ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к диспергируемой таблетке для пероральной суспензии, содержащей N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамид, метансульфонатную соль, представленную следующей формулой (I), известную как дабрафениба мезилат или Тафинлар®, и в дальнейшем называемая соединением A:
 (Соединение A).
(Соединение A).
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамид (далее соединение B) представляет собой соединение, которое раскрыто и заявлено в виде свободного основания, наряду с его фармацевтически приемлемыми солями и сольватами, в качестве ингибитора активности BRAF, особенно при лечении рака, в международной заявке № PCT/US2009/042682, поданной 4 мая 2009 г.; Международной публикации № WO/2009/137391, поданной 12 ноября 2009 г., полное раскрытие которых включено в данный документ путем ссылки. Соединение B представляет собой соединение Примера 58a.
Соединение B можно получать описанным в международной заявке № PCT/US2009/042682 путем. Соединение B можно получать, как описано в патентной публикации США № US 2011/0172215, опубликованной 14 июля 2011 г., полное раскрытие которой включено в данный документ путем ссылки.
Соответственно, соединение В получают в форме метансульфонатной соли, или соединения А, или дабрафениба мезилата, как определено в данном документе. Другие подходящие фармацевтически приемлемые формы солей соединения B включают сульфатную, гидрохлоридную и натриевую соли. Специалист в данной области может получать данные соли, например, используя описание Международной заявки № PCT/US2009/042682 или патентную публикацию США № US 2011/0172215. Соединение А получают в Примерах 58d-e патентной публикации США № US 2011/0172215.
Твердые пероральные фармацевтические лекарственные формы являются популярными и полезными формами лекарств для введения фармацевтически активных соединений. Известно множество таких форм, включая таблетки, капсулы, драже, пастилки и порошки.
Однако в промышленном масштабе составление приемлемой твердой пероральной фармацевтической лекарственной формы не является простой задачей. При введении in vivo фармакокинетические свойства фармацевтически активных соединений могут существенно различаться в зависимости от состава. Композиция должна быть способна доставлять определенное количество фармацевтически активного соединения, достаточное для достижения желаемых уровней терапевтического лекарственного средства, а также минимизировать нежелательные эффекты (например, токсичность), связанные с неоптимальным терапевтическим содержанием лекарственного средства. Кроме того, состав и способ изготовления должны быть такими, которые обеспечивают целостность лекарственной формы, сохраняющуюся неизменной до момента использования. Лекарственная форма также должна обладать приемлемыми свойствами распадаемости и растворения, обеспечивающие желаемый профиль при использовании.
При изготовлении высококачественных лекарственных форм таких фармацевтически активных соединений, как соединение А, возникают определенные проблемы. Хотя и было обнаружено, что форма мезилатной соли улучшает биодоступность соединения A, соединение A представляет собой соединение с высокой проницаемостью и низкой растворимостью, которое очень слабо растворяется в сильнокислых водных средах и практически нерастворимо в слабокислых, pH-нейтральных и щелочных средах. Разработчик должен сбалансировать уникальные химические свойства препарата со свойствами каждого наполнителя, чтобы приготовить безопасную, эффективную и простую в использовании твердую пероральную фармацевтическую лекарственную форму.
Твердые лекарственные формы Соединения A, такие как таблетки и капсулы, раскрыты в Международной заявке № PCT/US2009/042682. Соединение A в виде капсул по 50 мг и 75 мг было одобрено Управлением США по надзору за пищевыми продуктами и лекарственными веществами (FDA) для лечения метастатической меланомы с положительной мутацией BRAF V600E в качестве монотерапии или в комбинации с траметинибом. Комбинация Соединения А и траметиниба также была одобрена FDA для лечения немелкоклеточного рака легких с мутацией BRAF V600E. Безопасность и эффективность Соединения А в настоящее время оценивается для педиатрического лечения солидных опухолей с мутацией BRAF в недавно проведенном исследовании I фазы. Однако целевые дозы Соединения А, прогнозируемые для популяции педиатрических пациентов, могут быть существенно ниже, чем те, которые предоставляются существующими составами капсул.
Хотя таблетки и капсулы могут быть приемлемы для применения взрослыми, такие составы могут быть нежелательным или нецелесообразным для детей или людей, с трудом глотающих таблетки и капсулы. В педиатрических популяциях часто более желательно предоставлять диспергируемую композицию для перорального введения, такую как порошок или таблетка, которую можно сначала диспергировать в проглатываемой водной среде перед употреблением пациентом. В отличие от порошка для пероральной суспензии, диспергируемая таблетированная композиция обычно обеспечивает более короткое время разведения при суспендировании в водной среде без потерь лекарственного средства.
Было бы желательно предоставить Соединение А в диспергируемой твердой композиции, в частности, в фармацевтической диспергируемой таблетке для перорального введения в виде суспензии (в настоящем документе также называемой «диспергируемой таблеткой для пероральной суспензии») в коммерческом масштабе, которая удобна для приема детьми, и которая обеспечивает суточную дозу соединения А.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтической диспергируемой таблетке для пероральной суспензии соединения A, которая годится для разведения в воде. Кроме того, настоящее изобретение относится к способу приготовления диспергируемой таблетки, а также к способу применения диспергируемой таблетки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном варианте осуществления настоящее изобретение направлено на пероральные фармацевтические лекарственные формы, которые содержат соединение A, предпочтительные лекарственные формы представляют собой диспергируемые таблетированные формы, предпочтительно изготавливаемые в промышленном масштабе.
Используемый в данном контексте термин «диспергируемая таблетка» означает фармацевтическую композицию в форме таблетки, которая диспергируется в водной фазе, предпочтительно в воде, для перорального введения в виде водной суспензии.
В одном варианте осуществления настоящее изобретение относится к диспергируемой таблетке, содержащей массовую долю соединения А от 5 до 40% в пересчете на общую массу таблетки.
В одном варианте осуществления настоящее изобретение относится к диспергируемой таблетке, содержащей терапевтически эффективное количество соединения А, составляющее массовую долю от 5 до 40% в пересчете на общую массу таблетки.
Используемый в данном контексте термин «улучшенные свойства» и другие его формы подразумевает несколько преимуществ в фармакокинетическом профиле высвобождения соединения А in vivo из диспергируемой таблетки, в которой используется аспект настоящего изобретения, по сравнению с препаратом, который не использует такой аспект настоящего изобретения, причем подходящая композиция производится в промышленном масштабе. Примеры улучшенных свойств включают: повышенную пероральную биодоступность, улучшенную физическую и химическую стабильность, улучшенную фотостабильность, устойчиво воспроизводимый фармакокинетический профиль, улучшенный фармакокинетический профиль, устойчиво воспроизводимую скорость растворения и стабильную пероральную фармацевтическую композицию, при смешивании диспергируемой таблетки с водным носителем.
Известно, что соединение А обладает высокой проницаемостью и плохой растворимостью в воде. Соединение А в воде образует пересыщенный раствор и склонно к быстрому преобразованию, идущему в растворе, или осаждению в виде свободного основания дабрафениба. Замедление осаждения соединения А и поддержание пересыщенного раствора в течение продолжительного периода времени имеет особое значение для обеспечения большей абсорбции in vivo, и приводит к более высокой биодоступности Соединения А.
Было обнаружено, что относительная биодоступность соединения А зависит от характеристик растворения композиции. В частности, было обнаружено, что на скорость диссоциации составов капсул Соединения А влияет наличие материала оболочки капсулы из полимерной гипромеллозы (ГПМЦ) (Ouellet et al., J. Pharm. Sci., 102(9): 3100-3109). Исследования растворения in vitro, где сравнивали составы желатиновых и гипромеллозных капсул соединения А, показали более высокий процент растворения с гипромеллозой (составляющей более 20% от общего веса капсулы) по сравнению с желатиновыми капсулами, замедление осаждение дабрафениба, поддержание перенасыщенного раствора в течение длительного периода времени, что приводило к более высокой пероральной биодоступности (Ouellet et al., J. Pharm. Sci., 102(9): 3105-3107).
В одном варианте осуществления настоящее изобретение также относится к диспергируемой таблетке, содержащей:
(а) Соединение A,
(b) гипромеллозу и
(c) по меньшей мере одно фармацевтически приемлемое формообразующее, подходящее для приготовления диспергируемых таблеток, в которых содержание соединения А или его фармацевтически приемлемой соли, рассчитанное как массовая доля активного начала в процентах от общей массы диспергируемой таблетки, составляет от примерно 5% до 40%, предпочтительно - массовую долю примерно 15% в пересчете на общую массу диспергируемой таблетки; содержание гипромеллозы в массовых долях может варьироваться от примерно 1% до 25%, предпочтительно от примерно 5% до 10% в пересчете на общую массу диспергируемой таблетки.
Определенную трудность при составлении диспергируемой таблетки соединения А представляет собой высокое содержание гипромеллозы. Высокое содержание гипромеллозы может отрицательно влиять на профиль растворения диспергируемых таблетированных составов соединения А, увеличивая время полного распада и диспергирования таблеток, делая лекарственную форму неудобной и требующей длительного времени для приготовления достаточно диспергированного в водной среде препарата перед приемом пациентом. Помимо оптимизации содержания гипромеллозы, выбор разрыхлителей имеет решающее значение для ускорения разрушения таблетки на мелкие частицы в присутствии водной среды.
В диспергируемых таблетках могут присутствовать один или несколько обычно используемых фармацевтически приемлемых формообразующих, например, таких как по меньшей мере один наполнитель, например, лактоза, этилцеллюлоза, микрокристаллическая целлюлоза, по меньшей мере один разрыхлитель, например, сшитый поливинилпирролидинон, например, кросповидон, по меньшей мере одно вещество, способствующее скольжению, например, коллоидный диоксид кремния, по меньшей мере одно смазывающее вещество, например, стеарат магния.
Ссылка делается на обширную литературу по этому вопросу для этих и других фармацевтически приемлемых наполнителей и процедур, упомянутых в данном документе, см., в частности, Handbook of Pharmaceutical Excipients, Eighth Edition, под редакцией Paul J. Sheskey, American Pharmaceutical Association, Washington, USA и Pharmaceutical Press, London; и Lexikon der Hilfsstoffe für Pharmazie, Kosmetik and angrenzende Gebiete под редакцией H.P. Fiedler, 4th Edition, Edito Cantor, Aulendorf и более ранние издания, которые включены в данный документ путем ссылки.
Другая определенная проблема, связанная с приготовлением диспергируемой таблетки соединения А, заключается в выборе разрыхлителя, используемого для увеличения площади поверхности продукта и смягчения связующего вещества, которое удерживает вместе твердые частицы таблетки. В начале распада гипромеллоза может использовать свободную воду для гидратации с образованием геля и ингибировать поглощение воды разрыхлителем путем впитывания (капиллярного действия) или набухания, тем самым продлевая распад таблетки. Это явление еще более выражено при более высоком содержании гипромеллозы в составе, что приводит к еще более длительному времени распада таблеток.
Подходящие разрыхлители согласно изобретению включают, но не ограничиваются ими, крахмалы, целлюлозы, сшитые полимеры и шипучие агенты, такие как кукурузный крахмал, картофельный крахмал, предварительно желатинизированный крахмал, модифицированный кукурузный крахмал, кроскармеллоза натрия, кросповидон, гликолят крахмала натрия, метилцеллюлоза, карбоксиметилцеллюлоза и их соли, такие как кальциевая и натриевая. В одном варианте осуществления настоящего изобретения разрыхлитель представляет собой кроскармеллозу натрия или кросповидон, предпочтительно кросповидон. Было обнаружено, что кроскармеллоза натрия в высоких концентрациях может превращаться в гель и, таким образом, увеличивать время полного распада (распадаемость).
