Область техники, к которой относится изобретение
Изобретение относится к медицине и фармакологии, конкретно к фармацевтическим композициям для введения с замедленным высвобождением активного ингредиента (фармацевтической форме), содержащим по меньшей мере один фармакологически активный пептид, способу их получения, набору, включающему лиофилизированный пептид и водный раствор неорганической соли или соли уксусной кислоты, и способу лечения заболеваний у млекопитающих и человека с использованием такой фармацевтической формы.
Уровень техники
В области техники известны следующие фармацевтические формы для введения с замедленным высвобождением фармацевтически активного пептида.
1. Фармацевтические формы для введения с микроинкапсулированными и/или инкорпорированными и/или конъюгированными фармацевтически активными пептидами в биоразрушаемой полимерной матрице (например, как описано в статьях Maulding H.V., J.Controlled Release, 6, 167-76, (1987), Siegel R.A., Langer R., Pharm. Res., 1,2-10, (1984), Патент WO 9832423, Патент WO 2001078687).
2. Фармацевтические формы для введения, включающие плохорастворимые в воде комплексы фармацевтически активного пептида и органической молекулы носителя, такие как, например, полисахариды (как, например, описано в Патенте WO 20000473234).
В обоих случаях ферментное разложение матрицы или комплекса приводит к замедленному высвобождению пептида.
Однако изготовление известных микрокапсул или частиц и нерастворимых комплексов пептидных соединений требует очень сложных процедур, направленных на получение форм для введения с замедленным высвобождением активного ингредиента. Обычно нерастворимые и плохорастворимые соединения получают путем осаждения пептидного соединения с помощью противоиона. Осадок собирают фильтрацией и центрифугированием, промывают водой и высушивают. В большинстве случаев твердый материал затем превращают в порошок. Все отдельные стадии способа получения должны осуществляться в условиях GMP в асептическом рабочем помещении, чтобы было возможно таким образом обеспечить стерильность конечного продукта.
В способах получения микрокапсул используют более или менее токсичные органические растворители с целью растворения, биоразрушаемой полимерной матрицы. Затем растворенную активную субстанцию и полимеры матрицы эмульгируют. После выпаривания органического растворителя частицы или микрокапсулы отделяют, промывают и высушивают.
Настоящее изобретение направлено на расширение ассортимента пролонгированных лекарственных средств на основе фармакологически активных пептидов, оно также позволяет упростить способ их получения и использования.
Сущность изобретения
Неожиданно было обнаружено, что формы для введения с замедленным высвобождением активного ингредиента для фармацевтически активных пептидов получают при восстановлении лиофилизированного пептидного соединения с помощью низкоконцентрированного раствора неорганической соли перед введением, причем количество лиофилизированного пептидного соединения выбирается таким образом, чтобы раствор или суспензия пептида после восстановления были высококонцентрированными.
В качестве возможного объяснения допускают, что в данных условиях происходит контролируемое образование агрегатов пептидных соединений, который или которые демонстрируют задержанное растворение. Результатом этого является обнаруженное замедленное высвобождение данного активного ингредиента в систему кровообращения.
В этом случае образование агрегатов приводит к формированию коллоидной дисперсии, на вязкость которой влияет концентрация пептидного соединения, концентрация соли и время выдерживания после восстановления.
Согласно данному изобретению фармацевтические гелевые препараты, включающие по меньшей мере одно фармацевтически активное ионное пептидное соединение, смешивают в заданном количестве величины Xoptimum (в мг пептида/мл препарата) с водным раствором соли неорганической или уксусной кислоты в заданной концентрации величины Yoptimum (в мас./об.%), причем показано, что введение возможно сразу после смешивания или после периода выдерживания до приблизительно 120 минут, и величина Хoptimum может быть выбрана тест-способом А, включающим стадии применения различных количеств Хn (число различных количеств n, где n≥1) (в мг) пептида в виде смеси с изотоническим водным раствором маннита на или в тест-системе и выбора количества Хoptimum (в мг пептида/мл смеси), которое обеспечивает в эксперименте наиболее благоприятные уровни пептида в плазме крови в тест-системе относительно Сmax (максимальная концентрация в плазме крови) и tmax (время достижения Сmax), и концентрация Yoptimum может быть выбрана тест-способом В, включающим стадии применения количества Xoptimum (в мг пептида/мл смеси) пептида в виде смеси с водными растворами, которые отличаются по концентрации Yn (число различных концентраций n, где n≥1) (в мас./об.%) на или в тест-системе, и выбор концентрации Yoptimum (в мас./об.%) определен как концентрация, при которой эксперимент приводит в результате в самому высокому значению для концентрации в плазме Cactive, где Cmin<Cactive<Cmax (Cmin=самая низкая концентрация пептида в плазме, при которой пептид еще имеет адекватный фармацевтический эффект в эксперименте). В то же самое время это влияет на время tactive до тех пор, пока не будет достигнута активная концентрация в плазме, при которой получают tactive>tmax.
