ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способам защиты нервных клеток, в частности клеток центральной нервной системы млекопитающих, от повреждений при ишемическом инсульте, в том числе от токсического действия глютамата и от апоптоза. Способы, являющиеся предметом настоящего изобретения, заключаются в применении агониста альфа-2 рецепторов адреналина бримонидина для предотвращения повреждения и гибели нервных клеток при болезни Паркинсона и болезни Альцгеймера.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтическим композициям, включающим соединения, которые способны воздействовать на альфа-2 адренергические рецепторы. Кроме того, настоящее изобретение включает в себя способы лечения различных заболеваний и состояний, в том числе заболеваний и состояний, вызванных нарушениями центральной нервной системы.
Адренергические рецепторы человеческого организма являются интегральными мембранными белками и подразделяются на два больших класса: альфа- и бета-адренорецепторы. Оба типа рецепторов обеспечивают работу периферического отдела симпатической нервной системы, связывая катехоламины, норэпинефрин и эпинефрин.
Норэпинефрин выделяется адренергическими нервными окончаниями, а эпинефрин - мозговым веществом надпочечников. Сродство адренергических рецепторов к данным веществам является одним из принципов классификации: альфа-адренорецепторы связывают норэпинефрин более активно, чем эпинефрин, и гораздо активнее, чем синтетическое соединение изопротеренол. Обратная закономерность характерна для бета-адренорецепторов. Во многих тканях реакции, такие как сокращение гладкой мускулатуры, на активацию альфа-адренорецепторов и бета-адренорецепторов являются разнонаправленными.
Впоследствии функциональное различие между альфа- и бета-адренорецепторами было детально изучено в ходе фармакологических исследований с использованием различных лабораторных животных и источников тканей. Альфа- и бета-адренорецепторы были дополнительно разделены на подтипы: альфа-1, альфа-2, бета-1 и бета-2.
Кроме того, было установлено, что эти рецепторы также могут существовать в различных вариантах: альфа-2 рецепторы подразделяются на подтипы альфа-2А, альфа-2В и альфа-2С.
Были установлены функциональные различия между альфа-1 и альфа-2 рецепторами и описаны соединения, обладающие специфическим сродством к ним.
Так, публикация WO 92/00073 содержит описание R(+) энантиомера теразозина, избирательно взаимодействующего с адренергическими рецепторами подтипа альфа-1. Избирательность действия данного соединения на альфа-1 и альфа-2 рецепторы имеет большое значение, поскольку стимуляция альфа-2 рецепторов подавляет секрецию эпинефрина и норэпинефрина, а подавление активности альфа-2 рецепторов повышает секрецию этих гормонов. Таким образом, использование неселективных альфа-адреноблокаторов (например, феноксибензамина и фентоламина) ограничено их способностью посредством стимуляции α2-адренорецепторов повышать уровень катехоламинов в плазме крови, что приводит к известным физиологическим изменениям (увеличение частоты сердечных сокращений и стимуляция сокращения гладкомышечных клеток). Важно, что избирательность действия веществ в отношении альфа-1 и альфа-2 рецепторов определяется константой KD, которая позволяет лишь сопоставить степень сродства веществ к рецепторам без учета фактического биологического эффекта от активации сравниваемых рецепторов.
Напротив, способ определения избирательности действия агонистов альфа-рецепторов включает в себя испытание RSAT (Технология отбора и амплификации рецепторов), описанное Messier с соавторами. High Throughput Assays Of Cloned Adrenergic, Muscarinic, Neurokinin And Neurotrophin Receptors In Living Mammalian Cells, Pharmacol. Toxicol. 76:308-11 (1995), и адаптированное для изучения альфа-2 рецепторов. Данная работа включена в данное описание посредством ссылки. Данный способ позволяет определить вклад рецептора в уменьшение контактного торможения, которое приводит к избирательной пролиферации клеток, несущих на себе данный рецептор, в смешанной популяции всех клеток. Увеличение числа клеток определяется по содержанию маркерного гена, внесенного в клетки путем трансфекции (например, гена бета-галактозидазы, активность которой можно легко определить на пластине с 96 лунками). Рецепторы, активирующие G-белок, Gq, усиливают этот ответ. Альфа-2 рецепторы, которые обычно связаны с Gi-белком, активируют ответ согласно RSAT при взаимодействии с гибридным Gq-белком, который несет на себе домен узнавания рецептора Gi, т.н. Gq/i5 2 [Conklin и соавт., Substitution Of Three Amino Acids Switches Receptor Specificity Of Gqa To That Of Gia, Nature 363: 274-6. (1993)]. Данная работа включена в данное описание посредством ссылки.
Агонисты альфа-адренорецепторов эффективны для лечения различных заболеваний и патологических состояний. Такие вещества, как, например, клонидин, применяются в качестве системных и глазных антигипертензивных агентов, агентов для лечения аддиктивных состояний, таких как курение и токсикомания, а также для лечения дисменореи. Другой агонист альфа-адренорецепторов - тизанидин - используется для уменьшения спастического компонента при рассеянном склерозе посредством снижения мышечного тонуса. По данным некоторых исследований вещества этой группы обладают и некоторой анальгетической активностью.
Несмотря на эффективность, использование этих средств часто сопровождается выраженными побочными эффектами, включающими в себя седацию, сердечно-сосудистые расстройства (гипотензия и урежение пульса), а также головокружение, что ограничивает показания к их назначению. В частности, кривые терапевтической и седативной дозы этих препаратов часто перекрещиваются, т.е. седативный эффект начинает проявляться при той же дозе, что и терапевтический (например, гипотензивный или анальгетический) in vivo.
Такие соединения, как клонидин, тизанидин и дексмедетомидин описываются в литературе как агонисты альфа-2 рецепторов адреналина, что во многом основано на результатах исследования рецепторного связывания, см. Hieble et al., J.Med Chem. 38: 3415 (September 1, 1995); Ruffolo, et al., J.Med. Chem. 38: 3681 (September 15, 1995) - обе работы включены в данное описание посредством ссылки. Хотя эти вещества являются агонистами альфа-2 рецепторов, однако то, что эти вещества также обладают значительной активностью в отношении альфа-1 рецепторов, не является принятым. Взаимное влияние активации альфа-1 рецепторов и альфа-2 рецепторов также не является общепризнанным или общеизвестным.
Напротив, вещество бримонидин и его 2-инидазолин-2-илиминовые производные, обладающие схожими свойствами (см. ниже), относятся к агонистам альфа-2 рецепторов и гораздо активнее взаимодействуют с альфа-2 рецепторами, чем с альфа-1 рецепторами.
Расстройства ЦНС относятся к неврологическим заболеваниям. Причиной некоторых из них считают дефицит холинергических, дофаминергических, адренергичесих и/или серотонинергических эффектов. Наиболее распространенными расстройствами ЦНС являются пресенильная деменция (начало болезни Альцгеймера), сенильная деменция (деменция Альцгеймеровского типа) и паркинсонизм, включая болезнь Паркинсона.
Существующее представление о болезни Альцгеймера основано на том, что в некоторых отделах мозга больных (например, в гиппокампе и в коре больших полушарий) наблюдается гибель нервных клеток. Исследованиями, проведенными в 1970-х гг., установлено, что некоторые из погибающих нейронов являются холинергическими (т.е. взаимодействуют посредством нейротрансмиттера ацетилхолина, который разрушается под действием фермента ацетилхолинэстеразы [Jones, et al., Intern. J.Neurosci. 50: 147 (1990); Perry, Br. Med. Bull. 42: 63 (1986); Sitaram, et al., Science 201: 274 (1978)].
Препараты, появившиеся на рынке в течение последнего десятилетия (например, такрин и донепезил), относятся к ингибиторам ацетилхолинэстеразы. Предотвращая распад ацетилхолина, эти вещества замедляют прогрессирование болезни Альцгеймера в начальной стадии. Однако после окончательной гибели холинергических нейронов и отсутствии выработки ацетилхолина эти препараты становятся неэффективными.
Помимо гибели нейронов в головном мозге больных происходит образование белковых кластеров. Последние могут накапливаться как внутри нейронов, так и в межклеточном пространстве. Внутриклеточные кластеры также именуются нейрофибриллярными клубочками и выглядят как спирально сплетенные парные волокна. Исследования показали, что эти клубочки состоят из тау-протеина. Этот белок играет значительную роль, так как связывается с тубулином, из которого образуются микротрубочки. Содержание нейрофибриллярных клубочков коррелирует с тяжестью заболевания.
