Предлагаемое изобретение относится к новому способу получения катионных циклопентадиенильных комплексов палладия общей формулы [CpPd(L)2]BF4 и [CpPd(L∧L)]BF4, где Ср - циклопентадиенил, L - монодентатные фосфиновые лиганды, такие как трифенилфосфин (PPh3), трис(о-метоксифенил)фосфин (ТОМРР), трис(пара-толил)фосфин (Р(р-То1)3), трис(2-фурил)фосфин (TFP), трис(2-тиенил)фосфин (ТТР), дифенил(циклогексил)фосфин (P(Ph)2Cy); L∧L - бидентатные фосфорорганические лиганды, такие как 1,3-бис(дифенилфосфино)пропан (dppp)) L4-бис(дифенилфосфино)бутан (dppb), 1,5-бис(дифенилфосфино)пентан (dpppent), 1,6-бис(дифенилфосфино)гексан (dpphex) и 1,1'-бис(дифенилфосфино)ферроцен (dppf); которые могут быть использованы как в индивидуальном виде в качестве катализаторов, так и в качестве компонентов каталитических систем реакций селективной димеризации стирола, аддитивной полимеризации норборнена и его производных, теломеризации диеновых углеводородов с ОН- и NH-нуклеофилами.
В научной литературе, посвященной синтезу катионных циклопентадиенильных комплексов палладия, встречается относительно малое количество работ, что связано с ограничениями (и недостатками) существующих способов синтеза. Известные из литературы способы синтеза таких соединений можно классифицировать по природе реагирующих веществ с целью формирования многоцентровой связи Pd-Cp:
1) взаимодействие дигалогенидных диеновых комплексов палладия состава (диен)PdBr2 с циклопентадиенильными комплексами железа CpFe(CO)2X, приводящее к образованию катионных комплексов состава [(диен)PdCp][FeBr4], где диен=Ph4C4, C8H12, X=Br, CpFe(CO)2[l,2];
2) взаимодействие галогенидных ен-ильных комплексов палладия [(ен-ил)PdX]2 с циклопентадиенилом таллия (или натрия), с последующей атакой образовавшихся комплексов-интермедиатов (ен-ил)PdCp электрофилами (например, Ph3C+BF4-), приводящее к образованию [(C8H12)PdCp]BF4, где ен-ил=C8H12acac или C8H12OMe, X=С1 или Br [3,4];
3) взаимодействие дигалогенидных фосфиновых комплексов палладия общей формулы Pd2(PR3)2X4 с циклопентадиенилом таллия, с последующим добавлением либо олефина и солей серебра AgY [5], либо фосфина (например, PEt3), где R=Et, Ph, Bu; X=CI, Br, 1; Y=CIO4, BF4 [6];
4) взаимодействие дихлоридных комплексов палладия PdCl2L2 с солями серебра AgY и последующим добавлением циклопентадиена, где Y=PF6, NO3, L или L2 - монодентатные или бидентатные фосфор-, мышьяк- и сурьма-содержащие соединения [7, 8, 9].
Среди вышеперечисленных методов, встречаются способы синтеза катионных циклопентадиенильных комплексов палладия с фосфиновыми лигандами. Один из таких способов базируется на превращении в катионные комплексы соединений общей формулы CpPd(PR3)X, где R=Et, X=CI, Br, I, либо R=Ph, Bu, X=Br. Такие соединения получают путем взаимодействия (R3P)2Pd(μ-X)2PdX2 с циклопентадиенилом таллия в тетрагидрофуране (ТГФ) при комнатной температуре с выходом 80% [10]. В зависимости от добавляемых (для формирования катионного комплекса) реагентов можно получать различные продукты. Например, при добавлении к раствору исходного комплекса в бензоле 1,5-кратного избытка PEt3 наблюдается выпадение темно-красных кристаллов катионного комплекса [CpPd(PEt3)2]X, с выходом 75% [6]. При добавлении к раствору CpPd(PR3)Br в CH2CI2 эквимолярного количества AgY, растворенного в ацетоне, в присутствии стабилизирующего олефина или лабильного лиганда при 0°С происходит образование катионного комплекса состава [CpPd(PR3)(L)]Y, где R=Et, Bu, Ph; Y=ClO4, BF4; L=PhCN, орто-СН3(С6Н4)CN, C=O, PhSCN, CH2=CH2, СН3СН2=СН2, PhCH=CH2, пара-СН3(С6Н4)СН=СН2, пара-СН3О(С6Н4)СН=СН2, пара-С1(С6Н4)СН=СН2, пара-NO2(С6Н4)СН=СН2, пара-СН3СО(С6Н4)СН=СН2 [5].
