2420-175089RU/051
ПРИМЕНЕНИЕ ОПИОИДОВ ИЛИ МИМЕТИКОВ ОПИОИДОВ ДЛЯ ЛЕЧЕНИЯ ПАЦИЕНТОВ С УСТОЙЧИВОЙ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛЬЮ
Изобретение относится к новым способам лечения пациентов с устойчивой злокачественной опухолью.
Способы противораковой терапии часто являются неэффективными из-за устойчивости клеток опухолей к радио- и/или химиотерапии. Когда во время терапии развивается устойчивость, она часто проявляется либо как уменьшенное значение регрессии опухоли при такой же дозе (либо радиоактивного излучения, либо цитотоксического вещества), либо как увеличенная доза, которая является необходимой для такого же значения регрессии опухоли. Когда устойчивость является врожденной, т.е. не приобретенной или индуцированной из-за противоракового лечения, у клеток опухолей уже изначально отсутствует чувствительность к одному или нескольким противораковым лекарственным средствам или ионизирующему излучению.
Чувствительность к химиотерапии клеток злокачественных опухолей часто меняется индивидуально. Например, для карциномы поджелудочной железы известно, что только приблизительно для 25% всех пациентов благоприятно противораковое лекарственное средство гемцитабин. Другие 75% обладают врожденной устойчивостью к этой химиотерапии. Дополнительными примерами клеток опухолей с врожденной химио- и радиоустойчивостью являются клетки глиобластомы или меланомы.
Врожденная или приобретенная устойчивость (или отсутствие ответа) клеток опухолей к радио- и/или химиотерапии может иметь множество причин и может - как проиллюстрировано выше - меняться индивидуально. Несмотря на интенсивные исследования, точные механизмы еще остаются неясными. Однако, известно, что любая одиночная мутация, например, в участке связывания лекарственного вещества или в рамках процесса детоксикации клеток, может являться ответственной за отсутствующую или уменьшенную химиочувствительность. Проявление перекрестной устойчивости к нескольким противораковым лекарственным средствам также часто ограничивает эффективность способов противоракового лечения.
Значительной клинической важностью обладает феномен так называемой множественной лекарственной устойчивости (MDR). Согласно этой концепции, увеличивается экспрессия мембранных белков, а именно, членов АТФ-связывающей кассеты (ABC) белков-транспортеров, таких как P-гликопротеин, или белков, ассоциированных с множественной лекарственной устойчивостью (MRP), что приводит к увеличенному выведению лекарственных веществ посредством активного транспорта через мембрану клеток. Пациенты, обладающие множественной лекарственной устойчивостью, наиболее часто являются устойчивыми к широкому спектру цитотоксических лекарственных средств.
Устойчивость не является ограниченной химиотерапевтическими средствами или противораковыми лекарственными средствами; пациенты со злокачественной опухолью могут также обладать врожденной или приобретенной устойчивостью к ионизирующему излучению, применяемому при радиотерапии. Врожденная радиоустойчивость известна, например, для клеток меланомы и глиобластомы.
Радиоустойчивость может также являться индуцированной воздействием малых или фракционированных доз ионизирующего излучения. В нескольких исследованиях документирован этот эффект in vitro даже для клеток человека, так же как для нескольких моделей на животных. Могут быть вовлечены различные клеточные механизмы защиты от радиоактивного излучения, такие как изменения уровней некоторых цитоплазматических и ядерных белков, усиление процессов экспрессии генов или репарации ДНК.
Таким образом, в онкологии существует большая необходимость в новых способах, делающих лечение злокачественных опухолей более эффективным. В частности, целью настоящего изобретения является предоставление новых способов лечения пациентов со злокачественной опухолью, являющихся устойчивыми к общепринятым способам противораковой терапии, таким как противораковые лекарственные средства (химиотерапия) или радиотерапия, или для лечения пациентов со злокачественной опухолью с устойчивыми к апоптозу клетками.
Этой цели достигают применением опиоидов или миметиков опиоидов для лечения пациентов со злокачественной опухолью, устойчивой к радиотерапии и/или химиотерапии, поскольку в настоящее время обнаружено, что опиоиды, способные ингибировать пролиферацию клеток и/или рост клеток злокачественных опухолей, могут преодолевать устойчивость этих клеток злокачественных опухолей. Таким образом, эти опиоиды обеспечивают новые способы лечения пациентов, которых до сих пор считали не поддающимися лечению или не поддающимися эффективному лечению общепринятыми способами противораковой терапии. Эту группу предположительно не поддающихся лечению пациентов со злокачественной опухолью можно называть также «не отвечающими», «плохо отвечающими» или «нехимиочувствительными» или «нерадиочувствительными» пациентами со злокачественной опухолью.
Более того, обнаружено, что опиоиды и миметики опиоидов могут преодолевать устойчивость к апоптозу клеток злокачественных опухолей, и, таким образом, их можно эффективно применять в клинике в качестве противораковых веществ. В частности, наиболее неожиданно, обнаружено, что опиоиды - в частности, метадон - являлись настолько эффективными, как общепринятая химиотерапия (например, доксорубицином) и способы радиотерапии против неустойчивых (т.е. чувствительных) лейкозных клеток, и что нормальные лимфоциты выживали после этого лечения. Таким образом, в одном варианте осуществления изобретения опиоиды также являются эффективными для уничтожения клеток опухолей, но по существу не действуют на нормальные здоровые клетки пациента.
В контексте настоящего изобретения термин «опиоид» определяют как химически гетерогенную группу природных, синтетических или полусинтетических веществ, действующих как агонисты или антагонисты, которые все могут связываться с хорошо известными опиоидными рецепторами, предпочтительно с опиоидным рецептором μ, и которые способны к аресту пролиферации клеток злокачественных опухолей. Группа опиоидов включает природные опиаты, такие как алкалоиды, подобные морфину, дигидрокодеину, кодеину и тебаину, так же как полусинтетические опиаты, полученные из природных опиатов (например, гидроморфон, гидрокодон, оксикодон, оксиморфон, дезоморфин, диацетилморфин (героин), никоморфин, дипропаноилморфин, бензилморфин и этилморфин), или полностью синтетические опиоиды, такие как фентанил, петидин и метадон, трамадол или пропоксифен. Она включает также эндогенные опиоидные пептиды, которые могут продуцироваться естественным образом в организме, как эндорфины, динорфины или энкефалины, но которые также можно синтезировать.
Опиоиды известны благодаря их использованию в качестве анальгетиков. Тот факт, что опиоидные рецепторы, особенно опиоидные рецепторы μ, вовлечены в активацию путей передачи сигнала, приводящих к апоптозу, был известен ранее (Polakiewicz et al. 1998). В последнее десятилетие обнаружено, что опиоиды могут стимулировать апоптоз (Hatsukari et al. 2003). Кроме того, обсуждали применение опиоидов для индукции апоптоза в клетках мелкоклеточного рака легкого (Heusch & Maneckjee 1999). Однако лежащие в основе механизмы не являются известными, и эти результаты не позволяют предполагать применение опиоидов для преодоления устойчивости к общепринятым способам противоракового лечения.
По изобретению опиоид является способным ингибировать пролиферацию и/или рост клеток злокачественных опухолей. Эта активность может включать, например, цитостатическую или цитотоксическую активность, так же как арест роста клеток и/или опухолей. Пролиферация клеток злокачественной опухоли является результатом ингибирования деления клеток. В частности, опиоиды или миметики опиоидов индуцируют гибель клеток опухолей. Гибель клеток в контексте изобретения включает все типы гибели клеток. Это может включать как некротическую, так и апоптотическую гибель клеток или аутофагию. В одном варианте осуществления изобретения гибель клеток индуцирована активацией зависимого от каспаз или не зависимого от каспаз пути. Однако опиоиды могут индуцировать гибель клеток через различные пути. В предпочтительном варианте осуществления изобретения опиоиды индуцируют апоптоз в клетках злокачественных опухолей.
В общем, известно, что апоптоз можно индуцировать через два главных биохимических пути. «Путь рецептора смерти» (или внешний путь) включает индуцированную рецептором TNF (фактора некроза опухоли) модель и индуцированную рецептором Fas модель (рецептор Fas известен также как Apo-1 или CD95). Связывание с этими рецепторами приводит к формированию в клетке индуцирующих гибель путей передачи сигнала, включая активацию каспаз-8. «Митохондриальный путь» (или внутренний путь) включает высвобождение цитохрома С из митохондрий, связывание Apaf-1 и активацию прокаспазы-9. Известно, что некоторые регуляторы активируют или дезактивируют пути апоптоза, такие как проапоптотические белки Bax и Bak или антиапоптотические белки Bcl-2, BclXL или XIAP.
В контектсе изобретения термин «миметики опиоидов» определяют как вещество, которое способно напрямую или опосредованно индуцировать в клетках злокачественных опухолей по существу такой же эффект, как опиоиды, в частности, принимая во внимание эффекты связывания опиоидов с опиоидным рецептором (например, рецептором μ) и/или индукцию гибели клеток, в частности, апоптоз посредством митохондриального пути. Термин «миметики опиоидов» включает также вещество, которое приводит к сверхэкспрессии опиоидных рецепторов, например, такое как кокаин, и вследствие этого опосредованно индуцирует гибель клеток.
В одном варианте осуществления изобретения опиоиды или миметики опиоидов индуцируют апоптоз посредством одного или нескольких из следующих механизмов:
i. расщепление каспазы-3 и PARP в клетках опухолей;
ii. расщепление каспазы-9 и понижающая регуляция XIAP;
iii. понижающая регуляция BclXL.
В предпочтительном варианте осуществления изобретения опиоид является членом группы метадона, включающей D-/L-метадон, левометадон, левацетилметадол и пиритрамид. Все эти опиоиды можно использовать в форме солей. Рацемическую форму D-/L-метадона предпочтительно предоставляют в форме гидрохлорида. В предпочтительном варианте осуществления изобретения опиоид метадон индуцирует апоптоз в клетках злокачественных опухолей через митохондриальный путь.
По изобретению термины «устойчивость», «радиоустойчивость» или «химиоустойчивость» определяют как уменьшенную чувствительность клеток злокачественных опухолей по меньшей мере к одной общепринятой терапии злокачественных опухолей, т.е. к противораковому лекарственному средству или радиотерапии. Пациента, страдающего такой злокачественной опухолью, определяют как «устойчивого» пациента со злокачественной опухолью. Поскольку устойчивость может являться врожденной или приобретенной, наблюдаемое уменьшение чувствительности присутствует по сравнению с полностью чувствительными «нормальными» клетками злокачественных опухолей, которые отвечают на терапевтически эффективную дозу применяемого противоракового лекарственного средства и/или радиоактивного излучения, или по сравнению с исходной чувствительностью в начале терапии. В последнем случае устойчивость проявляется либо как уменьшенное значение регрессии опухоли при такой же дозе (либо радиоактивного излучения, либо цитотоксического вещества), либо как увеличенная доза, которая является необходимой для такого же значения регрессии опухоли.
В особенно предпочтительном варианте осуществления опиоиды или миметики опиоидов используют для лечения пациентов со злокачественной опухолью, обладающих одним или несколькими из следующих видов устойчивости:
устойчивость к апоптозу;
множественная лекарственная устойчивость;
устойчивость к противораковому лекарственному средству;
устойчивость к цитотоксическому лекарственному средству;
устойчивость к реакционноспособным формам кислорода;
устойчивость к повреждающим ДНК средствам;
устойчивость к токсическим антителам;
устойчивость к доксорубицину;
одиночная или перекрестная устойчивость, в частности, к одному или нескольким из следующих лекарственных веществ: метотрексат, цитарабин, цисплатин, этопозид, винкристин, паклитаксел (таксол), карбоплатин, тенипозид, дексаметазон, преднизолон, циклофосфамид, ифосфамид, доксорубицин, эпирубицин, даунорубицин, меркаптопурин, флударабин, 5-фторурацил;
устойчивость к облучению (например, альфа, бета, гамма или электроны Оже).