Внеклеточные белковые кластеры, или бляшки, состоят из отложений β-амилоидного белка. Расположенные рядом с ними нейроны часто выглядят набухшими и деформированными, а амилоидные бляшки окружены воспалительной микроглией. Микроглия, являющаяся частью иммунной системы головного мозга, вероятно, предназначена для разрушения погибших нейронов и удаления их фрагментов, а, возможно, и самих бляшек.
Неизвестно, нормально ли функционируют нейроны в области бляшки или рядом с ней, поскольку плотность бляшек лишь слабо коррелирует с тяжестью деменции. Кроме того, бляшки обнаруживаются у большинства пожилых людей, независимо от того, есть ли у них болезнь Альцгеймера. Тем не менее, их высокое содержание в гиппокампе и коре больших полушарий специфично для болезни Альцгеймера, и они появляются задолго до обнаружения нейрофибриллярных клубочков.
Бета-амилоидные бляшки содержат фрагмент интегрального мембраного белка, который называют белком-предшественником бета-амилоида (ВАРР), содержащий 42 аминокислоты. Этот фрагмент образуется в ходе двухэтапного протеазного расщепления белка ВАРР: сначала бета-секретазой, а затем - гамма-секретазой. Обычным продуктом расщепления белка бета-секретазой и гамма-секретазой является кислый пептид из 40 аминокислотных остатков, который, в отличие от 42-аминокислотного кислого пептида, не участвует в развитии болезни Альцгеймера.
Болезнь Паркинсона (БП) - это инвалидизирующее заболевание неизвестной этиологии, которое характеризуется тремором и ригидностью мышц. Особенностью данного заболевания является дегенерация дофаминергических нейронов (т.е., нейронов, выделяющих дофамин), особенно в черной субстанции и в вентральных отделах покрышки моста среднего мозга [Rinne, et al., Brain Res. 54: 167 (1991) and Clark, et al., Br.J.Pharm. 85: 827 (1985)]. Черная субстанция участвует в координации нейрональных сигналов, контролирующих движения и положение тела. В вентральных отделах покрышки моста содержатся нейроны, окончания которых проецируются на префронтальную кору, которая связана с высшими когнитивными функциями.
Было создано несколько препаратов для лечения БП. Одним из них является SINEMET®, выпускающийся в форме таблеток с замедленным высвобождением, содержащий смесь карбидопа и леводопа [DuPont Merck Pharmaceutical Co.]. Другим препаратом для лечения БП является ELDEPRYL® - таблетки, содержащие селефилина гидрохлорид [Somerset Pharmaceuticals, Inc.]. Еще одно средство - PARLODEL® - таблетки бромокриптина мезилата [Sandoz Pharmaceuticals Corporation]. Berliner и соавт. (патент США №5210076) предложили способ лечения БП и других нейродегенеративных заболеваний с помощью меланина. Однако ни один их этих препаратов не предотвращает гибель нервных клеток.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Несмотря на известный нейропротекторный эффект бримонидина и его производных в отношении зрительного нерва, клеток сетчатки и нейронов спинного мозга в зоне повреждения при его местном или системном введении, эти препараты ранее не предлагалось использовать для лечения нейродегенеративных заболеваний головного мозга, таких как болезнь Альцгеймера и болезнь Паркинсона, отчасти из-за существования гематоэнцефалического барьера, а также из-за значительного седативного эффекта, характерного для существующих агонистов альфа-адренорецепторов (клонидина, тизанидина и дексметатомидина). Таким образом, седативный эффект этих препаратов при системном введении в терапевтической дозе существенно ограничивает их практическое использование в качестве средств для парентерального или перорального введения.
Авторы изобретения неожиданно обнаружили, что бримонидин и его производные при системном введении обладают нейропротекторным эффектом в отношении нейронов головного мозга. Бримонидин и его производные характеризуются гораздо большим терапевтическим окном между нейропротекторной и седативной активностью по сравнению с большинством существующих агонистов альфа-адренорецепторов.
Бримонидин и его терапевтический эффект описаны в работе Danielewicz и соавт. (патенты США №3890319 и 4029792). В этих патентах бримонидин раскрыт как регулятор сердечно-сосудистой системы, имеющий следующую формулу:
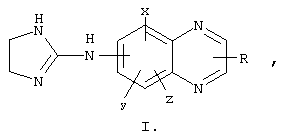
где 2-имидазолин-2-иламиногруппа может быть присоединена к хиноксалину в положении 5-, 6-, 7- или 8-; х, у и z могут быть присоединены в положениях 5-, 6-, 7- или 8- и могут представлять собой водород, галоген, C1-5алкил, C1-5алкокси или трифторметил; R является водородом, С1-5алкилом или C1-5алкокси в положении 2- или 3-, или может отсутствовать. Указанные соединения могут быть синтезированы в соответствии с методами, описанными в патентах США 3890319 и 4029792, которые включены в данное описание посредством ссылки. В работе "Влияние агониста альфа-2 адренорецепторов с относительной избирательностью действия на зрительную систему у кошек, кроликов и обезьян", Current Eye Rsrch., 5, (9), pp.665-676 (1986), J.A.Burke и соавт. показали, что производное хиноксалина бримонидин, формула которого представлена ниже, эффективно снижает внутриглазное давление у кроликов, кошек и обезьян. Исследуемые вещества наносились местно на роговицу животных.
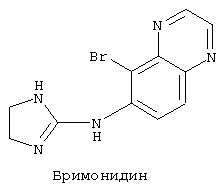
Известно, что агонист альфа-2 адренорецепторов бримонидин защищает клетки сетчатки глаза, в том числе фоторецепторы и ганглионарные клетки, от повреждения при глаукоме, пигментной дистрофии и возрастной дегенерации желтого пятна при местном или системном применении (патент США 6194415).
Первый аспект настоящего изобретения относится к способам лечения нейродегенеративных заболеваний головного мозга, включающим введение в головной мозг млекопитающего терапевтически эффективного количества бримонидина.
Бримонидин обладает менее выраженной активностью в отношении альфа-1 рецепторов, чем другие хорошо известные агонисты альфа-2 рецепторов. Хотя эти агонисты рассматриваются как селективные агонисты альфа-2 рецепторов, на самом деле все эти соединения обладают более высокой активностью в отношении альфа-1А рецепторов, чем в отношении какого-либо подтипа альфа-2 рецепторов. В отличие от них, бримонидин по меньшей мере в 5,5 раз более активен в отношении каждого из подтипов альфа-2 рецепторов, чем в отношении альфа-1А рецептора. Представленные ниже данные были получены с помощью вышеупомянутого испытания RSAT.
Авторы изобретения также обнаружили, что для бримонидина характерно широкое терапевтическое окно (т.е. большой диапазон между минимальной дозой, вызывающей терапевтический эффект за счет связывания с альфа-2-адренорецепторами, и дозой, вызывающей седативный эффект) по сравнению с другими соединениями, обладающими более высокой активностью по отношению к альфа-1-адренорецептором.
Так, например, терапевтический и седативный эффекты тизанидина и клонидина проявляются при дозе около 100 мкг/кг при внутрибрюшинном введении. Напротив, у бримонидина терапевтическая доза составляет от 10 мкг/кг, а седативный эффект проявляется при дозе от 30 мкг/кг. Таким образом, бримонидин может использоваться в качестве нейропротектора для системного введения с меньшим риском чрезмерного седативного действия.
Эффективность действия, опосредуемого рецепторами того или иного подтипа, в соответствии с изобретением определяли с помощью описанного выше способа RSAT.
Авторы предполагают, что уменьшение стимуляции альфа-1 рецепторов вызывает снижение 50% эффективной концентрации (ЕС50) бримонидина, что обеспечивает терапевтический эффект при более низкой концентрации препарата, т.е. данный препарат обладает более выраженной активностью в отношении альфа-1 рецепторов по сравнению с другими аналогичными соединениями без изменения зависимости седативного эффекта от дозы.
В другом аспекте настоящее изобретение относится к способам предотвращения гибели или дегенерации нервных клеток, которые подходят к области головного мозга, включающей голубое пятно, или отходят от нее, включающим введение терапевтического количества бримонидина в указанные клетки.