Один из вариантов осуществления изобретения представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение является катионным.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение является анионным.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение является одно-, двух- или поливалентным катионным или анионным пептидом.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение является одно-, двух- или полиамфолитнымм пептидом.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение имеет длину от 5 до 20 аминокислот.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение имеет длину от 8 до 12 аминокислот.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение представляет собой аналог GnRH (рилизинг-гормон гонадотропина).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение представляет собой антагонист GnRH.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение выбрано из группы, состоящей из цетрореликса, тевереликса, абареликса, ганиреликса, азалина В, антида, детиреликса, рамореликса, дегареликса, D-63153 или их фармацевтически активных солей или смесей.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что фармацевтически активное ионное пептидное соединение представляет собой антагонист GnRH D-63153.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что неорганическая соль или соль уксусной кислоты представляет собой физиологически переносимую соль.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что водная неорганическая соль или соль уксусной кислоты выбрана из группы, состоящей из хлорида натрия, хлорида кальция, хлорида магния, ацетата натрия, ацетата кальция и ацетата магния.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что смесь фармацевтически активного ионного пептидного соединения и водного раствора неорганической соли или соли уксусной кислоты представляет собой жидкую суспензию или полутвердую дисперсию.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что количество Х фармацевтически активного ионного пептидного соединения лежит в интервале от приблизительно 5 до приблизительно 50 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что количество Х фармацевтически активного ионного пептидного соединения лежит в интервале от приблизительно 10 до приблизительно 50 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что количество Х фармацевтически активного ионного пептидного соединения лежит в интервале от приблизительно 20 до приблизительно 30 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что количество Х фармацевтически активного ионного пептидного соединения находится в области приблизительно 25 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что D-63153 является фармацевтически активным ионным пептидным соединением и количество Х лежит в интервале от приблизительно 5 до приблизительно 50 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что D-63153 является фармацевтически активным ионным пептидным соединением и количество Х лежит в интервале от приблизительно 10 до приблизительно 50 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что D-63153 является фармацевтически активным ионным пептидным соединением и количество Х лежит в интервале от приблизительно 20 до приблизительно 30 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что D-63153 является фармацевтически активным ионным пептидным соединением и количество Х находится в области приблизительно 25 мг/мл общего количества фармацевтического препарата.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что концентрация Y водного раствора соли неорганической или уксусной кислоты равна или меньше 0,9% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что концентрация Y водного раствора соли неорганической или уксусной кислоты лежит в интервале от приблизительно 0,01% до приблизительно 0,9% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что концентрация Y водного раствора соли неорганической или уксусной кислоты лежит в интервале от приблизительно 0,05% до приблизительно 0,5% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что концентрация Y водного раствора соли неорганической или уксусной кислоты составляет приблизительно 0,1% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что неорганическая соль является хлоридом натрия и что концентрация Y равна или меньше приблизительно 0,9% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что неорганическая соль является хлоридом натрия и что концентрация Y лежит в интервале от приблизительно 0,01% до приблизительно 0,9% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что неорганическая соль является хлоридом натрия и что концентрация Y лежит в интервале от приблизительно 0,05% до приблизительно 0,5% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что неорганическая соль является хлоридом натрия и что концентрация Y составляет приблизительно 0,1% (мас./об.).
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что по меньшей мере одно фармацевтически активное ионное пептидное соединение представляет собой D-63153 и неорганическая соль является хлоридом натрия.
Следующий вариант осуществления представляет собой фармацевтический препарат, отличающийся тем, что по меньшей мере одно фармацевтически активное ионное пептидное соединение представляет собой D-63153 и его количество Х составляет приблизительно 25 мг/мл препарата, и тем, что неорганическая соль является хлоридом натрия и ее концентрация Y составляет приблизительно 0,1% (мас./об.).
Следующий аспект изобретения представляет собой фармацевтический препарат, отличающийся тем, что смесь фармацевтически активного ионного пептидного соединения и водного раствора неорганической соли или соли уксусной кислоты является молекулярно-дисперсионной или коллоидной смесью, которая может иметь жидкую или полутвердую консистенцию.
Следующий аспект изобретения представляет собой фармацевтический препарат, отличающийся тем, что коллоидная дисперсия образуется посредством восстановления.
Следующий аспект изобретения представляет собой фармацевтический препарат, отличающийся тем, что коллоидная дисперсия образуется путем хранения или выдерживания после восстановления, и изменения ее вязкости как функции времени, и при этом улучшается воспроизводимость задержанного высвобождения активного ингредиента
Следующий аспект изобретения представляет собой способ получения фармацевтического препарата, включающий стадии А) объединения количества Хoptimum (в мг/мл конечного препарата) по меньшей мере одного фармацевтически активного ионного пептидного соединения в лиофилизированной форме и водного раствора соли неорганической или уксусной кислоты в концентрации со значением Yoptimum (мас./об.%) и В) смешивания компонентов.
