Настоящее изобретение относится к новому применению альфа-2-дельта лигандов и их фармацевтически приемлемых производных. В частности, оно относится к новому использованию габапентина и прегабалина.
Симптомы нижних мочевыводящих путей (СНМП) включают три группы симптомов, а именно ирритативные, обструктивные симптомы и симптомы неудобства после мочеиспускания. К обструктивным симпомам относятся безотлагательное, учащенное мочеиспускание и никтурия, которые могут быть связаны с: гиперактивным мочевым пузырем (ГМП) и доброкачественной гиперплазией предстательной железы (ДГПЖ).
Гиперактивный мочевой пузырь (ГМП) диагностируют по безотлагательному мочеиспусканием в сочетании с недержанием мочи или без нее, обычно с учащенным мочеиспусканием и никтурией [Abrams et al., Neurourology and Urodynamics 21:167-178 (2002)]. Симптом ГМП одинаково распространен как у мужчин, так и у женщин, и приблизительно 16% населения США страдают этим заболеванием [Stewart et al., Prevalence of Overactive Bladder in the United State: Result from the NOBLE Program; Abstract Presented at the 2nd International Consultation on Incontinence, July 2001, Paris, France].
Термины ГМП в сочетании с недержанием мочи и ГМП без недержания относятся к больным, у которых при ГМП наблюдается недержание мочи или оно отсутствует, соответственно. До недавнего времени основным симптомом ГМП считался симптом недержания мочи. Однако появление новых данных означает, что у большого количества больных отсутствует недержание мочи (то есть больные с ГПМ без недержания). Таким образом, Liberman et al [Health Related Quality of Life Among Adults with Symptoms of Overactive Bladder: Results From A US Community-Based Survey; Urology 57 (6), 1044-1050, 2001] недавно исследовали влияние всех симптомов ГМП на качество жизни пациента в США. Это исследование показало, что у пациентов, страдающих ГМП без очевидного недержания мочи, снижается качество жизни по сравнению с контрольными группами.
ДГПЖ представляет собой хроническое прогрессирующее заболевание, которое может быть причиной таких осложнений, как острая задержка мочи, повторяющиеся инфекции мочевыводящих путей, камни мочевого пузыря и почечная дисфункция. Частота заболеваемости и средняя тяжесть СНМП, связанных с ДГПЖ, увеличивается у мужчин с возрастом.
ДГПЖ приводит к увеличению размера предстательной железы, что приводит к синдрому обструкции пузырно-уретрального отдела, а также к вторичным изменениям функции мочевого пузыря. Такое увеличение предстательной железы и изменение функций мочевого пузыря проявляются в виде симптомов накопления (ирритативные симтпомы) и опорожнения (обструктивные симптомы).
Было описано, что альфа-2-дельта лиганды имеют большое количество показаний. Самый известный альфа-2-дельта лиганд, габапентин (I), известный как Neurontin®, 1-(аминометил)-циклогексилуксусная кислота, впервые был описан в патентной литературе в семействе патентов, к которым относится патент США № 4024175.
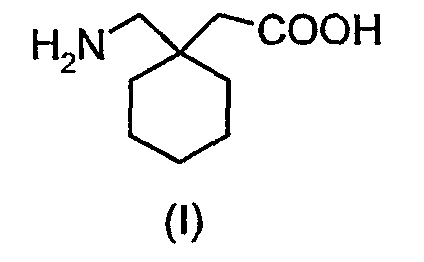
Соединение было одобрено для лечения эпилепсии и нейропатической боли.
Второй альфа-2-дельта лиганд, прегабалин (II), (S)-(+)-4-амино-3-(2-метилпропил)бутановая кислота, был описан в европейской патентной заявке номер EP641330 для противосудорожной терапии, применяемой при лечении эпилепсии, и в EP0934061 для лечения боли.

Совсем недавно, в международной патентной публикации №WO02/085839, были описаны альфа-2-дельта лиганды нижеприведенной формулы, которые могут использоваться при большом количестве заболеваний, включая боль:

где каждый R1 и R2 независимо выбран из H, прямого или разветвленного алкила из 1-6 атомов углерода, циклоалкила из 3-6 атомов углерода, фенила и бензила, при условии, за исключением трициклооктанового соединения формулы (XVIII), что R1 и R2 одновременно не могут быть водородом.
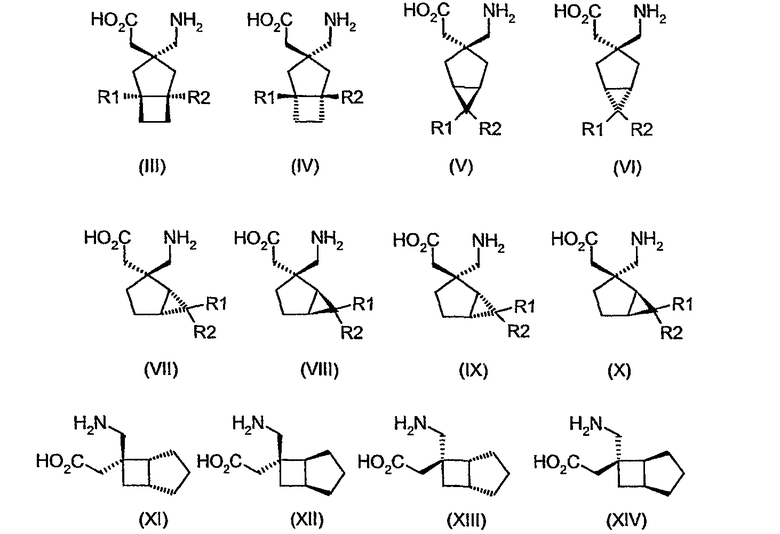
Другие примеры альфа-2-дельта лигандов представлены ниже:

Циклические альфа-2-дельта лиганды, которые могут использоваться по настоящему изобретению, проиллюстрированы следующей формулой (I):

где X представляет собой карбоновую кислоту или биоизостер карбоновой кислоты;
n равно 0, 1 или 2; и
R1, R1a, R2, R2а, R3, R3а, R4 и R4а независимо выбраны из H и C1-C6 алкила, или
R1 и R2 или R2 и R3 вместе образуют С3-С7 циклоалкильное кольцо, которое необязательно замещено одним или двумя заместителями, выбранными из C1-C6 алкила,
или его фармацевтически приемлемая соль.
В формуле (I), соответственно, R1, R1a, R2a, R3а, R4 и R4a представляют собой H, а R2 и R3 независимо выбраны из H и метила, или R1a, R2а, R3a и R4a представляют собой H, и R1 и R2 или R2 и R3 вместе образуют С3-С7 циклоалкильное кольцо, которое необязательно замещено одним или двумя метильными заместителями. Соответствующий биоизостер карбоновой кислоты выбран из тетразолила и оксадиазолонила. X предпочтительно представляет карбоновую кислоту.
В формуле (I), предпочтительно, R1, R1a, R2а, R3a, R4 и R4a представляют собой H, а R2 и R3 независимо выбраны из H и метила, или R1a, R2а, R3a и R4a представляют собой H, и R1 и R2 или R2 и R3 вместе образуют С4-C5 циклоалкильное кольцо, или, если n равно 0, то R1, R1a, R2а, R3a, R4 и R4а представляют собой H, а R2 и R3 образуют циклопентильное кольцо, или, если n равно 1, то R1, R1a, R2a, R3a, R4 и R4a представляют собой H, а R2 и R3 оба являются метилом, или R1, R1a, R2а, R3a, R4 и R4a представляют собой H, а R2 и R3 образуют циклобутильное кольцо или, если n равно 2, то R1, R1a, R2, R2a, R3, R3a, R4 и R4а представляют собой H, или, если n равно 0, R1, R1a, R2a, R3a, R4 и R4а представляют собой H, а R2 и R3 образуют циклопентильное кольцо. Ациклические альфа-2-дельта лиганды, которые используются по настоящему изобретению, проиллюстрированы следующей формулой (II):

где:
n равно 0 или 1, R1 представляет собой водород или (С1-С6)алкил; R2 представляет собой водород или (С1-С6)алкил; R3 представляет собой водород или (С1-С6)алкил; R4 представляет собой водород или (С1-С6)алкил; R5 представляет собой водород или (С1-С6)алкил, и R2 представляет собой водород или (С1-С6)алкил,
или их фармацевтически приемлемая соль.
В соответствии с формулой (II), предпочтительно, R1 представляет собой С1-С6 алкил, R2 представляет собой метил, R3-R6 представляют собой водород, а n равно 0 или 1. Более предпочтительно, R1 представляет собой метил, этил, н-пропил или н-бутил, R2 представляет собой метил, R3-R6 представляют собой водород, и n равно 0 или 1. Если R2 представляет собой метил, R3-R6 представляют собой водород, а n равно 0, то R1, предпочтительно, представляет собой этил, н-пропил или н-бутил. Если R2 представляет собой метил, R3-R6 представляют собой водород, а n равно 1, то R1, предпочтительно представляет собой метил или н-пропил. Предпочтительно, соединения формулы (II) находятся в 3S,5R конфигурации.
Примеры альфа-2-дельта лигандов, которые используются по настоящему изобретению, являются соединения, описанные, в целом или конкретно, в патенте США №4024175, в частности габапентин, в EP641330, в частности прегабалин, в патенте США №5563175, WO9733858, WO9733859, WO9931057, W09931074, W09729101, WO02085839, в частности [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусная кислота, в WO9931075, в частности, 3-(1-аминометил-циклогексилметил)-4H-[1,2,4]оксадиазол-5-он и C-[1-(1H-тетразол-5-илметил)циклогептил]метиламин, в WO9921824, в частности, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусная кислота, в WO0190052, WO0128978, в частности, (1α,3α,5α)(3-амино-метил-бицикло[3.2.0]гепт-3-ил)уксусная кислота, в EP0641330, W09817627, WO0076958, в частности, (3S,5R)-3-аминометил-5-метилоктановая кислота, в PCT/IB03/00976, в частности, (3S,5R)-3-амино-5-метилгептановая кислота, (3S,5R)-3-амино-5-метилнонановая кислота и (3S,5R)-3-амино-5-метилоктановая кислота, в EP1178034, EP1201240, WO9931074, WO03000642, WO0222568, WO0230871, WO0230881, WO02100392, WO02100347, WO0242414, WO0232736 и WO0228881 или их фармацевтически приемлемые соли, каждый из которых приведен здесь в качестве ссылки.
Предпочтительные альфа-2-дельта лиганды по настоящему изобретению включают: габапентин, прегабалин, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6-ил]уксусную кислоту, 3-(1-аминометилциклогексилметил)-4H-[1,2,4]оксадиазол-5-он, C-[1-(1H-тетразол-5-илметил)циклогептил]метиламин, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусную кислоту, (1α,3α,5α)(3-амино-метил-бицикло[3.2.0]гепт-3-ил)уксусную кислоту, (3S, 5R)-3-аминометил-5-метилоктановую кислоту, (3S,5R)-3-амино-5-метилгептановую кислоту, (3S,5R)-3-амино-5-метилнонановую кислоту и (3S,5R)-3-амино-5-метилоктановую кислоту, или их фармацевтически приемлемые соли. В частности, предпочтительные альфа-2-дельта лиганды по настоящему изобретению выбраны из габапентина, прегабалина и (1α,3α,5α)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты, или их фармацевтически приемлемых солей.
В WO 00/01135 описано использование аналогов глутаминовой кислоты и гамма-аминомасляной кислоты для лечения недержания мочи.
Неожиданно было обнаружено, что альфа-2-дельта лиганды, такие как описано выше, могут использоваться при лечении СНМП, кроме использования для лечения недержания мочи, связанных с ГМП и/или ДГПЖ. Более конкретно, было обнаружено, что альфа-2-дельта лиганды могут использоваться при лечении частых позывов при СНМП, связанного с ГМП и/или ДГПЖ. Это обнаружение было неожиданным, так как предполагалось, что соединение, которое, как известно, используется для лечение недержания мочи (то есть нежелательное и часто неосознанное упускание мочи вследствие нарушения мышечного контроля) может снижать симптомы частых позывов, которые наблюдаются у больных с ГМП и ДГПЖ.
Таким образом, в настоящем изобретении предлагается применение альфа-2-дельта лиганда, или его фармацевтически приемлемой соли или сольвата, для лечения СНМП, кроме применения для лечения недержания мочи, связанных с ГМП и/или ДГПЖ.
Предпочтительно, СНМП представляет частые позывы к мочеиспусканию. Предпочтительно, СНМП связаны с ДГПЖ. Предпочтительно, если СНМП связаны с ДГПЖ, то ГМП представляет собой ГМП без недержания мочи.
Предпочтительно, альфа-2-дельта-1-лиганд выбран из:


или его фармацевтически приемлемой соли или сольвата,
где каждый R1 и R2 независимо выбран из H, прямого или разветвленного алкила из 1-6 атомов углерода, циклоалкила из 3-6 атомов углерода, фенила и бензила, при условии, за исключением трициклооктанового соединения формулы (XVIII), что R1 и R2 одновременно не могут быть водородом; или выбран из

