Изобретение относится к новой композиции для лечения или предупреждения рака простаты, применению соединения для производства фармацевтической композиции для лечения или предупреждения рака простаты и способу лечения или предупреждения рака простаты посредством введения эффективного количества соединения теплокровным животным.
Ингибитор тестостерон-5α-редуктазы известен как терапевтическое средство для лечения доброкачественной гиперплазии (гипертрофии) простаты, которое уменьшает размер простаты, ингибируя рост клеток простаты, вызванный избытком мужских гормонов (главным образом дигидротестостерона). К тому же ингибиторы тестостерон-5α-редуктазы, такие как финастерид, продают как терапевтическое средство для лечения доброкачественной гиперплазии простаты в Соединенных Штатах Америки и Европе. С некоторыми ингибиторами тестостерон-5α-редуктазы проводят клинические исследования с целью разработки на их основе терапевтических средств для лечения рака простаты.
Восстановительное воздействие на простату соответствует величине активности тестостерон-5α-редуктазы. Однако, в отличие от доброкачественной гиперплазии простаты, терапевтическое действие при раке простаты не соответствует только величине активности ингибитора тестостерон-5α-редуктазы, так как большое число факторов участвует в пролиферации при раке простаты.
Не все из соединений, включенных в данную заявку, являются неизвестными соединениями; например, N-[1-метил-1-(4-метоксифенил)этил)-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид представляет собой соединение, описанное в заявке на патент Японии (Kokai) Hei 8-73492 и в патенте Японии (Kokoku) Hei 8-19151, и является ингибитором тестостерон-5α-редуктазы.
В течение многих лет заявители данного изобретения проводят серьезные исследования синтеза производных, характеризующихся активностью ингибитора тестостерон-5α-редуктазы, и их фармакологической активности. Удалось установить, что соединения, подобные вышеупомянутым соединениям, имеющие специфическую структуру, оказывают прекрасное лечебное и профилактическое действие при раке простаты, что и представляют в данном изобретении.
Целью данного изобретения является композиция для лечения или предупреждения рака простаты. Кроме того, другим объектом данного изобретения является применение вышеупомянутого соединения для получения фармацевтической композиции для лечения или предупреждения рака простаты, и способ для лечения или предупреждения рака простаты, включающий введение эффективного количества вышеупомянутого соединения теплокровным животным.
Новая композиция данного изобретения для лечения или предупреждения рака простаты содержит в качестве активного ингредиента соединение формулы (I), изображенное ниже, его фармакологически приемлемые соли или другое его производное, предпочтительно содержит в качестве активного ингредиента N-[1-метил-1-(4-метоксифенил)этил] -3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид, и назначают его наиболее предпочтительно перорально.
Кроме того, предлагается новое применение соединения формулы (I), предствленного ниже, его фармакологически приемлемых солей или другого его производного для получения фармацевтической композиции для лечения или предупреждения рака простаты, предпочтительно применение N-[1-метил-1-(4-метоксифенил)этил]-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида для производства фармацевтической композиции для лечения или предупреждения рака простаты, и назначают его предпочтительно перорально.
Новый способ лечения или профилактики данного изобретения представляет собой способ лечения или предупреждения рака простаты, состоящий из введения эффективного количества соединения формулы (I), изображенного ниже, его фармакологически приемлемых солей или другого его производного теплокровным животным, предпочтительно способ лечения или предупреждения рака простаты, состоящий из введения эффективного количества N-[1-метил-1-(4-метоксифенил)этил] -3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида теплокровным животным, наиболее предпочтительно назначают его перорально.

где R1 и R2 одинаковые или различные и представляют собой атом водорода, гидроксильную группу или группу низшего алкокси.
В формуле (I),
термин "низшая алкокси группа" означает алкокси группу с линейной или разветвленной цепью, содержащую от 1 до 6 атомов углерода, такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изо-бутокси, s-бутокси, трет-бутокси, н-пентокси, изопентокси, 2-метилбутокси, неопентокси, н-гексилокси, 4-метилпентокси, 3-метилпентокси, 2-метилпентокси, 3,3-диметилбутокси, 2,2-диметилбутокси, 1,1-диметилбутокси, 1,2-диметилбутокси, 1,3-диметилбутокси и 2,3-диметилбутокси, предпочтительно алкокси группу с линейной или разветвленной цепью, содержащую от 1 до 4 атомов углерода, более предпочтительно метокси группу.