Следующий вариант осуществления изобретения представляет собой способ получения фармацевтического препарата, отличающийся тем, что фармацевтически активное ионное пептидное соединение представляет собой D-63153 и неорганическая соль является хлоридом натрия.
Следующий вариант осуществления изобретения представляет собой способ получения фармацевтического препарата, отличающийся тем, что фармацевтически активное ионное пептидное соединение представляет собой D-63153 и его количество составляет приблизительно 25 мг/мл, и тем, что неорганическая соль является хлоридом натрия и ее концентрация составляет приблизительно 0,1% (мас./об.).
Следующий вариант осуществления изобретения представляет собой способ получения фармацевтического препарата, отличающийся тем, что далее включает стадию стерилизации пептидного препарата путем облучения γ-лучами или пучком электронов.
Следующий вариант осуществления изобретения представляет собой способ получения фармацевтического препарата, отличающийся тем, что продукция пептидного препарата осуществляется с использованием асептических процедур.
Следующий аспект изобретения представляет собой набор для получения фармацевтического препарата, включающий предварительно заданное количество Х (в мг/мл конечного препарата) фармацевтически активного ионного пептидного соединения в лиофилизированной форме и водный раствор соли неорганической или уксусной кислоты в предварительно заданной концентрации Y% (мас./об.).
Следующий вариант осуществления изобретения представляет собой набор для получения фармацевтического препарата, отличающийся тем, что фармацевтически активное пептидное соединение является D-63153 в лиофилизированной форме.
Следующий вариант осуществления изобретения представляет собой набор для получения фармацевтического препарата, отличающийся тем, что лиофилизат D-63153 дополнительно содержит маннит.
Следующий вариант осуществления изобретения представляет собой набор для получения фармацевтического препарата, отличающийся тем, что неорганическая соль является хлоридом натрия.
Следующий вариант осуществления изобретения представляет собой набор для получения фармацевтического препарата, отличающийся тем, что количество XD-63153 составляет приблизительно 25 мг в конечном препарате и концентрация водного раствора хлорида натрия составляет приблизительно 0,1 мас./об.%.
Следующий аспект изобретения представляет собой набор, отличающийся тем, что лиофилизированный фармацевтически активный пептид, например D-63153, используют при необходимости вместе с одним или более фармацевтически приемлемыми наполнителями или дополнительными нейтральными компонентами и низкоконцентрированным водным раствором неорганической соли, предпочтительно хлоридом натрия.
Следующий аспект изобретения представляет собой способ лечения заболевания путем введения пациенту фармацевтического препарата, содержащего фармацевтически активное пептидное соединение, отличающийся тем, что в качестве такового используют фармацевтический препарат по п.1, который вводят пациенту подкожно или внутримышечно.
Следующий вариант осуществления изобретения представляет собой способ лечения пациента с помощью фармацевтически активного пептидного препарата, отличающийся тем, что введенный фармацевтический препарат демонстрирует замедленную фармацевтическую активность.
Следующий вариант осуществления изобретения представляет собой способ лечения пациента с помощью фармацевтически активного пептидного препарата, отличающийся тем, что введенный фармацевтический препарат демонстрирует замедленную фармацевтическую активность в течение по меньшей мере 4 недель.
Следующий вариант осуществления изобретения представляет собой способ лечения пациента с помощью фармацевтически активного пептидного препарата, отличающийся тем, что введенный фармацевтический препарат демонстрирует замедленную фармацевтическую активность в течение по меньшей мере 8 недель.
Следующий вариант осуществления изобретения представляет собой способ лечения пациента с помощью фармацевтически активного пептидного препарата, отличающийся тем, что введенный фармацевтический препарат демонстрирует замедленную фармацевтическую активность в течение по меньшей мере 12 недель.
Следующий вариант осуществления изобретения представляет собой способ лечения гормонозависимого нарушения у пациента посредством подкожного или внутримышечного введения вышеупомянутых фармацевтических препаратов нуждающемуся в этом пациенту.
Следующий аспект изобретения представляет собой способ лечения рака простаты у пациента посредством подкожного или внутримышечного введения фармацевтического препарата, соответствующего вышеописанному изобретению, нуждающемуся в этом пациенту.
Следующий аспект изобретения представляет собой способ лечения рака молочной железы у пациента посредством подкожного или внутримышечного введения фармацевтического препарата, соответствующего вышеописанному изобретению, нуждающемуся в этом пациенту.
Следующий аспект изобретения представляет собой способ лечения миом матки у пациента посредством подкожного или внутримышечного введения фармацевтического препарата, соответствующего вышеописанному изобретению, нуждающемуся в этом пациенту.
Следующий аспект изобретения представляет собой способ лечения эндометриоза у пациента посредством подкожного или внутримышечного введения фармацевтического препарата, соответствующего вышеописанному изобретению, нуждающемуся в этом пациенту.