Соответственно, в контексте настоящего изобретения «устойчивость» может являться полной или частичной; иными словами, пациенты, которых рассматривают как поддающихся лечению по изобретению, могут проявлять уменьшенную чувствительность или даже полное отсутствие чувствительности к общепринятым способам противоракового лечения. Этих пациентов можно также определять как «не отвечающих» или «слабо отвечающих».
Дополнительным синонимом для «устойчивой» злокачественной опухоли или опухоли является «рефрактерный» тип злокачественной опухоли, который также может являться полностью или частично рефрактерным. Врожденную устойчивость можно таким образом определить как «первично рефрактерная злокачественная опухоль». Особой формой рефрактерных или устойчивых клеток злокачественных опухолей являются так называемые «кинетически рефрактерные клетки»; феномен, известный, например, для клеток лейкоза, когда клетки сначала уничтожают, но потом они быстро воспроизводятся, так что эффективное лечение вряд ли возможно.
Как применяют в контексте настоящего изобретения, термин «общепринятое» лечение или терапия относится к принятому в настоящее время и широко используемому терапевтическому лечению конкретного типа злокачественной опухоли, основанному на результатах прошлых исследований и/или разрешении регуляторного органа.
Общепринятые противораковые лекарственные средства включают цитотоксические и цитостатические средства, которые уничтожают клетки злокачественных опухолей или уменьшают и/или останавливают их рост или пролиферацию. Механизмы действия этих противораковых лекарственных средств могут варьировать; примерами являются антиметаболиты (например, цитарабин, метотрексат, меркаптопурин или клофарабин), сшивающие ДНК средства (например, цисплатин и его производные), интеркалирующие в ДНК вещества (например, доксорубицин), блокаторы топоизомеразы (например, этопозид), ингибиторы киназы (например, цетуксимаб), стероиды (например, дексаметазон) или ингибиторы митоза (например, винкристин). Одним из примеров общепринятого противоракового лечения лейкоза является введение доксорубицина.
Общепринятая радиотерапия может включать также терапию радиоактивным излучением, что означает применение излучения высоких энергий из x-лучей, альфа, бета и гамма-лучей, электронов Оже, ультрафиолетовых лучей, нейтронов, протонов и других источников для уничтожения клеток злокачественных опухолей и уменьшения размера опухолей. Радиоактивное излучение может происходить из устройства вне организма (наружная дистанционная радиотерапия), или может происходить из источников радиоактивного излучения, помещенных в организме поблизости от клеток злокачественных опухолей (внутренняя радиотерапия). В системной радиотерапии применяют радиоактивное вещество, такое как радиоактивно меченое моноклональные антитело, проходящее по кровотоку к ткани-мишени. Радиоустойчивые клетки злокачественных опухолей не отвечают или только частично отвечают на эти виды лечения.
Как подробно описано выше, согласно одному варианту осуществления изобретения опиоиды или миметики опиоидов применяют для преодоления или «прерывания» врожденной или приобретенной устойчивости клеток злокачественных опухолей к общепринятым способам противоракового лечения и/или лечения радиоактивным излучением или устойчивости к апоптозу. В одном варианте осуществления изобретения клетки злокачественных опухолей, рассматриваемые как поддающиеся лечению по изобретению, экспрессируют опиоидный рецептор, в частности, опиоидный рецептор μ.
В следующем варианте осуществления группа злокачественных опухолей включает, но не ограничивается ими, лейкоз, рак груди, глиобластому, рак предстательной железы, рак легкого, немелкоклеточный рак легкого (NSCLC), злокачественную опухоль мозга, рак толстого кишечника, колоректальный рак.
Примерами типов злокачественных опухолей, рассматриваемых как поддающиеся лечению по изобретению, с врожденной устойчивостью к облучению, являются клетки глиобластомы, меланомы или злокачественной опухоли поджелудочной железы. Рак груди, рак мочевого пузыря или лейкоз часто являются устойчивыми к химиотерапевтическим средствам. Примерами типов злокачественных опухолей, у которых часто развивается устойчивость, являются меланома, карцинома толстого кишечника, опухоли мозга, глиобластома, злокачественная опухоль мозга, рак поджелудочной железы, рак печени, рак яичника, рак молочной железы, рак легкого, хронический лейкоз или остеосаркома.
В следующем варианте осуществления изобретения опиоиды или миметики опиоидов можно применять в комбинации - т.е. в качестве сочетания - с общепринятыми противораковыми веществами или способами лечения, например, с цитостатическими или цитотоксическими веществами или радиотерапией. Опиоиды или миметики опиоидов можно комбинировать, например, с природными и/или синтетическими противораковыми веществами, природными и/или синтетическими цитотоксическими веществами, антибиотиками, цитотоксическими антителами, гормонами, психотропными лекарственными средствами, естественным образом или генетически модифицированными организмами и веществами из организмов (например, растений, микроорганизмов, плодов), анальгетиками и/или различными видами радиоактивного излучения, не связанного или связанного с веществами (например, антителами). Пациент может являться устойчивым или не устойчивым к этому лечению.
«Сочетание» обозначает фармацевтический препарат, содержащий терапевтически эффективное количество любого из опиоидов или миметиков опиоидов (компонент A), как определено по изобретению, и по меньшей мере одно дополнительное противораковое вещество (компонент B). Это «сочетание» может составлять одну композицию или по меньшей мере две композиции, которые можно вводить пациентам одновременно или последовательно. Вышеупомянутые вещества предпочтительно комбинируют с метадоном.
Сочетание по изобретению может являться преимущественным для эффективного лечения клеток злокачественных опухолей, поскольку оно может оказывать синергические эффекты по сравнению с отдельными композициями. В частности, возможно сочетание с метадоном в качестве компонента A и одним из следующих средств в качестве компонента B: метотрексат, цитарабин, цисплатин, этопозид, винкристин. Более того, возможно комбинированное лечение, включающее также обработки облучением.
В предпочтительном варианте осуществления изобретения опиоиды применяют для лечения либо устойчивых, либо чувствительных несолидных злокачественных опухолей, т.е. всех гематологических злокачественных новообразований, поражающих кровь, костный мозг и лимфатические узлы, включая острый лимфобластный лейкоз, B-клеточный лимфатический лейкоз, острый миелоидный лейкоз, хронический миелоидный лейкоз, хронический лимфоцитарный лейкоз и все предшествующие лейкозу формы, волосатоклеточный лейкоз, болезнь Ходжкина, неходжкинскую лимфому и множественную миелому.
ПРИМЕР 1: Применение метадона для лечения клеток лейкоза и особенно для лечения клеток лейкоза, которые не удалось уничтожить с помощью противораковых лекарственных средств, обычно используемых в общепринятых способах терапии
Лекарственные средства и реагенты
Гидрохлорид D,L-метадона (метадон, Sigma, Taufkirchen, Germany) являлся свежерастворенным в стерильной дистиллированной воде перед каждым экспериментом для обеспечения постоянного качества препаратов.
Культура клеток
Линию клеток миелоидного лейкоза человека HL-60 и линию T-клеток лимфобластного лейкоза человека CEM выращивали на RPMI 1640 (GIBCO, Invitrogen, Karlsruhe, Germany), содержащей 10% эмбриональную телячью сыворотку (Biochrom, Berlin, Germany), 10 мМ HEPES, pH 7,3 (Biochrom), 100 ед./мл пенициллина (GIBCO), 100 мкг/мл стрептомицина (GIBCO) и 2 мМ L-глутамин (Biochrom) при 37°C и 5% CO2. CEMCD95R являются устойчивыми к 1 мкг/мл анти-CD95 (Friesen et al. 1996), и CEMCDOXOR являются устойчивыми к 0,1 мкг/мл доксорубицина (Friesen et al. 2004). CEMCD95R являются устойчивыми к апоптозу и обладающими множественной лекарственной устойчивостью. CEMCD95R обладает перекрестной устойчивостью к нескольким противораковым лекарственным средствам, таким как метотрексат, цитарабин, цисплатин, этопозид, винкристин, и к гамма- и бета-излучению (Friesen et al. 1996, Los et al. 1997, Friesen et al. 2007). Все линии клеток, используемые в этом исследовании, являлись свободными от микоплазмы.
Индукция апоптоза
Клетки лейкоза (1×105 клеток/мл) обрабатывали 30, 20, 15, 10 мкМ метадоном в 150-мл флаконах или 96-луночных планшетах. Через 24 ч и 48 ч количественные характеристики апоптоза измеряли проточной цитометрией, как описано (Carbonari et al. 1994, Friesen et al. 2003). Кратко, для определения апоптоза, клетки лизировали буфером Николетти, содержащим 0,1% цитрат натрия плюс 0,1% Тритон X-100 и 50 мкг/мл йодида пропидия, как описано Nicoletti et al. (1991). Процент апоптотических клеток измеряли анализом гиподиплоидной ДНК (subG1) или FSC/SSC (Nicoletti et al. 1991, Carbonari et al. 1994). Окрашенные йодидом пропидия (PI) ядра или профили прямого светорассеяния/бокового светорассеяния (FSC/SSC) клеток анализировали проточной цитометрией (FACSCalibur, Becton Dickinson, Heidelberg, Germany).
Выделение лимфоцитов периферической крови
Лимфоциты периферической крови (PBL) выделяли из свежей крови здоровых индивидов. PBL (1×106 клеток в 1 мл) обрабатывали 30, 20, 15, 10 мкМ метадоном в 96-луночных планшетах. Через 24 ч и 48 ч количественные характеристики апоптоза измеряли проточной цитометрией, как описано (Carbonari et al. 1994).
Ингибирование метадона индуцирует активацию каспаз посредством zVAD.fmk
Ингибирование активации каспаз осуществляли, как описано ранее (Friesen et al. 2007). Кратко, трипептидный ингибитор каспаз с широким спектром действия zVAD.fmk (бензоилкарбонил-Val-Ala-Asp-фторметилкетон, Enzyme Systems Products, Dubli, USA) использовали в концентрации 50 мкмоль/л. Клетки HL-60 и CEM предварительно инкубировали с zVAD.fmk 1 ч до обработки метадоном. Через 24 ч и 48 ч процент апоптотических клеток измеряли анализом гиподиплоидной ДНК (subG1) или FSC/SSC. Окрашенные йодидом пропидия (PI) ядра (Nicoletti et al. 1991) или профиль прямого светорассеяния/бокового светорассеяния (FSC/SSC) клеток (Carbonari et al. 1994) анализировали проточной цитометрией (FACSCalibur, Becton Dickinson, Heidelberg, Germany).
Анализ Вестерн-блоттингом
Анализы Вестерн-блоттингом выполняли, как описано (Friesen et al. 2004, Friesen et al. 2003). Иммунодетекцию PARP, каспазы-3, каспазы-9, каспазы-8, XIAP, CD95, CD95-L, Bax, Bcl-XL и β- актина выполняли с использованием поликлонального антитела кролика анти-PARP (1:5000, Roche), моноклонального антитела мыши против каспазы-3 (1:1000, Cell-Signalling), моноклонального антитела мыши против каспазы-8 (1:1000, Cell-Signalling), поликлонального антитела кролика против активной каспазы-9 (1:1000, Cell-Signalling), моноклонального антитела мыши анти-XIAP (1:1000, Transduction-Laboratories, Lexington, Kentucky), моноклонального антитела мыши анти-Fas (анти-CD95) (1:1000, Transduction-Laboratories), моноклонального антитела мыши против лиганда Fas (анти-CD95-лиганд) (1:250, BD, Pharmingen), поликлонального антитела кролика анти-Bax (1:250, Oncogene, Cambridge), поликлонального антитела кролика анти-Bcl-Xs/L (1:1000, Santa-Cruz Biotechnology, Santa-Cruz, CA), поликлонального антитела кролика анти-p21 (1:1000, Santa-Cruz) и моноклонального антитела мыши против β-актина (Sigma). Конъюгированное с пероксидазой антитело козы против IgG мыши или конъюгированное с пероксидазой антитело козы против IgG кролика (1:5000, Santa-Cruz) в качестве вторичного антитела использовали для усиленной системы хемилюминесценции (ECL, Amersham- Pharmacia, Freiburg, Germany). Равное нанесение белка контролировали детекцией β-актина.
