Настоящее изобретение относится к способам получения пептидов и промежуточных соединений, использованных для получения указанных пептидов.
Один объект настоящего изобретения включает:
(А) Способ получения соединений формулы I
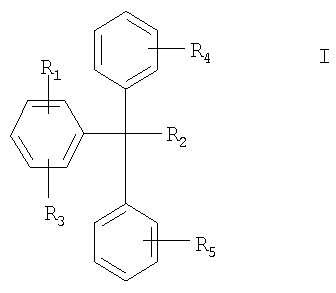
где R1 означает реакционноспособный заместитель или место присоединения к твердой фазе;
R2 означает реакционноспособный заместитель, а
R3, R4 и R5 каждый независимо означает водород или один или более заместителей, которые присоединены к каждому бензольному кольцу и которые выбирают из группы, включающей гидрокси, амино, C1-С10алкил, C1-С10алкокси, C1-С10алкиламино, ди(С1-С10)алкиламино, карбамоил, C1-С10алкилкарбамоил, ди(С1-С10)алкилкарбамоил, галоген(С1-С10)алкил, галоген и нитро;
в свободной форме или в форме соли; причем способ включает
(а) взаимодействие соединения формулы VI с электрофильным соединением:
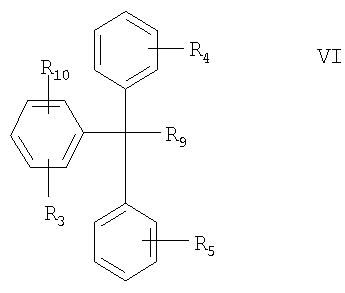
где R3, R4 и R5 определены выше;
R9 означает -ОН, -ОМ или -ОМХ, где М означает металл, а Х означает нуклеофильный заместитель;
R10 означает -М или -MX, где М означает металл, а Х означает нуклеофильный заместитель;
в свободной форме или в форме соли;
и гидролиз полученного соединения с образованием соединения формулы I, где
R2 означает гидроксигруппу;
(б) необязательное превращение соединения формулы I, где R2 означает гидроксигруппу, в соединение формулы I, где R2 означает другую группу, кроме гидрокси;
(в) необязательное превращение R1 в составе соединения формулы I в другую группу R1;
(г) необязательное удаление защитной группы в соединении формулы I, содержащем защитную группу; и
(д) при необходимости превращение соединения формулы I, полученного в свободной форме, в требуемую соль или наоборот;
(Б) способ получения твердофазной системы, включающий получение соединения формулы I по способу, описанному выше, и конденсацию соединения с модифицированным или содержащим соответствующие функциональные группы твердофазным материалом;
(В) соединение формулы V в свободной форме или в форме соли
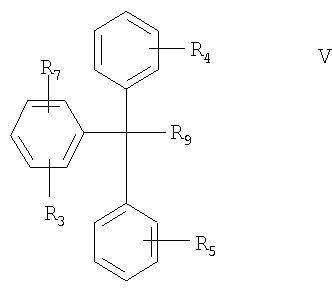
где R3, R4, R5 и R9 определены выше, а
R7 означает нуклеофильный заместитель;
(Г) соединение формулы VI в свободной форме или в форме соли
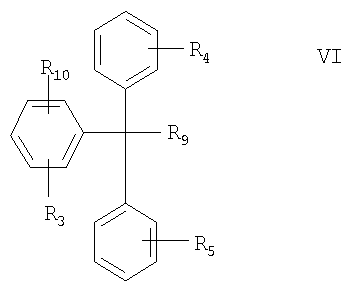
где R3, R4, R5 и R9 определены выше, а
R10 означает -М или -MX, где М означает металл, а Х означает нуклеофильный заместитель.
В настоящем изобретении предлагается простой способ получения соединений формулы I, который используют для твердофазного химического синтеза. Согласно способу по настоящему изобретению можно непосредственно получить соединение формулы I, присоединенное к твердой фазе, или, если R1 означает реакционноспособный заместитель, то соединение формулы I можно простым способом присоединить к твердой фазе на более поздней стадии. Наличие реакционноспособного заместителя R2 позволяет использовать соединения формулы I в качестве связующих звеньев при синтезе олигомеров и полимеров, таких как гликопептиды, нуклеотиды и белки, прежде всего при твердофазном синтезе пептидов. Соединения формулы I, прежде всего те, в которых R2 означает галоген, используют в химическом синтезе в качестве защитных агентов для защиты функциональных групп, таких как, например, амино- и гидроксигруппы.
Соединения формул V и VI используют в качестве промежуточных соединений для получения соединений формулы I.
Соединение формулы VI получают при взаимодействии соединения формулы V с металлом или металлоорганическим соединением:
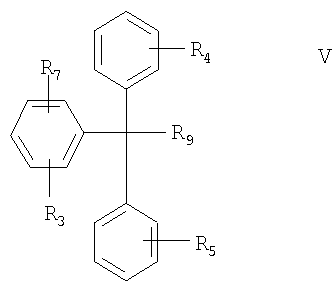
где R3, R4, R5 и R9 определены выше, а
R7 означает нуклеофильный заместитель.
Соединение формулы V получают следующим способом:
(i) взаимодействие соединения формулы II с металлом или металлоорганическим соединением
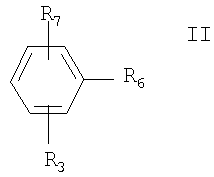
где R6 и R7 каждый означает нуклеофильный заместитель, а R3 определен выше и при необходимости защищен удаляемой защитной группой; и
(ii) взаимодействие соединения, полученного согласно п.(i), с соединением формулы III
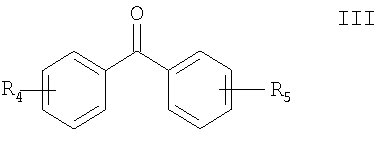
где R4 и R3 определены выше и при необходимости защищены удаляемой защитной группой.
Способ по настоящему изобретению проводят в одном реакционном сосуде без выделения промежуточных соединений.
Термины, используемые в настоящем описании, имеют следующие значения:
«Алкил» означает прямой или разветвленный радикал, предпочтительно С1-С4алкил.
