Область техники
Настоящее изобретение относится к фторированным производным 4-азастероида, их синтезу и их применению в качестве модуляторов рецептора андрогена. В частности, соединения настоящего изобретения представляют собой тканеселективные модуляторы рецептора андрогена и, в связи с этим, являются полезными для лечения состояний, вызванных недостаточностью андрогенов или интенсивность симптомов которых может быть уменьшена введением андрогена, таких как остеопороз, периодонтальная болезнь, перелом (трещина)кости, хрупкостьи саркопения.
Предшествующий уровень техники
Рецептор андрогена (AR) относится к надсемейству ядерных рецепторов стероидных/тиреоидных гормонов, другие представители которого включают рецептор эстрогена (ER), рецептор прогестерона (PR), рецептор глюкокортикоида (GR) и рецептор минералокортикоида (MR). AR экспрессируется во многих тканях тела и представляет собой рецептор, посредством которого экспрессируются физиологические, а также патофизиологические действия эндогенных лигандов андрогена, таких как тестостерон (Т) и дигидротестостерон (DHT). Структурно AR состоит из трех основных функциональных доменов: домена, связывающего лиганд (LBD); ДНК-связывающего домена и аминоконцевого домена. Соединение, которое связывается с AR и имитирует действия эндогенного лиганда AR, называют агонистом AR, в то время как соединение, которое ингибирует действия эндогенного лиганда AR, называют антагонистом AR.
Связывание лиганда андрогена с AR дает комплекс лиганд/рецептор, который после транслокации внутри ядра клетки связывается со специфическими регуляторными последовательностями ДНК (называемыми элементами ответа андрогена или ARE) в пределах областей промотора или энхансера гена или генов мишени, присутствующих в ядре клетки. Затем наращиваются другие белки, называемые кофакторами, которые связываются с аминоконцевым доменом или лигандсвязывающим доменом (LBD) рецептора, что приводит к транскрипции и последующей трансляции гена с получением белка(ов), кодируемого указанным геном или генами.
Андрогенотерапию использовали в клинике для лечения целого ряда нарушений у мужчин, таких как репродуктивные нарушения и первичный или вторичный мужской гипогонадизм. Кроме того, различные природные или синтетические агонисты AR были клинически исследованы при лечении мышечно-скелетных нарушений, таких как болезнь костной ткани, гемопоэтические расстройства, нервно-мышечная болезнь, ревматическая болезнь, кахексия, а также при заместительной гормональной терапии (HRT), как, например, в случае недостаточности андрогенов у женщин. Кроме того, антагонисты AR, такие как флутамид и бикалутамид, были использованы для лечения рака предстательной железы. Поэтому было бы полезно иметь доступные соединения, которые могут активировать («агонизировать») функцию AR тканеселективным способом, что обеспечило бы проявление желательных благотворных остео- и миоанаболических действий андрогенов, но без проявления негативных андрогенных свойств, таких как вирилизация и индуцирование атерогенного (способствующего развитию атеросклероза) профиля липида, которые могут привести к развитию сердечно-сосудистого заболевания.
Роль андрогенов в остеогенезе подтверждается документами. Например, анаболические стероиды, такие как нандролон деканоат или станозолол, как было показано, увеличивают костную массу у женщин в постменопаузальном периоде. Благотворные воздействия андрогенов на костную ткань при постменопаузальном остеопорозе были зарегистрированы в последних исследованиях, использующих комбинированное введение тестостерона и эстрогена [Hofbauer, et al., "Androgen effects on bone metabolism: recent progress and controversies," Eur. J. Endocrinol.140: 271-286 (1999)]. Комбинированное лечение значительно увеличивало скорость повышения плотности минеральных компонентов кости (BMD) и ее величину в поясничной области и области бедра, по сравнению с лечением только одним эстрогеном. Кроме того, комбинации эстроген-прогестин, которые содержали андрогенный прогестин (такой как норетиндрон), а не медроксипрогестерон ацетат, давали лучшую положительную динамику для BMD тазобедренного сустава. Эти результаты недавно были подтверждены при сравнительном исследовании двойным слепым методом на протяжении более 2 лет, в котором, как было показано, пероральные комбинации сопряженных эстрогенов (СЕЕ) и метилтестостерона оказываются эффективными в стимуляции наращивания костной массы в позвоночнике и тазобедренном суставе, в то время как лечение только сопряженными эстрогенами предотвращало разрежение кости (остеопороз) ["A two-year, double-blind comparison of estrogen-androgen and conjugated estrogens in surgically menopausal women: Effects on bone mineral density, symptoms and lipid profiles," J. Reprod. Med., 44: 1012-1020 (1999)]. Несмотря на благоприятные воздействия андрогенов на женщин в постменопаузальном периоде, использование андрогенов было ограничено из-за нежелательного вирилизирующего и метаболического действия андрогенов. Данные Watts и коллег демонстрируют, что «приливы» уменьшаются у женщин, подвергнутых лечению CEE и метилтестостероном; однако 30% из вышеуказанных женщин страдали от существенного увеличения (появления) угрей и волос на лице, осложнение всех современных методов фармакотерапии с использованием андрогена [Watts, et al., "Comparison of oral estrogens and estrogens plus androgen on bone mineral density, menopausal symptoms, and lipid-lipoprotein profiles in surgical menopause," Obstet. Gynecol., 85: 529-537 (1995)]. Кроме того, как видно из других исследований, добавление метилтестостерона к СЕЕ заметно снижало уровни HDL. Поэтому не селективные к ткани агонисты AR могут увеличить риск возникновения сердечно-сосудистого заболевания. Таким образом, возможность вирилизации и негативные воздействия на липидный профиль современных методов андрогенотерапии предоставляют существенное логическое обоснование для разработки тканеселективных агонистов рецептора андрогена для кости. Делается ссылка на J. A. Kanis, "Other agents for generalized osteoporosis," in Osteoporosis, Blackwell Science, Ch. 8, pp 196-227 (1994), где обсуждаются неселективные анаболические стероиды для лечения остеопороза.
Также было установлено, что андрогены играют важную роль в костном метаболизме у мужчин, которая соответствует роли эстрогенов у женщин [Anderson, et al., "Androgen supplementation in eugonadal men with osteoporosis - effects of six months of treatment on bone mineral density and cardiovascular risk factors," Bone, 18: 171-177 (1996)]. Даже у эугонадных мужчин с установленным остеопорозом терапевтическая реакция на лечение тестостероном предоставляла дополнительное доказательство того, что андрогены оказывают важные остеоанаболические воздействия. Средняя BMD в поясничной области возрастает от 0,799 до 0,839 г/см2, за период от 5 до 6 месяцев в ответ на лечение 250 мг сложного эфира тестостерона, вводимого внутримышечно каждые две недели. Обычный сценарий для проявления андрогенной недостаточности имеет место у мужчин с раком простаты стадии D (метастатический), которые подвергаются терапии, направленной на подавление выработки андрогена (ADT). Эндокринная орхиэктомия (удаление яичек) достигается длительным воздействием агонистов GnRH, тогда как блокада рецептора андрогена осуществляется действием флутамида, нилутамида, бикалутамида или RU 58841 (антагонисты AR). В ответ на выключение эндокринной функции вышеуказанные мужчины страдают от «приливов», существенного остеопороза (разрежение кости), слабости и утомления. В последнем экспериментальном исследовании мужчин с раком простаты стадии D нарушение остеогенеза (50% против 38%) и остеопороз (38% против 25%) встречались более часто у мужчин, которые подвергались ADT на протяжении более чем одного года, чем у пациентов, которые не подвергались лечению ADT [Wei, et al., "Androgen deprivation therapy for prostate cancer results in significant loss of bone density," Urology, 54: 607-611 (1999)]. BMD поясничной области позвоночника оказалась значительно ниже у мужчин, которые подверглись ADT. Таким образом, помимо использования тканеселективных агонистов AR для лечения остеопороза, тканеселективные антагонисты AR в простате, которые не обладают антагостическим действием на костную и мышечную ткань, могут быть полезными средствами для лечения рака простаты, либо сами по себе, либо в качестве вспомогательного лечебного средства при традиционной ADT, как, например, комбинация агонист/антагонист GnRH[См. также A. Stoch, et al., J. Clin. Endocrin. Metab., 86: 2787-2791 (2001)]. Тканеселективные антагонисты AR могут также быть полезными для лечения синдрома поликистоза яичников у женщин в постменопаузальном периоде [см. С.A. Eagleson, et al., "Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone," J. Clin. Endocrinol. Metab,, 85: 4047-4052 (2000) и E. Diamanti-Kandarakis, "The Effect of a Pure Antiandrogen Receptor Blocker, Flutamide, on the Lipid Profile in the Polycystic Ovary Syndrome," Int. J. Endocrinol. Metab., 83: 2699-2705 (1998)].
Существует потребность в более эффективных средствах для лечения нарушения остеогенеза и остеопороза как для мужчин, так и для женщин. Остеопороз характеризуется разрежением костной ткани вследствие нарушения равновесия между резорбцией (разрушением) и образованием костной ткани, которое начинается в четвертом десятилетии и продолжается на всем протяжении жизни со скоростью около 1-4% в год [Eastell, "Treatment of postmenopausal osteoporosis," New Engl. J. Med., 338: 736 (1998)]. В Соединенных Штатах в настоящее время имеется приблизительно 20 миллионов человек с выявленными трещинами и переломами позвонков вследствие остеопороза. Кроме того, имеет место около 250000 трещин (переломов) тазобедренного сустава (бедра) в год вследствие остеопороза, ассоциируемых с 12-20% коэффициентом смертности в пределах первых двух лет, тогда как 30% из вышеуказанных пациентов требуют помощи медсестер на дому после перелома, и многие из указанных пациентов никогда не станут полностью амбулаторными больными снова. У женщин в постменопаузальном периоде дефицит эстрогена ведет к повышенной резорбции кости, приводящей к потере костной ткани в позвонках около 5% в год, сразу после менопаузы. Поэтому первым направлением лечения и профилактики указанного выше состояния является ингибирование резорбции кости бисфосфонатами, эстрогенами, селективными модуляторами рецептора эстрогена (SERM) и кальцитонином. Однако ингибиторов резорбции кости недостаточно для восстановления костной массы у пациентов, которые уже потеряли значительную часть костной массы. Увеличение BMD позвоночника, достигнутое лечением бисфосфонатами, может достигнуть 11% после лечения алендронатом в течение 7 лет. Кроме того, поскольку скорость обновления костной ткани различна в зависимости от места поражения костной ткани, более высокая в трабекулярной кости позвонков, чем в корковом слое длинных костей, то ингибиторы резорбции кости менее эффективны в повышении BMD тазобедренного сустава и предотвращении перелома бедра. Поэтому остеоанаболические средства, которые увеличивают кортикальный остеогенез и костную массу длинных костей, стимулируя периостальный остеогенез, вероятно, можно направить на решение неудовлетворенной потребности в лечении остеопороза, особенно для пациентов с высоким риском перелома бедра. Остеоанаболические средства также дополняют ингибиторы резорбции кости, которые атакуют трабекулярную оболочку, приводя к образованию биомеханически благоприятной костной структуры(Schmidt, et al., "Anabolic steroid: Steroid effects on bone in women," In: J. P. Bilezikian, et al., Ed., Principles of Bone Biology, San Diego: Academic Press, 1996). Тканеселективные агонисты AR с уменьшенными вредными воздействиями на сердечно-сосудистую систему и ограниченным вирилизирующим потенциалом могут быть использованы в качестве монотерапии для профилактики и/или лечения остеопороза у женщин. Кроме того, соединение с остеоанаболическими свойствами по отношению к костной и мышечной ткани, но с пониженной активностью в простате и придаточных тканях половых органов, может быть использовано для профилактики и/или лечения остеопороза у мужчин и нарушения остеогенеза (остеопении) у мужчин, особенно пожилых мужчин.
Кроме того, селективные модуляторы рецептора андрогена могут быть использованы для лечения некоторых гемопоэтических нарушений. Известно, что андрогены стимулируют почечную гипертрофию и продуцирование эритропоэтина (EPO). До введения рекомбинантного человеческого EPO андрогены использовали для лечения анемии, вызванной хронической почечной недостаточностью. Кроме того, было установлено, что андрогены при фармакологических дозах повышают сывороточные уровни EPO у пациентов с нетяжелой апластической анемией и миелодиспластическими синдромами, но не у пациентов без анемии. Лечебное воздействие на анемию нуждается в избирательном действии, таком которое может быть обеспечено селективными модуляторами рецептора андрогена.
Кроме того, селективные модуляторы рецептора андрогена могут также иметь клиническое значение как вспомогательное средство при лечении ожирения. Указанный подход к снижению жира в теле подтверждается опубликованными наблюдениями, согласно которым введение андрогена уменьшало подкожный и висцеральный абдоминальный жир у тучных людей [J.C. Lovejoy, et al., "Oral anabolic steroid treatment, but not parenteral androgen treatment, decreases abdominal fat in obese, older men," Int. J. Obesity, 19: 614-624 (1995)]. Поэтому SARM, лишенные андрогенных воздействий на простату, могут быть полезными для лечения тучных мужчин. В отдельном исследовании введение андрогена привело к потере подкожного абдоминального жира у тучных женщин в постменопаузальном периоде[J.C. Lovejoy, et al., "Exogenous Androgens Influence Body Composition and Regional Body Fat Distribution in Obese Postmenopausal Women - A Clinical Research Center Study," J. Clin. Endocrinol. Metab., 81: 2198-2203 (1996)]. В последнем исследовании было установлено, что нандролон деканоат, слабое андрогенное и анаболическое средство, повышает массу тела у тощих людей и интенсивность обмена веществ в покое у тучных женщин в постменопаузальном периоде, соблюдающих диету, способствующую снижению массы тела.
Нестероидные соединения, обладающие свойством модуляциирецептора андрогена, были раскрыты в патентах США №№ 5688808; 5696130; 6017924; 6093821; WO 01/16139 (опубликованной 8 марта 2001); и WO 01/16108 (опубликованной 8 марта 2001), все переуступлены Ligand Pharmaceuticals, и в WO 01/27086, переуступленной Kaken Pharm. Co. Дополнительная характеристика известного уровня техники, имеющего отношение к развитию селективных модуляторов рецептора андрогена, имеется в L. Zhi and E. Martinborough in Ann. Rep. Med. Chem. 36: 169-180 (2001). Нестероидные SARM были раскрыты в J.P. Edwards, "New Nonsteroidal Androgen Receptor Modulators Based on 4-(Trifluoromethyl)-2(1H)-Pyrrolidino[3,2-g]quinolinone," Bioorg. Med. Chem. Lett., 8: 745-750 (1998) и в L. Zhi et al., "Switching Androgen Receptor Antagonists to Agonists by Modifying C-ring Substituents on Piperidino[3,4-g]quinolinone," Bioorg. Med. Chem. Lett., 9: 1009-1012 (1999).
В клинической области существует потребность в более эффективных средствах, которые могут выявлять положительные реакции заместительной андрогенной терапии, но без нежелательных побочных действий, присущих неселективным к ткани агонистам AR. Требуются именно такие соединения, которые могут обеспечивать такие же положительные реакции, как заместительная андрогенная терапия, но без нежелательных побочных действий. Также требуются андрогенные соединения, которые проявляют селективные действия на различные ткани тела. В данном изобретении идентифицированы соединения, которые функционируют как селективные модуляторы рецептора андрогена (SARM), с использованием ряда in vitro клеточных проб, которые профилируют опосредованную лигандом активацию AR, а именно (i) N-C взаимодействие, (ii) подавление транскрипции и (iii) активацию транскрипции. Соединения-SARM в настоящем изобретении, идентифицированные перечисленными выше методами, демонстрируют тканеселективный агонизм AR in vivo, т.е. агонизм в кости (стимуляция остеогенеза на модели остеопороза у грызунов) и антагонизм в простате (минимальные воздействия на рост простаты у кастрированных грызунов и антагонизм роста простаты, индуцированного агонистами AR).
Соединения настоящего изобретения, идентифицированные как SARM, используют для лечения заболеваний или состояний, вызванных недостаточностью андрогена, интенсивность симптомов которых может быть уменьшена введением андрогена. Такие соединения являются идеальными для лечения остеопороза у женщин и мужчин в качестве монотерапии или в комбинации с ингибиторами резорбции кости, такими как бисфосфонаты, эстрогены, SERM, ингибиторы катепсина К, антагонисты рецептора интегрина αvβ3, кальцитонин и ингибиторы протонного насоса. Они могут быть также использованы со средствами, которые стимулируют остеогенез, такими как паратиреоидный гормон или его аналоги. Соединения-SARM по данному изобретению могут также быть использованы для лечения заболеваний простаты, таких как рак простаты и доброкачественная гиперплазия простаты (BPH). Кроме того, соединения данного изобретения демонстрируют минимальные воздействия на кожу (прыщи и рост волос на лице) и могут быть полезны для лечения гирсутизма. Дополнительно, соединения данного изобретения могут стимулироватьрост мышечной ткани и могут быть полезны для лечения саркопении и хрупкости. Вышеуказанные соединения можно использовать для уменьшения подкожного и висцерального абдоминального жира при лечении ожирения. Кроме того, соединения данного изобретения могут проявлять андрогенный агонизм в центральной нервной системе и могут быть использованы для лечения вазомоторных симптомов («приливы») и для повышения энергии и либидо, особенно у женщин в период менопаузы. Соединения настоящего изобретения могут быть использованы для лечения рака простаты, либо как таковые, либо как вспомогательное лечебное средство при традиционной терапии с использованием агониста/антагониста GnRH, из-за их способности восстанавливать кость, либо как замена при антиандрогенной терапиииз-за их способности антагонизировать андроген в простате и свести к минимуму истощение костной ткани в скелетной системе. Далее, соединения настоящего изобретения могут быть использованы, из-за их способности восстанавливать кость, при лечении рака поджелудочной железы в качестве вспомогательного средства для лечения антиандрогеном, или как монотерапия из-за их антиандрогенных свойств, при этом демонстрируя преимущество перед традиционными антиандрогенами, заключающееся в способности восстанавливать кость. Дополнительно, соединения данного изобретения могут увеличивать количество клеток крови, таких как эритроциты и тромбоциты, и могут быть полезны для лечении гемопоэтических расстройств, таких как апластическая анемия. Наконец, соединения данного изобретения оказывают минимальные воздействия на липидный метаболизм. Таким образом, вследствие своего тканеселективного агонизмак рецептору андрогена, изложенного выше, соединения данного изобретения являются идеальными для заместительной гормональной терапии для мужчин, страдающих от гипогонадизма (недостаточности секреции андрогена).
Поэтому целью настоящего изобретения является разработка фторированных производных 4-азастероида, которые используют в качестве селективных модуляторов рецептора андрогена.
Другой целью настоящего изобретения является разработка фармацевтических композиций, содержащих фторированные производные 4-азастероида по данному изобретению в сочетании с фармацевтически приемлемым носителем.
Другой целью настоящего изобретения является разработка фармацевтических композиций, содержащих фторированные производные 4-азастероида, для использования в качестве селективных модуляторов рецептора андрогена.
Другой целью настоящего изобретения является разработка способов лечения заболеваний или состояний, вызванных недостаточностью андрогена, интенсивность симптомов которых может быть уменьшена введением андрогена.
Другой целью настоящего изобретения является разработка способов лечения заболеваний или состояний, вызванных недостаточностью андрогена, интенсивность симптомов которых может быть уменьшена введением андрогена в комбинации с другими средствами.
Другой целью настоящего изобретения является разработка фторированных производных 4-азастероида и их фармацевтических композиций для использования в качестве лекарственного средства при лечении заболеваний или состояний, вызванных недостаточностью андрогена, интенсивность симптомов которых может быть уменьшена введением андрогена.
Другой целью настоящего изобретения является разработка фторированных производных 4-азастероида и их фармацевтических композиций для получения лекарственного средства для лечения заболеваний или состояний, вызванных недостаточностью андрогена, интенсивность симптомов которых может быть уменьшена введением андрогена.
Эти и другие цели настоящего изобретения становятся очевидными из подробного описания, которое следует ниже.
Краткое описание изобретения
Настоящее изобретение относится к соединениям структурной формулы I:
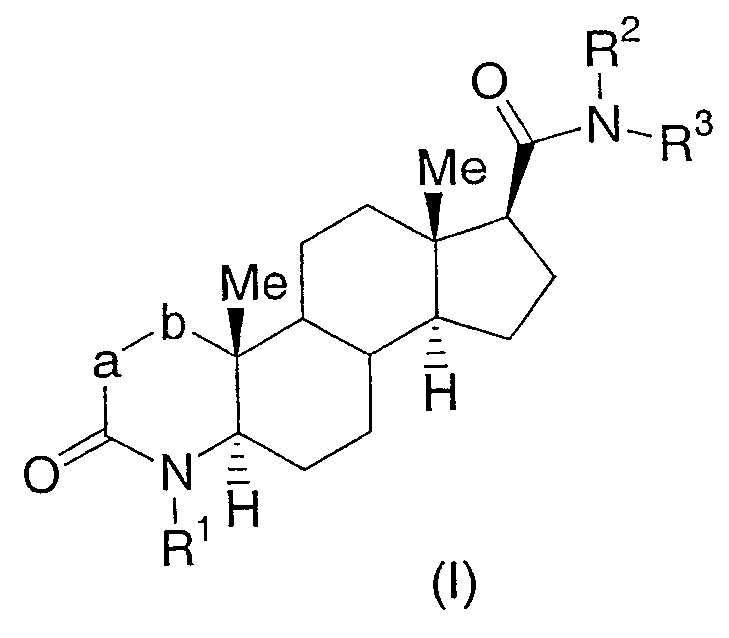
или их фармацевтически приемлемой соли или их энантиомеру; где
n равно 0, 1 или 2;
a-b означает CF=CH, CHFCH2 или CF2CH2;
R1 представляет собой водород, гидроксиметил или C1-3алкил, где алкил является незамещенным или замещен одним-семью атомами фтора;
R2 представляет собой водород или C1-4алкил;
R3 выбран из
C1-4алкила,
(CH2)n-циклогетероалкила и
(CH2)n-арила, где арил выбран из
(1) фенила,
(2) нафтила,
(3) бензимидазолила,
(4) бензофуранила,
(5) бензотиофенила,
(6) бензоксазолила,
(7) бензотиазолила,
(8) бензодигидрофуранила,
(9) 1,3-бензодиоксолила,
(10) 2,3-дигидро-1,4-бензодиоксинила,
(11) индолила,
(12) хинолила,
(13) изохинолила,
(14) фуранила,
(15) тиенила,
(16) имидазолила,
(17) оксазолила,
(18) тиазолила,
(19) изоксазолила,
(20) изотиазолила,
(21) пиразолила,
(22) пирролила,
(23) пиридила,
(24) пиримидила,
(25) пиразинила,
(26) тиадиазолила,
(27) оксадиазолила,
(28) триазолила,
(29) тетразолила и
(30) инданила;
где алкильная группа или циклогетероалкильная группа является незамещенной или замещена одним-тремя заместителями, независимо выбранными из галогена, гидрокси и С1-4алкокси; арильная группа, определенная в пунктах (1)-(30), является незамещенной или замещена одной-тремя группами, независимо выбранными из галогена, фенила, С1-8алкила, С3-8циклоалкила, С3-8циклогетероалкила, фенил-С1-6алкила, амино-С0-6алкила, С1-6алкиламино-С0-6алкила, (C1-6алкил)2амино-C0-6алкила, фенил-C0-6алкиламино-С0-6алкила, (фенил-C0-6алкил)2амино-C0-6алкила, C1-6алкилтио, фенил-C0-6алкилтио, C1-6алкилсульфинила, фенил-C0-6алкилсульфинила, C1-6алкилсульфонила, фенил-C0-6алкилсульфонила, C1-6алкокси-C0-6алкила, фенил-C0-6алкокси-C0-6алкила, гидроксикарбонил-C0-6алкила, C1-6алкоксикарбонил-C0-6алкила, фенил-C0-6алкоксикарбонил-C0-6алкила, гидроксикарбонил-C1-6алкилокси, гидрокси-C0-6алкила, циано, нитро, перфтор-C1-4алкила, перфтор-C1-4алкокси, оксо, C1-6алкилкарбонилокси, фенил-C0-6алкилкарбонилокси, С1-6алкилкарбониламино, фенил-C0-6алкилкарбониламино, С1-6алкилсульфониламино, фенил-C0-6алкилсульфониламино, C1-6алкоксикарбониламино, фенил-C0-6алкоксикарбониламино, C1-6алкиламинокарбониламино, фенил-C0-6алкиламинокарбониламино, (C1-6алкил)2аминокарбониламино, (фенил-C0-6алкил)2аминокарбониламино, (C1-6алкил)2аминокарбонилокси и (фенил-C0-6алкил)2аминокарбонилокси; и где углеродный атом любого метилена (СН2) в (CH2)n является незамещенным или замещен одной-двумя группами, независимо выбранными из галогена, гидрокси и С1-4алкила; или два заместителя, когда они принадлежат одной и той же метиленовой (СН2) группе, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопропильную группу;
или R2 и R3 вместе образуют 5- или 6-членное насыщенное кольцо, конденсированное с 5- или 6-членной ароматической кольцевой системой, имеющей 0, 1 или 2 гетероатома, выбранных из N, O и S.