Этот способ особенно эффективен для профилактики заболевания, т.е. до начала гибели нейронов и в стадии, предшествующей прогрессированию болезни Альцгеймера или болезни Паркинсона. Не желая ограничиваться какой-либо конкретной теорией механизма нейропротективного эффекта соединений изобретения, авторы предполагают, что бримонидин при использовании его в способах изобретения может стимулировать образование определенных факторов группы bcl-2; повышенная экспрессия этих факторов регистрируется как повышенная экспрессия мРНК, кодирующей их образование; эти факторы (bcl-2 и bcl-XL) могут подавлять процесс апоптоза и уравновешивать эффект факторов апоптоза из группы bcl-2 (bad и bax), которые образуются в результате губительных воздействий на нервные клетки. Таким образом, считают, что соединения изобретения, которые обладают нейропротективным эффектом, могут использоваться в комбинации с другими соединениями, которые ингибируют гибель клетки. Эти соединения, ингибирующие гибель клетки, включают в себя антагонисты NMDA-рецепторов, особенно мемантин, блокирующий цитотоксический эффект повышенной концентрации глутамата; ингибиторы синтетазы оксида азота; ингибиторы свободных радикалов и блокаторы кальциевых каналов.
Для введения бримонидина в мозг млекопитающего может использоваться любой подходящий путь введения. Среди млекопитающих предпочтительным является человек. При выборе пути введения необходимо стремиться к тому, чтобы достигнуть максимального положительного эффекта, т.е. низкой эффективной концентрации и малой частоты побочных эффектов.
В соответствии с предложенной схемой бримонидин может назначаться перорально, парентерально, внутривенно, подкожно и посредством других путей системного введения. Препараты назначают в эффективной терапевтической дозе изолированно или в комбинации с другими фармацевтически приемлемыми носителями или наполнителями.
В зависимости от предполагаемой схемы лечения терапевтическая доза бримонидина может содержаться в любой приемлемой лекарственной форме (таблетки, свечи, пилюли, капсулы, порошки, микстуры, растворы, растворы для внутривенного введения, настои, суспензии, эмульсии, аэрозоли и пр.). Предпочтительным является однократный режим введения точной дозы препарата, а также введение лекарственной формы с замедленным высвобождением, обеспечивающей длительное и управляемое действие. Желательно, чтобы лекарственная форма включала в себя фармацевтически приемлемый наполнитель, активное соединение или соединения, а также другие лекарственные средства, носители, адъюванты и пр.
В твердых лекарственных формах могут использоваться нетоксичные носители, например маннитол, лактоза, крахмал, стеарат магния, натриевая соль сахарина, полиаклиленгликоли, тальк, целлюлоза, глюкоза, сахароза и карбонат магния, обладающие соответствующей степенью очистки. Примером твердой лекарственной формы по изобретению является суппозиторий, содержащий пропиленгликоль в качестве носителя.
Жидкие лекарственные формы могут представлять собой растворы или суспензии, содержащие одно или несколько активных соединений, а также адъюванты и носители, такие как вода, солевой раствор, водный раствор декстрозы, глицерин, этанол и пр. При необходимости лекарственная композиция может включать в себя небольшое количество вспомогательных веществ, таких как увлажнители, эмульгаторы, буферные агенты и пр. Примерами таких вспомогательных веществ являются ацетат натрия, монолаурат сорбитана, триэтаноламин, ацетат натрия, триэтаноламина олеат и пр. Способы приготовленя подобных лекарственных форм хорошо известны специалистам, например из Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 16th Edition, 1980. Состав композиции во всех случаях должен содержать одно или несколько соединений изобретения в количестве, достаточном для достижения необходимого терапевтического эффекта.
Парентеральное введение как правило осуществляется путем инъекций (подкожных, внутримышечных или внутривенных). Формы для инъекций представляют собой либо обычные формы (растворы или суспензии, порошки для приготовления растворов или суспензий), либо эмульсии или растворы для вливаний. Подходящими наполнителями для них являются вода, солевой раствор, декстроза, глицерин, этанол и пр. Кроме того, при необходимости фармацевтическая композиция для инъекций или вливаний может содержать небольшое количество нетоксичных вспомогательных веществ, таких как увлажнители, эмульгаторы, буферные вещества и пр.
Количество вводимого бримонидина зависит от эффекта, который следует достичь, вида млекопитающего, тяжести и природы заболевания, способа введения, фармакодинамики используемого вещества или веществ, а также от мнения лечащего врача. В общем случае терапевтически эффективная доза составляет от 0,5-1 до 100 мг/кг/сутки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способам лечения нейродегенеративных заболеваний головного мозга, включающим введение в головной мозг млекопитающего терапевтически эффективного количества бримонидина или его фармацевтически активной соли.
Авторы изобретения обнаружили, что бримонидин обладает необычно высокой способностью предотвращать повреждение и гибель нейронов, в том числе апоптоз, по сравнению с веществами, являющимися агонистами альфа-1 адренорецепторов. Не желая ограничиваться теорией, авторы считают, что стимуляция альфа-1 адренорецепторов накладывается на нейропротекторное действие альфа-2 адренорецепторов и снижает седативный эффект таких агентов, как клонидин и тизанидин, так, что величина ЕС50 таких препаратов является такой же или отличается не более, чем в 3 раза от эффективной нейропротекторной дозы этих веществ. Так, нейропротекторный эффект неселективных агентов, таких как клонидин, тизанидин, дексметатомидин, наблюдается при концентрации, которая способна вызвать седацию или является токсической.
Отчасти по этой причине препараты, действующие на альфа-адренорецепторы, ранее широко не применялись в качестве нейропротекторов и использовались только для местного (локального) применения, например в офтальмологии, что исключало их системное действие.
Авторы объясняют нейропротекторный эффект бримонидина стимуляцией альфа-2В и/или альфа-2С рецепторов. Считается, что количество рецепторов данного типа в головном мозге невелико. Однако авторы обнаружили, что способ, являющийся предметом данного изобретения, обеспечивает нейропротекторный эффект в отношении нейронов, окончания которых подходят к голубому пятну - области, которая в первую очередь поражается болезнью Альцгеймера. Таким образом, применение бримонидина будет полезным при предотвращении гибели нейронов при таких нейродегенеративных состояниях, как болезнь Альцгеймера и болезнь Паркинсона.
Связь глаукомы с болезнью Альцгеймера
Не желая ограничиваться теорией, авторы изобретения представляют гипотезу, объясняющую эффективность бримонидина как нейропротектора при болезни Альцгеймера и болезни Паркинсона.
По данным последних исследований, при болезни Альцгеймера (БА) происходит гибель ганглионарных клеток сетчатки (ГКС) и их аксонов. Амилоидные β-пептиды и фосфорелированный тау-протеин играют роль в очаговой гибели нейронов и накопления белка при БА. В погибших при глаукоме ГКС такой белок отсутствует. Механизмом гибели нейронов при БА и при глаукоме является апоптоз, хотя пути передачи сигнала для деградации нейронов при этих состояниях различны. При БА происходит гибель норадренергических нейронов голубого пятна, аксоны которых оканчиваются в различных отделах мозга. Это приводит к очаговому снижению концентрации норадреналина (НА) и нейрональных альфа-2 рецепторов. Активация альфа-2 рецепторов подавляет нейрональный апоптоз посредством механизмов передачи сигнала, опосредованных протеинкиназой В. Снижение иннервации НА может способствовать нейрональному апоптозу как при БА, так и при глаукоме. Агонисты альфа-2 рецепторов адреналина обладают способностью замедлять процесс гибели нейронов при обоих состояниях благодаря компенсированию снижения иннервации НА.