Метадон индуцирует уничтожение клеток для лейкозных клеток CEM и HL-60 посредством апоптоза с нетоксическими эффектами на нелейкозные PBL
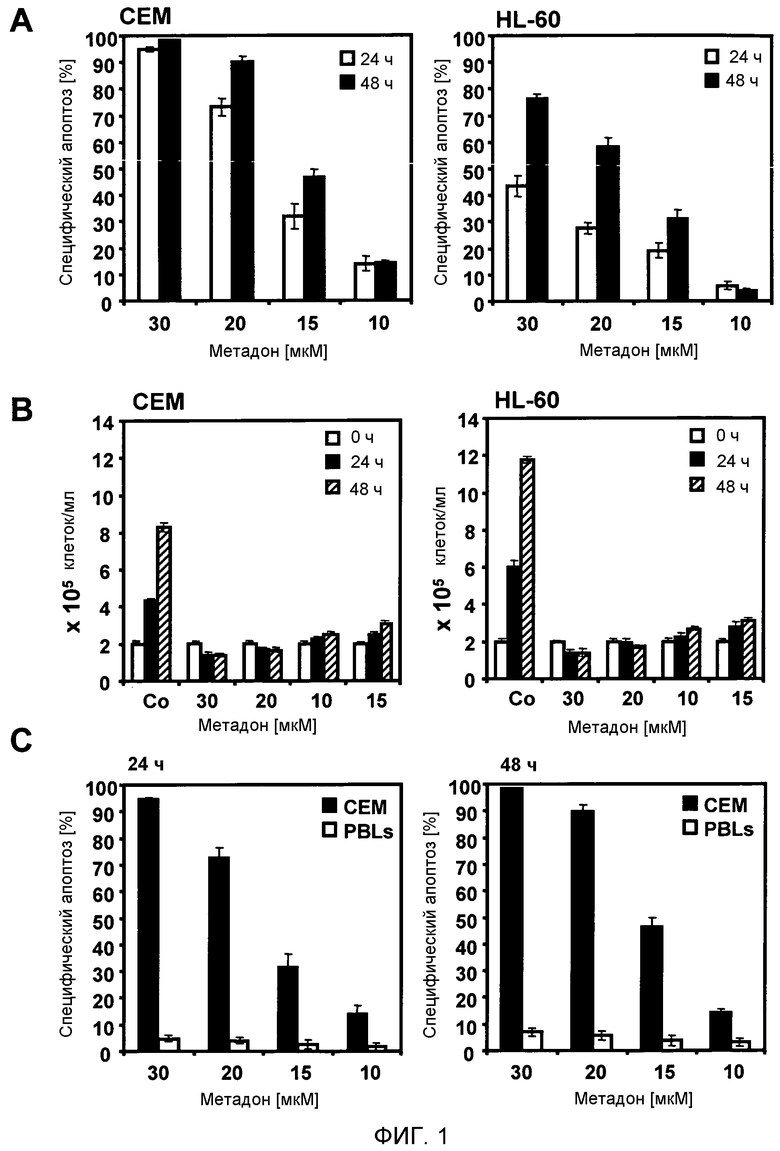
Показано, что при лейкозах и солидных опухолях противораковые лекарственные средства индуцируют апоптоз и ингибируют пролиферацию (Kaufmann & Earnshaw 2000). Таким образом, анализировали, может ли терапевтическое опиоидное лекарственное средство метадон также ингибировать пролиферацию и запускать апоптоз в линии T-клеток лимфобластного лейкоза человека CEM и линии клеток миелоидного лейкоза человека HL-60, сравнимо с хорошо известными общепринятыми противораковыми лекарственными средствами (фиг. 1A, B). Через 24 ч и 48 ч после обработки различными концентрациями метадона (30, 20, 15, 10 мкМ) сильную индукцию апоптоза (фиг. 1A) и сильное ингибирование роста (фиг. 1B) детектировали для клеток CEM и HL-60. Затем анализировали, индуцирует ли метадон апоптоз также в нелейкозных лимфоцитах периферической крови (PBL) (фиг. 1C). Выделенные PBL инкубировали с различными концентрациями метадона (30, 20, 15, 10 мкМ). Через 24 ч и 48 ч после обработки метадоном обнаружили, что метадон не может уничтожать PBL в концентрациях, сравнимых с концентрациями, используемыми для лечения клеток лейкоза, таких как CEM (фиг. 1C). Это показывает, что метадон индуцирует апоптоз в клетках лейкоза с нетоксическими эффектами на нелейкозные PBL.
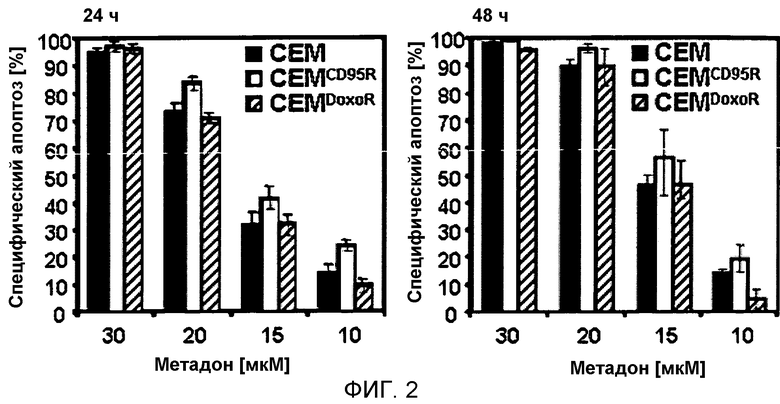
Метадон прерывает устойчивость к доксорубицину, устойчивость к CD95 и множественную лекарственную устойчивость в лейкозных клетках
Устойчивость к противораковым лекарственным средствам является лимитирующим фактором в лечении пациентов с лейкозом и опухолями (Bergman & Harris 1997, Friesen et al. 2003). Обнаружено, что метадон обладает сильной противолейкозной активностью и эффективно уничтожает лейкозные клетки. Таким образом, анализировали, может ли метадон также индуцировать гибель клеток в устойчивых к доксорубицину лейкозных клетках, которые являются устойчивыми к апоптозу. Устойчивые к доксорубицину лейкозные клетки CEM (CEMDoxoR) обрабатывали различными концентрациями метадона (30, 20, 15, 10 мкМ). Через 24 ч и 48 ч после обработки метадоном гибель клеток измеряли проточной цитометрией. После обработки 30, 20, 15 мкМ метадона в устойчивых к доксорубицину клетках CEMDoxoR измеряли сильную индукцию апоптоза, которая являлась сходной с индукцией чувствительных лейкозных клеток CEM (фиг. 2), что указывает на то, что метадон преодолевает устойчивость к доксорубицину и устойчивость к апоптозу.
Затем исследовали, может ли метадон также уничтожать лейкозные клетки, которые обладали множественной лекарственной устойчивостью. Таким образом, устойчивые к CD95 лейкозные клетки CEMCD95R, являвшиеся устойчивыми к таким противораковым лекарственные средствам, как этопозид, цисплатин, метотрексат, цитарабин, доксорубицин, винкристин, обрабатывали различными концентрациями метадона (30, 20, 15, 10 мкМ). После обработки 30, 20, 15 мкМ метадона сильную индукцию апоптоза в устойчивых к CD95 лейкозных клетках CEMCD95R измеряли через 24 ч и 48 ч, которая являлась сходной с индукцией чувствительных лейкозных клеток CEM (фиг. 2). Это позволяет предполагать, что метадон не только вызывает сильную индукцию апоптоза в чувствительных лейкозных клетках, но также уничтожает устойчивые к доксорубицину и CD95 лейкозные клетки, которые не смогли уничтожить общепринятые противораковые лекарственные средства (Friesen et al. 1996, Los et al. 1997).
Метадон индуцирует зависимую от каспаз гибель клеток и активирует митохондрии в чувствительных, устойчивых к доксорубицину, устойчивых к CD95 и обладающих множественной лекарственной устойчивостью лейкозных клетках
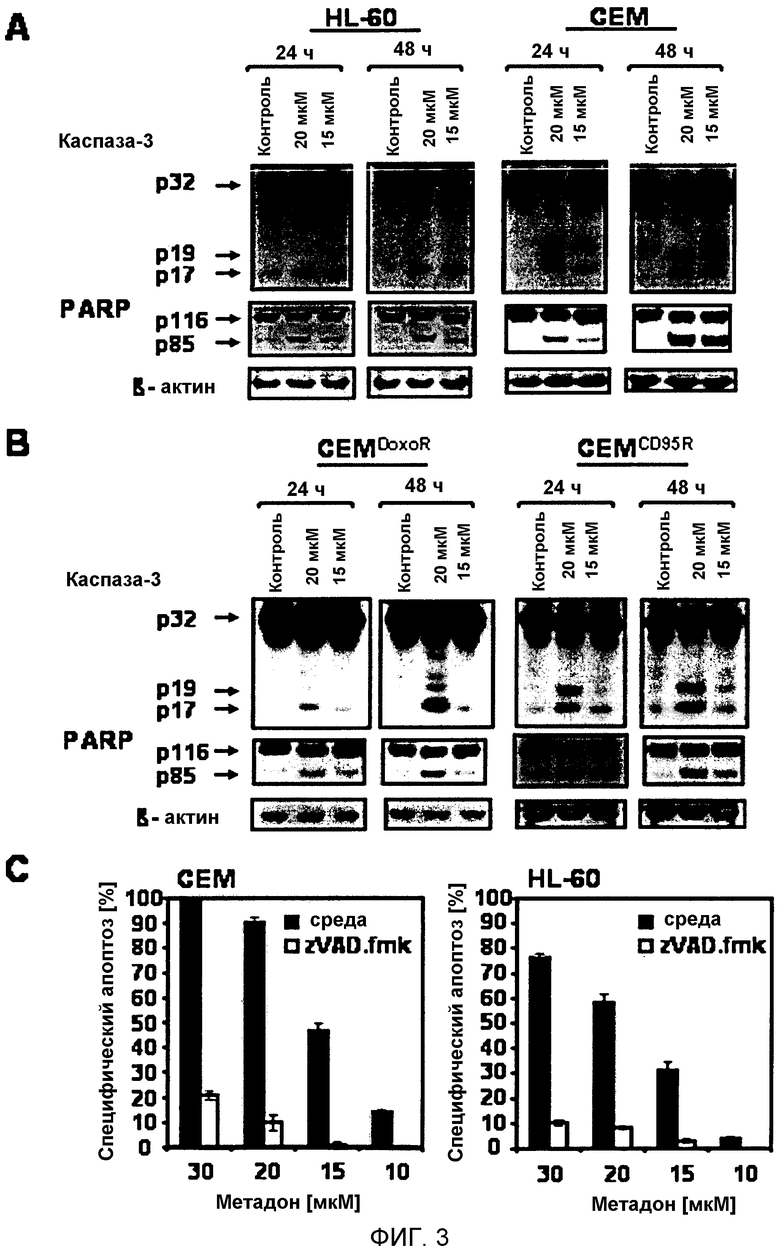
Метадон индуцирует апоптоз в чувствительных и в устойчивых лейкозных клетках с помощью неизвестных молекулярных механизмов. Таким образом, необходимо исследовать механизм и эффекторные молекулы, которые могут изменяться при запускаемой метадоном гибели клеток в лейкозных клетках.
