Настоящее изобретение относится к диспергируемым таблеткам, например, включающим 4-[3,5-бис(2-гидроксифенил)-[1,2,4]триазол-1-ил] бензойную кислоту или ее фармацевтически приемлемую соль, которая в дальнейшем в контексте настоящего изобретения будет называться соединением I.
Соединение I при пероральном введении является активным хелатором железа, который показан при лечении перегрузки железом при анемиях, вызванных переливанием крови, в частности при большой талассемии, промежуточной талассемии и серповидно-клеточной анемии, для снижения связанных с избытком железа заболеваемости и смертности. Соединение I может применяться также при лечении гемохроматоза.
Клинические талассемии (большая или промежуточная) являются наследственными заболеваниями, характеризуемыми недостаточным продуцированном гемоглобина, что приводит к снижению образования и увеличению разрушения эритроцитов.
Серповидно-клеточная анемия вызвана мутацией гена гемаглобина-бета, приводящей к образованию аномального гемоглобина S. Нормальные эритроциты погибают после 120 дней, в то время как серповидные клетки (эритроциты с гемоглобином S) разрушаются быстрее (от 10 до 20 дней), вызывая анемию. Это анемия и определяет общеизвестное название этого заболевания: серповидно-клеточная анемия.
Гемохроматоз, наиболее часто встречающаяся форма заболевания, связанного с перегрузкой железом, является наследственным заболеванием, вызывающим излишнее усвоение и накопление железа в организме. Железо накапливается в органах и повреждает их. В отсутствие лечения такое заболевание выводит эти органы из строя.
Пациенты с серповидно-клеточной анемией или талассемией, которым делают многократное переливание крови, и пациенты с гемохроматозом нуждаются в специальной терапии для выведения железа из организма, называемой хелаторной терапией.
Соединение I имеет следующую формулу:
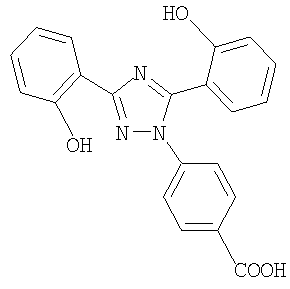
Соединение I в форме свободной кислоты, ее солей и их кристаллических формах описано в международной заявке WO 97/49395, которая, таким образом, включена как ссылка (дата публикации 31 декабря 1997 года).
Обычная суточная доза соединения I, назначаемая для лечения талассемии, высока, например, от 5 до 40 мг/кг массы тела в день у взрослых и детей. У детей суточная доза предпочтительно составляет от 5 до 30 мг/кг массы тела в день. Из-за большой дозировки размеры таблетки делают невозможным приготовление обычных таблеток. Таким образом, существует необходимость в создании пероральной лекарственной формы, которая была бы удобна при введении взрослым и детям и обеспечивала бы фармакологически эффективную суточную дозу соединения I.
Авторы изобретения неожиданно обнаружили, что получение соединения I в форме диспергируемых таблеток позволяет создать пероральную лекарственную форму с очень высокой лекарственной нагрузкой, которая удобна при введении, например детям и пожилым людям, и стабильна. Под "диспергируемой таблеткой" в контексте настоящего изобретения понимается таблетка, которая перед приемом распадается в водной фазе, например в воде.
Таким образом, в настоящем изобретении представлена диспергируемая таблетка с высокой лекарственной нагрузкой, включающая соединение I в качестве действующего вещества, причем действующее вещество присутсвует в количестве от примерно 5 до 40 мас.%, например, в количестве по меньшей мере примерно 10, 15, 20, или 25%, предпочтительно более 25 мас.% в пересчете на общую массу диспергируемой таблетки. В частности, содержание соединения I можно варьировать от 25 до 40%, например от 28 до 32 мас.% в пересчете на общую массу диспергируемой таблетки.
Настоящее изобретение относится к диспергируемой таблетке, включающей хелирующее железо, фармакологически эффективное количество соединения I или его фармацевтически приемлемой соли в количестве от 5% до 40 мас.% в пересчете на общую массу таблетки.
В одном из вариантов осуществления изобретения представлена диспергируемая таблетка, включающая соединение I или его фармацевтически приемлемую соль в количестве от 5% до 40 мас.% в пересчете на общую массу таблетки.
