Область техники
Настоящее изобретение относится к способу получения ароматического поликарбоната.
Уровень техники
Ароматические поликарбонаты (здесь и далее в настоящем документе сокращенно обозначаемые как РС) в общем случае получают в результате проведения реакции полимеризации между дифенилкарбонатом (здесь и далее в настоящем документе сокращенно обозначаемым как DPC) и бисфенолом А (здесь и далее в настоящем документе сокращенно обозначаемом как ВРА).
Переработка являющегося побочным продуктом фенола
При проведении реакции полимеризации в качестве побочного продукта образуется фенол (здесь и далее в настоящем документе сокращенно обозначаемый как "PL"). Данный являющийся побочным продуктом PL в качестве примесей содержит DPC, BPA, олигомеры, полученные в результате прохождения реакции с участием от одной до семи молекул DPC и от одной до семи молекул ВРА, и тому подобное. Способ, по которому данный являющийся побочным продуктом PL возвращают на стадию получения ВРА либо на стадию получения DPC, известен.
А именно, способ, по которому являющийся побочным продуктом PL возвращают на стадию получения ВРА как таковой либо после проведения очистки до низкой степени чистоты, описывается в патентном документе 1. Причина того, почему полученный в качестве побочного продукта PL может характеризоваться низкой степенью чистоты, заключается в том, что даже если DPC и олигомеры будут гидролизованы до получения PL и ВРА, и данные соединения поступят на стадию получения ВРА, это не будет составлять какой-либо проблемы.
Кроме того, способ, по которому являющийся побочным продуктом PL очищают до высокой степени чистоты, а после этого возвращают на стадию получения DPC, описывается в патентном документе 2, патентном документе 3 и последующих. Причина того, почему полученный в качестве побочного продукта PL должен характеризоваться высокой степенью чистоты, заключается в том, что необходимо предотвратить попадание ВРА и тому подобного на стадию получения DPC и возникновение засорения.
Соединение стадий получения
Обычно используют твердый ВРА, который получают в результате охлаждения ВРА в расплавленном состоянии после проведения очистки. Однако в случае, если аппарат для получения ВРА устанавливают поблизости от аппарата для получения РС, ВРА в расплавленном состоянии как таковой либо в виде раствора, образованного смесью BPA/PL, характеризующегося определенным составом, можно подавать в аппарат для получения РС и полимеризовать. Данный способ устраняет необходимость проведения повторного нагревания либо очистки и приводит к достижению улучшенного термического коэффициента полезного действия.
Переработка отработанного раствора
Кроме того, каждая из стадий получения DPC и ВРА и стадии получения РС в результате приводят к получению отработанного раствора, содержащего большое количество органических соединений.
Говоря конкретно, во-первых, на стадии получения РС образование отработанного раствора происходит следующим образом. На основной стадии, на которой DPC и ВРА используют в качестве исходных материалов при получении РС в результате проведения стадии полимеризации, ингредиенты, превращенные в пар на стадии полимеризации, ожижают и подвергают воздействию стадии перегонки для извлечения PL, а получающийся в результате остаток после перегонки представляет собой отработанный раствор. Данный остаток после перегонки содержит PL, DPC, BPA, олигомеры, полученные из нескольких молекул, скомбинированных из DPC и ВРА, и тому подобное, и извлечение данных ингредиентов оказывает значительное влияние на выход РС.
В данной связи способ, по которому остаток после перегонки возвращают на стадию полимеризации, описывается в патентном документе 3 и патентном документе 4, а способ, по которому остаток после перегонки подвергают перегонке еще раз для извлечения ингредиентов, а получающийся в результате остаток после извлечения/перегонки используют в качестве топлива, описывается в патентном документе 1.
На стадии получения DPC на основной стадии, на которой PL и карбонильное соединение используют в качестве исходных материалов при получении DPC, в результате проведения стадии реакции и стадии перегонки остаток после перегонки, получающийся в результате проведения стадии перегонки, представляет собой отработанный раствор. Данный остаток после перегонки содержит DPC, и его извлечение оказывает значительное влияние на выход DPC.
В данной связи способ, по которому остаток после перегонки подвергают перегонке еще раз для извлечения DPC, а данный DPC возвращают в жидкую реакционную смесь, получающуюся в результате проведения стадии реакции, описывается в патентном документе 5.
Кроме того, на стадии получения ВРА основная стадия, на которой PL и ацетон используют в качестве исходных материалов при получении ВРА в результате проведения стадии реакции синтеза, стадии кристаллизации и стадии разделения твердой/жидкой фаз, приводит к получению маточной жидкости, получающейся в результате проведения стадии разделения твердой/жидкой фаз. Как описывается в патентном документе 6, данная маточная жидкость помимо большого количества PL и ВРА содержит побочные продукты, такие как 2,4-изомер, трисфенолы и производные хромана, а, кроме того, в небольшом количестве содержит окрашенные примеси и окрашивающие примеси. Поскольку данная маточная жидкость содержит PL и ВРА, которые являются исходными материалами при получении ВРА, организуют ее циркуляцию по всем стадиям и повторное использование. Однако в том случае, если все количество маточной жидкости будет циркулировать без переработки, то тогда будут накапливаться побочные продукты и окрашенные примеси и окрашивающие примеси. Поэтому данные побочные продукты и примеси необходимо удалять.
Вакуумное устройство
На стадии получения DPC подвергают очистке в результате проведения перегонки при кипячении в условиях флегмообразования. На стадии получения РС PL удаляют, проводя его отделение от DPC в результате перегонки. Данные операции по перегонке проводят при пониженном давлении при использовании вакуумного устройства для того, чтобы уменьшить температуру перегонки (см. патентный документ 7).
[Патентный документ 1] JP-A-2000-53759
[Патентный документ 2] JP-A-10-60106
[Патентный документ 3] JP-A-9-255772
[Патентный документ 4] JP-A-9-165443
[Патентный документ 5] JP-A-2002-322130
[Патентный документ 6] JP-A-5-331088
[Патентный документ 7] JP-A-9-38402
Описание изобретения
[Переработка являющегося побочным продуктом фенола; проблема интеграции трех стадий получения]
Однако в каждом из случаев, описанных выше, не проводили никаких исследований в отношении уровня содержания воды, несмотря на то, что на уровни содержания примесей, отличных от воды, таких как, например, ВРА и DPC, внимание обращали. Являющемуся побочным продуктом фенолу сопутствуют вода, содержащаяся в DPC и ВРА, используемых в качестве исходных материалов при полимеризации, в особенности в тех веществах, которые были отверждены и, таким образом, содержат воду, и вода, подаваемая совместно с катализатором полимеризации. На стадии получения ВРА присутствие воды приводит к уменьшению каталитической активности, что уменьшает конверсию. На стадии получении DPC присутствие воды приводит не только к уменьшению каталитической активности, но также и к гидролизу получаемого DPC.
Кроме того, являющийся побочным продуктом фенол, полученный на стадии полимеризации при получении РС, содержит указанные выше примеси. Данные побочные продукты включают примеси, которые не являются проблематичными даже в случае их подачи на стадию получения DPC, но приводят к возникновению проблемы в случае их подачи на стадию получения ВРА. Побочные продукты, кроме того, включают примеси, которые действуют обратным образом.
Кроме того, как указывается выше, необходимо проводить стадию удаления воды из ингредиентов, выделенных в виде дистиллята на стадии полимеризации при получении РС и включающих в качестве основного компонента фенол. Однако в том случае, если стадию получения DPC, стадию получения ВРА и стадию получения РС интегрируют таким образом, чтобы проводить их в одном месте, то тогда организуют две либо более чем две стадии для удаления воды, потому что стадия получения ВРА также включает точно такую же стадию.
Соединение стадий получения
На стадии кристаллизации полученного ВРА на стадии получения ВРА твердая фаза склонна образовывать отложения на тех частях кристаллизатора и других элементах, которые попадают в контакт с жидкостью. Таким образом, необходимо будет прекращать проведение данной стадии и выполнять чистку с интервалами в несколько месяцев. По этой причине данные стадии на стадии получения ВРА, которая заключена в пределах между стадией реакции синтеза и стадией кристаллизации, проводят поочередно.
В противоположность этому стадии получения DPC несвойственна проблема, описанная выше, и DPC можно получать непрерывно. По этой причине в случае хранения ВРА в необходимом количестве при одновременном выдерживании ВРА в расплавленном состоянии полимеризацию при получении РС можно проводить непрерывно.
Однако, если ВРА выдерживать в расплавленном состоянии, он склонен подвергаться пожелтению, разложению и тому подобному, а это оказывает влияние на качество получаемого РС.
Переработка отработанного раствора
Кроме того, поскольку остаток после перегонки, получающийся в результате проведения стадии получения РС, содержит PL, возврат всего количества остатка на стадию полимеризации будет оказывать влияние на скорость начальной полимеризации, поскольку PL присутствует там от начала полимеризации. Кроме того, поскольку остаток после перегонки, получающийся в результате проведения стадии получения РС, в общем случае является окрашенным, отправление данного остатка на рецикл без проведения какой-либо переработки приводит к получению окрашенного продукта РС. Даже, несмотря на то, что остаток после перегонки подвергают перегонке еще раз, получающийся в результате остаток после извлечения/перегонки также содержит данные ингредиенты в небольших количествах. Следовательно, отбрасывание данного остатка без проведения какой-либо переработки не только оказывает влияние на эффективность получения, но и становится источником проблемы, связанной с появлением нагрузки для окружающей среды.
В дополнение к этому остаток после перегонки, получающийся в результате проведения стадии получения DPC, обычно как таковой отбрасывают. Поскольку данный остаток после перегонки все еще содержит DPC, отбрасывание остатка без проведения какой-либо переработки не только оказывает влияние на эффективность получения, но также может стать и источником проблемы, связанной с появлением нагрузки для окружающей среды.
Вакуумное устройство
Кроме того, если перегонку проводят при пониженном давлении, то тогда бывают случаи, когда ингредиенты дистиллята, такие как PL и DPC, отводят при использовании вакуумного устройства с получением жидкой массы, отчасти остающейся в трубопроводе, ведущем к вакуумному устройству, либо также остающиеся ингредиенты дистиллята будут затвердевать, что сделает невозможным поддержание состояния вакуума. В дополнение к этому в случае, когда ингредиенты, которые выделяют в виде дистиллята, такие как PL и DPC, подаются в виде рефлюкса насосом, появляется вероятность того, что трубопровод для рефлюкса может быть засорен, например, потому что такие ингредиенты дистиллята в трубопроводе затвердевают.
В соответствии с этим цель изобретения заключается в предложении способа того, как справляться с являющимся побочным продуктом фенолом, по которому уровень содержания воды в являющемся побочным продуктом PL, полученном на стадии получения PC, ограничивают величиной в заданном диапазоне, обеспечивая, таким образом, эффективность получения на стадии получения ВРА и стадии получения DPC, куда подают являющийся побочным продуктом PL, в результате чего поддерживается совокупная эффективность получения РС.
Еще одна цель заключается в обеспечении экономии трудовых затрат на проведение переработки в виде очистки в отношении являющегося побочным продуктом фенола, полученного на стадии полимеризации при получении ароматического поликарбоната, в результате подачи являющегося побочным продуктом фенола на стадию получения дифенилкарбоната либо стадию получения бисфенола А в соответствии с примесями, содержащимися в феноле.
И еще одна цель относится к проблеме интеграции трех стадий получения и заключается в обеспечении экономии трудовых затрат на проведение переработки в виде очистки ингредиентов дистиллята, которые в качестве основного ингредиента содержат фенол и образуются на стадии полимеризации при получении ароматического поликарбоната, в результате использования существующих стадий, используемых для получения дифенилкарбоната и бисфенола А.
Дополнительная цель относится к соединению стадий получения и заключается в предложении способа соединения стадий получения, который способен обеспечить создание способа получения РС, обладающего достаточным качеством.
Еще одна дополнительная цель относится к способу переработки отработанного раствора и заключается в улучшении совокупной эффективности и уменьшении нагрузки для окружающей среды в результате возврата остатка после перегонки, получающегося в результате проведения стадии получения DPC, и остатка после перегонки, получающегося в результате проведения стадии получения РС, на специальные участки стадий получения РС.
И еще одна дополнительная цель относится к стадиям перегонки и заключается в подавлении образования жидкой массы либо затвердевания в трубопроводах в аппарате, в котором PL либо DPC выделяют в виде дистиллята.
В качестве способа того, как справляться с являющимся побочным продуктом фенолом, изобретение предлагает способ получения ароматического поликарбоната (РС), который включает стадию получения дифенилкарбоната (DPC), на которой фенол (PL) и, по меньшей мере, одно карбонильное соединение используют в качестве исходных материалов при получении дифенилкарбоната (DPC), и/или стадию получения бисфенола А (ВРА), на которой фенол (PL) и ацетон используют в качестве исходных материалов при получении бисфенола А (ВРА), и стадию получения ароматического поликарбоната (РС), на которой дифенилкарбонат (DPC) и бисфенол А (ВРА) используют в качестве исходных материалов при получении ароматического поликарбоната (РС) в результате проведения стадии полимеризации при получении РС, а являющийся побочным продуктом фенол извлекают, отличающийся тем, что количество воды, содержащейся в являющемся побочным продуктом феноле, извлеченном на стадии получения ароматического поликарбоната (РС), выдерживают на уровне 0,2 мас.% либо менее, а данный фенол используют в качестве части исходного материала на стадии получения дифенилкарбоната (DPC) и/или стадии получения бисфенола А (ВРА).
Фенол, используемый в качестве исходного материала на стадии получения дифенилкарбоната (DPC), может являться фенолом (PL), содержащим крезол и/или ксиленол в количестве 20-1000 ч/млн (мас.), а фенол (PL), получаемый на стадии полимеризации на стадии получения ароматического поликарбоната (РС), можно использовать в качестве, по меньшей мере, части фенола, используемого в качестве исходного материала на стадии получения бисфенола А (ВРА).
Кроме того, способ может отличаться тем, что 50-95 мас.% фенола, полученного в качестве побочного продукта на стадии получения ароматического поликарбоната, используют в качестве, по меньшей мере, части фенола, используемого на стадии получения дифенилкарбоната, а 50-5 мас.% его используют в качестве, по меньшей мере, части исходного материала для стадии получения бисфенола А.
Что касается соединения стадий, то способ можно охарактеризовать тем, что до и/или после проведения стадии перегонки PL проводят стадию хранения при получении РС, на которой хранят ожиженные, превращенные в пар ингредиенты при получении РС, предназначенные для проведения стадии перегонки PL, и/или являющийся побочным продуктом фенол, извлеченный на стадии перегонки PL, тем, что после проведения стадии перегонки при получении DPC проводят стадию хранения DPC, на которой хранят дифенилкарбонат, полученный на стадии перегонки при получении DPC, и/или тем, что стадию хранения ВРА, на которой хранят смесь бисфенола А и фенола, проводят в промежутке между стадией кристаллизации/выделения ВРА и стадией полимеризации при получении РС. Данный способ отличается тем, что резервуары для хранения, используемые на соответствующих стадиях хранения, в соответствии с потребностью характеризуются удельными вместимостями, продемонстрированными далее.
10(Vc/Fc)(1)
(В выражении (1) Vc обозначает вместимость (м3) резервуара для хранения при получении РС, а Fc обозначает расход (м3/час) ожиженных, превращенных в пар ингредиентов при получении РС либо являющегося побочным продуктом фенола)
10(Vd/Fd)(2)
(В выражении (2) Vd обозначает вместимость (м3) резервуара для хранения при получении DPC, а Fd обозначает расход (м3/час) дифенилкарбоната)
10(Vb/Fb)(3)
(В выражении (3) Vb обозначает вместимость (м3) резервуара для хранения при получении BPA, а Fb обозначает расход (м3/час) бисфенола А, подаваемого на стадию полимеризации при получении РС)
Что касается способов переработки отработанных вод, то способ можно охарактеризовать тем, что остаток после перегонки PL подают на стадию перегонки при получении DPC и/или стадию извлечения/перегонки при получении DPC, тем, что остаток после перегонки PL и/или, по меньшей мере, любого представителя, выбираемого из остатка после перегонки при получении DPC и остатка после извлечения/перегонки при получении DPC, подают на стадию переработки маточной жидкости при получении ВРА, либо тем, что остаток после перегонки PL подают на стадию перегонки при получении DPC и/или стадию извлечения/перегонки при получении DPC, а впоследствии остаток после перегонки при получении DPC и/или остаток после извлечения/перегонки при получении DPC подают на стадию переработки маточной жидкости при получении ВРА.
Кроме того, что касается стадий перегонки, то способ можно охарактеризовать тем, что перегонную колонну на стадии перегонки при получении DPC либо стадии перегонки PL снабжают конденсатором для конденсации дистиллята, вакуумным устройством для уменьшения давления в системе и вакуумным трубопроводом, который соединяет конденсатор с вакуумным устройством, и тем, что вакуумный трубопровод наклонен книзу от стороны конденсатора к стороне вакуумного устройства, а совокупная высота частей, поднимающихся кверху от стороны конденсатора к стороне вакуумного устройства, составляет 1 м либо менее.
Краткое описание чертежей
Фиг.1 представляет собой схему технологического процесса, демонстрирующую пример стадии получения DPC, соответствующей изобретению.
Фиг.2 представляет собой схему технологического процесса, демонстрирующую пример стадии получения ВРА, соответствующей изобретению.
Фиг.3 представляет собой схему технологического процесса, демонстрирующую пример стадии выделения воды (стадии (b-2)) на стадии получения ВРА, соответствующей изобретению.
Фиг.4 представляет собой схему технологического процесса, демонстрирующую пример стадии переработки маточной жидкости (стадии (g)) на стадии получения ВРА, соответствующей изобретению.
Фиг.5 представляет собой схему технологического процесса, демонстрирующую пример стадии получения РС, соответствующей изобретению.
Фиг.6 представляет собой схему технологического процесса, демонстрирующую еще один пример стадии получения РС, соответствующей изобретению.
Фиг.7 представляет собой схему технологического процесса, демонстрирующую пример случая, в котором стадия получения DPC, стадия получения ВРА и стадия получения РС, соответствующие изобретению, включают стадию хранения при получении DPC, стадию хранения при получении ВРА и стадию хранения при получении РС.
Фиг.8 представляет собой схему технологического процесса, демонстрирующую пример варианта реализации, в котором на стадии получения DPC, стадии получения ВРА и стадии получения РС, соответствующих изобретению, на указанную стадию подают остаток после перегонки PL (X2), остаток после перегонки при получении DPC (Х1) и остаток после извлечения/перегонки при получении DPC (Х1').
Фиг.9 представляет собой изображение, иллюстрирующее рефлюксный аппарат в соответствии с изобретением.
Номера позиций и символы на чертежах имеют нижеследующие значения: 1 обозначает реактор получения DPC, 2 - колонну дегидрохлорирования, 3 - смесительную емкость, 4 - емкость щелочной нейтрализации, 5 - емкость водной промывки, 6 - колонну первой перегонки при получении DPC, 7 - колонну второй перегонки при получении DPC, 8 - колонну извлечения/перегонки при получении DPC, 11 - резервуар для извлеченного PL при получении ВРА, 12 - колонну выделения PL, 13 - испаритель фенола, 14 - реактор переработки остатка, 15 - регенерирующий реактор, 21 - смесительную емкость, 22 - первый реактор полимеризации, 23 - второй реактор полимеризации, 24 - третий реактор полимеризации, 25 - четвертый полимеризатор, 26 - теплообменник, 27 - теплообменник, 28 - конденсатор, 29 - резервуар для извлеченного PL при получении РС, 29а - первый резервуар для извлеченного PL при получении РС, 29b - второй резервуар для извлеченного PL при получении РС, 30 - колонну первой перегонки PL, 31 -колонну второй перегонки PL, 32 - экструдер, 33а и 33b - рефлюксный аппарат, 41 - перегонную колонну, 42 - конденсатор, 43 - вакуумное устройство, 44 - вакуумный трубопровод, 45 - резервуар для конденсата, 46 - насос для подачи жидкости, 47 - рефлюксный трубопровод, 48 - туманоуловитель, 49 - дренажное отверстие, 50 - питающее отверстие, 51 - клапан, 52 - клапан, 53 - дренажное отверстие, 54 - дренажное отверстие, 55 - питающее отверстие, 56 - клапан, 57 - выпускное отверстие, А - ацетон, ВРА - бисфенол А, C1 - щелочной катализатор, С2 - основный катализатор, CDC - фосген, D1 - газообразная хлористоводородная кислота, D2 - нейтрализованные отработанные воды, D3 - отработанные воды, D4 - низкокипящий дистиллят при получении ВРА, D5 - выпускаемый газ, D6 - низкокипящий дистиллят при получении РС, D7 - отработанный раствор, AW - смесь вода/ацетон, DPC - дифенилкарбонат, Е1 - щелочной водный раствор, F - смешанный газ, I - кислоту, J - добавку, р - превращенные в пар ингредиенты при получении РС, р1 - первые превращенные в пар ингредиенты при получении РС, р2 - вторые превращенные в пар ингредиенты при получении РС, PL - фенол, s-PL - фенол, являющийся побочным продуктом, W - воду, Х1 - остаток после перегонки при получении DPC, Х1' - остаток после выделения/перегонки, Х2 - остаток после перегонки PL, символ а - жидкую реакционную смесь, содержащую DPC, b - дегидрохлорированную жидкость, d - извлеченную жидкость, содержащую DPC, е - нейтрализованную жидкость, f - жидкость, подвергнутую водной промывке, g - остаток после первой перегонки, k - извлеченный жидкий PL, р - превращенные в пар ингредиенты при получении РС, а q - остаток первой стадии.
Наилучший способ реализации изобретения
Далее подробно будут описываться варианты осуществления изобретения.
Способ получения ароматического поликарбоната (РС), соответствующий изобретению, представляет собой способ, по которому для получения полимера полимеризуют дифенилкарбонат (DPC) и бисфенол А (ВРА).
Стадия получения DPC
DPC получают из PL и, по меньшей мере, одного карбонильного соединения в качестве исходных материалов. На данное используемое карбонильное соединение ограничений не накладывается до тех пор, пока оно может образовывать карбонильную группу DPC. Примеры данного карбонильного соединения включают фосген (здесь и далее в настоящем документе сокращенно обозначаемый как "CDC"), монооксид углерода, диалкилкарбонаты и тому подобное. Далее приводятся пояснения в отношении стадии, на которой в качестве карбонильного соединения при получении DPC в результате проведения стадии промывки DPC и стадии перегонки при получении DPC после прохождения реакции используют CDC.
Стадию получения DPC составляет способ, продемонстрированный на фиг.1. А именно, проводят стадию реакции при получении DPC, на которой PL и CDC используют в качестве исходных материалов и вводят в реактор получения DPC 1 совместно с щелочным катализатором (С1), например пиридином. Несмотря на то, что на условия проведения реакции на данной стадии особенных ограничений не накладывается, реакцию предпочитается проводить в условиях при 50-180°С и обычном давлении, когда PL находится в расплавленном состоянии. Соотношение между количествами PL и CDC в смеси (молярное соотношение) предпочтительно является таким, что количество CDC составляет 0,40-0,49 моль на один моль PL с точки зрения полного расходования CDC.
Жидкую реакционную смесь, содержащую DPC, а, полученную в результате проведения стадии реакции при получении DPC, подают в колонну дегидрохлорирования 2, где проводят стадию дегидрохлорирования. Газообразную хлористоводородную кислоту (D1), полученную в реакторе получения DPC 1 и колонне дегидрохлорирования 2, извлекают и подают на стадию переработки хлористоводородной кислоты (не показана).
После этого полученную дегидрохлорированную жидкость b подвергают воздействию стадии промывки DPC. Данную стадию промывки DPC составляют стадия нейтрализации и стадия водной промывки, описанные далее. А именно, проводят стадию нейтрализации, на которой дегидрохлорированную жидкость b подают в смесительную емкость 3, а после этого в емкость щелочной нейтрализации 4 для проведения нейтрализации под действием щелочного водного раствора (Е1) хлористоводородной кислоты, остающейся не удаленной в колонне дегидрохлорирования. Выпускаемые здесь нейтрализованные отработанные воды (D2) подают на стадию переработки отработанных вод (не показана) для извлечения эффективных органических ингредиентов, содержащихся в них, а после этого подвергают переработке активированного осадка.
После этого проводят стадию водной промывки DPC, на которой полученную нейтрализованную жидкость, е, подают в емкость водной промывки 5 и промывают, используя воду (W). Отработанные воды (D3), выпускаемые на данной стадии водной промывки DPC, можно повторно использовать в качестве щелочного разбавителя при получении щелочного водного раствора (Е1), предназначенного для использования на стадии нейтрализации.
Жидкость, подвергнутую водной промывке, f, полученную на стадии водной промывки DPC, подают в перегонную колонну, где проводят стадию перегонки при получении DPC. Несмотря на то, что на фиг.1 используют три перегонные колонны, стадия данным режимом не ограничивается. В случае использования трех перегонных колонн колонну первой перегонки при получении DPC 6 используют для извлечения смешанного газа (F), содержащего воду, PL и щелочной катализатор. Данный смешанный газ (F) можно разделить на данные ингредиенты, которые можно повторно использовать в реакционной системе.
Остаток после первой перегонки g, выпускаемый из колонны первой перегонки при получении DPC 6, еще раз подвергают перегонке в колонне второй перегонки при получении DPC 7 для извлечения в качестве дистиллята очищенного DPC, который является продуктом.
На условия проведения перегонки для колонны первой перегонки при получении DPC 6 особенных ограничений не накладывается до тех пор, пока вода, щелочной катализатор и PL будут отгоняться, а DPC - оставаться. Предпочтительным является давление 1,3-13 кПа. Используемой температурой является температура кипения при данном давлении. С другой стороны, на условия проведения перегонки для колонны второй перегонки при получении DPC 7 особенных ограничений не накладывается до тех пор, пока DPC будет отгоняться, а примеси, характеризующиеся температурами кипения, более высокими по сравнению с температурой кипения DPC, - оставаться. Предпочтительными являются давление 1,3-6,5 кПа и температура 150-220°С.
Между прочим, остаток после перегонки при получении DPC Х1, выпускаемый из колонны второй перегонки при получении DPC 7, содержит примеси, в основном содержащие метилзамещенные производные DPC, полученные в результате прохождения реакции с метилфенолом, который является фенолсодержащей примесью, и бромзамещенные производные DPC, полученные в результате прохождения реакции с бромом, остающимся в CDC. Однако данный остаток после перегонки Х1, кроме того, содержит и сам DPC. Следовательно, данный остаток после перегонки при получении DPC X1 можно еще раз перегнать для извлечения дифенилкарбоната (DPC). В данном случае остаток после перегонки при получении DPC X1 подвергают воздействию стадии извлечения/перегонки при использовании колонны извлечения/перегонки при получении DPC 8, как это продемонстрировано на фиг.1. Таким образом, в результате проведения перегонки можно извлекать извлеченную жидкость, содержащую DPC, d. Из кубового остатка в перегонной колонне извлекают остаток после извлечения/перегонки при получении DPC (X1'), в котором сконцентрированы метилзамещенные и бромзамещенные производные DPC.
На условия проведения перегонки для колонны извлечения/перегонки при получении DPC 8 особенных ограничений не накладывается до тех пор, пока DPC будет отгоняться, а примеси, характеризующиеся температурой кипения, более высокой по сравнению с температурой кипения DPC, - оставаться. Предпочтительными являются давление 1,3-6,5 кПа и температура 150-220°C.
Поскольку извлеченная жидкость, содержащая DPC, d, которая представляет собой дистиллят из колонны извлечения/перегонки при получении DPC, представляет собой ингредиент, в большом количестве содержащий DPC, ее подают в смесительную емкость 3. Таким образом, DPC можно отправить на рецикл на стадию промывки/перегонки, а эффективность извлечения дифенилкарбоната (DPC) можно дополнительно улучшить.