Термин "его фармакологически приемлемые соли" подразумевает соединение (I) данного изобретения, которое превращают в его соли, примеры таких солей предпочтительно включают соли щелочных металлов, такие как соль натрия, соль калия или соль лития, соли щелочноземельных металлов, такие как соль кальция и соль магния, и соли металлов, такие как соль алюминия, соль железа и соль цинка.
Находясь на воздухе, соединение (I) данного изобретения поглощает некоторое количество влаги и в результате присоединяет абсорбированную воду или превращается в соответствующий гидрат. Данное изобретение включает также и такие соединения.
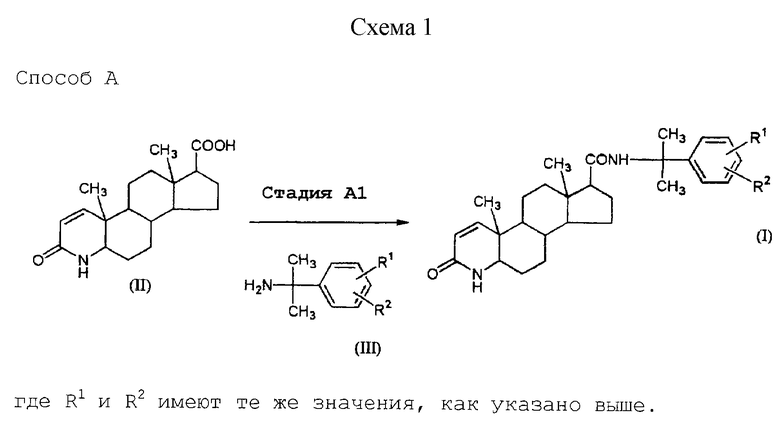
Соединение (I) данного изобретения получают по способу, представленному в схеме 1 (см. в конце описание).
Способ А представляет собой способ получения требуемого соединения (I) путем конденсации производного карбоновой кислоты (II) с аминопроизводным (III).
Стадия А1 представляет собой получение соединения (I) реакцией соединения (II) или его реактивного производного с соединением (III). Реакцию проводят в соответствии с общепринятыми способами пептидного синтеза, например, азидным способом, способом активированных сложных эфиров, способом смешанных ангидридов или способом конденсации.
Из вышеупомянутых способов, азидный способ выполняют следующим образом: соединение (II) или его эфир взаимодействует с гидразином в инертном растворителе (например, в диметилформамиде) приблизительно при комнатной температуре, чтобы получить гидразид аминокислоты. Гидразид аминокислоты реагирует с соединением азотистой кислоты до получения производного азида, за которым следует обработка производного азида аминопроизводным (III).
Используемое в изобретении соединение азотистой кислоты включает, например, нитриты щелочных металлов, такие как нитрит натрия или нитриты алкилов, такие как изоамиловый нитрит.
Реакцию предпочтительно проводят в инертном растворителе, а используемый в изобретении растворитель включает, например, амиды, такие как диметилформамид и диметилацетамид, сульфоксиды, такие как диметилсульфоксид, и пирролидоны, такие как N-метилпирролидон. Две стадии реакции (получение азида и производного амида (I)) обычно проводят в одном реакционном тигле. Температура реакции колеблется от -50oС до 0oС для первой реакции и от -10oС до 10oС для второй реакции, а время реакции составляет от 5 минут до 1 часа для первой реакции и от 10 часов до 5 дней для второй реакции.
Согласно способу активированных эфиров реакцию проводят между соединением (II) и агентом для получения активированных эфиров с последующей реакцией активированного эфира с аминопроизводным (III).
Обе реакции предпочтительно проводят в инертном растворителе, а используемый в изобретении растворитель включает, например, галоидзамещенные углеводороды, такие как хлористый метилен и хлороформ, простые эфиры, такие как диэтиловый эфир и тетрагидрофуран, амиды, такие как диметилформамид и диметилацетамид, и нитрилы, такие как ацетонитрил.
Применяемый в изобретении агент этерификации включает, например, N-гидроксильные соединения, такие как N-гидроксисукцинимид, 1-гидроксибензотриазол и N-гидрокси-5-норборнен-2,3-дикарбоксимид или дисульфидные соединения, такие как дипиридилдисульфид. Реакцию активированной этерификации предпочтительно проводят в присутствии агента конденсации, такого как дициклогексилкарбодиимид, карбонилдиимидазол или трифенилфосфин.
