Изобретение относится к фармакологии и медицине, в частности к фармацевтическим композициям, обеспечивающим пролонгированные периоды доставки фармацевтически активного пептидного соединения.
Уровень техники
Ряд заболеваний и клинических нарушений лечат введением фармацевтически активного пептида. Одним из таких примеров является рак простаты, который представляет собой рак, зависимый от полового гормона, и который можно лечить введением аналога лютеинизирующий гормон-высвобождающего гормона (LHRH), нарушающего образование лютеинизирующего гормона (LH), который регулирует синтез мужских гормонов. В частности, для снижения образования LH были использованы пептидные аналоги LHRH, действующие как суперагонисты рецептора лютеинизирующий гормон-высвобождающего гормона, такие как лейпролид и госерелин.
Во многих случаях терапевтическая эффективность фармацевтически активного пептида зависит от его непрерывного присутствия in vivo в течение пролонгированного периода времени. Для получения непрерывной доставки пептида in vivo, с целью избежания необходимости повторных введений, желательным является препарат с задержанным выходом или задержанной доставкой. Один из подходов к задержанной доставке лекарственного вещества представляет собой микроинкапсулирование, при котором активный ингредиент заключают в полимерную мембрану с целью получения микрочастиц. Например, суперагонисты LHRH, такие как лейпролид и госерелин, обычно инкапсулируют в микрочастицу, содержащую сополимер полилактида/полигликолида для приготовления препаратов, пригодных для депо-инъекции, которая обеспечивает задержанную доставку суперагонистов в течение нескольких недель или месяцев (см., например, Патенты США 4675189, 4677191, 5480656 и 4728721).
Дополнительные препараты с задержанной доставкой для введения фармацевтически активных пептидов in vivo непрерывно в течение пролонгированного периода времени являются необходимыми.
Сущность изобретения
В данном изобретении представлены фармацевтические композиции, содержащие стабильный нерастворимый в воде комплекс, состоящий из пептидного соединения (например, пептида, полипептида, белка, пептидомиметика и т.п.), предпочтительно фармацевтически активного пептидного соединения, и макромолекулы-носителя, которая обеспечивает задержанную доставку пептидного соединения in vivo при введении комплекса. Соответственно комплекс, соответствующий изобретению, может обеспечить непрерывную доставку фармацевтически активного пептидного соединения субъекту в течение пролонгированных периодов времени, например, в течение одного месяца. Более того, ассоциация пептидного соединения и макромолекулы-носителя в прочном стабильном комплексе позволяет загружать в препарат высокие концентрации пептидного соединения.
Комплекс, соответствующий изобретению, образуется при смешивании пептидного соединения и макромолекулы-носителя в условиях, при которых формируется практически нерастворимый в воде комплекс, например, водные растворы пептидного соединения и макромолекулы-носителя смешивают до выпадения комплекса в осадок. Комплекс может находиться в форме твердого вещества (например, в виде пасты, гранул, порошка или лиофилизата) или комплекс в форме порошка может быть подвергнут достаточно тонкому распылению с образованием стабильных жидких суспензий или полутвердых дисперсий.
В предпочтительном варианте осуществления пептидное соединение нерастворимого в воде комплекса представлено аналогом LHRH, более предпочтительно антагонистом LHRH, а макромолекула-носитель является анионным полимером, предпочтительно карбоксиметилцеллюлозой. Комплекс, соответствующий изобретению, может быть подвергнут стерилизации, например, γ-облучением или облучением пучком электронов, перед введением in vivo.
Предложен также способ лечения субъекта при состоянии, поддающемся воздействию аналогов LHRH, путем введения ему соответствующей изобретению композиции, содержащей аналог LHRH. В предпочтительном варианте осуществления соответствующие изобретению способы лечения используют для лечения рака простаты.
Перечень фигур чертежей
На фиг.1 приведены графики, представляющие уровни тестостерона в плазме (в нг/мл; белые квадраты) и уровни РРI-149 в плазме (в нг/мл; черные квадраты) у крыс (левый график) и собак (правый график) спустя некоторое время после внутримышечной инъекции комплекса РРI-149 и карбоксиметилцеллюлозы.
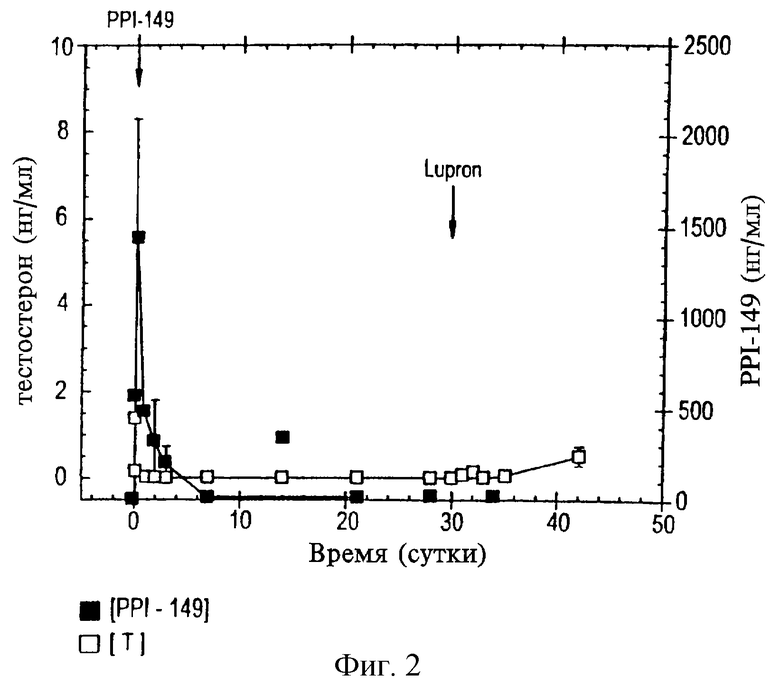
На фиг. 2 приведен график, представляющий уровни тестостерона в плазме (нг/мл, белые квадраты) и уровни РРI-149 в плазме (в нг/мл, черные квадраты) у крыс спустя некоторое время после внутримышечной инъекции комплекса антагониста LHRH PPI-149 и карбоксиметилцеллюлозы в день 0 и инъекции агониста LHRH LupronТМ в день 30, который демонстрирует супрессию LupronТМ - индуцированного выброса тестостерона при предварительном введении PPI-149.
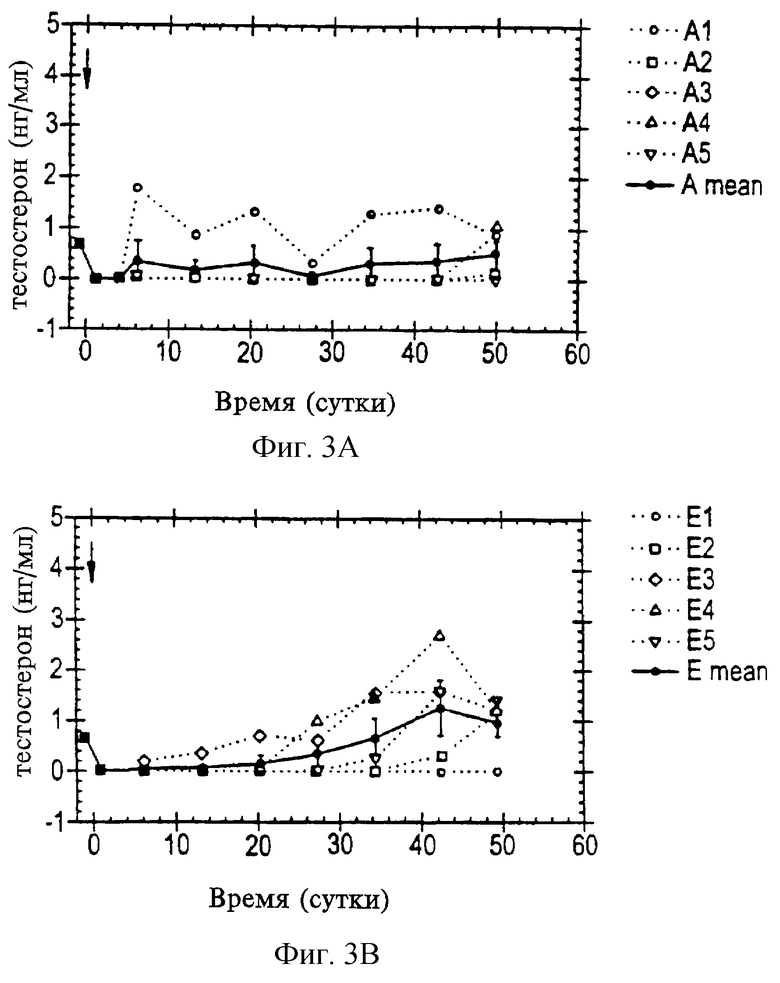
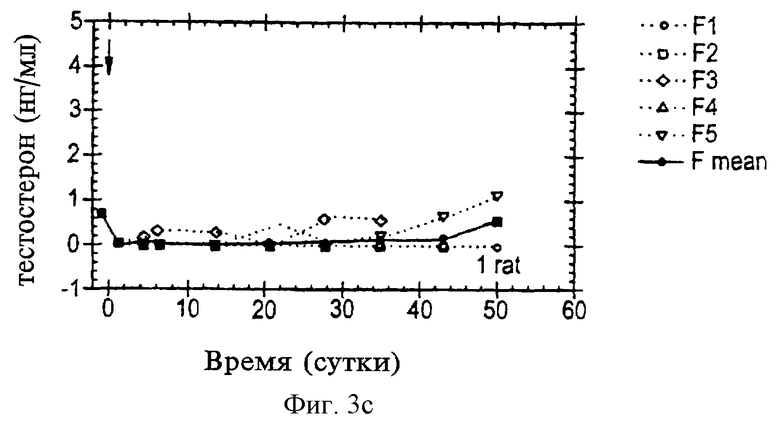
На фиг. 3А-3С приведена серия графиков, представляющих уровни тестостерона в плазме (в нг/мл) у самцов крыс Sprague-Dawley в течение времени после внутримышечной инъекции РРI-149-СМС (фиг.3А), PPI-258-CMC (фиг.3В) или CetrorelixТМ-CMC (фиг.3С) (СМС-карбоксиметилцеллюлоза).
