Настоящее изобретение относится к способу получения цианоборатов щелочных металлов, к их дальнейшему превращению в соли, включающие цианоборатные анионы и органические катионы, к этим солям, и к их применению в качестве ионных жидкостей.
Ионные жидкости или жидкие соли представляют собой ионные частицы, состоящие из органического катиона и в основном неорганического аниона. Они не содержат нейтральных молекул, и в основном имеют температуру плавления ниже, чем 373 K. Из предшествующего уровня техники известен целый ряд соединений, которые используются в качестве ионных жидкостей. В частности, они являются также объектом ряда патентов и патентных заявок.
Таким образом, ионные жидкости, не содержащие растворителя, были впервые раскрыты Hurley и Wier в ряде американских патентов (US 2446331, US 2446339 и US 2446350). Эти "соли, которые являются расплавленными при комнатной температуре", включали AlCl3 и целый ряд галогенидных соединений н-алкилпиридиниума.
В последние годы на эту тему было опубликовано несколько обзорных статей (R.Sheldon "Catalytic reactions in ionic liquids", Chem. Commun., 2001, 2399 - 2407; M.J.Earle, К.R.Seddon "Ionic liquids. Green solvent for the future", Pure Appl. Chem., 72 (2000), 1391-1398; P.Wasserscheid, W.Keim "lonische Flüssig-keiten - neue Lösungen für die Übergangsmetallkatalyse" [Ionic Liquids -Novel Solutions for Transition-Metal Catalysis], Angew. Chem., 112 (2000), 3926-3945; Т.Welton "Room temperature ionic liquids. Solvents for synthesis and catalysis", Chem. Rev., 92 (1999), 2071-2083; R. Hagiwara, Ya. Ito "Room temperature ionic liquids of alkylimidazolium cations and fluoroanions". Journal of Fluorine Chem., 105 (2000), 221-227).
Свойства ионных жидкостей, например точка плавления, тепловая и электрохимическая стабильность, вязкость, значительно зависят от природы аниона. В отличие от этого, полярность и гидрофильность или липофильность могут быть различными в зависимости от соответствующего выбора пары катион/анион. Поэтому существует основное требование к новым ионным жидкостям, имеющим различные свойства, которые обеспечивают дополнительные возможности относительно их применения.
Решающие достижения в области ионных жидкостей были достигнуты с открытием 1-этил-3-метилимидазолиум хлоралюмината. Эта соль имеет широкий жидкостной диапазон и электрохимическое окно больше, чем 3 В и, таким образом, является интересной для электрохимических и синтетических целей. Однако ее использование ограничено химической неустойчивостью, особенно к влажности. После открытия более устойчивого к гидролизу 1-этил-3-метилимидазолиум тетрафторбората были исследованы комбинации катионов алкилимидазолиума с неорганическими или органическими анионами, из которых 1-этил-3-метилимидазолиум тетрафторборат характеризуется как лучший.
Стабильность имидазолиумового катиона является относительно высокой и его температура разложения в высокой степени определяется анионом. Таким образом, 1-этил-3-метилимидазолиумовые соли с трифлатным и бис(трифторметилсульфонил)имидным анионами являются стабильными аж до 400°С, в то время как 1-этил-3-метилимидазолиум тетрафторборат является стабильным только до 300°С.
Предшествующий уровень техники описывает боратные анионы, в которых фторные лиганды заменены на цианидные (Е.Bernhardt, G.Henkel, H.Willner, Z.Anorg. Allg. Chem. 626 (2000) 560; D.Williams, В.Pleune, J.Kouvetakis, M.D.Williams, R.A.Andersen, J.Amer. Chem. Soc. 122 (2000) 7735; Е.Bernhardt, M.Berkei, M.Schurmann, H.Willner, Z.Anorg. Allg. Chem. 628 (2002) 1734) и трифторметильные лиганды (Е.Bernhardt, G.Henkel, H.Willner, G.Pawelke, H.Burger, Chem. Eur. J. 7 (2001) 4696; G.Pawelke, H. Burger, Coord. Chem. Rev. 215 (2001) 243). Трифторметил бораты синтезированы в этих статьях, исходя из цианоборатов, но цианобораты получают только с большими сложностями и в маленьких количествах. Синтез [B(CN4)]- является трудоемким и может быть выполнен только в маленьком препаративном масштабе. Кроме того, исходные материалы достаточно дорогие.
Цель настоящего изобретения заключается в обеспечении новых устойчивых соединений, имеющих ценные свойства, которые могут использоваться как ионные жидкости, и способа их получения. В частности, цель изобретения состоит в том, чтобы обеспечить соли с боратными анионами, которые имеют более высокую стабильность, чем соли с тетрафторборатными анионами.
Еще одна цель настоящего изобретения состоит в том, чтобы обеспечить эффективный и экономичный способ получения этих боратных солей и их прекурсоров.
В соответствии с изобретением эта цель достигается с помощью отличительных признаков независимого пункта Формулы изобретения и зависимых пунктов.
Поэтому настоящее изобретение относится, во-первых, к способу получения цианоборатов щелочных металлов общей формулы (1)

в которой M выбирают из группы, которая включает Li, Na, K, Rb и Cs, в которой легкодоступные исходные вещества тетрафторборат щелочного металла M[BF4] (M=Li, Na, K, Rb, Cs) и цианид щелочного металла MCN (M=Li, Na, K, Rb, Cs) вводят в реакцию один с другим в твердофазной реакции.
Тетрафторборат щелочного металла, используемый в соответствии с настоящим изобретением, представляет собой предпочтительно тетрафторборат калия K[BF4] или тетрафторборат натрия Na[BF4], и цианид щелочного металла, используемый в соответствии с настоящим изобретением, представляет собой предпочтительно цианид калия KCN или цианид натрия NaCN.
В предпочтительном варианте способа в соответствии с настоящим изобретением, тетрафторборат щелочного металла вводят в реакцию с цианидом щелочного металла в присутствии галогенида лития. При этом галогенид лития выбирают из LiCl, LiBr и LiI, особенно предпочтительным является хлорид лития LiCl.
В каждом отдельном случае цианид щелочного металла и галогенид лития могут использоваться в избытке одного из двух реагентов. Однако цианид щелочного металла и галогенид лития предпочтительно вводят в реакцию приблизительно в молярном соотношении 1:1.
Тетрафторборат щелочного металла и цианид щелочного металла предпочтительно используют в молярном соотношении от 1:4 до 1:12, особенно предпочтительно в молярном соотношении приблизительно 1:9.
Поэтому особенно предпочтительно используется молярное соотношение тетрафторборат щелочного металла: цианид щелочного металла: галогенид лития, составляющее приблизительно 1:9:9.
Исходные материалы, используемые для этой реакции в соответствии с настоящим изобретением, особенно предпочтительно представляют собой тетрафторборат калия K[BF4] в качестве тетрафторбората щелочного металла и цианид калия KCN в качестве цианида щелочного металла.
Твердофазную реакцию в соответствии с настоящим изобретением выполняют при температурах в интервале между 100°С и 500°С. Предпочтение отдается температурам от 250 до 400°С, особенно предпочтительно 280-340°С.
Не подразумевая никакого ограничения, объект твердофазной реакции в соответствии с настоящим изобретением объясняется со ссылкой на общий пример: K[BF4], KCN и LiCl смешивают в молярном соотношении 1:9:9 и далее вводят в реакцию при плавлении. Температуру реакции выбирают таким образом, что, с одной стороны, смесь KCN/LiCl образует эвтектическое плавление при 270-290°С, и, с другой стороны, только что образованные тетрацианоборатные соли разлагаются медленно (<400-500°С). Анализ диффрактограмм порошков охлажденного расплава KCN с LiCl (молярное соотношение 1:1) позволяет определить смешанные кристаллы K (Cl, CN) типа (a=6,34 Å, F m3m) и дополнительное не идентифицированное соединение (d=4,958, 2,878, 2,728, 2,482, 2,175 Å). Выход K[B(CN)4] фактически не зависит от температуры в интервале 280-340°С и составляет приблизительно 40-60%, на основе K[BF4]. В дополнительных исследованиях найдено, что понижение в молярном соотношении K[BF4] к KCN/LiCl от 1:9 до 1:4,5 приводит к снижениям в выходе продукта. Рамановские спектры реакционных смесей показывают, что тетрацианоборат после реакции находится в форме литиевой соли (ν(CN)=2263 см-1).
