Изобретение относится к фармацевтическим таблеткам, содержащим 4-(4-метилпиперазин-1-илметил)-N-[4-метил-3-(4-пиридин-3-ил)пиримидин-2-иламино)фенил]бензамид, или его фармацевтически приемлемые соли, которое в дальнейшем называется как соединение I.
Соединение I имеет формулу (I)
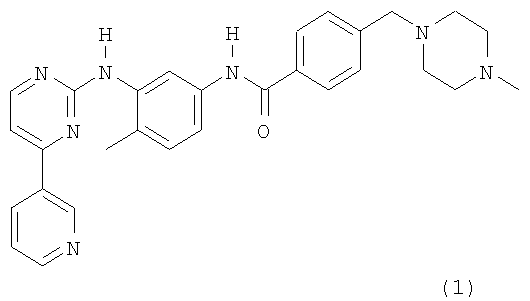
Соединение I в виде свободного основания и его приемлемые соли раскрыты в заявке European Patent 0564409. Мезилат соединения I и альфа- и бета-кристаллические формы мезилата соединения I раскрыты в международной заявке на патент WO 99/03854.
Назначенные при лечении лейкемии суточные дозировки мезилата соединения I обычно являются высокими, например, 400-800 мг для взрослых. Таким образом, существует потребность в оральной дозировочной форме, которая является удобной для введения и обеспечивает количество соединения I, равное суточной дозировке.
Соответственно, настоящее изобретение обеспечивает таблетки с высоким содержанием лекарственного препарата, которые включают в себя фармакологически эффективное количество соединения I или его фармацевтически приемлемой соли, присутствующих в количестве приблизительно от 30% до 80%, например, по меньшей мере, приблизительно от 35, 40, 45, 50 или 55% до 60, 65, 70, 75 или 80%, предпочтительно, больше чем 55%. В частности, количество соединения I может изменяться от 45 до 80%, например, от 50 до 70 мас.% в расчете на общую массу таблетки.
Соединение I может находиться в форме свободного основания или его фармацевтически приемлемых солей, например, в форме мономезилата. Активный компонент соответствует соединению I в форме свободного основания. Например, 119,5 мг мезилатной соли соединения I соответствует 100 мг активного компонента - свободного основания соединения I.
Кроме того, настоящее изобретение предоставляет таблетки, включающие в себя
(а) фармакологически эффективное количество соединения I и
(б) по меньшей мере, один фармацевтически приемлемый наполнитель, подходящий для получения таблеток, в которых количество соединения I или его фармацевтически приемлемой соли, выраженное в виде процентного содержания (по массе) активного компонента в расчете на всю таблетку, составляет приблизительно от 30% до 80%, например, по меньшей мере, приблизительно от 35, 40, 45, 50 или 55% до 60, 65, 70, 75 или 80%, предпочтительно, больше чем 55%. В частности, количество соединения I может изменяться от 45 до 80%, например, от 50 до 70 мас.% активного компонента в расчете на общую массу таблетки.
В другом замысле изобретение предоставляет таблетку, в которой соединение I находится в кристаллической форме.
В дополнительном замысле изобретения применяется мономезилатная соль соединения I.
В предпочтительном варианте осуществления изобретения мономезилатная соль соединения I находится в кристаллической форме, например, альфа- или бета-кристаллической форме, наиболее предпочтительной является мономезилатная соль соединения I в бета-кристаллической форме.
В таблетках может присутствовать один или несколько фармацевтически приемлемых наполнителей, например те, которые применяются традиционно, например, (1.1) по меньшей мере, один связующий материал, например, микрокристаллическая целлюлоза, гидроксипропилметилцеллюлоза, (1.2) по меньшей мере, один агент, вызывающий дезинтеграцию, например, сшитый поливинилпирролидон, например Crospovidone®, (1.3) по меньшей мере, один агент для скольжения, например, коллоидный диоксид кремния, (1.4) по меньшей мере, один смазочный материал, например, стеарат магния и/или (1.5) основное покрытие. В таблетке согласно настоящему изобретению микрокристаллическая целлюлоза применяется в качестве связующего материала.