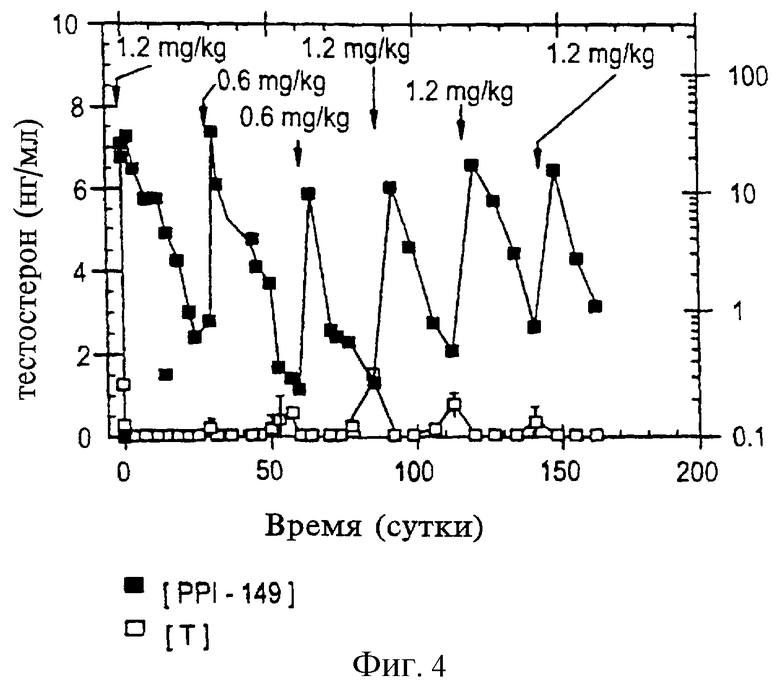
На фиг.4 приведен график, представляющий уровни тестостерона в плазме (в нг/мл; белые квадраты) и уровни PPI-149 в плазме (в нг/мл; черные квадраты) у собак спустя некоторое время после подкожной инъекции РРI-149-СМС в назначенных дозах с интервалами 28 дней, который демонстрирует пролонгированную супрессию уровней тестостерона в плазме.
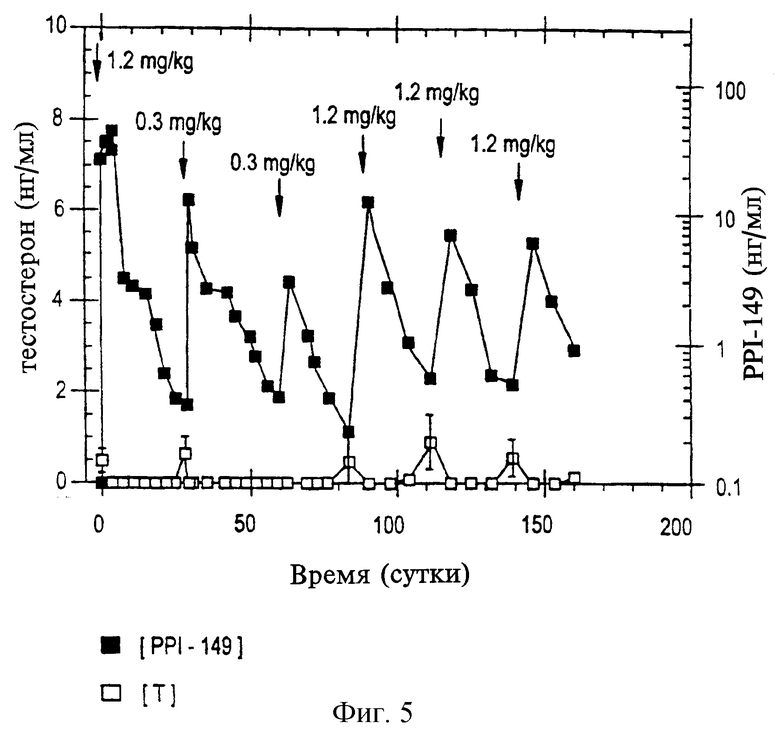
На фиг.5 приведен график, представляющий уровни тестостерона в плазме (в нг/мл; белые квадраты) и уровни PPI-149 в плазме (в нг/мл; черные квадраты) у собак спустя некоторое время после внутримышечной инъекции РРI-149-СМС в назначенных дозах с интервалами 28 дней, который демонстрирует пролонгированную супрессию уровней тестостерона в плазме.
Сведения, подтверждающие возможность осуществления изобретения
Данное изобретение касается фармацевтических композиций, содержащих стабильный нерастворимый в воде комплекс, состоящий из пептидного соединения (например, пептида, полипептида, белка, пептидомиметика и т.п.) и макромолекулы-носителя, способов получения данных композиций и способов применения данных композиций. Преимущества фармацевтических композиций, соответствующих изобретению, включают возможность доставки фармацевтически активного пептидного соединения (систематически или местно) в течение пролонгированных периодов времени (например, нескольких недель, одного месяца или нескольких месяцев) и возможность введения высоких концентраций пептидного соединения в комплекс.
Для лучшего понимания изобретения сначала дают определения ряда терминов.
Как используют в данном контексте, термин "пептидное соединение" предназначен для определения соединений, состоящих, по крайней мере частично, из остатков аминокислот, связанных амидными связями (т.е. пептидными связями). Термин "пептидное соединение" охватывает пептиды, полипептиды и белки. Как правило, пептид будет состоять из менее чем приблизительно 100 остатков аминокислот, более часто из менее чем 50 остатков аминокислот и даже более часто из менее чем 25 остатков аминокислот. Термин "пептидное соединение" далее предполагает охват аналогов пептидов, производных пептидов и пептидомиметиков, которые имитируют химическую структуру пептида, состоящего из существующих в естественных условиях аминокислот. Примеры аналогов пептидов включают пептиды, содержащие одну или более неприродных аминокислот. Примеры производных пептидов включают пептиды, в которых боковая цепь аминокислот, пептидный скелет или амино- или карбокси-конец был дериватизирован (например, пептидные соединения с метилированными амидными связями). Примеры пептидомиметиков включают пептидные соединения, в которых пептидный скелет замещен одной или более бензодиазепиновыми молекулами (см., например, статью James G. L и соавт. (1993) Science, т. 260, стр. 1937-1942), "инверсо"-пептиды, в которых все L-аминокислоты замещены соответствующими D-аминокислотами, "ретро-инверсо"-пептиды (см. Патент США No 4522752, выданный Sisto), в которых последовательность аминокислот перевернута ("ретро") и все L-аминокислоты замещены соответствующими D-аминокислотами ("инверсо"), и другие изостерические соединения, такие как миметики пептидного скелета (т.е. амидной связи), включающие модификации амидного азота, α-углерода, карбонила амида, полное замещение амидной связи, удлинения, делеции или перекрестные связи в скелете. Известен ряд модификаций пептидного скелета, в том числе ψ[CH2S], ψ[CH2NH], ψ[CSNH2], ψ[NHCO], ψ[COCH2] и ψ[(Е) или (Z) СН=СН]. В вышеиспользованной номенклатуре ψ означает отсутствие амидной связи. Структура, замещающая амидную группу, приведена в скобках. Другие возможные модификации включают N-алкил- (или арил-)замещение (ψ[CONR]), перекрестное связывание скелетов для создания лактамов или других циклических структур или другие производные, в том числе производные по С-концевому гидроксиметилу, О-модифицированные производные и производные, модифицированные по N-концу, включая замещенные амиды, такие как алкиламиды и гидразиды.
Как используют в данном контексте, термин "фармацевтически активное пептидное соединение" предназначен для определения пептидного соединения, которое проявляет фармакологическую активность либо в существующей до введения форме, либо после процессинга in vivo (т.е. фармацевтически активные пептидные соединения включают пептидные соединения с конститутивной фармакологической активностью и пептидные соединения в форме "пролекарства", которые должны после введения каким-либо образом метаболизироваться или процессироваться in vivo для проявления фармакологической активности).
Как используют в данном контексте, термины "многовалентное катионное пептидное соединение" и "многовалентное анионное пептидное соединение" предназначены для определения пептидных соединений, содержащих множество положительных или отрицательных зарядов, соответственно. Термины "двухвалентное катионное" или "двухвалентное анионное" пептидное соединение относятся к пептидному соединению, содержащему два положительных или отрицательных заряда, соответственно. Термины "трехвалентное катионное" или "трехвалентное анионное" пептидное соединение относятся к пептидному соединению, содержащему три положительных или отрицательных заряда, соответственно.
Как используют в данном контексте, термин "аналог LHRH" предусматривает охват пептидных соединений, которые имитируют структуру лютеинизирующий гормон-высвобождающего гормона. Аналог LHRH может быть агонистом LHRH или антагонистом LHRH.
Как используют в данном контексте, термин "агонист LHRH" предназначен для определения соединения, которое стимулирует рецептор лютеинизирующий гормон-высвобождающего гормона (LHRH-R), стимулируя таким образом высвобождение лютеинизирующего гормона, а термин "антагонист LHRH" относится к соединению, которое ингибирует LHRH-R, ингибируя таким образом высвобождение лютеинизирующего гормона. Примеры агонистов LHRH включают лейпролид (торговое название Lupron® (лапрон); Abbot/TAP), госерелин (торговое название Zoladex® (золадекс), Zeneca); бусерелин (Hoechst), трипторелин (известный также, как Decapeptyl (декапептил), D-Trp-6-LHRH и Debiopharm® (дебиофарм); Ipsen (Beaufour), нафарелин (торговое название Synarel® (синарел); Syntex), лутрелин (Wyeth), цисторелин (Hoechst), гонадорелин (Ayerst) и гистрелин (Ortho).
Как используют в данном контексте, термин "антагонист LHRH" предназначен для определения соединения, которое ингибирует рецептор лютеинизирующий гормон-высвобождающего гормона, ингибируя таким образом высвобождение лютеинизирующего гормона. Примеры антагонистов LHRH включают Antide (антид), Cetrorelix (цетрореликс), соединения, описанные в Патенте США 5470947, выданном Folkers и соавт.; Публикации РСТ No WO 89/01944 Folkers и соавт.; Патенте США 5413990, выданном Haviv; Патенте США 5300492, выданном Haviv; Патенте США 5371070, выданном Koerber и соавт.; Патенте США 5296468, выданном Hoeger и соавт.; Патенте США 5171835, выданном Janaky и соавт.; Патенте США 5003011, выданном Coy и соавт.; Патенте США 4431635, выданном Coy и соавт.; Патенте США 4992421, выданном De и соавт.; Патенте США 4851385, выданном Roeske и соавт. ; Патенте США 4801577, выданном Nestor Jr. соавт., и Патенте США 4689396, выданном Roeske и соавт., а также соединения, описанные в Патентной заявке США No 08/480494, имеющей название "Пептиды-антагонисты LHRH", и в соответствующей международной заявке (заявка РСТ No PCT/US96/09852), также имеющей название "Пептиды-антагонисты LHRH", полное содержание обеих заявок специально введено в данное описание в виде ссылки. Особенно предпочтительный антагонист LHRH содержит структуру Ac-D-Nal1, 4-Cl-D-Phe2, D-Pal3, N-Me-Tyr5, D-Asn6, Lys(iPr)8, D-Ala10-LHRH, обозначаемую в данном контексте PPI-149.