Вышеуказанные соединения эффективны в качестве агонистов рецептора андрогена и, в частности, эффективны в качестве селективных агонистов рецептора андрогена (SARM). Поэтому указанные соединения используют для лечения состояний, вызванных недостаточностью андрогена или интенсивность симптомов которых может быть уменьшена введением андрогена.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения настоящего изобретения и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способам лечения состояний, вызванных недостаточностью андрогена или интенсивность симптомов которых может быть уменьшена введением андрогена, у млекопитающего, нуждающегося в таком лечении, путем введения соединений и фармацевтических композиций по данному изобретению.
Настоящее изобретение также относится к способам лечения остеопороза, нарушения остеогенеза, глюкокортикоид-индуцированного остеопороза, периодонтальной болезни, перелома кости, повреждения кости после реконструктивной костной хирургии, саркопении, хрупкости, старения кожи, мужского гипогонадизма, постменопаузальных симптомов у женщин, атеросклероза, гиперхолестеринемии, гиперлипидемии, ожирения, апластической анемии и других гемопоэтических нарушений, артритных состояний, таких как, например, воспалительный артрит и восстановление поврежденного сустава, ВИЧ-истощения, рака простаты, раковой кахексии, мышечной дистрофии, преждевременного угасания функции яичников и аутоиммунного заболевания, путем введения соединений и фармацевтических композиций настоящего изобретения как таковых или в комбинации с терапевтически эффективным количеством другого средства, которое, как известно, используют для лечения таких состояний.
Подробное описание изобретения
Настоящее изобретение относится к соединениям, которые используют в качестве агонистов рецептора андрогена, в частности в качестве селективных агонистов рецептора андрогена. Соединения настоящего изобретения представляют собой соединения, описываемые структурной формулой I:
или их фармацевтически приемлемую соль или их энантиомер; где
n равно 0, 1 или 2;
a-b означает CF=CH, CHFCH2 или CF2CH2;
R1 представляет собой водород, гидроксиметил или C1-3алкил, где алкил является незамещенным или замещен одним-семью атомами фтора;
R2 представляет собой водород или C1-4алкил;
R3 выбран из
C1-4алкила,
(CH2)n-циклогетероалкила и
(CH2)n-арила, где арил выбран из
(1) фенила,
(2) нафтила,
(3) бензимидазолила,
(4) бензофуранила,
(5) бензотиофенила,
(6) бензоксазолила,
(7) бензотиазолила,
(8) бензодигидрофуранила,
(9) 1,3-бензодиоксолила,
(10) 2,3-дигидро-1,4-бензодиоксинила,
(11) индолила,
(12) хинолила,
(13) изохинолила,
(14) фуранила,
(15) тиенила,
(16) имидазолила,
(17) оксазолила,
(18) тиазолила,
(19) изоксазолила,
(20) изотиазолила,
(21) пиразолила,
(22) пирролила,
(23) пиридила,
(24) пиримидила,
(25) пиразинила,
(26) тиадиазолила,
(27) оксадиазолила,
(28) триазолила,
(29) тетразолила и
(30) инданила;
где алкильная группа или циклогетероалкильная группа является незамещенной или замещена одним-тремя заместителями, независимо выбранными из галогена, гидрокси и С1-4алкокси; арильная группа, определенная в пунктах (1)-(30), является незамещенной или замещена одной-тремя группами, независимо выбранными из галогена, фенила, С1-8алкила, С3-8циклоалкила, С3-8циклогетероалкила, фенил-С1-6алкила, амино-С0-6алкила, С1-6алкиламино-С0-6алкила, (C1-6алкил)2амино-C0-6алкила, фенил-C0-6алкиламино-С0-6алкила, (фенил-C0-6алкил)2амино-C0-6алкила, C1-6алкилтио, фенил-C0-6алкилтио, C1-6алкилсульфинила, фенил-C0-6алкилсульфинила, C1-6алкилсульфонила, фенил-C0-6алкилсульфонила, C1-6алкокси-C0-6алкила, фенил-C0-6алкокси-C0-6алкила, гидроксикарбонил-C0-6алкила, C1-6алкоксикарбонил-C0-6алкила, фенил-C0-6алкоксикарбонил-C0-6алкила, гидроксикарбонил-C1-6алкилокси, гидрокси-C0-6алкила, циано, нитро, перфтор-C1-4алкила, перфтор-C1-4алкокси, оксо, C1-6алкилкарбонилокси, фенил-C0-6алкилкарбонилокси, С1-6алкилкарбониламино, фенил-C0-6алкилкарбониламино, С1-6алкилсульфониламино, фенил-C0-6алкилсульфониламино, C1-6алкоксикарбониламино, фенил-C0-6алкоксикарбониламино, C1-6алкиламинокарбониламино, фенил-C0-6алкиламинокарбониламино, (C1-6алкил)2аминокарбониламино, (фенил-C0-6алкил)2аминокарбониламино, (C1-6алкил)2аминокарбонилокси и (фенил-C0-6алкил)2аминокарбонилокси; и где углеродный атом любого метилена (СН2) в (CH2)n является незамещенным или замещен одной-двумя группами, независимо выбранными из галогена, гидрокси и С1-4алкила; или два заместителя, когда они принадлежат одной и той же метиленовой (СН2) группе, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопропильную группу;
или R2 и R3 вместе образуют 5- или 6-членное насыщенное кольцо, конденсированное с 5- или 6-членной ароматической кольцевой системой, имеющей 0, 1 или 2 гетероатома, выбранных из N, O и S.
В одном варианте соединений настоящего изобретения R1 представляет собой водород или метил. В классе указанного выше варианта R1 представляет собой метил.
Во втором варианте соединений настоящего изобретения a-b означает CF=CH.
В третьем варианте соединений настоящего изобретения a-b означает CHFCH2.
В четвертом варианте соединений настоящего изобретения R2 представляет собой водород и R3 представляет собой (CH2)n-арил. В классе указанного выше варианта n равно 0 или 1.
В пятом варианте соединений настоящего изобретения R1 представляет собой метил, a-b означает CF=CH, R2 представляет собой водород и R3 представляет собой (CH2)n-арил. В классе указанного выше варианта n равно 0 или 1.
В шестом варианте соединений настоящего изобретения R1 представляет собой метил, a-b означает CHFCH2, R2 представляет собой водород и R3 представляет собой (CH2)n-арил. В классе указанного выше варианта n равно 0 или 1.
В другом варианте R1 выбран из водорода и метила, a-b выбран из CHFCH2, R2 представляет собой водород и R3 представляет собой (CH2)n-циклогетероалкил. В классе указанного выше варианта R1 представляет собой метил и a-b означает CF=CH.
В еще одном варианте осуществления данного изобретения R3 выбран из:
C1-4алкила и
(CH2)n-арила, где арил выбран из
(1) фенила,
(2) нафтила,
(3) бензимидазолила,
(4) бензофуранила,
(5) бензотиофенила,
(6) бензоксазолила,
(7) бензотиазолила,
(8) бензодигидрофуранила,
(9) 1,3-бензодиоксолила,
(10) 2,3-дигидро-1,4-бензодиоксинила,
(11) индолила,
(12) хинолила,
(13) изохинолила,
(14) фуранила,
(15) тиенила,
(16) имидазолила,
(17) оксазолила,
(18) тиазолила,
(19) изоксазолила,
(20) изотиазолила,
(21) пиразолила,
(22) пирролила,
(23) пиридила,
(24) пиримидила,
(25) пиразинила,
(26) тиадиазолила,
(27) оксадиазолила,
(28) триазолила,
(29) тетразолила и
(30) инданила;
где алкильная группа или циклогетероалкильная группа является незамещенной или замещена одним-тремя заместителями, независимо выбранными из галогена, гидрокси и С1-4алкокси; арильная группа, определенная в пунктах (1)-(30), является незамещенной или замещена одной-тремя группами, независимо выбранными из галогена, фенила, С1-8алкила, С3-8циклоалкила, С3-8циклогетероалкила, фенил-С1-6алкила, амино-С0-6алкила, С1-6алкиламино-С0-6алкила, (C1-6алкил)2амино-C0-6алкила, фенил-C0-6алкиламино-С0-6алкила, (фенил-C0-6алкил)2амино-C0-6алкила, C1-6алкилтио, фенил-C0-6алкилтио, C1-6алкилсульфинила, фенил-C0-6алкилсульфинила, C1-6алкилсульфонила, фенил-C0-6алкилсульфонила, C1-6алкокси-C0-6алкила, фенил-C0-6алкокси-C0-6алкила, гидроксикарбонил-C0-6алкила, C1-6алкоксикарбонил-C0-6алкила, фенил-C0-6алкоксикарбонил-C0-6алкила, гидроксикарбонил-C1-6алкилокси, гидрокси-C0-6алкила, циано, нитро, перфтор-C1-4алкила, перфтор-C1-4алкокси, оксо, C1-6алкилкарбонилокси, фенил-C0-6алкилкарбонилокси, С1-6алкилкарбониламино, фенил-C0-6алкилкарбониламино, С1-6алкилсульфониламино, фенил-C0-6алкилсульфониламино, C1-6алкоксикарбониламино, фенил-C0-6алкоксикарбониламино, C1-6алкиламинокарбониламино, фенил-C0-6алкиламинокарбониламино, (C1-6алкил)2аминокарбониламино, (фенил-C0-6алкил)2аминокарбониламино, (C1-6алкил)2аминокарбонилокси и (фенил-C0-6алкил)2аминокарбонилокси; и где углеродный атом любого метилена (СН2) в (CH2)n является незамещенным или замещен одной-двумя группами, независимо выбранными из галогена, гидрокси и С1-4алкила; или два заместителя, когда они принадлежат одной и той же метиленовой (СН2) группе, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопропильную группу.
Иллюстративными, но неограничивающими примерами соединений настоящего изобретения, которые полезны в качестве модуляторов рецептора андрогена, являются следующие:
N-(2,2,2-трифторэтил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(3-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2-трифторметилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2-хлорфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(4-метоксифенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(3-метоксифенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2-метилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(3-метилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(3-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(4-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(4-хлор-2-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2,4-дифторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(α-метилфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(фенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(4-хлор-2-трифторметилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(5-метилпиридин-2-ил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тиофен-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тиофен-3-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2-трифторметилфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(бензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(1-метилбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(1-метил-5-трифторметилбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(5-хлорбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(5-метоксибензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(бензтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2,3-дигидро-1,4-бензодиоксин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(4-метилтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тиазол-4-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(1-метилимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тетрагидро-2Н-пиран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тетрагидро-2H-пиран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2,3-дигидро-1,4-бензодиоксин-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2,3-дигидро-1,4-бензодиоксин-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тетрагидрофуран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(тетрагидрофуран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(3Н-имидазо[4,5-b]пиридин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(2-трифторметилфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(3-метоксифенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(4-метоксифенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(2-трифторметилфенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(2-хлорфенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(бензимидазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(1-метилбензимидазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(тиазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
N-(фуран-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид и
N-(тиофен-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид;
их фармацевтически приемлемые соли и их энантиомеры.
В еще одном варианте осуществления соединения настоящего изобретения выбраны из:
N-(2-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-хлорбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-метоксибензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(бензтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2Н-пиран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2H-пиран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3Н-имидазо[4,5-b]пиридин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(тиазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(фуран-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида и
N-(тиофен-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
В еще одном варианте соединения настоящего изобретения выбраны из:
N-(тетрагидро-2Н-пиран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2H-пиран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3H-имидазо[4,5-b]пиридин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
В одном варианте воплощения данного изобретения, соединения выбраны из:
N-(2-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-хлорбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-метоксибензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(бензтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(тиазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(фуран-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида и
N-(тиофен-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
Соединения настоящего изобретения могут иметь асимметрические центры, хиральные оси и хиральные плоскости (описанные в: E.L. Eliel and S.H. Wilen, Stereochemistry of Carbon Compounds, John Wiley & Sons, New York, 1994, pages 1119-1190) и встречаются в виде рацематов, рацемических смесей и в виде индивидуальных диастереомеров, причем все их возможные изомеры и их смеси, включая оптические изомеры, входят в объем настоящего изобретения. Кроме того, раскрытые в данном описании соединения могут существовать в виде таутомеров, и при этом подразумевается, что обе таутомерные формы охватываются объемом данного изобретения, даже если описана только одна таутомерная структура. Например, имеется в виду, что любой пункт, имеющий отношение к соединению А ниже, включает и таутомерную структуру В, и наоборот, а также смеси таутомерных форм. В данном описании Х представляет остаток фторированных производных 4-азастероида настоящего изобретения.
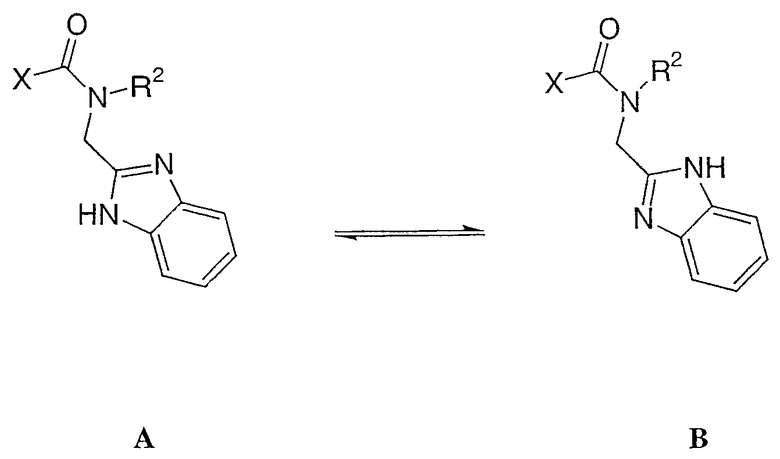
Используемый в данном описании термин «алкил» означает алканы с прямой или разветвленной цепью, содержащие от одного до десяти углеродных атомов или любое число углеродных атомов в пределах указанного диапазона (т.е. метил, этил, 1-пропил, 2-пропил, н-бутил, втор-бутил, трет-бутил и т.д.). Термин «C0алкил» (как и в «C0-8алкилариле») подразумевает отсутствие алкильной группы.
Термин «алкенил» означает алкены с прямой или разветвленной цепью, содержащие суммарно от двух до десяти углеродных атомов или любое число углеродных атомов в пределах указанного диапазона.
Термин «алкинил» означает алкины с прямой или разветвленной цепью, содержащие суммарно от двух до десяти углеродных атомов или любое число углеродных атомов в пределах указанного диапазона.
Термин «алкилиден» означает алкилиденовую группу с прямой или разветвленной цепью, содержащую суммарно от одного до десяти углеродных атомов или любое число углеродных атомов в пределах указанного диапазона.
Термин «циклоалкил» означает циклические кольца алканов, содержащие суммарно от трех до восьми углеродных атомов или любое число углеродных атомов в пределах указанного диапазона (т.е. циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил).
Используемый в данном описании термин «циклогетероалкил» означает (3-8)-членное полностью насыщенное гетероциклическое кольцо, содержащее один или два гетероатома, выбранных из N, O или S. Примеры циклогетероалкильных групп включают, но не ограничиваются ими, пиперидинил, пирролидинил, азетидинил, морфолинил, оксациклопентан, оксациклогексан и пиперазинил. В одном варианте осуществления настоящего изобретения циклогетероалкил выбран из пиперидинила, пирролидинила, оксациклопентана, оксациклогексана и морфолинила.
Используемый в данном описании термин «алкокси» относится к алкоксидам с прямой или разветвленной цепью, содержащим определенное число углеродных атомов (например, С1-5алкокси) или любое число углеродных атомов в пределах указанного диапазона (т.е. метокси, этокси и т.д.).
Используемый в данном описании термин «арил» относится к моноциклической или бициклической системе, включающей, по меньшей мере, одно ароматическое кольцо, причем эта моноциклическая или бициклическая система содержит 0, 1, 2, 3 или 4 гетероатома, выбранных из N, O или S, и эта моноциклическая или бициклическая система является либо незамещенной или замещена одной или несколькими группами, независимо выбранными из галогена, фенила, C1-8алкила, C3-8циклоалкила, C3-8циклогетероалкила, фенил-C1-6алкила, амино-C0-6алкила, C1-6алкиламино-C0-6алкила, (C1-6алкил)2амино-С0-6алкила, фенил-C0-6алкиламино-C0-6алкила, (фенил-C0-6алкил)2амино-C0-6алкила, C1-6алкилтио, фенил-C0-6алкилтио, C1-6алкилсульфинила, фенил-C0-6алкилсульфинила, C1-6алкилсульфонила, фенил-C0-6алкилсульфонила, C1-6алкокси-C0-6алкила, фенил-C0-6алкокси-C0-6алкила, гидроксикарбонил-C0-6алкила, C1-6алкоксикарбонил-C0-6алкила, фенил-C0-6алкоксикарбонил-C0-6алкила, гидроксикарбонилC1-6алкилокси, гидрокси-C0-6алкила, циано, нитро, перфторC1-4алкила, перфторС1-4алкокси, оксо, C1-6алкилкарбонилокси, фенил-C0-6алкилкарбонилокси, C1-6алкилкарбониламино, фенил-C0-6алкилкарбониламино, C1-6алкилсульфониламино, фенил-C0-6алкилсульфониламино, C1-6алкоксикарбониламино, фенил-C0-6алкоксикарбониламино, C1-6алкиламинокарбониламино, фенил-C0-6алкиламинокарбониламино, (C1-6алкил)2аминокарбониламино, (фенил-C0-6алкил)2аминокарбониламино, (C1-6алкил)2аминокарбонилокси и (фенил-C0-6алкил)2аминокарбонилокси. Предпочтительно, арильная группа является незамещенной, моно-, ди- или тризамещенной одним-тремя заместителями, выбранными из вышеназванных; более предпочтительно арильная группа является незамещенной, моно- или дизамещенной одним-двумя выбранными из вышеназванных заместителями.
Всякий раз, когда термин «алкил» или «арил» или любой из их корней появляется как префикс в названии заместителя (например, арил C0-8алкил), следует иметь в виду, что он включает все определенные выше ограничения для «алкила» и «арила». Обозначенное число углеродных атомов (например, С0-8) относится независимо к числу углеродных атомов в алкиле или циклоалкильной части, или к алкильной части большего заместителя, в котором алкил появляется в виде префикса.
Термины «арилалкил» и «алкиларил» включают алкильную часть, где алкил такой, как определено выше, и включают арильную часть, где арил такой, как определено выше. Примеры арилалкила включают, но не ограничиваются ими, бензил, фторбензил, хлорбензил, фенилэтил, фенилпропил, фторфенилэтил, хлорфенилэтил, тиенилметил, тиенилэтил и тиенилпропил. Примеры алкиларила включают, но не ограничиваются ими, толуол, этилбензол, пропилбензол, метилпиридин, этилпиридин, пропилпиридин и бутилпиридин.
Используемый в данном описании термин «галоген» включает йод, бром, хлор и фтор.
Термин «окси» означает атом кислорода (О). Термин «тио» означает атом серы (S). Термин «оксо» означает «=O». Термин «карбонил» означает «C=O».
Следует иметь в виду, что термин «замещенный» включает варианты многократного замещения названным заместителем. В тех случаях, когда множественные заместители описаны или заявлены, замещенное соединение может быть независимо замещено одним или несколькими из описанных или заявленных заместителей, однократно или многократно. Под независимо замещенным подразумевается, что заместители (два или несколько) могут быть одинаковыми или различными.
Когда любая переменная (например, R2, R3 и т.д.) встречается более чем один раз в любом заместителе или формуле I, ее определение в каждом случае является независимым от ее определения в каждом другом случае. Кроме того, комбинации заместителей и/или переменных допустимы только тогда, когда такие комбинации приводят к получению стабильным соединением.
Согласно стандартной химической номенклатуре, используемой на протяжении описания данного изобретения, сначала указывается концевая часть обозначенной боковой цепи, за которой следует соседняя функциональность по направлению к точке присоединения. Например, заместитель C1-5алкилкарбониламиноС1-6алкил, эквивалентен
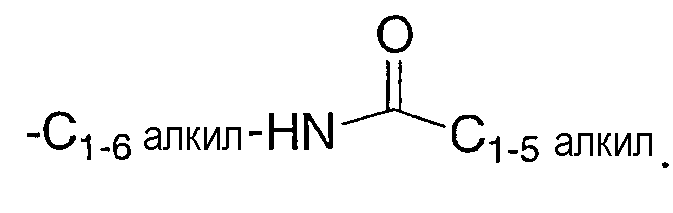
При выборе соединений настоящего изобретения для специалиста в данной области очевидно, что различные заместители, т.е. R1, R2, R3 и т.д., должны выбираться в соответствии с известными принципами связности химической структуры.