В литературе имеется множество ссылок, относящихся к этим и другим наполнителям и к упомянутым в них способам, смотрите, например, Handbook of Pharmaceutical Excipients (Справочник фармацевтических наполнителей, 3-е издание), Third Edition, п/ред. Arthur H. Kibbe, American Pharmaceutical Association, Washington, USA and Pharmaceutical Press, London; и Lexikon der Hilfsstoffe für Pharmazie, Kosmetik and angrenzende Gebiete edited by H.P. Fiedler, 4th Edition, Edito Cantor, Aulendorf и более ранние издания, которые включены в это изобретение как ссылки.
Связующие материалы (1.1) включают, но не ограничиваются, крахмалы, например, картофельный, пшеничный или кукурузный крахмал; микрокристаллическую целлюлозу, например, такие продукты, как Avicel®, Filtrak®, Heweten® или Pharmacel®; гидроксипропилцеллюлозу; гидроксиэтилцеллюлозу; гидроксипропилметилцеллюлозу, например, гидроксипропилметилцеллюлоза Type 2910 USP, гипромелоза, и поливинилпирролидон (ПВП), например, Povidone® K30 от фирмы BASF. Предпочтительно, применяется гидроксипропилметилцеллюлоза Type 2910 USP.
Подходящие агенты, вызывающие дезинтеграцию (1.2), согласно изобретению включают, но не ограничиваются, кукурузный крахмал; Ca-карбоксиметилцеллюлозу (КМЦ)-; Na-КМЦ; микрокристаллическую целлюлозу; сшитый ПВП, например, известный и имеющийся в продаже под торговыми названиями Crospovidone®, Polyplasdone®, поступающий в продажу от фирмы ISP, или Kollidon® XL; альгиновую смолу; альгинат натрия и гуаровую смолу. Предпочтительно, применяется сшитый ПВП, например, Crospovidone®.
В качестве агента для скольжения (1.3) может быть использовано одно или несколько следующих веществ: диоксид кремния; коллоидный диоксид кремния, например, безводный коллоидный диоксид кремния, например, Aerosil® 200, трисиликат магния, порошкообразная целлюлоза, крахмал и тальк. Предпочтительно, используют безводный коллоидный диоксид кремния или/и коллоидный диоксид кремния.
В качестве смазочного материала (1.4) может быть использовано одно или несколько следующих веществ: стеарат Mg, Al или Ca, PEG 4000-8000 и/или тальк. Предпочтительно, применяется стеарат магния.
Один или несколько из этих наполнителей могут быть выбраны и использованы с учетом конкретных желаемых свойств таблетки обычным экспериментальным методом.
Согласно настоящему изобретению количество связующего материала (1.1) может изменяться в диапазоне приблизительно от 1 до 40%, предпочтительно, от 1 до 30%, особенно от 1 до 25 мас.% в расчете на общую массу таблетки.
Количество агента, вызывающего дезинтеграцию (1.2), может изменяться в диапазоне от 5 до 40%, например от 10 до 35 мас.% в расчете на общую массу таблетки.
Количество агента для скольжения (1.3) может изменяться в диапазоне от 0,1 до 10%, особенно от 0,1 до 5%, например от 0,5 до 3 мас.% в расчете на общую массу таблетки или от 2 до 4 мас.%в расчете на общую массу таблетки.
Количество смазочного материала (1.4) может изменяться в диапазоне от 0,1 до 5%, например от 0,5 до 2 мас.%в расчете на общую массу таблетки.
Количество основного покрытия (1.5) может изменяться от 1 до 10%, предпочтительно, от 1,5 до 5 мас.%в расчете на общую массу таблетки.
Можно признать, что любой выбранный наполнитель может выполнять несколько функций, например, агента, вызывающего дезинтеграцию, связующего материала, агента для скольжения, и/или смазочного материала.
В предпочтительном замысле изобретения, таблетка включает в себя следующие наполнители, один или несколько связующих материалов в общем количестве приблизительно от 1% до 25 мас.% в расчете на общую массу таблетки, один или несколько агентов, вызывающих дезинтеграцию в общем количестве приблизительно от 10% до 35 мас.%в расчете на общую массу таблетки, один или несколько агентов для скольжения в общем количестве приблизительно от 0,5% до 3 мас.% в расчете на общую массу таблетки, и/или один или несколько смазочных материалов в общем количестве приблизительно от 0,5% до 2 мас.%в расчете на общую массу таблетки.