Стадия получения ВРА
Стадию получения ВРА составляет способ, продемонстрированный на фиг.2. А именно, PL и ацетон (А) в качестве исходных материалов при получении ВРА в результате проведения стадии реакции при получении ВРА (стадии (а)), стадии удаления низкокипящих соединений при получении ВРА (стадии (b)), стадии кристаллизации/выделения ВРА (стадии (с)), стадии нагревания/плавления (стадии (d)), стадии удаления PL (стадии (е)) и стадии гранулирования (стадии (f)).
Далее по раздельности будут разъяснены индивидуальные стадии.
Стадия (а) представляет собой стадию, на которой PL и ацетон (А) подвергают реакции конденсации в присутствии кислотного катализатора с получением ВРА. PL и ацетон (А), используемые в данном случае в качестве исходных материалов, вводят в реакцию при таких условиях, когда количество PL будет превышать стехиометрическое количество. Молярное соотношение между PL и ацетоном (А), выраженное через соотношение количеств PL/ацетон (А), находится в диапазоне 3-30, предпочтительно 5-20. Реакцию проводят при температуре в общем случае в диапазоне 30-100°С, предпочтительно 50-90°С, а давление в общем случае находится в диапазоне от обычного давления до 5 кг/см2 избыточного давления.
В качестве кислотного катализатора возможно использование неорганической кислоты, например хлористоводородной кислоты, органической кислоты, ионообменной смолы и тому подобного. В том случае, если в качестве кислотного катализатора будут использовать ионообменную смолу, то тогда подходящей будет являться катионообменная смола в форме сульфоновой кислоты, относящаяся к типу геля, которая характеризуется степенью сшивания 1-8%, предпочтительно 2-6%. Однако на ионообменную смолу особенных ограничений не накладывается. В качестве кислотного катализатора также возможно использование и хлористоводородной кислоты.
Несмотря на то, что катионообменную смолу в форме сульфоновой кислоты можно использовать как таковую, возможно использование и катионообменной смолы с сульфоновой кислотой, которую подвергли модифицированию в соответствии с потребностью. Примеры соединений, используемых для модифицирования, включают соединения, имеющие меркаптогруппу.
В качестве такого соединения, имеющего меркаптогруппу, возможно использование любого из соединений, которые известны своей применимостью в данном приложении, таких как аминоалкантиолы, например 2-аминоэтантиол, ω-пиридилалкантиолы, например 2-(4-пиридил)этантиол, и тиазолидиновые соединения, которые легко приобретают меркаптогруппу при проведении гидролиза и тому подобного, например 2,2-диметилтиазолидин.
Реакционная смесь, получающаяся в результате проведения стадии реакции (а), в дополнение к ВРА в общем случае содержит не вступивший в реакцию PL, не вступивший в реакцию ацетон (А), катализатор и воду (W), полученную в ходе реакции, а кроме того, содержит побочные продукты, включающие окрашенные вещества.
Стадия (b) представляет собой стадию, на которой из жидкой реакционной смеси, полученной на стадии (а), удаляют низкокипящие ингредиенты при получении ВРА и катализатор, например хлористоводородную кислоту. Низкокипящие ингредиенты при получении ВРА включают воду W, полученную в результате проведения реакции, не вступивший в реакцию ацетон (А) и вещества, характеризующиеся температурой кипения, близкой к их температурам кипения. На данной стадии данные низкокипящие ингредиенты удаляют из реакционной смеси в результате проведения, например, вакуумной перегонки, а твердые ингредиенты, например катализатор, удаляют, например, в результате проведения фильтрования. В случае использования реактора, включающего неподвижный слой катализатора, удаление катализатора не является особенно необходимым. При проведении вакуумной перегонки предпочитается использовать давление и температуру в диапазонах 50-300 мм ртутного столба и 70-130°С соответственно. Существуют случаи, в которых не вступивший в реакцию PL образует азеотроп, и часть его удаляется из системы.
Низкокипящий дистиллят при получении ВРА (D4), превращенный в пар на стадии (b), содержит воду и небольшое количество ацетона (А) и фенола (PL). Как продемонстрировано на фиг.3, данный низкокипящий дистиллят при получении ВРА (D4) подают в колонну выделения PL 12, где необязательно при использовании экстрагента проводят стадию выделения воды (стадию (b-2)), на которой через куб колонны извлекают PL. Извлеченный жидкий PL k, полученный на данной стадии выделения воды, хранят в резервуаре для извлеченного PL 11 при получении ВРА. Переработку смеси вода/ацетон AW, извлеченной через верх колонны выделения PL 12, проводят отдельно.
Стадия (с) представляет собой стадию, на которой жидкую смесь, полученную на стадии (b), охлаждают до выпадения в осадок смеси BPA и PL и, таким образом, выделения смеси. Перед проведением данной стадии (с) концентрацию ВРА в жидкой смеси, полученной на стадии (b), в результате отгонки либо добавления PL можно выдерживать на уровне 10-50 мас.%, предпочтительно 20-40 мас.%. Данная операция является предпочтительной, поскольку она увеличивает выход аддукта, и эффективной при регулировании кажущейся вязкости смеси суспензии для улучшения перерабатываемости. Примеры смеси BPA и PL включают кристаллы аддукта ВРА и PL и простые смеси между кристаллами ВРА и кристаллами PL.
Охлаждение на стадии (с) в общем случае проводят до температуры 45-60°С, в результате чего происходит выделение кристаллов аддукта ВРА/PL либо кристаллов каждого соединения, и система становится суспензией. Данное охлаждение проводят при использовании теплообменника, расположенного вне системы, либо в результате теплоотвода с использованием скрытой теплоты парообразования воды, добавляемой в кристаллизатор. После этого данную жидкую суспензию подвергают фильтрованию, разделению по способу центрифугирования и тому подобному для ее разделения на кристаллы и маточную жидкость, содержащую побочные продукты. Кристаллы подвергают воздействию следующей стадии. Для обеспечения дополнительного улучшения выхода часть либо все количество выделенной маточной жидкости отправляют на рецикл на стадию (а) через стадию переработки маточной жидкости при получении ВРА (g), которая будет описываться далее, и используют в качестве части либо всего количества PL, используемого в качестве исходного материала.
Стадия (d) представляет собой стадию, на которой кристаллы, полученные на стадии (с), нагревают и расплавляют. Кристаллы аддукта в общем случае характеризуются составом, при котором уровень содержания ВРА находится в диапазоне 45-70 мас.%, а уровень содержания PL находится в диапазоне 55-30%. Данные кристаллы плавят в результате нагревания до 100-160°С, а после этого подвергают воздействию следующей стадии.
Стадия (е) представляет собой стадию, на которой PL удаляют из расплава, полученного на стадии (d), получая, таким образом, расплав ВРА. Для удаления PL из расплава, полученного на стадии (d), и, таким образом, диссоциации аддукта используют вакуумную перегонку либо другую методику. Таким образом, можно извлечь высокочистый ВРА. Предпочтительно, чтобы данную вакуумную перегонку проводили бы при давлении 10-100 мм ртутного столба и температуре, которая находится в диапазоне 150-220°С и, по меньшей мере, на 10°С превышает температуру плавления смеси бисфенол А (ВРА)/фенол (PL), присутствующей в системе. Также была предложена и методика, в которой для удаления остаточного PL в дополнение к вакуумной перегонке проводят и отгонку с водяным паром.
Стадия (f) представляет собой стадию, на которой расплавленный ВРА, полученный на стадии (е), охлаждают/отверждают и гранулируют с получением гранулированного продукта. При использовании гранулятора, например, распылительной сушилки, расплавленный ВРА формуют в капли и охлаждают/отверждают до получения продукта. Капли получают в результате распыления, скапывания, разбрызгивания и тому подобного, а охлаждение проводят обычно с использованием азота, воздуха и тому подобного.
На данной стадии получения ВРА, в особенности на стадии (а), побочные продукты, включающие 2,4'-бисфенол А (здесь и далее в настоящем документе обозначаемые как "побочные продукты при получении ВРА") синтезируют одновременно с ВРА. Данные побочные продукты при получении ВРА в основном содержатся в маточной жидкости, полученной на стадии (с), и они циркулируют по стадии получения ВРА. По этой причине побочные продукты при получении ВРА имеют тенденцию накапливаться в данной системе циркуляции. Существует тенденция к тому, что, когда накопление побочных продуктов при получении ВРА произойдет свыше определенной степени, разделение на стадии (с) станет недостаточным, и побочные продукты начнут отчасти присутствовать и на стороне ВРА, что в результате приведет к ухудшению качества продукта ВРА. По этой причине часть либо все количество маточной жидкости, полученной на стадии (с), подвергают воздействию стадии переработки маточной жидкости при получении ВРА (стадии (g)), тем самым, выделяя/удаляя из маточной жидкости побочные продукты при получении ВРА. Данную маточную жидкость, в которой таким образом уменьшили количество побочных продуктов при получении ВРА, используют в качестве исходного материала при получении ВРА, благодаря чему можно обеспечивать качество продукта ВРА.
Стадия (g) представляет собой стадию, по которой в результате проведения перегонки извлекают PL, либо стадию, на которой маточную жидкость нагревают в присутствии основного вещества в целях разложения побочных продуктов при получении ВРА, присутствующих в маточной жидкости, и, таким образом, получения PL и производных PL, а данные соединения при использовании кислотного катализатора либо щелочного катализатора впоследствии вводят в реакцию для получения ВРА, который извлекают.
Говоря конкретно, как продемонстрировано на фиг.4, часть либо все количество маточной жидкости сначала вводят в испаритель PL 13, а одновременно с этим вводят основное вещество, такое как гидроксид натрия либо гидроксид калия. После этого содержимое нагревают до температуры, не меньшей, чем температура кипения PL, для превращения PL в пар, который выпускают через верхнюю часть испарителя PL 13. Получающийся в результате остаток после выпаривания, который в качестве основных компонентов содержит ВРА и побочные продукты при получении ВРА, выпускают через нижнюю часть испарителя PL 13 и подают в реактор переработки остатка 14. Остаток нагревают до 180-300°С, таким образом вызывая реакции разложения ВРА и побочных продуктов при получении ВРА с получением продуктов разложения, включающих изопропенилфенол, который является промежуточным соединением при разложении ВРА. Данные продукты разложения отгоняют через верх колонны. С другой стороны, кубовый остаток, получаемый в данном реакторе переработки остатка 14, в виде отработанного раствора, содержащего большое количество органических соединений, подают на стадию переработки отработанного раствора (не показана), такую как, например, утилизация в результате сжигания.
PL и производные PL, полученные в качестве продуктов разложения побочных продуктов при получении ВРА, отгоняют через верхнюю часть реактора переработки остатка 14 и подают в регенерирующий реактор 15. Если в данной операции PL и производные PL будут отводить через верхнюю часть реактора переработки остатка 14, то тогда их будут перемешивать с PL, выпускаемым через верхнюю часть испарителя PL 13. В результате концентрация производных PL уменьшается, и протекание нежелательных побочных реакций может быть подавлено.
После этого в регенерирующем реакторе 15 PL и производные PL, которые представляют собой продукты разложения побочных продуктов при получении ВРА, еще раз вводят в реакцию, используя кислотный катализатор, тем самым получая ВРА и тому подобное. Данный продукт реакции совместно с не вступившим в реакцию PL перемешивают с PL, используемым в качестве исходного материала, и подают на стадию (а). Поскольку в результате проведения стадии (с) ВРА извлекают как таковой, а PL используют в качестве исходного материала, эффективность получения ВРА может быть улучшена.
Стадия получения РС
Стадию получения РС составляет способ, продемонстрированный на фиг.5. А именно, DPC и ВРА, полученные по способам, описанным выше, используют в качестве исходных материалов и вводят в смесительную емкость 21 совместно с основным катализатором (С2), таким как, например, водный щелочной раствор. Получающуюся в результате смесь подают в реактор полимеризации для проведения стадии полимеризации при получении РС. На реактор полимеризации особенных ограничений не накладывается до тех пор, пока в нем можно будет проводить конденсационную полимеризацию при одновременной отгонке фенола, образующегося в качестве побочного продукта полимеризации. Реактор полимеризации может представлять собой реактор любого типа, выбираемый из вертикального реактора, горизонтального реактора и реактора колонного типа.
На количество таких реакторов полимеризации особенных ограничений не накладывается. Однако, поскольку реакция полимеризации представляет собой конденсационную полимеризацию, сопровождаемую отщеплением фенола, предпочитается использовать два либо более чем два реактора полимеризации для того, чтобы условия проведения полимеризации можно было бы изменять в соответствии со степенью полимеризации. На фиг.5 продемонстрирована группа реакторов полимеризации, которые составляют три вертикальных реактора полимеризации (первый реактор полимеризации 22, второй реактор полимеризации 23 и третий реактор полимеризации 24) и один горизонтальный полимеризатор (четвертый полимеризатор 25), соединенные последовательно. Условия проведения полимеризации в данном случае, например, включают 200-250°С и 50-200 торр для первого реактора полимеризации 22, 230-280°С и 10-50 торр для второго реактора полимеризации 23, 250-300°С и 0,2-5 торр для третьего реактора полимеризации 24 и 260-320°С и 0,05-2 торр для четвертого полимеризатора 25. Если будут использовать такие условия проведения полимеризации, то по мере прохождения полимеризации будут проводить отгонку являющегося побочным продуктом фенола (здесь и далее в настоящем документе сокращенно обозначаемого как "s-PL"), благодаря чему можно будет получить РС, характеризующийся желательной степенью полимеризации.
Превращенные в пар ингредиенты при получении РС р, полученные на стадии полимеризации при получении РС и включающие в качестве основного компонента s-PL, ожижают с использованием теплообменников 26 и 27 и конденсатора 28 и подают в резервуар для извлеченного PL 29 при получении РС. Выпускаемый газ (D5), остающийся неожиженным, отводят в сторону вакуумного устройства и подают на стадию переработки (не показана).
РС, полученный на стадии полимеризации при получении РС, подают в экструдер 32. В данном экструдере 32 присутствующие летучие соединения удаляют в виде выпускаемого газа (D5) и для проведения нейтрализации катализатора и тому подобного добавляют кислоту I и различные добавки J. Полимер после этого подвергают переработке (не показана), такой как, например, гранулирование, для получения в качестве продукта РС.
Как утверждается выше, превращенные в пар ингредиенты при получении РС р ожижают и подают в резервуар для извлеченного PL 29 при получении РС. Несмотря на то, что превращенные в пар ингредиенты при получении РС р включают в качестве основного компонента s-PL, они, кроме того, включают DPC и ВРА, которые использовали в качестве исходных материалов, олигомеры, полученные в результате прохождения конденсационной полимеризации между одной либо несколькими молекулами DPC и одной либо несколькими молекулами ВРА, воду, полученную из щелочного катализатора, и тому подобное. По этой причине превращенные в пар ингредиенты при получении РС р для извлечения s-PL подвергают стадии перегонки PL. Примеры данной стадии перегонки PL включают способ, по которому используют перегонные колонны, скомбинированные в виде двухстадийной компоновки, такой как та, которая продемонстрирована на фиг.5. Во-первых, в колонне первой перегонки PL 30 ингредиенты, характеризующиеся температурой кипения, более низкой по сравнению с температурой кипения PL, которые включают воду, превращают в пар и отгоняют, а низкокипящий дистиллят при получении РС (D6), содержащий в качестве основного компонента воду и содержащий PL, удаляют и извлекают. После этого получающийся в результате остаток после перегонки первой стадии q в колонне первой перегонки PL 30 подают в колонну второй перегонки PL 31, где для извлечения s-PL в результате проведения перегонки высококипящие соединения отсекают, а остаток после перегонки PL (X2), который является высококипящими соединениями, извлекают.
Переработка являющегося побочным продуктом фенола (s-PL) 1; уровень содержания воды в s-PL
Уровень содержания воды в s-PL, извлеченном на стадии перегонки PL в результате проведения перегонки, желательно составляет 0,2 мас.% либо менее, предпочтительно 0,1 мас.% либо менее, более предпочтительно 0,05 мас.% либо менее, еще более предпочтительно 0,01 мас.% либо менее. В том случае, если уровень ее содержания будет превышать 0,2 мас.%, то тогда данный s-PL, будучи поданным на стадию получения DPC либо стадию получения ВРА, будет приводить к уменьшению каталитической активности на стадии получения DPC и на стадии получения ВРА и будет склонен к тому, чтобы на стадии получения DPC вызывать прохождение гидролиза и тому подобного, что будет описываться далее. По этой причине использование такого s-PL склонно приводить в результате к ухудшению эффективности получения на стадии получения DPC и стадии получения ВРА. С другой стороны, нижний предел уровня содержания воды составляет 0 мас.%, поскольку чем ниже будет уровень содержания воды, тем лучше.
Если предпринять попытку отогнать воду из превращенных в пар ингредиентов при получении РС р в колонне первой перегонки PL 30, то тогда, если перегонку проводить при обычных условиях, воде будет сопутствовать значительное количество PL, что в результате приведет к потерям. Это объясняется тем, что вода и PL обладают свойством образовывать азеотроп. Методики уменьшения данных потерь включают проведение десорбции перегонкой, использование условий вакуума для устранения азеотропии и увеличение количества теоретических тарелок и флегмового числа. В любом случае с экономической точки зрения нежелательно проводить полное разделение. По этой причине возможно использование способа, по которому низкокипящий дистиллят при получении РС (D6), полученный в результате отгонки воды совместно с частью PL в колонне первой перегонки PL 30 при обычных условиях проведения перегонки, подают на стадию промывки DPC на стадии получения DPC и/или на стадию выделения воды (стадию (b-2)) на стадии получения ВРА.
Условия проведения перегонки для колонны первой перегонки PL 30 предпочтительно включают давление в диапазоне от атмосферного давления до пониженного давления величиной в несколько десятков торр и температуру верха колонны, которая будет не меньше температуры кипения воды при данном давлении и не больше температуры кипения фенола при данном давлении. Газ, таким образом выпускаемый через верх данной колонны первой перегонки PL 30, представляет собой смешанный газ вода/PL, и температуру верха колонны предпочитается доводить до температуры кипения смешанного газа, характеризующегося целевым составом. В том случае, если температура верха колонны будет более низкой по сравнению с температурой кипения воды, то тогда уровень содержания воды в остатке первой стадии q увеличится, что приведет в результате к появлению возможности того, что s-PL, полученный в колонне второй перегонки PL 31, может начать характеризоваться повышенным уровнем содержания воды, выходящим за пределы данного диапазона. С другой стороны, в том случае, если температура верха колонны будет больше температуры кипения PL, то тогда количество s-PL, содержащегося в низкокипящем дистилляте при получении РС (D6), увеличится, что в результате приведет к необходимости использования большого количества энергии для его извлечения. Таким образом, такая температура является неэкономичной.
Говоря конкретно, уровень содержания PL в газе, выпускаемом через верх колонны первой перегонки PL 30, является таким, что концентрация PL предпочтительно будет составлять 50 мас.% либо более, а в особенности предпочтительно 70 мас.% либо более и 99,8 мас.% либо менее.
Таким образом полученный s-PL используют в качестве части исходного материала на стадии получения DPC либо стадии получения ВРА.
Говоря конкретно, на стадии получения DPC s-PL используют на стадии реакции при получении DPC. В случае подачи на стадию реакции при получении DPC s-PL можно использовать в качестве части либо всего количества PL, используемого в качестве исходного материала. Поскольку в данном случае уровень содержания воды в нем находится в пределах указанного выше диапазона, вода, содержащаяся в s-PL, будет оказывать незначительное влияние на стадию реакции, и эффективность получения DPC может быть обеспечена.
Далее, на стадии получения ВРА s-PL используют на стадии реакции при получении ВРА (стадии (а)). На стадии реакции при получении ВРА s-PL можно использовать в качестве части либо всего количества PL, используемого в качестве исходного материала. Поскольку в данном случае уровень содержания воды в нем находится в пределах указанного выше диапазона, вода, содержащаяся в s-PL, будет оказывать незначительное влияние на стадию реакции синтеза, и эффективность получения ВРА может быть обеспечена.
Переработка низкокипящего дистиллята при получении РС (D6)
Низкокипящий дистиллят (D6), превращенный в пар при отгонке в колонне первой перегонки PL 30, возвращают на стадию промывки DPC на стадии получения DPC либо на стадию выделения воды (стадию (b-2)) на стадии получения ВРА, как утверждалось выше, в результате чего можно будет извлечь PL в низкокипящем дистилляте при получении РС (D6).
Говоря конкретно, если низкокипящий дистиллят при получении РС (D6) возвращают на стадию промывки DPC, например, в емкость щелочной нейтрализации 4, на стадии получения DPC, как это продемонстрировано на фиг.1, то тогда PL, содержащийся в низкокипящем дистилляте при получении РС (D6), экстрагируют в органическую фазу (жидкую реакционную смесь), извлекают в виде смешанного газа (F) в колонне первой перегонки при получении DPC 6 на следующей стадии и, в заключение, используют в качестве исходного материала при получении DPC.
С другой стороны, если низкокипящий дистиллят при получении РС (D6) возвращают на стадию водной промывки (стадию (b-2)) на стадии получения BPA, говоря более конкретно, например, в колонну выделения PL 12, как это продемонстрировано на фиг.2 либо 3, то тогда PL, содержащийся в низкокипящем дистилляте при получении РС (D6), извлекают в виде извлеченного жидкого PL k через куб колонны и, в заключение, используют в качестве исходного материала при получении ВРА.
Переработка являющегося побочным продуктом фенола (s-PL) 2; переработка превращенных в пар ингредиентов при получении РС на стадии полимеризации при получении РС
Как продемонстрировано на фиг.5, превращенные в пар ингредиенты при получении РС р, которые включают фенол, полученный в качестве побочного продукта на стадии полимеризации, ожижают при использовании теплообменников 26 и 27 и конденсатора 28 и подают в резервуар для извлеченного PL 29 при получении РС. Выпускаемый газ (D5), остающийся неожиженным, подают на стадию переработки (не показана).
Между прочим, смеси превращенных в пар ингредиентов, полученные соответственно в аппаратах от первого реактора полимеризации 22 до третьего реактора полимеризации 24, отличаются друг от друга по примесям, отличным от являющегося побочным продуктом фенола. Говоря конкретно, превращенные в пар ингредиенты, выпускаемые со стадии полимеризации на раннем этапе, в дополнение к являющемуся побочным продуктом фенолу включают примеси, такие как примеси, характеризующиеся температурой кипения, более низкой по сравнению с температурой кипения PL, небольшое количество карбонильного соединения, дифенилкарбонат и тому подобное. Данные примеси представляют собой исходный материал, используемый на стадии получения DPC, и продукты реакции, получаемые на данной стадии, а высококипящие соединения, такие как, например, ВРА, в них не содержатся. Следовательно, превращенные в пар ингредиенты, которые включают данные примеси, можно использовать в качестве части PL, используемого на стадии получения DPC, без проведения очистки либо после проведения очистки до низкой степени чистоты.
С другой стороны, превращенные в пар ингредиенты, выпускаемые со стадии полимеризации на позднем этапе стадии полимеризации при получении РС, в дополнение к являющемуся побочным продуктом фенолу включают примеси, характеризующиеся температурой кипения, более высокой по сравнению с температурой кипения PL, такие как DPC, BPA и олигомеры, полученные из DPC и BPA. В них почти не содержится каких-либо примесей, характеризующихся температурой кипения, более низкой по сравнению с температурой кипения PL. Данные примеси подвергаются гидролизу на стадии получения ВРА и становятся исходным материалом и продуктами реакции для данной стадии получения. Почти не содержится каких-либо спиртов и тому подобного, что могло бы стать причиной уменьшения каталитической активности на стадии получения ВРА. По этой причине превращенные в пар ингредиенты, которые содержат данные примеси, можно использовать в качестве части PL, используемого на стадии получения ВРА, без проведения очистки либо после проведения очистки до низкой степени чистоты.
Следовательно, превращенные в пар ингредиенты, выпускаемые на стадии полимеризации при получении РС, можно извлекать в виде двух порций, как это продемонстрировано на фиг.6. А именно, превращенные в пар ингредиенты, выпускаемые на первом из двух этапов стадии полимеризации, то есть первые, превращенные в пар ингредиенты при получении РС р1, извлеченные из первого реактора полимеризации 22 либо из первого реактора полимеризации 22 и второго реактора полимеризации 23 и содержащие являющийся побочным продуктом фенол, можно подавать в первый резервуар для извлеченного PL 29а при получении РС. Кроме того, превращенные в пар ингредиенты, выпускаемые на втором из двух этапов стадии полимеризации, то есть вторые, превращенные в пар ингредиенты при получении РС р2, извлеченные из второго реактора полимеризации 23 и последующих реакторов полимеризации либо из третьего реактора полимеризации 24 и последующего реактора полимеризации и содержащие являющийся побочным продуктом фенол, можно подавать во второй резервуар для извлеченного PL при получении РС.
Таким образом, извлеченные первые превращенные в пар ингредиенты при получении РС р1 можно использовать в качестве части PL, используемого в качестве исходного материала на стадии получения DPC без проведения стадии перегонки PL, продемонстрированной на фиг.5. С другой стороны, вторые превращенные в пар ингредиенты при получении РС р2 можно использовать в качестве части PL, используемого в качестве исходного материала на стадии получения ВРА без проведения стадии перегонки PL, продемонстрированной на фиг.5.
Предпочтительно, чтобы реакторы полимеризации (реакторы), из которых получают первые превращенные в пар ингредиенты при получении РС р1, которые содержат являющийся побочным продуктом фенол, используемый на стадии получения DPC, то есть первый реактор полимеризации 22 либо первый реактор полимеризации 22 и второй реактор полимеризации 23, были бы снабжены рефлюксным аппаратом 33а и 33b для ожижения и кипячения в условиях флегмообразования части превращенных в пар ингредиентов. В числе ингредиентов дистиллята, отбираемых из первого реактора полимеризации 22 либо из первого реактора полимеризации 22 и второго реактора полимеризации 23, ингредиенты, которые характеризуются температурой кипения, более высокой по сравнению с температурой кипения PL, можно возвращать в соответствующие реакторы полимеризации при использовании рефлюксного аппарата 33а и 33b. В результате в полученных первых превращенных в пар ингредиентах при получении РС р1 можно дополнительно уменьшить долю ингредиентов, характеризующихся температурой кипения, более высокой по сравнению с температурой кипения PL.
Между прочим, превращенные в пар ингредиенты, извлеченные из второго реактора полимеризации 23, подают в любой резервуар, выбираемый из первого резервуара для извлеченного PL 29а при получении РС и второго резервуара для извлеченного PL 29b при получении РС, как утверждается выше. Что касается выбора резервуара для извлеченного продукта, то может иметься только трубопровод для любого из двух резервуаров для извлеченного продукта. В альтернативном варианте могут иметься трубопроводы, соответственно соединенные с двумя резервуарами для извлеченного продукта, и на соответствующих трубопроводах можно расположить клапаны 34а и 34b для того, чтобы подходящим образом производить переключение между трубопроводами, как это продемонстрировано на фиг.6. Причинами этого являются нижеследующие. Количество примесей, отличных от являющегося побочным продуктом фенола, которые содержатся в превращенных в пар ингредиентах, извлеченных из второго реактора полимеризации 23, является относительно небольшим. Таким образом, данные превращенные в пар ингредиенты можно использовать либо в качестве первых превращенных в пар ингредиентов при получении РС р1, либо в качестве вторых превращенных в пар ингредиентов при получении РС р2.
