Область техники, к которой относится изобретение
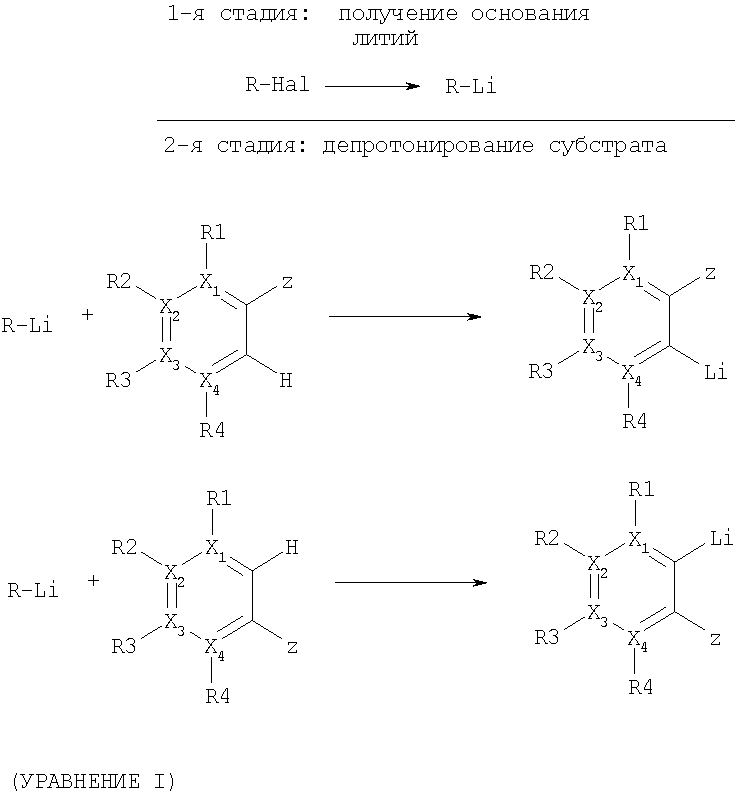
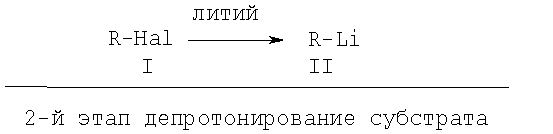
Изобретение относится к способу получения органических соединений путем образования соединений ариллития и их превращения с соответствующими электрофильными соединениями, причем сначала в результате взаимодействия галогенных соединений с металлическим литием получают соединение лития, которое затем с целью депротонирования ароматических соединений подвергают превращению с образованием требуемого ароматического соединения лития, причем при необходимости последнее приводят в реакцию с соответствующим электрофильным соединением (уравнение I):
Уровень техники
Развитие металлорганической химии, в частности химии лития, при получении соединений для фармацевтической и агрохимической промышленностей, а также для многочисленных других случаев применения в последние годы приобрело почти экспоненциальный характер, если при этом по оси времени откладывать число случаев применения или количество соответственно произведенных продуктов. Основными причинами этого являются постоянно усложняющиеся структуры необходимых высококачественных препаратов для фармацевтического и аграрного секторов, с одной стороны, и почти неограниченный потенциал синтеза литийорганических соединений при образовании сложных органических структур, с другой стороны.
Почти каждое литийорганическое соединение может быть легко получено с помощью современного арсенала металлорганической химии и превращено почти с каждым электрофильным соединением в соответствующий требуемый продукт. При этом большинство литийорганических соединений может быть получено одним из следующих способов:
(1) несомненно важнейшим способом является реакция обмена "галоген-металл", при которой в большинстве случаев ароматические соединения брома подвергают превращению воздействием n-бутиллития при низких температурах;
(2) в результате взаимодействия ароматических соединений брома с металлическим литием могут быть также получены очень многие литийорганические соединения;
(3) весьма существенное значение имеет кроме того депротонирование органических соединений посредством алкиллития (напр., BuLi) или амидов лития (напр., LDA или LiNSi).
