ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к смесям, содержащим липиды и антиоксидант.
Настоящее изобретение также относится к предшественникам составов (предварительным составам), в которых при воздействии воды или водных сред, таких как физиологические жидкости, самопроизвольно происходит фазовый переход, в результате чего образуется матрица с контролируемым высвобождением. В частности, изобретение относится к смесям, предварительным составам и композициям, имеющим улучшенную стойкость к окислению.
УРОВЕНЬ ТЕХНИКИ
Многие биоактивные агенты, включая фармацевтические средства, питательные вещества, витамины и т.д., имеют «функциональное окно». То есть существует диапазон концентраций, при которых может наблюдаться некоторый биологический эффект указанных агентов. Если концентрация в соответствующей части организма (например, местная или отражаемая концентрацией в сыворотке) падает ниже определенного уровня, то благоприятное действие не может быть приписано агенту. Аналогично, как правило, существует верхняя концентрация, выше которой увеличение концентрации не проводит к дополнительному усилению действия. В некоторых случаях увеличение концентрации выше определенного уровня приводит к нежелательным или даже опасным эффектам.
Некоторые биоактивные агенты имеют продолжительный биологический период полувыведения и/или широкое функциональное окно, и, таким образом, их можно вводить по мере необходимости для поддержания функциональной биологической концентрации в течение значительного периода времени (например, от 6 часов до нескольких дней). В других случаях скорость клиренса высока и/или функциональное окно является узким, и, таким образом, для поддержания биологической концентрации в пределах этого окна требуется регулярное (или даже непрерывное) введение доз небольшого размера. Это может быть особенно трудным, если желательно или необходимо применение способов введения, отличных от перорального (например, парентеральное введение), так как самостоятельное введение может быть затруднено, что, таким образом, вызывает неудобства и/или плохое соблюдение схемы лечения. В таких случаях желательным было бы однократное введение, которое обеспечивало бы терапевтический уровень активного агента в течение всего периода, на протяжении которого необходима активность.
Некоторым пациентам, проходящим лечение, как правило, требуется поддержание терапевтической дозы в течение значительного периода и/или проведение лечения в течение многих месяцев или лет. Таким образом, система депо, позволяющая вводить и высвобождать контролируемым образом повышенную дозу в течение продолжительного периода времени, может обеспечивать значительные преимущества по сравнению с традиционными системами доставки.
Определенные составы согласно настоящему изобретению образуют неламеллярную жидкокристаллическую фазу после введения. Применение структур с неламеллярной фазой (таких как жидкокристаллические фазы) при доставке биоактивных агентов в настоящее время относительно хорошо изучено. Наиболее эффективная система липидного депо описана в WO 2005/117830, и особенно предпочтительное липидное депо описано в указанном документе. Тем не менее, остается задача получения составов депо, имеющих улучшенные характеристики в нескольких аспектах.
Липидные системы доставки с контролируемым высвобождением были разработаны для активных агентов, включая GLP-1 (WO 2006/131730), аналоги соматостатина (WO 2006/075124), аналоги РФЛГ (WO 2006/075125), а также непептидные агенты, такие как бупренорфин (WO 2014/016428). Липидные системы также представляют ценность для лечения сами по себе и не обязательно должны включать активные агенты. Например, одобренный FDA жидкий пероральный препарат episil® облегчает боль, вызванную оральным мукозитом и другими воспалительными состояниями полости рта, за счет образования липидного барьера в полости рта, но не требует добавления какого-либо активного агента.
Особенно универсальной комбинацией липидов является глицерилдиолеат (GDO) и фосфатидилхолин (PC). Тем не менее, составы с замедленным высвобождением могут быть получены с применением широкого спектра других липидных компонентов, включая токоферол (WO 2006/075123), производные сорбита (WO 2016/102683), триглицериды (WO 2016/066655) и различные фосфолипидные компоненты, включая фосфатидилэтаноламины (WO 2013/083459 и WO 2013/083460).
Как липидные компоненты, в частности ненасыщенные липиды, так и любые активные агенты, содержащиеся в предварительном составе или композиции с замедленным высвобождением, подвержены окислению при хранении или in vivo. Желательно уменьшить уровень окисления, так как окислительные процессы могут уменьшать содержание активного агента и/или способствовать образованию нежелательных продуктов разложения. Это, в свою очередь, уменьшает срок годности продукта.
Одним конкретным фактором, способствующим окислению в липидных композициях, является присутствие следовых количеств ионов металлов, в частности переходных металлов, таких как железо (Fe). Даже если липидные компоненты имеют высокий класс чистоты, часто трудно полностью удалить следы указанных ионов. Считается, что оборудование, применяемое для получения липидных составов, обычно включает нержавеющую сталь, из которой небольшие количества ионов металлов (в частности, Fe) могут попадать в смесь. Таким образом, в липидные составы обычно включают антиоксиданты. Они обычно действуют за счет образования хелатов с ионами какого- либо металла, что тем самым затрудняет их участие в окислительных процессах.
Обязательным условием является то, что любой антиоксидант должен быть растворим в липидной смеси, например, в предварительном составе. В WO 2012/160213 описано, что тщательно контролируемое количество воды может быть включено в предварительные липидные составы и не вызывает фазовые изменения в жидкокристаллической фазе. В предварительные составы, содержащие значительное количество воды, можно включать эффективное количество водорастворимого антиоксиданта, такого как аскорбиновая кислота, неорганические соли агентов, образующих хелаты с металлами, таких как этилендиаминтетрауксусная кислота (ЭДТА) (например, соли натрия или кальция) и лимонная кислота. Тем не менее, для определенных активных агентов необходимо избегать длительного воздействия воды при хранении (например, если активный агент чувствителен к влаге) или может быть получен более желательный профиль высвобождения без включения воды в предварительный состав. Предотвращение контакта с водой также может уменьшать количество следовых металлов, которые могут присутствовать, так как ионы металлов обычно имеют более высокую растворимость в воде по сравнению с органическим растворителем или липидной средой. В липидных составах, имеющих низкое содержание воды, невозможно применять традиционные водорастворимые антиоксиданты, так как они могут не иметь необходимую растворимость в по существу безводной липидной среде. Следовательно, было бы предпочтительно обеспечить антиоксидант, который растворим в по существу безводной липидной среде и который ограничивает или предотвращает окислительное разложение липидных компонентов смеси, например, предварительного состава, и любого содержащегося активного агента. Это особенно важно для агентов, образующих хелаты с металлами, таких как ЭДТА, стандартные неорганические соли (натриевые или кальциевые) которой не растворимы или имеют пренебрежительно малую растворимость в неводных средах (например, в липидных матрицах).
В WO 2010/020794 описаны тиолированные антиоксиданты, которые обеспечивают конкретные преимущества в липидных системах и также подходят для неводных липидных систем. Тем не менее, для определенных конечных применений присутствие тиолированного антиоксиданта может быть неприемлемым. Это, в частности, относится, например, к пептидам или белкам, содержащим тиолированные группы или дисульфидные мостики. В WO 2010/020794 также упоминается возможность включения ЭДТА или натриевых, динатриевых и кальций-динатриевых солей ЭДТА в качестве хелатообразующего агента, но этот вариант не проиллюстрирован в примерах. Авторы настоящего изобретения установили, что ЭДТА или ее обычные соли не растворимы в сколько-нибудь значимой степени в типах липидных составов, описанных в WO 2010/020794, т.е. в составах на основе GDO, SPC и органического растворителя, такого как этанол.
В настоящем изобретении неожиданно было установлено, что эффективные количества алкиламмониевых солей ЭДТА могут растворяться в неводной липидной среде, и полученные смеси, например, предварительные составы, обладают высокой стойкостью к окислительному разложению при хранении. Кроме того, хотя и считается, что алкиламмониевые соли ЭДТА влияют на уменьшение разложения посредством ожидаемого механизма хелатного связывания ионов металлов, настоящее изобретение позволяет в некоторых вариантах реализации улучшать стойкость к окислению сверх того уровня, который можно было бы ожидать при наличии только такого механизма. Авторы изобретения установили, что включение алкиламмониевых солей ЭДТА может предотвращать или существенно понижать скорость окисления разнообразных липидных компонентов и/или содержащихся в них активных агентов. Авторы изобретения обнаружили, что включение алкиламмониевых солей ЭДТА может существенно уменьшать потери активного агента в образцах лекарственного средства, изучаемых в исследованиях стабильности, и, таким образом, увеличивать срок годности лекарственного продукта. Преимущество солей ЭДТА заключается в их дешевизне, простоте получения с использованием разнообразных катионов и в том, что они, как правило, считаются безопасными (и широко применяются, например, для фармацевтического применения).
Обнаруженный авторами изобретения для алкиламмониевых солей ЭДТА эффект стабилизации и продления срока годности может быть связан не только с предотвращением или уменьшением окислительных реакций, но также с предотвращением или уменьшением других реакций химического разложения, например, гидролиза, ацилирования, дезамидирования.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно первому аспекту в изобретении предложена смесь:
i) по меньшей мере одного липида и/или по меньшей мере одного масла; и
ii) алкиламмониевой соли ЭДТА (например, содержащей анион этилендиаминтетрауксусной кислоты или ее аналога);
где смесь имеет содержание воды в диапазоне от 0 до 1,0 масс.%.
Согласно всем аспектам аналоги этилендиаминтетрауксусной кислоты и их соответствующие анионы, как правило, являются такими, как описано ниже в настоящем документе.
В настоящем изобретении также предложен фармацевтический состав, содержащий соответствующую комбинацию липидных вспомогательных веществ, органический растворитель и алкиламмониевую соль ЭДТА, который можно применять в качестве состава-предшественника депо (для краткости называемого в настоящем документе предварительным составом) для удовлетворения одной или более потребностей, описанных выше.
Согласно второму аспекту, таким образом, в изобретении предложен предварительный состав, содержащий:
i) липидную смесь, содержащую:
а) по меньшей мере одно из моно-, ди- или триациллипида и/или токоферола;
b) необязательно по меньшей мере один фосфолипид;
c) по меньшей мере один биосовместимый органический растворитель; и
ii) алкиламмониевую соль ЭДТА (например, содержащую анион этилендиаминтетрауксусной кислоты или ее аналога); и
где предварительный состав имеет содержание воды в диапазоне от 0 до 1,0 масс.%.
В предпочтительном варианте реализации предварительный состав образует или может образовывать по меньшей мере одну структуру с жидкокристаллической фазой при контакте с избытком водной жидкости.
При использовании в настоящем описании «липидная смесь» может представлять собой «липидную матрицу с контролируемым высвобождением».
Особенно предпочтительной комбинацией компонентов в некоторых вариантах реализации является глицерилдиолеат (GDO), фосфатидилхолин (PC), этанол и тетракис-(этаноламмоний)ЭДТА. Предварительный состав согласно всем вариантам реализации может дополнительно содержать активный агент, такой как описано в настоящем документе.
Предварительные составы особенно подходят для контролируемого и замедленного высвобождения активного агента, в частности, агента, для которого требуется крайне плоский профиль высвобождения и/или минимальный «скачок» концентрации после введения или действие которого усиливается в указанных условиях. В соответствующем варианте реализации в изобретении, таким образом, предложена смесь из:
i) липидной смеси, содержащей:
а) по меньшей мере одно из моно-, ди- или триациллипида и/или токоферола;
b) необязательно по меньшей мере один фосфолипид;
c) по меньшей мере один биосовместимый органический растворитель;
d) активный агент; и
ii) алкиламмониевой соли ЭДТА (например, содержащей анион этилендиаминтетрауксусной кислоты или ее аналога); и
где смесь имеет содержание воды в диапазоне от 0 до 1,0 масс.%;
для получения предварительного состава для применения для замедленного высвобождения указанного активного агента. В предпочтительном варианте реализации предварительный состав образует или способен образовывать по меньшей мере одну структуру с жидкокристаллической фазой при контакте с избытком водной жидкости.
«Биоактивные агенты» или «активные агенты», описываемые в настоящем документе, могут представлять собой любое соединение, обладающее желаемым биологическим или физиологическим действием, такое как пептид, белок, лекарственное средство, антиген, питательное вещество, косметическое средство, отдушка, вкусоароматическая добавка, диагностическое средство, фармацевтическое средство, витамин или диетическое вещество, и включены в количестве, достаточном для обеспечения концентрации in vivo на функциональном уровне (включая локальные концентрации в случае композиций для местного применения). В одном из вариантов реализации «активный агент» представляет собой натуральный или синтетический пептидный или непептидный активный фармацевтический ингредиент (АФИ), который обеспечивает терапевтическое, паллиативное и/или профилактическое действие при введении подходящему субъекту (как правило, нуждающемуся в указанном действии).
В дополнительном варианте реализации, таким образом, в изобретении предложен способ лечения субъекта, представляющего собой человека или млекопитающее, отличное от человека, включающий введение указанному субъекту предварительного состава, такого как описано в настоящем документе. Указанный способ может быть предназначен для лечения нуждающегося в этом субъекта, представляющего собой человека или млекопитающее, не являющееся человеком, для борьбы (например, излечения, улучшения, предупреждения или ослабления симптомов) по меньшей мере с одним состоянием, выбранным из акромегалии, раковых заболеваний, карцином, меланом, опухолей, экспрессирующих по меньшей мере один рецептор соматостатина, sst(2)-положительных опухолей, sst(5)-положительных опухолей, рака предстательной железы, эндокринных опухолей гастроэнтеропанкреатической системы, нейроэндокринных опухолей (ГЭП-НЭО) гастроэнтеропанкреатической системы (ГЭП), нейроэндокринных опухолей легкого (НЭО легкого), карциноидных опухолей, инсулином, TSH-секретирующих аденом гипофиза, гастрином, опухолей с образованием вазоактивного интестинального пептида (ВИП) и глюкаго ном, повышенного уровня гормона роста (GH), повышенного уровня инсулиноподобного фактора роста I (IGF-I), варикозного кровотечения (особенно в пищеводе), проблем с желудочно-кишечным трактом, вызванных химиотерапией (таких как диарея), лимфореи, диабетической ретинопатии, офтальмопатии при поражениях щитовидной железы, ожирения, панкреатита и родственных состояний. Указанные способы, в частности, можно применять, если компонент d) представляет собой по меньшей мере один аналог соматостатина, такой как описано в настоящем документе. Предварительные составы, такие как описано в настоящем документе, для применения в указанных способах составляют дополнительный аспект изобретения.
Соответственно, согласно дополнительному аспекту в настоящем изобретении предложено применение имеющей низкую вязкость смеси из:
i) липидной смеси, содержащей:
а) по меньшей мере одно из моно-, ди- или триациллипида и/или токоферола;
b) по меньшей мере один фосфолипид;
c) по меньшей мере один биосовместимый органический растворитель; и
ii) алкиламмониевой соли ЭДТА (например, содержащей анион этилендиаминтетрауксусной кислоты или ее аналога);
где смесь имеет содержание воды в диапазоне от 0 до 1,0 масс.%;
для получения предварительного лекарственного состава с низкой вязкостью для применения в образовании in vivo депо для лечения по меньшей мере одного состояния, выбранного из акромегалии, раковых заболеваний, карцином, меланом, опухолей, экспрессирующих по меньшей мере один рецептор соматостатина, sst(2)-положительных опухолей, sst(5)-положительных опухолей, рака предстательной железы, эндокринных опухолей гастроэнтеропанкреатической системы, нейроэндокринных опухолей гастроэнтеропанкреатической системы (ГЭП-НЭО), НЭ опухолей легкого (НЭО легкого), карциноидных опухолей, инсулином, гастрином, опухолей с образованием вазоактивного интестинального пептида (ВИП) и глюкагоном, TSH-секретирующих аденом гипофиза, повышенного уровня гормона роста (GH), повышенного уровня инсулиноподобного фактора роста I (IGF-I), варикозного кровотечения (особенно в пищеводе), проблем в желудочно-кишечном тракте, вызванных химиотерапией (таких как как диарея), лимфореи, диабетической ретинопатии, офтальмопатии при поражениях щитовидной железы, ожирения, панкреатита и родственных состояний. Указанные способы применения, в частности, можно использовать, если компонент d) представляет собой по меньшей мере один аналог соматостатина, такой как описано в настоящем документе.
Определенные активные агенты (например, определенные пептиды) имеют благоприятные эффекты, которые по природе являются косметическими, но не терапевтическими (или дополняют их). Указанные эффекты включают снижение массы тела и/или подавление чувства голода, а также контролирование пигментации кожи или волос, роста волос и т.д. В настоящем изобретении, таким образом, дополнительно предложен способ косметического лечения субъекта, представляющего собой человека или млекопитающее, отличное от человека, включающий введение указанному субъекту предварительного состава, такого как описано в настоящем документе.
Указанный косметический способ в общем случае не является способом терапии (т.е. не оказывает терапевтическое или медицинское благоприятное действие).
Одно из преимуществ составов согласно настоящему изобретению по сравнению со многими другими композициями с контролируемым высвобождением заключается в том, что они стабильны при хранении в конечной форме, и, таким образом, перед введением они практически или совсем не требуют дополнительной подготовки. Это позволяет получать готовые к введению предварительные составы и также поставлять их в удобной готовой к введению форме. Согласно дополнительному аспекту, таким образом, в изобретении предложено предварительно заполненное устройство для введения, содержащее предварительный состав, такой как описано в настоящем документе. Указанное устройство в общем случае позволяет проводить однократное введение или несколько введений композиции, что будет обеспечивать доставку, например, дозы активного агента в диапазоне от 1 мкг до 15 мг/день, например, от 0,1 мг до 15 мг/день или от 1 мкг до 5 мг/день.
Согласно дополнительному аспекту в изобретении предложен набор, содержащий указанное устройство для введения согласно изобретению.
Набор может необязательно содержать инструкции по подкожному или внутримышечному введению указанного предварительного состава. Все предварительные составы, описанные в настоящем документе, подходят для применения в указанном наборе и, таким образом, могут содержаться в нем.
Наборы согласно настоящему изобретению могут необязательно включать дополнительные компоненты для введения, такие как иглы, тампоны и т.д., и необязательно содержат инструкции по введению.
Согласно дополнительному аспекту в изобретении предложена алкиламмониевая соль ЭДТА, содержащая по меньшей мере один катион алкиламмония формулы NR1R2R3R4n+, такой как определено в настоящем описании, при условии, что катион алкиламмония не является триметиламмонием, тетраметиламмонием, триэтиламмонием или тетраэтиламмонием.
КРАТКОЕ ОПИСАНИЕ ПРИЛАГАЕМЫХ ЧЕРТЕЖЕЙ
Фигура 1. Зависимость уровня октреотида в образцах 53-54 от времени при хранении в условиях 25°C/60% отн.вл. и 40°C/75% отн.вл.
Фигура 2. Зависимость уровня октреотида в образцах 55-60 от концентрации ЭДТА (0-750 ppm или 0-0,075 масс.%) для трех временных интервалов (0, 1 и 2 месяца) после хранения при 40°C/75% отн.вл.
Фигура 3. Зависимость уровня OCT в составах на основе SPC/GDO/EtOH/PG без ЭДТА (образец 61) и с добавкой 100 ppm ЭДТА (образец 62) от времени при 25°C/60% отн.вл. Составы хранили в предварительно заполненных стеклянных шприцах.
Фигура 4. Зависимость уровня OCT в составах на основе SPC/GDO/EtOH/PG от концентрации Fe3+ в присутствии 0,25, 100 и 250 ppm ЭДТА (образцы 63-78), полученная через 1 месяц хранения при 40°C/75% отн.вл. Составы хранили в пробирках, свободное пространство которых занимал воздух окружающей среды.
Фигура 5. Зависимость уровня OCT с составах SPC/GDO/EtOH/PG от мольного отношения ЭДТА:Fe3+ через 1 месяц хранения при 40°C/75% отн.вл. Составы хранили в пробирках, свободное пространство которых занимал воздух окружающей среды. Фигура 6. Зависимость значений уровня (a) и коэффициента стабильности (b) OCT в составах SPC/GDO/EtOH/PG от времени при 40°C/75% отн.вл.: без добавок (образец 79, контрольный), с ЭДТА(Na) (образец 80), с ЭДТА(Na)/ЭТА (образец 81), с ЭДТА (образец 82) и с ЭДТА/ЭТА (образец 83). Составы хранили в пробирках, свободное пространство которых занимал обычный воздух. За исключением контрольного образца 79 все составы также содержали 5 ppm Fe3+.
Фигура 7. Зависимость уровня OCT в составах на основе SPC/GDO/EtOH/PG без ЭДТА (образец 79) и с добавками 100 ppm ЭДТА, растворенной в липидном составе с использованием ЭТА (образец 84), ди-ЭТА (образец 85) или этилендиамина (образец 86) от времени при 40°C/75% отн.вл. Составы хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 8. Зависимость уровня OCT в составах на основе SPC/GDO/EtOH/PG (образец 79 - контрольный без ЭДТА, и образец 84 с добавкой 100 ppm ЭДТА) от времени при 40°C/75% отн.вл. Составы хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 9. Анализ уровня SOM в составах на основе SPC/GDO/EtOH/PG (образец 89 - контрольный без ЭДТА, и образец 90 с добавкой 100 ppm ЭДТА) от времени при 40°C/75% отн.вл. (a) и 25°C/60% отн.вл. (b). Составы хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 10. Зависимость уровня (a) и коэффициента стабильности (b) для GOS в составах SPC/GDO/EtOH/ДМСО без добавок ЭДТА (образец 93) и с добавкой 100 ppm ЭДТА (образец 94) от времени при 40°C/75% отн.вл. Оба состава содержали 5 ppm Fe3+, и их хранили в пробирках, свободное пространство которых занимал обычный воздух. Фигура 11. Зависимость уровня (a) и коэффициента стабильности (b) для OXY в составах SPC/GDO/EtOH/ДМСО без добавок ЭДТА (образец 95) и с добавкой 100 ppm ЭДТА (образец 96) от времени при 40°C/75% отн.вл. Оба состава содержали 5 ppm Fe3+, и их хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 12. Зависимость уровня (a) и коэффициента стабильности (b) для GRN в составах SPC/GDO/EtOH/ДМСО без добавок ЭДТА (образец 97) и с добавкой 100 ppm ЭДТА (образец 98) от времени при 40°C/75% отн.вл. Оба состава содержали 5 ppm Fe3+, и их хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 13. Зависимость уровня (a) и коэффициента стабильности (b) для GOS в составах SPC/GMO/EtOH/ДМСО без добавок ЭДТА (образец 99) и с добавкой 100 ppm ЭДТА (образец 100) от времени при 40°C/75% отн.вл. Оба состава содержали 5 ppm Fe3+, и их хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 14. Зависимость уровня (a) и коэффициента стабильности (b) для GOS в составах SPC/SbOil/EtOH/ДМСО без добавок ЭДТА (образец 101) и с добавкой 100 ppm ЭДТА (образец 102) от времени при 40°C/75% отн.вл. Оба состава содержали 5 ppm Fe3+, и их хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 15. Зависимость концентрации кислорода в свободном пространстве пробирки для составов на основе SPC/GDO (50/50 (масс./масс.)) без добавок ЭДТА (образцы 103 и 104) и с добавкой 100 ppm ЭДТА (105 и 106) без (a) и в присутствии 5 ppm Fe3+ (b) от времени при 60°C/отн.вл. окружающей среды. Составы хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 16. Зависимость концентрации кислорода в свободном пространстве пробирки для составов на основе SPC/GDO (35/65 (масс./масс.)) без добавок ЭДТА (образцы 107 и 108) и с добавкой 100 ppm ЭДТА (109 и 110) без (a) и в присутствии 5 ppm Fe3+ (b) от времени при 60°C/отн.вл. окружающей среды. Составы хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 17. Зависимость концентрации кислорода в свободном пространстве пробирки для составов на основе SPC/GDO (50/50 (масс./масс.)) без добавок ЭДТА (образцы 103 и 104) и с добавкой 100 ppm ЭДТА (105 и 106) без (a) и в присутствии 5 ppm Fe3+ (b) от времени при 40°C/75% отн.вл. Составы хранили в пробирках, свободное пространство которых занимал обычный воздух.
Фигура 18. Зависимость концентрации кислорода в свободном пространстве пробирки для составов на основе SPC/GDO (35/65 (масс./масс.)) без добавок ЭДТА (образцы 107 и 108) и с добавкой 100 ppm ЭДТА (109 и 110) без (a) и в присутствии 5 ppm Fe3+ (b) от времени при 40°C/75% отн.вл. Составы хранили в пробирках, свободное пространство которых занимал обычный воздух.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Липиды и масла, особенно те, которые содержат ненасыщенные группы, склонны к окислению. Поэтому в смесях, которые содержат липиды или масла, постепенно со временем снижается чистота, например, при хранении или применении. Это нежелательно и может приводить к нежелательным изменениям физических и/или химических свойств смеси. Особенно важно минимизировать количество продуктов распада в смесях, применяемых в фармацевтике, так как продукты распада могут быть вредными для пациента, и их содержание в любом случае часто необходимо поддерживать в строго контролируемых пределах.
