Настоящее изобретение относится к новому способу получения α-кетокислот, прежде всего α-кетометионина, и их производных.
α-Кетокислоты являются важными продуктами и используются помимо прочего дополнительно к аминокислотам в терапии хронической почечной недостаточности (Jungers и др.. Blood Purification, 6, 1988, ее. 299-314; Clasen и др., Med. Klin., 73, 1978, сс. 1403-1408).
В литературе описаны самые разнообразные способы синтеза α-кетокислот, в том числе, в частности, путем реакции реактива Гриньяра с диалкилоксалатом и последующего гидролиза образовавшегося сложного эфира до свободной кислоты (Rambaud и др., Synthesis, 564, 1988; Macritchie и др., Tetrahedron: Asymmetry, 8, 1997, с.3895). α-Кетокислоты можно также получать путем проводимого при кислотном катализе гидролиза ацилцианидов (Nozaki и др., Tetrahedron: Asymmetry, 4, 1993, с.2179). В публикациях автора Biliek содержится информация о получении целого ряда α-кетокислот. При этом алкилиденгидантоины подвергают омылению в основных реакционных условиях (Biliek, Monath. Chem., 92, 1961, сс. 343-352).
α-Кетокислоты можно также получать из соответствующего альдегида и диоксида углерода путем реакции, приводящей к изменению (инверсии) полярности. Одна из возможностей изменения полярности альдегида заключается в образовании циклического дитиана, из которого путем депротонирования сильным основанием можно образовать карбанион. Этот карбанион можно затем подвергать взаимодействию с диоксидом углерода. В завершение циклический дитиан расщепляют в окислительных условиях, например, взаимодействием с солями ртути, с образованием требуемой α-кетокислоты
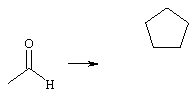
В соответствии с работами Согеу и Seebach (Corey и др., Angew. Chem., 77, 1965, сс. 1134-1136; Seebach и др., J. Org. Chem., 40, 1975, сс. 231-237) для синтеза циклических дитиоацеталей (дитианов) обычно используют дитиолы. Подобный синтез отличается особой простотой, поскольку из-за высокой термодинамической устойчивости образовавшегося пяти- соответственно шестичленного цикла образование циклических дитианов носит преимущественный характер. Помимо этого циклизация взаимодействием с дитиолами предпочтительна с кинетической точки зрения. Однако именно подобная высокая термодинамическая устойчивость является существенным недостатком, поскольку значительно затрудняет расщепление дитианового кольца, в связи с чем выделение требуемой α-кетокислоты в свободном виде возможно лишь в химически жестких условиях. Обычно дитиан расщепляют проведением окислительного процесса (Seebach, Synthesis, 1969, ее. 17-36). По этой причине отделение и повторное использование дитиола более невозможны, поскольку серу для отделения окисляют и затем взаимодействием с солями ртути осаждают в виде труднорастворимой соли. К другим недостаткам этого способа получения α-кетокислот относятся высокая стоимость пригодных для его осуществления дитиолов, таких, например, как 1,2-этандитиол или 1,3-пропандитиол, а также их наличие в малых количествах, которые были бы доступны для промышленного применения. Указанные недостатки делают описанный выше способ получения α-кетокислот, основанный на изменении полярности, малопривлекательным для реализации в промышленном масштабе.
В литературе описано множество примеров по превращению короткоцепных тиолов, таких, например, как метилмеркаптан, путем их взаимодействия с альдегидами в соответствующие тиоацетали (Трофимов и др., "Журнал органической химии", 8, 1972, с.2036; Rothstein и др., J. Chem. Soc. 1940, с.1563). Помимо этого известно применение таких дитиоацеталей в приводящих к изменению полярности реакциях с различными электрофилами. Однако в литературных источниках имеется лишь единственный пример успешного химического превращения ароматического тиоацеталя взаимодействием с СО2 по реакции, приводящей к изменению полярности (Micetich и др., Heterocycles, 23, 1985, сс. 585-592).
α-Кетометионин представляет собой особую α-кетокислоту, поскольку она промежуточно образуется в организме в процессе превращения D-метионина в L-метионин. α-Кетометионин можно получать многочисленными способами его синтеза, и он широко представлен в литературе. Помимо этого из литературных источников известны некоторые его соли и иные производные, такие, например, как эфир кетометионина. Способы получения α-кетометионина при этом принципиально можно подразделить на химические и биохимические.
а) Биохимические способы
По методу Meister путем катализируемого L-аминооксидазами окисления метионина удалось получить натриевую соль α-кетометионина с выходом 77% (Meister, Journ. Biol. Chem., 197, 1952, с.309). Ранее Waelsch и др. установили, что содержащиеся в печени аминооксидазы способны превращать метионин в α-кетометионин (Waelsch и др., Journ. Am. Chem. Soc., 61, 1938, с.2252).
У Mosbach и др. также описано получение α-кетометионина путем катализируемого L-аминооксидазами окисления метионина. При этом используют иммобилизованные клетки Providencia sp.PCM 1298 (Mosbach и др., Enzyme Microb. Technol., 4, 1982, с.409).
Недостатки получения α-кетометионина, соответственно его производных с помощью биологических систем с использованием либо очищенных ферментов, либо целых клеток обычно состоят в пониженном выходе продукта с единицы объема в единицу времени, а также в технически сложных выделении и очистке продукта. При использовании высокочистых ферментов к этим недостаткам добавляется еще и недостаток, связанный с высокой стоимостью и сложностью разработки, производства и очистки ферментов, а также с невозможностью в большинстве случаев повторного применения уже использованных ферментов.
б) Химические способы
В 1957 г. Sakurai и др. впервые опубликовали способ химического синтеза α-кетометионина. Ключевой стадией при этом являлся гидролиз метил-α-метоксалил-γ-метилмеркаптопропионата до α-кетометионина действием разбавленной соляной кислотой (Sakurai и др., J. of Biochem., 44, 9, 1957, с.557).
Почти в то же время Yamada и др. опубликовали такой же способ синтеза, в соответствии с которым в первых экспериментах по получению α-кетометионина через промежуточно образующийся α-оксимовый эфир α-кетометионин удавалось получать лишь с пониженным выходом (Chibata и др., Bull. Agr. Chem. Soc. Japan, 21, 1957, с.336).
Существенный недостаток опубликованного Sakurai и Yamada способа состоит в образовании солей в значительных количествах, из-за чего промышленное получение α-кетометионина этим способом невозможно. Помимо этого такой способ синтеза α-кетометионина характеризуется неэкономным расходованием атомов, поскольку часть молекулы отщепляется на одной из стадий синтеза в виде диоксида углерода и тем самым необратимо утрачивается. Поэтому промышленный синтез α-кетометионина подобным способом был бы слишком дорогим и экономически неэффективным.
Сравнительно недавно в WO 2006/072711 был опубликован способ получения α-кетометионина исходя из бутадиена. При этом бутадиен селективно окисляют до ненасыщенного моноэпоксида, который затем путем катализируемого кислотой раскрытия цикла взаимодействием с водой превращают в соответствующий 1,2-диол. Путем последующего окисления 1,2-диола получают α,β-ненасыщенную α-кетокислоту, из которой затем путем 1,4-присоединения MeSH получают α-кетометионин.