Соединения настоящего изобретения, как было установлено, являются тканеселективными модуляторами рецептора андрогена (SARM). В одном аспекте соединения настоящего изобретения могут быть использованы для активации функции рецептора андрогена у млекопитающего и, в частности, для активации функции рецептора андрогена в костной и/или мышечной ткани и блокирования или ингибирования («антагонизирования») функции рецептора андрогена в простате индивидуума мужского пола или матке индивидуума женского пола. Активацию AR в костной ткани можно анализировать через обнаружение стимуляции остеогенеза на модели остеопороза у грызунов, а антагонизм AR в простате можно анализировать наблюдением минимальных воздействий на рост простаты у кастрированных грызунов и антагонизм роста простаты, индуцированный агонистами AR, как это подробно описано в Примерах.
Другой аспект настоящего изобретения связан с соединениями структурной формулы I, которые блокируют функцию рецептора андрогена в простате индивидуума мужского пола или в матке индивидуума женского пола, индуцируемую агонистами AR, но не в волосистой части кожи или в голосовых связках, и активируют функцию рецептора андрогена в костной и/или мышечной ткани, но не в органах, которые контролируют уровни липидов в крови (например, печень).
Соединения настоящего изобретения могут быть использованы для лечения состояний, которые вызваны недостаточностью андрогена или интенсивность которых может быть уменьшена возмещением андрогена, включая, но не ограничиваясь ими, остеопороз, нарушение остеогенеза, глюкокортикоид-индуцированный остеопороз, периодонтальную болезнь, перелом (трещину) кости, повреждение кости после реконструктивной костной хирургии, саркопению, хрупкость, старение кожи, мужской гипогонадизм, постменопаузальные симптомы у женщин, атеросклероз, гиперхолестеринемию, гиперлипидемию, ожирение, апластическую анемию и другие гемопоэтические нарушения, артритные состояния, такие как, например, воспалительный артрит и восстановление сустава, ВИЧ-истощение, рак простаты, раковую кахексию, мышечную дистрофию, преждевременное угасание функции яичников и аутоиммунное заболевание, как таковыми или в комбинации с другими активными средствами. Лечение осуществляют введением млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения структурной формулы I. Кроме того, вышеуказанные соединения используют в качестве компонентов в фармацевтических композициях как таковые или в комбинации с другими активными средствами.
В одном варианте осуществления соединения настоящего изобретения могут быть использованы для лечения состояний у индивидуума мужского пола, которые вызваны недостаточностью андрогена или которые можно улучшить восполнением андрогена, включая, но не ограничиваясь ими, остеопороз, нарушение остеогенеза, глюкокортикоид-индуцированный остеопороз, периодонтальную болезнь, ВИЧ-истощение, рак простаты, раковую кахексию, ожирение, апластическую и другие анемии, и мышечные дистрофии, как таковыми или в комбинации с другими активными средствами. Лечение осуществляют введением терапевтически эффективного количества соединения структурной формулы I индивидууму мужского пола, нуждающемуся в таком лечении.
«Артритное состояние» или «артритные состояния» относятся к заболеванию, при котором воспаление локализуется в суставах, или к любым воспалительным состояниям суставов, чаше всего остеоартриту и ревматоидному артриту (Academic Press Dictionary of Science Technology; Academic Press; 1st edition, January 15, 1992). Соединения формулы I также полезны, как таковые или в комбинации, для лечения или профилактики артритных состояний, таких как болезнь Бехчета; бурсит и тендинит; CPPD deposition disease; запястный синдром; синдром Элерса-Данлоса; фибромиалгия; подагра; инфекционный артрит; воспалительное заболевание кишечника; ювенильный артрит; красная волчанка; болезнь Лайма; синдром Марфана; миозит; остеоартрит; несовершенный остеогенез; остеонекроз; полиартериит; полимиалгия ревматическая; псориатический артрит; феномен Рейно; рефлекс синдрома дистрофии симпатической нервной системы; синдром Рейтера; ревматоидный артрит; склеродерма и синдром Шегрена. Вариант осуществления данного изобретения охватывает лечение или профилактику артритного состояния, которые включают введение терапевтически эффективного количества соединения формулы I. Подвариант осуществления представляет собой лечение или профилактику остеоартрита, которые включают введение терапевтически эффективного количества соединения формулы I. См.: Cutolo M, Seriolo B, Villaggio B, Pizzorni C, Craviotto C, Sulli A. Ann. N.Y. Acad. Sci. 2002 Jun;966:131-42; Cutolo, M. Rheum Dis Clin North Am 2000 Nov; 26(4):881-95; Bijlsma JW, Van den Brink HR. Am J Reprod Immunol 1992 Oct-Dec;28(3-4):231-4; Jansson L, Holmdahl R.; Arthritis Rheum 2001 Sep; 44(9):2168-75; and Purdie DW. Br Med Bull 2000; 56(3):809-23. Также см. Merck Manual, 17th edition, pp. 449-451.
При использовании в комбинации для лечения артритных состояний соединения формулы I могут использоваться с любым из раскрытых в данном описании лекарственных средств, полезных для комбинированной терапии, или могут использоваться с другими, известными для лечения или профилактики артритных состояний лекарственными средствами, такими как кортикостероиды, цитотоксические лекарственные средства (или другие лекарственные средства, корректирующие болезнь или вызывающие ее ремиссию), лечение золотом, метотрексат, NSAID и ингибиторы СОХ-2.
В другом варианте осуществления соединения настоящего изобретения могут быть использованы для лечения состояний у индивидуума женского пола, которые вызваны недостаточностью андрогена или которые можно улучшить восполнением андрогена, включая, но не ограничиваясь ими, остеопороз, нарушение остеогенеза, глюкокортикоид-индуцированный остеопороз, постменопаузальные симптомы, периодонтальную болезнь, ВИЧ-истощение, раковую кахексию, ожирение, апластическую и другие анемии, мышечные дистрофии, преждевременное угасание функции яичников и аутоиммунное заболевание, как таковые или в комбинации с другими активными средствами. Лечение осуществляют введением терапевтически эффективного количества соединения структурной формулы I индивидууму женского пола, нуждающемуся в таком лечении.
Соединения структурной формулы I также можно использовать при лечении рака простаты в качестве вспомогательных лечебных средств при традиционной терапии, направленной на снижение андрогенов для восстановления кости, сведения к минимуму остеопороза и поддержания или повышения плотности минеральных компонентов кости. Таким образом, вышеуказанные соединения могут быть использованы вместе с традиционной терапией депривацией андрогенов, включая агонисты/антагонисты GnRH, такие как описано в P. Limonta, et al., "LHRH analogues as anticancer agents: pituitary and extrapituitary sites of action," Exp. Opin. Invest. Drugs, 10: 709-720 (2001); H.J. Strieker, "Luteinizing hormone-releasing hormone antagonists," Urology, 58 (Suppl. 2A): 24-27 (2001); R.P. Millar, et al., "Progress towards the development of non-peptide orally-active GnRH antagonists," British Medical Bulletin, 56: 761-772 (2000); and A.V. Schally et al., "Rational use of agonists and antagonists of LH-RH in the treatment of hormone-sensitive neoplasms and gynecologic conditions," Advanced Drug Delivery Reviews, 28: 157-169 (1997). Кроме того, возможно, что соединения структурной формулы I могут быть использованы в комбинации с антиандрогенами, такими как флутамид, 2-гидроксифлутамид (активный метаболит флутамида), нилутамид и бикалутамид (Casodex™), для лечения рака простаты.
Далее, соединения настоящего изобретения могут быть также использованы для лечения рака поджелудочной железы, либо из-за их свойств антагониста андрогена, либо как вспомогательное лечебное средство к антиандрогену, такому как флутамид, 2-гидроксифлутамид (активный метаболит флутамида), нилутамид и бикалутамид (Casodex™).
Соединения структурной формулы I оказывают минимальные негативные воздействия на липидный обмен. Поэтому, с учетом их тканеселективных свойств агониста андрогена, соединения данного изобретения имеют преимущества перед существующими подходами к заместительной гормональной терапии для индивидуумов мужского пола, страдающих от гипогонадизма (пониженная активность андрогена).
Дополнительно, соединения настоящего изобретения могут повышать число клеток крови, таких как эритроциты и тромбоциты, и могут быть использованы для лечения гемопоэтических нарушений, таких как апластическая анемия.
Типичные соединения настоящего изобретения обычно демонстрируют субмикромолярное связывающее сродство по отношению к рецептору андрогена. Поэтому соединения данного изобретения используют для лечения млекопитающих, страдающих от расстройств, связанных с функцией рецептора андрогена. Фармакологически эффективные количества соединения, включая их фармацевтически приемлемые соли, вводят млекопитающему для лечения расстройств, связанных с функцией рецептора андрогена или которые можно улучшить введением дополнительного андрогена, таких как остеопороз, периодонтальная болезнь, перелом (трещина) кости, повреждение кости после реконструктивной костной хирургии, саркопения, хрупкость, старение кожи, мужской гипогонадизм, постменопаузальные симптомы у женщин, атеросклероз, гиперхолестеринемия, гиперлипидемия, ожирение, апластическая анемия и другие гемопоэтические нарушения, рак поджелудочной железы, артритные состояния, например воспалительный артрит и восстановление сустава.
Как правило, предпочтительно вводить соединения настоящего изобретения в их энантиомерно чистой форме. Рацемические смеси можно разделить на их индивидуальные энантиомеры любым из целого ряда обычных методов. Такие методы включают хиральную хроматографию, получение функциональных производных с хиральным вспомогательным веществом с последующей хроматографией или кристаллизацией и фракционную кристаллизацию диастереомерных солей.
Как рассматривается в данном описании, соединение настоящего изобретения, которое функционирует как «агонист» рецептора андрогена, может связываться с рецептором андрогена и инициировать физиологический или фармакологический ответ, характерный для указанного рецептора. Термин «тканеселективный модулятор рецептора андрогена» относится к лиганду рецептора андрогена, который имитирует действие природного лиганда в некоторых тканях, но не в других. «Частичный агонист» представляет собой агонист, который не способен индуцировать максимальную активацию популяции рецептора, независимо от количества применяемого соединения. «Полный агонист» индуцирует полную активацию популяции рецептора андрогена при определенной концентрации. Соединение настоящего изобретения, которое функционирует как «антагонист» рецептора андрогена, может связываться с рецептором андрогена и блокировать или ингибировать андрогенассоциированные ответы, обычно индуцируемые природным лигандом рецептора андрогена.
Термин «фармацевтически приемлемые соли» относится к солям, получаемым из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Особенно предпочтительны соли аммония, кальция, лития, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиамины, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Когда соединение настоящего изобретения является основным, соли можно получить из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, муравьиную, фумаровую, глюконовую, глутаминовую, бромисто-водородную, хлористо-водородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, малоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, пропионовую, янтарную, серную, винную, п-толуолсульфоновую, трифторуксусную кислоты и т.п. Особенно предпочтительными являются лимонная, фумаровая, бромисто-водородная, хлористо-водородная, малеиновая, фосфорная, серная и винная кислоты.
Термин «терапевтически эффективное количество» означает количество соединения структурной формулы I, которое вызывает биологический или лечебный ответ ткани, системы, животного или человека, к которому стремится исследователь, ветеринар, лечащий врач или другой клиницист.
Используемый в данном описании термин «композиция», как предполагается, охватывает продукт, содержащий конкретные ингредиенты в заданных количествах, а также любой продукт, который является следствием, непосредственно или косвенно, комбинации конкретных ингредиентов в заранее определенных количествах.
Под термином «фармацевтически приемлемый» подразумевается, что носитель, разбавитель или эксципиент должен быть совместимым с другими ингредиентами, входящими в состав препарата, и не быть вредным для реципиента.
Следует иметь в виду, что термины «назначение (прием) соединения» и «введение соединения» означают обеспечение индивидуума, нуждающегося в лечении, соединением данного изобретения или пролекарством соединения данного изобретения.
Под термином «модуляция функции, опосредованной рецептором андрогена, тканеселективным способом» подразумевается модуляция функции, опосредованной рецептором андрогена, селективно (или отлично от других) в анаболической (костной и/или мышечной) ткани (костной или мышечной) в отсутствие такой модуляции в андрогенной (репродуктивной) ткани, такой как простата, яичко, семенные пузырьки, яичник, матка и другие придаточные ткани половых органов. В одном варианте осуществления активируется функция рецептора андрогена в анаболической ткани, тогда как функция рецептора андрогена в андрогенной ткани блокируется или подавляется.
Введение соединения структурной формулы I для осуществления на практике предлагаемых способов лечения выполняют введением эффективного количества соединения структурной формулы I пациенту, нуждающемуся в таком лечении или профилактике. Необходимость в профилактическом введении в соответствии со способами настоящего изобретения определяется с помощью использования известных факторов риска. Эффективное количество конкретного соединения определяется, в конечном счете, лечащим врачом, но зависит от таких факторов, как конкретное заболевание, подлежащее лечению, тяжесть заболевания и наличие других заболеваний или состояний, от которых страдает пациент; выбранный путь введения соединения, другие сопутствующие (одновременно принимаемые) лекарственные средства и методы лечения, в которых может нуждаться пациент, и другие факторы, принимаемые во внимание врачом.
Как правило, суточная доза соединения структурной формулы I для взрослого человека может варьироваться в широком диапазоне от 0,01 до 1000 мг в день. Наиболее предпочтительно дозы варьируются в пределах от 0,1 до 200 мг/день. В случае перорального введения предпочтительно предусматриваются композиции в форме таблеток, содержащих от 0,01 до 1000 мг, в частности, 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 3,0, 5,0, 6,0, 10,0, 15,0, 25,0, 50,0, 75, 100, 125, 150, 175, 180, 200, 225 и 500 миллиграмм активного ингредиента для симптоматической корректировки дозы для млекопитающего, подлежащего лечению.
Доза может быть введена в виде разовой суточной дозы, или общую суточную дозу можно вводить в виде разделенных доз два, три или четыре раза в день. Кроме того, исходя из свойств конкретного соединения, выбранного для введения, дозу можно вводить не так часто, например еженедельно, раз в две недели, раз в месяц и т.д. Конечно, в случае менее частого введения разовая доза должна быть соответственно больше.
При введении интраназальными путями, трансдермальными путями, посредством ректальных или вагинальных суппозиториев или при помощи внутривенного раствора, введение дозы должно быть, конечно, непрерывным, а не периодическим, на всем протяжении схемы приема лекарственного средства.
Иллюстрацию изобретения представляет фармацевтическая композиция, содержащая любое из соединений, описанных выше, и фармацевтически приемлемый носитель. Кроме того, иллюстрацией изобретения является фармацевтическая композиция, полученная объединением любого из соединений, описанных выше, и фармацевтически приемлемого носителя. Иллюстрацией изобретения является способ получения фармацевтической композиции, включающий объединение любого из соединений, описанных выше, и фармацевтически приемлемого носителя.
Препараты тканеселективного модулятора рецептора андрогена, используемые в настоящем способе в лечебных целях, включают соединение структурной формулы I вместе с приемлемым для него носителем и, необязательно, другие терапевтически активные ингредиенты. Носитель должен быть фармацевтически приемлемым в том смысле, что он должен быть совместимым с другими ингредиентами препарата и не быть вредным для реципиента, подвергаемого воздействию препарата.
Поэтому настоящее изобретение далее предлагает фармацевтический препарат, содержащий соединение структурной формулы I вместе с его фармацевтически приемлемым носителем.
Препараты включают препараты, подходящие для перорального, ректального, внутривагинального, местного или парентерального (включая подкожное, внутримышечное и внутривенное) введения. Предпочтительными препаратами являются препараты, подходящие для перорального введения.
Препараты могут быть представлены в дозированной лекарственной форме, и их можно получить любым из способов, известных в области фармации. Все способы включают стадию смешения активного соединения с носителем, который составляет один или несколько ингредиентов. В общем, препараты получают однородным и тщательным смешением активного соединения с жидким носителем, восковым твердым носителем или тонкоизмельченным твердым носителем, а затем, если требуется, формованием полученного продукта в желательную дозированную форму.
Препараты, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, саше, таблетки или леденцы, причем каждая содержит заранее заданное количество активного соединения; в виде порошка или гранул; либо суспензии, либо раствора в водной жидкостиили неводной жидкости, например сиропа, эликсира или эмульсии.
Таблетку можно получить прессованием или формованием, необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки можно получить, сжимая в подходящей машине активное соединение в свободно текучей форме, например в виде порошка или гранул, необязательно смешанное со вспомогательными ингредиентами, такими как, например, связующие вещества, смазки, инертные разбавители, дезинтегрирующие средства или красители. Формованные таблетки можно получить формованием в подходящей машине смеси активного соединения, предпочтительно в порошковой форме, с подходящим носителем. Подходящие связующие вещества включают, без ограничения, крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подслащивающие вещества, природные и синтетические камеди, такие как аравийская камедь, трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и т.п. Смазки, используемые в таких лекарственных формах, включают, без ограничения, олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Дезинтегрирующие средства включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п.
Пероральные жидкие формы, такие как сиропы или суспензии в соответствующим образом ароматизированных суспендирующих или диспергирующих средствах, таких как синтетические и природные камеди, например трагакант, аравийская камедь, метилцеллюлоза и т.п., могут быть получены добавлением активного соединения в раствор или суспензию. Дополнительные диспергирующие средства, которые могут быть использованы, включают глицерин и т.п.
Препараты для вагинального или ректального введения могут быть представлены в виде суппозитория с обычным носителем, т.е. основой, которая нетоксична и не вызывает раздражение слизистых оболочек, совместима с соединением структурной формулы I и стабильна при хранении и не связывает или не препятствует высвобождению соединения структурной формулы I. Подходящие основы включают: масло какао (теоброминовое масло), полиэтиленгликоли (такие как карбовоск и полигликоли), комбинации гликоль-поверхностно-активное вещество, полиоксил 40 стеарат, эфиры полиоксиэтилированного сорбитана и жирных кислот (такие как Tween, Myrj и Arlacel), глицеринизированный желатин и гидрогенизированные растительные масла. Когда используют суппозитории на основе глицеринизированного желатина, может быть использован консервант, такой как метилпарабен или пропилпарабен.
Препараты местного применения, содержащие активный лекарственный компонент, могут быть получены смешением активного компонента с рядом веществ-носителей, известных в данной области, таких как, например, спирты, гель из aloe vera, аллантоин, глицерин, масла витамина А и Е, минеральное масло, PPG2 миристилпропионат и т.п., с получением, например, спиртовых растворов, очищающих средств местного применения, очищающих кремов, гелей для кожи, лосьонов для кожи и шампуней в составах крема или геля.
Соединения настоящего изобретения можно также вводить в форме липосомных систем доставки, таких как маленькие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из ряда фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Соединения настоящего изобретения могут быть также доставлены с использованием моноклональных антител в качестве отдельных носителей, к которым присоединяются молекулы соединения. Соединения настоящего изобретения также можно сочетать с растворимыми полимерами в качестве носителей, способных целенаправленно доставлять лекарственные средства. Такие полимеры включают поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксид полилизин, замещенный пальмитоильными остатками. Кроме того, соединения настоящего изобретения можно сочетать с классом биоразлагаемых полимеров, используемых для достижения контролируемого высвобождения лекарственного средства, например полимер молочной кислоты, поли-эпсилон-капролактон, полимер оксимасляной кислоты, сложные полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатические блоксополимеры гидрогелей.
Препараты, подходящие для парентерального введения, охватывают препараты, которые включают стерильный водный препарат активного соединения, который предпочтительно является изотоническим по отношению к крови реципиента. Такие препараты соответственно включают раствор или суспензию соединения, которые являются изотоническими по отношению к крови реципиента. Такие препараты могут содержать дистиллированную воду, 5% декстрозы в дистиллированной воде или физиологическом растворе и активное соединение. Часто полезно использовать фармацевтически и фармакологически приемлемую аддитивную соль кислоты активного соединения, которая имеет соответствующую растворимость в используемых растворителях. Применяемые препараты также охватывают концентрированные растворы или твердые вещества, содержащие активное соединение, которые при разбавлении соответствующим растворителем дают раствор, подходящий для парентерального введения.
Соединения настоящего изобретения можно сочетать с классом биоразлагаемых полимеров, используемых для достижения контролируемого высвобождения лекарственного средства, например полимер молочной кислоты, поли-эпсилон-капролактон, полимер оксимасляной кислоты, сложные полиортоэфиры, полиацетали, полидигидропираны, полицианоакрилаты и сшитые или амфипатические блоксополимеры гидрогелей.
Фармацевтическая композиция и способ настоящего изобретения могут дополнительно включать другие терапевтически активные соединения, обычно применяемые для лечения вышеуказанных состояний, включая остеопороз, периодонтальную болезнь, перелом (трещину) кости, повреждение кости после реконструктивной костной хирургии, саркопению, хрупкость, старение кожи, мужской гипогонадизм, постменопаузальные симптомы у женщин, атеросклероз, гиперхолестеринемию, гиперлипидемию, ожирение, апластическую анемию и другие гемопоэтические нарушения, рак поджелудочной железы, артритные состояния, такие как воспалительный артрит и регенерация пораженного сустава.
Для лечения и профилактики остеопороза соединения настоящего изобретения можно вводить в комбинации с укрепляющим костную ткань средством, выбранным из антирезорбционных средств, остеоанаболических средств и других средств, оказывающих благоприятное воздействие на скелет через механизмы, которые точно не установлены, таких как кальциевые добавки, флавоноиды и аналоги витамина D. Такое комбинированное лечение может также оказать благоприятное воздействие на такие состояния, как периодонтальная болезнь, перелом (трещину) кости и повреждение кости после реконструктивной костной хирургии. Например, соединения настоящего изобретения можно эффективно вводить в комбинации с эффективными количествами других средств, таких как эстрогены, бисфосфонаты, SERM, ингибиторы катепсина К, антагонисты рецептора интегрина αvβ3, ингибиторы вакуолярной АТФазы, полипептидный остеопротегерин, антагонисты VEGF, тиазолидиндионы, кальцитонин, ингибиторы протеинкиназы, паратиреоидный гормон (PTH) и его аналоги, антагонисты рецептора кальция; средства, усиливающие секрецию гормона роста; высвобождающий гормон гормона роста, инсулиноподобный фактор роста, костный морфогенетический белок (BMP), ингибиторы антагонизма BMP, производные простагландина, факторы роста фибробласта, витамин D и его производные, витамин K и его производные, соевые изофлавоны, соли кальция и фторидные соли. Состояния периодонтальной болезни, перелома (трещины) кости и повреждения кости после реконструктивной костной хирургии могут также улучшиться от таких комбинированных методов лечения. В одном варианте осуществления соединение настоящего изобретения может быть эффективно введено в комбинации с эффективным количеством укрепляющего костную ткань средства, выбранного из эстрогена или производного эстрогена, одного или в комбинации с прогестином или производным прогестина; бисфосфонатом; антиэстрогеном или селективным модулятором рецептора эстрогена; антагонистом рецептора интегрина αvβ3; ингибитором катепсина К; ингибитором вакуолярной АТФазы (аденозинтрифосфатаза) остеокласта; кальцитонином и остеопротегерином.