Следующий аспект изобретения представляет собой способ лечения преждевременного полового созревания у пациента посредством подкожного или внутримышечного введения фармацевтического препарата, соответствующего вышеописанному изобретению, нуждающемуся в этом пациенту.
Следующий аспект изобретения представляет собой способ модификации репродуктивной функции у пациента посредством подкожного или внутримышечного введения фармацевтического препарата, соответствующего вышеописанному изобретению, нуждающемуся в этом пациенту.
В предпочтительном варианте осуществления пептидное соединение формы для введения представляет собой аналог GnRH, даже более предпочтительно антагонист GnRH, и неорганическая соль является хорошо растворимой физиологической солью, предпочтительно хлоридом натрия.
Вследствие парентерального введения необходимо, чтобы пептидное соединение находилось в виде порошка и раствор для восстановления были стерильными.
Данное изобретение делает возможным легко получить суспензии с замедленным высвобождением активного ингредиента пептидного соединения, предпочтительно антагониста GnRH. Их получают посредством восстановления высококонцентрированного лиофилизата пептидного соединения, содержащего маннит, с использованием разбавленного раствора неорганической соли (например, раствора хлорида натрия).
Образование фармацевтического препарата, соответствующего изобретению, кроме того, зависит от следующих параметров.
1) Концентрация пептидного соединения в растворе после восстановления
2) Концентрация неорганической соли в растворе, используемом для восстановления
3) Время выдерживания раствора после восстановления и полученная при этом степень агрегации, которая отражается в повышении вязкости.
Высокая концентрация пептидного соединения приводит к его агрегации, которая может контролироваться добавлением раствора неорганической соли. Растворимость пептидного соединения снижается по мере повышения концентрации соли. Коллоидные свойства становятся более преобладающими, чем свойства раствора, как это ясно из повышения вязкости даже в такой степени, что приводит к образованию геля. "Гель" в этой связи представляет собой бикогерентную систему, состоящую из агрегата пептида в качестве твердой фазы и воды в качестве жидкой фазы.
Соответствующие изобретению формы для введения для фармацевтически активных пептидов с замедленным высвобождением активного ингредиента всегда перед введением находятся в форме геля.
При идеальном интервале концентрации соли в комбинации с подходящим количеством пептидных соединений может быть получено замедленное высвобождение активного ингредиента в течение 4 недель или больше.
Для получения раствора неорганической соли можно использовать любую физиологически переносимую неорганическую соль, предпочтительно хлорид натрия.
Восстановление происходит при использовании низкоконцентрированного раствора соли. В данном случае концентрация должна быть равна или меньше приблизительно 0,9% (мас./об.), предпочтительно находиться в интервале от приблизительно 0,01% до приблизительно 0,9%, особенно предпочтительно в интервале от приблизительно 0,05% до приблизительно 0,5% (мас./об.), очень предпочтительно составлять приблизительно 0,1% (мас./об.).
Низкоконцентрированный раствор хлорида натрия с концентрацией хлорида натрия, лежащей в интервале от приблизительно 0,05% до приблизительно 0,5% (мас./об.), предпочтительно приблизительно 0,1% (мас./об.) является предпочтительным.
Пептид в препарате представляет собой соединение фармакологически активного пептида, который может быть одно-, двух- или поливалентным катионным или анионным пептидом. Пептид может состоять из от 5 до 20 аминокислот в длину, более предпочтительно из 8-12 аминокислот в длину. Более конкретно пептидное соединение представляет собой аналог GnRH, и аналог GnRH является антагонистом GnRH. Примерами аналогов GnRH являются цетрореликс, тевереликс (см. статью Deghenghi и соавт., Biomed & Pharmacother, 47, 107, (1993)), абареликс (см. статью Molineaux и соавт., Molecular Urology, 2, 265, (1998)), ганиреликс (см. статью Nestor и соавт., J.Med. Chem., 35, 3942, (1992)), азалин В, антид, А-75998 (см. статью Cannon и соавт., J.Pharm. Sci., 84, 953, (1995)), детереликс (см. статью Andreyko и соавт., J.Clin. Endocrinol. Metab., 74, 399, (1992)), RS-68439, рамореликс (см. статью Stockemann и Sandow, J.Cancer Res. Clin. Oncol., 119, 457, (1993)), дегареликс (см. статью Broqua P., Riviere и соавт., JPET, 301, 95), D-63153 (международная заявка РСТ: ЕР 00/02165).
Структуры вышеупомянутых аналогов GnRH приведены, например, в вышеупомянутых ссылках и следующих обзорных статьях: Behre и соавт., Обзор по антагонистам GnRH (GnRH antagonists: an overview), Proceedings of the 2nd World Conference on Ovulation Induction, The Parthenon Publishing Group Ltd, UK; Kutsher и соавт., Angew. Chem., 109, 2240, (1997).