Липиды и масла плохо смешиваются с водой, и, таким образом, содержание воды в липидах и маслах обычно является низким. Поэтому трудно получать составы липидов или масел с использованием водорастворимых антиоксидантов. Следовательно, было бы желательно найти антиоксидант, который можно было бы включать в составы совместно с липидами или маслами для предотвращения окисления смеси. Настоящее изобретение решает эти проблемы.
Смеси согласно настоящему изобретению являются по существу неводными и включают по меньшей мере один липид и/или масло (компонент i) и по меньшей мере одну алкиламмониевую соль ЭДТА (компонент ii). Согласно предпочтительному аспекту смесь представляет собой предварительный состав. Предварительные составы согласно настоящему изобретению получены на основе липидов, являются по существу неводными и образуют композицию депо при контакте с водной жидкостью. При использовании в настоящем описании термины «состав» или «предварительный состав» относятся к смеси компонентов (i) и (ii) (где компонент (i) содержит компоненты (a), (c) и необязательно (b) и (d)), которая, как правило, имеет низкую вязкость. Термин «депо» относится к композиции, которая образуется при взаимодействии предварительного состава с избытком водной жидкости, например, которое происходит при разнообразных способах парентерального введения. Не желая быть связанными теорией, полагают, что это изменение вызвано по меньшей мере отчасти заменой растворителя (с) на водную жидкость. Депо, как правило, имеет гораздо более высокую вязкость по сравнению с соответствующим предварительным составом и обеспечивает постепенное высвобождение какого-либо активного агента, содержащегося в депо.
Согласно предпочтительному аспекту составы согласно настоящему изобретению образуют неламеллярную фазу (например, неламеллярную жидкокристаллическую фазу) после введения. Применеие структур с неламеллярной фазой (таких как жидкокристаллические фазы) при доставке биоактивных агентов в настоящее время относительно хорошо изучено. Наиболее эффективная система депо с использованием липидов описана в WO 2005/117830, и подходящая липидная матрица для применения в настоящем изобретении описана в этом документе, содержание которого включено в настоящий документ во всей полноте посредством ссылки. Описание наиболее эффективных фазовых структур для указанных составов можно найти в обсуждении WO 2005/117830 и в частности на странице 29 указанной публикации. Предпочтительно предварительный состав согласно настоящему изобретению имеет структуру фазы L2 (жидкая фаза) или представляет собой жидкий раствор или молекулярный раствор.
Все значения в % указаны в настоящем документе по массе, если не указано иное. Значение в процентах (%) по массе может быть сокращенно описано как, например, масс.%. Кроме того, указанное значение в % по массе соответствует % от всего предварительного состава, включая все компоненты, указанные в настоящем документе, если не указано иное. Если содержание в процентах по массе указано для компонента (d), то масса относится к количеству свободного основания (например, в случае использования соли), если не указано иное. В определенных примерах приведено содержание конкретных солей в масс.%, но это значение указывается по мере необходимости и может быть легко переведено в соответствующую массу свободного основания.
Предварительные составы могут необязательно состоять по существу только из компонентов, указанных в настоящем документе (включая, при необходимости, дополнительные необязательные компоненты, указанные ниже в настоящем документе и в прилагаемой формуле изобретения), и согласно одному из аспектов полностью состоят из таких компонентов.
Предварительные составы на липидной основе, описанные в настоящем документе, содержат липидную смесь (i), которая включает липидные компоненты (а), органический растворитель (с) и необязательно (b) и (d), и алкиламмониевую соль ЭДТА (ii).
Авторы настоящего изобретения неожиданно установили, что при соответствующем выборе антиоксиданта стойкость к окислению липида и/или масла, а также в случае предварительных составов любого активного агента, содержащегося в предварительном составе, может быть значительно улучшена.
Хотя известны различные алкиламмониевые соли ЭДТА, например, описанные в Scott and Kyffin (Biochem.J. (1978) 169, 697-701), их использование в качестве антиоксиданта в липидных системах и совместимость с указанными составами на момент подачи заявки были не известны. Скотт и Киффин описывают использование растворимых солей ЭДТА при деминерализации образцов костей, где ЭДТА действует как хелатообразующий агент. Утверждается, что особенно подходящим раствором является 80% водный этанол, содержащий 0,2 М ЭДТА триметиламмония. Его использование в липидных составах или растворимость в липидах не рассматриваются. Задачей соли ЭДТА согласно настоящему изобретению является действие в качестве консервирующего или увеличивающего стабильность агента в липидных составах, и она значительно отличается от того, что было описано ранее.
Компонент i) - Липид и/или масло
Во всех вариантах реализации изобретения смесь содержит по меньшей мере один липид и/или масло (компонент i)и имеет содержание воды 0-1,0 масс.%. Смеси липидов, смеси масел или смеси липидов и масел можно применять в качестве компонента i).
При использовании в настоящем описании термин «масло» относится к насыщенным или ненасыщенным C5-C70 углеводородам, которые являются жидкими при комнатных температуре и давлении. Предпочтительными маслами для применения в изобретении являются насыщенные или ненасыщенные C10-C60 углеводороды, предпочтительно насыщенные или ненасыщенные C10-C40 углеводороды.
В одном из вариантов реализации компонент i) представляет собой масло, которое подходит для применения в качестве смазывающего вещества. Указанные масла, как правило, представляют собой насыщенные С10-С40 углеводороды. Желательно, чтобы смазывающие вещества были устойчивы к окислению, так как окисление приводит к увеличению вязкости смазывающего вещества.
В одном из вариантов реализации компонент i) содержит, состоит по существу или состоит из по меньшей мере одной жирной кислоты или сложного эфира жирной кислоты (липида). Жирные кислоты/липиды отличаются от «масел» тем, что они содержат полярную «головную группу» карбоновой кислоты или сложного эфира, а углеводородная цепь образует неполярную «хвостовую» группу. Сложные эфиры жирных кислот представляют собой этерифицированные жирные кислоты. Жирные кислоты или сложные эфиры, применяемые в настоящем изобретении, могут быть твердыми или жидкими при комнатных температуре и давлении, предпочтительно жидкими.
Примеры неполярных «хвостовых» групп включают C6-С32 алкильные и алкенильные группы, которые, как правило, содержатся в виде длинноцепочечных карбоновых кислот или их сложных эфиров. Их часто описывают при помощи количества атомов углерода и количества ненасыщенных участков в углеродной цепи. Таким образом, CX:Z обозначает углеводородную цепь, содержащую X атомов углерода и Z ненасыщенных участков. Примеры, в частности, включают лауроильную (C12:0), миристоильную (C14:0), пальмитоильную (C16:0), фитаноильную (C16:0), пальмитолеоильную (C16:1), стеароильную (C18:0), олеоильную (C18:1), элаидоильную (C18:1), линолеоильную (C18:2), линоленоильную (C18:3), арахидоноильную (C20:4), бегеноильную (C22:0) и лигноцероильную (C24:9) группы. Во избежание сомнений, если в настоящем документе описано количество атомов углерода в «цепи» или «хвосте», то указанное количество включает атом углерода фрагмента -C(O)O-, как принято в данной области техники.
Таким образом, типичные неполярные цепи основаны на жирных кислотах природных сложноэфирных липидов, включая капроновую, каприльную, каприновую, лауриновую, миристиновую, пальмитиновую, фитановую, пальмитоловую, стеариновую, олеиновую, элаидиновую, линолевую, линоленовую, арахидоновую, бегеновую или лигноцериновую кислоты, или соответствующих спиртах.
Предпочтительными неполярными цепями являются пальмитиновая, стеариновая, олеиновая и линолевая кислоты, в частности, олеиновая кислота.
Липид(-ы) может(-гут) быть насыщенным(-и) или ненасыщенным(-и), но предпочтительно содержит(-ат) по меньшей мере 1 масс.% ненасыщенного липида (в пересчете на общее содержание липидов), например, по меньшей мере 5 масс.% (5-100%), по меньшей мере 15 масс.% ( 15-100%), по меньшей мере 30 масс.% (30-100%), по меньшей мере 50 масс.% (50-100%) или по меньшей мере 80 масс.% (80-100%).
В одном из вариантов реализации компонент i) представляет собой отдельную жирную кислоту/сложный эфир жирной кислоты или смесь жирных кислот/сложных эфиров жирных кислот. Как правило, компонент i) содержит смесь насыщенных и ненасыщенных жирных кислот. В предпочтительном варианте реализации липид(-ы) и/или масло(-а) экстрагируют из природного источника.
В одном из вариантов реализации компонент i) представляет собой пищевой липид, такой как миндальное масло, масло авокадо, сливочное масло, масло канолы, касторовое масло, кокосовое масло, кукурузное масло, хлопковое масло, льняное масло, топленое масло, свиной жир, льняное масло, масло макадамии, маргарин, горчичное масло, оливковое масло, пальмовое масло, арахисовое масло, масло тыквенных семечек, масло рисовых отрубей, сафлоровое масло, кунжутное масло, соевое масло, подсолнечное масло, масло чайных семян, растительное масло или масло грецкого ореха. Во избежание сомнений, вышеуказанные пищевые липиды являются «липидами», но не «маслами», если рассматривать компонент (i), так как они содержат жирные кислоты, в частности, в виде сложных эфиров жирных кислот, но не углеводороды.
В одном из вариантов реализации компонент i) является таким, как определено для компонента а) или b), что описано в последующих разделах.
В особенно предпочтительном варианте реализации смесь состоит по существу или состоит из компонентов i) и ii).
Предварительные составы
Предварительный состав представляет собой подгруппу «смесей», описанных выше, в которых компонент i) представляет собой липидную смесь и содержит по меньшей мере один нейтральный липидный «компонент а)» и необязательно по меньшей мере один фосфолипидный «компонент b)». Предварительные составы дополнительно содержат компонент с) и необязательно компонент d), как описано ниже.
Компонент а) - Нейтральный липид
Предпочтительные диапазоны содержания компонента a) составляют 20-90 масс.% от предварительного состава, предпочтительно 30-70 масс.%, более предпочтительно 33-60% (например, 43-60%), в частности, от 38 до 43%.
Компонент «а», такой как указано в настоящем описании, представляет собой по меньшей мере один моно-, ди- или триациллипид, содержащий полярную «головную» группу и по меньшей мере одну неполярную «хвостовую» группу. В качестве альтернативы компонент а) может содержать или состоять из токоферола(-ов). Согласно предпочтительному аспекту компонент а) содержит по меньшей мере один нейтральный диациллипид (не имеющий суммарного заряда при физиологическом рН). При использовании в настоящем описании термин «ациллипид» относится к липидному компоненту, содержащему полиольную «головную» группу и одну или более неполярных «хвостовых групп». В определенных вариантах реализации полиол может представлять собой глицерин, сахар или гекситан, такой как сорбитан. Термин «гекситан» обозначает гексит формулы HOCH2(СНОН)4СН2ОН, который циклизовался в результате формального удаления одного эквивалента воды с образованием пяти- или шестичленного кольца, предпочтительно пятичленного фуранозного кольца. Сорбитан является особенно подходящей «головной группой», в частности, в качестве компонента моноациллипидного компонента в определенных вариантах реализации.
В случае ди- и триациллипидов липидный компонент наиболее предпочтительно должен содержать глицериновую головную группу и две или три неполярные хвостовые группы. Две или три неполярные группы могут содержать одинаковое или различное количество атомов углерода, и каждая из них независимо может быть насыщенной или ненасыщенной. Примеры неполярных групп включают C6-С32 алкильные и алкенильные группы, которые, как правило, присутствуют в виде сложных эфиров длинноцепочечных карбоновых кислот. Их часто описывают при помощи количества атомов углерода и количества ненасыщенных участков в углеродной цепи. Таким образом, CX:Z обозначает углеводородную цепь, содержащую X атомов углерода и Z ненасыщенных участков. Примеры, в частности, включают лауроильную (C12:0), миристоильную (C14:0), пальмитоильную (C16:0), фитаноильную (C16:0), пальмитолеоильную (C16:1), стеароильную (C18:0), олеоильную (C18:1), элаидоильную (C18:1), линолеоильную (C18:2), линоленоильную (C18:3), арахидоноильную (C20:4), бегеноильную (C22:0) и лигноцероильную (C24:9) группы. Таким образом, типичные неполярные цепи основаны на жирных кислотах природных сложноэфирных липидов, включая капроновую, каприльную, каприновую, лауриновую, миристиновую, пальмитиновую, фитановую, пальмитоловую, стеариновую, олеиновую, элаидиновую, линолевую, линоленовую, арахидоновую, бегеновую или лигноцериновую кислоты, или соответствующих спиртах. Предпочтительными неполярными цепями являются пальмитиновая, стеариновая, олеиновая и линолевая кислоты, в частности, олеиновая кислота.
Смеси любого количества моноацил-, диацил- и/или триациллипидов можно применять в качестве компонента а). Указанный компонент предпочтительно включает по меньшей мере часть С18 липидов (например, диацилглицерин (DAG), содержащий одну или более С18:0, С18:1, С18:2 или С18:3 неполярных групп), такой как глицерилдиолеат (GDO) и/или глицерилдилинолеат (GDL). Крайне предпочтительным примером является DAG, содержащий по меньшей мере 50%, предпочтительно по меньшей мере 80% и даже содержащий по существу 100% GDO.
Так как GDO и другие диацилглицерины могут быть получены из природных источников, обычно существует определенная доля «примесного» липида, имеющего цепи другой длины, и т.д. В этом контексте «чистый» GDO представляет собой сложный диэфир глицерина и двух C18:1 жирных кислот. Любой другой диацилглицерин рассматривают как примесь. Согласно одному из аспектов GDO используют в настоящем описании для обозначения любого коммерческого класса GDO с сопутствующими примесями (т.е. GDO коммерческого класса чистоты). Указанные примеси могут быть отделены и удалены путем очистки, но при условии того, что степень чистоты является одинаковой, это редко требуется. Тем не менее, при необходимости «GDO» может представлять собой по существу химический чистый GDO, такой как GDO, имеющий чистоту по меньшей мере 70%, предпочтительно по меньшей мере 75% и более предпочтительно по меньшей мере 80%. Соответственно, содержание С18:1 в GDO, описанном в настоящем документе, может составлять примерно 80%, предпочтительно по меньшей мере 85% и более предпочтительно по меньшей мере 90%.
Следует понимать, что любой применяемый материал, включая компонент а), может включать неудаляемые следовые примеси металлов, необязательно включая тяжелые металлы. Согласно сертификатам анализа коммерчески доступного GDO (например, производства Croda), типичная максимальная концентрация тяжелых металлов (или элементных примесей) в GDO составляет 5 ppm. Не будучи связанными теорией, полагают, что частое присутствие указанных металлических компонентов и их связывание согласно различным аспектам настоящего изобретения могут по меньшей мере частично определять наблюдаемую дополнительную стабильность. Тем не менее, более распространенной проблемой может являться присутствие ионов железа, которые могут абсорбироваться из сплавов на основе железа, используемых при обработке/хранении материалов.
Компонент b) - Фосфолипид
Необязательный компонент «b» в предпочтительных липидных матрицах согласно настоящему изобретению представляет собой по меньшей мере один фосфолипид. Согласно WO 2016/066655 известно, что липидные матрицы с медленным высвобождением на основе триациллипидов могут образовывать композиции депо при воздействии водных жидкостей и не требуют присутствия фосфолипидного компонента, хотя фосфолипид также может присутствовать. Таким образом, в одном из вариантов реализации компонент a) содержит, состоит или состоит по существу из триациллипида(-ов), и компонент b) является необязательным. Тем не менее, если компонент а) содержит более 50% моноацил- или диациллипидов или токоферола или смесей любых из указанных компонентов, то фосфолипидный компонент b) предпочтительно будет содержаться. В одном из вариантов реализации компонент a) содержит менее 50% (например, от 0 до 45%) триациллипида (в пересчете на общее количество компонента a)), и компонент b) присутствует (например, составляет от 20 до 80 масс.% предварительного состава).
Предпочтительные диапазоны содержания компонента b), если он присутствует, составляют 20-80 масс.% от предварительного состава, предпочтительно 30-70 масс.%, более предпочтительно 33-55% (например, 35-55%), в частности, от 38 до 43%. Если компонент b) присутствует, то отношение a:b, как правило, составляет от 40:60 до 70:30, предпочтительно от 45:55 до 55:45 и более предпочтительно от 40:60 до 54:46, например, от 45:55 до 54:46 или от 47:53 до 53:47. Отношения примерно 50:50 (например, от 49:51 до 51:49) являются крайне эффективными в определенных вариантах реализации.
Предпочтительные фосфолипидные полярные «головные» группы включают фосфатидилхолин, фосфатидилэтаноламин, фосфатидилсерин и фосфатидилинозитол.
Наиболее предпочтительными являются фосфатидилхолин (PC) и фосфатидилэтаноламин (PE), особенно, PC. Как и в случае компонента а), этот компонент содержит полярную головную группу и по меньшей мере одну неполярную хвостовую группу. Различие между компонентами а) и b) определяется, главным образом, полярной группой. Таким образом, неполярные фрагменты могут быть получены подходящим способом из жирных кислот или соответствующих спиртов, рассмотренных выше для компонента а). Фосфолипид содержит две неполярные группы. И снова C18 группы являются предпочтительными и могут быть объединены с любой другой подходящей неполярной группой, в частности, с C16 группами.
Фосфолипидный фрагмент может быть получен из природного источника. В случае PC подходящие источники фосфолипидов включают яйца, сердце (например, крупного рогатого скота), головной мозг, печень (например, крупного рогатого скота) и растительные источники, включая сою. Указанные источники могут обеспечивать один или более ингредиентов компонента b, который может содержать любую смесь фосфолипидов. Можно применять любой отдельный PC или смесь PC из указанных или других источников, но наиболее подходят смеси, содержащие соевый PC или яичный PC. Компонент PC предпочтительно содержит по меньшей мере 50% соевого PC или яичного PC, более предпочтительно по меньшей мере 75% соевого PC или яичного PC и наиболее предпочтительно по существу чистый соевый PC или яичный PC.
В одном из вариантов реализации, относящемся ко всем аспектам изобретения, компонент b) содержит PC. Предпочтительно PC получают из сои. Предпочтительно PC содержит 18:2 жирные кислоты в качестве основного компонента жирных кислот и 16:0 и/или 18:1 в качестве вторичных компонентов жирных кислот. Они предпочтительно присутствуют в PC в отношении от 1,5:1 до 6:1. PC, содержащий примерно 60-65% 18:2, от 10 до 20% 16:0, 5-15% 18:1, где оставшуюся часть составляют, главным образом, другие содержащие 16 атомов углерода и 18 атомов углерода жирные кислоты, является предпочтительным и типичным соевым PC.
В альтернативном, но в равной степени предпочтительном варианте реализации компонент PC может содержать синтетический диолеоил-PC (DOPC). Применение DOPC может обеспечивать повышенную стабильность и, таким образом, может быть особенно предпочтительным для композиций, которые должны сохранять стабильно сть при длительном хранении и/или имеют продолжительный период высвобождения in vivo. В указанном варианте реализации компонент PC предпочтительно содержит по меньшей мере 50% синтетического диолеоил-PC, более предпочтительно по меньшей мере 75% синтетического диолеоил-PC и наиболее предпочтительно по существу чистый синтетический диолеоил-PC. Любой оставшийся PC предпочтительно представляет собой соевый или яичный PC, как указано выше.
Так как предварительные составы согласно изобретению необходимо вводить субъекту, возможно с включением активного агента, важно, чтобы компоненты были биосовместимыми. В этом отношении предпочтительные липидные матрицы для применения в предварительных составах согласно настоящему изобретению являются особенно предпочтительными, так как токоферол, РС и ацилглицерины, в частности DAG, хорошо переносятся и разрушаются in vivo на компоненты, которые обычно присутствуют в организме млекопитающих.
Следует понимать, что компонент b) может включать неудаляемые следовые примеси тяжелых металлов. Согласно сертификатам анализа коммерчески доступного соевого PC (например, производства Lipoid), типичная максимальная концентрация тяжелых металлов (или элементных примесей) в соевом PC составляет 10 ppm.
Синтетические или высокочистые PC, такие как диолеоилфосфатидилхолин (DOPC), являются особенно эффективными в качестве компонента b) в целом или его части. Синтетический диолеоил-PC наиболее предпочтительно представляет собой 1,2-диолеоил-sn-глицеро-3-фосфохолин, и другие синтетические компоненты PC включают DDPC (1,2-дидеканоил-sn-глицеро-3-фосфохолин); DEPC (1,2-диэрукоил-sn-глицеро-3-фосфохолин); DLOPC (1,2-дилинолеоил-sn-глицеро-3-фосфохолин); DLPC (1,2- дилауроил-sn-глицеро-3-фосфохолин); DMPC (1,2-димиристоил-sn-глицеро-3-фосфохолин); DOPC (1,2-диолеоил-sn-глицеро-3-фосфохолин); DPPC (1,2-дипальмитоил-sn-глицеро-3-фосфохолин); DSPC (1,2-дистеароил-sn-глицеро-3-фосфохолин); MPPC (1-миристоил-2-пальмитоил-sn-глицеро-3-фосфохолин); MSPC (1-миристоил-2-стеароил-sn-глицеро-3-фосфохолин); PMPC (1-пальмитоил-2-миристоил-sn-глицеро-3-фосфохолин); POPC (1-пальмитоил-2-олеоил-sn-глицеро-3-фосфохолин); PSPC (1-пальмитоил-2-стеароил-sn-глицеро-3-фосфохолин); SMPC (1-стеароил-2-миристоил-sn-глицеро-3-фосфохолин); SOPC (1-стеароил-2-олеоил-sn-глицеро-3-фосфохолин); и SPPC (1-стеароил-2-пальмитоил-sn-глицеро-3-фосфохолин) или любую их комбинацию.
Особенно предпочтительной комбинацией компонентов a) и b) являются GDO и PC, в частности, GDO и соевый PC и/или DOPC. Соответствующие количества каждого компонента, подходящего для комбинации, представляют собой те количества, которые указаны в настоящем документе для отдельных компонентов в любой комбинации. Это относится также к любым комбинациям компонентов, указанным в настоящем документе, если это допускается по контексту.
Компонент с) - биосовместимый органический растворитель
Компонент с) в предварительных составах согласно изобретению представляет собой по меньшей мере один биосовместимый органический растворитель. Так как предварительный состав предназначен для образования композиции депо после введения (например, in vivo), как правило, при контакте с избытком водной жидкости, желательно, чтобы указанный растворитель переносился субъектом и мог смешиваться с водной жидкостью и/или диффундировать или растворяться из предварительного состава в водной жидкости. Растворители, имеющие по меньшей мере умеренную растворимость в воде, таким образом, являются предпочтительными. Как будет описано ниже, компонент с) может включать полярный сорастворитель.
Компонент с) содержит или состоит по меньшей мере из одного растворителя, выбранного из группы, состоящей из: спиртов, аминов, амидов или сложных эфиров.
Предпочтительно компонент с) содержит по меньшей мере одноатомный спирт в качестве растворителя. Наиболее предпочтительно компонент с) содержит этанол, пропанол, изопропанол или их смеси. Особенно предпочтительно компонент с) содержит или состоит из этанола. Компонент с) может содержать или состоять из одноатомного спирта в качестве растворителя, предпочтительно этанола, и полярного сорастворителя. Смеси, содержащие или состоящие из этанола и пропиленгликоля, также являются крайне предпочтительными.
Количество компонента с) в предварительном составе оказывает значительное влияние на некоторые отличительные признаки. В частности, вязкость и скорость (и продолжительность) высвобождения могут значительно изменяться в зависимости от уровня растворителя. Таким образом, количество растворителя является по меньшей мере достаточным для обеспечения смеси с низкой вязкостью, но также, в соответствии с дополнительным определением, для обеспечения желаемой скорости высвобождения.
Как правило, уровень растворителя от 1 до 30%, в частности от 2 до 20%, обеспечивает подходящие свойства высвобождения и вязкости. В некоторых вариантах реализации предпочтительными являются уровни от 2 до 18%, такие как от 2 до 16%, в частности, от 2 до 15%.
Как указано выше, количество компонента с) в предварительных составах согласно изобретению по меньшей мере является достаточным для обеспечения смеси с низкой вязкостью (например, молекулярного раствора) компонентов а), с) и ii) (компонентов b) и d), которые являются необязательными, что описано в настоящем документе), и может быть легко определено для любой конкретной комбинации компонентов стандартными способами.
Фазовое поведение может быть проанализировано способами, такими как визуальное наблюдение в комбинации с поляризованной световой микроскопией, способы рассеяния и дифракции рентгеновского излучения, ядерный магнитный резонанс и крио-просвечивающая электронная микроскопия (крио-ПЭМ) для обнаружения растворов, L2 или L3 фаз или жидкокристаллических фаз или, как в случае крио-ПЭМ, дисперсных фрагментов указанных фаз. Вязкость может быть измерена напрямую стандартными средствами. Как описано выше, подходящей вязкостью для практического применения является вязкость, которая позволяет проводить эффективное введение при помощи шприца и, в частности, стерильное фильтрование. Она может быть определена легко, как указано в настоящем документе.