Если превращенные в пар ингредиенты, полученные в качестве дистиллята из второго реактора полимеризации 23, подают в первый резервуар для извлеченного PL 29а при получении РС либо во второй резервуар для извлеченного PL 29b при получении РС, то тогда количество подаваемых ингредиентов регулируют в соответствии с количествами превращенных в пар ингредиентов из других реакторов полимеризации, в частности с количеством являющегося побочным продуктом фенола, содержащегося в данных превращенных в пар ингредиентах, и количеством примесей, содержащихся в данных превращенных в пар ингредиентах.
Количество данного являющегося побочным продуктом фенола в являющемся побочным продуктом феноле, содержащемся в превращенных в пар ингредиентах, полученных на стадии полимеризации при получении РС, которое необходимо подать на стадию получения DPC и использовать в качестве части являющегося исходным материалом PL, то есть количество являющегося побочным продуктом фенола, содержащегося в первых превращенных в пар ингредиентах при получении РС р1, желательно составляет 50-95 мас.%, предпочтительно 50-70 мас.%, при расчете на все количество являющегося побочным продуктом фенола, содержащегося в превращенных в пар ингредиентах, полученных на стадии полимеризации при получении РС, то есть при расчете на совокупное количество являющегося побочным продуктом фенола, содержащегося в первых превращенных в пар ингредиентах при получении РС р1 и содержащегося во вторых превращенных в пар ингредиентах при получении РС р2.
С другой стороны, количество являющегося побочным продуктом фенола, содержащегося во вторых превращенных в пар ингредиентах при получении РС р2, в желательном варианте составляет 50-5 мас.%, предпочтительно 50-30 мас.%, при расчете на все количество являющегося побочным продуктом фенола, содержащегося в превращенных в пар ингредиентах, полученных на стадии полимеризации при получении РС.
В том случае, если количество являющегося побочным продуктом фенола, содержащегося в первых превращенных в пар ингредиентах при получении РС р1, будет меньшим 50 мас.%, то тогда примеси, характеризующиеся температурой кипения, более низкой по сравнению с температурой кипения DPC, в особенности спирты и тому подобное, будут иметь тенденцию к попаданию во вторые превращенные в пар ингредиенты при получении РС р2. Использование таких превращенных в пар ингредиентов р2 без проведения какой-либо переработки в качестве исходного материала при получении ВРА имеет тенденцию приводить в результате к уменьшению активности в реакции. Таким образом, такие количества являющегося побочным продуктом фенола нежелательны. С другой стороны, в том случае, если его количество будет превышать 95 мас.%, то тогда появится возможность того, что ВРА и олигомеры, которые характеризуются температурой кипения, более высокой по сравнению с температурой кипения DPC, смогут попадать в первые превращенные в пар ингредиенты при получении РС р1, вызывая засорение трубы при получении DPC.
Содержание высококипящих соединений, характеризующихся температурой кипения, более высокой по сравнению с температурой кипения DPC, таких как ВРА и олигомеры, полученные из DPC и ВРА, в превращенных в пар ингредиентах, используемых на стадии получения DPC, то есть в первых превращенных в пар ингредиентах при получении РС р1, предпочтительно составляет 1,0 мас.% либо менее, более предпочтительно 0,1 мас.% либо менее. В том случае, если уровень их содержания будет превышать 1,0 мас.%, то тогда появится возможность того, что при получении DPC может произойти засорение трубы.
Кроме того, уровень содержания низкокипящих соединений, характеризующихся температурой кипения, более низкой по сравнению с температурой кипения DPC, например, карбонильного соединения и спирта, полученного в качестве побочного продукта из карбонильного соединения, в превращенных в пар ингредиентах, используемых на стадии получения ВРА, то есть во вторых превращенных в пар ингредиентах при получении РС р2, предпочтительно составляет 100 ч/млн (мас.) либо менее, более предпочтительно 50 ч/млн (мас.) либо менее. Между прочим, PL, используемый в качестве исходного материала на стадии получения ВРА, содержит побочный продукт - фенол, содержащийся в первых превращенных в пар ингредиентах при получении РС р1 и во вторых превращенных в пар ингредиентах при получении РС р2, и дополнительно включает коммерческий фенол для покрытия недостатка и PL, циркулирующий по стадии получения ВРА. По этой причине уровень содержания низкокипящих соединений в данном PL, используемом в качестве исходного материала, является более низким по сравнению с уровнем содержания низкокипящих соединений в являющемся побочным продуктом феноле и, в общем случае, составляет 20 ч/млн (мас.) либо менее, предпочтительно 5 ч/млн (мас.) либо менее. В том случае, если уровень их содержания будет более высоким, чем 20 ч/млн (мас.), то тогда появится возможность того, что низкокипящие соединения смогут уменьшить каталитическую активность при получении ВРА, что приведет к уменьшению производительности.
В том случае, если карбонильное соединение будет представлять собой диалкилкарбонат и/или алкиларилкарбонат, то тогда спирт, полученный в качестве побочного продукта из карбонильного соединения, будет представлять собой алкиловый спирт, полученный из диалкилкарбоната и/или алкиларилкарбоната.
В результате такого извлечения превращенных в пар ингредиентов при получении РС р в виде двух порций и подачи двух порций на стадию получения DPC и стадию получения ВРА в соответствии с типами примесей стадию перегонки PL на стадии получения РС можно опустить, и может быть сделан вклад в улучшение эффективности получения.
Переработка являющегося побочным продуктом фенола (s-PL) 3; классификация по примесям для способов использования превращенных в пар ингредиентов при получении РС р и коммерческого PL
Между прочим, в том случае, если стадия получения DPC и стадия получения ВРА и стадия получения РС будут иметь один и тот же масштаб, то тогда количество s-PL, получаемого на стадии получения РС, продемонстрированной на фиг.5, теоретически будет составлять приблизительно половину от совокупного количества PL, используемого в качестве исходного материала на стадии получения DPC и стадии получения ВРА, а количество PL, используемого в качестве исходного материала на стадии получения DPC, теоретически будет составлять ту же самую величину, что и количество PL, используемого в качестве исходного материала на стадии получения ВРА. Недостаток покрывают, используя коммерческий PL (здесь и далее в настоящем документе обозначаемый как "коммерческий PL"). По этой причине бывают случаи, когда возникает проблема того, как использовать s-PL и коммерческий PL.
В общем случае коммерческий PL в некотором количестве содержит примеси, такие как крезол и/или ксиленол и гидроксиацетон, в то время как превращенные в пар ингредиенты при получении РС р, из которых s-PL не был извлечен в результате проведения перегонки, характеризуются пониженным уровнем содержания примесей, таких как крезол и/или ксиленол и гидроксиацетон. По этой причине то, как использовать s-PL, можно определить, исходя из разницы в уровне содержания примесей, таких как крезол и/или ксиленол и гидроксиацетон.
Говоря конкретно, предпочитается, чтобы PL, используемый в качестве исходного материала на стадии получения DPC, представлял бы собой фенол, содержащий крезол и/или ксиленол в количестве 20-1000 ч/млн (мас.) (здесь и далее в настоящем документе обозначаемый как "крезолсодержащий PL"), и чтобы PL, используемый в качестве исходного материала на стадии получения ВРА, представлял бы собой фенол, содержащий крезол и/или ксиленол в количестве, меньшем 20 ч/млн (мас.) (здесь и далее в настоящем документе обозначаемый как "не содержащий крезола PL").
Примеры крезолсодержащего PL включают коммерческий PL. Данный коммерческий PL в дополнение к крезолу и ксиленолу содержит примеси, вызывающие окрашивание, такие как гидроксиацетон, в количестве нескольких десятков ч/млн (мас.).
Как утверждалось выше, данный крезолсодержащий PL можно использовать как таковой на стадии реакции при получении DPC. Количество каждой из примесей в данном крезолсодержащем PL, таких как крезол, ксиленол и примеси, вызывающие окрашивание, например гидроксиацетон, находится в диапазоне, допустимом для стадии реакции при получении DPC. В результате проведения переработки в колонне первой перегонки при получении DPC 6, которая будет описана далее, такие примеси отгоняют в виде части смешанного газа F либо удаляют в виде остатка после перегонки. Следовательно, даже если DPC, полученный при использовании данного крезолсодержащего PL, будут использовать на стадии получении РС, это не будет оказывать влияния на качество полученного РС.
Примеры не содержащего крезола PL включают превращенные в пар ингредиенты при получении РС р, из которых s-PL в результате проведения перегонки не был извлечен. Уровень содержания примесей, являющихся каталитическими ядами, например гидроксиацетона, в превращенных в пар ингредиентах при получении РС р предпочтительно является более низким чем 10 ч/млн, более предпочтительно более низким чем 5 ч/млн, в особенности предпочтительно более низким чем 1 ч/млн. Уровень их содержания, не меньший чем 10 ч/млн, в результате приведет к значительному укорачиванию срока службы катализатора.
В дополнение к примесям, указанным выше, таким как крезол, ксиленол и примеси, являющиеся каталитическими ядами, либо примеси, вызывающие окрашивание, такие как гидроксиацетон, примеры примесей, содержащихся в превращенных в пар ингредиентах при получении РС р, включают DPC, BPA и олигомеры, полученные в результате прохождения реакции между одной либо несколькими молекулами DPC и одной либо несколькими молекулами ВРА.
Уровень содержания крезола и/или ксиленола в превращенных в пар ингредиентах при получении РС р предпочтительно составляет 20 ч/млн (мас.) либо менее, более предпочтительно 10 ч/млн (мас.) либо менее. В том случае, если уровень их содержания будет большим, чем 20 ч/млн (мас.), то тогда появится возможность того, что смогут образоваться алкилзамещенные производные ВРА, приводящие к уменьшению степени чистоты ВРА.
Превращенные в пар ингредиенты при получении РС р содержат воду. Присутствие воды на стадии получения ВРА вызывает уменьшение каталитической активности и, таким образом, приводит к уменьшению степени превращении при получении ВРА. Поэтому необходимо проводить стадию удаления воды. Для того чтобы этого добиться, предпочитается подвергать превращенные в пар ингредиенты при получении РС р воздействию стадии удаления воды, а после этого использовать ингредиенты р на стадии (а) стадии получения ВРА.
В качестве стадии удаления воды возможно использование стадии выделения воды (стадии (b-2)) как таковой. А именно, на фиг.3 превращенные в пар ингредиенты при получении РС р подают в качестве исходного материала в колонну выделения PL 12 для проведения стадии удаления воды. Смесь вода/ацетон AW, которую на данной операции в ходе отгонки превращают в пар, подвергают отдельной переработке. С другой стороны, остаток после перегонки в колонне выделения PL 12 подают в колонну удаления высококипящих соединений, несмотря на то, что это не продемонстрировано на фиг.2 и 3. После этого можно провести стадию удаления высококипящих соединений для выделения ингредиентов, характеризующихся температурой кипения, более высокой по сравнению с температурой кипения PL, в результате чего высококипящие ингредиенты выделяют/удаляют в виде кубового остатка, получающегося в результате проведения перегонки, а PL извлекают в виде дистиллята.
Таким образом, извлеченный PL без проведения какой-либо переработки в качестве являющегося исходным материалом PL можно подвергать воздействию стадии реакции при получении ВРА (стадии (а)) на стадии получения ВРА. В альтернативном варианте извлеченный PL можно временно хранить в резервуаре для извлеченного PL 11 при получении ВРА, а после этого подвергать воздействию стадии (с) и по мере надобности воздействию стадии реакции при получении ВРА (стадии (а)) через стадию переработки маточной жидкости (стадию (g)), как это продемонстрировано на фиг.2. Одна из причин того, почему извлеченный PL подвергают воздействию стадии кристаллизации/выделения ВРА (стадии (с)), заключается в том, что в качестве очищающей жидкости для синтезированного ВРА предпочитается использовать чистый PL. Еще одна причина заключается в том, что, даже если извлеченный PL будет подвергнут воздействию стадии кристаллизации/выделения ВРА (стадии (с)), примеси, которые попадают на данную стадию, будут представлять собой 2,4'-изомеры ВРА и тому подобное (здесь и далее в настоящем документе обозначаемые как "побочные продукты при получении ВРА"), полученные на стадии реакции при получении ВРА, и не будут оказывать влияния на стадию реакции синтеза. С другой стороны, высококипящие ингредиенты, выпускаемые со стадии удаления высококипящих соединений, подают на описанную ранее стадию переработки маточной жидкости (g), на которой можно извлекать эффективные ингредиенты.
В результате таких подач превращенных в пар ингредиентов при получении РС р на стадию получения ВРА и использования коммерческого PL на стадии получения DPC можно уменьшить негативное влияние присутствующих примесей, а стадию перегонки PL на стадии получения РС можно опустить. Таким образом, можно внести вклад в улучшение эффективности получения.
Соединение стадий получения; наличие стадий хранения
1. Стадия получения РС
На стадии получения РС стадию хранения при получении РС (стадию первого хранения при получении РС либо стадию второго хранения при получении РС), предназначенную для хранения жидкости, полученной в результате ожижения превращенных в пар ингредиентов при получении РС р, которые необходимо подвергать стадии перегонки PL, и/или для хранения s-PL, извлеченного на стадии перегонки PL, можно расположить до и/или после стадии перегонки PL, как это продемонстрировано на фиг.7. Наличие стадии хранения при получении РС оказывает нижеследующее действие. Даже если проведение стадии полимеризации при получении РС временно прекращать либо осуществлять периодически, превращенные в пар ингредиенты при получении РС р либо s-PL, хранящиеся на данной стадии хранения при получении РС, можно непрерывно подавать в качестве являющегося исходным материалом PL на последующую стадию, то есть стадию перегонки PL либо стадию реакции при получении DPC либо стадию реакции при получении ВРА, в результате чего последующую стадию можно проводить непрерывно.
Вместимость резервуара для хранения при получении РС, используемого на стадии хранения при получении РС, можно определить, принимая во внимание время работы и время простаивания для стадии полимеризации при получении РС. Говоря конкретно, резервуар предпочтительно характеризуется вместимостью, удовлетворяющей требование, представленное в нижеследующем выражении (1):

В выражении (1) Vc обозначает вместимость (м3) резервуара для хранения при получении РС, а Fc обозначает расход (м3/час) ожиженных, превращенных в пар ингредиентов при получении РС либо являющегося побочным продуктом фенола.
Если величина Vc/Fc будет меньше 10, то тогда появятся случаи, когда будет трудно производить непрерывную подачу на последующую стадию превращенных в пар ингредиентов при получении РС р либо s-PL. Кроме того, трудно будет проводить регулирование тех флуктуаций в составе превращенных в пар ингредиентов при получении РС р, которые сопровождают изменения марки продукта РС. С другой стороны, величина Vc/Fc может быть большей 100. Однако чрезмерно большие вместимости резервуаров являются нежелательными, поскольку необходимость хранения такого большого количества невелика с точки зрения эффективности получения, что в результате приведет скорее к появлению отходов, чем к улучшению эффективности получения, и поскольку чрезмерно продолжительное время выдерживания является нежелательным с точки зрения термостойкости.
Между прочим, данный резервуар для хранения при получении РС можно располагать только на одной стадии, выбираемой из стадии первого хранения при получении РС и стадии второго хранения при получении РС, или же можно располагать на каждой из двух стадий. На одной стадии хранения при получении РС можно располагать один резервуар для хранения при получении РС того типа, что описывался ранее, или же последовательно либо параллельно можно располагать два либо более чем два резервуара для хранения при получении РС. В том случае, если располагать будут два либо более чем два резервуара Vc в выражении (1) будет обозначать совокупную вместимость данных имеющихся резервуаров.
2. Стадия получения DPC
На стадии получения DPC стадию хранения при получении DPC, предназначенную для хранения DPC, полученного в результате проведения стадии перегонки при получении DPC, можно расположить после стадии перегонки при получении DPC, как это продемонстрировано на фиг.7. Наличие данной стадии хранения при получении DPC оказывает нижеследующее действие. Даже если проведение стадии получения DPC временно прекращать либо осуществлять периодически, DPC, хранящийся на данной стадии хранения при получении DPC, можно непрерывно подавать в качестве являющегося исходным материалом DPC на последующую стадию, то есть стадию получения РС, в результате чего РС можно получать непрерывно.
Вместимость резервуара для хранения при получении DPC, используемого на стадии хранения при получении DPC, можно определить, принимая во внимание время работы и время простаивания для стадии получения DPC. Говоря конкретно, резервуар предпочтительно характеризуется вместимостью, удовлетворяющей требование, представленное в нижеследующем выражении (2):
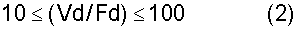
В выражении (2) Vd обозначает вместимость (м3) резервуара для хранения при получении DPC, а Fd обозначает расход (м3/час) дифенилкарбоната.
Если величина Vd/Fd будет меньше 10, то тогда появятся случаи, когда будет трудно производить непрерывную подачу DPC на последующую стадию. С другой стороны, величина Vd/Fd может быть большей 100. Однако чрезмерно большие вместимости резервуаров являются нежелательными, поскольку необходимость хранения такого большого количества невелика с точки зрения эффективности получения, что в результате приведет скорее к появлению отходов, чем к улучшению эффективности получения, и поскольку чрезмерно большие вместимости резервуаров являются нежелательными также и с точки зрения термостойкости.
Между прочим, можно располагать один резервуар для хранения при получении DPC того типа, что описывался ранее, или же последовательно либо параллельно можно располагать два либо более чем два резервуара для хранения при получении DPC. В том случае, если располагать будут два либо более чем два резервуара, Vd в выражении (2) будет обозначать совокупную вместимость данных имеющихся резервуаров.
3. Стадия получения ВРА
На стадии получения ВРА стадию хранения при получении ВРА, предназначенную для хранения смеси бисфенола А (ВРА) и фенола (PL), располагают после стадии удаления PL (стадии (е)), как это продемонстрировано на фиг.7, либо между стадией кристаллизации/выделения ВРА (стадией (с)) и стадией удаления PL (стадией (е)), несмотря на то, что второй случай на фиг.7 не продемонстрирован.
Наличие данной стадии хранения при получении ВРА оказывает нижеследующее действие. Даже если проведение любой одной стадии на стадии получения ВРА временно прекратить, а стадии, предшествующие остановленной стадии, проводить периодически, смесь, хранящуюся на данной стадии хранения при получении ВРА, можно подавать на стадию получения РС, благодаря чему РС можно получать непрерывно.
В частности, стадия кристаллизации/выделения ВРА (стадия (с)) имеет тенденцию приводить к образованию твердых отложений на тех частях использованного кристаллизатора, включающих кристаллизационную емкость и теплообменник, которые вступают в контакт с жидкостью. Таким образом, необходимо прекращать проведение данной стадии и осуществлять чистку через интервалы в несколько месяцев. По этой причине стадии, заключенные в диапазоне от стадии реакции при получении ВРА (стадии (а)) до стадии кристаллизации/выделения ВРА (стадии (с)), имеют тенденцию к тому, чтобы их проводили периодически. Следовательно, расположение стадии хранения при получении ВРА после стадии удаления PL (стадии (е)) либо в промежутке между стадией кристаллизации/выделения ВРА (стадией (с)) и стадией удаления PL (стадией (е)), несмотря на то, что второй случай на фигуре не продемонстрирован, делает возможным непрерывное проведение стадии получения РС, даже если стадии, заключенные в пределах от стадии реакции при получении ВРА (стадии (а)) до стадии кристаллизации/выделения ВРА (стадии (с)), и проводить периодически.
Примеры формы смеси, предназначенной для хранения на стадии хранения при получении ВРА, включают кристаллы аддукта ВРА и PL, суспензию, содержащую кристаллы аддукта BPA и PL, жидкую смесь ВРА и PL и тому подобное.
Смесь ВРА/PL в общем случае характеризуется составом, при котором уровень содержания ВРА находится в диапазоне 45-70 мас.%, а уровень содержания PL находится в диапазоне 55-30 мас.% По этой причине, если температура во время хранения будет составлять 0-95°С, то тогда аддукт будет кристаллическим. В том случае, если доля фенола в смеси будет высока, PL, который не будет находиться в форме аддукта с ВРА, будет переходить в расплавленное состояние тогда, когда температура хранения возрастет до 40°С либо более. В данном случае смесь представляет собой суспензию либо раствор. Кроме того, если температура хранения превысит 95°С, то тогда аддукт расплавится и, таким образом, перейдет в состояние расплава.
Температура хранения предпочтительно находится в диапазоне 45-150°С. Желательно, чтобы смесь ВРА/PL находилась бы в состоянии суспензии либо раствора. Для целей предотвращения разложения либо окрашивания ВРА предпочтительно выдерживать смесь при минимально возможной температуре. В данных условиях можно ингибировать образование 4-изопропенилфенола, который, как представляется, является веществом, вызывающим окрашивание и получающимся в результате разложения ВРА.
Также важно, чтобы атмосфера в резервуаре для хранения представляла бы собой атмосферу инертного газа, такого как газообразный азот, во избежание образования включений воздуха. Несмотря на то, что в качестве материала резервуара для хранения можно использовать обычную аустенитную нержавеющую сталь либо ферритную нержавеющую сталь, предпочитается использовать материал, восстановленный при растворении железа, что вызывает ослабление цветового тона. В частности, предпочтительным является материал стали, характеризующийся уровнем содержания хрома, равным 16% или более, и уровнем содержания углерода, равным 0,03% или более. Например, предпочитается использование SUS304, а не SUS316 и использование SUS316 либо SUS304, а не SUS316L либо SUS304L. Само собой разумеется, что более предпочтительными являются SUS309S и SUS310S, которые характеризуются более высоким уровнем содержания хрома.
Вместимость резервуара для хранения смеси ВРА/PL можно определить, принимая во внимание время работы и время простаивания для стадии получения ВРА. Говоря конкретно, вместимость резервуара предпочтительно удовлетворяет требованию, представленному в нижеследующем выражении (3):
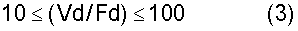
В выражении (3) Vb обозначает вместимость (м3) резервуара для хранения при получении BPA, а Fb обозначает расход (м3/час) бисфенола А, подаваемого на стадию полимеризации при получении РС.
Если величина Vb/Fb будет меньше 10, то тогда появятся случаи, когда будет трудно проводить стадию полимеризации при получении РС. С другой стороны, величина Vb/Fb может быть большей 1000. Однако чрезмерно большие вместимости резервуаров являются нежелательными, поскольку необходимость хранения такого большого количества невелика с точки зрения эффективности получения, что в результате приведет скорее к появлению отходов, чем к улучшению эффективности получения, и поскольку долговременное выдерживание является нежелательным также и с точки зрения качества.
Между прочим, можно располагать один резервуар для хранения данного типа или же последовательно либо параллельно можно располагать два либо более чем два резервуара для хранения. В том случае, если располагать будут два либо более чем два резервуара, Vb в выражении (3) будет обозначать совокупную вместимость данных имеющихся резервуаров.
Поскольку на стадии хранения при получении ВРА хранят аддукт, который находится в форме, показанной выше, реакция разложения ВРА имеет тенденцию протекать тогда, когда величина показателя рН будет смещена в кислотную сторону либо щелочную сторону. Для того чтобы этого не допустить, в резервуаре для хранения при получении ВРА, находящемся в промежутке между стадией кристаллизации/выделения ВРА (стадией (с)) и стадией полимеризации при получении РС, либо на стадии, предшествующей данным стадиям, предпочитается организовать стадию нейтрализации при получении ВРА (не показана). На стадии нейтрализации при получении ВРА можно нейтрализовать кислотный ингредиент либо основный ингредиент, присутствующие в смеси, в результате чего можно будет ингибировать прохождение разложения ВРА в смеси.
Переработка отработанного раствора
(Переработка остатка после перегонки PL (X2))
Остаток после перегонки PL (X2) содержит PL, DPC, BPA, олигомеры, полученные в результате прохождения конденсационной полимеризации между одной либо несколькими молекулами DPC и одной либо несколькими молекулами ВРА, и тому подобное. В их числе в больших количествах содержатся PL, BPA и DPC. Следовательно, для того чтобы эффективно использовать данные эффективные ингредиенты, остаток после перегонки PL (X2) подают на стадию перегонки либо стадию извлечения/перегонки на стадии получения DPC либо на стадию переработки маточной жидкости (стадию (g)) на стадии получения ВРА, как это продемонстрировано на фиг.8.
Если стадия получения DPC не будет включать стадию извлечения/перегонки, а остаток после перегонки PL (Х2) будут подавать на стадию перегонки на стадии получения DPC, то тогда остаток после перегонки PL (X2) специально будут подавать в колонну первой перегонки при получении DPC 6, как это продемонстрировано на фиг.1. В результате остаток после перегонки PL (X2) подвергают перегонке в колонне первой перегонки при получении DPC 6 и колонне второй перегонки при получении DPC 7, а PL и DPC извлекают. В их числе PL извлекают в виде одного компонента смешанного газа (F) в колонне первой перегонки при получении DPC 6, в то время как DPC извлекают в колонне второй перегонки при получении DPC 7.
Если стадия получения DPC будет включать стадию извлечения/перегонки, а остаток после перегонки PL (Х2) будут подавать на стадию извлечения/перегонки на стадии получения DPC, то тогда остаток после перегонки PL X2 специально будут подавать в колонну извлечения/перегонки при получении DPC 8, как это продемонстрировано на фиг.1. В результате остаток после перегонки PL (X2) будут подвергать перегонке в колонне извлечения/перегонки при получении DPC 8, а PL и DPC - извлекать и подавать в смесительную емкость 3. В их числе PL извлекают в виде одного компонента смешанного газа (F) в колонне первой перегонки при получении DPC 6, в то время как DPC извлекают в колонне второй перегонки при получении DPC 7.
Кроме того, если остаток после перегонки PL (X2) будут подавать на стадию переработки маточной жидкости (стадию (g)) на стадии получения ВРА, то тогда остаток после перегонки PL (X2) специально будут подавать в реактор переработки остатка 14, как это продемонстрировано на фиг.4. В результате PL как таковой отгоняют, в то время как другие ингредиенты разлагаются и в регенерирующем реакторе 15 опять образуют ВРА и другие соединения. Данные полученные соединения подают в реактор получения ВРА (не показан) на стадии получения ВРА. Таким образом, PL используют в качестве исходного материала, и ВРА движется совместно с синтезированным ВРА.
(Переработка остатка после перегонки (Х1) либо остатка после извлечения/перегонки (X1'))
На стадии получения DPC остаток после перегонки (Х1), полученный тогда, когда стадия получения DPC не будет включать стадию извлечения/перегонки, либо остаток после извлечения/перегонки (X1'), полученный тогда, когда стадия получения DPC будет включать стадию извлечения/перегонки, будет содержать примеси в виде производных DPC, такие как DPC, метилзамещенные производные DPC и бромзамещенные производные DPC, и другие примеси. Если остаток после перегонки PL (Х2) будут вводить на стадию перегонки либо стадию извлечения/перегонки, то тогда остаток (Х1) либо (Х1') дополнительно будет содержать ВРА, олигомеры, полученные в результате прохождения конденсационной полимеризации между одной либо несколькими молекулами DPC и одной либо несколькими молекулами ВРА, и другие примеси. В числе данных примесей в больших количествах содержатся ВРА и DPC. Следовательно, для того чтобы эффективно использовать данные эффективные ингредиенты, остаток после перегонки (Х1) либо остаток после извлечения/перегонки (X1') подают на стадию переработки маточной жидкости (стадию (g)) на стадии получения ВРА.
В данном случае остаток после перегонки (Х1) либо остаток после извлечения/перегонки (X1') специально подают в реактор переработки остатка 14, как это продемонстрировано на фиг.4. В результате ингредиенты разлагаются, а часть продуктов разложения в регенерирующем реакторе 15 превращается опять в ВРА. Полученные ВРА и фенол подают в реактор получения ВРА (не показан) на стадии получения ВРА.
