Настоящее изобретение относится к тетрагидро-4H-пиридо[l,2-a]пиримидинам, родственным соединениям и их фармацевтически приемлемым солям, синтезу и применению этих соединений в качестве ингибиторов фермента ВИЧ-интегразы. Соединения и их фармацевтически приемлемые соли по настоящему изобретению полезны для профилактики или лечения ВИЧ-инфекции и для лечения или задержки проявления СПИДа.
Уровень техники
Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), является этиологическим фактором комплексного заболевания, включающего прогрессирующее разрушение иммунной системы (синдром приобретенного иммунодефицита; СПИД) и дегенерацию центральной и периферической нервной системы. Этот вирус ранее был известен как LAV, HTLV-III или ARV. Общим признаком репликации ретровирусов является вставка ДНК провируса, кодирующей вирусную интегразу в геном клетки-хозяина, обязательная стадия в репликации ВИЧ в T-лимфоцитах и моноцитарных клетках человека. Считается, что интеграция под действием интегразы происходит в три стадии: сборка стабильного нуклеопротеидного комплекса с последовательностями вирусной ДНК; отщепление двух нуклеотидов от 3' концов линейной ДНК провируса; ковалентное связывание освобожденных 3' OH-концов ДНК провируса с ДНК клетки-хозяина на ступенчато расположенных сайтах расщепления. Четвертая стадия процесса, репаративный синтез образовавшегося двухнитевого разрыва, может быть выполнена с помощью клеточных ферментов.
Секвенирование нуклеиновой кислоты ВИЧ свидетельствует о наличии pol-гена в одной открытой рамке считывания [Ratner, L. et al., Nature, 313, 277(1985)]. Гомология последовательностей аминокислот свидетельствует о том, что pol-последовательность кодирует обратную транскриптазу, интегразу и ВИЧ-протеазу [Toh, H. et al., EMBO J. 4, 1267 (1985); Power, M.D. et al., Science, 231, 1567 (1986); Pear1, L.H. et al., Nature, 329, 351 (1987)]. Установлено, что все три фермента необходимы для репликации ВИЧ.
Известно, что некоторые противовирусные соединения, действующие как ингибиторы репликации ВИЧ, являются эффективными средствами в лечении СПИДа и схожих заболеваний, включая ингибиторы обратной транскриптазы, такие как азидотимин (AZT) и эфавиренз, и ингибиторы протеазы, такие как индинавир и нелфинавир. Соединения по настоящему изобретению являются ингибиторами ВИЧ-интегразы и ингибиторами репликации ВИЧ. Ингибирование интегразы in vitro и репликации ВИЧ в клетках является прямым следствием ингибирования реакции переноса цепи, катализируемой рекомбинантной интегразой in vitro в клетках, зараженных ВИЧ. Особое преимущество настоящего изобретения состоит в высокоспецифичном ингибировании ВИЧ-интегразы и репликации ВИЧ.
Сущность изобретения
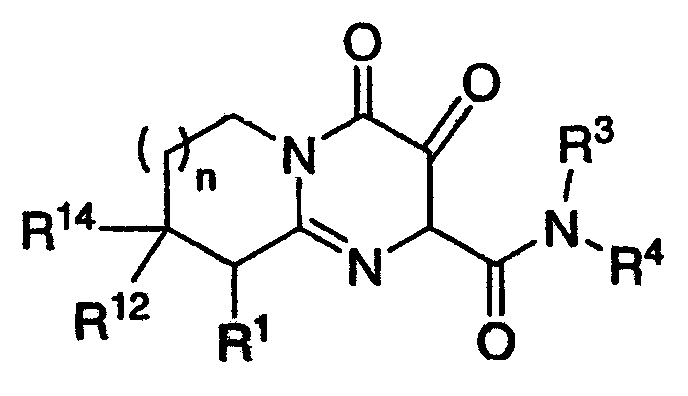
Настоящее изобретение касается новых пиридопиримидиновых производных и родственных соединений. Эти соединения полезны в ингибировании ВИЧ-интегразы, профилактике ВИЧ-инфекции и в профилактике, лечении или задержке проявления СПИДа и/или СПИД-ассоциированного комплекса (ARC), либо в виде их соединений, либо в их виде фармацевтически приемлемых солей или гидратов (когда это целесообразно), либо в качестве ингредиентов фармацевтических композиций, в комбинации с другими ВИЧ/СПИД-противовирусными средствами, противоинфекционными средствами, иммуномодуляторами, антибиотиками или вакцинами, либо в отсутствие указанных средств. Более конкретно, настоящее изобретение включает соединения формулы I и их фармацевтически приемлемые соли:
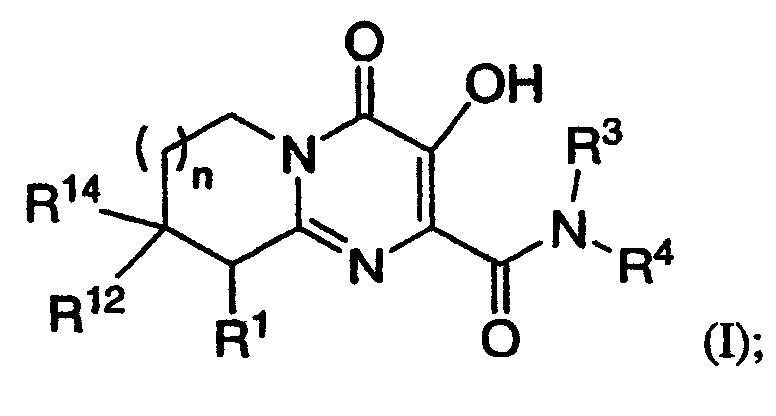
R1 означает H или NR2R5;
R2 означает CH3;
R5 означает
1) C(O)CH2SO2CH3,
2) C(O)C(O)N(CH3)2,
3) SO2N(CH3)2 или
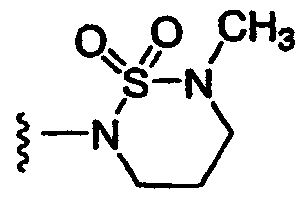
4) SO2R20, где R20 означает:

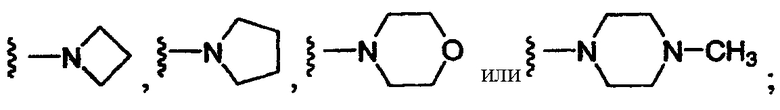
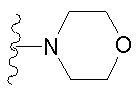
либо альтернативно, R2 и R5 вместе с атомом азота, к которому присоединены, образуют
или
R3 означает водород;
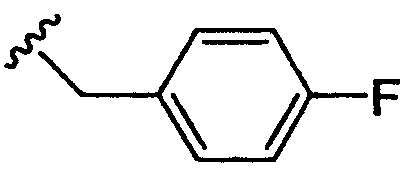
R4 означает:
1) п-фторбензил,
2) 4-фтор-3-метилбензил,
3) 3-хлорбензил или
4) 3-хлор-4-метилбензил;
R12 и R14 оба означают H, за исключением, что когда R5 означает C(O)C(O)N(CH3)2, и R4 означает п-фторбензил, и n равно 1, то R12 и R14 либо оба означают H, либо оба означают CH3; и
n означает целое число, равное нулю, 1 или 2.
Настоящее изобретение также включает фармацевтические композиции, содержащие соединение по данному изобретению, и способы получения таких фармацевтических композиций. Кроме того, настоящее изобретение включает способы лечения СПИДа, способы задержки проявления СПИДа, способы профилактики СПИДа, способы профилактики ВИЧ-инфекции и способы лечения ВИЧ-инфекции.
Другие варианты осуществления, аспекты и отличительные признаки настоящего изобретения либо указаны ниже в приведенном описании, либо становятся очевидными из последующего описания, примеров и формулы изобретения.
Подробное описание изобретения
Настоящее изобретение включает соединения приведенной выше формулы I и их фармацевтически приемлемые соли. Эти соединения и соответствующие фармацевтически приемлемые соли являются ингибиторами ВИЧ-интегразы.
Вариантом осуществления настоящего изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 означает NR2R5; n означает целое число, равное 1 или 2; и все другие переменные принимают первоначально указанные значения.
Другим вариантом осуществления настоящего изобретения является соединение формулы I или его фармацевтически приемлемая соль, где R1 означает NR2R5; R2 означает CH3; R5 означает 1) C(O)C(O)N(CH3)2 или 2) SO2R20, где R20 означает
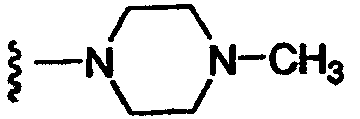
R3 означает водород; R4 означает п-фторбензил или 4-фтор-3-метилбензил; R12 и R14 оба означают H, за исключением, что когда R5 означает C(O)C(O)N(CH3)2, и R4 означает п-фторбензил, и n равно 1, то R12 и R14 либо оба означают H, либо оба означают CH3; и n означает целое число, равное 1 или 2.
Настоящее изобретение также включает соединение формулы II или его фармацевтически приемлемую соль:
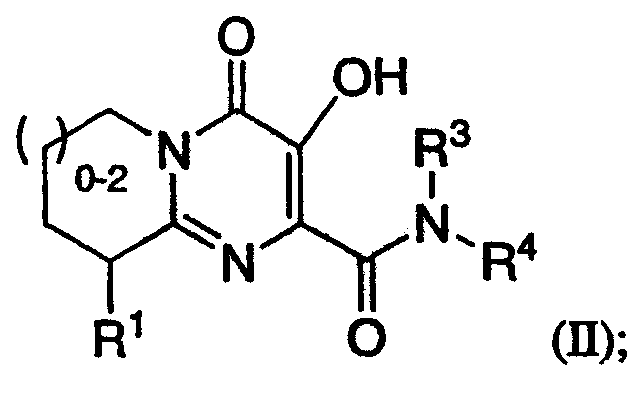
где R1 означает водород, NR2R5, OR2, SR2, SOR2, SO2R2, SO2NR2R5 или OC(O)NR2R5; R3 означает водород; R4 означает
R2 означает 1) водород, 2) CH3 или 3)
и
R5 означает 1) C(O)CH3, 2) C(O)CH2SO2CH3, 3) CH3, 4) C(O)C(O)N(CH3)2, 5) SO2CH3, 6) SO2N(CH3)2, 7) C(O)CH2N(CH3)2, 8) SO2CH2SO2CH3, 9) C(O)CF3, 10)
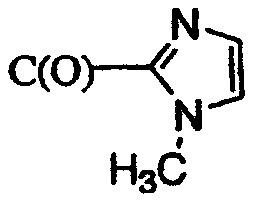
11)
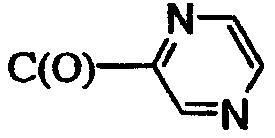
или 12)
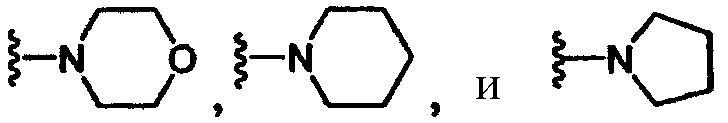
либо R2 и R5, вместе с атомом азота, к которому присоединены, образуют гетероциклическое кольцо, выбираемое из группы, включающей
Другим вариантом осуществления настоящего изобретения является соединение или его фармацевтически приемлемая соль, выбираемые из группы, включающей соединения, перечисленные в приведенной ниже таблице.
Дополнительные варианты осуществления настоящего изобретения включают следующее:
(a) Фармацевтическую композицию, содержащую соединение формулы (I) фармацевтически приемлемый носитель.
(b) Фармацевтическую композицию, содержащую продукт, полученный комбинированием (например, смешиванием) эффективного количества соединения формулы (I) и фармацевтически приемлемого носителя.
(c) Фармацевтическую композицию по п. (a) или (b), дополнительно содержащую терапевтически эффективное количество средства для лечения ВИЧ-инфекции/СПИДа, выбираемого из группы, включающей ВИЧ/СПИД-противовирусные средства, иммуномодуляторы и противоинфекционные средства.
(d) Фармацевтическую композицию по п. (с), где средство для лечения ВИЧ-инфекции/СПИДа представляет собой противовирусное средство, выбираемое из группы, включающей ингибиторы ВИЧ-протеазы, ненуклеозидные ингибиторы обратной транскриптазы ВИЧ и нуклеозидные ингибиторы обратной транскриптазы ВИЧ.
(e) Комбинацию, полезную для ингибирования ВИЧ-интегразы, лечения или профилактики ВИЧ-инфекции, или профилактики, лечения или задержки проявления СПИДа, представляющую собой терапевтически эффективное количество соединения формулы (I) и терапевтически эффективное количество средства для лечения ВИЧ-инфекции/СПИДа, выбираемого из группы, включающей ВИЧ/СПИД-противовирусные средства, иммуномодуляторы и противоинфекционные средства.
(f) Комбинацию по п. (e), где средство для лечения инфекции ВИЧ/СПИДа представляет собой противовирусное средство, выбираемое из группы, включающей ингибиторы ВИЧ-протеазы, ненуклеозидные ингибиторы обратной транскриптазы ВИЧ и нуклеозидные ингибиторы обратной транскриптазы ВИЧ.
(g) Способ ингибирования ВИЧ-интегразы у нуждающегося в этом пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I).
(h) Способ профилактики или лечения ВИЧ-инфекции у нуждающегося в этом пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I).
(i) Способ по п. (h), где соединение формулы (I) вводят в комбинации с терапевтически эффективным количеством, по меньшей мере, одного противовирусного средства, выбираемого из группы, включающей ингибиторы ВИЧ-протеазы, ненуклеозидные ингибиторы обратной транскриптазы ВИЧ и нуклеозидные ингибиторы обратной транскриптазы ВИЧ.
(j) Способ профилактики, лечения или задержки проявления СПИДа у нуждающегося в этом пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I).
(k) Способ по п. (j), где соединение вводят в комбинации с терапевтически эффективным количеством, по меньшей мере, одного противовирусного средства, выбираемого из группы, включающей ингибиторы ВИЧ-протеазы, ненуклеозидные ингибиторы обратной транскриптазы ВИЧ и нуклеозидные ингибиторы обратной транскриптазы ВИЧ.
(l) Способ ингибирования ВИЧ-интегразы у нуждающегося в этом пациента, включающий введение пациенту фармацевтической композиции по п. (a), (b), (c) или (d) либо комбинации по п. (e) или (f).
(m) Способ профилактики или лечения ВИЧ-инфекции у нуждающегося в этом пациента, включающий введение пациенту фармацевтической композиции по п. (a), (b), (c) или (d) либо комбинации по п. (e) или (f).
(n) Способ профилактики, лечения или задержки проявления СПИДа у нуждающегося в этом пациента, включающий введение пациенту фармацевтической композиции по п. (a), (b), (c) или (d) либо комбинации по п. (e) или (f).
Настоящее изобретение также включает соединение (i) для применения с целью, (ii) для применения в качестве лекарственного средства с целью, или (iii) для применения в получении лекарственного средства, используемого с целью: (a) ингибирования ВИЧ-интегразы, (b) профилактики или лечения ВИЧ-инфекции или (c) профилактики, лечения или задержки проявления СПИДа. При применении в указанных целях соединения по настоящему изобретению, необязательно, могут быть использованы в комбинации с одним или более средствами для лечения ВИЧ/СПИДа, выбираемыми из группы, включающей ВИЧ/СПИД-противовирусные средства, противоинфекционные средства и иммуномодуляторы.
Соединения по настоящему изобретению могут иметь асимметрические центры и могут существовать, за исключением особо указанных случаев, в виде смесей стереоизомеров или в виде индивидуальных диастереомеров или энантиомеров, при этом все изомерные формы входят в объем настоящего изобретения.
N-замещенные гидроксипиримидиноновые соединения по настоящему изобретению могут также существовать в виде таутомеров, таких как приведенный ниже таутомер соединения формулы I:
Само собой разумеется, что настоящее изобретение включает все таутомеры гидроксипиримидиноновых соединений формулы I (или II), как в отдельности, так и в смесях.
Соединения по настоящему изобретению полезны в ингибировании ВИЧ-интегразы, для профилактики или лечения инфекции вируса иммунодефицита человека (ВИЧ) и профилактики, лечения или задержки проявления последующих патологических состояний, таких как СПИД. Профилактика СПИДа, лечение СПИДа, задержка проявления СПИДа, или профилактика или лечение ВИЧ-инфекции определяются, как включающие, но не в порядке ограничения, лечение широкого ряда состояний при ВИЧ-инфекции: СПИДа, ARC (СПИД-ассоциированный комплекс), как симптоматических, так и асимптоматических, и существующего либо потенциального контакта с ВИЧ-инфекцией. Например, соединения по настоящему изобретению полезны в лечении ВИЧ-инфекции после предполагаемого контакта с ВИЧ, произошедшего при переливании крови, замене жидкости организма, укусах, случайном уколе зараженной иглой или контакте с кровью пациента во время хирургической операции.