В аналогичной реакции, используя NaCN/LiCl смесь, смешанные кристаллы (Li, Na) (Cl, CN) типа (a=5,50 Å F m3m) образуются в расплаве NaCN с LiCl (молярное соотношение 1:1), кроме того, с небольшим количеством LiCN (d=5,216, 3,626 Å, температура плавления = 160°С). Эвтектика (120-140°С) образуется между NaCN и LiCl, в отличие от KCN/LiCl, но только смешанные кристаллы плавятся при 360-540°С; возможно, это причина низких выходов (приблизительно 25%) Na[B(CN)4].
В течение обработки продуктов реакции, сначала должен быть удален избыток цианида. Найдено, что окисление цианида, используя водный 30% Н2О2 раствор, является лучшим способом обработки. Низкая навеска соли и полное и быстрое разложение цианида, оставшегося в реакционной смеси, так же, как и хорошие выходы, перевешивают единственный недостаток, часто бурное и сложно контролируемое прохождение реакции цианида. Тетрацианоборат затем экстрагируют из водного раствора и превращают в К или Na соль с помощью реэкстракции.
Альтернативным способом, доступным для обработки продуктов твердофазной реакции, является окисление непрореагировавшего цианида, используя водный NaOCl раствор, которое происходит в течение нескольких минут в очень мягких условиях, т.е. без нагревания или вспенивания реакционной смесь. Далее обработку выполняют аналогично обработке с помощью Н2О2. Однако эта дополнительная обработка является более трудоемкой и занимает много времени из-за большей навески соли.
Более того, настоящее изобретение относится к способу получения цианоборатов щелочных металлов общей формулы (2)
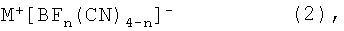
в которой n=0, 1, 2 или 3 и
М выбирают из группы, которая включает Li, Na, K, Rb и Cs,
в которой цианид щелочного металла MCN, где М=Li, Na, K, Rb, Cs, вводят в реакцию с бортрифторидэтератом BF3·OEt2.
При использовании крупнозернистого цианида калия KCN и BF3·OEt2 эквимолярные количества K[BF4] и K[BF2(CN)2] также образуются в реакции в соответствии с настоящим изобретением параллельно с первичным аддуктом K[BF3(CN)], в соответствии со следующими уравнениями:
K[BF3(CN)]+BF3·OEt2↔K[BF4]+BF2(CN)·OEt2
BF2(CN)·OEt2+KCN→K[BF2(CN)2]+Et2O
Кроме того, две соли K[BF(CN)3] и K[B(CN)4] образуются в меньшем количестве, первая из которых в особенности, если реакционную смесь поддерживают при температурах, больших, чем комнатная температура.
В соответствии с настоящим изобретением бортрифторидэтерат вводят в реакцию с цианидом щелочного металла в присутствии апротонного растворителя. Не подразумевая никакого ограничения, в качестве апротонного растворителя могут использоваться, например, ацетонитрил, диэтиловый эфир, тетрагидрофуран и/или диметоксиэтан.
Цианид щелочного металла, используемый для способа в соответствии с настоящим изобретением, представляет собой предпочтительно цианид калия KCN.
Исходные материалы предпочтительно вводят в реакцию в соответствии с настоящим изобретением при температурах от - 80 до 100°С, особенно предпочтительно при комнатной температуре.
В течение реакции могут образовываться летучие побочные продукты, которые удаляют при пониженном давлении. Главным образом, однако, побочные продукты, которые являются нерастворимыми в используемых растворителях, отделяют с помощью фильтрования. Растворитель, при желании, удаляют при пониженном давлении вместе с летучими побочными продуктами, и полученные цианобораты щелочных металлов, при желании, могут быть отделены и очищены с помощью обычных способов, известных специалистам, квалифицированным в данном уровне техники.
Третьим и четвертым объектом настоящего изобретения является способ получения солей с цианоборатными анионами общей формулы (3) и соответствующих солей общей формулы (3)
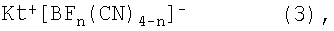
в которой n=0, 1, 2 или 3, и Kt+ представляет собой органический катион, при условии, что катион Kt+ не обозначает [N(C4H9)4]+ для n=0.
Для получения этих солей цианоборат щелочного металла общей формулы М+ [B(CN)4]-, в которой М выбирают из группы, которая включает Li, Na, K, Rb и Cs, или цианоборат щелочного металла общей формулы М+ [BFn(CN)4-n]-, в которой n=0, 1, 2 или 3 и М выбирают из группы, которая включает Li, Na, K, Rb и Cs, вводят в реакцию с Kt+ X-, где Х представляет собой атом галогена, выбранный из Cl, Br и I, и Kt+ представляет собой органический катион, при условии, что катион Kt+ не обозначает [N(C4H9)4]+ для n=0.
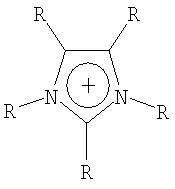
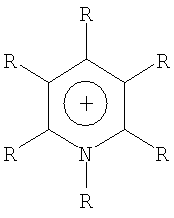
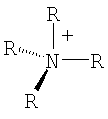
Органический катион Kt+ предпочтительно выбирают из группы
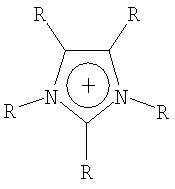
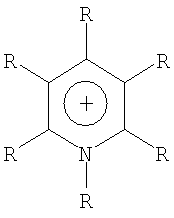
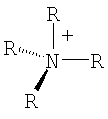
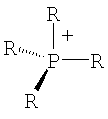
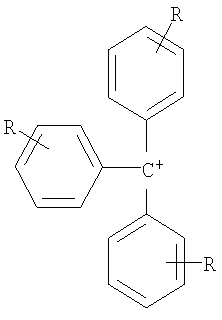
где R=Н, при условии, что, по крайней мере, один R на гетероатоме отличается от Н,
линейный или разветвленный алкил, который имеет 1-20 атомов углерода, линейный или разветвленный алкенил, который имеет 2-20 атомов углерода и одну или более двойных связей и линейный или разветвленный алкинил, который имеет 2-20 атомов углерода и одну или более тройных связей,
насыщенный, частично или полностью ненасыщенный циклоалкил, который имеет 3-7 атомов углерода,
галоген, в особенности фтор или хлор, при условии, что не присутствует ни одной связи галоген - гетероатом,
-NO2, при условии, что не присутствует ни одной связи с положительно заряженным гетероатомом, и, по крайней мере, один R отличается от NO2,
-CN, при условии, что не присутствует ни одной связи с положительно заряженным гетероатомом, и, по крайней мере, один R отличается от CN,
где R, в каждом случае, являются одинаковыми или различными,
где R могут быть прикреплены один к другому в парах простой или двойной связью,
где один или более R могут быть частично или полностью замещены атомами галогена, в частности -F и/или -Cl, или частично -CN или -NO2, при условии, что не все R полностью галогенированны,
и где один или два атома углерода радикала R могут быть заменены на гетероатомы и/или группы атомов, выбранных из группы -O-, -С(O)-, С(O)O-, -S-, -S(O)-, -SO2-, -S(O)2O-, -N=, -P=, -NR'-, -PR'-, -P(O)(OR'), -P(O)(OR')O-, -P(O)(NR'R'), -P(O)(NR'R')O-, -P(O)(NR'R')NR'-, -S(O)NR'- и -S(O)2NR'-, где R'=H, не-, частично или перфторированный C1 до С6-алкил или не-, частично или перфторированный фенил.
Для целей настоящего изобретения, полностью ненасыщенные заместители также подразумевают ароматические заместители.
Помимо водорода подходящими заместителями R органического катиона в соответствии с настоящим изобретением являются: C1 до С20-, в особенности C1- до С12-алкильные группы, C2- до С20-, в особенности С2- до С12-, алкенильные или алкинильные группы, насыщенные или ненасыщенные, т.е. также ароматические, С3- до С7-циклоалкильные группы, NO2, CN или галогены. Однако ограничивающий фактор для галогенов заключается в том, что они встречаются только как заместители на атомах углерода, но не на гетероатомах.