Недостаток указанного способа состоит в необходимости использования дорогого бутадиена в качестве исходного материала. Вполне вероятно, что стоимость бутадиена в будущем продолжит возрастать в прямой зависимости от цен на нефть. Еще один недостаток описанного в WO 2006/0727211 способа состоит в том, что для его осуществления невозможно было бы использовать ни одну из уже существующих установок для производства метионина, соответственно его гидроксианалога (ГАМ) и поэтому для проведения каждой отдельной стадии описанного в указанной публикации способа потребовалось бы сооружать новые промышленные установки. Описанным в WO 2006/072711 способом всегда синтезируют свободный α-кетометионин, который нестабилен и который очень сложно выделять.
α-Кетометионин, соответственно его производные, такие, например, как его соли или эфиры, представляют собой особо важные соединения, поскольку они в качестве добавок к кормам являются альтернативной заменой метионину, соответственно его гидроксианалогу (ГАМ).
В появившейся в 1942 г. публикации Rudolph и др. говорилось, что натриевая соль α-кетометионина может использоваться в качестве замены D,L-метионину и благодаря этому способна ускорять рост молодых крыс (Rudolph и др., J. Biol. Chem., 145, 1942, с.210).
Во многих публикациях неоднократно приводились подтверждения тому, что α-кетометионин может служить источником серы, необходимой для биосинтеза L-метионина и L-цистеина (Sizer и др., Poultry Sci., 44, 1964, с.673; Baker и др., Poultry Sci., 54, 1975, с.584; Baker, J. Nutr., 106, 1976, с.1376).
Помимо этого по результатам исследований, проведенных Baker и Harter, удалось установить, что относительная биологическая доступность кальциевой соли α-кетометионина при ее применении в качестве добавки к кормам для кур, способствующей их росту, составляет 83% в сравнении с L-метионином (100%) и ГАМ (53%) (Baker и Harter, Proceedings of the Society for Experimental Biology and Medicine, 156, 1977, с.201). Данные об относительной биологической доступности различных производных метионина, в том числе и α-кетометионина (90% в сравнении с L-метионином), опубликованы Baker в "Utilization of Precursors for L-Amino Acids" на с.39.
Применение α-кетометионина и его солей, эфиров и амидов в качестве кормовой добавки описано в WO 2006/072711.
Исходя из описанных выше недостатков, присущих уровню техники, в основу настоящего изобретения была положена задача разработать усовершенствованный, экологичный и пригодный для реализации в промышленном масштабе способ получения кетокислот, прежде всего α-кетометионина и его производных, которые в качестве кормовых добавок являются альтернативной заменой метионину, соответственно ГАМ.
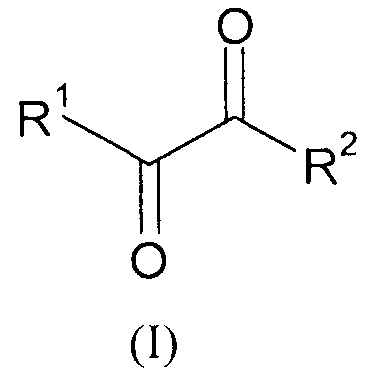
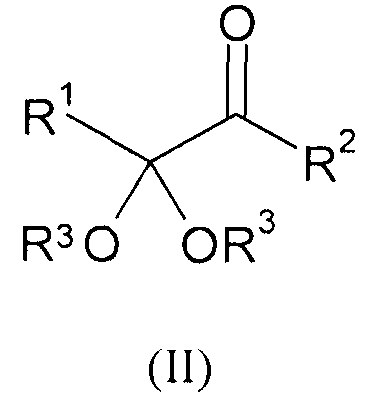
Указанную задачу позволяет решить предлагаемый в изобретении способ. Объектом изобретения в соответствии с этим является способ получения α-кетокислот, а также их производных общей формулы (I) или (II)
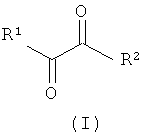 ,
, 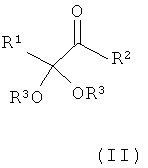 ,
,
где
R1 обозначает разветвленную либо линейную С1-С18алкильную, С5-С8циклоалкильную, винильную, аллильную, С6-С10арил-С1-С4алкиленовую, предпочтительно бензильную, или С4-С9гетероарил-С1-С4алкиленовую группу, предпочтительно 4-имидазолилметиленовую или 3-индолилметиленовую группу, и необязательно замещен,
R2 обозначает -OR''' или -NR'R'', где R' и R'' могут иметь одинаковые или разные значения и представляют собой атом водорода или разветвленную либо линейную C1-С6алкильную группу, а R''' представляет собой атом водорода или разветвленную либо линейную С1-С8алкильную, С5-С8циклоалкильную, аллильную или бензильную группу, при этом алкил в R', R'' и/или R''' необязательно замещен, или R'" представляет собой ион щелочного, щелочноземельного или одно- либо двухвалентного переходного металла,
R3 имеют одинаковые или разные значения и представляют собой атом водорода или разветвленную либо линейную С1-С8алкильную, аллильную или бензильную группу, при этом алкил и/или бензил необязательно замещены/замещен, или оба остатка R3 совместно представляют собой С2-С8алкандиил и совместно образуют кольцо либо оба остатка R3 и R''' совместно являются частью С3-С8алкантриильной группы общей формулы -R3(CH-)R'''- и совместно образуют бициклическую группу, заключающийся в том, что
а) альдегид R1СНО подвергают взаимодействию с двумя одинаковыми или разными тиолами формулы R4-SH, где R4 обозначает разветвленную либо линейную, необязательно замещенную С1-С6алкильную, С5-С8циклоалкильную, аллильную или бензильную группу, с получением соответствующего дитиоацеталя,
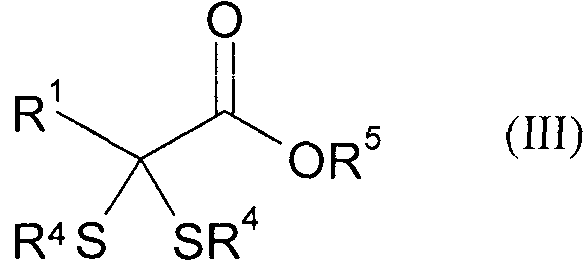
б) образовавшийся дитиоацеталь подвергают в присутствии сильного основания взаимодействию с карбонилсодержащими электрофилами, такими как диоксид углерода, фосген, эфир хлормуравьиной кислоты, эфир ортомуравьиной кислоты или диалкилкарбонат, и последующему гидролизу с получением α,α-(дитио)карбоновой кислоты или ее производных формулы (III)
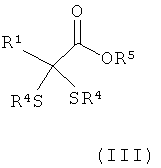 ,
,
где R5 может представлять собой атом водорода или разветвленную либо линейную C1-С6алкильную группу, а остатки R4 и R1 имеют указанные выше значения, и
в) α,α-(дитио)карбоновую кислоту или ее производные формулы (III) превращают путем проводимого при кислотном катализе сольволиза в присутствии по меньшей мере 1 молярного эквивалента воды в α-кетокислоту или ее производные общей формулы (I) или (II) с выделением тиолов формулы R4SH.
Указанные в качестве значений R1 остатки необязательно могут быть замещены -SH, -SСН3, -СООН, -CONH2, -СНО, гуанидилом, -ОН, -NR'R'' или -SS-CH2-C(H)NH2-C2H, где R' и R'' имеют указанные выше значения.