или его фармацевтически приемлемой соли или сольвата.
Более предпочтительно, альфа-2-дельта лиганд представляет собой габапентин (I), прегабалин (II) или (1α, 3α, 5α)(3-амино-метил-бицикло[3.2.0]гепт-3-ил)уксусную кислоту (III')
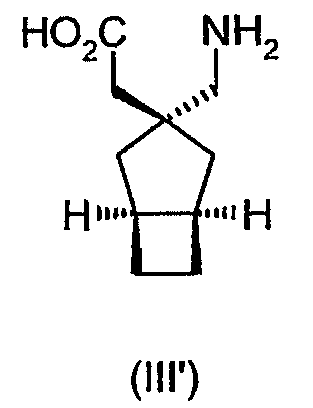
или его фармацевтически приемлемую соль или сольват.
Альфа-2-дельта лиганд, или его фармацевтически приемлемое производное, может вводиться самостоятельно или в любой удобной фармацевтической форме. Пероральное введение является предпочтительным. В этом случае соответствующая дозировка альфа-2-дельта лиганда, или активной группы его фармацевтически приемлемого производного, составляет от около 5 до около 50 мг/кг массы тела и, предпочтительно, от около 0,1 до около 200 мг/кг.
Изобретение также относится к способу лечения СНМП, исключая способ лечения недержания мочи, связанных с ГМП и/или ДГПЖ, который заключается во введении альфа-2-дельта лиганда, или его фармацевтически приемлемой соли или сольвата, больному при необходимости такого лечения.
Альфа-2-дельта лиганды, в частности, соединения, описанные выше, могут использоваться в сочетании с другими соединениями. Например, они могут использоваться в сочетании с антагонистами α1-адренергического рецептора.
Таким образом, еще одним аспектом настоящего изобретения является применение сочетания альфа-2-дельта лиганда и антагониста α1-адренергического рецептора, или его фармацевтически приемлемых солей или сольватов, для получения лекарственного средства для лечения СНМП, связанных с ГМП и/или ДГПЖ.
Кроме того, настоящее изобретение относится к продукту, содержащему альфа-2-дельта лиганд и антагонист α1-адренергического рецептора, или его фармацевтически приемлемые соли или сольваты, в виде объединенного препарата для одновременного, отдельного или последовательного введения при лечение СНМП, связанных с ГМП и/или ДГПЖ.
Предпочтительно, СНМП не является симптомом недержания мочи. Более предпочтительно, СНМП являются частыми позывами к мочеиспусканию. Предпочтительно, СНМП связаны с ДГПЖ. Предпочтительно, если СНМП связаны с ДГПЖ, то ГМП представляет собой ГМП без недержания мочи.
Антагонисты α1-адренергического рецептора, которые могут использоваться в сочетании с альфа-2-дельта лигандами, включают, но ими не ограничиваются,
(i) теразозин (патент США №4026894);
(ii) доксазозин (патент США №4188390);
(iii) празозин (патент США №3511836);
(iv) буназозин (патент США №3920636);
(v) альфузозин (патент США №4315007);
(vi) нафтопидил (патент США №3997666);
(vii) тамсулозин (патент США №4703063);
(viii) силодозин (патент США №5387603) или
(ix) соединения, описанные в опубликованной международной патентной заявке No. WO 98/30560 (в частности, в примере 19 и их мезилатная соль).
Содержание патентных заявок и, в частности, общие формулы терапевтически активных соединений по пунктам формулы изобретения и примеры этих соединений приведены здесь в качестве ссылки в полном объеме.
Альтернативно, альфа-2-дельта лиганды могут использоваться в сочетании с соединением, которое демонстрирует NRI и/или SRI активность. Таким образом, дополнительным аспектом настоящего изобретения является применение сочетания альфа-2-дельта лиганда и соединения, демонстрирующего NRI и/или SRI активность, или его фармацевтически приемлемых солей или сольватов, для получения лекарственного средства для лечения СНМП, связанных с ГМП и/или ДГПЖ.
Кроме того, изобретение относится к продукту, содержащему альфа-2-дельта лиганд и соединение, демонстрирующее NRI и/или SRI активность, или его фармацевтически приемлемые соли или сольваты, в виде объединенного препарата для одновременного, отдельного или последовательного применения при лечении СНМП, связанных с ГМП и/или ДГПЖ.
Предпочтительно, СНМП не является симптомом недержания мочи. Более предпочтительно, СНМП являются частыми позывами к мочеиспусканию. Предпочтительно, СНМП связаны с ДГПЖ. Предпочтительно, если СНМП связаны с ДГПЖ, то ГМП представляет собой ГМП без недержания мочи.
Соединения, обладающие NRI и/или SRI активностью, которые могут быть эффективны для применения по настоящему изобретению, включают, но ими не ограничиваются, ниже представленные соединения:
(i) Флуоксетин, флувоксамин, пароксетин, сертралин, циталопрам, ванлафаксин, нефазодон, тразодон, дулоксетин и ребоксетин (и в рацемически и энантиомерно чистых формах, например, S,S-ребоксетин) и
(ii) Соединения, описанные в европейской патентной публикации № 1220831 и 1154984, в патенте США №4018830, международной патентной публикации WO 97/17325, в патенте США №5190965, 5430063, 4161529 и временной заявке США № 60/121313.
Содержание патентных заявок и, в частности, общие формулы терапевтически активных соединений пунктов формулы изобретения и иллюстрируемых соединений, приведены здесь в качестве ссылки в полном объеме.
Альтернативно, альфа-2-дельта лиганды могут использоваться в сочетании с ингибитором HMG-CoA редуктазы. Таким образом, дополнительным аспектом настоящего изобретения является использование сочетания альфа-2-дельта лиганда и ингибитора HMG-СоА редуктазы, или его фармацевтически приемлемой соли или сольвата, для получения лекарственного средства для лечения СНМП, связанного с ГМП и/или ДГПЖ.
Кроме того, изобретение относится к продукту, содержащему альфа-2-дельта лиганд и ингибитор HMG-СоА редуктазы, или его фармацевтически приемлемую соль или сольват, в виде объединенного препарата для одновременного, отдельного или последовательного применения при лечении СНМП, связанного с ГМП и/или ДГПЖ.
Предпочтительно, СНМП не является симптомом недержания мочи. Более предпочтительно, СНМП являются частыми позывами к мочеиспусканию. Предпочтительно, СНМП связаны с ДГПЖ. Предпочтительно, если СНМП связаны с ДГПЖ, то ГМП представляет собой ГМП без недержания мочи.
Ингибиторы HMG-СоА редуктазы, эффективные для применения по настоящему изобретению, включают, но ими не ограничиваются, соединения представленные ниже:
(i) Флувастатин натрий, [R*,S*,-(E)]-(±)-7-[3-(4-фторфенил)-1-(1-метилэтил)-H-индол-2-ил]-3,5-дигидрокси-6-гептеновая кислота, мононатриевая соль;
(ii) Церивастатин натрий, [S-[R*,S*-(E)]-7-[4-(4-фторфенил)-5-метоксиметил)-2,6-бис(1-метилэтил)-3-пиридинил]-3,5-дигидрокси-6-гептеноат;
(iii) Аторвастатин кальций, тригидрат кальциевой соли (2:1) [R-(R*,R*)]-2-(4-фторфенил)-бета, дельта-гидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1H-пиррол-1-гептановой кислоты;
(iv) Ловастатин, {S-[1-альфа(R*), 3-альфа, 7-бета, 8-бета (2S*, 4S*), 8a бета]}-1,2,3,7,8,8a-гексагидро-3,7-диметил-8-[2-(тетрагидро-4-гидрокси-6-оксо-2H-пиран-2-ил)этил]-1-нафталенил-2-метилбутаноат;
(v) Правастатин натрия, 1-нафтален-гептановая кислота, 1,2,6,7,8,8a-гексагидро-бета, дельта, 6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-, мононатриевая соль, {1S-[1альфа(бета*, дельта*), 2 альфа, 6 альфа, 8 бета (R*), 8a альфа]} и
(vi) Симвастатин, бутановая кислота, 2,2-диметил-1,2,3,7,8,8a-гексагидро-3,7-диметил-8-[2-(тетрагидро-4-гидрокси-6-оксо-2H-пиран-2-ил)-этил]-1-нафталениловый эфир, {1S-[1 альфа, 3 альфа, 7 бета, 8 бета (2S*, 4S*), -8а бета]}.
Альтернативно, альфа-2-дельта лиганды могут использоваться в сочетании с ингибиторами PDEV. Таким образом, дополнительным аспектом настоящего изобретения является применение сочетания альфа-2-дельта лиганда и ингибитора PDEV, или его фармацевтически приемлемых солей или сольватов, для получения лекарственного средства для лечения СНМП, связанного с ГМП и/или ДГПЖ.
Кроме того, настоящее изобретение относится к продукту, содержащему альфа-2-дельта лиганд и ингибитор PDEV, или его фармацевтически приемлемые соли или сольваты, в виде объединенного препарата для одновременного, отдельного или последовательного применения при лечении СНМП, связанных с ГМП и/или ДГПЖ.
Предпочтительно, СНМП не является симптомом недержания мочи. Более предпочтительно, СНМП являются частыми позывами к мочеиспусканию. Предпочтительно, СНМП связаны с ДГПЖ. Предпочтительно, если СНМП связаны с ДГПЖ, то ГМП представляет собой ГМП без недержания мочи.
Ингибиторы PDEV, которые могут использоваться для объединения с альфа-2-дельта лигандами, включают, но ими не ограниваются:
(i) ингибиторы PDE5, указанные в международных публикациях патентных заявок №№ WO03/000691; WO02/64590; WO02/28865; WO02/28859; WO02/38563; WO02/36593; WO02/28858; WO02/00657; WO02/00656; WO02/10166; W002/00658; WO01/94347; WO01/94345; WOOO/15639 и WOOO/15228;
(ii) ингибиторы PDE5, указанные в патентах США №6143746; 6143747 и 6043252;
(iii) пиразоло[4,3-d]пиримидин-7-оны, описанные в EP-А-0463756; пиразоло[4,3-d] пиримидин-7-оны в EP 0526004; пиразоло[4,3-d]пиримидин-7-оны, описанные в опубликованной международной патентной заявке WO 93/06104; изомеры пиразоло[3,4-d]пиримидин-4-онов, описанные в опубликованной международной патентной заявке WO 93/07149; хиназолин-4-оны, описанные в опубликованной международной патентной заявке WO 93/12095; пиридо[3,2-d]пиримидин-4-оны, описанные в опубликованной международной патентной заявке WO 94/05661; пурин-6-оны, описанные в опубликованной международной патентной заявке WO 94/00453; пиразоло[4,3-d]пиримидин-7-оны, описанные в опубликованной международной патентной заявке WO 98/49166; пиразоло[4,3-d]пиримидин-7-оны, описанные в опубликованной международной патентной заявке WO 99/54333; пиразоло [4,3-d] пиримидин-4-оны, описанные в EP 0995751; пиразоло[4,3-d]пиримидин-7-оны, описанные в опубликованной международной патентной заявке WO 00/24745; пиразоло[4,3-d]пиримидин-4-оны, описанные в EP 0995750; гексагидропиразино[2'1':6,1]пиридо[3,4-b]индол-1,4-дионы, описанные в опубликованной международной заявке WO95/19978; пиразоло[4,3-d]пиримидин-4-оны, описанные в WO00/27848; имидазо[5,1-f] [1,2,4]триазин-оны, описанные в EP 1092719 и в опубликованной международной заявке WO, 99/24433 и бициклические соединения, описанные в опубликованной международной заявке WO 93/07124; пиразоло[4,3-d]пиримидин-7-оны, описанные в опубликованной международной заявке WO 01/27112; пиразоло[4,3-d]пиримидин-7-оны, описанные в опубликованной международной заявке WO 01/27113; соединения описанные в EP-А-1092718 и соединения, описанные в EP-А-1092719; трициклические соединения, описанные в EP-А-1241170; алкилсульфоновые соединения, описанные в опубликованной международной заявке WO 02/074774; соединения, описанные в опубликованной международной заявке WO 02/072586; соединения, описанные в опубликованной международной заявке WO, 02/079203 и соединения, описанные в WO 02/074312.
(iv) предпочтительно, 5-[2-этокси-5-(4-метил-1-пиперазинилсульфонил)фенил]-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (силденафил, например, продаваемый как Viagra®), также известный как 1-[[3-(6,7-дигидро-1-метил-7-оксо-3-пропил-1H-пиразоло[4,3-d]пиримидин-5-ил)-4-этоксифенил]сульфонил]-4-метилпиперазин (см. EP-A-0463756); 5-(2-этокси-5-морфолиноацетилфенил)-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (см. EP-A-0526004); 3-этил-5-[5-(4-этилпиперазин-1-илсульфонил)-2-н-пропоксифенил]-2-(пиридин-2-ил)метил-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (см. WO98/49166); 3-этил-5-[5-(4-этилпиперазин-1-илсульфонил)-2-(2-метоксиэтокси)пиридин-3-ил]-2-(пиридин-2-ил)метил-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (см. WO99/54333); (+)-3-этил-5-[5-(4-этилпиперазин-1-илсульфонил)-2-(2-метокси-1(R)-метилэтокси)пиридин-3-ил]-2-метил-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, также известный как 3-этил-5-{5-[4-этилпиперазин-1-илсульфонил]-2-([(1R)-2-метокси-1-метилэтил]окси)пиридин-3-ил}-метил-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (см. WO99/54333); 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, также известный как 1-{6-этокси-5-[3-этил-6,7-дигидро-2-(2-метоксиэтил)-7-оксо-2H-пиразоло[4,3-d]пиримидин-5-ил]-3-пиридилсульфонил}-4-этилпиперазин (см., WO 01/27113, пример 8); 5-[2-изобутокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-(1-метилпиперидин-4-ил)-2,6-дигидро-7H-пиразоло [4,3-d] пиримидин-7-он (см., WO 01/27113, пример 15); 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-фенил-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (см., WO 01/27113, пример 66); 5-(5-ацетил-2-пропокси-3-пиридинил)-3-этил-2-(1-изопропил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d|пиримидин-7-он (см., WO 01/27112, пример 124); 5-(5-ацетил-2-бутокси-3-пиридинил)-3-этил-2-(1-этил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (см., WO 01/27112, пример 132); (6R,12aR)-2,3,6,7,12,12a-гексагидро-2-метил-6-(3,4-метилендиоксифенил)пиразино[2',1':6,1]пиридо[3,4-b]индол-1,4-дион (тадалафил, IC-351, Cialis ®), то есть соединение примеров 78 и 95 из опубликованной международной заявки WO95/19978, так же, как соединение примеров 1, 3, 7 и 8; 2-[2-этокси-5-(4-этил-пиперазин-1-ил-1-сульфонил)-фенил]-5-метил-7-пропил-3H-имидазо [5,1-f] [1,2,4] триазин-4-он (варденафил, LEVITRA®) также известный как 1-[[3-(3,4-дигидро-5-метил-4-оксо-7-пропилимидазо[5,1-f]-аз-триазин-2-ил)-4-этоксифенил]сульфонил]-4-этилпиперазин, то есть соединение примеров 20, 19, 337 и 336 из опубликованной международной заявки WO99/24433; соединение примера 11 из опубликованной международной заявки WO93/07124 (EISAI); соединения 3 и 14 от Rotella D P, J. Med. Chem., 2000, 43, 1257; 4-(4-хлорбензил)амино-6,7,8-триметоксихиназолин; N-[[3-(4,7-дигидро-1-метил-7-оксо-3-пропил-1H-пиразоло[4,3-d]-пиримидин-5-ил)-4-пропоксифенил]сульфонил]-1-метил-2-пирролидинпропанамид ["DA-8159" (пример 68 из WO00/27848)]; и 7,8-дигидро-8-оксо-6-[2-пропоксифенил]-1H-имидазо [4,5-g] хиназолин и 1-[3-[1-[(4-фторфенил)метил]-7,8-дигидро-8-оксо-1H-имидазо[4,5-g]хиназолин-6-ил]-4-пропоксифенил]карбоксамид.
(v) 4-бром-5-(пиридилметиламино)-6-[3-(4-хлорфенил)-пропокси]-3(2H)пиридазинон; 1-[4-[(1,3-бензодиоксол-5-илметил)амино]-6-хлор-2-хинозолинил]-4-пиперидин-карбоновая кислота, мононатриевая соль; (+)-цис-5,6а,7,8,9,9а-гексагидро-2-[4-(трифторметил)-фенилметил-5-метил-циклопент-4,5]имидазо[2,1-b]пурин-4(3H)-он; фуразлоциллин; цис-2-гексил-5-метил-3,4,5,6a,7,8,9,9a-октагидроциклопент[4,5]-имидазо[2,1-b]пурин-4-он; 3-ацетил-1-(2-хлорбензил)-2-пропилиндол-6-карбоксилат; 3-ацетил-1-(2-хлорбензил)-2-пропилиндол-6-карбоксилат; 4-бром-5-(3-пиридилметиламино)-6-(3-(4-хлорфенил)пропокси)-3-(2H)пиридазинон; 1-метил-5(5-морфолиноацетил-2-н-пропоксифенил)-3-н-пропил-1,6-дигидро-7H-пиразоло(4,3-d]пиримидин-7-он; мононатриевая соль 1-[4-[(1,3-бензодиоксол-5-илметил)амино]-6-хлор-2-хиназолинил]-4-пиперидинкарбоновой кислоты; Pharmaprojects № 4516 (Glaxo Wellcome); Pharmaprojects № 5051 (Bayer); Pharmaprojects №5064 (Kyowa Hakko; см. WO 96/26940); Pharmaprojects № 5069 (Schering Plough); GF-196960 (Glaxo Wellcome); E-8010 и E-4010 (Eisai); Bay-38-3045 и 38-9456 (Bayer); FR229934 и FR226807 (Fujisawa); и Sch-51866.
Содержание патентных заявок и журнальных статей и, в частности, общие формулы терапевтически активных соединений по пунктам формулы изобретения и примеры этих соединений приведены здесь в качестве ссылки в полном объеме.
Предпочтительно, ингибитор PDEV выбран из синденафила, тадалафила, варденафила, DA-8159 и 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-она.
Наиболее предпочтительно, ингибитор PDE5 представляет собой силденафил и его фармацевтически приемлемые соли. Силденафил цитрат предпочтительно находится в виде соли.
Альтернативно, альфа-2-дельта лиганды могут быть объединены с мускариновыми антагонистами. Таким образом, дополнительным аспектом настоящего изобретения является применение сочетания альфа-2-дельта лиганда и мускариновых антагонистов, или их фармацевтически приемлемых солей или сольватов, для получения лекарственного средства для лечения СНМП, связанного с ГМП и/или ДГПЖ.
Кроме того, изобретение относится к продукту, содержащему альфа-2-дельта лиганд и мускариновый антагонист, или его фармацевтически приемлемые соли или сольваты, в виде объединенного препарата для одновременного, отдельного или последовательного применения при лечении СНМП, связанного с ГМП и/или ДГПЖ.
Предпочтительно, СНМП не является симптомом недержания мочи. Более предпочтительно, СНМП являются частыми позывами к мочеиспусканию. Предпочтительно, СНМП связаны с ДГПЖ. Предпочтительно, если СНМП связаны с ДГПЖ, то ГМП представляет собой ГМП без недержания мочи.
Мускариновый антагонист может быть селективным в отношении рецепторов М3, или он может быть неселективным антагонистом в отношении М1, М2 и М3. Антагонисты, селективные в отношении рецептора М3, являются предпочтительными.
Мускариновые антагонисты, которые могут использоваться для объединения с альфа-2-дельта лигандами включают, но ими не ограничиваются:
(i) Атропин, флувоксат, гиосцин, оксибутинин, толтеродин и соединения, описанные в международной патентной публикации № WO 89/06644, пропантелин, пропиверин, троспиум и соединения, описанные в международной патентной публикации № WO 98/05641, а также их фармацевтически приемлемые соли.
(ii) Особенно предпочтительными являются дарифенацин, оксибутинин, толтеродин и соединения, описанные в международной патентной публикации № WO 89/06644, и соединения, описанные в международной патентной публикации № WO 98/05641, а также их фармацевтически приемлемые соли.
Интерес, в частности, представляет толтеродин.
Содержание патентных заявок и журнальных статей и, в частности, общие формулы терапевтически активных соединений по пунктам изобретения, и примеры этих соединений приведены здесь в качестве ссылки в полном объеме.
Альтернативно, альфа-2-дельта лиганды могут быть объединены с ингибиторами циклооксигеназы. Таким образом, дополнительным аспектом настоящего изобретения является применение сочетания альфа-2-дельта лиганда и ингибитора COX, или его фармацевтически приемлемых солей или сольватов, для получения медикамента для лечения СНМП, связанного с ГМП и/или ДГПЖ.
Кроме того, настоящее изобретение относится к продукту, содержащему альфа-2-дельта лиганд и ингибитор COX в виде объединенного препарата для одновременного, отдельного или последовательного применения при лечении СНМП, связанного с ГМП и/или ДГПЖ.
Предпочтительно, СНМП не является симптомом недержания мочи. Более предпочтительно, СНМП являются частыми позывами к мочеиспусканию. Предпочтительно, СНМП связаны с ДГПЖ. Предпочтительно, если СНМП связаны с ДГПЖ, то ГМП представляет собой ГМП без недержания мочи. Предпочтительно ингибитор COX представляет собой ингибитор COX2.
Ингибиторы COX, которые могут использоваться для объединения с альфа-2-дельта лигандами, включают, но ими не ограниваются:
(i) ибупрофен, напроксен, беноксапрофен, флурбипрофен, фенопрофен, фенбуфен, кетопрофен, индопрофен, пирпрофен, карпрофен, оксапрозин, прапопрофен, миропрофен, тиоксапрофен, супрофен, альминопрофен, тиапрофеновая кислота, флупрофен, буклоксиновую кислоту, индометацин, сулиндак, толметин, зомепирак, диклофенак, фенклофенек, алклофенак, ибуфенак, изоксепак, фурофенак, тиопинак, зидометацин, ацетилсалициловую кислоту, индометацин, пироксикам, теноксикам, набуметон, кеторолак, азапропазон, мефенамовую кислоту, толфенамовую кислоту, дифлунисал, производные подофиллотоксина, ацеметацин, дроксикам, флоктафенин, оксифенбутазон, фенилбутазон, проглуметацин, ацеметацин, фентиазак, клиданак, оксипинак, мефенамовую кислоту, меклофенамовую кислоту, флуфенамовую кислоту, нифлумовую кислоту, флуфенизал, судоксикам, этодолак, пипрофен, салициловую кислоту, трисалицилат магний холина, салицилат, бенорилат, фентиазак, клопинак, фепразон, изоксикам и 2-фтор-a-метил[1,1'-дифенила]-4-уксусная кислота, 4-(нитроокси)бутиловый эфир (см. Wenk, et al., Europ. J. Pharmacol. 453:319-324 (2002));
(ii) мелоксикам, (CAS регистрационный номер 71125-38-7; описанный в патенте США № 4233299), или его фармацевтически приемлемая соль или пролекарство;
(iii) замещенные производные бензопирана, которые описаны в патенте США № 6271253. Также производные бензопирана, описанные в патенте США № 6034256 и 6077850, а также в международных публикациях WO 98/47890 и WO 00/23433;
(iv) селективные ингибиторы хромена COX2, описанные в патенте США № 6077850 и в патенте США № 6034256;
(v) соединения, описанные в международных публикациях патентных заявок WO 95/30656, WO 95/30652, WO 96/38418 и WO 96/38442, и соединения, описанные в европейской публикации патентной заявки № 799823, а также их фармацевтически приемлемые производные;
(vi) целекоксиб (патент США № 5466823), валдекоксиб (патент США № 5633272), деракоксиб (патент США № 5521207), рофекоксиб (патент США № 5474995), эторикоксиб (международная публикация патентной заявки № WO 98/03484), JTE-522 (японская публикация патентной заявки № 9052882), или его фармацевтически приемлемая соль или пролекарство;
(vii) парекоксиб (описанный в патенте США № 5932598), который является терапевтически эффективным пролекарством трициклического селективного ингибитора COX2 валдекоксиба (описан в патенте США № 5633272), в частности, парекоксиб натрия;
(viii) ABT-963 (описанный в международной публикации патентной заявки № WO 00/24719)
(ix) нимесулид (описанный в патенте США № 3840597), флосулид (описанный в J. Carter. Exp. Qpin. Ther. Patents, 8(1), 21-29 (1997)), NS-398 (описанный в патенте США № 4885367), SD 8381 (описанный в патенте США № 6034256), BMS 347070 (описанный в патенте США № 6180651), S-2474 (описанный в европейской патентной публикации № 595546) и MK-966 (описанный в патенте США № 5968974);
(x) соединения и фармацевтически приемлемые производные, описанные в патенте США № 6395724, патенте США № 6077868, патенте США № 5994381, патенте США № 6362209, патенте США № 6080876, патенте США № 6133292, патенте США № 6369275, патенте США № 6127545, патенте США № 6130334, патенте США № 6204387, патенте США № 6071936, патенте США № 6001843, патенте США № 6040450, международной публикации патентной заявки No WO 96/03392, международной публикации патентной заявки No WO 96/24585, патенте США № 6340694, патенте США № 6376519, патенте США № 6153787, патенте США № 6046217, патенте США № 6329421, патенте США № 6239137, патенте США № 6136831, патенте США № 6297282, патенте США № 6239173, патенте США № 6303628, патенте США № 6310079, патенте США № 6300363, патенте США № 6077869, США, патенте № 6140515, патенте США № 5994379, патенте США № 6028202, патенте США № 6040320, патенте США № 6083969, патенте США № 6306890, патенте США № 6307047, патенте США № 6004948, патенте США № 6169188, патенте США № 6020343, патенте США № 5981576, патенте США № 6222048, патенте США № 6057319, патенте США № 6046236, патенте США № 6002014, патенте США № 5945539, патенте США № 6359182, международной публикации патентной заявки № WO 97/13755, международной публикации патентной заявки № WO 96/25928, международной публикации патентной заявки № WO 96/374679, международной публикации патентной заявки № WO 95/15316, международной публикации патентной заявки № WO 95/15315, международной публикации патентной заявки, № WO, 96/03385, международной патентной заявки № WO 95/00501, международной патентной заявке № WO 94/15932, международной публикации патентной заявке, № WO 95/00501, международной публикации патентной заявки, № WO 94/27980, международной публикации патентной заявки, № WO 96/25405, международной публикации патентной заявки, № WO 96/03388, международной публикации патентной заявки, № WO 96/03387, патенте США № 5344991, международной публикации патентной заявки № WO 95/00501, международной публикации патентной заявки № WO 96/16934, международной публикации патентной заявки № WO 96/03392, международной публикации патентной заявки № WO 96/09304, международной публикации патентной заявки № WO 98/47890, и международной публикации патентной заявки № WO 00/24719.
Содержание любой патентной заявки и, в частности, общие формулы терапевтически активных соединений по пунктам формулы изобретения, и примеры этих соединений приведены здесь в качестве ссылки в полном объеме.
При лечении ДГПЖ альфа-2-дельта лиганды могут быть объединены с соединением, которое замедляет рост предстательной железы. Например, в композиции могут быть объединены альфа-2-дельта лиганд и ингибитор человеческой 5-α редуктазы [смотри международную патентную заявку WO 95/28397].
Таким образом, дополнительным аспектом настоящего изобретения является применение сочетания альфа-2-дельта лиганда и ингибитора человеческой 5-α редуктазы или его фармацевтически приемлемой соли или сольватов для получения лекарственного средства для лечения СНМП, который не является недержанием мочи, связанных с ДГПЖ.