Характерные соединения по настоящему изобретению были проверены на ингибирование в исследовании активности интегразы в реакции переноса цепей. Испытания проводят способом, описанным в WO 02/30930. Испытание также соответствует приведенному в Wolfe, A.L. et al., J. Virol. 1996, 70: 1424-1432 для рекомбинантной интегразы, за тем исключением, что: (i) для анализа используют предварительно собранные комплексы переноса интеграза-цепь; (ii) реакцию переноса цепи осуществляют в присутствии ингибитора в 2,5 мМ MgCl2, используя от 0,5 до 5 нМ 3'-ФИТЦ-меченого субстрата ДНК-мишени, и (iii) продукты реакции переноса цепи распознают, используя анти-ФИТЦ, антитело, конъюгированное со щелочной фосфатазой, и хемилюминесцентный субстрат для щелочной фосфатазы. Характерные соединения по настоящему изобретению, согласно данному испытанию оказывают ингибирующее действие на активность в реакции переноса цепи. Например, соединения, приведенные в представленной ниже таблице, исследованы методом анализа активности интегразы и дают значения IC50 порядка 5 мкмоль/л или ниже. Дополнительно описание проведения испытания с использованием предварительно собранных комплексов может быть найдено в Hazuda et al., J. Virol. 1997, 71. 7005-7011; Hazuda et al., Drug Design and Discovery 1997,15: 17-24 и Hazuda et al., Science 2000, 287: 646-650.
Некоторые характерные соединения по настоящему изобретению были также изучены в испытании на ингибирование острой ВИЧ-инфекции T-лимфоцитов, осуществляемом согласно Vacca, J.P. et al., Proc. Natl. Acad. Sci. USA 1994,91.: 4096. Эти соединения, включающие соединения, приведенные ниже в таблице, дают значения IC95 порядка 20 мкмоль/л или ниже.
Соединения по настоящему изобретению могут также действовать как ингибиторы ВИЧ-рибонуклеазы H (РНКазы H). Обратная транскриптаза (RT) вируса иммунодефицита человека типа 1 (ВИЧ-1) катализирует конверсию геномной РНК в двухцепочечную ДНК провируса после проникновения в клетку, используя РНК- и ДНК-зависимые полимеразные активности и активность РНКазы H. RT ВИЧ-l является асимметрическим димером, состоящим из полипептидов p66 и p51. Каталитические активности RT управляются отдельными сайтами в субъединице p66; т.е. N-конец p66 отвечает за РНК- и ДНК-зависимую ДНК-полимеразную активность, и домен pl5 на С-конце отвечает за активность РНКазы H. РНКаза H требуется для расщепления РНК-цепи РНК:ДНК гетеродуплексных промежуточных продуктов в обратной транскрипции. Соединения по настоящему изобретению могут селективно связываться с доменом РНКазы H RT ВИЧ-1 и ингибировать активность указанного домена. Ингибирующая активность соединений по отношению к РНКазе H может быть измерена с применением подходящих испытаний, известных из уровня техники, таких как испытание, описанное в Shaw-Reid et al., J. Biol. Chem. 2003, 278 (5): 2777-2780. Таким образом, настоящее изобретение включает способ ингибирования ВИЧ-РНКазы H у нуждающегося в этом пациента, который состоит во введении пациенту эффективного количества соединения по изобретению. Настоящее изобретение включает также соединение (i) для применения в целях, (ii) для применения в виде лекарственного средства в целях или (iii) для применения в получении лекарственного средства, используемого в целях ингибирования ВИЧ-РНКазы H.
Соединения по настоящему изобретению могут быть введены в форме фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" означает соль, обладающую эффективностью исходного соединения, которая не является биологически или иным образом неприемлемой (например, не является ни токсичной, ни иным образом вредной по отношению к реципиенту). Подходящие соли включают кислотно-аддитивные соли, которые могут, например, быть получены смешением раствора соединения по настоящему изобретению с раствором фармацевтически приемлемой кислоты, такой как хлористоводородная кислота, серная кислота, уксусная кислота, трифторуксусная кислота или бензойная кислота. Когда соединения по изобретению содержат кислотную составляющую, подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов (например, соли натрия или калия), соли щелочно-земельных металлов (например, соли кальция или магния) и соли, образованные с подходящими органическими лигандами, такие как четвертичные аммониевые соли. Также, в случае наличия кислотной (-COOH) или спиртовой группы, фармацевтически приемлемые сложные эфиры могут быть использованы для изменения растворимости или гидролизных характеристик соединения.
В целях ингибирования ВИЧ-интегразы или ВИЧ-РНКазы H, профилактики или лечения ВИЧ-инфекции или профилактики, лечения или задержки проявления СПИДа соединения по настоящему изобретению могут быть введены перорально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, подложечные инъекции или инфузии), посредством аэрозольной ингаляции или ректально, в форме стандартной дозы фармацевтической композиции, содержащей терапевтически эффективное количество соединения и общепринятые нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и растворители.
Термин "введение" и варианты этого термина (например, "прием" соединения) по отношению к соединению по изобретению означают, что нуждающемуся в лечении индивидууму вводят соединение или пролекарство соединения. Когда соединение по изобретению или пролекарство вводят в комбинации с одним или более другими активными средствами (например, противовирусными средствами, полезными для лечения инфицирования вирусом ВИЧ или СПИДа), понятно, что термин "введение" и каждый из вариантов указанного термина включают одновременное и последовательное введение соединения или пролекарства и других средств.
Подразумевается, что термин "композиция", как использован здесь, касается продукта, включающего указанные ингредиенты в указанных количествах, а также любого продукта, образующегося прямо или косвенно, при комбинировании указанных ингредиентов в указанных количествах.
"Фармацевтически приемлемые" означает, что ингредиенты фармацевтической композиции должны быть совместимы друг с другом и не наносить вреда реципиенту.
Термин "субъект" (альтернативно называемый здесь "пациент"), как использован здесь, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, являющемуся объектом лечения, наблюдения или исследования.
Термин "терапевтически эффективное количество", как использован здесь, означает такое количество активного соединения или фармацевтического средства, которое вызывает биологический или фармакологический отклик в ткани, системе, организме животного или человека, которого добивался исследователь, ветеринар, врач или другой клиницист, что включает ослабление или профилактику симптомов излечиваемых или предупреждаемых болезни или состояния. Термин также включает количество активного соединения, достаточное для ингибирования ВИЧ-интегразы и/или РНКазы H, и тем самым вызывающее ожидаемый отклик. Когда активное соединение (т.е. активный ингредиент) вводят в виде соли, указанное количество активного ингредиента относится к соединению в форме свободной кислоты или свободного основания.
Фармацевтические композиции могут быть в форме перорально-вводимых суспензий либо таблеток или капсул, назальных спреев, стерильных препаратов для инъекции, например, в виде стерильных водных или масляных суспензий для инъекции, или суппозиториев. Эти композиции могут быть получены хорошо известными из уровня техники способами и содержат общепринятые наполнители. Подходящие способы и ингредиенты описаны в Remington's Pharmaceutical Sciences, 18th edition, edited by A. R. Gennaro, Mack Publishing Co., 1990, который полностью включен сюда в качестве ссылки.
Соединения по настоящему изобретению могут быть введены перорально в пределах доз от 0,001 до 1000 мг/кг массы тела млекопитающего (например, человека) в день в виде единой дозы или нескольких доз. Предпочтительный интервал доз составляет от 0,01 до 500 мг/кг массы тела в день, перорально, в виде единой дозы или нескольких доз. Другой предпочтительный интервал доз составляет от 0,1 до 100 мг/кг массы тела в день, перорально, в виде единой дозы или нескольких доз. Для перорального приема композиции могут быть представлены в форме таблеток или капсул, содержащих 1,0-500 миллиграмм активного ингредиента, в частности 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400 и 500 миллиграмм активного ингредиента для симптоматического подбора дозировки проходящему лечение пациенту. Конкретный уровень доз и частота введения дозы для каждого отдельного пациента могут варьироваться в зависимости от факторов, включающих активность конкретного используемого соединения, метаболическую устойчивость и длительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, диету, способ и время введения, скорость выведения из организма, лекарственную комбинацию, тяжесть конкретного состояния и проходящего терапию реципиента.
Как указано выше, настоящее изобретение также касается использования ингибирующих ВИЧ-интегразу соединений по настоящему изобретению с одним или более средствами, полезными в терапии ВИЧ-инфекции или СПИДа. Например, соединения по настоящему изобретению могут быть эффективно применены, независимо, при предварительном воздействии и/или последующем воздействии, в комбинации с эффективными количествами одного или более ВИЧ/СПИД-противовирусных средств, иммуномодуляторов, противоинфекционных средств или вакцин, полезных для терапии ВИЧ-инфекции или СПИДа, таких как средства, приведенные в таблице 1 в WO 01/38332 или в таблице в WO 02/30930, оба документа полностью включены сюда в качестве ссылок. Очевидно, что рамки объема комбинаций соединений по настоящему изобретению с ВИЧ/СПИД-противовирусными средствами, иммуномодуляторами, противоинфекционными средствами или вакцинами не ограничиваются списком, приведенным в вышеуказанных таблицах в WO 01/38332 и WO 02/30930, и включают, в принципе, любую комбинацию с любой фармацевтической композицией, полезной для лечения СПИДа. ВИЧ/СПИД-противовирусные средства и другие средства обычно используют в этих комбинациях на уровне общепринятых доз и согласно известным из уровня техники схемам приема, включая, например, дозировки, указанные в Physicians' Desk Reference, 57th edition, Thomson PDR, 2003. Диапазон доз соединения по настоящему изобретению в таких комбинациях соответствует указанному выше.
Сокращения, используемые в данном описании, в частности, на схемах и в примерах, включают следующие: AIDS=синдром приобретенного иммунодефицита, ARC=СПИД-ассоциированный комплекс, Bn=бензил, CBZ (или Cbz)=бензилоксикарбонил, DBU=1,8-диазабицикло[5.4.0]ундец-7-ен, DMAD=диметилацетилендикарбоксилат, ДМФА=N,N-диметилформамид, ДМСО=диметилсульфоксид, EtOAc=этилацетат, FIA-MS=масс-спектрометрия с анализом методом впрыскивания в поток, h=час(ы), ВИЧ=вирус иммунодефицита человека, ВЭЖХ=высокоэффективная жидкостная хроматография, IPA=изопропанол, LDA=литийдиизопропиламид, Me=метил, MeOH=метанол, NMP=N-метилпирролидинон, ЯМР=ядерный магнитный резонанс, Pd/C=палладиевый катализатор на угле, ОФ-ВЭЖХ=ВЭЖХ с обращенной фазой, TFA=трифторуксусная кислота, THF=тетрагидрофуран.
Соединения по настоящему изобретению легко могут быть получены по следующим реакционным схемам и примерам или модификацией указанных приемов с использованием легкодоступных исходных материалов и реагентов. В приведенных ниже реакционных схемах возможно также использование вариантов, которые сами по себе известны специалисту в данной области, но не приведены подробно. Кроме того, другие способы получения соединений по изобретению станут очевидными для специалиста в данной области в свете приведенных ниже схем и примеров. Если не оговорено особо, переменные, перечисленные в схемах 1, A, B, C и D, имеют следующие значения:
PΛ означает водород или защитную группу, например сложный эфир, такой как, но не в порядке ограничения, бензоат и пивалат, или простой эфир, такой как, но не в порядке ограничения, бензиловый эфир, который обычно удаляют в условиях, используемых для превращения сложного метилового эфира в амид, или удаляют на другой стадии. Защитную группу обычно используют для синтеза и/или очистки.
RΛ означает водород или C1-6-алкил.
Y означает водород или NRsaRsb.
Rsa означает C1-6-алкил, C(O)Rsc, C(O)C(O)NRscRsd, SO2RSC, SO2NRscRsd, C(O)CH2SO2RSC, C(O)CH2NRscRsd, SO2CH2SO2RSC или CH(CH3)RSC либо Rsa и Rsb, вместе с атомом азота, к которому присоединены, образуют гетероциклическое кольцо, содержащее 1 или 2 гетероатома.
Rsb означает водород, C1-6-алкил или C(O)CF3, либо Rsa и Rsb, вместе с атомом азота, к которому присоединены, образуют гетероциклическое кольцо, содержащее 1 или 2 гетероатома.
Rsc означает C1-6-алкил, арил, 5- или 6-членное гетероарильное кольцо, которое не замещено или замещено C1-6-алкилом, C(O)CH2SO2C1-6-алкилом или (CH2)1-6-арилом.
Rsd означает C1-6-алкил.
RS5 означает Rsc, C(O)NRscRsd, CH2SO2RSC, CH2NRscRsd, NRscRsd или CH2SO2RSC.
Rs1 означает водород.
Rs2 означает CH2Rse, где Rse означает незамещенный арил или арил, замещенный галогеном.
Соединения по настоящему изобретению могут быть получены сочетанием соответствующих аминов с подходящим замещенным алкил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксилатом (или карбоновыми кислотами или галогенидами) или алкил-3-гидрокси-4-оксо-4,6,7,8-тетрагидропирроло[1,2-a]пиримидин-2-карбоксилатом (или карбоновыми кислотами или галогенидами), или алкил-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксилатом (или карбоновыми кислотами или галогенидами), или алкил-3-гидрокси-4-оксо-6,7,8,9,10,11-гексагидро-4Н-пиримидо[1,2-a]азепин-2-карбоксилатом (или карбоновыми кислотами или галогенидами), как представлено на схеме 1.
Схема 1
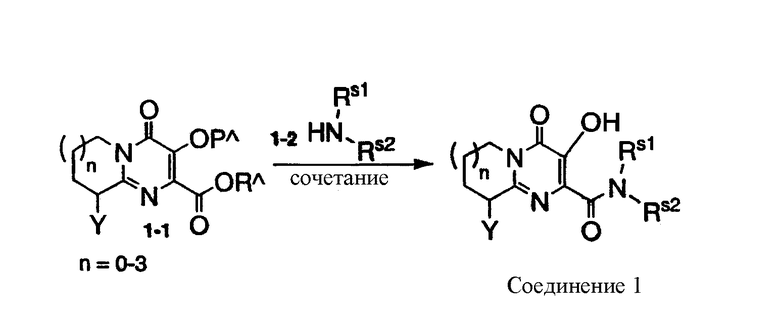
Способы сочетания производных карбоновых кислот с аминами с образованием карбоксамидов хорошо известны из уровня техники. Подходящие способы описаны, например, в Jerry March, Advanced Organic Chemistry, 3rd edition, John Wiley & Sons, 1985, pp. 370-376. Амины формулы 1-2 могут быть получены с использованием способов, описанных в Richard Larock, Comprehensive Organic Transformations, VCH Publishers Inc, 1989, pp 385-438, либо стандартных вариантов указанных способов.
Схема A представляет общий синтез карбоксамидов A-5. Метиловый эфир A-4 может быть подвергнут взаимодействию с амином 1-2 в растворителях, таких как ДМФА, метанол, этанол, толуол, NMP, при соответствующей температуре (например, от 20 до 150°C), что дает конечное соединение A-5. Метиловый эфир A-4 может быть синтезирован одним из трех синтетических способов. Согласно первому способу амидингидрохлорид A-la (sX=H; sY=H) может быть подвергнут взаимодействию с диметил-2-(бензилокси)-3-оксосукцинатом в присутствии основания, что дает промежуточный защищенный метиловый эфир A-2, защита которого легко может быть снята, что приводит к получению метилового эфира A-4. Согласно второму способу амидоксин A-lb (sX=OH; sY=H), полученный в три стадии из третбутилбензилоксикарбамата, может быть подвергнут взаимодействию с DMAD, что дает циклическое промежуточное соединение A-3a, которое может быть подвергнуто перегруппировке путем нагревания в соответствующем растворителе (например, ксилоле), что приводит к образованию метилового эфира A-5. Согласно третьему способу амидоксим A-1c (sX=H; sY=OH) может быть подвергнут взаимодействию с DMAD в соответствующем растворителе (например, ацетонитриле), что приводит к образованию промежуточного соединения A3-b, которое может быть подвергнуто перегруппировке в метиловый эфир A-4 путем нагревания в соответствующем растворителе (например, ксилоле). Все три приведенных способа могут быть применены к амидинам и амидоксимам, содержащим заместители в кольце. Схема A иллюстрируется примером 1.