Наиболее близким предлагаемому нами является способ синтеза, описанный в работе [7]. Двухстадийный метод синтеза подразумевает на первом этапе взаимодействие PdCl2L2 с двумя эквивалентами AgPF6 в ацетоне (Ме2СО), с образованием, по мнению авторов, бикатионного комплекса [(Me2C=0)2PdL2] [PF6]2, устойчивого в растворе ацетона (но не выделяемого). На втором этапе взаимодействие раствора, полученного после фильтрования, со свежеперегнанным циклопентадиеном (пятикратный избыток) приводит к образованию комплексов [Pd(η5-C5H5)L2]PF6, где L=PMe2Ph, PPh3, AsMe2Ph, AsPh3, SbPh3 и L2=(SS)-, (RR,SS)-, (RS)-C6H4(PMePh)2-o или (SS)-, (RR,SS)-, (RS)-C6Hi(AsMePh)2-o. Также по данному методу был осуществлен синтез комплексов [Pd(η5-C5H5)(cod)]PF6 [7] и [(η5-C5H5)Pd(PMe2Ph)2]Cl04 [8]. Кроме того, по данному методу возможен и другой способ синтеза, основанный на взаимодействии PdCl2L2 с AgNO3 в водно-метанольной смеси (1:1). После добавления к реакционной смеси циклопентадиена, раствор фильтруют от AgCl и обрабатывают избытком водного раствора NH4PF6, что приводит к осаждению кристаллических продуктов [7, 9].
Общими недостатками существующих методов синтеза являются: использование токсичных солей таллия или дорогостоящих солей серебра; дополнительная стадия отделения продукта реакции от образовавшихся галогенидов таллия и/или серебра (фильтрование или центрифугирование); необходимость проведения синтеза исходных реагентов (например, в первом методе [11]), протекающего не всегда с хорошими выходами.
Технической задачей предлагаемого изобретения является создание нового способа, позволяющего упростить синтез катионных циклопентадиенильных комплексов палладия с моно- и бидентатными фосфинами, не требующего использования циклопентадиенила таллия и солей таллия и/или серебра, содержащих слабокоординирующий анион, и исключающего дополнительную стадию отделения продуктов реакции (катионных комплексов палладия) от галогенидов таллия и/или серебра. Поставленная задача достигается благодаря способности катионных ацетилацетонатных комплексов палладия состава [(acac)Pd(L)2]BF4 и [(acac)Pd(L∧L)]BF4 вступать в реакцию непосредственно с циклопентадиеном в полярном растворителе (метанол или хлористый метилен, а также смесь метанола и хлористого метилена) в присутствии эфирата трифторида бора при комнатной температуре с образованием катионных циклопентадиенильных комплексов состава [CpPd(L)2]BF4 и [CpPd(L∧L)]BF4, где асас- ацетилацетонат; Ср - циклопентадиенил; L=PPh3, ТОМРР, Р(р-То1)3, TFP, ТТР, P(Ph)2Cy; L∧L=dppp, dppb, dpppent, dpphex и dppf.
Исходные ацетилацетонатные комплексы могут быть получены практически с количественными выходами способом лигандного замещения ацетонитрила в комплексе [(acac)Pd(MeCN)2]BF4 [12, 13]. Для синтеза комплекса-прекурсора не требуется использование солей таллия или серебра, он осуществляется в хлористом метилене при комнатной температуре путем взаимодействия коммерчески доступных реактивов: Pd(acac)2 с двумя эквивалентами BF3⋅OEt2 в присутствии ацетонитрила [14].
Хотя механизм синтеза катионных циклопентадиенилпалладиевых комплексов из ацетилацетонатных предшественников неизвестен, в ходе изучения результатов практических испытаний было предположено, что процесс происходит ступенчато, ранним этапом является реакция молекул трифторида бора с ацетилацетонатным лигандом с образованием кl-C-acac-Pd, которые затем реагируют с циклопентадиеном Эфират трифторида бора BF3⋅OEt2 используется в качестве реагента, элиминирующего анионный лиганд из координационной сферы палладия(II).