Соединение D-63153 описано в числе других в немецкой патентной заявке No DE 19911771.3. Физико-химические данные сведены на Фиг.6.
Концентрация фармацевтически активного пептида может лежать в интервале от приблизительно 5 мг/мл до приблизительно 50 мг/мл, предпочтительно от приблизительно 10 мг/мл до приблизительно 50 мг/мл, особенно предпочтительно от приблизительно 20 мг/мл до приблизительно 30 мг/мл и очень особенно предпочтительно составлять приблизительно 25 мг/мл (мл = общий объем конечной формы для введения).
Все фармацевтически активные пептиды могут быть использованы в указанных концентрациях. Пептид D-63153 является особенно предпочтительным.
Следует отметить, что данное изобретение направлено на получение форм для введения для фармацевтически активных пептидов с замедленным высвобождением активного ингредиента.
В соответствии с изобретением соль уксусной кислоты основания пептидного соединения полностью растворяют в водном растворе уксусной кислоты до образования прозрачного раствора. Раствор разбавляют водой для инъекций, которая содержит необходимое количество маннита для того, чтобы образовался изотонический раствор, который можно вводить.
После стерилизации путем фильтрования раствора его разливают во флаконы и лиофилизируют.
Раствор хлорида натрия (например, 0,1%) используют для восстановления перед введением для того, чтобы, таким образом, контролировать агрегацию пептида и, вследствие этого, также растворимость. Восстановление осуществляют по мере необходимости при осторожном кручении или встряхивании, что нужно для того, чтобы избежать вспенивания.
Фармацевтические формы для введения, соответствующие изобретению, обеспечивают замедленную доставку пептидного соединения после введения формы для введения субъекту. Продолжительность и степень высвобождения можно варьировать путем изменения концентраций пептидного соединения и концентрации используемой соли.