Схема A
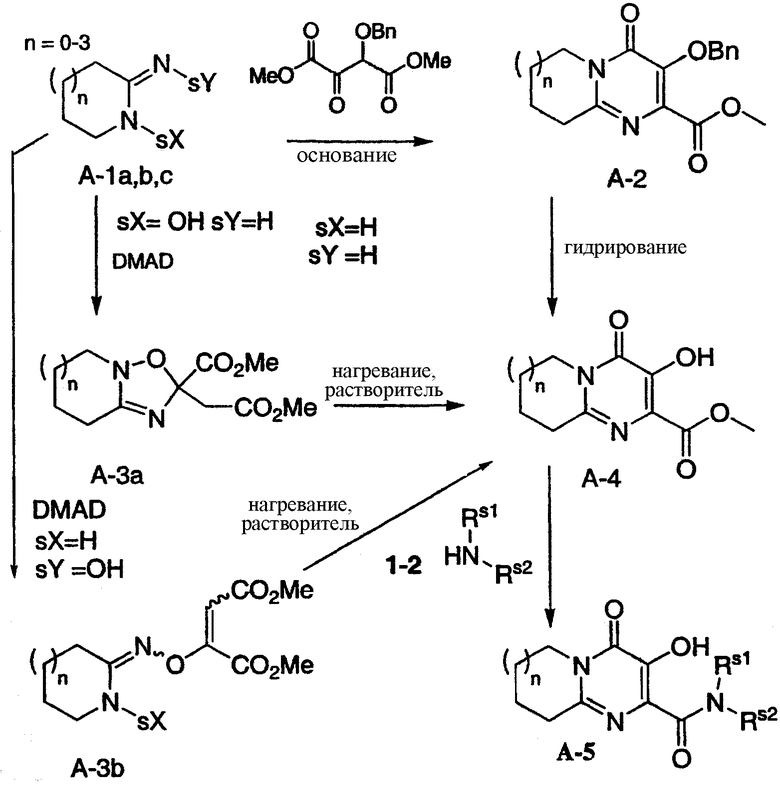
Схема B представляет способ получения соединений по настоящему изобретению, содержащих амино-, простую эфирную, простую тиоэфирную, сульфоксидную или сульфоновую группу в положении 9 ядра 3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксамида. Бром-производное B-l может быть получено из метилового эфира A-4 с помощью первоначальной защитой гидроксигруппы на A-4 подходящей защитной группой (например, превращением в бензоат, или пивалат, или в бензилокси) и последующим осуществлением контакта защищенного A-4 с бромирующим агентом (например, NBS). Бром-производное B-l может быть затем обработано нуклеофильным реагентом ("Nu"; например, амином, тиолом или алкоголятом), что дает, при выделении или без выделения, промежуточный метиловый эфир B-2, который подвергают взаимодействию с заданным амином, что приводит к образованию конечного продукта B-3. Если нуклеофильный реагент представляет собой тиол или содержит окисляемую серу, в схему может быть включена стадия окисления с целью получения сульфоксида или сульфона. Если нуклеофильный реагент включает сложный эфир, сложный эфир может быть превращен в амид, использованием стандартных химических превращений после синтеза B-3. Схема B иллюстрируется примером 2.
Схема B
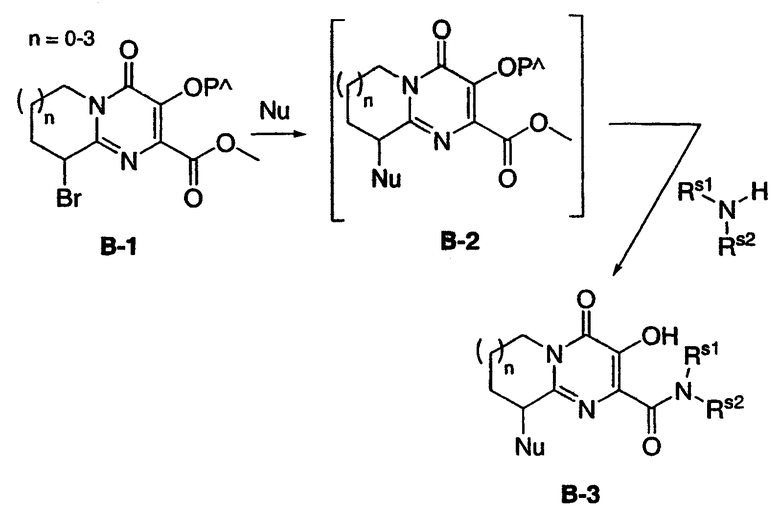
Схема С представляет общий синтез производных C-3 или C-4, содержащих в кольце алифатический заместитель, такой как амид, сульфонамид, сульфонилмочевина, карбамат или мочевина. Бром-производное B-1 может быть обработано бензиламином С-1 и затем гидрировано или непосредственно подвергнуто взаимодействию с амином C-1a, что дает промежуточное соединение C-2, которое затем может быть обработано амином 1-2, с выделением или без выделения, и затем подвергнуто реакции сочетания с карбоновой кислотой или взаимодействию с карбонилхлоридом (или сульфонилхлоридом либо сульфамоилхлоридом) или изоцианатом, что дает конечный продукт C-3. Если C-3 содержит RS4=Rs3О(CO)CO, то C-3 может быть в дальнейшем подвергнуто взаимодействию с нуклеофильным реагентом, таким как амин, что приводит к образованию продукта C-4. Схема С иллюстрируется примерами 3, 4 и 6. Последние две стадии можно поменять местами.
Схема С
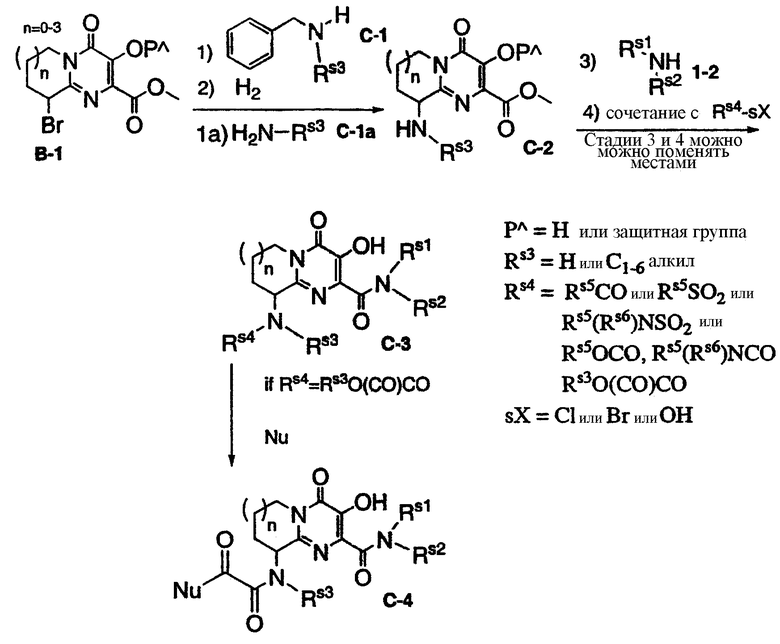
1. сочетание с RS4-sX, 2. стадии 3 и 4 можно поменять местами, 3. PΛ=Н или защитная группа, 4. RS3=Н или C1-6-алкил, or=или, if=если.
Схема D представляет синтез гомохиральных соединений C-3, C3a,b и C-4. Осуществляют замещение бром-производного B-l хиральными аминами D-l, приводящее к смеси диастереоизомеров, с последующим или одновременным удалением защитной группы. Проводят восстановительное алкилирование аминогруппы в положении 9 с помощью альдегидов или кетонов D-2, что дает промежуточное соединение D-3. Смесь диастереоизомеров может быть разделена путем кристаллизации или хроматографии, что приводит к отдельным диастереоизомерам D-3a,b. Rs6 может быть удален гидрированием, что дает гомохиральное промежуточное соединение C-2a,b. Последующее взаимодействие с амином 1-2 и сочетание с карбоновой кислотой или обработка карбонилхлоридом (или сульфонилхлоридом либо сульфамоилхлоридом) или изоцианатом приводит к конечному гомохиральному продукту C-3a,b. Как и в случае рацемических соединений C-3 на схеме C, может быть проведена дополнительная стадия для получения гомохиральных соединений C-4. Схема D иллюстрируется примерами 5 и 7.
Схема D
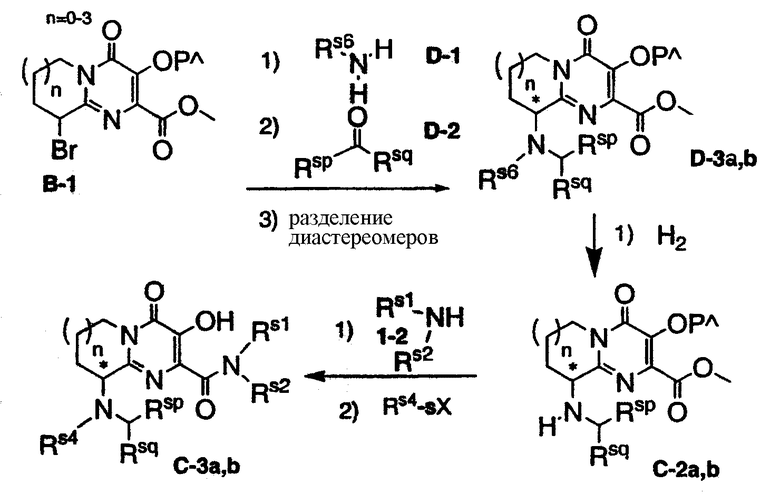
PΛ=Н или защитная группа
Rs6=хиральный алкильный остаток (например, (S)-a-метилбензил)
Rsp, Rsq= Н или C1-6-алкил
Rs4=Rs5CO или Rs5SO2 или Rs5(Rs6)NSO2 или Rs5OCO,Rs3O(CO)CO Rs5(Rs6)NCO
sX=Cl или Br или ОН
Следующие примеры служат исключительно целям иллюстрации изобретения и его практического осуществления. Примеры не следует рассматривать как ограничивающие объем или сущность изобретения.
ПРИМЕР 1
N-(4-Фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид
Стадия 1a: Третбутилбензилокси(4-цианобутил)карбамат (Bergeron,
R. J., McManis, J. S., Tetrahedron 45 (16), 4939-4944 (1989).
К раствору третбутилбензилоксикарбамата в безводном диметилформамиде добавляют 5 моль% иодида натрия и порциями 1,36 экв. гидрида натрия (60% дисперсия в минеральном масле). Смесь перемешивают при комнатной температуре в течение 15 мин, после чего добавляют 1,05 экв. 4-хлорвалеронитрила. Смесь нагревают до 85°C и перемешивают в течение 3,5 ч. После охлаждения до комнатной температуры смесь гасят водой и экстрагируют диэтиловым эфиром. Объединенные органические фазы концентрируют и промывают полунасыщенным водным тиосульфатом натрия, водой и насыщенным раствором соли. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Маслянистый остаток промывают петролейным эфиром и сушат при высоком вакууме, получая третбутилбензилокси(4-цианобутил)карбамат в виде светло-желтого масла.
1Н-ЯМР(400 МГц, CDCl3) δ:7.38(м, 5Н), 4.84(с, 2Н), 3.45(т, J=6.4 Гц, 2Н), 2.34(т, J=6.8 Гц, 2Н), 1.70(м, 4Н), 1.52(с, 9Н). МС m/z: 271(М+Н)+.
Стадия 2a: l-(Бензилокси)пиперидин-2-имингидрохлорид
Третбутилбензилокси-(4-цианобутил)карбамат растворяют в растворе 4 M HCl в 1,4-диоксане и смесь перемешивают в течение 18 ч при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток обрабатывают этилацетатом и диэтиловым эфиром. Полученное твердое вещество, промытое диэтиловым эфиром, фильтруют и сушат при высоком вакууме, что дает 1-(бензилокси)пиперидин-2-имингидрохлорид в виде бледно-желтого твердого вещества.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:9.53(с, 1Н), 8.97(с, 1Н), 7.57-7.41(м, 5Н), 5.05(с, 2Н), 3.67(т, J=6.0 Гц, 2Н), 2.64(т, J=6.4 Гц, 2Н), 1.90-1.84(м, 2Н), 1.69-1.63(м, 2Н).
МС m/z: 205(М+Н)+.
Стадия 3a: 2-Иминопиперидин-1-ол гидрохлорид
Раствор 1-(бензилокси)пиперидин-2-имин гидрохлорида в метаноле, содержащем палладий на активированном угле (10%, мас./мас.) перемешивают в среде водорода при атмосферном давлении в течение 3 ч. Катализатор отделяют фильтрованием и раствор концентрируют досуха при пониженном давлении. Остаток растирают с диэтиловым эфиром, фильтруют и сушат при высоком вакууме, получая 2-иминопиперидин-1-ол гидрохлорид в виде бледно-желтого твердого вещества.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:11.76(с, 1Н), 8.82(с, 1Н), 8.49(с, 1Н), 3.63(т, J=6.0 Гц, 2Н), 2.63(т, J=6.0 Гц, 2Н), 1.87(м, 2Н), 1.66(м, 2Н). 13С-ЯМР (150 МГц, ДМСО-d6) δ:159.06, 50.92, 25.76, 22.01, 17.22.
МС m/z: 115(М+Н)+.
Стадия 4a: Метил-2-(2-метокси-2-оксоэтил)-5,6,7,8-тетрагидро-2Н-
[1,2,4]оксадиазоло[2,3-а]пиридин-2-карбоксилат
К раствору 2-иминопиперидин-1-ол гидрохлорида в хлороформе добавляют триэтиламин. Смесь перемешивают в течение 5 мин при комнатной температуре, затем охлаждают до 0°C и добавляют по каплям при перемешивании 1,2 экв. диметилацетилендикарбоксилата. Охлаждающую баню убирают и смесь перемешивают при комнатной температуре в течение одного часа. Растворитель удаляют при пониженном давлении и раствор распределяют между этилацетатом и полунасыщенным водным хлоридом аммония. Водную фазу дополнительно экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и фильтруют через силикагель. Растворитель удаляют в вакууме, получая метил-2-(2-метокси-2-оксоэтил)-5,6,7,8-тетрагидро-2Н-[1,2,4]оксадиазоло[2,3-а]пиридин-2-карбоксилат в виде светло-желтого масла.
1Н-ЯМР(400 МГц, CDCl3) δ:3.82(с, 3Н), 3.70(с, 3Н), 3.51(м, 1Н), 3.36(м, 1Н) 3.31 (д, J=16.6 Гц, 1Н), 2.98(д, J=16.6 Гц, 1Н), 2.53(м, 2Н), 1.94(м, 2Н), 1.74(м, 2Н). 13С-ЯМР (100 МГц, CDCl3) δ:169.15, 168.88, 164.97, 103.27, 55.71, 52.97, 51.84, 42.26, 26.06, 23.49, 22.83.
МС m/z: 257(М+Н)+.
Стадия 5a: Метил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-
пиридо[1,2-а]пиримидин-2-карбоксилат
Раствор метил-2-(2-метокси-2-оксоэтил)-5,6,7,8-тетрагидро-2H-[1,2,4]оксадиазоло[2,3-а]пиридин-2-карбоксилата в безводном o-ксилоле помещают в двугорлую круглодонную колбу. Колбу снабжают термометром и закрывают мембраной. Смесь нагревают до 148-150°C в течение 5 ч. Нагревающую баню убирают и смесь оставляют стоять при комнатной температуре в течение 16 ч. К смеси, содержащей осадок, добавляют диэтиловый эфир. Спустя 5 мин осадок отделяют фильтрованием, промывают диэтиловым эфиром и сушат в вакууме. Продукт, метил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксилат, получают в виде бледно-коричневого твердого вещества.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:10.03(с, 1Н), 3.86(т, J=6.0 Гц, 2Н), 3.80 (с, 3Н), 2.75(т, J=6.8 Гц, 2Н), 1.90-1.70(м, 4Н). 13С-ЯМР (150 МГц, ДМСО-d6) δ:165.81, 158.65, 148.60, 143.10, 127.07, 51.98, 42.87, 30.32, 20.91, 18.40.
МС m/z: 115(М+Н)+.
Следующая методика представляет собой альтернативный способ синтеза метил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксилата.