Следующие примеры иллюстрируют данное изобретение:
Пример 1. Синтез [CpPd(PPh3)2]BF4.
К раствору комплекса [(acac)Pd(PPh3)2]BF4 (0,400 г, 0,490 ммоль) в 10 мл метанола добавили 0,3 мл BF3⋅OEt2 (2,24 ммоль) и перемешивали в течение 1 часа при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,22 мл свежеперегнанного циклопентадиена (2,45 ммоль) и перемешивали в течение 8 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈5 мл и высадили добавкой 20 мл диэтилового эфира порошок фиолетового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.), выход составил 0,327 г (85,3% от теоретического). Элементный анализ: рассчитано для C41H35BF4P2Pd: С, 62.90; Н, 4.51; экспериментально: С, 63.03; Н, 4.44. ЭСИ-МС

соответствует естественному содержанию изотопов 10В и 11В, соответственно).
Пример 2. Синтез [CpPd(P(p-Tol)3)2]BF4 (в хлористом метилене).
К раствору комплекса [(acac)Pd(P(p-Tol)3)2]BF4 (0,150 г, 0,167 ммоль) в 5 мл CH2Cl2 добавили 0,23 мл 10% раствора BF3⋅OEt2 в CH2Cl2 (0,183 ммоль) и перемешивали в течение 30 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,08 мл свежеперегнанного циклопентадиена (0,832 ммоль) и перемешивали в течение 1 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈3 мл и высадили добавкой 10 мл диэтилового эфира порошок фиолетового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Полученный порошок растворили в 5 мл ацетонитрила, отфильтровали, упарили в вакууме и сушили (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,104 г (71,5% от теоретического). Элементный анализ: рассчитано для C47H47BF4P2Pd: С, 65.11; Н, 5.46; экспериментально: С, 65.22; Н, 5.58. ЭСИ-МС (режим 
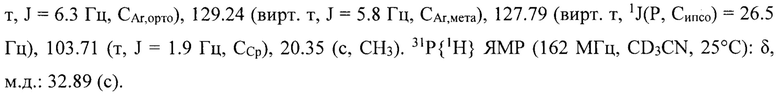
Пример 3. Синтез [CpPd(P(p-Tol)3)2]BF4 (в метаноле).
К раствору комплекса [(acac)Pd(P(p-Tol)3)2]BF4 (0,150 г, 0,167 ммоль) в 10 мл метанола добавили 0,1 мл BF3⋅OEt2 (0,84 ммоль) и перемешивали 15 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,14 мл свежеперегнанного циклопентадиена (1,67 ммоль) и перемешивали в течение 8 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈3 мл и высадили добавкой 10 мл диэтилового эфира порошок фиолетового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт. ст.) в течение 8 ч, выход составил 0,120 г (82,5% от теоретического). Элементный анализ: рассчитано для C47H47BF4P2Pd: С, 65.11; Н, 5.46; экспериментально: С, 65.25; Н, 5.53. ЭСИ-МС (режим съемки положительных ионов, MeCN): m/z 779.22 [М+]. 1Н ЯМР (60 МГц, CD3CN, 40°С): δ, м.д.: 7.27-7.09 (м, 24Н, HAr), 5.50 (т, J=2.1 Гц, 5Н, НСр), 2.36 (с, 18Н, НСн3).