Каспазы играют критическую роль в индукции апоптоза противораковыми лекарственными средствами (Kaufmann & Earnshow 2000, Hengartner 2000). Таким образом, анализы Вестерн-блоттингом использовали для исследования, активирует ли метадон каспазы в лейкозных клетках HL-60 и CEM, так же как в устойчивых к доксорубицину лейкозных клетках CEMDoxoR, являвшихся устойчивыми к апоптозу, и в устойчивых к CD95 лейкозных клетках CEMCD95R, обладавших множественной лекарственной устойчивостью и являвшихся устойчивыми к апоптозу. После обработки различными концентрациями метадона (20, 15 мкМ) каспаза-3 и PARP являлись расщепленными в лейкозных клетках HL-60 и CEM (фиг. 3A), так же как в устойчивых к доксорубицину лейкозных клетках CEMDoxoR и в устойчивых к CD95 лейкозных клетках CEMCD95R (фиг. 3B). Активация каспазы-8, которую, как показано, противораковые лекарственные средства индуцируют в лейкозных клетках, не обнаружена после обработки метадоном. Для исследования критической роли метадона в активации каспаз, клетки CEM и HL-60 предварительно инкубировали с ингибитором каспаз широкого спектра действия zVAD.fmk. Инкубация с zVAD.fmk почти полностью ингибировала индуцированный метадоном апоптоз (фиг. 3C), позволяя предполагать, что каспазы занимают центральное место в индуцированном метадоном апоптозе лейкозных клеток.
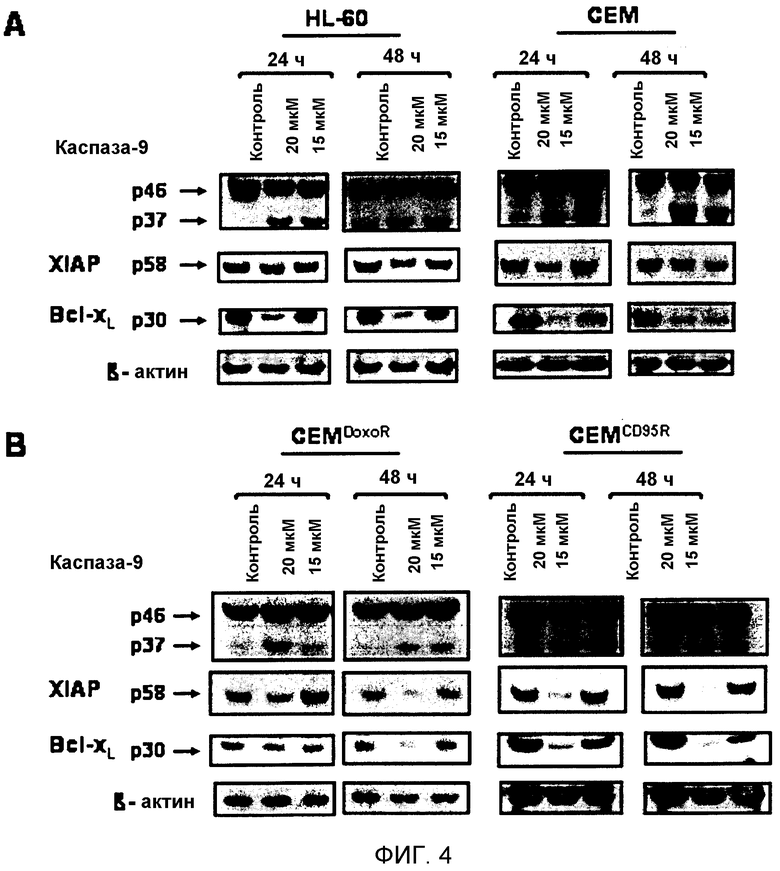
Показано, что противораковые лекарственные средства активируют митохондриальный путь, так же как путь лиганд/рецептор в лейкозных клетках и клетках опухолей (Kaufmann & Earnshow 2000). Исследовали, могут ли митохондрии также играть роль в индуцированном метадоном апоптозе лейкозных клеток. CEM, HL-60, устойчивые к доксорубицину лейкозные клетки CEMDoxoR и устойчивые к CD95 лейкозные клетки CEMCD95R обрабатывали различными концентрациями метадона (20, 15 мкМ) (фиг. 4). Через 24 ч и 48 ч сильное расщепление (фрагмент 37 кДа) каспазы-9 и сильную понижающую регуляцию ингибирующего каспазы белка XIAP (X-сцепленного ингибирующего апоптоз белка) обнаружили в лейкозных клетках HL-60 и CEM (фиг. 4A), так же как в устойчивых к доксорубицину лейкозных клетках CEMDoxoR и в устойчивых к CD95- лейкозных клетках CEMCD95R (фиг. 4B), обладавших множественной лекарственной устойчивостью и являвшихся устойчивыми к апоптозу.
Изменения митохондрий регулировали про- и антиаоптотические члены семейства Bcl-2. Через 24 ч и 48 ч сильную понижающую регуляцию Bcl-xL обнаружили в лейкозных клетках HL-60 и CEM (фиг. 4A), так же как в устойчивых к доксорубицину лейкозных клетках CEMDoxoR и в устойчивых к CD95 лейкозных клетках CEMCD95R (фиг. 4B) после обработки различными концентрациями метадона (20, 15 мкМ). Повышающую регуляцию Bax не обнаружили после обработки метадоном в лейкозных клетках.
Более того, повышающую регуляцию индуцирующих смерть лигандов и индуцирующих смерть рецепторов, таких как CD95, повышающую регуляцию которых, как показано, вызывают противораковые лекарственные средства, не обнаружили после обработки метадоном в лейкозных клетках (данные не представлены). Это показывает, что метадон индуцирует апоптоз непосредственной активацией врожденного митохондриального пути в чувствительных, так же как в устойчивых лейкозных клетках.
ПРИМЕР 2: Метадон индуцирует апоптоз в клетках глиобластомы
Глиобластома представляет собой наиболее агрессивный и наиболее распространенный тип первичных опухолей мозга. Известно, что клетки глиобластомы являются высоко устойчивыми к химиотерапевтическим средствам и облучению. Лечение может включать в себя химиотерапию и радиотерапию, но они представляют собой только паллиативные меры и не обеспечивают излечения. Более того, многие лекарственные средства не могут пересекать гематоэнцефалический барьер и, таким образом, являются бесполезными для лечения глиобластомы. Метадон способен пересекать гематоэнцефалический барьер. Исследовали эффект метадона на клетки глиобластомы. Более того, тестировали эффект на клетки глиобластомы метадона в сочетании с терапевтическими концентрациями доксорубицина.
Линию клеток глиобластомы человека A172 выращивали в DMEN (Invitrogen, Karlsruhe, Germany), содержащей 10% эмбриональную телячью сыворотку (Biochrom, Berlin, Germany), 10 мМ HEPES, pH 7,3 (Biochrom), 100 ед./мл пенициллина (Invitrogen), 100 мкг/мл стрептомицина (Invitrogen) и 2 мМ L-глутамин (Biochrom) при 37°C и 5% CO2.
Перед обработкой метадоном клетки глиобластомы высевали с плотностью 7000 клеток/см2 и обработку проводили через 24 ч после высева клеток.
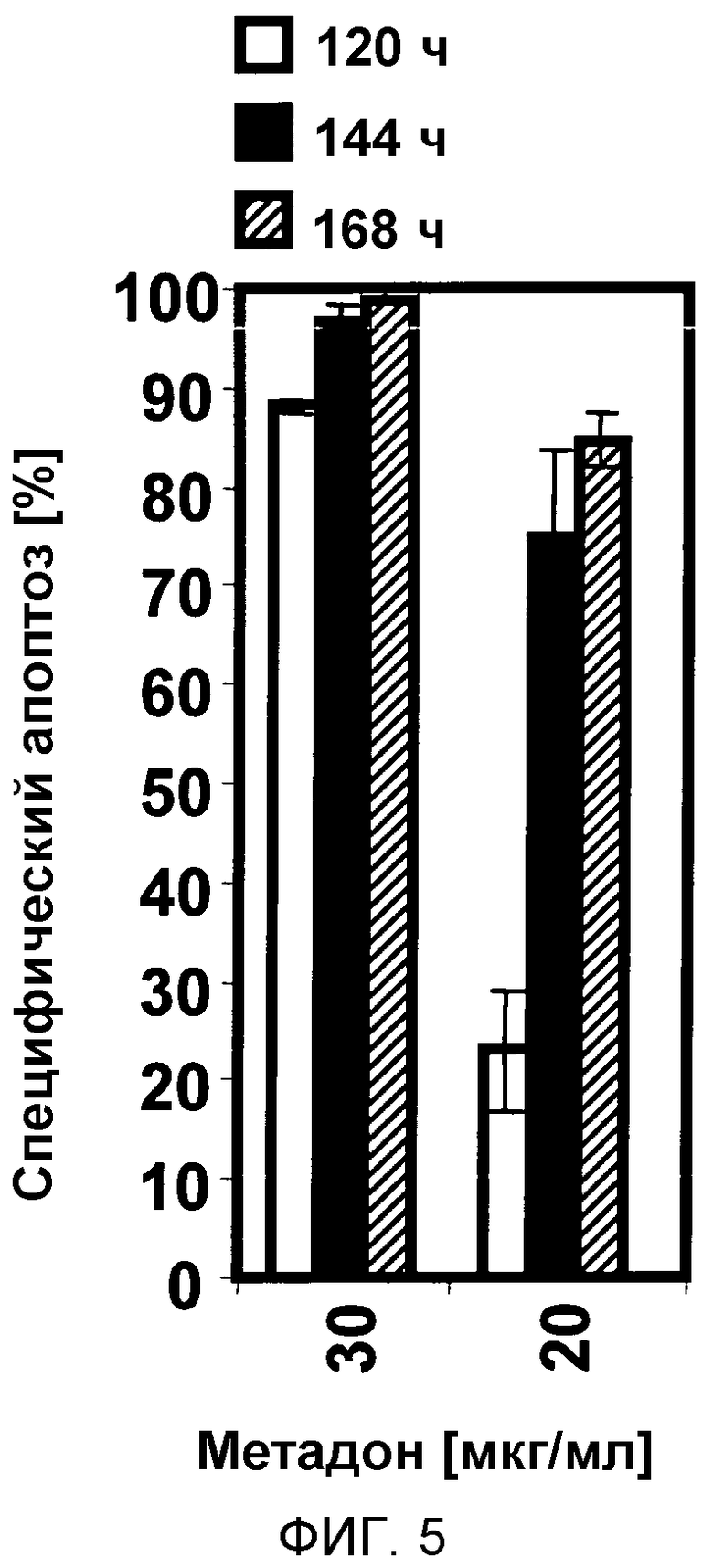
Клетки глиобластомы A172 (7000 клеток/см2) обрабатывали 30, 20 мкг/мл метадона в 75-см2 флаконах. Через 120 ч, 144 ч и 168 ч количественные характеристики апоптоза измеряли проточной цитометрией. Для определения апоптоза клетки лизировали буфером Николетти, содержащим 0,1% цитрат натрия плюс 0,1% Тритон X-100 и 50 мкг/мл йодида пропидия. Окрашенные йодидом пропидия (PI) ядра анализировали проточной цитометрией (FACSCalibur, Becton Dickinson, Heidelberg, Germany).
Через 120 ч, 144 ч и 168 ч процент апоптотических клеток измеряли анализом гиподиплоидной ДНК. Процент специфической гибели клеток рассчитывали следующим образом: 100 x (погибшие в результате эксперимента клетки (%) - спонтанно погибшие клетки в культуральной среде (%)/(100% - спонтанно погибшие клетки в среде (%)). Обработка метадоном приводила к более 20% погибших клеток через 120 ч и более 80% погибших клеток через 168 ч после обработки 20 мкг/мл метадона. Обработка метадоном приводила к более 85% погибших клеток через 120 ч и почти 100% погибших клеток через 168 ч после обработки 30 мкг/мл метадона (фиг. 5). Сходные результаты получены в трех независимых экспериментах. Это показывает, что метадон индуцирует высокие уровни апоптоза в клетках глиобластомы.