«Алкокси» означает прямой или разветвленный радикал, предпочтительно С1-С4алкокси.
«Ациламино» означает группу формулы -NH-C(O)-R, где R означает прямой или разветвленный C1-С10алкил, циклоалкил или арил, предпочтительно R означает С1-С4алкил.
«Ацилокси» означает группу формулы -O-C(O)-R, где R определен выше.
«Арил» предпочтительно означает С6-С10арил, например фенил.
«Галоген» означает фтор, хлор, бром или иод.
«Галогеналкил» означает прямой или разветвленный C1-С10алкил, замещенный одним или более, например, одним, двумя или тремя атомами галогена, предпочтительно атомами фтора или хлора. Предпочтительно галогеналкил означает С1-С4алкил, замещенный одним, двумя или тремя атомами фтора или хлора.
«Металлоорганическое соединение» означает соединение, в котором атом углерода органической группы связан с металлом. Металлоорганическое соединение предпочтительно означает металлалкил, например алкиллитий, например прямой или разветвленный C1-С10алкиллитий, или в другом варианте означает арилметалл, например ариллитий.
Более предпочтительно алкиллитий означает С3-С6алкиллитий, такой как бутиллитий или гексиллитий.
В другом варианте металлоорганическое соединение означает органическое соединение магния, например прямой или разветвленный алкилмагний или арилмагний, предпочтительно C1-С6алкилмагний. Органические соединения магния известны под названием реагенты Гриньяра. Органическое соединение магния предпочтительно означает галогенид органического соединения магния, предпочтительно иодид или бромид.
В других вариантах воплощения настоящего изобретения металлоорганическое соединение может означать соединение алкил- или арилцинка, например соединение C1-С6алкилцинка или соединение C1-С6алкил- или арилолова.
М предпочтительно означает литий или магний.
R1 означает реакционноспособный заместитель, пригодный для присоединения соединения к твердой фазе. R1 означает -C(O)R', -C(O)-OR', -C(O)-NR'R'', -R12-NR'R", -R12-OR', -NR'R'' или -С(O)Х, где R' и R'' каждый независимо означает водород или прямой или разветвленный C1-С10алкил, например, С1-С4алкил, R12 означает прямой или разветвленный C1-С10алкил, например, С1-С4алкил, а Х означает нуклеофильный заместитель, предпочтительно галоген, например хлор. Заместитель R1 может находиться в пара-, орто- или мета-положении, предпочтительно пара-положении.
В другом варианте R1 означает группу, через которую соединения присоединены к твердофазному материалу, а твердофазный материал, например, означает полистирол. Предпочтительно указанная группа означает -С(O)-Р, -С(O)-ОР, -C(O)-NR'-P, -R12-NR'-P, -R12-OP, -NR'-P, -C(O)R12P, -C(O)-OR12P, -C(O)-NR'R12P, -R12NR'R12P, -R12OR12P, -NR'R12-P или -R12-P, где R', R'' и R12 определены выше, а Р означает твердофазный материал. Более предпочтительно R1 означает -С(O)-ОР, -C(O)-OR12-P, -C(O)-NH-P, -C(O)-NH-R12-P, -NH-R12-P или -R12-P, где R12 означает метил, например -СН2-Р.
R2 предпочтительно означает реакционноспособный заместитель, пригодный для присоединения соединения к биологическому олигомеру или полимеру или к их мономерному звену, например к аминокислоте или полипептиду. R2 может означать группы гидрокси, ациламино, ацилокси, амино, галоген, сульфгидрил, C1-С10алкокси или С6-С10арилокси, предпочтительно галоген.
Каждое бензольное кольцо, представленное формулами I-VII, может содержать в качестве заместителей одну или более групп. Например, R3 означает от одного до четырех заместителей, предпочтительно один или два заместителя, присоединенных к бензольному кольцу, как представлено формулами I, II и IV-VII. R4 и R5 могут означать от одного до пяти заместителей, предпочтительно от одного до трех заместителей, присоединенных к каждому бензольному кольцу, как представлено формулами I, II и IV-VII. Каждый заместитель может находиться в любом положении бензольного кольца, к которому они присоединены. Более предпочтительно R4 и/или R5 означают заместители в орто- или пара-положении бензольного кольца, к которому они присоединены.
Каждый из заместителей R3, R4 и R5 при необходимости защищен удаляемой защитной группой, например, если они содержат группу -ОН или -NH2, которые не участвуют в реакции. Защитные группы, их введение и удаление, описано, например, в книге «Protective Groups in Organic Synthesis», T.W.Greene и др., John Willey & Sons Inc; Second Edition, 1991. Предпочтительно каждый из заместителей R3, R4 или R5 означает группу, для которой не требуется защита, например любую из перечисленных выше групп за исключением гидрокси, амино или нитро.
Если заместители R3, R4 или R5 означают галоген, то он предпочтительно означает фтор или хлор. Если заместители R3, R4 или R5 означают галогеналкил, то он предпочтительно означает трифторметил. Предпочтительно R3 означает С1-С4алкил, галоген или водород. Предпочтительно R4 и R5 каждый независимо означает С1-С4алкилкарбамоил, ди(С1-С4)алкилкарбамоил, карбамоил, трифторметил, фтор или хлор. Предпочтительно R4 и R5 являются одинаковыми.
Предпочтительно нуклеофильные заместители R6 и R7 каждый независимо означает галоген, более предпочтительно бром или иод и наиболее предпочтительно R6 и R7 каждый означает бром. R7 может находиться в пара-, орто- или мета-положении, предпочтительно в пара-положении.
В одном варианте воплощения настоящего изобретения соединение формулы II сначала взаимодействует с металлом или металлоорганическим соединением с образованием соединения формулы IV:
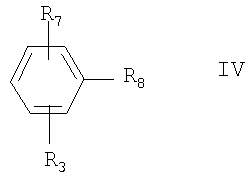
где R3 и R7 определены выше, a R8 означает -М или -MX, где М означает металл, а Х означает нуклеофильный заместитель, предпочтительно галоген.