NO2 и CN не встречаются как заместители положительно заряженного гетероатома; кроме того, не все заместители одновременно принимают значение NO2 или CN.
Заместители R также могут быть скреплены в пары таким путем, что образуются циклический, би- или полициклический катионы. Эти заместители могут быть частично или полностью замещены атомами галогена, в особенности атомом F и/или Cl, или частично группой CN или NO2 и содержат один или два гетероатома или группы атомов, выбранных из группы О, (О), С(O)O, S, S(O), SO2, SO2O, N, P, NH, PH, NR', PR', P(O)(OR'), P(O)(OR')O, P(O)(NR'R'), P(O)(NR'R')O, P(O)(NR'R')NR', S(O)NR' и S(O)2NR'. В случае полного галогенирования, однако, не все присутствующие заместители R могут быть полностью галогенированны, т.е. по крайней мере, один R не является пергалогенированным.
Не подразумевая никакого ограничения, примеры заместителей в соответствии с настоящим изобретением органического катиона представляют собой:
-F, -Cl, -Br, -I, -СН3, -С2Н5, -С3Н7, -СН(СН3)2, -С4Н9, -С(СН3)3, -С5Н11, -С6Н13, -С6Н13, -C7H15, -C8H17, -C9H19, -С10Н21, -C12H25, -C20H41, -ОСН3, -ОСН(СН3)2, -СН2OCH3, -С2Н4OCH(СН3)2, -SCH3, -SCH(СН3)2, -C2H4SC2H5, -С2Н4SCH(СН3)2, -S(O)CH3, -SO2СН3, -SO2C2H5, -SO2С3Н7, -SO2СН(СН3)2, -CH2SO2CH3, -OSO2СН3, -OSO2CF3, -CH2N(H)C2H5, -C2H4N(H)C2H5, -CH2N(CH3)CH3, -C2H4N(CH3)CH3, -N(CH3)2, N(СН3)С3Н5, -N(СН3)CF3, O-C4H8-O-C4H9, -S-C2H4-N(C4H9)2, -OCF3, -S(O)CF3, -SO2CF3, -CF3, -C2F5, -С3F7, -C4F9, -С(CF3)3, -CF2SO2CF3, -C2F4N(C2F5)C2F5, -CF=CF2, -С(CF3)=CFCF3, -CF2CF=CFCF3, -CF=CFN(CF3)CF3, -CFH2, -CHF2, -СН2CF3, -С2F2Н3, -С3FH6, -СН2С3F7, -С(CFH2)3, -СНО, -С(O)ОН, -СН2С(O)ОН, -СН2С(O)СН3, -СН2С(O)С2Н5, -СН2С(O)ОСН3, СН2С(O)ОС2Н5, -С(O)СН3, -С(O)ОСН3,


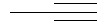

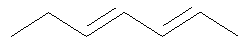

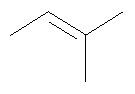
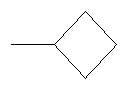
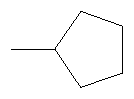
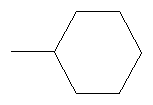
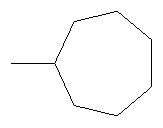
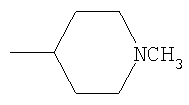
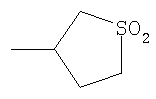
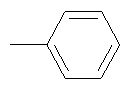
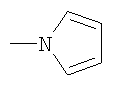
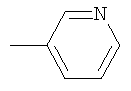
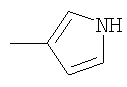
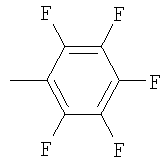
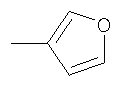
Не подразумевая никакого ограничения, следующие органические катионы являются особенно предпочтительными как соли в соответствии с настоящим изобретением:
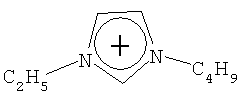
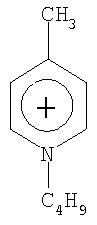
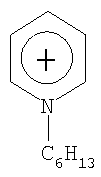
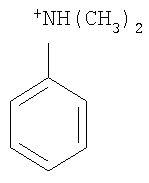
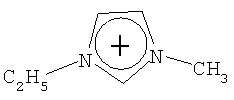
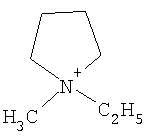
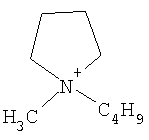
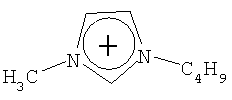
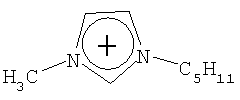

N(C2H5)4 + N(C4H9)4 + P(C2H5)4 + P(C4H9)4 + P(C6H13)3(C14H29)+
Эти соли в соответствии с настоящим изобретением преимущественно являются очень легко растворимыми в органических растворителях. В сравнении с известными жидкими солями, эти соли в соответствии с настоящим изобретением, неожиданно, имеют низкую вязкость. Эти соли в соответствии с настоящим изобретением преимущественно являются стабильными. Они могут быть выделены и могут храниться при комнатной температуре. Кроме того, эти соли в соответствии с настоящим изобретением относительно легко получаются, и требуют легко доступных исходных материалов.
Все соединения в соответствии с настоящим изобретением и соединения формулы [N(C4H9)4]+[B(CN)4]- имеют солеподобный характер, относительно низкие точки плавления (обычно ниже 100°С) и могут использоваться в качестве ионных жидкостей.
Эти соли в соответствии с настоящим изобретением и соли формулы [N(C4H9)4]+[B(CN)4]- могут использоваться в качестве растворителей для многих синтетических или каталитических реакций, таких как, например, ацилирование и алкилирование Фриделя-Крафтса, реакции циклоприсоединения Дильса-Альдера, реакции гидрогенирования и окисления, реакции Хека. Более того, например, могут быть синтезированны фторированные растворители для вторичной и первичной батарей.
Эти соли в соответствии с настоящим изобретением и соли формулы [N(C4H9)4]+[B(CN)4]- являются подходящими в качестве прекурсоров для получения жидкокристаллических соединений и активных ингредиентов, в числе других для лекарственных средств и средств для защиты сельскохозяйственных культур.
Возможно также использовать соединения в соответствии с настоящим изобретением и соли формулы [N(C4H9)4]+[B(CN)4]- в качестве неводного электролита, не обязательно в комбинации с другими электролитами, известными специалисту, квалифицированному в данном уровне техники.
Кроме того, эти соли в соответствии с настоящим изобретением и соли формулы [N(C4H9)4]+[B(CN)4]- являются интересными соединениями в качестве безводных, полярных веществ в подходящих реакциях в качестве катализатора фазового переноса или в качестве среды для гетерогенизации гомогенных катализаторов.
Полное раскрытие содержания всех заявок, патентов и публикаций, упомянутых выше и ниже, включено в эту заявку посредством ссылки.
Даже без дополнительных комментариев, предполагается, что специалист, квалифицированный в данном уровне техники, будет в состоянии использовать вышеупомянутое описание в самом широком объеме. Поэтому предпочтительные воплощения и примеры просто должны быть расценены как описательное раскрытие сущности изобретения, которые в любом случае абсолютно не ограничивают изобретение.
ЯМР спектры измеряли на растворах в дейтерированных растворителях при 20°С на спектрометре Bruker Avance DRX-300 с 5 мм 1Н/ВВ широкополосной головкой с дейтерированным затвором. Измерительные частоты различных ядер составляют: 1Н: 300,13 МГц, 11В: 96,92 МГц, 13С: 75,47 МГц, 19F: 282,41 МГц и 15N: 30,41 МГц. Метод сравнений показан отдельно для каждого спектра или каждого набора данных.