ПРИМЕР 3: Показаны синергические эффекты метадона в сочетании с доксорубицином для индукции апоптоза в клетках глиобластомы
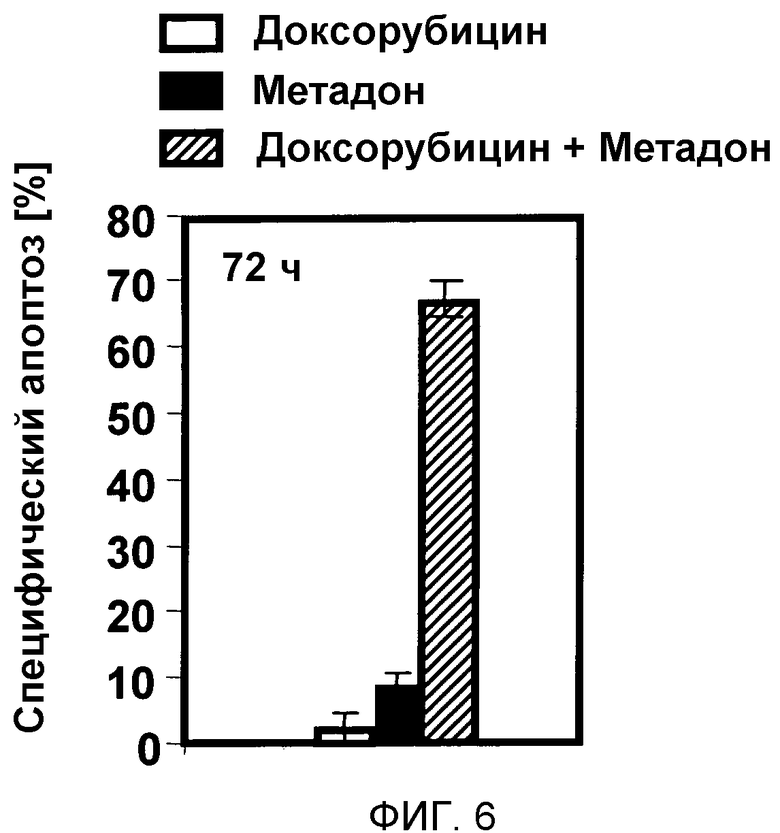
Клетки глиобластомы A172 (7000 клеток/см2) обрабатывали терапевтической концентрацией 0,1 мкг/мл доксорубицина (доксорубицин, белые столбцы), низкой концентрацией 1 мкг/мл метадона (метадон, черные столбцы) и 0,1 мкг/мл доксорубицина в дополнение к 1 мкг/мл метадона (доксорубицин + метадон, заштрихованные столбцы). Через 72 ч количественные характеристики апоптоза измеряли проточной цитометрией. Для определения апоптоза клетки лизировали буфером Николетти, содержащим 0,1% цитрат натрия плюс 0,1% Тритон X-100 и 50 мкг/мл йодида пропидия. Окрашенные йодидом пропидия (PI) ядра анализировали проточной цитометрией (FACSCalibur, Becton Dickinson, Heidelberg, Germany).
Комбинация терапевтической концентрации доксорубицина с низкими концентрациями метадона оказывала сильный апоптотический эффект на тестированные клетки глиобластомы (фиг. 6). Этот анализ показывает, что метадон оказывает синергические эффекты при применении вместе с другими химиотерапевтическими средствами, в данном случае, доксорубицином.
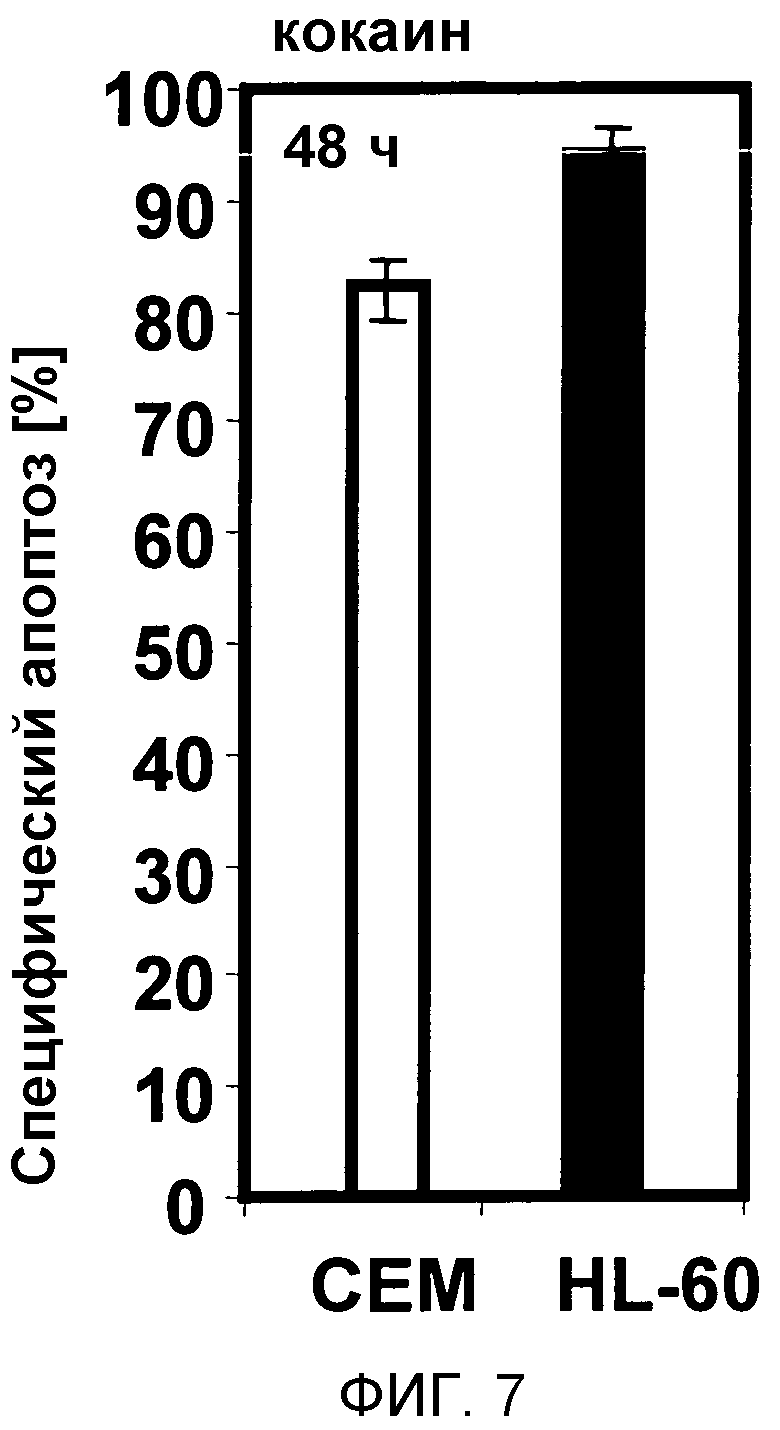
ПРИМЕР 4: Индукция апоптоза в лейкозных клетках с использованием кокаина
Клетки CEM (белый столбец) и HL-60 (черный столбец) обрабатывали 1000 мкг/мл кокаина. Через 48 ч процент апоптотических клеток измеряли анализом гиподиплоидной ДНК. Процент специфической гибели клеток рассчитывали следующим образом: 100×(погибшие в результате эксперимента клетки (%) - спонтанно погибшие клетки в среде (%))/(100% - спонтанно погибшие клетки в среде (%)). Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
Результаты показывают, что кокаин способен индуцировать апоптоз в клетках CEM, как показано на фиг. 7.
ПРИМЕР 5: Индукция апоптоза в клетках злокачественных опухолей, выделенных от пациентов ex vivo, с использованием D,L-метадона отдельно или в сочетании с флударабином
Клетки B-ALL (B-клеточного лимфатического лейкоза) и CLL (хронического лимфатического лейкоза) выделяли от пациентов ex vivo. Эти клетки обрабатывали различными концентрациями D,L-метадона или D,L-метадона в дополнение к флударабину.
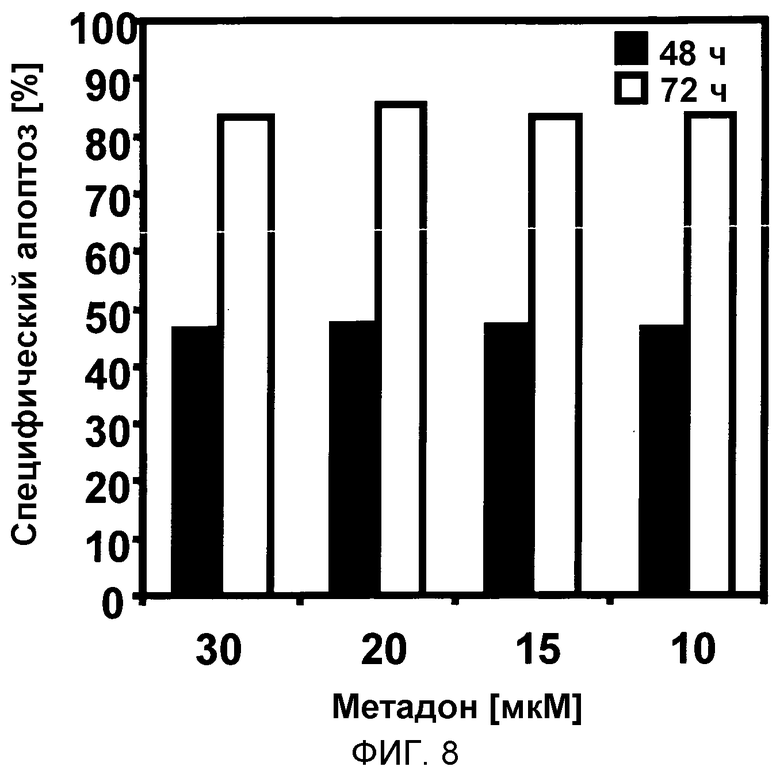
В первом исследовании клетки B-ALL (B-клеточного лимфатического лейкоза) (50000 клеток/100 мкл) обрабатывали 30, 20, 15, 10 мкМ метадона. Через 48 ч и 72 ч количественные характеристики апоптоза измеряли проточной цитометрией. Результаты показывают, что уже 10 мкМ метадон являлся достаточным для индукции апоптоза в пределах до более 80% клеток, обрабатываемых в течение 72 час (смотри фиг. 8).
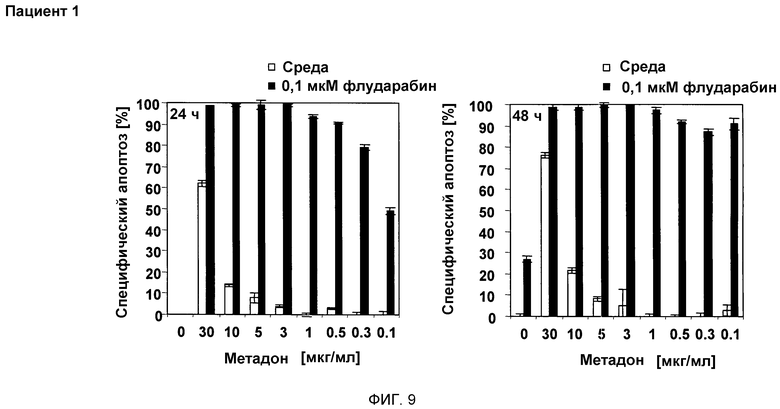
Во втором исследовании клетки CLL (хронического лимфатического лейкоза) (50000 клеток/200 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3 и 0,1 мкг/мл метадона отдельно или в дополнение к 0,1 мкМ флударабина. Через 24 ч и 48 ч количественные характеристики апоптоза измеряли проточной цитометрией. Результаты показывают, что апоптоз был индуцирован в 100% клеток уже через 24 ч, при сочетании 3 мкг/мл метадона с 0,1 мкМ флударабина (смотри фиг. 9).