Нейродегенеративные заболевания чаще приводят к инвалидизации, чем к смерти больного. Их длительное и прогрессирующее течение имеет большое социально-экономическое значение в нашем обществе, что во многом объясняется числом специалистов, участвующих в лечении таких больных. Глаукома соответствует критериям типичного нейродегенеративного заболевания. При этом заболевании происходит постепенная и прогрессирующая гибель ганглионарных клеток сетчатки (ГКС), часто сопровождающаяся повышением внутриглазного давления (ВГД). Гибель нейронов при глаукоме не ограничивается только областью сетчатки и может захватывать латеральное коленчатое ядро и даже зрительную кору. Патогенез нейродегенерации при глаукоме до конца не ясен. Существуют данные о снижении скорости аксонального транспорта белков в ГКС вследствие компрессии аксонов зрительного нерва на уровне решетчатой пластинки. Трофические факторы (например, мозговой нейротрофический фактор - МНТФ) посредством ретроградного транспорта поступают из окончаний аксонов ГКС в тела нейронов, что является необходимым условием жизнедеятельности ГКС. Сдавление или разрушение аксонов ГКС может снижать концентрацию трофических факторов, вызывая гибель ГКС вследствие их недостаточного питания. В других исследованиях установлено, что возможными факторами, способствующими гибели нейронов при глаукоме, являются локальная ишемия (гипоксия), активация рецепторов глутамата, усиленное образование окислительных радикалов под действием оксида азота, активация рецепторов иммунной системы.
Связь глаукомы и болезни Альцгеймера (БА)
Как и при глаукоме, возможными факторами, способствующими гибели нейронов при БА, являются трофическая недостаточность, повреждающий эффект свободных радикалов, гипоксия и иммунные механизмы. Главным образом при БА страдают нейроны коры, гиппокампа, септальной и таламической областей, а также ствола мозга, но может наблюдаться и гибель ГКС. Гистологические исследования сетчатки и зрительных нервов свидетельствуют об атрофии и гибели ГКС и их аксонов. По данным ряда исследований, потеря аксонов зрительного нерва у больных БА становится значимой при среднем числе аксонов 68/1000 мкм2, что составляет меньше половины от нормального показателя (116/1000 мкм2). Значение имеет и размер аксона: как при БА, так и при глаукоме главным образом страдают крупные аксоны. В других исследованиях зрительных нервов при БА не было обнаружено признаков дегенерации аксонов ГКС. Различные результаты этих исследований трудно адекватно оценить из-за небольшого числа больных и малого размера контрольной группы (7 и 10 человек); степень прогрессирования БА в этих исследованиях не была стандартизирована и, вероятнее всего, различалась; кроме того, различались и способы гистологической оценки и подсчета аксонов. Исследования отличались и по способам прижизненного измерения толщины волокон и деформации нейрональных окончаний у больных БА. Результаты варьировали от полного отсутствия глаукоматозных изменений до выраженных изменений, степень которых коррелировала с уровнем когнитивных нарушений. Наиболее убедительное доказательство наличия связи между БА и глаукомой было получено в крупном исследовании, проведенном Bayer и соавт. Авторы показали, что приблизительно у 26% больных БА (n=112) имелись глаукоматозные изменения сетчатки, в частности сужение полей зрения и деформация нейронов, а в контрольной группе (n=116) аналогичные изменения наблюдались лишь в 5% случаев.
Нейрофибриллярные клубочки (НФК) и амилоидные бляшки являются патологическими признаками нейродегенерации при БА. Белок-предшественник амилоида (БПА) является мембранным протеином, который при расщеплении альфа-секретазой и гидролазой бета-амилоида (ГБА) образует пептиды sAPPα и Aβ1-40. Основным производным БПА в большинстве клеток является sAPPα. Более крупная форма Аβ-пептида - Аβ1-42 - склонна к агрегации и является основным компонентом ядра амилоидных бляшек. Нарушение процессов модификации и деградации БПА, вероятно, способствует повышению концентрации Аβ1-42 пептида при БА. Пептид Аβ1-42 токсичен для клеток в культуре и, возможно, вызывает гибель нейронов при БА. Генетическими исследованиями установлено, что мутации БПА являются вероятным пусковым фактором в патогенезе БА. Другие гены (например, презенилин 1 и 2 - PS 1/2 или аполипопротеин Е (АПО-Е) также способствуют прогрессированию БА за счет повышения уровня белка Аβ.
Основным компонентом НФК является тау-протеин (белок, связанный с микротрубочками). При БА происходит накопление тау-протеина высокой степени фосфорелирования, который образует агрегаты в филаментах тела нейрона. Фосфорелированный тау-протеин, по-видимому, обладает низкой способностью связываться с микротрубочками и образует агрегаты с белками нейрофиламентов, что приводит к образованию НФК. Фосфорелированный тау-протеин вероятно играет решающую роль в токсическом действии Аβ, и пептид Аβ1-42, возможно, способствует агрегации тау-протеина и гиперфосфорелированию тау-протеинкиназой II. Оба белка, пептид Аβ1-42 и фосфорелированный тау-протеин, таким образом, способствуют образованию другого продукта, обладающего нейротоксичным действием.
По данным исследований, в сетчатке при возрастной атрофии, пигментной ретинопатии и возрастной дегенерации желтого пятна отмечается некоторое повышение иммунореактивности в отношении тау-протеина, БПА и Аβ-пептида. Эти сдвиги не сопровождаются формированием НФК или амилоидных бляшек. Аналогичным образом, иммуноцитохимическое повышение иммунореактивности в отношении БПА в ГКС было отмечено в исследовании с моделированием внутриглазной гипертензии у крысы. При иммуноблоттинге сетчатки крыс с внутриглазной гипертензией было обнаружено, что повышенная иммунореактивность сопровождается снижением длины БПА и увеличением фрагментов, содержащих Аβ. Было высказано предположение о том, что компрессия аксонов ГКС при глаукоме способствует развитию аномальных белков нейрофиламентов и повреждению нейронов, вызванному Aβ-пептидом. Несмотря на аномалии БПА, Аβ-пептида и тау-протеина при моделировании глаукомы у крыс и при других заболеваниях сетчатки, потеря аксонов зрительного нерва и дегенерация ГКС при БА не приводит к формированию НФК или амилоидных бляшек. Таким образом, остается неясным, является ли дегенерация ГКС при БА результатом аномалий БПА или тау-белка, которые, по-видимому, лежат в основе гибели нейронов центральной нервной системы при БА.
Апоптоз как возможный механизм гибели нейронов при глаукоме и БА.
Процесс гибели клетки, называемый апоптозом, по-видимому, вносит вклад в гибель нейронов при глаукоме и БА.
Апоптоз - это процесс постепенного разрушения клетки, который заключается в уменьшении объема ядра и цитоплазмы за счет расщепления нуклеиновых кислот и белков цитоскелета эндонуклеазами и протеазами. Несмотря на то, что результаты проведенных исследований свидетельствуют о роли апоптоза в процессе гибели нейронов при глаукоме или БА, ряд авторов оспаривали этот тезис. Различие во взглядах во многом объясняется различными морфологическими критериями апоптоза и/или различной интерпретацией результатов расщепления ядерной ДНК по данным исследования с маркировкой концевых участков деоксиуридин трифосфатом с помощью деоксинуклеотидил трансферазы (TUNEL-способа). Разрушение ядра при апоптозе трудно идентифицировать при микроскопическом исследовании вследствие сморщивания и разрушения пораженных нейронов. Процесс разрушения занимает около 24 часов в культуре клеток и несколько дней в нативной нервной ткани. Таким образом, у больных с длительно текущими заболеваниями при одномоментном исследовании лишь небольшое число нейронов содержит признаки апоптоза.
Для выявления расщепления ядерной ДНК как маркера апоптозной деградации можно использовать электрофорез ДНК в геле или электрофорез в переменном поле. Эти способы позволяют оценить состояние ДНК в гомогенизированных образцах тканей. При этом необходимо, чтобы в образцах содержалось 105 и более клеток в стадии апоптоза. С учетом длительного течения БА или глаукомы каждый день лишь небольшое число нейронов будет содержать разрушающиеся ядра, т.е. в гомогенизате ткани мозга или сетчатки фрагментированная ДНК будет присутствовать в недостаточном количестве. Маркировка 3'-конца ДНК с помощью d-UTP, соединенного с флуорохромом применяется для выявления признаков фрагментации ДНК in situ в срезах тканей или в образцах мозга и сетчатки, полученных при патологоанатомическом исследовании. Ядра ГКС, визуализируемые при TUNEL-исследовании, обнаруживаются в сетчатке человека при глаукоме, а также при моделировании глаукомы у животных. Небольшой процент ядер, визуализируемых при TUNEL-исследовании, по-видимому, объясняется постепенной гибелью ГКС при глаукоме. Количество TUNEL-положительных нейронов при БА варьирует по данным различных исследований: некоторые авторы обнаруживали большое число таких нейронов при БА, другие не отмечали увеличения их процентного содержания по сравнению с контрольной группой больных, соответствующих опытной группе по возрасту. Кроме того, в ряде работ обнаруживался необычно высокий уровень TUNEL-положительных нейронов в головном мозге больных БА, например, в коре головного мозга до 25% нейронов визулизировались при TUNEL-исследовании.