Пример 4. Синтез [CpPd(TOMPP)2]BF4
К раствору комплекса [(acac)Pd(TOMPP)2]BF4 (0,338 г, 0,339 ммоль) в 10 мл метанола добавили 0,2 мл BF3⋅OEt2 (1,70 ммоль) и перемешивали 15 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,14 мл свежеперегнанного циклопентадиена (1,70 ммоль) и перемешивали в течение 2 ч при комнатной температуре. Добавкой 40-50 мл диэтилового эфира к полученному раствору высадили порошок зеленого цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт. ст.) в течение 8 ч, выход составил 0,231 г (71,0% от теоретического). Элементный анализ: рассчитано для C47H47BF4O6P2Pd: С, 58.62; Н, 4.92; экспериментально: С, 59.09; Н, 5.15. ЭСИ-МС (режим

Пример 5. Синтез [CpPd(TFP)2]BF4
К раствору комплекса [(acac)Pd(TFP)2]BF4 (0,338 г, 0,339 ммоль) в смеси растворителей 5 мл МеОН и 2 мл CH2Cl2 добавили 0,14 мл BF3⋅OEt2 (1,15 ммоль) и перемешивали 30 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,10 мл свежеперегнанного циклопентадиена (1,15 ммоль) и перемешивали в течение 2 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈3 мл и высадили добавкой 10 мл н-пентана порошок фиолетового цвета. Осадок отфильтровали и дважды промыли н-пентаном порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,123 г (74,1% от теоретического). Элементный анализ: рассчитано для C29H23BF4O6P2Pd: С, 48.02; Н, 3.21;

Пример 6. Синтез [CpPd(TTP)2]BF4
К раствору комплекса [(acac)Pd(TTP)2]BF4 (0,132 г, 0,154 ммоль) в 7 мл метанола добавили 0,08 мл свежеперегнанного циклопентадиена (0,8 ммоль) и перемешивали в течение 15 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,10 мл BF3⋅OEt2 (0,772 ммоль) и перемешивали в течение 72 ч при комнатной температуре. Образовавшийся осадок коричневого цвета отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,021 г (16,5% от теоретического). ЭСИ-МС (режим

Пример 7. Синтез [CpPd(PPh2Cy)2]BF4
К раствору комплекса [(acac)Pd(PPh2Cy)2]BF4 (0,250 г, 0,302 ммоль) в смеси растворителей 5 мл МеОН и 3 мл CH2Cl2 добавили 0,18 мл BF3⋅OEt2 (1,508 ммоль) и перемешивали 30 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,13 мл свежеперегнанного циклопентадиена (1,508 ммоль) и перемешивали в течение 4 ч при комнатной температуре. Добавили еще 0,26 мл циклопентадиена (3,016 ммоль) и перемешивали в течение 15 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈3 мл и высадили добавкой 25 мл диэтилового эфира порошок фиолетового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,183 г (76,4% от теоретического). Элементный анализ: рассчитано для C41H47BF4P2Pd: С, 61.94; Н, 5.96; экспериментально: С, 61.87; Н, 6.01.

Пример 8. Синтез [CpPd(dppf)]BF4
К раствору комплекса [(acac)Pd(dppf)]BF4 (0,300 г, 0,354 ммоль) в 10 мл метанола добавили 0,22 мл BF3⋅OEt2 (1,77 ммоль) и перемешивали 15 мин при комнатной температуре. После охлаждения до 0°С данного раствора добавили 0,15 мл свежеперегнанного циклопентадиена (1,77 ммоль) и перемешивали в течение 8 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈3 мл и высадили добавкой 10 мл диэтилового эфира порошок темно-красного цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,180 г (62,0% от теоретического). Элементный анализ: рассчитано для C39H33BF4FeP2Pd: С, 57.64; Н, 4.09; экспериментально: С, 57.05; Н, 4.27. ЭСИ-МС (режим съемки положительных ионов, MeCN): m/z 525.04 [М+]. 1Н ЯМР

Пример 9. Синтез [CpPd(dppp)]BF4
К раствору комплекса [(acac)Pd(dppp)]BF4 (0,100 г, 0,142 ммоль) в смеси растворителей 5 мл МеОН и 2 мл CH2Cl2 добавили 0,09 мл BF3⋅OEt2 (0,709 ммоль) и перемешивали 30 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,06 мл свежеперегнанного циклопентадиена (0,709 ммоль) и перемешивали в течение 2 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈3 мл и высадили добавкой 10 мл н-пентана порошок розового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,071 г (74,2% от теоретического). Элементный анализ: рассчитано для C32H31BF4P2Pd: С, 57.30; Н, 4.66; экспериментально: С, 56.95; Н, 4.78. ЭСИ-МС (режим съемки положительных ионов,

Пример 10. Синтез [CpPd(dppb)]BF4
К раствору комплекса [(acac)Pd(dppb)]BF4 (0,100 г, 0,139 ммоль) в 5 мл метанола добавили 0,09 мл BF3⋅OEt2 (0,70 ммоль) и перемешивали 30 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,06 мл свежеперегнанного циклопентадиена (0,70 ммоль) и перемешивали в течение 2 ч при комнатной температуре. Образовавшийся раствор упарили в вакууме до ≈3 мл и высадили добавкой 10 мл диэтилового эфира порошок фиолетового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,060 г (60,0% от теоретического). Элементный анализ: рассчитано для C33H33BF4P2Pd: С, 57.88; Н, 4.86; экспериментально: С, 57.36; Н, 4.96.