При лечении остеопороза активность соединений настоящего изобретения отличается от активности антирезорбционных средств: эстрогены, бисфосфонаты, SERM, кальцитонин, ингибиторы катепсина К, ингибиторы вакуолярной АТФазы; средства, препятствующие пути RANK/RANKL/остеопротегерин, ингибиторы р38 или любые другие ингибиторы генерации остеокластов или активации остеокластов. Соединения структурной формулы I скорее стимулируют остеогенез, чем ингибируют резорбцию костной ткани, действуя предпочтительно на кортикальный слой кости, который ответственен за существенную часть прочности кости. Уплотнение кортикального слоя кости существенно способствует снижению риска перелома (трещины), особенно переломов бедра. Комбинация тканеселективных модуляторов рецептора андрогена структурной формулы I с антирезорбционными средствами, такими как эстроген, бисфосфонаты, антиэстрогены, SERM, кальцитонин, антагонисты рецептора интегрина αvβ3; ингибиторы редуктазы HMG-CoA, ингибиторы вакуолярной АТФазы и ингибиторы катепсина К, особенно полезна из-за комплементарности анаболических и антирезорбционных действий на костную ткань.
Антирезорбционные средства представляют собой средства, которые известны в данной области для ингибирования резорбции кости, и включают, например, эстроген и производные эстрогена, которые включают стероидные соединения, обладающие эстрогенной активностью, такие как, например, 17β-эстрадиол, эстрон, сопряженные эстрогены (PREMARIN®), конский эстроген, 17β-этинилэстрадиол и т.п. Эстроген или производное эстрогена может быть использовано одно или в комбинации с прогестином или производным прогестина. Неограничивающими примерами производных прогестина являются норетиндрон и медроксипрогестерон-ацетат.
Бисфосфонаты также являются антирезорбционными средствами. Бисфосфонатные соединения, которые могут быть также использованы в комбинации с соединением структурной формулы I по данному изобретению, включают:
(а) алендроновую кислоту: (4-амино-1-гидроксибутилиден)бисфосфоновая кислота;
(b) алендронат (также известный как натрий алендронат или мононатрий тригидрат): (4-амино-1-гидроксибутилиден)бисфосфонат мононатрий тригидрат (алендроновая кислота и алендронат описаны в патентах США 4922007, Kieczykowski et al., выданном 1 мая 1990; и 5019651, Kieczykowski, выданном 28 мая 1991, описания которых включены в настоящее описание в полном объеме в виде ссылок);
(c) [(циклогептиламино)метилен]бисфосфонат (инкадронат), который описан в патенте США 4970335, Isomura et al., выданном 13 ноября 1990, который включен в настоящее описание в полном объеме в виде ссылки;
(d) (дихлорметилен)бисфосфоновая кислота (клодроновая кислота) и динатриевая соль (клодронат), которые описаны в патенте Бельгии 672205 (1966) и J. Org. Chem 32, 4111 (1967), содержание которых включено в настоящее описание в полном объеме в виде ссылок;
(е) [1-гидрокси-3-(1-пирролидинил)пропилиден]бисфосфонат (EB-1053);
(f) (1-гидроксиэтилиден)бисфосфонат (этидронат);
(g) [1-гидрокси-3-(метилпентиламино)пропилиден]бисфосфонат (ибандронат), который описан в патенте США № 4927814, выданном 22 мая 1990, который включен в настоящее описание в полном объеме в виде ссылки;
(h) (6-амино-1-гидроксигексилиден)бисфосфонат (неридронат);
(i) [3-(диметиламино)-1-гидроксипропилиден]бисфосфонат (олпадронат);
(j) (3-амино-1-гидроксипропилиден)бисфосфонат (памидронат);
(k) [2-(2-пиридинил)этилиден]бисфосфонат (пиридронат), описанный в патенте США № 4761406, который включен в настоящее описание в полном объеме в виде ссылки;
(l) [1-гидрокси-2-(3-пиридинил)этилиден]бисфосфонат (ризедронат);
(m) {[(4-хлорфенил)тио]метилен}бсфосфонат (тилудронат), описанный в патенте США 4876248, Breliere et al., 24 октября 1989, который включен в настоящее описание в полном объеме в виде ссылки;
(n) [1-гидрокси-2-(1H-имидазол-1-ил)этилиден]бисфосфонат (цоледронат) и
(o) [1-гидрокси-2-имидазопиридин-(1,2-a)-3-илэтилиден]бисфосфонат (минодронат).
В одном варианте способов и композиций настоящего изобретения бисфосфонат выбирают из алендроната, клодроната, этидроната, ибандроната, инкадроната, минодроната, неридроната, олпадроната, памидроната, пиридроната, ризедроната, тилудроната и цоледроната и их фармацевтически приемлемых солей и их смесей. В классе указанного варианта осуществления бисфосфонат выбирают из алендроната, ризедроната, цоледроната, ибандроната, тилудроната и клодроната. В подклассе вышеуказанного класса бисфосфонат представляет собой алендронат, его фармацевтически приемлемые соли и его гидраты, и их смеси. Конкретной фармацевтически приемлемой солью алендроната является мононатрий алендронат. Фармацевтически приемлемые гидраты мононатрий алендроната включают моногидрат и тригидрат. Конкретной фармацевтически приемлемой солью ризедроната является мононатрий ризедронат. Фармацевтически приемлемые гидраты мононатрий ризедроната включают гемипентагидрат.
Используемый на всем протяжении описания и формулы изобретения термин «алендроновая кислота» включает родственные формы бисфосфоновых кислот, фармацевтически приемлемые солевые формы и равновесные смеси вышеуказанных соединений. Он включает в себя кристаллические, кристаллогидратные и аморфные формы алендроновой кислоты и ее фармацевтически приемлемых солей. Он, в частности, включает безводный мононатрий алендронат, моногидрат мононатрий алендронат и тригидрат мононатрий алендронат.
Далее, антиэстрогенные соединения, такие как ралоксифен (см., например, патент № 5393763), кломифен, цукломифен, энкломифен, нафоксиден, CI-680, CI-628, CN-55945-27, Mer-25, U-11555A, U-100A и их соли, и т.п. (см., например, патенты США №№ 4729999 и 4894373), могут быть использованы в комбинации с соединением структурной формулы I в способах и комбинациях по данному изобретению. Указанные средства также известны как SERM или селективные модуляторы рецептора эстрогена, средства, которые, как известно в данной области, предотвращают остеопороз, ингибируя резорбцию кости путями, которые, как считают, аналогичны путям эстрогена. Такие средства могут быть использованы в комбинации с соединениями настоящего изобретения для успешноголечения нарушений костной ткани, включая остеопороз. Указанные средства включают, например, тамоксифен, ралоксифен, лазофоксифен, торемифен, азорксифен, EM-800, EM-652, TSE 424, кломифен, дролоксифен, идоксифен и левормелоксифен [Goldstein, et al., "A pharmacological review of selective estrogen receptor modulators," Human Reproduction Update, 6: 212-224 (2000), и Lufkin, et al., "The role of selective estrogen receptor modulators in the prevention and treatment of osteoporosis, "Rheumatic Disease Clinics of North America, 27: 163-185 (2001)]. SERM также обсуждаются в "Targeting the Estrogen Receptor with SERMs," Ann. Rep. Med. Chem. 36: 149-158 (2001).
Антагонисты рецептора интегрина αvβ3 подавляют резорбцию кости и могут быть использованы в комбинации с тканеселективными модуляторами рецептора андрогена структурной формулы I для лечения нарушений кости, включая остеопороз. Пептидилированные, а также пептидомиметитические антагонисты рецептора интегрина αvβ3 были описаны как в научной, так и патентной литературе. Например, упоминается W.J. Hoekstra and B.L. Poulter, Curr. Med. Chem. 5: 195-204 (1998) и материалы, использованные при экспертизе заявки; WO 95/32710; WO 95/37655; WO 97/01540; WO 97/37655; WO 98/08840; WO 98/18460; WO 98/18461; WO 98/25892; WO 98/31359; WO 98/30542; WO 99/15506; WO 99/15507; WO 00/03973; EP 853084; EP 854140; EP 854145; Патенты США №№ 5204350; 5217994; 5639754; 5741796; 5780426; 5929120; 5952341; 6017925 и 6048861. Были представлены данные о способности антагонистов рецептора интегрина αvβ3 предотвращать резорбцию кости in vitro и in vivo (см. V.W. Engleman et al., "A Peptidomimetic Antagonist of the αvβ3 Integrin Inhibits Bone Resorption In Vitro and Prevents Osteoporosis In Vivo," J. Clin. Invest. 99: 2284-2292 (1997); S.B. Rodan et al., "A High Affinity Non-Peptide αvβ3 Ligand Inhibits Osteoclast Activity In Vitro and In Vivo," J. Bone Miner. Res. 11: S289 (1996); J.F. Gourvest et al., "Prevention of OVX-Induced Bone Loss With a Non-peptidic Ligand of the αvβ3 Vitronectin Receptor," Bone 23: S612 (1998); M.W. Lark et al., "An Orally Active Vitronectin Receptor αvβ3 Antagonist Prevents Bone Resorption In Vitro and In Vivo in the Ovariectomized Rat," Bone 23: S219 (1998)). Другие антагонисты αvβ3 описаны в R.M. Keenan et al., "Discovery of Potent Nonpeptide Vitronectin Receptor (αvβ3) Antagonists," J. Med. Chem. 40: 2289-2292 (1997); R.M. Keenan et al., "Benzimidazole Derivatives As Arginine Mimetics in 1,4-Benzodiazepine Nonpeptide Vitronectin Receptor (αvβ3) Antagonists," Bioorg. Med. Chem. Lett. 8: 3165-3170 (1998); и R.M. Keenan et al., "Discovery of an Imidazopyridine-Containing 1,4-Benzodiazepine Nonpeptide Vitronectin Receptor (αvβ3) Antagonist With Efficacy in a Restenosis Model," Bioorg. Med. Chem. Lett. 8: 3171-3176 (1998). Другие антагонисты рецептора интегрина αvβ3, молекулы которых содержат бензазепин, бензодиазепин и бензоциклогептен, описаны в нижеследующих патентных публикациях: WO 96/00574, WO 96/00730, WO 96/06087, WO 96/26190, WO 97/24119, WO 97/24122, WO 97/24124, WO 98/14192, WO 98/15278, WO 99/05107, WO 99/06049, WO 99/15170, WO 99/15178, WO 99/15506, патент США № 6159964 и WO 97/34865. Антагонисты рецептора интегрина αvβ3, молекулы которых содержат фрагменты дибензоциклогептена, дибензоциклогептана и дибензоксазепина, были описаны в WO 97/01540, WO 98/30542, WO 99/11626, WO 99/15508, WO 00/33838, патентах США №№ 6008213 и 6069158. Другие антагонисты рецептора интегрина остеокластов, молекулы которых имеют конформационные циклические ограничения, были описаны в патентной литературе. Опубликованные патентные заявки или выданные патенты, описывающие антагонисты, молекулы которых имеютограничение за счет фенила, включаютWO 98/00395, WO 99/32457, WO 99/37621, WO 99/44994, WO 99/45927, WO 99/52872, WO 99/52879, WO 99/52896, WO 00/06169, EP 0820988, EP 0820991, патенты США №№ 5741796; 5773644; 5773646; 5843906; 5852210; 5929120; 5952381; 6028223 и 6040311. Опубликованные патентные заявки или выданные патенты, описывающие антагонисты, имеющие ограничение благодаря наличию моноциклической структуры, включают WO 99/26945, WO 99/30709, WO 99/30713, WO 99/31099, WO 99/59992, WO 00/00486, WO 00/09503, EP 0796855, EP 0928790, EP 0928793, патенты США №№ 5710159; 5723480; 5981546; 6017926 и 6066648. Опубликованные патентные заявки или выданные патенты, раскрывающие антагонисты, имеющие ограничение благодаря наличию бициклической структуры, включают WO 98/23608, WO 98/35949, WO 99/33798, EP 0853084, патенты США №№ 5760028; 5919792 и 5925655. В качестве дополнительной научной и патентной литературы также ссылаются на нижеследующие обзоры, в которых рассматриваются антагонисты интегрина альфа v: M. E. Duggan, et al, "Ligands to the integrin receptor αvβ3, Exp. Opin. Ther. Patents, 10: 1367-1383 (2000); M. Gowen, et al., "Emerging therapies for osteoporosis," Emerging Drugs, 5: 1-43 (2000); J.S. Kerr, et al., "Small molecule αv integrin antagonists: novel anticancer agents," Exp. Opin. Invest. Drugs, 9: 1271-1291 (2000); and W.H. Miller, et al., "Identification and in vivo efficacy of small-molecule antagonists of integrin αvβ3 (the vitronectin receptor)," Drug Discovery Today, 5: 397-408 (2000).
Катепсин K, прежде известный как катепсин O2, представляет цистеиновую протеазу и описан в публикации международной заявки PCT WO 96/13523, опубликованной 9 мая 1996; патенте США № 5501969, выданном 3 марта 1996; и патенте США № 5736357, выданном 7 апреля 1998, описания которых включены в настоящее описание в полном объеме в виде ссылок. Цистеиновые протеазы, в частности катепсины, связаны с рядом состояний заболеваний, таких как метастазирование опухоли, воспаление, артрит и ремоделирование костной ткани. При кислых рН катепсины могут разрушать коллаген типа-I. Ингибиторы катепсиновых протеаз могут ингибировать разрушение остеокластов костной ткани, ингибируя деградацию волокон коллагена, и поэтому являются полезными при лечении болезней резорбции костной ткани, таких как остеопороз.
Представители класса ингибиторов редуктазы HMG-CoA, известные как «статины», как было установлено, инициируют рост новой костной ткани, замещая костную массу, потерянную в результате остеопороза (См. The Wall Street Journal, Friday, December 3, 1999, page Bl). Поэтому статины имеют перспективу для лечения резорбции кости. Примеры ингибиторов редуктазы HMG-CoA включают статины в их лактонизированных формах или дигидроксильных раскрытых кислотных формах и их фармацевтически приемлемые соли и их сложные эфиры, включая, но не ограничиваясь ими, ловастатин (см. патент США № 4342767); симвастатин (см. патент США № 4444784); симвастатин в дигидроксильной раскрытой кислотной форме, в частности его аммонийной или кальциевой соли; правастатин, в частности, его натриевую соль (см. патент США № 4346227); флувастатин, в частности его натриевую соль (см. патент США № 5354772); аторвастатин, в частности его кальциевую соль (см. патент США № 5273995); церивастатин, в частности его натриевую соль (см. патент США № 5177080), росувастатин, также известный как ZD-4522 (см. патент США № 5260440) и питавастатин, также упоминаемый как NK-104, итавастатин или нисвастатин (см. публикацию международной заявки PCT WO 97/23200).
Ингибиторы вакуолярной АТФазы остеокласта, также называемые ингибиторами протонного насоса, могут быть также использованы вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. Протонная АТФаза, которая обнаружена на апикальной мембране остеокласта, как сообщалось, играет значительную роль в процессе резорбции кости. Поэтому указанный протонный насос представляет заманчивую мишень для разработки ингибиторов резорбции кости, которые являются потенциально полезными для лечения или профилактики остеопороза и родственных метаболических заболеваний [см. C. Farina et al., "Selective inhibitors of the osteoclast vacuolar proton ATPase as novel bone antiresorptive agents," DDT, 4: 163-172 (1999)].
Ангиогенный фактор VEGF, как было показано, стимулирует костно-резорбирующую активность выделенных остеокластов зрелого кролика посредством связывания с его рецепторами на остеокластах [см. M. Nakagawa et al., "Vascular endothelial growth factor (VEGF) directly enhances osteoclastic bone resorption and survival of mature osteoclasts," FEBS Letters, 473: 161-164 (2000)]. Поэтому разработка антагонистов связывания VEGF с рецепторами остеокластов, таких как KDR/Flk-1 и Flt-1, может обеспечивать дополнительный подход к лечению или профилактике резорбции кости.
Активаторы рецепторов-γ, активируемых пероксисомальным пролифератором (PPARγ), такие как тиазолидиндионы (TZD), ингибируют образование остеокластподобных клеток и резорбцию кости in vitro. Результаты, сообщаемые R. Okazaki et al. в Endocrinology, 140: 5060-5065 (1999), указывают на локальный механизм на уровне клеток костного мозга, а также системный механизм на уровне метаболизма глюкозы. Неограничивающие примеры активаторов PPARγ включают глитазоны, такие как троглитазон, пиоглитазон, росиглитазон, и BRL 49653.
Кальцитонин можно также использовать вместе с тканеселективным модулятором рецептора андрогена структурной формулы I. Предпочтительно используют лососевый кальцитонин в виде назального аэрозоля (Azra et al., Calcitonin. 1996. In: J. P. Bilezikian, et al., Ed., Principles of Bone Biology, San Diego: Academic Press; and Silverman, "Calcitonin," Rheumatic Disease Clinics of North America, 27: 187-196, 2001).
Ингибиторы протеинкиназы можно также использовать вместе с тканеселективными модуляторами структурной формулы I. Ингибиторы киназы включают ингибиторы, описанные в WO 01/17562, и в одном варианте осуществления они выбраны из ингибиторов р38. Отдельные варианты осуществления ингибиторов р38, используемые в настоящем изобретении, включают SB 203580 [Badger et al., "Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock, and immune function," J. Pharmacol. Exp. Ther., 279: 1453-1461 (1996)].
Остеоанаболические средства представляют собой средства, которые, как известно в данной области, участвуют в построении костной ткани путем увеличения продуцирования костного белкового матрикса. Указанные остеоанаболические средства включают, например, различные формы паратиреоидного гормона (PTH), такие как PTH (1-84), PTH (1-34) природного происхождения, его аналоги, природные или с замещениями, и, в частности, подкожную инъекцию паратиреоидного гормона. PTH, как было установлено, повышает активность остеобластов, клеток, которые образуют костную ткань, тем самым стимулируя синтез новой костной ткани (Modern Drug Discovery, Vol. 3, No. 8, 2000). В исследованиях, сообщенных на Первом Всемирном Конгрессе по остеопорозу, проведенном в Чикаго в июне 2000г, женщины, подвергнутые комбинированному лечению PTH-эстроген, демонстрировали увеличение позвоночной костной массы на 12,8% и увеличение общей массы бедра на 4,4%. Другое исследование, представленное на том же конгрессе, показало, что PTH может способствовать увеличению объема кости, а также плотности кости. Клиническое испытание воздействия фрагмента (1-34) человеческого паратиреоидного гормона [hPTH(1-34)] на женщин в постменопаузальном состоянии, страдающих остеопорозом, привело к ≥65% снижению возникновения переломов (трещин) позвоночника и к 54% снижению возникновения переломов (трещин) в областях, не относящихся к позвонкам, после, в среднем, 22 месяцев лечения [см. J.M. Hock, Bone, 27: 467-469 (2000) and S. Mohan, et al., Bone, 27: 471-478 (2000), и материалы, использованные при экспертизе заявки]. Таким образом, можно считать, что PTH и его фрагменты, такие как hPTH(1-34), эффективны при лечении остеопороза, сами по себе или в комбинации с другими средствами, такими как тканеселективные модуляторы рецептора андрогена по данному изобретению. Инъецируемая рекомбинантная форма человеческого PTH, Forteo (терипаратид), получила законное одобрение в США для лечения остеопороза.
Также в комбинации с SARM по данному изобретению используют антагонисты рецептора кальция, которые индуцируют секрецию PTH, как это описано Gowen et al. в "Antagonizing the parathyroid calcium receptor stimulates parathyroid hormone secretion and bone formation in osteopenic rats," J. Clin. Invest. 105: 1595-604 (2000).
Средства, стимулирующие секрецию гормона роста; гормон роста, высвобождающий гормон гормона роста и т.п., также представляют собой остеоанаболические средства, которые могут быть использованы с соединениями структурной формулы I для лечения остеопороза. Типичные средства, стимулирующие секрецию гормона роста, описаны в патенте США № 3239345; патенте США № 4036979; патенте США № 4411890; патенте США № 5206235; патенте США № 5283241; патенте США № 5284841; патенте США № 5310737; патенте США № 5317017; патенте США № 5374721; патенте США № 5430144; патенте США № 5434261; патенте США № 5438136; патенте США № 5494919; патенте США № 5494920; патенте США № 5492916; патенте США № 5536716; патентных публикациях EPO 0144230; 0513974; патентных публикациях PCT WO 94/07486; WO 94/08583; WO 94/11012; WO 94/13696; WO 94/19367; WO 95/03289; WO 95/03290; WO 95/09633; WO 95/11029; WO 95/12598; WO 95/13069; WO 95/14666; WO 95/16675; WO 95/16692; WO 95/17422; WO 95/17423; WO 95/34311; 96/02530; Science, 260, 1640-1643 (June 11, 1993); Ann. Rep. Med. Chem.. 28: 177-186 (1993); Bioorg. Med. Chem. Lett., 4: 2709-2714 (1994); and Proc. Natl. Acad. Sci. USA, 92: 7001-7005 (1995).
Инсулиноподобный фактор роста (IGF) может быть также использован вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. Инсулиноподобные факторы роста могут быть выбраны из инсулиноподобного фактора роста I, одного или в комбинации с белком 3, связывающим IGF, и IGF II [См. Johannson and Rosen, "The IGFs as potential therapy for metabolic bone diseases," 1996, In: Bilezikian, et al., Ed., Principles of Bone Biology, San Diego: Academic Press; and Ghiron et al., "Effects of recombinant insulin-like growth factor-I and growth hormone on bone turnover in elderly women," J. Bone Miner. Res. 10: 1844-1852 (1995)].
Костный морфогенетический белок (BMP) может быть также использован вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. Костный морфогенетический белок включает BMP 2, 3, 5, 6, 7, а также родственные молекулы TGF бета и GDF 5 [Rosen et al., "Bone morphogenetic proteins," 1996. In: J. P. Bilezikian, et al., Ed., Principles of Bone Biology, San Diego: Academic Press; and Wang EA, "Bone morphogenetic proteins (BMPs): therapeutic potential in healing bony defects," Trends Biotechnol., 11: 379-383 (1993)].
Ингибиторы антагонизма BMP могут быть также использованы вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. В одном варианте ингибиторы антагонистов BMP выбраны изингибиторов BMP антагонистовSOST, noggin, chordin, gremlin и dan [Massague and Chen, "Controlling TGF-beta signaling," Genes Dev., 14: 627-644, 2000; Aspenberg et al., "The bone morphogenetic proteins antagonist Noggin inhibits membranous ossification," J. Bone Miner. Res. 16: 497-500, 2001; Brunkow et al., "Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein," Am. J. Hum. Genet. 68: 577-89 (2001)].