В настоящем изобретении предложена диспергируемая таблетка, имеющая время полного распада (распадаемость), например, в водной среде, в воде, не более 3 минут, предпочтительно - 3 минуты или меньше, как измерено с помощью устройства для измерения распадаемости согласно испытанию на распадаемость в European Pharmacopoeia 2.9.1 (т.е. время распада таблетки в воде при температуре от 15oC до 25oC). Соответственно, диспергируемая таблетка обеспечивает быстрое время растворения и, следовательно, удобна для приема, например, детьми. Это приводит к более аккуратному соблюдению режима приема средства пациентами.
Под «временем распада» подразумевается время, в течение которого диспергируемая таблетка распадается в воде при температуре от 15oC до 25oC в приборе для измерения времени распада.
Диспергируемая таблетка настоящего изобретения приспособлена диспергировать в водной фазе, предпочтительно в воде.
За ходом дисперсии можно наблюдать визуально. Считается, что таблетка полностью распалась, если на экране не остается никакого остатка, или если остаток есть, то он состоит из мягкой массы, не имеющей ощутимо твердой, не увлажненной сердцевины, или на экране остаются только фрагменты покрытия (таблетки).
Известно, что с увеличением твердости диспергируемой таблетки увеличивается продолжительность распада и уменьшается истираемость. Следовательно, короткое время распада обычно имеют относительно мягкие таблетки, которым потенциально не хватает механической прочности (т.е. высокие значения истираемости). Однако, недостаточно твердые таблетки обычно крошатся, разламываются или разрушаются до того, как это требуется (т.е. в ходе упаковки, перемещения, хранения или в любое другое время до внесения таблетки в проглатываемую водную среду для приема таблетки).
Настоящее изобретение направлено на создание диспергируемой таблетки соединения А с высокими уровнями содержания гипромеллозы, коротким временем распада для удобного получения суспензии и низкой истираемостью, способной выдерживать обычную обработку сыпучих продуктов и первичную упаковку. Неожиданно было обнаружено, что состав, содержащий суммарную массовую долю соединения А и гипромеллозы примерно от 1% до 13% от общего веса диспергируемой таблетки, может дать быстро диспергирующую композицию, время распада которой не превышает 3 минуты, как измерено с помощью устройства для измерения распадаемости согласно испытанию на распадаемость в European Pharmacopoeia 2.9.1 (т.е. время распада таблетки в воде при температуре от 15oC до 25oC), и низкую истираемость менее 1% после 100 оборотов.
Используемый в данном контексте термин «лекарственное средство» или «активное начало» и их формы, если не указано иное, означает соединение А или N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамида метансульфонат.
Используемый в данном контексте термин «Соединение В» или N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамид представляет собой свободное или не представляющее собой соль и несольватированное соединение. Соединение B также относится к содержанию свободного или не представляющего собой соль и несольватированного соединения в количестве соединения A.
Под термином «промышленный масштаб» и его формами в данном контексте подразумевается приготовление партии в размере более чем примерно 20 кг смеси для прямого прессования, соответственно - более 50 кг, соответственно - более 75 кг или размера партии, подходящей для приготовления по меньшей мере примерно 50000 таблеток, соответственно - по меньшей мере 75000 таблеток, соответственно - по меньшей мере 100000 таблеток.
Термин «примерно» означает в области, приблизительно или около. Когда термин «примерно» используется в сочетании с числовым интервалом, он изменяет этот интервал, расширяя границы выше и ниже приведенных числовых значений. В общем, термин «примерно» используется в данном документе для изменения числового значения выше и ниже заявленного значения с отклонением на 10%.
Термин «эффективное количество» и его формы означает такое количество лекарственного средства или активного начала, которое вызывает биологический или медицинский ответ ткани, системы, животного или человека, который ожидает получить, например, исследователь или клиницист. Кроме того, термин «терапевтически эффективное количество» означает любое количество, которое по сравнению с соответствующим субъектом, который не получил такого количества, приводит к улучшенному лечению, выздоровлению, профилактике или ослаблению заболевания, расстройства или побочного эффекта или снижению скорости развития заболевания или расстройства. Термин также включает в себя количества, эффективные для усиления нормальной физиологической функции.
Соответственно, диспергируемую таблетку, содержащую Соединение А, можно использовать для лечения новообразования, в частности чувствительного новообразования (рака или опухоли) у млекопитающего. Настоящее изобретение также относится к способу лечения новообразования, в частности восприимчивого новообразования, у млекопитающего, нуждающегося в этом, который включает введение млекопитающему терапевтически эффективного количества Соединения А в диспергируемой таблетке данного изобретения.
«Восприимчивое новообразование» в данном контексте к новообразованиям, которые чувствительны к лечению ингибитором киназы, и, в частности, к новообразованиям, которые чувствительны к лечению ингибитором Raf. Новообразования, связанные с ненормальной активностью одной или нескольких киназ семейства Raf и, в частности, новообразования, которые несут в себе мутацию киназы семейства Raf, сверхэкспрессию киназы семейства Raf или мутацию активатора киназы семейства Raf выше по сигнальному пути, или сверхэкспрессию активатора киназы семейства Raf выше по сигнальному пути и, следовательно, восприимчивы к лечению ингибитором Raf, известны в данной области и включает как первичные, так и метастатические опухоли и рак. См. Catalogue of Somatic Mutations in Cancer (COSMIC), the Wellcome Trust Sanger Institute, http://www.sanger.ac.uk/genetics/CGP/cosmic/ и те ссылки, которые приведены в нем.
Конкретные примеры восприимчивых новообразований, которые охвачены изобретением, включают, но не ограничиваются ими:
аденокарциному Баррета;
рак желчных протоков;
рак молочной железы;
рак шейки матки;
холангиокарциному;
опухоли центральной нервной системы, включая первичные опухоли ЦНС, такие как глиобластомы, астроцитомы (включая мультиформную глиобластому) и эпендимомы, и вторичные опухоли ЦНС (то есть метастазы в центральную нервную систему опухолей, возникших вне центральной нервной системы),
рак толстой и прямой кишки, включая карциному толстой кишки;
рак желудка;
карциному головы и шеи, включая плоскоклеточный рак головы и шеи;
гематологические раковые заболевания, включая лейкемии и лимфомы, такие как острый лимфобластный лейкоз, острый миелогенный лейкоз (ОМЛ), миелодиспластические синдромы, хронический миелогенный лейкоз, лимфома Ходжкина, лимфома неходжкинского типа, мегакариобластный лейкоз, множественная множественная миелома и эритролейкоз;
гепатоцеллюлярную карциному;
рак легких, включая мелкоклеточный рак легких и немелкоклеточный рак легких;
рак яичников;
рак эндометрия;
рак поджелудочной железы;
аденому гипофиза;
рак простаты;
рак почки;
саркому;
раковые заболевания кожи, включая меланомы; и
раковые заболевания щитовидной железы.
Приведенный выше список предназначен для раскрытия каждого из перечисленных новообразований в отдельности. В одном определенном варианте осуществления восприимчивое новообразование представляет собой новообразование, которое несет в себе мутацию в B-Raf.
В другом варианте осуществления предложена диспергируемая таблетка соединения А для применения при лечении восприимчивого новообразования (например, аденокарциномы Баррета; карциномы желчных путей; рака молочной железы; рака шейки матки; холангиокарциномы; опухолей центральной нервной системы, включая первичные опухоли ЦНС, такие как глиобластомы, астроцитомы (например, мультиформная глиобластома) и эпендимомы, и вторичные опухоли ЦНС (то есть метастазы в центральную нервную систему опухолей, возникших вне центральной нервной системы), рака толстой и прямой кишки, включая карциному толстой кишки; рака желудка; рака головы и шеи, в том числе плоскоклеточного рака головы и шее; гематологического рака, включая лейкозы и лимфомы, такие как острый лимфобластный лейкоз, острый миелобластный лейкоз (ОМЛ), миелодиспластические синдромы, хронический миелолейкоз, лимфому Ходжкина, неходжкинскую лимфому, мегакариобластный лейкоз, множественную миелому и эритролейкемиу; гепатоцеллюлярной карциномы, рака легкого, включая мелкоклеточный рак легкого и немелкоклеточный рак легкого; рака яичников; рака эндометрия; рака поджелудочной железы; аденомы гипофиза; рака простаты; рака почки; саркомы; раковых заболеваний кожи, включая меланомы; и раковых заболеваний щитовидной железы) у млекопитающего (например, человека), нуждающегося в этом.
Используемый здесь термин «совместное введение» означает либо одновременное введение, либо любой способ раздельного последовательного введения диспергируемой таблетки, содержащей соединение А, и другого активного средства (агента) или средств (агентов), о которых известно, что они полезны для лечения рака, включая химиотерапию и лучевую терапию. Термин «дополнительное(ый) активное(ый) средство (агент) или средства (агенты)» в данном контексте включает в себя любое соединение или терапевтическое средство, которые известны или которые демонстрируют полезные свойства при введении пациенту, нуждающемуся в лечении рака. Используемый здесь термин «дополнительный активный агент или агенты» используется взаимозаменяемо с дополнительным противоопухолевым средством или средствами. Если введение не является одновременным, то предпочтительно соединения вводить как можно ближе одно за другим. Кроме того, не имеет значения, вводят ли соединения в одной и той же лекарственной форме, например одно соединение можно вводить путем инъекции, а другое соединение можно вводить перорально. Соответственно, «совместное введение» будет состоять, по существу, из диспергируемой таблетки, содержащей соединение А, и второй фармацевтической лекарственной формы, содержащей дополнительное активное средство. Соответственно, «совместное введение» будет состоять, по существу, из диспергируемой таблетки, содержащей соединение А, второй фармацевтической лекарственной формы, содержащей дополнительное активное средство, и третьей фармацевтической лекарственной формы, содержащей еще одно дополнительное активное средство.
Как правило, любое противоопухолевое средство, обладающее активностью против чувствительной опухоли, подвергающейся лечению, можно вводить совместно при лечении рака, как описано в настоящем изобретении. Примеры таких средств можно найти в Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6th edition (February 15, 2001), Lippincott Williams & Wilkins Publishers. Специалист в данной области техники сможет определить, какие комбинации средств будут полезны на основании конкретных характеристик лекарств и ракового заболевания, требующего лечения. Типичные противоопухолевые средства, используемые в настоящем изобретении, включают, но не ограничиваются ими, средства против микротрубочек, такие как дитерпеноиды и алкалоиды барвинка; координационные комплексы платины; алкилирующие агенты, такие как азотистые иприты, оксазафосфорины, алкилсульфонаты, нитрозомочевины и триазены; антибиотики, такие как антрациклины, актиномицины и блеомицины; ингибиторы топоизомеразы II, такие как эпиподофиллотоксины; антиметаболиты, такие как пуриновые и пиримидиновые аналоги и антифолатные соединения; ингибиторы топоизомеразы I, такие как камптотецины; гормоны и гормональные аналоги; ингибиторы сигнальных путей; нерецепторные ингибиторы ангиогенеза тирозинкиназы; иммунотерапевтические средства; проапоптотические средства; ингибиторы передачи сигналов клеточного цикла; ингибиторы протеасом; и ингибиторы метаболизма рака.
Примерами дополнительного активного средства или средств (противоопухолевого средства) для использования в комбинации или совместно с предлагаемой в данном изобретении фармацевтической лекарственной формой являются химиотерапевтические средства.
Средства против микротрубочек или антимитотические средства представляют собой фазоспецифические агенты, действующие против микротрубочек опухолевых клеток в ходе фазы М или фазы митоза клеточного цикла. Примеры средств против микротрубочек включают, но не ограничиваются ими, дитерпеноиды и алкалоиды барвинка.