DSC измерения выполняли на приборе Netzsch DSC 204. Температуру и чувствительность калибровали, используя нафталин, бензойную кислоту, KNO3, AgNO3, LiNO3 и CsCl. В каждом случае 5-20 мг веществ взвешивали в алюминиевом тигле и запечатывали алюминиевыми крышками с маленьким отверстием. Исследование выполняли в температурном интервале от 25 до 500°С. Если не указано иначе, скорость нагрева составляет 10 К мин-1. В течение измерения пространство над образцом обрабатывали сухим азотом. Образцы веществ, чувствительных к воздуху, получали в вытяжном шкафу и переносили к аналитическим приборам в виале, заполненной аргоном. Оценку данных выполняли, используя программу Netzsch Protens 4.0.
Элементный анализ выполняли с помощью методов микроанализа сжиганием (microanalysis combustion methods), используя прибор Euro EA3000 компании HEKA-Tech GmbH. Образцы веществ, чувствительных к воздуху, получали в вытяжном шкафу и переносили к аналитическим приборам в виале, заполненной аргоном. Пределы погрешностей для записанных атомов составляют: С: ±0,3%, Н: ±0,1%, N: ±0,2%.
Пример 1: Синтез K[B(CN)4]
KCN, LiCl и K[BF4] размалывают на крупные гранулы и смешивают одно с другим в ступке в вытяжном шкафу (MBraun, Munich). Эту смесь размалывают мелкодисперсно, используя коммерчески доступную кофемолку. Реакционную смесь затем переносят в никелевый тигель (⊘внутренний = 101 мм, dстенки = 2 мм, h=85 мм). Этот тигель неплотно накрывают железной крышкой, переносят из вытяжного шкафа в муфельную печь (VMK 93, Kontron Material und Strukturanalyse GmbH) и нагревают. Когда реакция завершилась, тигель с металлической крышкой вынимают из все еще горячей муфельной печи и охлаждают до комнатной температуры на воздухе.
Охлажденную серую/черную пористую реакционную смесь переносят из тигля в ступку и крупнодисперсно размалывают. Затем к раздробленному твердому веществу добавляют 150 мл воды в 3 л химический стакан, и добавляют в общем 350 мл Н2О2 (30% водный раствор, приблизительно 3 моль) порциями приблизительно по 30 мл в течение получаса с постоянным перемешиванием. Реакцию, которая начинается экзотермически с бурным выделением газа, контролируют добавлением льда. Эту реакционную смесь (V=2,3 л) делят на два 3 л химических стакана и подкисляют, используя концентрированную HCl (приблизительно 300 мл, приблизительно 3,6 моль) (рН 5-7), до того, пока не перестанет наблюдаться выделение газа. Далее проверяют, присутствуют ли еще в этой смеси цианидные осадки (тест на цианид, Merck KGaA, Darmstadt, Germany). Затем эту смесь отфильтровывают и при перемешивании к этому желтому раствору добавляют 28 мл (0,34 моль) конц. HCl. Далее добавляют 47 г (63 мл, 0,33 моль) трипропиламина. Эту реакционную смесь перемешивают в течение 15 минут и экстрагируют дихлорметаном (250, 150 и 50 мл). Объединенные органические фазы промывают с помощью 200 мл Н2О, и промывные воды реэкстрагируют с помощью 25 мл дихлорметана. Объединенные дихлорметановые фазы сушат над MgSO4 и фильтруют через стеклянный фильтр (D4). 35 г (0,63 моль) КОН растворяют в маленьком количестве воды и добавляют к органическому раствору при интенсивном перемешивании. Бежевое маслянистое вещество сразу же выпадает в осадок и образует комки на дне сосуда после дополнительного перемешивания (30 минут). Смесь дихлорметан/трипропиламин отфильтравывают декантированием и продукт экстрагируют из осадка с помощью THF (200, 100 и 50 мл). Собранные THF фазы сушат, используя K2СО3, и наконец все летучие составляющие удаляют в роторном испарителе. Белый продукт промывают дихлорметаном и сушат при комнатной температуре при пониженном давлении.
13С{1H}-ЯМР: δ=123,3 ppm (q, 4C, CN), 1Δ13C(10/11B)=0,0021 ppm, 1J(11B, 13C)=70,9 Гц; 11B-ЯМР: δ=-38,6 ppm, 1J(11B, 13C)=71,2 Гц; растворитель: CD3CN; эталонные вещества: 13С-ЯМР пик растворителя (относительно TMS) и 11В-ЯМР BF3-Et2O/CD3CN в качестве внешнего стандарта.
ЯМР данные являются идентичными с теми, которые известны из предшествующего уровня техники (Е.Bernhardt, G.Henkel, H.Willner, Z.Anorg. Allg. Chem. 626 (2000) 560). Результаты элементного анализа:
В соответствии с DSC измерениями соль разлагается при температуре выше, чем 450°С.
Пример 2: Синтез Na[B(CN)4]
170,3 г (2,62 моль) KCN, 116,1 г (2,74 моль) LiCl и 37,2 г (0,30 моль) K[BF4] взвешивают, крупнодисперсно размалывают в ступке и смешивают одно с другим. Дальнейшая процедура соответствует процедуре, описанной в Примере 1 (температура реакции 300°С, время реакции 1,5 часов), как получение дихлорметанового экстракта.
2 эквивалента NaOH (приблизительно 25 г, 0,63 моль) растворяют в как только можно меньшем количестве воды (приблизительно 10-20 мл) и по каплям добавляют к органическому раствору при интенсивном перемешивании. Бежевое маслянистое вещество сразу же выпадает в осадок и образует комки на дне сосуда после дополнительного перемешивания (30 минут). Смесь дихлорметан/трипропиламин отфильтравывают декантированием и продукт экстрагируют из осадка с помощью THF (200, 100 и 50 мл). Если этот бежевый осадок становится жидким благодаря экстракции, его вязкая консистенция может быть восстановлена осторожным добавлением Na2CO3 или Na2SO4.
Собранные THF фазы сушат, используя К2СО3 или Na2SO4, и наконец все летучие составляющие удаляют в роторном испарителе. Белый продукт промывают дихлорметаном для того, чтобы удалить аминные осадки и сушат при 60°С при пониженном давлении. Выход составляет 25,3 г (62%, 0,18 моль).
13С{1Н}-ЯМР: δ=123,3 ppm (q, 4C, CN), 1Δ13С(10/11В)=0,0021 ppm, 1J(11B, 13C)=70,9 Гц; 11В-ЯМР: δ=-38,6 ppm, 1J(11В, 13С)=71,2 Гц; растворитель: CD3CN; эталонные вещества: С-ЯМР пик растворителя (относительно TMS) и 11В-ЯМР BF3-Et2O/CD3CN в качестве внешнего стандарта.
ЯМР данные являются идентичными с теми, которые известны из предшествующего уровня техники (Е.Bernhardt, G.Henkel, H.Willner, Z.Anorg. Allg. Chem. 626 (2000) 560).
Пример 3: Тетрацианоборат лития, Li[B(CN)4]
5 г (32 ммоль) K[B(CN)4] растворяют в 20 мл воды и вводят в реакцию с 8 мл 37% соляной кислоты (96 ммоль) и 8 мл nPr3Н (42 ммоль). Эту смесь затем экстрагируют дважды с помощью 50 мл СН2Cl2 каждый раз, органическую фазу сушат, используя MgSO4, и добавляют раствор 3 г LiOH-Н2О (72 ммоль) в 20 мл воды и смесь перемешивают энергично в течение одного часа. Все летучие продукты удаляют при пониженном давлении. Li[B(CN)4] экстрагируют из осадка с помощью 50 мл СН3CN в аппарате Сокслета. Органическую фазу выпаривают в роторном испарителе. Сырой продукт перекристаллизовывают из воды, промывают с помощью 50 мл CH2Cl2 и освобождают от осадков растворителя при пониженном давлении. Выход составляет 3,5 г (80%, 29 ммоль).
В соответствии с DSC измерениями соль разлагается при температуре выше, чем 470°С.