Соединение I может находиться в форме свободной кислоты или ее фармацевтически приемлемых солей, предпочтительно в форме свободной кислоты. В контексте данного изобретения понятие "действующая часть" относится к соединению I в форме свободной кислоты, понятие соединения I включает как соединение I в форме свободной кислоты или одной из ее фармацевтически приемлемых солей, так и любые ее кристаллические формы, включая гидраты и сольваты, если не указано иное, там, где это уместно и целесообразно.
В настоящем изобретении представлены также диспергируемые таблетки, включающие:
(а) Соединение I или его фармацевтически приемлемую соль, и
(б) по крайней мере, один из фармацевтически приемлемых эксципиентов, подходящий для приготовления диспергируемых таблеток, где количество соединения I или его фармацевтически приемлемой соли, рассчитанное как процентное отношение массы содержащейся действующей части к общей массе диспергируемой таблетки, составляет от 5 до 40 мас.% например, не менее чем примерно 10, 15, 20, или 25%, предпочтительно более 25 мас.%, в пересчете на общую массу диспергируемой таблетки. В частности, количество соединения I как действующего вещества может варьировать от 25 до 40%, например от 28 до 32 мас.% в пересчете на общую массу диспергируемой таблетки.
В предпочтительном варианте осуществления настоящего изобретения в настоящем изобретении представлена диспергируемая таблетка, в которой соединение I находится в форме свободной кислоты (соединение I в форме свободной кислоты).
В наиболее предпочтительном варианте осуществления настоящего изобретения, соединение I в форме свободной кислоты находится в кристаллической форме.
В указанной таблетке может быть представлен один или несколько фармацевтически приемлемых эксципиентов, например общепринятые эксципиенты, например, (1.1), по крайней мере, один наполнитель, например лактоза, этилцеллюлоза, микрокристаллическая целлюлоза, (1.2) по крайней мере, один разрыхляющий агент, например поливинилпирролидинон с поперечными связями, например Crospovidone®, (1.3) по крайней мере, одно связующее вещество, например поливинилпиридон, гидроксипропилметилцеллюлоза, (1.4), по крайней мере одно ПАВ, например лаурилсульфат натрия, (1.5) по крайней мере, одно антиадгезивное вещество, например коллоидный оксид кремния, (1.6) по крайней мере, одно смазывающее вещество, например стеарат магния.
Упомянутые в настоящем изобретении, а также другие фармацевтически приемлемые экципиенты и методики широко освещены в специальной литературе, имеющей отношение к предмету настоящего изобретения, в особенности см. Handbook of Pharmaceutical Excipients, третье Изадние, под ред. Arthur H. Kibbe, American Pharmaceutical Association, Washington, USA и Pharmaceutical Press, London; и Lexikon der Hilfsstoffe für Pharmazie, Kosmetik and angrenzende Gebiete под ред. Н.Р. Fiedler, 4-ое издание, Edito Cantor, Aulendorf и более ранние издания, включенные настоящей ссылкой.
Наполнителем (1.1) в соответствии с настоящим изобретением является лактоза, особенно моногидрат лактозы, предпочтительно моногидрат лактозы (200 меш) и сухая аэрозоль лактозы, микрокристаллическая целлюлоза, особенно РН 102, РН 101.
Подходящие разрыхлители (1.2) в соответствии с настоящим изобретением представляют собой кукурузный крахмал, СМС-Са, CMC-Na, микрокристаллическую целлюлозу, поливинилпирролидинон с поперечными связями, известный под коммерческими названиями Crospovidone®, Polyplasdone® и про изводимый компаниями ISP, или Kollidon® XL, альгиновую кислоту, альгинат натрия и гуаровую камедь, но не ограничиваются ими. Предпочтительно используется поливинилпирролидинон с поперечными связями, например Crospovidone®.
Связующие вещества (1.3) представляют собой, но не ограничены, крахмал, например картофельный, пшеничный или кукурузный, микрокристаллическую целлюлозу, например такие продукты, как Avicel, Filtrak, Heweten или Pharmacel; гидроксиропилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилметилцеллюлозу, например гидроксипропилметилцеллюлозу - Туре 2910 USP, гипромеллозу и поливинилпирролидон, например Povidone® K30 производтсва BASF. Предпочтительно использование поливинилпирролидона, наиболее предпочтительно использование PVP K.30.