Стадия 1b: Диметил-2-(бензилокси)-3-оксосукцинат
Раствор метил(бензилокси)ацетата (1 экв.) и диметилоксалата (1,2 экв.) в сухом ТГФ охлаждают до -78°C и добавляют по каплям LDA (2M в смеси ТГФ-гептан, 1,2 экв). После перемешивания в течение одного часа охлаждающую баню убирают и перемешивание продолжают еще один час. Реакцию гасят при 0°C, выливая в охлажденную 1 н. водную HCl, и водную фазу экстрагируют EtOAc; органический слой промывают насыщенным раствором соли, сушат и концентрируют, что дает сырой продукт, который используют без дополнительной очистки.
Стадия 2b: Метил-3-(бензилокси)-4-оксо-6,7,8,9-тетрагидро-4H-
пиридо[1,2-а]пиримидин-2-карбоксилат
Промышленно выпускаемый 2-иминопиперидингидрохлорид (1,5 экв.) добавляют при комнатной температуре к раствору оксосукцината, полученного на стадии lb, (1 экв.) в MeOH. После добавления по каплям чистого DBU (4,5 экв.) реакционную смесь перемешивают в течение 2 дней. Выпаривание растворителя дает остаток, который поглощают EtOAc и промывают 1 н. HCl и насыщенным раствором соли; органический слой сушат над Na2SO4 и растворитель удаляют. Сырой продукт используют без дополнительной очистки.
Аналитический образец этого продукта очищают флэш-хроматографией (смесь петролейный эфир/EtOAc 1:2-1:5), получены следующие спектроскопические данные:
1Н-ЯМР(400 МГц, CDCl3) δ:7.49-7.30(м, 5Н), 5.25(с, 2Н), 4.00(т, J=6.2 Гц, 2Н), 3.86 (с, 3Н), 2.94(т, J=6.6 Гц, 2Н), 2.01-1.95(м, 2Н), 1.92-1.87(м, 2Н). 13С-ЯМР (75 МГц, CDCl3) δ:164.1, 159.3, 154.1, 141.1, 140.6, 136.0, 128.1, 127.8, 127.7, 73.8, 52.2, 42.7, 30.9, 21.0, 18.4.
МС m/z: 315(М+Н)+.
Стадия 3b: Метил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-
пиридо[1,2-а]пиримидин-2-карбоксилат
Промежуточный метил-3-(бензилокси)-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксилат, полученный на стадии 2b, растворяют в MeOH и добавляют при комнатной температуре каталитический 10% Pd/C. Смесь перемешивают в атмосфере H2 в течение 3,5 ч. Фильтрование катализатора и выпаривание метанола дает остаток, к которому добавляют диэтиловый эфир; фильтрование приводит к получению метил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксилата в виде бледно-коричневого твердого вещества со спектроскопическими характеристиками, идентичными характеристикам соединения, описанного на стадии 5a.
Стадия 6: N-(4-Фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-
4Н-пиридо[1,2-а]пиримидин-2-карбоксамид
Раствор метил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксилата, полученного на стадии 3b или стадии 5a, и 4-фтор-бензиламина (2 экв.) в метаноле перемешивают и нагревают до 65°C в течение 22 ч. Растворитель удаляют при пониженном давлении и указанный в заголовке продукт получают препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют, получая указанное в заголовке соединение в виде рыхлого белого материала.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:12,12(с, 1Н), 9.35(м, 1Н) 7,36 (м, 2Н), 7.15(м, 2Н), 4.44 (м, 2Н), 3.84(т, J=6.4 Гц, 2Н), 2.80 (т, J=6.8 Гц, 2Н), 1.90-1.73(м, 4Н).
МС m/z: 318(М+Н)+.
ПРИМЕР 2
N-(4-Фторбензил)-3-гидрокси-9-морфолин-4-ил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид гидрохлорид
Стадия 1: Метил-3-(бензоилокси)-4-оксо-6,7,8,9-тетрагидро-4Н-
пиридо[1,2-а]пиримидин-2-карбоксилат
К раствору метил-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксилата (полученного согласно примеру 1) в пиридине добавляют бензойный ангидрид (1,55 экв.). Смесь перемешивают при комнатной температуре в течение 16 ч. Растворитель удаляют при пониженном давлении и остаток распределяют между этилацетатом и 0,5 M водным HCl. Водную фазу экстрагируют этилацетатом и объединенные органические фазы промывают 0,5 M водным HCl, водой и насыщенным раствором соли. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют досуха в вакууме. Указанное в заголовке соединение получают после флэш-хроматографии (элюент - смесь петролейный эфир/этилацетат, 1:2) в виде неокрашенного твердого вещества.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:8.07(м, 2Н), 7.78(м, 1Н), 7.62(м, 2Н), 3.86 (т, J=6.0 Гц, 2Н), 3.74 (с, 3Н), 2.92 (т, J=6.4 Гц, 2Н), 1.93-1.81(м, 4Н).
МС m/z: 329(М+Н)+.
Стадия 2: Метил-3-(бензоилокси)-9-бром-4-оксо-6,7,8,9-
тетрагидро-4Н-пиридо[1,2-a]пиримидин-2-карбоксилат
Смесь метил-3-(бензоилокси)-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксилата, N-бромсукцинимида (1,2 экв.) и дибензоилпероксида (70%, 0,13 экв.) в четыреххлористом углероде перемешивают при нагревании до температуры кипения с обратным холодильником в течение одного часа. Смесь охлаждают до комнатной температуры, сукцинимид отделяют фильтрованием и растворитель удаляют при пониженном давлении. Метил-3-(бензоилокси)-9-бром-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксилат получают после флэш-хроматографии (элюент смесь петролейный эфир/этилацетат, 1:1) в виде бледно-желтого масла.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:8.08(м, 2Н), 7.79 (м, 1Н), 7.63(м, 2Н), 5.58 (м, 1Н), 4.24 (м, 1Н), 3.77 (с, 3Н), 3,72(м, 1Н), 2.43-2.35 (м, 1Н), 2.30-2.05 (м, 3Н).
МС m/z: 409/407(М+Н)+.
Стадия 3: N-(4-Фторбензил)-3-гидрокси-9-морфолин-4-ил-4-оксо-
6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-
карбоксамид.
К раствору метил-3-(бензоилокси)-9-бром-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-a]пиримидин-2-карбоксилата в ДМФА добавляют морфолин (3 экв.) и смесь перемешивают при комнатной температуре в течение 1 ч. Растворитель удаляют при пониженном давлении и остаток растирают с диэтиловым эфиром. Сырой продукт растворяют в метанол, добавляют 4-фторбензиламин (3 экв.) и смесь перемешивают в течение 1,5 ч при 65°C. Растворитель удаляют при пониженном давлении и продукт очищают препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют и вновь растворяют в 1 н. HCl. Растворитель удаляют при пониженном давлении и остаток лиофилизуют из смеси вода/ацетонитрил, получая гидрохлорид N-(4-фторбензил)-3-гидрокси-9-морфолин-4-ил-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксамида в виде слегка розоватого рыхлого материала.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:12.34(с, 1Н), 10.99(с, 1Н), 10.47(с, 1Н), 7.44 (м, 2Н), 7.16(м, 2Н), 4.85(м, 1Н), 4.60-4.40(м, 3Н), 4.10-3.85(м, 4Н), 3.60-3.05(м, 5Н замаскированный сигналом воды), 2.35-2.15(м, 2Н), 2.03-1.80(м, 2Н).
МС m/z: 403(М+Н)+.
ПРИМЕР 3
(+/-)-9-[[(Диметиламино)сульфонил](метил)амино]-N-(4-фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксамид C-3
Стадия 1: Метил-9-[бензил(метил)амино]-3-гидрокси-4-оксо-6,7,8,9
-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксилат
гидрохлорид
К перемешиваемому раствору бром-производного, метил-3-(бензоилокси)-9-бром-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксилата (полученного согласно примеру 2, стадия 2), в безводном диметилформамиде добавляют N-бензил-N-метиламин (3 экв.). Смесь перемешивают в течение 1,5 ч при комнатной температуре, после чего добавляют диэтиловый эфир и 2 M HCl в диэтиловом эфире. Смесь перемешивают в течение 5 мин, образовавшийся осадок отделяют фильтрованием и промывают диэтиловым эфиром. Осадок растворяют в безводном метаноле и раствор концентрируют досуха при пониженном давлении. Указанный в заголовке сырой продукт, полученный в виде бледно-желтого масла, содержащего избыток N-бензил-N-метиламингидрохлорида, используют без дополнительной очистки.
МС m/z: 344 (M+H)+.
Стадия 2: Метил-3-гидрокси-9-(метиламино)-4-оксо-6,7,8,9-
тетрагидро-4H-пиридо[1,2-a]пиримидин-2-карбоксилат
Полученный на стадии 1 раствор сырого продукта в метаноле, содержащий палладий на активированном угле (10% мас./мас.) перемешивают в среде водорода при атмосферном давлении в течение 3 ч. Катализатор отделяют фильтрованием и раствор концентрируют досуха при пониженном давлении. Остаток растирают с диэтиловым эфиром, фильтруют и сушат при высоком вакууме, получая сырой продукт в виде желтого твердого вещества, который используют на следующей стадии без дополнительной очистки.
МС m/z: 254 (M+H)+.
Стадия 3: N-(4-Фторбензил)-3-гидрокси-9-(метиламино)-4-оксо-6,7,
8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксамид
К раствору полученного на стадии 2 сырого продукта в сухом метаноле добавляют избыток триэтиламина. Растворитель удаляют при пониженном давлении и затем при высоком вакууме. Маслянистый остаток растворяют в безводном метаноле и добавляют 4-фторбензиламин (3,1 экв., теор.). Смесь перемешивают и нагревают до 60°C в течение ночи. После охлаждения до комнатной температуры растворитель удаляют при пониженном давлении. Остаток растирают с диэтиловым эфиром и оставляют при высоком вакууме на 15 мин. Указанный в заголовке сырой продукт получают в виде желтого твердого вещества, которое содержит избыток 4-фторбензиламина (около 3,5 экв.), и используют без дополнительной очистки.
МС m/z: 347 (M+H)+.
Стадия 4: (+/-)-9-[[(Диметиламино)сульфонил](метил)амино]-N-
(4-фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-
4H-пиридо[1,2-a]пиримидин-2-карбоксамид
К раствору полученного на стадии 3 неочищенного соединения в смеси 2:1 тетрагидрофурана и 2 M водного гидроксида натрия добавляют N,N-диметилсульфамоилхлорид (4.6 экв.). Смесь перемешивают при комнатной температуре в течение 16 ч. Смесь концентрируют при пониженном давлении и продукт выделяют препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют, получая указанное в заголовке соединение в виде рыхлого, слегка розоватого вещества.
1Н-ЯМР(300 МГц, CDCl3) δ:11.95(с, 1Н). 9.13(м, 1Н), 7.38(м, 2Н), 7.04(м, 2Н), 4.98 (м, 1Н), 4.56(м, 2Н), 4.36(м, 1Н), 3.62(м, 1Н), 2.84(с, 6Н), 2.57(с, 3Н), 2.38-1.85(м, 4Н). 13С-ЯМР (100 МГц, CDCl3) δ:167.53, 162.55, 160.11, 157.74, 145.76, 144.11, 132.70, 128.89, 128.81, 124.82, 114.60, 114.39, 58.06, 42.93, 41.53, 37.00, 29.03, 23.89, 20.09.
МС m/z: 454(М+Н)+.
ПРИМЕР 4
(+/-)-N1-(2-{[(4-Фторбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-9-ил)-N1,N2,N2-триметилэтандиамид
Стадия 1: (+/-)-N1-(2-{[(4-Фторбензил)амино]карбонил}-3-
гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]
пиримидин-9-ил)-N1,N2,N2-триметилэтандиамид
К перемешиваемому раствору сырого N-(4-фторбензил)-3-гидрокси-9-(метиламино)-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксамида (синтезированному, как описано в примере 3, стадия 3) в дихлорметане добавляют 6 экв. триэтиламина и 6 экв. метилхлороксоацетата. Смесь перемешивают при комнатной температуре в течение 2 ч, растворитель удаляют при пониженном давлении и остаток растворяют в растворе диметиламина (2 M) в тетрагидрофуране. Смесь перемешивают при 57°C в течение ночи. После охлаждения до комнатной температуры растворитель удаляют при пониженном давлении и продукт выделяют препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют, получая указанное в заголовке соединение в виде рыхлого, слегка розоватого вещества. Согласно ЯМР продукт представляет собой смесь ротамеров.
1Н-ЯМР(400 МГц, ДМСО-d6) δ:12.05(с, 0,2Н), 11.89(с, 0,8Н), 9.21(м, 0,8Н), 8.74(м, 0,2Н), 7.40-7.28(м, 2Н), 7.20-7.10(м, 2Н), 5.17(м, 0,8Н), 4.63-4.35 (м, 2,2Н), 4.13-4.00(м, 1Н), 3.65-3.53(м, частичное наложение сигнала воды), 2.95-2.75(м, 9Н), 2.15-1.80(м, 4Н). 13С-ЯМР (100 МГц, ДМСО-d6) δ:167.87, 167.73, 165.92, 165.46, 164.51, 164.30, 162.42, 160.01, 157.50, 157.41, 146.27, 146.18, 145.76, 145.49, 134.44, 129.43, 129.35, 129.08, 129.00, 125.17, 125.05, 115.07, 114.85, 57.47, 53.60, 43.14, 41.37, 36.49, 35.95, 32.92, 32.64, 32.36, 28.19, 23.88, 22.12, 19.67, 19.35.
МС m/z: 115(М+Н)+.
ПРИМЕР 5
(+)-N1-(2-{[(4-Фторбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-9-ил)-N1,N2,N2-триметилэтандиамид
Стадия 1: (+)3-Гидрокси-2-(метоксикарбонил)-N-метил-4-оксо-N-
[(1S)-1-фенилэтил]-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-9-аммония трифторацетат
К смеси 7:3 (об/об) метанола и воды при -30°C, содержащей (1S)-1-фенилэтиламин (4,5 экв.), добавляют бром-производное, метил-3-(бензоилокси)-9-бром-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-a]пиримидин-2-карбоксилат (синтезированный, как указано в примере 2, стадия 2) (1,0 экв.). Смесь энергично перемешивают в течение 1,5 ч при -30°C. Охлаждающую баню убирают и перемешивание продолжают в течение 1 ч при комнатной температуре. PH доводят приблизительно до 5 с помощью уксусной кислоты, после чего добавляют 37% водный формальдегид (11,5 экв.) и цианоборгидрид натрия (3,25 экв.). После перемешивания при комнатной температуре в течение 20 мин объем снижают приблизительно до 1/4 при пониженном давлении. Образовавшийся белый осадок отделяют фильтрованием и фильтрат подкисливают до pH 2-3 с помощью трифторуксусной кислоты. Раствор наносят на картриджи с катионообменной смолой (Varian MEGA BOND ELUTE SCX), картрижды промывают метанолом и сырой продукт вымывают с помощью 2 M аммиака в метаноле. Объединенные элюенты концентрируют досуха при пониженном давлении, маслянистый остаток растворяют в метаноле и нейтрализуют трифторуксусной кислотой. После удаления растворителя получают маслянистый остаток. Полученные в соотношении 1:3 диастереоизомеры разделяют путем очистки препаративной ОФ-ВЭЖХ (колонка: С18), элюенты: вода (0,1% TFA), ацетонитрил (0,1% TFA). Основной диастереоизомер выходит вторым пиком, и после лиофилизации указанное в заголовке соединение получают в виде слегка розоватого твердого вещества.
1Н-ЯМР(500 МГц, пиридин-d5) δ:7.53(м, 2Н), 7.39(м, 2Н), 7.29(м, 1Н), 4.45(м, 1Н), 4.38(м, 1Н), 4.14(м, 1Н), 3.91(с, 3Н), 3.80(м, 1Н), 2.13(с, 3Н), 1.95-1.82(м, 3Н), 1.70-1.60(м, 1Н), 1.49(д, J=6.4 Гц, 3Н).
МС m/z: 358(М+Н)+.
Стадия 2: (-)3-Гидрокси-2-(метоксикарбонил)-N-метил-4-оксо-
6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-9-
аммония трифторацетат
Раствор (+)-3-гидрокси-2-(метоксикарбонил)-N-метил-4-оксо-N-[(1S)-1-фенилэтил]-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-9-аммония трифторацетата в метаноле, содержащем палладий на активированном угле (10%, мас./мас.) перемешивают в среде водорода при атмосферном давлении в течение 1,5 ч. Катализатор отделяют фильтрованием и раствор концентрируют досуха при пониженном давлении, получая указанное в заголовке соединение в виде слегка розоватого масла.
