Изобретение относится к биохимии, конкретнее к биологически активным пептидам, обладающим антиапоптотической активностью, которые могут найти применение в медицине и фармакологии для профилактики и/или лечения функциональных нарушений, возникающих при патологическом развитии процесса апоптоза.
Апоптоз представляет собой регулируемый способ программируемой гибели клеток, который происходит в нормальных физиологических условиях [R.S. Hotchkiss, A. Strasser, J.E. McDunn, Р.Е. Swanson, Cell death, N Engl J. Med. 361 (2009) 1570-1583; A.H. Wyllie, «Where, о death, is thy sting?» a brief review of apoptosis biology, Mol. Neurobiol. 42 (2010) 4-9; S. Fulda, A.M. Gorman, O. Hori, A. Samali, Cellular stress responses: cell survival and cell death, Int. J. Cell Biol. (2010) 214074]. Апоптоз крайне важен для выживания многоклеточных организмов, позволяя избавляться от поврежденных или инфицированных клеток, которые могут влиять на нормальное функционирование [A. Ben Younes, М.С. Maiuri, S. Lavandero, G. Kroemer, Senescence, apoptosis or autophagy? When a damaged cell must decide its path-a mini-review, Gerontology 54 (2008) 92-99; K. Labbe, M. Saleh, Cell death in the host response to infection, Cell Death Differ. 15 (2008) 1339-1349]. Апоптоз чаще всего участвует в процессах нормального возобновления клеток, тканевого гомеостаза, эмбриогенеза, развитии нервной системы и эндокринзависимой атрофии тканей. Клетки, подвергающиеся апоптозу, проявляют характерные морфологические и биохимические особенности. Эти особенности включают агрегацию хроматина, ядерную и цитоплазматическую конденсацию, разделение цитоплазмы и ядра связанные с мембраной пузырьки (апоптотические тела), которые содержат рибосомы, морфологически интактные митохондрии и ядерный материал. In vivo эти апоптотические тела быстро распознаются и фагоцитируются либо макрофагами, либо соседними эпителиальными клетками. Благодаря этому эффективному механизму удаления апоптотических клеток in vivo не возникает воспалительной реакции. In vitro апоптотические тела, а также оставшиеся фрагменты клеток в конечном итоге набухают и лизируются. Поддержание баланса между пролиферацией клеток и клеточной смертью имеет решающее значение для гомеостаза тканей и органов. В общих чертах сдвиг в сторону клеточной пролиферации приводит к новообразованиям, в то время как избыточная гибель клеток вызывает воспалительные реакции.
В настоящее время относительно хорошо изучены два пути развития апоптотического процесса: внешний и внутренний, они стимулируются внеклеточными и внутриклеточными сигналами соответственно [L. Duprez, Е. Wirawan, Т. Vanden Berghe, P. Vandenabeele, Major cell death pathways at a glance, Microbes Infect. 11 (2009) 1050-1062; M.R. Sprick, H. Walczak, The interplay between the Bcl-2 family and death receptor-mediated apoptosis, Biochim. Biophys. Acta 1644 (2004) 125-132]. Внешний путь опосредуется подгруппой рецепторов фактора некроза опухоли (TNFR) - надсемейством, которое включает TNFR, Fas и TRAIL. Активация этих так называемых «рецепторов смерти» приводит к рекрутированию и активации инициирующих каспаз, таких как каспазы 8 и 10. Процесс включает образование и активацию таких комплексов, как смерть индуцирующий сигнальный комплекс (DISC). Это приводит к активации эффекторной каспазы, как правило каспазы 3. Активированная каспаза 3 отвечает за расщепление ряда так называемых субстратов смерти, это приводит к появлению характерных маркеров апоптотической клетки, таких как фрагментация ДНК, ядерная фрагментация, мембранный блеббинг и другие морфологические и биохимические изменения. Недавние данные свидетельствуют о еще большей сложности и разнообразии внешних путей развития апоптоза, которые могут включать также перекрестную активацию других апоптотических путей, таких как внутренние пути апоптоза, а также некротические пути [R.S. Whelan, V. Kaplinskiy, R.N. Kitsis, Cell death in the pathogenesis of heart disease: mechanisms and significance, Annu. Rev. Physiol. 72 (2010) 19-44].
Автономный, или внутренний, путь в основном регулируется митохондриями [S. Gupta, G.E. Kass, Е. Szegezdi, В. Joseph, The mitochondrial death pathway: apromising therapeutic target in Diseases, J. Cell. Mol. Med. 13 (2009) 1004-1033; J.K. Brunelle, A. Letai, Control of mitochondrial apoptosis by the Bcl-2 family, J. Cell Sci. 122 (2009) 437-441; L. Galluzzi, E. Morselli, О. Kepp, I. Vitale, A. Rigoni, E. Vacchelli, M. Michaud, H. Zischka, M. Castedo, G. Kroemer, Mitochondrial gateways to cancer, Mol. Aspects Med. 31 (2010) 1-20]. Наиболее хорошо изученная форма внутреннего пути апоптоза инициируется стресс-опосредованным выделением цитохрома с из митохондрий, что приводит к формированию апоптосомы. Затем апоптосома активирует инициирующую каспазу, обычно каспазу 9, что приводит к активации каспазы 3. Это приводит к развитию того же типа апоптотического ответа, какой наблюдается при внешнем пути активации апоптоза. В ответ на апоптотические стимулы, проапоптотические члены семейства белков Вс1-2 (Вах и Bak) активируются и действуют на митохондрии, индуцируя выделение цитохрома С. Другие проапоптотические белки, также выделяемые митохондриями, такие как Smac/Diablo (второй митохондриальный активатор каспаз/прямой IAP-связывающий белок с низкой pi), сериновая протеаза Omi/HtrA2, эндонуклеаза G (EndoG) и апоптоз-индуцирующий фактор (AIF) [А.В. Gustafsson, R.A. Gottlieb, Heart mitochondria: gates of life and death, Cardiovasc. Res. 77 (2008) 334-343; D. Brenner, T.W. Mak, Mitochondrial cell death effectors, Curr. Opin. Cell Biol. 21 (2009) 871-877; C. Wang, R.J. Youle, The role of mitochondria in apoptosis, Annu. Rev. Genet. 43 (2009) 95-118]. Эти примеры демонстрируют центральную роль митохондрий в регулируемом и сложном процессе запрограммированной гибели клеток [L. Galluzzi, Е. Morselli, О. Керр, G. Kroemer, Targeting post-mitochondrial effectors of apoptosis for neuroprotection, Biochim. Biophys. Acta 1787 (2009) 402-413; W.C. Cheng, K.M. Leach, J.M. Hardwick, Mitochondrial death pathways in yeast and mammalian cells, Biochim. Biophys. Acta 1783 (2008) 1272-1279; P. Nagley, G.C. Higgins, J.D. Atkin, P.M. Beart, Multifaceted deaths orchestrated by mitochondria in neurones, Biochim. Biophys. Acta 1802 (2010) 167-185]. Как упоминалось выше, активация митохондриального пути может также возникать после активации внешнего пути апоптоза. Это происходит через опосредованное каспазой 8 расщепление члена проапоптотического семейства Вс1-2 - белка Bid -приводящее к его активированной форме tBid [J.K. Brunelle, A. Letai, Control of mitochondrial apoptosis by the Bcl-2 family, J. Cell Sci. 122 (2009) 437-441].