Тканеселективные модуляторы рецептора андрогена по данному изобретению могут быть также объединены с полипептидным остеопротегерином для лечения состояний, связанных с разрежением костной ткани, таким как остеопороз. Предпочтительно остеопротегерином является остеопротегерин млекопитающего и более предпочтителен человеческий остеопротегерин. Полипептидный остеопротегерин, представитель надсемейства рецепторов фактора некроза опухолей, используют для лечения болезней костной ткани, характеризующихся повышенным разрежением кости, таким как остеопороз. Упоминается патент США № 6288032, который включен в настоящее описание в полном объеме в виде ссылки.
Производные простагландина также могут быть использованы вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. В одном варианте производные простагландина выбраны из агонистов простагландиновых рецепторов EP1, EP2, EP4, FP и IP или их производных [Pilbeam et al., "Prostaglandins and bone metabolism," 1996. In: Bilezikian, et al. Ed. Principles of Bone Biology, San Diego: Academic Press; Weinreb et al., "Expression of the prostaglandin E(2) (PGE(2)) receptor subtype EP(4) and its regulation by PGE(2) in osteoblastic cell lines and adult rat bone tissue," Bone, 28: 275-281 (2001)].
Факторы роста фибробластов могут быть также использованы вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. Факторы роста фибробластов включают aFGF, bFGF и родственные пептиды с активностью FGF [Hurley Florkiewicz, "Fibroblast growth factor and vascular endothelial growth factor families," 1996. In: J. P. Bilezikian, et al., Ed. Principles of Bone Biology, San Diego: Academic Press].
Помимо ингибиторов резорбции кости и остеоанаболических средств имеются также другие средства, которые, как известно, оказывают благоприятное воздействие на скелет посредством механизмов, которые точно не установлены. Указанные средства могут быть также с пользой объединены с тканеселективными модуляторами рецептора андрогена структурной формулы I.
Витамин D и производные витамина D могут быть также использованы вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. Витамин D и производные витамина D включают природный витамин D, 25-OH-витамин D3, 1α,25(OH)2 витамин D3, 1α-OH-витамин D3, 1α-OH-витамин D2, дигидротахистерол, 26,27-F6-1α,25(OH)2 витамин D3, 19-нор-1α,25(OH)2 витамин D3, 22-оксакальцитриол, кальципотриол, 1α,25(OH)2-16-ен-23-ин-витамин D3 (Ro 23-7553), EB1089, 20-эпи-1α,25(OH)2 витамин D3, KH1060, ED71, 1α,24(S)-(OH)2 витамин D3, 1α,24(R)-(OH)2 витамин D3 [см., Jones G., "Pharmacological mechanisms of therapeutics: vitamin D and analogs," 1996. In: J. P. Bilezikian, et. al. Ed. Principles of Bone Biology, San Diego: Academic Press].
Витамин K и производные витамина K могут быть также использованы вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I. Витамин K и производные витамина K включают менатетренон (витамин K2) [см. Shiraki et al., "Vitamin K2 (menatetrenone) effectively prevents fractures and sustains lumbar bone mineral density in osteoporosis," J. Bone Miner. Res. 15: 515-521 (2000)].
Соевые изофлавоны, включая иприфлавон, могут быть использованы вместе с тканеселективными модуляторами рецептора андрогена структурной формулы I.
Фторидные соли, включая фторид натрия (NaF) и мононатрий фторфосфат (MFP), могут быть также использованы с тканеселективными модуляторами рецептора андрогена структурной формулы I. Кроме того, с тканеселективными модуляторами рецептора андрогена структурной формулы I могут быть использованы пищевые кальциевые добавки. Пищевые кальциевые добавки включают карбонат кальция, цитрат кальция и природные соли кальция (Heaney. Calcium. 1996. In: J. P. Bilezikian, et al., Ed., Principles of Bone Biology, San Diego: Academic Press).
Диапазоны суточных доз для ингибиторов резорбции кости, остеоанаболических средств и других средств, которые могут быть использованы для оказания благоприятного воздействия на скелет, при использовании в комбинации с соединением структурной формулы I, представляют собой диапазоны доз, которые известны в данной области. В таких комбинациях, как правило, диапазон суточных доз для тканеселективного модулятора рецептора андрогена структурной формулы I составляет для взрослого человека от 0,01 до 1000 мг в день, более предпочтительно от 0,1 до 200 мг/день. Однако возможны корректировки в сторону уменьшения дозы каждого средства вследствие повышенной эффективности действия комбинированного средства.
В частности, когда используют бисфосфонат, дозы от 2,5 до 100 мг/день (измеренные в расчете на свободную бисфосфоновую кислоту) являются подходящими для лечения, более предпочтительно от 5 до 20 мг/день, особенно около 10 мг/день. Профилактически следует использовать дозы от около 2,5 до около 10 мг/день и особенно около 5 мг/день. Для уменьшения побочных действий желательно применять комбинацию соединения структурной формулы I и бисфосфоната один раз в неделю. В случае введения один раз в неделю, дозы от около 15 мг до 700 мг в неделю бисфосфоната и от 0,07 до 7000 мг соединения структурной формулы I могут быть использованы либо раздельно, либо в комбинированной лекарственной форме. Соединение структурной формулы I можно удобно вводить, используя устройство для доставки с контролируемым высвобождением лекарственного средства, особенно в случае введения один раз в неделю.
Для лечения атеросклероза, гиперхолестеринемии и гиперлипидемии соединения структурной формулы I можно эффективно вводить в комбинации с одним или несколькими дополнительными активными средствами. Дополнительное активное средство или средства могут представлять собой липидизменяющие соединения, такие как ингибиторы редуктазы HMG-CoA, или средства, имеющие другие виды фармацевтической активности, или средства, которые имеют как липидизменяющие действия, так и другие виды фармацевтической активности. Примеры ингибиторов редуктазы HMG-CoA включают статины в их лактонизированных формах или дигидроксильных раскрытых кислотных формах и их фармацевтически приемлемые соли и их сложные эфиры, включая, но не ограничиваясь ими, ловастатин (см. патент США № 4342767); симвастатин (см. патент США № 4444784); симвастатин в дигидроксильной раскрытой кислотной форме), особенно его аммонийные или кальциевые соли; правастатин, особенно его натриевую соль (см. патент США № 4346227); флувастатин, особенно его натриевую соль (см. патент США № 5354772); аторвастатин, особенно его кальциевую соль (см. патент США № 5273995); церивастатин, особенно его натриевую соль (см. патент США № 5177080) и нисвастатин, также известный как NK-104 (см. публикацию международной заявки PCT WO 97/23200). Дополнительные активные средства, которые могут быть использованы в комбинации с соединением структурной формулы I, включают, но не ограничиваются ими, ингибиторы синтазы HMG-CoA; ингибиторы сквален-эпоксидазы; ингибиторы сквален-синтетазы (также известные как ингибиторы сквален-синтазы), ингибиторы ацил-кофермент A:холестерин-ацилтрансферазы (ACAT), включая селективные ингибиторы ACAT-1 или ACAT-2, а также двойственные ингибиторы ACAT-1 и -2; ингибиторы микросомального белка, ответственного за перенос триглицеридов(MTP); пробукол; ниацин; ингибиторы абсорбции холестерина, такие как SCH-58235, также известные как эзетимиб и 1-(4-фторфенил)-3(R)-[3(S)-(4-фторфенил)-3-гидроксипропил)]-4(S)-(4-гидроксифенил)-2-азетидинон, который описан в патентах США №№ 5767115 и 5846966; вещества усиливающие экскрецию желчных кислот; индукторы рецептора ЛНП (LDL) (липопротеин низкой плотности); ингибиторы агрегации тромбоцитов, например антагонисты рецептора гликопротеинового фибриногена IIb/IIIa и аспирин; агонисты человеческих гамма-рецепторов, активируемых пероксисомальным пролифератором (PPARγ), включая соединения, обычно называемые глитазонами, например троглитазон, пиоглитазон и розиглитазон, и включая соединения, составляющие структурный класс, известный как тиазолидиндионы, а также соединения-агонисты PPARγ за пределами структурного класса тиазолидиндионов; агонисты PPARα,такие как клофибрат, фенофибрат, включая тонкоизмельченный фенофибрат, и гемфиброзил; двойственные агонисты PPARα/γ; витамин B6 (также известный как пиридоксин) и его фармацевтически приемлемые соли, такие как соль HCl; витамин B12 (также известный как цианокобаламин); фолиевая кислота или ее фармацевтически приемлемая соль или ее сложный эфир, такие как натриевая соль и соль метилглюкамина; антиоксидантные витамины, такие как витамин С и Е и бета-каротин; бета-блокаторы; антагонисты ангиотензина II, такие как лосартан; ингибиторы ангиотензин-превращающего фермента, такие как эналаприл и каптоприл; блокаторы кальциевого канала, такие как нифедипин и дилтиазем; антагонисты эндотелина; средства, такие как лиганды LXR, которые повышают экспрессию гена ABC1; бисфосфонатные соединения, такие как натрий алендронат; и ингибиторы циклооксигеназы-2, такие как рофекоксиб и целекоксиб, а также другие средства, которые, как известно, используют для лечения вышеуказанных состояний.
Диапазоны суточных доз для ингибиторов редуктазы HMG-CoA, при использовании в комбинации с соединениями структурной формулы I, соответствуют дозам, которые известны в данной области. Аналогично, диапазоны доз для ингибиторов синтазы HMG-CoA; ингибиторов сквален-эпоксидазы; ингибиторов сквален-синтетазы (также известные как ингибиторы сквален-синтазы), ингибиторов ацил-кофермента А:холестерин-ацилтрансферазы (ACAT), включая селективные ингибиторы ACAT-1 или ACAT-2, а также двойственные ингибиторы ACAT-1 и -2; ингибиторов микросомального белка, ответственного за перенос триглицеридов(MTP); пробукола; ниацина; ингибиторов абсорбции холестерина, включая эзетимиб; веществ, стимулирующих экскрецию желчных кислот; индукторов рецептора ЛНП (LDL) (липопротеин низкой плотности); ингибиторов агрегации тромбоцитов, включая антагонисты рецептора гликопротеинового фибриногена IIb/IIIaи аспирин; агонистов человеческих гамма-рецепторов, активируемых пероксисомальным пролифератором (PPARγ); агонистов PPARα; двойственных агонистов PPARα/γ); витамина B6; витамина B12; фолиевой кислоты; антиоксидантных витаминов; бета-блокаторов; антагонистов ангиотензина II; ингибиторов ангиотензинпревращающего фермента; блокаторов кальциевого канала; антагонистов эндотелина; средств, таких как лиганды LXR, которые повышают экспрессию гена ABC1; бисфофонатных соединений; и ингибиторов циклооксигеназы-2, также соответствуют дозам, известным в данной области, хотя вследствие совместного действия с соединениями структурной формулы I, при введении в комбинации, доза может быть несколько ниже.
Один вариант изобретения представляет способ действия маркера костного обновления у млекопитающего, включающий введение терапевтически эффективного количества соединения структурной формулы I. Неограничивающие примеры маркеров костного обновления могут быть выбраны из продуктов разложения коллагена типа I, С-телопептиды (CTX), обнаруживаемые в моче; продуктов разрушения поперечных (межмолекулярных) связей коллагена типа I, N-телопептиды (NTX), обнаруживаемые в моче; DXA и DPD.
В соответствии со способом настоящего изобретения индивидуальные компоненты комбинации можно вводить раздельно в различные моменты времени во время курса лечения или одновременно в раздельных или единых комбинированных формах. Поэтому необходимо иметь в виду, что настоящее изобретение охватывает все вышеуказанные схемы одновременного или альтернативного лечения, и термин «введение» следует интерпретировать соответствующим образом. Будет правильно считать, что комбинации соединений данного изобретения с другими средствами, применимыми для лечения заболеваний, вызванных недостаточностью андрогена или интенсивность симптомов которых может быть уменьшена введением андрогена, не выходят за рамки объема настоящего изобретения.
Аббревиатуры, используемые в описании получения соединений настоящего изобретения:
Получение соединений данного изобретения
Соединения настоящего изобретения могут быть получены в соответствии со способами, представленными в нижеследующих схемах реакций и примерах, или их модификациями, используя легкодоступные исходные продукты, реагенты, и обычными способами или их вариациями, известными обычному специалисту-практику в области синтетической органической химии. Конкретные обозначения переменных на схемах представлены только в целях иллюстрации, и при этом подразумевается, что они не ограничивают описанные способы.
Селективные модуляторы рецептора андрогена (SARM) структурной формулы 1-5 получали, как описано в общих чертах на Схеме 1. Исходный продукт представлял 17β-карбоксилат 1-1, который описан G.H. Rasmusson et al, J. Med. Chem. 27: 1690-1701 (1984).
Схема 1
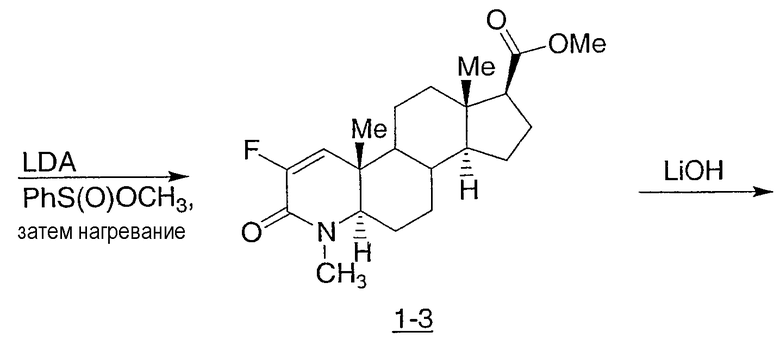
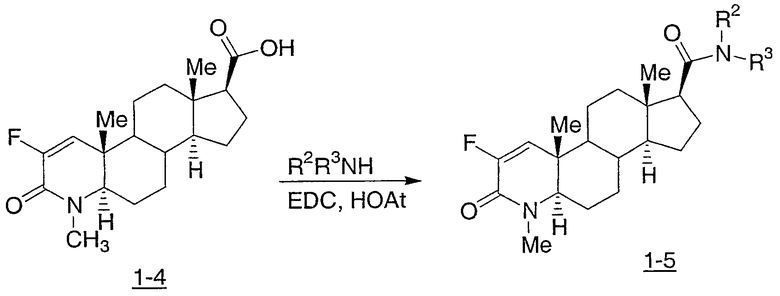
Альтернативно, соединения структурной формулы 1-5 получали из промежуточного продукта 1-3, как показано на Схеме 2:
Схема 2
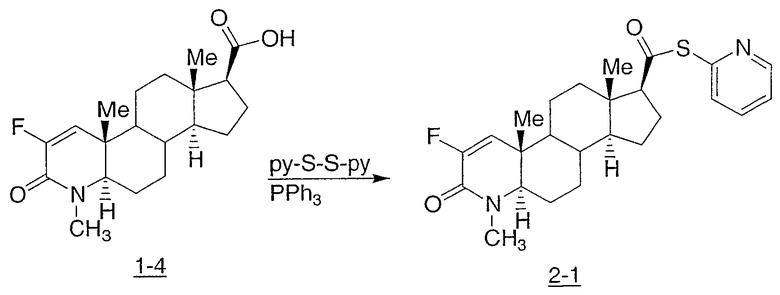
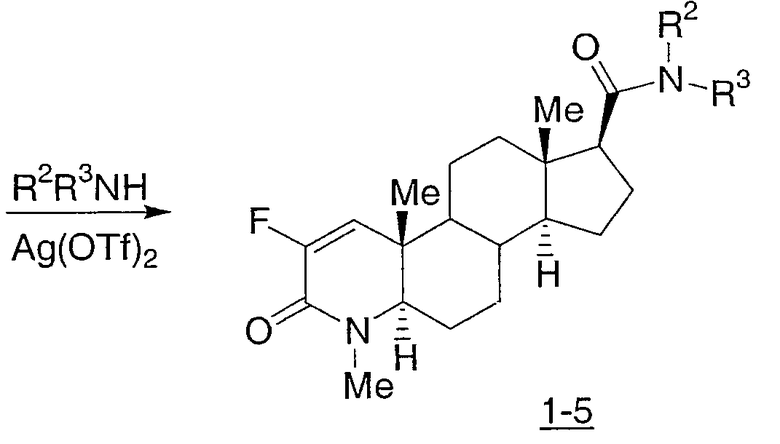
Соединения структурной формулы 3-6 на Схеме 3 получали, как описано в общих чертах на Схеме 3, приведенной ниже. Исходный продукт представлял 17β-карбоновую кислоту 3-1, которая описана в G.H. Rasmusson et al, J. Med. Chem., 29: 2298-2315 (1986) и R.L. Tolman, et al., J. Steroid Biochem. Mol. Biol., 60: 303-309 (1997).
Схема 3
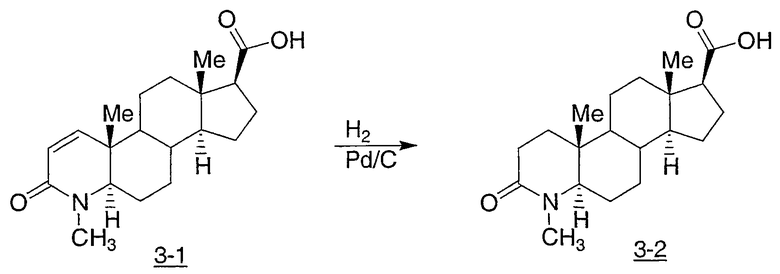
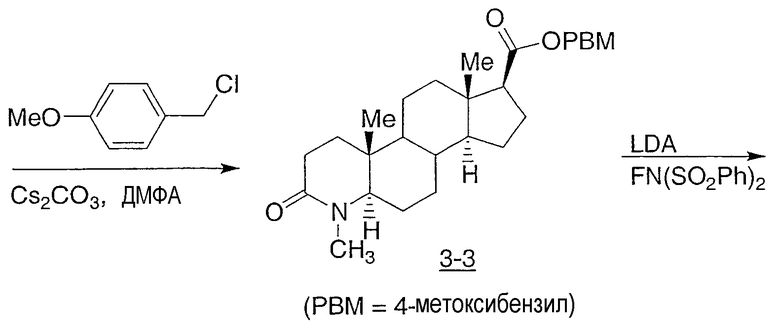
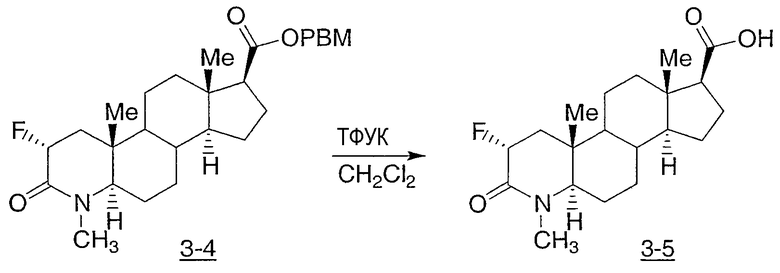
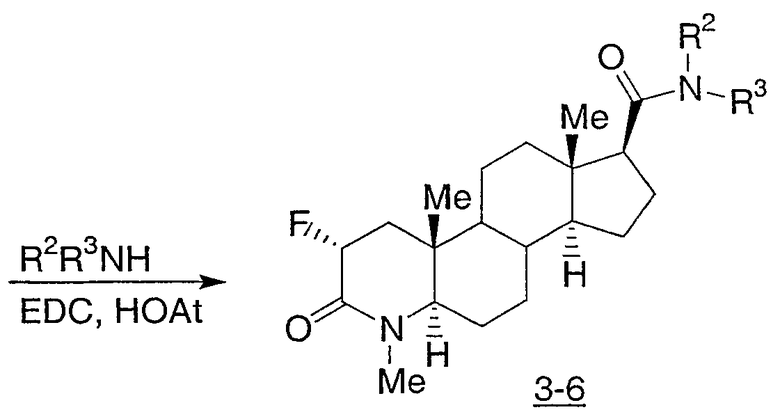
Альтернативно, соединения структурной формулы 3-6 получали из промежуточного продукта 3-5, как показано на Схеме 4:
Схема 4
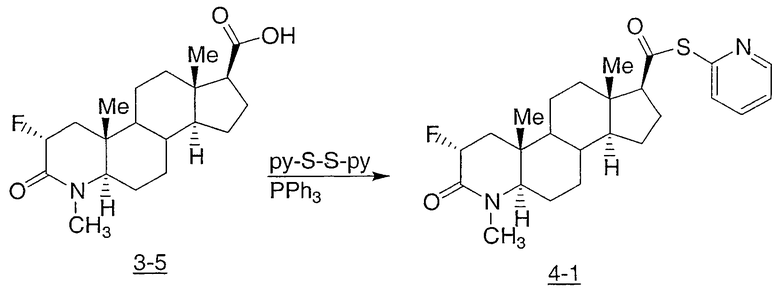
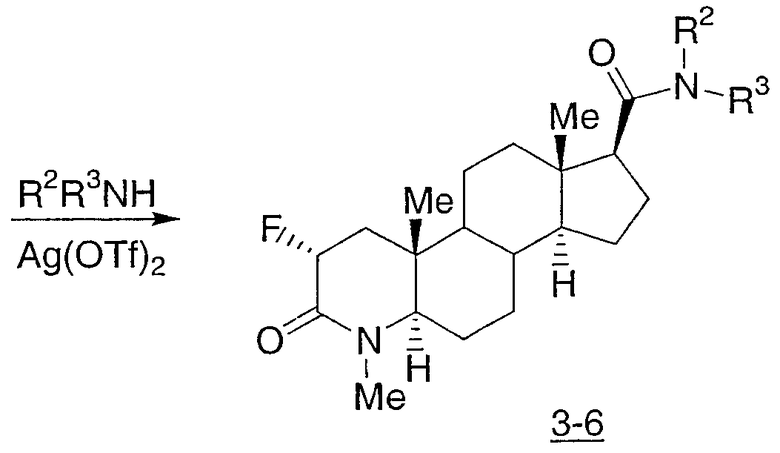
Нижеследующие примеры, как предполагается, дополнительно иллюстрируют детали получения и использования соединений настоящего изобретения. Подразумевается, что приведенные ниже примеры никоим образом не ограничивают объем настоящего изобретения, и их не следует истолковывать таким образом. Кроме того, следует иметь в виду, что соединения, описанные в нижеследующих примерах, не образуют единственный тип соединений, который рассматривается в качестве изобретения, и что любая комбинация соединений или их частей может, сама по себе, образовывать тип соединений. Специалистам в данной области очевидно, что для получения указанных соединений могут быть использованы известные вариации условий и способов нижеследующих методик получения. Все температуры представлены в градусах Цельсия, если не указано особо.