В предпочтительном варианте R1 обозначает винил или разветвленный или линейный, необязательно замещенный С1-С8алкил, более предпочтительно винил или разветвленный либо линейный, необязательно замещенный С1-С4алкил, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил и трет-бутил.
В особенно предпочтительном варианте R обозначает винил, н-пропил или 2-(метилтио)этил, прежде всего 2-(метилтио)этил.
В предпочтительном варианте R2 обозначает -OR''', где R''' представляет собой атом водорода или разветвленную либо линейную С1-С4алкильную группу.
В предпочтительном варианте R3 независимо друг от друга обозначают атом водорода или разветвленную либо линейную, необязательно замещенную С1-С4алкильную группу, прежде всего изопропил или изобутил, или оба остатка R3 совместно обозначают С2-С4алкандиил и совместно образуют кольцо либо оба остатка R3 и R''' совместно являются частью С3-С6алкантриильной группы общей формулы -R3(СН-)R'''- и совместно образуют бициклическую группу.
В предпочтительном варианте R обозначает метил или этил, прежде всего метил.
При осуществлении предлагаемого в изобретении способа сначала получают нециклический дитиоацеталь (стадия а)).
Затем полученный на стадии а) дитиоацеталь по реакции, приводящей к изменению полярности, подвергают в присутствии основания взаимодействию с карбонилсодержащим электрофилом, таким, например, как диоксид углерода, фосген, эфир хлормуравьиной кислоты, эфир ортомуравьиной кислоты или диалкилкарбонат (стадия б)), с получением α,α-(дитио)карбоновой кислоты или ее производных. В качестве электрофила предпочтительно использовать диоксид углерода, который можно применять либо в виде газа, либо в виде твердого вещества (например, сухого льда), либо в виде жидкости (надкритической или в виде растворителя).
Для применения при первоначальном на стадии б) депротонировании наиболее пригодны основания, соответствующая которым кислота имеет показатель рКа более 20. К предпочтительным основаниям при этом относятся алкиллитиевые соединения, прежде всего н-бутиллитий и трет-бутиллитий, амиды и гидриды металлов, прежде всего амид натрия и гидрид натрия, а также гидроксиды или карбонаты металлов индивидуально либо в сочетании с хелатообразующими лигандами, такими, например, как краун-эфиры, предпочтительно гидроксид калия или карбонат калия в сочетании с 18-краун-6.
Депротонирование на стадии б) в присутствии вышеуказанных оснований предпочтительно проводить в апротонных органических растворителях, в жидком надкритическом диоксиде углерода или в жидком аммиаке. Особенно предпочтительны при этом простые эфиры, такие как тетрагидрофуран, алкилсульфоксиды, такие как диметилсульфоксид (ДМСО), ароматические соединения, такие как толуол, алифатические и циклические алканы, такие как н-гексан и циклогексан, а также алкиламиды, такие как диметилформамид.
Реакцию предпочтительно проводить при температуре в пределах от -80 до 100°С, более предпочтительно от -30 до 25°С.
Гидролиз предпочтительно проводить в водных растворах при значении рН в пределах от 1 до 14, более предпочтительно в пределах от 1 до 4 или от 9 до 13.
В целом гидролиз проводят при температуре в пределах от -20 до 100°С, предпочтительно от 0 до 30°С.
После этого на стадии в) образовавшуюся на стадии б) α,α-(дитио)карбоновую кислоту или ее производные подвергают проводимому при кислотном катализе сольволизу с выделением тиолов или их солей, при этом продукт может в каждом случае в зависимости от применяемого растворителя представлять собой, например, свободную α-кетокислоту, а также ее соли, эфир α-кетокислоты, амид α-кетокислоты, кетальное производное α-кетокислоты или кетальное производное эфира α-кетокислоты (см. также приведенную ниже схему).
Под "сольволизом" подразумевается реакция α,α-(дитио)карбоновой кислоты с растворителем, например гидролиз (реакция с водой).
В целом сольволиз проводят при температуре в пределах от 20 до 200°С, предпочтительно от 50 до 150°С, и при кислотном катализе. К предпочтительным катализаторам относятся, например, паратолуолсульфоновая кислота, СF3SO3Н, а также минеральные кислоты, такие как НСl или H2SO4, или сильные органические кислоты, которые не являются нуклеофильными.
Образовавшиеся тиолы формулы R4SH можно удалять из реакционной смеси путем вакуумирования, путем подачи инертного газа или иным пригодным для этой цели метолом разделения, таким, например, как разделение фаз, кристаллизация, комплексообразование или осаждение, возвращать в технологический процесс и тем самым вновь использовать затем в реакции, приводящей к изменению полярности.
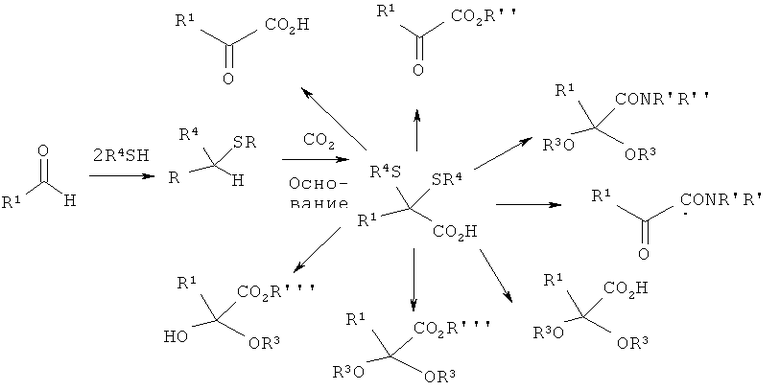
В одном из вариантов в процессе проводимого при кислотном катализе сольволиза на стадии в) в качестве растворителя используют воду с получением при этом свободной α-кетокислоты формулы (I), в которой R2 обозначает OR''', где R''' представляет собой атом водорода, а R1 имеет указанные выше значения.
В другом варианте в процессе проводимого при кислотном катализе сольволиза на стадии в) в качестве растворителя используют спирт R'''OH, где R''' представляет собой разветвленную либо линейную С1-С8алкильную, С5-С8циклоалкильную, аллильную или бензильную группу, а алкил необязательно замещен, с получением при этом эфира α-кетокислоты формулы (I), в которой R2 обозначает OR''', где R''' имеет указанные выше в данном абзаце значения, а R1 имеет указанные выше значения.
В еще одном варианте в процессе проводимого при кислотном катализе сольволиза на стадии в) в качестве растворителя используют диол HO-R3-ОН, где R3 представляет собой разветвленную либо линейную, необязательно замещенную С2-С8алкандиильную группу, с получением при этом циклического кеталя формулы (II), в которой R2 обозначает OR''', где R''' представляет собой H, R3 имеет указанные выше в данном абзаце значения, a R1 имеет указанные выше значения.
В следующем варианте в процессе проводимого при кислотном катализе сольволиза на стадии в) в качестве растворителя используют триол формулы HOR(CHOH)R''OH, где остатки R3 и R''' совместно являются частью разветвленной либо линейной, необязательно замещенной С3-С8алкантриильной группы общей формулы -R3(CH-)R'''-, с получением при этом бициклического эфира в виде кеталя формулы (II), в которой R2 обозначает OR''', где R''' имеют указанные выше в данном абзаце значения, R3 имеет указанные выше в данном абзаце значения, а R1 имеет указанные выше значения.