Реакция этерификации протекает при температуре от -10oС до 100oС и приблизительно при комнатной температуре протекает реакция активированного эфирного соединения с аминопроизводным (III), а время реакции составляет от 30 минут до 80 часов для обеих реакций.
При проведении реакции активированного эфира с амином в реакционную систему добавляют 4-диметиламинопиридин.
При осуществлении способа смешанных ангидридов получают смешанный ангидрид соединения (II) с последующей реакцией смешанного ангидрида с аминопроизводным.
Реакцию получения производного смешанного ангидрида выполняют посредством проведения реакции соединения (II) с агентом, для образования производного смешанного ангидрида (например, галоидзамещенные угольные кислоты низших (C1-C4) алкилов, таких как этиловый эфир хлоругольной кислоты и изобутил хлоругольной кислоты, галоидное соединение низшего алканоила, такое как хлористый пивалоил, низший алкил или диарил цианфосфорных кислот, такие как диэтиловый эфир цианфосфорной кислоты и дифенилцианфосфат, или сульфонил-галоидные соединения, такие как 2,4,6-триизопропилбензолсульфонилхлорид, паратолуолсульфонилхлорид и метансульфонилхлорид) в инертном растворителе (например, галоидзамещенных углеводородах, амидах и простых эфирах, описанных выше). Предпочтительно реакцию проводят в присутствии органических аминов, таких как триэтиламин и N-метилморфолин, а температура реакции колеблется от -10oС до 50oС, время реакции составляет от 30 минут до 20 часов.
Реакцию производного смешанного ангидрида с аминопроизводным (III) предпочтительно проводят в инертном растворителе (например, галоидзамещенных углеводородах, амидах и простых эфирах, описанных выше) в присутствии органических аминов. Температура реакции располагается в области от 0oС до 80oС, а необходимое для реакции время составляет от 1 часа до 48 часов.
Реакцию проводят при одновременном присутствии соединения (II), соединения (III) и агента для образования производного смешанного ангидрида без изоляции производного смешанного ангидрида.
Способ конденсации осуществляют посредством реакции между соединением (II) и аминопроизводным (III) непосредственно в присутствии агента конденсации, такого как дициклогексилкарбодиимид, карбонилдиимидазол или йодид 2-хлор-1-метил-пиридиния/триэтиламин. Данную реакцию проводят по способу, аналогичному способу, описанному для получения активированного эфира.
В случае, когда защищенная гидроксильная группа находится в R1 и R2, защитную группу удаляют согласно общепринятым способам.
Неочищенное соединение (II) или его активированный эфир известны или их получают в соответствии с описанными способами (например, J. Med. Chem., 27, 1690 (1984); J. Med. Chem., 29, 2298 (1986).
К тому же, соединение (III) является известным или получают по способам [например:
Synthesis, 593 (1976);
J. Org. Chem., 36, 305 (1971);
Angew. Chem., 82, 138 (1970);
Synthesis, 24 (1978);
Synthetic Commun., 18, 777 (1988);
Synthetic Commun., 18, 783 (1988);
Organic Reaction, 3, 337 (1946);
Org. Synthesis, 51, 48 (1971);
Tetrahedron. 30, 2151 (1974); and
J. Org. Chem. , 37, 188 (1972)], и, например, неочищенное соединение данного изобретения, содержащее H2N=C(Me)(Me)-Ph(R1)(R2) компонент, получают по способу, описанному в Synthesis, P. 24 (1978). Далее см. схему 2 реакции (см. в конце описания), которая включает реакцию Гриньяра (Grignard), реакцию азидирования гидроксильной группы и реакцию восстановления.
Фиг. 1 в примере 1 демонстрирует противоопухолевое действие при раке простаты человека.
Вертикальная ось: относительное соотношение объема опухоли (выраженное в логарифме).
Горизонтальная ось: число дней после первоначального введения.
Пунктир с крестиками: контрольная группа.
Черные точки: группа введения финастерида.
Пунктир с кругами: группа введения соединения 1.
Соединение 1: N-[1-метил-1-(4-метоксифенил)этил)-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид.
В дальнейшем данное изобретение подробно описывают, ссылаясь на примеры и цитируемый пример, но не ограничивают этим сферу данного изобретения.