Дитерпеноиды, полученные из природных источников, являются фазоспецифическими противораковыми средствами, которые действуют на G2/M фазы клеточного цикла. Считается, что дитерпеноиды стабилизируют субъединицу β-тубулина микротрубочек путем связывания с этим белком. Разрушение структуры белка, по-видимому, ингибируется митозом и последующей гибелью клеток. Примеры дитерпеноидов включают, но не ограничиваются ими, паклитаксел и его аналог доцетаксел.
Паклитаксел - это 5β,20-эпокси-1,2α,4,7β,10β,13α-гекса-гидрокситакс-11-ен-9-она 4,10-диацетат 2-бензоат 13-сложный эфир с (2R,3S)-N-бензоил-3-фенилизосерином; представляет собой натуральный дитерпеновый продукт, выделенный из тихоокеанского тиса Taxus brevifolia и поступающий в продажу в виде раствора для инъекций TAXOL. Это терпен - член семейства таксанов. Паклитаксел был одобрен для клинического использования в лечении рефрактерного рака яичников и рака молочной железы в Соединенных Штатах.
Доцетаксел - это (2R,3S)-N-карбокси-3-фенилизосерина N-трет-бутиловый сложный эфир, 13-сложный эфир с 5β-20-эпокси-1,2α,4,7β,10β,13α-гексагидрокситаксакс-11-ен-9-она 4-ацетатом 2-бензоатом, тригидрат; поступает в продажу в виде раствора для инъекций TAXOTERE. Доцетаксел показан для лечения рака молочной железы. Доцетаксел является полусинтетическим производным паклитаксела q.v., полученного с использованием природного предшественника - 10-деацетилбаккатина III, экстрагированного из хвои европейского тисового дерева. Дозолимитирующей токсичностью для доцетаксела является нейтропения.
Алкалоиды барвинка являются фазоспецифическими противоопухолевыми агентами, полученными из растения барвинка. Алкалоиды барвинка действуют на М-фазу (митоз) клеточного цикла, специфически связываясь с тубулином. Как следствие, связанная молекула тубулина не способна полимеризоваться в микротрубочки. Считается, что митоз останавливается в метафазе с последующей гибелью клеток. Примеры алкалоидов барвинка включают, но не ограничиваются ими, винбластин, винкристин и винорелбин.
Винбластин - винкалейкобластин сульфат - поступает в продажу в виде раствора для инъекций VELBAN. Хотя этот препарат потенциально показан в качестве терапии второй линии для различных солидных опухолей, он в первую очередь показан при лечении рака яичка и различных лимфом, включая болезнь Ходжкина; и лимфоцитарных и гистиоцитарных лимфом. Миелосупрессия является дозолимитирующим побочным эффектом винбластина.
Винкристин - винкалейкобластин, 22-оксо-, сульфат - поступает в продажу в виде раствора для инъекций ONCOVIN. Винкристин показан для лечения острых лейкозов, а также применяется в схемах лечения злокачественных лимфомы Ходжкина и неходжкинской лимфомы. Алопеция и неврологические эффекты являются наиболее распространенным побочным действием винкристина и в меньшей степени проявляются эффекты миелосупрессии и желудочно-кишечного мукозита.
Винорелбин - 3',4'-дидегидро-4'-дезокси-C'-норвинкалеукобластина [R-(R*,R*)-2,3-дигидроксибутандиоат (1:2)(соль)] - поступает в продажу в виде раствора для инъекций винорелбина тартрата (NAVELBINE) и является полусинтетическим алкалоидом барвинка. Винорелбин показан в качестве отдельного средства или в сочетании с другими химиотерапевтическими средствами, такими как цисплатин, при лечении различных солидных опухолей, в частности немелкоклеточного рака легкого, прогрессирующего рака молочной железы и гормонально-рефрактерного рака предстательной железы. Миелосупрессия является наиболее часто встречающимся дозолимитирующим побочным эффектом винорелбина.
Координационные комплексы платины представляют собой нефазоспецифические противораковые средства, которые взаимодействуют с ДНК. Комплексы платины проникают в опухолевые клетки, подвергаются гидратации и образуют внутри- и межцепочечные перекрестные связи с ДНК, вызывая неблагоприятные биологические эффекты для опухоли. Примеры координационных комплексов платины включают, но не ограничиваются ими, цисплатин и карбоплатин.
Цисплатин - цис-диаминдихлороплатин - поступает в продажу в виде раствора для инъекций PLATINOL. Цисплатин в первую очередь показан при лечении метастатического рака яичек и яичников и запущенного рака мочевого пузыря. Основными дозолимитирующими побочными эффектами цисплатина являются нефротоксичность, которая может контролироваться гидратацией и диурезом, а также ототоксичность.
Карбоплатин - [1,1-циклобутан-дикарбоксилато(2-)-O, O']диаминплатина - поступает в продажу в виде раствора для инъекций PARAPLATIN. Карбоплатин в первую очередь показан для первой и второй линии лечения прогрессирующего рака яичников. Подавление функции костного мозга является дозолимитирующей токсичностью карбоплатина.
Алкилирующие агенты являются нефазоспецифическими противораковыми средствами и сильными электрофилами. Обычно алкилирующие агенты образуют ковалентные связи с ДНК путем алкилирования нуклеофильных фрагментов молекулы ДНК, таких как фосфатные, амино-, сульфгидрильные, гидроксильные, карбоксильные и имидазольные группы. Такое алкилирование нарушает функцию нуклеиновой кислоты, приводя к гибели клеток. Примеры алкилирующих агентов включают, но не ограничиваются ими, азотистые иприты, такие как циклофосфамид, мелфалан и хлорамбуцил; алкилсульфонаты, такие как бусульфан; нитрозомочевины, такие как кармустин; и триазены, такие как дакарбазин.
Циклофосфамид - 2-[бис(2-хлорэтил)амино]тетрагидро-2H-1,3,2-оксазафосфорин-2-оксид моногидрат - поступает в продажу в виде раствора для инъекций или таблеток CYTOXAN®. Циклофосфамид показан в качестве отдельного средства или в сочетании с другими химиотерапевтическими средствами при лечении злокачественных лимфом, множественной миеломы и лейкемии. Алопеция, тошнота, рвота и лейкопения являются наиболее распространенными дозолимитирующими побочными эффектами циклофосфамида.
Мелфалан - 4-[бис(2-хлорэтил)амино]-L-фенилаланин - поступает в продажу в виде раствора для инъекций или таблеток ALKERAN®. Мелфалан показан для паллиативного лечения множественной миеломы и неоперабельной эпителиальной карциномы яичника. Подавление функции костного мозга является наиболее распространенным дозолимитирующим побочным эффектом мелфалана.
Хлорамбуцил - 4-[бис(2-хлорэтил)амино]бензолбутановая кислота - поступает в продажу в виде таблеток LEUKERAN®. Хлорамбуцил показан для паллиативного лечения хронического лимфатического лейкоза и злокачественных лимфом, таких как лимфосаркома, гигантская фолликулярная лимфома и болезнь Ходжкина. Подавление функции костного мозга является наиболее распространенным дозолимитирующим побочным эффектом хлорамбуцила.
Бусульфан - 1,4-бутандиолдиметансульфонат - поступает в продажу в виде таблеток MYLERAN®. Бусульфан показан для паллиативного лечения хронического миелогенного лейкоза. Подавление функции костного мозга является наиболее распространенными дозолимитирующими побочными эффектами бусульфана.
Кармустин - 1,3-[бис(2-хлорэтил)-1-нитрозомочевина - поступает в продажу в виде отдельных флаконов с лиофилизированным материалом под названием BiCNU®. Кармустин показан как отдельное средство или в сочетании с другими средствами для паллиативного лечения опухолей головного мозга, множественной миеломы, болезни Ходжкина и неходжкинских лимфом. Развивающаяся впоследствии миелосупрессия является наиболее часто встречающимся дозолимитирующим побочным действием кармустина.
Дакарбазин - 5-(3,3-диметил-1-триазено)имидазол-4-карбоксамид - поступает в продажу в виде отдельных флаконов с материалом под названием DTIC-Dome®. Дакарбазин показан для лечения метастатической злокачественной меланомы, а в сочетании с другими средствами - для второй линии лечения болезни Ходжкина. Тошнота, рвота и анорексия являются наиболее распространенными дозолимитирующими побочными эффектами дакарбазина.
Антибиотики-антинеопластики представляют собой нефазоспецифические средства, которые связываются с ДНК или вставляются в нее. Как правило, такое воздействие приводит к стабильным комплексам ДНК или разрыву цепей, что нарушает обычную функцию нуклеиновых кислот, приводя к гибели клеток. Примеры антибиотиков-антинеопластиков включают, но не ограничиваются ими, актиномицины, такие как дактиномицин, антроциклины, такие как даунорубицин и доксорубицин; и блеомицины.
Дактиномицин, также известный как Актиномицин D, поступает в продажу в форме для инъекций под названием COSMEGEN®. Дактиномицин показан для лечения опухоли Вильмса и рабдомиосаркомы. Тошнота, рвота и анорексия являются наиболее распространенными дозолимитирующими побочными эффектами дактиномицина.
Даунорубицин - (8S-цис-)-8-ацетил-10-[(3-амино-2,3,6-тридезокси-α-L-ликсогексопиранозил)окси]-7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12-нафтацендиона гидрохлорид - поступает в продажу в виде липосомальной формы для инъекций DAUNOXOME® или формы для инъекций CERUBIDINE®. Даунорубицин показан для индукции ремиссии при лечении острого нелимфоцитарного лейкоза и запущенной ВИЧ-ассоциированной саркомы Капоши. Миелосупрессия является наиболее часто встречающимся дозолимитирующим побочным эффектом даунорубицина.
Доксорубицин - (8S,10S)-10-[(3-амино-2,3,6-тридезокси-α-L-ликсогексопиранозил)окси]-8-гликолоил-7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12-нафтацендиона гидрохлорид - поступает в продажу в виде формы для инъекций под названием RUBEX® или ADRIAMYCIN RDF®. Доксорубицин в первую очередь показан для лечения острого лимфобластного лейкоза и острого миелобластного лейкоза, но также является полезным компонентом в лечении некоторых солидных опухолей и лимфом. Миелосупрессия является наиболее часто встречающимся дозолимитирующим побочным эффектом доксорубицина.
Блеомицин представляет собой смесь цитотоксических гликопептидных антибиотиков, выделенных из штамма Streptomyces verticillus, он поступает в продажу в виде BLENOXANE®. Блеомицин показан как отдельное средство или в сочетании с другими средствами в качестве паллиативного лечения плоскоклеточной карциномы, лимфом и карцином яичка. Легочная и кожная токсичность являются наиболее распространенными дозолимитирующими побочными эффектами блеомицина.
Ингибиторы топоизомеразы II включают, но не ограничиваются этим, эпиподофиллотоксины.
Эпиподофиллотоксины являются фазоспецифическими противоопухолевыми агентами, полученными из растения мандрагоры. Эпиподофиллотоксины обычно поражают клетки в S и G2 фазах клеточного цикла, образуя тройной комплекс с топоизомеразой II и ДНК, вызывая разрывы цепи ДНК. Разрывы нитей накапливаются, и наступает гибель клеток. Примеры эпиподофиллотоксинов включают, но не ограничиваются ими, этопозид и тенипозид.