В следующем варианте в процессе проводимого при кислотном катализе сольволиза на стадии в) в качестве растворителя используют амин HNR'R'', где R' и R'' могут иметь одинаковые или разные значения и представляют собой атом водорода или разветвленную либо линейную С1-С6алкильную группу, при условии, что R' и R'' не могут одновременно обозначать атом водорода. При этом получают амид формулы (I), в которой R2 обозначает NR'R'', где R' и R'' имеют указанные выше в данном абзаце значения.
При получении α-кетокислот, а также их производных предлагаемым в изобретении способом образующийся на стадии а) дитиоацеталь выполняет также функцию защитной группы, которую после присоединения диоксида углерода (стадия б)) можно вновь удалять в присутствии кислоты путем сольволиза (стадия в)) в вакууме или путем подачи инертного газа, такого, например, как азот. Связанное с этим преимущество состоит в возможности последующего возврата выделяющегося при этом тиола в технологический цикл и его повторного применения в круговом процессе.
При получении α-кетометионина может оказаться предпочтительным получать дитиоацеталь на стадии а) либо взаимодействием 3-(метилтио)пропаналя (3-метилмеркаптопропионового альдегида (ММП)) с метилтиолом (MeSH), либо непосредственно из акролеина путем трехкратного присоединения метилтиола:
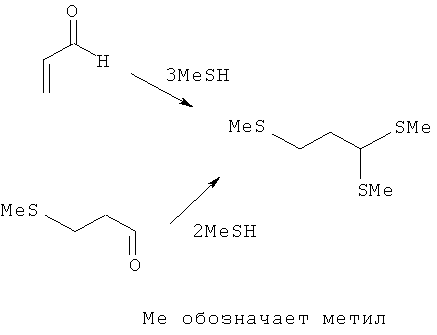
По сравнению с уровнем техники предлагаемый в изобретении способ позволяет получать α-кетокислоты, соответственно их производные, с повышенным выходом, в чем состоит существенное экономическое преимущество.
Помимо этого для реализации предлагаемого в изобретении способа можно в отличие от описанного в WO 2006/072711 способа и далее использовать уже существующие установки для получения 3-(метилтио)пропаналя, т.е. установки для получения акролеина, а также метилтиола.
Предлагаемый в изобретении способ, кроме того, экологичен, поскольку в качестве структурного фрагмента С-1 преимущественно используют диоксид углерода, что вносит свой вклад в охрану окружающей среды (Киотский протокол).
В отличие от описанного в WO 2006/072711 способа получения α-кетометионина исходя из бутадиена предлагаемый в изобретении способ обладает высокой гибкостью, поскольку позволяет непосредственно получать не только саму α-кетокислоту, но и все ее производные, такие, например, как эфиры, кетали и другие.
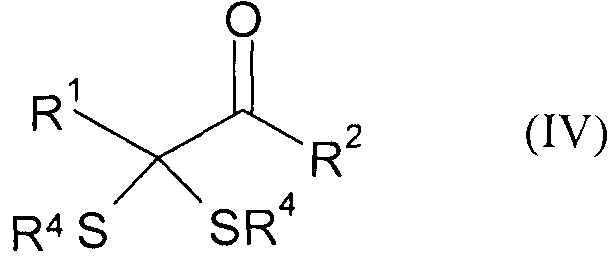
Следующим объектом изобретения является промежуточный продукт формулы (IV)
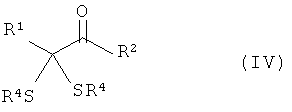
в которой
R1 обозначает разветвленную либо линейную С1-С18алкильную, С5-С8циклоалкильную, винильную, аллильную, С6-С10арил-С1-С4алкиленовую, предпочтительно бензильную, или С4-С9 гетероарил-С1-С4алкиленовую группу, предпочтительно 4-имидазолилметиленовую или 3-индолилметиленовую группу, и необязательно замещен,
R2 обозначает -OR''' или -NR'R'', где R' и R'' могут иметь одинаковые или разные значения и представляют собой атом водорода или разветвленную либо линейную C1-С6алкильную группу, а R''' представляет собой атом водорода или разветвленную либо линейную С1-С8алкильную, С5-С8циклоалкильную, аллильную или бензильную группу, при этом алкил в R', R'' и/или R''' необязательно замещен, или R''' представляет собой ион щелочного, щелочноземельного или одно- либо двухвалентного переходного металла, и
R4 имеют одинаковые или разные значения и могут представлять собой разветвленную либо линейную, необязательно замещенную C1-С6алкильную, С5-С8циклоалкильную, аллильную или бензильную группу.
Предпочтительны предлагаемые в изобретении промежуточные продукты формулы (IV), в которой R1 обозначает необязательно замещенный С1-С4алкил, прежде всего СН3SСН2СН2.
Следующим объектом изобретения является применение тиола формулы R4-S-H, где R4 представляет собой разветвленную либо линейную, необязательно замещенную C1-С6алкильную, С5-С8циклоалкильную, аллильную или бензильную группу, для изменения полярности алифатических и/или ароматических альдегидов.
Примеры
Пример 1: Получение 1,1.3-трис-(метилтио)пропана (2) из 3-(метилтио)пропаналя (1)
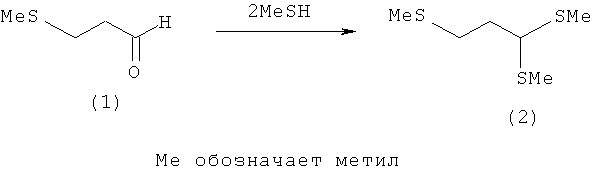
3-(Метилтио)пропаналь (1) (1,44 моля, 150 г) при 0°С насыщают НСl (газообразным) и затем при 0°С в течение 30 мин непосредственно добавляют по каплям к MeSH (6,25 моля, 300 г). После этого реакционную смесь нагревают до 20°С и в течение 24 ч перемешивают при 20°С. Полученный после удаления избыточного MeSH под вакуумом продукт по данным его анализа газовой хроматографией (ГХ) на 91% состоит из тиоацеталя (2) и на 8% - из 3-(метилтио)пропаналя (1). Для дальнейшей очистки эту смесь растворяют в диэтиловом эфире и промывают 30%-ным водным раствором пиросульфита натрия (Na2S2O5). После разделения фаз органическую фазу сушат над MgSO4. После удаления растворителя получают тиоацеталь (2) в виде желтоватого масла (168 г, выход: 64%, чистота по данным ГХ-анализа: 98%).
1H-ЯМР 1,1,3-трис-(метилтио)пропана (2) (500 МГц, СDСl3): δ=2,01-2,05 (m, 2Н, СН2), 2,11 (s, 9Н, 3×SСН3), 2,71 (t, 3J=7,3 Гц, 2Н, СН2), 3,83 (t, 3J=7,3 Гц, 1Н, СН).
13С-ЯМР 1,1,3-трис-(метилтио)пропана (2) (125,8 МГц, CDCl3): δ=12,6 (2×SСН3), 15,5 (SCH3), 32,0 (CH2), 33,9 (СH2), 53,0 (С(SСН3)2).