Пример 11. Синтез [CpPd(dppent)]BF4
К раствору комплекса [(acac)Pd(dppent)]BF4 (0,100 г, 0,136 ммоль) в 6 мл метанола добавили 0,08 мл BF3⋅OEt2 (0,68 ммоль) и перемешивали 30 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,06 мл свежеперегнанного циклопентадиена (0,70 ммоль) и перемешивали в течение 2 ч при комнатной температуре. Добавкой 30 мл диэтилового эфира к полученному раствору высадили порошок фиолетового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,045 г (48,0% от теоретического). Элементный анализ: рассчитано для C34H35BF4P2Pd: С, 58.44; Н, 5.05; экспериментально: С, 59.96; Н, 4.95. ЭСИ-МС (режим съемки положительных ионов, MeCN): m/z 611.12 [М+], полученную изотопную картину можно интерпретировать как [M1]+:[М2]2+:[М3]3+= 9.5:0.1:0.4. 1Н ЯМР (400 МГц, CD3CN, 25°С): δ, м.д.: 7.61-7.26 (м, 20Н,Hph), 5.79 (т, J=2.0 Гц, 0.3Н, изомер (димер), НСр), 5.72 (т, J=2.0 Гц, 0.7Н, изомер (тример), НСр), 5.44 (т, J=2.0 Гц, 4Н, основной изомер (мономер), НСр), 2.54 (тт, J=9.6, 4.8 Гц, 4Н, СН2), 2.30-2.10 (м, 2Н, СН2, перекрывается с примесями в CD3CN), 1.84-1.69 (м, 4Н, СН2). 13С{1Н} ЯМР (101 МГц, CD3CN, 25°С): δ, м.д.: 133.57 (вирт. т, J=23.7 Гц, CPh,ИНСО), 132.35 (вирт. т, J =5.7 Гц, СРh.ОРТО), 131.34 (с, СРh.пара), 129.03 (вирт. т, J= 5.4 Гц, СРh,МЕТА), 103.32 (т, J=2.0 Гц, основной изомер, ССр), 103.02 (с, ушир, минорный изомер, ССр), 26.74 (вирт.т, J=15.8 Гц, Р-СН2), 22.56 (т, J=5.9 Гц, СН2), 22.16 (с, СН2). 31Р{1Н} ЯМР (162 МГц, CD3CN, 25°С): δ, м.д.: 28.76 (с, 0.04Р), 28.25 (с, 0.16Р), 19.08 (с, 0.8Р).
Пример 12. Синтез {[CpPd(dpphex)]BF4}n (где n=2, 3)
К раствору комплекса {[(acac)Pd(dpphex)]BF4}n (0,227 г, 0,304 ммоль, n=2) в 10 мл метанола добавили 0,19 мл BF3⋅OEt2 (1,52 ммоль) и перемешивали 30 мин при комнатной температуре. После охлаждения до 0°С данного раствора желтого цвета добавили 0,14 мл свежеперегнанного циклопентадиена (1,52 ммоль) и перемешивали в течение 2 ч при комнатной температуре. Добавкой смеси 5 мл диэтилового эфира и 15 мл н-пентана к полученному раствору высадили порошок фиолетового цвета. Осадок отфильтровали и дважды промыли диэтиловым эфиром порциями по 10 мл. Сушили в вакууме (0,5 мм рт.ст.) в течение 8 ч, выход составил 0,178 г (82,0% от теоретического). По данным 1Н ЯМР в полученном образце детектируется аддукт с диэтиловым эфиром ([Et2O]:[Pd] ≈ 1:3). Элементный анализ: рассчитано для C35H37BF4P2Pd: С, 58.97; Н, 5.23; экспериментально: С, 59.25; Н, 5.48. ЭСИ-МС (режим съемки положительных ионов, MeCN): m/z 625.14 [М+], полученную изотопную картину можно интерпретировать как [М1]+:[М2]2+:[М3]3+=0:4:6. 1Н ЯМР (400 МГц, CD3CN, 25°С): δ, м.д.: 7.72-7.21 (м, 20Н, HРh), 5.78 (т, J=2.0 Гц, 3Н, основной изомер (тример), Нср), 5.76-5.69 (м, 1.5Н, минорный изомер (димер), Нср), 5.61 (т, J=2.0 Гц, 0.5Н, минорный изомер, НСр), 2.34-2.06 (м, 3Н, СН2), 1.99-1.46 (м, 3Н, СН2, перекрывается примесями в CD3CN), 1.41-1.21 (м, 6Н, СН2). 13С{1Н} ЯМР (101 МГц, CD3CN, 25°С): δ, м.д.: (сигналы от ипсо-С фрагмента PPh2 не детектирутся) 131.93 (вирт. т, J=5.6 Гц, CРh,орто), 130.57 (с, CPh.napa), 128.11 (с, CРh,мета), 102.18 (с, минорный изомер (димер), CCр), 101.51 (с, основной изомер (тример), CCр), 29.16 (с, СН2), 25.46 (с, СН2).