Настоящее изобретение также относится к продукту, содержащему альфа-2-дельта лиганд и ингибитор человеческой 5-α редуктазы, или его фармацевтически приемлемые соли или сольваты, в качестве объединенного препарата для одновременного, отдельного или последовательного применения при лечении СНМП, который не является недержанием мочи, связанных с ДГПЖ.
Предпочтительно СНМП представляет собой частые позывы к мочеиспусканию.
Содержание любой из опубликованных патентных заявок и, в частности, общие формулы терапевтически активных соединений по пунктам формулы изобретения и примеры этих соединений приведены здесь в качестве ссылки в полном объеме.
Применение описанных здесь соединений и сочетаний может иметь преимущество, заключающееся в более высокой эффективности, более длительной продолжительности действия, меньшем количестве побочных эффектов, улучшенной избирательности или других полезных свойств, достигаемых при таком применении, по сравнению с предшествующим уровнем техники при лечении СНМП, связанных с ГМП и/или ДГПЖ, в частности, когда СНМП представляет частые позывы к мочеиспусканию.
Соединения по настоящему изобретению получают способами, хорошо известными специалистам в данной области. В частности, в патентах, патентных заявках и публикациях, указанных выше, каждая из которых приведена здесь в качестве ссылки, проиллюстрированы соединения, которые могут использоваться в сочетании, в фармацевтических композициях, в способах и наборах в соответствии с настоящими изобретениями, и способы получения указанных соединений.
Фармацевтически приемлемые соли соединений, подходящие для использования по изобретению, включают соли добавления кислот и оснований.
Соответствующие соли добавления кислот получают из кислот, которые образуют нетоксические соли. Примеры включают ацетат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, эдисилат, эсилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, хибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напзилат, пикотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, сахарат, стеарат, сукцинат, тартрат, тозилат и соль трифторуксусной кислоты.
Соответствующие соли основания получают из оснований, которые образуют нетоксические соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглюмина, оламина, калия, натрия, трометамина и цинка.
Обзор подходящих солей представлен в «Handbook of Pharmaceutical Salts: Properties, Selection, and Use» Stahl Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Фармацевтически приемлемая соль соединения, которая может использоваться в соответствии с настоящим изобретением может быть легко получена путем смешения раствора соединений и соответствующей желаемой кислоты или основания. Соль, которая может выпасть в осадок из раствора, можно собрать фильтрацией или восстанавлением упариванием растворителя. Степень ионизации этой соли может изменяться от полностью ионизированной до практически неионизированной.
Соединения, подходящие для применения по настоящему изобретению, могут существовать как в нерастворимой, так и в растворимой форме. Термин «сольват» используется здесь для описания молекулярного комплекса, содержащего соединение по изобретению в одной или нескольких молекулах фармацевтически приемлемого растворителя, например этанола. Термин «гидрат» используется, если растворителем является вода.
В рамках изобретения находятся комплексы, такие как хлатраты, комплексы включения препарат-хозяин, где, в противоположность вышеуказанным сольватам, препарат и хозяин находятся в стехиометрическом или нестехиометрическом количествах. Изобретение также включает комплексы препарата, содержащие два или несколько органических и/или неорганических компонента, которые могут быть в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизированными, частично ионизированными или неионизированными. Обзор таких комплексов представлен в J Pharm Sci, 64 (8), 1269-1288, Haleblian (август 1975).
Все приведенные здесь ссылки соединений, подходящих для применения по настоящему изобретению, включают ссылки на их соли, сольваты и комплексы и сольваты и комплексы их солей.
Соединения, подходящие для применения по настоящему изобретению, включают соединения, определенные выше, их полиморфы, пролекарства и изомеры (включая оптические, геометрические и таутомерные изомеры), как определено здесь и соединения, меченные изотопами.
Как указано, изобретение включает все полиморфы соединений, подходящих для применения по настоящему изобретению, как определено здесь выше.
Также в рамках настоящего изобретения находятся так называемые «пролекарства» соединений, подходящих для применения по настоящему изобретению. Такие определенные производные соединений, подходящих для применения по настоящему изобретению, которые сами могут обладать небольшой или не обладать фармакологической активностью, при введении в или при нанесении на организм могут преобразовываться в соединения с желательной активностью, например, путем гидролиза. Такие производные называются «пролекарствами». Дополнительную информацию по использованию пролекарств можно найти в «Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) and Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (ed. E B Roche, American Pharmaceutical Association).
Пролекарства в соответствии с настоящим изобретением могут, например, быть получены путем замены соответствующих функциональных групп соединений, подходящих для применения по настоящему изобретению, определенными группами, известными специалисту в данной области как «предгруппы», как описано, например, в «Design of Prodrugs», H Bundgaard (Elsevier, 1985).
Некоторые примеры пролекарств в соответствии с изобретением включают:
(i) соединение содержит функциональную группу карбоновой кислоты (-COOH), ее сложного эфира, например, замену водорода на (C1-C8)алкил;
(ii) соединение содержит спиртовую функциональную группу (-OH), его простой эфир, например, замену водорода на (C1-С6)алканоилоксиметил; и
(iii) соединение содержит первичную или вторичную функциональную аминогруппу (-NH2 или -NHR, где R не является H), ее амид, например, замену одного или обоих водородов (C1-C10)алканоилом.
Другие примеры замещающих групп в соответствии с вышеуказанными примерами и примеры других пролекарств могут быть найдены в вышеуказанным ссылках.
Наконец, определенные соединения, подходящие для применения по настоящему изобретению могут сами действовать как пролекарства других соединений, подходящих для применения по настоящему изобретению.
Соединения, подходящие для применения по настоящему изобретению, содержащие один или несколько ассиметричных атомов углерода, могут существовать в виде двух или более стереоизомеров. Если соединение содержит алкенильную или алкениленовую группу, то возможны геометрические изомеры цис/транс (или Z/E). Если соединения содержат, например, кето или оксим группу или ароматическую группу, то может возникать таутомерный изомеризм («таутомеризм»). Отсюда следует, что единичное соединение может обладать более чем одним типом изомеризма.
В рамках настоящего изобретения находятся все стереоизомеры, геометрические изомеры и таутомерные формы соединений, подходящих для применения по настоящему изобретению, как описано выше, включая соединения, обладающие более чем одним типом изомеризма и смеси одного или нескольких из них. Настоящее изобретение также включает все соли добавления кислот или оснований, в которых каунтер-ион является оптически активным, например, D-лактат или L-лизин, или рацемические смеси, например, DL-тартрат или DL-аргинин.
Настоящее изобретение включает все фармацевтически приемлемые соединения, меченные изотопами, подходящие для применения по настоящему изобретению, как описано выше, если один или несколько атомов заменены атомами с одинаковыми атомными номерами, но атомная масса или массовое число отличается от атомной массы или массового числа, обычно существующего в природе.
Примеры изотопов, подходящих для введения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15О, 17O и 18O, фосфора, такие как 32P, и серы, такие как 35S.
Фармацевтически приемлемые сольваты в соответствии с изобретением включают сольваты, где растворитель кристаллизации может быть изотопно замещен, например, D2O, d6-ацетон, d6-DMSO.
Соединения по изобретению, предназначенные для фармацевтического применения, могут вводиться как кристаллические или аморфные продукты. Они могут вводиться самостоятельно или в сочетании с одним или несколькими другими соединениями по изобретению или в сочетании с одним или несколькими другими лекарственными препаратами (или с любым их сочетанием). Обычно их вводят в виде композиции в сочетании с одной или несколькими фармацевтически приемлемыми наполнителями. Термин «наполнитель» используется здесь для описания любого ингредиента, отличного от соединения(й) по изобретению. Выбор наполнителя зачастую зависит от факторов, таких как конкретный способ введения, действие наполнителя на растворимость и стабильность, и природу лекарственной формы.
Фармацевтические композиции, подходящие для доставки соединений по настоящему изобретению, и способы их получения хорошо известны специалисту в данной области. Такие композиции и способы их получения могут быть найдены, например, в «Remington's Pharmaceutical Sciences», 19th Edition (Mack Publishing Company, 1995).
Соединения по изобретению могут вводиться перорально. Пероральное введение может включать в себя глотание, так чтобы соединение достигло желудочно-кишечного тракта, или может использоваться защечное или подъязычное введение, при котором соединение попадает в кровоток непосредственно из ротовой полости.
Композиции, подходящие для перорального применения, включают твердые композиции, такие как таблетки, капсулы, содержащие частицы, жидкости или порошки, лепешки (включая лепешки с жидким наполнением), жевательные конфеты, мульти- и наночастицы, гели, твердые растворы, липосомы, пленки (включая мукоадгезивные пленки), вагинальные свечи, спреи и жидкие композиции.
Жидкие композиции включают суспензии, растворы, сиропы и эликсиры. Такие композиции могут использоваться в качестве наполнителей в мягких или твердых капсулах и обычно включают носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или несколько эмульгирующих агентов и/или суспендирующих агентов. Жидкие композиции могут также быть получены восстановлением твердого продукта, например саше.
Соединения по изобретению могут также использоваться в быстрорастворимой, быстрораспадающейся формах, таких как описаны в Expert Opinion in Therapeutic Patents, 11 (6), 981-986 by Liang and Chen (2001).
В случае лекарственной формы в виде таблетки, в зависимости от дозы, лекарственный препарат может составлять от 1 мас.% до 80 мас.% лекарственной формы, обычно от 5 мас.% до 60 мас.% лекарственной формы. Кроме лекарственного препарата, таблетки обычно содержат дезинтегратор. Примеры дезинтеграторов включают натрий крахмал гликолат, натрий карбоксиметилцеллюлозу, кальций карбоксиметилцеллюлозу, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу, замещенную низшим алкилом, крахмал, предварительно клейстеризованный крахмал и натрий альгинат. Обычно дезинтегратор содержит от 1 мас.% до 25 мас.%, предпочтительно, от 5 мас.% до 20 мас.% лекарственной формы.
Связывающие агенты обычно используются для придания свойств склеивания композиции в таблетке. Подходящие связывающие агенты включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические смолы, поливинилпирролидон, предварительно клейстеризованный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, моногидрат, высушенный распылением, безводную лактозу и тому подобное), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и двухосновной кальций фосфат дигидрат.
Таблетки также могут необязательно содержать поверхностно-активные вещества, такие как натрий лаурилсульфат и полисорбат 80, и агенты скольжения, такие как диоксид силикона и тальк. В случае их присутствия, поверхностно-активные вещества могут содержать от 0,2 мас.% до 5 мас.% таблетки, и агенты скольжения могут содержать от 0,2 мас.% до 1 мас.% таблетки.
Таблетки также обычно содержат смачивающие агенты, такие как стеарат магния, стеарат кальция, стеарат цинка, натрий стеарил фумарат и смеси магния стеарата и натрия лаурил сульфата. Смачивающие агенты содержат от 0,25 мас.% до 10 мас.%, предпочтительно, от 0,5 мас.% до 3 мас.% таблетки.
Другими возможными ингредиентами являются антиоксиданты, красители, ароматизаторы, консерванты и исправляющее вкус лекарственного средства вещество.
Примеры таблеток содержат около 80% лекарственного препарата, от около 10 мас.% до около 90 мас.% связывающего агента, от около 0 мас.% до около 85 мас.% разбавителя, от около 2 мас.% до около 10 мас.% дезинтегратора, и от около 0,25 мас.% до около 10 мас.% смачивающего агента.
Таблеточные смеси могут быть спрессованы непосредственно или с помощью барабана с образованием таблетки. Таблеточные смеси или части смесей могут, альтернативно, подвергаться сухому, влажному гранулированию и гранулированию плавлением, плавлению-застыванию и экструдированию перед таблетированием. Окончательная композиция может содержать один или несколько слоев и может быть покрыта или не покрыта; она даже может быть инкапсулирована.
Композиция для таблеток описана в «Pharmaceutical Dosage Forms: Tablets, Vol. 1», H. Lieberman and L. Lachman, Marcel Dekker, N.Y., N.Y., 1980 (ISBN 0-8247-6918-X).
Твердые композиции для перорального введения могут быть получены для немедленного и/или отсроченного высвобождения. Композиции с измененным высвобождением включают композиции с отсроченным, длительным, импульсным, контролируемым, направленным и программируемым высвобождением.
Подходящим образом модифицированные композиции для целей изобретения описаны в патенте США № 6106864. Подробное описание других подходящих способов получения таких композиций, например высокоэнергетичных дисперсий и осмотических и покрытых частиц, могут быть найдены в Verma et al., Pharmaceutical Technology On-line, 25(2), 1-14 (2001). Применение жевательных резинок для обеспечения контролируемого высвобождения описано в WO 00/35298.
Соединения по изобретению могут также вводиться непосредственно в кровоток, в мышцы или во внутренние органы. Подходящие способы для парентерального введения включают внутривенное, внутриартериальное, внутрибрюшинное, интретекальное, внутрижелудочковое, интрауретальное, интрастернальное, внутричерепное, внутримышечное и подкожное. Подходящие устройства для парентерального введения включают игольчатые (включая микроигольчатые) шприцы, безыгольчатые шприцы и способы инфузии.
Парентеральные композиции обычно представляют собой водные растворы, которые могут содержать наполнители, такие как соли, углеводороды и буферные агенты (предпочтительно до pH 3-9), но, в некоторых случаях применения, они могут быть получены с более подходящим рецептурным составом, например, в виде стерильного безводного раствора или в виде сухой формы для применения для соединения с подходящим носителем, таким как стерильная, апирогенная вода.
Получение парентеральных композиций в стерильных условиях, например путем лиофилизации, может быть легко осуществлено, используя стандартные фармацевтические свойства, хорошо известные специалисту в данной области.
Растворимость соединений по настоящему изобретению, используемых при получении парентеральных растворов, может повышаться путем применения соответствующих подходов получения композиций, таких как введение агентов, повышающих растворимость.
Композиции для парентерального введения могут быть получены для немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают композиции с отсроченным, длительным, импульсным, контролируемым, направленным и программируемым высвобождением. С такими соединениями по изобретению могут быть получены твердые, полутвердые композиции или тиксотропные жидкие композиции для введения в виде имплантируемого депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких композиций включают стенты, покрытые лекарственным препаратом, и микросферы PGLA.
Соединения по изобретению могут также применяться местно на коже или слизистой, то есть кожно или чрескожно. Обычные композиции для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, опудривающее средство, повязки, пены, пленки, кожные пластыри, облатки, имплантанты, губки, волокна, бандажи и микроэмульсии. Также могут использоваться липосомы. Обычные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. В композицию могут вводиться вещества, способствующие проникновению, см., например, J Pharm Sci, 88 (10), 955-958, Finnin and Morgan (October 1999).
Другие способы местного применения включают доставку электропорацией, ионофорезом, фонофорезом, сонофорезом и шприцами с микроиголками или безигольными шприцами (например Powderject™, Bioject™ и тому подобное).
Композиции для местного введения могут быть получены для немедленного и/или модифицированного высвобождения. Композиции для модифицированного высвобождения включают композиции с отсроченным, длительным, импульсным, контролируемым, направленным и программируемым высвобождением.
Соединения по изобретению могут также вводиться интраназально или ингаляционно, обычно в форме сухого порошка (либо самостоятельно, либо в смеси, например, в сухой смеси с лактозой, или в виде смеси частиц компонентов, например, в смеси с фосфолипидами, например, фосфатидилхолином) из ингалятора для сухого порошка или в виде аэрозольного спрея из контейнера, в котором он находится под давлением, насоса, срея, пульверизатора (предпочтительно, пульверизатора с использованием электрогидродинамической аппаратуры с получением мелкой пыли), или распылителя, с или без применения подходящего пропеллента, такого как 1,1,1,2-тетрафторэтана или 1,1,1,2,3,3,3-гептафторпропана.
Для интраназального применения порошок может содержать биоадгезивный агент, например хитозан или циклодекстрин.
Контейнер, в котором смесь находится под давлением, насос, спрей, распылитель или пульверизатор содержит раствор или суспензию соединения(й) по изобретению, содержащий, например, этанол, водный этанол или подходящий альтернативный агент для диспергирования, растворения или увеличения высвобождения активного вещества, пропеллента(ов) в качестве растворителя и, необязательно, поверхностно-активного вещества, такого как триолеат сорбита, олеиновая кислота или олигомолочная кислота.
Перед использованием в композиции сухого порошка или суспензии лекарственное средство тонкоизмельчают до размера, подходящего для доставки ингалятором (обычно менее 5 микрон). Это может быть достигнуто любым соответствующим способом измельчения, таким как спиральное измельчение, измельчение в псевдосжиженном слое, обработка сверхкритической жидкости с получением наночастиц, гомогенизация в высоком давлении или сушка распылением.
Рецептурная смесь для капсул (получение, например, из желатина или HPMC), блистеров и картриджей для использования в ингаляторах или инсуффляторе может содержать порошковую смесь соединения по изобретению, подходящее порошковое основание, такое как лактоза или крахмал и модификатор свойств, такой как l-лейцин, маннит или стеарат магния. Лактоза может быть безводной или образовывать моногидрат, последний является предпочтительным. Другие подходящие наполнители включают дестран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу.
Рецептурный состав подходящего раствора для применения в пульверизаторе с электрогидродинамическими аппаратами для получения мелкой пыли может содержать от 1 мкг до 20 мг соединения по изобретению на пшик, и объем пшика может изменяться от 1 мкл до 100 мкл. Обычная композиция может содержать соединение по изобретению пропиленгликоль, стерильную воду, этанол и хлорид натрия. Другие варианты растворителей, которые могут использоваться вместо пропиленгликоля, включают глицерин и полиэтиленгликоль.
В композиции по изобретению, предназначенные для ингаляционного/интраназального введения, могут быть добавлены подходящие вкусовые добавки, такие как ментол и левоментол или подсластители, такие как сахарин или сахарин натрия.
Композиции для ингаляционного/интраназального введения могут быть получены для незамедлительного и/или модифицированного высвобождения, используя, например, поли(DL)-молочную-когликолевую кислоту (PGLA).
Композиции с измененным высвобождением включают композиции с отсроченным, длительным, импульсным, контролируемым, направленным и программируемым высвобождением.
Соединения по изобретению могут быть введены ректально или вагинально, например, в виде свечей, пессарий или клизмой. Масло какао является обычной основой для свечей, однако могут использоваться различные подходящие альтернативы.
Композиции для ректального/вагинального введения могут быть получены для незамедлительного и/или модифицированного высвобождения. Композиции с измененным высвобождением включают композиции с отсроченным, длительным, импульсным, контролируемым, направленным и программируемым высвобождением.
Соединения по изобретению могут также вводиться непосредственно в глаз или ухо, обычно в виде капель тонкодисперстной суспензии или раствора в изотоническом, pH-установленном, стерильном физиологическом растворе. Другие композиции, подходящие для глазного и ушного введения, включают мази, биодеградируюмые (например, рассасывающиеся гелевые тампоны, коллагеновые) и небиодеградируемые (например, силиконовые) имплантаты, облатки, линзы и системы, состоящие из частиц или везикул, такие как ниосомы или липосомы. Полимер, такой как поперечносвязанная полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза, или метилцеллюлоза, или гетерополисахаридный полимер, например гелановая смола, может быть включен вместе с консервантом, таким как бензалконий хлорид. Такие композиции могут также быть доставлены с помощью ионофореза.
Композиции для глазного/ушного введения могут быть получены для незамедлительного и/или модифицированного высвобождения. Композиции с измененным высвобождением включают композиции с отсроченным, длительным, импульсным, контролируемым, направленным и программируемым высвобождением.
Соединения по изобретению могут быть объединены с растворимыми макромолекулярными веществами, такими как циклодекстрин и его подходящие производные или полимеры, содержащие полиэтиленгликоль, для улучшения их растворимости, скорости растворения, улучшения вкуса, биодоступности и/или стабильности для использования в любом вышеуказанном пути введения.
Было обнаружено, что комплексы, например содержащие лекарственный препарат и циклодекстрин, обычно используются в большинстве лекарственных форм и путях введения. Могут использоваться оба типа комплексов включения и не включения. В качестве варианта непосредственного комплекса образования с лекарственным препаратом в качестве дополнительной добавки, то есть в качестве носителя, растворителя или солюбилизирующего агента может использоваться циклодекстрин. Для этих целей наиболее часто используется альфа-, бета-, и гамма-циклодекстрины, примеры которых можно найти в международных патентных заявках WO 91/11172, WO 94/02518 и WO 98/55148.
Поскольку, например, для лечения определенных заболеваний или состояний может быть желательным введение сочетания активных соединений, то в рамках настоящего изобретения две или более фармацевтических композиций, по крайней мере, одно из которых содержит соединение по изобретению, могут быть подходящим образом объединены с образованием набора для одновременного введения композиций.
Таким образом, набор по изобретению содержит две или несколько отдельных композиций, по крайней мере одна из которых содержит соединение, описанное в настоящем изобретении, и приспособление для раздельного хранения указанных композиций, таких как контейнер, разделенная бутылочка или разделенный пакет из фольги. Примером такого набора является широко используемая блистерная упаковка, используемая для упаковки таблеток, капсул и тому подобное.
Набор по изобретению обычно подходит для введения различных лекарственных форм, например пероральных или парентеральных, для введения отдельных композиций с различными интервалами введения или для титрования отдельных композиций друг против друга. Для удобства, набор обычно включает руководства для введения и может включать так называемую памятку.
Во избежание неопределенности, при указании «лечения» оно включает лечебное, паллиативное и профилактическое лечение.
Химические примеры:
Пример 1:
1-Трет-бутиловый эфир 2-метиловый эфир (2S,4S)-4-(3-хлорфенокси)пирролидин-1,2-дикарбоновой кислоты