Крайне предпочтительной комбинацией компонентов a), b) и c) является GDO, соевый PC и этанол, в частности, GDO, соевый PC и смеси этанола и пропиленгликоля. Как указано выше, соответствующие количества каждого компонента, подходящего для комбинации, представляют собой те количества, которые указаны в настоящем документе для отдельных компонентов в любой комбинации.
Предпочтительно галогензамещенные углеводороды должны составлять небольшую часть компонента с) или отсутствовать, так как они имеют более низкую биосовместимость.
Компонент c) при использовании в настоящем описании может представлять собой отдельный растворитель или смесь подходящих растворителей, но в общем случае имеет низкую вязкость. Вязкость компонента-растворителя с «низкой вязкостью» c) (отдельного растворителя или смеси), как правило, должна составлять не более 18 мПа⋅с при 20°C. Предпочтительно она составляет не более 15 мПа⋅с, более предпочтительно не более 10 мПа⋅с и наиболее предпочтительно не более 7 мПа⋅с при 20°С.
Согласно описанию WO 2012/160213 добавление полярного растворителя к растворителю на основе одноатомного спирта обеспечивает многочисленные преимущества, включая пониженную вязкость и профиль с пониженным мгновенным высвобождением активного агента. В дополнение к предпочтительным аспектам, описанным ранее для компонента с), в одном особенно предпочтительном варианте реализации компонент с) содержит растворитель на основе одноатомного спирта и полярный сорастворитель. Термин «полярный сорастворитель» при использовании в настоящем описании определяет растворитель, имеющий диэлектрическую проницаемость (diel) по меньшей мере 28 при 25°С, более предпочтительно по меньшей мере 30 при 25°С, но который не является водой или какой-либо водной жидкостью. К наиболее подходящим примерам относятся пропиленгликоль (diel ~ 32) и N-метил-2-пирролидон (NMP, diel ~ 32). Предпочтительные уровни компонента с), приведенные в настоящем описании, в равной степени применимы к смесям растворителя на основе одноатомного спирта и полярного сорастворителя, если по контексту не допускается иное.
В особенно предпочтительном варианте реализации компонент с) содержит, состоит по существу из или состоит из смеси растворителя на основе одноатомного спирта и полярного сорастворителя. В одном из вариантов реализации полярный сорастворитель может представлять собой органический растворитель на основе двухатомного C3-C6 спирта, т.е. C3-C6 органический растворитель, содержащий две гидроксигруппы.
Растворитель на основе двухатомного спирта предпочтительно представляет собой пропиленгликоль. Полярный сорастворитель, если присутствует, содержится в количестве от 2 до 12 масс.% в предварительном составе, например, от 3 до 10 масс.%, в частности, от 4 до 9 масс.%. Этот уровень рассматривают как часть диапазонов, указанных выше для компонента с). В одном из вариантов реализации компонент c) содержит, состоит по существу из или состоит из смеси этанола и пропиленгликоля (PG).
Если одновременно содержатся органический растворитель на основе одноатомного спирта и полярный сорастворитель, например, этанол и PG, то отношение растворителя на основе одноатомного спирта к полярному сорастворителю предпочтительно находится в диапазоне от 20:80 до 70:30, предпочтительно от 30:70 до 70:30 (масс./масс.), более предпочтительно от 40:60 до 60:40. Примерно равные количества компонентов на основе одно- и двухатомных спиртов являются крайне эффективными.
В особенно предпочтительном варианте реализации компонент с) присутствует в количестве от 1 до 30% и содержит, состоит или состоит по существу из смеси этанола и PG, где отношение этанола к PG (масс./масс.) находится в диапазоне от 30:70 до 70:30, предпочтительно от 40:60 до 60:40. Более предпочтительно компонент с) присутствует в количестве в диапазоне от 5 до 15 масс.% или от 8 до 18 масс.%, наиболее предпочтительно 8-18 масс.% и представляет собой смесь этанола и PG в соотношении от 40:60 до 60:40 (масс./масс.).
Во избежание сомнений, даже если в предварительных составах согласно настоящему изобретению присутствует полярный сорастворитель, общее содержание воды сохраняется на уровне, описанном в различных вариантах реализации в настоящем документе (например, от 0,1 до 1,0 масс.%).
Компонент ii) - алкиламмониевая соль
Компонент ii) представляет собой алкиламмониевую соль, содержащую анион ЭДТА («этилендиаминтетрауксусная кислота» или «эдетовая кислота») или анион аналога ЭДТА, такой как описано ниже, и по меньшей мере один катион алкиламмония формулы (I):
NR1R2R3R4 n+(I)
где каждый R1-R4 независимо представляет собой H или линейную или разветвленную C1-10 группу (как описано в настоящем документе) при условии, что по меньшей мере один из R1-R4 не является H.
Как правило, и предпочтительно n = 1. Тем не менее, для солей аммония, содержащих более одного атома азота, таких как этилендиамин (NH2CH2CH2NH2), может существовать смесь катионов с зарядом +1 и +2 (т.е. NH2CH2CH2NH3+ и NH3CH2CH2NH32+). Образование поликатионных фрагментов может предотвращаться до определенной степени за счет обеспечения избытка предшественника амина, как описано ниже. Тем не менее, специалистам в данной области техники будет понятно, что возможно образование смешанных катионов.
Каждый из R1-R4 может быть одинаковым или различным при условии, что по меньшей мере один из R1-R4 не является H. Предпочтительно все группы-заместители R1-R4, которые не являются H, являются одинаковыми. Таким образом, предпочтительными катионами являются NRH3+, NR2H2+ и NR3H+ или NR4+, где группы «R» являются одинаковыми. Первичные, вторичные и третичные аммониевые катионы являются более предпочтительными по сравнению с четвертичными катионами, так как первые можно легко получать путем объединения соответствующего амина с ЭДТА, как описано ниже.
Каждый из R1-R4 независимо представляет собой H или линейную или разветвленную C1-10 алкильную, алкенильную или алкинильную группу, предпочтительно C1-C5. Наиболее предпочтительно каждый из R1-R4 представляет собой линейную или разветвленную C1-5 алкильную группу, в частности, линейную C1-C5 или C1-C3 алкильную группу.
Каждый R1-R4 независимо может быть дополнительно замещен одной или более группами OH или NH2 (или NH3+). В одном из вариантов реализации заместитель R, содержащий m атомов углерода, может максимально содержать m-1 групп OH и/или NH2. Например, если R1 представляет собой С8, то R1 может содержать до 7 групп ОН, в частности по одной группе ОН, присоединенной к каждому атому углерода, отличному от атома углерода, непосредственно присоединенного к атому N аммония. Указанный вариант реализации является особенно важным в случае, где катион алкиламмония получен из аминополиола (например, меглумина (MeNHCH2(CHOH)4CH2OH)). В качестве альтернативного примера, если R1 представляет собой С3, то R1 может содержать до 2 групп ОН, например, серинол (NH2СН(СН2ОН)2). В одном из вариантов реализации по меньшей мере один из R1-R4 представляет собой линейную C1-C6 группу, замещенную по меньшей мере одной группой OH или NH2.
В одном из вариантов реализации любые две группы R1-R4, взятые вместе, образуют C4-C8, предпочтительно C4-C6 кольцо, которое может необязательно содержать одну или более экзоциклических групп OH или NH2. Если любые две группы R1-R4, взятые вместе, образуют кольцо, то также может содержаться одно эндоциклическое звено O или NH. В частности, предполагается, что можно применять соли морфолина (т.е., если любые два из R1-R4, взятые вместе, образуют шестичленное C4 кольцо, содержащее один эндоциклический атом O). В указанном варианте реализации две группы R1-R4 совместно с N образуют морфолиновое кольцо, где оставшиеся группы R1-R4 имеют приведенные выше определения.
Особенно предпочтительные катионы алкиламмония включают катионы, полученные в результате N-протонирования или, в менее предпочтительном варианте реализации, N- алкилирования амина, выбранного из:
этаноламина «ЭТА» (NH2(CH2CH2OH));
диэтаноламина «ди-ЭТА» (NH(CH2CH2OH)2);
меглумина (NH(CH3)CH2(CHOH)4CH2OH));
трис-гидроксиметиламина «TRIS» (N(CH2OH)3);
этилендиамина (NH2CH2CH2NH2); или
серинола (NH2CH(CH2OH)2).
Предпочтительно масса катиона алкиламмония формулы (I) должна быть ниже 500 а.е.м., предпочтительно ниже 350 а.е.м., в частности, ниже 250 а.е.м. Соли ЭДТА, содержащие ион этаноламмония (HOCH2CH2NH3+), являются особенно предпочтительными согласно настоящему изобретению. Наиболее предпочтительно соль ЭДТА должна представлять собой соль ЭДТА с этаноламином (ЭТА), предпочтительно соль ЭДТА только с ЭТА.
В одном из вариантов реализации изобретение относится к солям ЭДТА, содержащим анион ЭДТА и по меньшей мере один катион алкиламмония формулы (I), как описано выше, при условии, что катион алкиламмония не является триметиламмонием, тетраметиламмонием, триэтиламмонием или тетраэтиламмонием.
Считается, что катион алкиламмония способствует увеличению растворимости в липидах соли ЭДТА по сравнению с традиционной солью ЭДТА с металлом (неорганическая соль), такой как ЭДТА динатрия. Так как ЭДТА содержит четыре звена карбоновой кислоты, алкиламмониевая соль может содержать до четырех катионов аммония и тетраанионный анион ЭДТА.
При использовании в настоящем описании термин «ЭДТА» может представлять собой этилендиаминтетрауксусную кислоту как таковую. В качестве альтернативы, ЭДТА, как указано в настоящем документе, может включать как саму этилендиаминтетрауксусную кислоту, так и аналоги ЭДТА. Таким образом, «ЭДТА» в данном документе включает «ЭДТА и ее аналоги», если это допускает контекст.
Подходящими аналогами ЭДТА являются те, которые содержат по меньшей мере одно глицинатное звено (т.е. звено -NCH2COO-) в молекуле, предпочтительно по меньшей мере 2, по меньшей мере 3 или по меньшей мере 4 глицинатных звена. Подходящие аналоги ЭДТА включают:
иминодиуксусную кислоту (IDA) - (NH(CH2CO2H)2;
нитрилотриуксусную кислоту (NTA) - N(CH2CO2H)3;
пентетовую кислоту* - N(CH2CO2H)2CH2CH2N(CH2CO2H)CH2CH2N(CH2CO2H)2;
эгтазиновую кислоту - N(CH2CO2H)2CH2CH2OCH2CH2OCH2CH2N(CH2CO2H)2
NOTA - [N(CH2CO2H)CH2CH2]3
DOTA - [N(CH2CO2H)CH2CH2]4
* Также известна как «DTPA»
В одном из вариантов реализации аналог ЭДТА имеет структуру, указанную ниже на формуле (II):
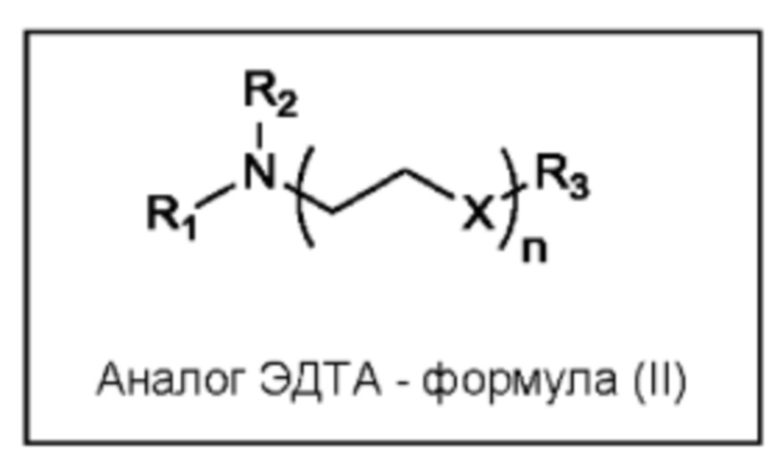
где n равен 1-10, предпочтительно 1-5, в частности, 1, 2 или 3;
X представляет собой CH2, O или NR4
каждый R1, R2, R3 и R4 по отдельности представляет собой H или CH2CO2H, предпочтительно CH2CO2H; или
R1 и R3 совместно представляют собой ковалентную связь (т.е. аналог ЭДТА является циклическим), и каждый R2 и R4 по отдельности представляет собой H или CH2CO2H, предпочтительно CH2CO2H.
Количества ЭДТА и отношения ЭДТА к (d), определенные в настоящем документе, в равной мере относятся к ЭДТА и аналогам ЭДТА. Во всех вариантах реализации ЭДТА предпочтительно применяют в качестве противоиона в компоненте (ii).
Получение соли ЭДТА
Соль ЭДТА можно получать предварительно и растворять или диспергировать в одном или более компонентах перед получением смеси, например, предварительного состава, или она может образовываться in situ. Образование in situ обычно является предпочтительным, благодаря простоте реализации. Подходящий способ получения алкиламмониевой соли ЭДТА включает растворение ЭДТА (в форме кислоты) и необходимого алкиламина (основание) в компоненте-растворителе (с) или в растворителе, который является предшественником (или подкомпонентом) компонента-растворителя (c), и обеспечение перемешивания до полного растворения твердых веществ. В случае смесей, отличных от предварительных составов, таких как определено в настоящем документе, соль ЭДТА можно получать предварительно и растворять или диспергировать в компоненте i).
Авторы изобретения установили, что, как правило, в случае моноамина требуется по меньшей мере 3,0, предпочтительно по меньшей мере 3,5 (например, от 3,5 до 10) мольных эквивалента амина (который является предшественником соли аммония) относительно количества ЭДТА для растворения соли в компоненте-растворителе (с). Как описано в примерах, минимальное отношение амина к ЭДТА, необходимое для растворения соли, может быть различным в зависимости от конкретного выбора алкиламмониевой соли. Тем не менее, соответствующее мольное отношение может быть определено экспериментально путем простого наблюдения, при каком мольном избытке алкиламина твердая ЭДТА полностью растворяется в растворителе. В одном из вариантов реализации амин добавляют в количестве, превышающем стехиометрическое количество амина, которое формально требуется для получения тетрааммониевой соли ЭДТА. Например, как описано в следующих примерах, для эффективного растворения ЭДТА с применением TRIS может требоваться 5,0 эквивалента амина или более.
Для определенных диаминов или триаминов мольное отношение для обеспечения достаточной растворимости соли ЭДТА может быть не таким высоким, как в случае моноамина. Для полиаминов (диаминов, триаминов и т.д.), таких как NH2CH2CH2NH2, требуемое мольное отношение может быть ниже по сравнению с моноамином.
Подходящие уровни для полиаминов могут составлять 2,0 или более (например, от 2,0 до 4,0) или 2,5 или более. Снова, подходящие уровни могут быть определены путем оптимизации. В качестве руководства, мольные эквиваленты амина, обсуждаемые выше, могут представлять собой мольное отношение моноамина к ЭДТА или отношение аминогрупп к ЭДТА, если амин (или смесь аминов) содержит более одной аминогруппы в молекуле (в отдельной молекуле или в среднем по смеси).
Не существует верхнего предела для количества эквивалентов амина, который может присутствовать, хотя следует понимать, что, как правило, не следует включать амин в количестве, превышающем необходимое для обеспечения эффективного растворения.
Типичный практический предел может составлять 20 эквивалентов, предпочтительно 10 эквивалентов.
Авторы изобретения установили, что для получения алкиламмониевой соли ЭДТА необходимо использовать кислую форму ЭДТА в качестве исходного вещества, но не применяемый традиционно ЭДТА динатрия (ЭДТА(Na)). Ни ЭДТА (эдетовая кислота), ни ЭДТА(Na) не растворяются в подходящих/предпочтительных растворителях (например, EtOH/PG) в отсутствие алкиламина (например, ЭТА) даже после нескольких месяцев перемешивания. Удивительно, что ЭДТА(Na) не растворяется в EtOH/PG даже в присутствии ЭТА.
Таким образом, типичная процедура получения соли включает растворение свободной тетракислоты ЭДТА (которая может иметь форму гидрата) в растворителе (с) или в растворителе, который является предшественником компонента-растворителя (с), который включает по меньшей мере растворитель на основе одноатомного спирта, такой как этанол, и может также содержать полярный сорастворитель, такой как описано выше, предпочтительно в смеси этанола и PG. Затем добавляют необходимое количество эквивалентов алкиламина и перемешивают смесь, например, путем перемешивания вращением с донышка на крышку или магнитного перемешивания, до растворения ЭДТА, которое может быть установлено путем визуального наблюдения.
24-часовое перемешивание, как правило, является достаточным периодом времени для обеспечения эффективного растворения, например, в случае получения соли ЭТА/ЭДТА.
Также в объем изобретения включено получение соли в растворителе, который является предшественником компонента-растворителя (с). Под «предшественником» понимают, что растворитель, в котором образуется соль ЭДТА, не идентичен конечному составу компонента-растворителя (с), но содержание растворителя(-ей) в предшественнике можно регулировать для обеспечения конечного состава растворителя (с) в предварительном составе. В качестве примера, можно получать соль в смеси EtOH:PG (1:2), и дополнительное количество этанола добавляют во время или после образования соли для достижения конечного отношения EtOH:PG (1:1) в компоненте (c).
Отношение алкиламина к ЭДТА
Авторы изобретения неожиданно установили, что сверх определенного отношения алкиламин:ЭДТА химическая стабильность активного агента в предварительном составе начинает уменьшаться. Это может происходить в результате взаимодействия избытка алкиламина с активным агентом, которое может происходить напрямую или через продукты разрушения. Соответственно, предпочтительно выбранное количество алкиламина должно быть достаточным для полного растворения ЭДТА в компоненте- растворителе (с), но не должно значительно превышать этот уровень. Предпочтительно количество включаемого алкиламина не более чем в 2 раза превышает уровень, требуемый для полного растворения, предпочтительно не более чем в 1,5 раза, предпочтительно не более чем в 1,2 раза. Количество алкиламина, необходимое для полного растворения ЭДТА в компоненте-растворителе (c), может быть установлено способами, описанными выше.
В одном из вариантов реализации компонент (ii) содержит противоион алкиламмония, содержащий только одну амино- или алкиламиногруппу, и отношение ЭДТА:общее содержание указанного противоиона алкиламмония и любого амина в форме свободного основания в предварительном составе составляет 1:≥3,0; предпочтительно 1:≥3,5, наиболее предпочтительно находится в диапазоне от 1:3,0 до 1:10.
В одном из вариантов реализации компонент (ii) содержит противоион алкиламмония, содержащий две или более амино- и/или алкиламиногрупп, где отношение ЭДТА:общее содержание указанного противоиона алкиламмония и любого амина в форме свободного основания в предварительном составе составляет 1:≥2,0; предпочтительно находится в диапазоне от 1:2,0 до 1:4,0.
Согласно особенно предпочтительному аспекту соль ЭДТА представляет собой соль ЭТА ЭДТА. Авторы изобретения установили, что в указанном варианте реализации для полного растворения ЭДТА в компоненте-растворителе (с) (например, в смеси EtOH/PG 50:50) необходимо включать примерно по меньшей мере 3,5 мольных эквивалента ЭТА относительно количества ЭДТА. Соответственно, отношение ЭТА к ЭДТА предпочтительно составляет не более 7:1. Отношение эквивалентов ЭТА к ЭДТА предпочтительно находится в диапазоне от 3,5 до 7 (моль/моль), предпочтительно от 3,5 до 5, наиболее предпочтительно от 3,5 до 4,5. Наиболее предпочтительно применяют 4 эквивалента ЭТА относительно количества ЭДТА (моль/моль).
Количество соли ЭДТА
Уровень алкиламмониевой соли ЭДТА выбирают таким образом, чтобы обеспечивать соответствующую стабильность компонентов липидного носителя и активного агента (если он содержится) в течение требуемого срока хранения и при выбранных условиях хранения. Факторы, которые следует учитывать при определении соответствующих количеств алкиламмониевой соли ЭДТА, включают: реакционную способность липидных компонентов и активного агента (если он содержится), содержание активного агента (если он содержится), молекулярную массу активного агента, условия хранения (содержание кислорода, влажность, температура), требуемую продолжительность защиты от окисления и концентрацию ионов металлов, присутствующих в предварительном составе (которые могут катализировать процессы разложения).
Для подавления каталитической активности металлов, например, Fe, предварительный состав, как правило, включает соль ЭДТА на таком уровне, что отношение соли ЭДТА к металлу (например, Fe, особенно в форме ионов Fe(II) и Fe(III)) составляет по меньшей мере примерно 2:1 (моль/моль), т.е. соль ЭДТА присутствует по меньшей мере в 2-кратном мольном избытке. В типовом способе мольное отношение определяют для максимальной расчетной концентрации иона металла (в частности, иона Fe) и ЭДТА, обеспеченной в отношении примерно 2:1 к указанном максимальному расчетному значению. На практике мольное отношение ЭДТА к металлу (например, ионам Fe) составляет 2:3 или более.
Авторы изобретения установили, что существует предпочтительный уровень ЭДТА, выше которого не происходит увеличение стойкости смеси, например, предварительного состава, к окислению, и фактически стабильность может в некоторой степени уменьшаться. На это влияет количество ионов металлов (например, ионов Fe), присутствующих в составе, что подробно обсуждается в разделе «экспериментов». Тем не менее, в общем случае подходящее количество соли ЭДТА в предварительном составе (вычисленное в пересчете на свободную кислоту ЭДТА) составляет 0,001-0,02 масс.% (10-200 ppm), предпочтительно 0,001-0,015 масс.% (10-150 ppm), в частности, 0,002-0,015 масс.% (20-150 ppm). Особенно предпочтительный уровень составляет 0,005-0,015 масс.% (50-150 ppm), наиболее предпочтительно 0,008-0,012 масс.% (80-120 ppm).Уровень 100 ppm подходит для защиты от действия 10 ppm металла (эквивалент железа), что является приемлемым для обеспечения соответствующей стойкости лекарственного продукта.
В определенных вариантах реализации уровень ЭДТА (в пересчете на массу то лько ЭДТА, исключая катионы аминов) может находиться в диапазоне от 0,001 до 0,8 масс.% (от 10 до 8000 ppm), от 0,002 до 0,5 масс.% (от 20 до 5000 ppm), от 0,005 до 0,2 масс.% (от 50 до 2000 ppm) или от 0,01 до 0,1 масс.% (от 100 до 1000 ppm) предварительного состава. В определенных вариантах реализации уровень ЭДТА может находиться в диапазоне от 0,001 до 0,050 масс.% (от 10 до 500 ppm) смеси, например, предварительного состава, предпочтительно от 0,002 до 0,030 масс.% (от 20 до 300 ppm) смеси.
Уровень добавляемого алкиламина может быть установлен после определения оптимального отношения алкиламина к ЭДТА, как описано в предыдущих разделах.
В одном из вариантов реализации отношение (ii) к (d) находится в диапазоне от 1:1 до 1:5000 (масс./масс.), предпочтительно от 1:1 до 1:500 (масс./масс.), предпочтительно находится в диапазоне от 1:50 до 1:300.
Содержание воды
Включение солей ЭДТА, содержащих ион алкиламмония формулы (I), позволяет включать антиоксидант в смесь, например, в предварительный состав, при низком содержании воды. Тем не менее, исключительно трудно полностью удалить все следы воды (особенно из сырья). Даже несмотря на то, что можно получать составы, по существу не содержащие воду, предварительные составы, как правило, хранят в готовой к применению форме, например, в шприцах, и возможно в охлажденном состоянии. Шприцы часто не полностью герметичны для воздуха, и это означает, что уровень воды в предварительном составе может увеличиваться до заметного уровня со временем, например, в течение нескольких месяцев, даже если начальный уровень воды был незначительным.
Начальный абсолютный уровень воды в смеси, например, в предварительном составе, составляет от 0 до 1,0 масс.%. Предпочтительно содержание воды составляет менее 1,0 масс.%, предпочтительно менее 0,8 масс.%, предпочтительно менее 0,5 масс.%. Наиболее предпочтительно уровень воды находится в диапазоне от 0,1 до 0,9 масс.%, в частности, от 0,2 до 0,8 масс.%. Указанные уровни относятся к абсолютному уровню воды, а не к уровню добавляемой воды. Любые неудаляемые следы воды, присутствующие в компонентах а), b) или c), включены в указанный уровень воды. После 3 месяцев хранения абсолютный уровень воды предпочтительно составляет не более 1,5 масс.%. Абсолютный уровень воды может быть измерен способами, хорошо известными в данной области техники, такими как титрование по Карлу Фишеру. В частности, содержание воды предпочтительно измеряют согласно способу, описанному в Фармакопее США (USP 40 - NF 35, USP <921>, определение воды, способ Ia).
Компонент d) - активный агент
Предварительные составы согласно настоящему изобретению могут содержать один или более пептидных или непептидных активных агентов. Подчеркивается, что неожиданное открытие того, что окисление липидных предварительных составов, имеющих низкое содержание воды (не более 1,0%), и необязательно любого активного агента, содержащегося в указанных составах, может быть понижено в результате включения конкретных солей ЭДТА, описанных в настоящем документе, обеспечивает крайне широкую применимость, и, таким образом, природа биоактивного агента особенно не важна для реализации изобретения. Действительно, так как окисление липидных компонентов уменьшается в способе согласно изобретению, преимущества настоящего изобретения могут быть достигнуты независимо от природы или даже присутствия какого-либо активного агента.