Подходящими ПАВ (1.4) согласно изобретению являются: лаурилсульфат натрия, бетаин, соли четвертичного аммония, полисорбаты, сложные эфиры сорбита и полоксамер. Предпочтительным ПАВ является лаурилсульфат натрия.
В качестве антиадгезивных веществ (1.5), можно использовать одно или несколько из перечисленных ниже веществ: кремний; коллоидный кремний, например безводный коллоидный силикагель, например Aerosil® 200, трисиликат магния, порошкообразную целлюлозку, крахмал и тальк. Предпочтительно используется коллоидный диоксид кремния.
В качестве смазывающих веществ (1.6) могут использоваться одно или более из перечисленных ниже веществ: стеарат магния, алюминия или кальция; PEG 4000-8000, тальк, бензоат натрия, сложные моноэфиры жирных кислот и глицерина, например моностеарат глицерина например, производства Danisco, UK), дибегенат глицерина (например, Compritol ATO888™, Gattefossé, Франция), пальмитиново-стеариновый сложный эфир глицерина (например, Precirol™, Gattefossé, Франция полиоксиэтиленгликоль (PEG, BASF), гидрогенизированное хлопковое масло (Lubitrab, Edward Mendell Co Inc), касторовое масло (Cutina HR, Henkel). Предпочтительно использовать стеарат магния.
Один или более из этих фармацевтически приемлемых эксципиентов может быть выбран и применен в зависимости от конкретных желаемых свойств таблетки с помощью обычных экспериментальных процедур.
Согласно настоящему изобретению количество наполнителя (1.1) можно варьировать в пределах от примерно 35 до 55%, в особенности от 40 до 50 мас.% в пересчете на общую массу диспергируемой таблетки.
Количество разрыхлителя (1.2) можно варьировать в пределах от 5 до 40%, например от 10 до 35 мас.% в пересчете на общую массу диспергируемой таблетки.
Количество связующего вещества (1.3) можно варьировать от 1 до 10%, предпочтительно от 1,5 до 5 мас.% в пересчете на общую массу диспергируемой таблетки.
Количество ПАВ (1.4) может варьировать от 0,1 до 2%, предпочтительно от 0,2 до 1%.
Количество антиадгезивного вещества (1.5) может варьировать в пределах от 0,1 до 5%, предпочтительно от 0,1 до 2,5 мас.% в пересчете на общую массу диспергируемой таблетки.
Количество смазывающего вещества (1.6) может быть ниже 1 мас.% в пересчете на общую массу диспергируемой таблетки, предпочтительно менее 0,4% и еще более предпочтительно количество смазывающего вещества находится в пределах от 0,01 до 0,4%. Крайне предпочтительно количество смазывающего вещества более 0,02 и менее 0,4 мас.% в пересчете на общую массу диспергируемой таблетки.
Желательно, чтобы каждый из данных эксципиентов выполнял более одной функции, например наполнителя, разрыхлителя, связующего вещества, антиадгезивного вещества и/или смазывающего вещества.
В одном из вариантов осуществления изобретения смазывающее вещество составляет менее 1 мас.% в пересчете на общую массу диспергируемой таблетки, предпочтительно менее 0,4%.
Изобретение относится также и к диспергируемой таблетке, в которой смазывающее вещество представляет собой стеарат магния.
В предпочтительном варианте осуществления изобретения диспергируемая таблетка включает следующие фармацевтически приемлемые эксципиенты: один или более наполнитель, в целом составляющий примерно от 40 до 50 мас.% в пересчете на общую массу диспергируемой таблетки, одно или более связующее вещество, в целом составляющее примерно от 1,5 до 5 мас.% в пересчете на общую массу диспергируемой таблетки, один или более разрыхлитель, в целом составляющий примерно от 10 до 35 мас.%, в пересчете на общую массу диспергируемой таблетки, одно или более антиадгезивное вещество, в целом составляющее примерно от 0,1 до 0,5 мас.% в пересчете на общую массу диспергируемой таблетки, и/или одно или более смазывающее вещество, которое в целом составляет примерно от 0,01 до 0,4 мас.% в пересчете на общую массу диспергируемой таблетки.
Абсолютные количества каждого фармацевтически приемлемого эксципиента и их количества относительно других фармацевтически приемлемых эксципиентов также зависят от конкретных желаемых свойств таблетки и могут быть выбраны с помощью обычных экспериментальных процедур.