К перемешиваемому раствору 1-трет-бутилового эфира 2-метилового эфира (2S,4R)-4-гидроксипирролидин-1,2-дикарбоновой кислоты (CAS Reg 74844-91-0) (6,1 кг, 24,87 моль), 3-хлорфенола (3,52 кг, 27,39 моль) и трифенилфосфина (7,18 кг, 27,37 моль) в трет-бутил метиловом эфире (30,5 л) при 0°C по каплям добавляли диизопропилазодикарбоксилат (5,53 кг, 27,35 моль) в трет-бутилметиловом эфире (15 л). Смесь перемешивали в течение ночи при 20°C. Реакционную смесь фильтровали и жидкость промывали 0,5M гидроксидом натрия (вод.) (2x12,5 л) и водой (12,2 л). Растворитель трет-бутилметиловый эфир заменяли на н-гептан (42,7 л) с помощью дистилляции при атмосферном давлении и охлаждали для кристаллизации сырого продукта, который собирали фильтрацией (11,1 кг, 125% объединенные с 35% восстановленным диизопроилдикарбоксилатом и оксидом трифенилфосфина - уточненный выход = 86%).
1H ЯМР (400 МГц, CDCl3): δ = 1,46, 1,49 (2 x с, 9H), 2,47 (2H, м), 3,71 (5H, м), 4,42 (1H, м), 4,42, 4,54 (1H, 2 x м), 4,87 (1H, м), 6,68 (1H, м), 6,79 (1H, с), 6,92 (1H, м), 7,18 (1H, м).
LRMS (Электрораспыление): m/z 378 (MNa+).
1-Трет-бутиловый эфир (2S,4S)-4-(3-хлорфенокси)пирролидин-1,2-дикарбоновой кислоты
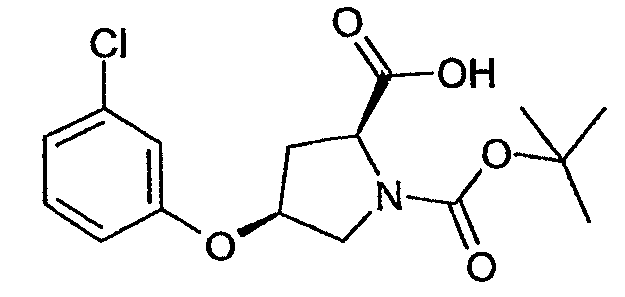
К 1-трет-бутиловому эфиру 2-метиловому эфиру (2S,4S)-4-(3-хлорфенокси)пирролидин-1,2-дикарбоновой кислоты (11,1 кг, 20,28 моль) в ТГФ (26,6 л) добавляли раствор LiOH·H2O (4,86 кг, 115,4 моль) в воде (55,5 л). Смесь перемешивали в течение ночи при 25°C. ТГФ удаляли дистилляцией и полученный водный раствор экстрагировали дихлорметаном (33,3 л и 16,7 л). Объединенные слои дихлорметана экстрагировали водой (33 л и 16,7 л). Объединенную водную фазу доводили до pH 3-3,5 с помощью 1M соляной кислоты (водный раствор) и экстрагировали дихлорметаном (2x22,2 л). Объединенные фазы дихлорметана заменяли на толуол (33,3 л), который охлаждали для кристаллизации продукта, который собирали фильтрацией (6,1 кг, 98%).
1H ЯМР (400 МГц, CDCl3): δ 1,42, 1,48 (2 x с, 9H), 2,30-2,70 (м, 2H), 3,60-3,80 (м, 2H), 4,40-4,60 (м, 1H), 4,86 (м, 1H), 6,71 (м, 1H), 6,82 (м, 1H), 6,94 (м, 1H), 7,16 (м, 1H).
LRMS (Электрораспыление): m/z [MNa+] 364, 340 [M-1] 340.
(2S,4S)-4-(3-Хлорфенокси)пирролидин-2-карбоновая кислота (XXVIII)