Этопозид - 4'-диметил-эпиподофиллотоксин 9[4,6-0-(R)-этилиден-β-D-глюкопиранозид] - поступает в продажу в виде раствора для инъекций или капсул под названием VePESID и широко известен как VP-16. Этопозид показан как отдельное средство или в сочетании с другими химиотерапевтическими средствами при лечении рака яичка и немелкоклеточного рака легкого. Миелосупрессия является наиболее часто встречающимся побочным действием этопозида. Случаи лейкопении имеют тенденцию к более тяжелому течению, чем тромбоцитопения.
Тенипозид - 4'-диметил-эпиподофиллотоксин 9[4,6-0-(R)-тенилиден-β-D-глюкопиранозид] - поступает в продажу в виде раствора для инъекций под названием VUMON и широко известен как VM-26. Тенипозид показан как отдельное средство или в сочетании с другими химиотерапевтическими средствами при лечении острого лейкоза у детей. Миелосупрессия является наиболее часто встречающимся дозолимитирующим побочным эффектом тенипозида. Тенипозид может вызывать как лейкопению, так и тромбоцитопению.
Антиметаболиты онкогенов представляют собой фазоспецифическими противоопухолевые агенты, которые действуют на S-фазу (синтез ДНК) клеточного цикла, ингибируя синтез ДНК или ингибируя синтез пуринового или пиримидинового основания и тем самым ограничивая синтез ДНК. Вследствие этого S-фаза останавливается, и впоследствии клетка гибнет. Примеры антиметаболитных противоопухолевых агентов включают, но не ограничиваются ими, фторурацил, метотрексат, цитарабин, мекаптопурин, тиогуанин и гемцитабин.
5-Фторурацил-5-фтор-2,4-(1Н,3Н)пиримидиндион - поступает в продажу под названием фторурацил. Введение 5-фторурацила приводит к ингибированию синтеза тимидилата, он также включается в структуры и РНК, и ДНК. Результатом обычно является гибель клеток. 5-Фторурацил показан как отдельное средство или в сочетании с другими химиотерапевтическими средствами при лечении рака молочной железы, толстого кишечника, прямой кишки, желудка и поджелудочной железы. Миелосупрессия и мукозит являются дозолимитирующими побочными действиями 5-фторурацила. Другие аналоги фторпиримидина включают 5-фтордезоксиуридин (флоксуридин) и 5-фтордезоксиуридина монофосфат.
Цитарабин - 4-амино-1-β-D-арабинофуранозил-2(1H)-пиримидинон - поступает в продажу под названием CYTOSAR-U и широко известен как Ара-С. Считается, что цитарабин фазоспецифичен в отношении S-фазы клетки, ингибируя удлинение цепи ДНК за счет терминального включения цитарабина в растущую цепь ДНК. Цитарабин показан как отдельное средство или в сочетании с другими химиотерапевтическими средствами при лечении острого лейкоза. Другими аналогами цитидина являются 5-азацитидин и 2',2'-дифтордезоксицитидин (гемцитабин). Цитарабин вызывает лейкопению, тромбоцитопению и мукозит.
Меркаптопурин - 1,7-дигидро-6Н-пурин-6-тиона моногидрат - поступает в продажу под названием PURINETHOL. Меркаптопурин фазоспецифичен в отношении S-фазы клетки, ингибируя синтез ДНК по еще не установленному механизму. Меркаптопурин показан как отдельное средство или в сочетании с другими химиотерапевтическими средствами при лечении острого лейкоза. Ожидаемыми побочными действиями при высоких дозах меркаптопурина являются миелосупрессия и желудочно-кишечный мукозит. Азатиоприн - это полезный аналог меркаптопурина.
Тиогуанин - 2-амино-1,7-дигидро-6Н-пурин-6-тион - поступает в продажу под названием TABLOID. Тиогуанин фазоспецифичен в отношении S-фазы клетки, ингибируя синтез ДНК по еще не установленному механизму. Тиогуанин показан как отдельное средство или в сочетании с другими химиотерапевтическими средствами при лечении острого лейкоза. Миелосупрессия, включая лейкопению, тромбоцитопению и анемию, является наиболее распространенным дозолимитирующим побочным действием при введении тиогуанина. Однако проявляется и побочные желудочно-кишечные действия, которые могут быть дозолимитирующими. Другие аналоги пурина включают пентостатин, эритрогидроксинониладенин, флударабина фосфат и кладрибин.
Гемцитабин - 2'-дезокси-2', 2'-дифторцитидина моногидрохлорид (β-изомер) - поступает в продажу под названием GEMZAR. Гемцитабин фазоспецифичен в отношении S-фазы клетки, блокируя клетки на границе фаз G1/S. Гемцитабин показан в комбинации с цисплатином при лечении местно-распространенного немелкоклеточного рака легкого и сам по себе при лечении местно-распространенного рака поджелудочной железы. Миелосупрессия, включая лейкопению, тромбоцитопению и анемию, является наиболее распространенным дозолимитирующим побочным действием при введении гемцитабина.
Метотрексат - N-[4[[(2,4-диамино-6-птеридинил)метил]метиламино]бензоил]-L--глутаминовая кислота - поступает в продажу в виде метотрексата натрия. Метотрексат специфично действует на S-фазу, ингибируя синтез ДНК, репарацию и/или репликацию ДНК путем ингибирования редуктазы дигидрофолиевой кислоты, которая необходима для синтеза пуриновых нуклеотидов и тимидилата. Метотрексат показан в виде отдельного средства или в сочетании с другими химиотерапевтическими средствами при лечении хориокарциномы, менингеальной лейкемии, неходжкинской лимфомы и карцином молочной железы, головы, шеи, яичника и мочевого пузыря. Ожидаемым побочным действием при введении метотрексата являются миелосупрессия (лейкопения, тромбоцитопения и анемия) и мукозит.
Камптотецины в качестве ингибиторов топоизомеразы I, включая камптотецин и производные камптотецина, доступны или находятся в стадии разработки. Считается, что цитотоксическая активность камптотецинов связана с его действием, ингибирующим топоизомеразу I. Примеры камптотецинов включают, но не ограничиваются ими, иринотекан, топотекан и различные оптические формы 7-(4-метилпиперазинометилен)-10,11-этилендиокси-20-камптотецина, описанные ниже.
Иринотекан HCl - (4S)-4,11-диэтил-4-гидрокси-9-[(4-пиперидинопиперидино) карбонилокси]-1H-пирано[3',4',6,7]индолизино[1,2-b]хинолин-3,14(4H,12Н)-диона гидрохлорид - поступает в продажу в виде раствора для инъекций CAMPTOSAR.
Иринотекан является производным камптотецина, который вместе со своим активным метаболитом SN-38 связывается с комплексом топоизомераза I - ДНК. Считается, что цитотоксичность возникает в результате невосстанавливаемых разрывов двойных цепей, вызванных взаимодействием топоизомераза I: ДНК : иринтекан или тройной комплекс SN-38 с ферментами репликации. Иринотекан показан для лечения метастатического рака толстого кишечника или прямой кишки. Дозолимитирующими побочными эффектами иринотекана HCl являются миелосупрессия, включая нейтропению, и воздействие на желудочно-кишечный тракт, включая диарею.
Топотекан HCl - (S)-10-[(диметиламино)метил]-4-этил-4,9-дигидрокси-1H-пирано[3',4',6,7]индолизино[1,2-b]хинолин-3,14-(4H,12H)-диона моногидрохлорид - поступает в продажу в виде раствора для инъекций HYCAMTIN®. Топотекан является производным камптотецина, который связывается с комплексом топоизомераза I - ДНК и предотвращает повторное лигирование одноцепочечных разрывов, вызванных топоизомеразой I, в ответ на напряжение при скручивании молекулы ДНК. Топотекан показан для второй линии лечения метастатического рака яичника и мелкоклеточного рака легкого. Дозолимитирующими побочными эффектами топотекана HCl является миелосупрессия, в первую очередь - нейтропения.
Представляет также интерес следующее производное камптотецина, имеющее Формулу A, включая форму рацемической смеси (R, S), а также энантиомеры R и S:

известное под химическим названием "7-(4-метилпиперазинометилен)-10,11-этилендиокси-20(R, S)-камптотецин (рацемическая смесь), или "7-(4-метилпиперазинометилен)-10,11-этилендиокси-20(R)-камптотецин (R-энантиомер), или 7-(4-метилпиперазинометилен)-10,11-этилендиокси-20(S)-камптотецин (S-энантиомер). Такое соединение, а также родственные соединения описаны, включая способы получения, в патентах США №№ 6063923; 5342947; 5559235; и 5491237.
Гормоны и гормональные аналоги - это полезные соединения для лечения раковых заболеваний, при которых существует связь между гормоном(ами) и ростом и/или отсутствием роста опухоли. Примеры гормонов и гормональных аналогов, полезных при лечении рака, включают, но не ограничиваются ими, адренокортикостероиды, такие как преднизон и преднизолон, которые полезны при лечении злокачественной лимфомы и острого лейкоза у детей; аминоглутетимид и другие ингибиторы ароматазы, такие как анастрозол, летразол, воразол и экземестан, полезные при лечении адренокортикальной карциномы и гормонзависимой карциномы молочной железы, содержащей рецепторы эстрогена; прогестрины, такие как ацетат мегестрола, пригодный для лечения гормонозависимого рака молочной железы и карциномы эндометрия; эстрогены, андрогены и антиандрогены, такие как флутамид, нилутамид, бикалутамид, ципротерона ацетат и 5α-редуктазы, такие как финастерид и дутастерид, полезные при лечении рака предстательной железы и доброкачественной гипертрофии предстательной железы; антиэстрогены, такие как тамоксифен, торемифен, ралоксифен, дролоксифен, йодоксифен, а также селективные модуляторы рецептора эстрогена (SERMS), такие как описаны в патентах США № 5681835; 5877219; и 6207716, полезные для лечения гормонозависимой карциномы молочной железы и других восприимчивых форм рака; и гонадотропин-рилизинг-гормон (GnRH) и его аналоги, которые стимулируют высвобождение лейтинизирующего гормона (LH) и/или фолликулостимулирующего гормона (FSH) для лечения рака предстательной железы, например, агонисты и антагонисты LHRH, такие как ацетат госерелина и лупролид.
Ингибиторы пути передачи сигнала - это ингибиторы, которые блокируют или ингибируют химический процесс, вызывающий внутриклеточные изменения. В данном контексте это изменение представляет собой пролиферацию или дифференцировку клеток. Ингибиторы передачи сигнала, используемые в настоящем изобретении, включают ингибиторы рецепторных тирозинкиназ, нерецепторных тирозинкиназ, блокаторов доменов SH2/SH3, серин/треонинкиназ, фосфотидилинозитол-3-киназ, сигнального пути мио-инозитола и онкогенов Ras.
Некоторые протеинтирозинкиназы катализируют фосфорилирование специфических тирозильных остатков в различных белках, участвующих в регуляции роста клеток. Такие протеинтирозинкиназы можно подразделить на рецепторные или нерецепторные киназы.