Как используют в данном контексте, термин "макромолекула-носитель" предназначен для обозначения макромолекулы, которая может комплексироваться с пептидным соединением с образованием нерастворимого в воде комплекса. Перед комплексированием с пептидным соединением макромолекула-носитель обычно является растворимой в воде. Предпочтительно макромолекула имеет мол. массу по меньшей мере 5 кД, более предпочтительно 10 кД. Термин "анионная макромолекула-носитель" предусматривает включение отрицательно заряженных молекул с высокой молекулярной массой, таких как анионные полимеры. Термин "катионная макромолекула-носитель" предусматривает включение положительно заряженных молекул с высокой молекулярной массой, таких как катионные полимеры.
Как используют в данном контексте, термин "нерастворимый в воде комплекс" предназначен для определения физически и химически стабильного комплекса, который образуется при соответствующем смешивании пептидного соединения и макромолекулы-носителя согласно описанным здесь способам. Комплекс обычно представляет собой осадок, который получают при смешивании водных препаратов пептидного соединения и макромолекулы-носителя. Не предполагая ограничений по механизму, образование предпочтительных нерастворимых в воде комплексов, соответствующих изобретению, предусматривает вовлечение (т.е., по меньшей мере, частично обусловлено) ионными взаимодействиями в случаях, когда пептидное соединение является катионным, а молекула-носитель - анионной, и наоборот. Дополнительно или альтернативно образование нерастворимого в воде комплекса, соответствующего изобретению, может включать (т.е. быть, по меньшей мере, частично обусловленным) гидрофобные взаимодействия. Более того, образование нерастворимого в воде комплекса, соответствующего изобретению, может включать (т. е. быть, по меньшей мере, частично обусловленным) ковалентные взаимодействия. Описание комплекса, как "нерастворимого в воде" служит для указания на то, что комплекс не растворяется в воде практически или легко, как видно по его осаждению из водного раствора. Однако следует понимать, что "нерастворимый в воде" комплекс, соответствующий изобретению, может обладать ограниченной растворимостью (т.е. частичной растворимостью) в воде либо in vitro, либо в водной физиологической среде in vivo.
Как используют в данном контексте, термин "задержанная доставка" предназначен для определения непрерывной доставки фармацевтического агента in vivo в период времени после введения, предпочтительно в течение, по меньшей мере, нескольких дней, недели или нескольких недель. Задержанная доставка агента может быть продемонстрирована, например, непрерывным терапевтическим эффектом агента в течение периода времени (например, для аналога LHRH задержанная доставка аналога может быть продемонстрирована непрерывной супрессией синтеза тестостерона в течение периода времени). Альтернативно задержанная доставка агента может быть продемонстрирована детекцией присутствия агента in vivo спустя некоторое время.
Как используют в данном контексте, термин "субъект" предусматривает включение теплокровных животных, предпочтительно млекопитающих, более предпочтительно приматов и наиболее предпочтительно человека.
Как используют в данном контексте, термин "введение субъекту" предназначен для определения приготовления, доставки и введения композиции (т.е. фармацевтического препарата) субъекту каким-либо приемлемым способом с целью доставки композиции в желаемый участок организма субъекта, включая доставку парентеральным или пероральным путем, внутримышечной инъекцией, подкожной/внутрикожной инъекцией, внутривенной инъекцией, защечным введением, чрескожной доставкой и ректальным, вагинальным, интраназальным введением или введением через толстую кишку или дыхательные пути.
Как используют в данном контексте, термин "состояние, поддающееся воздействию аналога LHRH" предусматривает включение заболеваний, нарушений и других состояний, при которых введение агониста LHRH или антагониста LHRH имеет желаемый эффект, например, терапевтически благоприятный эффект. Примеры состояний, которые лечат аналогом LHRH, включают гормонозависимые раки (в том числе рак простаты, рак молочной железы, рак яичников, рак матки и рак яичка), доброкачественную гипертрофию простаты, преждевременное половое созревание, эндометриоз, фиброму матки, бесплодие (посредством оплодотворения in vitro) и оплодотворяющую способность (т.е. использование в контрацепции).
Один из аспектов данного изобретения касается фармацевтической композиции, содержащей нерастворимый в воде комплекс фармацевтически активного пептидного соединения и макромолекулы-носителя. В предпочтительном варианте осуществления образование нерастворимого в воде комплекса обусловлено, по меньшей мере частично, ионными взаимодействиями между фармацевтически активным пептидным соединением и макромолекулой-носителем. В данных вариантах осуществления либо фармацевтически активное пептидное соединение является катионным, а макромолекула-носитель - анионной, либо фармацевтически активное пептидное соединение является анионным, а макромолекула-носитель - катионной. В другом варианте осуществления образование нерастворимого в воде комплекса обусловлено, по меньшей мере частично, гидрофобными взаимодействиями между фармацевтически активным пептидным соединением и макромолекулой-носителем. В предпочтительном варианте осуществления пептидное соединение, используемое в комплексе, представляет собой многовалентное катионное пептидное соединение, а макромолекула-носитель является анионной макромолекулой.
Фармацевтические композиции, соответствующие изобретению, обеспечивают задержанную доставку пептидного соединения субъекту in vivo после введения субъекту композиции. При этом продолжительность задержанной доставки может варьировать в зависимости от концентрации пептидного соединения и от макромолекулы-носителя, использованной для получения комплекса. Например, в одном варианте осуществления одна доза нерастворимого в воде комплекса обеспечивает задержанную доставку пептидного соединения субъекту в течение, по меньшей мере, одной недели после введения фармацевтической композиции субъекту. В другом варианте осуществления одна доза нерастворимого в воде комплекса обеспечивает задержанную доставку пептидного соединения субъекту в течение, по меньшей мере, двух недель после введения фармацевтической композиции субъекту. В еще одном варианте осуществления одна доза нерастворимого в воде комплекса обеспечивает задержанную доставку пептидного соединения субъекту в течение, по меньшей мере, трех недель после введения фармацевтической композиции субъекту. В другом варианте осуществления одна доза нерастворимого в воде комплекса обеспечивает задержанную доставку пептидного соединения субъекту в течение, по меньшей мере, четырех недель после введения фармацевтической композиции субъекту. Препараты, которые обеспечивают задержанную доставку в течение более длительных или более коротких периодов времени, также охватываются изобретением, например, препараты, которые обеспечивают непрерывную доставку в течение 1 дня, 1-7 дней, одного месяца, двух месяцев, трех месяцев и т. п. Непрерывная доставка пептидного соединения в течение нескольких месяцев может быть осуществлена, например, путем повторного введения месячных доз, каждая из которых обеспечивает задержанную доставку пептидного соединения в течение приблизительно одного месяца (см., например, пример 14).
Пептидное соединение любого размера может быть пригодным для использования в комплексе, пока пептидное соединение обладает способностью образовывать нерастворимый в воде нековалентный комплекс с макромолекулой-носителем при смешивании пептидного соединения и макромолекулы-носителя. Однако в некоторых предпочтительных вариантах осуществления пептидное соединение представляет собой пептид длиной приблизительно от 5 до приблизительно 20 аминокислот, длиной приблизительно от 8 до приблизительно 15 аминокислот или длиной приблизительно от 8 до приблизительно 12 аминокислот. В препаратах может быть использован ряд фармацевтически активных пептидов, неограничивающие примеры которых включают аналоги LHRH (обсуждаются ниже), аналоги брадикинина, гормон паращитовидной железы, адренокортикотропный гормон (АСТН), кальцитонин и аналоги вазопрессина (например, 1-дезамино-8-D-аргинин-вазопрессин (DDAVP)).
Несмотря на то, что многие макромолекулы-носители могут быть использованы для образования нерастворимых в воде комплексов, соответствующих изобретению, предпочтительные макромолекулы представляют собой полимеры, предпочтительно водорастворимые полимеры. В предпочтительном варианте осуществления макромолекула-носитель является анионным полимером, таким как анионное производное многоатомного спирта, или его фрагментом и их солями (например, натриевыми солями). Анионные структуры, которыми может быть дериватизирован многоатомный спирт, включают, например, карбоксилатную, фосфатную или сульфатную группы. Особенно предпочтительный анионный многоатомный спирт представляет собой анионное производное полисахарида или его фрагмент или их соли (например, натриевые соли). Макромолекула-носитель может содержать молекулы одного вида (например, один тип полимера) или молекулы двух или более различных видов (например, смесь двух типов полимеров). Примеры специфических анионных полимеров включают карбоксиметилцеллюлозу, альгин, альгинат, анионные ацетатные полимеры, анионные акриловые полимеры, ксантановые смолы, гликолат натрия крахмала и их фрагменты, производные и фармацевтически приемлемые соли, а также анионные производные карагинана, анионные производные полигалактуроновой кислоты и сульфатированные и сульфонированные производные полистирола. Предпочтительным анионным полимером является натриевая соль карбоксиметилцеллюлозы. Примеры катионных полимеров включают поли-L-лизин и другие полимеры основных аминокислот.
В особенно предпочтительном варианте осуществления изобретения пептидное соединение нерастворимого в воде комплекса является аналогом LHRH, например агонистом LHRH или, более предпочтительно, антагонистом LHRH. Данные аналоги LHRH обычно имеют длину 10 аминокислот. Предпочтительные антагонисты LHRH включают антагонисты LHRH, которые содержат пептидное соединение, в котором остаток пептидного соединения, соответствующий аминокислоте в положении 6 природного LHRH млекопитающих, содержит структуру D-аспарагина (D-Asn). Как используют в данном контексте, термин "структура D-аспарагина" предусматривает включение D-Asn и его аналогов, производных и миметиков, которые сохраняют функциональную активность D-Asn. Другие предпочтительные антагонисты LHRH включают антагонисты LHRH, которые содержат пептидное соединение, содержащее структуру A-B-C-D-E-F-G-H-I-J, где
А - пиро-Glu, Ac-D-Nal, Ac-D-Qal, Ac-Sar или Ac-D-Pal;
В - His или 4-Cl-D-Phe;
С - Trp, D-Pal, D-Nal, L-Nal, D-Pal(N-O) или D-Trp;
D - Ser;
E - N-Me-Ala, Tyr, N-Me-Tyr, Ser, Lys(iPr), 4-Cl-Phe, His, Asn, Met, Ala, Arg или lle;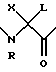
в которой
R и X независимо представлены Н или алкилом и
L содержит небольшой полярный фрагмент;
G - Leu или Trp;
Н - Lys(iPr), Gln, Met или Arg;
I - Pro и
J - Gly-NH2 или D-Ala-NH2,
или их фармацевтически приемлемые соли.