Различные результаты TUNEL-исследования при БА вероятно объясняются методологическими трудностями в процессе маркировки ДНК, особенно при работе с образцами нервной ткани, полученными при патологоанатомическом исследовании: 1) помимо эндонуклеаз, повреждение ДНК вызывают кислородные радикалы, что также может давать положительное окрашивание при TUNEL-исследовании; 2) ложноположительные результаты могут быть следствием слишком длительной или недостаточной фиксации тканей; 3) клетки, входящие в фазу митоза, могут окрашиваться положительно; 4) характер расщепления ДНК зависит от типа эндонуклеаз, что заметно отражается на результатах исследования; 5) результаты TUNEL-исследования зависят от концентрации бивалентных катионов (например, Mg2+ или Со2+). Таким образом, несмотря на то, что TUNEL-исследование применяется для выявления признаков апоптоза, его результаты необходимо оценивать с осторожностью. TUNEL-исследование ДНК может применяться в комбинации с другими маркерами гибели клеток (в частности, с красителями, обладающими сродством к ДНК и позволяющими визуализировать конденсацию хроматина, а также с иммуноцитохимическими способами выявления белков, являющихся маркерами распада клеток.
Процессы передачи сигнала при апоптозе у больных глаукомой и БА
Процессы передачи сигнала при апоптозе могут быть условно разделены на три этапа: 1) премитохондриальный этап, 2) митохондриальный этап и 3) постмитохондриальный этап (этап разрушения). Как и при расщеплении ядерной ДНК, распад ядра в процессе апоптоза включает в себя 1) отделение гистонов и ламинов от ядерной ДНК, 2) конденсацию или уплотнение фрагментированной ДНК и 3) образование ядерных телец, покрытых мембраной и содержащих фрагментированную и укороченную ДНК. По данным исследований, степень распространения, характер течения и длительность процесса дегенерации ядер при апоптозе варьируют в зависимости от фенотипа клетки и от механизма повреждения. В нашей работе была продемонстрирована вариабельность изменений в клеточном ядре при апоптозе по данным патологоанатомического исследования сетчатки больных, страдающих глаукомой, сетчатки животных при моделировании глаукомы, нейронов черной субстанции мышей при МРТР-воздействии, нейронов гиппокампа свиней при гипоксическом повреждении, а также нейронов черной субстанции, содержащих нейромеланин, у больных БП. Таким образом, на основании морфологических изменений клеточного ядра трудно отделить апоптоз от других причин повреждения клетки.
Известно, что распад клеток при апоптозе опосредуется несколькими путями передачи сигналов. Путь передачи сигнала зависит от механизма повреждения и от фенотипа клетки и определяет различные морфологические изменения ядер, в том числе конденсацию хроматина и/или фрагментацию ДНК. Во многих путях передачи сигнала об апоптозе ведущую роль играют каспазы (семейство протеаз). У млекопитающих описано более десятка различных каспаз. Каспазы синтезируются в виде неактивных проферментов, которые активируются путем протеолиза и приобретают способность гидролизовать другие виды каспаз, белки цитоскелета, ядерные белки и белки, участвующие в механизмах подавления апоптоза. Существует четыре различных пути передачи сигнала, которые реализуются в процессе конденсации хроматина и фрагментации ДНК. В двух из них участвуют каспазы: 1) путь, включающий в себя цитохром С; фактор, активирующий протеазу апоптоза-1 (Apaf-1) и прокаспазу-9; а также 2) путь, включающий в себя митохондиральный активатор каспаз (SMAC)/прямой IAP-связывающий белок (Diablo). Два других пути передачи сигнала не зависят от каспаз: 1) путь, включающий в себя фактор, инициирующий апоптоз (AIF), и 2) путь, включающий эндонуклеазу-G.
Впервые показано, что цитохром-С, высвобождаемый из митохондрий, взаимодействует с Apaf-1 и дАТФ, превращая прокаспазу-9 в активированную каспазу-9. Активированная каспаза-9 также высвобождается из митохондрий. Этот фермент играет ключевую роль в некоторых формах апоптоза за счет превращения прокаспазы-3 в активированную каспазу-3. Активированная каспаза-3 гидролизует молекулу ингибитора каспаз-активируемой ДНК-азы (ICAD) до каспаз-активированной ДНК-азы (ICAD), которая катализирует фрагментацию ДНК. Активированная каспаза-3 запускает другие механизмы разрушения клетки, в том числе конденсацию хроматина по действием протеаз, расщепление актина цитоскелета желозином и расщепление ядерного ламина каспазой-6.
По данным ряда исследований, активация каспазы-3 играет роль в процессе гибели ГКС при глаукоме. Хотя в ряде исследований была подтверждена активация каспазы-3 при БА, по данным других исследований путь передачи сигнала, включающий в себя цитохром С, каспазу-3 и CAD, не участвует в фрагментации ДНК нейронов у больных БА. Каспаза-3, как и другие каспазы, ингибируется одним или несколькими видами конститутивно активированных белков-ингибиторов апоптоза (IAP). Белки IAP связываются с молекулами каспаз, вызывая их инактивацию. Тем самым предотвращается или замедляется гибель нейронов, опосредованная путем передачи сигнала через цитохром С, каспазу-3 и CAD. В свою очередь, высвобождаемый из митохондрий фактор SMAC/Diablo также может связываться и инактивировать белки IAP, позволяя каспазам 9, 3 и 6 передать сигнал о начале апоптоза. Несмотря на то, что по данным исследования белки IAP играют роль в развитии нейродегенеративных состояний, подобных болезни Альцгеймера (например, синдром Дауна, гибель ГКС у крыс после аксонотомии), остается неизученным вопрос об участии белков IAP и SMAC/Diablo в механизмах апоптоза, опосредованных каспазой 3 при БА и глаукоме.
При некоторых формах апоптоза растворимый флавопротеин - фактор, инициирующий апоптоз (AIF) - поступает из межмембранного пространства митохондрий в ядро, где вызывает фрагментацию ДНК и стимулирует конденсацию хроматина. Высвобождение AIF из митохондрий может происходить независимо от цитохрома С и каспазы 9. Причина селективного поступления AIF из митохондрий остается неизвестной. Микроинъекция клеток, содержащих рекомбинантный фактор AIF, вызывает конденсацию хроматина только по краю ядра, в то время как микроинъекция активированной каспазы 3 или его субстрата - CAD - вызывает конденсацию хроматина во всем объеме клеточного ядра. При других формах апоптоза фактор AIF способствует выходу цитохрома С из митохондрий. Таким образом, фрагментация ядерной ДНК и конденсация хроматина может индуцироваться различным распределением каспазы 3 внутри ядра, действием фактора AIF или последовательным действием AIF и каспазы 3. Митохондриальная ДНК-аза - эндонуклеаза G, которая в обычных условиях вызывает фрагментацию ДНК, также может выходить из митохондрий при некоторых формах апоптоза и непосредственно разрушать ДНК. Фрагментация ядерной ДНК под действием эндонуклеазы G не зависит от активности каспазы или CAD. Эндонуклеаза G может поступать в клетку из митохондрий под действием фактора BID и является маркером апоптоза, опосредованного лигандом FAS (см. ниже). Процесс фрагментации ДНК под действием эндонуклеазы G на ядерном уровне изучен недостаточно, однако он, вероятнее всего, вносит дополнительные коррективы в механизм разрушения ядра при апоптозе с участием каспазы 3 и/или фактора AIF.
Учитывая растущий объем знаний, касающихся путей передачи сигнала при апоптозе, не удивительно, что выделено несколько морфологических форм разрушения ядра. Согласно существующим представлениям апоптоз ГКС при глаукоме происходит под влиянием (или, по меньшей мере, с участием) каспазы 3, а апоптоз при БА может опосредоваться каспазой или не зависеть от нее, тем самым отражая путь передачи сигнала через AIF и/или эндонуклеазу G, а также через активацию каспазы 3.