Пример 4: Тетрацианоборат аммония, NH4[B(CN)4]
0,31 г (2,0 ммоль) K[B(CN)4] растворяют в 8 мл воды, затем вводят в реакцию с раствором 0,20 г (1,1 ммоль) (NH4)2[SiF6] в 8 мл воды. Все летучие составляющие удаляют при пониженном давлении. NH4[B(CN)4] экстрагируют из осадка с помощью 10 мл СН3CN. Органическую фазу выпаривают в роторном испарителе. Сырой продукт промывают с помощью 10 мл CH2Cl2 и сушат при пониженном давлении. Выход составляет 0,25 г (93%, 1,9 ммоль).
В соответствии с DSC измерениями соль разлагается при температуре выше, чем 300°С.
Пример 5: Тритил тетрацианоборат, [Ph3С][В(CN)4]
500 мг (2,3 ммоль) Ag[B(CN)4] и 726 мг (2,3 ммоль) (С6Н5)3CBr в безводном ацетонитриле приводят во взаимодействие в 250 мл стеклянной колбе с ПТФЭ крышкой (Young, London). Ацетонитрил удаляют при пониженном давлении через 4 часа и затем добавляют 100 мл дихлорметана. Суспензию фильтруют через Celite® - покрытый фильтр в колбу Шленка (Schlenk flask). Реакционную колбу промывают дважды дихлорметаном (20 мл и 10 мл). Раствор выпаривают до 10 мл при пониженном давлении, и, после добавления 70 мл безводного гексана, выпадает в осадок оранжевое твердое вещество. Это вещество отфильтровывают через фильтр Шленка и промывают дополнительными 10 мл гексана. Оранжевый [Ph3С][В(CN)4] сушат при пониженном давлении и хранят в вытяжном шкафу. Выход составляет 408 мг (51%, 1,3 ммоль).
1Н-ЯМР: δ=7,73 ppm (m, 6H, o-H), δ=7C94 ppm (m, 6H, м-Н), δ=8,31 ppm (tt, 3H, n-H); 13C{1H}-ЯМР: δ=122,7 ppm (q, 4C, CN), 1J(11B, 13C)=71,5 Гц, δ=131,0 ppm (s, 6C, m-c), δ=140,2 ppm (s, 3С, i-С), δ=143,0 ppm (s, 6C, o-C), δ=143,8 ppm (s, 3С, n-С), δ=211,2 ppm (s, 1C, С+); 11В-ЯМР: δ=-38,6 ppm, 1J(11B, 13C)=71,3 Гц; растворитель: CDCl3; эталонные вещества: 1Н- и 13С-ЯМР сигнал растворителя (относительно TMS) и 11В-ЯМР BF3-Et2O/CD3CN в качестве внешнего стандарта.
Результаты элементного анализа [Ph3С][B(CN)4]:
[Ph3С][В(СН)4] плавится при 158°С с разложением.
Пример 6: [HNPhMe2][B(CN)4]
1,50 г (9,7 ммоль) K[B(CN)4] растворяют в 50 мл воды. Сначала 3 мл (36 ммоль) конц. HCl раствора и затем 1,23 мл (9,7 ммоль) N,N-ди-метиланилина добавляют к раствору при перемешивании, вследствие чего выпадает в осадок белое твердое вещество. Раствор экстрагируют дважды дихлорметаном (100 мл и 30 мл), органическую фазу сушат, используя MgSO4, и дихлорметан удаляют при пониженном давлении, что приводит к получению белого [HNPhMe2][B(CN)4], который очищают промыванием с пентаном. Выход составляет 2,12 г (92%, 8,9 ммоль).
1H-ЯМР: δ=3,23 ppm (s, 6H, СН3), 1Δ1H(12/13С)=-0,0023, 1J(1Н, 13С)=145,48 Гц, δ=7,64-7,58 ppm (m, 5H, С6Н5); 13С{1H}-ЯМР: δ=47,8 ppm (s, 2C, СН3), δ=121,5 ppm (s, 2C, С6Н5), δ=123,2 ppm (s, 4C, CN), 1J(11В, 13С)=71,3 Гц, 1Δ13C(10/11B)=-0,0020 ppm, δ=131,5 ppm (s, 2C, С6Н5), δ=131,6 ppm (s, 1C, C6H5), δ=143,1 ppm (s, 1C, C6H5); 11В-ЯМР: δ=-38,6 ppm, 1J(11B, 13C)=71,3 Гц; 15Н-ЯМР: δ=103,2 ppm (q, 4N, CN), 1J(11B, 15N)=0,73 Гц; растворитель: CD3CN; эталонные вещества: 1Н- и 13С-ЯМР сигнал растворителя (относительно TMS), 11В-ЯМР BF3Et2O/CD3CN в качестве внешнего стандарта и 15N-ЯМР 80% СН3NO2 в CD3CN в качестве внешнего стандарта.
Результаты элементного анализа [HNPhMe2][B(CN)4]:
[HNPhMe2][B(CN)4] плавится при 101°С и разлагается экзотермически при температуре выше, чем 246°С.
Пример 7: Тетраэтиламмониум тетрацианоборат, [Et4N][B(CN)4]
7 г (46 ммоль) K[B(CN)4] растворяют в 300 мл воды и 8,4 г (46 ммоль) [Et4N]Cl-H2O растворяют в 130 мл воды. Эти два раствора объединяют, вследствие чего выпадает в осадок белое твердое вещество. После перемешивания в течение 30 минут добавляют 250 мл ди-хлорметана, в котором растворяется осажденное вещество. Эти две фазы разделяют, и органическую фазу сушат над MgSO4. Дихлорметан удаляют в роторном испарителе, и белое твердое вещество промывают несколько раз пентаном и затем сушат при пониженном давлении. Выход составляет 10,5 г (96%, 43 ммоль).
1H-ЯМР: δ=1,22 ppm (tt, 12H, СН3), 1Δ1H(12/13c)=-0,0019 ppm, 1J(1H, 13C)=128,78 Гц, 3J(1H, 1H)=7,27 Гц; δ=3,13 ppm (q, 8H, CH2), 1Δ1H(12/13C)=-0,0034 ppm, 1J(1Н, 13С)=140,30 Гц, 2J(1H, 14N)=1,89 Гц; 3J(1Н, 1Н)=7,28 Гц; 13С{1H}-ЯМР: δ=7,8 ppm (s, 4С, СН3); δ=53,2 ppm (t, 4C, CH2), 1J(13C, 15N)=3,1 Гц; δ=123,3 ppm (q, 4C, CN), 1Δ13C(10/11B)=0,0021 ppm, 1J(11B, 13C)=70,9 Гц; 11В-ЯМР: δ=-38,6 ppm, 1J(11B, 13C)=71,2 Гц; растворитель: CD3CN эталонные вещества: 1Н- и 13С-ЯМР пик растворителя (относительно TMS) и 11В-ЯМР BF3-Et2O/CD3CN в качестве внешнего стандарта.
Результаты элементного анализа [Et4N][B(CN)4]:
[Et4N][B(CN)4] плавится при 230°С. Дальнейшее обратимое фазовое превращение происходит при температуре 145°С. Соль разлагается при температуре выше, чем 360°С.
Пример 8: 1-Бутил-3-метилимидазолиум тетрацианоборат [C8H15N2][B(CN)4]
0,35 г (2,3 ммоль) K[B(CN)4] растворяют в 20 мл воды. 0,53 г (3,0 ммоль) [C8H15N2]Cl в 20 мл воды добавляют при перемешивании. Раствор экстрагируют дважды дихлорметаном (30 мл и 20 мл), органическую фазу промывают водой (20 мл) и сушат, используя MgSO4, и затем дихлорметан удаляют при пониженном давлении. Выход составляет 0,50 г (87%, 2,0 ммоль).
Результаты элементного анализа [C8H15N2][B(CN)4]:
[C8H15N2][B(CN)4] плавится при температуре ниже, чем - 50°С, и разлагается эндотермически при температуре выше, чем 410°С.
Пример 9: 1-Этил-3-метилимидазолиум тетрацианоборат [C6H11N2][B(CN)4]
[С6Н11N2][В(CN)4] получают аналогично [C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [C6H11N2][B(CN)4]:
[С6Н11N2C[В(CN)4] плавится при температуре ниже, чем - 50°С, и разлагается эндотермически при температуре выше, чем 420°С.