Предполагается, что изобретение применимо к липидным предварительным составам, содержащим любой представляющий интерес биоактивный агент. Биоактивные агенты могут представлять собой любое соединение, обладающее желаемым биологическим или физиологическим действием, такое как пептид, белок, лекарственное средство, антиген, питательное вещество, косметическое средство, отдушка, вкусоароматическая добавка, диагностическое средство, фармацевтическое средство, витамин или диетическое вещество, и включены в количестве, достаточном для обеспечения концентрации in vivo на функциональном уровне (включая локальные концентрации в случае местных композиций). Наиболее предпочтительными активными агентами являются фармацевтические агенты, включая лекарственные средства, вакцины и диагностические агенты. Особенно предпочтительным классом активных агентов являются соматостатины и аналоги соматостатина.
Примеры лекарственных средств, которые могут доставляться в композиции согласно настоящему изобретению, включают, но не ограничиваются ими, антибактериальные агенты, иммуномодуляторы, включая иммуностимуляторы и иммунодепрессанты, противораковые и/или противовирусные лекарственные средства, такие как аналоги нуклеозидов, паклитаксел и его производные, противовоспалительные лекарственные средства/агенты, такие как нестероидные противовоспалительные лекарственные средства и кортикостероиды, сердечно-сосудистые лекарственные средства, включая средства для снижения уровня холестерина и кровяного давления, обезболивающие, противорвотные средства, включая антагонисты гистаминовых H1, NK1 и 5-HT3 рецепторов, кортикостероиды и каннабиноиды, антипсихотические средства и антидепрессанты, включая ингибиторы захвата серотонина, простагландины и производные, вакцины и модуляторы костной ткани. Диагностические агенты включают меченные радионуклидами соединения и контрастные вещества, включая рентгеновские, ультразвуковые и МРТ-контрастные агенты. Питательные вещества включают витамины, коферменты, пищевые добавки и т.д.
Особенно подходящие активные агенты включают агенты, которые обычно имеют низкую продолжительность удерживания в организме из-за быстрого расщепления или выведения, а также агенты с плохой пероральной биодоступностью. Они включают активные агенты на основе пептидов, белков и нуклеиновых кислот, гормоны и другие встречающиеся в природе агенты в исходной или модифицированной форме. При введении указанных агентов в виде композиции депо, полученной из предварительного состава согласно настоящему изобретению, устойчивый уровень агентов обеспечивается в течение периода времени, который может составлять до нескольких дней, недель или даже нескольких месяцев, несмотря на то, что агенты имеют высокую скорость клиренса. Это обеспечивает очевидные преимущества с точки зрения стабильности и соблюдения пациентом схемы лечения по сравнению с введением доз ежедневно несколько раз в день в течение того же периода. В одном предпочтительном варианте реализации активный агент, таким образом, имеет биологический период полувыведения (при попадании в кровоток) менее 1 дня, предпочтительно менее 12 часов и более предпочтительно менее 6 часов. В некоторых случаях указанный период может составлять не более 1-3 часов или менее. Подходящими агентами также являются агенты, которые обладают плохой пероральной биодоступностью по сравнению с инъекцией, где активный агент также или в качестве альтернативы имеет биодоступность менее 20% или предпочтительно менее 2%, в частности, менее 0,2% и наиболее предпочтительно менее 0,1% при применении пероральных составов. Количество биологически активного агента, которое можно включать в предварительные составы согласно настоящему изобретению, зависит от действующей дозы и периода, в течение которого композиция депо, образующаяся при введении, должна обеспечивать замедленное высвобождение. Как правило, доза, определенная для конкретного агента, примерно равна нормальной суточной дозе, умноженной на количество дней, в течение которых предварительный состав должен обеспечивать высвобождение. Очевидно, что указанное количество необходимо регулировать с учетом любых нежелательных эффектов большой дозы в начале лечения, и, таким образом, в общем случае оно соответствует максимальной применяемой дозе. Точное подходящее количество для любого случая может быть легко определено в рамках подходящей экспериментальной работы.
В одном из вариантов реализации предварительный состав согласно изобретению может содержать один или более пептидных активных агентов. Пептидные активные агенты могут содержать от 5 до 60 природных и/или синтетических аминокислот, в частности, от 5 до 50 или от 5 до 40 аминокислот.
Активные агенты на основе пептидов и белков включают лекарственные средства для человека и ветеринарные препараты, выбранные из группы, состоящей из адренокортикотропного гормона (АКТГ) и его фрагментов, ангиотензина и родственных ему пептидов, антител и их фрагментов, антигенов и их фрагментов, предсердных натрийуретических пептидов, биоадгезивных пептидов, брадикининов и родственных им пептидов, кальцитониновых пептидов, включая кальцитонин и амилин и родственные им пептиды, вазоактивных интестинальных пептидов (ВИП), включая соматотропин-рилизинг-гормон (GHRH), глюкагона и секретина, опиоидных пептидов, включая проопиомеланокортиновые (POMC) пептиды, энкефалиновых пептидов, продинорфиновых пептидов и родственных пептидов, пептидов, родственных панкреатическому полипептиду, таких как нейропептид (NPY), пептид YY (PYY), панкреатический полипептид (PPY), фрагментов рецепторных белков клеточной поверхности, хемотаксических пептидов, циклоспоринов, цитокинов, динорфинов и родственных им пептидов, эндорфинов и фрагментов P-липотропина, энкефалина и родственных ему белков, ингибиторов ферментов, иммуностимулирующих пептидов и полиаминокислот, фрагментов фибронектина и родственных им пептидов, пептидов желудочно-кишечного тракта, агонистов и антагониста гонадотропин-рилизинг- гормона (GnRH), глюкагоноподобных пептидов 1 и 2, рилизинг-пептидов гормона роста, иммуностимулирующих пептидов, инсулинов и инсулиноподобных факторов роста, интерлейкинов, рилизинг-гормонов лютенизирующего гормона (LHRH) и родственных им пептидов (которые эквивалентны агонистам GnRH, таким как описано ниже), агонистов и антагонистов меланокортинового рецептора, меланоцит- стимулирующих гормонов и родственных им пептидов, пептидов, связанных с сигналом ядерной локализации, нейротензинов и родственных им пептидов, нейротрансмиттерных пептидов, опиоидных пептидов, окситоцинов, вазопрессинов и родственных им пептидов, паратиреоидного гормона и его фрагментов, протеинкиназ и родственных им пептидов, соматостатинов и родственных им пептидов, вещества P и родственных ему пептидов, трансформирующих факторов роста (TGF) и родственных им пептидов, фрагментов фактора некроза опухолей, токсинов и токсоидов и функциональных пептидов, таких как противоопухолевые пептиды, включая ангиостатины, антигипертензивных пептидов, пептидов, препятствующих свертыванию крови, и антимикробных пептидов; выбранных из группы, состоящей из белков, таких как иммуноглобулины, ангиогенины, морфогенные белки костей, хемокины, колониестимулирующие факторы (КСФ), цитокины, факторы роста, интерфероны (типа I и II), интерлейкины, лептины, факторы ингибирования лейкемии, факторы стволовых клеток, трансформирующие факторы роста и факторы некроза опухоли.
Интересным классом биоактивных агентов, подходящих для изобретения, являются пептидные гормоны, включая: семейство гликопротеиновых гормонов (гонадотропины (ЛГ, ФСГ, ХГЧ), тиреостимулирующий гормон (ТСГ); семейство проопиомеланокортинов (POMC), адренокортикотропный гормон (АКТГ); гормоны задней доли гипофиза, включая вазопрессин и окситоцин, семейство гормонов роста, включая гормон роста (GH), хорионический соматомаммотропин человека (hCS), пролактин (PRL), семейство панкреатических полипептидов, включая PP, PYY и NPY; меланин-концентрирующий гормон, (MCH); орексины; гормоны и пептиды желудочно- кишечного тракта, включая GLP-1 и GIP; грелин и обестатин; гормоны и цитокины жировой ткани, включая лептин, адипонектин и резистин; натрийуретические гормоны; паратиреоидный гормон (ПТГ); семейство кальцитонинов, включая кальцитонин и амилин; панкреатические гормоны, включая инсулин, глюкагон и соматостатин. Все синтетические пептиды, разработанные для обеспечения схожих спектров аффинности к рецепторам, что и указанные выше пептиды, также особенно подходят для настоящего изобретения.
Дополнительное значительное преимущество композиций депо согласно настоящему изобретению заключается в том, что активные агенты высвобождаются постепенно в течение продолжительных периодов времени и не требуют повторного введения. Композиции, таким образом, особенно подходят для ситуаций, когда соблюдение пациентом схемы лечения затруднительно, ненадежно, или когда очень важна дозировка, как в случае активных веществ, изменяющих настроение, активных веществ с узким терапевтическим окном и агентов, которые вводят детям или людям, чей образ жизни несовместим с надежным соблюдением режима дозирования, и агентов, «улучшающих качество жизни», для которых неудобства от повторяющегося введения могут перевешивать благоприятное действие активного вещества. Конкретные классы активных веществ, для которых этот аспект обеспечивает особые преимущества, включают противозачаточные средства, гормоны, включая противозачаточные гормоны, и, в частности, гормоны, применяемые у детей, такие как гормон роста, антиаддиктивные агенты и лекарственные средства, применяемые при лечении популяций, плохо соблюдающих схему лечения, таких как пациенты, страдающие от шизофрении, болезни Альцгеймера или болезни Паркинсона, антидепрессанты и противосудорожные средства.
Катионные пептиды и белки особенно подходят для применения, если часть предварительного состава содержит анионный амфифил, такой как жирная кислота или анионный липид, включая фосфатидную кислоту, фосфатидилглицерин, фосфатидилсерин. В указанном варианте реализации предпочтительные пептиды или белки включают октреотид, ланреотид, кальцитонин, окситоцин, интерферон-бета и - гамма, интерлейкины 4, 5, 7 и 8 и другие пептиды или белки, имеющие изоэлектрическую точку выше рН 7, в частности, выше рН 8.
Согласно одному предпочтительному аспекту настоящего изобретения композиция согласно изобретению является такой, что при воздействии водных жидкостей образуется обращенная мицеллярная кубическая (I2) фаза или смешанная фаза, включающая фазу I2, и в композицию включен полярный активный агент. Особенно подходящие полярные активные агенты включают пептидные и белковые активные вещества, олигонуклеотиды и низкомолекулярные водорастворимые активные вещества, включая те, что перечислены выше. Особый интерес согласно указанному аспекту представляет пептид октреотид и другие родственные соматостатину пептиды, интерфероны альфа и бета, агонисты рецептора глюкагоноподобного пептида 1 и глюкагоноподобного пептида 2, лейпрорелин и другие агонисты GnRH, абареликс и другие антагонисты GnRH, гранисетрон и ондансетон и другие антагонисты 5-НТ3 рецепторов.
Аналоги GnRH
Аналоги GnRH образуют один конкретный класс активных агентов, которые можно включать в составы согласно настоящему изобретению.
Агонисты гонадотропин-рилизинг-гормона (агонисты GnRH) представляют собой синтетические пептиды, полученные по образцу гипоталамического нейрогормона GnRH, который взаимодействует с рецептором гонадотропин-рилизинг-гормона и индуцирует его биологический ответ, т.е. высвобождение гормонов гипофиза, включая фолликулостимулирующий гормон (ФСГ) и лютеинизирующий гормон (ЛГ). Агонисты GnRH подходят для лечения раковых заболеваний, которые являются восприимчивыми к действию гормонов, при которых гипогонадальное состояние уменьшает вероятность их повторного появления. Таким образом, их обычно применяют для медикаментозного сдерживания рака предстательной железы и используют у пациентов с раком молочной железы. Другие показания включают лечение для задержки полового созревания у индивидуумов с преждевременным половым созреванием, борьбы с женскими расстройствами, которые зависят от выработки эстрогена. Кроме того, женщинам с меноррагией, эндометриозом, аденомиозом или фибромой матки можно вводить агонисты GnRH для подавления активности яичников и индуцирования гипоэстрогенного состояния.
Агонисты рецепторов гонадотропин-рилизинг-гормона (GnRH-RA), такие как леупролид (или лейпрорелин), гозерелин, гистрелин, трипторелин, бусерелин, деслорелин, нафарелин и родственные пептиды, применяются или показаны для лечения различных состояний, при которых их, как правило, вводят в течение продолжительного периода времени.GnRH-RA образуют предпочтительную группу активных агентов для применения в настоящем изобретении.
GnRH, как таковой, представляет собой посттрансляционно модифицированный декапептид со структурой пиро-Glu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2 (GnRH-I). Известны также два природных варианта: GNRH-II, имеющий замены 5-His, 7-Trp, 8-Tyr, и GnRH_III, содержащий 7-Trp, 8-Leu. Известно несколько пептидных аналогов со свойствами агонистов, в большинстве из которых 10-Gly-NH2 заменен на N-Et-NH2. Фертирелин имеет только одну замену 10-Gly на N-Et-NH2, при этом аналоги, имеющие дополнительные замены по сравнению с GnRH-I, включают лейпрорелин (леупролид), (6-D-Leu), бусерелин (6-Ser(But)), гистрелин (6-d-His(Imbzl)), деслорелин (6-d-Trp). Другим распространенным нонапептидным агонистом является гозерелин, который замещен 6-Ser (But) и имеет замену 10-Gly-NH2 на AzaGly-NH2. В нарафелине (6-d-Nal) и трипторелине (6-d-Trp) группа 10-Gly-NH2 сохранена. Структуры двух наиболее распространенных агонистов GnRH (леупролида и гозерелина) показаны ниже для ацетатных солей.
Леупролид: пиро-Glu-His-Trp-Ser-Tyr-D-Leu-Leu-Arg-Pro-N-Et-NH2 (ацетат) Гозерелин: пиро-Glu-His-Trp-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-Azgly-NH2 (ацетат) Известно небольшое количество антагонистов GnRH, которые также основаны на структуре GnRH-I. Они включают абареликс (D-Ala-D-Phe-D-Ala-Ser-Tyr-D-Asp-Leu- Lys(iPr)-Pro-D-Ala), антареликс (D-Nal-D-Phe-D-Pal-Ser-Phe-D-Hcit-Leu-Lys(iPr)-Pro-D- Ala); цетрореликс (D-Nal-D-Phe-D-Pal-Ser-Tyr-D-Cit-Leu-Arg-Pro-D-Ala), ганиреликс (D-Nal-D-Phe-D-Pal-Ser-Tyr-D-hArg-Leu-HArg-Pro-D-Ala), итреликс (D-Nal-D-Phe-D- Pal-Ser-NicLys-D-NicLys-Leu-Lys(iPr)-Pro-D-Ala) и Nal-Glu (D-Nal-D-Phe-D-Pal-Ser-D-Glu-D-Glu-Leu-Arg-Pro-D-Ala).
Введение однократных доз агониста GnRH, такого как леупролид, стимулирует высвобождение гонадотропинов гипофизом (т.е. ЛГ и ФСГ), что приводит к увеличению концентрации ЛГ и ФСГ в сыворотке и стимуляции стероидогенеза в яичниках и яичках. Временное увеличение уровня тестостерона и дигидротестостерона (ДГТ) в сыворотке у мужчин и концентраций эстрона и эстрадиола в сыворотке у женщин в пременопаузе наблюдается во время начальной терапии с однократным ежедневным введением лекарственного средства.
Несмотря на то, что действие высокоактивного агониста GnRH при краткосрочной и/или прерывающейся терапии заключается в стимуляции стероидогенеза, основным эффектом лекарственного средства у животных и человека при долгосрочном введении является ингибирование секреции гонадотропина и подавление стероидогенеза в яичниках и яичках. Точный(-е) механизм(-ы) действия выяснен(-ы) не полностью, но непрерывная терапия агонистом GnRH очевидно приводит к снижению числа GnRH в гипофизе и/или рецепторов ЛГ в яичках, что вызывает десенсибилизацию гипофиза и/или яичек, соответственно. Лекарственное средство, вероятно, не влияет на аффинность рецепторов к гонадотропинам. Механизм действия леупролида может также включать ингибирование и/или индуцирование ферментов, которые контролируют стероидогенез. Другие механизмы действия могут включать секрецию молекулы ЛГ с измененной биологической активностью или нарушение нормальных пульсирующих профилей секреции ЛГ и ФСГ.
Ряд серьезных медицинских показаний связан с концентрацией гонадных стероидных гормонов и/или зависит от нее. Они включают определенные неопластические заболевания, включая рак, в частности, молочной железы и предстательной железы, и доброкачественную гипертрофию предстательной железы; преждевременное или отсроченное половое созревание у подростков; гирсутизм; болезнь Альцгеймера; и определенные состояния, связанные с репродуктивной системой, такие как гипогонадизм, ановуляция, аменорея, олигоспермия, эндометриоз, лейомиома (фиброма матки), предменструальный синдром и поликистоз яичников. Контролирование указанной системы также важно для способов оплодотворения in vitro.
Несмотря на то, что в соответствии с ожиданиями лечение агонистом GnRH может усугублять состояния, на которые влияет концентрация гонадного стероидного гормона, эффект понижающей регуляции, обсуждаемый выше, приводит к уменьшению концентрации указанных гормонов до кастрационного уровня, если терапию проводят в течение примерно 2 недель или более. В результате долгосрочной терапии агонистом GnRH может происходить улучшение состояния или облегчение при опухолях, чувствительных к действию гормонов, таких как определенные формы рака предстательной железы и рака молочной железы, а также при преждевременном половом созревании и многих других состояниях, упомянутых выше.
В одном из вариантов реализации предварительные составы согласно настоящему изобретению содержат один или более аналогов GnRH. Так как GnRH представляет собой пептидный гормон, то типовые аналоги GnRH представляют собой пептиды, в частности, содержащие 12 аминокислот или менее. Предпочтительно указанные пептиды структурно родственны GnRH I, II и/или III и/или одному или более из известных аналогов, включая те, что перечислены в настоящем документе. Пептиды могут содержать только аминокислоты, выбранные из 20 α-аминокислот, включенных в генетический код, или более предпочтительно могут содержать их изомеры и другие природные и не встречающиеся в природе аминокислоты (в общем случае, α-, β- или γ- аминокислоты) и их аналоги и производные. Предпочтительные аминокислоты включают те, что перечислены выше в качестве компонентов известных аналогов GnRH.
Производные аминокислот особенно подходят для применения на концах пептидов, где концевая амино- или карбоксилатная группа может быть замещена на или содержать в качестве заместителя любую другую функциональную группу, такую как гидрокси, алкокси, карбокси, сложный эфир, амид, тио, амидо, амино, алкиламино, ди- или три- алкиламино, алкил (под которым в настоящем документе подразумевается C1-С12 алкил, предпочтительно С1-С6 алкил, например, метил, этил, н-пропил, изопропил, н-бутил, изо-, втор- или трет-бутил и т.д.), арил (например, фенил, бензил, нафтил и т.д.) или другие функциональные группы, предпочтительно содержащие по меньшей мере один гетероатом, и предпочтительно содержащие в целом не более 10 атомов, более предпочтительно не более 6.
Особенно предпочтительными аналогами GnRH являются конформационно ограниченные пептиды из 6-12 альфа-аминокислот, конкретные примеры которых включают те, что указаны выше, и, в частности, леупролид и гозерелин, имеющие последовательности, указанные выше.
«Аналоги GnRH» при использовании в настоящем описании обозначают любой агонист или антагонист GnRH, предпочтительно пептиды, производные пептидов или аналоги пептидов. Наиболее предпочтительными являются агонисты GnRH, полученные из пептидов, такие как те, что указано выше, и, в частности, леупролид или гозерелин. Аналог GnRH, если присутствует, в общем случае составляет от 0,02 до 12% по массе от общего количества предварительного состава (в пересчете на количество свободного основания). Типичные значения составляют от 0,1 до 10%, предпочтительно от 0,2 до 8% и более предпочтительно от 0,5 до 6%. Содержание аналога GnRH примерно 1-5% является наиболее предпочтительным.
Дозы аналога GnRH, подходящие для включения в предварительный состав, и, таким образом, объем применяемого состава, зависят от скорости высвобождения (которая контролируется, например, типом и количеством используемого растворителя) и продолжительности высвобождения, а также от желаемого терапевтического уровня, активности конкретного агента и скорости клиренса конкретного выбранного активного вещества. Обычно количество от 0,1 до 500 мг на дозу подходит для обеспечения терапевтического уровня в течение периода от 7 до 180 дней.
Предпочтительно оно составляет от 1 до 200 мг. Для леупролида или гозерелина уровень, как правило, составляет от 1 до 120 мг (например, для периода времени от 30 до 180 дней). Предпочтительно количество леупролида составляет от 0,02 до 1 мг в день при инъекционном введении для депо, предназначенного для высвобождения в течение периода от 30 дней до 1 года, предпочтительно от 3 до 6 месяцев. Очевидно, что стабильность активного вещества и линейность скорости высвобождения означают, что взаимосвязь содержания активного вещества и продолжительности действия может быть нелинейной. Депо, вводимое каждые 30 дней, может содержать, например, от 2 до 30 мг, или 90-дневное депо содержит от 6 до 90 мг активного вещества, такого как один из указанных в настоящем документе аналогов GnRH.
Аналоги соматостатина
Аналоги соматостатина образуют один конкретный класс активных агентов, которые можно включать в составы согласно настоящему изобретению. Соматостатины (факторы ингибирования высвобождения гормона роста, SST) представляют собой природные пептидные гормоны, которые широко распространены у животных, действующие в качестве нейротрансмиттеров в центральной нервной системе и имеющие различные паракринные/аутокринные регуляторные эффекты в некоторых тканях. У высших видов известны два биологически активных продукта, SST-14 и SST-28, причем последний является конгенером SST-14, удлиненным по N-концу.
SST-14 представляет собой циклический пептидный гормон из 14 остатков, имеющий последовательность Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys, где два остатка цистеина связаны посредством дисульфидного мостика с образованием β- изгиба II типа в ключевой связывающей последовательности Phe-Trp-Lys-Thr. Биологический период полувыведения природного SST-14 является очень коротким (1-3 минуты), и таким образом, он сам по себе не является подходящим терапевтическим средством в существующих составах, но становится доступным все большее число агонистов соматостатиновых рецепторов с более высокой активностью и/или увеличенным периодом клиренса in vivo.
Агонисты соматостатиновых рецепторов (SRA), такие как SST-14, SST-28, октреотид, ланреотид, вапреотид, пасиреотид (SOM 230) и родственные пептиды, применяются или показаны при лечении различных состояний, при которых их, как правило, вводят в течение продолжительного периода. SRA образуют предпочтительную группу активных агентов для применения в настоящем изобретении.
Октреотид, например, представляет собой синтетический октапептид, имеющий последовательность D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr-ол (2-7 дисульфидный мостик), и его, как правило, вводят в виде ацетатной соли. В указанном производном SST-14 сохранен ключевой β-изгиб Phe-(D)Trp-Lys-Thr, требуемый для проявления SST-подобной активности in vivo, но, в отличие от природного гормона, он имеет терминальный период полувыведения примерно 1,7 часа. Октреотид применяют для лечения состояний, включая карциноидные опухоли и акромегалию, и, как правило, вводят в течение продолжительного периода, составляющего несколько недель, а чаще нескольких месяцев или лет. Агонисты соматостатиновых рецепторов представляют особый интерес для лечения множества различных типов рака, поскольку обнаружено, что в широком спектре опухолей экспрессируются соматостатиновые рецепторы (SSTR). Существует пять известных типов SSTR (SSTR1-SSTR5), имеющих одинаково высокую аффинность к SST-14. Наиболее подробно изученные агонисты соматостатиновых рецепторов, включая октреотид, имеют высокую селективность к SSTR2 и SSTR5; таким образом, октреотид представляет особый интерес для лечения опухолей, экспрессирующих рецепторы указанных типов.
Самым распространенным «простым» составом октреотида является «Sandostatin» (RTM) производства Novartis. Он представляет собой водный раствор для подкожной (п.к.) инъекции, и для 100 мкг дозы максимальная концентрация 5,2 нг/мл достигается через 0,4 часа после инъекции. Продолжительность действия может составлять до 12 часов, но п.к. введение обычно проводят каждые 8 часов. Очевидно, что п.к. инъекция 3 раза в день в течение нескольких месяцев или лет не является идеальным режимом введения.
После однократного подкожного введения пасиреотида его уровень в плазме человека, как правило, быстро достигает максимального значения, примерно с интервалом от 15 минут до 1 часа после введения дозы, причем начальный период полувыведения составляет 2-3 часа после достижения указанного максимального значения. Хотя период полувыведения и увеличивается на более поздних фазах уменьшения концентрации, очевидно, что отношение Cmax/Cср для указанного способа доставки является достаточно высоким.