Время выдерживания после восстановления также является важным в плане высвобождения пептидного активного ингредиента. Время выдерживания составляет от приблизительно 0 до приблизительной 120 минут, предпочтительно от приблизительно 10 до приблизительной 120 минут, особенно предпочтительно от приблизительно 15 до 60 минут. Было обнаружено, что коллоидная система образуется путем изменений агрегации в период времени выдерживания и при этом повышается вязкость. При времени выдерживания, превышающем приблизительно 120 минут, никакого существенного дальнейшего изменения вязкости не наблюдается.
Фармацевтические формы для введения, соответствующие изобретению, могут быть предпочтительно введены подкожно (s.c.) или внутримышечно (i.m.). В случае внутримышечного введения инъекцию осуществляют, например, в большую ягодичную мышцу, предпочтительно в верхний наружный квадрант большой ягодичной мышцы. В случае подкожного введения инъекцию делают, например, в подкожный слой живота.
Перечень чертежей
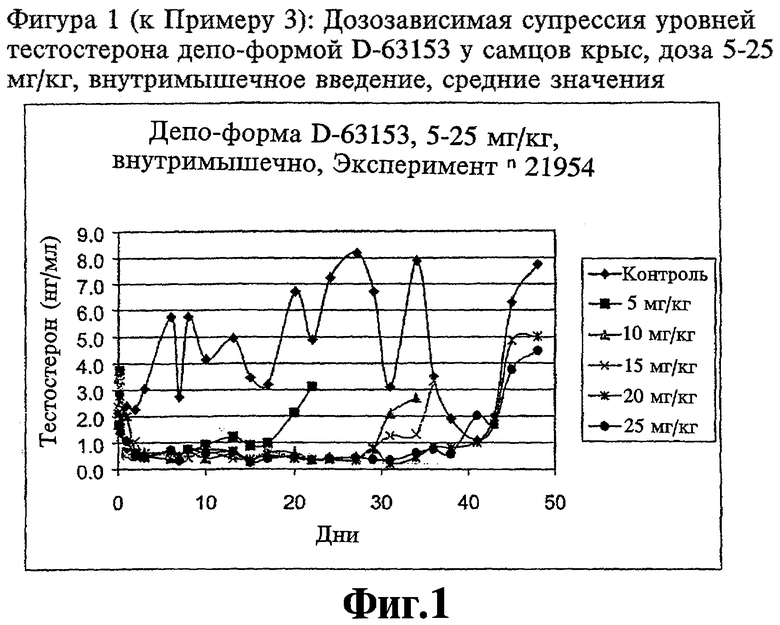
Фиг.1 (к Примеру 3): Дозозависимая супрессия уровней тестостерона депо-формой D-63153 у самцов крыс, доза 5-25 мг/кг, внутримышечное введение;
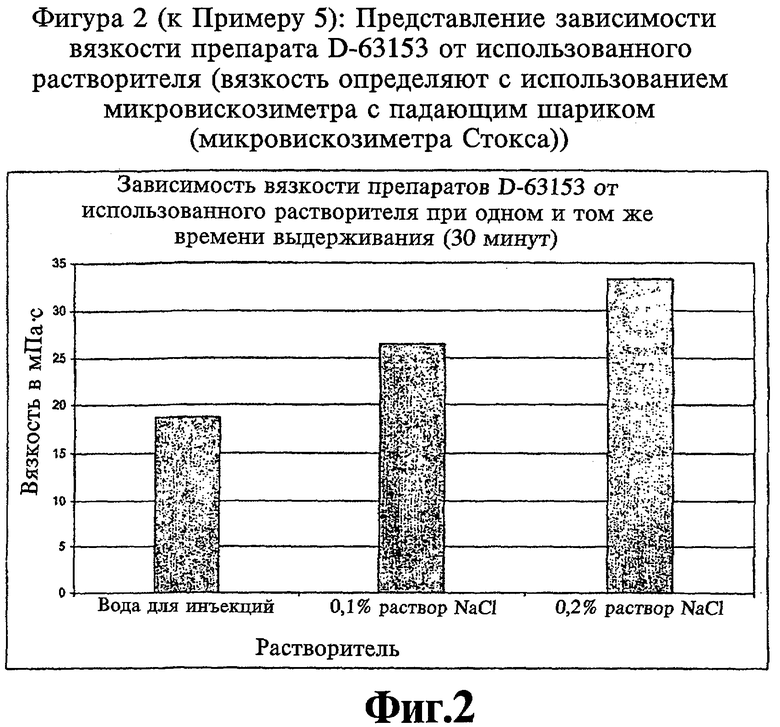
Фиг.2 (к Примеру 5): Представление зависимости вязкости препарата D-63153 от используемого растворителя.
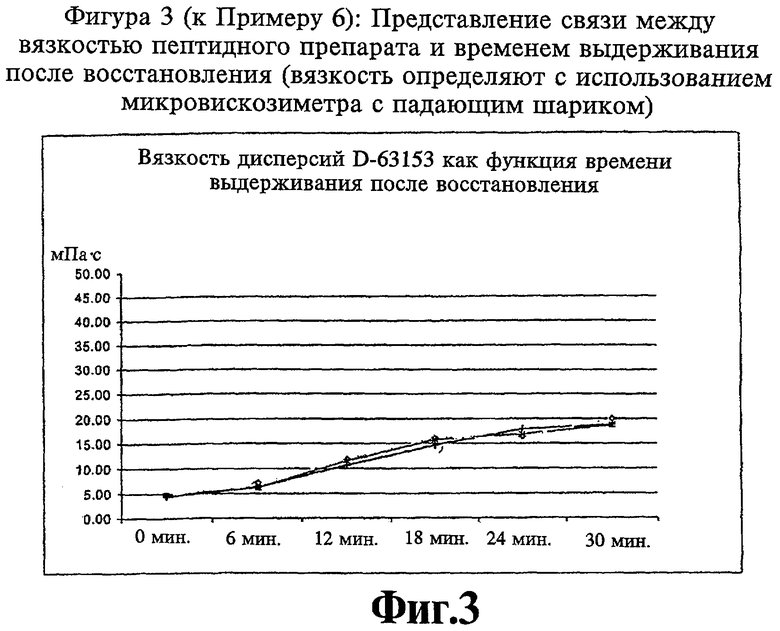
Фиг.3 (к Примеру 6): Представление связи между вязкостью пептидного препарата и временем выдерживания после восстановления.
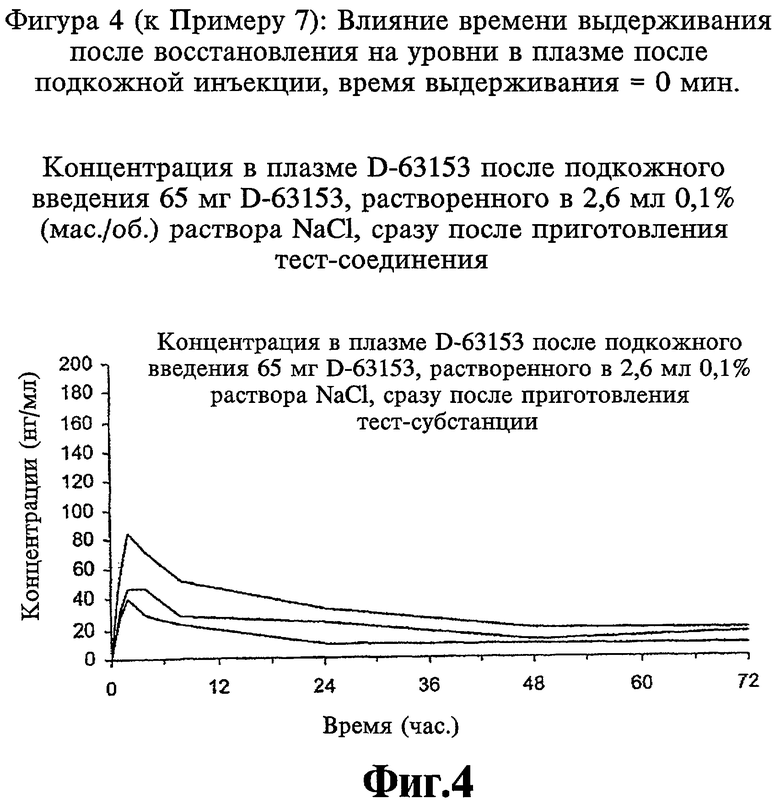
Фиг.4 (к Примеру 7): Концентрация в плазме D-63153 после подкожного введения 65 мг D-63153, растворенного в 2,6 мл 0,1% (мас./об.) раствора NaCl, сразу после приготовления тест-соединения.