Абсолютное количество каждого наполнителя и количество относительно других наполнителей аналогично зависит от желаемых свойств таблетки и также могут быть выбраны обычным экспериментальным методом. Например, таблетки могут быть выбраны таким образом, чтобы они проявляли ускоренное и/или замедленное выделение соединения I с количественным контролем выделения активного вещества, или без контроля. Предпочтительно, таблетку выбирают таким образом, чтобы она обеспечивала немедленное выделение соединения I, например, в виде мономезилатной соли бета-кристаллической формы соединения I.
Авторы настоящего изобретения столкнулись с проблемами при получении таблеток соединения I, в связи с показателями высокой хрупкости и низкой прочности на истирание. Кроме того, гибкость по количеству наполнителей, например агентов, вызывающих дезинтеграцию, является ограниченной из-за высокого содержания лекарственного препарата в продукте. Таким образом, все же существует потребность в промышленно приемлемых дозировочных формах соединения I для орального назначения пациенту при достаточном удобстве приема.
Согласно настоящему изобретению неожиданно было обнаружено, что получаются стабильные и фармацевтически приемлемые таблетки, содержащие соединение I. Авторы настоящего изобретения установили, что фармацевтически приемлемые твердые оральные формы дозировки в виде таблеток, которые особенно удобны для приема и являются стабильными, могут быть получены, используя способы прессования таблеток. Более конкретно, таблетки изобретения могут быть получены путем гранулирования, предпочтительно влажного гранулирования, с последующими способами прессования. Частицы соединения I, особенно его мезилатной соли, обладают большими размерами, например, 60% частиц исходного материала соединения I имеют размер, больше или равный 100 мкм, например, 90% частиц меньше или равны 420 мкм. Обычно способ влажного гранулирования осуществляют, используя частицы исходного материала размером меньше 100 мкм.
Отличительным свойством таблетки согласно изобретению является то, что она имеет высокое содержание соединения I при заданном относительно небольшом количестве наполнителя. Это обеспечивает получение таблеток небольшого размера. Общее количество наполнителей в унифицированной дозировке может составлять приблизительно 70 мас.% или меньше в расчете на общую массу таблетки, более конкретно, приблизительно 50% или меньше. Предпочтительно, содержание наполнителя находится в диапазоне приблизительно от 30 до 55%, более конкретно от 35 до 50 мас.% в расчете на общую массу таблетки.
Неожиданно согласно изобретению обеспечивается введение заданной унифицированной дозы соединения I при меньшем размере таблетки, чем это было возможно для таблетки уровня техники. Несмотря на высокое содержание лекарственного препарата, таблетки согласно изобретению имеют малый размер и поэтому они удобны при введении. Это приводит к улучшению схемы лечения пациента.
В другом варианте осуществления это изобретение обеспечивает таблетки, содержащие от 50 мг до 600 мг соединения I, например, приблизительно от 100 мг до 400 мг. Наиболее предпочтительно, таблетки согласно изобретению представляют собой таблетки, содержащие 100 мг и/или 400 мг соединения I.
Соответственно, настоящее изобретение предоставляет таблетки, содержащие мезилат соединения I, например, альфа-кристаллическую форму мезилата соединения I и/или бета-кристаллическую форму мезилата соединения I, в количестве, равном 100 мг и/или 400 мг свободного основания соединения I. Наиболее предпочтительной формой мезилата соединения I, применяемой для таблеток согласно изобретению, является бета-кристаллическая форма.
Согласно изобретению, способ получения таблеток заключается в формировании внутренней фазы, смешивании этой фазы вместе с внешней фазой, прессовании полученной смеси и, необязательно, покрытии таблеток.
Внутренняя фаза включает в себя соединение I. Предпочтительно, внутренняя фаза содержит соединение I и один или несколько наполнителей, более предпочтительно один или несколько связующих материалов, и наиболее предпочтительно количество одного или нескольких связующих материалов во внутренней фазе находится в диапазоне приблизительно от 1 до 30%, предпочтительно от 1 до 20% и более предпочтительно от 1 до 15%. Предпочтительно, связующие материалы внутренней фазы согласно изобретению представляют собой микрокристаллическую целлюлозу и гидроксипропилметилцеллюлозу. Количество микрокристаллической целлюлозы во внутренней фазе может изменяться приблизительно от 10 до 29%, в частности от 12 до 14 мас.%в расчете на общую массу таблетки. Количество гидроксипропилметилцеллюлозы во внутренней фазе может изменяться от 1 до 5%, предпочтительно, от 1 до 2 мас.% в расчете на общую массу таблетки. Соединение I и фармацевтически приемлемые наполнители внутренней фазы смешиваются вместе с водой, и смесь обрабатывают для гранулирования, например, используя влажный гранулятор с высокой степенью сдвига и получая влажные грануляты. Затем влажные грануляты могут быть высушены, например, в устройстве сушки с флюидным слоем гранул.