Пасиреотид LAR представляет собой состав пасиреотида долгосрочного действия, в котором решены некоторые из вышеуказанных проблем. Тем не менее, он представляет собой систему на основе полимерных микрочастиц и имеет все ограничения, присущие указанным системам, известные в данной области техники и описанные выше в настоящем документе.
Карциноидные опухоли представляют собой опухоли кишечника, образующиеся из специфических клеток с паракринными функциями (клетки APUD). Первичная опухоль обычно появляется в аппендиксе, где она является клинически доброкачественной. Во вторичных метастатических карциноидных опухолях кишечника секретируются избыточные количества вазоактивных веществ, включая серотонин, брадикинин, гистамин, простагландины и полипептидные гормоны. Клиническим результатом является карциноидный синдром (синдром эпизодического покраснения кожи, цианоз, колики в животе и диарея у пациента с пороком клапана сердца и, реже, с астмой и артропатией). Указанные опухоли могут расти в любом участке желудочно-кишечного тракта (и в легких), но примерно 90% возникают в аппендиксе. Оставшаяся часть приходится на подвздошную кишку, желудок, толстую кишку или прямую кишку. В настоящее время лечение карциноидного синдрома начинают с в.в. инъекции болюса, затем проводят в.в. инфузию. При достижении достаточного действия на симптомы начинают лечение составом депо октреотида в микросферах поли-молочной-гликолевой кислоты (PLGA). Тем не менее, в течение первых двух недель или более после инъекции депо, рекомендуется проводить ежедневные п.к. инъекции октреотида для компенсации его медленного высвобождения из сфер PLGA.
Определенные предварительные составы согласно настоящему изобретению содержат соли одного или более агонистов соматостатиновых рецепторов (которые являются предпочтительными примерами пептидных активных веществ, которые, в свою очередь, подразумеваются при описании каких-либо «активных агентов» в настоящем документе). Так как SST-14 представляет собой пептидный гормон, то типовые агонисты соматостатиновых рецепторов представляют собой пептиды, в частности, содержащие 14 аминокислот или менее. Предпочтительно указанные пептиды имеют структурные ограничения, такие как наличие цикличности и/или по меньшей мере одной внутримолекулярной поперечной связи. Амидные, сложноэфирные или, в частности, дисульфидные поперечные связи являются особенно подходящими. Предпочтительные ограниченные пептиды имеют β-изгиб 2 типа. Указанный изгиб представляет собой ключевой участок соматостатина. Пептиды могут содержать только аминокислоты, выбранные из 20 α-аминокислот, включенных в генетический код, или более предпочтительно могут содержать их изомеры и другие природные и не встречающиеся в природе аминокислоты (в общем случае, α-, β- или γ-, L- или D- аминокислоты) и их аналоги и производные. Термин «агонист соматостатинового рецептора» при использовании в настоящем описании может необязательно также включать SST-14 и/или SST-28, так как они представляют собой эффективные пептидные активные вещества при введении в виде солей в составах с очень высокой эффективностью замедленного высвобождения, описанных в настоящем до кументе. Производные аминокислот и аминокислоты, которые обычно не применяют для синтеза белков, особенно подходят для применения на концах пептидов, где концевая амино- или карбоксилатная группа может быть замещена на или содержать в качестве заместителя любую другую функциональную группу, такую как гидрокси, алкокси, сложный эфир, амид, тио, амино, алкиламино, ди- или три-алкиламино, алкил (под которым в настоящем документе подразумевается C1-С18 алкил, предпочтительно С1-С8 алкил, например, метил, этил, н-пропил, изопропил, н-бутил, изо-, втор- или трет-бутил и т.д.), арил (например, фенил, бензил, нафтил и т.д.) или другие функциональные группы, предпочтительно содержащие по меньшей мере один гетероатом, и предпочтительно содержащие в целом не более 10 атомов, более предпочтительно не более 6.
Особенно предпочтительными агонистами соматостатиновых рецепторов являются конформационно ограниченные пептиды, содержащие от 6 до 10 α-аминокислот, конкретные примеры которых включают октреотид, ланреотид (имеющий последовательность NH2-(D)Naph-Cys-Tyr-(D)Trp-Lys-Val-Cys-Thr-CONH2, и его циклическое производное, имеющее последовательность NH2-(D)Naph-Cys-Tyr-(D)Phe-Lys-Val-Cys-Thr-CONH2, оба из которых имеют внутримолекулярную дисульфидную поперечную связь Cys-Cys), пасиреотид (также известный как SOM 230) и вапреотид. Агонист соматостатиновых рецепторов, если присутствует, в общем случае составляет от 0,1 до 12% по массе от общего количества состава (в пересчете на количество свободного основания). Типичные значения составляют от 0,1 до 10%, от 0,5 до 9%, предпочтительно от 1 до 8% и более предпочтительно от 1 до 7%. Содержание агониста соматостатинового рецептора 2-6% является наиболее предпочтительным.
Дозы агониста соматостатинового рецептора, подходящие для включения в состав, и, таким образом, объем применяемого состава, зависят от скорости высвобождения (которая контролируется, например, типом и количеством используемого растворителя) и продолжительности высвобождения, а также от желаемого терапевтического уровня, активности и скорости клиренса конкретного выбранного активного вещества. Обычно количество от 1 до 500 мг на дозу подходит для обеспечения терапевтического уровня в течение периода от 7 до 90 дней.
Предпочтительно оно составляет от 5 до 300 мг. Для октреотида уровень, как правило, составляет от 10 до 180 мг (например, для периода времени от 30 до 90 дней). Предпочтительно количество октреотида составляет от примерно 0,2 до 3 мг в день при инъекционном введении. Таким образом, депо, вводимое каждые 30 дней, содержит от 6 до 90 мг, или 90-дневное депо содержит от 18 до 270 мг октреотида.
Для пасиреотида доза, как правило, составляет от примерно 0,05 до 40 мг в неделю во время введения депо, предпочтительно от 0,1 до 20 мг в неделю (например, от 1 до 5 мг в неделю) в течение периода от 1 до 24 недель, предпочтительно от 2 до 16 (например, 3, 4, 8, 10 или 12) недель. В альтернативном варианте реализации предварительный состав может быть приготовлен для еженедельного введения (например, каждые 7 ± 1 день). Общее количество от 0,05 до 250 мг пасиреотида на дозу может подходить для обеспечения терапевтического уровня в течение периода от 7 до 168 дней. Предпочтительно оно составляет от 0,1 до 200 мг, например, от 0,2 до 150 мг, от 0,1 до 100 мг, от 20 до 160 мг и т.д. Очевидно, что стабильность активного вещества и влияние на скорость высвобождения означают, что взаимосвязь содержания активного вещества и продолжительности действия может быть нелинейной. Депо, вводимое каждые 30 дней, может содержать, например, от 0,2 до 20 мг пасиреотида, или 90- дневное депо может содержать от 30 до 60 мг пасиреотида.
Если в составах согласно настоящему изобретению применяют соль пептидного активного агента, такого как SRA, то она представляет собой биологически переносимую соль. Подходящие соли включают ацетатные, памоатные, хлоридные или бромидные соли. Хлоридная соль является наиболее предпочтительной.
Другие активные агенты
В другом варианте реализации предварительный состав содержит активный агент, который не является соматостатином или аналогом соматостатина. Например, активный пептидный агент может представлять собой пептид, который не взаимодействует как агонист или антагонист ни с одним из рецепторов SST(1) - SST(5) (в частности, с соответствующими рецепторами человека).
Как правило, указанные предварительные составы не содержат какие-либо активные агенты на основе соматостатина или аналога соматостатина. То есть, содержится активный агент, который не включен в объем описания аналогов соматостатина, приведенного в предыдущем разделе. В частности, в указанном варианте реализации предварительный состав может содержать активный агент, который не выбран из эндогенных соматостатинов, SST-14, SST-28, октреотида, ланреотида, вапреотида или пасиреотида или их солей. Указанные пептиды предпочтительно не включены в предварительные составы согласно указанному варианту реализации. Предварительный состав предпочтительно не должен содержать соматостатины, агонисты соматостатиновых рецепторов и аналоги соматостатина.
Другие активные агенты, которые могут содержаться в предварительных составах согласно изобретению, включают:
антагонисты GnRH, например цетрореликс, ганиреликс, абареликс, дегареликс;
GLP-1 и его аналоги, например, GLP-1(7-37), GLP-1(7-36)-амид, лираглутид, семаглутид, эксенатид и ликсисенатид (AVE0010);
агонисты глюкагоноподобного пептида 2 (GLP-2) и их аналоги, например, GLP-2 и элсиглутид (ZP1846);
ингибиторы DPPIV; ингибиторы натрий-глюкозного котранспортера 2 (SGLT2).
Другие пептиды, подходящие для изобретения, включают: ангиопептин, ангиотензин I, II, III, антилейкинат, противовоспалительный пептид 2, апротинин, брадикинин, бомбезин, кальцитонин, кальцитриол, холецистокинин (CCK), колониестимулирующий фактор, кортикотропин-рилизинг-фактор, C-пептид, DDAVP, тетрапептид, выделенный из дерморфина (TAPS), динорфин, эндорфины, эндостатин, эндотелин, эндотелин-1, энкефалины, эпидермальный фактор роста, эритропоэтин, фактор роста фибробластов, фолликулостимулирующий гормон, фоллистатин, фоллитропин, галанин, галаниноподобный пептид, галектин-1, гастрин, гастрин-рилизинг-пептид, Г-КСФ, грелин, глиальный нейротрофический фактор, ГМ-КСФ, гранулоцитарный колониестимулирующий фактор, гормон роста, рилизинг-фактор гормона роста, фактор роста гепатоцитов, инсулин, инсулиноподобные факторы роста-I и I, интерфероны, интерлейкины, лептин, фактор ингибирования лейкемии, меланокортин 1, 2, 3, 4, меланоцит-стимулирующий гормон метастин, мотоцитарный хемотаксический белок-1 (MCP-1), морфицептин, NEP1-40, нейропептид Y, нейропептид W, орексин-A и орексин-B, окситоцин p21-Cip1/WAF-1, гибридный белок TAT, паратиреоидный гормон, фактор роста, выделенный из пигментного эпителия (PEDF), пептид, пептид, область "ручки" проренина, пептид YY (3-36), фактор активации тромбоцитов, фактор роста тромбоцитов, декапептид проренина, протегрин-1, PR39, пролактин, релаксин, секретин, вещество P, фактор некроза опухолей, урокортин, фактор роста эндотелия сосудов, вазоактивный интестинальный полипептид, вазопрессин.
Короткий период полувыведения опиоидов, таких как морфин, гидроморфон и оксикодон, требует частого введения указанных агентов для круглосуточного обезболивания, что делает их превосходными кандидатами для составов с долгосрочным высвобождением. Фентанил и бупренорфин подвергаются значительному метаболизму первого прохождения и не имеют достаточной биодоступности после перорального введения. Благодаря высокой активности фентанил и бупренорфин являются превосходными кандидатами для инъекционных составов депо долгосрочного действия согласно изобретению. Суфентанил, ремифентанил, оксиморфон, диморфон, дигидроэторфин, диацетилморфин являются другими высокоактивными агонистами опиоидных рецепторов, подходящими для применения в изобретении.
Бупренорфин также применяют для поддерживающего лечения опиоидной зависимости, а также потенциально кокаиновой и амфетаминовой и метамфетаминовой зависимости, так как существующие подъязычные составы бупренорфина имеют низкую биодоступность, крайне непостоянную эффективность и ограниченную продолжительность действия, что приводит к проблемам с непредсказуемым ответом на дозу и синдрому отмены, в частности, по утрам. Эти проблемы могут быть эффективно решены при применении инъекционного состава депо согласно изобретению, как и проблемы с неправильным применением и неправильно направленным действием, причем потребность в высоких подъязычных дозах устраняется за счет использования инъекций, при которых эффект от введения равной дозы значительно выше, что способствует улучшению применения лекарственного средства. Аналогично, опиоидные антагонисты можно применять для лечения зависимости с применением эффективной системы депо для инъекций, такой как предложено в изобретении. Подходящими опиатными антагонистами для применения в изобретении являются налоксон, налмефен и налтрексон.
Антипсихотические средства, включая рисперидон, илоперидон, палиперидон, оланзапин, азенапин, зипразидон и арипипразол, также особенно подходят для изобретения с точки зрения потенциала для улучшения соблюдения пациентом режима лечения, а также обеспечения стабильного уровня в плазме со временем. Аналогично, изобретение подходит для лечения деменции, болезни Альцгеймера и болезни Паркинсона, которые нежелательным образом влияют на когнитивные функции. Подходящие активные ингредиенты включают донепезил, ривастигмин, галантамин и эмантин, разагилин и прамипексол.
Другой группой активных агентов, которые могут содержаться в предварительных составах согласно изобретению, являются антагонисты 5HT3. Если активный агент содержит антагонист 5HT3 или антагонист 5HT3 второго поколения, то он предпочтительно выбран из ондансетрона, трописетрона, гранисетрона, доласетрона, палоносетрона, алосетрона, цилансетрона и/или рамосетрона или их смесей. Дозы антагониста 5HT3, подходящие для включения в состав, и, таким образом, объем применяемого состава, зависят от скорости высвобождения (которая контролируется, например, типом и количеством используемого растворителя) и продолжительности высвобождения, а также от желаемого терапевтического уровня, активности конкретного агента и скорости клиренса конкретного выбранного активного вещества. Обычно количество от 1 до 500 мг на дозу подходит для обеспечения терапевтического уровня в течение периода от 5 до 90 дней. Предпочтительно оно составляет от 1 до 300 мг. Для гранисетрона уровень, как правило, составляет от 10 до 180 мг (например, для периода времени от 3 до 60 дней). Предпочтительно количество гранисетрона составляет от 0,2 до 3 мг в день при инъекционном введении для депо, предназначенного для высвобождения в течение периода от 30 дней до 1 года, предпочтительно от 3 до 6 месяцев. Очевидно, что стабильность активного вещества и линейность скорости высвобождения означают, что взаимосвязь содержания активного вещества и продолжительности действия может быть нелинейной. Депо, вводимое каждые 30 дней, может содержать, например, от 2 до 30 мг, или 90-дневное депо содержит от 6 до 90 мг активного вещества.
В предпочтительном варианте реализации предварительный состав содержит по меньшей мере один активный агент, который не является агонистом соматостатинового рецептора. Предпочтительно предварительный состав не содержит агонисты соматостатиновых рецепторов. Таким образом, предварительный состав может не содержать активный агент, который взаимодействует как агонист или антагонист с любым из рецепторов SST(1) - SST(5) (в частности, у человека).
Термин «предварительный состав» согласно настоящему описанию представляет собой фармацевтическую композицию, предпочтительно представляет собой парентеральную фармацевтическую композицию, более предпочтительно инъекционную парентеральную фармацевтическую композицию, еще более предпочтительно представляет собой инъекционную парентеральную фармацевтическую композицию для подкожного или внутримышечного введения, еще более предпочтительно представляет собой инъекционную парентеральную фармацевтическую композицию для подкожного введения.
Необязательные дополнительные компоненты
В одном особенно предпочтительном варианте реализации настоящего изобретения композиции (предварительные составы и образующиеся депо) не включают фрагментирующие агенты, такие как фрагментирующий агент на основе полиэтиленоксида или поли(этиленгликоля) (ПЭГ), например, липид и/или поверхностно-активное вещество с привитым ПЭГ.
Например, композиции предпочтительно не включают фрагментирующие агенты, такие как полисорбат 80 (P80) или другие полисорбаты (например, полисорбат 20), ПЭГилированные фосфолипиды (ПЭГ-липиды, такие как DSPE-PEG(2000), DSPE- PEG(5000), DOPE-PEG(2000) и DOPE-PEG(5000), Solutol HS 15, ПЭГилированные жирные кислоты (например, ПЭГ-олеат), блок-сополимеры, такие как Pluronic® F127 и Pluronic® F68, этоксилированные производные касторового масла (например, кремофоры), ПЭГилированные сложные эфиры глицерина и жирных кислот (такие как TMGO-15 производства Nikko Chemicals) и ПЭГилированные токоферолы (такие как d-альфа-токоферил-поли(этиленгликоля)1000 сукцинат, известный как витамин Е TPGS производства Eastman).
Тем не менее, активный агент в виде порошка (например, в наборе согласно изобретению), а также активный агент, растворенный в липидном составе, могут приобретать стабильность (стабильность при хранении и in vivo) за счет применения определенных стабилизирующих добавок. Указанные добавки включают сахара (например, сахарозу, трегалозу, лактозу и т.д.), полимеры (например, полиолы, такие как карбоксиметилцеллюлоза), аминокислоты (такие как метионин, глутамат, лизин и т.д.), растворимые в липидах кислотные компоненты, такие как HCl, анионные липиды и/или поверхностно-активные вещества (такие как диолеоилфосфатидилглицерин (DOPG), пальмитоилолеоилфосфатидилглицерин (POPG) и олеиновая кислота (ОА)). Формы с одноразовой дозой должны сохранять стабильность и высокую активность при хранении перед применением, но их необходимо утилизировать после разового применения. Примечательно, что неводные предварительные составы, содержащие алкиламмониевую соль ЭДТА, обладают повышенной стабильностью при хранении при повышенных температурах, таких как 25°C или даже 40°C. Это обеспечивает преимущества с точки зрения простоты транспортировки и хранения (нет необходимости в охлаждении). Форма с одноразовой дозой предпочтительно должна обладать такой стабильностью, чтобы после хранения в течение 2 месяцев при 25°C (при наличии воздуха в свободном пространстве) концентрация исследуемого активного агента составляла по меньшей мере 95% от начальной концентрации исследуемого активного агента, и через 3 месяца концентрация исследуемого активного агента составляла по меньшей мере 90% от начальной концентрации исследуемого активного агента.
В одном предпочтительном варианте реализации предварительные составы, устройства и/или наборы согласно настоящему изобретению хранят при температуре выше 10°С (например, от 15 до 40°С), предпочтительно выше 20°С, например, при температуре окружающей среды. В соответствующем варианте реализации предварительные составы, устройства и/или наборы согласно настоящему изобретению не хранят при температурах охлаждения (например, ниже 10°C или ниже 5°C), таких как 1-6°C.
Форма с одноразовой дозой предпочтительно должна обладать такой стабильностью, чтобы после хранения в течение 2 месяцев при 40°C (при наличии воздуха в свободном пространстве) концентрация исследуемого активного агента составляла по меньшей мере 85% от начальной концентрации исследуемого активного агента, и через 3 месяца концентрация исследуемого активного агента составляла по меньшей мере 80% от начальной концентрации исследуемого активного агента.
Формы с многоразовыми дозами должны не только сохранять стабильность и высокую активность при хранении перед применением, но также должны сохранять стабильность, высокую активность, из них должны быть относительно/эффективно удалены бактерии на период введения в режиме многоразового применения после первого применения, при котором нарушается герметичность. По этой причине формы с многоразовыми дозами часто требуют применения противомикробного или микробиостатического агента, например, бактериостатического агента, консерванта.
Тем не менее, получение консервированных фармацевтических препаратов, содержащих белковые или пептидные активные вещества, часто оказывается затруднительным, так как при использовании консервантов возникают проблемы со стабильностью. Часто белки инактивируются, и образуются агрегаты, что может иногда приводить к появлению непереносимости в месте инъекции или иммуногенности к активному веществу. Это может дополнительно усугубляться при использовании дополнительных вспомогательных веществ или компонентов состава.
Согласно одному из аспектов каждый из вариантов реализации настоящего изобретения необязательно может содержать противомикробный или микробиостатический агент, который включает бактериостатические агенты и консервант. Указанные агенты включают хлорид бензалкония, м-крезол, бензиловый спирт или другие фенольные консерванты. Можно использовать типичные концентрации, известные в данной области техники.
Дополнительные компоненты, помимо упомянутых в качестве компонентов i) (включая компоненты a) и c), компоненты b) и d), которые являются необязательными) и ii), если они присутствуют, предпочтительно содержатся в количестве от 0 до 5% (например, от 0,01% до 5%) по массе, предпочтительно не более 2% по массе и более предпочтительно не более 1% по массе.
В одном из вариантов реализации компоненты а) и b) (с учетом любых примесей, неизбежно присутствующих в указанных компонентах) могут составлять по меньшей мере 95% липидных компонентов предварительных составов. Предпочтительно по меньшей мере 99% общего содержания липидов в предварительном составе составляют компоненты а) и b). Предпочтительно липидный компонент предварительного состава состоит по существу из компонентов а) и b).
Введение
Предварительные составы согласно настоящему изобретению в общем случае предназначены для парентерального введения. Указанное введение в общем случае не проводят внутрисосудистым способом, но предпочтительно является подкожным (п.к.), внутриполостным или внутримышечным (в.м.). Как правило, введение проводят путем инъекции, и этот термин используют в настоящем документе для обозначения любого способа, при котором состав вводят через кожу, например, при помощи иглы, катетера или безыгольного (не содержащего иглу) устройства для инъекций. Тем не менее, можно использовать преимущества высокого содержания лекарственного средства и другие благоприятные характеристики предложенного состава в непарентеральных способах введения, включая местное или системное нанесение на кожу, слизистые мембраны, в полость носа, щечную полость и/или полость рта. Предпочтительно указанное непарентеральное введение предназначено для местного применения. Предпочтительно парентеральное введение проводят путем в.м. или п.к. инъекции, наиболее предпочтительно путем глубокой п.к. инъекции. Важный отличительный признак композиции согласно настоящему изобретению заключается в том, что ее можно вводить в.м. и п.к. и другими способами в отсутствие токсичности или значительных местных эффектов. Она также подходит для внутриполостного введения. Глубокая п.к. инъекция имеет преимущество, заключающееся в том, что она является менее глубокой и менее болезненной для субъекта по сравнению с (глубокой) в.м. инъекцией, которую используют в некоторых существующих депо, и с технической точки зрения наиболее подходит в данном случае, так как сочетает в себе простоту инъекции и низкий риск местных побочных эффектов. Авторы настоящего изобретения неожиданно обнаружили, что составы обеспечивают замедленное высвобождение активного агента в течение предсказуемого периода времени как при подкожной, так и при внутримышечной инъекции. Таким образом, это позволяет в значительной степени изменять место введения и вводить дозу без подробного изучения глубины ткани в месте инъекции.
В одном из вариантов реализации липидные предварительные составы согласно настоящему изобретению обеспечивают неламеллярные жидкокристаллические композиции депо при воздействии водных жидкостей, в частности, in vivo. В настоящем описании термин «неламеллярный» используют для обозначения нормальной или обращенной жидкокристаллической фазы (такой как кубическая или гексагональная фаза) или фазы L3 или любой их комбинации. Термин «жидкокристаллический» обозначает все гексагональные, все кубические жидкокристаллические фазы и/или все их смеси. Гексагональная форма при использовании в настоящем документе обозначает «нормальную» или «обращенную» гексагональную (предпочтительно обращенную) форму, а «кубическая» фаза обозначает любую кубическую жидкокристаллическую фазу, если не указано иное.
Специалисту не составит труда идентифицировать те композиции, которые имеют подходящее фазовое поведение, после изучения описания и примеров, приведенных в настоящем документе, и WO 2005/117830, но наиболее предпочтительное фазовое поведение наблюдают для композиции, имеющей отношение компонентов a:b в диапазоне от 40:60 до 70:30, предпочтительно от 45:55 до 55:45 и более предпочтительно от 40:60 до 54:46. Отношения примерно 50:50 (например, от 49:51 до 51:49) являются наиболее предпочтительными, наиболее предпочтительно примерно 50:50.
Важно понимать, что предварительные составы согласно настоящему изобретению имеют низкую вязкость. В результате указанные предварительные составы не должны находиться в какой-либо объемной жидкокристаллической фазе, так как все жидкокристаллические фазы имеют вязкость, значительно превышающую значение, при котором их можно вводить с применением шприца или схожего инъекционного устройства введения. Предварительные составы согласно настоящему изобретению, таким образом, находятся в нежидкокристаллическом состоянии, таком как раствор, L2 или L3 фаза, в частности, раствор или L2. L2 фаза при использовании в настоящем документе предпочтительно представляет собой «набухшую» L2 фазу, содержащую более 5 масс.%, предпочтительно более 7% и наиболее предпочтительно более 9% органического растворителя на основе одноатомного спирта (компонент с), обладающего эффектом снижения вязкости.
Предварительные составы, описанные в настоящем документе, предпочтительно имеют «низкую вязкость». Она может определяться, например, возможностью введения из 1 мл одноразового шприца через иглу малого калибра. Предпочтительно смеси с низкой вязкостью можно вводить через иглу 19 калибра (по стандарту AWG), предпочтительно менее чем 19 калибра, более предпочтительно 23 калибра AWG (или наиболее предпочтительно даже 27 калибра) вручную. В особенно предпочтительном варианте реализации смесь с низкой вязкостью должна представлять собой смесь, способную проходить через стандартную мембрану для стерильного фильтрования, такую как шприцевой фильтр 0,22 мкм. Типичный диапазон подходящей вязкости для предварительных составов согласно изобретению может составлять, например, от 1 до 1000 мПа⋅с, предпочтительно от 10 до 800 мПа⋅с, более предпочтительно от 50 до 750 мПа⋅с и наиболее предпочтительно от 50 до 600 мПа⋅с при 20°C.