Рецепторные тирозинкиназы представляют собой трансмембранные белки, имеющие внеклеточный лиганд-связывающий домен, трансмембранный домен и тирозинкиназный домен. Рецепторные тирозинкиназы участвуют в регуляции роста клеток и обычно называются рецепторами факторов роста. Было показано, что несоответствующая или неконтролируемая активация многих из этих киназ, то есть ненормальная активность киназ рецептора фактора роста, например, при избыточной экспрессии или мутации, приводит к неконтролируемому росту клеток. Соответственно, ненормальная активность таких киназ была связана со злокачественным ростом ткани. Следовательно, ингибиторы таких киназ могут обеспечить способы лечения рака. Рецепторы фактора роста включают, например, рецептор эпидермального фактора роста (EGFr), рецептор фактора роста тромбоцитов (PDGFr), erbB2, erbB4, рецептор фактора роста эндотелия сосудов (VEGFr), тирозинкиназу с иммуноглобулиноподобными и с гомологичными эпидермальному фактору роста доменами (TIE-2), рецептор инсулиноподобного фактора роста -I (IGFI), макрофагальный колониестимулирующий фактор (cfms), BTK, ckit, cmet, рецепторы фактора роста фибробластов (FGF), рецепторы Trk (TrkA, TrkB и TrkC), рецепторы эфрина (eph) и протоонкоген RET. Некоторые ингибиторы рецепторов факторов роста находятся в стадии разработки и включают антагонисты лигандов, антитела, ингибиторы тирозинкиназы и антисмысловые олигонуклеотиды. Рецепторы факторов роста и агенты, ингибирующие функцию рецептора фактора роста, описаны, например, у Kath, John C., Exp. Opin. Ther. Patents (2000) 10(6):803-818; Shawver et al DDT Vol 2, No. 2 February 1997; и Lofts, F. J. et al, "Growth factor receptors as targets", New Molecular Targets for Cancer Chemotherapy, ed. Workman, Paul and Kerr, David, CRC press 1994, London.
Тирозинкиназы, которые не являются рецепторными киназами факторов роста, называются нерецепторными тирозинкиназами. Нерецепторные тирозинкиназы для использования в настоящем изобретении, которые являются мишенями или потенциальными мишенями для противораковых лекарственных средств, включают cSrc, Lck, Fyn, Yes, Jak, cAbl, FAK (киназа фокальной адгезии), тирозинкиназа Брутона и Bcr-Abl. Такие нерецепторные киназы и агенты, которые ингибируют нерецепторную функцию тирозинкиназы, описаны у Sinh, S. and Corey, S.J., (1999) Journal of Hematotherapy and Stem Cell Research 8 (5): 465-80; и Bolen, J.B., Brugge, J.S., (1997) Annual review of Immunology. 15: 371-404.
Блокаторы домена SH2/SH3 - это агенты, которые нарушают связывание домена SH2 или домена SH3 в различных ферментах или адапторных белках, включая субъединицу PI3-K p85, киназы семейства Src, молекулы-адаптеры (Shc, Crk, Nck, Grb2) и Ras-GAP. Домены SH2/SH3 в качестве мишеней для противораковых препаратов обсуждаются у Smithgall, TE (1995), Journal of Pharmacological and Toxicological Methods. 34(3) 125-32.
Ингибиторы серин/треонинкиназ, включая MAP каскадные блокаторы MAP-киназы, которые включают блокаторы митогенной или внеклеточно регулируемой киназы (MEK) и внеклеточно регулируемые киназы (ERK); и блокаторы членов семейства протеинкиназы С, в том числе блокаторы PKC (альфа, бета, гамма, эпсилон, мю, лямбда, йота, дзета). Семейство киназ IkB (IKKa, IKKb), семейство киназ PKB, члены семейства киназ akt, киназы бета-рецепторов PDK1 и TGF. Такие серин/треонинкиназы и их ингибиторы описаны у Yamamoto, T., Taya, S., Kaibuchi, K., (1999), Journal of Biochemistry. 126 (5) 799-803; Brodt, P, Samani, A., and Navab, R. (2000), Biochemical Pharmacology, 60. 1101-1107; Massague, J., Weis-Garcia, F. (1996) Cancer Surveys. 27:41-64; Philip, P.A., and Harris, A.L. (1995), Cancer Treatment and Research. 78: 3-27, Lackey, K. et al. Bioorganic and Medicinal Chemistry Letters, (10), 2000, 223-226; Патент США № 6268391; Pearce, L.R et al. Nature Reviews Molecular Cell Biology (2010) 11, 9-22; и Martinez-Iacaci, L., et al. Int. J. Cancer (2000), 88(1), 44-52.
Соответственно, фармацевтически активное соединение данного изобретения используют в комбинации с ингибитором MEK. Предусмотрен N-{3-[3-циклопропил-5-(2-фтор-4-иод-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]фенил}ацетамид или его фармацевтически приемлемая соль или сольват, который раскрыт и заявлен в Международной патентной заявке № PCT/JP2005/011082, зарегистрированной 10 июня 2005 г., полное раскрытие которой включено в данный документ ссылкой. N-{3-[3-циклопропил-5-(2-фтор-4-иод-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2H-пиридо[4,3-d]пиримидин-1-ил]фенил}ацетамид можно получать, как описано в Международной патентной заявке № PCT/JP2005/011082
Соответственно, фармацевтически активное соединение данного изобретения используют в комбинации с ингибитором Akt. Предусмотрен N-{(1S)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1H-пиразол-5-ил)-2-тиофенкарбоксамид или его фармацевтически приемлемая соль, который раскрыт и заявлен в Международной патентной заявке № PCT/US2008/053269, зарегистрированной 7 февраля 2008 г.; публикации Международной заявки № WO 2008/098104, зарегистрированной 14 августа 2008 г., полное раскрытие которых включено в данный документ ссылкой. N-{(1S)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1H-пиразол-5-ил)-2-тиофенкарбоксамид представляет собой соединение примера 96, и его можно получать согласно описанию в Международной патентной заявке № PCT/US2008/053269. Соответственно, N-{(1S)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1H-пиразол-5-ил)-2-тиофенкарбоксамид имеет форму гидрохлоридной соли. Специалист в данной области может получать данную соль, используя описание в Международной заявке № PCT/US2010/022323, зарегистрированной 28 января 2010 г.
Ингибиторы членов семейства фосфотидилинозитол-3-киназы, включая блокаторы PI3-киназы, ATM, ДНК-ПК и Ku, также могут быть полезны в настоящем изобретении. Такие киназы обсуждаются у Abraham, R.T. (1996), Current Opinion in Immunology. 8 (3) 412-8; Canman, C.E., Lim, D.S. (1998), Oncogene 17 (25) 3301-3308; Jackson, S.P. (1997), International Journal of Biochemistry and Cell Biology. 29 (7):935-8; и Zhong, H. et al. Cancer res, (2000) 60(6), 1541-1545.
В настоящем изобретении также представляют интерес ингибиторы передачи сигналов мио-инозитола, такие как блокаторы фосфолипазы С и аналоги мио-инозитола. Такие ингибиторы сигнальных путей описаны у Powis, G., and Kozikowski A., (1994) New Molecular Targets for Cancer Chemotherapy ed., Paul Workman and David Kerr, CRC press 1994, London.
Другой группой ингибиторов пути передачи сигнала являются ингибиторы Ras Oncogene. Такие ингибиторы включают ингибиторы фарнезилтрансферазы, геранилгеранилтрансферазы и протеаз CAAX, а также антисмысловые олигонуклеотиды, рибозимы и иммунотерапию. Было показано, что такие ингибиторы блокируют активацию ras в клетках, содержащих мутантный ras дикого типа, тем самым они действуют как антипролиферативные средства. Ингибирование онкогена Ras обсуждается у Scharovsky, O.G., Rozados, V.R., Gervasoni, S.I. Matar, P. (2000), Journal of Biomedical Science. 7(4) 292-8; Ashby, M.N. (1998), Current Opinion in Lipidology. 9 (2) 99-102; и BioChim. Biophys. Acta, (19899) 1423(3):19-30.
Как упомянуто выше, антагонисты антител к связыванию рецепторной киназы с лигандами также могут служить ингибиторами передачи сигнала. Эта группа ингибиторов пути передачи сигнала включает использование гуманизированных антител к внеклеточному лиганд-связывающему домену рецепторных тирозинкиназ. Например, C225 EGFR-специфичное антитело Imclone (см. Green, M.C. et al, Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev., (2000), 26(4), 269-286); антитело Herceptin® к erbB2; и 2CB VEGFR2-специфичное антитело (см. Brekken, R.A. et al, Selective Inhibition of VEGFR2 Activity by a monoclonal Anti-VEGF antibody blocks tumor growth in mice, Cancer Res. (2000) 60, 5117-5124).
Ингибиторы ангиогенеза нерецепторных киназ также могут быть полезны в настоящем изобретении. Ингибиторы VEGFR и TIE2, связанные с ангиогенезом, обсуждались выше в связи с ингибиторами передачи сигнала (оба рецептора являются рецепторными тирозинкиназами). Ангиогенез в целом связан с сигнальным путем erbB2/EGFR, поскольку об ингибиторах erbB2 и EGFR известно, что они подавляют ангиогенез - прежде всего экспрессию VEGF. Соответственно, ингибиторы нерецепторных тирозинкиназ можно использовать в комбинации с соединениями настоящего изобретения. Например, антитела к VEGF, которые не распознают VEGFR (рецепторную тирозинкиназу), но связываются с лигандом; низкомолекулярные ингибиторы интегрина (alpha(v) beta3), которые ингибируют ангиогенез; эндостатин и ангиостатин (не-РТК) также могут оказаться полезными в сочетании с раскрываемыми соединениями.
Средства, используемые в иммунотерапевтических схемах, также могут быть полезны в комбинации с соединениями Формулы (I). Существует ряд иммунологических стратегий для генерации иммунного ответа. Эти стратегии обычно относятся к вакцинации против опухолей. Эффективность иммунологических подходов можно значительно повысить путем комбинированного ингибирования сигнальных путей с использованием низкомолекулярного ингибитора. Обсуждение подхода иммунологической/противоопухолевой вакцины против erbB2/EGFR можно найти у Reilly RT et al. (2000), Cancer Res. 60: 3569-3576; и у Chen Y, Hu D, Eling DJ, Robbins J, and Kipps TJ. (1998), Cancer Res. 58: 1965-1971.
Средства, используемые в проапоптотических схемах (например, bcl-2 антисмысловые олигонуклеотиды), также можно использовать в комбинации по настоящему изобретению. Члены семейства белков Bcl-2 блокируют апоптоз. Поэтому повышенную регуляцию bcl-2 связывают с устойчивостью к химическому воздействию. Исследования показали, что эпидермальный фактор роста (EGF) стимулирует антиапоптотические члены семейства bcl-2 (т.е., mcl-1). Таким образом, стратегии, разработанные для подавления экспрессии bcl-2 в опухолях, продемонстрировали клиническое преимущество, а именно, антисмысловой олигонуклеотид Genta G3139 bcl-2. Такие проапоптотические стратегии с использованием антисмысловой олигонуклеотидной стратегии для bcl-2 обсуждаются в Water JS et al. (2000), J. Clin. Oncol. 18: 1812-1823; и Kitada S et al. (1994), Antisense Res. Dev. 4: 71-79.
Ингибиторы сигнальных путей клеточного цикла ингибируют молекулы, участвующие в регуляции клеточного цикла. Семейство протеинкиназ, называемое циклинзависимыми киназами (CDK), и их взаимодействие с семейством белков, называемых циклинами, регулирует протекание клеточного цикла эукариот. Скоординированная активация и инактивация различных комплексов циклин/CDK необходимы для нормального протекания клеточного цикла. Несколько ингибиторов сигнальных путей клеточного цикла находятся в стадии разработки. Например, примеры циклинзависимых киназ, включая CDK2, CDK4 и CDK6, и их ингибиторы приведены, например, в Rosania et al, Exp. Opin. Ther. Patents (2000) 10(2):215-230. Кроме того, p21WAF1/CIP1 был описан как сильный и универсальный ингибитор циклинзависимых киназ (Cdks) (Ball et al., Progress in Cell Cycle Res., 3: 125 (1997)). Соединения, которые известны тем, что они индуцируют экспрессию p21WAF1/CIP1, вовлечены в подавление пролиферации клеток, и была отмечена их активность, направленная на подавления опухолей (Richon et al., Proc. Nat Acad. Sci. U.S.A. 97(18): 10014-10019 (2000)), и они включены в качестве ингибиторов передачи сигналов клеточного цикла. Ингибиторы гистондеацетилазы (HDAC) вовлечены в транскрипционную активацию p21WAF1/CIP1 (Vigushin et al., Anticancer Drugs, 13(1): 1-13 (Jan 2002)), и представляют собой подходящие ингибиторы сигнальных путей клеточного цикла для использования в данном изобретении.