Авторы настоящего изобретения столкнулись с рядом трудностей при производстве диспергируемых таблеток, включающих соединение I, которые могут быть вызваны низкой плотностью действующего вещества и его электростатическими характеристиками, которые могут привести к плохой текучести и тенденции к слипанию.
В соответствии с настоящим изобретением было обнаружено, что фармацевтически приемлемые пероральные твердые лекарственные формы в виде диспергируемых таблеток, удобных для приема пациентом и диспергирующих в течение 5 и менее минут, предпочтительно 3 и менее минут, могут быть получены путем изготовления таблеток способами прессования. Более конкретно представленные в изобретении диспергируемые таблетки могут быть приготовлены методом гранулирования, предпочтительно влажного гранулирования, с последующим прессованием, предпочтительно при смазывании с помощью опрыскивания.
В общем влажное гранулирование может быть использовано для улучшения текучести и тенденции к слипанию, однако влажное гранулирование нежелательно в тех случаях, когда фармацевтическая композиция должна представлять собой диспергируемую таблетку. Действительно, влажное гранулирование увеличивает когезию частиц действующего вещества и увеличивает время дезинтеграции таблетки, что не соответствует правилам режима пациентов или Европейской Фармакопее, которая требует, чтобы время дезинтеграции таблетки не превышало 3 минут. Более того, было обнаружено, что после влажной грануляции действующее вещество остается липким и его трудно обрабатывать в машине для производства таблеток. Авторы настоящего изобретения неожиданно обнаружили, что проблема слипания может быть разрешена путем добавления смазывающего вещества в композицию таблетки, что не вызывает увеличения времени дезинтеграции, например, свыше приемлемых пределов, например свыше 5 минут.
Время дезинтеграции диспергируемых таблеток, представленных в настоящем изобретении, например, в водной среде, например в воде, составляет 5 минут или менее 5 минут. Представленные в настоящем изобретении диспергируемые таблетки несмотря на высокую лекарственную нагрузку являются диспергируемыми, например, в водной среде, например в воде, в течение менее чем 5 минут, предпочтительно менее чем 3 минут, и, таким образом, удобны для приема, например, детьми или пожилыми людьми. Это приводит к лучшему соблюдению пациентами режима лечения.
В другом варианте осуществления изобретения представлены диспергируемые таблетки, содержащие в качестве действующего вещества от 100 мг до 800 мг соединения I, например от 100 мг до 600 мг. Наиболее предпочтительно представленные в изобретении диспергируемые таблетки являются диспергируемыми таблетками, содержащими в качестве действующего вещества 125 мг, 250 мг или 500 мг соединения I.
Таким образом, в настоящем изобретении представлены диспергируемые таблетки, например диспергируемые таблетки, содержащие соединения I в количестве, равном 125 мг, 250 мг или 500 мг соединения I в форме свободной кислоты. Наиболее предпочтительно соединение I в форме свободной кислоты в соответствии с изобретением находится в кристаллической форме, особенно предпочтительно в кристаллической форме, получение которой описано в примере 5 международной заявки WO 97/49395, которая, таким образом, включена в виде ссылки.
В соответствии с настоящим изобретением способ получения диспергируемых таблеток состоит из гранулирования внутренней фазы, ее смешивания (вместе) с одним или более фармацевтически приемлемыми эксципиентами и прессования полученной смеси в условиях смазывания путем опрыскивания.