1Н-ЯМР(400 МГц, CD3OD) δ:4,41(м, 1Н), 4,14(м, 1н), 3,99(с, 3Н), 3,91(м, 1Н), 2,86(с, 3Н), 2,50(м, 1Н), 2,26(м, 1Н), 2,08(м, 1Н), 1,86(м, 1Н). МС m/z: 254(М+Н)+.
Стадия 3: (+)-N1-(2-{[(4-Фторбензил)амино]карбонил}-3-
гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]
пиримидин-9-ил)-N1,N2,N2-триметилэтандиамид
Раствор (-)-3-гидрокси-2-(метоксикарбонил)-N-метил-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-9-аммония трифторацетата, п-фторбензиламин (2,2 экв.) и триэтиламин (1,3 экв.) в метаноле перемешивают и нагревают до 65°C в течение 3 ч. Растворитель удаляют в вакууме и остаток растворяют в безводном дихлорметане. Добавляют метилхлороксоацетат (5 экв.) и триэтиламин (5 экв.) и смесь перемешивают при комнатной температуре в течение 50 мин. Растворитель удаляют при пониженном давлении и остаток растворяют в 2 M растворе диметиламина в тетрагидрофуране. Смесь перемешивают при 57°C в течение ночи. После охлаждения до комнатной температуры растворитель удаляют при пониженном давлении, и продукт выделяют препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют, получая указанный в заголовке продукт в виде рыхлого белого материала (энантиомерный избыток ee 94,4%).
Соединение растворяют в смеси этилацетат/гептан (смесь 3:2,5 (об./об.)) и оставляют стоять при комнатной температуре в течение четырех дней. Удаляют супернатант с образовавшегося осадка, концентрируют при пониженном давлении и остаток лиофилизуют из смеси вода/ацетонитрил, получая указанный в заголовке энантиомерно чистый продукт, e.e. 100% (e.e. определяют с помощью Chiral HPLC Chiralpak AS, подвижная фаза смесь 0,2% TFA-н-гекс./IPA), спектроскопические характеристики которого идентичны соответствующим характеристикам соединения, описанного в примере 4, стадия 1.
[а]20 D=+36,5+2,5° (C=0,63 в этаноле).
ПРИМЕР 6
(+/-)-N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-10-ил)-N,N',N'-триметилэтандиамид
Указанное в заголовке соединение получают согласно синтетической последовательности, приведенной в ПРИМЕРЕ 4, со следующими изменениями:
Стадия 1: l-(Бензилокси)азепан-2-имин
Третбутил(бензилокси)-(5-цианопентил)карбамат (синтезированный по методике, описанной в ПРИМЕРЕ 1 - стадия 1a, исходя из 6-бромгексаннитрила) растворяют в насыщенном растворе HCl в EtOH и смесь перемешивают в течение 45 минут. Через раствор барботируют азот для удаления избытка HCl. Растворитель удаляют при пониженном давлении и остаток, растворенный в 1,4-диоксане, обрабатывают триэтиламином, доводя pH до 10. Этанол удаляют и указанное в заголовке соединение, содержащее избыток триэтиламмонийхлорида и этил-6-[(бензилокси)амино]гексаноат, используют без дополнительной очистки. Аналитический образец очищают препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют.
1Н-ЯМР(400 МГц, ДМСО-d6, 300К) δ:9.39(с, 1Н), 8.81(с, 1Н), 7.61-7.52(м, 2Н), 7.48-7.38(м, 3Н), 5.03 (с, 2Н), 4.02-3.93(м, 2Н), 2.70-2.59(м, 2Н), 1.69-1.54(м, 6Н).
МС m/z: 219(М+Н)+.
Стадия 2: (+/-)-N-(2-{[(4-Фторбензил)амино]карбонил}-3-гидрокси-
4-оксо-4-6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-10-ил)-N,N',N'-триметилэтандиамид
К перемешиваемому раствору сырого N-(4-фторбензил)-3-гидрокси-10-(метиламино)-4-оксо-4,6,7,8,9,10-гексагидропиримидо[l,2-]азепин-2-карбоксамида (синтезированного, исходя из 1-(бензилокси)азепан-2-имина по методике, используемой в аналогичных 6-членных сериях (ПРИМЕР 3 - Стадия 3)) в дихлорметане, добавляют 3 экв. триэтиламина, 2 экв. калий(диметиламино)(оксо)ацетата, 2,2 экв. 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида и 2,2 экв. 1-гидроксибензотриазола. Смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют при пониженном давлении и остаток распределяют между этилацетатом и 1 M водным HCl. Водную фазу экстрагируют этилацетатом и объединенные органические фазы сушат над сульфатом натрия, фильтруют и концентрируют досуха в вакууме. Указанный в заголовке продукт выделяют препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют, получая указанное в заголовке соединение в виде рыхлого, белого материала. Согласно 1H-ЯМР продукт представляет собой смесь ротамеров.
1Н-ЯМР(400 МГц, ДМСО-d6, 300К) δ:12.29(уш.с, 0.1Н), 11.95(уш.с, 0.9Н), 9,30(уш.с, 0,9Н), 8.45(уш.т, 0.1Н), 7.38(дд, J=8.33, 5.5 Гц, 1.8Н), 7.33(дд, J=8.33, 5.5 Гц, 0.2Н), 7.15(т, J=9.0 Гц, 2Н), 5.45-5.25(м, 0.9Н), 4.94(дд, J=14, 5.7 Гц, 1.0Н), 4.84-4.79(м, 0.1Н), 4.57-4.43(м, 2Н), 3.54(дд, J=14,11 Гц, 0.9Н), 3.28-3.18(м, 0.1Н), 3.05(с, 0.3Н), 2.92(с, 2.7Н), 2.90(с, 5.4Н), 2.81(с, 0.3Н), 2.76(с, 0.3Н), 2.19-1.78(м, 5Н), 1.41-1.27(м, 1Н).
МС m/z: 460(М+Н)+.
ПРИМЕР 7
(-)-N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-10-ил)-N,N',N'-триметилэтандиамид
Стадия 1: Диметил-(2E)-2-[(азепан-2-илиденамино)окси]бут-2-
ендиоат и диметил-(2Z)-2-[(азепан-2-илиденамино)окси]
бут-2-ендиоат
К суспензии азепан-2-оноксима в ацетонитриле добавляют по каплям при перемешивании 1,1 экв. диметилацетилендикарбоксилата. Смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют при пониженном давлении, получая смесь 8/1 диметил-(2Е)-2-[(азепан-2-илиденамино)окси]бут-2-ендиоата и диметил-(2Z)-2-[(азепан-2-илиденамино)окси]бут-2-ендиоата в виде желтого масла. Для лучшей идентификации указанных в заголовке соединений небольшое количество сырого продукта очищают препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют. Изомер E:
1Н-ЯМР(400 МГц, ДМСО-d6, 300К) δ:7.08(уш.с, 1Н), 5,63(с, 1Н), 3.77(с, 3Н), 3.59(с, 3Н), 3.19-3.11(м, 2Н), 2.29-2.21(м, 2Н), 1.66-1.42(м, 6Н).
13С-ЯМР (125 ГМц, ДМСО-d6, 300К) δ:166.20, 162.81, 161.90, 161.61, 92.38, 52.42, 50.92, 42.28, 29.84, 29.32, 28.81, 25.40.
МС m/z: 271(М+Н)+.
Изомер Z: 1Н-ЯМР(400 МГц, ДМСО-d6, 300К) δ:6.66(уш.с, 1Н), 5,63(с, 1Н), 3.74(с, 3Н), 3.61(с, 3Н), 3.24-3.16(м, 2Н), 2.20-2.12(м, 2Н), 1.65-1.44(м, 6Н).
13С-ЯМР (125 МГц, ДМСО-d6, 300К) δ:165.01, 163.01, 161.45, 154.11, 101.09, 52.32, 51,05, 42.24, 29.93, 29.31, 28.45, 25.14.
МС m/z: 271(М+Н)+.
Стадия 2: Метил-3-гидрокси-4-оксо-4,6,7,8,9,10-
гексагидропиримидо[1,2-a]азепин-2-карбоксилат
Смесь диметил-(2E)-2-[(азепан-2-илиденамино)окси]бут-2-ендиоата и диметил-(2Z)-2-[(азепан-2-илиденамино)окси]бут-2-ендиоата в соотношении 8/1 растворяют в o-ксилоле и нагревают до температуры кипения с обратным холодильником. Спустя 16 ч растворитель удаляют при пониженном давлении и остаток, растворенный в этилацетате, экстрагируют насыщенным раствором NaHCO3 в воде. PH водной фазы доводят приблизительно до 3 добавлением 6 M водного HCl и раствор экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом натрия и концентрируют.
1Н-ЯМР(400 МГц, ДМСО-d6, 300К) δ:10.12(с, 1Н), 4.29-4.16(м, 2Н), 3.80(с, 3Н), 2.95-2.78(м, 2Н), 1.79-1.41(м, 6Н).
13С-ЯМР (100 МГц, ДМСО-d6, 300К) δ:165.79, 158.54, 153.41, 143.55, 126.61, 52.04, 43.02, 35.75, 28.76, 26.94, 24.58.
МС m/z: 239(М+Н)+.
Стадия 3: Метил-3-(бензоилокси)-4-оксо-4,6,7,8,9,10-
гексагидропиримидо[1,2-a]азепин-2-карбоксилат
К раствору метил-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксилата в пиридине добавляют бензойный ангидрид (1,1 экв.) и каталитическое количество диметиламинопиридина. Смесь перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют при пониженном давлении и остаток распределяют между дихлорметаном и 1 M водн. HC1. Водную фазу экстрагируют дихлорметаном и объединенные органические фазы промывают 1 M водным HCl и насыщенным раствором соли. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют досуха в вакууме. Указанное в заголовке соединение получают после флэш-хроматографии (элюенты петролейный эфир/этилацетат, смесь 6:4) в виде коричневого твердого вещества.
1Н-ЯМР(400 МГц, ДМСО-d6, 300К) δ:8.07(дд, J=8.6, 1.3 Гц, 2Н), 7.78(т, J=7.5 Гц, 1Н), 7.62(т, J=7.9 Гц, 2Н), 4.31-4.29(м, 2Н), 3.74(с, 3Н), 3.06-3.04(м, 2Н), 1.82-1.65(м, 6Н).
13С-ЯМР (75 МГц, ДМСО-d6, 300К) δ:162.78, 162.65, 162.43, 157.01, 140.29, 135.23, 134.18, 129.63, 128.86, 127.59, 52.46, 43.13, 36.07, 28.47, 26.12, 23.68.
МС m/z: 343(М+Н)+.
Стадия 4: Метил-3-(бензоилокси)-10-бром-4-оксо-4,6,7,8,9,10-
гексагидропиримидо[1,2-a]азепин-2-карбоксилат
Смесь метил-3-(бензоилокси)-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксилата, N-бромсукцинимида (2 экв.) и а,а'-азоизобутиронитрила (0,45 экв.) в четыреххлористом углероде перемешивают при нагревании до температуры кипения с обратным холодильником в течение 14 ч. Смесь охлаждают до комнатной температуры, сукцинимид отделяют фильтрованием и растворитель удаляют при пониженном давлении. Метил-3-(бензоилокси)-10-бром-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксилат получают после флэш-хроматографии (элюенты петролейный эфир/этилацетат, смесь 8:2) в виде бледно-желтого твердого вещества.
1Н-ЯМР(400 МГц, ДМСО-d6, 300К) δ:8.07,(дд, J=8.3, 0.9 Гц, 2Н), 7.79(т, J=7.5 Гц, 1Н), 7.63(т, J=7.9 Гц, 2H),5.63(дд,J=5.9,2.2 Гц, 1H),4.98(дд,J=14.3), 6.1 Гц, 1Н), 3.97(дд, J=14.3, 11.0 Гц, 1Н), 3.76(с, 3Н), 2.31-2.13(м, 2Н), 2.10-1.79(м, 3Н), 1.-1.61-1.48(м, 1Н).
13С-ЯМР (75 МГц, ДМСО-d6, 300К) δ:162.65, 162.14, 157.22, 157.10, 139.45, 136.59, 134.37, 129.72, 128.94, 127.36, 53.56, 52.67, 42.37, 31.52, 25.78, 24.40.
МС m/z: 423/421(М+Н)+.
Стадия 5: Метил-3-гидрокси-4-оксо-10-{[(1R)-1-фенилэтил]амино}
-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-
карбоксилат
Метил-3-(бензоилокси)-10-бром-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксилат (1,0 экв.) добавляют к раствору (1R)-1-фенилэтиламина (2,2 экв.) и триэтиламина (1 экв), растворенных в N,N-диметилформамиде. Смесь энергично перемешивают в течение 2 часов при комнатной температуре и затем при 50°C в течение 30 минут. Растворитель удаляют при пониженном давлении, и указанный в заголовке сырой продукт (смесь диастереоизомеров 1:1) пригоден для применения без дополнительной очистки. Согласно альтернативной методике (стадия 5A) твердый метил-3-(бензоилокси)-10-бром-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксилат (1,0 экв.) добавляют к раствору (1R)-1-фенилэтиламина (4,5 экв.), растворенного в смеси 7:3 метанол/вода при -30°C. Взаимодействие осуществляют в течение ночи, затем температуру поднимают до комнатной температуры и растворитель концентрируют, получая белое твердое вещество, которое отделяют фильтрованием и отбрасывают. Указанное в заголовке соединение (в виде смеси диастереоизомеров 7:3) экстрагируют дихлорметаном из маточного раствора в целях применения на следующей стадии без дополнительной очистки.
МС m/z: 358 (M+H)+.
Стадия 6: N-(4-фторбензил)-3-гидрокси-4-оксо-10-{[(1R)-1-
фенилэтил]амино}-4,6,7,8,9,10-гексагидропиримидо
[1,2-a]азепин-2-карбоксамид
п-Фторбензиламин (3 экв.) добавляют к метил-3-гидрокси-4-оксо-10-{[(1R)-1-фенилэтил]амино}-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксилату (полученному, как описано на стадии 5 или стадии 5A), растворенному в метаноле. Смесь перемешивают при 70°C в течение ночи, затем охлаждают до комнатной температуры для непосредственного применения на стадии 7. Согласно альтернативной методике (стадия 6A) растворитель удаляют и сырой продукт несколько раз кристаллизуют из ацетонитрила, что приводит к получению отдельного диастереоизомера указанного в заголовке соединения в виде 4-фторбензиламмониевой соли с d.e.>95%.
Стадия 7: (+)N-(4-фторбензил)-3-гидрокси-10-{метил-[(1R)-1-
фенилэтил]амино}-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксамид
N-(4-фторбензил)-3-гидрокси-4-оксо-10-{[(1R)-1-фенилэтил]амино}-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-2-карбоксамид, полученный на стадии 6, растворяют в метаноле и pH доводят приблизительно до 5 с помощью уксусной кислоты, после чего добавляют 37% водный формальдегид (6 экв.) и цианоборгидрид натрия (6,25 экв.). Смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют при пониженном давлении и остаток, растворенный в минимальном количестве метанола, подкисливают до pH 2-3 с помощью трифторуксусной кислоты. Раствор наносят на картриджи с катионообменной смолой (Varian MEGA BOND ELUTE SCX), картриджи промывают метанолом и сырой продукт вымывают с помощью 2 M аммиака в метаноле. Объединенные элюенты концентрируют досуха при пониженном давлении, получая маслянистый остаток. Продукт, смесь диастереоизомеров, разделяют путем очистки препаративной ОФ-ВЭЖХ (колонка: С18), элюенты: вода (0,1% TFA), ацетонитрил (0,1% TFA). Указанный в заголовке диастереоизомер выходит первым пиком, и после лиофилизации указанное в заголовке соединение получают в виде белого твердого вещества (TFA-соль).
Согласно альтернативной методике (стадия 7A) продукт стадии 6A подвергают взаимодействию тем же способом, что и продукт со стадии 6 при получении отдельного диастереомера без очистки методом ВЭЖХ.
1Н-ЯМР(300 МГц, ДМСО-d6-TFA, 300К) δ:9.42(т, J=6.2 Гц, 1Н), 9.20(уш.с, 1Н), 7.60(уш.д, J=7.3 Гц, 2Н), 7.51-7.29(м, 5Н), 7.21(т, J=8.9 Гц, 2Н), 4.98-4.75(м, 3Н), 4.69(дд, J=15.5, 6.9 Гц, 1Н), 4.47(дд, J=15.5, 5.5 Гц, 1Н), 3.66(т, J=12.8 Гц, 1Н), 2.94-2.81(м, 3Н), 1.97-1.81(уш.м, 2Н), 1.79-1.33(м, 7Н).