Примеры таких ингибиторов HDAC включают:
1. Вориностат, включая его фармацевтически приемлемые соли. Marks et al., Nature Biotechnology 25, 84-90 (2007); Stenger, Community Oncology 4, 384-386 (2007).
Вориностат имеет следующую химическую структуру и название:
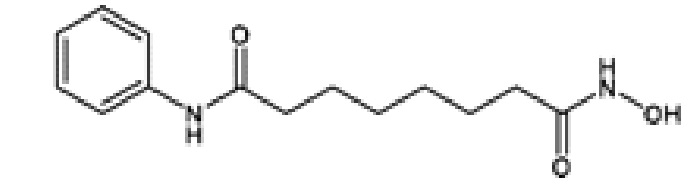
N-гидрокси-N'-фенил-октандиамид
2. Ромидепсин, включая его фармацевтически приемлемые соли.
Vinodhkumar et al., Biomedicine & Pharmacotherapy 62 (2008) 85-93.
Ромидепсин имеет следующую химическую структуру и название:

(1S,4S,7Z,10S,16E,21R)-7-этилиден-4,21-ди(пропан-2-ил)-2-окса-12,13-дитиа-5,8,20,23-тетрабицикло[8.7.6]трикоз-16-ен-3,6,9,19,22-пентон
3. Панобиностат, включая его фармацевтически приемлемые соли. Drugs of the Future 32(4): 315-322 (2007).
Панобиностат имеет следующую химическую структуру и название:

(2E)-N-гидрокси-3-[4-({[2-(2-метил-1H-индол-3-ил)этил]амино}метил)фенил]акриламид
4. Вальпроевая кислота, включая ее фармацевтически приемлемые соли. Gottlicher, et al., EMBO J. 20(24): 6969-6978 (2001).
Вальпроевая кислота имеет следующую химическую структуру и название:

2-пропилпентановая кислота
5. Моцетиностат (MGCD0103), включая его фармацевтически приемлемые соли. Balasubramanian et al., Cancer Letters 280: 211-221 (2009).
Моцетиностат имеет следующую химическую структуру и название:
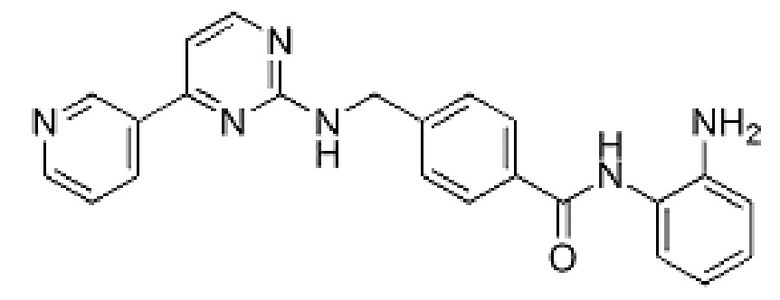
N-(2-аминофенил)-4-[[(4-пиридин-3-илпиримидин-2-ил)амино]метил]бензамид
Дополнительные примеры таких ингибиторов HDAC включены в Bertrand European Journal of Medicinal Chemistry 45, (2010) 2095-2116, в частности соединения, приведенные в следующей таблице, как указано ниже.

Протеасомные ингибиторы - это лекарства, которые блокируют активность протеасом - клеточных комплексов, которые разрушают белки, такие как белок p53. Некоторые ингибиторы протеасом поступают в продажу или находятся в стадии исследования лечения онкологических заболеваний. Подходящие ингибиторы протеасом для использования в настоящем изобретении включают:
1. Бортезомиб (Velcade®), включая его фармацевтически приемлемые соли. Adams J, Kauffman M (2004), Cancer Invest 22 (2): 304-11.
Бортезомиб имеет следующую химическую структуру и название:

[(1R)-3-метил-1-({(2S)-3-фенил-2-[(пиразин-2-илкарбонил)амино]пропаноил}амино)бутил]бороновая кислота
2. Дисульфирам, включая его фармацевтически приемлемые соли.
Bouma et al. (1998). J. Antimicrob. Chemother. 42 (6): 817-20.
Дисульфирам имеет следующую химическую структуру и название:

1,1',1'',1'''-[дисульфандиилбис(карбонотиоилнитрило)]тетраэтан
3. Эпигаллокатехина галлат (EGCG), включая его фармацевтически приемлемые соли. Williamson et al., (December 2006), The Journal of Allergy and Clinical Immunology 118 (6): 1369-74.
Эпигаллокатехина галлат имеет следующую химическую структуру и название:

[(2R,3R)-5,7-дигидрокси-2-(3,4,5-тригидроксифенил)хроман-3-ил]3,4,5-тригидроксибензоат
4. Салиноспорамид А, включая его фармацевтически приемлемые соли. Feling et at., (2003), Angew. Chem. Int. Ed. Engl. 42 (3): 355-7.
Салиноспорамид А имеет следующую химическую структуру и название:

(4R,5S)-4-(2-хлорэтил)-1-((1S)-циклогекс-2-енил(гидрокси)метил) -5-метил-6-окса-2-азабицикло[3.2.0]гептан-3,7-дион
Ингибиторы метаболизма рака - у многих опухолевых клеток метаболизм заметно отличается от метаболизма нормальных тканей. Например, скорость гликолиза - метаболического процесса, который превращает глюкозу в пируват - увеличивается, и образующийся пируват превращается в лактат, а не подвергается дальнейшему окислению в митохондриях через цикл трикарбоновых кислот (ТСА). Этот эффект часто наблюдается даже в аэробных условиях и известен как эффект Варбурга.
Лактатдегидрогеназа А (ЛДГ-А) - изоформа лактатдегидрогеназы, экспрессируемая в мышечных клетках - играет ключевую роль в метаболизме опухолевых клеток, выполняя восстановление пирувата до лактата, который затем может быть экспортирован из клетки. Было показано, что этот фермент активируется во многих типах опухолей. Изменение метаболизма глюкозы, описываемое эффектом Варбурга, является критическим для роста и пролиферации раковых клеток, и, как было установлено на ксенотрансплантных моделях, нокдаун LDH-A с использованием RNA-i приводит к снижению пролиферации клеток и роста опухолей.
D. A. Tennant et. al., Nature Reviews, 2010, 267.
P. Leder, et. al., Cancer Cell, 2006, 9, 425.
Ингибиторы метаболизма рака, включая ингибиторы LDH-A, пригодны для использования в комбинации с составами данного изобретения.
Подходящие наполнители в соответствии с изобретением включают, но не ограничиваются ими, фосфат кальция (например, двух- и трехосновный, гидратированный или безводный), сульфат кальция, карбонат кальция, карбонат магния, каолин, высушенную распылением или безводную лактозу, целлюлозу (например, микрокристаллическую целлюлозу, порошкообразную целлюлозу), предварительно желатинизированный крахмал, крахмал, лактитол, маннит, сорбит, мальтодекстрин, сахарную пудру, прессованный сахар, сахарозу, декстрозу и инозит. Для таблеток настоящего изобретения подходят наполнители, которые содержат мало воды или совсем не содержат ее. В одном варианте осуществления настоящего изобретения наполнители включают одно из маннита и микрокристаллической целлюлозы или оба этих наполнителя.
Подходящие вещества, способствующие скольжению, согласно настоящему изобретению включают, но не ограничиваются ими, диоксид кремния; коллоидный диоксид кремния, например, коллоидный безводный диоксид кремния, например, Aerosil®, Cab-O-Sil®, и тальк, например, Luzenac Phama®. В одном варианте осуществления настоящего изобретения вещество, способствующее скольжению, представляет собой коллоидный диоксид кремния.
Подходящие смазывающие вещества согласно изобретению включают, но не ограничиваются ими, стеарат Mg, Al или Ca, PEG 4000-8000, бензоат натрия, глицерил моно-жирной кислоты, например, имеющей молекулярную массу от 200 до 800 Дальтон, например, глицерил моностеарат (например, Danisco, Великобритания), глицерил дибегенат (например, CompritolATO888TM, Gattefossé, Франция), глицерил пальмито-стеариновый эфир (например, PrecirolTM, Gattefossé, Франция), полиоксиэтиленгликоль (PEG, BASF), гидрогенизированное хлопковое масло (Lubitrab, Edward Mendell Co Inc.), касторовое масло (Cutina HR, Henkel). В одном варианте осуществления настоящего изобретения смазывающее вещество представляет собой стеарат магния.
Согласно настоящему изобретению содержание наполнителя может варьироваться в пределах массовой доли от примерно 35% до 70%, в частности, примерно 65% в пересчете на общий вес диспергируемой таблетки. Содержание разрыхлителя может варьироваться в пределах массовой доли от примерно 2,5% до 13%, в частности, от примерно 5% до 10% в пересчете на общий вес диспергируемой таблетки. Содержание гипромеллозы в массовых долях может варьироваться от примерно 1% до 13%, в частности, от примерно 5% до 10% в пересчете на общий вес диспергируемой таблетки. Содержание вещества, способствующего скольжению, может варьироваться в пределах массовой доли от примерно 0,1% до 2,5%, в частности, от примерно 0,1% до 0,5% в пересчете на общий вес диспергируемой таблетки. Содержание смазывающего вещества может составлять массовую долю от примерно 0,1% до 2% в пересчете на общую массу диспергируемой таблетки, предпочтительно - от примерно 0,1% до 1,5%.
В одном варианте осуществления настоящего изобретения диспергируемая таблетка содержит следующие фармацевтически приемлемые формообразующие: один или несколько наполнителей с суммарным содержанием (массовые доли) примерно 65% в пересчете на общую массу диспергируемой таблетки, гипромеллозу с суммарным содержанием (массовые доли) от примерно 5% до 10% в пересчете на общую массу диспергируемой таблетки, один или несколько разрыхлителей с суммарным содержанием (массовые доли) от примерно 5% до 10% в пересчете на общую массу диспергируемой таблетки, одно или несколько веществ, способствующих скольжению, с суммарным содержанием (массовые доли) от примерно 0,1% до 0,5% в пересчете на общую массу диспергируемой таблетки, и/или одно или несколько смазывающих веществ с суммарным содержанием (массовые доли) от примерно 0,1% до 1,5% в пересчете на общую массу диспергируемой таблетки.
Согласно изобретению способ приготовления диспергируемых таблеток включает гранулирование внутренней фазы, смешивание ее с одним или несколькими фармацевтически приемлемыми формообразующими и прессование полученной смеси.