Внутренняя фаза содержит соединение I. Предпочтительно внутренняя фаза содержит соединение I и один или более фармацевтически приемлемых эксципиентов. Предпочтительно фармацевтически приемлемые эксципиенты представляют собой один или более наполнителей, один или более разрыхлителей, одно или более связующих веществ и одно или более ПАВ. Предпочтительно количество одного или более наполнителей во внутренней фазе находится в пределах от примерно 5 до 35 мас.% в пересчете на общую массу диспергируемой таблетки, более предпочтительно от 10 до 30%, еще более предпочтительно от 15 до 25%. Наполнитель в соответствии с настоящим изобретением представляет собой предпочтительно моногидрат лактозы. Предпочтительным разрыхлителем является Crospovidone XL. Количество одного или более представленного во внутренней фазе разрыхлителя предпочтительно находится в пределах от 5 до 30 мас.%, более предпочтительно от 7 до 25 мас.% в пересчете на общую массу диспергируемой таблетки. Соединение I, один или более наполнитель и один или более разрыхлитель смешивают вместе со смачивающим раствором, включающим одно или более ПАВ, воду и одно или более связующее вещество. Предпочтительным связующим веществом является PVP K.30. Смесь обрабатывают для грануляции, например, с использованием высокоскоростного "влажного" гранулятора для создания влажных гранул. Влажные гранулы могут быть затем высушены, например, с использованием сушильного аппарата с кипящим слоем, и откалиброваны, например, с использованием виброгранулятора. Внешняя фаза состоит из одного и более фармацевтически приемлемого эксципиента, ее смешивают с внутренней фазой с использованием, например, смесителя свободного падения. Предпочтительно прибавляют один или более наполнителей и одно или более антиадгезивное вещество. Наиболее предпочтительно в качестве наполнителей добавляют микрокристаллическую целлюлозу и лактозу. Еще более предпочтительно микрокристаллическую целлюлозу добавляют в количестве, находящемся в диапазоне от 5 до 20 мас.% в пересчете на общую массу диспергируемой таблетки, и лактозу добавляют в количестве, находящемся в диапазоне от 5 до 20 мас.% в пересчете на общую массу диспергируемой таблетки. В соответствии с изобретением внешняя фаза также может содержать одно или более антиадгезивных веществ, наиболее предпочтительно им является диоксид кремния. В предпочтительном варианте осуществления изобретения количество антиадгезивного вещества во внешней фазе находится в пределах примерно от 0,1 до 5%, предпочтительно от 0,1 до 2,5%, наиболее предпочтительно от 0,1 до 0,5 мас.% в пересчете на общую массу диспергируемой таблетки.
В одном из вариантов осуществления изобретения одно или более смазывающих веществ вместо включения в смесь внутренней и внешней фазы осаждают слоем на пуансоне машины для производства таблеток перед началом прессования. В соответствии с изобретением одно или более смазывающих веществ распыляют на соприкасающиеся с материалом поверхности прессовочных инструментов, например, на пуансоны и матрицы машины для производства таблеток, перед началом прессования.
В одном из вариантов осуществления изобретения процесс изготовления диспергируемых таблеток включает
(а) формирование внутренней фазы, включающее
I) смешивание соединения I вместе с фармацевтически приемлемыми эксципиентами,
II) влажное гранулирование смеси, полученной на стадии (I);
б) формирование внешней фазы, включающее
III) добавление остальных фармацевтически приемлемых эксципиентов к внутренней фазе, полученной на стадии (II) и смешивание;
в) формирование диспергируемой таблетки путем:
IV) прессования смеси, полученной на стадии (III) в условиях смазывания путем распылениея.
В еще одном варианте осуществления изобретения представлен процесс, включающий:
I) смешивание соединения I и фармацевтически приемлемых эксципиентов, например одного или более наполнителей, например лактозы, и одного или более разрыхлителей, например Crospovidone XL, в высокоскоростном смесителе;
II) добавление раствора одного или более ПАВ и одного или более связующих веществ, смачивание и размешивание смеси, например, в высокоскоростном смесителе, влажное гранулирование с использованием, например, вращающейся лопатки, сушку, например, в сушильном аппарате с кипящим слоем, и калибровку, например, в виброгрануляторе, и
III) добавление фармацевтически приемлемых эксципиентов, например, просеянных эксципиентов, таких как один или более наполнитель, например микрокристаллическая целлюлоза или лактоза, одно или более антиадгезивное вещество, например коллоидный диоксид кремния, и смешивание, например, в смесителе свободного падения;
IV) изготовление таблеток из смеси, полученной на стадии III), путем прессования, например, на обычном прессе для таблеток, предпочтительно на ротационной машине, и опрыскивания смазывающим веществом поверхностей инструментов пресса, контактирующих с материалами.
Методики, которые могут быть использованы в связи с настоящим изобретением, представляют собой общепринятые способы или способы, известные в данной области техники, а также методы, основанные на подобных способах, описанные, например, в L.Lachman et al. The Theory and Practice of Industrial Pharmacy, 3-е издание, 1986, H.Sucker et al., Pharmazeutische Technologie, Thieme, 1991, Hagers Handbuch der pharmazeutischen Praxis, 4-e издание (Springer Verlag, 1971) и Remington′s Pharmaceutical Sciences, 13-e издание, (Mack PubL, Co., 1970) или в более поздних изданиях.