Пример 1 - исследование 1 противоопухолевой активности
Солидную опухоль, которая образуется под кожей, прививают посредством субкультивирования подкожно от пяти до шести раз самцам и самкам бестимусных мышей линии LNCaP рака простаты человека системы культивированных клеток, приобретенных у American Type Culture Collection (ATCC). В исследовании используют эту линию рака простаты человека. Фрагмент солидной раковой опухоли этой линии, квадрат 3 мм, трансплантируют под кожу в подмышечной области BALB/cA Jcl-nu бестимусным мышам (Nippon Clea, самцы, возраст 8 недель). Мышей, у которых достоверно определяют опухоль, приблизительно через 20 дней безвыборочно делят на группы по 8-10 животных в каждой. N-[1-метил-1-(4-метоксифенил)этил]-3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид (соединение 1, 100 мг/кг) или финастерид (100 мг/кг) вводят перорально мышам в дозе 0,1 мл/10 г веса тела один раз в день утром в течение 28 последовательных дней. Диаметр опухоли измеряют дважды в неделю, а объем опухоли V (V=ab2/2) рассчитывают по главной оси (а) и второстепенной оси (b). Относительное объемное соотношение для каждой мыши представляют как Vn/Vo. Степень ингибирования роста опухоли (%) определяют из относительного объемного соотношения для каждой мыши.
Степень ингибирования роста опухоли (%)=(1-Vn/Vo)x100.
Vn: Объем опухоли на n-й день.
Vo: Объем опухоли в первый день введения.
Результаты
Данные относительного соотношения объема представлены на чертеже.
Финастерид продают в Соединенных Штатах Америки и Европе в качестве лекарственного средства для лечения доброкачественной гиперплазии простаты, и в настоящее время проводят клинические исследования его в качестве лекарственного средства для лечения рака простаты.
Чертеж демонстрирует, что финастерид не проявляет никакой ингибирующей активности роста опухоли, в то время как соединение 1 проявляет ингибирующую активность роста опухоли, максимальная степень ингибирования роста опухоли которой в течение 28 дней составляет 30%. Таким образом, соединение 1 является полезным в качестве терапевтического средства для лечения рака простаты.
Пример 2 - исследование 2 противоопухолевой активности
В этом исследовании используют самцов бестимусных мышей (возраст 4-5 недель). Бестимусных мышей разводят в асептических условиях.
Клетки LNCaP рака простаты человека приобретают у American Type Culture Collection (ATCC). Клетки LNCaP (5х106) трансплантируют подкожно бестимусным мышам для образования солидной опухоли, а затем используют в исследовании.
Бестимусным мышам под анестезией делают разрез внизу живота приблизительно 2 см, чтобы обнажить простату. Оболочку простаты осторожно вскрывают и фрагмент опухоли LNCaP вводят внутрь. Отверстие в оболочке зашивают рассасывающимся швом. Простату возвращают в брюшную полость и разрез внизу живота зашивают рассасывающимся швом. Этих мышей безвыборочно определяют в группы по 20 животных в каждой. Эти группы обозначают: группа соединения 1, животным которой вводят соединение 1 (20 мг/кг); группа финастерида, животным которой вводят финастерид (20 мг/кг) и контрольная группа без дозы соединения.
Наличие трансплантируемой опухоли у мышей подтверждают, когда растущую опухоль можно измерить снаружи тела мыши, а затем начинают введение соединения (1) и финастерида. Соединение 1 и финастерид вводят перорально ежедневно в дозах, приведенных выше. Объем трансплантированной опухоли рассчитывают согласно формуле, представленной в примере 1. Кроме того, мышей, когда они погибают вследствие рака, немедленно вскрывают, а опухоль взвешивают. В то же время также устанавливают наличие метастазов. Тканевые срезы метастазов готовят пропитыванием парафином после фиксации 10% формалином. Тканевые срезы окрашивают гематоксилином и эозином с последующим исследованием метастазов.
По критерию Стьюдента осуществляют обработку данных относительно веса трансплантированных опухолей для всех групп. Кроме того, по критерию Фишера (Fisher) оценивают случаи метастазов в каждой из групп. Обработку данных по тесту-U Манн-Витней (Mann-Whitney) и критерию Стьюдента проводят для двух групп, которые сравнивают по продолжительности срока действия. В частности, частоту выживания оценивают по критерию хи-квадрат (χ2) согласно Пирсона (Pearson) на 63 день после трансплантации опухоли. Степень риска меньше 5% рассматривают как существенную для этих исследований.
Результаты представлены в таблице в конце описания.
Как видно из таблицы, группа соединения 1 показывает более высокую частоту выживания по сравнению с группой финастерида.
Цитируемый пример 1
N-[1-метил-1-(4-метоксифенил)этил] -3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамид.
1,0 г 3-оксо-4-аза-5α-андрост-1-ен-17β-карбоновой кислоты, 1,6 г трифенилфосфина и 1,4 г 2,2'-дипиридилдисульфида последовательно добавляют к 30 мл сухого толуола и смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь, как таковую, подвергают колоночной хроматографии на 35 г силикагеля и элюируют смесью ацетон/метиленхлорид (1:9 до 1:1), чтобы получить 1,11 г производного сложного 2-пиридилтиоэфира.
5,0 г производного сложного 2-пиридилтиоэфира синтезируют по вышеописанному способу и 5,0 г 1-(4-метоксифенил)-1-метилэтиламина последовательно добавляют к 30 мл сухого метиленхлорида и смесь перемешивают при комнатной температуре в течение 3 дней. Реакционную смесь разбавляют 100 мл метиленхлорида, последовательно промывают IN хлористоводородной кислотой, водой, водным бикарбонатом натрия и насыщенным солевым раствором. Слой метиленхлорида высушивают над сульфатом магния и затем концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии с использованием 15 г силикагеля и элюируют смесью ацетон/метиленхлорид (1:9 до 1:1), чтобы получить 5,2 г целевого соединения.
ЯМР спектр (СDСl3) δ частей на тысячу: 0,68 (3Н, с), 0,98 (3Н, с), 0,90-2,20 (16Н, м), 1,70 (3Н, с), 1,72 (3Н, с), 3,35 (1Н, т, J=9 Гц), 3,80 (3Н, с), 5,48 (1Н, шир.), 5,76 (1Н, шир.), 5,83 (1Н, д, J=10 Гц), 6,82 (1Н, д, J=10 Гц), 6,88 (2Н, д, J=9 Гц), 7,32 (2Н, д, J=9 Гц).
ИК спектр νmax см-1 (KBr): 2969, 2938, 1672, 1599, 1514, 1455, 1248, 1181, 1035, 825.
[Промышленное применение]
Соединение (1) данного изобретения характеризуется превосходной противоопухолевой активностью и слабой токсичностью. Таким образом, оно является полезным в качестве композиции для лечения и предупреждения рака простаты.
Соединение (1) или его фармакологически приемлемые соли данного изобретения применяют в качестве композиции для лечения или предупреждения рака простаты. Соединение (1) само или в смеси соединения (1) с соответствующими фармакологически приемлемыми наполнителями, разбавителями и подобными вводят перорально в виде таблеток, капсул, гранул, порошков или сиропов.
Эти фармацевтические композиций готовят стандартными способами, которые хорошо известны специалистам в данной области, используя добавки. Добавки представляют собой наполнители (например, органические наполнители, такие как производные сахаров, например, лактоза, сахароза, глюкоза, маннит и сорбит; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, α-крахмал, декстриновый и карбоксиметилированный крахмал; производные целлюлозы, такие как кристаллическая целлюлоза, низкозамещенная гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза, кальций карбоксиметилцеллюлоза и натрий карбоксиметилцеллюлоза с внутренними мостиками; аравийская камедь; декстран; и Пуллулан (Pullulan); и неорганические наполнители, такие как производные силиката, например, жидкий кремневый ангидрид, синтетический кремнекислый алюминий и магний-алюминат метакремниевой кислоты; фосфаты, например, фосфат кальция; карбонаты, например, карбонат кальция; и сульфаты, например, сульфат кальция); смазочные вещества (например, стеариновая кислота; металлические соли стеариновой кислоты, такие как стеариновокислый кальций и стеариновокислый магний; тальк; коллоидный кремниевый ангидрид; воски, такие как пчелиный воск и спермацет; борная кислота; адипиновая кислота; сульфаты, такие как сульфат натрия; гликоль; фумаровая кислота; бензойнокислый натрий; DL-лейцин; натриевые соли алифатических кислот; лаурилсульфаты, такие как лаурилсульфат натрия и лаурилсульфат магния; кремневые кислоты, такие как кремневый ангидрид и кремневый гидрат; и вышеупомянутые производные крахмала); связывающие вещества (например, поливинилпирролидон, макрогол и те же самые соединения, которые описывают в вышеупомянутых наполнителях); дезинтеграторы (например, те же самые соединения, которые описывают в вышеупомянутых наполнителях; и химически модифицированные крахмалы и целлюлозы, такие как натрий-кроскармелаза, натрий карбоксиметилкрахмал и поливинилпирролидон с внутренними мостиками); стабилизаторы (например, параоксибензоаты, такие как метилпарабензоат и пропилпарабензоат; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; хлористый бензалконий; фенолы, такие как фенол и крезол; тимерозал; дегидроуксусная кислота; и сорбиновая кислота); корригенты (например, подсластители, подкисляющие вещества и благовонные соединения, обычно используемые); и разбавители.
Доза колеблется в зависимости от состояния и возраста пациента. Например, желательно назначать 0,001 мг/кг веса тела (предпочтительно 0,01 мг/кг веса тела) в качестве нижнего предела и 20 мг/кг веса тела (предпочтительно 1 мг/кг веса тела) в качестве верхнего предела от одного до нескольких раз в день в зависимости от симптомов.
Примеры композиций представляют следующим образом. Однако эти примеры не ограничивают компетенцию изобретения.
Пример 1 композиции
Капсулы
Соединение цитируемого примера 1 - 20,0 мг
Лактоза - 158,7
Кукурузный крахмал - 70,0
Стеарат магния - 1,3 - 250 мг
Вышеописанные порошки смешивают и просеивают через сито 60 меш, а затем полученные в результате порошки капсулируют в желатиновые капсулы 3 по 250 мг для получения капсул.
Пример 2 композиции
Таблетки
Соединение цитируемого примера 1 - 20,0 мг
Лактоза - 154,0
Кукурузный крахмал - 25,0
Стеарат магния - 1,0 - 200 мг
Вышеописанные порошки смешивают и формируют в таблетки с помощью устройства для создания таблеток, чтобы получить таблетку 200 мг.
Если необходимо, таблетку покрывают сахаром.


Изобретение относится к медицине и касается новой композиции для лечения или предупреждения рака простаты, содержащей в качестве активного ингредиента N-[1-метил-1-(4-метоксифенил)этил] -3-оксо-4-аза-5α-андрост-1-ен-17β-карбоксамида, и способа лечения или предупреждения рака простаты с помощью указанной композиции. Композиция обладает повышенной активностью. 3 с. и 3 з.п. ф-лы, 1 ил., 1 табл.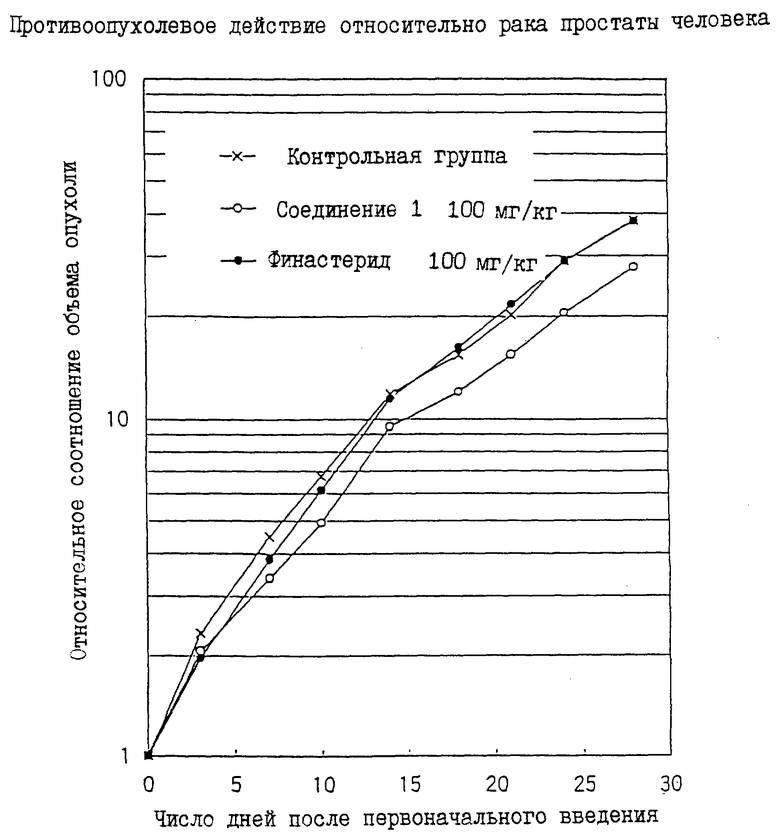
| JP 08073492 А, 19.03.1996 | |||
| US 4732897 А, 1985. |