Пример 10: п-Метилбутилпиридиниум тетрацианоборат [C10H16N][B(CN)4]
[C10H16N][B(CN)4] получают аналогично C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [C10H16N][B(CN)4]:
[C10H16N][B(CN)4] затвердевает при -25°С, плавится при 42°С и разлагается эндотермически при температуре выше, чем 390°С.
Пример 11: Получение K[BF2(CN)2]
Вариант А: 5,88 г (41 ммоль) BF3OEt2 и 30 мл CH3CN конденсируют на 4,12 г (63 ммоль) KCN в 50 мл колбе с ПТФЭ крышкой. Эту реакционную смесь перемешивают при комнатной температуре в течение 3 часов, и затем все летучие составляющие удаляют при пониженном давлении, и осадок растворяют в приблизительно 50 мл СН3CN и освобождают от KCN и K[BF4] фильтрованием. После удаления ацетонитрила при пониженном давлении, получают 2,66 г (19 ммоль) K[BF2(CN)2] (11B- и 19F-ЯМР: 93% [BF2(CN)2]-. 0,3% [BF3(CN)]- и приблизительно 7% неизвестных частиц). Выход: 92%. Чистый бесцветный K[BF2(CN)2] получают перекристаллизацией из воды. Выделенный выход: 2,08 г (72%, 15 ммоль).
Вариант В: 65 г (1,0 моль) KCN и 200 мл CH3CN сначала вводят в 500 мл круглодонную колбу с помощью капельной воронки. 50 мл (56 г, 0,4 моль) BF3OEt2 добавляют по каплям в течение получаса при перемешивании при комнатной температуре. В течение процесса добавления, температура поднимается до 50°С. После дополнительного перемешивания (1,5 часа) при комнатной температуре этот раствор отфильтровывают и осадок на фильтре (KCN и K[BF4]) промывают с помощью приблизительно 300 мл CH3CN. Объединенные ацетонитрильные фазы выпаривают в роторном испарителе, что приводит к получению 20 г чистого K[BF2(CN)2] в виде сырого продукта. Сырой продукт вводят в реакцию с 30 мл конц. HCl и 35 мл (25 г, 170 ммоль) трипропиламин в 200 мл воды и экстрагируют в виде трипропиламмониевой соли с помощью 200 мл дихлорметана. Эту дихлорметановую фазу сушат, используя MgSO4, и вводят в реакцию при интенсивном перемешивании с 25 г КОН, растворенного в как только можно меньшем количестве воды. Вязкую водную фазу отделяют и промывают дихлорметаном. Продукт экстрагируют из осадка с помощью приблизительно 300 мл СН3CN и раствор сушат, используя K2CO3, и выпаривают в роторном испарителе. Белый продукт промывают дихлорметаном и сушат при пониженном давлении. Выход: 17 г (60%, 120 ммоль). В соответствии с 11В-ЯМР, вещество содержит 98% [BF2(CN)2]-.
Пример 12: 1-Этил-3-метилимидазолиум трицианофторборат [C6H11N2][BF(CN)3]
[С6Н11N2][BF(СН)3] получают аналогично [C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [С6Н11N2][ВР(CN)3]:
При комнатной температуре [С6Н11N2][BF(CN)3] представляет собой жидкость.
Пример 13: 1-Бутил-3-метилимидазолиум трицианофторборат [C8H15N2][BF(CNC3]
[C8H15N2][BF(CN)3] получают аналогично [C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [C8H15N2][BF(CN)3]:
[C8H15N2][BF(CN)3] плавится при температуре ниже, чем - 50°С, и разлагается экзотермически при температуре выше, чем 300°С.
Пример 14: п-Метилбутилпиридиниум трицианофторборат [C10H16N][BF(CN)3]
[C10H16N][BF(CN)3] получают аналогично [C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [C10H16N][BF(CN)3]:
[C10H16N][BF(CN)3] плавится при температуре ниже, чем - 50°С, и разлагается экзотермически при температуре выше, чем 260°С.
Пример 15: 1-Этил-3-метилимидазолиум дицианодифторборат [C6H11N2][BF2(CN)2]
[C6H11N2][BF2(CN)2] получают аналогично [C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [С6Н11N2][BF2(CN)2]:
[С6Н11N2][BF2(CN)2] плавится при температуре ниже, чем - 50°С, и разлагается экзотермически при температуре выше, чем 200°С.
Пример 16: 1-Бутил-3-метилимидазолиум дицианодифторборат [C8H15N2C[BF2(CN)2]
[C8H15N2][BF2(CN)2] получают аналогично [C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [C8H15N2][BF2(CN)2]:
[C8H15N2][BF2(CN)2] плавится при температуре ниже, чем - 50°С, и разлагается экзотермически при температуре выше, чем 210°С.
Пример 17: п-Метилбутилпиридиниум дицианодифторборат [C10H16N][BF2(CN)2]
[C10H16N][BF2(CN)2] получают аналогично [C8H15N2][B(CN)4] с таким же выходом.
Результаты элементного анализа [C10H16N][BF2(CN)2]:
[C10H16N][BF2(CN)2] плавится при температуре ниже, чем - 50°С, и разлагается экзотермически при температуре выше, чем 190°С.
Пример 18: Тетрацианоборат калия
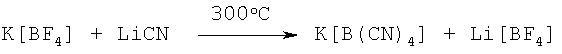
1,00 г (7,94 ммоль) тетрафторбората калия и 2,26 г (48,83 ммоль) цианида лития смешивают вместе в манипуляционной ручной камере в условиях инертной атмосферы. Смесь нагревается в никелевом тигеле в течение 20 минут от комнатной температуры до 300°С, и затем греется 40 минут при температуре 300°С. После охлаждения до комнатной температуры реакционная смесь обрабатывается 10 см3 воды и тремя порциями 40% пероксида водорода (10+5+5 см; в сумме 230 ммоль) и разбавляют дополнительными 20 см3 воды. Полученную в результате супензию обрабатываю 3,4 г (24,6 ммоль) карбоната калия и экстрагируют двумя порциями (80 см3 и 50 см3) тетрагидрофурана. Экстракт сушат над карбонатом калия. После фильтрации и выпаривания тетрагидрофурана, полученный продукт промывается дихлорметаном и снова растворяется в тетрагидрофуране. После фильтрации и выпаривания растворителя, остается 0,54 г твердого материала. Выход тетрацианобората калия составляет 44%.
Продукт характеризуется 11В-ЯМР спектром.
11В спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, ppm: -38,6 s.
Пример 19: 1-н-бутил-1-метилпирролидиний тетрацианоборат
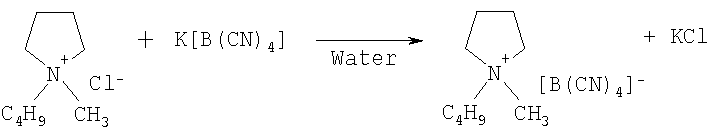
К перемешанному раствору 150,0 г (0,844 моль) 1-н-бутил-1-метилпирролидиний хлорида в 500 см3 воды добавляется по частям 130,0 г (0,844 моль) тетрацианобората калия. Реакционная смесь перемешивается магнитной мешалкой в течение 2 часов при комнатной температуре. Полученная эмульсия экстрагируется тремя объемами 200 см3 дихлорметана и экстракт промывается 5×200 см3 воды и обрабатывается оксидом алюминия (17,5 г) и активированный углеродом (5 г). После фильтрации и выпаривания дихлорметана остается 155 г прозрачной бесцветной жидкости. Выход продукта составляет 71%. Продукт характеризуется 1Н и 13С-ЯМР спектром.
1Н-ЯМР спектр (растворитель - ДМСО-d6, внешний стандарт - TMS), δ, ppm: 0,97 t (СН3); 1,36 t,q (CH2); 1,71 m (СН2); 2,11 m (2CH2); 2,99 s (СН3); 3,30 m (CH2); 3,46 m (2CH2); 3JH,H=7,2 Гц.
13C {Н}-ЯМР спектр (растворитель - ДМСО-d6, внешний стандарт - TMS), δ, ppm: 13,24, 19,24, 21,07, 24,89, 47,58, 63,06, 63,48, 121,80 q (CN), JC,B=71 Гц.