После введения многие из предпочтительных предварительных составов на липидной основе согласно настоящему изобретению претерпевают фазовый структурный переход от смеси с низкой вязкостью к композиции депо (обычно прикрепляющейся к ткани) с высокой вязкостью. В общем случае, происходит переход от молекулярной смеси с набухшей фазой L2 и/или L3 к одной или более жидкокристаллическим фазам (с высокой вязкостью), таким как нормальные или обращенные гексагональные или кубические жидкокристаллические фазы или их смеси. Дальнейшие фазовые переходы также могут проходить после введения. Очевидно, что полный фазовый переход не является необходимым для реализации изобретения, но по меньшей мере поверхностный слой вводимой смеси может образовывать жидкокристаллическую структуру. В общем случае, этот переход происходит быстро по меньшей мере на участке поверхности вводимого состава (который находится в прямом контакте с воздухом, поверхностью организма и/или физиологическими жидкостями). Наиболее предпочтительно он занимает несколько секунд или минут (например, от 1 секунды до 30 минут, предпочтительно до 10 минут, более предпочтительно 5 минут или менее). Фазовый переход оставшейся части композиции в жидкокристаллическую фазу может происходить медленнее за счет диффузии и/или диспергирования поверхностного участка.
Изобретение не ограничено составами, которые претерпевают фазовое превращение в жидкокристаллическую структуру после введения. Композиция депо может образовываться после введения посредством других механизмов, которые не требуют образования жидкокристаллической фазы. Например, в системе, описанной в WO 2016/066655, образование композиции депо не сопровождается превращением в жидкокристаллическую фазу.
Не связываясь с теорией, полагают, что при воздействии избытка водной жидкости предварительные составы согласно изобретению теряют часть или весь содержащийся органический растворитель (например, в результате диффузии) и захватывают водную жидкость из среды организма (например, среды in vivo). Для определенных липидных предварительных составов, таких как описано в настоящем документе, по меньшей мере из части состава предпочтительно образуется неламеллярная структура, в частности, с жидкокристаллической фазой. В большинстве случаев эти неламеллярные структуры являются очень вязкими, и их растворение или диспергирование в среде in vivo затруднено. Результатом является монолитное «депо», образующееся in vivo, в котором только незначительная область подвержена воздействию физиологических жидкостей. Кроме того, так как неламеллярная структура содержит крупные полярные, неполярные и граничные области, липидное депо крайне эффективно растворяет и стабилизирует активные агенты, такие как пептиды, и защищает их от механизмов деградации. Так как композиция депо, образующаяся из предварительного состава, постепенно разлагается в течение нескольких дней, недель или месяцев, активный агент постепенно высвобождается и/или диффундирует из композиции. Так как среда внутри композиции депо является относительно защищенной, предварительные составы согласно изобретению особенно подходят для активных агентов с относительно низким биологическим периодом полувыведения (см. выше).
Считается, что за счет включения сорастворителя в предварительные составы, которое было впервые описано в WO 2012/160213, скорость фазового перехода в неламеллярную (например, жидкокристаллическую) фазу на поверхности инъекционного предварительного состава может быть увеличена по сравнению с композициями, содержащими органические растворители по существу в отсутствие воды. Таким образом, улучшаются характеристики полученного депо и обеспечивается дополнительное контролирование высвобождения активного агента.
Системы депо, образующиеся из составов согласно настоящему изобретению, крайне эффективно защищают активный агент от разрушения и, таким образом, обеспечивают долгосрочный период высвобождения. Составы согласно изобретению, таким образом, могут обеспечивать in vivo депо пептидных активных агентов, которые необходимо вводить только раз каждые 5-90 дней, предпочтительно 5-60 дней, более предпочтительно 6-32 дня. Очевидно, что более длительный период стабильного высвобождения желателен для комфорта пациента и соблюдения схемы лечения, а также требует меньше времени от медицинских работников, если композиция не предназначена для самостоятельного введения. Когда композиция должна вводиться самостоятельно, соблюдению пациентом схемы лечения может способствовать введение раз в неделю (например, каждые 7 дней, необязательно ± 1 день), раз в две недели (например, каждые 14 дней, необязательно ± 2 дня) или раз в месяц (например, каждые 28 или 30 дней (необязательно ± 7 дней), таким образом, пациент не забывает о необходимости введения.
Значительным преимуществом предшественников-депо согласно настоящему изобретению является то, что они представляют собой стабильные гомогенные фазы.
То есть, их можно хранить в течение значительных периодов времени (предпочтительно по меньшей мере 6 месяцев) при комнатной температуре или в холодильнике в отсутствие разделения фаз. Помимо обеспечения удобного хранения и легкого введения, это позволяет выбирать дозу активного агента (например, аналога соматостатина, например, октреотида) с учетом вида, возраста, пола, веса и/или физического состояния индивидуального субъекта, путем инъекции выбранного объема.
Таким образом, в настоящем изобретении предложены способы, включающие выбор конкретного дозируемого количества для индивидуума, в частности, в зависимости от массы тела субъекта. Средством для выбора этой дозы является выбор вводимого объема.
Согласно одному предпочтительному аспекту в настоящем изобретении предложен предварительный состав, содержащий липидную смесь i), содержащую компоненты a), b), c) и необязательно d), компонент ii) и 0-1,0% воды. Диапазоны количеств указанных компонентов, как правило, составляют 20-60% а), 20-60% b), 1-30% c) и 0,001-0,8% ii). Предварительные составы согласно настоящему изобретению являются крайне предпочтительными, так как они сохраняют стабильность при долгосрочном хранении в конечной форме, «готовой к введению». В результате их можно легко поставлять для введения медицинскими работниками или пациентами или лицами, осуществляющими уход, которые необязательно должны являться полностью подготовленными медицинскими работниками и могут не иметь опыта или навыков получения сложных препаратов. Это особенно важно при длительных медленно протекающих заболеваниях, таких как диабет.
Устройства
Согласно дополнительному аспекту в настоящем изобретении предложено одноразовое устройство для введения (которое также должно включать компонент устройства), в которое была предварительно введена отмеренная доза предварительного состава согласно настоящему изобретению. Указанное устройство, как правило, содержит одноразовую дозу, готовую к введению, которая, в общем случае, упакована стерильно, и, таким образом, композиция хранится внутри устройства перед введением. Подходящие устройства включают картриджи, ампулы и, в частности, шприцы и цилиндры шприцев со встроенными иглами или со стандартными фитингами (например, Люэра), предназначенными для установки подходящей одноразовой иглы.
Наборы
Предварительно заполненные устройства согласно изобретению также могут быть подходящим образом включены в набор для введения, который также составляет дополнительный аспект изобретения. Согласно дополнительному аспекту в изобретении, таким образом, предложен набор для введения по меньшей мере одного активного агента, содержащий отмеренную дозу состава согласно изобретению и необязательно устройство для введения или его компонент. Предпочтительно доза удерживается внутри устройства или компонента, которое(-ый) подходит для в.м. или предпочтительно п.к. введения. Наборы могут включать дополнительные компоненты для введения, такие как иглы, тампоны и т.д., и необязательно и предпочтительно содержат инструкции по введению. Указанные инструкции, как правило, относятся к введению способом, таким как описано в настоящем документе, и/или к лечению заболевания, указанного выше в настоящем документе.
В изобретении предложено предварительно заполненное устройство для введения, такое как указано в настоящем документе, и набор, такой как указано в настоящем документе, содержащий предварительный состав, такой как описано в настоящем документе.
Согласно альтернативному аспекту настоящего изобретения «набор» может содержать по меньшей мере два сосуда, первый из которых содержит смесь с низкой вязкостью из i) липидной смеси, содержащей компоненты a), c) и необязательно b), и ii), такую как описано в настоящем документе, и второй содержит измеренную дозу по меньшей мере одного активного агента d), такого как описано в настоящем документе.
Указанный «двухкомпонентный набор» может содержать активный агент d) в виде порошкового состава в одной пробирке или предварительно заполненном шприце и компоненты i) и ii) во второй пробирке или предварительно заполненном шприце. В случае двух шприцев перед инъекцией предварительно заполненные шприцы объединяют и смешивают порошок, содержащий активный агент, с матричным составом, перемещая цилиндры шприца назад и вперед, в результате чего образуется раствор или суспензия, который(-ую) вводят путем инъекции. В качестве альтернативы жидкий липидный состав выливают из одной пробирки или предварительно заполняют в шприц и вводят путем инъекции в пробирку, содержащую порошковый активный агент (например, пептид). Указанный состав затем можно перемешивать вручную путем встряхивания или другим подходящим способом перерастворения (например, перемешивая на вортексе и т.д.). Компонент-растворитель может присутствовать в одном или обоих сосудах (например, пробирках или шприцах). Если в растворителе по меньшей мере отчасти растворен активный агент, то он в общем случае имеет форму раствора или суспензии.
Таким образом, согласно указанному аспекту в изобретении предложен двухкомпонентный набор, содержащий
1) первый сосуд, содержащий смесь с низкой вязкостью компонентов a) - c), такую как описано в настоящем документе;
2) необязательный второй сосуд, содержащий по меньшей мере один пептидный активный агент,
3) компонент-антиоксидант ii) необязательно в третьем сосуде, предпочтительно во втором сосуде или наиболее предпочтительно в первом сосуде;
4) необязательно и предпочтительно по меньшей мере одно из:
5) по меньшей мере одного шприца (который может представлять собой один или оба из указанных первого и второго сосудов);
6) иглы для введения, такой как описано в настоящем документе;
7) инструкций по получению композиции согласно изобретению из содержимого первого и второго сосудов;
8) инструкций по введению, в результате которого образуется депо, такое как описано в настоящем документе.
Предпочтительные отличительные признаки и комбинации
Помимо отличительных признаков и предпочтительных отличительных признаков, указанных в настоящем документе, смеси, например, предварительные составы, согласно изобретению могут иметь один или более следующих предпочтительных отличительных признаков, которые могут быть выбраны независимо или в комбинации:
Все указанные в настоящем документе отношения могут необязательно отличаться до 10% от указанного количества, необязательно и предпочтительно до 5%;
Компонент а) содержит, состоит по существу или предпочтительно состоит из GDO;
Компонент b) содержит, состоит по существу или предпочтительно состоит из соевого PC;
Компонент с) содержит, состоит по существу или предпочтительно состоит из спирта, содержащего 1, 2, 3 или 4 атома углерода, предпочтительно изопропанола или более предпочтительно этанола;
Компонент с) включает полярный сорастворитель, такой как пропиленгликоль; Предварительный состав не содержит какой-либо аналог соматостатина (такой как описано в настоящем документе);
Предварительный состав имеет низкую вязкость, такую как указано в настоящем документе;
Предварительный состав образует неламеллярную жидкокристаллическую фазу, такую как указано в настоящем документе, при введении in vivo;
Из предварительного состава после введения in vivo образуется депо, которое высвобождает по меньшей мере один активный агент на терапевтическом уровне в течение периода по меньшей мере 7 дней, предпочтительно по меньшей мере 21 день, более предпочтительно по меньшей мере 28 дней;
Помимо отличительных признаков и предпочтительных отличительных признаков, указанных в настоящем документе, способ(-ы) лечения согласно настоящему изобретению может(-гут) иметь один или более следующих предпочтительных отличительных признаков, которые могут быть выбраны независимо или в комбинации:
Способ включает введение по меньшей мере одного состава, имеющего один или более предпочтительных отличительных признаков, таких как указано выше;
Способ включает введение по меньшей мере одного состава, такого как указано в настоящем документе, путем в.м., п.к. (например, глубокой п.к.) инъекции;
Способ включает введение с применением предварительно заполненного устройства для введения, такого как указано в настоящем документе;
Способ включает введение через иглу не более чем 20 калибра, предпочтительно меньше 20 калибра и наиболее предпочтительно 22 калибра, 23 калибра или менее; Способ включает однократное введение каждые 5-90 дней, предпочтительно 6-32 дня (например, 7 дней или 28-31 день);
Помимо отличительных признаков и предпочтительных отличительных признаков, указанных в настоящем документе, применения предварительных составов, указанных в настоящем документе, для получения лекарственных средств может(-гут) иметь один или более следующих предпочтительных отличительных признаков, которые могут быть выбраны независимо или в комбинации;
Применение включает применение по меньшей мере одного состава, имеющего один или более предпочтительных отличительных признаков, таких как указано выше;
Применение включает получение лекарственного средства для введения по меньшей мере одного состава, такого как указано в настоящем документе, путем в.м. или п.к. инъекции;
Применение включает получение лекарственного средства для введения с применением предварительно заполненного устройства для введения, такого как указано в настоящем документе;
Применение включает получение лекарственного средства для введения через иглу не более чем 20 калибра, предпочтительно меньше 20 калибра и наиболее предпочтительно 22 калибра, 23 калибра или менее;
Применение включает получение лекарственного средства для введения один раз каждые 5-90 дней, предпочтительно 5-60 дней, более предпочтительно 6-32 дня; Помимо отличительных признаков и предпочтительных отличительных признаков, указанных в настоящем документе, предварительно заполненные устройства согласно изобретению могут иметь один или более следующих предпочтительных отличительных признаков, которые могут быть выбраны независимо или в комбинации:
Они содержат предпочтительный состав, такой как указано в настоящем документе;
Они содержат иглу менее чем 20 калибра, предпочтительно не более 22 калибра или не более 23 калибра;
Они содержат гомогенную смесь композиции согласно изобретению в готовой для инъекции форме.
Они содержат состав из компонентов i) (предпочтительно содержащий a), b) и c)) и ii) в комбинации с активным агентом.
Они имеют общий вводимый объем не более 5 мл, предпочтительно не более 3 мл, более предпочтительно не более 1,5 мл.
Помимо отличительных признаков и предпочтительных отличительных признаков, указанных в настоящем документе, наборы согласно изобретению могут иметь один или более следующих предпочтительных отличительных признаков, которые могут быть выбраны независимо или в комбинации:
Они содержат предпочтительный состав, такой как указано в настоящем документе;
Они содержат предварительно заполненное устройство, такое как указано в настоящем документе;
Они содержат иглу менее чем 20 калибра, предпочтительно не более 22 калибра или не более 23 калибра;
Они содержат пептидный активный агент;
Они имеют общий вводимый объем не более 5 мл, предпочтительно не более 3 мл, более предпочтительно не более 1,5 мл;
Они содержат инструкции по введению, касающиеся способа и/или частоты введения, такие как указано в настоящем документе;
Они содержат инструкции по введению для применения в способе лечения, таком как описано в настоящем документе;
Далее изобретение будет дополнительно проиллюстрировано в следующих неограничивающих примерах и на прилагаемых фигурах.
ПРИМЕРЫ
Материалы
Все материалы, которые использовали в примерах, были получены из коммерческих источников и имели фармакопейный класс чистоты, где это применимо, или самый высокий класс чистоты из доступных. В примерах используются следующие сокращения:
Общие способы
Получение растворов ЭДТА и ЭДТА(Na) в EtOH/PG
Образцы готовили путем взвешивания соответствующих количеств ЭДТА или ЭДТА(Na) и алкиламина в стеклянных пробирках, например, пробирках 15R, после чего добавляли органический растворитель или смесь растворителей (например, EtOH/PG (50/50 (масс./масс.)). Пробирки герметично закрывали и помещали в роликовый смеситель в режиме вращения с донышка на крышку при КТ или в магнитную мешалку. Во время растворения пробирки визуально проверяли на наличие нерастворенных частиц ЭДТА с использованием окружающего и кросс- поляризованного света.
Получение растворов FeCl 3 ×6H 2 O
Образцы готовили путем взвешивания соответствующего количества FeCl3×6H2O в стерилизованных стеклянных пробирках, после чего добавляли органический растворитель или смесь растворителей. Пробирки герметично закрывали и помещали в роликовый смеситель в режиме вращения с донышка на крышку при КТ до полного растворения FeCl3×6H2O.
Получение SOM(Cl)
Для способа ионного обмена примерно 120 г смолы Dowex 1x2 в хлоридной форме (50-100 меш) смешивали с равным количеством воды Millipore, добавляли в 200 мл стеклянную ионообменную колонку и оставляли для установления равновесия на ночь.
На следующий день перед ионообменом матрицу Dowex медленно промывали 900 мл воды Millipore и инициировали процесс ионного обмена. 3,743 г SOM(Ac) растворяли в 112,4 г воды Millipore. Свежеприготовленный (примерно за 30 мин) раствор SOM(Ac) помещали в верхнюю часть ионообменной колонки. Начинали элюирование (примерно 15 с/мл) и собирали фракции элюата по 50-250 мл, непрерывно промывая колонку водой Millipore. Объединяли фракции элюата с проводимостью более 50 мкСм/см, переносили в три 1000 мл круглодонные колбы, поверхностно замораживали в смеси EtOH/сухой лед на Rotavapor R-200, помещали охлаждаться при -80°C примерно на 1 ч и лиофилизировали в течение ночи примерно 36 ч. Полученное количество и выход SOM(Cl) составляли 3,096 г и 82,7%, соответственно. Полный обмен ацетата на хлорид подтверждали путем определения двух анионов с помощью УФ-ВЭЖХ с непрямым детектированием.
Получение GOS(Cl)
Для способа ионного обмена 34,7995 г смолы Dowex 1x2 в хлоридной форме (50-100 меш) смешивали с 36,6327 г воды Millipore, добавляли в 100 мл стеклянную ионообменную колонку и оставляли для установления равновесия на ночь. На следующий день перед ионообменом матрицу Dowex медленно промывали 700 мл воды Millipore и инициировали процесс ионного обмена. 0,8353 г GOS(Ac) растворяли в 13,9534 г воды Millipore. Свежеприготовленный (примерно за 30 мин) раствор GOS(Ac) помещали в верхнюю часть ионообменной колонки. Начинали элюирование (примерно 15 с/мл) и собирали фракции элюата по 15-50 мл, непрерывно промывая колонку водой Millipore. Объединяли фракции элюата с проводимостью более 35 мкСм/см, переносили в 500 мл круглодонную колбу, поверхностно замораживали в смеси EtOH/сухой лед на Rotavapor R-200, помещали охлаждаться при -80°C примерно на 1 ч и лиофилизировали в течение примерно 23 ч. Полученное количество и выход GOS(Cl) составляли 0,739 г и 88,5%, соответственно. Полный обмен ацетата на хлорид подтверждали путем определения двух анионов с помощью УФ-ВЭЖХ с непрямым детектированием.
Получение OXY(Cl)
Для способа ионного обмена 31,5493 г смолы Dowex 1x2 в хлоридной форме (50-100 меш) смешивали с 42,1860 г воды Millipore, добавляли в 100 мл стеклянную ионообменную колонку и оставляли для установления равновесия на ночь. На следующий день перед ионообменом матрицу Dowex медленно промывали 600 мл воды Millipore и инициировали процесс ионного обмена. 0,7933 г OXY(Ac) растворяли в 12,5166 г воды Millipore. Свежеприготовленный (примерно за 30 мин) раствор OXY(Ac) помещали в верхнюю часть ионообменной колонки. Начинали элюирование (примерно 15 с/мл) и собирали фракции элюата по 15-50 мл, непрерывно промывая колонку водой Millipore. Объединяли фракции элюата с проводимостью более 25 мкСм/см, переносили в 500 мл круглодонную колбу, поверхностно замораживали в смеси EtOH/сухой лед на Rotavapor R-200, помещали охлаждаться при -80°C примерно на 1 ч и лиофилизировали в течение ночи примерно 25 ч. Полученное количество и выход OXY(Cl) составляли 0,686 г и 86,5%, соответственно. Полный обмен ацетата на хлорид подтверждали путем определения двух анионов с помощью УФ-ВЭЖХ с непрямым детектированием.
Получение липидных составов
Липидные составы плацебо получали путем взвешивания соответствующих количеств SPC, GDO, раствора ЭДТА/алкиламин и раствора FeCl3×6H2O (при необходимости) в стерильных стеклянных пробирках. Затем закрытые пробирки помещали в роликовый смеситель при комнатной температуре до полного смешения с образованием прозрачного гомогенного жидкого раствора (<24 часов).
Составы, содержащие АФИ, получали путем добавления соответствующего количества порошка АФИ в липидные составы плацебо в стерилизованные стеклянные пробирки. Закрывали пробирки и помещали в роликовый смеситель при комнатной температуре до полного смешения с образованием прозрачного гомогенного жидкого раствора (примерно 24 часа).
В качестве примера, ЭДТА и ЭТА (в мольном отношении ЭДТА:ЭТА 1:4) растворяли в смеси EtOH/PG (50/50 (масс./масс.)). Затем соответствующие количества SPC, GDO (в массовом отношении SPC/GDO 50/50) и смеси EtOH/PG/ЭДТА/ЭТА взвешивали в стерилизованной стеклянной пробирке 20R. Затем закрытую пробирку помещали в роликовый смеситель при комнатной температуре до полного смешения с образованием прозрачного гомогенного жидкого раствора (<24 часов). Затем к липидным составам добавляли порошок OCT(Cl) в стерилизованной стеклянной пробирке 15R в концентрации 2,34 масс.%. Закрывали пробирку и помещали в роликовый смеситель при комнатной температуре до полного смешения с образованием прозрачного гомогенного жидкого раствора (24 часа).
Оценка стабильности октреотида в липидных составах (типовой способ)
Полученные липидные составы пептида (например, октреотида), такие как описано выше, размещали в стерилизованные стеклянные пробирки 2R (0,5 г состава на пробирку). Свободное пространство пробирок содержало воздух окружающей среды, т.е. свободное пространство не заполняли инертной атмосферой, такой как азот. Закрывали пробирки и помещали в камеры с контролируемыми условиями хранения при 25°C/60% отн.вл. и 40°C/75% отн.вл. В предварительно определенные моменты отбора проб (хранение до трех месяцев) удаляли две пробирки каждого состава и камеру для хранения, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Следует отметить, что процедура заполнения и условия хранения обеспечивали условия ускоренного разрушения, так как свободное пространство содержало воздух, а не инертную атмосферу, такую как азот.
Определение уровня пептидов в липидных составах путем УФ-ВЭЖХ
Определение уровня пептида (например, октреотида, такого как октреотида хлорид) в липидных составах проводили путем градиентной ВЭЖХ с УФ-детектированием.
Используемая аналитическая колонка HILIC представляла собой HALO Penta-HILIC 2,7 мкм, 150×3,0 мм. Количественную оценку проводили путем интерполяции значений площади пика пептида (например, октреотида), полученных в образцах липидного состава (полученных путем растворения липидного состава в растворителе образца при требуемой концентрации целевого пептида) в калибровочные кривые, полученные для стандартных растворов, содержащих известные концентрации соответствующего пептида.
Типичные используемые подвижные фазы (например, для октреотида) состояли из смеси вода: 2M хлорид натрия: ацетонитрил: трифторуксусная кислота 384:16:400:1 (об./об.) (подвижная фаза A) и вода: метанол: ацетонитрил: трифторуксусная кислота 20:30:950:1 (об./об.) (подвижная фаза B). Детектирование проводили при 220 нм. В качестве растворителя образца использовали смесь ацетонитрил: метанол (1:1 (об./об.)); октреотид элюировался примерно через 25,2 мин.
Представление данных
В разделе примеров помимо абсолютных значений уровня АФИ в некоторых случаях результаты также выражены как коэффициент стабильности исследуемого АФИ. Коэффициент стабильности вычисляли как отношение уровня АФИ в конкретном составе к уровню АФИ в составе сравнения. Если результаты выражены таким образом, то значения коэффициента стабильности более 1 обозначают увеличение стабильности АФИ по сравнению со стандартом сравнения.
Измерение концентрации кислорода в свободном пространстве пробирки Концентрацию кислорода в свободном пространстве пробирки измеряли при помощи микроволоконного оптического устройства для определения кислородопроницаемости PreSens Microx TX3 с компьютерным управлением, оборудованного игольчатым оптическим микродатчиком кислорода (NTH, плоский ломаный наконечник 140 мкм).
Измерения проводили путем введения микродатчика кислорода через резиновую пробку пробирки в свободное пространство и определения концентрации кислорода до получения стабильного показания (примерно 1 минуту).
Пример 1. Растворимость ЭДТА в присутствии алкиламина и без него
Готовили 0,08 масс.% растворы ЭДТА и ЭДТА(Na) в EtOH/PG (50/50 (масс./масс.)) в присутствии ЭТА и без него (таблица 1). Растворение ЭДТА и ЭДТА(Na) при перемешивании с донышка на крышку при КТ оценивали путем визуального осмотра (окружающий и кроссполяризованный свет) в течение 27 дней. Результаты показывают, что ни динатриевая соль (ЭДТА(Na)), ни кислотная форма ЭДТА не растворялись в EtOH/PG без использования ЭТА даже после 27-дневного перемешивания. Полученные результаты также показывают, что ЭДТА(Na) не растворялась в EtOH/PG даже в присутствии ЭТА, тогда как кислотная форма ЭДТА растворялась в EtOH/PG в присутствии 4 моль ЭТА на 1 моль ЭДТА уже после 24 часов перемешивания.