Настоящее изобретение относится к способу получения таблеток, содержащих внешнюю фазу. Эта внешняя фаза заключается в смеси внутренней фазы с одним или несколькими наполнителями. Внутренняя фаза и один или несколько наполнителей внешней фазы смешиваются вместе, например, используя диффузионный смеситель. Предпочтительно добавляют один или несколько связующих материалов. Наиболее предпочтительно добавляют микрокристаллическую целлюлозу. Еще более предпочтительно микрокристаллическую целлюлозу добавляют в количестве от 1% до 10 мас.%в расчете на общую массу таблетки. В предпочтительном варианте осуществления изобретения, количество микрокристаллической целлюлозы во внешней фазе составляет около 5 мас.% в расчете на общую массу таблетки. Кроме того, внешняя фаза согласно изобретению может содержать один или несколько агентов, вызывающих дезинтеграцию, наиболее предпочтительно Crospovidone®. В предпочтительном варианте осуществления, количество агента, вызывающего дезинтеграцию, во внешней фазе изменяется приблизительно от 10 до 30%, предпочтительно от 12 до 25%, наиболее предпочтительно около 15%.
В конкретном замысле изобретения во внешнюю фазу вводится один или несколько агентов для скольжения.
Согласно изобретению во внешнюю фазу вводится один или несколько смазочных материалов.
В дополнительном замысле изобретения, таблетки получают путем прессования смеси внутренней и внешней фаз, используя, например, пресс для таблетирования.
Таблетки могут быть необязательно покрыты, предпочтительно так, как описано в последующем.
В одном варианте осуществления изобретения, разработан способ получения таблетки, который заключается в том, что:
(а) формируется внутренняя фаза, которая включает в себя
(i) смешивание соединения I вместе с фармацевтически приемлемыми наполнителями,
(ii) влажное гранулирование,
(б) формируется внешняя фаза, которая включает в себя:
(iii) добавление дополнительных фармацевтически приемлемых наполнителей во внутреннюю фазу и смешивание;
(в) формируется таблетка путем
(iv) прессования смеси, полученной на стадии (iii), и, необязательно,
(г) покрытие.
Более конкретно, в одном аспекте настоящего изобретения разработан способ, заключающийся в том, что:
(i) смешивают соединение I и фармацевтически приемлемые наполнители, например, один или несколько связующих материалов, например микрокристаллическую целлюлозу, в смесителе интенсивного сдвига;
(ii) добавляют воду, подвергают смесь увлажнению/замешиванию, например, в смесителе интенсивного сдвига, просеивают с использованием механического сита и вращающегося лопастного колеса и сушат, например, в устройстве сушки с флюидным слоем;
(iii) добавляют фармацевтически приемлемые наполнители, например, просеянные наполнители, такие как один или несколько агентов, вызывающих дезинтеграцию, например, Crospovidone®, один или несколько связующих материалов, например, микрокристаллическую целлюлозу, один или несколько агентов для скольжения, например, коллоидный диоксид кремния, и смешивают, например, в диффузионном смесителе;
(iv) добавляют фармацевтически приемлемые наполнители, такие как один или несколько смазочных материалов, например, стеарат магния, просеивают, например, вручную с использованием сита, например, размером 900 мкм, и смешивают, например, в диффузионном смесителе;
(v) таблетируют смесь, полученную на стадии (iv), путем прессования, например, в традиционном таблеточном прессе, например, в устройстве таблетирования с эксцентриком ЕК-0 Korsch или в роторном устройстве таблетирования, предпочтительно, в роторном устройстве и
(vi) покрывают, например, в смазывающей форме, например, типа Glatt, Accela.
Термин "ядро" означает фазу гранулята (стадии (i) и (ii)), в том числе активный лекарственный препарат - соединение I и внешняя фаза, состоящая из наполнителей.
Термин "общая масса таблетки" означает массу таблетки, причем с внутренней и внешней фазами и покрытием (если оно имеется).