Если металл означает литий или металлоорганическое соединение означает органическое соединение лития, то R8 означает -Li. Если металл означает магний или металлоорганическое соединение означает реагент Гриньяра, то R8 означает -MgX, a X предпочтительно означает галоген. Затем соединение формулы IV взаимодействует с соединением формулы III с образованием соединения формулы V.
Нет необходимости в выделении и очистке соединений формул IV и V, их получают in situ.
Пригодные электрофильные соединения, использованные в данном способе, включают диоксид углерода, изоцианаты, нитрилы, ацилгалогениды (такие как фосген), приводящие к образованию, например, соединений формулы I, где R1 означает группы карбокси, карбамоил, алкилкарбамоил или ацил. В другом варианте электрофильное соединение может включать модифицированный твердофазный материал, например полимер Меррифильда, обеспечивающий прямое связывание соединений формулы VI с твердой фазой. В одном варианте воплощения электрофильное соединение означает соединение формулы X'-(СН2)n-Р, где X' означает нуклеофильный заместитель, например, галоген или тозилокси, n означает целое число от 1 до 4, предпочтительно 1, а Р означает твердофазный материал.
Если в качестве электрофильного соединения используется диоксид углерода, то способ предпочтительно включает взаимодействие соединений формулы V с металлом или металлоорганическим соединением с образованием соединения формулы VI, как описано выше, и последующее взаимодействие, предпочтительно in situ, соединения формулы VI с диоксидом углерода.
Если в качестве электрофильного соединения используется диоксид углерода, то предпочтительно образуется соединение формулы VII:
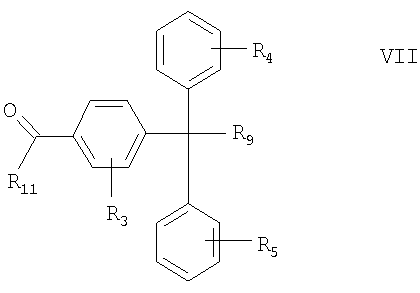
где R3, R4, R5 и R9 определены выше, а
R11 означает -ОН, -ОМ или -ОМХ, где М означает металл, а Х нуклеофильный заместитель, предпочтительно галоген, в форме соли или в свободной форме.
В другом варианте стадия карбоксилирования включает взаимодействие соединения формулы V с диоксидом углерода в присутствии металла или металлоорганического соединения с образованием соединения формулы VII.
Стадия гидролиза предпочтительно включает взаимодействие соединения формулы VII, где R11 означает -ОМ или -ОМХ и/или R9 означает -ОМ или -ОМХ, с водой или кислотой с образованием соединения формулы I, где R1 означает карбоксигруппу, a R2 означает гидроксигруппу, в форме соли или в свободной форме. Пригодные кислоты включают хлорид аммония, уксусную кислоту, серную кислоту и хлористоводородную кислоту. Используют также буферные растворы с определенным значением рН. Предпочтительно используют слабую кислоту и/или стадию проводят при рН от 4 до 7. Температуру реакции поддерживают в интервале от -50 до 50°С, предпочтительно в интервале от -10 до 10°С.
В другом варианте соединение формулы VII, где R11 означает -ОМ или -ОМХ, взаимодействует с нуклеофильным агентом, например амином или галогенидом, с образованием соединения формулы I, где R1 означает -С(O)-NR'R'' или -С(O)-Х, a R', R'' и Х определены выше.
Способ по настоящему изобретению проводят в инертном органическом растворителе, предпочтительно в эфире, например диэтиловом эфире, тетрагидрофуране или трет-бутилметиловом эфире. В качестве растворителя используют также углеводороды. Температуру реакции на стадии (а) поддерживают в интервале от -30 до +10°С, предпочтительно от -5 до 0°С. Реакцию проводят, например, с использованием от 0,5 до 2 экв., предпочтительно от 0,8 до 1,2 экв. и наиболее предпочтительно приблизительно 1 экв. металла или металлоорганического соединения в присутствии 1 экв. соединения формулы II. Используют также от 0,5 до 2 экв., предпочтительно от 0,8 до 1,2 экв. соединения формулы III в присутствии 1 экв. соединения формулы II.
Температуру реакции соединения формулы V с металлом или металлоорганическим соединением поддерживают в интервале от 0 до +50°С, предпочтительно от +20 до +30°С. Температуру реакции с электрофильным агентом (например, СО2) поддерживают в интервале от 0 до -30°С, предпочтительно от -5 до -10°С. Стадию гидролиза, например в присутствии кислоты, проводят при температуре в интервале от -10 до +10°С, предпочтительно от 0 до +5°С. Используют от 0,5 до 2 экв., более предпочтительно от 0,8 до 1,2 экв. металла или металлоорганического соединения в присутствии 1 экв. соединения формулы V.
Группы R1 и R2 превращают в другие группы R1 и R2, описанные выше, согласно известным методикам, таким как этерификация, амидирование или нуклеофильное замещение. Например, соединение формулы I, где R2 означает гидроксигруппу, можно превратить в соединение формулы I, где R2 означает галоген, при взаимодействии с ацилгалогенидом, например ацилхлоридом.
Предпочтительно соединение формулы I используют в свободной форме. Соединения в свободной форме или солевой форме получают в форме гидратов или сольватов, содержащих растворитель, используемый при кристаллизации.
Соединения формулы I можно выделить из реакционной смеси и очистить согласно известным методикам.
Исходные соединения для получения соединения формулы II или III известны в данной области техники или могут быть получены согласно методам, известным специалистам в данной области техники. Металлоорганические соединения получают по стандартным методикам, например при реакции алкил- или арилгалогенида с металлом, например с литием или магнием, суспендированным в диэтиловом эфире или тетрагидрофуране. Металлоорганическое соединение предпочтительно получают и используют в инертной (не содержащей кислорода) безводной атмосфере, например в атмосфере азота.