Под "внутренней фазой" в контексте настоящего изобретения следует понимать фазу гранулята, (стадии I и II), включающая действующее вещество (Соединение I) и один или более фармацевтически приемлемых эксципиентов.
Под "внешней фазой" в контексте настоящего изобретения понимается один или более фармацевтически приемлемых эксципиентов, добавленный к внутренней фазе (гранулятам) (стадия III).
Под "общей массой диспергируемой таблетки" понимается масса таблетки, состоящей из внутренней и внешней фазы.
Физическая и химическая стабильность может быть проверена общепринятыми способами, например может быть измерена растворимость, хрупкость, время дезинтеграции диспергируемых таблеток, анализ на продукты дезинтеграции соединения I, внешний вид и/или вид под микроскопом, например, после хранения при комнатной температуре, т.е. 25°С, и/или хранения при 40°С.
Диспергируемые таблетки могут иметь различную форму, например круглую, овальную, продолговатую, цилиндрическую, и любую другую подходящую форму. В одном из вариантов осуществления изобретения диспергируемые таблетки в соответствии с настоящим изобретением содержат небольшое количество стеарата магния, например примерно от 0,01 до 0,4 мас.%, в пересчете на общую массу диспергируемой таблетки в зависимости от количества содержащегося в ней соединения I, таким образом, достигается время дезинтеграции, соответствующее Спецификациям Европейской Фармакопеи.
В предпочтительном варианте осуществления изобретения таблетки, полученные описанным выше способом прессования, имеют круглую или овальную форму. Края таблеток могут быть скошены или закруглены. Наиболее предпочтительно диспергируемые таблетки имеют круглую форму со скошенными краями. Диспергируемые таблетки могут иметь насечки, тиснение или гравировку.
Диспергируемые таблетки в соответствии с изобретением предпочтительно круглые, плоские, со скошенными краями. 125-Миллиграммовые диспергируемые таблетки имеют диаметр от 10 до 20 мм, наиболее предпочтительно от 10 до 15 мм. Предпочтительный диаметр 125-миллиграммовых диспергируемых таблеток составляет 12 мм. Их толщина находится в пределах от 2,5 до 4,5 мм, предпочтительно от 3,2 до 3,9 мм. 250-Миллиграммовые диспергируемые таблетки имеют диаметр от 12 до 20 мм, наиболее предпочтительно от 14 до 18 мм, их наиболее предпочтительный диаметр составляет 15 мм. Их толщина находится в пределах от 3,5 до 5,5 мм, наиболее предпочтительно от 4 до 5 мм. 500-Миллиграммовые диспергируемые таблетки имеют диаметр от 15 до 30 мм, предпочтительно от 15 до 25 мм, наиболее предпочтительный диаметр составляет 20 мм. Их толщина находится в пределах от 4,5 до 6,5 мм, предпочтительно от 5 до 6 мм.
Представленные в настоящем изобретении диспергируемые таблетки, включающие в качестве действующего вещества примерно 125 мг соединения I, могут иметь твердость от примерно 50 до 125 N, предпочтительно от 60 до 100 N. Представленные в настоящем изобретении диспергируемые таблетки, включающие в качестве действующего вещества примерно 250 мг соединения I, могут иметь твердость от примерно 70 до 150 N, предпочтительно от 90 до 130 N. Представленные в настоящем изобретении диспергируемые таблетки, включающие в качестве действующего вещества примерно 500 мг соединения I, могут иметь твердость от примерно 80 до 190 N, предпочтительно от 110 до 160 N.
Предпочтительное время дезинтеграции, измеренное с помощью аппарата для измерения времени дезинтеграции, составляет не более 5 минут, наиболее предпочтительно менее 3 минут.
Под "временем дезинтеграции" в контексте настоящего изобретения понимается время, необходимое для того, чтобы диспергируемая таблетка распалась в воде при комнатной температуре в устройстве для измерения времени дезинтеграции.
Диспергируемая таблетка, представленная в настоящем изобретении, является диспергируемой в водной фазе, предпочтительно в воде.