Из одного этого уже следует, что для большей части данной химии требуется применение стандартных соединений алкиллития, причем здесь в большинстве случаев применяется n-BuLi. Синтез n-BuLi и родственных ему литийалифатических соединений является технически сложным и требует применения очень большого количества ноу-хау, вследствие чего n-бутиллитий, s-бутиллитий, трет.-бутиллитий и аналогичные молекулы - с учетом промышленного масштаба производства - имеют очень высокую продажную стоимость. В этом заключается наиболее крупный, но далеко не единственный недостаток такого обычно высоко эффективного и широко применяемого реактива.
Вследствие экстремальной чувствительности указанных литийалифатических соединений и пирофорных свойств их концентрированных растворов при необходимых объемах промышленного производства (годовые производственные объемы составляют от 5 до 500 т) требуется создание очень дорогостоящих систем логистики, обеспечивающих транспортировку, питание дозировочного приемника и дозирование, что требует больших вложений в основной капитал.
Кроме того во время превращений n-, s- и трет.бутиллития образуются либо бутаны (депротонирование), бутилгалогениды (реакция обмена "галоген-металл", эквивалент BuLi) или бутен и бутан (реакция обмена "галоген - металл", два эквивалента BuLi), которые при комнатной температуре являются газообразными и улетучиваются во время соответствующей гидролитической переработки реакционных смесей. В связи с этим требуются дополнительные и дорогостоящие мероприятия по очистке отходящих газов или соответствующие сжигательные устройства для соблюдения жестких законодательных актов о выбросах. Выходом из положения являются предлагаемые специализированными предприятиями альтернативы, такие, как, n-гексиллитий, которые хотя и не допускают образования бутанов, но значительно дороже бутиллития.
Еще одним недостатком служит образование сложных смесей растворителей после переработки. Вследствие высокой реакционной способности соединений алкиллития по отношению к простым эфирам, которые почти постоянно используются в качестве растворителей при последовательных превращениях, в большинстве случаев соединения алкиллития не применяются в таких растворителях. Хотя изготовители и предлагают широкий набор соединений алкиллития с самой разной концентрацией в самых разных углеводородах, однако, например, обменные реакции "галоген-металл" не протекают в чистых углеводородах, вследствие чего приходится принудительно работать со смесями из простых эфиров и углеводородов. Поэтому после гидролиза получают водосодержащие смеси из эфиров и углеводородов, переработка которых является дорогостоящей и в большинстве случаев не экономичной. При крупном промышленном производстве повторное использование растворителей является, конечно, непременным условием.
Поэтому в связи с названными выше причинами было бы очень желательно располагать способом, которым можно было бы получать применяемое для депротонирования соединение алкиллития на основе дешевого сырья: галогеналкана и металлического лития в простом эфире, и при этом одновременно или последовательно проводилось бы превращение посредством депротонируемого субстрата, так как при таком порядке действий можно было бы исключить все упомянутые выше недостатки, присущие "классическому" способу получения ароматических соединений лития.
Раскрытие изобретения
Настоящее изобретение решает все указанные задачи и касается способа получения соединений ариллития формул (IV) и (VI), а также при необходимости дополнительного превращения этих соединений посредством взаимодействия с соответствующими электрофильными соединениями, причем сначала в результате взаимодействия галогенных соединений (I) с металлическим литием получают соединение лития (II), на которое с целью превращения воздействуют ароматическими соединениями формулы (III) и/или (V) при депротонировании и образовании требуемых ароматических соединений лития (IV) и/или (VI) (уравнение I).