31Р{1H} ЯМР (162 МГц, CD3CN, 25°С): δ, м.д.: 28.61 (с, 0.6Р), 28.39-28.22 (м, 0.3Р), 27.11(с, 0.1Р).
Следует отметить, что промежуточное охлаждение раствора до 0°С после добавления эфирата трифторида бора и до добавления циклопентадиена не влияет на протекание последующей реакции. Охлаждение до 0 градусов Цельсия используется с целью замедлить самопроизвольную димеризацию циклопентадиена в дициклопентадиен (который не активен в данной реакции). Однако, как показали практические испытания, реакция протекает и без промежуточного охлаждения. Охлаждение раствора является скорее вспомогательным действием, нежели необходимостью. Поэтому охлаждение раствора до 0 градусов Цельсия не включено в формулу изобретения как существенный признак.
Пример 6 направлен на демонстрацию того, что даже при изменении последовательности ввода реагентов (вначале к раствору комплекса [(acac)Pd(TTP)2]BF4 в метаноле добавили свежеперегнанный циклопентадиен, и после перемешивания при комнатной температуре и последующего охлаждения до 0°С данного раствора добавили эфират трифторида бора) реакция будет протекать и приводить к образованию комплекса-продукта. Однако выход продукта в таком случае получен чуть ниже, чем в остальных примерах.
В примере 7 циклопентадиен добавлен в 2 этапа, поскольку добавление второй порции циклопентадиена приводит к увеличению его концентрации в реакционной смеси, которая снизилась в результате димеризации в дициклопентадиен (см. выше). Данное действие также не является существенным признаком изобретения, поскольку не влияет на химизм заявляемого способа, а просто направлено на некоторое повышение выхода конечного продукта.
Процентное соотношение при приготовлении смеси полярных растворителей метанола и хлористого метилена также не является существенным признаком изобретения, поскольку технический результат достигается в любом случае, в том числе и при использовании только одного из растворителей, как показано на вышеприведенных примерах.
Список литературы:
1. Maitlis P.М., Games М.L. A General Synthesis of Tetraphenylcyclobutadiene-Metal Complexes by Ligand-Transfer //Journal of the American Chemical Society. - 1963. - Vol. 85. -№. 12.-P. 1887-1888.
2. Maitlis P.M., Efraty A., Games M.L. Cyclobutadiene-Metal Complexes. IV. 1 (π-Cyclopentadienyl)(π-tetraphenylcyclobutadiene) palladium Halides and Related Complexes //Journal of the American Chemical Society. - 1965. - Vol. 87. - №. 4. - P. 719-724.
3. Johnson B. F. G., Lewis J., White D. A. Reactions of co-ordinated ligands. Part V. Reactions of triphenylmethyl tetrafluoroborate and fluoroboric acid with a variety of enyl metal complexes //Journal of the Chemical Society A: Inorganic, Physical, Theoretical. - 1970. - P. 1738-1745.
4. White D.A. Some hydroxycyclo-octenyl complexes of palladium (II) //Journal of the Chemical Society A: Inorganic, Physical, Theoretical. - 1971. - P. 145-147.
5. Kurosawa H., Majima Т., Asada N. Synthesis, structures, stabilities, and reactions of cationic olefin complexes of palladium (II) containing the. eta. 5-cyclopentadienyl ligand //Journal of the American Chemical Society. - 1980. - Vol. 102. - №. 23. - P. 6996-7003.
6. Cross R. J., Wardle R. Cyclopentadienyls of palladium and platinum //Journal of the Chemical Society A: Inorganic, Physical, Theoretical. - 1971. - P. 2000-2007.