Митохондриальные механизмы передачи сигнала при глаукоме и болезни Альцгеймера.
Как нейроны "решают" активировать сигнальные пути для апоптозной деградации? Изменения проницаемости митохондриальных мембран с высвобождением факторов, передающих сигнал об апоптозной деградации, являются решающим моментом во многих путях передачи сигнала при апоптозе. Один или несколько сигнальных факторов, участвующих в апоптозе (цитохром С, прокаспаза 9, AIF, SMAC/Diablo и эндонуклеаза G), поступают в межмембранное пространство митохондрий, расположенное между внутренней и наружной мембранами, из митохондриального матрикса. Существуют два возможных механизма, увеличивающих проницаемость мембран: 1) формирование новых или открытие существующих пор во внешней мембране и 2) открытие крупных пор, состоящих из нескольких белков (поры, увеличивающие проницаемость - РТРС) и соединяющих внутреннюю и наружные мембраны митохондрий.
Как показано на чертеже, белок ВАХ в комбинации с другим стимулятором апоптоза - ВАК, способствуют открытию пор РТРС и повышению проницаемости внешней мембраны, хотя возможные механизмы этих изменений проницаемости до конца не известны. Олигомеры белка ВАХ могут встраиваться в билипидный слой мембраны и вызывать его растворение. В некоторых моделях апоптоза фактор BID вызывал олигомеризацию ВАХ, который, в свою очередь, способствовал выходу цитохрома С и SMAC/Diablo из липосом или митохондрий. Фактор BID способствует встраиванию ВАХ во внешнюю мембрану митохондрий, также связыванию ВАХ с порами РТРС. При других формах апоптоза BID не участвует в процессе накопления ВАХ в митохондриях.
Открытие пор РТРС вызывает осмотические сдвиги на границах внутренней митохондриальной мембраны и, как следствие, отек митохондриального матрикса с разрывом наружной митохондриальной мембраны, что непосредственно приводит к выходу факторов апоптозной деградации из митохондрий. В некоторых случаях вещества, способствующие закрытию пор РТРС (например, циклоспорин А), препятствуют выходу цитохрома С из митохондрий и апоптозу. По данным исследований, фактор ВАХ связывается с транслокатором адениновых нуклеотидов (ANT) или с потенциал-зависимыми анионными каналами (VDAC), входящими в состав пор РТРС. Связывание ВАХ с ANT или VDAC приводит к выраженному повышению мембранной проводимости. Полученные данные свидетельствуют о том, что либо ANT либо VDAC вызывают повышение проницаемости внутренней митохондриальной мембраны при различных формах апоптоза. Цитохром С может находиться в митохондриальных кристах внутренней мембраны и попадает во внутриклеточную среду через поры РТРС при ее слиянии с наружной мембраной. Таким образом, открытие каналов РТРС наряду с образованием пор на наружной мембране обеспечивают высвобождение цитохрома С из митохондрий.
Иммуноцитохимическое патологоанатомическое исследование сетчатки больных глаукомой показало, что в некоторых ГКС происходило увеличение концентрации ВАХ, что соответствует результатам моделирования глаукомы у крыс. Повышение проницаемости внутренней митохондриальной мембраны под действием ВАХ приводит к снижению разности потенциалов на митохондриальной мембране, которая, в свою очередь, способствует уменьшению числа ГКС у крыс при моделировании глаукомы. Патологоанатомические исследования образцов ткани мозга больных БА показали, что увеличение концентрации ВАХ наблюдается лишь в некотором проценте нейронов, а в ряде случаев концентрация ВАХ не превышает аналогичный показатель в контрольной группе. В одном из исследований было обнаружено повышение концентрации ВАК при нормальной концентрации ВАХ. Таким образом, не ясно, способствует ли повышение проницаемости митохондриальных мембран под действием ВАХ апоптозу у больных БА. Возможно, существует несколько форм БА, одна из которых протекает с повышением концентрации ВАХ и активированной каспазы 3 (см. выше), а другая не связана с повышением проницаемости митохондриальных мембран.
Премитохондриальные механизмы передачи сигнала апоптоза при БА.
На чертеже представлена схема вероятных премитохондриальных и митохондриальных механизмов передачи сигнала апоптоза, способствующих гибели нейронов при глаукоме и БА. Оба сигнальных пути, представленных на чертеже, способствуют гибели ГКС при глаукоме: 1) путь р53-гицеральдегид-3-фосфат дегидрогеназа (GAPDH) - ВАХ; 2) путь FAS- или TNF-рецептор - FADD - каспаза 8 - ВАХ. Передача сигнала посредством факторов р53, Fas или TNF также является одним из механизмов развития БА.
Антиопухолевый протеин р53 играет роль во многих формах апоптоза и способен передавать сигнал апоптоза посредством транскрипционных и посттранскрипционных механизмов. Посредством транскрипционных механизмов протеин р53 вызывает повышение концентрации GAPDH и ВАХ при некоторых формах апоптоза. GAPDH - многофункциональный белок, основной функцией которого является гликолитическая. Кроме того, он играет роль сигнального белка апоптоза. Исследования с использованием антисмысловых олигонуклеотидов показали, что GAPDH является необходимым для развития апоптоза, который запускается в результате повреждения нейронов различными факторами. По-видимому, GAPDH подавляет транскрипцию антиапоптозных белков BCL-2 и BCL-XL. BCL-2 и BCL-ХL предотвращают увеличение проницаемости митохондриальной мембраны и высвобождение факторов апоптоза под действием ВАХ, тем самым подавляя апоптоз.
Рецепторы факторов некроза опухоли (TNF), включающие в себя TNF-рецептор и FAS-рецептор, стимулируют апоптоз с помощью механизмов передачи сигнала, опосредованных каспазой, без транскрипционных изменеий. Данные рецепторы связаны с адапторными белками (например, белками FADD), активирующими каспазы, в частности каспазу 8, которая, как показано на чертеже, способствует повышению проницаемости митохондриальной мембраны по действием ВАХ, а также непосредственно стимулирует апоптоз. Механизм апоптоза, опосредованный FAS и, возможно, TNF, связан с механизмом передачи сигнала через белок р53 посредством активации n-концевой киназы (JNK) (см. чертеж). Также исследования показали, что FAS-рецептор может активироваться посредством белка р53 после некоторого повреждения клетки, в частности вызванного агентами, разрушающими ДНК. Активация, вызванная белком р53, может стимулировать апоптоз посредством реакций FAS/лиганд FAS, а также белка р53 и иммунных/цитокиновых механизмов.
Роль норадреналина в апоптозе нейронов при БА и глаукоме.
Норадреналинэргические нейроны голубого пятна (ГП) быстро и интенсивно погибают при БА, и происходит образование НФК, являющихся характерным признаком заболевания. Аксоны нейронов ГП, проходящие в полушариях мозга, образуют густые разветвления в веществе переднего мозга (включая кору, септальную область, гиппокамп и таламус). В результате исчезновения норадреналинэргических нейронов голубого пятна в этих областях формируется выраженный дефицит НА при БА. Окончания нейронов голубого пятна содержат тирозингидроксилазу (ТГ), которая катализирует превращение тирозина в дофамин и дофамин-бета-гидроксилазу (ДБГ), которая преобразует дофамин в норадреналин (НА). Иммуногистохимическое исследование выявило существенное уменьшение количества нервных окончаний, содержащих ТГ и ДБГ, в дегенерирующих отделах мозга (кора и гиппокамп) у больных БА.
Первоначально считалось, что основные проявления гибели окончаний норадреналинэргических нейронов ГП при БА объясняются снижением уровня норадреналиновой стимуляции адренергических рецепторов. Аксоны нейронов ГП оканчиваются в микрососудах и каппилярах мозга и несут на себе альфа-адренорецепторы, чувствительность которых увеличивается при снижении регионарного кровотока.
В настоящее время общеизвестно, что окончания норадреналинэргических нейронов также могут проходить в непосредственной близости от других нейронов или подходить к ним. Высвобождаемый норадреналин активирует альфа-адренергические или альфа-адренергические рецепторы на поверхности нейронов. Альфа-2 адренорецепторы обнаруживаются во многих типах нейронов и/или их окончаниях, включая нейроны симпатических ганглиев, спинного мозга, ствола мозга, септальной области, гиппокампа и коры большого мозга. Альфа-адренорецепторы также присутствуют в головном мозге в большом количестве, но распределены неравномерно. Их содержание велико в коре, гиппокампе и некоторых ядрах гипоталамуса, однако они практически отсутствуют в таламических ядрах (кроме латерального и медиального коленчатых ядер) и ядрах септальной области.