Согласно изобретению способ покрытия может быть осуществлен при низкой температуре, например, между 30 и 40°С, предпочтительно, между 32 и 39°С, наиболее предпочтительно, при температуре в диапазоне приблизительно от 35 до 38°С. Способ покрытия может быть осуществлен при степени распыления предпочтительно в диапазоне от 30 до 105 г покрывающей дисперсии на 1 кг ядра в час, предпочтительно от 35 до 105 г/(кг.ч). Неожиданно было обнаружено, что набухание агента, вызывающего дезинтеграцию, например, Crospovidone®, также не приводит к прилипанию ядра в ходе распыления покрывающей смеси, проводимого при низкой температуре, как можно было ожидать на основании опыта специалиста в этой области техники.
Более того, таблетки проявляют повышенную прочность на истирание. Физическая и химическая стабильность может быть испытана традиционным образом, например, можно испытывать собственно таблетки путем измерения растворимости, хрупкости, времени разрушения, анализируя продукты разложения соединения I, внешний вид и/или данные микроскопии, например, после хранения при комнатной температуре, т.е. 25°С, и/или хранения при 40°С.
Ядро таблетки может иметь различную форму, например, круглую, овальную, прямоугольную, цилиндрическую или любую другую подходящую форму. Отличительным свойством таблеток согласно изобретению является их малый размер с учетом содержания соединения I или соли соединения I в этих таблетках.
В предпочтительном варианте осуществления изобретения описанные выше таблетки, полученные способом прессования, являются круглыми или овальными. Края таблеток могут иметь скошенную или закругленную кромку. Наиболее предпочтительно, таблетки являются овальными и/или круглыми. Таблетки согласно изобретению могут иметь зарубки на поверхности. Овальная таблетка может иметь небольшой размер в длину, например, от 10 до 20 мм, предпочтительно, от 15 до 20 мм, наиболее предпочтительно, от 17 до 19 мм; от 5 до 10 мм в ширину, предпочтительно, от 6,5 до 8 мм. Толщина таблетки составляет от 4 до 8 мм, предпочтительно, от 6 до 8 мм. Для получения прессованной таблетки применяются усилия сжатия между 10 и 20 кН, предпочтительно от 12 до 18 кН. Предпочтительно, овальная таблетка содержит 400 мг соединения I. Круглая таблетка может иметь следующие размеры, например, от 5 до 15 мм в диаметре, предпочтительно от 7 до 10 мм, наиболее предпочтительно около 9 мм. Толщина таблетки может составлять от 2 до 5 мм, предпочтительно от 2,5 до 4 мм. Для получения прессованной таблетки применяются усилия сжатия между 6 и 8 кН, предпочтительно от 8 до 14 кН. Предпочтительно, круглая таблетка содержит 100 мг соединения I. Предпочтительно, таблетка массой 100 мг представляет собой таблетку с зарубкой, наиболее предпочтительно, таблетка имеет с одной стороны зарубку с разрывом.
Кроме того, таблетки изобретения, содержащие приблизительно 100 мг соединения I, могут иметь твердость приблизительно от 30 до 140 Н, например, от 40 до 140 Н, от 30 до 100 Н, от 40 до 100 Н, предпочтительно от 50 до 80 Н. Таблетки изобретения, содержащие приблизительно 400 мг соединения I, могут иметь твердость приблизительно от 100 до 270 Н, например, от 100 до 250 Н, от 160 до 270 Н, от 160 до 250 Н, предпочтительно от 195 до 235 Н.
Время разрушения таблетки может составлять приблизительно от 20 минут или меньше. Предпочтительно, для таблетки со 100 мг соединения I время разрушения находится в диапазоне приблизительно от 2 до 10 минут, предпочтительно от 4 до 10 минут, например, от 4 до 8 минут. Предпочтительно, для таблетки с 400 мг соединения I время разрушения находится в диапазоне приблизительно от 7 до 15 минут, предпочтительно от 8 до 15 минут, например, от 8 до 14 минут.
Хрупкость таблеток измеряют по методу Фармакопеи США (USP). Хрупкость таблеток согласно изобретению, исследованная с применением рекомендаций Фармакопеи США, составляет 0% (см. чертеж).