Метод по настоящему изобретению может включать стадию присоединения соединения формулы I, где R1 означает реакционноспособный заместитель, к твердофазному материалу. Пригодные твердофазные материалы описаны, например, в DE 4306839 А1, и включают природные или синтетические органические или неорганические полимеры в стандартном виде, например в форме гранул или предпочтительно в виде поверхностного слоя на пригодной инертной подложке. Примеры подходящих полимерных материалов включают сшитый полистирол, например гранулы полистирола, Gly-HMD-MA/DMA и НЕМА. Соединения формулы I можно присоединить к твердофазному материалу при взаимодействии группы, содержащейся в твердой фазе, с заместителем R1. Таким образом, твердофазный материал предпочтительно содержит реакционноспособные группы, такие как аминогруппы. Предпочтительно соединение формулы I, где R1 означает карбоксигруппу или активированную карбоксигруппу, например в присутствии диизопропилкарбодиимида, взаимодействует с полимером, содержащим свободные аминогруппы.
Соединение формулы I можно использовать в качестве связующего звена. Таким образом, способ по настоящему изобретению включает стадию присоединения соединения формулы I, необязательно связанного с твердофазным материалом, с биологическим олигомером или полимером или с их мономерным звеном. Соединение можно конденсировать с биологической молекулой, например с аминокислотой или полипептидом, при взаимодействии группы в составе биологической молекулы с заместителем R2. Например, если R2 означает гидроксигруппу, а биологическая молекула означает полипептид или аминокислоту, то концевую карбоксильную группу биологической молекулы этерифицируют при взаимодействии с гидроксигруппой R2, необязательно через исходное взаимодействие соединения формулы I с ацилгалогенидом, которое приводит к замещению гидроксигруппы на галоген in situ.
В другом варианте в настоящем изобретении предлагается:
(Д) способ получения соединения формулы VIII
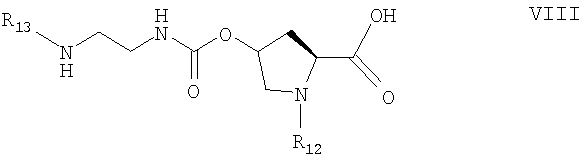
где R12 и R13 каждый означает удаляемую защитную группу, и группы R12 и R13 являются различными;
причем способ включает взаимодействие соединения формулы IX
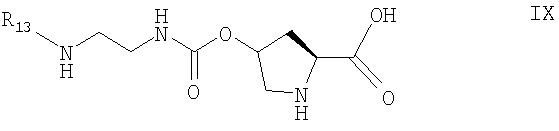
с пригодным соединением-донором R12;
(Е) промежуточное соединение, использованное в описанном выше способе, общей формулы XIV
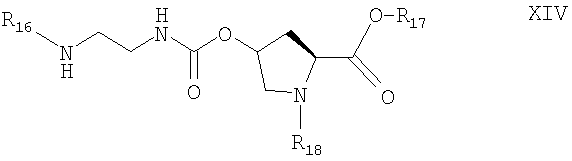
где R16 означает удаляемую защитную группу, за исключением группы флуоренилметоксикарбонил, и отличается от R18;
R17 означает водород или блокирующую группу, удаляемую с помощью гидролиза или гидрирования, а
R18 означает водород или удаляемую защитную группу, за исключением флуоренилметоксикарбонил.
В настоящем изобретении предлагается простой и эффективный способ получения соединений формулы VIII, который используют при синтезе пептидов, например, как описано в заявке WO 02/10192. Соединения формулы XIV используют в качестве промежуточных соединений при получении соединений формулы VIII.
Соединение формулы IX получают из соединения формулы Х
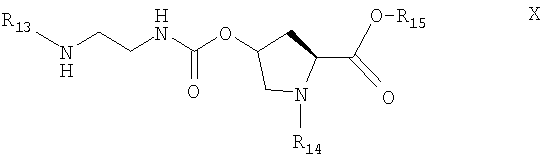
где R13 определен выше,
R14 означает удаляемую защитную группу и R14 отличается от R12 и R13, а
R15 означает блокирующую группу, удаляемую в условиях гидролиза или гидрирования.
Защитные группы, их введение и удаление описано, например, в книге «Protective Groups in Organic Synthesis», T.W.Greene и др., John Willey & Sons Inc; Second Edition, 1991. Пригодные донорные соединения для введения защитных групп, например аминозащитные агенты, известны специалистам в данной области техники, например ангидриды, галогениды, карбаматы или N-гидроксисукцинимиды, которые используют для введения защитных групп, описаны ниже.
Защитная группа R12 предпочтительно означает флуоренилметоксикарбонил. R13 или R16 предпочтительно означает защитную группу, отличающуюся от флуоренилметоксикарбонильной группы, и предпочтительно является более устойчивой в условиях гидролиза (например, в условиях гидролиза, катализируемого основаниями) и/или в условиях гидрирования по сравнению с R12 и/или R14, например, является более устойчивой по сравнению с флуоренилметоксикарбонильными и/или бензилоксикарбонильными группами. Более предпочтительно R13 или R16 означает трет-бутоксикарбонил.
Защитные группы R14 или R18 предпочтительно являются более устойчивыми в условиях гидролиза по сравнению с группой R12, например, более устойчивы по сравнению с флуоренилметоксикарбонильной группой. Группы R14 и R18 предпочтительно удаляют в условиях гидрирования.
Пригодные заместители R14 или R18 включают бензилоксикарбонил, 1,1-диметилпропинилоксикарбонил, винилоксикарбонил, N-гидроксипиперидинилоксикарбонил, 9-антрилметилоксикарбонил и фениламинотиокарбонил, аллил, нитробензил, трифенилметил, (пара-метоксифенил)дифенилметил, дифенил-4-пиридилметил или бензилсульфонил. Предпочтительные R14 или R18 означают защитные группы, содержащие оксикарбонил, например бензилоксикарбонил (карбобензокси).