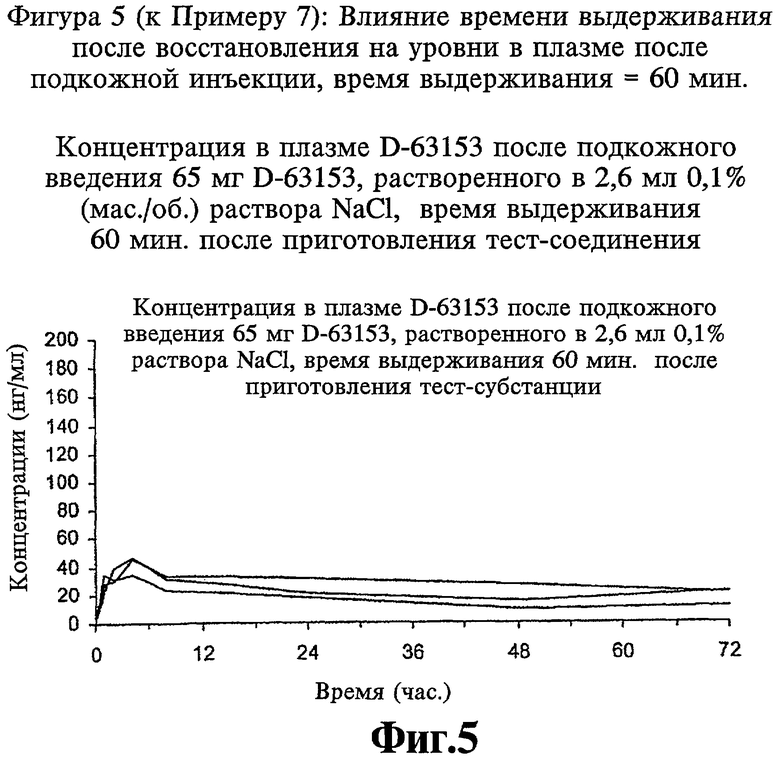
Фиг.5 (к Примеру 7): Концентрация в плазме D-63153 после подкожного введения 65 мг D-63153, растворенного в 2,6 мл 0,1% (мас./об.) раствора NaCl, через 1 ч (60 мин) после приготовления тест-соединения.
Фиг.6: Физико-химические характеристики D-63153.
Данное изобретение описано в деталях в примерах 1-7 без ограничения этим изобретения.
Сведения, подтверждающие возможность осуществления изобретения
Пример 1
200 г чистого D-63153 (в пересчете на свободное основание) растворяют в 3386,7 г водного раствора уксусной кислоты концентрации 30% с образованием прозрачного раствора. Добавляют 438,4 г маннита и растворяют при перемешивании. Раствор доводят до общего количества 20320 г водой для инъекций.
После стерилизации раствора фильтрованием его разливают порциями по 10 мл во флаконы для лиофилизации.
В результате, согласно способу, каждый флакон содержит 100 мг D-63153 (свободного основания) и 109,6 мг маннита.
Лиофилизат восстанавливают добавлением 4 мл раствора хлорида натрия в концентрации 0,1% и осторожно встряхивают (избегая вспенивания) для того, чтобы получить суспензию концентрации 25 мг/мл.
Пример 2
Лиофилизаты, которые содержат 75 мг D-63153, получают и восстанавливают с 3 мл растворителя (25 мг D-63153/мл). Восстановление осуществляют стерильной водой для инъекций (не-депо форма для введения, см. таблицу 1) или 0,1% NaCl (депо-форма для введения, см. таблицу 2). Дозу для однократного введения, 1,68 мг/кг, инъецируют подкожно собакам породы бигл. Уровни D-63153 в плазме измеряют в различные периоды времени после введения.
Путем использования депо-форм для введения можно снизить максимальные уровни в плазме (Сmax), тогда как площадь под кривой в основном поддерживается на стабильном уровне, что в результате приводит к депо-эффекту. Абсолютная биодоступность сохраняется в основном без изменений и по расчетам составляет 62% для не-депо форм для введения и 64,3% для депо-форм (см. статью Schwahn и Romeis, 1991).