Раствор 1-трет-бутилового эфира (2S,4S)-4-(3-хлорфенокси)пирролидин-1,2-дикарбоновой кислоты (29,25 моль) растворяли в ТГФ (20 л) и фильтровали. К этому раствору добавляли 4M HCl в диоксане (30 л) и перемешивали в течение ночи. К полученной суспензии добавляли трет-бутил метиловый эфир (70 л) и продукт собирали фильтрацией (7,06 кг, 86,7%).
1H ЯМР (400 МГц, CD3OD): δ = 2,65 (м, 2H), 3,60 (дд, 1H), 3,70 (д, 1H), 4,60 (дд, 1H), 5,02 (м, 1H), 6,88 (м, 1H), 6,97 (с, 1H), 7,03 (д, 1H), 7,29 (дд, 1H).
LRMS (Электрораспыление) [MH+] 242, [M-1] 240.
Микроанализ для С11Н12ClNO3·HCl 0,1 H2O:
Найдено: C 46,97; H 4,70; N 4,90.
Вычислено: C 47,20; H 4,75; N 5,00.
Пример 2:
1-Трет-бутиловый эфир 2-(2-изопропил-5-метилциклогексил)эфир 4-(3-фторбензил)пирролидин-1,2-дикарбоновой кислоты

1-Трет-бутиловый эфир 2-(2-изопропил-5-метилциклогексил)эфир 4-(3-фторбензилиден)пирролидин-1,2-дикарбоновой кислоты (1,20 г, 2,61 ммоль) растворяли в смеси этилацетат:толуол (1:1, 12 мл). Раствор подвергали гидрированию на оксиде платины (120 мг, 10% по массе) при 25°C и 15 фунт/кв.дюйм в течение 1 часа. Реакционную смесь фильтровали через волокна целлюлозы «Arbocel» и фильтрат восстанавливали под давлением. Осадок очищали флэш-хроматографией («Flashmaster»), элюируя гептаном:этилацетатом (15:1) с выходом соединения, указанного в заголовке в виде бесцветного масла (1,11 г, 91%).
1H ЯМР (400 МГц, CD3OD): δ = 0,72-1,37 (м, 13 H), 1,44 (д, 9H), 1,43-1,75 (м, 4H), 1,87-2,01 (м, 2H), 2,31-2,58 (м, 2H), 2,83 (д, 2H), 3,07 (т, 1H), 3,50-3,65 (м, 1H), 4,13-4,30 (дт, 1H), 4,71 (тд, 1H), 6,90 (д, 2H), 7,00 (д, 1H), 7,30 (кв, 1H).
LRMS (APCI): m/z [MH-BOC]+ 362.
Моногидрохлоридная соль (2S,4S)-4-(3-фторбензил)пирролидин-2-карбоновой кислоты (XXIX)