Термин "небольшой полярный фрагмент" относится к структуре, которая имеет маленький стерический объем и является относительно полярной. Полярность измеряют как гидрофильность по Р-шкале. Коэффициент распределения, Р, между 1-октанолом и водой использовали в качестве стандарта при определении гидрофильности соединения. Гидрофильность может быть выражена как logP, логарифм коэффициента распределения (см. статью Hansch и соавт. Nature (1962), т. 194, с. 178; Fujita и соавт. J. Am. Chem. Soc. (1964), т. 86, с. 5175). Были созданы стандартные таблицы гидрофильности для многих молекул и константы липофильности (гидрофобности) заместителей (обозначаемых п) для многих функциональных групп (см. , например, Hansch и Leo "Константы заместителей для корреляционного анализа в химии и биологии" ("Substituent Constants for Correlation Analysis in Chemistry and Biology", (1979) Wiley, New York, New York). Гидрофильность широкого круга проверяемых на гидрофильность структур может быть очень точно предсказана с помощью данных таблиц. Например, измеренный logP (октанол/вода) нафталина составляет 3,45. Константа заместителя р для -ОН составляет -0,67. Вследствие этого предсказанный logP для β-нафтола составляет 3,45+(-0,67)=2,78. Данное значение хорошо согласуется с измеренным logP для β-нафтола, который составляет 2,84. Как используют в данном контексте, термин "небольшой полярный фрагмент" относится к структурам, которые имеют logP в диапазоне между -1 и +2 и стерический объем которых не превышает стерический объем Тrp.
В ряде вариантов осуществления L содержит небольшой полярный фрагмент при условии, что F не является D-Cit, D-Hci или производным низшего алкила D-Cit или D-Hci. Предпочтительно F выбрано из группы, состоящей из D-Asn, D-Gln и D-Thr. Более предпочтительно F является D-Asn. Предпочтительно Е представлено тирозином (Тyr) или N-метил-тирозином (N-Ме-Тyr). В особенно предпочтительном варианте осуществления антагонист LHRH имеет следующую структуру: Ac-D-Nal1, 4-Cl-D-Phe2, D-Pal3, N-Me-Tyr5, D-Asn6, Lys(iPr)8, D-Ala10-LHRH (обозначаемую в данном контексте РРI-149). Особенно предпочтительный комплекс, соответствующий изобретению, содержит РРI-149 и карбоксиметилцеллюлозу.
В дополнение к нерастворимому в воде комплексу фармацевтические композиции, соответствующие изобретению, могут содержать дополнительные фармацевтически приемлемые носители и/или наполнители. Как используют в данном контексте, термин "фармацевтически приемлемый носитель" включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и антигрибковые агенты, изотонические и замедляющие всасывание агенты и т.п. при их физиологической совместимости. Предпочтительно, если носитель является пригодным для внутривенного, внутримышечного, подкожного или парентерального введения (например, путем инъекции). Наполнители включают фармацевтически приемлемые стабилизаторы и дизинтеграторы.
Дополнительно к фармацевтическим композициям аналогов LHRH, комплексированных с макромолекулой-носителем, изобретение далее охватывает упакованные препараты, содержащие данные комплексы, и шприцы, содержащие данные комплексы. Например, в изобретении представлен упакованный препарат для лечения субъекта при состоянии, поддающемся воздействию аналога LHRH. Препарат, содержащий нерастворимый в воде комплекс аналога LHRH (предпочтительно РРI-149) и макромолекулы-носителя (предпочтительно карбоксиметилцеллюлозы), упакован с инструкциями по применению нерастворимого в воде комплекса для лечения субъекта при состояниях, которые лечат аналогом LHRH. В другом варианте осуществления в изобретении представляют шприц, имеющий полость, отличающийся тем, что в полость введен нерастворимый в воде комплекс аналога LHRH (предпочтительно PPI-149) и макромолекулы-носителя (предпочтительно карбоксиметилцеллюлозы).
Комплекс, соответствующий изобретению, готовят путем смешивания пептидного соединения и макромолекулы-носителя в условиях образования нерастворимого в воде комплекса пептидного соединения и макромолекулы-носителя. Соответственно, другой аспект изобретения касается способов получения фармацевтических композиций. В одном варианте осуществления способ предусматривает:
получение пептидного соединения и макромолекулы-носителя;
смешивание пептидного соединения и макромолекулы-носителя в условиях образования нерастворимого в воде комплекса пептидного соединения и макромолекулы-носителя и
приготовление фармацевтической композиции, содержащей нерастворимый в воде комплекс.
Например, раствор пептидного соединения и раствор макромолекулы-носителя смешивают до тех пор, пока нерастворимый в воде комплекс пептидного соединения и макромолекулы-носителя не выпадет в осадок из раствора. В ряде вариантов осуществления растворы пептидного соединения и макромолекулы-носителя являются водными растворами. Альтернативно, если пептидная молекула или молекула-носитель (или обе) являются практически нерастворимыми в воде до их смешивания, то пептидная молекула и/или макромолекула-носитель могут быть растворены в смешивающемся с водой растворителе, таком как спирт (например, этанол) перед смешиванием двух компонентов комплекса. В другом варианте осуществления способа приготовления нерастворимого в воде комплекса раствор пептидного соединения и раствор макромолекулы-носителя смешивают и нагревают до тех пор, пока комплекс пептидного соединения и макромолекулы-носителя не выпадет в осадок из раствора. Количества пептидного соединения и макромолекулы-носителя, необходимые для получения нерастворимого в воде комплекса, могут изменяться в зависимости от определенных используемых пептидного соединения и макромолекулы-носителя, определенного растворителя(ей) и/или способа, используемого для получения комплекса. Обычно, однако, пептидное соединение будет находиться в избытке относительно макромолекулы-носителя по молярному соотношению. Часто пептидное соединение также будет находиться в избытке по массовому соотношению, как продемонстрировано в примерах. В определенных вариантах осуществления макромолекула-носитель, предпочтительно натриевая соль карбоксиметилцеллюлозы, и пептидное соединение, предпочтительно РРI-149, смешивают в соотношении 0,2:1 (мас./мас.) макромолекула-носитель : пептидное соединение. В различных других вариантах осуществления соотношение макромолекулы-носителя к пептидному соединению (мас./мас.) может составлять, например, 0,5:1, 0,4:1, 0,3:1, 0,25:1, 0,15:1 или 0,1:1. Не ограничивающие примеры условий и способов получения нерастворимого в воде комплекса, соответствующего изобретению, описаны далее в примерах 1-5 и 8-9.
После осаждения комплекса пептидное соединение/макромолекула из раствора осадок может быть удален из раствора известными в уровне техники средствами, такими как фильтрация (например, через нейлоновую мембрану 0,45 мкм), центрифугирование и т. п. Затем выделенная паста может быть высушена (например, под вакуумом или в печи при 70oС) и твердое вещество может быть перемолото или распылено с получением порошка известными в уровне техники средствами (например, размалыванием в клиновой или молотковой мельнице или растиранием пестиком в ступке). После перемалывания или распыления порошок может быть просеян через сито (предпочтительно сито с ячеей 90 мкм) для получения однородного распределения частиц. Более того, выделенная паста может быть заморожена и лиофилизирована до сухого состояния. Порошковая форма комплекса может быть диспергирована в растворе-носителе с образованием жидкой суспензии или полутвердой дисперсии, пригодной для инъекции. Соответственно в различных вариантах осуществления фармацевтическая композиция, соответствующая изобретению, представляет собой сухое твердое вещество, жидкую суспензию или полутвердую дисперсию. Примеры жидких носителей, пригодных для использования в жидких суспензиях, включают растворы солей, растворы глицерина и растворы лецитина.
В другом варианте осуществления фармацевтический препарат, соответствующий изобретению, является стерильным препаратом. Например, после образования нерастворимого в воде комплекса этот комплекс может быть простерилизован, оптимальной является стерилизация γ-облучением или облучением электронным пучком. Согласно этому соответствующий изобретению вышеописанный способ приготовления фармацевтической композиции может далее предусматривать стерилизацию нерастворимого в воде комплекса γ-облучением или облучением электронным пучком. Предпочтительно препарат стерилизуют γ-облучением с использованием дозы γ-облучения, по меньшей мере, 15 кГр (KGy). В других вариантах осуществления композицию стерилизуют γ-облучением с использованием дозы γ-облучения, по меньшей мере, 19 кГр или не менее 24 кГр. Как показано в примере 11, соответствующие изобретению препараты сохраняют приемлемый уровень стабильности при γ-облучении.
Альтернативно для приготовления стерильной фармацевтической композиции нерастворимый в воде комплекс может быть выделен с использованием принятых стерильных способов (например, с использованием стерильных исходных материалов и ведением процесса получения в асептических условиях). Соответственно, в другом варианте осуществления вышеописанного способа приготовления фармацевтического препарата нерастворимый в воде комплекс получают, используя асептические методы.
Способы образования нерастворимого в воде комплекса, соответствующего изобретению, описаны далее в примерах 1-5 и 8-9. Фармацевтические препараты, включая порошки, жидкие суспензии, полутвердые дисперсии, сухие твердые вещества (например, лиофилизированные твердые вещества) и их стерилизованные формы (например, с помощью γ-облучения), приготовленные согласно способам, соответствующим изобретению, также охватываются изобретением.
Еще один аспект изобретения касается способов использования фармацевтических препаратов, соответствующих изобретению, для лечения субъекта, страдающего от состояния, которое лечат фармацевтически активным пептидным соединением, входящим в нерастворимый в воде комплекс. Соответственно, в предпочтительном варианте осуществления изобретение представляет способ лечения субъекта от состояния, которое поддается воздействию аналога LHRH, предусматривающий введение субъекту фармацевтического препарата, содержащего нерастворимый в воде комплекс аналога LHRH и макромолекулы-носителя.
Фармацевтическая композиция может быть введена субъекту любым путем, подходящим для достижения желаемого терапевтического результата(ов), хотя предпочтительными способами введения являются парентеральные способы, в частности, внутримышечная (i.m.) инъекция и подкожная/внутрикожная (s.c./i. d. ) инъекция. Альтернативно препарат может быть введен субъекту перорально. Другие подходящие парентеральные способы введения включают внутривенную инъекцию, защечное введение, чрескожную доставку и ректальное, вагинальное, интраназальное введение, а также введение через дыхательные пути. Следует отметить, что когда препарат, который обеспечивает задержанную доставку в течение от недель до месяцев при i.m. или s.c./i.d. способе введения, вводят альтернативным путем, то может отсутствовать задержанная доставка агента в течение равного периода времени вследствие выведения агента с помощью других физиологических механизмов (т.е. лекарственная форма может быть выведена из области доставки таким образом, что пролонгированные терапевтические эффекты не наблюдаются в течение таких же по длительности периодов времени, как периоды времени, наблюдаемые при i.m. или s.c./i.d. инъекции).