Диспергируемые таблетки в соответствии с настоящим изобретением могут быть окрашены и/или помечены так, чтобы придать им уникальный внешний вид и сделать их легко узнаваемыми. Использование красителей может служить для улучшения внешнего вида и облегчения идентификации диспергируемых таблеток. Красители, пригодные для использования в фармакологии, обычно включают каротиноиды, оксиды железа или хлорофилл. Диспергируемые таблетки в соответствии с настоящим изобретением могут быть маркированы с использованием отпечатанных кодов.
Диспергируемые таблетки в соответствии с настоящим изобретением применяются при лечении перегрузки железом при анемиях, обусловленных переливаниями крови, в частности большой талассемии, промежуточной талассемии и серповидно-клеточной анемии, и при лечении гемохроматоза.
Активность и характеристики диспергируемых таблеток в соответствии с настоящим изобретением могут быть проверены в стандартных клинических испытаниях и/или испытаниях на животных.
Диспергируемые таблетки в соответствии с настоящим изобретением стабильны как в процесс приготовления, так и при хранении, например, в течение 2 или даже 3 лет в обычной упаковке, например в запечатанных алюминиевой фольгой блистерах или блистерах из триплекса. В соответствии с результатами стандартных тестов менее чем примерно 5%, например 2 или 3% соединения I как действующего вещества, может разлагаться в течение этого времени. Например, во флаконах, заполненных HDPE, за один год разложилось менее 1% соединения I как действующего вещества.
В зависимости от возраста, индивидуального состояния, и клинической картины эффективная суточная доза для пациентов с массой тела 70 кг составляет от 350 до 2800 мг соединения I.
Настоящее изобретение также относится к лекарственной упаковке, включающей диспергируемые таблетки в соответствии с настоящим изобретением, и печатную инструкцию, указывающую, что одна или более диспергируемые таблетки соединения I могут быть введены перорально.
Приведенные ниже примеры иллюстрируют, но никоим образом не ограничивают настоящее изобретение.
Пример 1: Состав диспергируемых таблеток (диспергируемые таблетки 125 мг, 250 мг и 500 мг), время дезинтеграции которых свыше 3 минут.
Пример 2: Состав диспергируемых таблеток (диспергируемые таблетки 125 мг, 250 мг и 500 мг), время дезинтеграции которых свыше 3 минут.
Диспергируемые таблетки, содержащие Соединение I в форме свободной кислоты, в соответствии с изобретением получают способом формирования внутренней фазы путем влажного гранулирования смеси ингредиентов фазы I и фазы II, фаза III образует внешнюю фазу, а смазывающее вещество (фаза IV) распыляется непосредственно на пуасоны машины для приготовления таблеток. *0,1 мас.% стеарата магния эквивалентно 1000 ppm.
Пример 3: Свойства диспергируемой таблетки (125 мг) по Примеру 2
20/20 в пределах ±10%
RSD ≤6,0%
отдельные ≤0,5%
общие 2,0%
Пример 4: Свойства диспергируемой таблетки (250 мг) по Примеру 2
Пример 5: Свойства диспергируемой таблетки (500 мг) по Примеру 2
20/20 в пределах ±10%
Пример 6: Анализ на стеарат магния
Л.Ф.: лабораторная фаза. Пилот.: пилотная фаза (х 2 размера объема лабораторной фазы), ПРФ: (партии промышленного размера), П.П.: предварительная проба, 0.1 мас.% стеарата магния эквивалентно 1000 ppm.