1-Трет-бутиловый эфир 2-(2-изопропил-5-метилциклогексил)эфир 4-(3-фторбензил)пирролидин-1,2-дикарбоновой кислоты (0,91 г, 1,96 ммоль) растворяли в толуоле (2 мл). Добавляли 6 н. хлористоводородную кислоту (50 мл) и перемешивали при кипячении с обратным холодильником в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и экстрагировали этилацетатом (3x20 мл). Водный слой концентрировали, упаривая при пониженном давлении с получением соединения, указанного в заголовке (417 мг, 81 %), в виде белого твердого вещества. С помощью 1H-ЯМР было показано, что соотношение цис:транс диастереоизомеров составляет 7:1, поэтому продукт перекристаллизовывали из изопропилового спирта с получением соединения, указанного в заголовке (170 мг, 65%) с соотношением цис:транс, равным 14:1, определенным с помощью ЯМР.
1H-ЯМР (400 МГц, CD3OD): (смесь диастереомеров 2S,4S:2S,4R (14:1)): 1,85 (кв, 1H), 2,51 (квин, 1H), 2,69-2,85 (м, 3H), 3,07 (т, 1H), 3,41 (дд, 1H), 4,38 и 4,48 (т, 1H), 6,90-7,04 (м, 3H), 7,32 (кв, 1H).
LRMS (APCI): m/z [MH]+ 224.
[a]D 25-1,27° (c=9,00 в метаноле).
Микроанализ для C12H14FNO2·HCl:
Найдено: C 55,56; H 5,81; N 5,34%.
Вычислено: C 55,50; H 5,82; N 5,39%.
Пример 3:
1-Трет-бутиловый эфир 2-(2-изопропил-5-метилциклогексил)эфир 4-(3-фторбензил)пирролидин-1,2-дикарбоновой кислоты

1-Трет-бутиловый эфир 2-(2-изопропил-5-метилциклогексил)эфир 4-(3-фторбензил)пирролидин-1,2-дикарбоновой кислоты получали способом, аналогичным способу получения 1-трет-третбутилового эфира 2-(2-изопропил-5-метилциклогексил)эфира 4-(3-фторбензил)пирролидин-1,2-дикарбоновой кислоты, используя соответствующий исходный продукт, алкениловый ментоловый эфир; [MH]480
Микроанализ C27H39·F2NO4 (смесь диастереомеров цис (основной) и транс):
Найдено: C 67,74; H 8,30; N 2,90%.
Вычислено: C 67,62; H 8,20; N 2,92%;
[α]D 25 -71,92° (c = 3,26 в метаноле)
Моногидрохлоридная соль (2S,4S)-4-(2,3-дифторбензил)пирролидин-2-карбоновой кислоты (XXX)
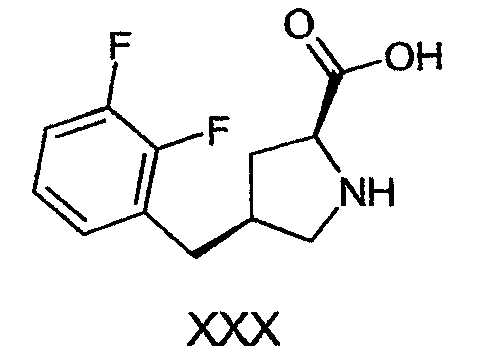
Соединение, указанное в заголовке, получали способом, описанным в примере 2, используя в качестве исходного продукта 1-трет-бутиловый эфир 2-(2-изопропил-5-метилциклогексил)эфир 4-(3-фторбензил)пирролидин-1,2-дикарбоновой кислоты, и очищали перекристаллизацией с ацетоном/простым эфиром с получением соединения, указанного в заголовке в виде смеси диастереоизомеров (2S,4S:2S,4R (12:1)), как было определено с помощью 1H-ЯМР (500 мг, 60 %), в виде белого твердого продукта.
1H-ЯМР (400 МГц, CD3OD) (смесь диастереоизомеров цис/транс (12:1)):δ 0,80-1,90 (м, 0,92H), 2,12-2,20 (м, 0,08H), 2,28-2,36 (м, 0,08H), 2,49-2,58 (кв, 0,92H), 2,66-2,81 (м, 1H), 2,83-2,95 (м, 2H), 3,02-3,13 (т, 1H), 3,46 (дд, 1H), 4,40 (дд, 0,92H), 4,48-4,54 (м, 0,08H), 7,03-7,20 (м, 3H).
LRMS (Электрораспыление): m/z [M + H]+ 242.
Микроанализ для С12H13NO2F2·HCl:
Найдено: C 51,42; H 5,08; N 5,01%.
Вычислено: C 51,90; H 5,08; N 5,04%.
Пример 4:
1,2-Ди-трет-бутиловый эфир (2S,4S)-пирролидин-1,2,4-трикарбоновой кислоты
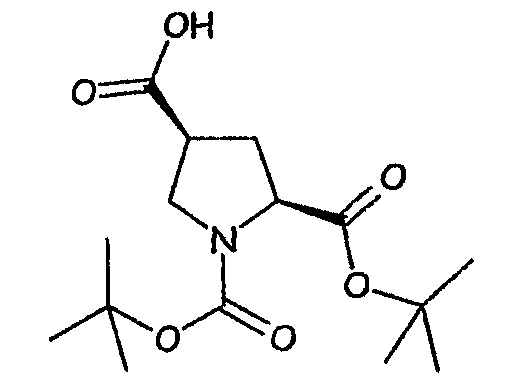
К смеси ди-трет-бутилового эфира 4-фенилпирролидин-1,2-дикарбоновой кислоты (CAS Reg. No. 344 286-69-7; J. Org. Chem., 2001, 3593-3596) (0,78 г, 2,24 ммоль) и периодата натрия (5,77 г, 27 ммоль), перемешиваемой при 0°C в атмосфере азота в этилацетате (5,5 мл), ацетонитриле (5,5 мл) и воды (8,5 мл) добавляли трихлорид рутения (10 мг, 0,05 ммоль) и перемешивали при комнатной температуре в течение 18 часов. Добавляли диэтиловый эфир (20 мл) и перемешивали еще 1 час. Добавляли 1M хлористоводородную кислоту (5 мл) и смесь экстрагировали этилацетатом (3x30 мл). Органические экстракты объединяли, сушили (MgSO4), фильтровали и упаривали при пониженном давлении. Осадок очищали хроматографией на силикагеле, элюируя смесью 50:50:1 этилацетата:гептана:ледяной уксусной кислоты с получением соединения, указанного в заголовке, в виде бесцветной смолы (501 мг, 78%).
1H-ЯМР (400 МГц, CDCl3): δ = 1,40-1,49 (м, 18H); 2,26-2,40 (м, 1H); 2,42-2,56 (м, 1H); 3,02-3,12 (м, 1H); 3,65-3,80 (м, 1,4H) и 3,80-3,88 (м, 0,6H) [ротамеры]; 4,09-4,20 (м, 0,7H) и 4,20-4,26 (м, 0,3H) [ротамеры].
LRMS (электрораспыление): [M-1] 314.
(2S,4S)-4-(3-Фторфеноксиметил)пирролидин-2-карбоновой кислоты (XXI)