Фармацевтический препарат содержит терапевтически эффективное количество аналога LHRH. Термин "терапевтически эффективное количество" относится к количеству, эффективному при дозах и в течение периодов времени, необходимых для достижения желаемого результата. Терапевтически эффективное количество аналога LHRH может изменяться в соответствии с такими факторами, как стадия заболевания, возраст и масса субъекта, и способностью аналога LHRH (в виде монотерапии или в комбинации с одним или более других лекарственных средств) вызывать у субъекта желаемую реакцию. Могут быть подработаны схемы лечения с целью обеспечения оптимального терапевтического ответа. Терапевтически эффективное количество является также количеством, при котором терапевтически благоприятные эффекты преобладают над какими-либо токсическими или вредными эффектами антагониста. Не ограничивающий диапазон для терапевтически эффективного количества аналога LHRH
составляет от 0,01 до 10 мг/кг. Предпочтительная доза аналога LHRH PPI-149 для пролонгированного снижения уровней тестостерона в плазме в течение 28 дней составляет приблизительно 0,1-10 мг/кг, более предпочтительно 0,3-1,2 мг/кг (по свободному пептиду) в жидкой суспензии объемом приблизительно 1 мл или менее. Следует отметить, что данные значения дозы могут изменяться в зависимости от остроты и тяжести облегчаемого состояния. Кроме того, необходимо понимать, что для какого-либо определенного субъекта следует разрабатывать специфические схемы приема в течение времени в соответствии с индивидуальными потребностями и профессиональным мнением лица, проводящего введение или руководящего введением композиций, и что приведенный в данном описании диапазон доз является только примером и не предназначен для ограничения объема или реализации заявленной композиции.
Способ лечения, соответствующий изобретению, может применяться для лечения различных состояний, заболеваний и нарушений, при которых введение аналога LHRH имеет желаемый клинический эффект. Примеры заболеваний и нарушений включают гормонозависимые раки, такие как рак простаты, рак молочной железы, рак яичников, рак матки и рак яичка, доброкачественную гипертрофию простаты, преждевременное половое созревание, эндометриоз и фиброз матки. Соответственно, в изобретении представлены способы лечения данных заболеваний и нарушений путем введения фармацевтической композиции, соответствующей изобретению. Кроме того, аналоги LHRH могут быть использованы для изменения оплодотворяющей способности. Согласно этому способы, соответствующие изобретению, также могут быть использованы для оплодотворения in vitro и с целью контрацепции.
В особенно предпочтительном варианте осуществления способ используют для лечения рака простаты, аналог LHRH, использованный в препарате, представляет собой антагонист LHRH, наиболее предпочтительно РРI-149, и способ обеспечивает задержанную доставку аналога LHRH in vivo в течение, по меньшей мере, четырех недель после внутримышечного или подкожного введения. Аналог LHRH, предпочтительно РРI-149, в виде соответствующей изобретению композиции может быть использован для ингибирования роста клеток рака простаты при введении аналога LHRH субъекту, страдающему от рака простаты. Более того, антагонист LHRH, предпочтительно РРI-149, в виде соответствующего изобретению препарата может быть использован для ингибирования выброса тестостерона, сопровождающего применение агониста LHRH, путем предварительного введения антагониста LHRH, предпочтительно РРI-149, субъекту, страдающему раком простаты, перед началом терапии агонистом LHRH. Способы ингибирования индуцированного агонистом LHRH выброса тестостерона и другие способы лечения рака простаты с использованием антагониста LHRH, в которых возможно применение препаратов, соответствующих данному изобретению, описаны далее в Патентной заявке США No 08/573109 под названием "Способы лечения простаты с использованием антагонистов LHRH", поданной 15 декабря 1995 г., и в частично продолжающейся Патентной заявке No 08/755593 также под названием "Способы лечения простаты с использованием антагонистов LHRH", поданной 25 ноября 1996 г., содержание которых включено в опубликованную заявку РСТ WO 97/22357. Полное содержание заявок США и опубликованной заявки РСТ специально введено в данное описание в виде ссылки.
Конкретные способы комплексирования фармацевтически активного пептидного соединения с макромолекулой-носителем приведены в примерах 1-5 и 8-9 ниже. Описаны также результаты анализов, которые демонстрируют, что комплекс, содержащий антагонист LHRH, может осуществлять задержанную доставку фармацевтически активного пептида in vivo (пример 6) и может ингибировать индуцируемый агонистом LHRH выброс тестостерона (пример 7). Следующие примеры, которые более подробно иллюстрируют изобретение, не следует рассматривать как ограничивающие. Содержание всех ссылок, патентов и опубликованных патентных заявок, приведенных в описании, включены в него в виде ссылок.
Пример 1
100 мл раствора антагониста LHRH PPI-149 готовят путем растворения 6,25 мг/мл PPI-149 в воде. Равный образец (минимум 100 мл) натриевой соли карбоксиметилцеллюлозы (CMC) USP (реагент фармацевтической чистоты в соответствии с требованиями фармакопеи США) (низкой вязкости, Hercules Chemical Co.) готовят как 0,125% (мас./об.) и перемешивают до растворения. Равные порции растворов PPI-149 и CMC смешивают (при соотношении СМС : пептид, 0,2:1 (мас. /мас.)) и получают твердое вещество. Твердый материал перемешивают в течение ночи, а затем собирают фильтрацией через нейлоновый фильтр 0,45 мкм. Анализ с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) фильтрата раствора показывает, что, по меньшей мере, 95% соединения РРI-149 превращается в твердый комплекс и удаляется из раствора. Выделенную белую пасту дважды промывают водой, а затем переносят во флакон и высушивают под вакуумом. После высушивания в течение 72 часов получают 633 мг белого порошка. Из твердого материала затем получают порошок перетиранием пестиком в ступке. Элементный анализ показывает, что комплекс содержит 57% пептида.
Пример 2
25 мг РРI-149 растворяют в 1 мл воды. К раствору добавляют 1 мл 0,5% раствора карбоксиметилцеллюлозы. При перемешивании смесь превращается в шелковистое белое твердое вещество. Смесь нагревают в колбе с обратным холодильником в течение пяти минут, при этом образуется хлопьевидный белый осадок. Данный материал выделяют центрифугированием/декантацией. Твердое вещество ресуспендируют в воде и собирают при повторном центрифугировании. Анализ с помощью ВЭЖХ фильтрата раствора показывает, что, по меньшей мере, 90% соединения РРI-149 превращается в твердый комплекс. Белый осадок высушивают под вакуумом и твердый материал измельчают перетиранием пестиком в ступке. Элементный анализ показывает, что комплекс содержит 77% пептида.
Пример 3
50 мг РРI-149 растворяют в 2 мл 5% маннита и смешивают с 2 мл 0,5% карбоксиметилцеллюлозы (низкой вязкости, USP, Spectrum Quality Chemicals). Смесь перемешивают и сразу же получают белый осадок. Суспензию замораживают и лиофилизируют до сухого состояния, получая комплекс РРI-149 с задержанной доставкой.
Пример 4
25 мг РРI-149 растворяют в 1 мл воды. К данному раствору добавляют 1 мл 0,5% альгината натрия, USP (Spectrum). При перемешивании смеси сразу же образуется белый осадок. Данный материал выделяют центрифугированием/декантацией. Твердое вещество ресуспендируют в воде и собирают при повторном центрифугировании. Белый осадок высушивают под вакуумом. При проведении элементного анализа получают значение содержания пептида 66%.
Пример 5
25 мг РРI-149 растворяют в 1 мл воды. Для подведения рН до 11,0 добавляют аммиак. К раствору добавляют 1 мл 0,5% альгиновой кислоты, USP (Spectrum). При перемешивании смеси сразу же образуется белый осадок. Данный материал выделяют центрифугированием/декантацией. Твердое вещество ресуспендируют в воде и собирают при повторном центрифугировании. Белый осадок высушивают под вакуумом. При проведении элементного анализа получают значение содержания пептида 79%.
Пример 6
Нерастворимый в воде комплекс антагониста LHRH PPI-149 и карбоксиметилцеллюлозы получают в соответствии с предшествующими примерами. Готовят суспензию комплекса РРI-149/СМС и разовую дозу вводят крысам и собакам внутримышечной инъекцией. Доза для крыс составляет 50 мкг/кг/день Х 60 дней, а доза для собак составляет 40 мкг/кг/день X 28 дней. Уровни тестостерона в плазме (в нг/мл) определяют в различных точках времени как отражение активности антагониста LHRH в организме животного. Репрезентативные результаты, представленные на графике на фиг. 1, демонстрируют, что внутримышечная инъекция комплекса РРI-149/СМС приводит к пролонгированной супрессии уровней тестостерона в плазме в течение, по меньшей мере, 42 дней у крыс и, по меньшей мере, 28 дней у собак (отмечено белыми квадратами на фиг. 1), что демонстрирует задержанную доставку антагониста LHRH. Проводят также мониторинг уровней PPI-149 (в нг/мл) в плазме животных (отмечено черными квадратами на фиг. 1). Исходный пик PPI-149 наблюдают в течение первых восьми дней, после чего PPI-149 практически не определяется в плазме, Несмотря на невозможность определения PPI-149 в плазме через приблизительно восемь дней, результаты определения уровня тестостерона показывают, что PPI-149 сохраняет терапевтическую активность in vivo в период эксперимента.
Пример 7
Нерастворимый в воде комплекс антагониста LHRH PPI-149 и карбоксиметилцеллюлозы получают в соответствии с предшествующими примерами. Готовят суспензию комплекса РРI-149/СМС и разовую дозу вводят крысам внутримышечной инъекцией в день 0. На день 30 крысам инъецируют агонист LHRH LupronТМ (лейпролид). Уровни тестостерона в плазме (в нг/мл, отмечено белыми квадратами на фиг. 2) определяют в различных точках времени как отражение активности антагониста LHRH в организме животного. Проводят также мониторинг уровней PPl-149 (в нг/мл) в плазме животных (отмечено черными квадратами на фиг. 2). Репрезентативные результаты, представленные на графике на фиг. 2, демонстрируют, что предварительное введение комплекса РРI-149/СМС быстро снижает тестостерон в плазме до кастрационных уровней и, более того, блокирует индуцированный агонистом LHRH выброс тестостерона. Несмотря на невозможность определения РРI-149 в плазме через приблизительно восемь дней, результаты определения уровня тестостерона показывают, что РРI-149 сохраняет терапевтическую активность in vivo в период эксперимента.
Пример 8
В данном примере нерастворимый комплекс получают из аналога LHRH PPl-258 и карбоксиметилцеллюлозы (CMC). PPl-258 имеет структуру ацетил-D-нафтилаланил-D-4-Сl-фенилаланил- D-пиридилаланил-L-серил-L-тирозил-D-аспарагинил-L-лейцил-L-Ne-изопропил-лизил-L-пролил-D-аланил-амид. Для получения депо-формы РРI-258/СМС 174,8 мг (148,6 мг нетто) PPl-258 добавляют к 29,72 мл воды и материал перемешивают с целью суспендирования и растворения пептида. К данному перемешанному раствору добавляют 1,85 мл 2% раствора натриевой соли CMC (Hercules). Сразу же выпадает осадок твердого вещества. При нагревании в колбе с обратным холодильником суспензия становится полупрозрачной, а затем появляется белый осадок. Через 5 минут нагревания в колбе с обратным холодильником реакцию охлаждают, а затем выпадает белый осадок. Твердое вещество промывают водой и высушивают под вакуумом в течение ночи. Высушенный порошок растирают пестиком в ступке и просеивают через сито из неокрашенной стали 90 мкм. Просеянный порошок (сито 90 мкм) собирают и охарактеризовывают. Общий выход составляет 198,4 мг сухого твердого вещества, что дает 110,8 мг просеянного порошка после стадии измельчения. Характеристика комплекса дает следующий композиционный состав: Пептид PPI-258 - 80%, CMC - 18,8%, вода - 6,6%.