(Остаток после перегонки PL (X2) и остаток после перегонки (Х1) либо остаток после извлечения/перегонки (Х1'))
Примеры способов переработки остатка после перегонки PL (X2), остатка после перегонки (Х1) и остатка после извлечения/перегонки (Х1') включают те, что были описаны выше. В их числе предпочтительным является способ, в котором остаток после перегонки PL (X2) подают на стадию перегонки при получении DPC либо на стадию извлечения/перегонки при получении DPC, а после этого остаток после перегонки при получении DPC (X1), получающийся в результате проведения стадии перегонки при получении DPC, либо остаток после извлечения/перегонки при получении DPC (X1') подают на стадию переработки маточной жидкости при получении ВРА (стадию (g)).
В результате проведения такой переработки остатков отработанные растворы, выпускаемые со стадии получения DPC, стадии получения ВРА и стадии получения РС, собирают воедино в один отработанный раствор (D7), получающийся в результате проведения стадии переработки маточной жидкости (стадии (g)) на стадии получения бисфенола А (ВРА). Таким образом, отработанные растворы, соответственно получающиеся в результате проведения трех стадий и в больших количествах содержащие органические соединения, можно собрать воедино, и количество всех выпускаемых отработанных растворов можно уменьшить. Следовательно, стадию переработки отработанного раствора (не показана) проводят эффективно, а нагрузка для стадии может быть уменьшена.
Вакуумное устройство
Перегонный аппарат, такой как перегонные колонны, и полимеризатор на стадии перегонки при получении DPC, стадии полимеризации при получении РС и стадии перегонки PL снабжают конденсатором для конденсации ингредиентов дистиллята, вакуумным устройством для уменьшения давления в системе и вакуумным трубопроводом, который соединяет конденсатор и вакуумное устройство. В отношении перегонного аппарата, описанного в качестве примера, пояснения приводятся далее. Как продемонстрировано на фиг.9, перегонный аппарат 41 снабжают конденсатором 42 для конденсации соединений дистиллята, таких как DPC и PL, вакуумным устройством 43 для уменьшения давления в системе и вакуумным трубопроводом 44, который соединяет конденсатор 42 с вакуумным устройством 43.
Данный перегонный аппарат 41 зачастую включает часть для кипячения в условиях флегмообразования. Устройства, которые составляют данную часть для кипячения в условиях флегмообразования, включают конденсатор 42 для конденсации соединений дистиллята, резервуар для конденсата 45 для сбора части конденсата, насос для подачи жидкости 46 для возврата конденсата в резервуаре для конденсата 45 в перегонный аппарат 41 и рефлюксный трубопровод 47, который соединяет насос для подачи жидкости 46 с перегонным аппаратом 41. Здесь и далее в настоящем документе данную часть для кипячения в условиях флегмообразования обозначают как "рефлюксный аппарат".
Вакуумное устройство 43 представляет собой устройство, которое используют для всасывания и нагнетания газа, присутствующего в перегонном аппарате 41 и рефлюксном аппарате, и, таким образом, создания в перегонном аппарате 41 состояния вакуума. Примеры данного вакуумного устройства 43 включают вакуумные насосы.
Вакуумный трубопровод 44, соединенный с данным вакуумным устройством 43, является наклоненным книзу от стороны конденсатора 42 к стороне вакуумного устройства 43. Желательно, чтобы в горизонтальных частях вакуумного трубопровода 44 части, имеющие данный наклон, были бы по возможности более длинными. На степень наклона особенных ограничений не накладывается до тех пор, пока угол наклона книзу, образуемый с горизонтальным направлением, будет больше 0° и не меньше 90°. Однако степень наклона книзу в желательном варианте составляет 1 см либо более, в более желательном варианте находится в диапазоне от 5 см до 1 м на 2 м по горизонтали в направлении вакуумного устройства. Более желательно, чтобы горизонтальные части, имеющие наклон, не имели бы части, которая была бы полностью горизонтальной либо поднималась бы кверху. Еще более желательно, чтобы наклон был бы постоянным по всему вакуумному трубопроводу 44. Термин "горизонтальные части" обозначает части, находящиеся в горизонтальном состоянии, и части, находящиеся в состоянии незначительного наклона по отношению к горизонтальному состоянию. Термин "полностью горизонтальные части" обозначает части, перпендикулярные к вертикальному направлению.
Наклонная часть может включать полностью горизонтальные части либо поднимающиеся части. Однако совокупная высота поднимающихся частей предпочтительно составляет 1 м либо менее, более предпочтительно 50 см либо менее, еще более предпочтительно 10 см либо менее. Наиболее предпочтительно, чтобы наклонная часть не включала бы таких частей. В том случае, если совокупная высота поднимающихся частей будет превышать 1 м, то появится возможность того, что тогда, когда дистиллят будет оставаться в поднимающихся частях, это может в результате привести к чрезмерно большим потерям давления и сделать невозможным проведение достаточного вакуумирования. Термин "поднимающиеся части" обозначает части, которые являются наклоненными кверху от стороны конденсатора 42 к стороне вакуумного устройства 43 противоположно наклону, описанному выше. В таких поднимающихся частях дистиллят склонен оставаться в виде жидкой либо твердой фазы. Если дистиллят будет оставаться, то это приведет в результате к потере давления. Чрезмерно большое количество остающегося дистиллята может привести к засорению самой трубы. Поэтому желательно, чтобы вакуумный трубопровод 44 не включал бы поднимающихся частей. По этой причине более желательно, чтобы вакуумный трубопровод был бы наклоненным только книзу от стороны конденсатора 42 к стороне вакуумного устройства 43.
Конденсатор 42 конденсирует PL, DPC и тому подобное. Конденсированный дистиллят выпускают из системы либо подают в резервуар для конденсата 45 в рефлюксном аппарате при подготовке с подачей в перегонный аппарат 41.
Давление в перегонной колонне 41 предпочтительно является пониженным давлением, более предпочтительно равным 1-200 торр, в особенности предпочтительно 5-100 торр. Если перегонная колонна 41 будет иметь такое внутреннее давление, то тогда давление в откачанном вакуумном трубопроводе 44, через который откачивают газ в перегонной колонне 41, в желательном варианте будет близким к давлению в перегонной колонне 41 либо меньшим его, а более желательно равным 1-100 торр.
Кроме того, трубопровод, проходящий от верха перегонной колонны 41 к конденсатору 42, в желательном варианте имеет конструкцию, которая удовлетворяет нижеследующим требованиям. Внутренний диаметр трубопровода в желательном варианте является таким, чтобы фактическая линейная скорость газа находилась бы в диапазоне 0,01-20 м/сек. Чем короче будет длина трубопровода, проходящего от верха перегонной колонны 41 к конденсатору 42, тем лучше. Его длина в желательном варианте составляет 10 м либо менее, в более желательном варианте 0 м. Кроме того, чем меньше будет количество изогнутых частей в трубопроводе, тем лучше. Их количество в желательном варианте равно 5 либо менее. С учетом данных требований наиболее желательно, чтобы конденсатор 42 располагали бы на верху перегонной колонны 41 в виде конденсатора для верхней части колонны. При удовлетворении данных требований в перегонной колонне 41 устанавливается более стабильное состояние вакуума, полученное при использовании вакуумного устройства 43.
Желательно, чтобы подачу газа, состоящего из превращенных в пар PL и DPC, проводили бы, создавая течение вниз через конденсатор 42. Также желательно, чтобы внутренняя часть и часть с выпускным отверстием у конденсатора 42 имела бы большой диаметр, что в результате приведет к получению низкой линейной скорости газа. В том случае, если линейная скорость газа будет высокой, это станет причиной падения давления, и появится возможность того, что в перегонном аппарате 41 нельзя будет сохранять состояние вакуума.
Желательно, чтобы туманоуловитель 48, предназначенный для улавливания тумана, располагали бы в промежутке между конденсатором 42 и вакуумным трубопроводом 44. Своей целью это имеет по возможности в большей степени предотвращение попадания тумана PL либо DPC в вакуумный трубопровод 44 и застаивания либо затвердевания его там.
Вакуумный трубопровод 44 в желательном варианте представляет собой устройство, предназначенное для нагревания и выдерживания его внутреннего пространства при температуре, не меньшей, чем температура плавления дистиллята. Примеры данного устройства включают устройства, в которых вакуумный трубопровод 44 имеет двухтрубную конструкцию или же конструкцию с паровым либо электрическим обогревом. В том случае, если дистиллят будет состоять из двух либо более чем двух ингредиентов, то тогда желательно, чтобы температура, при которой необходимо будет выдерживать внутреннее пространство трубопровода 44, не была бы более низкой по сравнению с температурой плавления соединения, характеризующегося температурой плавления, наиболее высокой в числе данных соединений. Несмотря на то, что внутреннее пространство вакуумного трубопровода 44 находится в состоянии вакуума, используемый в данном случае термин "температура плавления" обозначает температуру плавления в состоянии вакуума. Если внутреннее пространство вакуумного трубопровода 44 будут выдерживать при температуре, не меньшей, чем температура плавления дистиллята, то тогда PL либо DPC, которые не были сконденсированы в конденсаторе 42 и попали в вакуумный трубопровод 44, будут оставаться в жидком либо газообразном состоянии без затвердевания, в результате чего может быть дополнительно уменьшена вероятность того, что внутреннее пространство вакуумного трубопровода 44 может быть засорено. Поэтому с точки зрения проведения стадии внутреннее пространство вакуумного трубопровода 44 желательно выдерживать при температуре, не меньшей чем 80°С, что представляет собой температуру плавления DPC, и не большей, чем температура верха перегонного аппарата 41.
Желательно, чтобы вакуумный трубопровод 44 был бы снабжен, по меньшей мере, одним дренажным отверстием 49, обращенным книзу. Это обуславливается необходимостью того, что дистиллят, который в вакуумном трубопроводе 44 подвергся ожижению либо стал росой, нужно выпускать без застаивания в вакуумном трубопроводе 44. По меньшей мере, одно из таких дренажных отверстий 49 в желательном варианте располагают поблизости от той части вакуумного трубопровода 44, которая соединяется с вакуумным устройством 43. Причиной этого является нижеследующее. Поскольку вакуумный трубопровод 44 является наклоненным, выпуск дистиллята через отверстие, которое не располагается по возможности ближе к нижней части наклоненного трубопровода, в результате приводит к появлению возможности того, что дистиллят сможет оставаться в той части, которая располагается вне сферы действия отверстия. Данный выпуск жидкости можно проводить не во время вакуумирования при использовании вакуумного устройства 43, а после того, как работа всего аппарата будет остановлена. В том случае, если длина вакуумного трубопровода 44 будет превышать 3 м, желательно будет расположить дренажное отверстие 49 также и во внутренней части вакуумного трубопровода 44, поскольку это даст возможность легче предотвращать застаивание жидкости.
Кроме того, желательно, чтобы у вакуумного трубопровода 44 - в той его части, которая расположена на стороне конденсатора 42, - было бы предусмотрено наличие питающего отверстия 50, через которое можно было бы подавать нагретую текучую среду. Чем ближе будет позиция данного питающего отверстия 50 к конденсатору 42, тем более это будет желательно. Однако в том случае, если в промежутке между вакуумным трубопроводом 44 и конденсатором 42 будет присутствовать туманоуловитель 48, желательно, чтобы позиция питающего отверстия 50 располагалась бы не в промежутке между туманоуловителем 48 и конденсатором 42, а по возможности более близко к той части вакуумного трубопровода 44, которая будет соединена с туманоуловителем 48. В результате наличия питающего отверстия 50 нагретую текучую среду можно вводить через данное отверстие и выпускать через дренажное отверстие 49, благодаря чему можно проводить чистку вакуумного трубопровода 44. Поэтому желательно, чтобы часть, находящаяся в диапазоне от питающего отверстия 50 до дренажного отверстия 49, занимала бы по возможности более длинный участок вакуумного трубопровода 44. Вследствие наличия наклона более эффективной будет подача со стороны, соответствующей более высокой потенциальной энергии. Следовательно, желательно, чтобы питающее отверстие 50 на вакуумном трубопроводе 44 открывалось бы кверху.
Нагретая текучая среда представляет собой вещество, которое является текучей средой при температуре во внутреннем пространстве вакуумного трубопровода 44 и может представлять собой либо жидкость, либо газ. Примеры нагретой текучей среды включают водяной пар, PL, азот и тому подобное. Более желательно, чтобы нагретая текучая среда представляла бы собой водяной пар либо пар PL. Использование пара PL является еще более желательным, поскольку, даже если DPC и затвердеет в вакуумном трубопроводе 44, данную твердую фазу можно будет растворить. Нагретая текучая среда может состоять из одного из данных компонентов либо из смеси двух либо более чем двух данных компонентов. Однако желательно, чтобы нагретая текучая среда представляла бы ту среду, которая вряд ли бы вступала в реакцию с материалом вакуумного трубопровода 44, DPC и тому подобным в условиях температуры и давления в вакуумном трубопроводе 44. Более желательно, чтобы она представляла бы собой среду, которая совершенно бы не вступала в реакцию.
Желательно, чтобы вакуумный трубопровод 44 был бы снабжен клапанами 51 и 52, расположенными в его частях, соединенных с конденсатором 42, вакуумным устройством 43, туманоуловителем 48 и тому подобным. Это обуславливается тем, что при проведении чистки вакуумного трубопровода 44 под действием нагретой текучей среды, дозирование с использованием клапанов может предотвратить просачивание нагретой текучей среды из вакуумного трубопровода 44.
Также желательно, чтобы вакуумный трубопровод 44 был бы снабжен замораживающим конденсатором (не показан). Более желательно, чтобы параллельно были бы расположены два либо более чем два замораживающих конденсатора, что обеспечит возможность переключения. Наличие замораживающих конденсаторов (конденсатора) является желательным, поскольку можно бы принудительно отверждать и собирать ингредиенты дистиллята, которые не могут быть уловлены конденсатором 42, а в тех частях вакуумного трубопровода 44, которые располагаются по технологической схеме после замораживающих конденсаторов (конденсатора), подавлять образование засорения либо увеличение потерь давления.
В том случае, если из DPC будут отгонять низкокипящие соединения, такие как PL, то для того, чтобы ожижить газ, выпускаемый в качестве дистиллята из перегонного аппарата 41, эффективным будет также и использование двух либо более чем двух конденсаторов 42, соединенных последовательно. В данном случае температуры конденсаторов 42 предпочтительно регулируют таким образом, чтобы по мере того, как будет увеличиваться расстояние от перегонной колонны 41, температура бы постепенно уменьшалась. В частности, в первом конденсаторе 42 проводят регулировку таким образом, чтобы иметь температуру в диапазоне 80-150°С для того, чтобы уверенно сконденсировать высококипящие ингредиенты, такие как DPC, а для получающегося в результате конденсата организуют циркуляцию с подачей в перегонный аппарат 41. После этого несконденсированный газ, который не был подвергнут ожижению в первом конденсаторе 42, почти полностью ожижают во втором и последующих конденсаторах 42, где в результате регулировки имеют температуру 0-80°С. Конденсат, таким образом полученный в результате ожижения, частично подвергают кипячению в условиях флегмообразования с подачей в перегонный аппарат 41 по мере надобности, а оставшееся либо все количество конденсата выпускают в виде дистиллята. Наличие двух либо более чем двух конденсаторов 42 позволяет добиться нижеследующих преимуществ. Даже если в качестве дистиллята из перегонной колонны 41 будут выпускать большое количество DPC, DPC не будет ни затвердевать, ни вызывать засорение в конденсаторах 42, благодаря чему эксплуатация аппарата может происходить непрерывно, а перегонку можно стабильно проводить в течение длительного времени. Температура затвердевания DPC составляет 80°С, что превышает температуру затвердевания (40°С) PL. По этой причине, если в конденсаторе 42, функционирующем в условиях по температуре, подходящих для конденсации только одного обычного низкокипящего соединения, например PL, будет присутствовать большое количество DPC, то тогда появится возможность того, что внутри конденсатора 42 сможет затвердевать DPC. Следовательно, эффективной при предотвращении затвердевания и при стабильном выдерживании степени вакуумирования в перегонном аппарате 41 является методика, по которой два либо более чем два конденсатора 42 располагают и используют таким образом, чтобы DPC, который является высокоплавким соединением, принудительно бы конденсировали/удаляли перед тем, как будет сконденсирован несконденсированный газ. Кроме того, в том случае, если в дополнение к PL в отгоняемых низкокипящих соединениях будет присутствовать большое количество соединений, характеризующихся температурой кипения, более низкой по сравнению с температурой кипения PL, то тогда желательным будет использование таких конденсаторов 42 с конфигурацией многостадийной компоновки.
Далее со ссылкой на фиг.9 будут приведены пояснения в отношении описанного выше рефлюксного аппарата. В результате использования данного рефлюксного аппарата можно увеличить эффективность разделения в результате перегонки при одновременном подавлении превращения в пар высококипящих ингредиентов.
Горизонтальная часть трубопровода для кипячения в условиях флегмообразования 47, предназначенная для кипячения конденсата в условиях флегмообразования в резервуаре для конденсата с подачей в перегонный аппарат 41 при использовании насоса для подачи жидкости 46, в желательном варианте является наклоненной книзу от стороны перегонного аппарата 41 к стороне насоса для подачи жидкости 46. В желательном варианте данная горизонтальная часть, имеющая наклон, не включает какой-либо части, которая была бы полностью горизонтальной либо поднималась бы кверху. Более желательно, чтобы наклон был бы постоянным. В данном случае степень наклона книзу в желательном варианте составляет 1 см либо более, в более желательном варианте находится в диапазоне от 5 см до 1 м на 2 м по горизонтали в направлении насоса для подачи жидкости 46. Если будут иметься полностью горизонтальные части либо поднимающиеся части, то тогда будет необходимо, чтобы разница по высоте, образующаяся в результате наличия наклона, была бы больше, чем совокупная разница по высоте для поднимающихся частей, а совокупная высота поднимающихся частей в желательном варианте составляла бы 1 м либо менее, в более желательном варианте 10 см либо менее. Термин "поднимающиеся части" обозначает части, которые являются наклоненными кверху от стороны перегонного аппарата 41 к стороне насоса для подачи жидкости 46 противоположно наклону, описанному выше.
Та часть трубопровода для кипячения в условиях флегмообразования 47, которая располагается в промежутке между горизонтальной частью, характеризующейся наличием наклона, и насосом для подачи жидкости 46, может включать часть, которая является вертикальной либо является почти вертикально наклоненной, как это продемонстрировано на фиг.9, несмотря на то, что это зависит от позиции насоса для подачи жидкости 46. В данном случае дренажные отверстия 53 и 54 можно располагать в тех частях трубопровода, которые находятся в окрестности отверстия для всасывания и выпускного отверстия соответственно, у насоса для подачи жидкости 46 в целях выпуска жидкости, застаивающейся до и после насоса для подачи жидкости 46.
Рефлюксный трубопровод 47 в желательном варианте характеризуется наличием устройства для нагревания и выдерживания его внутреннего пространства при температуре, не меньшей, чем температура плавления дистиллята. В том случае, если дистиллят будет состоять из двух либо более чем двух ингредиентов, то тогда желательным будет нагревание и выдерживание внутреннего пространства трубопровода 47 при температуре, не меньшей, чем температура плавления соединения, характеризующегося температурой плавления, наиболее высокой в числе данных соединений. Если рефлюксный трубопровод выдерживают при температуре, не меньшей, чем температура плавления дистиллята, то тогда дистиллят будет оставаться в жидком либо газообразном состоянии без затвердевания, в результате чего может быть дополнительно уменьшена вероятность того, что внутреннее пространство может быть засорено дистиллятом. Примеры устройства для такого нагревания/выдерживания температуры включают устройства, в которых трубопровод имеет двухтрубную конструкцию или же конструкцию с паровым либо электрическим обогревом.
Желательно, чтобы данный рефлюксный трубопровод 47 имел бы питающее отверстие 55, расположенное на той его части, которая будет близко находиться от перегонной колонны 41. В том случае, если функционирование всего аппарата будет остановлено, то тогда нагретую текучую среду будут вводить через данное питающее отверстие 55 и выпускать через дренажное отверстие 54, благодаря чему можно будет проводить чистку рефлюксного трубопровода 47. В данном случае желательно, чтобы клапан 56 был бы расположен в промежутке между рефлюксным трубопроводом 47 и перегонным аппаратом 41 для того, чтобы предотвратить перетекание нагретой текучей среды в перегонный аппарат 41.
Между прочим, перегонный аппарат для выделения PL либо DPC в качестве дистиллята, такой как те, что описывались выше, можно использовать для извлечения PL либо DPC, например, на стадии перегонки при получении DPC, на которой из DPC удаляют примеси, включающие PL, для того, чтобы извлечь очищенный DPC, или же на стадии полимеризации при получении РС либо на стадии перегонки PL на стадии получения РС, на которой РС получают в результате проведения полимеризации за счет прохождения реакции между DPC и ВРА в вакууме при одновременном извлечении PL, полученного в качестве побочного продукта (s-PL). Перегонный аппарат, кроме того, является подходящим для использования на стадиях, на которых PL либо DPC точно так же выделяют в качестве дистиллята.
Кроме того, несмотря на то, что рефлюксный аппарат, PL либо DPC, такой как описанный выше, можно использовать на стадии, описанной выше в качестве примера, на которой PL и DPC, выпускаемые в качестве дистиллята из перегонного аппарата 41, возвращают в перегонный аппарат 41 для подавления превращения в пар высококипящих ингредиентов, содержащихся в подаваемых PL и DPC, и, таким образом, для увеличения эффективности разделения в результате проведения перегонки, он, кроме того, является подходящим и для использования на стадиях, на которых кипячение в условиях флегмообразования точно так же является необходимым. Между прочим, высококипящие ингредиенты можно по раздельности выпускать через нижнюю часть перегонного аппарата 41.
В том случае, если данные аппараты будут использовать для получения DPC, то тогда в желательном варианте рефлюксный трубопровод 47 - в той его части, которая является отличной от горизонтальной части, имеющей наклон, - будет иметь выпускное отверстие 57 для выпуска из системы DPC.
В результате использования описанных выше данных аппаратов соответственно, на данных стадиях засорение трубы, если такое произойдет, можно будет легко устранить в результате использования способа чистки, по которому через питающие отверстия 50 и 55, расположенные на соответствующем аппарате, подают нагретую текучую среду.
Примеры
Пример 1
Далее изобретение будет описываться более подробно в связи с переработкой s-PL.
Получение обезвоженного s-PL
Стадия полимеризации при получении РС
В атмосфере газообразного азота ВРА (произведенный в компании Mitsubishi Chemical Corp.; ВРА, полученный в справочном примере 2, приведенном далее), подаваемый при расходе, равном 34,3 кг/час, перемешивали в расплаве при 130°С с DPC (произведенным в компании Mitsubishi Chemical Corp.; DPC, полученным в справочном примере 1, приведенном далее), подаваемым при расходе, равном 33,5 кг/час. Данную смесь непрерывно подавали через трубу для ввода исходного материала, нагреваемую до 130°С, в первый вертикальный реактор полимеризации с перемешиванием, в которой в результате регулировки выдерживали температуру 210°С в атмосфере азота при обычном давлении. Уровень жидкости выдерживали постоянным, проводя регулировку степени открытости клапана, расположенного на линии выпуска полимера, соединенной с нижней частью емкости, таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам. Кроме того, одновременно с инициированием подачи смеси исходных материалов в качестве катализатора начинали непрерывно подавать карбонат цезия в форме водного раствора при расходе, равном 0,5·10-6 моль при расчете на один моль ВРА. После этого жидкую полимеризационную реакционную смесь, выпускаемую из нижней части емкости, непрерывно последовательно подавали во второй, третий и четвертый вертикальные реакторы полимеризации и пятый горизонтальный полимеризатор. Во время прохождения реакции уровень жидкости в каждой емкости регулировали таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам, и одновременно с этим отгоняли PL, полученный в качестве побочного продукта. Каждый из газов, получаемых в результате выпаривания и выпускаемых соответственно из емкостей в диапазоне от первого до третьего реакторов полимеризации, конденсировали и ожижали при использовании многостадийного конденсатора и для части конденсата в результате кипячения в условиях флегмообразования проводили подачу в реактор полимеризации, а оставшуюся часть извлекали и хранили в резервуаре для s-PL. С другой стороны, каждый из газов, полученных в результате выпаривания и выпускаемых из четвертого и пятого полимеризаторов, отверждали при использовании одного из двух замораживающих конденсаторов, расположенных параллельно. Получающуюся в результате твердую фазу расплавляли в результате переключения на другой замораживающий конденсатор, извлекали и хранили в резервуаре для s-PL.
Условия проведения для каждого реактора полимеризации представляли собой нижеследующее: первый реактор полимеризации (210°C, 100 торр), второй реактор полимеризации (240°С, 15 торр), третий реактор полимеризации (260°С, 0,5 торр) и четвертый реактор полимеризации (270°С, 0,5 торр). Кроме того, производительность при получении поликарбоната составляла 38,3 кг/час. Система функционировала в течение 400 часов.
Получающийся в результате полимер в расплавленном состоянии вводили в двухчервячный экструдер (произведенный в компании Kobe Steel, Ltd.; диаметр червяка 0,046 м; L/D=40,2) и сюда же непрерывно добавляли бутил-п-толуолсульфонат в количестве 5 ч/млн при расчете на количество поликарбоната. Бутил-п-толуолсульфонат находился в форме маточной смеси, полученной в результате диспергирования маточного раствора в хлопьях поликарбоната при использовании смесителя, и его подавали в экструдер в атмосфере азота при использовании массового питателя. Полимер подвергали гранулированию. Полученный поликарбонат характеризовался средневязкостной молекулярной массой (Mv), равной 21000, и начальным цветовым тоном (YI), соответствующим 1,7.
Измерение средневязкостной молекулярной массы (Mv)
Для измерения удельной вязкости (ηsp) при помощи вискозиметра Убеллоде при температуре 20°С использовали раствор в метиленхлориде, характеризующийся концентрацией РС (С), равной 0,6 г/дл, а молекулярную массу рассчитывали по значению вязкости при использовании следующих далее двух уравнений:
ηsp/С=[η](1+0,28ηsp)
[η]=1,23· 10-4(Mv)0,83
Измерение начального цветового тона (YI)
РС высушивали при 120°С в течение 6 часов в атмосфере азота, а после этого при использовании литьевой машины J-100, произведенной в компании The Japan Steel Works, Ltd., по способу литьевого формования при 360°С из него получали кусок, имеющий толщину 3 мм. Данный кусок исследовали при использовании прибора SC-1, произведенного в компании Suga Test Instruments Co., Ltd., получая значение YI (чем больше будет значение YI, тем больше РС будет окрашен).
Стадия очистки s-PL
Анализировали s-PL, извлекаемый на стадии полимеризации при производительности, равной 30,2 кг/час. В результате обнаружили 5,0 мас.% DPC, 0,5 мас.% ВРА, 0,3 мас.% олигомеров и 0,3 мас.% воды.
Данный s-PL непрерывно очищали при использовании следующих двух перегонных колонн. Колонну первой перегонки PL эксплуатировали при 200 торр и флегмовом числе 2, благодаря чему присутствующую воду отгоняли совместно с частью PL. Кубовый остаток подавали в колонну второй перегонки PL. Низкокипящий дистиллят при получении РС, выпускаемый из колонны первой перегонки при получении РС, характеризовался концентрацией фенола, приблизительно равной 90 мас.%. После этого колонну второй перегонки PL эксплуатировали при 50 торр и флегмовом числе 0,5 и получали очищенный s-PL через верх колонны при производительности, приблизительно равной 27 кг/час. С другой стороны, жидкую PL-содержащую смесь, содержащую DPC, BPA и олигомеры в количествах, равных 67 мас.%, 7 мас.% и 4 мас.% соответственно, непрерывно выпускали в качестве кубового остатка при производительности, приблизительно равной 2,2 кг/час.