Ди-трет-бутиловый эфир 4-(3-фторфеноксиметил)пирролин-1,2-дикарбоновой кислоты (475 мг, 1,2 ммоль) растворяли в растворе безводного хлористого водорода в диоксане (4M, 15 мл) и перемешивали при 50°C в атмосфере азота в течение 1 часа. Растворитель удаляли при пониженном давлении и полученный полутвердый продукт растирали в порошок с этилацетатом с получением твердого белого продукта, который перекристаллизовывали из этилацетата/изопропилового спирта с получением соединения, указанного в заголовке, в виде смеси диастереомеров (˜5:1 2S,4S:2S,4R) в виде белого гидрохлорида (90 мг, 35%).
1H-ЯМР (400 МГц, CD3OD): δ =2,04-2,09 (м, 0,8H); 2,33-2,47 (м, 0,4H); 2,65-2,75 (м, 0,8H); 2,88-3,00 (м, 1H); 3,33-3,40 (м, 1H); 3,52-3,60 (м, 0,8H); 3,60-3,68 (0,2H); 3,96-4,04 (м, 1H); 4,04-4,12 (м, 1H); 4,42-4,51 (м, 0,8H); 4,40-4,56 (м, 0,2H); 6,65-6,80 (м, 3H), 7,21-7,30 (м, 1H).
LRMS (электрораспыление): [M+1] 240; [M+23] 262; [M-1] 238.
Пример 5:
Гидрохлорид (R)-2,6-диметилнон-2-ена (3S,5R)-3-амино-5-метилоктановой кислоты
К (S)-цитронеллил бромиду (50 г, 0,228 моль) в ТГФ (800 мл) при 0°C добавляли LiCl (4,3 г), а затем CuCl2 (6,8 г). Через 30 минут добавляли хлорид метилмагния (152 мл 3M раствора в ТГФ, Aldrich) и раствор нагревали до комнатной температуры. Через 10 часов раствор охлаждали до 0°C и осторожно добавляли насыщенный водный раствор хлорида аммония. Полученные два слоя разделяли и водную фазу экстрагировали простым эфиром. Объединенные органические фазы сушили (MgSO4) и концентрировали с получением (R)-2,6-диметилнон-2-ена, 32,6 г; 93%. Использовали без дополнительной очистки.
1H-ЯМР (400 МГц, CDCl3): δ 5,1 (м, 1H), 1,95 (м, 2H), 1,62 (с, 3H), 1,6 (с, 3H), 1,3 (м, 4H), 1,2 (м, 2H), 0,8 (с, 6H).
(R)-4-Метилгептановая кислота
К (R)-2,6-диметилнон-2-ену (20 г, 0,13 моль) в ацетоне (433 мл) добавляли раствор CrO3 (39 г, 0,39 моль) в H2SO4 (33 мл)/H2O (146 мл) в течение 50 минут. Через 6 часов добавляли дополнительное количество CrO3 (26 г, 0,26 моль) в H2SO4 (22 мл)/H2O (100 мл). Через 12 часов раствор разбавляли насыщенным солевым раствором и раствор экстрагировали простым эфиром. Объединенные органические фазы сушили (MgSO4) и концентрировали. Флэш-хроматография (градиент от 6:1 до 2:1 гексан/EtOAc) давала (R)-4-метилгептановую кислоту в виде масла, 12,1 г; 65%. MS, m/z (относительная интенсивность): 143 [M-H, 100%].
(4R,5S)-4-Метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-он
К (R)-4-метилгептановой кислоте (19 г, 0,132 моль) и триэтиламину (49,9 г, 0,494 моль) в ТГФ (500 мл) при 0°C добавляли триметилацетилхлорид (20 г, 0,17 моль). Через 1 час LiCl (7,1 г, 0,17 моль) добавляли после (4R,5S)-(+)-4-метил-5-фенил-2-оксазолидининон) 3 (30 г, 0,17 моль). Смесь нагревали до комнатной температуры и через 16 часов фильтрат удаляли фильтрацией и раствор концентрировали при пониженном давлении. Флэш-хроматография (7:1 гексан/EtOAc) давала (4R,5S)-4-метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-он в виде масла, 31,5 г; 79%. [α]D=+5,5 (c 1 в CHCl3). MS, m/z (относительная интенсивность): 304 [M+H, 100%].
Трет-бутиловый эфир (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты
К (4R,5S)-4-метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-ону (12,1 г, 0,04 моль) в ТГФ (200 мл) при -50°C добавляли бис(триметилсилил)амид натрия (48 мл 1M раствора в ТГФ). Через 30 минут добавляли трет-бутилбромацетат (15,6 г, 0,08 моль). Раствор перемешивали в течение 4 часов при -50°C и затем нагревали до комнатной температуры. Через 16 часов добавляли насыщенный водный раствор хлорида аммония и два слоя разделяли. Водную фазу экстрагировали простым эфиром и объединенные органические фазы сушили (MgSO4) и концентрировали. Флэш-хроматография (9:1 гексан/EtOAc) давала трет-бутиловый эфир (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты в виде белого твердого продукта 12 г; 72%. [α]D=+30,2 (c 1 в CHCl3).
13C ЯМР (100 МГц; CDCl3) δ 176,47, 171,24, 152,72, 133,63, 128,87, 125,86, 80,85, 78,88, 55,34, 39,98, 38,77, 38,15, 37,58, 30,60, 28,23, 20,38, 20,13, 14,50, 14,28.
4-Трет-бутиловый эфир (S)-2-((R)-2-метилпентил)янтарной кислоты
К трет-бутиловому эфиру (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты (10,8 г, 0,025 моль) в H2O (73 мл) и ТГФ (244 мл) при 0°C добавляли заранее перемешанный раствор LiOH (51,2 мл 0,8 M раствора) и H2О2 (14,6 мл 30% раствора). Через 4 часа добавляли дополнительные 12,8 мл LiOH (0,8 M раствор) и 3,65 мл Н2О2 (30% раствор). Через 30 минут добавляли бисульфит натрия (7 г), сульфит натрия (13 г) и воду (60 мл), а затем добавляли гексан (100 мл) и простой эфир (100 мл). Два слоя разделяли и водный слой экстрагировали простым эфиром. Объединенные органические фазы концентрировали до масла, затем растворяли в гептане (300 мл). Полученный твердый продукт отфильтровали и фильтрат сушили (MgSO4) и концентрировали с получением 4-трет-бутилового эфира (S)-2-((R)-2-метилпентил)янтарной кислоты (6 г, 93%), которую использовали сразу, без дополнительной очистки. MS, m/z (относительная интенсивность): 257 [M+H, 100%].
Трет-бутиловый эфир (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты
Раствор 4-трет-бутилового эфира (S)-2-((R)-2-метил-пентил)янтарной кислоты (6,0 г, 23,22 ммоль) и триэтиламина (3,64 мл, 26,19 ммоль) в толуоле (200 мл) обрабатывали азидом дифенилфосфорила (5,0 мл, 23,22 мл) и перемешивали при комнатной температуре в течение 0,5 часов. После кипячения реакционной смеси с обратным холодильником в течение 3 часов и быстрого охлаждения, добавляли бензиловый спирт (7,2 мл, 69,7 ммоль) и раствор нагревали еще 3 часа. После того как реакционную смесь охлаждали, ее разбавляли этиловым эфиром (200 мл) и объединенный органический слой последовательно промывали насыщенным NaHCO3 и насыщенным солевым раствором и сушили (Na2SO4). Концентрированный органический компонент очищали хроматографией (MPLC), элюируя смесью 8:1 гексана:этилацетата с получением трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты (6,4 г, 75,8%). MS: M+1: 364,2, 308,2.
Трет-бутиловый эфир (3S,5R)-3-амино-5-метилоктановой кислоты
Раствор трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты (2,14 г, 5,88 ммоль) в ТГФ (50 мл) обрабатывали Pd/C (0,2 г) и H2 при 50 фунт/кв.дюйм в течение 2 часов. Реакционную смесь затем фильтровали и концентрировали до масла в вакууме с получением трет-бутилового эфира (3S,5R)-3-амино-5-метилоктановой кислоты с количественным выходом. MS: M+1: 230,2, 174,1.
Гидрохлорид (3S,5R)-3-амино-5-метилоктановой кислоты
Взвесь трет-бутилового эфира (3S,5R)-амино-5-метилоктановой кислоты (2,59 г, 11,3 ммоль) в 6н HCl (100 мл) кипятили с обратным холодильником в течение 18 часов, охлаждали и фильтровали через целит. Фильтрат концентрировали в вакууме до 25 мл и полученные кристаллы собирали и сушили с получением гидрохлорида (3S,5R)-3-амино-5-метилоктановой кислоты, т.пл. 142,5-142,7°C (1,2 г, 50,56%). Второй выход (0,91 г) получали из фильтрата.
Для C9H19NO2·HCl:
Вычислено: C 51,55, H 9,61, N 6,68, Cl 16,91.
Найдено: C 51,69, H 9,72, N 6,56, Cl 16,63.
Гидрохлорид (3S,5R)-3-амино-5-метилоктановой кислоты
4-Трет-бутиловый эфир 2S-(2R-метилпентил)янтарной кислоты (5,3 г), содержащийся в 30 мл метил трет-бутилового эфира взаимодействовал при комнатной температуре с 3,5 мл триэтиламином, а затем с дифенилфосфорилазидом (6,4 г). После достижения реакционной смеси экзотермической точки при 45°C и перемешивания в течение по меньшей мере 4 часов, реакционную смесь оставляли охлаждаться до комнатной температуры и оставляли разделяться на фазы. Нижний слой удаляли, а верхний слой промывали водой, а затем разбавленным водным раствором HCl. Самый нижний слой затем объединяли с 10 мл 6 н. водным раствором HCl, и перемешивали при 45-65°C. Реакционную смесь концентрировали дистилляцией в вакууме до около 10-14 мл и оставляли кристаллизоваться при охлаждении до около 5°C. После получения продукта фильтрации, продукт промывали толуолом и ресуспенидровали в толуоле. Продукт сушили нагреванием в вакуум с получением 2,9 г (67%) белого кристаллического продукта. Продукт может быть перекристаллизован из водной HCl. Т.пл. 137°C.
Пример 6:
(S)-3,7-диметил-окт-6-ениловый эфир метансульфоновой кислоты
К S-(-)-цитронеллолу (42,8 г, 0,274 моль) и триэтиламину (91 мл, 0,657 моль) в CH2Cl2 (800 мл) при 0°C добавляли хлорид метансульфонила (26 мл, 0,329 моль) в CH2Cl2 (200 мл). Через 2 часа при 0°C раствор промывали 1 н. HCl, затем насыщенным солевым раствором. Органическую фазу сушили (MgSO4) и концентрировали с получением соединения, указанного в заголовке в виде масла (60,5 г, 94%), которое затем использовали без дополнительной очистки. MS, m/z (относительная интенсивность): 139 [100%], 143 [100%].
(R)-2,6-Диметил-окт-2-ен
К (S)-3,7-диметил-окт-6-ениловому эфиру метансульфоновой кислоты (60 г, 0,256 моль) в ТГФ (1 л) при 0°C добавляли алюмогидрид лития (3,8 г, 0,128 моль). Через 7 часов, добавляли еще 3,8 г алюмогидрида лития и раствор нагревали до комнатной температуры. Через 18 часов добавляли еще 3,8 г алюмогидрида лития. Еще через 21 час реакционную смесь аккуратно гасили 1 н. лимонной кислотой и раствор еще раз разбавляли насыщенным солевым раствором. Две полученные фазы разделяли и органическую фазу сушили (MgSO4) и концентрировали с получением соединения, указанного в заголовке в виде масла, которое затем использовали без дополнительной очистки. MS, m/z (относительная интенсивность): 139 [M+H, 100%].
(R)-4-Метилгексановая кислота
Использовали способ, аналогичный способу синтеза (R)-4-метилгептановой кислоты, с получением кислоты, указанной в заголовке, в виде масла (9,3 г, 56%). MS, m/z (относительная интенсивность): 129 [M-H, 100%].
(4R,5S)-4-Метил-3-((R)-4-метилгексаноил)-5-фенилоксазолидин-2-он
Использовали способ, аналогичный способу синтеза (4R,5S)-4-метил-3-((R)-4-метилгептаноил)-5-фенилоксазолидин-2-она, с получением соединения, указанного в заголовке, в виде масла (35,7 г, 95%). MS, m/z (относительная интенсивность): 290 [M+H, 100%].
Трет-бутиловый эфир (3S,5R)-5-метил-3-[1-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-ил)метаноил]гептановой кислоты
Использовали способ, аналогичный способу синтеза трет-бутилового эфира (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)октановой кислоты, с получением соединения, указанного в заголовке, в виде масла (7,48 г; 31%). MS, m/z (относительная интенсивность): 178 [100%], 169 [100%]; [α]D=+21,6 (c 1 в CHCl3).
4-Трет-бутиловый эфир (S)-2-((R)-2-метилбутил)янтарной кислоты
Трет-бутиловый эфир (3S,5R)-5-метил-3-[1-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-ил)метаноил]гептановой кислоты (7,26 г, 0,018 моль) в H2O (53 мл) и ТГФ (176 мл) при 0°C добавляли заранее перемешанный раствор LiOH (37 мл 0,8 M раствора) и H2O2 (10,57 мл 30% раствора), и раствор нагревали до комнатной температуры. Через 2 часа добавляли бисульфит натрия (7 г), сульфит натрия (13 г) и воду (60 мл), и два слоя разделяли и водный слой экстрагировали простым эфиром. Объединенные органические фазы концентрировали до масла, которое растворяли в гептане (200 мл). Полученный твердый продукт отфильтровывали, и фильтрат сушили (MgSO4) и концентрировали с получением соединения, указанного в заголовке в виде масла (4,4 г), которое использовали без дополнительной очистки. MS, m/z (относительная интенсивность): 243 [100%].
Трет-бутиловый эфир (3S,5R)-3-бензиоксикарбониламино-5-метилгептановой кислоты
Это соединение получали как описано выше, используя в качестве исходного продукта 4-трет-бутиловый эфир (S)-2-((R)-2-метилбутил)янтарной кислоты с получением трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилгептановой кислоты в виде масла (выход 73,3%).
1H-ЯМР (400 МГц, CDCl3): δ 0,84 (т, 3H, J=7,33 Гц), 0,89 (д, 3H, J=6,60 Гц), 1,12-1,38 (м, 4H), 1,41 (с, 9H), 1,43-1,59 (м, 2H), 2,42 (м, 2H), 4,05 (м, 1H), 5,07 (т, 2H J=12,95 Гц), и 7,28-7,34 (м, 5H).
Трет-бутиловый эфир (3S, 5R)-амино-5-метилгептановой кислоты
Это соединение получали, как описано выше, используя в качестве исходного продукта трет-бутиловый эфир (3S,5R)-3-бензиоксикарбониламино-5-метилгептановой кислоты вместо трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты с получением соединения, указанного в заголовке.
1H ЯМР (400 МГц; CDCl3) δ 0,84 (перекрывающий т и д, 6H), 1,08-1,16 (м, 2H), 1,27-1,30 (м, 2H), 1,42 (с, 9H), 1,62 (ушир.с, 2H), 2,15 (дд, 1H, J=8,54 и 15,62 Гц), 2,29(дд, 1H, J=4,15 и 15,37 Гц) и 3,20(ушир.с, 2H).
Гидрохлорид (3S,5R)-амино-5-метилгептановой кислоты
Взвесь трет-бутилового эфира (3S,5R)-амино-5-метилгептановой кислоты (1,44 г, 6,69 ммоль) в 3 н. HCl кипятили с обратным холодильником в течение 3 часов, фильтровали горячим через целит и досуха концентрировали. После растирания полученного твердого продукта с этиловым эфиром получали гидрохлорид (3S,5R)-3-амино-5-метилгептановой кислоты (0,95 г, 85%). Т.пл. 126,3-128,3°C.
Пример 7:
(3S, 5R)-3-Амино-5-метилнонановая кислота
(R)-4-Метилоктановая кислота. Хлорид лития (0,39 г, 9,12 ммоль) и хлорид меди (I) (0,61 г, 4,56 ммоль) объединяли в 45 мл ТГФ при температуре окружающей среды и перемешивали 15 минут, затем охлаждали до 0°C и в это время добавляли бромид этилмагния (1M раствор в ТГФ, 45 мл, 45 ммоль). По каплям добавляли бромид (S)-цитронеллила (5,0 г, 22,8 ммоль), и раствор оставляли нагреваться до температуры окружающей среды при перемешивании в течение ночи. Реакционную смесь гасили, осторожно добавляя насыщенный NH4Cl (вод.), и перемешивали с Et2O и насыщенным NH4Cl (водный раствор) в течение 30 минут. Фазы разделяли и органическую фазу сушили (MgSO4) и концентрировали. Сырой (R)-2,6-диметил-дец-2-ен использовали без очистки. К раствору (R)-2,6-диметил-дец-2-ена (3,8 г, 22,8 ммоль) в 50 мл ацетона при 0°C добавляли реагент Джонса (2,7 M в H2SO4 (водный раствор), 40 мл 108 ммоль) и раствор оставляли медленно нагреваться до температуры окружающей среды, перемешивая в течение ночи. Смесь разделяли между Et2O и H2O, фазы разделяли и органическую фазу промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали. Остаток очищали флэш-хроматографией (8:1 гексан:EtOAc) с получением 2,14 г (59%) соединения, указанного в заголовке в виде бесцветного масла: LRMS: m/z 156,9 (M+). Реагент Джонса получали как 2,7M раствор, объединяя 26,7 г CrO3, 23 мл H2SO4, и разбавляли до 100 мл с помощью H2O.
(4R,5S)-4-Метил-3-((R)-4-метилоктаноил)-5-фенилоксазолидин-2-он
К (R)-4-метилоктановой кислоте (2,14 г, 13,5 ммоль) в 25 мл CH2Cl2 при 0°C добавляли 3 капли ДМФ, а затем оксалил хлорид (1,42 мл, 16,2 ммоль), что приводило к активному выделению газа. Раствор непосредственно нагревали до температуры окружающей среды, перемешивали 30 минут и концентрировали. В это время в раствор оксазолидинона (2,64 г, 14,9 ммоль) в 40 мл ТГФ по каплям добавляли н-бутиллитий (1,6 M раствор в гексане, 9,3 мл, 14,9 ммоль) при -78°C. Смесь перемешивали в течение 10 минут, в это время по каплям добавляли хлорангидрид в 10 мл ТГФ. Реакционную смесь перемешивали в течение 30 минут при -78°C, затем непосредственно нагревали до температуры окружающей среды и гасили насыщенным NH4Cl. Смесь разделяли между Et2О и насыщ. NH4Cl (водный раствор), фазы разделяли, и органическую фазу сушили (MgSO4), и концентрировали с получением 3,2 г соединения, указанного в заголовке, в виде бесцветного масла. LRMS: m/z 318,2 (M+).
Трет-бутиловый эфир (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)нонановой кислоты
К раствору диизопропиламина (1,8 мл, 12,6 ммоль) в 30 мл ТГФ при -78°C добавляли н-бутиллитий (1,6M раствор в гексане, 7,6 мл, 12,1 ммоль), и смесь перемешивали 10 минут при этом по каплям добавляли (4R,5S)-4-метил-3-((R)-4-метилоктаноил)-5-фенилоксазолидин-2-он (3,2 г, 10,1 ммоль) в 10 мл ТГФ. Раствор перемешивали в течение 30 минут, быстро добавляли по каплям трет-бутил бромацетат (1,8 мл, 12,1 ммоль) при -50°C, и смесь оставляли медленно нагреваться до 10°C в течение 3 часов. Смесь разделяли между Et2О и насыщ. NH4Cl (водный раствор), фазы разделяли и органическую фазу сушили (MgSO4) и концентрировали. Осадок очищали флэш-хроматографией (начиная с 16:1 и заканчивая 8:1 гексан:EtOAc) с получением 2,65 г (61%) соединения, указанного в заголовке, в виде бесцветного кристаллического твердого продукта, т.пл.=84-86°C. [δ]D 23+17,1 (c=1,00, CHCl3).
4-Трет-бутиловый эфир (S)-2-((R)-2-метилгексил)янтарной кислоты 4-трет-бутиловый эфир
К раствору трет-бутилового эфира (3S,5R)-5-метил-3-((4R,5S)-4-метил-2-оксо-5-фенилоксазолидин-3-карбонил)нонановой кислоты (2,65 г, 6,14 ммоль) в 20 мл ТГФ при 0°C добавляли заранее охлажденный (0°C) раствор моногидрата LiOH (1,0 г, 23,8 ммоль) и перекись водорода (30 мас.% водный раствор, 5,0 мл) в 10 мл H2O. Смесь энергично перемешивали в течение 90 минут, затем нагревали до температуры окружающей среды и перемешивали в течение 90 минут. Реакционную смесь гасили при 0°C, добавляя 100 мл 10% NaHSO3 (водный раствор), затем экстрагировали Et2O. Фазы разделяли, и органическую фазу промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали. Соединение, указанное в заголовке использовали без дополнительной очистки.
Трет-бутиловый эфир (3S,5R)-3-бензиоксикарбониламино-5-метилнонановой кислоты
Это соединение получали аналогичным образом, используя в качестве исходного продукта 4-трет-бутиловый эфир (S)-2-((R)-2-метилгексил)янтарной кислоты вместо 4-трет-бутилового эфира (S)-2-((R)-2-метилпентил)янтарной кислоты, с получением соединения, указанного в заголовке в виде масла (выход 71,6%).
1H ЯМР (400 МГц; CDCl3) δ 0,81 (т, 3H, J=4,40 Гц), 0,85 (д, 3H, J=6,55 Гц), 1,06-1,20 (м, 7H), 1,36 (с, 9H), 1,38-1,50 (м, 2H), 2,36 (м, 2H), 3,99 (м, 1H), 5,02 (м+с, 3H) и 7,28-7,28 (м, 5H).
Трет-бутиловый эфир (3S,5R)-3-амино-5-метилнонановой кислоты
Это соединение получали, как описано выше, используя в качестве исходного продукта трет-бутиловый эфир (3S,5R)-бензиоксикарбониламино-5-метилнонановой кислоты вместо трет-бутилового эфира (3S,5R)-3-бензиоксикарбониламино-5-метилоктановой кислоты. Выход = 97%.
1H ЯМР (400 МГц; CDCl3) δ 0,82 (перекрывающий д и т, 6H), 1,02-1,08 (м, 1H), 1,09-1,36 (м, 6H), 1,39 (с, 9H), 1,47 (ушир.с, 1H), 1,80 (с, 2H), 2,13 (дд, 1H, J=8,54 и 15,61 Гц), и 2,27 (дд, 1H, J=4,15 и 15,38 Гц).
Гидрохлорид (3S,5R)-3-амино-5-метилнонановой кислоты
Смесь трет-бутилового эфира (3S,5R)-3-амино-5-метилнонановой кислоты (1,50 г, 6,16 ммоль) в 3н HCl (100 мл) кипятили с обратным холодильником в течение 3 часов, фильтровали горячим через целит и концентрировали до 30 мл в вакууме. Полученные кристаллы собирали, промывали дополнительным количеством 3 н. HCl, и сушили с получением соединения, указанного в заголовке, т.пл. 142,5-143,3°C. Дополнительные выходы получали из фильтрата и они составляли 1,03 г (70,4%).
Для C10H21NO2·HCI:
Вычислено: C 53,68, H 9,91, N 6,26, Cl 15,85.
Найдено: C 53,89, H 10,11, N 6,13.
MS: M+1: 188,1.
Биологические примеры:
Биологическая активность соединений, подходящих для использования в соответствии с изобретением в качестве альфа-2-дельта лигандов может быть измерена в анализах связывания радиолиганда, используя [3H]габапентин и α-2-δ субъедицу, полученную из ткани мозга свиней (Gee N.S., Brown J.P., Dissanayake V.U.K., Offord J., Thurlow R., Woodruff G.N., J. Biol. Chem., 1996; 271:5879-5776).
Пример 1: Исследование эффектов альфа-2-дельта лиганда на рефлекс мочеиспускания у анестезированных крыс
1.0 Материалы и методы:
Животные
Четырех крыс, рожденных путем кесарева сечения (CD), весом приблизительно 225 г, анастезировали, используя уретан, и осуществляли продолжительную цистометрию.
Животных анастезировали, используя уретан (1,2 г/кг, в.в.). Глубину анастезии оценивали по стабильности давления крови и частоте сердечных ударов, и по отсутствию одергивания задней лапы в ответ на ее сжатие. При необходимости давали дополнительные дозы уретана (0,1 г кг-1, в.в.).
Трахею интубировали для поддержания свободного доступа воздуха, в наружную яремную вену вводили канюлю для введения препарата, и в общую сонную артерию вводили гепаринизированную канюлю (20 единиц/мл гепарина в 0,9% мас./об. раствора) для измерения артериального давления крови.
Давление крови измеряли, используя датчик давления и сердечный ритм (HR), получали на электронном табло в оперативном режиме на основании давления крови, используя PoneMah (Linton Pty Ltd UK). За температурой тела следили с помощью ректального температурного зонда и поддерживали между 36-38°C, используя гомеотермическое одеяло (Harvard, UK). На протяжении эксперимента животные самопроизвольно дышали воздухом.
На мочевом пузыре делали поперечный абдоминальный разрез. В верхушку мочевого пузыря вводили канюлю (0,52 мм внутренний и 1,2 мм внешний диаметр) для вливания раствора в мочевой пузырь, одновременно записывая внутрипузырное давление мочевого пузыря. В мочевой пузырь вливали физиологический раствор (0,9%) со скоростью 0,046 мл/мин, стимулируя максимальную скорость часового диуреза крыс (Klevmark B [1974] - Motility of the urinary bladder in cats during filling at physiological rates. I. Intravesical pressure patterns studied by a new method of cystometry. Acta Physiol Scand 90(3): 565-77). После хирургической операции животных оставляли стабилизироваться на приблизительно 30 минут. После периода стабилизации проводили цистометрию и наблюдали за функцией мочевого пузыря (интервал между мочеиспусканием) в ответ на габапентин-HCl (0,3, 1,3 и 10 мг/кг, вводимые внутривенно).
Статистический анализ
Различия между обрабатываемыми группами оценивали, используя ANOVA.
Соединение
Габапентин-HCl (Tocris, UK) растворяли в физиологическом растворе.
2.0 Результаты:
Интервалы времени между актами опорожнения
Интервалы между актами опорожнения увеличивались в ответ на габапентин (см. фиг. 1) относительно соответствующих периодов в контроле. Исследование показало, что габапентин значительно расширяет интвервал между актами опорожнения в зависимости от дозы. Действительно, 10 мг/кг габапентина, вводимого внутривенно, значительно продливал интервалы мочеиспускания по сравнению с соответствующими контрольными животными (3,1±0,29 против 9,89±2,72 мин P<0,05). В результате увеличения интервалов между актами мочеиспускания емкость мочевого пузыря значительно увеличилась, на 200% в ответ на габапентин. Интересно отметить, что габапентин, при введении в количестве 10 мг/кг, не имел каких-либо побочных эффектов на эффективность мочеиспускания, удержание мочи или сокращение мочевого пузыря при опорожнении.
Таким образом, было показано, что введение альфа-2-дельта лиганда повышало емкость мочевого пузыря и снижало СНМП у самок крыс, что указывает на эффективность такого подхода для лечения СНМП, связанного с ГМП.
Пример 2: Эффект альфа-2-дельта лиганда на частоту мочеиспускания и давление мочевого пузыря у самцов крыс co спонтанной гипертонией (SHR)
1.0 Материалы и методы:
Восемь (8) самцов крыс со спонтанной гипертонией получали хирургическим путем с помощью Data Sciences International (DSI) телеметрического транспондера (TL11M2-C50-PXT). За давлением мочевого пузыря постоянно следили с помощью катетера для измерения давления, введенного через верхушку мочевого пузыря.
Характеристики мочевого пузыря (давление мочевого пузыря, давление при опорожнении и число актов опорожнения) записывали в ответ на введение плацебо или лечение 60 мг/кг габапентина, вводимого подкожно (п/к) (см. фиг.2).
2.0 Результаты:
Как показано на фигуре, габапентин в количество 60 мг/кг п/к значительно снижал (P<0,05) интервалы между мочеиспусканиями и поэтому повышал емкость мочевого пузыря. Более того, и это важно для симптомов нижних мочевых путей (СНМП), среднее давление мочевого пузыря во время стадии наполнения цикла мочеиспускания было значительно ниже (P<0,05) в ответ на габапентин по сравнению с соответствующими периодами в контрольной группе (см. фиг.3).
Таким образом, было показано, что введение альфа-2-дельта лиганда увеличивает емкость мочевого пузыря и снижает СНМП у самцов животных, что указывает на эффективность такого подхода для лечения СНМП, связанного с ГМП.
Пример 3: Эффект прегабалина на модели определения функции мочевого пузыря с телеметрией
1.0 Материалы и методы
Животные:
В исследовании использовали крыс с самопроизвольной гипертензией (SHR), которым имплантировали зонды для телеметрии. Животные весом 200-250 г были получены от Charles River и содержались при цикле свет/тьма, 12:12, с доступом к пище и воде ab libitum.
Экспериментальные процедуры:
В каждый день эксперимента животным вводили подкожную инъекцию единичной дозы тестируемого соединения или носителя. Обычно оценку проводили через четыре дозы тестируемого соединения, разделяя каждый день исследования двумя, тремя днями промывки, то есть все животные получали полное лечение. На последнем этапе исследования из хвостовой вены брали 0,4 мл образца крови для РК анализа.
После введения дозы соединения животных размещали по отдельности в Metaboles для записи количества мочеиспусканий и измерения объема на акт мочеиспускания и общий объем мочеиспусканий. Давление мочевого пузыря постоянно регистрировали посредством телеметрии. Все параметры измеряли в течение 120 минут через 60 минут после введения дозы соединения.
В отдельном РК анализе 3-м животным вводили дозу тестируемого соединения. У животных на 15, 30 минуте и через 1 и 3 часа брали 4 образца пробы по 0,25 мл. По этим образцами, а также с образцами крови, полученными у животных на последнем этапе исследования, определяли РК соединений в SHR.
2.0 Результаты:
Эффект прегабалина на функцию мочевого пузыря исследовали на модели определения функции мочевого пузыря с телеметрией у крыс. В этой модели прегабалин продемонстрировал зависимое от дозы повышение среднего объема на акт мочеиспускания в течение двух часов (фиг.4) и соответствующее снижение частоты мочеиспускания. Общий объем при мочеиспускании в течение двух часов оставался неизменным для всех доз. Давление мочевого пузыря при мочеиспускании также оставалось неизменным по сравнению с контролем для всех доз. Таким образом, какой-либо эффект на давление в мочевом пузыре не был подвержен.
Вывод:
Результаты эксперимента показали, что прегабалин повышает средний объем на акт мочеиспускания и снижает частоту мочеиспускания не влияя на давление в мочевом пузыре.
Фиг.4 показывает эффект прегабалина на объем при акте мочеиспускания на модели определения функции мочевого пузыря с телеметрией.