7. Roberts N. K. et al. Cationic η5-cyclopentadienylpalladium (11) complexes. Compounds of type [Pd(η5-C5H5)L2]PF6 containing tertiary phosphines, arsines, and stibines. Crystal and molecular structure of [Pd(η5-C5H5)(SbPh3)2] PF6⋅CH2Cl2 //Journal of the Chemical Society, Dalton Transactions. - 1982. - №. 10. - P. 2093-2097.
8. Manojlović-Muir L., Cross R.J., Hoyle R.W. Structure of (η5-cyclopentadienyl) bis (dimethylphenylphosphine) palladium (II) perchlorate //Acta Crystallographica Section C: Crystal Structure Communications. - 1993. - Vol. 49. - №. 9. - P. 1603-1606.
9. Cross R.J. et al. Distortion of cyclopentadienyl rings in η5-c cyclopentadienyl-palladium complexes: Crystal structures of [Pd(C5H5)Cl (PMe2Ph)] and [Pd(C5H5)(Ph2PCH2CH2PPh2)][PF6] //Journal of organometallic chemistry. - 1994. - Vol. 468. -№.1-2.-P. 265-271.
10. Cross R.J., Wardle R. π-Cyclopentadienyls of palladium (II) and platinum (II) //Journal of Organometallic Chemistry. - 1970. - Vol. 23. - №. 1. - P. 4-6
11. King R.B. et al. Cyclopentadienyl metal carbonyls and some derivatives //Inorganic syntheses. - 1963.-Т. 7.-C. 99-115.
12. Пат. 2475492 Российская Федерация, МПК C07F15/00 C07F5/00. Способ получения катионных комплексов палладия с бидентатными фосфорорганическими лигандами / Д.С. Суслов, B.C. Ткач, М.В. Быков, М.В. Белова, О.И. Мисько; заявитель и патентообладатель Иркут. гос. ун-т.-№ 2011147269; заявл. 21.11.2011; опубл. 20.02.2013, Бюл. № 5. - [6 с.].
13. Пат. 2636741 Российская Федерация, МПК C07F 15/00 C01F 5/02 B01J 31/22. Способ получения катионных комплексов палладия с фосфиновыми лигандами /М.В. Пахомова, М.В. Быков, Д.С. Суслов, B.C. Ткач,; заявитель и патентообладатель Иркут. гос. ун-т.-№ 2017101937; заявл. 20.01.2017; опубл. 28.11.2017, Бюл. № 34.-[6 с.].
14. Пат. 2456295 Российская Федерация, МПК C07F 15/00 C07F 5/02 Способ получения (ацетилацетонато-к2-O,O')(бис-ацетонитрил)палладия тетрафторбората / Д.С.Суслов, B.C. Ткач, М.В. Быков, М.В. Белова; заявитель и патентообладатель Иркут. гос. ун-т.- № 2011112364; заявл. 31.03.2011; опубл. 20.07.2012, Бюл. № 20. - [4 с.].
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ (АЦЕТИЛАЦЕТОНАТО)(ЦИКЛООКТАДИЕН)ПАЛЛАДИЯ ТЕТРАФТОРБОРАТА | 2012 |
|
RU2508293C1 |
| СПОСОБ ПОЛУЧЕНИЯ (АЦЕТИЛАЦЕТОНАТО-κ-О,О')(БИС-АЦЕТОНИТРИЛ)ПАЛЛАДИЯ ТЕТРАФТОРБОРАТА | 2011 |
|
RU2456295C1 |
| СПОСОБ ПОЛУЧЕНИЯ РАЗНОЛИГАНДНЫХ КАТИОННЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ | 2009 |
|
RU2423373C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТИОННЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ С ДИИМИНОВЫМИ ЛИГАНДАМИ | 2011 |
|
RU2475491C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТИОННЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ С БИДЕНТАТНЫМИ ФОСФОРОРГАНИЧЕСКИМИ ЛИГАНДАМИ | 2011 |
|
RU2475492C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТИОННЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ С ФОСФИНОВЫМИ ЛИГАНДАМИ | 2017 |
|
RU2636741C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТИОННЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ С ДИИМИНОВЫМИ ЛИГАНДАМИ | 2014 |
|
RU2556224C1 |
| СПОСОБ АДДИТИВНОЙ ПОЛИМЕРИЗАЦИИ НОРБОРНЕНА И ЕГО ПРОИЗВОДНЫХ | 2015 |
|
RU2626745C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТИОННЫХ КОМПЛЕКСОВ ПАЛЛАДИЯ | 2011 |
|
RU2466134C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,3-ДИФЕНИЛБУТЕНА-1 | 2012 |