Экспериментальный пример 1
Получение DPC
Сначала будет приведено детальное разъяснение в отношении способа, по которому обезвоженный s-PL, выпускаемый в качестве дистиллята из колонны второй перегонки PL, продемонстрированной на фиг.5, использовали в качестве исходного материала для DPC при получении DPC, в то время как низкокипящий дистиллят при получении РС (D6), получающийся в результате обезвоживания s-PL в колонне первой перегонки PL 30, отправляли на рецикл на стадию промывки DPC.
Стадия реакции при получении DPC/стадия дегидрохлорирования
Стадию получения DPC проводили в соответствии со схемой технологического процесса, продемонстрированной на фиг.1. Между прочим, использованный реактор получения DPC 1 состоял из двух реакторов, соединенных последовательно.
Очищенный s-PL в расплавленном состоянии, характеризующемся температурой 50°С, при расходе, равном 30,0 кг/час (0,32 кмоль/час), непрерывно подавали в первый реактор получения DPC совместно с PL, содержащим в качестве катализатора пиридин, полученным в результате обезвоживания низкокипящих соединений, выпускаемых в качестве дистиллята из описанной далее колонны перегонки низкокипящих соединений. При проведении такой подачи очищенный s-PL нагревали до 150°С. В первый реактор получения DPC при достаточном перемешивании непрерывно подавали газообразный фосген (CDC) при расходе, равном 3,56 нм3/час (0,16 кмоль/час). Реакционную смесь, которая вытекала из первого реактора получения DPC и содержала фазу смеси газа/жидкости, подавали во второй реактор получения DPC через перепускную трубу. Для содержимого во втором реакторе получения DPC при достаточном перемешивании также проводили регулировку так, чтобы выдерживать температуру 150°С. Жидкую реакционную смесь подавали в колонну дегидрохлорирования 2. В колонне дегидрохлорирования 2 при 160°С осуществляли противоточное введение в контакт при использовании газообразного азота, для того чтобы обеспечить полное введение в реакцию фенилхлорформиата, полученного в качестве промежуточного соединения, и PL. Дегидрохлорированную жидкость b, характеризующуюся концентрацией DPC, приблизительно равной 89 мас.%, непрерывно выпускали из куба колонны дегидрохлорирования 2. Почти 100% подаваемого фосгена превращали в DPC. С другой стороны, выпускаемые газы, получающиеся в результате проведения синтеза DPC (D1; реакционный газ, выпускаемый из второго реактора получения DPC, и азотсодержащий газ, выпускаемый из колонны дегидрохлорирования 2), перемешивали друг с другом, а после этого охлаждали до 10°С. Получающийся в результате конденсат возвращали во второй реактор получения DPC, в то время как для хлористого водорода, полученного в виде несконденсированного газа, проводили нейтрализацию под действием водного щелочного раствора и выпуск.
Стадия промывки DPC/стадия водной промывки DPC
Полученную дегидрохлорированную жидкость, b, подавали в смесительную емкость 3 совместно с извлеченной жидкостью, содержащей DPC, d, извлеченной из описанной далее колонны извлечения/перегонки при получении DPC 8. После этого смесь подавали в емкость щелочной нейтрализации 4, снабженную тефлоновой футеровкой. В нейтрализационную емкость 4 подавали водный раствор гидроксида натрия с концентрацией, приблизительно равной 5 мас.% (жидкость, полученную в результате перемешивания водного раствора гидроксида натрия с концентрацией 25 мас.% с водной фазой, выделенной после проведения последующей стадии водной промывки, и с низкокипящим дистиллятом при получении РС, полученным на описанной выше стадии очистки s-PL). Содержимое перемешивали при 80°С в течение приблизительно 10 минут, а после этого проводили регулировку для того, чтобы получить значение рН, равное 8,5. Данную смесь оставляли отстаиваться для разделения и отделенную органическую фазу перепускали в емкость водной промывки 5. С другой стороны, водную фазу, остающуюся после разделения (которая содержит PL и поваренную соль), вводили в контакт с водяным паром, в результате чего присутствующий PL почти полностью извлекали в качестве низкокипящего дистиллята. Данный дистиллят подавали в емкость водной промывки 5 на последующей стадии. В емкости водной промывки 5 органическую фазу промывали, используя теплую воду в количестве, приблизительно равном 30 мас.% при расчете на количество органической фазы. Водную фазу (которую отправляли на рецикл в емкость нейтрализации/перемешивания) выделяли и получали жидкость, подвергнутую водной промывке, f, которая представляла собой "сырой" DPC (содержащий воду, пиридиновый катализатор и PL).
Стадия перегонки при получении DPC/стадия перегонки низкокипящих соединений
После этого жидкость, подвергнутую водной промывке, f, и водный раствор гидроксида натрия с концентрацией 0,1 н. непрерывно подавали на среднюю тарелку колонны первой перегонки при получении DPC 6 при расходах, приблизительно равных 42 кг/час и 70 мл/час соответственно. В качестве данной колонны первой перегонки при получении DPC 6 использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 150 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 220°С, температуры верха колонны 80-100°С и флегмового числа 1 для отгонки смешанного газа F, содержащего соединения, характеризующиеся температурой кипения, более низкой по сравнению с температурой кипения DPC, то есть воду, пиридиновый катализатор и не вступивший в реакцию PL. После обезвоживания смешанного газа F часть газа отводили, а остальное количество подавали в первый реактор получения DPC. С другой стороны, остаток после первой перегонки g, в основном состоящий из DPC, выпускали из куба колонны при производительности, приблизительно равной 37 кг/час. Воду в нем обнаружить было нельзя (10 ч/млн либо менее), нельзя было обнаружить и уровни содержания пиридина и PL (1 ч/млн либо менее) и 50 ч/млн соответственно.
Стадия перегонки при получении DPC/стадия перегонки высококипящих соединений
Кроме того, остаток после первой перегонки g непрерывно подавали в колонну второй перегонки при получении DPC 7. В качестве данной колонны второй перегонки при получении DPC 7 использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 200 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны, приблизительно равной 180°С, флегмового числа 0,5 и доли дистиллята, приблизительно равной 90%. В результате через верх колонны получали очищенный DPC при производительности, приблизительно равной 33,5 кг/час, а остаток после перегонки при получении DPC (X1), который представлял собой высококипящие соединения, отводили из куба колонны при производительности, приблизительно равной 4 кг/час. Данный очищенный DPC представлял собой высокочистый продукт, содержащий PL в количестве 80 ч/млн.
Стадия извлечения/перегонки при получении DPC
Кроме того, остаток после перегонки при получении DPC (X1), отводимый из куба колонны перегонки высококипящих соединений, одновременно подавали в колонну извлечения/перегонки при получении DPC 8 для проведения непрерывной перегонки при нижеследующих условиях. Извлеченную жидкость, содержащую DPC, d, извлекаемую через верх колонны при производительности, приблизительно равной 3,5 кг/час, отправляли на рецикл в смесительную емкость 3, в то время как кубовый остаток из колонны извлечения/перегонки при получении DPC 8 непрерывно отводили при производительности, приблизительно равной 0,2 кг/час. Что касается условий извлечения/перегонки при получении DPC, то использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 100 мм и высоту 3,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны 180°С и флегмового числа 0,5. В остатке после извлечения/перегонки при получении DPC (X1'), который представлял собой кубовый остаток из колонны извлечения/перегонки при получении DPC 8, обнаружили алкилзамещенные производные DPC и бромзамещенные производные DPC в количествах, равных приблизительно 7000 ч/млн (мас.) и приблизительно 800 ч/млн (мас.) соответственно.
Справочный пример 1
Получение DPC
DPC получали по тому же самому способу, что и в экспериментальном примере 1, за исключением того, что вместо очищенного s-PL использовали коммерческий PL (произведенный в компании Mitsubishi Chemical Corp.).
Результаты
Удостоверились в том, что, даже если использовали s-PL, получали тот же самый выход, что и в случае использования коммерческого PL, и эффективность получения была обеспечена.
Сравнительный пример 1
Получение DPC
DPC получали по тому же самому способу, что и в экспериментальном примере 1, за исключением того, что использовали s-PL (уровень содержания воды 0,3 мас.%), полученный в результате обхода по байпасу колонны первой перегонки PL на стадии очистки s-PL. В результате вследствие прохождения гидролиза на стадии реакции для DPC концентрация DPC в дегидрохлорированной жидкости b уменьшалась до 86 мас.%, что в результате приводило к значительному ухудшению эффективности получения.
Справочный пример 2
Получение ВРА
Стадия получения ВРА
ВРА получали в соответствии со схемами технологических процессов, продемонстрированными на фиг.2-4, следующим образом. В реактор получения ВРА проточного типа, снабженный термостатом, в количестве 60 л помещали набивку в виде кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.), в которой 15% сульфогрупп было нейтрализовано под действием 4-пиридинэтантиола. В данный реактор получения ВРА при температуре 80°С и расходе 68,2 кг/час вводили жидкую смесь PL и ацетона с молярным соотношением 10:1, обеспечивая протекание реакции между ними. Степень превращения ацетона составляла 80%. После того как низкокипящие соединения (не вступивший в реакцию ацетон, воду и часть PL) при производительности, равной 5,1 кг/час, удаляли, реакционную смесь охлаждали до 50°С, добиваясь осаждения кристаллов аддукта. Данную смесь отфильтровывали для ее разделения на кристаллы аддукта и маточную жидкость. Производительности при их получении составляли 16,5 кг/час и 46,5 кг/час соответственно. Десять массовых процентов данной маточной жидкости подавали на стадию переработки маточной жидкости, тогда как для остальной маточной жидкости как части исходного материала, вводимого в реактор получения ВРА, проводили циркуляцию.
Таким образом полученные кристаллы аддукта повторно растворяли в PL, подаваемом при расходе, равном 27,2 кг/час. Получающийся в результате раствор охлаждали до 50°С, добиваясь осаждения кристаллов, а после этого отфильтровывали для отделения кристаллов аддукта (11,3 кг/час) от маточной жидкости (32,5 кг/час). Выделенные кристаллы нагревали до 180°С при пониженном давлении 0,3 мм ртутного столба для удаления PL. Таким образом, при производительности, равной 7,7 кг/час, получали ВРА, характеризующийся степенью чистоты, равной 99,95% либо более.
Маточную жидкость, подаваемую на стадию переработки маточной жидкости, подвергали переработке с использованием испарителя PL, продемонстрированного на фиг.4, для отгонки части PL и концентрирования маточной жидкости. После этого к маточной жидкости добавляли 0,1 мас.% гидроксида натрия и затем маточную жидкость вводили в нижнюю часть реактора переработки остатка 14, в котором в результате регулировки выдерживали пониженное давление 50 мм ртутного столба и температуру 210°С. Данный реактор эксплуатировали при таких условиях, чтобы выдерживать в нижней части постоянство уровня жидкости (время пребывания 1 час), а нижний продукт в реакторе переработки остатка 14 отводили из системы при производительности, равной 0,5 кг/час. Кроме того, дистиллят, выпускаемый через верх реактора переработки остатка 14, перемешивали с PL, а данную смесь при расходе, равном 4,2 кг/час, вводили в регенерирующий реактор проточного типа 15, снабженный набивкой в виде 4 л кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.). Реакцию для смеси проводили в условиях 80°С. Для полученной жидкой реакционной смеси проводили циркуляцию с подачей в реактор получения ВРА, использованный на начальной стадии.
Низкокипящие соединения (не вступивший в реакцию ацетон, воду и часть PL), полученные в результате выделения из реакционной смеси, вводили в колонну извлечения PL при расходе, равном 5,1 кг/час. Одновременно с этим через верх колонны подавали этилбензол (добавка для разрушения азеотропа). В результате через верх колонны извлечения PL при производительности, равной 2,4 кг/час, выпускали жидкую смесь ацетона, воды и этилбензола, в то время как из куба колонны при производительности, равной 3,5 кг/час, выпускали PL. Кроме того, дистиллят (ацетон, воду и этилбензол), выпускаемый из верха колонны извлечения PL, вводили в колонну извлечения ацетона. Ацетон выпускали из верха колонны извлечения ацетона при производительности, равной 0,7 кг/час, в то время как смесь воды и этилбензола выпускали через куб колонны при производительности, равной 1,6 кг/час. Для PL, полученного через куб колонны извлечения PL, и ацетона, полученного через верх колонны извлечения ацетона, как части исходных материалов, вводимого в реактор синтеза, проводили циркуляцию.
Кроме того, ацетон и очищенный PL дополнительно подавали в реактор синтеза в количествах, соответствующих количествам, отводимым из системы, и количеству полученного ВРА. А именно, ацетон и очищенный PL подавали при расходах, равных 2,9 кг/час и 15 кг/час соответственно, обеспечивая непрерывное проведение реакции синтеза ВРА. Таким образом, вся система функционировала при непрерывном получении ВРА. ВРА, полученный в данном случае, вводили на описанную выше стадию полимеризации при получении РС, получая РС.
Использованный выше очищенный PL представлял собой вещество, полученное в результате переработки коммерческого PL для промышленного использования (концентрация воды 0,1 мас.%; концентрация примесей 0,05 мас.%; концентрация гидроксиацетона 20 ч/млн) за счет введения в контакт с материалом DIAION SK-104H, торговое наименование смолы, произведенной в компании Mitsubishi Chemical Corp., при 80°С в течение времени контактирования продолжительностью 50 минут, а после этого перегонки PL в перегонной колонне, характеризующейся температурой куба колонны 175°С и давлением верха колонны 560 мм ртутного столба, при извлечении PL через верх колонны. Уровень содержания гидроксиацетона в данном очищенном PL составлял 1 ч/млн либо менее.
Экспериментальный пример 2
Получение ВРА
Далее приводятся пояснения в отношении способа, в котором обезвоженный s-PL, выпускаемый в качестве дистиллята из колонны второй перегонки PL, продемонстрированной на фиг.5, использовали в качестве исходного материала для ВРА при получении ВРА, в то время как низкокипящий дистиллят при получении РС (D6), получающийся в результате обезвоживания s-PL, возвращали на стадию разделения ВРА/воды.
Низкокипящие соединения (не вступивший в реакцию ацетон, воду и часть PL), полученные в результате разделения реакционной смеси на стадии получения ВРА (справочный пример 2) (5,1 кг/час), перемешивали с низкокипящим дистиллятом, полученным через верх колонны первой перегонки на описанной выше стадии очистки s-PL. Данную смесь вводили в колонну извлечения PL и одновременно с этим через верх колонны проводили подачу этилбензола (добавки для разрушения азеотропа). В результате жидкую смесь ацетона, воды и этилбензола выпускали через верх колонны извлечения PL при производительности, равной 2,5 кг/час, в то время как PL выпускали из куба колонны при производительности, равной 4,0 кг/час. Кроме того, дистиллят (ацетон, воду и этилбензол), выпускаемые через верх колонны извлечения PL, вводили в колонну извлечения ацетона. Ацетон выпускали из верха колонны извлечения ацетона при производительности, равной 0,7 кг/час, в то время как смесь воды и этилбензола выпускали из куба колонны при производительности, равной 1,7 кг/час. Для PL, полученного через куб колонны извлечения PL, и ацетона, полученного через верх колонны извлечения ацетона, как части исходных материалов, вводимого в реактор синтеза, проводили циркуляцию.
Кроме того, ацетон и PL, который представлял собой очищенный s-PL, полученный через верх колонны второй перегонки PL на описанной выше стадии перегонки PL, дополнительно подавали в реактор получения ВРА в количествах, соответствующих количествам, отводимым из системы, и количеству полученного ВРА. А именно, ацетон и PL подавали при расходах 2,9 кг/час и 14,5 кг/час соответственно, обеспечивая непрерывное проведение реакции синтеза. Таким образом, вся система функционировала, обеспечивая непрерывное получение ВРА. Кроме того, ВРА, полученный в данном случае, вводили на описную выше стадию полимеризации, получая РС.
В результате вследствие отправления низкокипящего дистиллята, полученного через верх колонны первой перегонки на стадии очистки s-PL, на рецикл на существующую стадию на стадии получения ВРА почти все количество PL, содержащегося в низкокипящем дистилляте, можно эффективно извлекать в качестве исходного материала при получении ВРА. При проведении описанной выше операции ВРА и РС получали по способу, описанному выше. В результате полученным ВРА и РС проблема, связанная с качеством, совершенно несвойственна.
Сравнительный пример 2
Получение ВРА
ВРА получали по тому же самому способу, что и в экспериментальном примере 2, за исключением того, что вместо очищенного s-PL использовали s-PL (уровень содержания воды 0,3 мас.%, полученный в результате обхода по байпасу колонны первой перегонки PL на стадии очистки s-PL. В результате наблюдали уменьшение выхода ВРА. Причина этого, как представляется, заключается в том, что присутствующая вода вызывала уменьшение активности ионообменной смолы как катализатора реакции.
Сравнительный пример 3
Получение ВРА
ВРА получали по тому же самому способу, что и в справочном примере 2, за исключением того, что вместо очищенного PL в справочном примере 2 использовали коммерческий PL для промышленного использования. В результате выход ВРА постепенно уменьшался. Удостоверились в том, что присутствующий гидроксиацетон вызывал уменьшение каталитической активности.
Пример 2
При использовании экспериментальных примеров далее приводятся пояснения по переработке превращенных в пар ингредиентов при получении РС на стадии полимеризации при получении РС для случая переработки s-PL 2.
Пример получения DPC (1)
Пример, в котором DPC получают из коммерческого PL и CDC, продемонстрирован далее.
Стадия реакции
Во время непрерывной подачи в реактор расплавленного коммерческого PL и пиридинового катализатора при перемешивании при 150°С проводили непрерывную подачу газообразного фосгена. Газообразный хлористый водород, полученный в качестве побочного продукта в результате фосгенирования, охлаждали до 10°С. Получающийся в результате конденсат возвращали в реактор, а для несконденсировавшегося газа проводили нейтрализацию, используя водный щелочной раствор, и после этого выпуск. С другой стороны, жидкую реакционную смесь, содержащую приблизительно 91 мас.% DPC, непрерывно выпускали из реактора. Степень превращения фосгена на стадии реакции составляла почти что 100%.
Стадия промывки
Жидкую реакционную смесь и водный раствор гидроксида натрия с концентрацией, приблизительно равной 5 мас.%, подавали в емкость нейтрализации/перемешивания, снабженную тефлоновой футеровкой. Для содержимого проводили перемешивание при 80°С в течение приблизительно 10 минут и регулировку, обеспечивающую выдерживание значения рН, равного 8,5. После проведения нейтрализации органическую фазу отделяли, используя отстаивание, а после этого перепускали в емкость водной промывки/перемешивания. В емкости водной промывки/перемешивания органическую фазу промывали, используя теплую воду в количестве, приблизительно равном 30 мас.% при расчете на количество органической фазы. Водную фазу отделяли и получали "сырой" DPC (содержащий 1 мас.% воды, 2 мас.% пиридина, 8 мас.% PL и 89 мас.% DPC).
Стадия перегонки низкокипящих соединений
После этого "сырой" DPC непрерывно подавали на среднюю тарелку колонны перегонки низкокипящих соединений при расходе, приблизительно равном 30 кг/час. В качестве данной колонны перегонки низкокипящих соединений использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 150 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 220°С, температуры верха колонны 80-100°С, температуры средней тарелки 160°С и флегмового числа 1 для отгонки соединений, характеризующихся температурой кипения, более низкой по сравнению с температурой кипения DPC, то есть воды, пиридина и PL. Из куба колонны при производительности, приблизительно равной 26 кг/час, непрерывно выпускали DPC (уровни содержания воды 10 ч/млн (мас.) либо менее; пиридина 1 ч/млн (мас.) либо менее; PL 50 ч/млн (мас.)).
Стадия перегонки высококипящих соединений
Кроме того, данный DPC (кубовый остаток из колонны перегонки низкокипящих соединений) непрерывно подавали в колонну перегонки высококипящих соединений. В качестве данной колонны перегонки высококипящих соединений использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 200 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны, приблизительно равной 180°С, и флегмового числа 0,5. В результате через верх колонны при производительности, приблизительно равной 23,5 кг/час, получали очищенный DPC, а высококипящие соединения (DPC, содержащий приблизительно 350 ч/млн (мас.) алкилзамещенных производных DPC и приблизительно 40 ч/млн (мас.) бромзамещенных производных DPC) отводили из куба колонны при производительности, приблизительно равной 2,5 кг/час. Очищенный DPC представлял собой высокочистый продукт, содержащий PL в количестве, равном 80 ч/млн (мас.).
Пример получения DPC (2)
Пример, в котором DPC получали из коммерческого PL и диметилкарбоната, продемонстрирован далее.
Стадия реакции
Жидкий исходный материал, содержащий коммерческий PL, диметилкарбонат и тетрафеноксититан в качестве катализатора, подавали при расходе, равном 600 г/час (диметилкарбонат 390 г/час; PL 200 г/час; тетрафеноксититан 0,5 г/час), на десятую тарелку, отсчитывая от верха перегонной колонны тарельчатого типа (колонны первой реакции/перегонки), характеризующейся числом фактических тарелок, равным 50, и имеющей внутренний диаметр, равный 50 мм, и высоту, равную 5 м. Реакцию/перегонку проводили во время обогревания куба колонны с использованием обогревающего кожуха. Из верха колонны выпускали раствор диметилкарбоната, содержащий метанол, при кипячении раствора в условиях флегмообразования при флегмовом числе 12. Кубовый остаток, который содержал полученный метилфенилкарбонат и небольшое количество DPC, выпускали из куба колонны и подавали на десятую тарелку, отсчитывая от верха перегонной колонны тарельчатого типа (колонны второй реакции/перегонки), характеризующейся числом фактических тарелок, равным 50, и имеющей внутренний диаметр, равный 80 мм, и высоту, равную 4 м. В колонне второй реакции/перегонки реакция протекала дальше, а жидкость, содержащую DPC и полученный метилфенилкарбонат, выпускали из куба колонны. Основную часть диметилкарбоната, остающегося не вступившим в реакцию, и часть PL, остающегося не вступившим в реакцию, отгоняли через верх колонны второй реакции/перегонки и отправляли на рецикл в колонну первой реакции/перегонки.
Стадия отправки на рецикл
Метанолсодержащий раствор диметилкарборната, выпускаемый в качестве дистиллята из колонны первой реакции/перегонки, подавали на среднюю тарелку перегонной колонны (колонны азеотропной перегонки), характеризующейся числом фактических тарелок, равным 30, и имеющей внутренний диаметр, равный 32 мм, и высоту, равную 2,5 м. Перегонку проводили при флегмовом числе, равном 5. Жидкую смесь метанола и диметилкарбоната, которая имела состав, близкий к азеотропному составу, выпускали через верх колонны, а после этого подавали в экстракционную/перегонную колонну. В экстракционной/перегонной колонне из диметилкарбоната выделяли метанол. Метанол отводили из системы, в то время как диметилкарбонат отправляли на рецикл в колонну первой реакции/перегонки. Для кубового остатка из колонны азеотропной перегонки, который представлял собой диметилкарбонат, содержащий незначительное количество PL, проводили циркуляцию с подачей в колонну первой реакции/перегонки.
Стадия очистки
Высококипящую реакционную смесь, которую непрерывно выпускали из куба колонны второй реакции/перегонки и которая содержала катализатор и DPC, вводили в испаритель, из которого отводили конденсат, содержащий катализатор. С другой стороны, в колонну очистки дифенилкарбоната подавали превращенные в пар ингредиенты, включающие большое количество DPC, полученного в испарителе. Колонну очистки регулировали таким образом, чтобы выдерживать давление верха колонны 20 торр и температуру куба колонны 190°С. Низкокипящую смесь, содержащую фенол и метилфенилкарбонат, выпускали в качестве дистиллята через верх колонны. Часть данной смеси подвергали кипячению в условиях флегмообразования, а оставшуюся часть отправляли на рецикл в колонну второй реакции/перегонки. С другой стороны, из куба колонны очистки дифенилкарбоната отводили высококипящие примеси, а со средней тарелки колонны получали DPC.
Описанные выше операции непрерывно проводили до тех пор, пока на каждой стадии не достигали стационарного состояния. Тогда, когда на стадиях достигали стационарного состояния, отбирали и по методу высокоэффективной жидкостной хроматографии анализировали образец дифенилкарбоната. В результате в полученном дифенилкарбонате обнаружили 300 ч/млн (мас.) метилфенилкарбоната. Выход DPC составлял приблизительно 95% при расчете на количество PL.
Пример получения ВРА
Далее продемонстрирован пример, в котором из коммерческого PL и ацетона получали ВРА.
В реактор синтеза проточного типа, снабженный термостатом, в количестве 60 л помещали набивку в виде кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.), в которой 15% сульфогрупп было нейтрализовано под действием 4-пиридинэтантиола. В данный реактор синтеза при температуре 80°С и расходе 68,2 кг/час вводили жидкую смесь PL и ацетона с молярным соотношением 10:1, обеспечивая протекание реакции между ними. Степень превращения ацетона составляла 80%. После того, как низкокипящие соединения (не вступивший в реакцию ацетон, воду и часть PL) при производительности, равной 5,1 кг/час, отводили, реакционную смесь охлаждали до 50°С, добиваясь осаждения кристаллов аддукта. Данную смесь отфильтровывали для ее разделения на кристаллы аддукта и маточную жидкость. Производительности при их получении составляли 16,5 кг/час и 46,5 кг/час соответственно. Десять массовых процентов данной маточной жидкости подавали на стадию переработки маточной жидкости, тогда как для остальной маточной жидкости как части исходного материала, вводимого в реактор синтеза, проводили циркуляцию.
Таким образом полученные кристаллы аддукта повторно растворяли в PL, подаваемом при расходе, равном 27,2 кг/час. Получающийся в результате раствор охлаждали до 50°С, добиваясь осаждения кристаллов, а после этого отфильтровывали для отделения кристаллов аддукта (11,3 кг/час) от маточной жидкости (32,5 кг/час). Выделенные кристаллы нагревали до 180°С при пониженном давлении, равном 0,3 мм ртутного столба, для удаления PL. Таким образом, при производительности, равной 7,7 кг/час, получали ВРА, характеризующийся степенью чистоты, равной 99,95% либо более.
С другой стороны, маточную жидкость, подаваемую на стадию переработки маточной жидкости, концентрировали в результате отгонки части PL. После этого к маточной жидкости добавляли 0,1 мас.% гидроксида натрия и затем маточную жидкость вводили в куб колонны разложения/перегонки, в котором в результате регулировки выдерживали пониженное давление 50 мм ртутного столба и температуру 210°С. Данную колонну эксплуатировали при таких условиях, чтобы выдерживать в нижней части постоянство уровня жидкости (время пребывания 1 час), а кубовый остаток в колонне разложения/перегонки отводили из системы при производительности, равной 0,5 кг/час. Кроме того, дистиллят, выпускаемый через верх колонны разложения/перегонки, перемешивали с PL, а данную смесь при расходе, равном 4,2 кг/час, вводили в реактор проточного типа, снабженный набивкой в виде 4 л кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.). Реакцию для смеси проводили в условиях 80°С. Для полученной жидкой реакционной смеси проводили циркуляцию с подачей в реактор синтеза, использованный на начальной стадии.
В реактор синтеза дополнительно подавали коммерческий PL (18,5 кг/час) и ацетон (3,6 кг/час) в количествах, соответствующих количествам, отводимым из системы, и количеству полученного бисфенола А. Таким образом, реакцию синтеза проводили непрерывно, и вся система функционировала при непрерывном получении ВРА.
Пример получения РС (1)
Далее продемонстрирован пример, в котором при проведении стадий, продемонстрированных на фиг.6, из DPC, полученного в приведенном выше примере получения DPC (1), и ВРА, полученного в приведенном выше примере получения ВРА, получали РС.