Пример 2: Получение 1,1,3-трис-(метилтио)пропана (2) из акролеина (3)
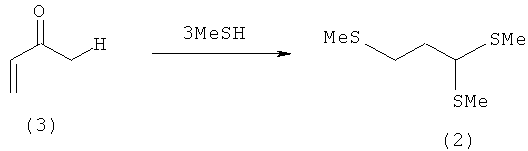
К MeSH (358 ммолей, 17,2 г), насыщенному газообразным НСl при 0°С, при перемешивании и при -78°С в течение 15 мин по каплям добавляют акролеин (3) (89 ммолей, 5,0 г). Полученную смесь медленно нагревают до 20°С и затем в течение 24 ч перемешивают при 20°С. После удаления избыточного MeSH под вакуумом эту смесь растворяют в диэтиловом эфире и промывают 30%-ным водным раствором пиросульфита натрия. После разделения фаз органическую фазу сушат над MgSO4. После дистилляции (88°С при давлении 1,2 мбара) получают чистый тиоацеталь (2) в виде желтоватого масла (11,4 г, выход: 70%, чистота по данным ГХ-анализа: 98%). Данные, полученные при анализе продукта ЯМР-спектроскопией, полностью совпадают с данными, полученными в примере 1.
Пример 3: Получение 2,2,4-трис-(метилтио)масляной кислоты (4) путем приводящей к изменению полярности реакции 1.1,3-трис-(метилтио)пропана (2) с сухим льдом (СО2)
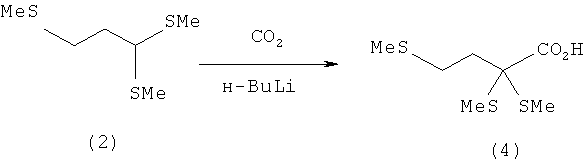
В трехгорлой колбе в атмосфере защитного газа 1,1,3-трис-(метилтио)пропан (2) (3,65 г, 20 ммолей) растворяют в 50 мл абсолютного тетрагидрофурана (ТГФ). После этого при -20°С и при перемешивании медленно, по каплям добавляют раствор бутиллития в н-гексане (14 мл, 1,6-молярный). При этом раствор приобретает ярко-желтую окраску. После перемешивания при этой температуре в течение 2 ч затем при -70°С порциями добавляют безводный сухой лед (СО2, 10 г), предварительно промытый в абсолютном ТГФ. Далее реакционный раствор медленно нагревают до 20°С и добавляют 10%-ный водный раствор КОН (80 мл). После разделения фаз органическую фазу промывают 10%-ным водным раствором КОН (2×50 мл). Объединенные КОН-фазы промывают диэтиловым эфиром (3×30 мл) и затем значение рН при охлаждении осторожно устанавливают на 1 добавлением концентрированной НСl. Продукт экстрагируют диэтиловым эфиром (3×50 мл). Объединенные эфирные фазы затем сушат над Na2SO4 и после фильтрации концентрируют на роторном испарителе. Таким путем получают 2,2,4-трис-(метилтио)масляную кислоту (4) в виде желтоватого масла (4,3 г, выход: 95%), которое медленно кристаллизуется при стоянии.
1Н-ЯМР 2,2,4-трис-(метилтио)масляной кислоты (4) (500 МГц, СDСl3): δ=2,12 (s, 6H, 2×SСН3), 2,14 (s, 3Н, SCH3), 2,22-2,26 (m, 2H, CH2), 2,67-2,70 (m, 2H, CH2).
13С-ЯМР 2,2,4- трис-(метилтио)масляной кислоты (4) (125,8 МГц, СDСl3): δ=12,5 (2×SСН3), 15,6 (С-5), 29,7 (С-3), 34,6 (С-4), 63,5 (С-2), 175,3 (CО2H).
Пример 4: Получение 2,2,4-трис-(метилтио)бутаноата кальция (5) из соответствующих α,α-(диалкилтио)карбоновых кислот (4)
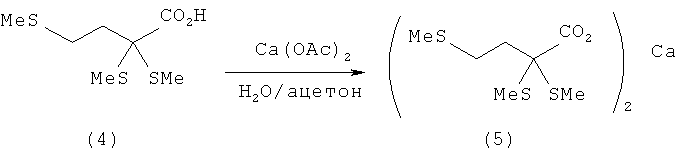
16,6 г 2,2,4-трис-(метилтио)масляной кислоты (4) (73,3 ммоля) растворяют в смеси из 140 мл Н2О и 35 мл ацетона. Затем при 20°С и при интенсивном перемешивании медленно, по каплям добавляют раствор 6,2 г ацетата кальция (93%-ный) и 22 мл Н2О. По истечении короткого промежутка времени выпадает белый осадок, который отфильтровывают через 30 мин. Полученное белое твердое вещество сначала дважды промывают Н2О порциями по 200 мл, а затем ацетоном порциями по 200 мл и диэтиловым эфиром порцией 200 мл и сушат в сушильном шкафу. Таким путем удается выделить в общей сложности 17,7 г 2,2,4-трис-(метилтио)бутаноата кальция (5) (М=490,8 г/моль, выход: 98%).
1Н-ЯМР 2,2,4-трис-(метилтио)бутаноата кальция (5) (500 МГц, ДМСО-D6): δ=1,93 (s, 6Н, 2SСН3), 1,95-1,99 (m, 2H, СН2), 2,05 (s, 3H, SCH3), 2,52-2,55 (m, 2H, CH2).
Пример 5: Получение 2,2,4-трис-(метилтио)масляной кислоты (4) путем приводящей к изменению полярности реакции 1,1,3-трис-(метилтио)пропана (2) с газообразным СО2
В трехгорлой колбе в атмосфере защитного газа 1,1,3-трис-(метилтио)пропан (2) (3,65 г, 20 ммолей) растворяют в 50 мл абсолютного ТГФ. После этого при -20°С и при перемешивании медленно, по каплям добавляют раствор бутиллития в н-гексане (14 мл, 1,6-молярный). При этом раствор приобретает ярко-желтую окраску. После перемешивания при этой температуре в течение 2 ч затем при -70°С через смесь в течение 30 мин пропускают подаваемый через фритту газообразный сухой СО2. Далее реакционный раствор медленно нагревают до температуры 20°С, при которой затем добавляют 10%-ный водный раствор КОН (80 мл). После разделения фаз органическую фазу промывают 10%-ным водным раствором КОН (2×50 мл). Объединенные КОН-фазы промывают диэтиловым эфиром (3×30 мл) и затем значение рН при охлаждении осторожно устанавливают на 1 добавлением концентрированной НСl. Продукт экстрагируют диэтиловым эфиром (3×50 мл). Объединенные эфирные фазы затем сушат над Na2SO4 и после фильтрации концентрируют на роторном испарителе. Таким путем получают 2,2,4-трис-(метилтио)масляную кислоту (4) в виде желтоватого масла (4,1 г, выход: 90%), которое медленно кристаллизуется при стоянии. Данные, полученные при анализе продукта ЯМР-спектроскопией, полностью совпадают с данными, полученными в примере 3.
