсией мягких, эластичных желатиновых капсул.
Капсулы можно изготовить экструдированием смешанных ингредиентов от (а) до (с) с использованием экструдера, затем сферонизацией и сушкой экструдата. Высушенные гранулы покрывают оболочкой, обеспечивающей контролируемое высвобождение (d), и наполняют ими состоящие из двух частей твердые желатиновые капсулы.
Композицию для энтеросолюбильных капсул можно изготовить экструдированием смешанных ингредиентов от (а) до (с) с использованием экструдера, затем сферонизацией и сушкой экструдата. Высушенные гранулы покрывают энтеросолюбильной оболочкой (d), содержащей пластификатор (е), и наполняют ими состоящие из двух частей твердые желатиновые капсулы.
Композиция G (энтеросолюбильная капсула с контролируемым высвобождением)
Энтеросолюбильные капсулы композиции Е можно изготовить нанесением на гранулы с контролируемым высвобождением покрытия в количестве 50 мг на капсулу из энтеросолюбильного полимера, такого как ацетофталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы или из анионоактивных полимеров метакриловой кислоты и метилового эфира метакриловой кислоты (Eudragit L). За исключением Eudragit L, указанные полимеры также должны включать 10% (от массы используемого полимера) пластификатора, чтобы предотвратить растрескивание оболочки во время нанесения или хранения. Подходящие пластификаторы включают диэтилфталат, трибутилцитрат и триацетин.
(iii) Композиция для внутривенной инъекции
Активный ингредиент растворяют в большей части фосфатного буфера при 35-40°С, затем доводят до нужного объема и фильтруют через стерильный микропористый фильтр в стерильные 10 мл стеклянные флаконы (типа 1), которые герметично запечатывают стерильными крышками и дополнительными укупорочными средствами.
(iv) Композиция для внутримышечной инъекции
Активный ингредиент растворяют в гликофуроле. Затем добавляют бензиловый спирт и растворяют, и добавляют воду до 3 мл. Смесь затем фильтруют через стерильный микропористый фильтр и герметично запечатывают в стерильные 3 мл стеклянные флаконы (типа 1).
(v) Композиция сиропа
Бензоат натрия растворяют в части очищенной воды и добавляют раствор сорбита. Добавляют активный ингредиент и растворяют. Полученный раствор смешивают с глицерином, а затем доводят до нужного объема очищенной водой.
(vi) Композиция суппозитория
Одну пятую Witepsol H15 расплавляют на поддоне с паровой рубашкой при максимальной температуре 45°С. Активный ингредиент просеивают через сито 200 lm и добавляют к расплавленной основе при перемешивании, с использованием Silverson с режущей головкой, до получения однородной дисперсии. Поддерживая температуру 45°С, добавляют оставшийся Witepsol H15 к суспензии, которую перемешивают до получения однородной смеси. Всю суспензию затем пропускают через сито из нержавеющей стали 250 lm и при перемешивании позволяют охладиться до 40°С. При температуре 38-40°С аликвотами смеси по 2,02 г наполняют подходящие пластиковые пресс-формы и суппозиториям позволяют остыть до комнатной температуры.
(vii) Композиция пессария
Указанные выше ингредиенты непосредственно смешивают, и изготавливают пессарии прессованием полученной смеси.
(viii) Трансдермальная композиция
Активный ингредиент и спирт USP желируют гидроксиэтилцеллюлозой и упаковывают в трансдермальное устройство с площадью поверхности 10 см2.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИИ АЛЬФА-2-ДЕЛЬТА-ЛИГАНДА С СЕЛЕКТИВНЫМ ИНГИБИТОРОМ ЦИКЛООКСИГЕНАЗЫ-2 | 2003 |
|
RU2286151C2 |
| АЛЬФА-2-ДЕЛЬТА ЛИГАНД ДЛЯ ЛЕЧЕНИЯ СИМПТОМОВ НИЖНИХ МОЧЕВЫВОДЯЩИХ ПУТЕЙ | 2003 |
|
RU2331438C2 |
| СОЛИ И КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2014 |
|
RU2654855C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ У МЛЕКОПИТАЮЩИХ | 2004 |
|
RU2344121C2 |
| ПРОИЗВОДНЫЕ ПРЕГАБАЛИНА ДЛЯ ЛЕЧЕНИЯ ПРИЛИВОВ | 2003 |
|
RU2353358C2 |
| БИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ГАММА-АМИНОКИСЛОТЫ | 2008 |
|
RU2446148C2 |
| ПОЛИМОРФНЫЕ ФОРМЫ | 2014 |
|
RU2670978C9 |
| СЕЛЕКТИВНЫЕ ОБРАТНЫЕ АГОНИСТЫ СЕРОТОНИНОВЫХ РЕЦЕПТОРОВ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ЗАБОЛЕВАНИИ | 2005 |
|
RU2442607C2 |
| АНАЛОГИ ГАММА-АМИНОМАСЛЯНОЙ КИСЛОТЫ (ГАМК) ДЛЯ ЛЕЧЕНИЯ БОЛИ И ДРУГИХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2679407C1 |
| 4-[2-(2-ФТОРФЕНОКСИМЕТИЛ)ФЕНИЛ]ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2515612C2 |
Настоящее изобретение относится к медицине и описывает комбинацию для терапевтического, профилактического или паллиативного лечения боли, состоящую, по существу, из (а) альфа-2-дельта лиганда, выбранного из габапентина, прегабалина, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты, 3-(1-аминометилциклогексилметил)-4Н-[1,2,4]оксадиазол-5-она, С-[1-(1Н-тетразол-5-илметил)циклогептил]метиламина, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты, (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты, (3S,5R)-3-аминометил-5-метилоктановой кислоты, (3S,5R)-3-амино-5-метилгептановой кислоты, (3S,5R)-3-амино-5-метилнонановой кислоты и (3S,5R)-3-амино-5-метилоктановой кислоты или фармацевтически приемлемой соли любого из них и (b) S,S-ребоксетина или его фармацевтически приемлемой соли, где альфа-2-дельта лиганд и S,S-ребоксетин присутствуют в интервале соотношений от 1:10 до 10:1 массовых частей. Благодаря данной комбинации появляется возможность уменьшения дозы каждого соединения, что приводит к уменьшению побочных эффектов и повышению клинической пригодности соединений. Описано также применение данной комбинации и набор, включающий данную комбинацию. 4 н. и 4 з.п. ф-лы.
(а) альфа-2-дельта лиганд, выбранный из габапентина, прегабалина, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусной кислоты, 3-(1-аминометилциклогексилметил)-4Н-[1,2,4]оксадиазол-5-она, С-[1-(1Н-тетразол-5-илметил)циклогептил]метиламина, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусной кислоты, (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты, (3S,5R)-3-аминометил-5-метилоктановой кислоты, (3S,5R)-3-амино-5-метилгептановой кислоты, (3S,5R)-3-амино-5-метилнонановой кислоты и (3S,5R)-3-амино-5-метилоктановой кислоты или фармацевтически приемлемой соли любого из них;
(b) (S,S)-ребоксетин или его фармацевтически приемлемую соль; и
(c) контейнер,
где вводимая комбинация используется для лечения боли, и где альфа-2-дельта лиганд и S,S-ребоксетин присутствуют в интервале соотношений от 1:10 до 10:1 мас. ч.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