На апоптоз клетки может влиять широкий спектр регуляторных стимулов. Отклонения в способности клеток к выживанию способствуют патогенезу ряда заболеваний человека, включая рак, вирусные инфекции, аутоиммунные заболевания, нейродегенеративные расстройства и СПИД. Средства, специфически меняющие апоптотический порог, могут иметь важное значение для лечения некоторых из этих заболеваний [Thompson, С. Apoptosis in the Pathogenesis and Treatment of Disease. Science, 1995, 267(5203):1456-1462]. Такие заболевания, как, например, рак, характеризуются накоплением клеток из-за повышения их выживаемости, связанного с ингибированием апоптоза. Другие заболевания характеризуются увеличением уровня апоптотической гибели клеток, например, СПИД, нейродегенеративные расстройства, такие как болезнь Альцгеймера, болезнь Паркинсона, или инфаркт миокарда и инсульт, а также токсин-индуцированные заболевания печени. Чрезмерная гибель клеток может быть вызвана действием приобретенных или генетических факторов, которые усиливают накопление сигналов, вызывающих апоптоз, или снижают пороговый уровень таких сигналов. Как в нормальном, так и в патологическом апоптозе участвуют одни и те же молекулярные механизмы, ведущие к дегенерации нейронов, однако основное различие заключается в недостаточном контроле над запуском апоптотических процессов в патологически измененной нервной ткани. Поэтому при неврологических заболеваниях важно снижать активность апоптотических процессов.
В настоящее время сигнальный путь активируемый протеинкиназой В (Akt/PKB) признан одним из наиболее важных путей регуляции выживаемости клеток. Активация этого пути позволяет клеткам противостоять апоптотическим стимулам [Yao R., Cooper G.M., Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor, 1995, Science, 267:2003-2006]. Akt/PKB, активированная Р13-киназой, является основным медиатором выживаемости клеток. Akt/PKB оказывает прямое влияние на сигнальные пути апоптоза, например, таргетирует проапоптотический белок семейства Bcl-2 - BAD. Она также изменяет транскрипционный ответ на апоптотические стимулы, например, воздействуя на факторы Forkhead и активность белков семейства р53.
Известно несколько типов ингибиторов апоптоза. Физиологические ингибиторы включают, например, факторы роста, внеклеточный матрикс, лиганд CD40, нейтральные аминокислоты, цинк, эстроген и андрогены. Известны такие вирусные гены, ингибирующие апоптоз, как аденовирусный Е1В, crmA вируса коровьей оспы, BHRF1 и LMP-1 вируса Эпштейна - Барр и герпесвирусный гамма(1)34.5. В качестве фармакологических агентов, которые ингибируют апоптоз, используют ингибиторы кальпаина, ингибиторы цистеиновой протеазы и опухолевые промоторы РМА, фенобарбитал и альфа-гексахлорциклогексан [Thompson, С. Apoptosis in the Pathogenesis and Treatment of Disease. Science, 1995, 267(5203):1456-1462]. Однако эти ингибиторы не подходят для лечения заболеваний, связанных с увеличением апоптоза, поскольку все они являются патогенными.
Из заявки US 2014323394 А1 (2014) известно использование трипептида (L-Lys)-(D-Pro)-(L-Thr) (KdPT) или его фармацевтически приемлемых солей для терапевтического, профилактического или косметического лечения заболевания с повышенным апоптозом, причем лечение имеет антиапоптотический эффект.
Из заявки JP 2002316929 А (2002) известны L-серин или глицин, способствующие экспрессии гена Bcl-w и используемые в качестве антиапоптотического агента и средства для ухода за кожей, а также препарат для наружного применения, состоящий из L-серина и/или глицина. Препарат может применяться для уменьшения симптомов старения кожи или слизистой оболочки или для заживления ран.
Из патента RU 2582247 (2016) известны биологически активные фрагменты из 17-24 последовательных аминокислот белка Daxx, включающих KKSRKEKKQTGSGPLG, а также их пептидомиметики и конъюгаты с проникающим в клетки пептидом. Упомянутые фрагменты позволяют эффективно ингибировать апоптоз клеток, в частности апоптоз клеток, опосредованный рецептором Fas, и могут быть использованы в медицине для лечения острого инфаркта миокарда, церебрального инфаркта, при трансплантации органов, операциях на сердце, острых нарушениях кровообращения, реперфузионных повреждениях и ишемии.
Из патента RU 2586772 (2014) известна фармацевтическая композиция для ингибирования апоптоза нейронов или нейродегенерации, включающая терапевтически эффективное количество соединения, выбранного из группы, состоящей из замещенного производного азола, его фармацевтически приемлемых солей, изомеров замещенного производного азола, сольватов замещенного производного азола, и их сочетания; и фармацевтически приемлемый носитель. Композиция может быть использована для изготовления лекарственного средства для предупреждения или лечения нейродегенеративных заболеваний
Из статьи: Бердалин А.Б., Гаврилова С.А., Голубева А.В., Буравков С.В., Кошелев В.Б. Влияние Семакса на апоптотическую гибель кардиомиоцитов крыс при необратимой ишемии и ишемии-реперфузии // Российский медико-биологический вестник им. академика И.П. Павлова. 2011;19(2):2-2 известно, что имеющийся на рынке пептидный препарат Семакс, представляющий собой модифицированный фрагмент АКТГ, способен снижать апоптотический индекс в областях, непосредственно не затронутых ишемией в двух моделях инфаркта миокарда: необратимой ишемии и ишемии-реперфузии.