Альфа-2 адренорецепторы, возможно, играют особую роль в развитии БА. По данным патологоанатомического исследования, количество альфа-2 адренорецепторов в коре и гиппокампе у больных БА снижено; гибель нейронов ГП и их норадреналинэргических окончаний, вероятно, сопровождается снижением числа некоторых подтипов альфа-2 адренорецепторов на поверхности иннервируемых нейронов. Кроме того, при БА происходит гибель нейронов, несущих на себе альфа-2 рецепторы адреналина. Фармакологическая активация альфа-2 адренорецепторов улучшает состояние больных и предотвращает гибель нейронов центральной нервной системы в ответ на повреждающее воздействие (например, ишемическое).
Альфа-2 адренорецепторы опосредуют ряд внутриклеточных реакций через пути передачи сигнала, в которых участвует G-белок, или через другие пути передачи сигнала, независимые от G-белка. По отношению к апоптозу активация этих рецепторов может индуцировать фосфорелирование протеинкиназы В (Akt) посредством пути, включающего в себя фосфатидилинозитол-3-киназу (PI3-киназу). Фосфорелированная киназа Akt подавляет апоптоз посредством транскрипционных и посттранскрипционных путей передачи сигнала. Основой действия фосфорелированной киназы Akt является предотвращение или уменьшение степени повышения проницаемости митохондриальной мембраны, которая является причиной некоторых форм апоптоза. Например, фосфорелированная киназа Akt поддерживает на постоянном уровне или увеличивает скорость синтеза BCL-2 и BCL-XL, а также предотвращает инактивацию этих протеинов под действием белка, который формирует гетеродимеры BCL-2/BAD или BCL-XL/BAD. BCL-2 и BCL-XL снижают проницаемость митохондриальных мембран, которая увеличивается под действием ВАХ, ВАК и BID, тем самым предотвращая или замедляя попадание митохондриальных факторов (сигнальных молекул апоптоза) в клеточную среду. Мы предполагаем, что норадреналин, агонисты альфа-2 рецепторов и BDNF способны подавлять апоптоз нейронов, на поверхности которых экспрессируются альфа-2 адренергические рецепторы или рецепторы тирозинкиназы В (нейротрофические рецепторы BDNF), а также защищать нейроны у больных БА от апоптоза. Также можно утверждать, что снижение высвобождения НА вследствие гибели нейронов ГП при БА снижает степень защищенности нейронов коры головного мозга; септальной области, гиппокампа и таламуса от апоптоза, вызванного А-пептидом и/или фосфорелированным тау-протеином, которые являются частью патогенеза БА.
Мы также предположили, что полная или частичная утрата механизмов подавления апоптоза ГКС, опосредованных НА, может объяснить гибель ГКС при БА и высокую частоту глаукомы у таких больных. Агонисты альфа-адренорецепторов (например, В-НТ920 и UK 14304 - бримонидин), а также антагонисты альфа-1 рецепторов (бетаксолол, метопролол и тимолол) используются для снижения внутриглазного давления при глаукоме благодаря своей способности уменьшать активную продукцию и пропотевание жидкости (по крайней мере, частично), действуя на ресничный эпителий. Помимо адренергических рецепторов ресничного эпителия, в сетчатке также присутствуют бета-2, альфа-1 и альфа-2 адренорецепторы.
Агонисты альфа-2 адренорецепторов (бримонидин, клонидин) снижают скорость гибели ГКС после ишемии сетчатки, сдавления зрительного нерва и повышения внутриглазного давления. Аналогичным образом антагонист альфа-1 рецепторов - бетаксолол - снижает скорость гибели ГКС после тромбоза артерий сетчатки и возбуждающего действия препарата. В ходе последних исследований, проведенных в культуре клеток сетчатки, было показано, что бримонидин и бетаксолол снижают скорость гибели ГКС на фоне действия возбуждающих токсинов. Таким образом, активация альфа-2 адренорецепторов, а не блокада альфа-адренорецепторов, может замедлить гибель нейронов ГКС. Исследование, проведенное нашей лабораторией, показало, что агонисты альфа-2 адренорецепторов способствуют снижению апоптоза в культурах нейроноподобных клеток, а также в первичных нейрональных культурах, выращиваемых на питательных средах на фоне введения ингибиторов киназ, митохондриальных токсинов и возбуждающих токсинов, а также, что антиапоптоз может быть полностью блокирован антагонистами альфа-2 адренорецепторов. Антиапоптозный эффект определяется степенью проницаемости митохондриальной мембраны, а также высоким уровнем BCL-2 и фосфорелированной киназы Akt.
Каковы общие механизмы апоптоза в нейронах центральной нервной системы и ГКС при БА? Несмотря на то, что роль НА в сетчатке глаза противоречива, иммуноцитохимическое исследование концентрации ДБГ позволило выявить признаки наличия норадреналинэргических аксонов во внутреннем слое сетчатки и ганглионарном слое у коров, обезьян и человека. На основании этих данных можно предположить, что отростки норадреналинэргических нейронов оканчиваются в непосредственной близости от тел и дендритов ГКС. Норадреналин обнаруживается во внутреннем сетчатом и внутреннем ядерном слоях сетчатки коров. Активация альфа-2 адренорецепторов играет роль в развитии изменений сетчатки у коров и обезьян. В совокупности эти данные свидетельствуют о том, что внутренние слои сетчатки содержат норадреналинэргические окончания, их которых НА поступает к рецепторам адреналина альфа-2 подтипа, расположенным на ГКС. Источник этих окончаний во внутреннем слое сетчатки неизвестен. В сетчатке отсутствуют тела нейронов, содержащие НА. Следовательно, данные аксоны сетчатки отходят от нейронов за пределами глаза. Возможно аксоны, содержащие НА, достигают сетчатки, располагаясь вдоль кровеносных сосудов или в составе зрительного нерва. Окрашиваемые при иммунологическом исследовании аксоны с ДБГ обнаруживались в зрительном нерве у коров. Следовательно, аксоны норадреналинэргических нейронов могут достигать сетчатки в составе зрительного нерва. Важно определить, существуют ли норадреналинэргические нейроны в области ГП, гипоталамуса или симпатических ганглиев, аксоны которых достигают сетчатки и обеспечивают ее иннервацию (данная проблема представляется особенно важной при полной гибели нейронов ГП и их аксонов, содержащих НА. Потеря норадреналинэргической иннервации ГКС и центральных нейронов при БА может объяснять гибель ГКС при глаукоме и БА. Кроме того, систематическое лечение больных БА с помощью агонистов альфа-2 адренорецепторов может привести к замедлению апоптоза ГКС и нейронов ЦНС при БА посредством действия на альфа-2 адренергические рецепторы, сохраняющиеся на жизнеспособных нейронах.
Пример
Методы
Тест "открытое поле"
Животных помещают в отдельные пластиковые емкости с двумя рядами фотоэлементов (вертикальным и горизонтальным) с целью регистрации горизонтальных и вертикальных движений (т.е. пройденного расстояния и "подъемов"). Исследование проводится при низком уровне шума и приглушенном освещении. Движения животного в емкости оцениваются в течение 5 минут и подсчитываются на основании числа срабатываний фотоэлементов и направления их расположения. В ходе последнего теста учитываются только те перемещения, при которых животное опирается на задние лапы. Все эпизоды классифицируются, как вставание "с опорой" и без "опоры". При вставании с опорой животное касается по меньшей мере одной передней лапой стенки контейнера. При вставании без опоры животное опирается только на задние лапы. Классификация эпизодов проводится в соответствии с видеозаписью эксперимента. Число эпизодов вставания без опоры является наиболее надежным показателем степени гибели дофаминовых нейронов.
Тест "Подвешивание за хвост"
Мышей подвешивают за хвост 3 раза по 10 минут. Каждую мышь удерживают за основание хвоста на высоте около 30 см над поверхностью стола до тех пор, пока мышь не поворачивается влево или вправо. Поворот влево оценивается как 0 баллов, вправо - 1 балл.