Пример 20: (доп.) 1-Этил-3-метилимидазолин тетрацианоборат
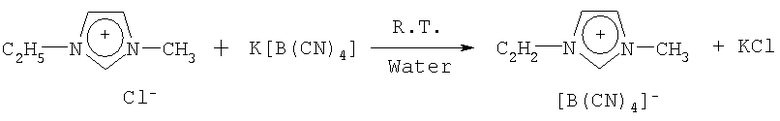
80,0 г (0,546 моль) 1-этил-метилимидазолин хлорида, растворенного в 200 см3 воды, добавляют к суспензии 84,1 г (0,546 моль) тетрацианобората калия в 800 см3 воды. Реакционную смесь смешивают в течение 10 часов при комнатной температуре и продукт экстрагируют 500 см3 дихлориетана (5×100 см3), промывают 4 порциями 100 см3 и 3 порциями 50 см3 воды. Дихлорметан извлекают в вакууме, а остаток сушат при температуре 90°С в вакууме при давлении 13,3 Па 20 часов. Получают 97 г жидкого материала, который содержит 18 частей на миллион примесей хлора и 30 частей на миллион воды. Выход продукта составляет 78,6%. Продукт характеризуется 1Н и 13В-ямр спектром.
1Н-ЯМР спектр (растворитель - CD3CN, внешний стандарт - TMS), δ, ppm: 1,45 t (СН3); 3,84 s (СН3); 4,18 q (CH2); 7,41 m (CH); 7,47 m (CH); 8,84 br. s. (CH); 3JH,H=7,3 Гц.
11В-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, ppm: - 38,6 s.
Пример 21: 1-Бутил-3-метилимидазолин тетрацианоборат получен по вышеуказанной методике.
Продукт характеризуется 1Н и 11В спектрами.
1Н-ЯМР спектр (растворитель - CD3CN, внешний стандарт - TMS), δ, ppm: 0,97 t (СН3); 1,36 t,q (СН3); 1,83 m (CH2); 3,85 s (СН3); 4,15 t (СН2); 7,35 m (CH); 7,39 m (CH); 8,41 br.s. (CH); 3JH,H=7,3 Гц.
11В-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, ppm: - 38,7 s.
Пример 22: Тетра-н-бутиламмония тетрацианоборат
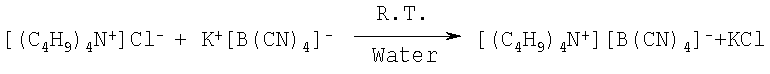
50,0 г (0,169 моль) тетрабутиламмония, растворенного в 200 см3 воды, добавляют к суспензии 26,0 г (0,169 моль) тетрацианобората калия в 400 см3 воды. Реакционную смесь смешивают в течение 2 часов при комнатной температуре и осаждают (белый твердый материал) на фильтре, промывают 2 порциями 75 см дистиллированной воды. Остаток сушат при температуре 60°С в вакууме при давлении 1,3 Па два дня. Выход продукта составляет 95%. Точка плавления равна 72°С. Продукт характеризуется 1Н и 11В-ЯМР спектром.
1Н-ЯМР спектр (растворитель - CD3CN, внешний стандарт - TMS), δ, ppm: 1,00 t (4СН3); 1,37 t,q (4CH2); 1,63 m (4СН2); 3,11 m (4СН2); 3JH,H=7,3 Гц.
11B-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, ppm: - 38,7 s.
Пример 23: Тетра-н-бутилфосфора тетрацианоборат
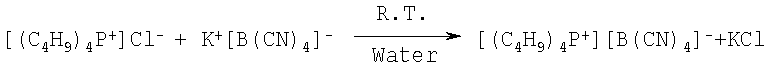
5,89 г (20 ммоль) хлорида тетрабутилфосфора, растворенного в 10 см воды, добавляют к суспензии 3,13 г (20 ммоль) тетрацианобората калия в 100 см воды. Реакционную смесь смешивают в течение 10 часов при комнатной температуре и осаждают (белый твердый материал) на фильтре, промывают 5 порциями 100 см3 дистиллированной воды. Остаток сушат при температуре 60°С в вакууме при давлении 1,3 Па два дня. Получают 5,65 г твердого материала. Выход продукта составляет 75%. Точка плавления равна 79°С. Продукт характеризуется 1Н и 11В-ЯМР спектром.
1Н-ЯМР спектр (растворитель - CD3CN, внешний стандарт - TMS), δ, ppm: 0,98 t (4СН3); 1,51 m (8CH2); 2,08 m (4СН2); 3JН,Н=7,0 Гц.
11В-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, ppm: - 38,7 s.
Пример 24: Три-н-гексил (н-тетрадецил)фосфора тетрацианоборат

51,4 г (99 ммоль) хлорида три-н-гексил (н-тетрадецил)фосфора, растворенного в 200 см3 дихлорметана, добавляют к раствору 16,07 г (104 ммоль) тетрацианобората калия в 400 см3 воды. Реакционную смесь смешивают магнитной мешалкой в течение 9 часов при комнатной температуре. Фаза дихлорметана отделяется, а водная фаза экстрагируется 2 порциями 100 см и 50 см3 дихлорметана. Объединенный органический раствор промывается 3 частями 100 см3 дистиллированной воды и сушится над сульфатом магния. После фильтрации и выпаривании дихлорметана остаток сушится при температуре 60°С в вакууме при давлении 1,3 Па один день. Получают 57,8 г маслянного материала. Выход продукта составляет 97,5% рассчитанный на хлорид три-н-гексил (н-тетрадецил)фосфора. Продукт характеризуется 1Н и 11В-ЯМР спектром.
1H-ЯМР спектр (растворитель - CD3CN, внешний стандарт - TMS), δ, ppm: 0,87-0,99 m (4СН3); 1,27-1,41 m (16CH2); 1,41-1.63 m (8СН2); 2.01-2.14 (4СН2).
11В-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, ppm: - 38,7 s.
Пример 25: 1-Бутил-4-метилпиридиний тетрацианоборат
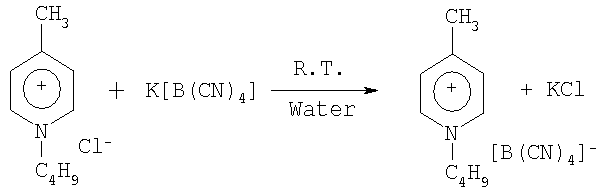
50,0 г (0,269 моль) хлорида 1-бутил-4-метилпиридиния, растворенного в 200 см3 дихлорметана, добавляют к суспензии 42,3 г (0,275 моль) тетрацианобората калия в 400 см3 воды. Реакционную смесь смешивают в течение 3 часов при комнатной температуре и продукт экстрагируют 3 частями 300 см3, 200 см3 и 100 см3 дихлорметана. Раствор дихлорметана отмывается 3 частями 200 см дистиллированной воды. Раствор сушится над сульфатом магния, фильтруется и дихлорметан выпаривается в вакууме. Остаток сушат при температуре 60°С в вакууме при давлении 1,3 Па один день. Получают 56,7 г желтоватого материала. Выход продукта составляет 79,2%. Продукт характеризуется 1H и 11В-ЯМР спектром.
1H-ЯМР спектр (растворитель - CD3CN, внешний стандарт - TMS), δ, ppm: 0,98 t (СН3); 1,38 t,q (CH2); 1,94 m (CH2); 2,65 s (СН3); 4,46 t (CH2); 7,84 d (2CH); 8,50 d (2CH); (CH); 3JН,Н=7,3 Гц, 3JН,Н=6,5 Гц.
11В-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, ppm: - 38,7 s.
Пример 26: Цианотрифторборат лития, Li[B(CN)F3]
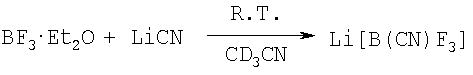
150 мг (3,5 ммоль) цианида лития растворяют в 0,5 см3 CD3CN, 0,25 см3 (2,0 моль) BF3-этерата добавляется при комнатной температуре. Реакционная смесь смешивается в течение 30 мин. 11В и 19F спектры показывают образование Li[B(CN)F3] (86,5% целевого продукта); Li[B(CN)2F2] (5,5%) и Li[BF4] (8,0%). После фильтрации и выпаривания растворителя Li[B(CN)F3] может быть отделен как твердый материал и очищен кристаллизацией или превращением в K[В(СН)F3] действием КОН в воде, аналогично получению K[B(CN)2F2] (Пример 11, вариант В).