Стадия полимеризации при получении РС
В атмосфере газообразного азота DPC и ВРА перемешивали в расплаве при соотношении 0,977 (мас.) с использованием смесительной емкости 21. Данную смесь в атмосфере азота непрерывно подавали в первый вертикальный реактор полимеризации с перемешиванием 22, в котором в результате регулировки выдерживали температуру 210°С и давление 100 торр. Уровень жидкости выдерживали постоянным, проводя регулировку степени открытости клапана, расположенного на линии выпуска полимера, соединенной с нижней частью емкости, таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам. Кроме того, одновременно с инициированием подачи смеси исходных материалов в качестве катализатора начинали непрерывно подавать карбонат цезия в форме водного раствора при расходе, равном 0,5·10-6 моль при расчете на один моль ВРА. После этого жидкую полимеризационную реакционную смесь, выпускаемую из нижней части емкости, непрерывно последовательно подавали во второй и третий вертикальные реакторы полимеризации 23 и 24 и четвертый горизонтальный полимеризатор 25. Во время прохождения реакции уровень жидкости в каждой емкости регулировали таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам, и одновременно с этим отгоняли PL, полученный в качестве побочного продукта. Газы, полученные в результате выпаривания и выпускаемые соответственно из первого и второго реакторов полимеризации 22 и 23, конденсировали и ожижали при использовании рефлюксного аппарата 33а и 33b соответственно и при использовании многостадийных конденсаторов 26 и 27 соответственно. Часть каждого из получающихся в результате конденсатов в результате кипячения в условиях флегмообразования подавали в реактор полимеризации, а оставшуюся часть извлекали и хранили в первом резервуаре для извлеченного PL 29а при получении РС. С другой стороны, газ, полученный в результате выпаривания и выпускаемый из третьего реактора полимеризации 24, отверждали при использовании одного из двух замораживающих конденсаторов, расположенных параллельно. Получающуюся в результате твердую фазу расплавляли в результате переключения на другой замораживающий конденсатор и извлекали и хранили во втором резервуаре для извлеченного PL 29b при получении РС. При проведении данной операции все количество превращенных в пар ингредиентов при получении РС, выпускаемых в качестве дистиллятов из первого и второго реакторов полимеризации 22 и 23, хранили в первом резервуаре для извлеченного PL 29a при получении РС, в то время как превращенные в пар ингредиенты при получении РС, выпускаемые в качестве дистиллята из третьего реактора полимеризации 24, хранили во втором резервуаре для извлеченного PL 29b при получении РС.
Условия проведения полимеризации для каждого реактора полимеризации представляли собой нижеследующее: первый реактор полимеризации 22 (210°C, 100 торр), второй реактор полимеризации 23 (240°С, 15 торр), третий реактор полимеризации 24 (260°С, 0,5 торр) и четвертый полимеризатор 25 (280°С, 0,5 торр).
Получающийся в результате полимер в расплавленном состоянии вводили в двухчервячный экструдер (произведенный в компании Kobe Steel, Ltd.; диаметр червяка 0,046 м; L/D=40,2) и гранулировали при одновременном добавлении сюда бутил-п-толуолсульфоната в количестве 5 ч/млн (мас.) при расчете на количество поликарбоната. Таким образом, полученный РС характеризовался величиной Mv, равной 21000, и начальным значением YI 1,7. Методы, использованные для измерения молекулярной массы (Mv) и начального цветового тона (YI), были описаны выше.
Превращенные в пар ингредиенты при получении РС
Для каждого из превращенных в пар ингредиентов при получении РС, извлеченных на стадии полимеризации, проводили исследование для определения его количества и состава. Полученные результаты представляли собой нижеследующее.
Первый резервуар для извлеченного PL 29а при получении РС
Количество фенола в извлеченных первых, превращенных в пар ингредиентах при получении РС р1 составляло приблизительно 60% при расчете на совокупное количество PL, отогнанного на стадии полимеризации при получении РС. В дополнение к PL обнаружили 1,1 мас.% DPC. ВРА и олигомерные ингредиенты не обнаружили.
Второй резервуар для извлеченного PL 29b при получении РС
Количество фенола в извлеченных вторых превращенных в пар ингредиентах при получении РС р2 составляло приблизительно 40% при расчете на совокупное количество PL, отогнанного на стадии полимеризации. В дополнение к PL обнаружили 6,0 мас.% DPC. Кроме того, обнаружили ВРА и олигомерные ингредиенты в количествах 1,2 мас.% и 0,3 мас.% соответственно.
Пример получения РС (2)
Далее продемонстрирован пример, в котором из DPC, полученного в приведенном выше примере получения DPC (2), и ВРА, полученного в приведенном выше примере получения ВРА, получали РС.
Стадия полимеризации при получении РС
Использовали ту же самую методику, что и в примере получения РС (1), приведенном выше, за исключением того, что в качестве DPC использовали DPC, полученный в примере получения DPC (2), приведенном выше. Полученный РС характеризовался величиной Mv, равной 21000, и начальным значением YI 1,7. Он имел то же самое качество, что и качество у РС, полученного в приведенном выше примере получения ароматического поликарбоната (1).
Превращенные в пар ингредиенты при получении РС
Для каждого из превращенных в пар ингредиентов при получении РС, извлеченных на стадии полимеризации, проводили исследование для определения его количества и состава. Полученные результаты представляли собой нижеследующее.
Первый резервуар для извлеченного PL 29а при получении РС
Количество фенола в извлеченных первых превращенных в пар ингредиентах при получении РС р1 составляло приблизительно 60% при расчете на совокупное количество PL, отогнанного на стадии полимеризации при получении РС. В дополнение к PL обнаружили 1,1 мас.% DPC и 95 ч/млн (мас.) метанола. ВРА и олигомерные ингредиенты не обнаружили.
Второй резервуар для извлеченного PL 29b при получении РС
Количество фенола в извлеченных вторых превращенных в пар ингредиентах при получении РС р2 составляло приблизительно 40% при расчете на совокупное количество PL, отогнанного на стадии полимеризации. В дополнение к PL обнаружили 6,0 мас.% DPC. Кроме того, обнаружили ВРА и олигомерные ингредиенты в количествах 1,2 мас.% и 0,3 мас.% соответственно. Метанол не обнаружили (5 ч/млн (мас.) либо менее).
Экспериментальный пример 1
Для получения DPC и ВРА использовали превращенные в пар ингредиенты при получении РС, содержащие являющийся побочным продуктом фенол и полученные в приведенном выше примере получения РС (1).
Получение DPC
DPC получали при использовании той же самой методики, что и в приведенном выше примере получения DPC (1), за исключением того, что 60% коммерческого PL, использованного в примере получения DPC (1), заменяли на первые превращенные в пар ингредиенты при получении РС р1, извлеченные в приведенном выше примере получения РС (1). В результате степень превращения фосгена на стадии реакции и качество полученного DPC были теми же самыми, что и в примере получения DPC (1), и были удовлетворительными.
Получение ВРА
ВРА получали при использовании той же самой методики, что и в приведенном выше примере получения ВРА, за исключением того, что 40% коммерческого PL, дополнительно непрерывно подаваемого в реактор синтеза в примере получения ВРА, заменяли на вторые, превращенные в пар ингредиенты при получении РС р2, извлеченные в приведенном выше примере получения РС (1). В результате степень превращения ацетона и качество бисфенола А были удовлетворительными.
Экспериментальный пример 2
Для получения DPC и ВРА использовали превращенные в пар ингредиенты при получении РС, содержащие являющийся побочным продуктом фенол и полученные в приведенном выше примере получения РС (2).
Получение DPC
DPC получали при использовании той же самой методики, что и в приведенном выше примере получения DPC (2), за исключением того, что 60% коммерческого PL, использованного в примере получения DPC (2), заменяли на первые превращенные в пар ингредиенты при получении РС р1, извлеченные в приведенном выше примере получения РС (2). В результате можно было получить DPC, обладающий тем же самым качеством. Удостоверились в том, что, даже если метанол, получаемый из метилфенилкарбоната, и попадал в систему, это не представляло из себя какой-либо проблемы.
Получение ВРА
ВРА получали при использовании той же самой методики, что и в приведенном выше примере получения ВРА, за исключением того, что 40% коммерческого PL, дополнительно непрерывно подаваемого в реактор синтеза в примере получения ВРА, заменяли на вторые превращенные в пар ингредиенты при получении РС р2, извлеченные в приведенном выше примере получения РС (2). В результате степень превращения ацетона и качество ВРА были удовлетворительными. Кроме того, концентрации метанола в исходных материалах, используемых для проведения реакции, не обнаружили.
Сравнительный экспериментальный пример 1
Для получения DPC использовали вторые, превращенные в пар ингредиенты при получении РС р2, извлеченные в приведенном выше примере получения РС (1).
Получение DPC
DPC получали при использовании той же самой методики, что и в приведенном выше примере получения DPC (1), за исключением того, что все количество коммерческого PL, использованного в примере получения DPC (1), заменяли на вторые превращенные в пар ингредиенты при получении РС р2, извлеченные в приведенном выше примере получения РС (1). В результате трубопровод для перепускания жидкой реакционной смеси на стадию промывки был подвержен засорению, что делало невозможным продолжение функционирования.
Сравнительный экспериментальный пример 2
Для получения ВРА использовали первые, превращенные в пар ингредиенты при получении РС р1, извлеченные в приведенном выше примере получения РС (2).
Получение ВРА
ВРА непрерывно получали по тому же самому способу, что и в примере получения ВРА, приведенном выше, за исключением того, что все количество коммерческого PL, дополнительно непрерывно подаваемого в реактор синтеза в примере получения ВРА, заменяли на первые превращенные в пар ингредиенты при получении РС р1 (содержащие 95 ч/млн (мас.) метанола), извлеченные в приведенном выше примере получения РС (2). В результате степень превращения ацетона значительно уменьшалась, то есть до 55,0%. Представляется, что присутствующий метанол вызывал ухудшение эксплуатационных характеристик катализатора. Концентрация метанола в являющемся исходным материалом феноле во время реакции синтеза составляла приблизительно 30 ч/млн (мас.) вследствие разбавления с использованием циркулирующей маточной жидкости.
Пример 3
При использовании экспериментальных примеров далее приводятся пояснения по классификации по примесям для способов использования превращенных в пар ингредиентов при получении РС р и коммерческого PL для случая переработки s-PL 3.
Экспериментальный пример 1
Получение DPC
Стадия реакции
Во время непрерывной подачи в реактор расплавленного коммерческого PL (произведенного в компании Mitsubishi Chemical Corp.; уровень содержания крезола 45 ч/млн (мас.); уровень содержания гидроксиацетона 27 ч/млн (мас.); здесь и далее в настоящем документе обозначаемого как "PL1") и пиридинового катализатора при перемешивании при 150°С проводили непрерывную подачу газообразного фосгена. Газообразный хлористый водород, полученный в качестве побочного продукта в результате фосгенирования, охлаждали до 10°С. Получающийся в результате конденсат возвращали в реактор, а для несконденсировавшегося газа проводили нейтрализацию, используя водный щелочной раствор, и после этого выпуск. С другой стороны, жидкую реакционную смесь, содержащую приблизительно 91 мас.% DPC, непрерывно выпускали из реактора.
Стадия промывки
Жидкую реакционную смесь и водный раствор гидроксида натрия с концентрацией, приблизительно равной 5 мас.%, подавали в емкость нейтрализации/перемешивания, снабженную тефлоновой футеровкой. Для содержимого проводили перемешивание при 80°С в течение приблизительно 10 минут и регулировку, обеспечивающую выдерживание значения рН, равного 8,5. После проведения нейтрализации органическую фазу отделяли, используя отстаивание, а после этого перепускали в емкость водной промывки/перемешивания. В емкости водной промывки/перемешивания органическую фазу промывали, используя теплую воду в количестве, приблизительно равном 30 мас.% при расчете на количество органической фазы. Водную фазу отделяли и получали "сырой" DPC (содержащий 1 мас.% воды, 2 мас.% пиридина, 8 мас.% PL и 89 мас.% DPC).
Стадия перегонки низкокипящих соединений
После этого "сырой" DPC непрерывно подавали на среднюю тарелку колонны перегонки низкокипящих соединений при расходе, равном приблизительно 30 кг/час. В качестве данной колонны перегонки низкокипящих соединений использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 150 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 220°С, температуры верха колонны 80-100°С, температуры средней тарелки 160°С и флегмового числа 1 для отгонки соединений, характеризующихся температурой кипения, более низкой по сравнению с температурой кипения DPC, то есть воды, пиридина и PL. Из куба колонны при производительности, приблизительно равной 26 кг/час, непрерывно выпускали DPC (уровни содержания воды 10 ч/млн (мас.) либо менее; пиридина 1 ч/млн (мас.) либо менее; PL 50 ч/млн (мас.)).
Стадия перегонки высококипящих соединений
Кроме того, данный DPC (кубовый остаток из колонны перегонки низкокипящих соединений) непрерывно подавали в колонну перегонки высококипящих соединений. В качестве данной колонны перегонки высококипящих соединений использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 200 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны, приблизительно равной 180°С, и флегмового числа 0,5. В результате через верх колонны при производительности, приблизительно равной 23,5 кг/час, получали очищенный DPC (содержащий 80 ч/млн (мас.) PL).
Справочный пример 1
DPC получали по тому же самому способу, что и в экспериментальном примере 1, за исключением того, что вместо коммерческого PL (PL1) использовали являющийся побочным продуктом фенол (уровень содержания крезола 5 ч/млн (мас.); уровень содержания гидроксиацетона 1 ч/млн (мас.) либо менее; здесь и далее в настоящем документе обозначаемый как "PL2"), полученный в результате проведения при нижеследующих условиях перегонки/очистки превращенных в пар ингредиентов при получении РС р, полученных на предприятии по производству РС в компании Mitsubishi Chemical Corp.
Условия перегонки/очистки
В первой перегонной колонне отгоняли присутствующую воду совместно с частью PL при 200 торр и флегмовом числе 2. Получающийся в результате кубовый остаток непрерывно подавали в колонну второй перегонки. В колонне второй перегонки через верх колонны при 50 торр и флегмовом числе 0,5 получали очищенный являющийся побочным продуктом фенол. В качестве кубового остатка непрерывно отводили жидкую PL-содержащую смесь, содержащую высококипящие ингредиенты, то есть DPC, BPA и олигомеры.
Результаты
Даже тогда, когда для получения DPC использовали коммерческий PL без очистки, получали высокочистый DPC, характеризующийся высокой степенью чистоты и отсутствием изменения окраски и тому подобным, как и в случае использования являющегося побочным продуктом фенола, который был подвергнут перегонке/очистке.
Экспериментальный пример 2 и сравнительный пример 1
Получение ВРА
В реактор помещали набивку в виде катионообменной смолы с сульфоновой кислотой (сшитый сополимер на основе сульфированного стирола/дивинилбензола (DIAION SK104Н, произведенный в компании Mitsubishi Chemical Corp.)), в которой 15 мас.% сульфогрупп было модифицировано под действием 4-(2-меркаптоэтил)пиридина. Являющуюся исходным материалом текучую среду, состоящую из любого PL (PL2 либо PL1) и ацетона (произведенного в компании Mitsubishi Chemical Corp.) (PL : ацетон = 13:1 (мольное)), при LHSV (часовой объемной скорости жидкости) 5 час-1 непрерывно пропускали через реактор, используя увлажненный фенолом катализатор, обеспечивающий прохождение реакции получения ВРА в течение 1008 часов при температуре реакции 70°С. В таблице продемонстрированы степень превращения ацетона, определенная спустя 120 часов после инициирования реакции, степень превращения ацетона, определенная по истечении 1008 часов после инициирования, и селективность получения ВРА. Кроме того, в качестве параметра сохранения активности рассчитывали соотношение между степенью превращения ацетона, определенной по истечении 1008 часов после инициирования реакции, и степенью превращения ацетона, определенной по истечении 120 часов после инициирования. Результаты данного вычисления также продемонстрированы в таблице.
Между прочим, степень превращения (%) и сохранение каталитической активности (%) рассчитывали при использовании следующих далее уравнений:
Степень превращения (%) = {[(количество поданного ацетона) - (количество не вступившего в реакцию ацетона)]/(количество поданного ацетона)}×100
Сохранение активности (%) = [(степень превращения ацетона по истечении 1008 часов)/(степень превращения ацетона по истечении 120 часов)]×100
Результаты
Если использовали PL, полученный в результате проведения перегонки/очистки PL-содержащего дистиллята со стадии полимеризации, то тогда в отличие от случая использования коммерческого PL добивались получения высоких степеней превращения и высокой степени сохранения каталитической активности.
Кроме того, использование PL, полученного в результате проведения перегонки/очистки PL-содержащего дистиллята со стадии полимеризации, приводило к получению ВРА, имеющего белый цветовой тон, в то время как использование коммерческого PL приводило к получению ВРА, который был несколько пожелтевшим. Предполагается, что использование данных продуктов ВРА при получении РС приводит к получению следующих далее результатов. В том случае, если для получения ВРА использовали PL, полученный в результате проведения перегонки/очистки PL-содержащего дистиллята со стадии полимеризации, то тогда, как представляется, получали белый РС, характеризующийся высокой степенью чистоты. В противоположность этому, если для получения ВРА использовали коммерческий PL, то тогда, как представляется, полученный РС был несколько пожелтевшим и характеризовался пониженной степенью чистоты.
Пример 4
После этого изобретение будет далее разъяснено более подробно в связи с соединением стадий получения в соответствии со схемой технологического процесса, продемонстрированной на фиг.7. В отличие от того, что приведено на фиг.7, в промежутке между стадией нагревания/плавления (стадией (d)) и стадией удаления PL (стадией (е)) располагали стадию хранения при получении ВРА.
Экспериментальный пример 1
Получение ВРА
В реактор получения ВРА проточного типа, снабженный термостатом, в количестве 60 л помещали набивку в виде кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.), в которой 15% сульфогрупп было нейтрализовано под действием 4-пиридинэтантиола. В данный реактор синтеза при температуре 80°С и расходе 68,2 кг/час вводили жидкую смесь PL и ацетона (А) с молярным соотношением 10:1, обеспечивая протекание реакции между ними. Степень превращения ацетона (А) составляла 80%. После того как низкокипящие соединения (не вступивший в реакцию ацетон, воду и часть PL) при производительности, равной 5,1 кг/час, отводили, реакционную смесь охлаждали до 50°С, добиваясь осаждения кристаллов аддукта. Данную смесь отфильтровывали для ее разделения на кристаллы аддукта и маточную жидкость. Производительности при их получении составляли 16,5 кг/час и 46,5 кг/час соответственно. 10 мас.% в данной маточной жидкости подавали на стадию переработки маточной жидкости, тогда как для остальной маточной жидкости как части исходного материала, вводимого в реактор синтеза, проводили циркуляцию.
Таким образом полученные кристаллы аддукта повторно растворяли в феноле, подаваемом при расходе, равном 27,2 кг/час. Получающийся в результате раствор охлаждали до 50°С, добиваясь осаждения кристаллов, а после этого отфильтровывали для отделения кристаллов аддукта (11,3 кг/час) от маточной жидкости (32,5 кг/час). Выделенные кристаллы перемешивали с очищенным PL, подаваемым при расходе, равном 1,5 кг/час, и получали смесь из 60 мас.% ВРА и 40 мас.% PL. Данную смесь при расходе, равном 12,8 кг/час, вводили в резервуар для хранения при получении ВРА и хранили в нем (при нижеследующих условиях), проводя непрерывную подачу на последующую стадию удаления PL при расходе, равном 12,0 кг/час.
Использованный резервуар для хранения при получении ВРА представлял собой резервуар, изготовленный из SUS304, характеризующийся вместимостью 150 л (у которого значение величины Vb/Fb, указанной в описании, составляло 22). Внутреннее пространство системы герметизировали, создавая атмосферу азота, а температуру в толще смеси BPA/PL в результате регулировки выдерживали на уровне 120°С. В дополнение к этому в смеси BPA/PL в количестве, приблизительно равном 10 ч/млн (мас.), обнаружили PL-сульфоновую кислоту, источником возникновения которой, как представляется, является ионообменная смола. По этой причине перед тем, как смесь перепускали в резервуар для хранения, к смеси для ее нейтрализации добавляли водный раствор каустической соды в количестве, необходимом для полной нейтрализации кислотного соединения. При инициировании операции уровень жидкости в резервуаре для хранения начинал немного и постепенно увеличиваться.
После этого смесь ВРА/PL в резервуаре для хранения при получении ВРА непрерывно перепускали на стадию удаления PL при расходе, равном 12,0 кг/час. Для удаления PL смесь нагревали до 180°С при пониженном давлении, равном 0,3 мм ртутного столба. Таким образом, при производительности, равной 7,2 кг/час (6,8 л/час), получали ВРА, характеризующийся степенью чистоты, равной 99,95% либо более. Полученный ВРА как таковой непрерывно подавали на стадию получения ароматического поликарбоната, которая будет описываться далее.
С другой стороны, маточную жидкость, подаваемую на стадию переработки маточной жидкости (не показана на фиг.7; см. фиг.4), подвергали переработке с использованием испарителя PL 13 для отгонки части PL и концентрирования маточной жидкости. После этого к маточной жидкости добавляли 0,1 мас.% гидроксида натрия, затем маточную жидкость вводили в нижнюю часть реактора переработки остатка 14, для которого проводили регулировку, обеспечивая выдерживание пониженного давления, равного 50 мм ртутного столба, и температуры, равной 210°С. Данный реактор функционировал при таких условиях, чтобы выдерживать постоянство уровня жидкости в нижней части (время пребывания 1 час), а нижний продукт реактора переработки остатка 14 отводили из системы при производительности, равной 0,5 кг/час. Кроме того, дистиллят, выпускаемый через верх реактора переработки остатка 14, перемешивали с PL, а данную смесь при расходе, равном 4,2 кг/час, вводили в регенерирующий реактор проточного типа 15, снабженный набивкой в виде 4 л кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.). Реакцию для смеси проводили в условиях 80°С. Для полученной жидкой реакционной смеси проводили циркуляцию с подачей в реактор получения ВРА, использованный на начальной стадии.
Дополнительно в реактор получения ВРА подавали коммерческий PL (18,5 кг/час) и ацетон (3,6 кг/час) в количествах, соответствующих количествам, отводимым из системы, и количеству полученного ВРА. Таким образом, реакцию синтеза проводили непрерывно, и вся система функционировала при непрерывном получении ВРА.
Получение РС
Далее продемонстрирован пример, в котором РС получали в соответствии со схемой технологического процесса, продемонстрированной на фиг.5.
В атмосфере газообразного азота непрерывно подаваемые ВРА и DPC перемешивали в расплаве при соотношении 1,024 (мас.) с использованием смесительной емкости 21. Данную смесь в атмосфере азота непрерывно подавали в первый вертикальный реактор полимеризации с перемешиванием 22, в котором в результате регулировки выдерживали температуру 210°С и давление 100 торр. Уровень жидкости выдерживали постоянным, проводя регулировку степени открытости клапана, расположенного на линии выпуска полимера, соединенной с нижней частью емкости, таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам. Кроме того, одновременно с инициированием подачи смеси исходных материалов в качестве катализатора начинали непрерывно подавать карбонат цезия в форме водного раствора при расходе, равном 0,5·10-6 моль при расчете на один моль ВРА. После этого жидкую полимеризационную реакционную смесь, выпускаемую из нижней части емкости, непрерывно последовательно подавали во второй и третий вертикальные реакторы полимеризации 23 и 24 и четвертый горизонтальный реактор полимеризации 25. Во время прохождения реакции уровень жидкости в каждом реакторе регулировали таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам, и одновременно с этим отгоняли PL, полученный в качестве побочного продукта. Превращенные в пар ингредиенты при получении РС р, выпускаемые из первого и второго реакторов полимеризации, конденсировали и ожижали при использовании соответствующих многостадийных конденсаторов. Часть каждого из получающихся в результате конденсатов в результате кипячения в условиях флегмообразования подавали в реактор полимеризации, а оставшуюся часть извлекали и хранили в резервуаре для PL 29 при получении РС. С другой стороны, каждый из газов, полученных в результате выпаривания и выпускаемых из третьего и четвертого реакторов полимеризации 24 и 25, отверждали при использовании одного из двух замораживающих конденсаторов, расположенных параллельно. Получающуюся в результате твердую фазу расплавляли в результате переключения на другой замораживающий конденсатор и извлекали и хранили в резервуаре для PL 29 при получении РС (не показана).
Условия проведения полимеризации для каждого реактора полимеризации представляли собой нижеследующее: первый реактор полимеризации (210°C, 100 торр), второй реактор полимеризации (240°С, 15 торр), третий реактор полимеризации (260°С, 0,5 торр) и четвертый реактор полимеризации (280°С, 0,5 торр).
Получающийся в результате полимер в расплавленном состоянии гранулировали при одновременном непрерывном добавлении бутил-п-толуолсульфоната в количестве 5 ч/млн (мас.) при расчете на количество РС. Таким образом полученный поликарбонат характеризовался величиной Mv, равной 21000, и начальным значением YI 1,8.
Измерение молекулярной массы (Mv)
Для измерения удельной вязкости (ηsp) при помощи вискозиметра Убеллоде при температуре 20°С использовали раствор в метиленхлориде, характеризующийся концентрацией РС (С), равной 0,6 г/дл, а молекулярную массу (Mv) рассчитывали по значению вязкости при использовании следующих далее двух уравнений:
ηsp/С=[η](1+0,28ηsp)
[η]=1,23Χ10-4(Mv)0,83
Измерение начального цветового тона (YI)
РС высушивали при 120°С в течение 6 часов в атмосфере азота, а после этого при использовании литьевой машины J-100, произведенной в компании The Japan Steel Works, Ltd., по способу литьевого формования при 360°С из него получали кусок, имеющий толщину 3 мм. Данный кусок исследовали при использовании прибора SC-1, произведенного в компании Suga Test Instruments Co., Ltd., получая значение YI (чем больше будет значение YI, тем больше РС будет окрашен).
Описанную выше операцию непрерывно проводили в течение одной недели. После этого проведение стадии реакции синтеза/кристаллизации временно прекращали приблизительно на 8 часов в целях очистки кристаллизатора ВРА. Однако в течение всего данного периода жидкую смесь, хранящуюся в резервуаре для хранения, использовали для обеспечения непрерывного проведения последующих стадий, благодаря чему можно было получать ВРА и ароматический поликарбонат. Качество данных продуктов, полученных в ходе непрерывного функционирования в течение одной недели, и качество продуктов, полученных при проведении последующих стадий после этого, были теми же самыми, что и качество в самом начале, и были удовлетворительными.
Экспериментальный пример 2
ВРА и ароматический поликарбонат непрерывно получали в результате использования той же самой методики, что и в экспериментальном примере 1, за исключением того, что вместимость резервуара для хранения при получении ВРА на стадии получения ВРА изменили на 2 м3 (Vb/Fb=294). Полученный ароматический поликарбонат характеризовался величиной Mv, равной 21000, и начальным значением YI 1,8.
Данную операцию непрерывно проводили в течение 2 месяцев. В течение всего данного периода ароматический поликарбонат продемонстрировал цветовой тон (начальное значение YI) 1,8-1,9, и полученный продукт был удовлетворительным. После этого проведение стадии реакции синтеза/кристаллизации ВРА временно прекращали приблизительно на 3 дня. Однако последующие стадии можно было проводить непрерывно при использовании жидкой смеси, хранящейся в резервуаре для хранения, и качество таким образом полученного продукта было удовлетворительным. После этого начальные стадии при получении ВРА (стадии, предшествующие резервуару для хранения при получении ВРА) проводили периодически при выполнении 3-дневной остановки с интервалами в 2 месяца, а стадии в диапазоне от конечных стадий получения ВРА до получения ароматического поликарбоната проводили непрерывно по способу, описанному выше. Таким образом, ароматический поликарбонат удовлетворительного качества можно было непрерывно получать в течение приблизительно полугода.