Таблица 1. Растворимость 0,08 масс.% ЭДТА и ЭДТА(Na) в EtOH/PG в присутствии ЭТА и без него.
Пример 2. Зависимость растворимости ЭДТА от мольного отношения ЭТА/ЭДТА
В таблице 2 сведены результаты зависимости растворимости ЭДТА в концентрации 0,38 масс.% в смесях растворителей EtOH/PG (1/1 (масс./масс.)) от мольного отношения ЭТА/ЭДТА. Единственный образец, в котором ЭДТА растворялась не полностью, имел наименьшее мольное отношение ЭТА/ЭДТА. Во всех других образцах ЭДТА растворялась после 24-часового перемешивания с донышка на крышку при КТ.
Полученные результаты показывают, что примерно 3,5 моль ЭТА на 1 мо ль ЭДТА близко к требуемому минимальному количеству, необходимому для растворения ЭДТА в используемом неводном растворителе.
Таблица 2. Зависимость растворимости 0,38 масс.% ЭДТА в EtOH/PG от мольного отношения ЭТА/ЭДТА.
Пример 3. Зависимость растворимости ЭДТА от мольного отношения ди-ЭТА/ЭДТА
В таблице 3 сведены результаты зависимости растворимости ЭДТА в смеси растворителей EtOH/PG (1/1 (масс./масс.)) в концентрации 0,38 масс.% ЭДТА от мольного отношения ди-ЭТА/ЭДТА после 24 ч перемешивания с донышка на крышку при КТ. Полученные результаты показывают, что примерно 4,5 моль ди-ЭТА на 1 моль ЭДТА близко к требуемому минимальному количеству, необходимому для растворения ЭДТА в используемом неводном растворителе.
Таблица 3. Зависимость растворимости 0,38 масс.% ЭДТА в EtOH/PG от мольного отношения ди-ЭТА/ЭДТА.
растворяется
Пример 4. Зависимость растворимости ЭДТА от мольного отношения этилендиамин/ЭДТА
В таблице 4 сведены результаты зависимости растворимости ЭДТА в смеси растворителей EtOH/PG (1/1 (масс./масс.)) в концентрации 0,38 масс.% ЭДТА от мольного отношения этилендиамин/ЭДТА после 24 ч перемешивания с донышка на крышку при КТ. Полученные результаты показывают, что примерно 2,5 моль этилендиамина на 1 моль ЭДТА близко к требуемому минимальному количеству, необходимому для растворения ЭДТА в используемом неводном растворителе.
Таблица 4. Зависимость растворимости 0,38 масс.% ЭДТА в EtOH/PG от мольного отношения этилендиамин/ЭДТА.
Пример 5. Зависимость растворимости ЭДТА от мольного отношения серинол/ЭДТА
В таблице 5 сведены результаты зависимости растворимости ЭДТА в смеси растворителей EtOH/PG (1/1 (масс./масс.)) в концентрации 0,38 масс.% ЭДТА от мольного отношения серинол/ЭДТА после 24 ч перемешивания с донышка на крышку при КТ. Полученные результаты показывают, что примерно 4 моль серинола на 1 моль ЭДТА близко к требуемому минимальному количеству, необходимому для растворения ЭДТА в используемом неводном растворителе.
Таблица 5. Зависимость растворимости 0,38 масс.% ЭДТА в EtOH/PG от мольного отношения серинол/ЭДТА.
Пример 6. Зависимость растворимости ЭДТА от мольного отношения TRIS/ЭДТА
В таблице 6 сведены результаты зависимости растворимости ЭДТА в смеси растворителей EtOH/PG (1/1 (масс./масс.)) в концентрации 0,38 масс.% ЭДТА от мольного отношения TRIS/ЭДТА после 7 дней перемешивания с донышка на крышку при КТ. Полученные результаты показывают, что примерно 5 моль TRIS на 1 моль ЭДТА близко к требуемому минимальному количеству, необходимому для растворения ЭДТА в используемом неводном растворителе.
Таблица 6. Зависимость растворимости 0,38 масс.% ЭДТА в EtOH/PG от мольного отношения TRIS/ЭДТА.
Пример 7. Стабильность OCT(Cl) в липидных составах в присутствии ЭДТА Липидные составы, содержащие 2,34 масс.% OCT(Cl) в присутствии 100 ppm ЭДТА или без нее, получали в соответствии с составами, приведенными в таблице 7.
Составы помещали в стерилизованные стеклянные пробирки 2R (0,5 г состава на пробирку), закрывали и помещали в камеры с контролируемыми условиями хранения при 40°C/75% отн.вл. или 25°C/60% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. В предварительно определенные моменты отбора проб (хранение до трех месяцев) удаляли две пробирки каждого состава и снимали условия хранения в камерах с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 7. Композиции составов FluidCrystal®, содержащих OCT(Cl) (в масс.%) совместно с ЭДТА и без нее.
Образцы двух составов отбирали для исследования стабильности, как описано в общих способах. Следует отметить, что процедура заполнения и условия хранения обеспечивали условия ускоренного разрушения, так как свободное пространство содержало воздух, а не инертную атмосферу, такую как азот. На фиг. 1 приведен уровень октреотида в различные моменты хранения и в различных условиях хранения. Как показано на фиг. 1, присутствие 0,01 масс.% (100 ppm) ЭДТА, растворенной в липидном составе при использовании 0,01 масс.% (100 ppm) ЭТА, значительно повышает стабильность пептида при обоих условиях хранения.
Пример 8. Влияние концентрации ЭДТА на стабильность пептида
Липидные составы, содержащие 2,27 масс.% OCT(Cl) и различные концентрации ЭДТА, получали в соответствии с составами, приведенными в таблице 8. Составы помещали в стерилизованные стеклянные пробирки 2R (0,5 г состава на пробирку), закрывали и помещали в камеры с контролируемыми условиями хранения при 40°C/75% отн.вл. или 25°C/60% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. В предварительно определенные моменты отбора проб (хранение до шести месяцев) удаляли две пробирки каждого состава и снимали условия хранения в камерах с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ - детектированием.
Таблица 8. Составы композиций с различными концентрациями ЭДТА (все компоненты в масс.%), содержащих 2,27 масс.% OCT(Cl).
Образцы шести составов отбирали для исследования стабильности, как описано в общих способах. Следует отметить, что процедура заполнения и условия хранения обеспечивали условия ускоренного разрушения, так как свободное пространство содержало воздух, а не инертную атмосферу, такую как азот. Результаты показаны на фигуре 2. Как показано, присутствие ЭДТА, растворенной в липидном составе при использовании ЭТА, значительно увеличивало стабильность пептида по сравнению с составом сравнения, не содержащим ЭДТА/ЭТА. Максимальный эффект стабилизации был достигнут для интервала концентраций 50-250 ppm (0,005-0,025 масс.%) ЭДТА.
ПРИМЕР 9. Долгосрочная стабильность OCT(Cl) в липидных составах в присутствии ЭДТА
Липидные составы, содержащие OCT(Cl) без ЭДТА или в присутствии 100 ppm ЭДТА, получали в соответствии с составами, приведенными в таблице 9. Составы помещали в 1 мл стерилизованные стеклянные шприцы 22G × 1/2'' (Schott AG) (0,5 г состава на шприц), герметично закрывали плунжером и помещали в камеру с контролируемыми условиями хранения при 25°C/60% отн.вл. В предварительно определенные моменты отбора проб (хранение до двенадцати месяцев) удаляли два шприца с каждым составом и извлекали из камер с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 9. Композиции липидных составов, содержащих OCT(Cl) (в масс.%) без ЭДТА и с добавками ЭДТА. Содержание октреотида соответствует 20 мг/мл свободного основания октреотида с поправкой на содержание пептида, чистоту и плотность состава.
На фигуре 3 приведен уровень октреотида в различные моменты хранения. Как показано, присутствие 0,01 масс.% (100 ppm) ЭДТА, растворенной в липидном составе при использовании ЭТА, значительно увеличивало долгосрочную стабильность пептида в предварительно заполненных шприцах в условиях долгосрочного хранения при 25°C/60% отн.вл.
ПРИМЕР 10. Стабильность OCT(Cl) в липидных составах в присутствии железа и ЭДТА
Липидные составы, содержащие OCT(Cl) и различные количества Fe3+ и ЭДТА, получали в соответствии с составами, приведенными в таблице 10. Составы помещали в стерилизованные стеклянные пробирки 2R (0,5 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 40°C/75% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. Через 1 месяц удаляли две пробирки каждого состава и снимали условия хранения в камерах с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 10. Составы композиций FluidCrystal® (в масс.%), содержащих OCT(Cl) и различные концентрации Fe3+ и ЭДТА. Содержание октреотида соответствует 20 мг/мл свободного основания октреотида с поправкой на содержание пептида, чистоту и плотность состава. Массовое отношение SPC/GDO составляет 50/50 для всех составов.
* 0,00097, 0,00242 и 0,00484 масс.% FeCl3×6H2O соответствуют 2, 5 и 10 ppm Fe3+, соответственно.
На фигуре 4 приведена зависимость уровня октреотида через 1 месяц от концентрации Fe3+ в присутствии различных количеств ЭДТА. Как видно, с увеличением концентрации Fe3+, необходимо все большее количество ЭДТА для защиты OCT от разрушения. Защита OCT от разрушения в присутствии Fe3+ усиливается при увеличении концентрации ЭДТА до 100 ppm, затем наблюдается некоторое снижение в диапазоне от 100 до 250 ppm. Также существует четкая связь между концентрацией Fe3+ и количеством ЭДТА, необходимым для подавления каталитической активности железа. Как показано на фигуре 5, максимальный эффект стабилизации достигается, начиная с мольного отношения ЭДТА:Fe3+ примерно 2:1. Это соответствует примерно 100 ppm ЭДТА при содержании Fe3+ 10 ppm.
ПРИМЕР 11. Стабильность OCT(Cl) в липидных составах, содержащих ЭДТА и железо, без ЭТА и в присутствии ЭТА
Липидные составы, содержащие ЭДТА или ЭДТА(Na) без ЭТА и в присутствии ЭТА, готовили в соответствии с композициями, приведенными в таблице 11. Как показано в примере 1, ни ЭДТА(Na), ни ЭДТА не растворяются в EtOH/PG без использования ЭТА. ЭДТА(Na) также не растворялся в EtOH/PG даже в присутствии ЭТА, что оценивали при визуальном осмотре. Соответственно, смеси, содержащие ЭДТА(Na), ЭДТА и ЭДТА(Na)/ЭТА в EtOH/PG, дополнительно фильтровали с использованием гидрофильного шприцевого фильтра PTFE Millex-LG 0,2 мкм для удаления нерастворенных частиц ЭДТА. После получения составы помещали в стерилизованные стеклянные пробирки 2R (0,5 г состава на пробирку), закрывали и помещали в камеры с контролируемыми условиями хранения при 40°C/75% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. В предварительно определенные моменты отбора проб (хранение до двух месяцев) удаляли две пробирки каждого состава и снимали условия хранения в камерах с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 11. Составы липидных композиций (в масс.%), содержащих OCT(Cl) и различные концентрации Fe3+ и ЭДТА. Содержание октреотида соответствует 20 мг/мл свободного основания октреотида с поправкой на содержание пептида, чистоту и плотность состава. Массовое отношение SPC/GDO составляло 50/50 для всех составов.
образца
**
* При получении указанных составов смеси ЭДТА в EtOH/PG фильтровали с использованием гидрофильного шприцевого фильтра PTFE Millex-LG 0,2 мкм для удаления нерастворимых частиц ЭДТА.
** 0,00242 масс.% FeCl3×6H2O соответствует 5 ppm Fe3+.
На фигуре 6 приведена зависимость значений уровня и коэффициента стабильности октреотида от времени, соответственно. Как видно, только ЭДТА, растворенная в липидном составе при использовании ЭТА, значительно увеличивала стабильность пептида по сравнению с составом сравнения в присутствии 5 ppm Fe3+. В тех же условиях для составов, содержащих ЭДТА(Na), ЭДТА или ЭДТА (Na)/ЭТА, было показано отрицательное влияние на стабильность OCT(Cl) (по сравнению с составом сравнения).
ПРИМЕР 12. Влияние различных алкиламинов и растворителей на стабильность OCT(Cl) в липидных составах, содержащих ЭДТА
Липидные составы получали в соответствии с составами, приведенными в таблице 12. Составы помещали в стерилизованные стеклянные пробирки 2R (0,5 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 40°C/75% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. В предварительно определенные моменты отбора проб (хранение до двух месяцев) удаляли две пробирки каждого состава и снимали условия хранения в камерах с контролируемой средо й, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ - детектированием.
Таблица 12. Композиции липидных составов, содержащих OCT(Cl) (в масс.%). Содержание октреотида соответствует 20 мг/мл свободного основания октреотида с поправкой на содержание пептида, чистоту и плотность состава. Массовое отношение SPC/GDO составляло 50/50, и мольные отношения ЭТА:ЭДТА, ди-ЭТА:ЭДТА и этилендиамин:ЭДТА составляли 4:1 для всех составов.
образца
На фигуре 7 приведен уровень октреотида в различные моменты хранения. Как показано, различные алкиламины (ЭТА, ди-ЭТА или этилендиамин), которые использовали для растворения 0,01 масс.% (100 ppm) ЭДТА в липидных составах, повышали стабильность пептида в схожей высокой степени по сравнению с составом сравнения. Полученные результаты также показывают, что положительное влияние ЭДТА на стабильность OCT(Cl) не зависит от смеси, используемой для получения липидных составов, что подтверждается данными для составов, содержащих EtOH/PG, на фигуре 8 по сравнению с фигурой 9.
ПРИМЕР 13. Стабильность SOM(Cl) в липидных составах в присутствии ЭДТА
Липидные составы, содержащие SOM(Cl) без ЭДТА и в присутствии 100 ppm ЭДТА и EtOH/PG, получали в соответствии с составами, приведенными в таблице 13. Составы помещали в стерилизованные стеклянные пробирки 2R (0,5 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 40°C/75% отн.вл. или 25°C/60% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. В предварительно определенные моменты отбора проб (хранение до трех месяцев) удаляли две пробирки каждого состава и снимали условия хранения в камерах с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 13. Композиции липидных составов, содержащих SOM(Cl) (в масс.%) без ЭДТА и с добавками ЭДТА. Массовое отношение SPC/GDO составляло 50/50 для всех составов.
На фигуре 9 приведен уровень SOM в различные моменты хранения и в различных условиях хранения. Как показано, присутствие 100 ppm ЭДТА, растворенной в липидном составе при использовании ЭТА, значительно увеличивало стабильность пептида в условиях хранения при 40°C/75% отн.вл. и 25°C/60% отн.вл.
ПРИМЕР 14. Стабильность GOS(Cl) в липидных составах в присутствии ЭДТА и железа
Липидные составы, содержащие GOS(Cl) без ЭДТА и в присутствии 100 ppm ЭДТА, получали в соответствии с составами, приведенными в таблице 14. Составы помещали в стерилизованные стеклянные пробирки 2R (0,9 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 40°C/75% отн.вл.
Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. Оба состава также содержали 5 ppm Fe3+ для обеспечения условий дополнительного окислительного воздействия. В предварительно определенные моменты отбора проб (хранение до двух месяцев) удаляли две пробирки каждого состава из камер с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 14. Композиции липидных составов, содержащих GOS(Cl) (в масс.%) без ЭДТА и с добавками ЭДТА. Массовое отношение SPC/GDO составляет 50/50 для всех составов.
* 0,00242 масс.% FeCl3×6H2O соответствует 5 ppm Fe3+
На фигуре 10 приведена зависимость значений уровня и коэффициента стабильности GOS от времени, соответственно. Как видно, применение 100 ppm ЭДТА, растворенной в липидном составе при использовании ЭТА, значительно увеличивало стабильность пептида в присутствии 5 ppm Fe3+. Данные указывают на то, что ЭДТА обеспечивает защиту пептида от металлов, содержащихся в низких или умеренных количествах, которые могут появляться в результате использования вспомогательных веществ, АФИ или технологического оборудования.
ПРИМЕР 15. Стабильность OXY(Cl) в липидных составах в присутствии ЭДТА и железа
Липидные составы, содержащие OXY(Cl) без ЭДТА и в присутствии 100 ppm ЭДТА, получали в соответствии с составами, приведенными в таблице 15. Составы помещали в стерилизованные стеклянные пробирки 2R (0,9 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 40°C/75% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. Оба состава также содержали 5 ppm Fe3+ для усиления условий окислительного воздействия. В предварительно определенные моменты отбора проб (хранение до двух месяцев) удаляли две пробирки каждого состава из камер с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 15. Композиции липидных составов, содержащих OXY(Cl) (в масс.%) без
ЭДТА и с добавками ЭДТА. Массовое отношение SPC/GDO составляет 50/50 для всех составов.
* 0,00242 масс.% FeCl3×6H2O соответствует 5 ppm Fe3+
На фигуре 11 приведена зависимость значений уровня и коэффициента стабильности OXY от времени, соответственно. Как видно, применение 100 ppm ЭДТА, растворенной в липидном составе при использовании ЭТА, значительно увеличивало стабильность пептида в присутствии 5 ppm Fe3+. Данные указывают на то, что ЭДТА обеспечивает защиту пептида от металлов, содержащихся в низких или умеренных количествах, которые могут появляться в результате использования вспомогательных веществ, АФИ или технологического оборудования.
ПРИМЕР 16. Стабильность GRN(0) в липидных составах в присутствии ЭДТА и железа
Липидные составы, содержащие GRN(0) без ЭДТА и в присутствии 100 ppm ЭДТА, получали в соответствии с составами, приведенными в таблице 16. Составы помещали в стерилизованные стеклянные пробирки 2R (1 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 40°C/75% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. Оба состава также содержали 5 ppm Fe3+ для обеспечения условий дополнительного окислительного воздействия. В предварительно определенные моменты отбора проб (хранение до двух месяцев) удаляли две пробирки каждого состава из камер с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 16. Композиции липидных составов, содержащих GRN(0) (в масс.%) без ЭДТА и с ЭДТА. При корректировке на уровень исследуемого вещества и содержание воды концентрация GRN составляла 9,94 мг/г для всех составов. Массовое отношение SPC/GDO составляет 50/50 для всех составов.
На фигуре 12 приведена зависимость значений уровня и коэффициента стабильности GRN от времени, соответственно. Как видно, 100 ppm ЭДТА, растворенная в липидном составе при использовании ЭТА, значительно увеличивала стабильность GRN в присутствии 5 ppm Fe3+. Данные указывают на то, что ЭДТА обеспечивает защиту активного вещества от металлов, содержащихся в низких или умеренных количествах, которые могут появляться в результате использования вспомогательных веществ, АФИ или технологического оборудования.
ПРИМЕР 17. Стабильность GOS(Cl) в составах на основе фосфолипида/моноглицерида (SPC/GMO) и фосфолипида/триглицерида (SPC/SbOil) в присутствии ЭДТА и железа
Липидные составы, содержащие GOS(Cl) без ЭДТА и в присутствии 100 ppm ЭДТА, получали в соответствии с составами, приведенными в таблице 17. Составы помещали в стерилизованные стеклянные пробирки 2R (1 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 40°C/75% отн.вл.
Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного разрушения, то есть не использовали инертную атмосферу, такую как азот. Все составы также содержали 5 ppm Fe3+ для усиления условий окислительного воздействия. В предварительно определенные моменты отбора проб (хранение до 9 недель) удаляли две пробирки каждого состава из камер с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали содержание (уровень) пептида путем градиентной ВЭЖХ с УФ-детектированием.
Таблица 17. Композиции липидных составов, содержащих GOS(Cl) (в масс.%) без ЭДТА и с ЭДТА. Массовые отношения SPC/GMO и SPC/SbOil составляли 50/50 для всех составов.
GMO
SbOil
* 0,00242 масс.% FeCl3×6H2O соответствует 5 ppm Fe3+
На фигуре 13 и фигуре 14 приведены зависимости значений уровня и коэффициента стабильности для GOS, растворенного в составах на основе SPC/GMO или SPC/SbOil, от времени, соответственно. Показано, что присутствие 100 ppm ЭДТА, растворенной в обеих основах составов при использовании ЭТА, значительно увеличивало стабильность пептида в присутствии 5 ppm Fe3+. Данные указывают на то, что ЭДТА обеспечивает защиту пептида от металлов, содержащихся в низких или умеренных количествах, которые могут появляться в результате использования вспомогательных веществ, АФИ или технологического оборудования.
ПРИМЕР 18. Окисление липидов в липидных составах плацебо в присутствии ЭДТА
Липидные составы плацебо без ЭДТА и в присутствии 100 ppm ЭДТА получали в соответствии с составами, приведенными в таблице 18. Составы помещали в стерилизованные стеклянные пробирки 2R (1 г состава на пробирку), закрывали и помещали в камеру с контролируемыми условиями хранения при 60°C/отн.вл. окружающей среды или 40°C/75% отн.вл. Свободное пространство пробирок содержало воздух окружающей среды для обеспечения условий ускоренного окисления липидов, то есть не использовали инертную атмосферу, такую как азот. Некоторые составы также содержали 5 ppm Fe3+ для усиления условий окислительного воздействия (таблица 18). В предварительно определенные моменты отбора проб (хранение до 9 дней при 60°C/отн.вл. окружающей среды и до 30 дней при 40°C/75% отн.вл.) удаляли две пробирки каждого состава из камер с контролируемой средой, устанавливали равновесие при комнатной температуре в течение 1 часа и анализировали концентрацию кислорода в свободном пространстве (расходование кислорода в настоящем примере используют как косвенную меру окисления липидов в липидных составах) с использованием игольчатого микродатчика кислорода.
Таблица 18. Композиции липидных составов (в масс.%) без ЭДТА и с ЭДТА. Массовое отношение SPC/GDO составляло 50/50 и 35/65 для образцов 103-106 и для образцов 107-110, соответственно.
*Мольное отношение ЭТА:ЭДТА составляет 5,5:1.
** 0,00242 масс.% FeCl3×6H2O соответствует 5 ppm Fe3+
Полученные результаты сведены на фигуре 15, фигуре 16, фигуре 17 и фигуре 18.
Данные, приведенные на фигурах, явным образом показывают, что независимо от условий хранения, отношения липидов, используемых при получении составов или присутствия Fe3+, добавление 100 ppm ЭДТА значительно уменьшало расходование кислорода (и, таким образом, окислительное разрушение липидов) для всех составов. Данные указывают на то, что ЭДТА обеспечивает защиту липидов от металлов, содержащихся в низких или умеренных количествах, которые могут появляться в результате использования вспомогательных веществ или технологического оборудования.
ПРИМЕР 19. Зависимость растворимости DTPA от мольного отношения ЭТА/DTPA
Готовили 0,08 масс.% растворы DTPA в EtOH/PG (50/50 (масс./масс.)) без ЭТА и в присутствии различных количеств ЭТА, добавляемых при различных мольных отношениях ЭТА/DTPA (таблица 19). Результаты показывают, что DTPA не растворяется в EtOH/PG без использования ЭТА. Полученные результаты также показывают, что примерно 4,3 моль ЭТА на 1 моль DTPA близко к требуемому минимальному количеству, необходимому для растворения DTPA в используемом неводном растворителе.