|
RU2487859C1 |
Изобретение относится к новому способу получения катионных циклопентадиенильных комплексов палладия с моно- и бидентатными фосфиновыми лигандами составов [CpPd(L)2]BF4 и [CpPd(L^L)]BF4. Способ осуществляют путем взаимодействия комплексов палладия(II) с элиминирующим анионный лиганд из координационной сферы палладия(II) реагентом в присутствии циклопентадиена при комнатной температуре в среде органического растворителя. В качестве исходных комплексов палладия(II) используются ацетилацетонатные комплексы [(acac)Pd(L)2]BF4 и [(acac)Pd(L^L)]BF4, а в качестве элиминирующего анионный лиганд из координационной сферы палладия(II) реагента используется эфират трифторида бора. Процесс проводят в присутствии циклопентадиена в среде дихлорметана, или метанола, или смеси дихлорметана и метанола. Указанные обозначения имеют следующие значения: асас – ацетилацетонат; Ср – циклопентадиенил; L - монодентатные фосфиновые лиганды, такие как трифенилфосфин (PPh3), трис(о-метоксифенил)фосфин (ТОМРР), трис(пара-толил)фосфин (Р(p-То1)3), трис(2-фурил)фосфин (TFP), трис(2-тиенил)фосфин (ТТР), дифенил(циклогексил)фосфин (P(Ph)2Cy); L^L - бидентатные фосфорорганические лиганды, такие как 1,3-бис(дифенилфосфино)пропан (dppp), 1,4-бис(дифенилфосфино)бутан (dppb), 1,5-бис(дифенилфосфино)пентан (dpppent), 1,6-бис(дифенилфосфино)гексан (dpphex) и 1,1’-бис(дифенилфосфино)ферроцен (dppf). Предложенный способ позволяет упростить синтез катионных циклопентадиенильных комплексов палладия с моно- и бидентатными фосфинами, не требует использования циклопентадиенила таллия и солей таллия и/или серебра и исключает дополнительную стадию отделения продуктов реакции от галогенидов таллия и/или серебра. 12 пр.
Способ синтеза катионных циклопентадиенильных комплексов палладия с моно- и бидентатными фосфиновыми лигандами составов [CpPd(L)2]BF4 и [CpPd(L∧L)]BF4, путем взаимодействия комплексов палладия(II) с элиминирующим анионный лиганд из координационной сферы палладия(II) реагентом в присутствии циклопентадиена при комнатной температуре в среде органического растворителя, отличающийся тем, что в качестве исходных комплексов палладия(II) используются ацетилацетонатные комплексы [(acac)Pd(L)2]BF4 и [(acac)Pd(L∧L)]BF4, а в качестве элиминирующего анионный лиганд из координационной сферы палладия(II) реагента используется эфират трифторида бора, процесс проводят в присутствии циклопентадиена в среде дихлорметана, или метанола, или смеси дихлорметана и метанола, где асас – ацетилацетонат; Ср – циклопентадиенил; L - монодентатные фосфиновые лиганды, такие как трифенилфосфин (PPh3), трис(о-метоксифенил)фосфин (ТОМРР), трис(пара-толил)фосфин (Р(p-То1)3), трис(2-фурил)фосфин (TFP), трис(2-тиенил)фосфин (ТТР), дифенил(циклогексил)фосфин (P(Ph)2Cy); L∧L - бидентатные фосфорорганические лиганды, такие как 1,3-бис(дифенилфосфино)пропан (dppp), 1,4-бис(дифенилфосфино)бутан (dppb), 1,5-бис(дифенилфосфино)пентан (dpppent), 1,6-бис(дифенилфосфино)гексан (dpphex) и 1,1'-бис(дифенилфосфино)ферроцен (dppf).
| ROBERTS N.K | |||
| et al., Cationic η5-cyclopentadienylpalladium (II) complexes | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Chem | |||
| Soc., Dalton Trans., 1982, no | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Передвижная кровать-ванна | 1925 |
|
SU2093A1 |
| KUROSAWA H | |||
| et al., Synthesis, Structures, Stabilities, | |||