Пример 9
В данном примере нерастворимый комплекс получают из аналога LHRH ЦетрореликсТМ (который известен так же, как SB-75) и карбоксиметилцеллюлозы (CMC). ЦетрореликсТМ имеет структуру ацетил-D-нафтилаланил-D-4-Сl-фенилаланил-D-пиридилаланил-L-серил-L-тирозил-D-цитрулил-L-лейцил-L-аргинил-L-пролил-D-аланил-амид. Для получения депо-формы Цетрореликс/СМС 102,8 мг (87 мг нетто) ЦетрореликсТМ добавляют к 17,4 мл воды и материал перемешивают с целью суспендирования и растворения пептида. К данному перемешанному раствору добавляют 1,1 мл 2% раствора натриевой соли CMC (Hercules). Сразу же выпадает белый осадок в виде комков. Суспензию нагревают в колбе с обратным холодильником в течение 5 минут и при охлаждении получают твердый белый осадок. Твердое вещество выделяют центрифугированием, промывают водой и высушивают под вакуумом в течение ночи. Высушенный порошок растирают пестиком в ступке и просеивают через сито из неокрашенной стали 90 мкм. Порошок собирают и охарактеризовывают. Общий выход составляет 95 мг сухого твердого вещества, что дает 60 мг просеянного порошка после стадии измельчения. Характеристика комплекса дает следующий композиционный состав: Пептид ЦетрореликсТМ - 75%, CMC - 20,7%, вода - 6,5%.
Пример 10
В данном примере исследуют in vivo задержанную доставку трех различных аналогов LHRH - PPl-149, PPl-258 и ЦетрореликсТМ, приготовленных в виде депо-форм с CMC, как описано в предшествующих примерах. Тестируют три различных носителя препарата - солевой раствор, глицерин (15% глицерина/4% декстрозы) и лецитин. Используют крыс Sprague-Dawley (25 самцов весом в интервале 300-325 г) и эффективность аналога LHRH определяют по снижению уровней тестостерона в плазме.
Дозировки и пути введения приведены в табл. 1.
Действующей дозой пептида является 300 мкг/кг/день на 30 дней, что составляет 2,7 мг/крысу при разовой внутримышечной (IМ) или подкожной (SC) инъекции в объеме 200 мкл. Общий объем, необходимый для инъекции группы из 5 крыс, составляет 1,3 мл при концентрации 13,5 мг/мл активного пептида. Объем инъекции остается постоянным, а массу порошка доводят до общего содержания пептида, как указано в табл. 2.
Разовую внутримышечную или подкожную инъекцию тест-препарата объемом 200 мкл делают в верхнюю поверхность левой задней конечности или под кожу между лопатками, соответственно, в день 0 при обезболивании.
Для анализа уровней тестостерона в плазме из ретроорбитального синуса берут приблизительно 0,4 мл крови в день 1 после введения препарата и в дни 3, 7, 14, 21, 28 и 35. При обработке крови получают плазму и замораживают на сухом льду для определения уровней тестостерона в плазме стандартными способами.
Репрезентативные результаты, приведенные на фиг. 3А-3С, показывают, что уровни тестостерона в плазме у самцов крыс Sprague-Dawley снижаются и поддерживаются на низких уровнях в течение, по меньшей мере, 28 дней и до 50 дней в ответ на задержанную доставку аналогов LHRH PPl-149, PPl-258 и ЦетрореликсТМ, приготовленных в виде депо-препаратов с CMC (представлено на фиг. 3А, 3В и 3С, соответственно). Данные результаты показывают, что все три препарата являются эффективными в отношении снижения уровней тестостерона в плазме in vivo и поддержания пониженных уровней тестостерона в плазме в течение времени.
Пример 11
В данном примере препараты РРI-149-СМС подвергают воздействию γ-облучения с целью стерилизации с последующей оценкой как физических, так и химических свойств облученных препаратов. Приведенные ниже данные показывают, что γ-облучение является приемлемым средством для стерилизации депо-формы РРI-149-СМС.
Стабильность пептида
Приблизительно по 40 мг каждой из двух различных партий PPl-149-СМС упаковывают раздельно (в воздушной среде) в ряд стеклянных флаконов типа 1, закрытых резиновыми пробками и алюминиевыми крышками. Затем флаконы подвергают воздействию ряда номинальных (условных) доз γ-облучения. Два флакона анализируют на чистоту пептида (выражаемую в %) на каждом уровне γ-облучения для каждой из партий. Результаты указывают на то, что при γ-облучении в дозах до 24 кГр включительно РРI-149-СМС стабильно проявляет не более чем 2%-ное уменьшение чистоты пептида (как определяют по профилю примесей с помощью ВЭЖХ). Второе исследование с использованием γ-облучения в повышенных дозах проводят с дополнительной лабораторной партией РРI-149-СМС. При обработке высокими дозами γ-лучей РРI-149-СМС демонстрирует исключительно хорошую химическую стабильность.
Последующее предварительное исследование препарата предпринимают с целью сравнения профиля деградации, полученного после γ-облучения РРI-149-СМС с профилем деградации, полученным после автоклавирования раствора РРI-149-СМС для инъекций (1 мг/мл). Готовят два образца: а) РРI-149-СМС, подвергнутый γ-облучению в дозе 19 кГр, и б) раствор РРI-149 (1 мг/мл), подвергнутый автоклавированию (121oС/20 мин). Хроматограммы, полученные при ВЭЖХ двух образцов, демонстрируют, что профили деградации двух образцов, очевидно, являются весьма близкими (дают близкие значения времени удерживания для основных пиков).
Стабильность при хранении в условиях воздействия неблагоприятных факторов после γ-облучения
Предварительное исследование препарата при хранении в условиях воздействия неблагоприятных факторов проводят также на флаконах после γ-облучения. Закупоренные флаконы из двух лабораторных партий РРI-149-СМС подвергают γ-облучению в дозе 19 кГр и хранят при 25oС, 37oС и 50oС сроком до одного месяца. Результаты по химической стабильности в данных предварительных исследованиях указывают на то, что γ-облучение в дозе 19 кГр с последующим хранением в условиях воздействия неблагоприятных факторов не приводит к значительной химической нестабильности даже в крайне неблагоприятных условиях хранения (например, в течение 1 недели при 50oС). Данные показывают, что при дозах γ-облучения до и включая 19 кГр при хранении РРI-149-СМС до 28 дней при или ниже 50oС стабильно проявляется менее чем 2%-ное снижение чистоты пептида (как определяют по профилю примесей с помощью ВЭЖХ). Несмотря на очевидное различие исходного содержания влаги в двух исследованных партиях, значительных различий в чистоте пептида, как при исходном предварительном исследовании стабильности образцов, так и после хранения сроком до месяца не устанавливают.
Анализ размера частиц PPl-149-СМС
Разрабатывают способ определения размера частиц с использованием рассеяния света лазера, который применим для исследований размеров частиц РРI-149-СМС. Для пояснения применения способа представляют предварительный эксперимент, который проводят для исследования эффекта γ-облучения на размер частиц PPl-149-СМС. Данный эксперимент основан на существующем понимании того, что в аморфных твердых материалах может происходить уплотнение частиц при хранении. Два образца лабораторной партии PPl-149-СМС упаковывают в стеклянные флаконы типа l, закрывают серыми бутилкаучуковыми пробками и запечатывают алюминиевыми крышками. Определение частиц проводят до и после γ-облучения в дозе 15,5 кГр. Определение частиц проводят с помощью рассеяния света лазера (с использованием Malvern Mastersizer SТМ, снабженного обратными линзами fourier). Образцы по 20 мг для анализа размера частиц с помощью рассеяния света лазера диспергируют в приблизительно 0,5 мл деионизированной воды путем сильного встряхивания, затем обрабатывают ультразвуком в бане при комнатной температуре в течение 5 минут. После определения фона проводят эксперимент для квалификации способа. Диспергированный образец добавляют по каплям в емкость с непрерывной подачей (номинальным объемом приблизительно 60 мл) до получения приблизительно 20% помутнения. Скорость вращения мешалки поддерживают во время эксперимента при значении 2700 об/мин (с проверкой фона). При данной скорости индуцируемые водоворотом пузырьки не образуются, но поддерживается соответствующая стабильная дисперсия. Сканирование проводят восемь раз, и анализ полученных данных свидетельствует о стандартном отклонении <0,03% как предельном для любой точки, взятой для получения результатов. Когда образец дисперсии выдерживают в резервуаре в течение 15 минут, а затем продолжают обработку, значительных изменений не отмечают, что указывает на отсутствие растворения частиц в ходе эксперимента.
Образцы анализируют с использованием вышеприведенных экспериментальных параметров. Сканирование проводят восемь раз и устанавливают среднее значение диаметра частиц. Отмечают распределение на два различных размера, при этом все имеют четкие границы в области большого размера частиц, что указывает на отсутствие агрегации. Одна партия РРI-149-СМС имеет очевидно более низкий средний диаметр объема частиц перед γ-облучением, чем образец после облучения. Данное предварительное исследование, вероятно, указывает на то, что во время стерилизации происходит некоторое уплотнение частиц.
Пример 12
В данном примере проводят различные предварительные эксперименты с целью исследования эффекта как γ-облучения, так и неблагоприятного воздействия температуры/влажности на твердую форму РРI-149-СМС.
Дифракция рентгеновских лучей на порошке
В исходном эксперименте два образца РРI-149-СМС по 60 мг упаковывают (в воздушной среде) в стеклянные флаконы типа I, закрытые серыми бутилкаучуковыми пробками и запечатанные алюминиевыми крышками. Затем один образец подвергают воздействию γ-облучения в дозе 19,0 кГр. Затем изучают состояние твердой формы двух образцов по 60 мг с помощью дифракции рентгеновских лучей на порошке. Сравнивают дифрактограммы, полученные перед и после воздействия γ-облучения в дозе 19,0 кГр.