Таблица 19. Зависимость растворимости 0,08 масс.% DTPA в EtOH/PG от мольного отношения ЭТА/DTPA.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Camurus AB
<120> MIXTURES AND FORMULATIONS
<130> 95.39.127583/03
<140> PCT/EP2017/074418
<141> 2017-09-26
<150> GB1616366.9
<151> 2016-09-27
<150> EP16190892.6
<151> 2016-09-27
<160> 21
<170> PatentIn version 3.5
<210> 1
<211> 10
<212> PRT
<213> Homo sapiens
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 1
Xaa His Trp Ser Tyr Gly Leu Arg Pro Xaa
1 5 10
<210> 2
<211> 10
<212> PRT
<213> Homo sapiens
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 2
Xaa His Trp Ser His Gly Trp Tyr Pro Xaa
1 5 10
<210> 3
<211> 10
<212> PRT
<213> Homo sapiens
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 3
Xaa His Trp Ser Tyr Gly Trp Tyr Pro Xaa
1 5 10
<210> 4
<211> 9
<212> PRT
<213> Artificial Sequence
<220>
<223> Фертирелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (9)..(9)
<223> Pro-N-Et-NH2
<400> 4
Xaa His Trp Ser Tyr Gly Leu Arg Xaa
1 5
<210> 5
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Лейпрорелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (6)..(6)
<223> D-Leu
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 5
Xaa His Trp Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 6
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Бусерелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (6)..(6)
<223> Ser(t-Bu)
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 6
Xaa His Trp Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 7
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Гистрелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (6)..(6)
<223> (D)-His(Imbzl)
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 7
Xaa His Trp Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 8
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Деслорелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (6)..(6)
<223> (D)-Trp
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 8
Xaa His Trp Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 9
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Госерелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (6)..(6)
<223> Ser(t-Bu)
<220>
<221> X
<222> (10)..(10)
<223> AzaGly-NH2
<400> 9
Xaa His Trp Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 10
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Нафарелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (6)..(6)
<223> (D)-Nal
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 10
Xaa His Trp Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 11
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Трипторелин
<220>
<221> X
<222> (1)..(1)
<223> пиро-Glu
<220>
<221> X
<222> (6)..(6)
<223> (D)-Trp
<220>
<221> X
<222> (10)..(10)
<223> Gly-NH2
<400> 11
Xaa His Trp Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 12
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Абареликс
<220>
<221> X
<222> (1)..(1)
<223> (D)-Ala
<220>
<221> X
<222> (2)..(2)
<223> (D)-Phe
<220>
<221> X
<222> (3)..(3)
<223> (D)-Ala
<220>
<221> X
<222> (6)..(6)
<223> (D)-Asp
<220>
<221> X
<222> (8)..(8)
<223> Lys(i-Pr)
<220>
<221> X
<222> (10)..(10)
<223> (D)-Ala
<400> 12
Xaa Xaa Xaa Ser Tyr Xaa Leu Xaa Pro Xaa
1 5 10
<210> 13
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Антареликс
<220>
<221> X
<222> (1)..(1)
<223> (D)-Nal
<220>
<221> X
<222> (2)..(2)
<223> (D)-Phe
<220>
<221> X
<222> (3)..(3)
<223> (D)-Pal
<220>
<221> X
<222> (6)..(6)
<223> (D)-Hcit
<220>
<221> X
<222> (8)..(8)
<223> Lys(i-Pr)
<220>
<221> X
<222> (10)..(10)
<223> (D)-Ala
<400> 13
Xaa Xaa Xaa Ser Phe Xaa Leu Xaa Pro Xaa
1 5 10
<210> 14
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Цетрореликс
<220>
<221> X
<222> (1)..(1)
<223> (D)-Nal
<220>
<221> X
<222> (2)..(2)
<223> (D)-Phe
<220>
<221> X
<222> (3)..(3)
<223> (D)-Pal
<220>
<221> X
<222> (6)..(6)
<223> (D)-Cit
<220>
<221> X
<222> (10)..(10)
<223> (D)-Ala
<400> 14
Xaa Xaa Xaa Ser Tyr Xaa Leu Arg Pro Xaa
1 5 10
<210> 15
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Ганиреликс
<220>
<221> X
<222> (1)..(1)
<223> (D)-Nal
<220>
<221> X
<222> (2)..(2)
<223> (D)-Phe
<220>
<221> X
<222> (3)..(3)
<223> (D)-Pal
<220>
<221> X
<222> (6)..(6)
<223> (D)-hArg
<220>
<221> X
<222> (8)..(8)
<223> HArg
<220>
<221> X
<222> (10)..(10)
<223> (D)-Ala
<400> 15
Xaa Xaa Xaa Ser Tyr Xaa Leu Xaa Pro Xaa
1 5 10
<210> 16
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Итреликс
<220>
<221> X
<222> (1)..(1)
<223> (D)-Nal
<220>
<221> X
<222> (2)..(2)
<223> (D)-Phe
<220>
<221> X
<222> (3)..(3)
<223> (D)-Pal
<220>
<221> X
<222> (5)..(5)
<223> NicLys
<220>
<221> X
<222> (6)..(6)
<223> (D)-NicLys
<220>
<221> X
<222> (8)..(8)
<223> Lys(i-Pr)
<220>
<221> X
<222> (10)..(10)
<223> (D)-Ala
<400> 16
Xaa Xaa Xaa Ser Xaa Xaa Leu Xaa Pro Xaa
1 5 10
<210> 17
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Nal-Glu
<220>
<221> X
<222> (1)..(1)
<223> (D)-Nal
<220>
<221> X
<222> (2)..(2)
<223> (D)-Phe
<220>
<221> X
<222> (3)..(3)
<223> (D)-Pal
<220>
<221> X
<222> (5)..(5)
<223> (D)-Glu
<220>
<221> X
<222> (6)..(6)
<223> (D)-Glu
<220>
<221> X
<222> (10)..(10)
<223> (D)-Ala
<400> 17
Xaa Xaa Xaa Ser Xaa Xaa Leu Arg Pro Xaa
1 5 10
<210> 18
<211> 14
<212> PRT
<213> Homo sapiens
<400> 18
Ala Gly Cys Lys Asn Phe Phe Trp Lys Thr Phe Thr Ser Cys
1 5 10
<210> 19
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Октреотид
<220>
<221> X
<222> (1)..(1)
<223> (D)-Phe
<220>
<221> X
<222> (4)..(4)
<223> (D)-Trp
<220>
<221> X
<222> (8)..(8)
<223> Thr-oл
<400> 19
Xaa Cys Phe Xaa Lys Thr Cys Xaa
1 5
<210> 20
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Лантреотид
<220>
<221> X
<222> (1)..(1)
<223> (D)-Naph
<220>
<221> X
<222> (4)..(4)
<223> (D)-Trp
<220>
<221> X
<222> (8)..(8)
<223> Thr-CONH2
<400> 20
Xaa Cys Tyr Xaa Lys Val Cys Xaa
1 5
<210> 21
<211> 8
<212> PRT
<213> Artificial Sequence
<220>
<223> Производное лантреотида
<220>
<221> X
<222> (1)..(1)
<223> (D)-Naph
<220>
<221> X
<222> (4)..(4)
<223> (D)-Phe
<220>
<221> X
<222> (8)..(8)
<223> Thr-CONH2
<400> 21
Xaa Cys Tyr Xaa Lys Val Cys Xaa
1 5
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЙ АНАЛОГ АЦИЛИРОВАННОГО ОКСИНТОМОДУЛИНА | 2018 |
|
RU2752787C1 |
| ПЕПТИДЫ С ЗАЩИТНОЙ АКТИВНОСТЬЮ ОТ ПОВРЕЖДЕНИЯ КЛЕТОК И ЛИПОСОМАЛЬНЫЕ СОСТАВЫ | 2020 |
|
RU2833316C1 |
| САМОСОБИРАЮЩИЕСЯ ПЕПТИДЫ В ПРОФИЛАКТИКЕ И ЛЕЧЕНИИ КАРИОЗНЫХ ПОРАЖЕНИЙ, СОПРОВОЖДАЮЩИХСЯ ОБРАЗОВАНИЕМ ПОЛОСТЕЙ | 2020 |
|
RU2828353C2 |
| ПЭГИЛИРОВАННЫЕ БИОЛОГИЧЕСКИ АКТИВНЫЕ ПЕПТИДЫ И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2748576C2 |
| Терапевтическое применение глюкагона и включающей его комбинации | 2019 |
|
RU2838895C2 |
| ДВОЙНОЙ АГОНИСТ РЕЦЕПТОРОВ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 И ГЛЮКАГОНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2021 |
|
RU2799327C1 |
| ПРОЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ С ВЫСОКОЙ СТЕПЕНЬЮ ПРОНИКНОВЕНИЯ НА ОСНОВЕ ПЕПТИДОВ И РОДСТВЕННЫХ ПЕПТИДАМ СОЕДИНЕНИЙ | 2010 |
|
RU2767051C2 |
| Новые производные оксинтомодулина и содержащая их фармацевтическая композиция для лечения ожирения | 2012 |
|
RU2739209C2 |
| Аналоги инсулина | 2017 |
|
RU2769476C2 |
| ПЕПТИДЫ И ПЕПТИДОМИМЕТИКИ ДЛЯ КОМБИНИРОВАННОГО ПРИМЕНЕНИЯ И ЛЕЧЕНИЯ В СУБПОПУЛЯЦИЯХ ПАЦИЕНТОВ С РАКОВЫМИ ЗАБОЛЕВАНИЯМИ | 2014 |
|
RU2732440C2 |

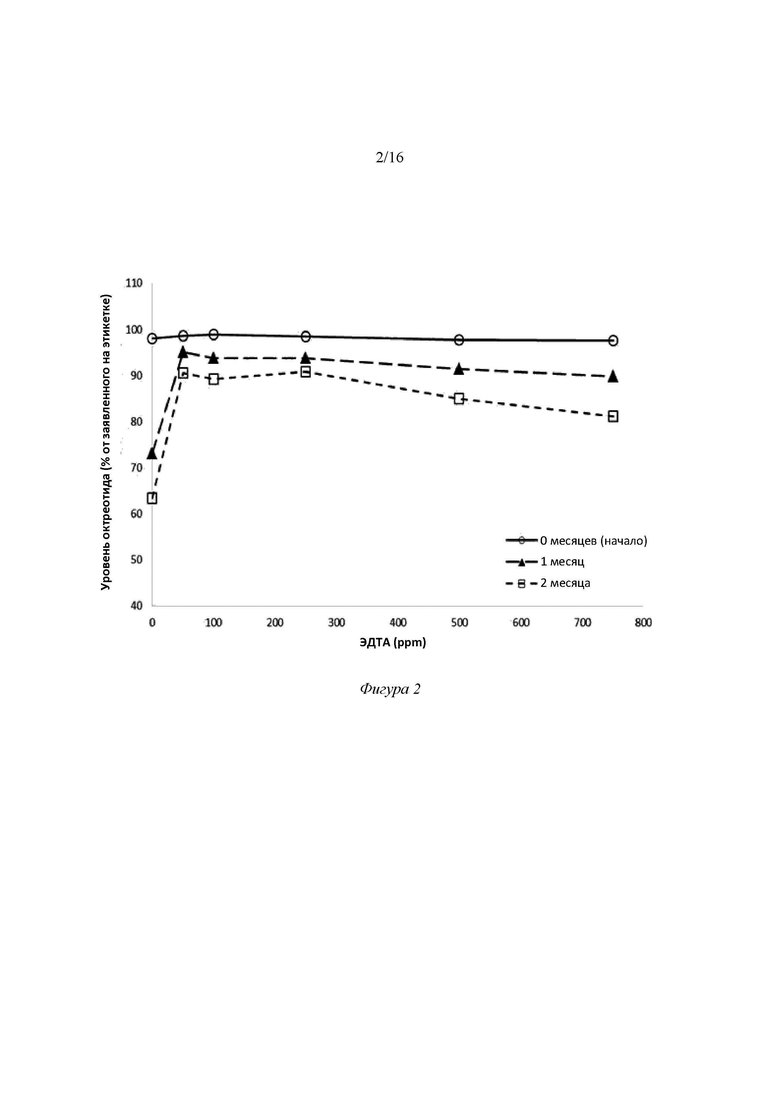



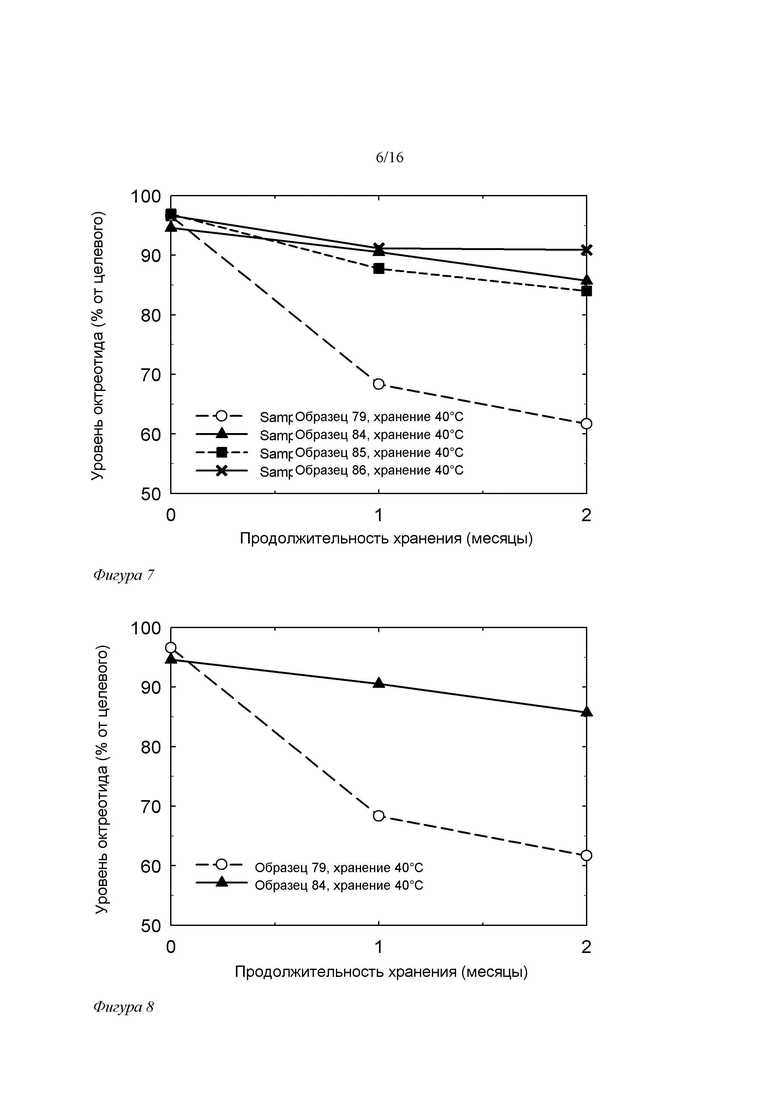
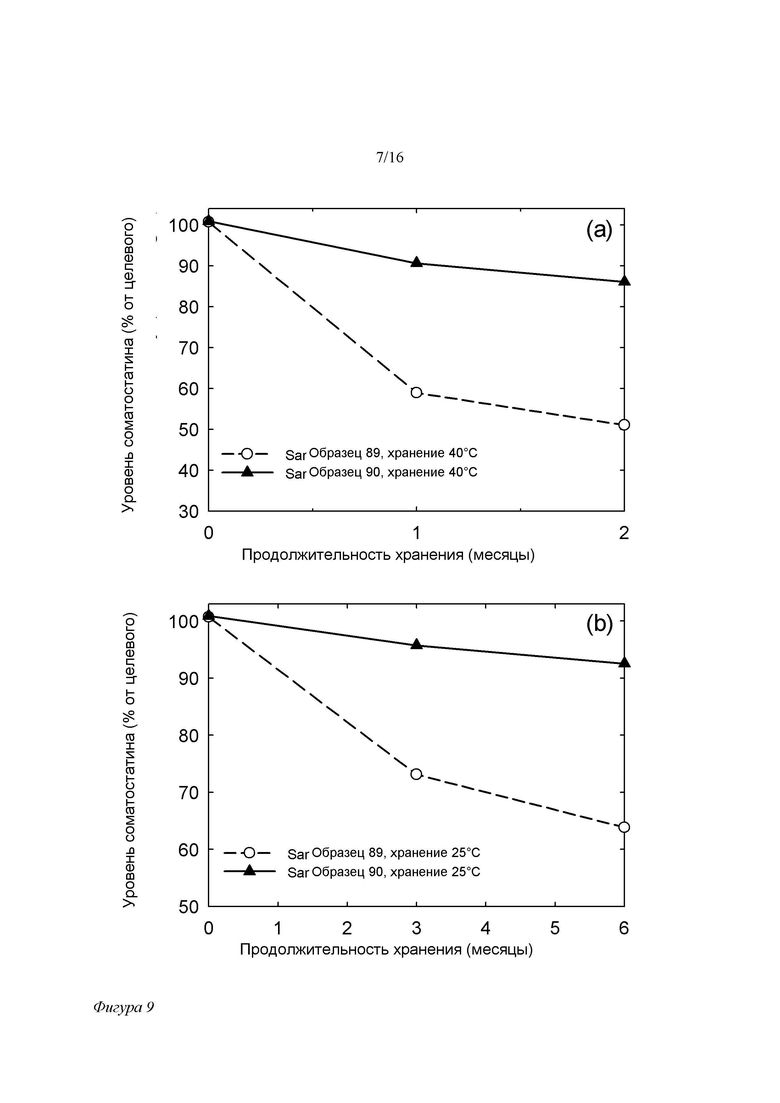





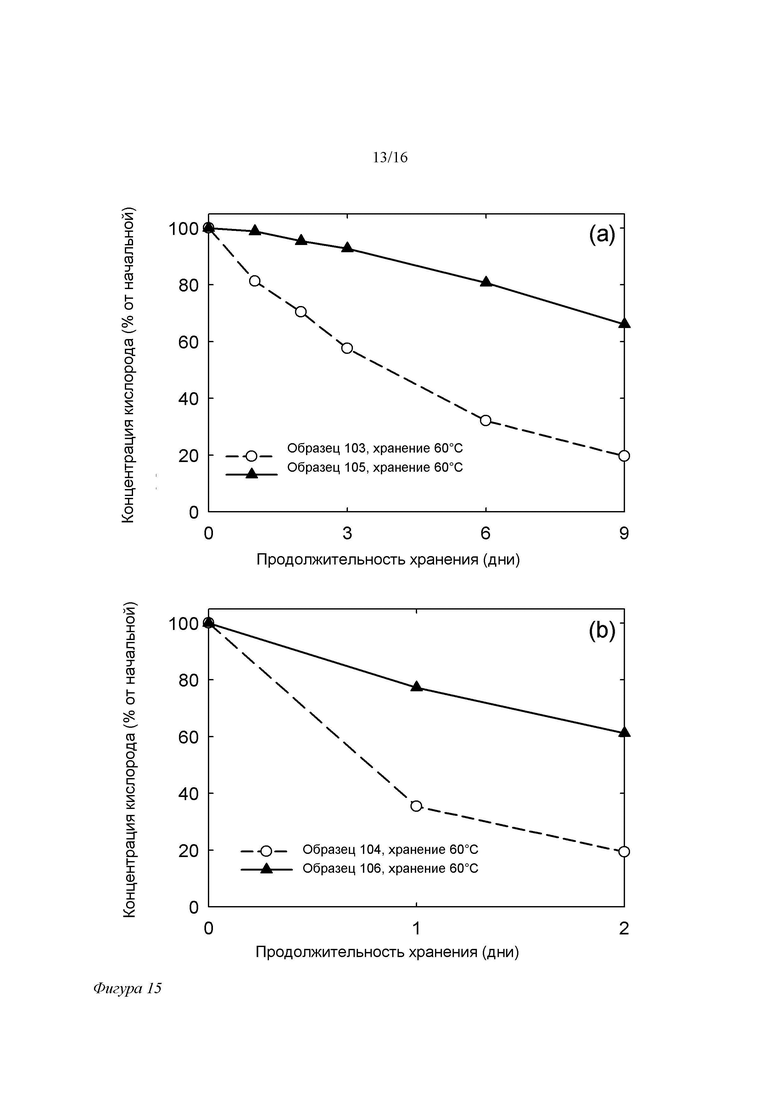

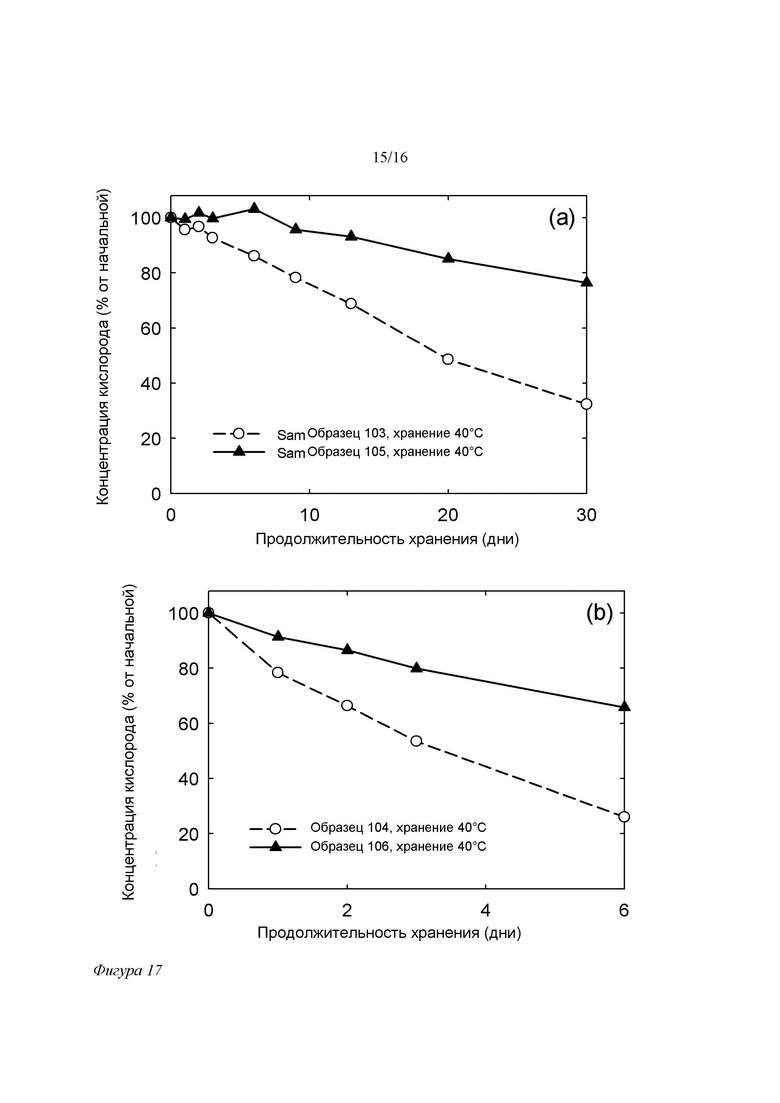

Настоящее изобретение относится к смесям, содержащим: i) по меньшей мере один липид и/или по меньшей мере одно масло; и ii) алкиламмониевую соль ЭДТА; где смесь имеет содержание воды в диапазоне от 0 до 1,0 масс. %. Изобретение дополнительно относится к предварительно заполненным устройствам для введения составов, к применению алкиламмониевой соли ЭДТА для уменьшения разрушения липидных компонентов и/или какого-либо активного агента, содержащегося в предварительном составе, и к алкиламмониевым солям ЭДТА, таким как описано в настоящем документе. 2 н. и 11 з.п. ф-лы, 18 ил., 19 табл., 19 пр.
1. Состав для улучшения стойкости к окислению липидов, содержащий:
i) липидную смесь, содержащую:
a) 33-60 масс. % по меньшей мере одного нейтрального диациллипида;
b) 33-60 масс. % по меньшей мере одного фосфолипида;
c) 1-30 масс. % по меньшей мере одного биосовместимого органического растворителя; и
ii) анион ЭДТА, где уровень ЭДТА составляет от 0,001 до 0,050 масс. % (10-500 ррm);
и катион амина, выбранного из:
этаноламина;
диэтаноламина;
меглумина;
трис-гидроксиметиламина;
этилендиамина; или
серинола (NH2CH(CH2OH)2);
где
когда амин имеет только одну амино- или алкиламиногруппу, молярное отношение ЭДТА: амин находится в диапазоне от 1:3,0 до 1:10;
когда амин имеет две амино- или алкиламиногруппы, молярное отношение ЭДТА: амин находится в диапазоне от 1:2,0 до 1:4,0;
и
где состав имеет содержание воды в диапазоне от 0 до 1,0 масс. %.
2. Состав по п. 1, отличающийся тем, что указанный катион представляет собой катион этаноламина (ЭТА) и указанный анион представляет собой анион ЭДТА.
3. Состав по п. 2, отличающийся тем, что отношение эквивалентов ЭТА к ЭДТА находится в диапазоне от 3,5 до 7 (моль/моль), предпочтительно от 3,5 до 5, наиболее предпочтительно от 3,5 до 4,5.
4. Состав по п. 1, отличающийся тем, что катион выбран из катионов:
этаноламина и
диэтаноламина.
5. Состав по пп. 1, 2 или 4, отличающийся тем, что с) содержится в количестве от 2 до 20 масс. %, предпочтительно от 5 до 18 масс. % или от 2 до 15 масс. %, более предпочтительно от 2 до 15 масс. %, и где с) содержит растворитель на основе одноатомного спирта.
6. Состав по пп. 1, 2 или 4, отличающийся тем, что с) содержится в количестве от 5 до 18 масс. % или от 2 до 15 масс. %, и содержит этанол.
7. Состав по пп. 1, 2 или 4, отличающийся тем, что компонент с) включает от 2 до 12 масс. % пропиленгликоля, предпочтительно от 3 до 10 масс. %, в частности от 3 до 8 масс. %; предпочтительно где оставшаяся часть компонента с) составляет этанол.
8. Состав по п. 1, дополнительно содержащий активный агент.
9. Состав по п. 8, отличающийся тем, что активный агент представляет собой активные агенты на основе пептидов, белков и нуклеиновых кислот, гормоны.
10. Состав по п. 9, отличающийся тем, что активный агент представляет собой пептид.
11. Состав по п. 10, отличающийся тем, что пептид содержит от 5 до 60 аминокислот.
12. Состав по п. 8 для применения в качестве лекарственного средства.
13. Предварительно заполненное устройство для введения активного агента субъекту, которое содержит состав по п. 8.
| WO 2010020794 A1, 25.02.2010 | |||
| JP 78013357 B, 09.05.1978 | |||
| WO 2005014777 A2, 17.02.2005 | |||
| JP 2002356464 A, 13.12.2002 | |||
| WO 2006131730 A1, 14.12.2006 | |||
| WO 2013174978 A1, 28.11.2013. |