Пример 6: Получение изопропилового эфира кетометионина (6) путем удаления тиола из 2,2,4-трис-(метилтио)масляной кислоты (4) в присутствии изопропанола
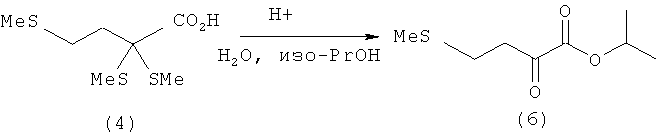
0,1 моля 2,2,4-трис-(метилтио)масляной кислоты (4) (22,6 г) растворяют в смеси из 200 мл толуола и 200 мл изопропанола. Затем добавляют 2,0 экв. Н2О (3,6 мл) и взятый на кончике шпателя моногидрат паратолуолсульфоновой кислоты. После этого всю смесь нагревают до температуры кипения и в течение 3 ч перемешивают при кипячении с обратным холодильником. После охлаждения добавляют 150 мл воды и трижды экстрагируют диэтиловым эфиром порциями по 100 мл. Объединенные эфирные фазы затем промывают разбавленным раствором гидрокарбоната натрия до нейтральной реакции и сушат над сульфатом магния. После фильтрации весь растворитель отгоняют на роторном испарителе. Таким путем получают изопропиловый эфир кетометионина (6) (14,5 г, выход: 76%) в виде желтоватого масла.
1Н-ЯМР изопропилового эфира кетометионина (6) (500 МГц, СDСl3): δ=1,35 (d, 3J=6,3 Гц, 6Н, 2СН3), 2,14 (s, 3Н, SСН3), 2,79 (t, 3J=7,2 Гц, 2Н, СН2), 3,15 (t, 3J=7,2 Гц, 2Н, СН2), 5,15 (quint, 3J=6,3 Гц, 1Н, СН).
Элементный анализ для С8Н14О3S (6) (М=190,26 г/моль): рассчитано: С 50,50, Н 7,43, S 16,85; обнаружено: С 50,66, Н 7,57, S 16,52.
Пример 7: Непосредственное получение кетометионинэтиленгликолькеталя (7) путем удаления тиола из 2,2,4-трис-(метилтио)масляной кислоты (4) в присутствии этиленгликоля
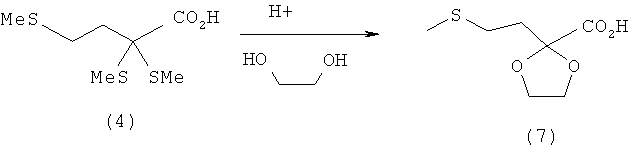
0,1 моля 2,2,4-трис-(метилтио)масляной кислоты (4) (22,6 г) растворяют в 200 мл этиленгликоля (1,2-этандиола). После этого добавляют 1,1 экв. Н2О (1,8 мл) и взятый на кончике шпателя моногидрат паратолуолсульфоновой кислоты. Затем всю смесь нагревают до 50°С и через раствор с постоянным расходом пропускают подаваемый через фритту поток азота. Через 4 ч реакционную смесь сливают в 300 мл воды и трижды экстрагируют диэтиловым эфиром порциями по 100 мл. Объединенные эфирные фазы сушат над MgSO4. После фильтрации диэтиловый эфир отгоняют на роторном испарителе. К защищенному в виде кеталя сложному эфиру затем добавляют 100 мл метанола и 100 мл 2-молярного раствора NaOH и перемешивают в течение 2 ч при 20°С. После этого раствор подкисляют концентрированной НСl до рН 1 и трижды экстрагируют диэтиловым эфиром порциями по 100 мл. Объединенные эфирные фазы затем сушат над MgSO4. После фильтрации диэтиловый эфир отгоняют и сырой продукт выкристаллизовывают из смеси метиленхлорида и н-гексана. Таким путем получают 13,0 г белого кристаллического твердого вещества (7) (выход: 68%, М=192,23 г/моль, температура плавления: 74°С (кристаллизация из смеси метиленхлорид/н-гексан)).
1Н-ЯМР 2-(2-(метилтио)этил)-1,3-диоксолан-2-карбоновой кислоты (7) (500 МГц, СDСl3): δ=2,11 (s, 3Н, SСН3), 2,24-2,28 (m, 2Н, СН2), 2,58-2,61 (m, 2Н, СН2), 4,07-4,14 (m, 4H, ОCH2СН2О).
13С-ЯМР 2-(2-(метилтио)этил)-1,3-диоксолан-2-карбоновой кислоты (7) (125,8 МГц, СDСl3): δ=15,5 (SCH3), 27,1 (СН2), 34,9 (СН2), 66,1 (2OСН2), 105,9 (С), 174,1 (COO).
Элементный анализ для C7H12O4S (7) (М=192,24 г/моль): рассчитано: С 43,74, Н 6,29, S 16,68; обнаружено: С 43,80, Н 6,25, S 16,61.
Пример 8: Непосредственное получение эфира кетометионина в виде кеталя (8а)/(8b) путем удаления тиола из α,α-(диалкилтио)карбоновой кислоты (4) в присутствии глицерина
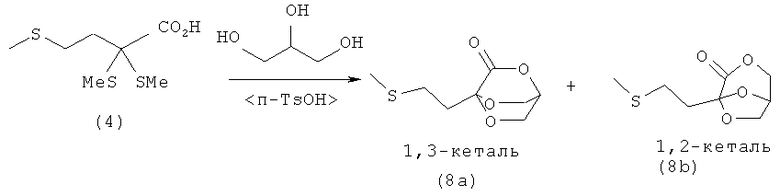
0,1 моля 2,2,4-трис-(метилтио)масляной кислоты (4) (22,6 г) растворяют в 100 мл глицерина (1,2,3-пропантриола). После этого добавляют 1,1 экв. Н2О (1,8 мл) и взятый на кончике шпателя моногидрат паратолуолсульфоновой кислоты. Затем всю смесь нагревают до 70°С и создают вакуум с остаточным давлением 750 мбар. Через 5,5 ч реакционную смесь сливают в 300 мл воды и трижды экстрагируют диэтиловым эфиром порциями по 100 мл. Объединенные эфирные фазы сушат над MgSO4. После фильтрации диэтиловый эфир удаляют на роторном испарителе и маслянистый сырой продукт (соотношение между соединениями (8а) и (8b) составляет 70:30) кристаллизуют из смеси метиленхлорид/н-гексан. Основной продукт (8а) кристаллизуется в виде бесцветных игольчатых кристаллов (9,2 г, выход: 45%, М=204,25 г/моль, температура плавления = 39,5°С (перекристаллизация из смеси метиленхлорид/н-гексан)).
1Н-ЯМР 4-(2-(метилтио)этил)-2,5,8-триоксабицикло[2.2.2]октан-3-она (8а) (500 МГц, CDCl3): δ=2,13 (s, 3H, SСН3), 2,17-2,20 (m, 2H, CH2), 2,65-2,68 (m, 2H, CH2), 4,12-4,13 (m, 4H, 2 СН2), 4,76 (s, 1H, CH).
13С-ЯМР 4-(2-(метилтио)этил)-2,5,8-триоксабицикло[2.2.2]-октан-3-она (8а) (125,8 МГц, CDCl3): δ=15,4 (SCH3), 26,9 (CH2), 33,2 (CH2), 66,5 (2OСН2), 70,9 (CH), 92,9 (С), 166,2 (СОО).
Элементный анализ для C8H12O4S (8а) (М=204,25 г/моль): рассчитано: С 47,04, Н 5,92, S 15,70; обнаружено: С 47,21, Н 5,93, S 15,69.