Схема 5
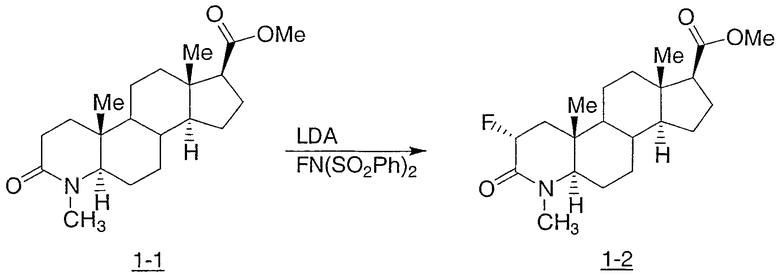
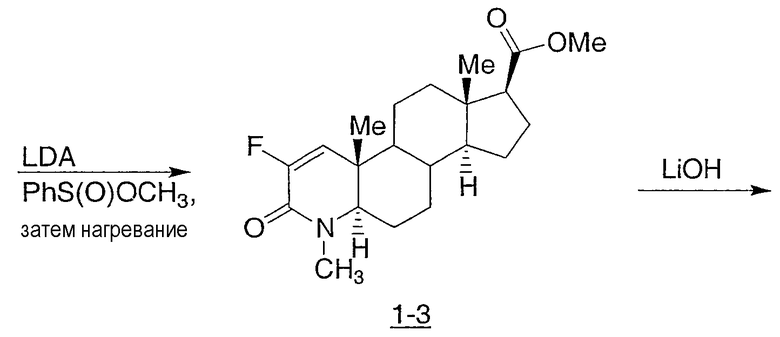
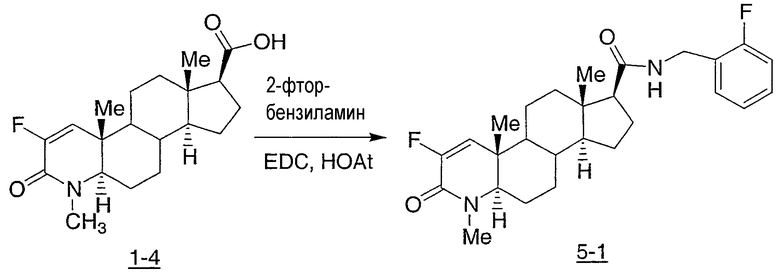
ПРИМЕР 1
Стадия A:Метиловый эфир 2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоновой кислоты (1-2)
К раствору 1-1 (7,5 г, 21,6 ммоль) в ТГФ (100 мл) при -78°C добавляют по каплям раствор 1,5M LDA в ТГФ (17,3 мл, 25,9 ммоль) на протяжении 20 мин и затем перемешивают 1 час. Затем добавляют раствор FN(SO2Ph)2 (10,2 г, 32,4 ммоль) в ТГФ (40 мл) на протяжении 20 мин. Спустя 30 мин охлаждающую баню убирают и реакционную смесь перемешивают в течение 14 ч. Добавляют Et2O и смесь промывают водой, насыщенным водным гидрокарбонатом натрия, насыщенным раствором соли, сушат (MgSO4)и затем концентрируют. Хроматография на силикагеле (гексаны до EtOAc в качестве элюента) дает 1-2(4,2 г) в виде бесцветного твердого вещества.
МС: [M+H] Выч.: 366, найдено: 366,1.
Стадия B:Метиловый эфир 2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновой кислоты (1-3)
К раствору 1-2(30 г, 82,1 ммоль) в ТГФ (400 мл) при -78°C добавляют по каплям раствор 1,5M LDA в ТГФ (71,1 мл, 107 ммоль) на протяжении 30 мин и затем перемешивают 1 ч. Затем добавляют метилбензолсульфинат (19,23 г, 123 ммоль) на протяжении 15 мин. Спустя 30 мин охлаждающую баню убирают и реакционную смесь перемешивают в течение 1 ч. Добавляют Et2O и смесь промывают водой, насыщенным водным гидрокарбонатом натрия, насыщенным раствором соли, сушат (MgSO4) и затем концентрируют. Остаток растворяют в толуоле (200 мл) и нагревают при температуре дефлегмации в течение 2 ч. Выпаривание растворителя и хроматография остатка на силикагеле (гексаны до смеси 50% EtOAc/гексаны в качестве элюента) дают 1-3 (20,4 г) в виде бледно-желтого твердого вещества.
МС: [M+H] Выч.: 364, найдено: 364,1.
Стадия C:2-Фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновая кислота (1-4)
К раствору 1-3(2,4 г, 6,6 ммоль) в 1,4-диоксане (50 мл) добавляют раствор гидроксида лития (0,41 г, 9,9 ммоль) в воде (20 мл) и смесь нагревают при 100°C в течение 3 ч. После охлаждения смесь разбавляют этилацетатом и затем промывают 1N HCl, насыщенным раствором соли, сушат (MgSO4) и затем концентрируют, получая 1-4 (2,2 г) в виде бледно-желтого твердого вещества.
МС: [M+H] Выч.: 350, найдено: 350.
Стадия D:N-(2-Фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид (5-1)
Смесь 1-4(0,12 г, 0,34 ммоль), EDC (0,079 г, 0,41 ммоль), HOAt (0,056 г, 0,41 ммоль), NMM (0,15 мл, 1,37 ммоль) и 2-фторбензиламина (0,52 г, 0,41 ммоль) в ДМФА (2 мл) перемешивают в течение 14 ч. Смесь разбавляют водой, фильтруют и твердое вещество промывают водой, затем диэтиловым эфиром и затем сушат ввакууме, получая 5-1 (0,12 г) в виде бледно-желтого твердого вещества.
МС: [M+H] Выч.: 457,2661, найдено: 457,2666.
Схема 6
ПРИМЕР 2
Стадия A:S-(Пиридин-2-ил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карботиоат (2-1)
Смесь 1-4 (0,80 г, 2,30 ммоль), 2,2'-дитиопиридина (1,01 г, 4,6 ммоль) и трифенилфосфина (1,2 г, 4,6 ммоль) в толуоле (10 мл) перемешивают 14 ч. После выпаривания растворителя твердое вещество суспендируют в толуоле (5 мл) и разбавляют диэтиловым эфиром. Твердое вещество отфильтровывают, промывают диэтиловым эфиром и затем сушат в вакууме, получая 2-1 (0,12 г) в виде бледно-желтого твердого вещества.
МС: [M+H] Выч.: 443, найдено: 443.
Стадия В:N-(2-Трифторметилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид (6-1)
Смесь 2-1 (0,25 г, 0,57 ммоль), 2-трифторметиланилина (0,18 г, 1,13 ммоль) и трифлата серебра (II) (0,15 г, 0,57 ммоль) в дихлорметане (3 мл) перемешивают 14 ч. После выпаривания растворителя хроматография остатка на силикагеле (гексаны до EtOAc в качестве элюента) дает 6-1 (0,12 г) в виде бледно-желтого твердого вещества.
МС: [M+H] Выч.: 493,2473, найдено: 493,2470.
Примеры 3-50, приведенные в Таблице, получают способом, аналогичным Примерам 1 и 2, но используя соответствующий амин для получения карбоксамида.
ТАБЛИЦА 1
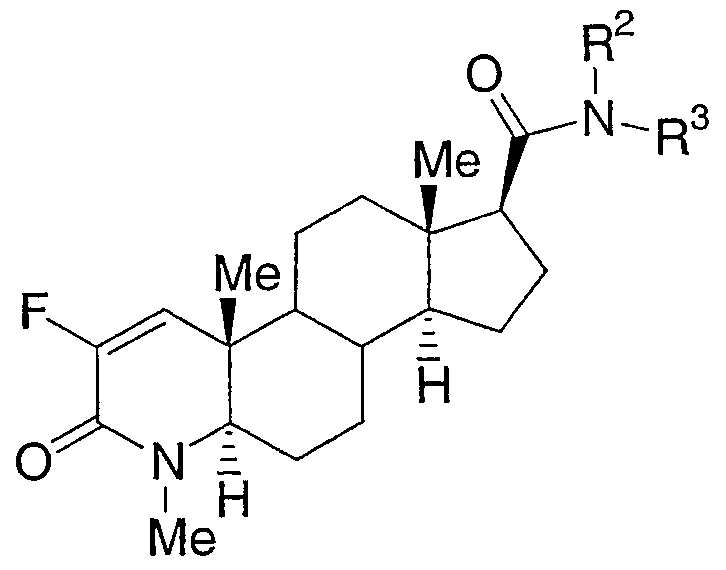
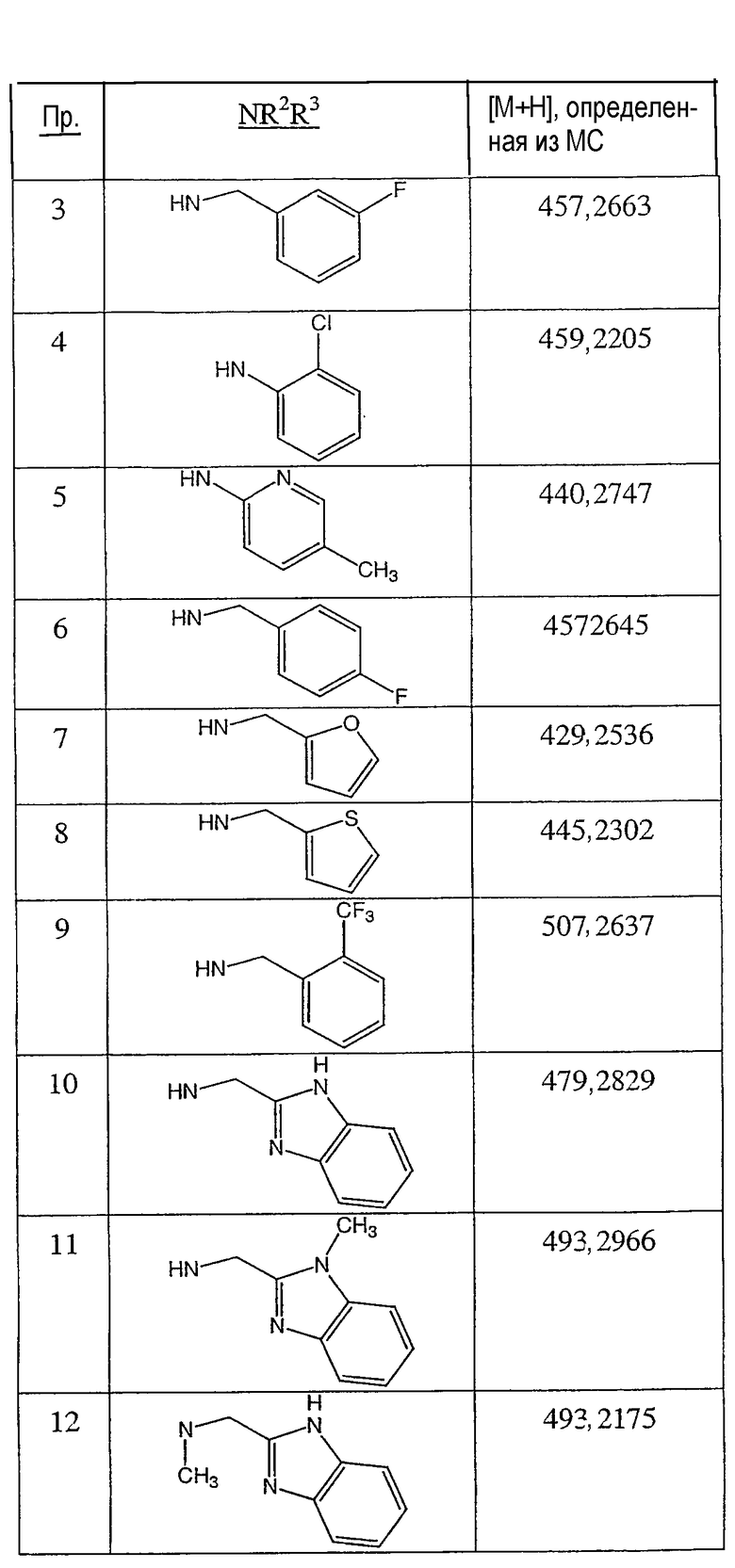
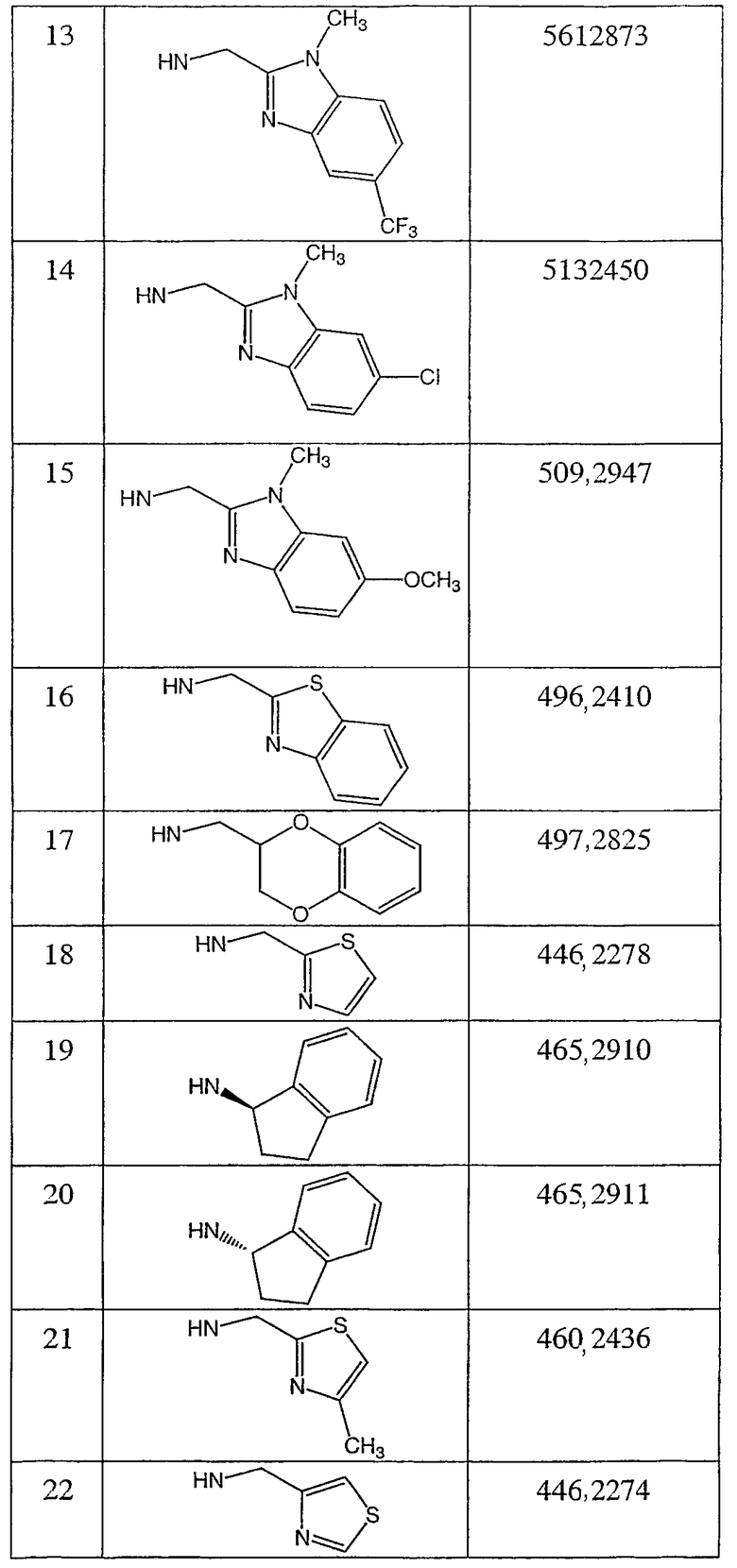
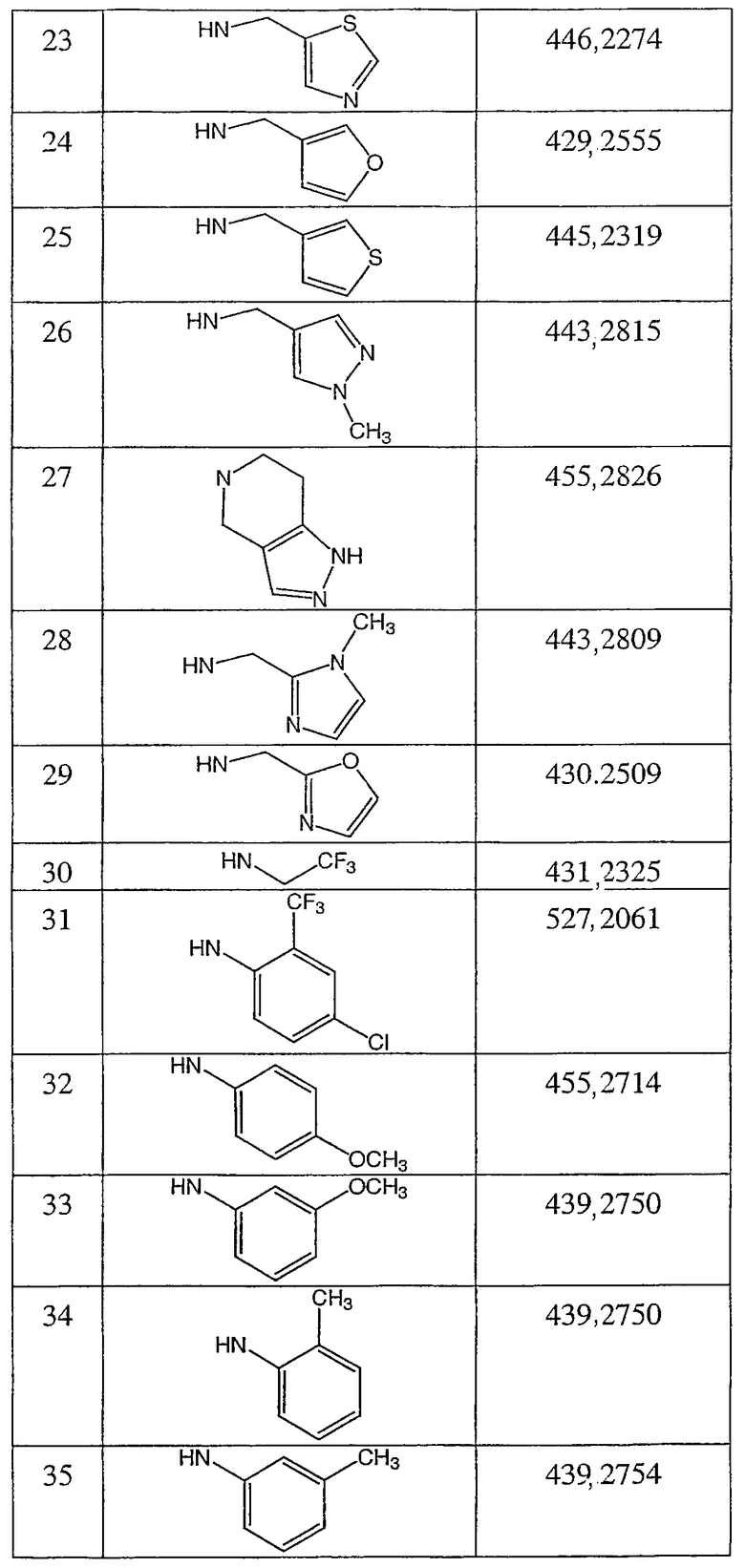
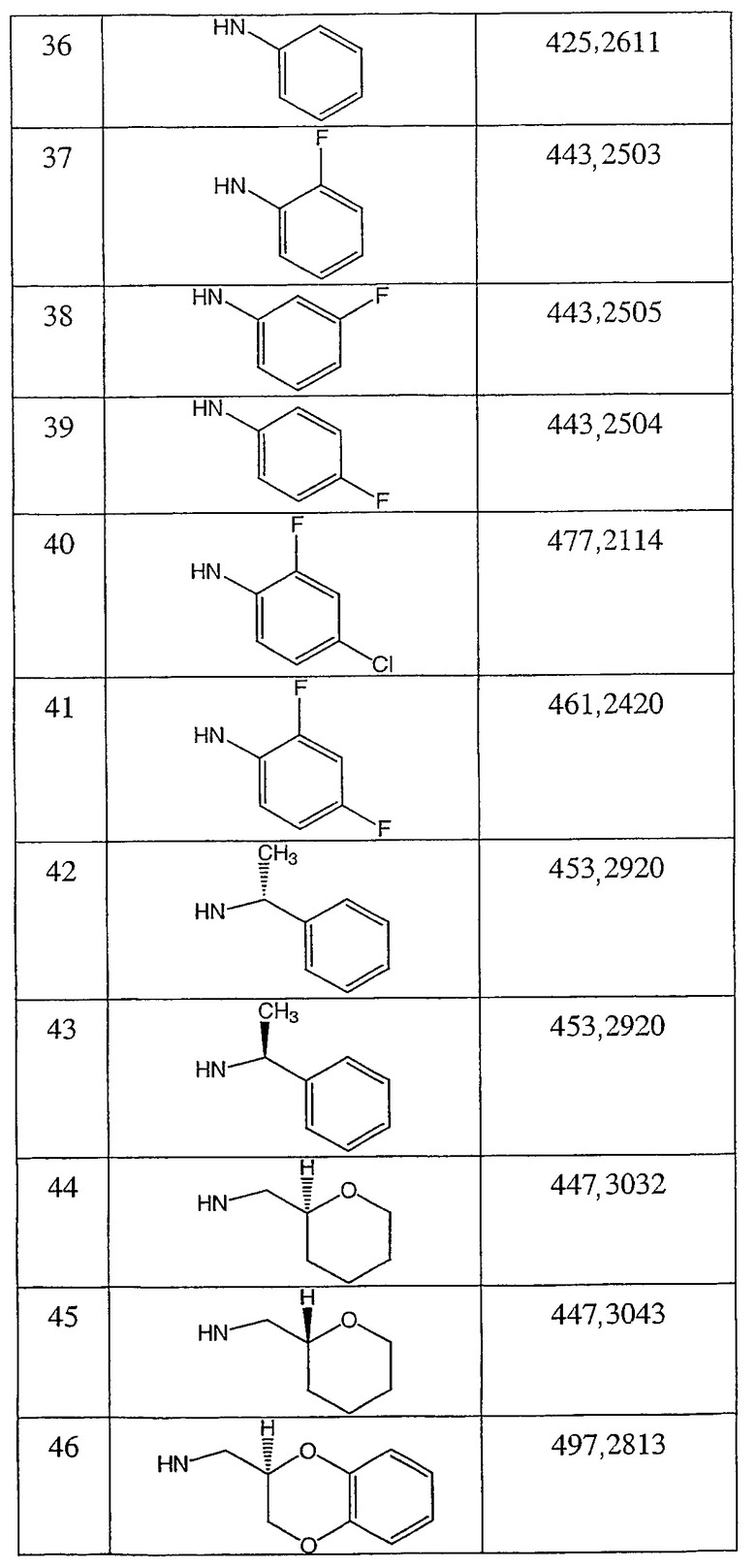
Схема 7
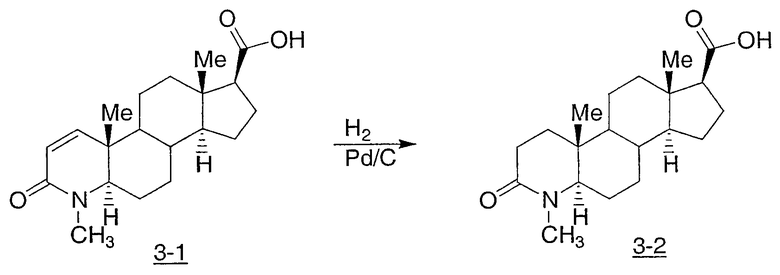
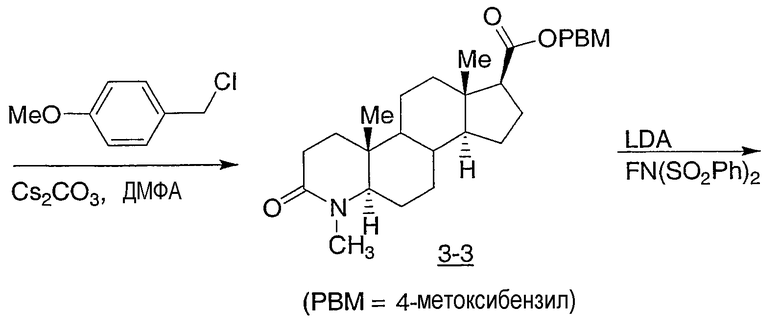
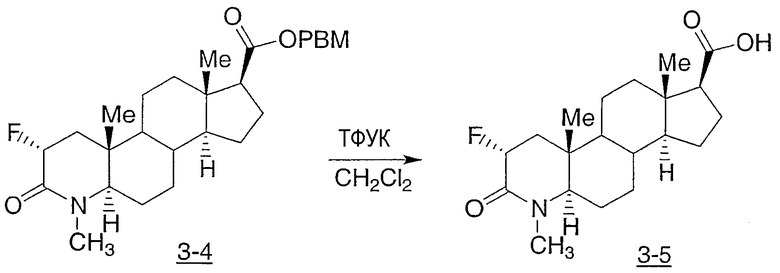
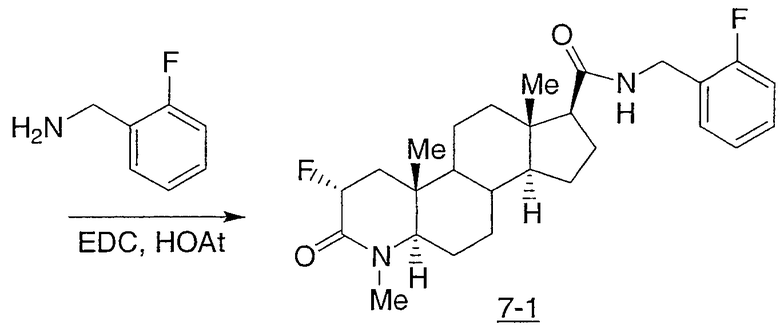
ПРИМЕР 51
Стадия A:4-Метил-3-оксо-4-аза-5α-андростан-17β-карбоновая кислота (3-2)
К суспензии 3-1 (13,2 г, 39,82 ммоль) и EtOH (200 мл) добавляют LiOH (2 г, 47,8 ммоль, растворенный в 20 мл H2O). К раствору добавляют 10% Pd/C (1 г) и затем смесь перемешивают при давлении Н2 1 атм в течение 4,0 часов. Реакционную смесь фильтруют через слой целита и затем концентрируют. К остатку добавляют 1N HCl. Твердое вещество, которое образовалось, отделяют, промывают Et2O и затем сушат в вакууме, получая 3-2 (10,5 г) в виде белого твердого вещества.