Кроме того, таблетки изобретения могут быть окрашены, и/или таблетки или покрытие могут быть помечены таким образом, чтобы они имели различный внешний вид и чтобы они были легко распознаваемы. Применение красителей может обеспечить улучшение внешнего вида, а также идентификации таблеток. Красители, подходящие для использования в лекарственных препаратах, обычно включают каротиноиды, оксиды железа или хлорофил. Таблетки изобретения могут быть помечены, используя отпечатанный код.
Способы, которые могут быть использованы, могут быть традиционными, или известными из уровня техники, или основанными на таких способах, например, таких, которые описаны в книге L. Lachman и др. The Theory и Practice of Industrial Pharmacy, 3rd Ed, 1986, H. Sucker et al, Pharmazeutische Technologie, Thieme, 1991, Hagers Handbuch der pharmazeutischen Praxis, 4th Ed. (Springer Verlag, 1971) и Remington's Pharmaceutical Sciences, 13th Ed., (Mack Publ., Co., 1970) или в более поздних изданиях.
Таблетки изобретения применяются для пациентов, которым прописано соединение I, например, для лечения опухолей, как показано в стандартных тестах. Активность и другие показатели таблеток изобретения могут быть охарактеризованы в стандартных клинических испытаниях и/или в испытаниях на животных.
В частности, таблетки изобретения применяются, например, для лечения незлокачественных и злокачественных пролиферативных нарушений, например, лейкемии, глиомы, саркомы, опухолей простаты, груди, легких, яичников и желудочно-кишечной системы.
Таблетки изобретения, содержащие фармакологически эффективное количество соединения I или соли соединения I, могут быть назначены в качестве единственного активного лекарственного препарата или может быть предусмотрено назначение вместе с другим активным лекарственным препаратом, например, с одновременным или раздельным введением других лекарственных препаратов.
Более того, полученные таблетки изобретения являются стабильными как в процессе получения, так и при хранении, например, в течение 2 лет или даже 3 лет в традиционной упаковке, например, в герметичной блистерной упаковке из алюминия. За это время может разложиться приблизительно меньше 5%, например, 2 или 3% или менее соединения I или соли соединения I, как определено в традиционных испытаниях.
Таблетки изобретения, например, таблетки по 100 и 400 мг, биологически эквивалентны имеющимся в продаже твердым желатиновым капсулам со 100 мг соединения I. Пациенты легко переносят введение 400 мг соединения I в твердых желатиновых капсулах (4×100 мг) в виде одной таблетки, покрытой пленкой.
В зависимости от возраста, индивидуального состояния, способа назначения и рассматриваемой клинической картины заболевания, для пациентов весом приблизительно 70 кг вводятся эффективные дозы, например, суточная доза таблеток изобретения, содержащих, например, 100-1000 мг, например, от 100 до 800 мг, предпочтительно, от 100 до 600 мг, особенно 400 мг соединения I.
Кроме того, это изобретение относится к способу введения пациенту-человеку, нуждающемуся в таком лечении, соединения I или его фармацевтически приемлемой соли в форме таблетки, один раз в день в течение периода больше чем 3 месяца. Это изобретение особенно относится к такому способу, в котором взрослому человеку вводится суточная доза от 100 до 1000 мг, предпочтительно от 100 до 800 мг, особенно от 200 до 600 мг, предпочтительно 400 мг соединения I. Следует признать, что уровень конкретной дозы для любого отдельного пациента будет зависеть от множества факторов, в том числе от возраста, веса пациента, общего здоровья, сочетания лекарственного препарата с одним или несколькими активными лекарственными препаратами, типом и тяжести заболевания.
Соответственно, в дополнительном аспекте настоящее изобретение предоставляет способ лечения субъекта, который заключается в приеме таблетки согласно изобретению, которая включает в себя фармакологически эффективное количество соли соединения I субъекту, нуждающемуся в таком лечении, необязательно с одновременным, последовательным или раздельным введением другого лекарственного препарата, например, циклоспорина, рапамицина, аскомицина, кортикостероидов, циклофосфамида, азатиоприна, метотрексата, бреквинара, лефлуномида, мизорибина, микофенольной кислоты и/или микофенолята мофетила.
Когда таблетки изобретения принимаются в рамках совместной терапии, дозировки мезилата соединения I могут быть снижены, например, от половины до одной трети от их дозировки, используемой при индивидуальном приеме.
Упаковка лекарственного препарата включает в себя таблетки согласно изобретению и напечатанные инструкции, предписывающие, что одну или несколько таблеток соединения I можно принимать орально.