Сравнительный пример 1
ВРА и ароматический поликарбонат непрерывно получали в результате использования той же самой методики, что и в экспериментальном примере 1, за исключением того, что вместимость резервуара для хранения при получении ВРА на стадии получения ВРА изменили на 50 л (Vb/Fb=7). Полученный ароматический поликарбонат характеризовался величиной Mv, равной 21000, и начальным значением YI 1,8.
Однако по истечении 2 дней, прошедших от момента инициирования операции, резервуар для хранения ВРА становился заполненным, и это уменьшало производительность на начальных стадиях на стадии получения ВРА (стадиях, предшествующих резервуару для хранения). Поэтому функционирование регулировали таким образом, чтобы выдерживать в резервуаре для хранения постоянство уровня жидкости. После этого прекращали проведение только начальных стадий. В результате уровень жидкости в резервуаре для хранения быстро понижался, и приблизительно через 4 часа резервуар становился пустым, что делало необходимым прекращение проведения последующих стадий и всех стадий получения РС.
Сравнительный экспериментальный пример 2
ВРА непрерывно получали в сравнительном экспериментальном примере 1, в котором производительность на начальных стадиях получения ВРА (стадиях, предшествующих резервуару для хранения) регулировали таким образом, чтобы выдерживать в резервуаре для хранения постоянство уровня жидкости. Полученный ВРА охлаждали, а после этого гранулировали и данные гранулы временно хранили во вновь установленном бункере для порошкообразного ВРА (вместимость 1 м3). После этого тогда, когда гранулы подавали и растворяли в растворе, содержащем DPC, при использовании порошкового массового питателя, РС непрерывно получали по способу, описанному выше. До тех пор, пока ВРА находился в бункере для порошка, временные прекращения проведения стадий получения ВРА не представляли собой никакой проблемы в отношении производительности при получении РС. Однако данный способ оказался невыгодным с точки зрения термического коэффициента полезного действия, поскольку было необходимо временно охлаждать расплавленный ВРА для его превращения в форму порошка, а после этого проводить нагревание для растворения. В дополнение к этому во время подачи РВА происходило пыление, что приводило к возникновению проблемы, связанной с засорением линии газа.
Сравнительный экспериментальный пример 3
ВРА непрерывно получали при производительности, равной 7,7 кг/час, в сравнительном экспериментальном примере 1, в котором производительность на стадиях, следующих после резервуара для хранения, была несколько повышенной для того, чтобы выдерживать постоянство уровня жидкости в резервуаре для хранения. Полученный ВРА индивидуально хранили во вновь установленном резервуаре, характеризующемся вместимостью 2 м3, где в результате регулировки выдерживали внутреннюю температуру, равную 160°С. После этого, как и в сравнительном экспериментальном примере 1, в качестве исходного материала для получения РС подавали ВРА при расходе, равном 7,2 кг/час. Полученный РС характеризовался начальным значением YI 1,8. Однако по мере того, как операция продолжалась, цветовой тон начинал отчетливо ухудшаться. Выдерживание одного только ВРА в расплавленном состоянии в результате приводило к значительному ухудшению качества.
Экспериментальный пример 3
Далее приводится экспериментальный пример, в котором стадию получения ВРА, стадию получения DPC и стадию получения РС соединяли друг с другом, а в данные стадии получения включали стадии хранения при получении ВРА, при получении DPC и при получении РС.
Получение ВРА
Получение ВРА проводили по тому же самому способу, что и в экспериментальном примере 1.
Получение DPC
Стадия реакции
Расплавленный фенол, имеющий температуру 50°С, при расходе, равном 6,4 кг/час, непрерывно подавали в первый реактор совместно с содержащим в качестве катализатора пиридин фенолом, полученным в результате обезвоживания низкокипящих соединений, выпускаемых в качестве дистиллята из описанной далее колонны перегонки низкокипящих соединений. Одновременно с проведением такой подачи расплавленный фенол нагревали до 150°С. Газообразный фосген при расходе, равном 0,75 нм3/час, непрерывно подавали в первый реактор при достаточном перемешивании. Реакционную смесь, которая вытекала из первого реактора и содержала фазу смеси газа/жидкости, подавали во второй реактор через перепускную трубу. Для содержимого второго реактора при достаточном перемешивании также проводили регулировку так, чтобы выдерживать температуру 150°С. Жидкую реакционную смесь подавали в колонну дегазации. В колонне дегазации при 160°С проводили противоточное введение в контакт при использовании газообразного азота, для того чтобы обеспечить полное введение в реакцию фенилхлорформиата, полученного в качестве промежуточного соединения, и фенола. Жидкую реакционную смесь, характеризующуюся концентрацией дифенилкарбоната, приблизительно равной 89 мас.%, непрерывно выпускали из куба колонны дегазации. Почти 100% подаваемого фосгена превращали в дифенилкарбонат. С другой стороны, выпускаемые газы, получающиеся в результате проведения синтеза дифенилкарбоната (реакционный газ, выпускаемый из второго реактора получения DPC, и азотсодержащий газ, выпускаемый из колонны дегазации) перемешивали друг с другом, а после этого охлаждали до 10°С. Получающийся в результате конденсат возвращали во второй реактор, в то время как для хлористого водорода, полученного в виде несконденсированного газа, проводили нейтрализацию под действием водного щелочного раствора и выпуск.
Стадия промывки
Полученную жидкую реакционную смесь, дифенилкарбонат, извлеченный из описанной далее колонны извлечения/перегонки, и водный раствор гидроксида натрия с концентрацией, приблизительно равной 5 мас.% (жидкость, полученную в результате перемешивания водного раствора гидроксида натрия с концентрацией 25 мас.% и водной фазы, выделенной после проведения последующей стадии водной промывки), подавали в емкость нейтрализации/перемешивания, снабженную тефлоновой футеровкой. Для содержимого проводили перемешивание при 80°С в течение приблизительно 10 минут и регулировку, обеспечивающую выдерживание значения рН, равного 8,5. После проведения нейтрализации органическую фазу отделяли, используя отстаивание, а после этого перепускали в емкость водной промывки/перемешивания. С другой стороны, водную фазу, остающуюся после разделения (которая содержит фенол и поваренную соль), вводили в контакт с водяным паром, в результате чего присутствующий фенол почти полностью извлекали в виде фенолсодержащей воды. Данную воду подавали в емкость водной промывки/перемешивания на последующей стадии. В емкости водной промывки/перемешивания органическую фазу промывали, используя теплую воду в количестве, приблизительно равном 30 мас.% при расчете на количество органической фазы. Водную фазу (которую отправляли на рецикл в емкость нейтрализации/перемешивания) выделяли и получали "сырой" дифенилкарбонат (содержащий воду, пиридиновый катализатор и фенол).
Стадия перегонки низкокипящих соединений
После этого "сырой" дифенилкарбонат и водный раствор гидроксида натрия с концентрацией 0,1 н. непрерывно подавали на среднюю тарелку колонны перегонки низкокипящих соединений при расходах, приблизительно равных 9 кг/час и 15 мл/час соответственно. В качестве данной колонны перегонки низкокипящих соединений использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 100 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 220°С, температуры верха колонны 80-100°С и флегмового числа 1 для отгонки соединений, характеризующихся температурой кипения, более низкой по сравнению с температурой кипения дифенилкарбоната, то есть воды, пиридинового катализатора и не вступившего в реакцию фенола. После обезвоживания низкокипящих соединений часть данных соединений отводили, а остальное количество подавали в первый реактор для фосгенирования. С другой стороны, дифенилкарбонат выпускали из куба колонны при производительности, приблизительно равной 7,8 кг/час. Воду в нем обнаружить было нельзя (10 ч/млн либо менее), нельзя было обнаружить и уровни содержания пиридина и фенола (1 ч/млн либо менее) и 50 ч/млн соответственно.
Стадия перегонки высококипящих соединений
Кроме того, данный дифенилкарбонат (кубовый остаток из колонны перегонки низкокипящих соединений) непрерывно подавали в колонну перегонки высококипящих соединений. В качестве данной колонны перегонки высококипящих соединений использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 100 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны, приблизительно равной 180°С, флегмового числа 0,5 и доли дистиллята, приблизительно равной 90%. В результате через верх колонны получали очищенный дифенилкарбонат при производительности, приблизительно равной 7,1 кг/час (7 л/час), а высококипящие соединения отводили из куба колонны при производительности, приблизительно равной 0,8 кг/час. Очищенный дифенилкарбонат представлял собой высокочистый продукт, содержащий фенол в количестве 80 ч/млн. Определенное количество (приблизительно 100 л) данного дифенилкарбоната хранили при 100°С в резервуаре, изготовленном из материала SUS316, характеризующемся вместимостью 200 л (у которого значение величины Vd/Fd, указанной в описании, составляло 29), а после этого в качестве исходного материала для получения РС подавали при расходе, равном 7 л/час.
Стадия извлечения/перегонки
Кроме того, кубовый остаток, отводимый из куба колонны перегонки высококипящих соединений, одновременно подавали в колонну извлечения/перегонки для проведения непрерывной перегонки при следующих далее условиях. Дифенилкарбонат извлекали через верх колонны при производительности, приблизительно равной 0,7 кг/час, и отправляли на рецикл в емкость нейтрализации/перемешивания, в то время как кубовый остаток непрерывно отводили при производительности, приблизительно равной 0,04 кг/час. Что касается условий извлечения/перегонки при получении дифенилкарбоната, то использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 50 мм и высоту 3,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны 180°С и флегмового числа 0,5. В кубовом остатке из колонны извлечения/перегонки обнаружили алкилзамещенные производные дифенилкарбоната и бромзамещенные производные дифенилкарбоната в количествах, равных приблизительно 7000 ч/млн (мас.) и приблизительно 800 ч/млн соответственно.
Получение РС
Стадия полимеризации
В атмосфере газообразного азота бисфенол А, полученный выше и подаваемый при расходе, равном 7,2 кг/час, перемешивали в расплаве при 130°С с дифенилкарбонатом, подаваемым при расходе, равном 7,1 кг/час. Данную смесь непрерывно подавали через трубу для ввода исходного материала, нагреваемую до 130°С, в первый вертикальный реактор полимеризации с перемешиванием, в которой в результате регулировки выдерживали температуру 210°С в атмосфере азота при обычном давлении. Уровень жидкости выдерживали постоянным, проводя регулировку степени открытости клапана, расположенного на линии выпуска полимера, соединенной с нижней частью емкости, таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам. Кроме того, одновременно с инициированием подачи смеси исходных материалов в качестве катализатора начинали непрерывно подавать карбонат цезия в форме водного раствора при расходе, равном 0,5·10-6 моль при расчете на один моль бисфенола А. После этого жидкую полимеризационную реакционную смесь, выпускаемую из нижней части емкости, непрерывно последовательно подавали во второй, третий и четвертый вертикальные реакторы полимеризации и пятый горизонтальный реактор полимеризации. Во время прохождения реакции уровень жидкости в каждой емкости регулировали таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам, и одновременно с этим отгоняли фенол, полученный в качестве побочного продукта. Каждый из газов, получаемых в результате выпаривания и выпускаемых соответственно из емкостей в диапазоне от первого до третьего реакторов полимеризации, конденсировали и ожижали при использовании многостадийного конденсатора и часть конденсата в результате кипячения в условиях флегмообразования подавали в реактор полимеризации, а оставшуюся часть извлекали и хранили в резервуаре для являющегося побочным продуктом фенола. С другой стороны, каждый из газов, полученных в результате выпаривания и выпускаемых из четвертого и пятого реакторов полимеризации, отверждали при использовании одного из двух замораживающих конденсаторов, расположенных параллельно. Получающуюся в результате твердую фазу расплавляли в результате переключения на другой замораживающий конденсатор и извлекали и хранили в резервуаре для являющегося побочным продуктом фенола.
Условия проведения полимеризации для каждой реакционной емкости представляли собой нижеследующее: второй реактор полимеризации (210°C, 100 торр), третий реактор полимеризации (240°С, 15 торр), четвертый реактор полимеризации (260°С, 0,5 торр) и пятый реактор полимеризации (270°С, 0,5 торр). Система функционировала при производительности при получении поликарбоната, равной 8,0 кг/час.
После этого получающийся в результате полимер в расплавленном состоянии вводили в двухчервячный экструдер (произведенный в компании Kobe Steel, Ltd.) и сюда же непрерывно добавляли бутил-п-толуолсульфонат в количестве 5 ч/млн при расчете на количество поликарбоната. Бутил-п-толуолсульфонат находился в форме маточной смеси, полученной в результате диспергирования маточного раствора в хлопьях поликарбоната при использовании смесителя, и его в атмосфере азота подавали в экструдер при использовании массового питателя. Полимер подвергали гранулированию. Полученный таким образом поликарбонат характеризовался величиной Mv, равной 21000, и начальным значением YI 1,7.
Молекулярная масса (Mv)
Для измерения удельной вязкости (ηsp) при помощи вискозиметра Убеллоде при температуре 20°С использовали раствор в метиленхлориде, характеризующийся концентрацией поликарбоната (С), равной 0,6 г/дл. Молекулярная масса представляла собой значение, рассчитанное по значению вязкости при использовании следующих далее двух уравнений:
ηsp/С=[η](1+0,28ηsp)
[η]=1,23Χ10-4(Mv)0,83
Начальный цветовой тон (YI)
Поликарбонатную смолу высушивали при 120°С в течение 6 часов в атмосфере азота, а после этого при использовании литьевой машины J-100, произведенной в компании The Japan Steel Works, Ltd., по способу литьевого формования при 360°С из него получали кусок, имеющий толщину 3 мм. Данный кусок исследовали при использовании прибора SC-1, произведенного в компании Suga Test Instruments Co., Ltd., получая значение YI (чем больше будет значение YI, тем больше РС будет окрашен).
Стадия очистки являющегося побочным продуктом фенола
Являющийся побочным продуктом фенол, извлеченный со стадии полимеризации при производительности, приблизительно равной 6,3 кг/час (6 л/час), в определенном количестве (приблизительно 100 л) хранили в резервуаре для являющегося побочным продуктом фенола (резервуаре, изготовленном из материала SUS304, характеризующемся вместимостью 200 л; резервуар демонстрировал значение величины Vc/Fc, указанной в описании, равное 33), а после этого подвергали нижеследующим перегонке/очистке. Получающийся в результате очищенный фенол в качестве исходного материала для получения DPC отправляли на рецикл. Недостаток отчасти покрывали, используя коммерческий фенол.
Первую перегонную колонну эксплуатировали при 200 торр и флегмовом числе 2, благодаря чему присутствующую воду отгоняли совместно с частью фенола. Кубовый остаток подавали в колонну второй перегонки. Колонну второй перегонки эксплуатировали при 50 торр и флегмовом числе 0,5 и через верх колонны получали очищенный фенол при производительности, приблизительно равной 5,8 кг/час. Данный очищенный фенол подавали на стадию получения DPC через резервуар для очищенного фенола. С другой стороны, жидкую фенольную смесь, содержащую дифенилкарбонат, бисфенол А и олигомеры в количествах, равных 67 мас.%, 7 мас.% и 4 мас.% соответственно, непрерывно выпускали в качестве кубового остатка.
Эксперимент, описанный выше, непрерывно проводили в течение 400 часов. В ходе эксперимента становилось необходимо временно прекращать проведение стадии получения DPC вследствие возникновения проблемы, связанной с регулировкой температуры и тому подобным. Однако, поскольку использовали жидкость, хранящуюся в резервуаре для хранения, никакой необходимости в прекращении функционирования всей системы не было, и можно было быстро принимать меры по устранению проблемы. В ходе проведения операции получали марку поликарбоната, характеризующуюся величиной Mv, равной 15000, что в результате приводило к возникновению временных изменений в композиции являющегося побочным продуктом фенола, извлеченного из реактора полимеризации. Однако изменения можно было уменьшить в резервуаре для хранения, а условия проведения операций на последующей стадии очистки являющегося побочным продуктом фенола можно было плавно изменять.
Пример 5
Далее изобретение будет подробно разъяснено в связи с переработкой отработанного раствора. Между прочим, методы, использованные для измерения средневязкостной молекулярной массы (Mv) и начального цветового тона (YI), описывались в настоящем документе выше.
Экспериментальный пример 1
Стадия получения DPC
Стадию получения DPC проводили в соответствии со схемой технологического процесса, продемонстрированной на фиг.1. Подробности представляли собой нижеследующее.
Стадия реакции при получении DPC
Во время непрерывной подачи в реактор получения DPC 1 расплавленного PL и пиридинового катализатора при перемешивании при 150°С сюда же непрерывно подавали газообразный фосген (CDC). После этого смесь подавали в колонну дегидрохлорирования 2. Газообразный хлористый водород (D1), полученный в качестве побочного продукта в результате фосгенирования, охлаждали до 10°С. Получающийся в результате конденсат возвращали в реактор, в то время как для газа, оставшегося несконденсированным, проводили нейтрализацию, используя водный щелочной раствор, и после этого выпуск. С другой стороны, дегидрохлорированную жидкость b, содержащую DPC в количестве, приблизительно равном 91 мас.%, непрерывно выпускали из колонны дегидрохлорирования 2.
Стадия промывки DPC/стадия водной промывки DPC
Гидрохлорированную жидкость b подавали в смесительную емкость 3, а после этого в емкость щелочной нейтрализации 4, снабженную тефлоновой футеровкой. Кроме того, в емкость щелочной нейтрализации 4 подавали водный раствор гидроксида натрия с концентрацией, приблизительно равной 5 мас.%. Для содержимого проводили перемешивание при 80°С в течение приблизительно 10 минут и регулировку, обеспечивающую выдерживание значения рН, равного 8,5. После проведения нейтрализации органическую фазу отделяли, используя отстаивание, а после этого перепускали в емкость водной промывки 5. В емкости водной промывки 5 органическую фазу промывали, используя теплую воду в количестве, приблизительно равном 30 мас.% при расчете на количество органической фазы. Водную фазу отделяли и получали жидкость, подвергнутую водной промывке, f, которая представляла собой "сырой" DPC (содержащий 1 мас.% воды, 2 мас.% пиридина, 8 мас.% PL и 89 мас.% DPC).
Стадия перегонки при получении DPC/стадия перегонки низкокипящих соединений
После этого жидкость, подвергнутую водной промывке, f, непрерывно подавали на среднюю тарелку колонны первой перегонки PL 6 при расходе, приблизительно равном 30 кг/час. В качестве данной колонны первой перегонки PL 6 использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 150 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 220°С, температуры верха колонны 80-100°С, температуры средней тарелки 160°С и флегмового числа 1 для отгонки смешанного газа F, содержащего соединения, характеризующиеся температурой кипения, более низкой по сравнению с температурой кипения DPC, то есть воду, пиридин и PL. Из куба колонны при производительности, приблизительно равной 26 кг/час, непрерывно выпускали остаток после первой перегонки g, который представлял собой кубовый остаток, содержащий DPC, (уровни содержания воды 10 ч/млн (мас.) либо менее; пиридина 1 ч/млн (мас.) либо менее; PL 50 ч/млн (мас.)).
Стадия перегонки при получении DPC/стадия перегонки высококипящих соединений
Кроме того, данный остаток после первой перегонки g непрерывно подавали в колонну второй перегонки PL 7. В качестве данной колонны второй перегонки PL 7 использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 200 мм и высоту 4,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны, приблизительно равной 180°С, и флегмового числа 0,5. В результате через верх колонны получали очищенный DPC при производительности, приблизительно равной 23,5 кг/час, а остаток после перегонки при получении DPC (X1), который представлял собой высококипящие соединения (DPC, содержащий алкилзамещенные производные DPC и бромзамещенные производные DPC в количествах, равных приблизительно 350 ч/млн (мас.) и приблизительно 40 ч/млн (мас.) соответственно), отводили из куба колонны при производительности, приблизительно равной 2,5 кг/час. Очищенный DPC представлял собой высокочистый продукт, содержащий PL в количестве 80 ч/млн (мас.).
Стадия получения РС
Стадия полимеризации при получении РС
РС получали в соответствии со схемой технологического процесса, продемонстрированной на фиг.5, следующим образом. В атмосфере газообразного азота очищенный DPC, полученный на стадии получения DPC, и ВРА перемешивали в расплаве при соотношении 0,977 (мас.) при использовании смесительной емкости 21. Данную смесь непрерывно подавали через трубу для подачи исходного материала, нагреваемую до 130°С, в первый вертикальный реактор полимеризации 22, в котором в результате регулировки выдерживали температуру 210°С в атмосфере азота при обычном давлении. Уровень жидкости выдерживали постоянным, проводя регулировку степени открытости клапана, расположенного на линии выпуска полимера, соединенной с нижней частью емкости, таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам. Кроме того, одновременно с инициированием подачи смеси исходных материалов в качестве катализатора начинали непрерывно подавать карбонат цезия в форме водного раствора при расходе, равном 0,5·10-6 моль при расчете на один моль ВРА. После этого жидкую полимеризационную реакционную смесь, выпускаемую из нижней части емкости, непрерывно последовательно подавали во второй и третий реакторы полимеризации и четвертый горизонтальный полимеризатор. Во время прохождения реакции уровень жидкости в каждой емкости регулировали таким образом, чтобы в результате получить среднее время пребывания, равное 60 минутам, и одновременно с этим отгоняли PL, полученный в качестве побочного продукта. Превращенные в пар ингредиенты при получении РС р, выпускаемые из емкостей в диапазоне от первого до третьего реакторов полимеризации, конденсировали и ожижали при использовании соответствующих многостадийных конденсаторов. Часть каждого из получающихся в результате конденсатов, проводя кипячение в условиях флегмообразования, подавали в реактор полимеризации, а оставшуюся часть извлекали и хранили в резервуаре для извлеченного PL 29 при получении РС. С другой стороны, газы, полученные в результате выпаривания и выпускаемые из четвертого полимеризатора, отверждали при использовании одного из двух замораживающих конденсаторов, расположенных параллельно. Получающуюся в результате твердую фазу расплавляли в результате переключения на другой замораживающий конденсатор и извлекали и хранили в резервуаре для извлеченного PL 29 при получении РС (не показан).
Условия проведения полимеризации для каждой реакционной емкости представляли собой нижеследующее: первый реактор полимеризации (210°C, 100 торр), второй реактор полимеризации (240°С, 15 торр), третий реактор полимеризации (260°С, 0,5 торр) и четвертый полимеризатор (270°С, 0,5 торр).
Получающийся в результате полимер в расплавленном состоянии вводили в двухчервячный экструдер (произведенный в компании Kobe Steel, Ltd.; диаметр червяка 0,046 м; L/D=40,2) 32 и гранулировали при одновременном добавлении сюда бутил-п-толуолсульфоната в количестве 5 ч/млн (мас.) при расчете на количество РС. Таким образом полученный РС характеризовался средневязкостной молекулярной массой (Mv), равной 21000, и начальным цветовым тоном (YI), соответствующим 1,7.
Стадия перегонки PL
Анализировали превращенные в пар ингредиенты при получении РС р, извлекаемые на стадии полимеризации при получении РС. В результате обнаружили 5,0 мас.% DPC, 0,5 мас.% ВРА, 0,3 мас.% олигомеров и 0,2 мас.% воды.
Превращенные в пар ингредиенты при получении РС р непрерывно очищали при использовании следующих двух перегонных колонн (колонна первой перегонки PL 30 и колонна второй перегонки PL 31). Колонну первой перегонки PL 30 эксплуатировали при 200 торр и флегмовом числе 2, благодаря чему присутствующую воду отгоняли совместно с частью PL в качестве низкокипящего дистиллята при получении РС (D6). Кубовый остаток непрерывно подавали в колонну второй перегонки PL 31 в качестве остатка первой стадии q. Колонну второй перегонки PL 31 эксплуатировали при 50 торр и флегмовом числе 0,5 и через верх колонны получали очищенный PL. Остаток после перегонки PL (X2), содержащий DPC, BPA и олигомеры в количествах, равных 67 мас.%, 7 мас.% и 4 мас.% соответственно, непрерывно выпускали в качестве кубового остатка.
(Пример, в котором остаток после перегонки Х2 на стадии получения РС подавали на стадию перегонки на стадии получения DPC)
Остаток после перегонки PL (X2; содержащий 22 мас.% PL, 67 мас.% DPC, 7 мас.% ВРА и 4 мас.% олигомеров), отводимый со стадии очистки являющегося побочным продуктом PL на описанной выше стадии получения РС, подавали в колонну первой перегонки при получении DPC 6 на описанной выше стадии получения DPC. Затем остаток после первой перегонки g, который представлял собой кубовый остаток из колонны первой перегонки при получении DPC 6, подавали в колонну второй перегонки при получении DPC 7. В результате через верх колонны первой перегонки при получении DPC 6 извлекали почти что 100% PL, содержащегося в остатке после перегонки PL (Х2), а через верх колонны второй перегонки при получении DPC 7 извлекали приблизительно половину DPC. Таким образом, эффективные ингредиенты можно было эффективно извлекать просто в результате отправления остатка после перегонки PL (X2) на рецикл на существующие указанные стадии.
DPC и РС получали по способам, описанным выше, при одновременном проведении продемонстрированной выше операции. В результате полученным DPC и РС проблема, связанная с качеством, совершенно была несвойственна.
Экспериментальный пример 2
Стадия получения DPC
Стадию получения DPC проводили в соответствии со схемой технологического процесса, продемонстрированной на фиг.1. Подробности представляли собой нижеследующее.
Стадия извлечения/перегонки
Остаток после перегонки при получении DPC (X1), отводимый со стадии перегонки высококипящих соединений на стадии получения DPC в экспериментальном примере 1, подавали в колонну извлечения/перегонки при получении DPC 8 и проводили непрерывную перегонку при нижеследующих условиях для извлечения в качестве дистиллята через верх колонны извлеченной жидкости, содержащей DPC, d. Извлеченный дистиллят отправляли на рецикл в смесительную емкость на стадии получения DPC, добиваясь достижения улучшенного выхода. С другой стороны, остаток после извлечения/перегонки при получении DPC (X1'), который представлял собой высококипящие соединения (DPC, содержащий алкилзамещенные производные DPC и бромзамещенные производные DPC в количествах, равных приблизительно 7000 ч/млн (мас.) и приблизительно 800 ч/млн (мас.) соответственно), непрерывно отводили в качестве кубового остатка из колонны извлечения/перегонки при получении DPC.
Для извлечения/перегонки при получении DPC использовали колонну непрерывной перегонки, характеризующуюся числом теоретических тарелок, равным 8, которая имела внутренний диаметр 100 мм и высоту 3,0 м, была снабжена сверху рефлюксным аппаратом, имела в своей середине часть для подачи исходного материала и имела участок концентрирования и участок извлечения, набивкой каждого из которых служила набивка Sulzer Packing (произведенная в компании Sumitomo Heavy Industries, Ltd.). Перегонку проводили в условиях степени вакуумирования 20 торр, температуры масла-теплоносителя, приблизительно равной 240°С, температуры верха колонны 180°С и флегмового числа 0,5.
Другие операции проводили по тому же самому способу, что и в экспериментальном примере 1. Таким образом, в виде высокочистого продукта получали очищенный DPC (содержащий PL в количестве, равном 80 ч/млн (мас.)) и синтезировали PC.
(Пример, в котором остаток после перегонки (Х2) на стадии получения РС подавали на стадию извлечения/перегонки на стадии получения DPC)
Остаток после перегонки PL (X2; содержащий 22 мас.% PL, 67 мас.% DPC, 7 мас.% ВРА и 4 мас.% олигомеров), отводимый со стадии перегонки PL на описанной выше стадии получения РС, подавали в колонну извлечения/перегонки при получении DPC 8 на стадии получения DPC. В результате через верх колонны извлечения/перегонки извлекали почти что 100% PL, содержащегося в остатке после перегонки, и приблизительно 80 мас.% DPC, содержащегося в остатке. Таким образом, эффективные ингредиенты можно было эффективно извлекать просто в результате отправления остатка после перегонки PL X2 на рецикл на существующие указанные стадии.