Однако все описанные средства предназначены для введения пациенту в то время, когда процесс апоптоза уже запущен, и возможностей повлиять на процесс апоптотической гибели клеток гораздо меньше. Поэтому актуальным является поиск новых средств, предназначенных для предотвращения и профилактики апоптотической гибели клеток, повышения их устойчивости к патологическому процессу апоптоза, тесно связанному с нейродегенеративными заболеваниями.
Задачей, на решение которой направлено настоящее изобретение, было расширение ассортимента эффективных средств, обладающих антиапоптотической активностью, средств, которые могут быть использованы для профилактики развития патологического апоптоза.
Техническая задача была решена в ходе исследования пептидов, входящих в состав природных гормонов, регулирующих жизнедеятельность высших организмов. Поставленная задача решается тем, что предложен синтетический пептид КК-1, проявляющий антиапоптотическую активность и имеющий формулу CH3CO-(D-Lys)-Lys-Arg-Arg-NH2.
Сущность изобретения заключается в том, что экспериментальным путем было установлено, что указанный пептид обладает выраженными антиапоптотическими свойствами. Заявленный пептид способен снижать уровень маркеров цитолиза (NSE и S-100), рилизинг которых прямо коррелируют с объемом очага некротической гибели нейронов при ишемии головного мозга. Снижение уровня фрагментированной ДНК, которое наблюдается при курсовом введении заявленного пептида в течение 5 суток до реперфузионного повреждения головного мозга, указывает на его антиапоптическую активность при использовании в качестве профилактического средства. Установлено также, что антиапоптотическая активность КК-1 опосредована активацией в интактных клетках киназы Akt1, способствующей их выживанию через ингибирование ряда ключевых элементов активации апоптоза, в частности, фосфорилированных форм каспазы 8 и белков Bad и р53. Вполне вероятно, что пептид КК-1 является позитивным регулятором внутриклеточного сигнального пути PI3K/Akt/mTOR.
Изобретение иллюстрируется следующими графическими материалами
На Фиг. 1. представлен график изменения усредненного клеточного индекса клеток линии SH-SY5Y от времени инкубирования с 5 мкМ активатора апоптоза (нижний график) и при действии комплекса 5 мкМ активатора апоптоза и 1 мкМ пептида КК-1 (верхний график). Момент добавления веществ соответствует 22 часам.
На Фиг. 2. представлена ящичная диаграмма определения флуоресценции активной формы каспазы-8 в лизатах клеток SH-SY5Y через 24 часа после добавления активатора апоптоза, КК-1 и при их совместном действии.
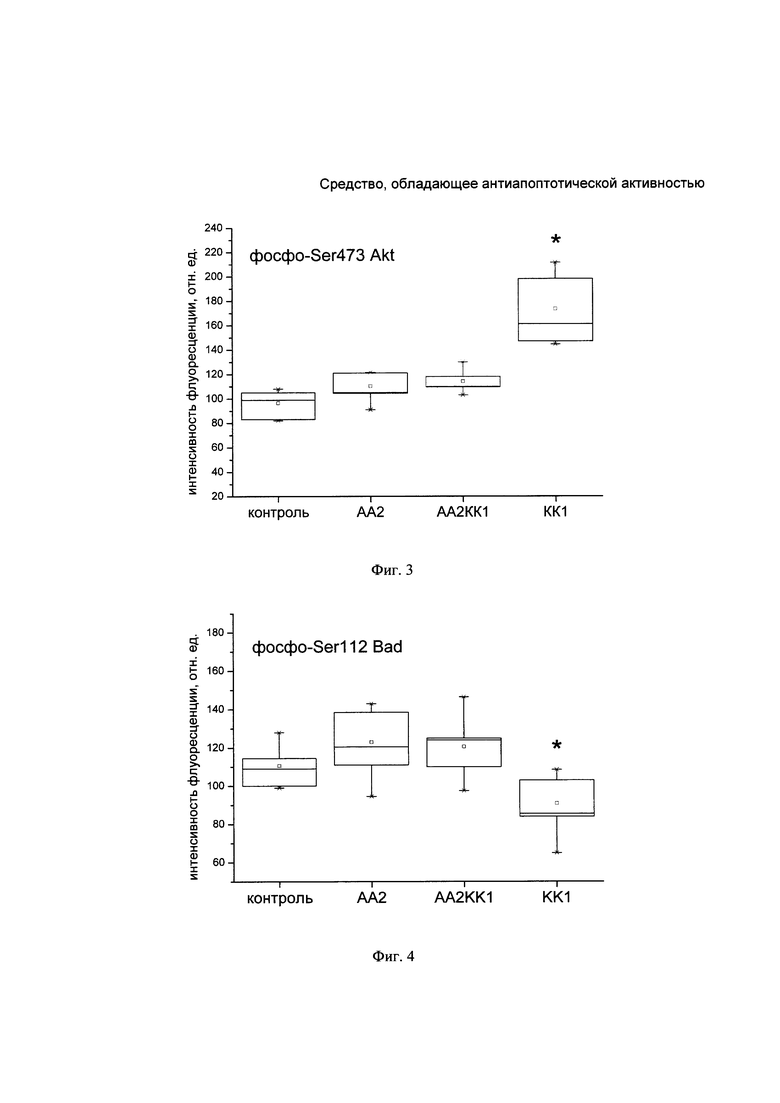
На Фиг. 3. представлена ящичная диаграмма определения флуоресценции активной формы Akt1 в лизатах клеток SH-SY5Y при действии активатора апоптоза, КК-1 и при их совместном действии.