Стадия B:4-Метоксибензиловый эфир 4-метил-3-оксо-4-аза-5α-андростан-17β-карбоновой кислоты (3-3)
К суспензии 3-2 (10,5 г, 31,48 ммоль) и ДМФА (200 мл) добавляют Cs2CO3 (10 г, 47,8 ммоль) и 4-метоксибензилхлорид (5,9 г, 37,78 ммоль). Смесь нагревают до 60°C на протяжении ночи. Добавляют EtOAc и смесь промывают 1N HCl, насыщенным раствором соли, сушат (MgSO4) и концентрируют. Остаток растирают с EtOAc и затем фильтруют. Раствор концентрируют и остаток подвергают хроматографии на силикагеле (гексаны до EtOAc в качестве элюента), получая 3-3 (7,22 г) в виде белого твердого вещества.
МС: [M+H] Выч.: 454,6, найдено: 454,3.
Стадия C:4-Метоксибензиловый эфир 2α-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновой кислоты (3-4)
К раствору 3-3 (7,2 г, 15,9 ммоль) в ТГФ (100 мл) при -78°C добавляют по каплям раствор 1,5M LDA в ТГФ (12,7 мл, 19,05 ммоль) на протяжении 20 мин и затем перемешивают в течение 1 ч. Затем на протяжении 20 мин добавляют раствор FN(SO2Ph)2 (6,0 г, 19,05 ммоль) в ТГФ (40 мл). Спустя 30 мин охлаждающую баню убирают и реакционную смесь перемешивают в течение 14 ч. Добавляют Et2O и затем промывают водой, насыщенным водным гидрокарбонатом натрия, насыщенным раствором соли, сушат (MgSO4) и затем концентрируют. Хроматография на силикагеле (гексаны до EtOAc в качестве элюента) дает 3-4 (3,1 г) в виде бесцветного твердого вещества.
МС: [M+H] Выч.: 472,6, найдено: 472,3.
Стадия D:2α-Фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновая кислота (3-5)
К раствору 3-4 (3,1 г, 6,57 ммоль) в CH2Cl2 (20 мл) добавляют ТФУК (10 мл). Спустя 30 минут раствор концентрируют и затем подвергают азеотропной перегонке с толуолом. Остаток растворяют в CH2Cl2 и затем промывают 1N HCl, насыщенным раствором соли, сушат (MgSO4) и затем концентрируют, получая 3-5 (2,1 г) в виде белого твердого вещества.
МС: [M+H] Выч.: 352,5, найдено: 352,2.
Стадия E: N-(2-Фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид (7-1)
Смесь 3-5 (0,10 г, 0,285 ммоль), EDC (0,066 г, 0,34 ммоль), HOAt (0,047 г, 0,34 ммоль), NMM (0,13 мл, 1,14 ммоль) и 2-фторбензиламина (0,52 г, 0,41 ммоль) в ДМФА (2 мл) перемешивают в течение 14 ч. Смесь разбавляют водой, фильтруют и твердое вещество промывают водой, затем диэтиловым эфиром и затем сушат в вакууме, получая 7-1(0,12 г) в виде бледно-желтого твердого вещества.
МС: [M+H] Выч.: 459,2818, найдено: 459,2812.
Схема 8
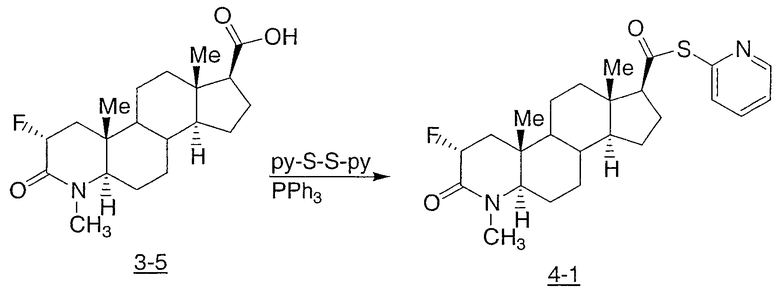
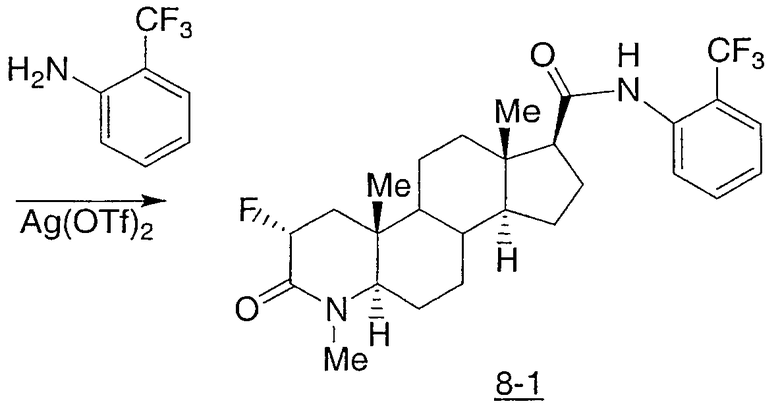
ПРИМЕР 52
Стадия A:S-(Пиридин-2-ил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карботиоат (4-1)
Используя способ, описанный на Схеме 4, 4-1 получают из 3-5.
МС: [M+H] Выч.: 445, найдено: 445,1.
Стадия B:N-(2-Трифторфенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамид (8-1)
Используя способ, описанный на Схеме 4, 8-1 получают из 4-1.
МС: [M+H] найдено:: 495,2624.
Примеры 53-67, приведенные в Таблице 2, получают способом, аналогичным Примерам 51 и 52, но используя соответствующий амин для получения карбоксамида.
ТАБЛИЦА 2
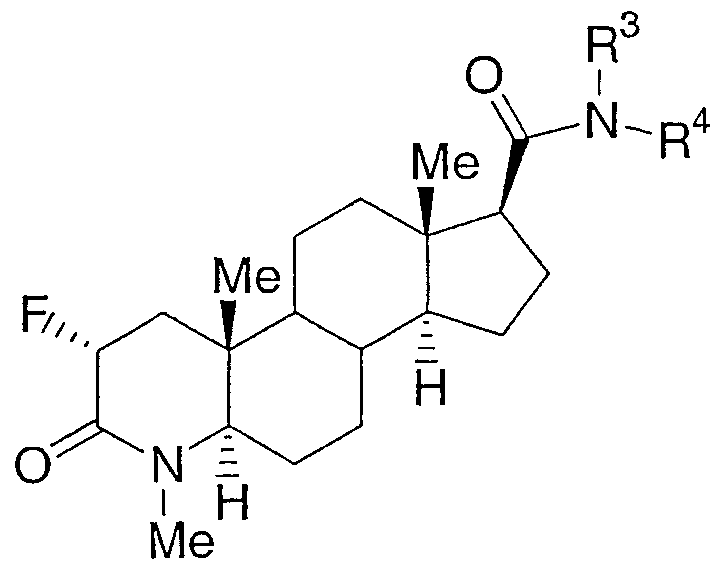
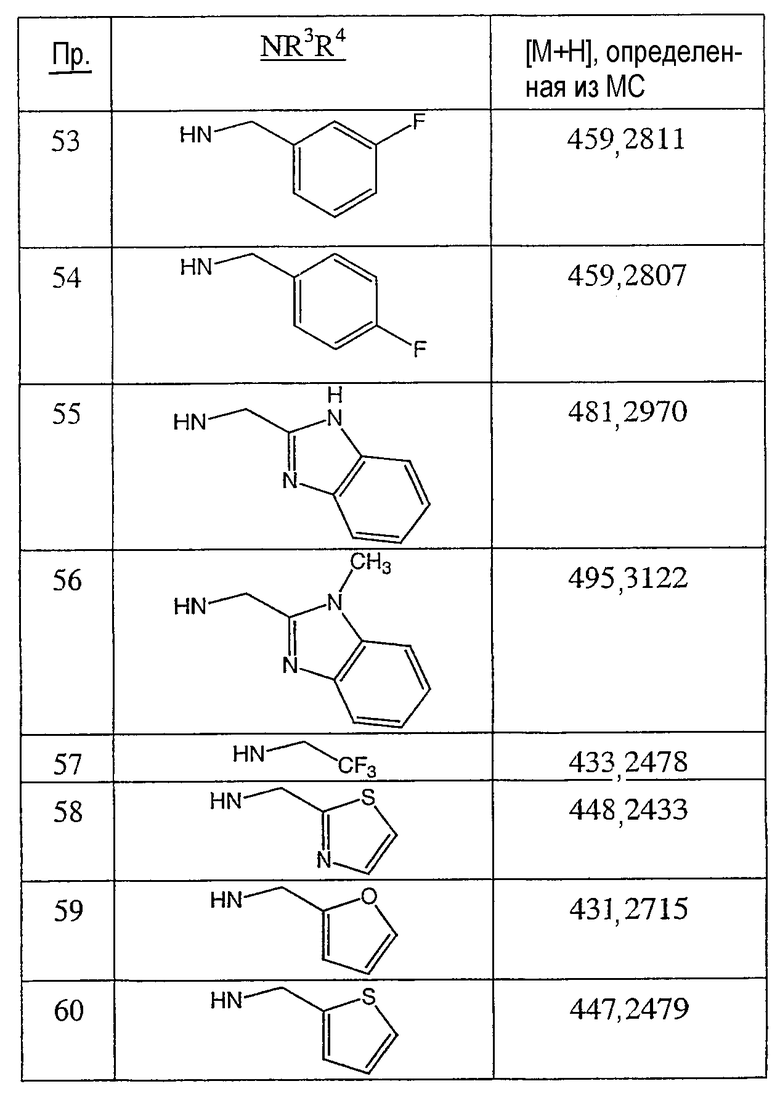
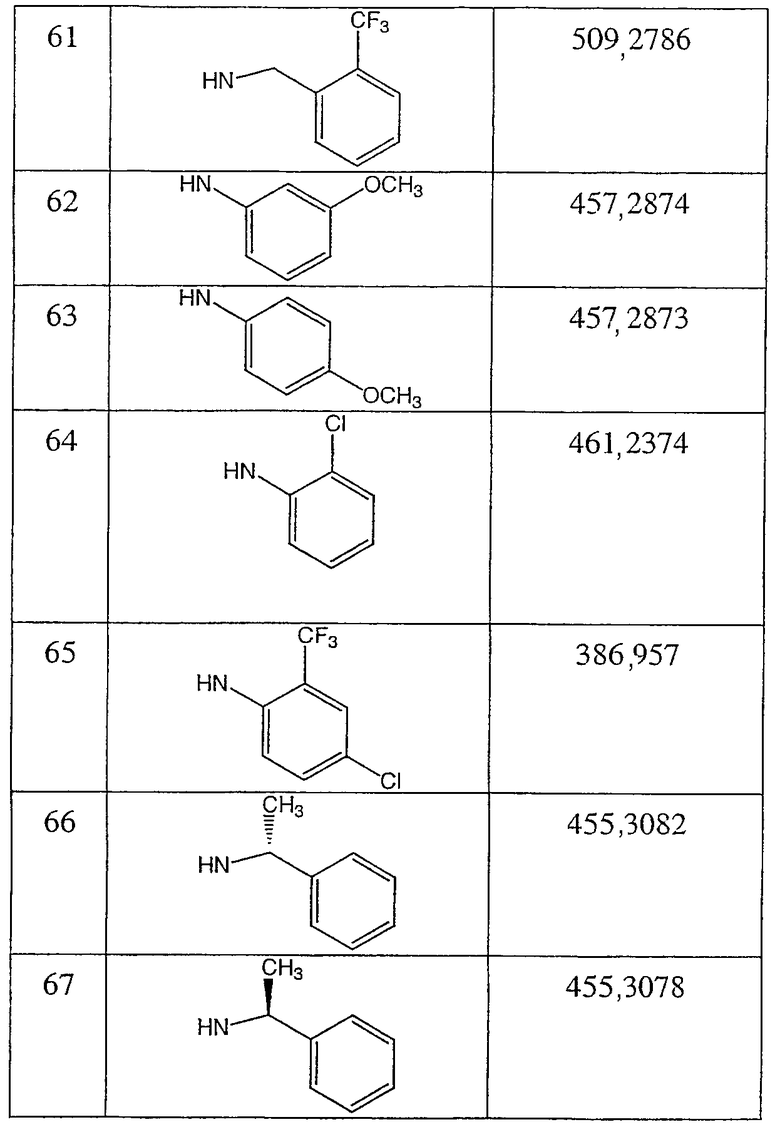
ПРИМЕР 68
ПЕРОРАЛЬНАЯ КОМПОЗИЦИЯ
В качестве конкретного варианта пероральной композиции соединения данного изобретения, 50 мг соединения настоящего изобретения формируют с достаточно тонкоизмельченной лактозой, чтобы получить общее количество от 580 до 590 мг, которым заполняют твердую желатиновую капсулу размера 0.
ПРИМЕР 69
Тансдермальный пластырь
Силиконовую жидкость и соединение структурной формулы I смешивают вместе и добавляют коллоидный диоксид кремния для повышения вязкости. Затем полученный продукт дозируют в полимерный ламинат, впоследствии герметизируемый нагреванием, состоящий из следующего: прокладка для высвобождения сложного полиэфира; адгезив для контакта с кожей, состоящий из силиконовых и акриловых полимеров, контрольной мембраны, которая представляет собой полиолефин (например, полиэтилен, поливинилацетат или полиуретан), и непроницаемой защитной мембраны, изготовленной из многослойного материала (мультиламината) на основе сложного полиэфира. Затем полученную ламинированную пластину режут на пластыри площадью 10 см2. Состав дан для 100 пластырей.
ПРИМЕР 70
Суппозиторий
Полиэтиленгликоль 1000 и полиэтиленгликоль 4000 смешивают и расплавляют. Соединение структурной формулы I вводят при перемешивании в расплавленную смесь, выливают в формы и полученной смеси дают охладиться. Состав дан для 1000 суппозиториев.
ПРИМЕР 71
Раствор для инъекций
Соединение структурной формулы I и буферные агенты растворяют в пропиленгликоле примерно при 50°C. Затем при перемешивании добавляют воду для инъекций и полученный раствор фильтруют, заполняют им ампулы, герметизируют и стерилизуют обработкой в автоклаве. Состав дан для 1000 ампул.
ПРИМЕР 72
Раствор для инъекций
Соединение структурной формулы I, гептагидрат сульфата магния и буферные агенты растворяют в воде для инъекций при перемешивании, полученный раствор фильтруют, заполняют им ампулы, герметизируют и стерилизуют обработкой в автоклаве. Состав дан для 1000 ампул.
Нижеследующие испытания используют для того, чтобы охарактеризовать активность тканеселективных модуляторов рецептора андрогена настоящего изобретения.
IN VITRO И IN VIVO ИСПЫТАНИЯ ДЛЯ ОПРЕДЕЛЕНИЯ СОЕДИНЕНИЙ, ОБЛАДАЮЩИХ АКТИВНОСТЬЮ SARM
1. Анализ сродства соединения к эндогенно экспрессируемому AR методом вытеснения меченного радиоактивным изотопом лиганда на основе гидроксилапатита
Буфер для связывания: TEGM (10 мМ Трис-HCl, 1 мМ EDTA, 10% глицерин, 1 мМ бета-меркаптоэтанол, 10 мМ молибдат натрия, pH 7,2)
50% Суспензия HAP: Calbiochem Hydroxylapatite, Fast Flow, в 10 мМ Трис, pH 8,0 и 1 мМ EDTA.
Буфер для промывки: 40 мМ Трис, pH 7,5, 100 мМ KCl, 1 мМ EDTA и 1 мМ EGTA.
95% EtOH
Метилтриенолон, [17α-метил-3H], (R1881*); NEN NET590
Метилтриенолон (R1881), NEN NLP005 (растворять в 95% EtOH)
Дигидротестостерон (DHT) [1,2,4,5,6,7-3H(N)] NEN NET453
Гидроксилапатит Fast Flow; Calbiochem Cat#391947
Молибдат = Молибденовая кислота (Sigma, M1651)
Культуральные среды клеток MDA-MB-453:
RPMI 1640 (Gibco 11835-055) w/23,8 мМ NaHCO3, 2 мМ L-глутамин
Gibco#15710-072)
Пассаж клеток:
Клетки (Hall R. E., et al., European Journal of Cancer, 30A: 484-490 (1994)) промывают дважды в PBS, свободную от фенолового красного смесь Трипсин-EDTA разбавляют в том же самом PBS 1:10. Клеточные слои промывают 1X трипсином, излишний трипсин сливают и клеточные слои инкубируют при 37°C в течение ˜2 мин. Постукивая по флакону, устанавливают момент наступления отслаивания клеток. Как только клетки начнут соскальзывать с сосуда, добавляют полную среду для уничтожения трипсина. В этот момент клетки подсчитывают, затем разбавляют до соответствующей концентрации и разделяют по сосудам или чашкам для дальнейшего культивирования (обычно разбавление от 1:3 до 1:6).
Получение лизата клеток MDA-MB-453
После того как клетки MDA достигнут от 70 до 85% слияния, их отслаивают, как описано выше, и собирают, центрифугируя при 1000g в течение 10 мин при 4°C. Осадок клеток промывают дважды TEGM (10 мМ Трис-HCl, 1 мМ EDTA, 10% глицерин, 1 мМ бета-меркаптоэтанол, 10 мМ молибдат натрия, pH 7,2). После конечной промывки клетки ресуспендируют в TEGM при концентрации 107 клеток/мл. Клеточную суспензию быстро замораживают в жидком азоте или на бане со смесью этанол/сухой лед и переносят в морозильную камеру при -80°C на сухой лед. До того как приступить к испытанию на связывание, замороженные образцы оставляют на смеси лед-вода только для оттаивания (˜1 час). Затем образцы центрифугируют при 12500g-20000g в течение 30 мин при 4°C. Супернатант используют для постановки испытания немедленно. При использовании 50 мкл супернатанта испытуемое соединение можно приготовить в 50 мкл буфера TEGM.
Способ множественного скрининга соединения:
Получают 1x TEGM буфер, и содержащую изотоп смесь для анализа получают в следующем порядке: EtOH (конечная концентрация в реакционной смеси 2%), 3H-R1881 или 3H-DHT (конечная концентрация в реакционной смеси 0,5 нМ) и 1х TEGM [например, для 100 образцов, 200 мкл (100 x 2) EtOH + 4,25 мкл 1:10 вещества 3H-R1881 + 2300 мкл (100 x 23) 1x TEGM]. Соединение последовательно разбавляют, например, если исходная конечная концентрация составляет 1 мкМ и соединение находится в 25 мкл раствора, для дубликатных образцов приготавливают 75 мкл 4 Х 1 мкМ раствор и 3 мкл 100 мкМ добавляют к 72 мкл буфера, и серийное разбавление 1:5.