Li[B(CN)F3]
11В-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2О), δ, -3,8 q ppm.
19F-ЯМР спектр (растворитель - CD3CN, внешний стандарт - CCI3F), δ, -141,0 q ppm, JB,F=22 Гц.
Li[B(CN)2F2]
11В-ЯМР спектр (растворитель - CD3CN, внешний стандарт - BF3-Et2O), δ, -7,4 t ppm.
19F-ЯМР спектр (растворитель - CD3CN, внешний стандарт - CCI3F), δ, -155,9 q ppm, JB,F=38 Гц.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРАЛКИЛЦИАНО- ИЛИ ПЕРФТОРАЛКИЛЦИАНОФТОРБОРАТОВ | 2011 |
|
RU2575352C2 |
| СПОСОБ СИНТЕЗА КАТИОННЫХ ЦИКЛОПЕНТАДИЕНИЛЬНЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ С ФОСФИНОВЫМИ ЛИГАНДАМИ | 2023 |
|
RU2831381C1 |
| ИОННЫЕ ЖИДКОСТИ НИЗКОЙ ВЯЗКОСТИ | 2006 |
|
RU2413732C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОНИЕВЫХ СОЛЕЙ С ТЕТРАФТОРБОРАТНЫМ АНИОНОМ, ИМЕЮЩИХ НИЗКОЕ СОДЕРЖАНИЕ ГАЛОГЕНИДОВ | 2005 |
|
RU2415843C2 |
| СПОСОБ ПОЛУЧЕНИЯ (АЦЕТИЛАЦЕТОНАТО-κ-О,О')(БИС-АЦЕТОНИТРИЛ)ПАЛЛАДИЯ ТЕТРАФТОРБОРАТА | 2011 |
|
RU2456295C1 |
| КВАТЕРНИЗОВАННЫЙ ПОЛИБЕНЗИМИДАЗОЛ | 2011 |
|
RU2575849C2 |
| СПОСОБ ПОЛУЧЕНИЯ (АЦЕТИЛАЦЕТОНАТО)(ЦИКЛООКТАДИЕН)ПАЛЛАДИЯ ТЕТРАФТОРБОРАТА | 2012 |
|
RU2508293C1 |
| ИОННЫЕ ЖИДКОСТИ, СОДЕРЖАЩИЕ АНИОНЫ [N(CF)] | 2003 |
|
RU2351601C2 |
| СПОСОБ АДДИТИВНОЙ ПОЛИМЕРИЗАЦИИ НОРБОРНЕНА И ЕГО ПРОИЗВОДНЫХ | 2015 |
|
RU2626745C2 |
| СПОСОБ АДДИТИВНОЙ СОПОЛИМЕРИЗАЦИИ НОРБОРНЕНА С 5-МЕТОКСИКАРБОНИЛНОРБОРНЕНОМ | 2017 |
|
RU2653060C1 |
Описывается способ получения цианоборатов щелочных металлов общей формулы M+[B(CN)4]-, где М - калий действием цианида щелочного металла на тетрафторборат калия в условиях твердофазной реакции, их дальнейшее превращение в соли, включающие цианоборатные анионы и органические катионы, указанные соли могут использоваться в качестве ионных жидкостей. 8 н. и 10 з.п. ф-лы, 1 табл.

в которой М выбирают из группы, которая включает К, отличающийся тем, что тетрафторборат калия вводят в реакцию с цианидом щелочного металла MCN, в котором M=Li, Na, К, в твердофазной реакции и реакция проходит при температурах в интервале между 100°С и 500°С.
в которой М выбирают из группы, которая включает Li, Na, К, отличающийся тем, что тетрафторборат калия вводят в реакцию с цианидом щелочного металла MCN, в котором М=Li, Na, К в твердофазной реакции в присутствии галогенида лития, выбранного из LiCl, LiBr, LiI, при температурах в интервале между 100°С и 500°С, и образующийся тетрацианборат калия при необходимости растворяют в воде и подкисляют, эту смесь экстрагируют путем обработки водным раствором NaOH или LiOH.
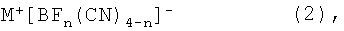
в которой n=0, 1, 2 или 3
и М выбирают из группы, которая включает Li, Na, К,
отличающийся тем, что цианид щелочного металла MCN вводят в реакцию с бортрифторидэтератом BF3·OEt2 в присутствии апротонного растворителя при температурах от -80 до 100°С.
отличающийся тем, что используемый цианид щелочного металла представляет собой цианид калия KCN.
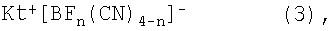
в которой n=0, 1, 2 или 3, и Kt+ представляет собой органический катион, при условии, что катион Kt+ не обозначает [N(C4H9)4]+ для n=0,
отличающийся тем, что цианоборат щелочного металла общей формулы M+[B(CN)4]-, в которой М выбирают из группы, которая включает Li, Na, К, полученный по любому из пп.1-7, или цианоборат щелочного металла общей формулы M+[BFn(CN)4-n]-, в которой n=0, 1, 2 или 3 и М выбирают из группы, которая включает Li, Na, К, полученный по одному из пп.8-11 вводят в реакцию с Kt+Х-,
где Х представляет собой галоген, выбранный из Cl, Br и I, и Kt+ представляет собой органический катион,
при условии, что катион Kt+ не обозначает [N(C4H9)4]+ для n=0 и где органический катион Kt+ выбирают из группы, которая включает
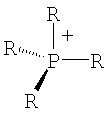
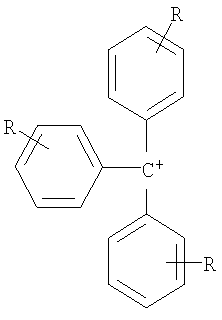
где R=H, при условии, что, по крайней мере, один R на гетероатоме отличается от Н,
линейный или разветвленный алкил, который имеет 1-20 атомов углерода, линейный насыщенный, частично или полностью ненасыщенный циклоалкил, который имеет 3-7 атомов углерода, где R в каждом случае являются одинаковыми или различными.
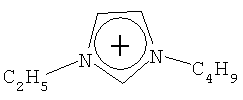
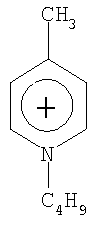
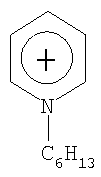
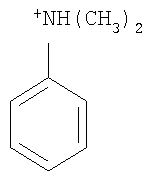
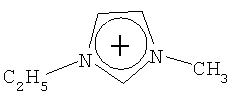
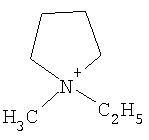
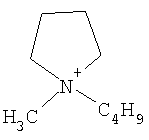
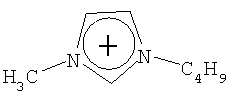
N(C2H5)4 +N(C4H9)4 +P(C2H5)4 +P(C4H9)4 +P(C6H13)3(C14H29)+
в которой n=0, 1, 2 или 3, и Kt+ представляет собой органический катион, при условии, что катион Kf+ не обозначает [N(C4H9)4] для n=0 и органический катион Kt+ выбирают из группы, которая включает
где R=H, при условии, что, по крайней мере, один R на гетероатоме отличается от Н,
линейный или разветвленный алкил, который имеет 1-20 атомов углерода, насыщенный, частично или полностью ненасыщенный циклоалкил, который имеет 3-7 атомов углерода,
где R в каждом случае являются одинаковыми или различными.
N(C2H5)4 +N(C4H9)4 +P(C2H5)4 +P(C4H9)4 +P(C6H13)3(C14H29)+.
| E.BERNARDT et al ZEITSHRIFT fur ANORGANISHE und ALLGEMEINE CHEMIE, 626(2), 560-568, 2000. |