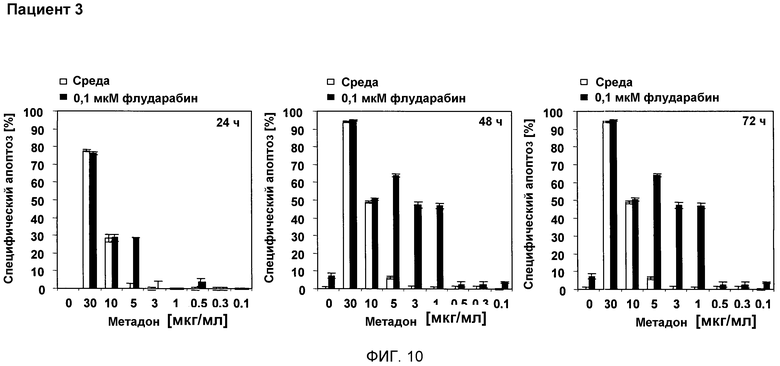
В третьем исследовании клетки CLL (хронического лимфатического лейкоза) (50000 клеток/200 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл метадона отдельно или в дополнение к 0,1 мкМ флударабина. Через 24 ч и 48 ч количественные характеристики апоптоза измеряли проточной цитометрией. Результаты показывают, что апоптоз был индуцирован почти в 50% клеток через 48 часа, при сочетании 3 мкг/мл метадона с 0,1 мкМ флударабина. Апоптоз был индуцирован в 90% клеток через 48 ч, при сочетании 30 мкг/мл метадона с 0,1 мкМ флударабина (смотри фиг. 10).
Таким образом, как позволяют предполагать эти исследования, обработка метадоном отдельно или в сочетании с цитостатическим средством является успешной для клеток злокачественных опухолей, выделенных от пациентов, ex vivo.
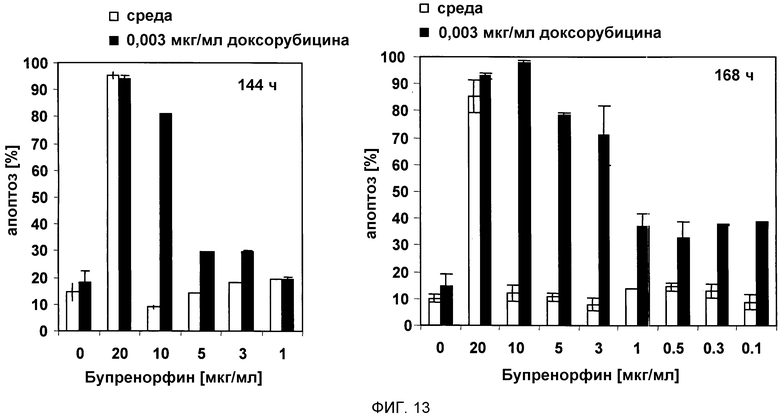
ПРИМЕР 6: Индукция апоптоза в лейкозных клетках с использованием бупренорфина в сочетании с доксорубицином
Бупренорфин успешно использовали в сочетании с доксорубицином для индукции апоптоза в лейкозных клетках HL-60 in vitro. Бупренорфин представляет собой полусинтетическое опиоидное лекарственное средство, применяемое также в качестве анальгетика. Он является частичным агонистом μ-опиоидного рецептора.
Линию клеток острого миелоидного лейкоза человека HL-60 (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл бупренорфина отдельно или в дополнение к 0,003 мкг/мл или 0,001 мкг/мл доксорубицина. Через 144 ч или 168 ч количественные характеристики апоптоза измеряли проточной цитометрией.
Результаты показывают, что 20 мкг/мл бупренорфина являлось достаточным для индукции апоптоза более чем в 90% клеток через 144 ч. Такой же результат получили при добавлении 0,003 мкг/мл доксорубицина. Апоптоз был индуцирован почти в 100% клеток при сочетании 10 мкг/мл бупренорфина с 0,003 мкг/мл доксорубицина и инкубацией клеток в течение 168 ч (смотри фиг. 13).
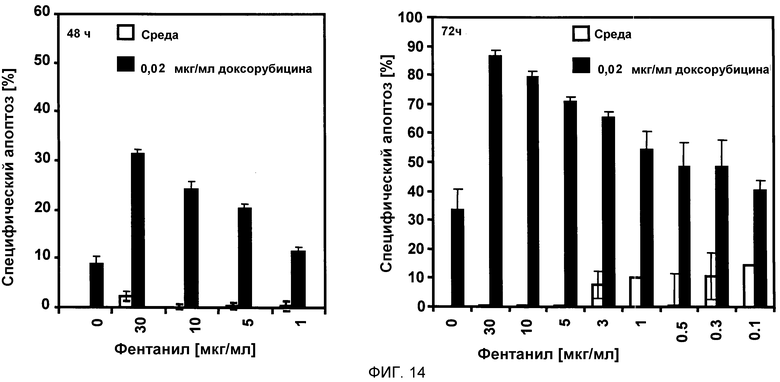
ПРИМЕР 7: Индукция апоптоза в лейкозных клетках с использованием фентанила в сочетании с доксорубицином
Фентанил успешно использовали в сочетании с доксорубицином для индукции апоптоза в лейкозных клетках CEM in vitro. Фентанил представляет собой синтетический опиоид, который часто используют в качестве анальгетика при лечении боли, связанной с злокачественными опухолями. Фентанил является агонистом опиоидного рецептора μ.
Линию клеток T-клеточного лейкоза человека CEM (10000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл фентанила отдельно или в дополнение к 0,02 мкг/мл доксорубицина. Через 48 ч и 72 ч количественные характеристики апоптоза измеряли проточной цитометрией.
Результаты показывают, что фентанил успешно индуцировал апоптоз более чем в 85% клеток через 72 ч, при сочетании 30 мкг/мл фентанила с 0,02 мкг/мл доксорубицина (смотри фиг. 14).
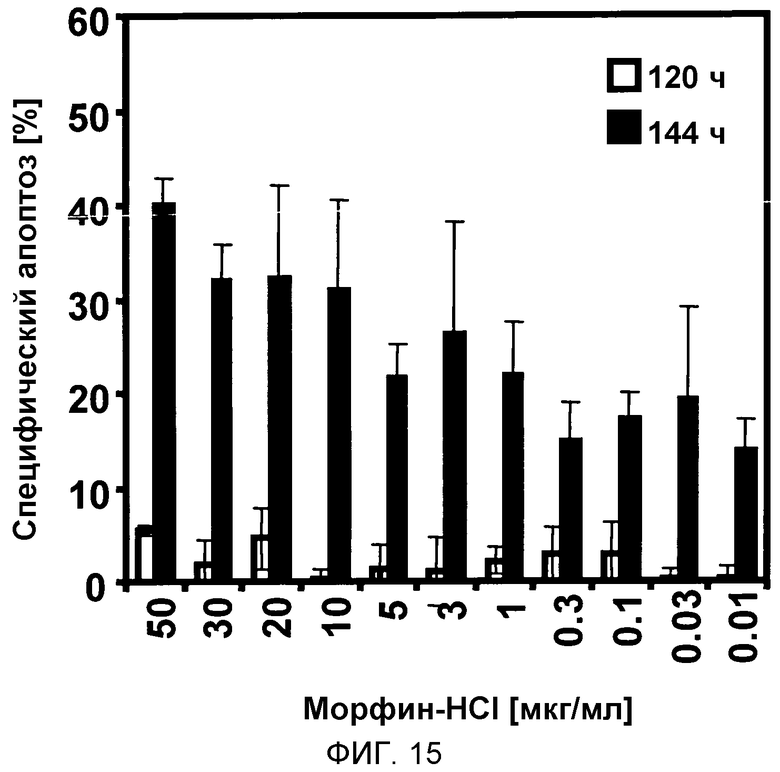
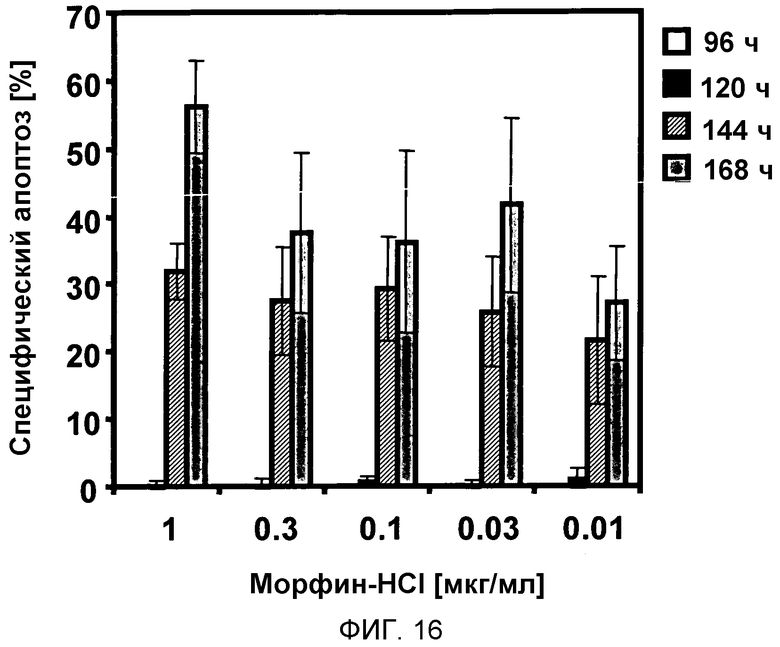
ПРИМЕР 8: Индукция апоптоза в лейкозных клетках с использованием морфина
Морфин успешно использовали отдельно и в сочетании с доксорубицином для индукции апоптоза в лейкозных клетках HL-60. Морфин представляет собой опиат, в настоящее время применяемый в качестве анальгетика для лечения боли, связанной со злокачественными опухолями. Морфин является агонистом опиоидного рецептора μ.
В первом исследовании клетки острого миелоидного лейкоза HL-60 (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1, 0,03, 0,01 мкг/мл морфина. Через 120 ч или 144 ч количественные характеристики апоптоза измеряли проточной цитометрией. Результаты показывают, что через 144 ч апоптоз был индуцирован в 40% клеток при применении 40 мкг/мл морфина (смотри фиг. 15).
Во втором исследовании клетки острого миелоидного лейкоза человека HL-60 (5000 клеток/100 мкл) обрабатывали 1, 0,5, 0,3, 0,1, 0,03, 0,01 мкг/мл морфина. Через 96 ч, 120 ч, 144 ч и 168 ч количественные характеристики апоптоза измеряли проточной цитометрией. Результаты показывают, что через 168 ч апоптоз был индуцирован более чем в 50% клеток при применении 1 мкг/мл морфина (смотри фиг. 16).
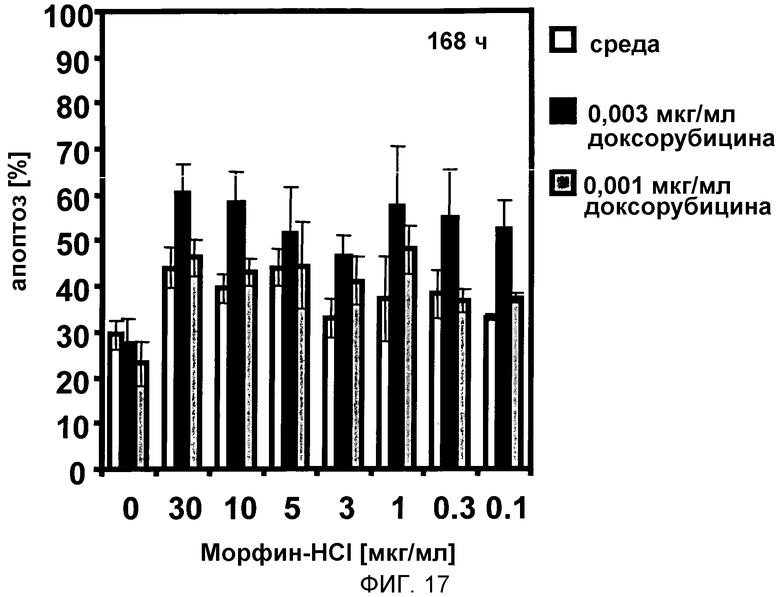
В третьем исследовании клетки острого миелоидного лейкоза человека HL-60 (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл морфина отдельно или в дополнение к 0,003 мкг/мл или 0,001 мкг/мл доксорубицина. Через 168 ч количественные характеристики апоптоза измеряли проточной цитометрией. Результаты показывают, что апоптоз был индуцирован более чем в 50% клеток при применении 1 мкг/мл морфина в сочетании с 0,001 мкг/мл доксорубицина (смотри фиг. 17).