В последующем исследовании 60 мг образца РРI-149-СМС (после γ-облучения) вносят в стеклянный флакон типа I и помещают в предварительно уравновешенный термостат при 50oС/относительной влажности 75% на 5 дней. Сразу после извлечения из термостата контейнер с образцом закрывают серой бутилкаучуковой пробкой и запечатывают алюминиевой крышкой. Полученную при облучении дифрактограмму рентгеновских лучей на порошке данного образца после воздействия неблагоприятных условий сравнивают с другим образцом той же партии, который выдерживают при комнатной температуре в закрытом контейнере. Образцы анализируют с помощью автоматического порошкового дифрактометра Siemens D500, оборудованного графитовым монохроматором и источником рентгеновских лучей Cu ( ), работающим при 50 кВ, 40 мА. Диапазон два-θ-сканирования составляет 4-40oС при использовании окна шага сканирования 0,05o/1,2 с. Щели луча представлены под No (1) 1o, (2) 1o, (3) 1o, (4) 0,15o и (5) 0,15o ширины. Два-θ-калибровку проводят с использованием стандарта слюды NBS (Национальное бюро стандартов США) (SRM 675). Образцы анализируют с помощью пластины образца с нулевым фоном.
), работающим при 50 кВ, 40 мА. Диапазон два-θ-сканирования составляет 4-40oС при использовании окна шага сканирования 0,05o/1,2 с. Щели луча представлены под No (1) 1o, (2) 1o, (3) 1o, (4) 0,15o и (5) 0,15o ширины. Два-θ-калибровку проводят с использованием стандарта слюды NBS (Национальное бюро стандартов США) (SRM 675). Образцы анализируют с помощью пластины образца с нулевым фоном.
Приведенные данные показывают, что перед γ-облучением PPl-149-СМС не имеет кристаллической или псевдокристаллической структуры. На самом деле он дает образец дифракции рентгеновских лучах на порошке, характерный для аморфного твердого вещества (широкое плато между 2-20o 20 при отсутствии значительных пиков на дифрактограмме). Образец PPl-149-СМС после облучения дает тип дифракции, очень близкий типу дифракции необлученного образца, указывая на то, что воздействие γ-облучения (в дозах до 19 кГр включительно) скорее всего не индуцирует полиморфный переход состояния твердого материала. Подобным образом образец PPl-149-СМС, подвергнутый неблагоприятному воздействию температуры/влажности, дает тип дифракции, очень близкий типу дифракции необлученного образца и облученного образца, что явно предполагает отсутствие излишней тенденции у РРI-149-СМС к индукции полиморфных переходов твердого состояния материала.
Гигроскопичность
Проводят предварительные исследования РРI-149-СМС (после облучения) для определения поглощения влаги в условиях равновесия (измеряют по увеличению массы) при постоянной температуре (25oС) в различных условиях относительной влажности. Анализ равновесия влаги (% воды) как функция относительной влажности (% ОВ) указывает на то, что содержание влаги постепенно повышается до приблизительно 80% относительной влажности. При высокой относительной влажности (95% ОВ) PPl-149-СМС способен к значительному поглощению влаги. Полагают, что при значениях относительной влажности не более 80% ОВ большая предосторожность в плане защиты от влаги не является необходимой, таким образом возможно осуществление стадий производства в условиях комнатной влажности (при избежании экстремальных значений влажности).
Пример 13
В данном примере проводят исследования растворимости PPl-149-СМС. Эксперименты проводят в условиях с погружением и без погружения. PPl-149-СМС имеет приблизительную растворимость 100 мкг/мл (измеряют и выражают по свободному пептиду) при 25oС в забуференном 0,1 М фосфатом солевом растворе при рН 7,3. В условиях с погружением (определенных как <10% коэффициента насыщения в системе при заданной температуре) даже в отсутствие перемешивания PPl-149-СМС растворяется быстро (измеряют и выражают по свободному пептиду). В подобном эксперименте равновесие растворимости РРI-149-СМС определяют (измеряют и выражают по свободному пептиду) при 25oС в забуференном 0,1 М фосфатом солевом растворе при рН 7,3, используя три образца: только РРI-149-СМС, РРI-149-СМС в присутствии 10% дополнительного (по массе) РРI-149 (выражают как свободный пептид, но вводят как РРI-149, связанный с ацетатом) и РРI-149-СМС в присутствии 50% дополнительной (по массе) натриевой соли карбоксиметилцеллюлозы USP. Все три образца явно имеют близкую равновесную растворимость пептида. Поскольку выбранная буферная система имитирует физиологические условия, присутствие дополнительных свободных карбоксиметилцеллюлозных или пептидных структур, имеющихся в РРI-149-СМС, как представляется, не влияет на растворимость.
Пример 14
В данном примере характеристики фармакокинетики, фармакодинамики и безопасности повторных подкожных (SC) и внутримышечных (IM) введений доз РРI-149-СМС получают на собаках.
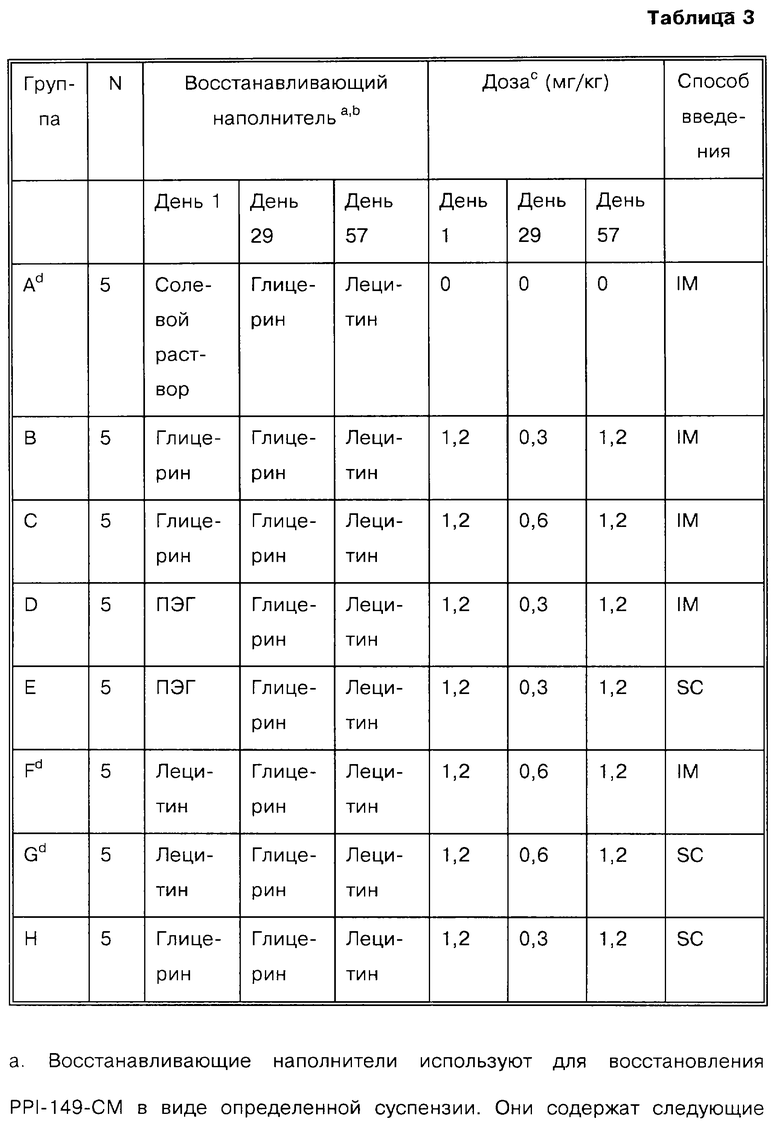
В первом исследовании, которое проводят в течение трех месяцев, анализируют сорок самцов собак-биглов, используя ежемесячные внутримышечные или подкожные инъекции РРI-149-СМС в дозах 1,2 мг/кг (день 1), 0,3 или 0,6 мг/кг (день 29) и 1,2 мг/кг (день 57) в ряде восстанавливающих наполнителей. Для исследования используют восемь групп по пять собак, как показано в табл. 3.
Данное исследование проводят таким образом, что эффективность PPl-149-СМС в исходной дозе в различных наполнителях оценивают в течение первого месяца лечения. В течение второго месяца исследований собаки получают уменьшенные дозы PPl-149-СМС с целью установления эффективной "поддерживающей "дозы. В течение третьего месяца планируют оценку безопасности в течение длительного времени и характеристик эффективности РРI-149-СМС.
Дозы для внутримышечной или подкожной инъекции, приготовленные в одном из восстанавливающих наполнителей, или дозы для внутримышечной инъекции контрольного препарата вводят в каждый из дней введения дозы во внешнюю поверхность правой задней конечности (внутримышечно) или в область между лопаток (подкожно). Материал набирают в туберкулиновый шприц объемом 1 м3 с короткой скошенной иглой размера 23. Область инъекции смачивают спиртовым тампоном непосредственно перед введением. Вводимый объем определяют в зависимости от специфической дозы пептида/кг массы тела. Следует отметить, что все дозы относятся к количеству вводимого пептида РРI-149.
Каждое животное осматривают, по меньшей мере, дважды в день в течение всего исследования на предмет явных признаков токсического или фармакологического эффекта и изменений в общем поведении и внешнем виде. Все аномальные клинические наблюдения регистрируют.
Кровь берут перед введением первой дозы и в различные моменты времени после введения дозы для полного анализа крови (СВС), химического анализа сыворотки и определения концентраций РРI-149 и тестостерона путем радиоиммуноанализа дважды в неделю.
После трехмесячного исследования девять животных умерщвляют и их ткани отбирают для расширенного патологического и гистопатологического анализа. Животных для умерщвления выбирают из группы с введением контрольного наполнителя, одной из групп с внутримышечным введением и одной из групп с подкожным введением. Ткани, отобранные для расширенного патологического и гистопатологического анализа при умерщвлении на 3 месяц, включают участок введения (подкожного или внутримышечного), надпочечники, аорту, кости, костный мозг, головной мозг, диафрагму, пищевод, глаза с глазным нервом, почки, толстый кишечник (слепую кишку, толстую кишку), печень с желчным пузырем, легкие с бронхами, лимфатические узлы, поджелудочную железу, гипофиз, простату с уретрой, слюнные железы, седалищный нерв, скелетную мышцу, кожу, тонкий кишечник (двенадцатиперстную кишку, тощую кишку, подвздошную кишку), спинной мозг, селезенку, желудок, яичко, тимус, щитовидную железу с паращитовидной железой, язык, трахею, мочевой пузырь и крупные повреждения.
Не имеется значительных изменений в гематологии и химии крови относительно нормы при исследовании леченых или контрольных животных. Расширенная и гистологическая оценка умерщвленных на третий месяц животных показывает отсутствие очевидных различий между леченными РРI-149-СМС собаками и контрольными (леченными наполнителем) животными за исключением изменений в яичках и простате, как ожидается при использовании данного антагониста LHRH.