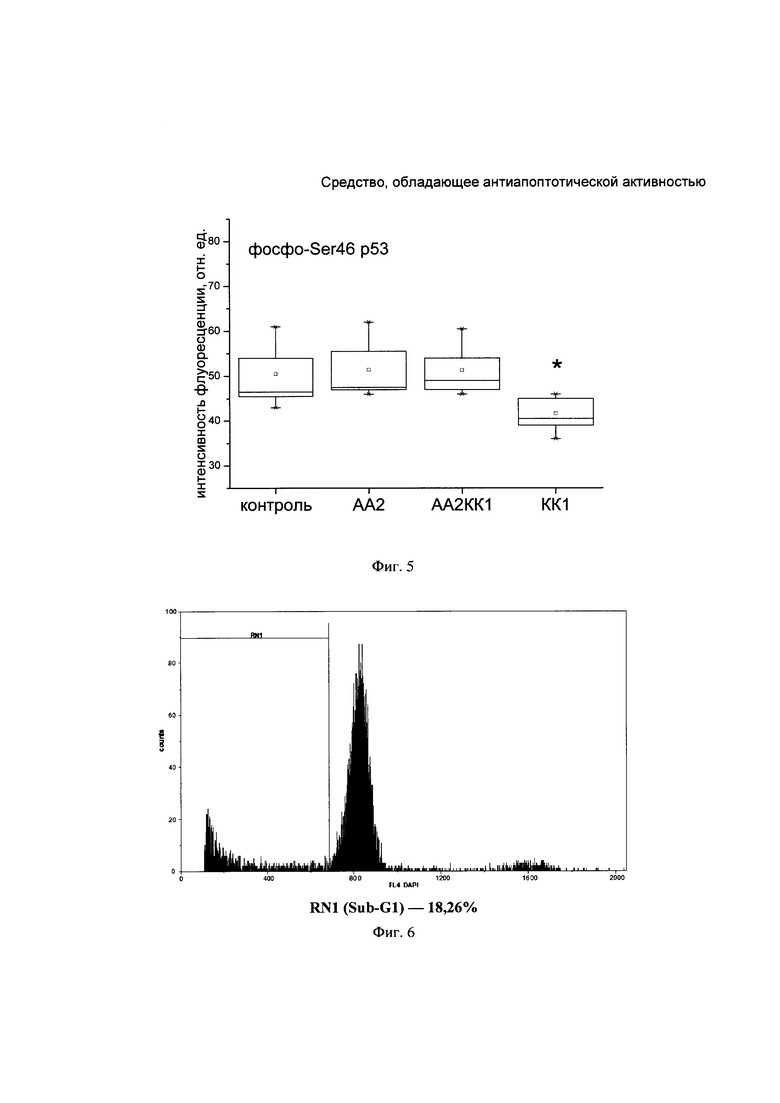
На Фиг. 4. представлена ящичная диаграмма определения флуоресценции активной формы Bad в лизатах клеток SH-SY5Y при действии активатора апоптоза, КК-1 и при их совместном действии.
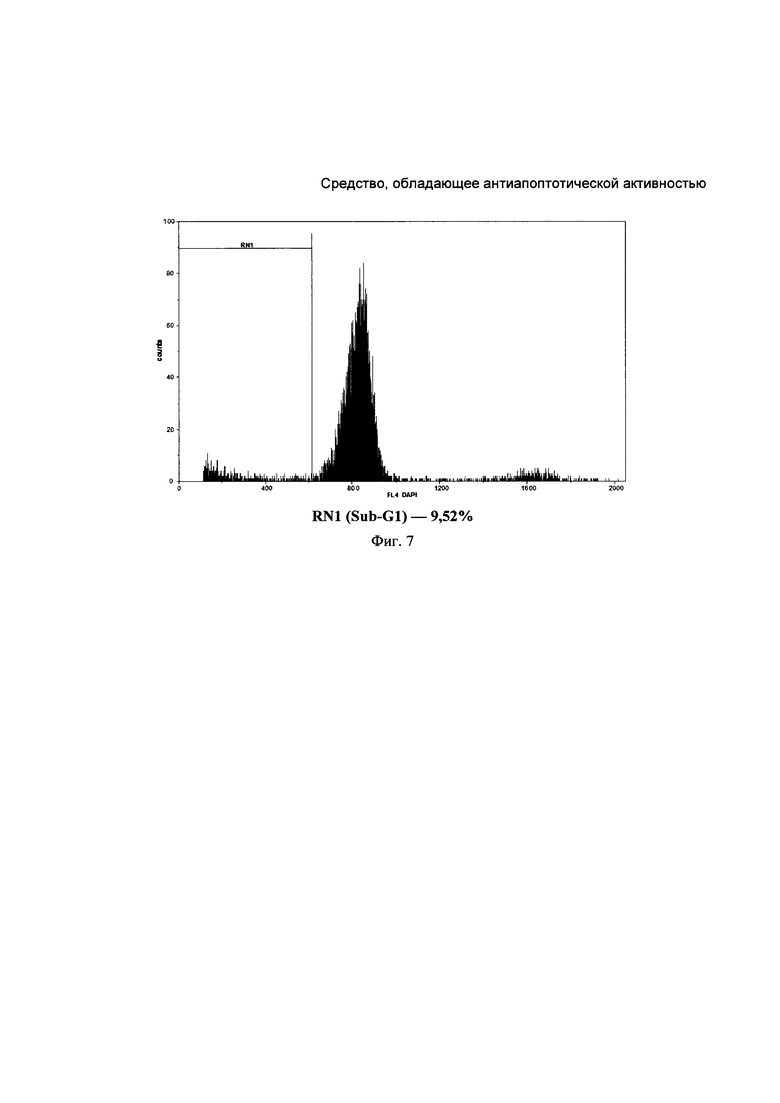
На Фиг. 5. представлена ящичная диаграмма определения флуоресценции активной формы белка р53 в лизатах клеток SH-SY5Y при действии активатора апоптоза, КК-1 и при их совместном действии.
На Фиг. 6. представлена типичная гистограмма распределения уровня фрагментированной ДНК в ядрах нейронов коры головного мозга крысы из группы контрольной патологии. Фрагментация ДНК составила 17,94%.
На Фиг. 7. представлена типичная гистограмма распределения уровня фрагментированной ДНК в ядрах нейронов коры головного мозга крысы из группы, получавшей КК-1. Фрагментация ДНК составила 6,78%.
Сущность и промышленная применимость изобретения поясняются следующими примерами.
Пример 1. Синтез пептида.
Пептидный гомолог фрагмента АКТГ15-18 CH3CO-(D-Lys)-Lys-Arg-Arg-NH2 под шифром КК-1 синтезирован в ФГУП «Государственный научно-исследовательский институт особо чистых препаратов» ФМБА России (Санкт-Петербург). Синтез пептида CH3CO-(D-Lys)-Lys-Arg-Arg-NH2 был проведен твердофазным методом с использованием Boc/Bzl стратегии на синтезаторе пептидов Coupler-250. Для временной защиты а-аминофункции использовали трет-бутилоксикарбонильную группу. Для блокирования боковых радикалов аргинина и лизина использовали мезитиленсульфонильную и 2-хлорбензилоксикарбонильную группировки соответственно.