Во время тестирования учитывают также положение передних лап, которое оценивается по 4-балльной шкале. Вытянутые или расположенные над головой передние лапы соответствуют 0 баллам. Сжатые или прижатые к туловищу лапы - 3 балла. При промежуточном положении лап результат теста оценивается как 1 или 2 балла. Положение задних лап также оценивается по балльной шкале.
Тест "Постройка гнезд"
Четырех мышей одного пола, взятых из одной группы, помещают в пластиковую коробку. Затем в коробку помещают комок из 8 полосок, изготовленных из бумажного полотенца. Через 24, 48, 72 и 96 часов после введения препарата оценивают наличие "гнезд", построенных из бумажных полосок. Результаты классифицируют следующим образом: 0 баллов = бумага разделена на клочки и образует гнездо, закрывающее животное целиком; 1 = бумага разделена на клочки и образует гнездо, не закрывающее животное сверху; 2 = отдельные полоски разделены на клочки, на бумаге следы зубов, полоски собраны кучно в одной области; 3 = на полосках бумаги имеются отдельные следы зубов, полоски не собраны в клубок; 4 = отсутствуют признаки постройки гнезда.
Подсчет количества нейронов
Через 55-60 дней после инъекции МРТР мышей выводят из эксперимента с помощью нембутала натрия. Головной мозг перфузируют раствором фосфатного буфера, а затем фиксатором Lana's (смесь параформальдегида и пикриновой кислоты). Головной мозг полностью извлекают и помещают в фиксатор Lana's на 7-10 дней. Затем готовят фронтальные срезы мозга с интервалом 50 мкм с помощью вибратома. Срезы окрашивают с помощью антител к тирозингидроксилазе, которая является лимитирующим фактором синтеза дофамина. Микроскопию срезов проводят при 100-кратном увеличении. Срезы тканей (2.9 и 3.6 мм кзади от теменной линии) используются для подсчета нейронов в черной субстанции и вентральной части покрышки мозга. Эти срезы проходят через среднюю часть ростральной половины и среднюю часть каудальной половины черной субстанции. Учитываются только нейроны с положительным окрашиванием на ТГ, от которых отходят 2-6 нейритов. Общее среднее число нейронов вычисляется по 4 срезам (ростральный, каудальный, левый и правый). Отдельно оценивают среднее число нейронов в черной субстанции и вентральной области покрышки мозга. Эти показатели анализируют с помощью дисперсионного способа, а различия между группами - по критерию Фишера.
Порядок проведения эксперимента.
У мышей, получающих инъекции пиридинового токсина 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (МРТР), наблюдаются очаги гибели дофаминергических нейронов в черной субстанции и вентральной области покрышки мозга. Уменьшение числа дофаминергических нейронов в черной субстанции клинически соответствует болезни Паркинсона. Тот же процесс в вентральной части покрышки мозга может приводить к когнитивному дефициту как при болезни Паркинсона, так и при болезни Альцгеймера, что объясняется проекцией проводящих путей в коре лобных долей.
30 мышей линии С57В1/В6 (возраст 8-12 недель) выдерживают в виварии 12-14 дней с целью адаптации. Затем животных случайным образом распределяют между группами: одна из группа получает МРТР и диметилсульфоксид; другая - только диметилсульфоксид, третья - МРТР и бримонидин (3 мг/кг/сутки).
В описываемых испытаниях каждую мышь подвергали тесту "открытое поле" и тесту "подвешивание за хвост". Тест "открытое поле" является наиболее распространенным способом оценки поведения мышей после введения МРТР и позволяет оценить нарушение транспорта дофамина в черной субстанции. Тест "подвешивание за хвост" позволяет оценить повреждение полосатого тела; тест с постройкой гнезда - недостаточную стимуляцию полосатого тела со стороны коры лобных долей.
Двум группам из трех вводят исследуемое вещество (или плацебо) путем инфузии. Введение осуществляют подкожно через осмотический микроирригатор в течение 14 дней со скоростью 0,25 мкл/ч. Через 3 дня после имплантации микроирригатора проводят тест "открытое поле" и тест с фиксацией животного за хвост. Аналогичные тесты проводят в контрольной группе мышей, которым не устанавливают микроирригатор. Непосредственно после тестирования животным опытной группы подкожно вводят МРТР в дозе 40 мг/кг. Затем во всех группах проводят тест "открытое поле" и тест с фиксацией за хвост через 10-12 дней и 30-40 дней после инъекции МРТР. Тест "открытое поле" и тест с фиксацией за хвост повторяют через 50-55 дней после введения МРТР.
При оценке поведения животных результаты тестирования анализируют с помощью дисперсионного способа, различия между группами оценивают по критерию Фишера.
Результаты
Тест "открытое поле"
Перед введением МРТР достоверных различий между 3 группами по пройденному расстоянию и по количеству вставаний на лапы не выявлено.
Через 10 и 30 дней после введения МРТР животные в группе плацебо были значительно более активными, чем в опытной группе. У мышей, получавших бримонидин, изменений активности не отмечено, т.е. отсутствовали значительные различия между данной группой и группой плацебо.
Инъекция МРТР вызывала снижение общего числа вставаний на задние лапы через 10 дней. Через 30 дней этот показатель был нормальным (по сравнению с группой плацебо) и был несколько ниже по сравнению с контрольной группой.
В группе плацебо число вставаний без опоры было ниже, чем у здоровых мышей. Влияния инъекций МРТР или других препаратов на число вставаний с опорой не выявлено.
Тест "Подвешивание за хвост"
Перед введением МРТР не наблюдалось различий между группами в тесте "подвешивание за хвост", и вообще не наблюдалось различий между группами по положению задних лап после введения МРТР. Введение МРТР значительно уменьшило число вытягиваний передних лап во время каждого из трех подвешиваний. Таким образом, это уменьшение не проходит с течением времени. Бримонидин не снизил это уменьшение во время введения. Однако после введения (через 14 дней после инъекции) препарат снизил это уменьшение.
Подсчет числа нейронов
В черной субстанции мышей, получавших МРТР, число нейронов было на 58% меньше, чем в контрольной группе. В группе бримонидина уменьшение числа нейронов было значительно менее выраженным.
В вентральной области покрышки мозга на фоне введения МРТР наблюдалось уменьшение числа нейронов в среднем на 28% по сравнению с контролем. Бримонидин уменьшал этот показатель приблизительно на 10%.
Приведенные выше примеры иллюстрируют возможные варианты осуществления изобретения и не ограничивают объем его правовой охраны. Изобретение определено в формуле, которая завершает описание.
Изобретение относится к способам лечения нейродегенеративного состояния головного мозга, такого как болезнь Паркинсона и болезнь Альцгеймера, и способам предотвращения гибели или дегенерации нервных клеток. Млекопитающему вводят терапевтически эффективное количество композиции, содержащей бримонидин или его фармацевтически приемлемую соль. Изобретение позволяет лечить или предотвращать дегенерацию нервных клеток за счет нейропротекторного действия бримонидина в отношении нейронов головного мозга. 2 н. и 4 з.п. ф-лы, 1 ил., 2 табл.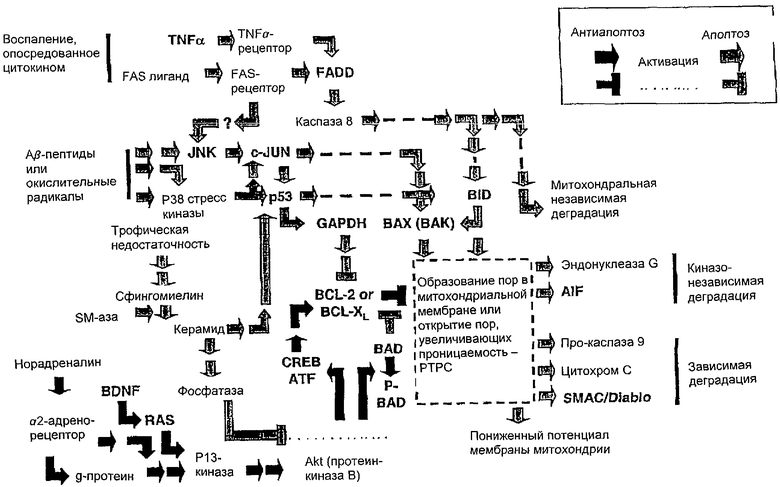
| CHOPIN P | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