В отношении фармакокинетики PPl-149-СМС отмечают, что у всех собак, леченных 1,2 мг/кг РРI-149-СМС, ресуспендированным в ряде восстанавливающих наполнителей, при внутримышечном или подкожном введении имеются близкие фармакокинетические профили PPl-149 в плазме при пике концентрации в плазме в первые 2 дня с последующим медленным снижением экспоненциальным образом в течение следующего месяца. PPI-149-СМС дает аналогичное распределение PPl-149 при суспендировании в каком-либо из трех восстанавливающих наполнителей, используемых в данном исследовании.
Касательно эффективности PPl-149-СМС в отношении эндокринной системы отмечают, что кастрационные уровни тестостерона (<0,6 нг/мл) наблюдают в течение 24 часов после начала введения PPl-149-СМС у всех собак, и уровни в основном сохраняются в кастрационном интервале в течение первого месяца независимо от способа введения или выбора восстанавливающего наполнителя. Двадцать шесть (26) из 35 собак (75%) имеют кастрационные уровни тестостерона в образце крови, полученном непосредственно перед введением второй дозы РРI-149-СМС на день 29. Данные результаты указывают на то, что исходная доза 1,2 мг/кг у собак успешно индуцирует быструю длительную супрессию (>28 дней) тестостерона в плазме. На второй месяц введения при исследовании эффективности "поддерживающей" дозы (дозы, более низкой, чем исходная) результаты указывают на то, что введение 0,3 или 0,6 мг/кг РРI-149-СМС поддерживает кастрационные уровни тестостерона в течение более чем 20 дней у 30 из 35 собак. В конце второго месяца лечения (день 57) у 21 из 35 собак (60%) сохраняется кастрационный уровень, тогда как 14 животных имеют уровень тестостерона в интервале нормы (>0,6% нг/мл). Дозу 1,2 мг/кг вводят в начале третьего месяца. Концентрации РРI-149 в плазме поддерживаются в течение следующего периода из 28 дней, тогда как уровни тестостерона в плазме снова являются "кастрационными". В конце третьего месяца (день 85) уровни тестостерона в плазме, как показывают, находятся в кастрационном интервале у 30 из 35 леченных РРI-149-СМС собак.
Вкратце, тридцать пять (35) собак получают 1,2 мг/кг PPl-149-СМС в день 1, 0,3 или 0,6 мг/кг РРI-149-СМС в день 29 и 1,2 мг/кг РРI-149-СМС в день 57 при использовании внутримышечного или подкожного введения с рядом восстанавливающих наполнителей. Из данных 35 собак 19 животных (54%) имеют уровни тестостерона в плазме, которые сохраняются на кастрационном уровне в течение всего курса терапии. Таким образом, введение PPl-149-СМС с интервалами 28 дней способно привести к полной супрессии тестостерона в плазме, которая является быстрой (все животные имеют кастрационные уровни через 24 часа) и длительной (поддерживается в течение курса введения).
Исследования, аналогичные вышеописанным, проводят в течение шести месяцев на собаках для дальнейшей оценки длительной безопасности и характеристик эффективности РРI-149-СМС. Животные получают исходную дозу 1,2 мг/кг РРI-149-СМС внутримышечно либо подкожно и пять последующих доз (при концентрациях либо 0,3 мг/кг, 0,6 мг/кг, либо 1,2 мг/кг) через интервалы 28 дней. Уровни тестостерона и РРI-149-СМС в плазме оценивают с помощью радиоиммуноанализа через регулярные промежутки времени. Репрезентативные результаты представлены на фиг. 4 (для лечения путем подкожного введения) и фиг. 2 (для лечения путем внутримышечного введения), которые иллюстрируют уровни тестостерона в плазме (белые квадраты) и РРI-149 (черные квадраты). Определенные дозы, используемые при каждом введении РРI-149-СМС, показаны на графиках. Результаты, представленные на фиг. 4 и 5, демонстрируют далее, что введение РРI-149-СМС с 28-дневными интервалами способно привести к полной супрессии тестостерона в плазме, которая является быстрой и длительной при поддержании пониженных уровней тестостерона в плазме в течение шести месяцев.
Эквивалентные решения
Специалистам известны или могут быть определены с помощью не более чем рутинных экспериментов многие эквивалентные решения специфических вариантов осуществления описанного изобретения. Предусматривается, что данные эквивалентные решения охватываются следующей формулой изобретения.

Изобретение относится к медицине. Изобретение характеризует препараты для задержанной доставки, содержащие нерастворимый в воде комплекс пептидного соединения (например, пептида, полипептида, белка, пептидомиметика и т.п. ) и макромолекулу-носитель. Композиции, соответствующие изобретению, позволяют загружать небольшой объем высокой концентрацией пептидного соединения, что обеспечивает пролонгированные периоды доставки фармацевтически активного пептидного соединения, например в течение одного месяца после введения комплекса. Комплексы, соответствующие изобретению, могут быть перемолоты или раздроблены до тонкоизмельченного порошка. В порошкообразном виде комплексы образуют стабильные водные суспензии и дисперсии, пригодные для инъекции. В предпочтительном варианте осуществления пептидное соединение комплекса представлено аналогом LHRH, предпочтительно антагонистом LHRH, а макромолекула-носитель является анионным полимером, предпочтительно карбоксиметилцеллюлозой. Описаны также способы получения комплексов, соответствующих изобретению, и способы использования комплексов, содержащих аналоги LHRH, для лечения состояний, которые лечат аналогами LHRH. Изобретение обеспечивает терапевтическую эффективность фармацевтической композиции, эффективность способа и шприца. 10 с. и 44 з.п. ф-лы, 3 табл., 5 ил.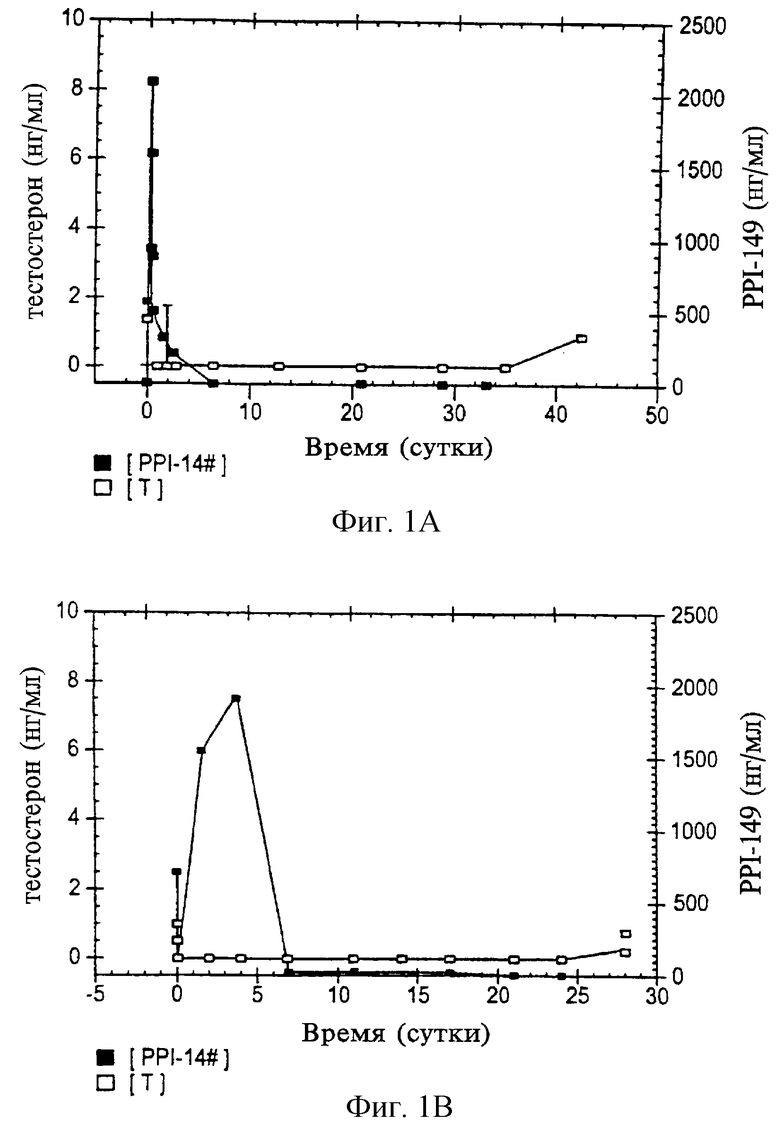
А-В-С-D-Е-F-G-Н-I-J,
в которой А-пиро-Glu, Ас-D-Nal, Ас-D-Qal, Ас-Sar или Ас-D-Pal;
В-Нis или 4-Cl-D-Phe;
С-Trp, D-Pal, D-Nal, L-Nal, D-Pal(N-О) или D-Trp;
D-Ser;
Е-N-Ме-Ala, Tyr, N-Ме-Tyr, Ser, Lys(iPr), 4-CI-Phe, Нis, Asn, Met, Ala, Arg или Ilе;
F-D-Asn, D-Gln или D-Thr;
G-Leu или Trp;
Н-Lys(iPr), Gln, Met или Arg;
I-Pro;
J - Gly-NH2, или D-Ala-NH2,
или его фармацевтически приемлемая соль.
Ас-D-Nаl-4-Cl-D-Phe-D-Pal-Ser-N-Ме-Tyr-D-Asn-Leu-Lys(iPr)-Pro-D-Ala.
Ас-D-Nal-4-Cl-D-Phe-D-Pal-Ser-N-Ме-Tyr-D-Asn-Leu-Lys(iPr)-Pro-D-Ala.
Ас-D-Nal-4-Cl-D-Phe-D-Pal-Ser-N-Ме-Tyr-D-Asn-Leu-Lys(iPr)-Pro-D-Ala.
Ас-D-Nal-4-Cl-D-Phe-D-Pal-Ser-N-Ме-Tyr-D-Asn-Leu-Lys(iPr)-Pro-D-Ala
и макромолекула-носитель является карбоксиметилцеллюлозой.
Ас-D-Nal-4-Cl-D-Phe-D-Pal-Ser-N-Me-Tyr-D-Asn-Leu-Lys(iPr)-Pro-D-Ala
и макромолекула-носитель является карбоксиметилцеллюлозой.
| US 5480656 A, 02.01.1996 | |||
| СПОСОБ ИЗГОТОВЛЕНИЯ ТВЕРДОЙ ЦИЛИНДРИЧЕСКОЙ ЛЕКАРСТВЕННОЙ ФОРМЫ С РЕГУЛИРУЕМЫМ ВЫДЕЛЕНИЕМ ВЕЩЕСТВА С ПРАКТИЧЕСКИ ПОСТОЯННОЙ СКОРОСТЬЮ | 1988 |
|
RU2072835C1 |
| RU 95113481 A, 27.11.1997 | |||
| СПОСОБ ПОЛУЧЕНИЯ МИКРОКАПСУЛЫ ПОЛИПЕПТИДА | 1991 |
|
RU2018306C1 |