МС m/z: 465(М+Н)+.
[a]20 D=+62±2(C=0.1 в хлороформе).
Стадия 8: (-)2-{[(4-Фторбензил)амино]карбонил}-3-гидрокси-N-
метил-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]
азепин-10-аммонийтрифторацетат
Раствор TFA-соли продукта со стадии 7 (или 7A) в метаноле, содержащий палладий на активированном угле (10%, мас./мас.) перемешивают в среде водорода при атмосферном давлении в течение 4 ч. Катализатор отделяют фильтрованием и раствор концентрируют досуха при пониженном давлении, получая указанное в заголовке соединение.
1Н-ЯМР(300 МГц, ДМСО-d6-TFA, 300К) δ:9.88(уш.с, 1Н), 9.56(уш.с, 1Н), 9.14(уш.с, 1Н), 7.41(дд, J=8.6, 5.7 Гц, 2Н), 7.17(т, J=8.8 Гц, 2Н), 4.92(дд, J=14.6, 4.6 Гц, 1Н), 4.72(уш.м, 1Н), 4.58-4.44(м, 2Н), 3.51(дд, J=13.9, 11.7 Гц, 1Н), 2.66(т, J=4.9 Гц, 3Н), 2.29(д, J=13.3 Гц, 1Н), 2.02-1.57(м, 4Н), 1.45-1.27(м, 1Н).
МС m/z: 361(М+Н)+.
[a]20 D=-4±2(C=0.4 в метаноле).
Стадия 9: (-)-N-(2-{[(4-Фторбензил)амино]карбонил}-3-гидрокси-
4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]
азепин-10-ил)-N,N',N'-триметилэтандиамид
Метилхлороксоацетат (2-6 экв.) и N-этилдиизопропиламин (4 экв.) добавляют к раствору аммонийтрифторацетатного соединения со стадии 8 в хлороформе. Смесь перемешивают при комнатной температуре в течение 1 ч. Растворитель удаляют при пониженном давлении и остаток, растворенный в 2 M растворе диметиламина в метаноле, перемешивают при комнатной температуре в течение 6 ч. Растворитель удаляют при пониженном давлении и остаток, растворенный в дихлорметане, промывают 1 M HCl в воде. Органическую фазу сушат над безводным Na2SO4 и растворитель удаляют при пониженном давлении. Указанный в заголовке продукт выделяют препаративной ОФ-ВЭЖХ, используя воду (0,1% TFA) и ацетонитрил (0,1% TFA) в качестве элюентов (колонка: С18). Содержащие продукт объединенные фракции лиофилизуют, получая указанный в заголовке продукт с энантиомерным избытком 99,5% (e.e. определяют с помощью Chiral HPLC Chiralpak AD, подвижная фаза смесь 0,2% TFA- н-гекс./0,2% TFA- этанол с 3% метанола). Аморфную калиевую соль указанного в заголовке соединения получают обработкой соединения, растворенного в ацетонитриле, водным KOH и последующей лиофильной сушкой.
Спектр 1Н-ЯМР идентичен соответствующему спектру соединения, описанного в примере 6.
13С-ЯМР(100 МГц, ДМСО-d6, 300К) δ:168.01, 165.80, 165.03, 161.30(д, JC-F=243 Гц), 157.68, 149.67, 145.94, 134.59, 129.56(д, JC-F=8.5 Гц), 124.72, 115.10(д, JC-F=21 Гц), 55.88, 42.42, 41.56, 36.24, 32.79, 32.34, 28.83, 27.10, 26.15.
МС m/z: 460(М+Н)+.
[a]20 D=-72±2(C=0.1 в хлороформе).
ПРИМЕР 8
Рацемический N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-10-ил)-N,N',N'-триметилэтандиамид
Стадия 1: Получение ω-гидрокси-N-метиламинонитрила 3

К 5% водному раствору H2SO4 добавляют 3,4-дигидро-2H-пиран (DHP; 21,1 г) при 20-35°C. Полученный раствор выдерживают при 20-35°C в течение 1 ч. Реакционную смесь охлаждают до 0-5°C и нейтрализуют до pH 6-7 посредством 40% водного метиламина. К реакционной смеси добавляют, в указанном порядке, метиламингидрохлорид и цианид натрия. Образовавшийся раствор выдерживают при комнатной температуре в течение 36 ч. Реакционную смесь экстрагируют IPAc (6x150 мл). Объединенные органические слои концентрируют до общего объема порядка 150 мл (выход в эксперименте порядка 91%) и используют на следующей стадии.
1Н-ЯМР(CDCl3, 400 МГц) δ:3.81(м, 1Н), 3.45(м, 2Н), 2.47(с, 3Н), 1.90-1.40(м, 6Н).
Стадия 2: Получение ω-гидрокси-N-метил-N-Boc-аминонитрила 4
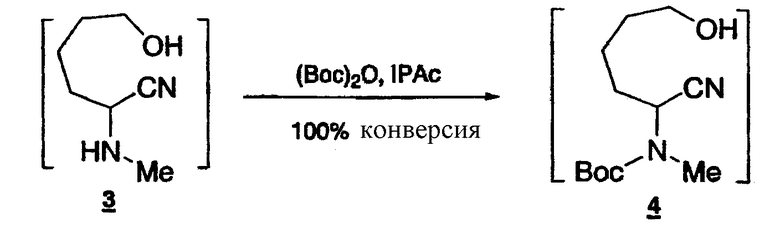
К раствору ω-гидрокси-N-метиламинонитрила 3 (0,2106 моль) в IPAc (со стадии 1) добавляют (Boc)2O (48,3 г) при комнатной температуре. Полученный раствор выдерживают при 30-35°C в течение 2 ч (100% конверсия по 1H-ЯМР). Реакционную смесь охлаждают до 0-5°C и добавляют смесь 5% NH2OH/10% NH4Cl (35 мл). Образовавшуюся смесь выдерживают при 10-20°C в течение 3 ч. После разделения фаз водный слой экстрагируют IPAc (80 мл), объединенные органические слои промывают насыщенным раствором соли (50 мл) и затем концентрируют и растворитель заменяют на IPA (общий объем 230 мл), который используют на следующей стадии.
1Н-ЯМР(CDCl3, 400 МГц) δ:5.18(м, 1Н), 3.64(кв, J=5.7 Гц, 2Н), 2.88(с, 3Н), 1.88-1.75(м, 3Н), 1.65-1.61(м, 2Н), 1.49-1.46(м, 1Н), 1.18(с, 9Н).
Стадия 3: Получение гидроксиамидина 5
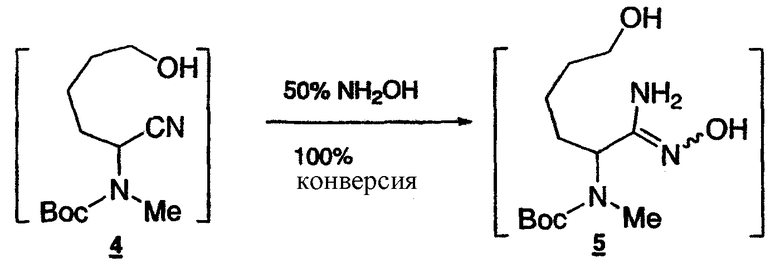
нитрил 4
К раствору N-Boc-аминонитрила 4 (0,2106 моль) в IPA (общий объем 230 мл) добавляют 50% гидроксиламин (16,2 мл) при температуре окружающей среды. Полученный раствор выдерживают при 60°C в течение 3 ч. Реакционную смесь затем концентрируют и растворитель заменяют, получая метанольный раствор (общий объем 230 мл), который используют на следующей стадии.
1Н-ЯМР(CDCl3, 400 МГц) δ:7.53(уш.с, 1Н), 4.84(уш.с, 2Н), 4.84(т, J=7.1 Гц, 1Н), 3.71-3.62(м, 2Н), 2.72(с, 3Н), 2.00(уш.с, 1Н), 1.92-1.82(м, 1Н), 1.76(1.55(м, 3Н), 1.49(с, 9Н), 1.42-1.23(м, 2Н).
Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1 мл/мин. Время удерживания: амидоксим - 6,152 минуты и 6,256 минуты (два изомера).
Стадия 4: Получение O-алкенамидоксима 6
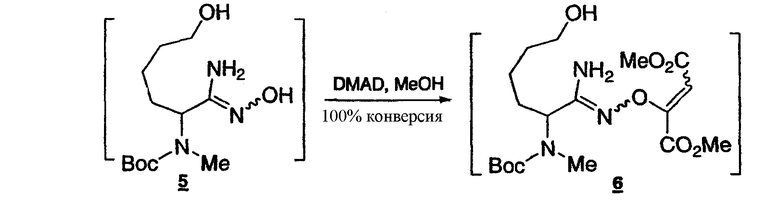
К раствору гидроксиамидина 5 (около 0,2106 моль) в метаноле (общий объем 230 мл) добавляют диметилацетилендикарбоксилат (27,10 мл) при комнатной температуре. Полученный раствор выдерживают при комнатной температуре в течение 16 ч. Реакционную смесь концентрируют и растворитель заменяют на кумол при 40-60°C (общий объем 430 мл). Раствор используют на следующей стадии.
1Н-ЯМР(CDCl3, 400 МГц) δ:5.82(с, 0.28Н), 5.73(с, 0.72Н), 5.44(уш.с, 1.77Н), 5.25(уш.с, 0.56Н), 4.61(м, 1Н), 3.89(с, 0.84Н), 3.84(с, 2.16Н), 3.72(с, 2.16Н), 3.68(с, 0.84Н), 3.65-3.58(м, 2Н), 2.73(с, 0.84Н), 2.71(с, 2.16Н), 1.90-1.52(м, 4Н), 1.47(с, 9Н), 1.43-1.30(м, 2Н).
Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1 мл/мин. Время удерживания: амидоксим 6-12,051, 12,315 минут, соотношение около 3,6:1.
Стадия 5: Получение пиримидина 7
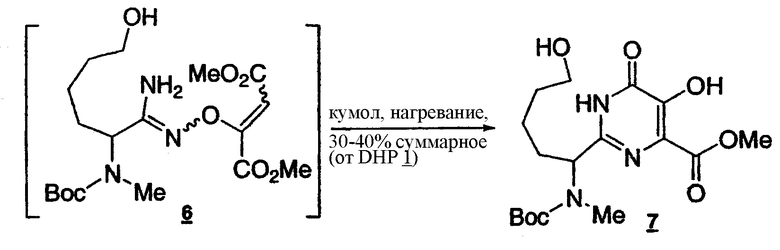
Раствор O-алкенамидоксима 6 (около 0,2106 моль) в кумоле (общий объем 430 мл) нагревают до 120°C (внутренняя температура) в течение 12 ч. Реакционную смесь затем охлаждают приблизительно до 60°C, концентрируют до общего объема 250 мл, затем разбавляют EtOAc (250 мл) и охлаждают до 25-35°C. Затем медленно добавляют 5% бикарбонат натрия (330 мл, около 1 эквив.) и полученный раствор выдерживают при 25-35°C в течение 0,5 ч. После разделения фаз органический слой вновь экстрагируют 5% бикарбонатом натрия (180 мл). Объединенные водные экстракты подкисливают с помощью 5 н. HCl до pH 2-3 и экстрагируют посредством EtOAc (3x250 мл). Объединенные органические слои промывают насыщенным раствором соли (150 мл). Органический раствор концентрируют и растворитель заменяют на ТГФ (порядка 30-40% общий выход, KF порядка 100-150 ч/млн).
1Н-ЯМР(CDCl3, 400 МГц) δ:10.66(уш.с, 2Н), 4.77(м, 1Н), 4.01(с, 3Н), 3.72-3.67(м, 2Н), 2.77(с, 3Н), 2.20-1.55(м, 5Н), 1.48(с, 9Н), 1.43-1.35(м, 1Н).
Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1 мл/мин. Время удерживания: пиримидин 7 - 9,905 минут.
Стадия 6: Получение бисмезилпиримидина 8
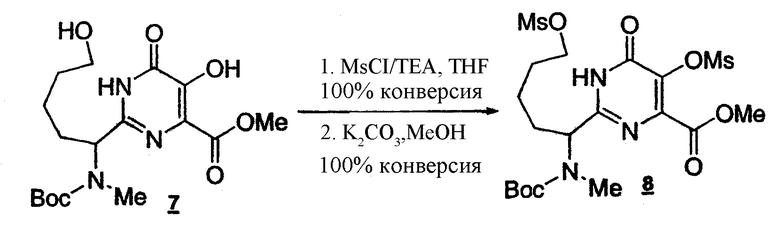
К раствору пиримидина 7(43,5 г, около 80% чистоты, 0,09029 моль) в ТГФ (275 мл) медленно добавляют TEA (37,8 мл) и MsCl (21,0 мл), одновременно, при 0-5°C за 1 ч. Полученный раствор выдерживают при той же температуре в течение 4 ч. Твердое вещество отделяют фильтрованием, промывают ТГФ (3x100 мл). Объединенные фильтраты концентрируют и растворитель заменяют на метанол (общий объем 200 мл). К раствору тримезилпиримидина в метаноле добавляют карбонат калия (12,5 г, 0,09029 моль) при 10-20°C. Полученный раствор выдерживают при той же температуре в течение 6-10 ч (мониторинг методом ВЭЖХ). Реакционную смесь нейтрализуют до pH 6-7 с помощью 5 н. HCl и концентрируют до общего объема порядка 100 мл. Добавляют 16% раствор соли (100 мл) и полученный раствор экстрагируют EtOAc (3x100 мл). Объединенные органические слои промывают насыщенным раствором соли (50 мл), концентрируют и растворитель заменяют на ДМФА. Побочный продукт (MeSO3Me), образующийся в количестве 1 эквив. при селективном гидролизе тримезилпиримидина, удаляют азеотропной перегонкой с ДМФА при 60-65°C (мониторинг методом 1H-ЯМР до <10 моль%). Концентрация бисмезилпиримидина 8 в ДМФА равна 0,3 M (общий объем 300 мл).
1Н-ЯМР(CDCl3, 400 МГц) δ:11.00(уш.с, 1Н), 4.78(д, J=7.8 Гц, 1н), 4.24-4.15(м, 2Н), 3.95(с, 3Н), 3.50(с, 3Н), 2.99(с, 3Н), 2.81(с, 3Н), 2.12-2.11(м, 1Н), 1.90-1.76(м, 2Н), 1.46(с, 9Н), 1.43-1.35(м, 2Н).
Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1 мл/мин. Время удерживания: тримезилпиримидин 14,140 минут; бисмезилпиримидин 12,760 минут.
Стадия 7: Получение пиримидинмезилата с семичленным циклом 9
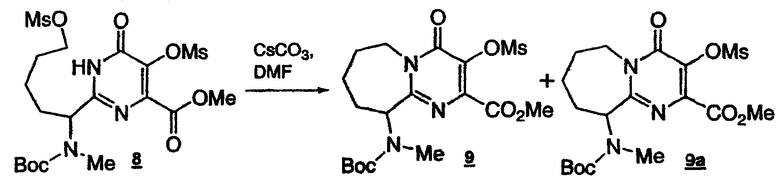
К раствору бисмезилпиримидина 8 (0,09029 моль) в ДМФА (общий объем 300 мл) добавляют карбонат цезия (35,30 г) при комнатной температуре. Полученную взвесь выдерживают при 55°C в течение 2-3 ч (76% конверсия по ВЭЖХ). После нейтрализации до pH 7, полученную смесь разбавляют 250 мл воды, экстрагируют IPAc (2x250 мл). Объединенные органические слои промывают насыщенным раствором соли (2x200 мл). Органический слой концентрируют, получая сырой продукт. Половину сырого продукта очищают, пропуская через короткую колонку (силикагель, смесь гексан:EtOAc 2:1), что приводит к получению требуемого продукта 9 (6,00 г, 98A% чистота) и 9a (2,3 г, 40A% чистота). Общий выход циклического продукта в расчете на DHP составляет, с учетом поправок, около 13%.
1Н-ЯМР(CDCl3, 400 МГц) Для соединения 9: δ:5.34(м, 1Н), 5.22(м, 1Н), 3.93(с, 3Н), 3.51(с, 3Н), 2.97(с, 3Н), 2.20-2.05(м, 3Н), 1.90-1.65(м, 2Н), 1.44(с, 9Н), 1.24(м, 1Н). Для соединения 9а: 11.86(уш.с, 1Н), 7.90-7.55(уш.с, 1Н), 7.31(дд, J=8.5, 5.4 Гц, 2Н), 7.06(т, J=8.5 Гц, 2Н), 5.40-4.90(м, 2Н), 4.53-4.40(м, 2Н), 3.45-3.23(м, 1Н), 2.23-2.05(м, 3Н), 1.86-1.76(м, 1Н); 1.74-1.64(м, 1Н), 1.47-1.37(м, 1Н), 1.30(с, 9Н). Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1 мл/мин. Время удерживания: пиримидинмезилат с семичленным циклом 9 13,969 минут; пиримидин с семичленным циклом 9a 13,141 минут.