Следующие, не ограничивающие примеры иллюстрируют изобретение.
Пример 1. Рецептура таблетки (таблетка 100 мг)
Состав унифицированной формы дозировки и количество в загрузке
Таблетки согласно изобретению со 100 мг соединения I в форме свободного основания и указанные выше таблетки получают путем влажного гранулирования смеси соли соединения I с (1.1), смешивания с 3(1.1), (1.2), (1.3) и (1.4), прессования и покрытия полученных таблеток водной дисперсией покрывающей смеси (1.5).
Процесс покрытия может быть осуществлен при низкой температуре, например, в диапазоне приблизительно от 35 до 38°С. Процесс покрытия может быть осуществлен при степени распыления предпочтительно в диапазоне от 30 до 105 г покрывающей дисперсии на 1 кг ядра в час ("ядро" соответствует спрессованной внутренней и внешней фазе), например, от 35 до 105 г на кг ядра в час.
Пример 2. Рецептура таблетки (таблетка 400 мг)
Таблетки согласно изобретению с 400 мг соединения I и указанные ниже таблетки получают путем влажного гранулирования смеси соли соединения I с (1.1), смешивания 3(1.1), (1.2), (1.3) и (1.4), прессования и покрытия полученных таблеток водной дисперсией покрывающей смеси (1.5).
Процесс покрытия может быть осуществлен при низкой температуре, например, в диапазоне приблизительно от 35 до 38°С. Процесс покрытия может быть осуществлен при степени распыления предпочтительно в диапазоне от 30 до 105 г покрывающей дисперсии на 1 кг ядра в час ("ядро" соответствует спрессованной внутренней и внешней фазе), например, от 35 до 105 г на кг ядра в час.
Пример 3. Размеры таблеток
Для таблеток с дозировкой, равной или менее 650 мг, был взят образец из общего числа таблеток с общим весом, соответствующим близко к 6,5 г (см. чертеж). Для таблеток с дозировкой более 650 мг был взят образец из 10 всех таблеток. Таблетки должны быть тщательно очищены от пыли перед их испытанием. Аккуратно определялся вес образца таблетки и затем эти таблетки помещали в барабан. Барабан вращали 100 раз и вынимали таблетки. Удаляли любые следы пыли с этих таблеток, как и перед их прокручиванием в барабане, и аккуратно взвешивали.
В результате проведения опыта получены таблетки практически в неизменном виде с высокой устойчивостью к истиранию и рыхлостью 0,0%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТАБЛЕТКА С ВЫСОКИМ СОДЕРЖАНИЕМ ЛЕКАРСТВЕННОГО ПРЕПАРАТА | 2009 |
|
RU2405540C1 |
| ДИСПЕРГИРУЕМЫЕ ТАБЛЕТКИ ДЕФЕРАСИРОКС | 2003 |
|
RU2338532C2 |
| НОВЫЙ СОСТАВ | 2006 |
|
RU2821230C2 |
| НОВЫЙ СОСТАВ | 2006 |
|
RU2483716C2 |
| ГАЛЕНОВЫ СОСТАВЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2005 |
|
RU2384328C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИМАТИНИБ И ЗАМЕДЛИТЕЛЬ ВЫСВОБОЖДЕНИЯ | 2006 |
|
RU2404775C2 |
| ГАЛЕНОВЫ СОСТАВЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2009 |
|
RU2438661C2 |
| ТВЕРДАЯ ПЕРОРАЛЬНАЯ ЛЕКАРСТВЕННАЯ ФОРМА И СПОСОБ ЛЕЧЕНИЯ | 2011 |
|
RU2480210C1 |
| ТВЕРДАЯ ДОЗИРОВАННАЯ ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ ОРАЛЬНОГО ВВЕДЕНИЯ, СОДЕРЖАЩАЯ КОМБИНАЦИЮ ВИЛДАГЛИПТИНА И ГЛИКВИДОНА | 2014 |
|
RU2585378C1 |
| МАТРИЧНАЯ ТАБЛЕТКА С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ НЕРАМЕКСАНА | 2006 |
|
RU2422135C2 |
Изобретение относится к области фармацевтики и касается таблетки с высоким содержанием лекарственного препарата, которая включает в качестве активного компонента соединение формулы (I) или его фармацевтически приемлемую соль в количестве приблизительно от 30% до 80 мас.% активного компонента, в расчете на общую массу таблетки и по меньшей мере одно связующее, которое содержит микрокристаллическую целлюлозу или гидроксипропилметилцеллюлозу. 4 н. и 12 з.п. ф-лы, 1 ил.