В качестве нерастворимой матрицы был использован 4-метил-бензгидриламинополимер (m-BHA-resin) с начальной емкостью 1 мМ/г. 2,9 г ВНА-полимера поместили в реакционный сосуд синтезатора и проводили наращивание полипептидной цепи по следующей программе присоединения одного аминокислотного остатка (Таблица 1). Удаление временной защитной группы проводили трифторуксусной кислотой, нейтрализацию - 5% раствором диизопропилэтиламина в диметилформамиде. Конденсацию осуществляли методом активированных эфиров, используя трехкратный избыток соответствующего активированного компонента. Предварительное активирование карбоксильной компоненты проводили в течение 30 минут, используя оксибензотриазол и диизопропилкарбодиимид. Для контроля полноты протекания реакции конденсации использовали нингидриновый тест. После присоединения аминокислотных остатков, соответствующих последовательности синтезируемого пептида, проводили ацетилирование N-концевой аминогруппы, используя 40 эквивалентов уксусного ангидрида (5 мл) в 25 мл диметилформамида. Затем пептидил-полимер высушивали в эксикаторе до постоянного веса. Выход защищенного пептидил-полимера 96%. При такой обработке удалялись все боковые защитные группировки, и пептид отщеплялся от высокомолекулярной матрицы, время деблокирования составляло один час.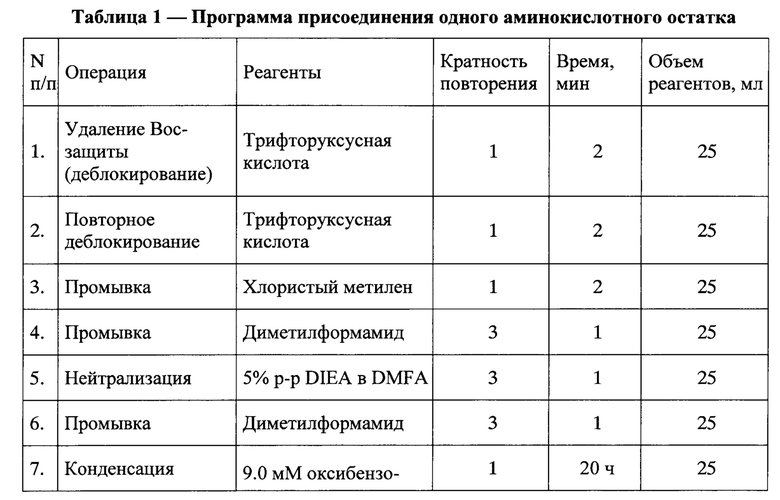
Удаление боковых защитных группировок и отщепление пептида от нерастворимой матрицы проводили под действием безводного жидкого фтористого водорода в присутствии скавенджеров. 3,5 г пептидил-полимера обрабатывали 27 мл фтористого водорода и 3 мл м-крезола при 0°С и перемешивании на магнитной мешалке в течение 1 часа. Затем фтористый водород упаривали и суспендировали остаток в 50 мл 50% раствора уксусной кислоты в воде. Раствор фильтровали и экстрагировали 3 раза по 10 мл эфиром. После лиофилизации водного раствора получено 1,4 г грубого продукта.
Синтезированный пептидный препарат был очищен с помощью препаративной обращенно-фазовой жидкостной хроматографии на колонке Prep-Nova-Pak HR С18, 6 мкм, 60  , (19×300 мм), Waters, США и охарактеризован данными аминокислотного анализа (аминокислотный анализатор Alpha Plus, LKB, Швеция) и масс-спектрометрии (масс-спектрометр Per Sepetive Biosystems «Voyager-DE Biospectrometry Workstation))). Аминокислотный состав конечного продукта соответствовал теоретическому. Чистота по данным аналитической ВЭЖХ составила не менее 98%. Выход конечного продукта - 50%.
, (19×300 мм), Waters, США и охарактеризован данными аминокислотного анализа (аминокислотный анализатор Alpha Plus, LKB, Швеция) и масс-спектрометрии (масс-спектрометр Per Sepetive Biosystems «Voyager-DE Biospectrometry Workstation))). Аминокислотный состав конечного продукта соответствовал теоретическому. Чистота по данным аналитической ВЭЖХ составила не менее 98%. Выход конечного продукта - 50%.
Перевод пептида в ацетатную форму осуществляли по следующей процедуре: 450 мг полученного пептида в форме трифторацетатной соли растворили в 45 мл деионизованной воды, добавили смолу IRA-68 в ацетатной форме и перемешивали в течение 30 минут. Затем полимер отфильтровали, промыли водой, фильтрат и промывные воды объединили и лиофилизировали. Получено 350 мг КК-1 в ацетатной форме (74%).
Пример 2. Влияние КК-1 на уровень маркеров апоптоза.
Клетки нейробластомы человека линии SH-SY5Y (ATCC,CRL-2266) культивировали во флаконах в среде DMEM/F12 (Биолот, Санкт-Петербург) с добавлением 10% эмбриональной сыворотки крупного рогатого скота и антибиотиков стрептомицина-пенициллина при температуре 37°С в СО2-инкубаторе в атмосфере 5% CO2.
Для определения интегрального клеточного ответа с помощью оборудования xCelligence RTCA (AceaBioscience, США) 20 тыс.клеток вносили в лунку специализированного планшета, позволяющего определять клеточный индекс в режиме реального времени, и культивировали в полной среде DMEM/F12. На следующий день после пассажа к клеткам добавляли равный объем среды, содержащей 2-кратные концентрации активатора апоптоза 2 и КК-1. Мониторинг клеточного индекса проводили в течение 3 дней после добавления ксенобиотика.
Для определения содержания молекулярных маркеров апоптоза клетки культивировали в 24-луночных планшетах, промывали на следующий день после пассажа и добавляли среду, содержащую активатор апоптоза 2 и КК-1. После 24-часовой инкубации определяли маркеры апоптоза в клеточных лизатах с использованием многопараметрической иммунофлуоресцентной технологии Luminex хМАР.