R15 или R17 означают:
(i) C1-С10алкил, например С1-С4алкил, предпочтительно метил, этил, пропил или бутил, отличающийся от трет-бутильной группы, более предпочтительно метил;
(ii) С3-С8циклоалкил, необязательно замещенный одним или более заместителями С1-С4алкил, например метил. Предпочтительный циклоалкил означает С3-С6циклоалкил;
(iii) С6-С10арил, необязательно замещенный одним или более стабилизирующих заместителей, например группами галоген или нитро;
предпочтительный арил означает фенил, необязательно замещенный одним, двумя или тремя галогенами, предпочтительно хлором;
(iv) (С6-С10арил)1-3(С1-С10)алкил, необязательно замещенный в арильной группе (i) одним или более стабилизирующих заместителей, например, галогеном или нитро, или (ii) двумя заместителями, которые вместе с атомами углерода в цикле, к которому они присоединены, образуют 5- или 6-членный цикл, необязательно содержащий один, или два атома азота, или кислорода. (C1-С10арил)1-3(С1-С10)алкил предпочтительно означает (i) (фенил)1-3-С1-С4алкил, более предпочтительно бензил, дифенилметил или трифенилметил, необязательно замещенный в каждом бензольном кольце одним, двумя или тремя атомами галогена, например хлором, (ii) антрилметил, например, 9-антрилметил, или (iii) пиперонил;
(v) С6-С10арил(С1-С4)алкокси(С1-С4)алкил, предпочтительно бензилоксиметил;
(vi) С6-С10арилкарбонил(С1-С4)алкил, предпочтительно фенацил.
Предпочтительные R15 или R17 означают группу, которую удаляют в условиях гидрирования, такую как бензил, бензилоксиметил, фенацил, трифенилметил, пиперонил или 9-антрилметил, предпочтительно бензил.
Соединение формулы IX получают (i) гидролизом сложного эфира соединения формулы Х с образованием соответствующей карбоновой кислоты и (ii) при удалении защитной группы R14. Предпочтительно стадию гидролиза проводят перед удалением защитной группы R14. Защитную группу R14 удаляют восстановительным гидрированием. Данный способ, включающий стадию гидролиза, является пригодным, если группа R15 не удаляется в условиях гидрирования. Стадию гидролиза предпочтительно проводят в присутствии основания в качестве катализатора, например в присутствии гидроксида натрия и в присутствии полярного растворителя, например в метаноле.
В другом варианте соединение формулы IX получают гидрированием соединения формулы X, где R15 означает группу, удаляемую в условиях гидрирования, например бензил. Стадию гидрирования проводят с использованием подходящего катализатора, например палладия на угле.
Соединение формулы Х получают при взаимодействии с соединением формулы XI
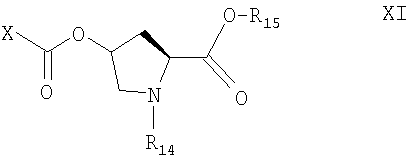
где Х означает нуклеофильный заместитель, a R14 и R15 определены выше, с соединением формулы XII
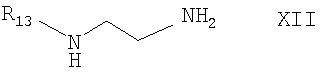
где R13 определен выше. Данную стадию проводят в любом пригодном органическом растворителе, предпочтительно в углеводородном растворителе, более предпочтительно в толуоле.
Соединение формулы XII представляет собой защищенный этилендиамин, в котором одна аминогруппа защищена удаляемой защитной группой. Нуклеофильный заместитель Х в соединении формулы XI предпочтительно означает галоген, такой как фтор, хлор, бром или иод, более предпочтительно хлор. Соединение формулы XI, где Х означает галоген, получают при взаимодействии соединения формулы XIII
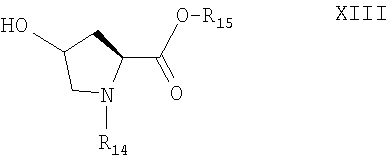
с ацилгалогенидом, например фосгеном, трифосгеном, фенилхлорформиатом или 4-нитрофенилхлорформиатом, предпочтительно 4-нитрофенилхлорформиатом. Данную стадию проводят в присутствии органического основания, например диметиламинопиридина, в неполярном растворителе, таком как толуол.
Соединение формулы XIII является коммерческим препаратом, например, если R15 означает метил, или его получают этерификацией 4-гидроксипролина согласно методам, известным в данной области техники, например при взаимодействии с бензиловым спиртом или метанолом. Полученный сложный эфир затем защищают при взаимодействии с пригодным соединением R14 для введения защитной группы, например с бензилксикарбонил-N-гидроксисукцинимидом.
Нет необходимости в выделении и очистке соединения формулы XI, поскольку соединение формулы XIII взаимодействует с ацилгалогенидом и продукт реакции затем взаимодействует с соединением формулы XII в одном и том же сосуде.
Введение защитной группы R12 в соединение формулы IX проводят в присутствии карбоната натрия/ацетонитрила.
Соединение формулы VIII выделяют из реакционной смеси и очищают согласно стандартным методикам.
В соединениях формул VIII-XI и XIII, описанных выше, оксигруппа в пролине может находиться в цис- или транс-положении, предпочтительно в транс-положении. Цис- и транс-изомеры получают в виде индивидуальных соединений с использованием в качестве исходного материала соответствующих цис- или транс-гидроксипролинов.
Несмотря на то, что в литературе отсутствует подобное описание методов получения исходных материалов, эти соединения являются известными или их можно получить согласно методикам, известным в данной области техники, или описанным ниже в настоящем описании.
Следующий объект настоящего изобретения относится к получению соединения формулы VIII, где R12 означает флуоренилметоксикарбонил, а R13 означает удаляемую защитную группу, в отличии от флуоренилметоксикарбонильной группы, причем способ включает взаимодействие соединения формулы VIII с соединением для введения флуоренилметоксикарбонильной группы, например флуоренилметоксикарбонил-N-гидроксисукциимидом.
Настоящее изобретение описано со ссылками на следующие соответствующие варианты его воплощения, в которых используются следующие сокращения:
Fmoc означает флуоренилметоксикарбонил;
Вос означает трет-бутоксикарбонил;
Cbo означает карбобензокси (бензилоксикарбонил);
OSu означает N-гидроксисукцинимид;
HPTF означает фракцию гептана;
ЖХВР означает жидкостную хроматографию высокого разрешения;
ТГФ означает тетрагидрофуран;
ТБМЭ означает трет-бутилметиловый эфир;
ДМФА означает диметилформамид.
Пример 1
Получение 4-(дифенилгидроксиметил)бензойной кислоты
1,4-Дибромбензол (47,2 г, 0,2 М) добавляли к ТГФ (240 мл). Прозрачный раствор охлаждали до -65°С. В течение 30 мин добавляли раствор бутиллития (0,22 М, 94 мл 20% раствора в циклогексане).