Также применяется альтернативная методика с использованием LiH: к раствору бисмезилпиримидина 8 (65 мг) в диоксане (1 мл) добавляют LiH в порошке при комнатной температуре. Полученную смесь выдерживают при 65°C в течение 4 ч. Затем реакционную смесь охлаждают до комнатной температуры и добавляют 1 н. HCl, чтобы погасить избыток LiH. Раствор экстрагируют EtOAc (2х5 мл). Объединенный органический слой промывают насыщенным раствором соли и затем концентрируют. Остаток очищают флэш-хроматографией (силикагель, смесь гексан: EtOAc=2:1), получая пиримидинмезилат с семичленным циклом 9 (45,6 мг, 85%).
1Н-ЯМР(CDCl3, 400 МГц) δ:5.34(м, 1Н), 5.22(м, 1Н), 3.93(с, 3Н), 3.51(с, 3Н), 3.47(м, 1Н), 2.97(с, 3Н), 2.20-2.05(м, 3Н), 1.90-1.65(м, 2Н), 1.44(с, 9Н), 1.24(м, 1Н).
Стадия 8: Получение пиримидинамида с семичленным циклом 10
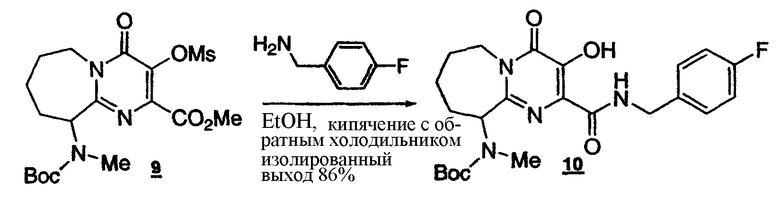
К раствору пиримидинмезилата с семичленным циклом 9(6 г) в EtOH (80 мл) добавляют 4-фторбензиламин (5,060 г). Полученный раствор кипятят с обратным холодильником в течение 8 ч (100% конверсия по ВЭЖХ). Реакционную смесь концентрируют до общего объема приблизительно 20 мл и добавляют 80 мл EtOAc. К полученному раствору добавляют 20% раствор соли (15 мл), 4 н. HCl (15 мл) и воду (10 мл). После разделения фаз водный слой опять экстрагируют EtOAc (25 мл). Объединенные органические слои промывают смесью 4 н. HCl:20% раствор соли (1:1, 3x15 мл), насыщенным раствором соли (15 мл). Органический раствор концентрируют до общего объема порядка 30 мл. К раствору в течение 1 ч медленно добавляют гексан (70 мл). Полученную взвесь выдерживают при 0-5°C в течение 1 ч. Кристаллическое твердое вещество отделяют фильтрованием, промывают смесью гексан:EtOAc (4:1, 50 мл), сушат в вакууме при продувании азотом и получают пиримидинамид с семичленным циклом 10 (5,30 г, 86%, ВЭЖХ>97A%).
1Н-ЯМР(CDCl3, 400 МГц) δ:11.85(уш.с, 1Н), 7.84(уш.с, 0.5Н), 7.68(уш.с, 0.5Н), 7.31(м, 2Н), 7.04(м, 2Н), 5.40-4.90(м, 2Н), 4.53(м, 2Н), 3.38(м, 1Н), 2.87(с, 3Н), 2.20-2.15(м, 3Н), 1.90-1.40(м, 3Н), 1.37(с, 9Н).
Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1 мл/мин. Время удерживания: пиримидин с семичленным циклом 10-15,467 минут.
Стадия 9: Получение гидрохлорида пиримидинамида с семичленным 11
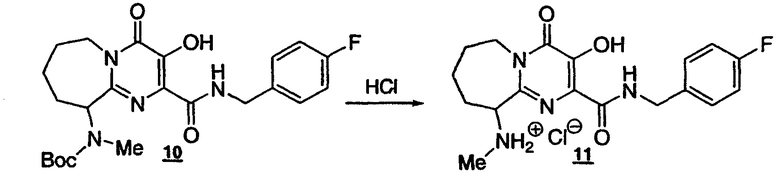
Через раствор этилацетата (3,5 мл) барботируют газообразный HCl (0,5389 г), при температуре от -30 до -20°C. В HCl-EtOAc-раствор вносят при температуре от -30 до -20°C пиримидинамид с N-Boc-семичленным циклом 10 (кристаллическое твердое вещество). Полученный раствор медленно нагревают до комнатной температуры за 2,5 ч и выдерживают при комнатной температуре в течение 0,5 ч (100% конверсия по ВЭЖХ). Реакционную смесь разбавляют EtOAc (7 мл). Полученную взвесь выдерживают при 0-5°C в течение 1 ч. Кристаллическое твердое вещество отделяют фильтрованием, промывают EtOAc, гексаном, сушат в вакууме при продувании азотом и получают требуемый продукт 11 (98% изолированный выход, >97A% чистота).
1Н-ЯМР(CDCl3, 400 МГц) δ:12.35(с, 1Н), 9.96(т, J=6.3 Гц, 1Н), 9.51(уш.с, 1Н), 9.19(уш.с, 1Н), 7.42(дд, J=8.5, 5.6 Гц, 2Н), 7.19(т, J=8.5 Гц, 2Н), 4.92(дд, J=14.5, 5.1 Гц, 1Н), 4.71(м, 1Н), 4.57-4.45(м, 2Н), 3.52(т, J=14.5 Гц), 2.65(т, J=5.0 Гц, 3Н), 2.30 (уш.д, J=12.6 Гц, 1Н), 1.99-1.92(м, 1Н), 1.90-1,75(м, 2Н), 1.68-1.60(м, 1Н), 1.41-1.33(м, 1Н).
Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1 мл/мин. Время удерживания: гидрохлорид пиримидинамида с семичленным циклом 8,118 минут.
Стадия 10: Получение рацемического N-(2-{[(4-фторбензил)амино]
карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидро-
пиримидо[1,2-a]азепин-10-ил)-N,N',N'-триметил-
этандиамида 14

К раствору кислоты 12 (122 мг) в ТГФ (3 мл) добавляют этилхлорформиат (92 мкл) при 0-5°C. Затем к реакционной смеси медленно добавляют при 0-5°C 4-NMM (106 мкл). Реакционную смесь выдерживают при той же температуре в течение 2 ч. К раствору смешенного ангидрида добавляют в виде твердого вещества при 0-5°C гидрохлорид пиримидина 11 (79,4 мг) и выдерживают при той же температуре в течение 5 ч и затем при 5-10°C в течение еще 2 ч (100% конверсия по ВЭЖХ).
К реакционной смеси добавляют водный диметиламин (40%, 158 мкл) и смесь выдерживают при 10-15°C в течение 1 ч, по истечении которого осуществляют мониторинг реакции методом ВЭЖХ для подтверждения полноты превращения. Реакционную смесь подкисливают 2 н. HCl до pH 3-4 при 5-15°C. Добавляют, в указанном порядке, EtOAc (6 мл) и насыщенный раствор соли (2 мл). После разделения фаз органический слой промывают 1 н. HCl (2 мл), насыщенным раствором соли (2х2 мл). Органический слой концентрируют до суммарного объема 1 мл. Медленно за 0,5 ч добавляют гексан (5 мл). Полученную взвесь выдерживают при 0-5°C в течение 1 ч. Кристаллическое твердое вещество отделяют фильтрованием, промывают смесью гексан/EtOAc (5:1), MTBE, сушат в вакууме при продувании азотом и получают указанное в заголовке соединение 14 (75,6 мг, 82%).
1Н-ЯМР(CDCl3, 400 МГц) δ:12.13(с, 1Н), 9.41(уш.с, 1Н), 7.38(дд, J=8.5, 5.4 Гц, 2Н), 7.00(т, J=8.5 Гц, 2Н), 5.40(уш.с, 1Н), 5.29(дд, J=14.5, 6.0 Гц, 1Н), 4.60(дд, J=14.5, 6.6 Гц, 1Н), 4.52(дд, J=14.5, 6.3 Гц, 1Н), 3.35(дд, J=14.5, 11.6 Гц, 1Н), 3.04(с, 3Н), 3.01(с, 3Н), 2.98(с, 3Н), 2.23-2.12(м, 3Н), 1.95-1.81(м, 2Н), 1.58-1.49(м, 1Н).
Условия ВЭЖХ: колонка: Zorbax, Rx C8 250x4,6 мм; температура: 30°C; детектирование при 210 нм; подвижная фаза: смесь 0,1% водн. H3PO4 (A)/MeCN (B); градиент: от 90:10 смеси (A)/(B) до 10:90 за 15 мин, 10:90 поддерживают в течение 5 мин, от 10:90 до 90:10 смеси (A)/(B) за 10 секунд; скорость потока: 1
мл/мин. Время удерживания: указанное в заголовке соединение 14-12, 191 минут.
В приведенной ниже таблице приведен список полученных соединений по настоящему изобретению. В таблице представлены структура и название каждого соединения, масса молекулярного иона плюс 1 (M+), установленные посредством FIA-MS, и синтетическая схема, используемая для получения каждого соединения.
(Пр.1)
(Пр.2)
(Пр.3)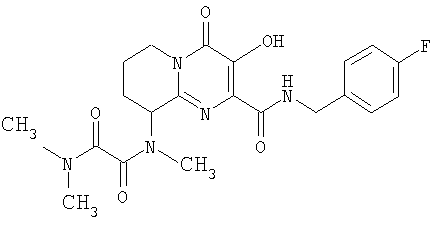
(Пр.4)
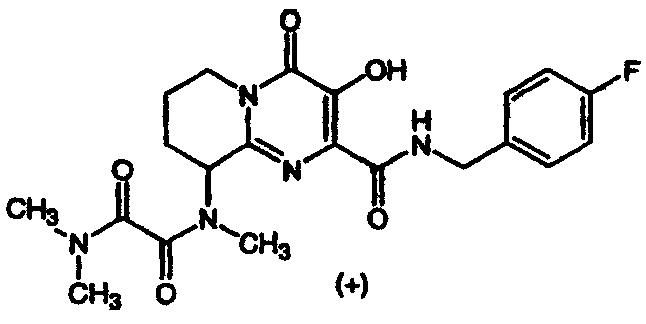
(Пр.5)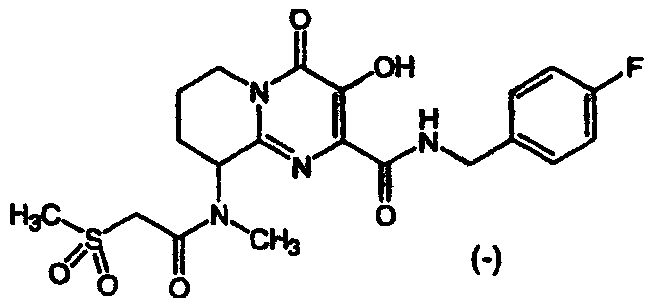
амино}-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксамид
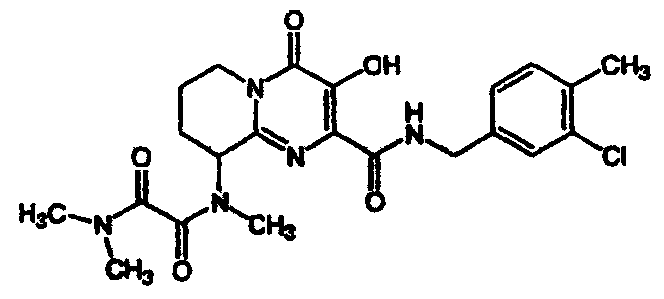
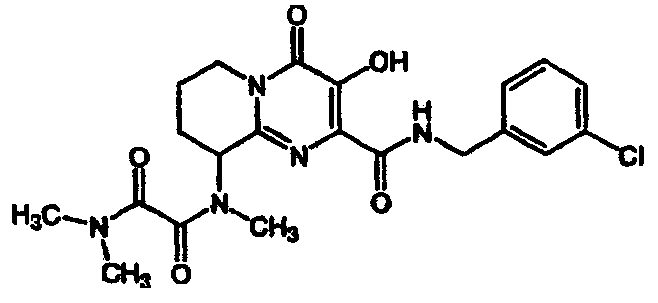
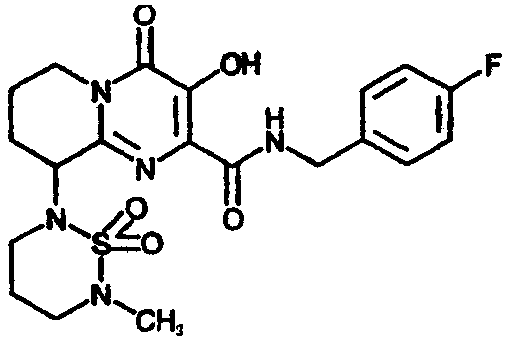
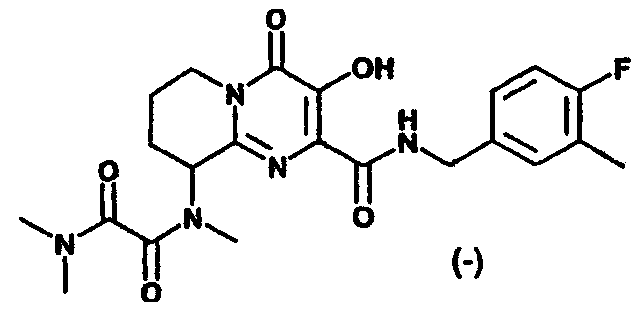
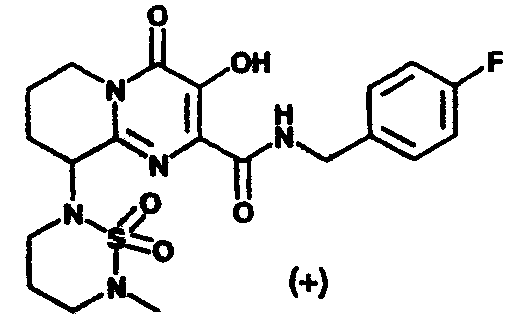
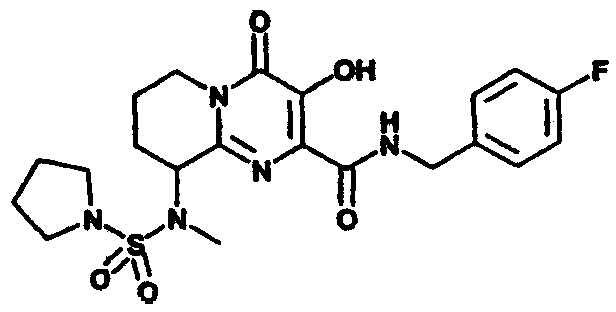
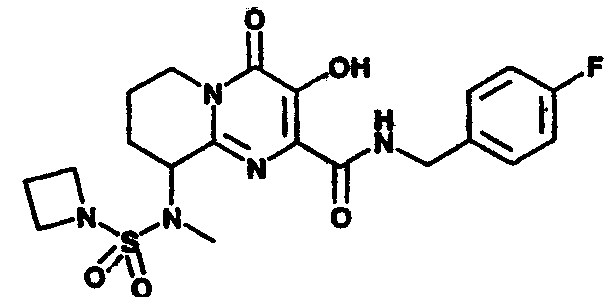

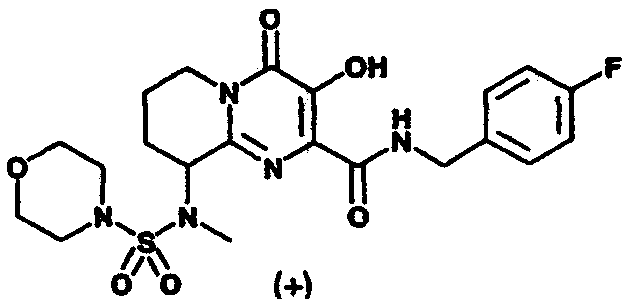
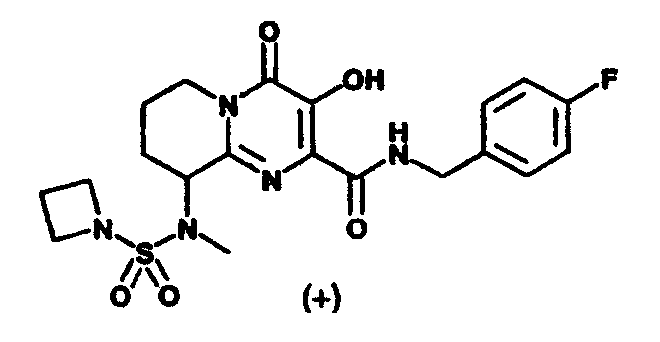
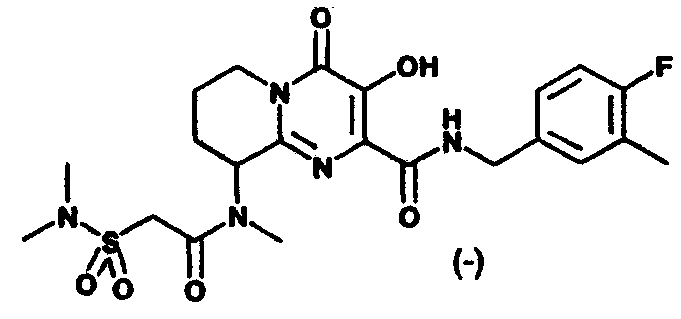
сульфонил]-ацетил}(метил)амино]-N-(4-фтор-3-метилбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксамид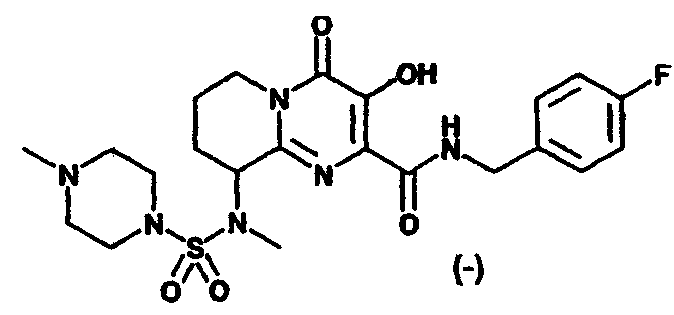
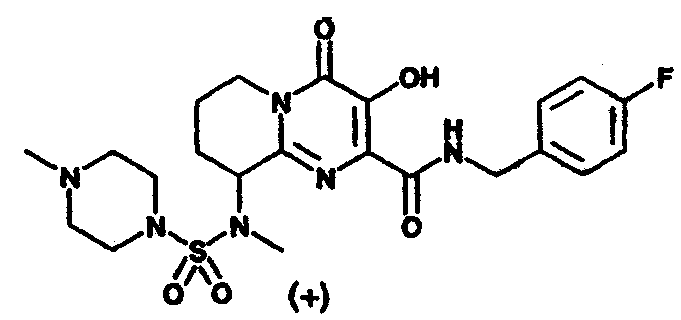
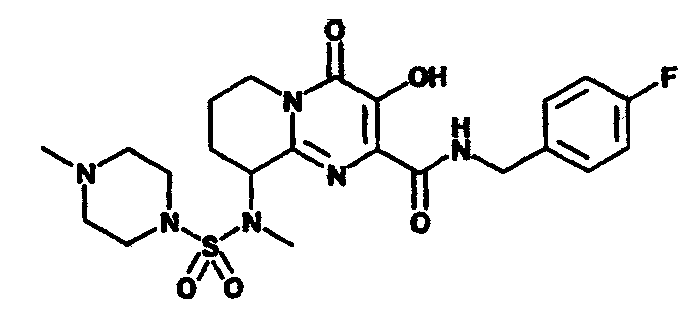
сульфонил]амино}-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-2-карбоксамид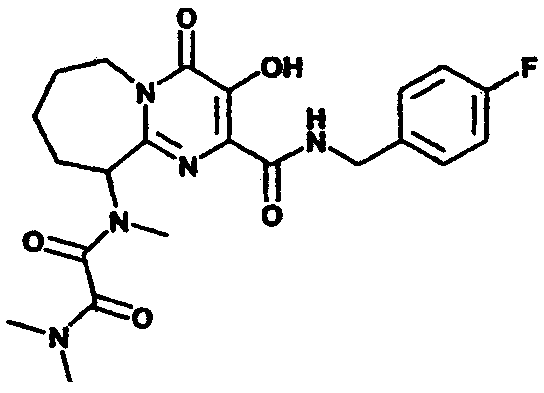
(Пр. 6)
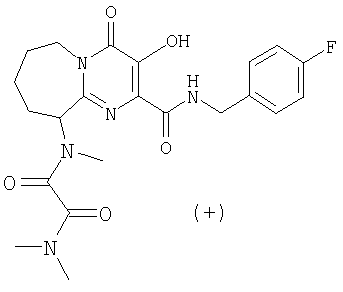
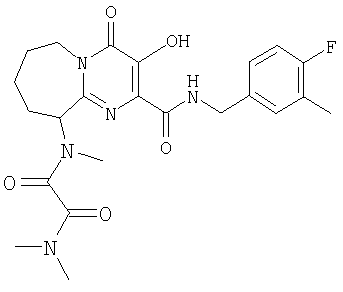
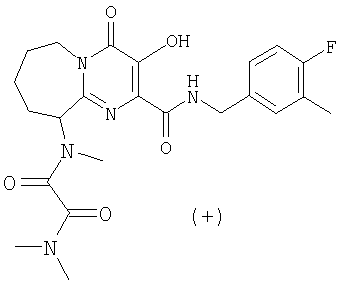
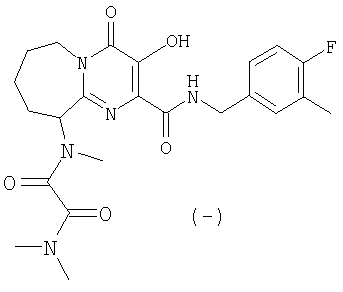
Хотя приведенное выше описание излагает основные принципы настоящего изобретения и сопровождается примерами, представленными в целях иллюстрации, практическое осуществление изобретения охватывает все возможные изменения, дополнения и/или модификации, охватываемые рамками объема приложенных пунктов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| СПОСОБ ПОЛУЧЕНИЯ 3-{2-[4-(6-ФТОРБЕНЗО[D]ИЗОКСАЗОЛ-3-ИЛ)ПИПЕРИДИН-1-ИЛ]ЭТИЛ}-2-МЕТИЛ-6,7,8,9-ТЕТРАГИДРО-4Н-ПИРИДО[1,2-А]ПИРИМИДИН-4-ОНА, ПРОМЕЖУТОЧНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО ПРОИЗВОДНОГО | 2003 |
|
RU2272037C9 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2008 |
|
RU2490265C2 |
| ИНГИБИТОРЫ Axl ДЛЯ ПРИМЕНЕНИЯ В КОМБИНИРОВАННОЙ ТЕРАПИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ, УСТРАНЕНИЯ ИЛИ ЛЕЧЕНИЯ МЕТАСТAЗИРУЮЩЕГО РАКА | 2010 |
|
RU2555326C2 |
| Производные гексагидропиримидо (1,2-а) азепины,обладающие антиангинозной активностью | 1980 |