Пример 3
Для тестирования D-63153 депо в отношении активности супрессии тестостерона его инъекционно вводят в 5 различных дозах (5-25 мг/кг) внутримышечно (i.m.) самцам крыс. Депо форму для введения получают путем ресуспендирования лиофилизата D-63153 в стерильном растворе NaCl в концентрации 0,1%. Уровень тестостерона измеряют до введения лекарственного препарата и в каждом случае через 4 часа, 8 часов и 24 часа после введения. Кроме того, уровень тестостерона определяют один раз в день в первую неделю после инъекции, а затем через день, в каждом случае до тех пор, пока уровень тестостерона не возвратится в пределы нормы. Контрольной группе вводят только раствор носителя (см. Фиг.1). Во всех группах определяют дозозависимую супрессию уровней тестостерона. Супрессия продолжается от 17 дней (5 мг/кг) до 43 дней (20 мг/кг). Затем уровни тестостерона в течение нескольких дней входят в пределы нормы.
Пример 4
10 мг лиофилизатов D-63153 восстанавливают в 4 мл стерильной воды для инъекций (не-депо форма для введения, 2,5 мг/мл D-63153, клиническая фаза 1а) и 100 мг лиофилизатов D-63153 растворяют в 4 мл 0,1% NaCl (депо-форма для введения, 25 мг/мл D-63153, клиническая фаза 1b). Добровольным участникам эксперимента мужского пола делают внутримышечные инъекции по 10 мг/субъекта. Уровни D-63153 в плазме измеряют в различные периоды времени после введения (см. Таблицу 3).
Результаты показывают, что депо эффект может быть подтвержден как посредством более низких уровней Сmax и AUC0-24 в плазме, так и посредством удлинения периода tmax, T1/2 и, в особенности, увеличения MRT (среднего времени удержания). Депо-форма для введения имеет почти такое же время AUC0-tlast, как не-депо форма для введения (887,44 нг·ч/мл по сравнению с 1165,93 нг·ч/мл), демонстрируя таким образом, что две композиции имеют близкую биодоступность. Высвобождение из депо формы для введения замедлено, на что указывают более низкий уровень Сmax и значение MRT, которое более, чем в два раза выше.
Пример 5
Получают лиофилизаты, содержащие 65 мг и 100 мг D-63153, и восстанавливают их растворителем с образованием в результате раствора, который имеет концентрацию 25 мг D-63153/мл. Используемые растворители представлены водой для инъекций, 0,1% раствором NaCl и 0,2% раствором NaCl. Степень изменений коллоидных свойств растворов исследуют по их вязкости. Результаты отображены на Фиг.2.
Пример 6
Получаютлиофилизаты, содержащие 100 мг D-63153, и восстанавливают их растворителем с образованием в результате раствора, который имеет концентрацию 25 мг/мл. С целью описания изменения коллоидной системы, которая образуется после восстановления, на Фиг.3 приведена вязкость, как функция времени выдерживания или времени хранения после восстановления.
Пример 7
Получают лиофилизаты, содержащие 65 мг D-63153, и восстанавливают их 2,6 мл раствора NaCl концентрации 0,1%, и полученный раствор в одном случае подкожно вводят собакам сразу (время выдерживания 0 мин) (см. Фиг.4) и в другом случае подкожно вводят собакам (см. Фиг.5) через один час после восстановления (время выдерживания 60 мин). Уровни D-63153 измеряют в течение 72 часов.
Коллоидную систему получают путем изменений агрегации в течение времени выдерживания, за которое его вязкость возрастает. Это связано с небольшим изменением в графиках уровней в плазме и в результате приводит к тому, что максимальная концентрация в плазме снижается и воспроизводимость графиков уровней в плазме улучшается.
Фармакокинетические параметры не-депо формы введения D-63153 у собак породы бигл, 1,68 мг/кг при подкожном введении
Фармакокинетические параметры депо-формы введения D-63153 у собак породы бигл, 1,68 мг/кг при подкожном введении
Фармакокинетические параметры D-63153: сравнение не-депо и депо-форм введения у добровольных участников эксперимента мужского пола, 10 мг/субъекта (0,14-0,17 мг/кг) при внутримышечном введении

Изобретение относится к химико-фармацевтической промышленности и касается фармацевтических форм для введения с замедленным высвобождением, содержащим, по меньшей мере, один фармакологически активный пептид. Изобретение относится также к способу их получения, набору, содержащему лиофилизированный пептид и водный раствор неорганической соли или соли уксусной кислоты, и применению водного раствора соли неорганической или уксусной кислоты для получения формы для фармацевтического введения, которая высвобождает пептиды непрерывным образом в течение длительного периода времени. Форма позволяет существенно продлить высвобождение активного пептида, используя эффект депо. 6 н. и 36 з.п. ф-лы, 4 табл., 6 ил.
Приоритет по пунктам:
| ЛИОФИЛИЗАТ НА ОСНОВЕ ПЕПТИДА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1994 |
|
RU2145234C1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