Смесь перемешивали в течение 5 мин, а затем в течение 30 мин добавляли раствор бензофенона (36,4 г, 0,2 М в 180 мл ТГФ, процесс экзотермический). Смесь перемешивали в течение 30 мин при -65°С. Затем через 30 мин температуру поднимали до -10°С и раствор перемешивали при данной температуре в течение 1 ч.
Реакционную смесь охлаждали до -65°С, затем в течение 30 мин добавляли раствор бутиллития (0,22 М, 94 мл 20% раствора в циклогексане).
Полученную суспензию разбавляли 200 мл ТГФ. Затем через раствор в течение 90 мин при -65°С пропускали газообразный диоксид углерода. Температуру поднимали до 20°С и смесь перемешивали в течение ночи. Смесь охлаждали до 0°С и в течение 30 мин добавляли водный раствор хлорида аммония (120 мл 10% раствора). На данной стадии получали 4-(дифенилгидроксиметил)бензойную кислоту.
Смесь упаривали в вакууме при 45°С. Величину рН остатка доводили до 4 с помощью уксусной кислоты и раствор смешивали с 400 мл Н2О. Экстракцию проводили с использованием этилацетата (2×150 мл). Органические фазы экстрагировали 100 мл воды, объединенные фазы встряхивали с 10% водным раствором гидроксида калия (2×120 мл). Объединенные водные фазы доводили до рН 1-2 хлористоводородной кислотой при 20°С и затем экстрагировали ТБМЭ (2×150 мл). Объединенные фазы в ТБМЭ смешивали с 50 мл воды и 50 мл насыщенного раствора Na2SO4, сушили над сульфатом магния и упаривали при 45°С в вакууме, при этом получали неочищенный продукт.
38,3 г Неочищенного продукта растворяли в ТБМЭ (300 мл) при 40°С. Прозрачный раствор желтого цвета концентрировали до объема 60 мл (240 мл ТБМЭ отгоняли). Смесь перемешивали в течение 1 ч при 40°С (кристаллизация). Добавляли 50 мл HPTF, смесь охлаждали до 0°С и перемешивали при 0°С в течение 1 ч. После упаривания, промывки (2×15 мл) HPTF и высушивания в течение ночи при 45°С в вакууме получали кристаллы белого цвета.
Присоединение 4-(дифенилгидроксиметил)бензойной кислоты к твердой фазе
15 г 4-(Дифенилгидроксиметил)бензойной кислоты и 7,54 г гидроксибензотриазола растворяли в 140 мл ДМФА при перемешивании в течение 15 мин. Затем добавляли 15,3 мл диизопропилкарбодиимида и раствор выдерживали при комнатной температуре в течение 30 мин. Раствор перемешивали в течение ночи при комнатной температуре в присутствии аминометилированного полистирола. После промывки ДМФА, метанолом и ТГФ носитель, содержащий связующее звено, сушили в вакууме.
Пример 2
Получение 4-(дифенилгидроксиметил)бензойной кислоты (альтернативный способ)
К 12 л ТБМЭ в тщательно высушенном реакторе Hastelloy объемом 100 л в течение 20 мин добавляли 3,0 кг н-бутиллития (20% в циклогексане; 9,37 моля) при температуре -5°С (прозрачный раствор). В течение 30 мин при температуре в интервале от -5 до 0°С добавляли 2,00 кг 1,4-дибромбензола (8,48 моля), растворенного в 16 л ТБМЭ. Контейнер, из которого добавляли реагент, промывали 3 л ТБМЭ.
Смесь перемешивали при -5°С в течение 30 мин, а затем в течение 20 мин при температуре в интервале от -5 до 0°С добавляли 1,55 кг бензофенона (8,50 моля) в 8 л ТБМЭ. Контейнер, из которого добавляли реагент, промывали 3 л ТБМЭ. При этом образовывалось небольшое количество твердого вещества белого цвета. После перемешивания в течение 15 мин при -5°С отбирали пробу для анализа ЖХВР (образец 1). Реакционную смесь перемешивали в течение 25 мин при -5°С и нагревали до +25°С.
К смеси в течение 25 мин при температуре в интервале от +25 до +27°С добавляли 3,2 кг н-бутиллития (20% в циклогексане, 10,00 моля). При добавлении наблюдалась слабо экзотермическая реакция, цвет смеси изменялся на светло-зеленый и образовалось некоторое количество осадка и пены. После перемешивания в течение 20 мин отбирали пробу для анализа ЖХВР (образец 2). В зависимости от результата анализа пробы 2 методом ЖХВЭ добавляли 0,3 кг н-бутиллития после перемешивания в течение 35 мин при 25°С. Затем после перемешивания в течение 15 мин отбирали пробу для анализа ЖХВР (образец 3). Шланги, через которые добавляли н-бутиллитий, промывали 1,5 л ТБМЭ и реакционную смесь охлаждали до -10°С. К смеси в течение 20 мин при температуре в интервале от -10 до -5°С порциями добавляли 1,99 кг сухого льда (твердый СО2). При этом наблюдалась экзотермическая реакция и образовался осадок светло-желтого цвета. После перемешивания в течение 15 мин при -10°С добавляли 11 л ТБМЭ и реакционную смесь нагревали до 0°С. В течение 15 мин при температуре в интервале от 0 до +5°С добавляли 5 л 18% водного раствора хлористоводородной кислоты. При этом наблюдались экзотермическая реакция и растворение осадка (pH≤1).