RSD: относительное стандартное отклонение
Пример 7: Свойства диспергируемой таблетки (250 мг) по Примеру 2
20/20 в пределах ±10%
RSD ≤6,0%
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ ПРЕПАРАТИВНАЯ ФОРМА ЦЕФАКЛОРА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2150276C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АЛПЕЛИСИБ | 2015 |
|
RU2690685C2 |
| ТАБЛЕТКА С ВЫСОКИМ СОДЕРЖАНИЕМ ЛЕКАРСТВЕННОГО ПРЕПАРАТА | 2009 |
|
RU2405540C1 |
| ТАБЛЕТКА С ВЫСОКИМ СОДЕРЖАНИЕМ ЛЕКАРСТВЕННОГО ПРЕПАРАТА | 2003 |
|
RU2363450C2 |
| ГАЛЕНОВЫ СОСТАВЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2005 |
|
RU2384328C2 |
| НОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2779429C2 |
| ГАЛЕНОВЫ СОСТАВЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2009 |
|
RU2438661C2 |
| Фармацевтическая композиция N-[5-[2-(3,5-диметоксифенил)этил]-2H-пиразол-3-ил]-4-[(3R,5S)-3,5-диметилпиперазин-1-ил]бензамида | 2013 |
|
RU2674978C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ МИКОФЕНОЛЬНУЮ КИСЛОТУ ИЛИ ЕЕ СОЛЬ (МИКОФЕНОЛЯТ) | 2007 |
|
RU2381026C2 |
| ТВЕРДАЯ ПЕРОРАЛЬНАЯ ЛЕКАРСТВЕННАЯ ФОРМА И СПОСОБ ЛЕЧЕНИЯ | 2011 |
|
RU2480210C1 |
Изобретение относится к области медицины и фармакологии касается диспергируемой таблетки, включающей соединений формулы (1) в количестве 5-40 мас.% и по меньшей мере один разрыхлитель в количестве 10-35 мас.%, и способа ее получения Таблетки обладают улучшенным профилем растворения. 2 н. и 12 з.п. ф-лы, 7 табл.
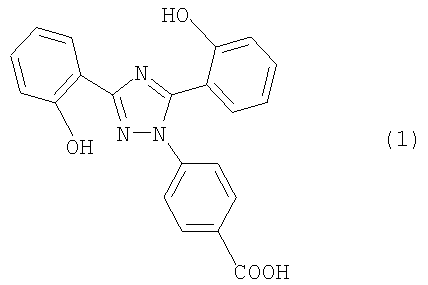
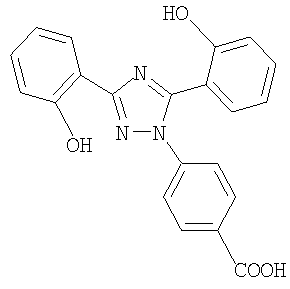
или его фармацевтически приемлемую соль в количестве от 5 до 40 мас.% в пересчете на общую массу таблетки и (б) по меньшей мере один разрыхлитель в общем количестве от 10 до 35 мас.% в пересчете на общую массу таблетки.
I) по меньшей мере один наполнитель, общее количество которого составляет примерно от 35 до 55 мас.%, в пересчете на общую массу таблетки,
II) по меньшей мере одно связующее вещество, общее количество которого составляет примерно от 1,5 до 5 мас.%, в пересчете на общую массу таблетки,
III) по меньшей мере одно поверхностно-активное вещество, общее количество которого составляет примерно от 0,2 до 1 мас.%, в пересчете на общую массу таблетки,
IV) по меньшей мере одно антиадгезивное вещество, общее количество которого составляет примерно от 0,1 до 0,5 мас.%, в пересчете на общую массу таблетки,
V) по меньшей мере, одно смазывающее вещество, общее количество которого составляет менее примерно от 0,4 мас.%, в пересчете на общую массу таблетки.
I) наполнитель присутствует в общем количестве 46,9 мас.% в пересчете на общую массу таблетки,
II) разрыхлитель присутствует в общем количестве 20 мас.% в пересчете на общую массу таблетки,
III) связующее вещество присутствует в общем количестве 3 мас.% в пересчете на общую массу таблетки,
IV) поверхностно-активное вещество присутствует в общем количестве 0,5 мас.% в пересчете на общую массу таблетки,
V) антиадгезивное вещество присутствует в общем количестве 0,2 мас.% в пересчете на общую массу таблетки,
VI) по меньшей мере одно смазывающее вещество присутствует в общем количестве менее чем 0,2 мас.% в пересчете на общую массу таблетки.
I) смешивание соединения I или его фармацевтически приемлемой соли с одним или более фармацевтически приемлемыми эксципиентами;
II) влажное гранулирование смеси, полученной на стадии (I);
III) смешивание гранулятов, полученных на стадии (II), с одним или более фармацевтически приемлемыми эксципиентами с образованием смеси; и
IV) распыление смазывающего вещества на материалы контактирующих поверхностей прессующих инструментов машины для изготовления таблеток и сжатие смеси, полученной на стадии (III), с получением таблетки.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), ПРОИЗВОДНЫЕ 3,5-ДИФЕНИЛ-1,2,4-ТРИАЗОЛОВ, В НЕЕ ВХОДЯЩИЕ | 1997 |
|
RU2208010C2 |