Таким образом, как позволяют предполагать эти исследования, опиат и агонист рецептора μ морфин можно успешно использовать для индукции апоптоза в лейкозных клетках.
Содержание следующих литературных источников, процитированных в настоящем документе, приведено в качестве ссылки:



ФИГУРЫ
Фигура 1:
На фигуре 1 показано, что опиоид метадон эффективно индуцирует апоптоз в лейкозных клетках, но не влияет на нелейкозные, здоровые клетки PBL.
1A: Клетки CEM и HL-60 обрабатывали различными концентрациями метадона, как указано. Через 24 ч (белые столбцы) и 48 ч (черные столбцы) процент апоптотических клеток измеряли анализом гиподиплоидной ДНК. Процент специфической гибели клеток рассчитывали следующим образом: 100 x (погибшие в результате эксперимента клетки (%) - спонтанно погибшие клетки в среде (%))/(100% - спонтанно погибшие клетки в среде (%)). Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
1B: Клетки CEM и HL-60 (2×105 клеток/мл) обрабатывали различными концентрациями метадона, как указано, или оставляли необработанными (Co, контроль). Через 0 ч (белые столбцы), 24 ч (черные столбцы) и 48 ч (заштрихованные столбцы) подсчитывали число клеток в 1 мл. Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
1C: Клетки CEM (черные столбцы) и PBL (белые столбцы) обрабатывали различными концентрациями метадона, как указано. Через 24 ч и 48 ч процент апоптотических клеток измеряли анализом гиподиплоидной ДНК. Процент специфической гибели клеток рассчитывали, как описано на фигуре 1A. Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
Фигура 2:
На фигуре 2 показано, что опиоид метадон индуцирует апоптоз в устойчивых к CD95 (CEMCD95R) и в устойчивых к доксорубицину (CEMDoxoR) клетках с частотой апоптоза, сравнимой с частотой в родительских чувствительных лейкозных клетках CEM.
Лейкозные клетки CEM (черные столбцы), CEMCD95R (белые столбцы) и CEMDoxoR (заштрихованные столбцы) обрабатывали различными концентрациями метадона, как указано. Через 24 ч и 48 ч процент апоптотических клеток измеряли анализом гиподиплоидной ДНК. Процент специфической гибели клеток рассчитывали, как описано для фигуры 1A. Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
Фигура 3:
На фигуре 3 показано, что опиоид метадон индуцирует зависимую от каспаз гибель чувствительных (HL-60, CEM), устойчивых к доксорубицину (CEMDoxoR) и устойчивых к CD95 (CEMCD95R), обладавших множественной лекарственной устойчивостью и являвшихся устойчивыми к апоптозу, лейкозных клеток.
3A: и 3B: Метадон индуцирует активацию каспазы-3 и расщепление PARP в клетках HL-60, CEM, CEMDoxoR и CEMCD95R. A, клетки HL-60, CEM, B, CEMDoxoR, CEMCD95R обрабатывали различными концентрациями метадона, как указано, или оставляли необработанными (Контроль). Через 24 ч и 48 ч проводили анализ Вестерн-блоттингом каспазы-3 и PARP. Активный фрагмент каспазы-3 детектировали на уровне ~19 и 17 кДа и продукт расщепления PARP на уровне ~85 кДа. Равное нанесение белка контролировали посредством антитела против бета-актина.
3C: Ингибирование активации каспазы с помощью zVAD.fmk блокирует индуцированный метадоном апоптоз в клетках CEM и HL-60. Клетки CEM и HL-60 обрабатывали различными концентрациями метадона, как указано, в отсутствие (черные столбцы, среда) или в присутствии (белые столбцы, 50 мкМ zVAD.fmk) 50 мкМ zVAD.fmk. Через 48 ч процент апоптотических клеток измеряли анализом гиподиплоидной ДНК. Процент специфической гибели клеток рассчитывали, как описано для фигуры 1 A. Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
Фигура 4:
На фигуре 4 показано, что опиоид метадон активирует митохондриальный путь в чувствительных (HL-60, CEM), в устойчивых к доксорубицину (CEMDoxoR) и в устойчивых к CD95 (CEMCD95R), обладавших множественной лекарственной устойчивостью и являвшихся устойчивыми к апоптозу, лейкозных клетках.
4A и 4B: Метадон индуцирует активацию каспазы-9, понижающую регуляцию XIAP и понижающую регуляцию Bcl-xL в клетках HL-60, CEM, CEMDoxoR и CEMCD95R.
Клетки HL-60, CEM (4A), CEMDoxoR, CEMCD95R (4B) обрабатывали различными концентрациями метадона, как указано, или оставляли необработанными (Контроль). Через 24 ч и 48 ч проводили анализ Вестерн-блоттингом каспазы-9, XIAP и Bcl-xL. Активный фрагмент каспазы-9 детектировали на уровне ~37 кДа, XIAP детектировали на уровне ~58 кДа и Bcl-xL детектировали на уровне ~30 кДа. Равное нанесение белка контролировали посредством антитела против бета-актина.
Фигура 5:
На фигуре 5 показано, что опиоид метадон индуцирует апоптоз в клетках глиобластомы.
Клетки глиобластомы A172 обрабатывали различными концентрациями метадона, как указано. Через 120 ч (белые столбцы), 144 ч (черные столбцы) и 168 ч (заштрихованные столбцы) процент апоптотических клеток измеряли анализом гиподиплоидной ДНК.
Процент специфической гибели клеток рассчитывали следующим образом: 100×(погибшие в результате эксперимента клетки (%) - спонтанно погибшие клетки в среде (%))/(100% - спонтанно погибшие клетки в среде (%)). Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
Фигура 6:
На фигуре 6 показано, что апоптоз можно успешно индуцировать в клетках глиобластомы с использованием сочетания терапевтических концентраций доксорубицина и низких концентраций метадона.
Клетки глиобластомы A172 (7000 клеток/см2) обрабатывали терапевтической концентрацией 0,1 мкг/мл доксорубицина (доксорубицин, белые столбцы), с низкой концентрацией 1 мкг/мл метадона (метадон, черные столбцы) и 0,1 мкг/мл доксорубицина в дополнение к 1 мкг/мл метадона (доксорубицин + метадон, заштрихованные столбцы). Через 72 ч количественные характеристики апоптоза измеряли проточной цитометрией. Для определения апоптоза клетки лизировали буфером Николетти, содержащим 0,1% цитрат натрия плюс 0,1% Тритон X-100 и 50 мкг/мл йодида пропидия. Окрашенные йодидом пропидия (PI) ядра анализировали проточной цитометрией (FACSCalibur, Becton Dickinson, Heidelberg, Germany).
Фигура 7:
На фигуре 7 показано, что апоптоз можно успешно индуцировать в клетках CEM с использованием опиоида кокаина.
Клетки CEM (белый столбец) и HL-60 (черный столбец) обрабатывали 1000 мкг/мл кокаина. Через 48 ч процент апоптотических клеток измеряли анализом гиподиплоидной ДНК. Процент специфической гибели клеток рассчитывали следующим образом: 100×(погибшие в результате эксперимента клетки (%) - спонтанно погибшие клетки в среде (%))/(100% - спонтанно погибшие клетки в среде (%)). Данные приведены как среднее от трех повторов со стандартным отклонением (SD) менее 10%. Сходные результаты получены в трех независимых экспериментах.
Фигура 8:
На фигуре 8 показано, что D,L-метадон индуцирует гибель клеток в клетках пре-B-ALL (B-клеточного лимфоцитарного лейкоза), выделенных от пациентов ex vivo.
Клетки B-ALL (B-клеточного лимфоцитарного лейкоза) (50000 клеток/100 мкл) обрабатывали 30, 20, 15, 10 мкМ метадона. Через 48 ч (черные столбцы) и 72 ч (белые столбцы) количественные характеристики апоптоза измеряли проточной цитометрией.
Фигура 9:
На фигуре 9 показано, что D,L-метадон в сочетании с флударабином индуцирует гибель клеток в устойчивых клетках CLL (хронического лимфоцитарного лейкоза), выделенных от пациентов ex vivo.
Клетки CLL (хронического лимфоцитарного лейкоза) (50000 клеток/200 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл метадона отдельно (белые столбцы) или в дополнение к 0,1 мкМ флударабина (черные столбцы). Через 24 ч и 48 ч количественные характеристики апоптоза измеряли проточной цитометрией.
Фигура 10:
На фигуре 10 показано, что D,L-метадон в сочетании с флударабином индуцирует гибель клеток в устойчивых клетках CLL (хронического лимфатического лейкоза), выделенных от пациентов ex vivo.
Клетки CLL (хронического лимфатического лейкоза) (50000 клеток/200 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл метадона отдельно (белые столбцы) или в дополнение к 0,1 мкМ флударабина (черные столбцы). Через 24 ч и 48 ч количественные характеристики апоптоза измеряли проточной цитометрией.
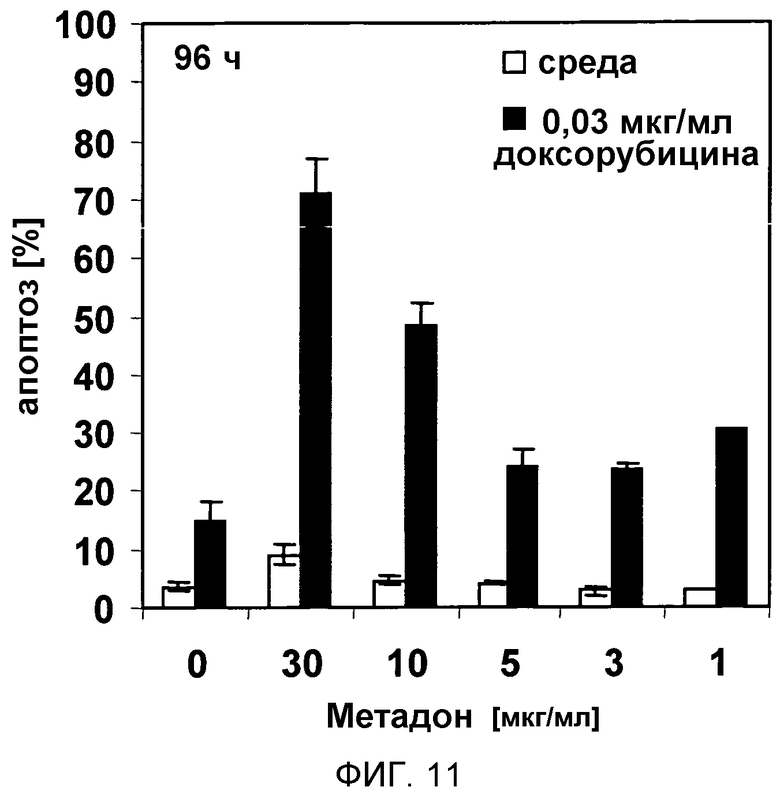
Фигура 11:
На фигуре 11 показана индукция апоптоза в линии клеток B-клеточного лейкоза человека Tanoue, обработанной доксорубицином + метадоном in vitro.
Линию клеток B-ALL (B-клеточного лимфатического лейкоза) Tanoue (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1 мкг/мл метадона отдельно (белые столбцы) или в дополнение к 0,03 мкг/мл доксорубицина (черные столбцы). Через 96 ч количественные характеристики апоптоза измеряли проточной цитометрией.