Внутренняя фаза содержит Соединение А. Предпочтительно, внутренняя фаза содержит Соединение А и один или несколько фармацевтически приемлемых формообразующих. Предпочтительно, фармацевтически приемлемые формообразующие внутренней фазы представляют собой один или несколько наполнителей, один или несколько разрыхлителей, гипромеллозу и одно или несколько веществ, способствующих скольжению. Предпочтительно, чтобы содержание одного или нескольких наполнителей составляло массовую долю в пределах от 5% до 30% в пересчете на общую массу диспергируемой таблетки, более предпочтительно - от примерно 10% до 25%, и особенно предпочтительно - примерно 20%. Наполнители согласно изобретению предпочтительно представляют собой маннит и микрокристаллическую целлюлозу. Разрыхлитель предпочтительно представляет собой кросповидон. Содержание разрыхлителя во внутренней фазе предпочтительно составляет массовую долю менее 10%, более предпочтительно - менее 7% в пересчете на общую массу диспергируемой таблетки. Содержание гипромелллозы во внутренней фазе предпочтительно составляет массовую долю менее 10%, более предпочтительно - менее 3% в пересчете на общую массу диспергируемой таблетки. Предпочтительно, чтобы вещество, способствующее скольжению, представляло собой коллоидный диоксид кремния. Содержание вещества, способствующего скольжению, во внутренней фазе предпочтительно составляет массовую долю в пределах от примерно 0,1% до 1%, более предпочтительно - менее 0,5% в пересчете на общую массу диспергируемой таблетки.
Соединение А и гипромеллоза, один или несколько наполнителей, один или несколько разрыхлителей и одно или несколько веществ, способствующих скольжению, смешивают вместе в смесителе. После внесения стеарата магния для смазывания смесь обрабатывают для сухого гранулирования, например, с использованием вальцевания и мельницы для гранулирования.
Внешнюю фазу, содержащую один или несколько фармацевтически приемлемых формообразующих, смешивают с внутренней фазой с использованием, например, диффузионного смесителя. Предпочтительно добавляют гипромеллозу, один или несколько наполнителей и один или несколько разрыхлителей. Наиболее предпочтительно во внешнюю фазу в качестве наполнителей добавлять маннит и микрокристаллическую целлюлозу. Еще более предпочтительно во внешнюю фазу добавлять маннит так, чтобы его массовая доля была в пределах от примерно 12% до 45% в пересчете на общую массу диспергируемой таблетки, а микрокристаллическую целлюлозу добавлять во внешнюю фазу так, чтобы ее массовая доля была в пределах от примерно 8% до 20% в пересчете на общую массу диспергируемой таблетки. Наиболее предпочтительно во внешнюю фазу добавлять кросповидон в качестве разрыхлителя. Еще более предпочтительно во внешнюю фазу добавлять кросповидон составляет массовую долю в пределах от примерно 1% до 5%, более предпочтительно - менее 5% в пересчете на общую массу диспергируемой таблетки.
Внешнюю фазу, содержащую гипромеллозу, один или несколько наполнителей и один или несколько разрыхлителей смешивают вместе с использованием, например, диффузионного смесителя, с гранулами из внутренней фазы. После смазывания стеаратом магния конечную смесь прессуют с использованием подходящего роторного пресса для получения диспергируемых таблеток.
В одном варианте осуществления изобретения способ приготовления диспергируемой таблетки включает в себя
(a) формирование внутренней фазы, которое включает
(i) смешивание Соединения А с фармацевтически приемлемыми формообразующими;
(ii) сухое гранулирование,
(b) формирование внешней фазы, которое включает
(i) внесение дополнительных фармацевтически приемлемых формообразующих во внутреннюю фазу и перемешивание;
(c) формирование диспергируемой таблетки путем
(i) прессования смеси, полученной на стадии b(i).
Более конкретно, в одном аспекте настоящее изобретение предлагает способ, включающий в себя:
(i) смешивание Соединения А, гипромеллозы и фармацевтически приемлемых формообразующих, например, одного или нескольких наполнителей, например, маннита и микрокристаллической целлюлозы, с одним или несколькими разрыхлителями, например, кросповидоном, и одним или несколькими веществами, способствующими скольжению, например, коллоидным диоксидом кремния, в диффузионном смесителе;
(ii) внесение в смесь одного или нескольких смазывающих веществ, например, стеарата магния, обработку смеси для сухого гранулирования, например, с использованием вальцевания и мельницы для гранулирования; и
(iii) внесение гипромеллозы и фармацевтически приемлемых формообразующих, например просеянных формообразующих, таких как один или несколько наполнителей, например, маннита и микрокристаллической целлюлозы, одного или нескольких разрыхлителей, например кросповидона, и смешивание, например, в диффузионном смесителе;
(iv) смазывание смеси стеаратом магния;
(v) таблетирование смеси, полученной на стадии (iv), прессованием, например, в обычном таблеточном прессе, предпочтительно с роторным устройством.
Под «внутренней фазой» подразумевается гранулированная фаза (стадии (i) и (ii)), включающая в себя активное начало - Соединение А - и один или несколько фармацевтически приемлемых формообразующих.
Под «внешней фазой» подразумевается один или несколько фармацевтически приемлемых формообразующих, добавляемых к внутренней фазе (грануляту) (стадия (iii) и (iv)).
Под «общей массой диспергируемой таблетки» подразумевается масса таблетки, состоящей из внутренней и внешней фазы.
Физическую и химическую стабильность можно проверять любым общепринятым способом, например, можно протестировать сами диспергируемые таблетки путем измерения растворимости, истираемости, распадаемости, размера частиц дисперсии, анализа содержания Соединения A, продуктов разложения и внешнего вида, например, после хранения при комнатной температуре, т.е. при 25°C/относительной влажности 60%, и/или хранения при 40°C/относительной влажности 75%.
Диспергируемые таблетки могут различаться по форме и иметь, например, круглую, овальную, продолговатую, цилиндрическую или любую другую подходящую форму. В варианте осуществления изобретения диспергируемые таблетки, полученные описанным выше способом прессования, имеют круглую или овальную форму. Края диспергируемых таблеток могут быть скошены или закруглены и могут иметь риску. Наиболее предпочтительно, чтобы диспергируемые таблетки были круглыми с двояковыпуклыми скошенными краями.
В варианте осуществления изобретения диспергируемая таблетка содержит дозу в размере от 10 мг до 25 мг Соединения A в качестве активного начала, предпочтительно дозу в размере 10 мг Соединения A в качестве активного начала.
Диспергируемая таблетка согласно изобретению предпочтительно является круглой, двояковыпуклой со скошенными краями. Диспергируемая таблетка имеет диаметр в пределах от 5 мм до 10 мм, предпочтительно - от 5 мм до 7 мм, и более предпочтительно - 6 мм.
Твердость или прочность на раздавливание таблеток согласно настоящему изобретению можно определять, используя стандартные испытания. Твердость таблеток предпочтительно определять, следуя стандартному испытанию, изложенному в European Pharmacopoeia 2.9.8. Можно использовать такое устройство, как испытательное устройство для таблеток Kraemer® 3S. Этим испытанием определяют прочность таблеток на раздавливание, которое измеряют по силе, которая требуется для разрушения таблеток раздавливанием.
Диспергируемая таблетка, содержащая дозу в размере 10 мг Соединения A в качестве активного начала может кроме того характеризоваться средним значением твердости в пределах от примерно 25 до 75 Н, предпочтительно - не более чем 55 Н.
Было обнаружено, что составы, содержащие Соединение A, примерно от 5% до 10% (вес/вес) кросповидона, примерно от 5% до 10% (вес/вес) гипромеллозы, где гипромеллоза характеризуется вязкостью в пределах от 4 мПа.с до 6 мПа.с, предпочтительно - 5 мПа.с, измеренной при 20°C в 2%-ном растворе в воде (вес/вес), и уровне замещения метоксильными группами, составляющем от 28% до 30%, или при вязкости от 80 мПа.с до 120 мПа.с, предпочтительно - 100 мПа.с, измеренной при 20°C в 2%-ном растворе в воде (вес/вес), и уровне замещения метоксильными группами, составляющем от 19% до 24%, и наполнители маннит и микрокристаллическую целлюлозу в массовом соотношении от 2.5:1 до 2:1, могут применяться для получения диспергируемых таблеток с низким значением истираемости и коротким периодом распадаемости, которые соответствуют спецификациям в European Pharmacopeia.
Диспергируемые таблетки данного изобретения, кроме того, могут быть окрашены и/или маркированы так, чтобы придать им индивидуальный внешний вид и сделать их мгновенно узнаваемыми. Использование красителей или лаковых пигментов может служить для улучшения внешнего вида, а также для идентификации диспергируемых таблеток. Диспергируемые таблетки данного изобретения можно маркировать с использованием отпечатанных кодов.
Диспергируемые таблетки данного изобретения полезны для лечения солидных опухолей с мутацией BRAF.
Действие и характеристики диспергируемых таблеток данного изобретения могут быть указаны в стандартных клинических испытаниях и/или испытаниях на животных.
Кроме того, полученные диспергируемые таблетки данного изобретения стабильны как в процессе производства, так и при хранении, например, в течение 2 лет или даже 3 лет в обычной упаковке, например в герметичных алюминиевых блистерных упаковках. Согласно результатам, полученным в общепринятых тестах, в течение этого времени может разлагаться примерно 5%, например, 2 или 3% или меньше, Соединения А (активного начала). Например, менее 1% Соединения А (активного начала) разлагается за один год в наполненных ПЭВД бутылках или блистерах.
Изобретение относится также к способу введения млекопитающему, предпочтительно человеку, нуждающемуся в таком лечении, Соединения А в форме диспергируемой таблетки. Изобретение также относится к применению Соединения А в форме диспергируемой таблетки для лечения млекопитающего, предпочтительно человека, при одном из вышеупомянутых заболеваний или расстройств. Изобретение относится, в частности, к такому способу, где пациенту вводят суточную дозу от 4,5 мг/кг до 5,25 мг/кг массы тела в день Соединения A в качестве активного начала. Следует понимать, что уровень дозы, определяемый для индивидуального пациента, будет зависеть от множества факторов, включая возраст, массу тела, общее состояние здоровья, комбинацию лекарств с одним или несколькими активными лекарственными средствами, тип и тяжесть заболевания.
Упаковка с лекарственным средством содержит диспергируемые таблетки согласно изобретению и печатные инструкции, рекомендующие пероральное введение одной или нескольких диспергируемых таблеток соединения А.
В другом варианте осуществления настоящего изобретения предложена диспергируемая таблетка Соединения А для применения в терапии.
В другом аспекте настоящего изобретения предложен способ введения фармацевтической композиции данного изобретения пациенту, нуждающемуся в такой терапии, который включает в себя (i) объединение композиции с водной средой, (ii) диспергирование композиции в водной среде с образованием дисперсии и (iii) пероральный прием дисперсии.
ПРИМЕРЫ
Используемые здесь символы и условные обозначения, используемые в этих процессах, схемах и примерах, соответствуют тем, которые используются в современной научной литературе, например, в журнале Американского химического общества (Journal of the American Chemical Society) или в Journal of Biological Chemistry. Если не указано иное, все температуры указаны в oC (градусы Цельсия).
Пример 1
Таблица 1 - Примеры композиций диспергируемых таблеток
b Полимерная гипромеллоза с уровнем замещения метоксильными группами 19% - 24% и уровнем замещения гидроксипропильными группами 7% - 12%/
Диспергируемые таблетки соединения A, представленные в таблице 1, обеспечивают быстро диспергирующие композиции, время полного распада которых составляет не более 3 минут, и которые отличаются низкой истираемостью: менее 0,5% после 100 оборотов. Были протестированы различные соотношения кросповидона и гипромеллозы. Составы с более низкой вязкостью, содержащие гипромеллозу, приводили к более длительной распадаемости. Составы с одинаковым содержанием гипромеллозы (7,5% вес./вес.) и содержанием кросповидона (7,5% вес./вес.), но с гипромеллозой другого сорта с точки зрения номинальной вязкости и уровня замещения метоксильными группами (Форма 2a и Форма 5a) приводили к более короткой распадаемости в случае состава с гипромеллозой более вязкого сорта (Форма 5a). Составы с более низким содержанием гипромеллозы (5% вес./вес.) и более высоким содержанием кросповидона (10% вес./вес.) показали сходную распадаемость (не более 1 мин) вне зависимости от вязкости и уровня замещения метоксильными группами используемого сорта гипромеллозы (Форма 1a и Форма 4a). Влияние номинальной вязкости гипромеллозы на продолжительность распадаемости оказывается более выраженным, если в диспергируемых таблетированных составах Соединения А используются более высокие уровни содержания гипромеллозы.