Для получения клеточных лизатов в лунку 24-луночного планшета добавляли 200 мкл лизирующего буфера (Merck/Millipore) с добавлением набора ингибиторов протеаз (Complete, Roche, Швейцария), ингибиторов фосфатаз (Phosphatase Inhibitor Cocktail 2, Sigma, США) и 25 Е/мл фермента бензоназы (Sigma, США) на 10 мин.
Экстракт центрифугировали при 20000 g при 40°С в течение 20 мин. В супернатанте определяли концентрацию белка по методу Лоури с использованием микропланшетного спектрофотометра Epoch (Biotek, США). Для дальнейшего анализа концентрацию белка в экстракте доводили до 0,4 мг/мл.
Анализ активированных фосфорилированных или протеолитически фрагментированных белков - маркеров апоптоза проводили с помощью иммунофлуоресцентного метода по технологии Luminex хМАР. Для анализа использовали набор реактивов для определения ранних маркеров апоптоза (7-Р1ех MILLIPLEX MAP Early Apoptosis Magnetic Bead Kit, Кат. №48-669 MAG, Merck/Millipore, США), который позволяет определять активированные фосфорилированные формы следующих белков: Akt1 (Ser473), р 53 (Ser46), BAD (Ser112), Bcl-2 (Ser70), JNK (Thr183/Tyr185), а также активных форм каспазы 8, гидролизованной по Asp384, и каспазы 9, гидролизованной по Asp315.
В культуры клеток нейробластомы в логарифмической фазе роста добавляли активатор апоптоза 2 в диапазоне значений конечной концентрации 1-20 мкМ, пептид КК-1 в конечной концентрации 1 мкМ, а также активатор апоптоза 2 совместно с КК-1.
Данные, полученные в ходе экспериментальных исследований, были статистически обработаны в программе Bio-Plex Data Pro Plus. Оценку различий средних значений проводили с использованием однофакторного дисперсионного анализа (ANOVA).
Полученные результаты показали, что КК-1 проявляет цитопротекторный эффект на фоне действия активатора апоптоза 2 (1-[(3,4-дихлорфенил)метил]-1H-индол-2,3-диона), который активирует апоптоз с IC50 4-9 мкМ, обеспечивая цитохром С-зависимую олигомеризацию белка Apaf-1 в зрелую апоптосому.
На Фигуре 1 представлен график изменения клеточного индекса от времени при действии на клетки активатора апоптоза (нижний график) и при действии комплекса активатор апоптоза - КК-1 (верхний график).
Активатор апоптоза в концентрации 5 мкМ способствует накоплению активной формы протеазы каспазы-8 (Фигура 2). Добавление 1 мкМ пептида в культуральную среду снижало уровень активной формы фермента до контрольного уровня. В интактных клетках КК-1 проявляет выраженную тенденцию к снижению уровня активной формы каспазы-8.
КК-1 достоверно увеличивает уровень активной формы белка Akt1 в интактных клетках (Фигура 3). На фоне активатора апоптоза содержание активной формы Akt1 в лизатах клеток SH-SY5Y также достоверно выше контрольного уровня.
В то же время КК-1 достоверно снижает уровень активной формы белка Bad и р53 относительно контрольного уровня (Фигуры 4 и 5).
Таким образом, КК-1 в концентрации 1 мкМ проявляет цитопротекторные и антиапоптотические свойства в клетках нейробластомы человека линии SH-SY5Y при действии 5 мкМ активатора апоптоза 2. Уровень клеточного индекса при действии комплекса активатор апоптоза 2 - пептид выше, чем при действии только активатора апоптоза 2. КК-1 снижает уровень активной формы каспазы-8, накопленной при действии активатора апоптоза 2, в клеточных лизатах SH-SY5Y. Кроме того, пептид значительно усиливает накопление в интактных клетках активной формы белка Akt1, фосфорилированной по Ser473, и снижает уровни активных форм белков Bad, фосфорилированного по Ser112, и р53, фосфорилированного по Ser46. Можно предположить, что в основе нейропротекторного действия КК-1 лежит антиапоптотическая активность, опосредованная активацией киназы Akt1, которая вовлечена в регуляцию роста и выживаемости клеток. Это свидетельствует о том, что заявляемый пептид КК-1 может быть использован в качестве профилактического средства при нейродегенеративных заболеваниях, тесно связанных с развитием патологического апоптоза.
Пример 3. Моделирование острого нарушения мозгового кровообращения (ОНМК): экспериментальная модель ишемии-реперфузии.
Эксперименты проводили на белых беспородных самцах крысы массой 160-170 г. Экспериментальную модель ишемии-реперфузии (ИР) воспроизводили путем наложения клипс на обе общие сонные артерии продолжительностью 20 минут под пропофоловым наркозом (60 мг/кг в/б).
Пример 4. Влияние КК-1 на уровень нейрон-специфической энолазы NSE и белка S-100.
20 белых беспородных самцов крысы массой 160-170 граммов рандомизировали на 4 группы по 5 особей:
1 группа - ложнооперированные;
2 группа - контрольная патология (КП);
3 группа - животные, получавшие цитиколин (Ferrer International S.A., Испания) в дозе 250 мг/кг внутрибрюшинно;
4 группа - животные, получавшие КК-1 в дозе 0,02 мг/кг интраназально.
ОНМК моделировали, как описано в примере 3.
КК-1 и препарат сравнения вводили один раз в день в -5 ÷ -1 дни до ОНМК. Псевдооперированные крысы претерпевали все этапы оперативного вмешательства, кроме наложения клипс. Животным группы КП интраназально вводили физиологический раствор.
Активность NSE и содержание белка S-100 в сыворотке крови измеряли на 4 сутки ИР (острая фаза ишемического повреждения) при помощи метода твердофазного иммуноферментного анализа с использованием наборов NSE ELISA KIT (DAI, США) и S 100 ELISA KIT (Fujirebio Diagnostics Inc., Швеция) на приборе компании «Hipson» (Чешская Республика).
Нейрон-специфическая энолаза (NSE) и белок S-100 являются специфическими маркерами нейродеструкции, возникающей на фоне ишемического и реперфузионного повреждения головного мозга [Буреш Я. Методики и основные эксперименты по изучению мозга и поведения / Я. Буреш, О. Бурешова, Дж. П. Хьюстон; пер. с англ. Е.Н. Живописцевой; под ред. проф. А.С. Батуева. - М.: Высшая школа, 1991.-399 с].