Сначала смешивают вместе 25 мкл 3H-R1881-содержащего раствора и 25 мкл раствора соединения, с последующим добавлением 50 мкл раствора рецептора. Реакционную смесь осторожно перемешивают, в течение короткого времени центрифугируют примерно при 200 об/мин и инкубируют при 4°C на протяжении ночи. Получают 100 мкл 50% суспензии HAP и добавляют к инкубированной реакционной смеси, которую затем интенсивно встряхивают и инкубируют на льду в течение от 5 до 10 минут. Реакционную смесь интенсивно встряхивают еще два раза, чтобы ресуспендировать HAP при инкубировании реакционной смеси. Затем образцы в 96-луночном планшете промывают буфером для промывки, используя универсальную машину для промывки планшета для сбора культивируемых клеток FilterMate™ (Packard). Процесс промывки переносит осадок HAP, содержащий связанный с лигандом экспрессированный рецептор, в Unifilter-96 GF/B фильтровальный планшет (Packard). Осадок HAP на фильтровальном планшете инкубируют с 50 мкл сцинтилляционной жидкости MICROSCINT (Packard) в течение 30 минут перед подсчетом на микросцинтилляционном счетчике TopCount (Packard). Рассчитывают значения IC50, используя R1881 в качестве эталона. Тканеселективные модуляторы рецептора андрогена настоящего изобретения показывают значения IC50 1 микромоль или меньше.
2. Подавление промотора MMP1, тест неустойчивой трансфекции (TRAMPS)
Клетки HepG2 культивируют в не содержащей фенолового красного MEM, содержащей 10% FCS, обработанную древесным углем, при 37°C с 5% СО2. Для трансфекции клетки помещают при 10000 клеток/лунку в 96-луночные планшеты с белыми чистыми лунками. Спустя двадцать четыре часа клетки переносят совместно с репортерной конструкцией промотор MMP1-люцифераза и экспрессирующей конструкцией макак резус (отношение 50:1), используя реагент для трансфекции FuGENE6, следуя протоколу, рекомендованному производителем. Репортерную конструкцию промотор MMP1-люцифераза генерируют включением фрагмента (-179/+63) человеческого промотора MMP1 в репортерную конструкцию (Promega) pGL2 люциферазы и конструкцию экспрессии AR макак резус генерируют в экспрессирующем векторе CMV-Tag2B (Stratagene). Затем клетки культивируют в течение 24 часов и затем обрабатывают испытуемыми соединениями в присутствии 100 нМ форбол-12-миристат-13-ацетата (PMA), используемого для увеличения базальной активности промотора MMP1. Соединения добавляют в указанный момент в диапазоне концентраций от 100 до 0,03 нМ, 10 разбавлений, при концентрации по 10Х, 1/10 объема (пример: 10 микролитров лиганда при 10X добавляют к 100 микролитрам среды, уже присутствующей в лунке). Далее клетки культивируют в течение дополнительных 48 часов. Затем клетки промывают дважды PBS и лизируют, добавляя в лунки 70 мкл буфера для лизиса (1x, Promega). Активность люциферазы измеряют в 96-луночном планшете, используя люменметр 1450 Microbeta Jet (Perkin Elmer). Активность испытуемых соединений представляют как подавление сигнала люциферазы исходя из контрольных уровней, стимулированных PMA. Приводятся значения EC50 и Eмакс. Тканеселективные модуляторы рецептора андрогена по данному изобретению активируют репрессию, как правило, при субмикромолярных значениях EC50 и значениях Eмакс выше, чем около 50%.
Ссылки:
a. Newberry EP, Willis D, Latifi T, Boudreaux JM, Towler DA, "Fibroblast growth factor receptor signaling activates the human interstitial collagenase promoter via the bipartite Ets-APl element," Mol. Endocrinol.11: 1129-44 (1997).
b. Schneikert J, Peterziel H, Defossez PA, Klocker H, Launoit Y, Cato AC, "Androgen receptor-Ets protein interaction is a novel mechanism for steroid hormone-mediated down-modulation of matrix metalloproteinase expression," J. Biol. Chem. 271: 23907-23913 (1996).
3. Двухгибридный тест на индуцированное лигандом взаимодействие между N-концевым и С-концевым доменами рецептора андрогена на млекопитающем (поведение агониста)
Этот тест оценивает способность агонистов AR индуцировать взаимодействие между N-концевым доменом (NTD) и С-концевым доменом (CTD) rhAR, которая отражает in vivo потенциал вирилизации, опосредованный активированными рецепторами андрогена. Взаимодействие NTD и CTD rhAR количественно определяют как индуцированную лигандом ассоциацию между слитым белком Gal4DBD-rhARCTD и слитым белком в двухгибридном тесте млекопитающего в клетках CV-1 почки обезьяны.
За день до трансфекции клетки CV-1 трипсинизируют и подсчитывают, а затем помещают при 20000 клеток/лунку в 96-луночные планшеты или в бульшие планшеты (в увеличенном масштабе, соответственно) в DMEM + 10% FCS. На следующее утро клетки CV-1 котрансфицируют с pCBB1 (конструкция слитого белка Gal4DBD-rhARLBD, экспрессируемая под контролем раннего промотора SV40), pCBB2 (конструкция слитого белка VP16-rhAR NTD, экспрессируемая под контролем раннего промотора SV40) и pFR (репортер, чувствительный к Gal4-люциферазе, Promega), используя реагент LIPOFECTAMINE PLUS (GIBCO-BRL), следуя методике, рекомендованной поставщиком. Кратко, смесь ДНК из 0,05 мкг pCBBl, 0,05 мкг pCBB2 и 0,1 мкг pFR смешивают в 3,4 мкл OPTI-MEM (GIBCO-BRL), смешанной с "PLUS Reagent" (1,6 мкл, GIBCO-BRL), и инкубируют при комнатной температуре (к.т.) в течение 15 мин, получая прекомплексованную ДНК.
Для каждой лунки 0,4 мкл Реагента LIPOFECTAMINE (GIBCO-BRL) разбавляют в 4,6 мклOPTI-MEM во второй пробирке и смешивают, получая разбавленный реагент LIPOFECTAMINE. Прекомплексованную ДНК (указанная выше) и разбавленный реагент LIPOFECTAMINE (указанный выше) объединяют, смешивают и инкубируют в течение 15 мин при к.т. Среду на клетках заменяют 40 мкл/лунку OPTI-MEM, и 10 мкл комплексов ДНК-липид добавляют в каждую лунку. Комплексы осторожно смешивают в среде и инкубируют при 37°C при 5% CO2 в течение 5 ч. После инкубации добавляют 200 мкл/лунку D-MEM и 13% FCS, очищенной древесным углем, с последующей инкубацией при 37°C при 5% CO2. Спустя 24 часа испытуемые соединения добавляют при желательной(ых) концентрации(ях) (1 нМ-10 нМ). Через сорок восемь часов измеряют активность люциферазы, используя систему LUC-Screen (TROPIX), следуя протоколу производителя. Анализ проводят непосредственно в лунках путем последовательного добавления, по 50 мкл каждого, сначала раствора 1 для анализа, а затем раствора 2 для анализа. После инкубации в течение 40 минут при комнатной температуре люминесценцию измеряют непосредственно с интервалом 2-5 секунд.
Активность испытуемых соединений рассчитывают как Emax относительно активности, получаемой с 3 нМ R1881. Типичные тканеселективные модуляторы рецептора андрогена настоящего изобретения в этом испытанииили не обнаруживают никакой активности агониста, или обнаруживают слабую активность агониста менее чем 50% при 10 микромолях.
Ссылка:
He B, Kemppainen JA, Voegel JJ, Gronemeyer H, Wilson EM, "Activation function in the human androgen receptor ligand binding domain mediates inter-domain communication with the NH(2)-terminal domain," J. Biol. Chem. 274: 37219-37225 (1999).
4. Двухгибридный тест на ингибирование взаимодействия между N-концевым и С-концевым доменами рецептора андрогена на млекопитающем (поведение антагониста)
Это испытание оценивает способность испытуемых соединений антагонизировать стимулирующие действия R1881 на взаимодействие между NTD и CTD rhAR в двухгибридном тестев клетках CV-1, как описано выше.
Через сорок восемь часов после трансфекции клетки CV-1 обрабатывают испытуемыми соединениями, как правило, при конечных концентрациях 10 мкМ, 3,3 мкМ, 1 мкМ, 0,33 мкМ, 100 нМ, 33 нМ, 10 нМ, 3,3 нМ и 1 нМ. После инкубации при 37°C при 5% CO2 в течение 10-30 минут агонист AR, метилтриенолон (R1881), добавляют до конечной концентрации 0,3 нМ и инкубируют при 37°C. Через сорок восемь часов измеряют активность люциферазы, используя систему LUC-Screen (TROPIX), следуя протоколу, рекомендованному производителем. Способность испытуемых соединений антагонизировать действие R1881 рассчитывают как относительную люминесценцию по сравнению со значением в случае использования одного R1881 при концентрации 0,3 нМ.
Соединения-SARM по данному изобретению обычно обнаруживают в этом испытании антагонистическую активность при значениях IC50 менее чем 1 микромоль.
5. Модуляция транс-активации рецептора андрогена (TAMAR)
Это испытание оценивает способность испытуемых соединений регулировать транскрипцию с репортерного гена MMTV-LUC в клетках MDA-MB-453, клеточная линия рака молочной железы человека, которая в природе экспрессирует человеческий AR. Испытание измеряет индукцию модифицированного MMTV LTR/промотора, связанного с репортерным геном LUC.
От 20000 до 30000 клеток/лунку помещают в белый 96-луночный планшет с чистыми лунками в «Среду экспоненциального роста», которая состоит из RPMI1640 без фенолового красного, содержащей 10% FBS, 4 мМ L-глутамин, 20 мМ HEPES, 10 мкг/мл человеческого инсулина и 20 мкг/мл гентамицина. Условия инкубирования составляют 37°C и 5% CO2. Трансфекцию проводят в периодическом режиме. Клетки трипсинизируют и подсчитывают до нужного числа клеток в соответствующем количестве свежей среды, затем осторожно смешивают с коктейлем Fugene/ДНК и помещают на 96-луночный планшет. Во все лунки добавляют 200 мкл среды + комплекс липид/ДНК и затем инкубируют при 37°C на протяжении ночи. Коктейль для трансфекции состоит из бессывороточного Optimem, реагента Fugene6 и ДНК. При компоновке коктейля следуют рекомендациям производителя (Roche Biochemical). Отношение липида (мкл) к ДНК (мкг) составляет приблизительно 3:2, и время инкубации составляет 20 мин при комнатной температуре. Через 16-24 часа после трансфекции клетки обрабатывают испытуемыми соединениями, так чтобы конечная концентрация ДМСО (носителя) составляла <3%. Клетки подвергают воздействию испытуемых соединений в течение 48 часов. Спустя 48 часов клетки лизируют лизирующим буфером для клеточных культур Promega в течение 30-60 минут и затем анализируют активность люциферазы в экстрактах в 96-луночном планшетном люменметре.
Активность испытуемых соединений рассчитывают как Eмакс относительно активности, полученной при 100 нМ R1881.
Ссылки:
a. R.E. Hall, et al., "MDA-MB-453, an androgen-responsive human breast carcinoma cell line with high androgen receptor expression," Eur. J. Cancer, 30A: 484-490 (1994).
b. R.E. Hall, et al., "Regulation of androgen receptor gene expression by steroids and retinoic acid in human breast-cancer cells," Int. J. Cancer., 52: 778-784 (1992).
6. In Vivo анализ простаты
Крыс-самцов линии Sprague-Dawley в возрасте 9-10 недель, самый ранний возраст половой зрелости, используют в режиме профилактики. Цель состоит в измерении степени замедления андрогенподобными соединениями резкой деградации (˜-85%) вентральной предстательной железы и семенных пузырьков, которая проявляется на седьмой день после удаления яичек (орхиэктомия [ORX]).
Крыс подвергают орхиэктомии (ORX). Каждую крысу взвешивают, затем анестезируют газом-изофлюраном, и это состояние поддерживают для осуществления операции. Делают 1,5-см переднезадний разрез в мошонке. Правое яичко временно выводят на поверхность тела. Семенную артерию и семявыводящий проток лигируют 4,0 шелковой нитью на 0,5 см проксимальнее яичка. Яичко отрезают маленькими хирургическими ножницами дистальнее лигатуры. Культю ткани возвращают в мошонку. Такую же операцию повторяют для левого яичка. После того как обе культи возвращают в мошонку, мошонку и вышележащую кожу сшивают 4,0 шелком. В случае (фиктивно)-ORX крыс осуществляются все процедуры, кроме лигирования и отрезания ножницами. Крысы полностью приходят в себя и полностью восстанавливают подвижность в пределах 10-15 минут.
Крысе вводят подкожно или перорально дозу испытуемого соединения сразу после наложения шва на хирургический разрез. Обработку испытуемым соединением продолжают в течение дополнительных шести последующих дней.
Вскрытие трупа и результаты вскрытия
Сначала крысу взвешивают, затем анестезируют в камере, наполненной СО2, до предсмертного состояния. Приблизительно 5 мл цельной крови отбирают кардиальной пункцией. Затем крысу обследуют на наличие признаков, подтверждающих гибель животного и завершенность ORX. Затем определяют местоположение вентральной части предстательной железы и аккуратно отслаивают. Вентральную простату сушат при помощи промокательной бумаги на протяжении 3-5 секунд и затем взвешивают (VPW). Наконец, определяют местоположение семенного пузырька и отслаивают его. Вентральный семенной пузырек сушат при помощи промокательной бумаги на протяжении 3-5 секунд и затем взвешивают (SVWT).
Первичными данными для этого анализа являются массы вентральной простаты и семенного пузырька. Вторичные данные включают сывороточный LH (лютеинизирующий гормон) и FSH (фолликулостимулирующий гормон) и возможные сывороточные маркеры остеогенеза (костеобразования) и вирилизации (андрогенизации). Данные анализируют с помощью ANOVA плюс Fisher PLSD post-hoc теста для идентификации различия между группами. Оценивают степень ингибирования испытуемыми соединениями потери VPW и SVWT, вызванной ORX.
7. In Vivo анализ остеогенеза
Крыс-самок линии Sprague-Dawley в возрасте 7-10 месяцев используют в режиме лечения, чтобы симулировать зрелых женщин. Крыс подвергают овариэктомии (OVX) за 75-180 дней до начала эксперимента, чтобы вызвать разрежение кости и симулировать недостаточность эстрогена, столь характерные для взрослых людей женского пола с нарушением остеогенеза (остеопения). Предварительное лечение низкой дозой сильнодействующего антирезорбционного средства, алендроната (0,0028mpk, подкожно, 2X/неделю) начинают на день 0. На день 15 начинают лечение испытуемым соединением. Лечение испытуемым соединением проводят по дням 15-31 со вскрытием трупа на день 32. Цель состоит в измерении степени увеличения остеогенеза андрогенподобными соединениями, обнаруживаемого по повышению мечения флуорохромом, на периостальной поверхности.
В обычном испытании исследуют девять групп из семи крыс каждая.
В дни 19 и 29 (пятый и пятнадцатый дни лечения) каждая крыса получала разовую подкожную инъекцию кальцеина (8 мг/кг).
Вскрытие трупа и результаты вскрытия
Сначала крысу взвешивают, затем анестезируют в камере, наполненной СО2, до предсмертного состояния. Приблизительно 5 мл цельной крови отбирают кардиальной пункцией. Затем крысу обследуют на наличие признаков, подтверждающих гибель животного и завершенность OVX. Сначала определяют местоположение матки и затем аккуратно отслаивают, сушат при помощи промокательной бумаги на протяжении 3-5 секунд и затем взвешивают (UW). Матку помещают в 10% забуференный до нейтральности формалин. Затем правую ногу ампутируют в области бедра. Бедренную кость и большую берцовую кость отделяют в области колена, по существу, очищают от плоти и затем помещают в 70% этанол.
1-см сегмент в центре правой бедренной кости, содержащий в центре проксимально-дистальную среднюю точку бедра, помещают в сцинтилляционную пробирку, дегидратируют и обезжиривают в спиртах высокой концентрации и ацетоне, затем вводят в растворы с возрастающими концентрациями метилметакрилата. Сегмент погружают в смесь 90% метилметакрилата:10% дибутилфталата, которой дают возможность полимеризоваться на протяжении (48-72)-часового периода. Сосуд раскалывают и пластиковый блок обрезают в форму, котораяхорошо захватывается тисочным держателем образцаLeica 1600 Saw Microtome,при этом продольную ось кости препарируют на поперечные срезы.Получают три поперечных среза толщиной 85 мкм и помещают на предметные стекла. От каждой крысы выбирают один срез, который аппроксимирует середину (центр) кости, и проводят слепое кодирование. Периостальную поверхность каждого среза оценивают на общую периостальную поверхность, несущую единственную флуорохромную метку, двойную флуорохромную метку, и на расстояние между метками.
Первичные данные для этого испытания представляют процент периостальной поверхности, несущей двойную метку, и степень аппозиции минерального матрикса (расстояние между метками (мкм)/10d), полунезависимые маркеры костеобразования (остеогенеза). Вторичные данные включают массу матки и гистологические особенности. Третичные данные могут включать сывороточные маркеры костеобразования и вирилизации. Данные анализируют при помощи ANOVA плюс Fisher PLSD post-hoc теста для идентификации различия между группами. Оценивают степень увеличения остеогенеза, обусловленную лечением испытуемыми соединениями.
Хотя приведенное выше описание раскрывает принципы настоящего изобретения с примерами, предназначенными для иллюстрации, подразумевается, что практика изобретения охватывает все из обычных вариаций, адаптаций или модификаций, которые не выходят за рамки объема нижеследующей формулы изобретения, и их эквиваленты.
Описывается соединение формулы I или его фармацевтически приемлемая соль или его энантиомер, где n равно 0 или 1; a-b означает CF=CH или CHFCH2; R1 представляет C1-3алкил, где алкил является незамещенным; R2 представляет собой водород; R3 выбран из С1-4алкила, (СН2)n-циклогетероалкила и (СН2)n-арила; или R2 и R3 вместе образуют 6-членное насыщенное кольцо, конденсированное с 5-членной ароматической кольцевой системой, имеющей 2 гетероатома, выбранных из N, фармацевтические композиции. Соединения формулы I представляют собой модуляторы рецептора андрогена (AR), при этом обладающие тканеселективным действием. Указанные соединения полезны в качестве агонистов рецептора андрогена в костной и/или мышечной ткани при антагонизировании AR в простате пациента мужского пола или в матке пациента женского пола. 18 н. и 18 з.п. ф-лы, 2 табл.
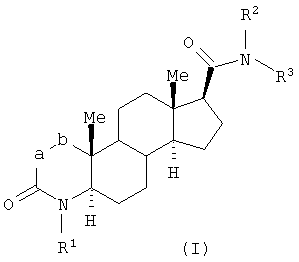
или его фармацевтически приемлемая соль или его энантиомер,
где n равно 0 или 1;
a-b означает CF=CH или CHFCH2;
R1 представляет C1-3алкил, где алкил является незамещенным;
R2 представляет собой водород;
R3 выбран из С1-4алкила, (СН2)n-циклогетероалкила и (СН2)n-арила, где арил выбран из
(1) фенила,
(2) бензимидазолила,
(3) бензотиазолила,
(4) 2,3-дигидро-1,4-бензодиоксинила,
(5) фуранила,
(6)тиенила,
(7) имидазолила,
(8) тиазолила,
(9) пиразолила,
(10) пиридила,
(11) инданила и
(12) имидазо [4,5-b] пиридинила;
где циклогетероалкильная группа является оксациклопентильной или оксациклогексильной группой, и
где алкильная группа или циклогетероалкильная группа является незамещенной или замещенной одним заместителем, выбранным из галогена; арильная группа, определенная в пп.1-12, является незамещенной или замещена одной-тремя группами, независимо выбранными из галогена, C1-8алкила и перфтор-С1-4алкила; и где углеродный атом любого метилена (CH2) в (СН2)n является незамещенным или замещен одной-двумя группами, независимо выбранными из С1-4алкила;
или R2 и R3 вместе образуют 6-членное насыщенное кольцо, конденсированное с 5-членной ароматической кольцевой системой, имеющей 2 гетероатома, выбранных из N.
N-(2-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-трифторметилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-хлорфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(4-метоксифенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-метоксифенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-метилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-метилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(4-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(4-хлор-2-фторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,4-дифторфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(α-метилфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N(фенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(4-хлор-2-трифторметилфенил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-метилпиридин-2-ил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тиофен-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тиофен-3-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-трифторметилфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(бензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(1-метилбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(1-метил-5-трифторметилбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-хлорбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-метоксибензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(бензтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(4-метилтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тиазол-4-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(1-метилимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2H-пиран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2H-пиран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3H-имидазо[4,5-b]пиридин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(2-трифторметилфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(3-метоксифенил)-2а-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(4-метоксифенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(2-трифторметилфенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(2-хлорфенил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(бензимидазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(1-метилбензимидазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(тиазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(фуран-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида; и
N-(тиофен-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
N-(2-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-хлорбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-метоксибензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(бензтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2H-пиран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2H-пиран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3H-имидазо[4,5-b]пиридин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(тиазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(фуран-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида и
N-(тиофен-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
N-(тетрагидро-2H-пиран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидро-2H-пиран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2,3-дигидро-1,4-бензодиоксин-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(S)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(тетрагидрофуран-2(R)-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N(3H-имидазо[4,5-b]пиридин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
N-(2-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-хлорбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(5-метоксибензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(бензтиазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-фторфенилметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(тиазол-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
N-(фуран-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида и
N-(тиофен-2-илметил)-2α-фтор-4-метил-3-оксо-4-аза-5α-андростан-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
N-(2,2,2-трифторэтил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-фторфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(2-трифторметилфенилметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(бензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(1-метилбензимидазол-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
N-(3H-имидазо[4,5-b]пиридин-2-илметил)-2-фтор-4-метил-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида;
их фармацевтически приемлемых солей и их энантиомеров.
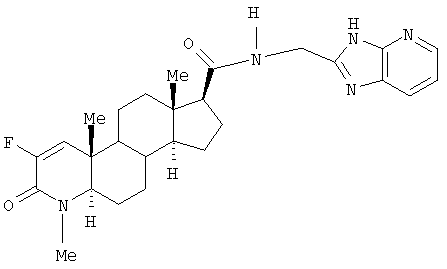
или его фармацевтически приемлемые соли.
| Brooks J.R | |||
| et al., 5 Alpha-reductase inhibitory and anti-androgenic activities of some 4-azasteroids in the rat | |||
| Steroids, 1986, 47(1), p.1-19 | |||
| Rasmusson G.H | |||
| et al., Azasteroids: structure-activity relationships for inhibition of 5 alpha-reductase and of androgen receptor binding | |||
| J Med Chem., 1986, Nov;29(11), p.2298-315 | |||
| US 5187278 A, 16.02.1993 | |||
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |