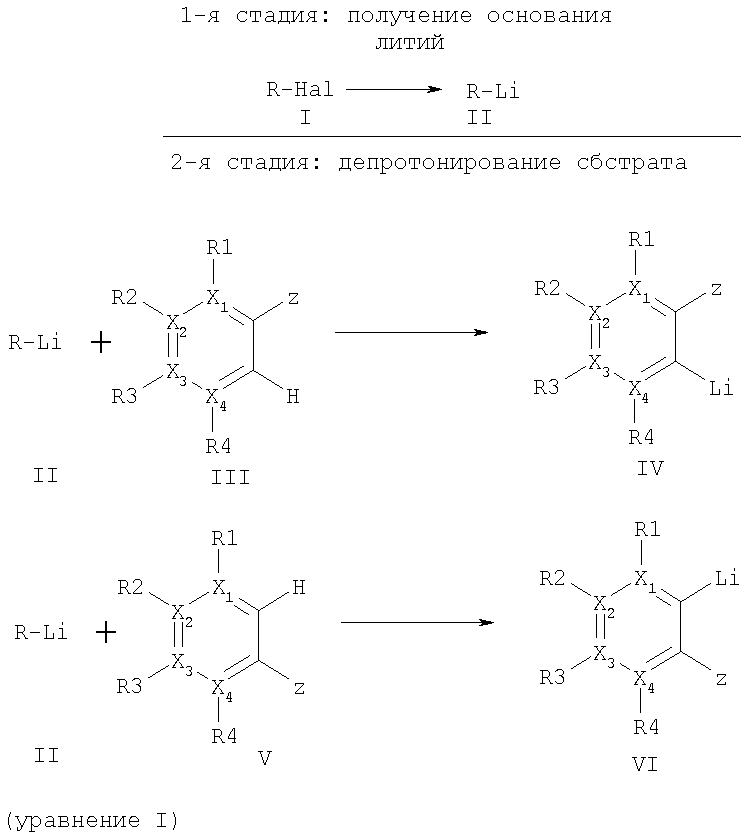
где R - метил, первичные, вторичные или третичные алкильные остатки с 2-12 атомами углерода, замещенный алкил, в котором один из заместителей выбирается из группы: фенил, замещенный фенил, арил, гетероарил, алкоксил, диалкиламин, алкилтио, замещенный алкил, замещенный или незамещенный циклоалкил с 3-8 атомами углерода, Hal - фтор, хлор, бром или йод,
X1-4 означают, независимо друг от друга, углерод, группа Х1-4 R1-4 может означать азот, или два расположенных рядом остатка X1-4 R1-4 могут вместе означать О (фураны), S (тиофены), NH или NR′, причем R′ означает алкил с 1-5 атомами углерода, SO2-фенил, SO2-р-толуил или бензоил.
Предпочтительными соединениями формулы (III), которые могут быть подвержены превращению способом согласно изобретению, являются, например, в числе прочих, бензолы, пиридины, пиридазины, пиримидины, пиразины, фураны, тиофены, N-замещенные пирролы, бензофураны, индолы или нафталины.
Остатки R1-4 и остаток Z означают заместители из группы: водород, метил, первичные, вторичные или третичные циклические или ациклические алкильные остатки с 2-12 атомами углерода, замещенные циклические или ациклические алкильные группы, алкокси, диалкиламино, алкиламино, ариламино, диариламино, фенил, замещенный фенил, алкилтио, диарилфосфино, диалкилфосфино, диалкил- или диариламинокарбонил, моноалкил- или моноариламинокарбонил, СО2 -, гидроксиалкил, алкоксиалкил, фтор или хлор, CN или гетероарил, при этом соответствующие два расположенных рядом остатка R1-4 вместе могут соответствовать ароматическому или алифатическому кольцу.
Полученные таким образом литийорганические соединения могут быть подвергнуты превращению посредством любых электрофильных соединений известньми из уровня техники способами. В результате взаимодействия с углеводородньми электрофилами могут быть получены, например, С, С-связи, в результате взаимодействия с соединениями бора могут быть получены бороновые кислоты, и в результате взаимодействия с посредством галоген- или алкоксисиланами обеспечивается очень эффективная возможность получения органосиланов.
В качестве галогеналифатических соединений могут применяться все имеющиеся или получаемые фтор-, хлор-, бром- или йодалифатические соединения литий в эфирных растворителях вступает в реакцию легко и почти во всех случаях со всеми галогеналифатическими соединениями с обеспечением количественного выхода. При этом предпочтительно применять хлор- или бромалифатические соединения, так как соединения йода часто являются дорогостоящими, а соединения фтора приводят к образованию LiF, который при последующей водной переработке в виде HF может вызвать дефекты в материале. В специальных случаях эффективным может оказаться применение и этих галогенидов.
В способе согласно изобретению предпочтительно применять такие алкилгалогениды, которые после депротонирования могут быть превращены в жидкие алканы. Особо предпочтительно применять хлор- или бромциклогексан, бензилхлорид, трет.-бутилхлорид, хлоргексаны или хлоргептаны.
Реакцию ведут в соответствующем органическом растворителе, при этом предпочтительными являются эфирные растворители, например, тетрагидрофуран, диоксан, диэтиловаый эфир, ди-n-бутиловый эфир, диизопропиловый эфир или анизол, предпочтительно применяется тетрагидрофуран.
Вследствие высокой реакционной способности соединений алкил- и ариллития, в частности, по отношению к используемым в качестве растворителей простьм эфирам, предпочтительные температуры реакции составляют от -100 до +25°С, особо предпочтительны температуры от -80 до -25°С.
Другое преимущество способа согласно изобретению состоит в том, что можно работать при довольно высоких концентрациях литийорганических соединений. Предпочтительными концентрациями алифатических или ароматических промежуточных продуктов формулы (II) являются 5-30 вес.%, в частности, 12-25 вес.%.
В предпочтительном варианте осуществления галогеналкан и ароматический субстрат добавляют к металлическому литию в эфире одновременно или в виде смеси. При таком однокомпонентном способе (одновременная добавка соединений (I), (III) и/или (IV) к литию в эфире) сначала образуется литийалифатическое соединение, которое немедленно депротонирует ароматические соединения. В отдельных случаях, особенно в том случае, когда между ароматическим соединением и металлическим литием могут протекать побочные реакции, возможно, сначала образовать посредством реакции между галогеналифатическим соединением и литием соединение алкиллития в среде эфира и лишь после этого добавлять ароматический субстрат.
Неожиданно было найдено, что при однокомпонентном предпочтительном варианте во многих случаях наблюдаются более высокие показатели выхода продукта по сравнению с тем, когда сначала получают RLi и только после этого вводят ароматический субстрат.
В настоящем способе литий может применяться в виде дисперсии, порошка, опилок, песка, гранул, кусков, слитков и в другом виде, причем размер частиц лития не является существенным для качества, а влияет только на продолжительность протекания реакции. Поэтому предпочтительны малые размеры частиц, например, гранулы, порошки или дисперсии. Количество вводимого лития в расчете на 1 моль превращаемого галогена составляет 1,95-2,5 моля, предпочтительно 1,98-2,15 моля.
В любом случае при добавке органических окислительно-восстановительных систем, таких, например, как бифенил, 4,4′-ди-трет. бутилбифенил или антрацен, во время превращения металлического лития на первой стадии отмечается заметное повышение скорости протекания реакции. Добавка таких систем оказывается эффективной прежде всего в том случае, когда время литиирования без такого катализа составляло более 12 ч.
Ароматическими соединениями, пригодньми для депротонирования, являются прежде всего любые соединения, являющиеся достаточно кислыми для обеспечения депротонирования в условиях согласно изобретению. Здесь следует, прежде всего, указать на все ароматические соединения, содержащие "орто-направленное замещенное" Z (ortho-directing substituents" Z), т.е., в частности, ароматические соединения, содержащие алкокси, F, Cl, замещенный амин, CN, гетероарил, аминоалкил, гидроксиалкил или аналогичные остатки. Механизм действия этих остатков состоит в том, что такие заместители обеспечивают координацию иона лития в алифатическом основании, в результате чего противоион R может быть очень легко депротонирован в орто-положении.
Кроме того, необходимо указать на все гетероциклы, такие как, например, фуран, являющиеся сильнокислыми в результате комбинации нескольких эффектов. В данном случае под действием, в числе прочего, индуктивного эффекта кислорода, а также sp2-гибридизации и углового напряжения на α-углероде протоны становятся достаточно подвижными ("кислыми") для обеспечения депротонирования. Это же относится и к другим гетероциклам.
В остальных случаях, когда замещаемые ароматические протоны не являются достаточно подвижными, депротонирование тем не менее возможно, для чего добавляются вспомогательные вещества, которые известны специалисту в данной области. Особенно зарекомендовал себя в этом случае калий-трет.бутилат, который уже при литировании галогеналифатического соединения добавляется в реакционную смесь в количестве от 0,05 до 1,2 эквивалента. В результате достигается литирование даже слабокислого бензола (в некоторых случаях при таком порядке действий образуется частично или даже полностью ароматическое соединение калия, и поскольку это не оказывает влияния на природу образующегося продукта реакции, то этим аспектом в данном случае можно пренебречь).
Полученные согласно изобретению ароматические соединения лития могут быть подвергнуты превращению электрофильными соединениями с помощью известных среднему специалисту методов, причем интерес представляют - с учетом промежуточных продуктов, необходимых для фармацевтической и агрохимической промышленностей, - в частности, углеродные, борные и кремниевые электрофилы. Превращение с помощью электрофила может проводиться либо после получения литированного соединения (IV) и/или (VI), либо, как уже упоминалось выше, при однокомпонентном способе в результате одновременной добавки в реакционную смесь.
Углеродные электрофилы происходят, в частности, из одной из следующих категорий (в скобках указаны соответствующие продукты):
арил- или алкилцианаты (бензонитрилы),
оксиран, замещенные оксираны (ArCH2CH2OH, ArCR2CR2OH) при R=R1 (одинаковые или разные),
азометины (ArCR1 2-NR′H),
нитроэнолаты (оксимы),
соли иммония (ароматические амины),
галогенное ароматическое соединение, арилтрифлаты, другие арилсульфонаты (биарилы),
диоксид углерода (ArCOOH),
моноксид углерода (Ar-СО-СО-Ar),
альдегиды, кетоны (ArCHR1-OH, ArCR1 2-OH),
ненасыщенные α,β-альдегиды/кетоны (ArCH(ОН)-винил, CR1(ОН)-винил),
кетены (АРС(=O)СН3 при использовании кетена, ArC(=O)-R1 при использовании замещенных кетенов),
соли щелочных и щелочноземельных металлов карбоновой кислоты (ArCHO при использовании формиатов, ArCOCH3 при использовании ацетатов, ArR1CO при использовании R1COOMet),
алифатические нитрилы (ArCOCH3 при использовании ацетонитрила, ArR1CO при использовании R1CN),
ароматические нитрилы (ArCOAr′),
амиды (ArCHO при использовании HCONR2, ArC(=O)R при использовании RCONTR′2),
сложные эфиры (Ar2C(OH)R′)
или средства алкилирования (Ar-алкил).
В качестве борных электрофилов применяются соединения формулы BW3, где W означает, независимо друг от друга, одинаковые или разные (алкокси с 1-6 атомами углерода), фтор, хлор, бром, йод, N (алкил с 1-6 атомами углерода)2 или S(алкил с 1-5 атомами углерода), при этом предпочтительными являются триалкоксибораны, BF3·OR2, BF3·ТНР, BCl3 или BBr3, особо предпочтительны триалкоксибораны.
В качестве кремниевых электрофилов применяются соединения формулы SiW4, где W, независимо друг от друга, означает одинаковые или разные (алкокси с 1-6 атомами углерода), фтор, хлор, бром, йод, N(алкил с 1-6 атомами углерода)2 или S(алкил с 1-5 атомами углерода), при этом предпочтительными являются тетраалкоксисиланы, тетрахлорсиланы или замещенные алкил- и арилгалогенсиланы или замещенные алкил- и арилалкоксисиланы.
Способ согласно изобретению является очень экономичным способом очень рентабельного превращения ароматического водорода в любые остатки.
Переработка проводится в основном водная, причем вводятся добавки как воды, так и водных минеральных кислот или реакционной смеси к воде или водным минеральным кислотам. Для достижения высоких показателей выхода продукции в данном случае задается показатель рН выделяемого продукта, т.е., как правило, слегка кислый, в случае с гетероциклами также и слегка щелочный, показатель рН. Продукты реакции получают, например, экстракцией и выпариванием органических фаз, альтернативно можно также производить отгонку органических растворителей из гидролизной смеси и затем фильтрацией получать выпавший в осадок продукт.
Степень чистоты продуктов, получаемых способом согласно изобретению, является, как правило, высокой, правда, для специальных случаев применения (фармацевтические полуфабрикаты) может оказаться необходимой дополнительная стадия очистки, производимая, например, перекристаллизацией с добавкой небольших количеств активированного угля. Выход продуктов реакции составляет 70-99%, типичные показатели выхода: 85-95%.
Ниже способ согласно изобретению поясняется - без ограничения изобретения - примерами.
Осуществление изобретения
Пример 1
Получение 2,6-диметоксифенил-бороновой кислоты из резорциндиметилового эфира и хлорциклогексана.
Смесь, приготовленную из 20,88 г хлорциклогексана (0,176 моля) и 22,1 г резорциндиметилового эфира (0,16 моля), вводили в виде капель в суспензию из 2,35 г гранул лития (0,34 моля) в 300 г тетрагидрофурана при -50°С, при этом продолжительность введения составила 2 часа. После превращения хлорциклогексана в количестве >97% (в целом после 9 часов), которое определяли газовой хроматографией, в течение 15 минут добавляли при той же температуре в виде капель 16,6 г триметилбората (0,16 моля). После перемешивания в течение 30 минут при -50°С реакционную смесь добавляли к 120 г воды, показатель рН задали 37%-ной соляной кислотой равным 6,3 и производили отгонку тетрагидрофурана и циклогексана при 35°С в условиях вакуума. В продукт в виде суспензии добавили 25 мл метилциклогексана, отсосали бесцветный продукт и промыли один раз 25 мл холодного метилциклогексана и один раз 25 мл холодной воды. После сушки получили 26,5 г 2,6-диметоксифенил-бороновой кислоты (0,146 моля, 91%, точка плавления 104-107°С) в виде бесцветных кристаллов, чистота, определенная высокоэффективной жидкостной хроматографией, составила более 99% (а/а).
Пример 2
Получение 5-формилфуран-2-бороновой кислоты из фурфурал-диэтилацетата и хлорциклогексана.
Смесь, приготовленную из 20,88 г хлорциклогексана (0,176 моля) и 27,2 г фурфурал-диэтилацетата (0,16 моля), вводили по капле в суспензию из 2,35 г гранул лития (0,34 моля) в 300 г тетрагидрофурана при -65°С, при этом время введения составило 2 часа. После превращения хлорциклогексана в количестве >97% (в целом после 10 часов), которое определяли газовой хроматографией, в течение 30 минут добавляли при той же температуре по капле 18,3 г триметилбората (0,176 моля). После перемешивания в течение 30 минут при -65°С реакционную смесь ввели в 120 г воды, показатель рН довели 37%-ной соляной кислотой до 6,3 и производили отгонку тетрагидрофурана и циклогексана при температуре не свыше 35°С в условиях вакуума. Затем рН довели до 1,5, перемешивали до полного выпадения продукта в осадок и фильтровали. После промывки слабохолодной водой и слабохолодным ацетоном получили после сушки 17,2 г 5-формил-2-фуранбороновой кислоты (0,123 моля, 77%) в виде тонкого порошка бежевого цвета, чистота, определенная высокоэффективной жидкостной хроматографией, составила более 99% (а/а).
Пример 3
Получение метилового эфира салициловой кислоты из анизола и хлорциклогексана.
Смесь, приготовленную из 20,88 г хлорциклогексана (0,176 моля) и 17,3 г анизола (0,16 моля), вводили по капле в суспензию из 2,35 г гранул лития (0,34 моля) в 300 г тетрагидрофурана при -50°С, при этом время введения составило 2 часа. После превращения хлорциклогексана в количестве >97% (в целом после 11 часов), которое определяли газовой хроматографией, при той же температуре вводили безводный диоксид углерода до насыщения им раствора. После перемешивания в течение 30 минут при -50°С реакционную смесь ввели в 100 г воды, показатель рН доводили 37%-ной соляной кислотой до 3,4 и производили отгонку растворителя при температуре не свыше 55°С в условиях вакуума. Отсосали прозрачный продукт и после сушки получили метиловый эфир салициловой кислоты (при выходе 79%) в виде бесцветных кристаллов, чистота, определенная высокоэффективной жидкостной хроматографией, составила более 99% (а/а). Экстракцией маточной щелочи дихлорметаном, сушкой сульфатом натрия и выпариванием можно дополнительно получить метиловый эфир салициловой кислоты, суммарный выход: 93%.
Пример 4
Получение 2,6-дифторацетофенона из 1,3-дифторбензола и ангидрида уксусной кислоты
Сначала приготовили раствор из трет.-бутиллития в тетрагидрофуране, причем 9,25 г трет.-бутилхлорида подвергали превращению с помощью 1,4 г гранул лития в 100 г тетрагидрофурана при -78°С. При достижении степени превращения свыше 97% (газовая хроматография, а/а) добавили 1,3-дифторбензол (11,4 г) и перемешивали сначала в течение 30 мин при -78°С и затем в течение 2 ч при -65°С. Полученный раствор 2,6-дифтор-1-литийбензола добавляли по капле в охлажденный до -5°С раствор из 22 г ангидрида уксусной кислоты в 35 г тетрагидрофурана. После обычной водной переработки получили 2,6-дифторацетофенон при выходе 92%.
Пример 5
Получение 2-(тиофенил)-этанола из тиофена и 1-хлоргептана
Смесь, приготовленную из 145 г 1-хлоргептана (1,1 моля) и 84,0 г тиофена (1,0 моля), вводили по капле в течение 3 часов в суспензию из 14,5 г гранул лития (2,1 моля) в 500 г тетрагидрофурана при -50°С. После превращения хлорциклогексана в количестве >97% (в целом после 9 часов), которое определяли газовой хроматографией, ввели при той же температуре 48 г окиси этилена (1,1 моля). После перемешивания в течение 30 минут при -50°С реакционную смесь ввели в 120 г воды, показатель рН доводили 37%-ной соляной кислотой до 5,9 и производили отгонку низкокипящих фракций при температуре не свыше 55°С в условиях вакуума. После трехкратной экстракции с применением соответственно 175 г дихлорметана, сушки сульфатом натрия, отделения фильтрацией сушильного средства и выпаривания до сухого остатка получили тиофенилэтанол при выходе 83%.
Пример 6
Получение бензойной кислоты из бензола и хлорциклогексана (депротонирование бензола)
Смесь, приготовленную из 0,2 моля хлорциклогексана и 0,2 моля бензола, добавляли по капле в суспензию из 0,4 моля гранул лития, 0,21 моля калий-трет.-бутилата и 35 мг бифенила в 300 г тетрагидрофурана при -72°С. После превращения хлорциклогексана в количестве >97% (в целом после 24 часов), которое определяли газовой хроматографией, вводили диоксид углерода до насыщения. Переработку вели аналогично примеру 3; бензойную кислоту получили при выходе 79%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СИНТЕЗА ПЕПТИДОВ, СОДЕРЖАЩИХ ПОДСТРУКТУРУ 4-ГИДРОКСИПРОЛИНА | 2003 |
|
RU2392265C2 |
| IN-SITU ПОЛИМЕРНАЯ СМЕСЬ ДЛЯ ШИН | 2018 |
|
RU2779347C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2003 |
|
RU2360903C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЕТОКИСЛОТ И ИХ ПРОИЗВОДНЫХ | 2008 |
|
RU2536046C2 |
| Способ синтеза поликарбонатов в присутствии биметаллического катализатора и регулятора степени полимеризации | 2012 |
|
RU2630688C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ТРИПТАЗЫ | 2005 |
|
RU2378271C2 |
| СПОСОБ СИНТЕЗА 5-(МЕТИЛ-1Н-ИМИДАЗОЛ-1-ИЛ)-3-(ТРИФТОРМЕТИЛ)БЕНЗАМИДА | 2006 |
|
RU2444520C2 |
| ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОНАФТАЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2194696C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ЗАМЕЩЕННЫХ САЛИЦИЛАМИДОВ | 2005 |
|
RU2411234C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ О-ХЛОРМЕТИЛФЕНИЛГЛИОКСИЛОВОЙ КИСЛОТЫ | 1997 |
|
RU2177472C2 |
Описывается улучшенный способ получения литийорганических соединений, обеспечивающий in situ стадию получения ариллитиевых соединений формулы IV и/или VI, которые затем подвергают действию электрофильных реагентов, при этом соединения IV и/или V получают действием галогенидов формулы (I) с металлическим литием с получением соединения II с последующим его взаимодействием с соединениями формул III и/или V с депротонированием и образованием соединений лития IV и/или VI (уравнение I), где R-СН3, первичный, вторичный или третичный С2-8-алкил, C5-10 - незамещенный циклоалкил, X1-4 - Н, два остатка рядом X1-4 R1-4 вместе образуют О или S, R1-4 и остаток Z-H, СН3, алкокси, алкоксиалкил, замещенный ациклический алкил или F, Hal - F, Br, Cl. Способ дешев и обладает, высокими выходами и степенью чистоты целевых продуктов. 8 з.п. ф-лы.
1-я стадия: получение основания
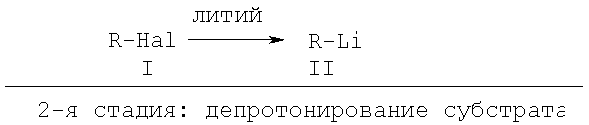
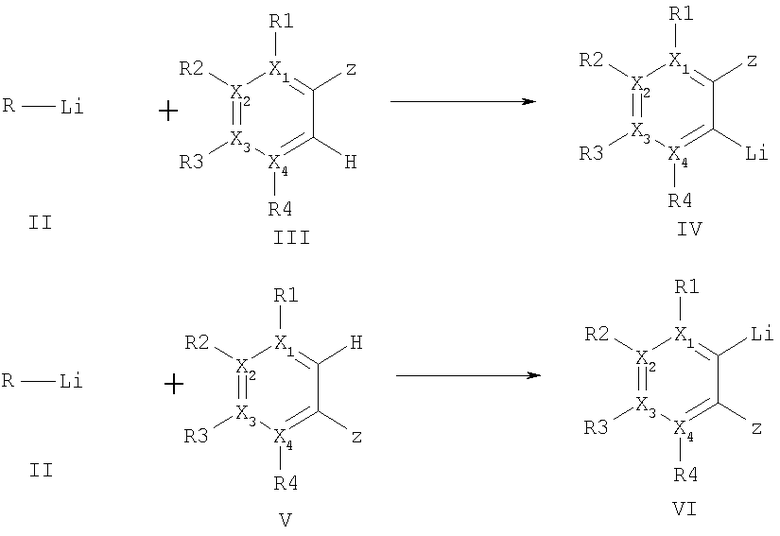
(уравнение I),
1-й этап получение основания
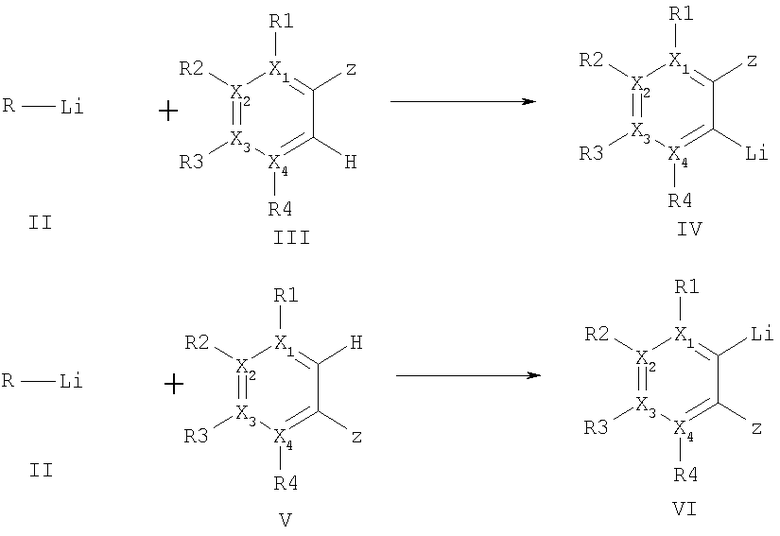
(уравнение I),
где R - метил, первичный, вторичный или третичный C2-8 - алкильный остаток, С5-10 - незамещенный циклоалкил,
Х1-4 - углерод, два расположенных рядом остатка X1-4 R1-4 вместе означают О или S,
R1-4 и остаток Z - означают водород, метил, алкокси, алкоксиалкил, замещенная ациклическая алкильная группа или фтор,
Hal - фтор, хлор или бром.
(III) или (V) в орто-положении по отношению к депротонируемому водороду несут в себе остатки алкоксила, F, или гидроксиалкила.
(V), либо одностадийным способом с одновременной добавкой в реакционную смесь.
| УСТРОЙСТВО ДЛЯ ПРЕДОТВРАЩЕНИЯ НЕПРАВИЛЬНОЙ РАБОТЫ БЫСТРОДЕЙСТВУЮЩЕЙ ДИСТАНЦИОННОЙ ЗАЩИТЫ ПРИ МЕЖДУФАЗОВЫХ ПОВРЕЖДЕНИЯХ В ЦЕПЯХ НАПРЯЖЕНИЯ | 1940 |
|
SU64905A1 |
| Способ получения -алкилированных органических соединений | 1971 |
|
SU667547A1 |