Прозрачный раствор переносили в сепаратор и реактор промывали 5 л ТБМЭ. После отделения водной фазы органическую фазу промывали 20 л воды. После разделения двух слоев органическую фазу экстрагировали 13 л 5% водного раствора КОН. Щелочную водную фазу отделяли и органический слой снова экстрагировали 13 л 5% водного раствора КОН. Объединенные щелочные водные слои переносили в реактор Hastelloy объемом 100 л. В течение 20 мин при температуре в интервале от 0 до 5°С добавляли 22 л ТБМЭ и 6 л 18% водного раствора хлористоводородной кислоты. При этом наблюдались экзотермическая реакция и образование осадка белого цвета, который растворялся при низком значении рН (рН≤1 после добавления HCl). Смесь перемешивали в течение 10 мин и переносили в сепаратор. Слои разделяли и водную фазу экстрагировали 16 л ТБМЭ. После разделения слоев объединенные органические фазы концентрировали при давлении 500 мбар, температуре в бане 45°С до объема 4-5 л (32 л ТБМЭ отгоняли) и добавляли кристаллы для затравки. Температуру поднимали до 50°С и при интенсивном перемешивании медленно добавляли 20 л HPTF. Осадок белого цвета перемешивали в течение 2 ч при температуре в бане 50°С. Температуру в бане снижали до 0°С и перемешивание продолжали в течение ночи (16 ч), при этом температуру суспензии поддерживали при 0°С. Суспензию белого цвета отфильтровывали и реактор промывали 5 раз 5 л маточного раствора. Осадок сушили при температуре в бане 45°С в вакууме (≥10 мбар) до постоянного веса (в течение ночи).
Пример 3
Получение Fmoc-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH из Cbo-(2S,4R)-Pro(4-OH)-OMe
1. Диметиламинопиридин (30,5 г, 250 ммолей) и Cbo-(2S,4R)-Pro(4-OH)-ОМе (34,9 г, 125 ммолей) растворяли в толуоле (870 мл). К полученному раствору при температуре от 0 до 5°С в течение 20 мин по каплям добавляли раствор 4-нитрофенилхлорформиата (31,5 г, 157 ммолей) в толуоле (206 мл) и перемешивали в течение 2 ч. Затем добавляли раствор Вос-этилендиамина (80,1 г, 500 ммолей) в толуоле (205 мл) и перемешивали при температуре окружающей среды в течение 12 ч. К смеси при температуре от 20 до 25°С добавляли раствор концентрированной серной кислоты (43,7 г, 450 ммолей) в воде (873 мл). Суспензию белого цвета фильтровали в вакууме и промывали толуолом (30 мл). Фазу в толуоле промывали водой (450 мл), карбонатом натрия (10 мас./мас.%, 450 мл) и три раза водой (по 450 мл). Фазу в толуоле сушили при перегонке азеотропной смеси (300 мл), которую заменяли на сухой толуол (2×300 мл). К раствору в сухом толуоле при 50°С добавляли гептан (130 мл) и охлаждали до 0°С в течение 2 ч. Образовавшийся осадок фильтровали, промывали два раза толуолом/гептаном (1:2 об/об, 70 мл) и сушили при 50°С в вакууме, при этом получали Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe в виде твердого вещества белого цвета.
2. Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe (20,0 г, 43,0 ммоля) растворяли в смеси тетрагидрофурана и метанола (1:1, 380 мл). К раствору добавляли 1М раствор гидроксида натрия (51,6 мл) и полученную смесь перемешивали в течение 4 ч при температуре окружающей среды. Величину рН смеси доводили до 3 добавлением серной кислоты (50 мл, 1М). Тетрагидрофуран и метанол отгоняли при 50°С и 50 мбар до прекращения перегонки растворителей. Полученный раствор молочного цвета разбавляли изопропилацетатом (113 мл) и водой (57 мл), фазы разделяли и фазу в изопропилацетате промывали раствором хлорида натрия (10%, 113 мл). Растворитель отгоняли (50°С, 50 мбар), при этом получали Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH (19,8 г) в виде пены, который использовали на следующей стадии без дополнительной очистки.
3. Палладий на угле (10%, 1,94 г, 0,042 ммоля) добавляли к раствору Cbo-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH (19,4 г, 43,0 ммоля) в изопропаноле (350 мл) и воде (37 мл). Через полученную смесь пропускали газообразный водород в течение 4 ч, катализатор отфильтровывали и остаток промывали смесью изопропанола (50 мл) и воды (50 мл). Фазу в смеси изопропанола и воды сушили при перегонке азиотропной смеси до уменьшения объема на 2/3, который непрерывно заменяли на смесь толуола и изопропанола (1:1 об/об). Полученный безводный раствор концентрировали в вакууме досуха (50°С, 200 мбар), при этом получали (2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-ОН в виде твердого вещества коричневатого цвета, который использовали на следующей стадии без дополнительной очистки.
4. (2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH (5,0 г, 15 ммолей) растворяли в смеси воды (25 мл) и триэтиламина (1,5 г, 15 ммолей) при 40°С. К прозрачному раствору добавляли раствор Fmoc-OSu (4,65 г, 14 ммолей) в ацетонитриле (25 мл) в течение 30 мин и перемешивали в течение 2 ч. Затем рН смеси доводили до 3 с использованием хлористоводородной кислоты (1 М, 13 мл) и перемешивали в течение 1 ч. Ацетонитрил отгоняли (40°С, 80 мбар) и заменяли на изопропилацетат, при этом получали двухфазную смесь. Нижнюю водную фазу отделяли и полученную органическую фазу промывали водой, два раза перегоняли с заменой на изопропилацетат и концентрировали до пены коричневатого цвета. Полученную пену растворяли в изопропилацетате (25 мл) и по каплям добавляли к гептану (200 мл), при этом получали осадок. Твердое вещество отфильтровывали, промывали изопропилацетатом/гептаном и сушили в вакууме при 40°С, при этом получали Fmoc-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH.
Пример 4
Получение Fmoc-(2S,4R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH из Cbo-(2S,4R)-Pro(4-OH)-OBzl
Синтез Cbo-(2S,4R)-Pro(4-OH)-OBzl описан в работе T.Makoto, H.Guoxia, V.J.Hruby, J.Org.Chem., 66, 1038-1042 (2001). Использовали способ синтеза, описанный в примере 3, но вместо Cbo-(2S,4R)-Pro(4-OH)-OMe использовали Cbo-(2S,4R)-Pro(4-OH)-OBzl и выполняли только стадии 1, 3 и 4 (исключая стадию 2).