Пример 9: Получение 1,1-бис-(метилтио)бутана (10)
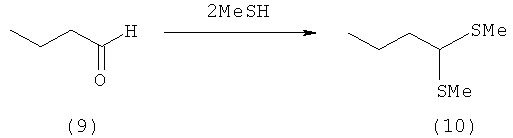
10,0 г бутаналя (9) (139 ммолей) при 0°С насыщают НСl (газообразным) и затем при 0°С в течение 25 мин непосредственно добавляют по каплям к MeSH (624 ммоля, 30,0 г). Далее реакционную смесь нагревают до 20°С и в течение 17 ч перемешивают при 20°С. Для дальнейшей очистки эту смесь растворяют в диэтиловом эфире и промывают 30%-ным водным раствором пиросульфита натрия. После разделения фаз органическую фазу сушат над Na2SO4. После удаления растворителя получают тиоацеталь (10) в виде прозрачного бесцветного масла (17,3 г, выход: 83%, чистота по данным ГХ-анализа: 98%). Данные ЯМР-спектроскопии совпадают с данными, представленными в литературе.
Пример 10: Получение 1,1-бис-(метилтио)пентановой кислоты (11) путем приводящей к изменению полярности реакции 1,1-бис-(метилтио)бутана (10) с сухим льдом (СО2)
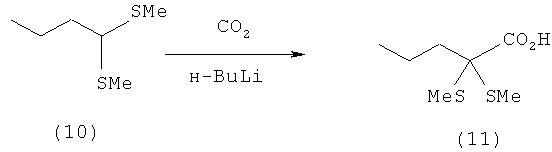
10 ммолей 1,1-бис-(метилтио)бутана (10) (1,26 г) в атмосфере азота добавляют в 25 мл сухого ТГФ и охлаждают до -20°С. После этого при -20°С в течение 5 мин по каплям добавляют 11 ммолей 1,6-молярного раствора н-BuLi в н-гексане (7,0 мл) и перемешивают в течение 2 ч. Прозрачный желтоватый реакционный раствор при -78°С по каплям добавляют к 15 г сухого льда и перемешивают в течение 36 ч, при этом температура повышается в течение ночи до 20°С. Затем реакционную смесь смешивают с 40 мл 10%-ного раствора КОН и фазы разделяют. Органическую фазу дважды промывают 10%-ным раствором КОН порциями по 25 мл и объединенные КОН-фазы трижды промывают метил-трет-бутиловый эфиром (МТБЭ) порциями по 25 мл. КОН-фазу затем подкисляют концентрированной НСl (водной) (до рН 1) и четырежды экстрагируют МТБЭ порциями по 25 мл. Объединенные органические фазы сушат над MgSO4 и концентрируют на роторном испарителе. Таким путем получают 2,2-(диметилтио)пентановую кислоту (11) в виде белого твердого вещества (1,0 г, выход: 51,5%, температура плавления: 85°С (перекристаллизация из смеси хлороформа, метанола и уксусной кислоты)).
1Н-ЯМР 2,2-(диметилтио)пентановой кислоты (11) (500 МГц, СDСl3): δ=0,96 (t, 3J=7,4 Гц, 3Н, С5H3), 1,50-1,60 (m, 2H, С4H2), 1,88-1,94 (m, 2H, С3H2), 2,10 (s, 6H, 2×SСН3), 11,5 (шир. s, 1H, C1OOH).
13С-ЯМР 2,2-(диметилтио)пентановой кислоты (11) (125,8 МГц, СDСl3): δ=12,5 (2×5СН3), 14,0 (С-5), 15,1 (С-4), 36,9 (С-3), 64,6 (С-2), 176,3 (С-1).
Пример 11: Получение этилового эфира 2-оксовалериановой кислоты (12) путем удаления тиола из 2,2-(диметилтио)пентановой кислоты (11) в присутствии воды и этанола
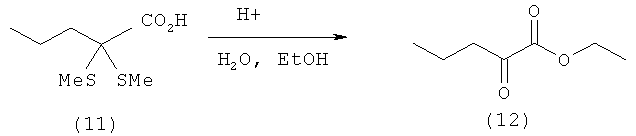
0,1 моля 2,2-(диметилтио)пентановой кислоты (11) (19,4 г) растворяют в 150 мл толуола и смешивают с 2,0 экв. H2O (3,6 мл) и взятым на кончике шпателя моногидратом паратолуолсульфоновой кислоты. После этого смесь нагревают до температуры кипения и через раствор пропускают подаваемый через фритту поток азота. После 3-часового кипячения с обратным холодильником реакционную смесь охлаждают до 80°С и смешивают с 200 мл этанола и еще одной порцией взятого на кончике шпателя моногидрата паратолуолсульфоновой кислоты. Далее реакционную смесь еще в течение 3 ч кипятят с обратным холодильником, после чего охлаждают до 20°С, добавляют 200 мл воды и трижды экстрагируют диэтиловым эфиром порциями по 100 мл. Объединенные эфирные фазы затем промывают разбавленным раствором гидрокарбоната натрия до нейтральной реакции и сушат над сульфатом магния. После фильтрации весь растворитель отгоняют на роторном испарителе. Таким путем получают этиловый эфир 2-оксовалериановой кислоты (12) (10,2 г, выход: 71%) в виде желтоватого масла.
1Н-ЯМР этилового эфира 2-оксовалериановрой кислоты (12) (500 МГц, СDСl3): δ=0,97 (t, 3J=7,3 Гц, 3Н, С5H3), 1,37 (t, 3J=7,3 Гц, 3Н, ОСН2СН3), 1,67 (sext, 3J=7,3 Гц, 2Н, С4H2), 2,81 (t, 3J=7,3 Гц, 2Н, С3H3), 4,32 (q, 3J=7,3 Гц, 2Н, ОСН2СН2).
13С-ЯМР этилового эфира 2-оксовалериановой кислоты (12) (125,8 МГц, СDСl3): δ=13,5 (ОСН2СН3), 14,0 (С-5), 16,6 (С-4), 41,2 (С-3), 62,3 (ОСН2СН3), 161,4 (C-1), 194,7 (С-2).
Пример 12: Получение 2-оксовалериановой кислоты (13) путем удаления тиола из 2,2-(диметилтио)пентановой кислоты (11) в присутствии воды
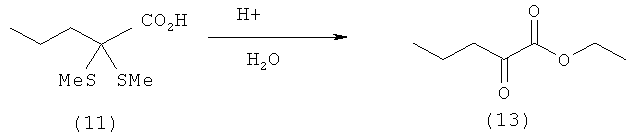
10,0 ммолей 2,2-(диметилтио)пентановой кислоты (11) (1,94 г) суспендируют в 30 мл дымящей соляной кислоты (37%-ной) и смешивают со взятым на кончике шпателя моногидратом паратолуолсульфоновой кислоты. После этого смесь нагревают до температуры кипения и перемешивают в течение 5 ч. При этом создают вакуум с остаточным давлением 750 мбар. По завершении реакции смесь разбавляют 100 мл воды и трижды экстрагируют диэтиловым эфиром порциями по 80 мл. Объединенные органические фазы трижды промывают раствором гидрокарбоната натрия порциями по 100 мл. Объединенные водные фазы затем подкисляют концентрированной соляной кислотой до рН 1 и трижды экстрагируют диэтиловым эфиром порциями по 100 мл. Объединенные эфирные фазы сушат над MgSO4, фильтруют и фильтрат концентрируют досуха при комнатной температуре в вакууме. Таким путем получают 2-оксовалериановую кислоту (13) в виде бесцветного масла (0,72 г, выход: 62%).