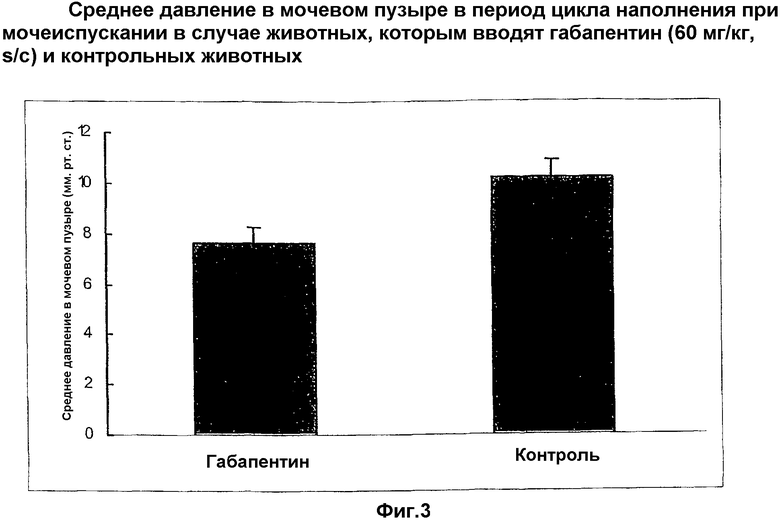

Изобретение относится к области фармакологии и медицины и касается применения альфа-2-дельта лиганда для получения лекарственного средства для лечения симптомов нижних мочевыводящих путей, отличных от недержания мочи, связанного с ГМП и/или ДГПЖ. Средство обладает повышенной эффективностью 11 з.п. ф-лы, 4 ил.










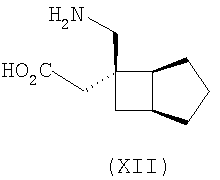







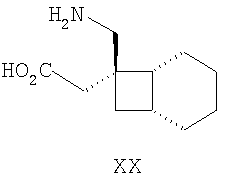
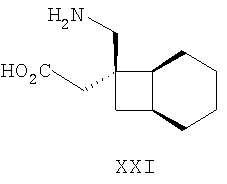





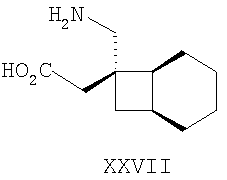

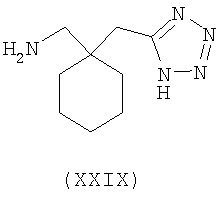
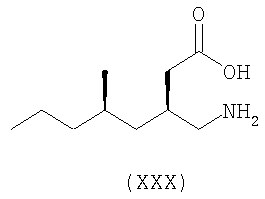

 ; или его фармацевтически приемлемой соли или сольвата;
; или его фармацевтически приемлемой соли или сольвата;
где каждый R1 и R2 независимо выбран из Н, прямого или разветвленного алкила из 1-6 атомов углерода, циклоалкила из 3-6 атомов углерода, фенила и бензила, при условии, кроме трициклооктанового соединения формулы (XVIII), что R1 и R2 одновременно не могут быть водородом; или выбран из



или его фармацевтически приемлемой соли или сольвата.
или его фармацевтически приемлемой соли или сольвата.
или его фармацевтически приемлемой соли или сольвата.
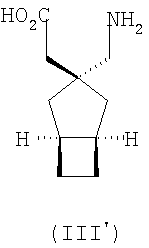
или его фармацевтически приемлемой соли или сольвата.
или ее фармацевтически приемлемая соль или сольват.
| WO 00/61135 А, 19.10.2000 | |||
| ГЕТЕРОБИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ПОЛУЧЕНИЯ ГЕТЕРОБИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ | 1993 |
|
RU2128656C1 |
| Разбрасыватель жидких удобрений | 1974 |
|
SU533352A1 |