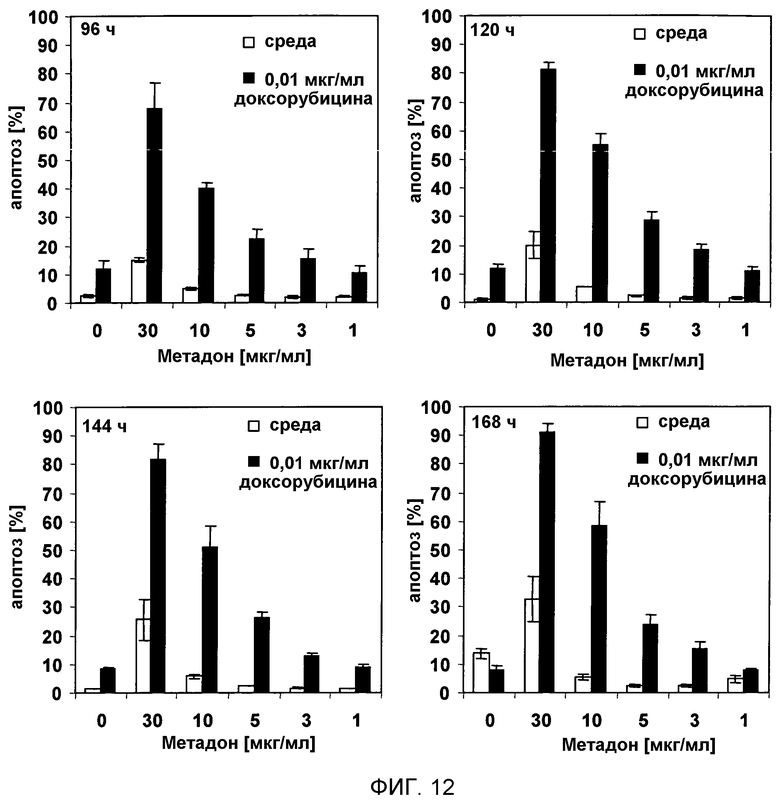
Фигура 12:
На фигуре 12 показана индукция апоптоза в линии предшественников клеток B-клеточного лейкоза человека Nalm6, обработанной доксорубицином + метадоном in vitro.
Линию клеток B-ALL (B-клеточного лимфатического лейкоза) Nalm6 (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1 мкг/мл метадона отдельно (белые столбцы) или в дополнение к 0,01 мкг/мл доксорубицина (черные столбцы). Через 96 ч, 120 ч, 144 ч, 168 ч количественные характеристики апоптоза измеряли проточной цитометрией.
Фигура 13:
На фигуре 13 показана индукция апоптоза в линии клеток острого миелоидного лейкоза человека HL-60, обработанной доксорубицином + бупренорфином in vitro.
Линию клеток острого миелоидного лейкоза HL-60 (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл бупренорфина отдельно (белые столбцы) или в дополнение к 0,003 мкг/мл или 0,001 мкг/мл доксорубицина (черные столбцы). Через 144 ч или 168 ч количественные характеристики апоптоза измеряли проточной цитометрией.
Фигура 14:
На фигуре 14 показана индукция апоптоза в линии клеток T-клеточного лейкоза человека CEM, обработанной доксорубицином + фентанилом in vitro.
Линию клеток T-клеточного лейкоза человека CEM (10000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл фентанила отдельно (белые столбцы) или в дополнение к 0,02 мкг/мл доксорубицина (черные столбцы). Через 48 ч и 72 ч количественные характеристики апоптоза измеряли проточной цитометрией.
Фигура 15:
На фигуре 15 показана индукция апоптоза в линии клеток острого миелоидного лейкоза человека HL-60, обработанной морфином in vitro.
Линию клеток острого миелоидного лейкоза человека HL-60 (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1, 0,03, 0,01 мкг/мл морфина. Через 120 ч (белые столбцы) или 144 ч (черные столбцы) количественные характеристики апоптоза измеряли проточной цитометрией.
Фигура 16:
На фигуре 16 показана индукция апоптоза в линии клеток острого миелоидного лейкоза человека HL-60, обработанной морфином in vitro.
Линию клеток острого миелоидного лейкоза человека HL-60 (5000 клеток/100 мкл) обрабатывали 1, 0,5, 0,3, 0,1, 0,03, 0,01 мкг/мл морфина. Через 96 ч (белые столбцы), 120 ч (черные столбцы), 144 ч (заштрихованные столбцы) и 168 ч (серые столбцы) количественные характеристики апоптоза измеряли проточной цитометрией.
Фигура 17:
На фигуре 17 показана индукция апоптоза в линии клеток острого миелоидного лейкоза человека HL-60, обработанной доксорубицином + морфином in vitro.
Линию клеток острого миелоидного лейкоза человека HL-60 (5000 клеток/100 мкл) обрабатывали 30, 10, 5, 3, 1, 0,5, 0,3, 0,1 мкг/мл морфина отдельно (белые столбцы) или в дополнение к 0,003 мкг/мл (черные столбцы) или 0,001 мкг/мл доксорубицина (серые столбцы). Через 168 ч количественные характеристики апоптоза измеряли проточной цитометрией.
| название | год | авторы | номер документа |
|---|---|---|---|
| НЕРЕПЛИЦИРУЕМЫЕ ПРОИСХОДЯЩИЕ ОТ ВИРУСОВ ЧАСТИЦЫ И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2705556C2 |
| Бифункциональные композиции для лечения рака | 2019 |
|
RU2764175C1 |
| ИНГИБИТОР МИШЕНИ РАПАМИЦИНА В КЛЕТКАХ МЛЕКОПИТАЮЩИХ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2768583C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ PAC-1 | 2016 |
|
RU2720509C2 |
| АКТИВАЦИЯ ПРОКАСПАЗЫ-3 С ПОМОЩЬЮ КОМБИНИРОВАННОЙ ТЕРАПИИ | 2013 |
|
RU2659936C2 |
| АНТИТЕЛО, СЕЛЕКТИВНОЕ В ОТНОШЕНИИ РЕЦЕПТОРА ЛИГАНДА, ИНДУЦИРУЮЩЕГО АПОПТОЗ И СВЯЗАННОГО С ФАКТОРОМ НЕКРОЗА ОПУХОЛИ, И ЕГО ПРИМЕНЕНИЕ | 2002 |
|
RU2313537C2 |
| СПОСОБЫ ПЕРСОНАЛИЗИРОВАННОГО МЕДИЦИНСКОГО ТЕСТИРОВАНИЯ EX VIVO НА ГЕМАТОЛОГИЧЕСКИЕ НОВООБРАЗОВАНИЯ | 2010 |
|
RU2636614C2 |
| КЛЕТОЧНАЯ ЛИНИЯ А4 Т-ЛИМФОБЛАСТНОГО ЛЕЙКОЗА ЧЕЛОВЕКА, ИСПОЛЬЗУЕМАЯ ДЛЯ СКРИНИНГА ПРОТИВООПУХОЛЕВЫХ ПРЕПАРАТОВ | 2004 |
|
RU2267532C1 |
| КОМПОЗИЦИЯ АЛЬФА-ФЕТОПРОТЕИНА И ИНДУКТОРОВ АПОПТОЗА ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2438695C2 |
| ПРИМЕНЕНИЕ ПАРАЗИТОВ И ВНЕКЛЕТОЧНЫХ ВЕЗИКУЛ, ПОЛУЧЕННЫХ ИЗ ПАРАЗИТОВ, ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ | 2020 |
|
RU2814990C2 |
Изобретение относится к медицине, а именно, к онкологии, и касается применения опиоидов для лечения пациентов с устойчивой злокачественной опухолью. Для этого вводят лекарственные средства, содержащие опиоиды способные ингибировать пролиферацию клеток злокачественных опухолей, в эффективных количествах.14з.п.ф-лы, 17ил.,8пр.
1. Применение опиоидов, способных ингибировать пролиферацию клеток злокачественных опухолей, для получения лекарственного средства для лечения пациентов с устойчивой злокачественной опухолью.
2. Применение по п.1, где пациент со злокачественной опухолью обладает по меньшей мере одним видом устойчивости из следующей группы: устойчивость к апоптозу, химиоустойчивость, радиоустойчивость.
3. Применение по п.1, где пациент обладает либо врожденной, либо приобретенной устойчивостью.
4. Применение по п.1, где пациент обладает одним или несколькими из следующих видов устойчивости:
i. устойчивость к апоптозу;
ii. множественная лекарственная устойчивость;
iii. устойчивость к противораковому лекарственному средству;
iv. устойчивость к цитотоксическому лекарственному средству;
v. устойчивость к реакционноспособным формам кислорода;
vi. устойчивость к повреждающим ДНК средствам;
vii. устойчивость к токсическим антителам;
viii. устойчивость к доксорубицину;
ix. одиночная или перекрестная устойчивость, в частности, к одному или нескольким из следующих лекарственных веществ: метотрексат, цитарабин, цисплатин, этопозид, винкристин, паклитаксел (таксол), карбоплатин, тенипозид, дексаметазон, преднизолон, циклофосфамид, ифосфамид, доксорубицин, эпирубицин, даунорубицин, меркаптопурин, флударабин, 5-фторурацил;
х. устойчивость к облучению (например, альфа, бета, гамма или электроны Оже).
5. Применение по любому из п.п.1-4, где пациент страдает по меньшей мере одной из злокачественных опухолей: лейкоз, злокачественная опухоль мозга, меланома, рак поджелудочной железы, рак груди, рак мочевого пузыря, карцинома толстого кишечника, рак печени, рак яичника, рак молочной железы, рак легкого, хронический лейкоз или остеосаркома.
6. Применение по любому из п.п.1-4, где указанный опиоид является способным индуцировать митохондриальный путь апоптоза в клетках злокачественных опухолей.
7. Применение по п.5, где указанный опиоид является способным индуцировать митохондриальный путь апоптоза в клетках злокачественных опухолей.
8. Применение по любому из п.п.1-4, где указанный опиоид принадлежит к группе метадона.
9. Применение по п.5, где указанный опиоид принадлежит к группе метадона.
10. Применение по любому из п.п.1-4, где указанный опиоид представляет собой форму гидрохлорида D-/L-метадона.
11. Применение по п.5, где указанный опиоид представляет собой форму гидрохлорида D-/L-метадона.
12. Применение по любому из п.п.1-4, где указанный опиоид выбран из группы, состоящей из фентанила, бупренорфина и морфина.
13. Применение по п.5, где указанный опиоид выбран из группы, состоящей из фентанила, бупренорфина и морфина.
14. Применение по п.6, где указанный опиоид является способным индуцировать апоптоз посредством по меньшей мере одной из следующих реакций:
i. расщепление каспазы-3 и PARP в клетках опухолей;
ii. расщепление каспазы-9 и понижающая регуляция XIAP;
iii. понижающая регуляция BclXL.
15. Применение по п.1, где указанный опиоид является способным индуцировать апоптоз посредством по меньшей мере одной из следующих реакций:
i. расщепление каспазы-3 и PARP в клетках опухолей;
ii. расщепление каспазы-9 и понижающая регуляция XIAP;
iii. понижающая регуляция BclXL.
| ПРОИЗВОДНЫЕ ДИБЕНЗ(B, F) (1,4)ОКСАЗЕПИН-11-ОНА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ УМЕНЬШЕНИЯ МУЛЬТИЛЕКАРСТВЕННОЙ РЕЗИСТЕНТНОСТИ РАКОВЫХ КЛЕТОК К ЦИТОТОКСИЧЕСКОМУ ЛЕКАРСТВУ | 1992 |
|
RU2086545C1 |
| WO 2006124413 A2 23.11.2006 | |||
| СЕНЬКОВА А.В | |||
| и др | |||
| "Моделирование синдрома множественной лекарственной устойчивости" Бюллетень сибирской медицины, 2008, Приложение 3,[ найдено 03.06.2013], найдено из Интернет: ssmu.ru›bull/08/p3/18.pdf | |||
| YOSHIDA A et al | |||
| “Opioid analgesic-induced apoptosis and caspase-independent | |||