Пример 2
Были подготовлены вальцевание, таблетки, содержащие Соединение А и ингредиенты, приведенные в таблице 1.
Таблица 2а - Интрагранулированные и экстрагранулированные композиции диспергируемых таблеток
Таблица 2b - Интрагранулированные и экстрагранулированные композиции диспергируемых таблеток
Способ изготовления таблетки
Смешивание - просеивание - смешивание
Компоненты внутренней фазы таблетки готовят для вальцевания. Соединение А, микрокристаллическую целлюлозу, ацесульфам калия, кросповидон, коллоидный диоксид кремния, ароматизатор, гипромеллозу и маннит смешивают в смесителе подходящего размера и перемешивают. Перемешанный материал просеивают через сито подходящего размера и переносят в смеситель подходящего размера и перемешивают.
Стеарат магния просеивают через сито подходящего размера и переносят в смеситель подходящего размера, содержащий перемешанный материал, и затем перемешивают в течение дополнительного времени.
Вальцевание и размол
Смесь с внесенным в нее смазывающим веществом подвергают сухому гранулированию, используя вальцы, с получением полос. Спрессованные полосы пропускают через грохот с получением гранул подходящего размера.
Смешивание - просеивание - смешивание
Компоненты внешней фазы таблетки готовят для таблетирования. Дополнительные количества микрокристаллической целлюлозы, маннита, гипромеллозы и кросповидона смешивают в смесителе подходящего размера и перемешивают. Перемешанный материал просеивают через сито подходящего размера и переносят в смеситель подходящего размера вместе с гранулами внутренней фазы и перемешивают. Смесь перемешивают, объединяя материалы внутренней и внешней фаз.
Стеарат магния просеивают через сито подходящего размера и переносят в смеситель подходящего размера, содержащий перемешанный материал, и затем перемешивают в течение дополнительного времени.
Прессование
Смесь со внесенным в нее смазывающим веществом прессуют в роторном таблеточном прессе, оборудованном круглой пресс-формой диаметром 6 мм со скошенными краями с заданным весом, равным 80 мг и получают диспергируемые таблетки по 10 мг. Образцы прессованных таблеток отбирают для технологического контроля отклонений индивидуального веса, внешнего вида, твердости, толщины, истираемости и распадаемости.
Таблетки упаковывают согласно требованиям в контейнеры из ПЭВП с влагопоглотителем или в блистерные упаковки (ПВХ/ПВДК, покрытые термосвариваемой алюминиевой фольгой) по 10 таблеток.
Пример 3
Свойства 10 мг диспергируемых таблеток
Хотя предпочтительные варианты осуществления изобретения проиллюстрированы вышеизложенным, следует понимать, что изобретение не ограничено точными раскрытыми здесь инструкциями, и что право на все модификации, входящие в объем следующей формулы изобретения, сохраняется.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИСПЕРГИРУЕМЫЕ КОМПОЗИЦИИ | 2017 |
|
RU2826218C2 |
| ПЕДИАТРИЧЕСКИЕ ТАБЛЕТКИ, СОДЕРЖАЩИЕ КАПЕЦИТАБИН | 2007 |
|
RU2455979C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ОМЕГАКАРБОКСИАРИЛЗАМЕЩЕННУЮ ДИФЕНИЛМОЧЕВИНУ, ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2420283C2 |
| КОМБИНАЦИЯ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ И ИНГИБИТОРА ПРОТЕИНКИНАЗЫ И ЕЕ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2770754C1 |
| Твёрдые лекарственные формы палбоциклиба | 2016 |
|
RU2686840C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ВИЧ-ИНФЕКЦИИ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ЛЕЧЕНИЯ | 2013 |
|
RU2543322C1 |
| ДИСПЕРГИРУЮЩИЕСЯ ТАБЛЕТКИ ДЕФЕРАСИРОКСА | 2005 |
|
RU2396954C2 |
| ГАЛЕНОВЫ ПРЕПАРАТЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2010 |
|
RU2535090C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ АЛКИЛСУЛЬФАТ НАТРИЯ | 2019 |
|
RU2759746C1 |
| ПРОТИВОТУБЕРКУЛЕЗНАЯ СТАБИЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ В ФОРМЕ ДИСПЕРГИРУЕМОЙ ТАБЛЕТКИ, СОДЕРЖАЩЕЙ ГРАНУЛИРОВАННЫЙ ИЗОНИАЗИД И ГРАНУЛИРОВАННЫЙ РИФАПЕНТИН, И СПОСОБ ЕЕ ПРИГОТОВЛЕНИЯ | 2014 |
|
RU2694056C2 |
Группа изобретений относится к области фармацевтики и предназначена для получения пероральной суспензии дабрафениба мезилата. Диспергируемая таблетка содержит метансульфонатную соль N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамида, гипромеллозу, маннит, микрокристаллическую целлюлозу, по меньшей мере одно фармацевтически приемлемое формообразующее, подходящее для приготовления таблеток. Компоненты используются в заявленных количествах. Таблетка характеризуется распадаемостью, измеренной в соответствии с испытанием на распадаемость в European Pharmacopoeia 2.9.1, причем время полного распада таблетки в воде при температуре от 15°C до 25°C составляет 3 минуты и меньше. В другом воплощении представлен способ получения указанной диспергируемой таблетки. Также обеспечивается диспергируемая таблетка, содержащая внутреннюю фазу и внешнюю фазу, где а) внутренняя фаза включает: метансульфонатную соль N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6–дифторбензолсульфонамида, микрокристаллическую целлюлозу, маннит, гипромеллозу, ацесульфам калия, кросповидон, коллоидный диоксид кремния, стеарат магния и (b) внешняя фаза включает: микрокристаллическую целлюлозу, маннит, гипромеллозу, кросповидон, стеарат магния. Использование группы изобретения позволяет получить стабильную диспергируемую таблетку дабрафениба мезилата, которая имеет короткое время дезинтеграции для удобного восстановления суспензии и низкую ломкость. 3 н. и 13 з.п. ф-лы, 4 табл., 3 пр.
1. Диспергируемая таблетка, содержащая (a) метансульфонатную соль N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамида в количестве от 5% до 40% масс. в пересчете на общую массу таблетки, (b) гипромеллозу в количестве от 1% до 13% масс. в пересчете на общую массу таблетки и (c) маннит и микрокристаллическую целлюлозу в количестве от 35% до 70% масс. в пересчете на общую массу таблетки, где массовое соотношение содержания маннита и микрокристаллической целлюлозы составляет примерно от 2,5:1 до 2:1, и d) по меньшей мере одно фармацевтически приемлемое формообразующее, подходящее для приготовления таблеток, где таблетка характеризуется распадаемостью, измеренной в соответствии с испытанием на распадаемость в European Pharmacopoeia 2.9.1, причем время полного распада таблетки в воде при температуре от 15°C до 25°C составляет 3 минуты и меньше.
2. Диспергируемая таблетка согласно п. 1, где содержание гипромеллозы составляет от примерно 5% до примерно 10% масс. в пересчете на общую массу таблетки.
3. Диспергируемая таблетка согласно п. 2, где значение номинальной вязкости гипромеллозы находится в пределах от 4 мПа⋅с до 6 мПа⋅с, которое измерено при 20°C при содержании в воде 2% масс. и уровне замещения метоксильными группами, составляющем от 28% до 30%.
4. Диспергируемая таблетка согласно п. 2, где значение вязкости гипромеллозы находится в пределах от 80 мПа⋅с до 120 мПа.с, которое измерено при 20°C при содержании в воде 2% масс. и уровне замещения метоксильными группами, составляющем от 19% до 24%.
5. Диспергируемая таблетка согласно п.1, где таблетка имеет среднее значение твердости, измеренное в соответствии с испытанием прочности на раздавливание в Европейской Фармакопее 2.9.8, не более 55 Н.
6. Диспергируемая таблетка согласно п. 1, где фармацевтически приемлемые формообразующие включают:
(i) по меньшей мере один разрыхлитель с суммарным содержанием от примерно 2,5% до 13% масс. в пересчете на общую массу таблетки,
(ii) по меньшей мере одно смазывающее вещество с суммарным содержанием от примерно 0,1% до 2% масс. в пересчете на общую массу таблетки и
(iii) по меньшей мере одно вещество, способствующее скольжению, с суммарным содержанием от примерно 0,1% до 2,5% масс. в пересчете на общую массу таблетки.
7. Диспергируемая таблетка согласно п. 6, где разрыхлитель представляет собой кросповидон.
8. Диспергируемая таблетка согласно п. 7, где содержание кросповидона составляет примерно от 5% до 10% масс. в пересчете на общую массу таблетки.
9. Диспергируемая таблетка согласно пп. 6-8, где способствующее скольжению вещество представляет собой коллоидный диоксид кремния.
10. Диспергируемая таблетка согласно пп. 6-9, где смазывающее вещество представляет собой стеарат магния.
11. Диспергируемая таблетка согласно п.1 для применения в лечении онкологического заболевания.
12. Диспергируемая таблетка согласно п.11 для применения в лечении онкологического заболевания, где онкологическое заболевание представляет собой солидную опухоль с мутацией BRAF.
13. Способ получения диспергируемой таблетки согласно пп. 1-14, который включает в себя:
(i) смешивание метансульфонатной соли N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамида и по меньшей мере одного фармацевтически приемлемого формообразующего;
(ii) гранулирование смеси, полученной в (i);
(iii) смешивание гранулятов, полученных в (ii), по меньшей мере с одним фармацевтически приемлемым формообразующим для образования смеси и
(iv) прессование смеси, полученной на стадии (iii), формируя таблетку.
14. Способ согласно п.13, где стадия гранулирования (ii) представляет собой сухое гранулирование.
15. Способ согласно п.14, где сухое гранулирование осуществляют с помощью вальцевания в мельнице для гранулирования.
16. Диспергируемая таблетка, содержащая внутреннюю фазу и внешнюю фазу, где
а) внутренняя фаза включает:
(i) метансульфонатную соль N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6 -дифторбензолсульфонамида;
(ii) микрокристаллическую целлюлозу;
(iii)маннит;
(iv) гипромеллозу;
(v) ацесульфам калия;
(vi) кросповидон;
(vii) коллоидный диоксид кремния;
(viii) стеарат магния; и
(b) внешняя фаза включает:
(i) микрокристаллическую целлюлозу;
(ii) маннит;
(iii) гипромеллозу;
(iv) кросповидон;
(v) стеарат магния; и
где (а) метансульфонатная соль N-{3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-
дифторбензолсульфонамида присутствует в количестве от 5% до 40% масс. в расчете на общую массу таблетки, (b) гипромеллоза присутствует в количестве от 1% до 13% масс. в расчете на общую массу таблетки, (c) маннит и микрокристаллическая целлюлоза присутствуют в количестве от 35% до 70% масс. в расчете на общую массу таблетки, где маннит и микрокристаллическая целлюлоза присутствуют в массовом соотношении примерно 2,5:1 до 2:1.
| CN 103751140 A, 30.04.2014 | |||
| US 2016046615 A1, 18.02.2016 | |||
| WO 03028705 A1, 10.04.2003 | |||
| CN 106539777 A, 29.03.2017 | |||
| WO 2013093655 A1, 27.06.2013. |