Пример 5
Получение Fmoc-(2R,R)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
Использовали способ синтеза, описанный в примере 3 или 4, но вместо Cbo-(2S,4R)-Pro(4-OH)-OMe или Cbo-(2S,4R)-Pro(4-OH)-OBzl использовали Cbo-(2R,4R)-Pro(4-OH)-OMe или Cbo-(2R,4R)-Pro(4-OH)-OBzl.
Пример 6
Получение Fmoc-(2S,4S)-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
Использовали способ синтеза, описанный в примере 3 или 4, но вместо Cbo-(2S,4R)-Pro(4-OH)-OMe или Cbo-(2S,4R)-Pro(4-OH)-OBzl использовали Cbo-(2S,4S)-Pro(4-OH)-OMe или Cbo-(2S,4S)-Pro(4-OH)-OBzl.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОЧЕВИНА И СУЛЬФАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TAFIA | 2007 |
|
RU2459619C2 |
| АЛЬТЕРНАТИВНЫЕ СПОСОБЫ СИНТЕЗА ИНГИБИТОРОВ РЕНИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2005 |
|
RU2411230C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| ЗАМЕЩЕННЫЕ 3-ПИРИДИЛМЕТИЛАМИНЫ И ФОКУСИРОВАННАЯ БИБЛИОТЕКА | 2003 |
|
RU2228930C1 |
| НОВЫЕ ЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЗОРБЦИИ КОСТИ И АНТАГОНИСТОВ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2180331C2 |
| ИНГИБИТОРЫ КАСПАЗ И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2382780C2 |
| КОМБИНАЦИИ АКТИВАТОРА (АКТИВАТОРОВ) РЕЦЕПТОРА, АКТИВИРУЕМОГО ПРОЛИФЕРАТОРОМ ПЕРОКСИСОМ (РАПП), И ИНГИБИТОРА (ИНГИБИТОРОВ) ВСАСЫВАНИЯ СТЕРИНА И ЛЕЧЕНИЕ ЗАБОЛЕВАНИЙ СОСУДОВ | 2008 |
|
RU2483724C2 |
| НОВЫЕ КОНЪЮГАТЫ АНАЛОГОВ СС-1065 И БИФУНКЦИОНАЛЬНЫЕ ЛИНКЕРЫ | 2011 |
|
RU2578719C9 |
| НОВЫЕ КОНЪЮГАТЫ АНАЛОГОВ СС-1065 И БИФУНКЦИОНАЛЬНЫЕ ЛИНКЕРЫ | 2011 |
|
RU2730502C2 |
| ПРОИЗВОДНЫЕ БИЦИКЛИЧЕСКИХ ИМИНОКИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ МАТРИКСНЫХ МЕТАЛЛОПРОТЕИНАЗ | 2004 |
|
RU2335494C2 |
Настоящее изобретение относится к способу получения соединения формулы VIII, где R12 и R13 каждый означает удаляемую защитную группу, где защитная группа R12 является более устойчивой к удалению путем гидролиза или гидрирования, чем R13; включающий взаимодействие соединения формулы IX, с пригодным соединением - донором R12, выбранным из ангидридов, галогенидов, карбаматов и N-гидроксисукцинимидов. 2 н. и 2 з.п. ф-лы.
1. Способ получения соединения формулы VIII
где R12 и R13 каждый означает удаляемую защитную группу, где защитная группа R12 является более устойчивой к удалению путем гидролиза или гидрирования, чем R13;
включающий взаимодействие соединения формулы IX
с пригодным соединением - донором R12, выбранным из ангидридов, галогенидов, карбаматов и N-гидроксисукцинимидов.
2. Способ по п.1, где соединение формулы IX получают путем:
(i) гидролиза соединения формулы Х
где R13 определен в п.1,
R14 означает удаляемую защитную группу, выбранную из группы, включающую бензилоксикарбонил, 1,1-диметилпропинилоксикарбонил, винилоксикарбонил, N-гидроксипиперидинилоксикарбонил, 9-антрилметилоксикарбонил, фениламинотиокарбонил, аллил, нитробензил, трифенилметил, пара-метоксифенил, дифенилметил, дифенил-4-пиридилметил и бензилсульфонил, и
R15 означает блокирующую группу, удаляемую в условиях гидролиза или гидрирования, выбранную из C1-С10алкила; С3-С8циклоалкила, необязательно замещенного одним или более С1-С4алкилом; С6-С10арила, необязательно замещенного одним или более галогеном или нитро; (С6-С10арил)1-3(С1-С10)алкила, необязательно замещенного в арильной группе (i) одним или более галогеном или нитро, или (ii) двумя заместителями, которые вместе атомами углерода в цикле, к которым они присоединены, образуют 5- или 6-членный цикл, необязательно содержащий один или два атома азота или кислорода; С6-С10арил(С1-С4)алкокси(С1-С4)алкила и С6-С10арилкарбонил(С1-С4)алкила;
где гидролиз или гидрирование представляют собой гидролиз, катализируемый основаниями, который может быть осуществлен в полярном растворителе, для получения соответствующей карбоновой кислоты, и
(ii) удаление защитной группы R14 путем восстановительного гидрирования в полученной карбоновой кислоте.
3. Способ получения соединения формулы VIII по п.1, где R12 означает флуоренилметоксикарбонил, a R13 означает удаляемую защитную группу, другую чем флуоренилметоксикарбонил, включающий взаимодействие соединения формулы IX с соединением-донором группы флуоренилметоксикарбонил.
4. Соединение формулы XIV
где R16 означает трет-бутоксикарбонил;
R17 означает водород или блокирующую группу, удаляемую с помощью гидролиза или гидрирования, выбранную из С1-С10алкила; С3-С8циклоалкила, необязательно замещенного одним или более С1-С4алкилом; С6-С10арила; (С6-С10арил)1-3(С1-С10)алкила;
R18 означает водород или удаляемую защитную группу, представляющую собой бензилоксикарбонил.
| ПРОИЗВОДНЫЕ ПРОЛИНА, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ЭЛАСТАЗЫ ЛЕЙКОЦИТОВ ЧЕЛОВЕКА | 1996 |
|
RU2159249C2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| T.W.GREEN "PROTECTIVE GROUPS IN ORGANIC SYNTHESIS", JOHN WILLEY @ SONS INC, 1981. | |||