Влияние КК-1 на динамику экспрессии этих нейрональных маркеров позволяет оценить выраженность возникающего при ишемии некроза ГМ, степень глиоцитарного замещения мозговой ткани, а также эффективность церебропротекторной терапии.
Как следует из данных, приведенных в Таблица 2, на 4 сутки реперфузионного периода уровень NSE в сыворотке крови крыс контрольной группы возрастал на 139% по сравнению с уровнем NSE в сыворотке крови ложнооперированных животных (р<0,05).

ЛО - группа ложнооперированных животных (группа 1).
Статистически значимые различия (р≤0,05): * - с группой ложнооперированных животных, ^ - с группой цитиколина, ! - с группой модельной патологии.
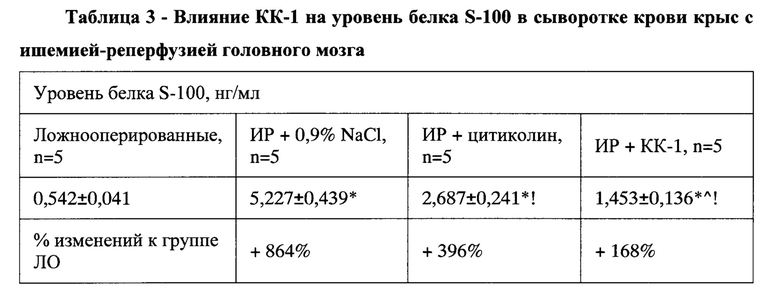
В этих же условиях уровень другого маркера нейродеструкции - белка S-100 - повышался в 9,6 раза (р<0,05) (
Таблица 3). Это свидетельствует в пользу активно развивающейся очаговой гибели ткани головного мозга и превалировании некротического типа гибели нейронов над апоптотическим [Буреш Я. Методики и основные эксперименты по изучению мозга и поведения / Я. Буреш, О. Бурешова, Дж. П. Хьюстон; пер. с англ. Е.Н. Живописцевой; под ред. проф. А.С. Батуева. - М.: Высшая школа, 1991.-399 с; Рациональная нейропротекция / И.Ф. Беленичев, В.И. Черний, Ю.М. Колесник и др. -Донецк: Издатель Заславский А.Ю., 2009 - 262 с; Manual of Stroke Models in Rats / Edited by Yanling Wang-Fisher. - London, New York: CRC Press, 2009. -332 p.].
JIO - группа ложнооперированных животных (группа 1).
Статистически значимые различия (р≤0,05): * - с группой ложнооперированных животных, ^ - с группой цитиколина, ! - с группой модельной патологии.
Установлено, что курсовое введение пептида до ОНМК препятствует резкому возрастанию уровней NSE и S-100. Так, уровень NSE возрастал всего на 19% по сравнению с показателем у условно интактных животных. Это в 7,3 и 2,4 раза меньше показателей у группы КП и группы, получавшей препарат сравнения цитиколин, соответственно (р<0,05). Также наблюдался гораздо менее выраженный рост уровня белка S-100 в сыворотке крови крыс с реперфузионным повреждением головного мозга. Он оказался на 168% выше уровня белка S-100 у ложнооперированных крыс и был в 5,1 и 2,4 раза меньшим от показателей у группы КП и группы, получавшей препарат сравнения цитиколин, соответственно (Р<0,05).
Снижение уровня маркеров нейродеструкции на фоне профилактического применения КК-1 свидетельствует об уменьшении очага некротической гибели нейронов в зоне ишемии, а также об угасании процесса глиоцитарного замещения погибших нейронов. Такое действие КК-1 объясняет его способность повышать выживаемость, снижать неврологический и когнитивный дефицит при ОНМК, оказывая, таким образом, интегральный защитный эффект при ишемии головного мозга.
Пример 5. Влияние КК-1 на уровень фрагментации ДНК. 20 белых беспородных самцов крысы массой 160-170 граммов рандомизировали на 4 группы по 5 особей:
1 группа - ложнооперированные;
2 группа - контрольная патология (КП);
3 группа - животные, получавшие цитиколин (Ferrer International S.A., Испания) в дозе 250 мг/кг внутрибрюшинно;
4 группа - животные, получавшие КК-1 в дозе 0,02 мг/кг интраназально.
ОНМК моделировали, как описано в примере 3.
КК-1 и препарат сравнения вводили один раз в день в -5 ÷ -1 дни до ОНМК. Псевдооперированные крысы претерпевали все этапы оперативного вмешательства, кроме наложения клипс. Животным группы КП интраназально вводили физиологический раствор.
После 72 часов реперфузионного периода крыс декапитировали и извлекали лобные части головного мозга (ГМ). Фрагментацию ДНК в ядрах нейронов лобной части ГМ крыс исследовали методом проточной цитометрии. Суспензию ядер получали путем добавления к ткани мозга специального раствора для исследования ядерной ДНК: CyStain DNA (Partec, Германия). Этот раствор позволяет одновременно экстрагировать ядра и метить ядерную ДНК диамидинофенилиндолом (DAPI). В процессе приготовления ядерных суспензий использовали специальные одноразовые фильтры CellTrics 50 мкм (Partec, Германия). Ядерные суспензии биоптата мозга готовили сразу после забора материала и промывки холодным, от +4° до +8°С, фосфатно-солевым буфером рН 7,4 (Sigma, США). Анализ проводили на многофункциональном научно-исследовательском проточном цитометре «Partec PAS» (Partec, Германия). Для возбуждения флуоресценции DAPI использовали ультрафиолетовую лампу. Из каждого образца ядерной суспензии проводили анализ 10 тыс.событий.
Распределение ДНК у разных животных оценивали на гистограммах с использованием линейной шкалы. Проточный анализ фрагментации ДНК выполняли с использованием программного обеспечения FloMax (Partec, Германия) путем выделения Sub-G1 участка на ДНК-гистограммах.