1Н-ЯМР 2-оксовалериановой кислоты (13) (500 МГц, ДМСО-D6): δ=0,88 (t, 3J=7,4 Гц, 3Н, С5H3), 1,53 (sext, 3J=7,4 Гц, 2Н, С4H2), 1,96 (t, 3J=7,4 Гц, 2Н, С3H3), 13,5 (шир. s, 1H, СООН).
13С-ЯМР 2-оксовалериановой кислоты (13) (125,8 МГц, ДМСО-D6): δ 13,64 (С-5), 16,37 (С-4), 40,56 (С-3), 163,28 (С-1), 196,93 (С-2).
Изобретение относится к способу получения α-кетокислот, а также их производных общей формулы (I) или (II), где R1 обозначает разветвленную либо линейную C1-C18алкильную группу и, где R1 необязательно замещен -SH или -SCH3, R2 обозначает -OR”', где R”' представляет собой атом водорода или разветвленную либо линейную C1-C8алкильную группу, оба остатка R3 совместно представляют собой C2-C8алкандиил и совместно образуют кольцо либо оба остатка R3 и R”' совместно являются частью C3-C8алкантриильной группы общей формулы -R3(CH-)R”'- и совместно образуют бициклическую группу, заключающемуся в том, что а) альдегид R1 CHO подвергают взаимодействию с одинаковыми или разными тиолами формулы R4-S-H, где R4 обозначает разветвленную либо линейную, необязательно замещенную C1-C6алкильную группу, с получением соответствующего дитиоацеталя, б) образовавшийся дитиоацеталь подвергают в присутствии сильного основания взаимодействию с карбонилсодержащим электрофилом и последующему гидролизу с получением α,α-(дитио)карбоновой кислоты или ее производных формулы (III), где R5 представляет собой атом водорода или разветвленную либо линейную C1-C6алкильную группу, а остатки R4 и R1 имеют указанные выше значения, и в) α,α-(дитио)карбоновую кислоту или ее производные формулы (III) превращают путем проводимого при кислотном катализе сольволиза в присутствии по меньшей мере 1 молярного эквивалента воды в α-кетокислоту или ее производные общей формулы (I) или (II) с выделением тиолов формулы R4SH. Изобретение также относится к промежуточному продукту формулы (IV). Усовершенствованный способ является экологичным и пригодным для реализации в промышленном масштабе. 2 н. и 12 з.п. ф-лы, 12 пр.
1. Способ получения α-кетокислот, а также их производных общей формулы (I) или (II)
, ,
где R1 обозначает разветвленную либо линейную C1-C18алкильную группу и, где
R1 необязательно замещен -SH или -SCH3,
R2 обозначает -OR”', где R”' представляет собой атом водорода или
разветвленную либо линейную C1-C8алкильную группу, оба остатка R3 совместно представляют собой C2-C8алкандиил и совместно образуют кольцо либо оба остатка R3 и R”' совместно являются частью C3-C8алкантриильной группы общей формулы -R3(CH-)R”'- и совместно образуют бициклическую группу,
заключающийся в том, что
а) альдегид R1 CHO подвергают взаимодействию с одинаковыми или разными тиолами формулы R4-S-H, где R4 обозначает разветвленную либо линейную, необязательно замещенную C1-C6алкильную группу, с получением соответствующего дитиоацеталя,
б) образовавшийся дитиоацеталь подвергают в присутствии сильного основания взаимодействию с карбонилсодержащим электрофилом и последующему гидролизу с получением α,α-(дитио)карбоновой кислоты или ее производных формулы (III)
,
где R5 представляет собой атом водорода или разветвленную либо линейную C1-C6алкильную группу, а остатки R4 и R1 имеют указанные выше значения, и
в) α,α-(дитио)карбоновую кислоту или ее производные формулы (III) превращают путем проводимого при кислотном катализе сольволиза в присутствии по меньшей мере 1 молярного эквивалента воды в α-кетокислоту или ее производные общей формулы (I) или (II) с выделением тиолов формулы R4SH.
2. Способ по п.1, отличающийся тем, что используемое на стадии б) для депротонирования сильное основание имеет измеренный на соответствующей ему кислоте показатель pKa более 20.
3. Способ по п.1 или 2, отличающийся тем, что депротонирование на стадии б) проводят в апротонных органических растворителях, в сжиженном надкритическом диоксиде углерода или в жидком аммиаке.
4. Способ по п.1 или 2, отличающийся тем, что стадию б) проводят при температуре в пределах от -80 до 100°С.
5. Способ по п.1 или 2, отличающийся тем, что выделяющийся на стадии в) тиол формулы R4SH удаляют из реакционной смеси, после чего возвращают в технологический цикл и вновь используют на стадии а).
6. Способ по п.5, отличающийся тем, что в качестве тиола формулы R4SH используют C1-C6алкилтиол, который удаляют из реакционной смеси путем создания вакуума или путем пропускания через нее инертного газа.
7. Способ по п.1 или 2, отличающийся тем, что на стадии а) в качестве тиола используют метилтиол.
8. Способ по п.1 или 2, отличающийся тем, что в качестве растворителя в процессе проводимого при кислотном катализе сольволиза на стадии в) используют воду.
9. Способ по п.1 или 2, отличающийся тем, что в качестве растворителя в процессе проводимого при кислотном катализе сольволиза на стадии в) используют спирт R”'OH, где R”' представляет собой разветвленную либо линейную C1-C8алкильную группу.
10. Способ по п.1 или 2, отличающийся тем, что в качестве растворителя в процессе проводимого при кислотном катализе сольволиза на стадии в) используют диол HO-R3-ОН, где R3 представляет собой разветвленную либо линейную C2-C8алкандиильную группу.
11. Способ по п.1 или 2, отличающийся тем, что в качестве растворителя в процессе проводимого при кислотном катализе сольволиза на стадии в) используют триол HOR3(CHOH)R”'OH, где остатки R3 и R”' совместно являются частью разветвленной либо линейной C3-C8алкантриильной группы общей формулы -R3(CH-)R”'-.
12. Способ по п.1 или 2, отличающийся тем, что на стадии а) используют альдегид R1CHO, где R1 представляет собой CH3SCH2CH2-.
13. Способ по п.1 или 2, отличающийся тем, что α-кетокислота общей формулы (I) представляет собой α-кетометионин, а также его производные, где R1 обозначает CH3SCH2CH2, при этом на стадии а) альдегид R1CHO, где R1 обозначает CH3SCH2CH2-, подвергают взаимодействию с метилтиолом.
14. Промежуточный продукт формулы (IV)
,
в которой
R1 обозначает разветвленную либо линейную C1-C18алкильную группу и, где
R1 замещен -SH или -SCH3,
R2 обозначает -OR”', где R”' представляет собой атом водорода или разветвленную либо линейную C1-C8алкильную группу или R”' представляет собой ион щелочного или щелочноземельного металла, и
R4 имеют одинаковые или разные значения и представляют собой разветвленную либо линейную C1-C6алкильную группу.
| A I MEYERS ET AL.: "The Use of Dithioesters as Acyl Anion Equivalents | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ очищения сернокислого глинозема от железа | 1920 |
|
SU47A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Видоизменение прибора с двумя приемами для рассматривания проекционные увеличенных и удаленных от зрителя стереограмм | 1919 |
|
SU28A1 |
| DATABASE BEILSTEIN | |||