|
SU981319A1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-4(3H)-ОНА В КАЧЕСТВЕ АНТАГОНИСТОВ TRPV4 | 2021 |
|
RU2840769C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3-{2-/4-(6-ФТОРБЕНЗО [D] ИЗОКСАЗОЛ-3-ИЛ)ПИПЕРИДИН-1-ИЛ /ЭТИЛ}-2-МЕТИЛ-6,7,8,9-ТЕТРАГИДРО-4H-ПИРИДО /1,2-А/ ПИРИМИДИН-4-ОНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 1995 |
|
RU2123004C1 |
| ПРОИЗВОДНЫЕ 1,2,4,5-ТЕТРАГИДРОБЕНЗО[D]АЗЕПИНОВ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2240317C2 |
Изобретение относится к соединениям формулы (I) и формулы (II) и их фармацевтически приемлемым солям, к их применению в качестве ингибитора ВИЧ-интегразы и к фармацевтической композиции на их основе. Соединения могут найти применение для лечения ВИЧ-инфекции. В формуле (I)
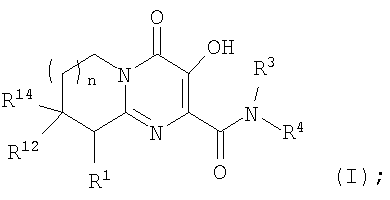
R1 означает Н или NR2R5; R2 означает СН3; R5 означает 1) С(O)СН2SO2СН3, 2) С(O)С(O)N(СН3)2, 3) SO2Н(СН3)2 или 4) SO2R20, где R20 означает


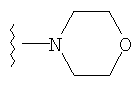 или
или 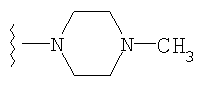 либо альтернативно, R2 и R5 вместе с атомом азота, к которому присоединены, образуют или
либо альтернативно, R2 и R5 вместе с атомом азота, к которому присоединены, образуют или 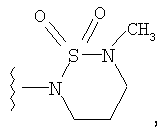 R3 означает водород; R4 означает 1) п-фторбензил, 2) 4-фтор-3-метилбензил, 3) 3-хлорбензил или 4) 3-хлор-4-метилбензил; R12 и R14 оба означают Н, за исключением, что когда R5 означает С(O)С(O)N(СН3)2, и R4 означает п-фторбензил, и n равно 1, то R12 и R14 либо оба означают Н, либо оба означают СН3; и n означает целое число, равное нулю, 1 или 2. 4 н. и 8 з.п. ф-лы, 5 сх., 1 табл.
R3 означает водород; R4 означает 1) п-фторбензил, 2) 4-фтор-3-метилбензил, 3) 3-хлорбензил или 4) 3-хлор-4-метилбензил; R12 и R14 оба означают Н, за исключением, что когда R5 означает С(O)С(O)N(СН3)2, и R4 означает п-фторбензил, и n равно 1, то R12 и R14 либо оба означают Н, либо оба означают СН3; и n означает целое число, равное нулю, 1 или 2. 4 н. и 8 з.п. ф-лы, 5 сх., 1 табл.

где R1 означает Н или NR2R5;
R2 означает СН3;
R5 означает
1) C(O)CH2SO2CH3,
2) С(O)С(O)N(СН3)2,
3) SO2N(СН3)2 или
4) SO2R20, где R20 означает
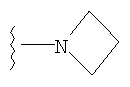
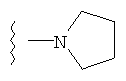
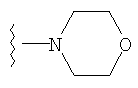 или
или 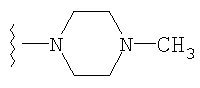
либо, альтернативно, R2 и R5 вместе с атомом азота, к которому присоединены, образуют
или 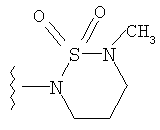
R3 означает водород;
R4 означает:
1) п-фторбензил,
2) 4-фтор-3-метилбензил,
3) 3-хлорбензил или
4) 3-хлор-4-метилбензил;
R12 и R14 оба означают Н, за исключением, что когда R5 означает С(O)С(O)N(СН3)2 и R4 означает п-фторбензил, и n равно 1, то R12 и R14 либо оба означают Н, либо оба означают СН3; и
n означает целое число, равное нулю, 1 или 2.
R1 означает NR2R5;
R2 означает СН3;
R5 означает С(O)С(O)N(СН3)2 или SO2R20, где R20означает
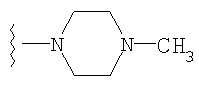
R3означает водород;
R4 означает п-фторбензил или 4-фтор-3-метилбензил;
R12 и R14 оба означают Н, за исключением, что когда R5 означает С(O)С(O)N(СН3)2 и R4 означает п-фторбензил, и n равно 1, то R12 и R14 либо оба означают Н, либо оба означают СН3; и
n означает целое число, равное 1 или 2.

где R1 означает водород, NR2R5, OR2, SR2, SOR2, SO2R2, SO2NR2R5 или OC(O)NR2R5;
R3 означает водород;
R4 означает
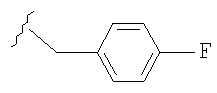
R2 означает
1)водород,
2) СН3 или
3)
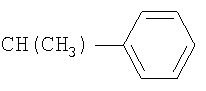
и R5 означает
1) С(O)СН3,
2) С(O)СН2SO2СН3,
3)СН3,
4) C(O)C(O)N(CH3)2,
5) SO2CH3,
6) SO2N(CH3)2,
7) C(O)CH2N(CH3)2,
8) SO2CH2SO2CH3,
9)С(O)CF3,
10)

11)
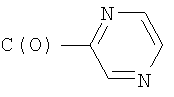
или
12) 
либо R2 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклический цикл, выбираемый из группы, включающей
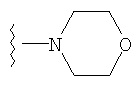
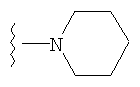 и
и 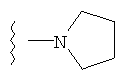
N-(4-фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N-(4-фторбензил)-3-гидрокси-9-морфолин-4-ил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
9-[[(диметиламино)сульфонил](метил)амино]N-(4-фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N1-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-9-ил)-N1,N2,N2-триметилэтандиамид;
(+)-N1-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-а]пиримидин-9-ил)-N1,N2,N2-триметилэтандиамид;
(-)-N-(4-фторбензил)-3-гидрокси-9-{метил[(метилсульфонил)ацетил]амино}-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-9-ил)-N,N',N'-триметилэтандиамид;
N-(2-{[(3-хлор-4-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-9-ил)-N,N',N'-триметилэтандиамид;
N-(2-{[(3-хлорбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-9-ил)-N,N',N'-триметилэтандиамид;
N-(4-фторбензил)-3-гидрокси-9-(6-метил-1,1-диоксидо-1,2,6-тиадиазинан-2-ил)-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(-)-N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-9-ил)-N,N',N'-триметилэтандиамид;
(+)-N-(4-фторбензил)-3-гидрокси-9-(6-метил-1,1-диоксидо-1,2,6-тиадиазинан-2-ил)-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N(4-фторбензил)-3-гидрокси-9-[метил(пирролидин-1-илсульфонил)амино]-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
9-[(азетидин-1-илсульфонил)(метил)амино]-N-(4-фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(-)-N-(4-фторбензил)-3-гидрокси-9-[метил(морфолин-4-илсульфонил)амино]-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(+)-N-(4-фторбензил)-3-гидрокси-9-[метил(морфолин-4-илсульфонил)амино]-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(+)-9-[(азетидин-1-илсульфонил)(метил)амино]-N-(4-фторбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(-)-9-[{[(диметиламино)сульфонил]ацетил}(метил)амино]-N-(4-фтор-3-метилбензил)-3-гидрокси-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(-)-N-(4-фторбензил)-3-гидрокси-9-{метил[(4-метилпиперазин-1-ил)сульфонил]амино}-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(+)-N-(4-фторбензил)-3-гидрокси-9-{метил[(4-метилпиперазин-1-ил)сульфонил]амино}-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N-(4-фторбензил)-3-гидрокси-9-{метил[(4-метилпиперазин-1-ил)сульфонил]амино}-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(-)N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(+)-N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-a]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(+)-N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N,N'-триметилэтандиамид;
(-)-N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-8,8-диметил-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-9-ил)-N,N',N'-триметилэтандиамид.
N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(+)-N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(-)-N-(2-{[(4-фтор-3-метилбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(-)-N-(4-фторбензил)-3-гидрокси-9-{метил[(4-метилпиперазин-1-ил)сульфонил]амино}-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
(+)-N-(4-фторбензил)-3-гидрокси-9-{метил[(4-метилпиперазин-1-ил)сульфонил]амино}-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N-(4-фторбензил)-3-гидрокси-9-{метил[(4-метилпиперазин-1-ил)сульфонил]амино}-4-оксо-6,7,8,9-тетрагидро-4Н-пиридо[1,2-а]пиримидин-2-карбоксамид;
N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-8,8-диметил-4-оксо-6,7,8,9-тетрагидро-4H-пиридо[1,2-a]пиримидин-9-ил)-N,N',N'-триметилэтандиамид;
N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(-)N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(+)-N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид.
N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид;
(-)N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид; и
(+)N-(2-{[(4-фторбензил)амино]карбонил}-3-гидрокси-4-оксо-4,6,7,8,9,10-гексагидропиримидо[1,2-а]азепин-10-ил)-N,N',N'-триметилэтандиамид.
Приоритеты от 12.12.2003 и 27.12.2002 в равной степени относятся к пп.1-12 формулы изобретения.
| RU 2001111746 А, 27.11.2003 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