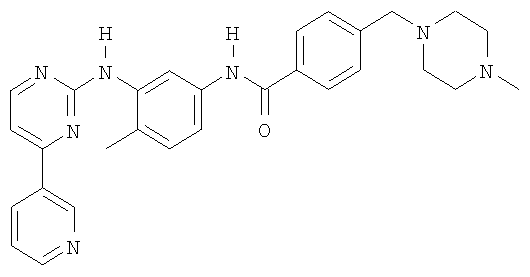
1. Таблетка, содержащая фармакологически эффективное количество соединения формулы (I)
или его фармацевтически приемлемой соли в количестве приблизительно от 30 до 80 мас.% активного компонента в расчете на общую массу таблетки, и где указанная таблетка содержит по меньшей мере одно связующее, которое содержит микрокристаллическую целлюлозу или гидроксипропилметилцеллюлозу, или их смеси.
2. Таблетка, содержащая
(a) фармакологически эффективное количество соединения I и
(b) один или более фармацевтически приемлемых эксципиентов, пригодных для приготовления таблеток, в которой соединение формулы I или его фармацевтически приемлемая соль присутствуют в количестве приблизительно от 30 до 80 мас.% активного компонента в расчете на общую массу таблетки, и где указанная таблетка содержит по меньшей мере одно связующее, которое содержит микрокристаллическую целлюлозу или гидроксипропилметилцеллюлозу, или их смеси.
3. Таблетка по п.1, в которой соединение формулы (I) или его фармацевтически приемлемая соль присутствует в количестве приблизительно от 50 до 80 мас.% активного компонента в расчете на общую массу таблетки.
4. Таблетка по п.1 или 2, в которой соединение формулы (I) представляет собой мономезилатную солевую форму.
5. Таблетка по п.4, в которой мономезилат соединения формулы (I) находится в бета-кристаллической форме.
6. Таблетка по п.1, которая дополнительно содержит разрыхлитель.
7. Таблетка по п.6, в которой разрыхлитель содержит поперечно-сшитый поливинилпирролидон.
8. Таблетка по п.1, которая дополнительно содержит агент скольжения.
9. Таблетка по п.8, в которой агент скольжения содержит коллоидный диоксид кремния и/или коллоидный безводный диоксид кремния.
10. Таблетка по п.1, которая дополнительно содержит связующий агент.
11. Таблетка по п.10, в которой связующий агент содержит стеарат магния.
12. Таблетка по п.1, в которой наполнитель включает в себя:
по меньшей мере, один связующий агент в общем количестве приблизительно от 1 до 25 мас.% в расчете на общую массу таблетки,
по меньшей мере, один разрыхлитель в общем количестве приблизительно от 10 до 35 мас.% в расчете на общую массу таблетки,
по меньшей мере, один агент скольжения в общем количестве приблизительно от 0,5 до 3 мас.% в расчете на общую массу таблетки, и/или
по меньшей мере, один смазочный агент в общем количестве приблизительно от 0,5 до 2 мас.% в расчете на общую массу таблетки.
13. Способ получения таблетки по одному из предшествующих пунктов, который заключается в том, что:
(i) смешивают соединение формулы (I) или его фармацевтически приемлемые соли с фармацевтически приемлемыми наполнителями, (ii) проводят влажное гранулирование, (iii) смешивают с фармацевтически приемлемыми наполнителями с образованием смеси; и
(iv) прессуют смесь, полученную на стадии (iii), с получением таблетки.
14. Способ по п.13, в котором таблетку покрывают.
15. Таблетка по любому пп.1-12, полученная способом влажного гранулирования.
16. Способ лечения субъекта, который заключается в введении таблетки по любому пп.1-12 или 15, которая включает в себя фармакологически эффективное количество соли соединения формулы (I) субъекту, нуждающемуся в таком лечении.
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА ИЛИ ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ОБЛАДАЮЩАЯ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2125992C1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Муравьев И.А | |||
| Технология лекарств | |||
| - М.: Медицина, 1980, т.1, с.342-345, 350-354. | |||