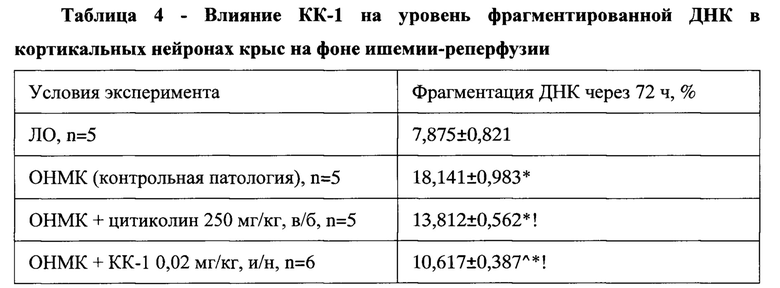
Экспериментальные данные, приведенные в Таблица 4, свидетельствуют о статистически достоверном возрастании уровня фрагментированной ДНК в группе контрольной патологии в 2,7 раза через 72 часа после моделирования ОНМК. Такая динамика указывает на формирование очага повреждения мозговой ткани, в котором превалирует апоптотическая гибель нейронов.
ЛО - группа ложнооперированных животных (группа 1).
Статистически значимые различия (р≤0,05): * - с группой ложнооперированных животных, ^ - с группой цитиколина, ! - с группой модельной патологии.
Типичные гистограммы распределения уровня фрагментированной ДНК в ядрах нейронов коры головного мозга крысы из группы контрольной патологии и группы, получавшей КК-1, приведены на Фигурах 6 и 7. КК-1 и препарат сравнения цитиколин при пятикратном ежедневном введении до нанесения травмы статистически достоверно уменьшали количество фрагментированной ДНК в нейронах коры лобной доли ГМ крыс с моделью ОНМК. На фоне их введения этот показатель снижался относительно группы КП в 1,7 и 1,3 раза соответственно (р<0,05).
Поскольку фрагментация ядерной ДНК служит маркером ранних стадий апоптоза, можно сделать вывод, что КК-1 реализует церебропротекторное действие за счет предотвращения апоптотической гибели нейронов. Антиапоптотическая активность пептида во многом объясняет его положительное влияние на гистологическую структуру коры ГМ при экспериментальном ОНМК.
Таким образом, заявленный пептид КК-1 - аналог фрагмента АКТГ15.18, имеющий формулу CH3CO-(D-Lys)-Lys-Arg-Arg-NH2, обладает выраженными апоптотическими свойствами при профилактическом использовании. Заявленный пептид достоверно снижает уровень маркеров цитолиза (NSE и S-100), рилизинг которых прямо коррелирует с объемом очага некротической гибели нейронов при ишемии головного мозга. Снижение уровня фрагментированной ДНК, которое наблюдается при профилактике реперфузионного повреждения головного мозга пептидом, указывает на его профилактическую антиапоптическую активность. Уменьшая зону некроза и препятствуя апоптической гибели нейронов в зоне пенумбры, КК-1 оказывает интегральное нейропротекторное действие.
Установлено, что профилактическая антиапоптотическая активность КК-1 опосредована активацией киназы Akt1, способствующей выживанию клеток через ингибирование ряда ключевых элементов активации апоптоза, в частности, фосфорилированных форм каспазы 8 и белков Bad и р53. Вполне вероятно, что пептид КК-1 является позитивным регулятором внутриклеточного сигнального пути PI3K/Akt/mTOR.
Заявитель просит рассмотреть представленные материалы заявки «Средство, обладающее антиапоптотическим действием» на предмет выдачи патента РФ на изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ВЫДЕЛЕНИЯ БЕЛКОВОГО ВЫСОКОМОЛЕКУЛЯРНОГО КОМПЛЕКСА АКТИВАЦИИ КАСПАЗЫ-2 ЧЕЛОВЕКА | 2016 |
|
RU2671510C1 |
| ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ПЕМФИГУСА, СОДЕРЖАЩИЕ АНТИТЕЛА К Fas-ЛИГАНДУ | 2008 |
|
RU2558260C2 |
| СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ПУЗЫРЧАТКИ, СОДЕРЖАЩИЕ АНТИТЕЛА ПРОТИВ ЛИГАНДА Fas | 2009 |
|
RU2556818C2 |
| ПРИМЕНЕНИЕ ПЕПТИДНОГО СОЕДИНЕНИЯ ДЛЯ ИНДУКЦИИ АПОПТОЗА В ОПУХОЛЕВОЙ КЛЕТКЕ | 2022 |
|
RU2796104C1 |
| Метод ингибирования апоптоза в клетках млекопитающих и композиция для его реализации | 2022 |
|
RU2825455C2 |
| ПЕПТИДНЫЕ СОЕДИНЕНИЯ ДЛЯ ИНДУКЦИИ АПОПТОЗА В ОПУХОЛЕВОЙ КЛЕТКЕ | 2021 |
|
RU2785794C2 |
| СПОСОБ УМЕНЬШЕНИЯ ОКИСЛИТЕЛЬНОГО ПОВРЕЖДЕНИЯ (ВАРИАНТЫ) | 2005 |
|
RU2376028C2 |
| ПРИМЕНЕНИЕ (2-ИМИДАЗОЛИН-2-ИЛАМИНО) ХИНОКСАЛИНОВ ДЛЯ ЛЕЧЕНИЯ ДЕМЕНЦИИ И БОЛЕЗНИ ПАРКИНСОНА | 2003 |
|
RU2332218C2 |
| ИНГИБИТОРЫ СИГНАЛЬНОГО ПУТИ CD95 ДЛЯ ЛЕЧЕНИЯ МДС | 2013 |
|
RU2652348C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ МОДУЛЯЦИИ АКТИВНОСТИ АТ2R | 2015 |
|
RU2721241C2 |

Изобретение относится к применению пептида общей формулы CH3CO-(D-Lys)-Lys-Arg-Arg-NH2 для профилактики апоптотической гибели нейронов, где пептид вводят до повреждения головного мозга в течение 5 суток. 7 ил., 4 табл., 5 пр.
Применение пептида общей формулы CH3CO-(D-Lys)-Lys-Arg-Arg-NH2 для профилактики апоптотической гибели нейронов, где пептид вводят до повреждения головного мозга в течение 5 суток.
| Дейко Р.Д | |||
| и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| ТЕТРАПЕПТИД И СРЕДСТВО, ОБЛАДАЮЩЕЕ ЦЕРЕБРОПРОТЕКТОРНОЙ И АНТИАМНЕСТИЧЕСКОЙ АКТИВНОСТЯМИ (ВАРИАНТЫ) | 2013 |
|
RU2537560C2 |