DPC и РС получали по способам, описанным выше, при одновременном проведении продемонстрированной выше операции. В результате полученным DPC и РС проблема, связанная с качеством, совершенно была несвойственна.
Экспериментальный пример 3
Стадия получения ВРА
Стадию получения ВРА проводили в соответствии со схемами технологических процессов, продемонстрированными на фиг.2-4. В реактор получения ВРА проточного типа, снабженный термостатом, в количестве 60 л помещали набивку в виде кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.), в которой 15% сульфогрупп было нейтрализовано под действием 4-пиридинэтантиола. В данный реактор получения ВРА при температуре 80°С и расходе 68,2 кг/час вводили жидкую смесь PL и ацетона с молярным соотношением 10:1, обеспечивая протекание реакции между ними. Степень превращения ацетона составляла 80%. После того как низкокипящие соединения (не вступивший в реакцию ацетон, воду и часть PL) при производительности, равной 5,1 кг/час, удаляли, реакционную смесь охлаждали до 50°С, добиваясь осаждения кристаллов аддукта. Данную смесь отфильтровывали для ее разделения на кристаллы аддукта и маточную жидкость. Производительности при их получении составляли 16,5 кг/час и 46,5 кг/час соответственно. Десять массовых процентов данной маточной жидкости подавали на стадию переработки маточной жидкости, тогда как для остальной маточной жидкости как части исходного материала, вводимого в реактор синтеза, проводили циркуляцию.
Таким образом полученные кристаллы аддукта повторно растворяли в феноле, подаваемом при расходе, равном 27,2 кг/час. Получающийся в результате раствор охлаждали до 50°С, добиваясь осаждения кристаллов, а после этого отфильтровывали для отделения кристаллов аддукта (11,3 кг/час) от маточной жидкости (32,5 кг/час). Выделенные кристаллы нагревали до 180°С при пониженном давлении 0,3 мм ртутного столба для удаления PL. Таким образом, при производительности, равной 7,7 кг/час, получали ВРА, характеризующийся степенью чистоты, равной 99,95% либо более.
С другой стороны, маточную жидкость, подаваемую на стадию переработки маточной жидкости, подвергали переработке с использованием испарителя PL 13, продемонстрированного на фиг.4, для отгонки части PL и концентрирования маточной жидкости. После этого к маточной жидкости добавляли 0,1 мас.% гидроксида натрия и затем маточную жидкость вводили в нижнюю часть реактора переработки остатка 14, выступающий в роли колонны разложения/перегонки, в котором в результате регулировки выдерживали пониженное давление 50 мм ртутного столба и температуру 210°С. Данный реактор эксплуатировали при таких условиях, чтобы выдерживать в нижней части постоянство уровня жидкости (время пребывания 1 час), а кубовый остаток в колонне разложения/перегонки отводили из системы при производительности, равной 0,5 кг/час. Кроме того, дистиллят, выпускаемый через верх реактора переработки остатка 14, перемешивали с PL, а данную смесь при расходе, равном 4,2 кг/час, вводили в регенерирующий реактор проточного типа 15, снабженный набивкой в виде 4 л кислотной катионообменной смолы в форме сульфоновой кислоты (торговое наименование DIAION SK-104, произведенной в компании Mitsubishi Chemical Corp.). Реакцию для смеси проводили в условиях 80°С. Для полученной жидкой реакционной смеси проводили циркуляцию с подачей в реактор получения ВРА, использованный на начальной стадии.
Ацетон (3,6 кг/час) и PL (18,5 кг/час) дополнительно подавали в реактор получения ВРА в количествах, соответствующих количествам, отводимым из системы, и количеству полученного ВРА. Таким образом, реакцию синтеза проводили непрерывно, и вся система функционировала при непрерывном получении ВРА.
(Пример, в котором остаток после перегонки при получении DPC (Х1) на стадии получения DPC и остаток после перегонки PL (Х2) на стадии получения РС подавали на стадию переработки маточной жидкости на стадии получения ВРА)
Остаток после перегонки при получении DPC (Х1; содержащий приблизительно 350 ч/млн (мас.) алкилзамещенных производных DPC, приблизительно 40 ч/млн (мас.) бромзамещенных производных DPC и DPC, составляющий остаток), выпускаемый со стадии получения DPC, и остаток после перегонки PL (X2; содержащий 22 мас.% PL, 67 мас.% DPC, 7 мас.% ВРА и 4 мас.% олигомеров), отводимый со стадии очистки являющегося побочным продуктом PL на стадии получения РС, подавали при расходах, равных 0,11 кг/час и 0,15 кг/час соответственно, на стадию переработки маточной жидкости (стадию (g)) на стадии получения ВРА. В ходе проведения данной операции реактор переработки остатка 14 функционировал при таких условиях, которые позволяли выдерживать постоянство уровня жидкости в нижней части (время пребывания 1 час) и отводить из системы нижний продукт при производительности, равной 0,6 кг/час.
В результате через верх реактора переработки остатка 14 на стадии переработки маточной жидкости извлекали почти что 100% PL, содержащегося в данных остатках после перегонки и продуктах разложения ВРА. Таким образом, эффективные ингредиенты можно было эффективно извлекать просто в результате отправления остатков после перегонки на рецикл на существующую указанную стадию.
ВРА, DPC и РС получали по способам, описанным выше, при одновременном проведении продемонстрированной выше операции. В результате полученным ВРА, DPC и РС проблема, связанная с качеством, совершенно была несвойственна.
Экспериментальный пример 4
(Пример, в котором остаток после извлечения/перегонки при получении DPC (Х1') на стадии получения DPC и остаток после перегонки PL (Х2) на стадии получения РС подавали на стадию переработки маточной жидкости на стадии получения ВРА)
Остаток после извлечения/перегонки при получении DPC (Х1'; содержащий приблизительно 7000 ч/млн (мас.) алкилзамещенных производных DPC, приблизительно 800 ч/млн (мас.) бромзамещенных производных DPC и DPC, составляющий остаток), выпускаемый со стадии получения DPC, и остаток после перегонки PL (X2; содержащий 22 мас.% PL, 67 мас.% DPC, 7 мас.% ВРА и 4 мас.% олигомеров), отводимый со стадии очистки являющегося побочным продуктом PL на стадии получения РС, подавали при расходах, равных 0,11 кг/час и 0,15 кг/час соответственно, на стадию переработки маточной жидкости (стадию (g)) на стадии получения ВРА. В ходе проведения данной операции реактор переработки остатка 14 функционировал при таких условиях, которые позволяли выдерживать постоянство уровня жидкости в нижней части (время пребывания 1 час) и отводить из системы нижний продукт при производительности, равной 0,6 кг/час.
В результате через верх реактора переработки остатка 14 на стадии переработки маточной жидкости извлекали почти что 100% PL, содержащегося в данных остатках после перегонки и продуктах разложения ВРА. Таким образом, эффективные ингредиенты можно было эффективно извлекать просто в результате отправления остатков после перегонки на рецикл на существующую указанную стадию.
ВРА, DPC и РС получали по способам, описанным выше, при одновременном проведении продемонстрированной выше операции. В результате полученным ВРА, DPC и РС проблема, связанная с качеством, совершенно была несвойственна.
Экспериментальный пример 5
(Пример, в котором остаток после перегонки PL (Х2) на стадии получения PC подавали на стадию извлечения/перегонки на стадии получения DPC, а остаток после извлечения/перегонки при получении DPC (Х1') подавали на стадию переработки маточной жидкости на стадии получения ВРА)
Остаток после перегонки PL (X2; содержащий 22 мас.% PL, 67 мас.% DPC, 7 мас.% ВРА и 4 мас.% олигомеров), отводимый со стадии очистки являющегося побочным продуктом PL на стадии получения РС, подавали в колонну извлечения/перегонки при получении DPC 8 на стадии получения DPC. Эффективный ингредиент, выпускаемый в качестве дистиллята из колонны извлечения/перегонки при получении DPC 8, отправляли на рецикл на стадию промывки на стадии получения DPC, а остаток после извлечения/перегонки при получении DPC (Х1') в качестве кубового остатка из колонны извлечения/перегонки при получении DPC 8 подавали на стадию переработки маточной жидкости (стадию (g)) на стадии получения ВРА. Эффективный ингредиент, извлеченный на данной стадии переработки, использовали в качестве исходного материала для синтеза на стадии получения ВРА. ВРА, DPC и РС непрерывно получали в течение 400 часов при одновременном проведении продемонстрированной выше операции. В результате полученным ВРА, DPC и РС проблема, связанная с качеством, совершенно была несвойственна. Высококипящие отходы можно было интегрировать в один поток, выпускаемый со стадии переработки маточной жидкости на стадии получения ВРА. Количество отходов значительно уменьшалось, а выход был значительно улучшен.
Несмотря на то, что изобретение было описано подробно и с использованием ссылок на конкретные варианты его реализации, специалисту в соответствующей области должно быть очевидно, что без отклонения от объема и сущности изобретения могут быть реализованы различные его изменения и модификации.
Данная заявка в своей основе имеет японскую патентную заявку, поданную 21 августа 2003 года (заявка № 2003-297844), японскую патентную заявку, поданную 21 августа 2003 года (заявка № 2003-297719), японскую патентную заявку, поданную 21 августа 2003 года (заявка № 2003-297832), японскую патентную заявку, поданную 12 ноября 2003 года (заявка № 2003-382667), японскую патентную заявку, поданную 12 ноября 2003 года (заявка № 2003-382773) и японскую патентную заявку, поданную 12 ноября 2003 года (заявка № 2003-382646), содержание которых во всей его полноте включается в настоящий документ для справки.
Применимость в промышленности
В соответствии с изобретением уровень содержания воды в являющемся побочным продуктом феноле ограничивают величиной в заданном диапазоне. По этой причине, даже если данный являющийся побочным продуктом фенол и используют в качестве части исходного материала, используемого на стадии получения дифенилкарбоната либо на стадии получения бисфенола А, то эффективность получения будет ухудшаться незначительно и оставаться почти что на прежнем уровне.
Кроме того, примеси, содержащиеся в являющемся побочном продукте феноле, извлеченном на раннем этапе полимеризации на стадии получения ароматического поликарбоната, представляют собой исходный материал, используемого на стадии получения дифенилкарбоната, и продукты реакции, получаемые на стадии. Следовательно, данный являющийся побочным продуктом фенол, который содержит данные примеси, без проведения очистки можно использовать в качестве, по меньшей мере, части фенола, используемого на стадии получения дифенилкарбоната.
С другой стороны, примеси, содержащие в являющемся побочном продуктом феноле, извлеченном на позднем этапе полимеризации на стадии получения ароматического поликарбоната, подвергаются гидролизу на стадии получения бисфенола А и становятся исходным материалом и продуктами реакции для данной стадии. В дополнение к этому в являющемся побочным продуктом феноле не содержится почти что никаких спиртов и тому подобного, что вызывало бы ухудшение каталитической активности на стадии получения бисфенола А. Следовательно, данный являющийся побочным продуктом фенол, который содержит данные примеси, без проведения очистки можно использовать в качестве, по меньшей мере, части фенола, используемого на стадии получения бисфенола А.
Кроме того, в результате реализации интегрального варианта проведения стадии получения дифенилкарбоната, стадии получения бисфенола А и стадии получения ароматического поликарбоната возможными становятся использование коммерческого фенола в качестве исходного материала при получении дифенилкарбоната и использование ингредиентов дистиллята, полученных на стадии полимеризации на стадии получения ароматического поликарбоната, в качестве исходного материала при получении бисфенола А.
Кроме того, если до и/или после проведения стадии перегонки PL проводить стадию хранения при получении РС, на которой хранят ожиженные превращенные в пар ингредиенты при получении РС, подвергаемые воздействию стадии перегонки PL, и/или являющийся побочным продуктом фенол, извлеченный на стадии перегонки PL, если после проведения стадии перегонки при получении DPC проводить стадию хранения при получении DPC, на которой хранят дифенилкарбонат, полученный на стадии перегонки при получении DPC, и/или, если в промежутке между стадией кристаллизации/выделения ВРА и стадией полимеризации при получении РС проводить стадию хранения при получении ВРА, на которой хранят смесь бисфенола А и фенола, то тогда качество соединения, полученного на каждой стадии, можно будет выдерживать в определенном диапазоне, а более поздние стадии можно будет проводить непрерывно независимо от предшествующих стадий.
Кроме того, если остаток после перегонки PL, остаток после перегонки при получении DPC и/или остаток после извлечения/перегонки при получении DPC подавать на указанную стадию, то тогда эффективные ингредиенты, содержащиеся в данных остатках, можно будет использовать полностью. Следовательно, совокупная эффективность может быть улучшена, а нагрузка для окружающей среды может быть уменьшена.
В дополнение к этому, если вакуумный трубопровод, который соединяет конденсатор с вакуумным устройством и является наклоненным, откорректировать таким образом, чтобы совокупная высота частей, поднимающихся кверху противоположно наклону, составляла бы 1 м либо менее, то тогда количество жидких и твердых веществ, остающихся в поднимающихся частях, может быть сведено к минимуму, и можно будет не допустить возникновения полного засорения трубопровода либо появления в нем чрезмерно больших потерь давления.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ И УСТАНОВКА ДЛЯ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТА | 2013 |
|
RU2622641C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА-А | 1994 |
|
RU2119906C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКОКСИДОВ АЛКИЛОЛОВА | 2005 |
|
RU2338749C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО ПОЛИКАРБОНАТА | 2007 |
|
RU2380382C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 2004 |
|
RU2329250C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА | 2013 |
|
RU2627266C2 |
| Способ получения бисфенола-А | 2019 |
|
RU2799337C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТА С ИСПОЛЬЗОВАНИЕМ СТРИППИНГОВОГО УСТРОЙСТВА | 2017 |
|
RU2752321C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТНОЙ СМОЛЫ | 2007 |
|
RU2430935C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 2005 |
|
RU2358967C2 |
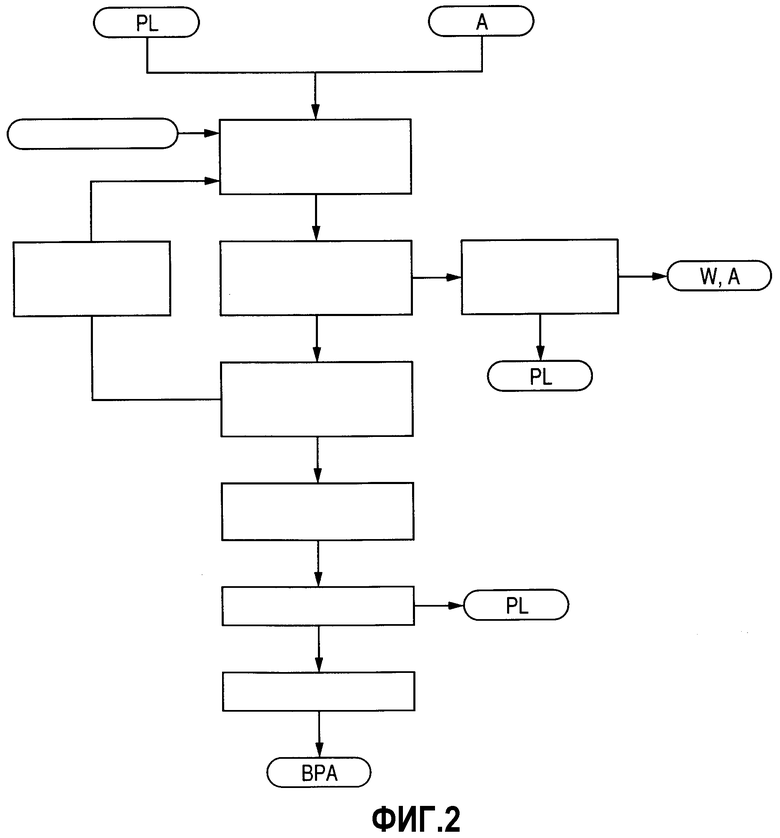


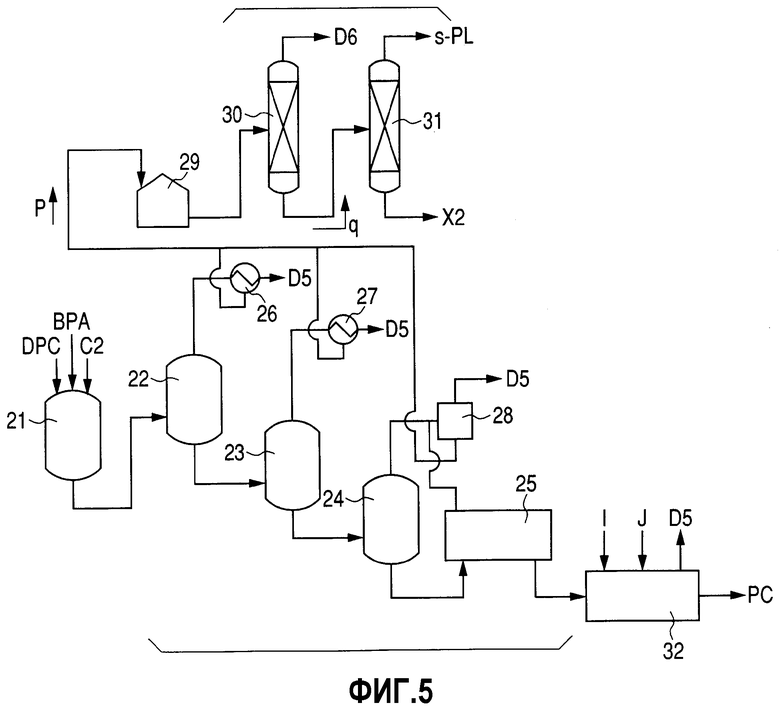
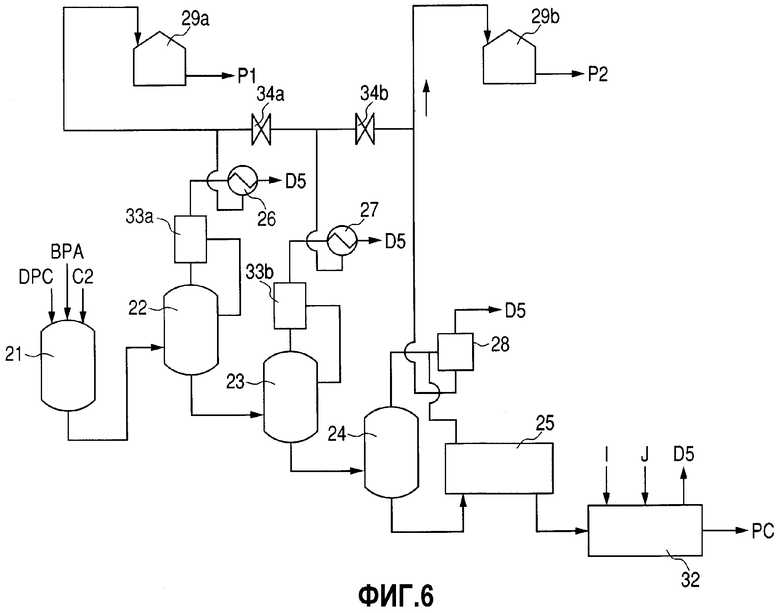
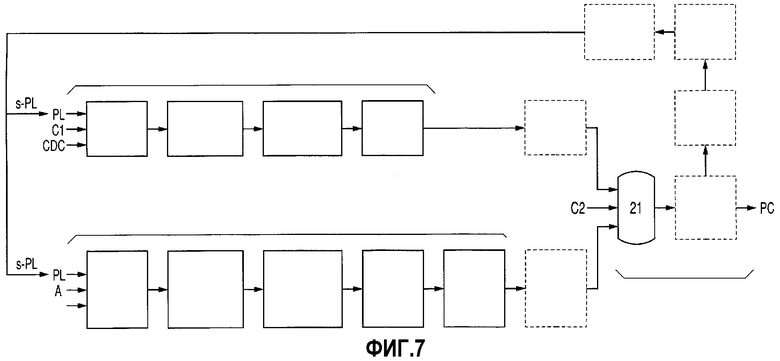
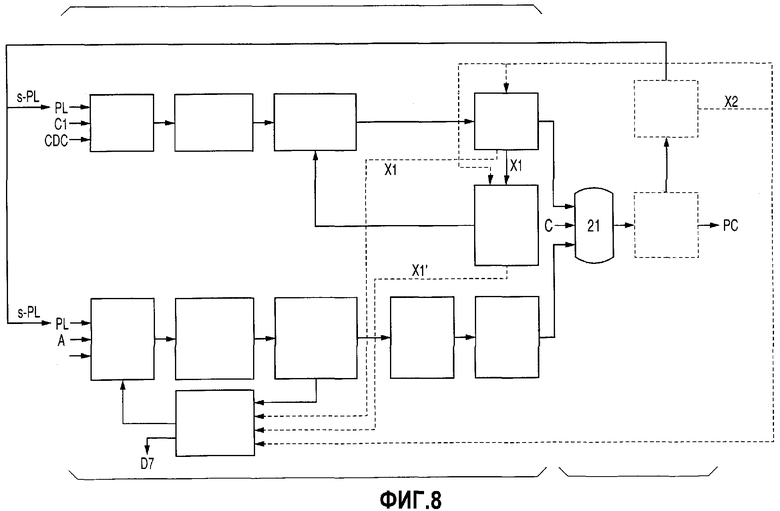
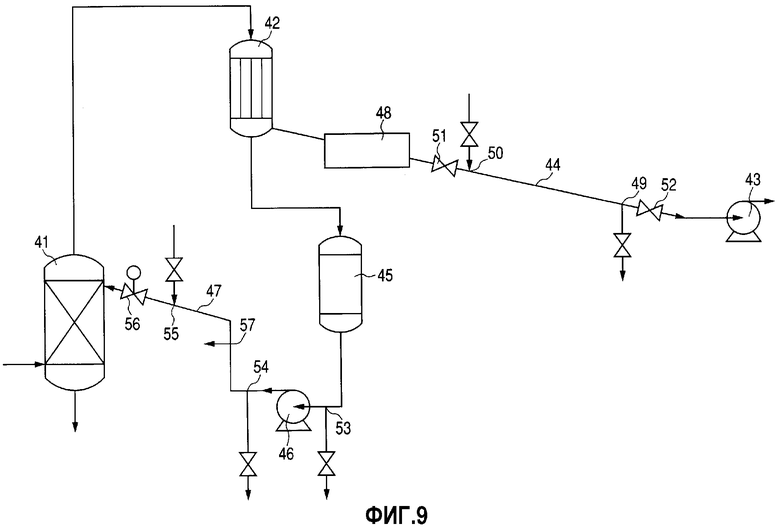
Изобретение относится к способу получения ароматического поликарбоната. Описан способ получения ароматического поликарбоната, который включает стадию получения дифенилкарбоната, на которой фенол и, по меньшей мере, одно карбонильное соединение используют в качестве исходных материалов при получении дифенилкарбоната, и/или стадию получения бисфенола А, на которой фенол и ацетон используют в качестве исходных материалов при получении бисфенола А, и стадию получения ароматического поликарбоната, на которой дифенилкарбонат и бисфенол А используют в качестве исходных материалов при получении ароматического поликарбоната в результате проведения стадии полимеризации ароматического карбоната, а побочный продукт фенол извлекают, отличающийся тем, что количество воды, содержащейся в побочном продукте феноле, извлеченном на стадии получения ароматического поликарбоната, устанавливают на уровне 0,2% мас.% либо менее, а данный фенол используют в качестве исходного материала на стадии получения дифенилкарбоната и/или стадии получения бисфенола А, и карбонильное соединение представляет собой фосген, окись углерода или диалкилкарбонат. Технический результат - решение проблемы побочного продукта фенола, полученного на стадии получения поликарбоната, на которой уровень воды ограничивают величиной в заданном диапазоне, а также эффективность получения ароматического поликарбоната. 42 з.п. ф-лы, 9 ил., 1 табл.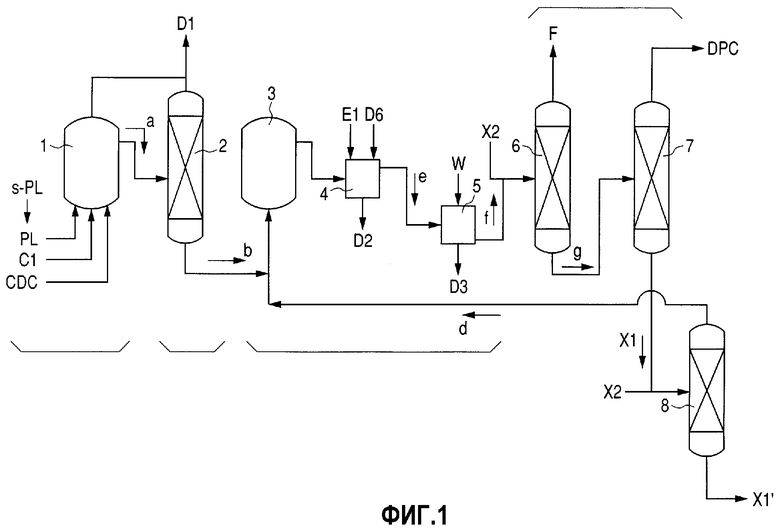
и стадию получения ароматического поликарбоната, на которой дифенилкарбонат и бисфенол А используют в качестве исходных материалов при получении ароматического поликарбоната в результате проведения стадии полимеризации ароматического карбоната, а побочный продукт фенол извлекают,
отличающийся тем, что количество воды, содержащейся в побочном продукте феноле, извлеченном на стадии получения ароматического поликарбоната, устанавливают на уровне 0,2 мас.% либо менее, а данный фенол используют в качестве исходного материала на стадии получения дифенилкарбоната и/или стадии получения бисфенола А, и карбонильное соединение представляет собой фосген, окись углерода или диалкилкарбонат.
низкокипящий дистиллят ароматического карбоната, получаемый на стадии перегонки фенола, подают на стадию промывки дифенилкарбоната.

(где Vc обозначает вместимость (м3) резервуара для хранения ароматического карбоната, a Fc обозначает расход (м3/ч) ожиженных испарившихся ингредиентов ароматического карбоната или побочного продукта фенола).

(где Vd обозначает вместимость (м3) резервуара для хранения дифенилкарбоната, a Fd обозначает расход (м3/ч) дифенилкарбоната).

(где Vb обозначает вместимость (м3) резервуара для хранения бисфенола А, а Fb обозначает расход (м3/ч) бисфенола А, подаваемого на стадию полимеризации ароматического карбоната).
вакуумный трубопровод наклонен книзу от стороны конденсатора к стороне вакуумного устройства, а совокупная высота частей, поднимающихся кверху от стороны конденсатора к стороне вакуумного устройства, составляет 1 м или менее.
вакуумный трубопровод наклонен книзу от стороны конденсатора к стороне вакуумного устройства, а совокупная высота частей, поднимающихся кверху от стороны конденсатора к стороне вакуумного устройства, составляет 1 м или менее.
стадия удаления воды представляет собой стадию выделения воды на стадии получения бисфенола А.
| US 6277945 B1, 21.08.2001 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТОВ | 0 |
|
SU219188A1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКОГО КАРБОНАТА | 1991 |
|
RU2041869C1 |
| СПОСОБ ПОЛУЧЕНИЯ БИСФЕНОЛА-А | 1994 |
|
RU2119906C1 |
| US 5747609 A, 05.05.1998 | |||
| JP 9255772 A, 30.09.1997. | |||

