Изобретение относится к новому способу получения производных эпотилона, содержащих не только 2-метилтиазольный заместитель, но и, например, другие гетероарильные или арильные заместители или гетероциклические радикалы, конденсированные с бензольным циклом, и метил в качестве заместителя в положении 12 в структуре эпотилона, а также к новым производным эпотилона и промежуточным соединениям для реализации способа и к методам их получения. Указанные промежуточные соединения являются новыми соединениями и входят в объем изобретения.
Эпотилоны (16-членные макролиды, которые впервые были выделены из микобактерии Sorangium cellulosum) представляют собой новый класс перспективных противоопухолевых агентов. Было установлено, что эпотилоны проявляют активность в отношении различных линий опухолевых клеток, включая клеточные линии опухоли молочной железы.
Эти агенты действуют по тому же биологическому механизму, что и таксол, противоопухолевое средство, используемое в качестве первичной терапии при лечении опухоли молочной железы. Было установлено, что они более эффективны, чем таксол.
Возможные применения эпотилонов включают лечение болезни Альцгеймера, малярии и заболеваний, вызванных грамотрицательными бактериями. Эпотилоны, прежде всего, пригодны для лечения пролиферативных заболеваний.
Термин "пролиферативные заболевания" означает, прежде всего, заболевания в виде солидных опухолей, жидких опухолей, например, лейкоза, и псориаза.
Термин "заболевание в виде солидной опухоли" означает, прежде всего, опухоль молочной железы, опухоль ободочной кишки и в общем случае желудочно-кишечного тракта, включая рак желудка, гематому, рак легких, прежде всего мелкоклеточный рак легких и немелкоклеточный рак легких, рак почки, мезотелиому, глиому, плоскоклеточный рак кожи, рак головы и шеи, рак мочеполовых путей, например рак шейки матки, рак матки, рак яичника, рак яичек, рак предстательной железы или мочевого пузыря, болезнь Ходжкина, карциноидный синдром или саркому Капоши.
Производные эпотилона описаны, например, в WO 97/19086. Эти производные получают из природных эпотилонов А и В.
Общий синтез эпотилона А описан Schinzer и др., Chem. Eur. J., 2, 11, 1477-1482 (1996). Другие методы синтеза эпотилона А и В и их производных описаны K.C.Nicolaou и др., Angew. Chem., 109, 170-172 (1997), и Nature, 387, 268-272 (1997).
В статье K.C.Nicolaou и др., Chem. Commun, 2343-2344 (1997), описан общий синтез 26-гидроксиэпотилона В и родственных соединений с использованием в качестве ключевых стадий реакции селективного олефинирования по Виттигу, альдольной реакции и макролактонизации. Более подробно общий синтез 26-гидроксиэпотилона В и родственных аналогов с использованием макролактонизации описан в статье K.C.Nicolaou и др., Tetrahedron, 54, 7127-7166 (1998).
Кроме того, в WO 98/25029 K.C.Nicolaou и др. описан и заявлен синтез эпотилона А, эпотилона В, аналогов эпотилона, библиотеки аналогов эпотилона с использованием твердофазных и жидкофазных химических методов.
Объектом и целью настоящего изобретения является преодоление всех недостатков известных способов и разработка более простого и улучшенного способа получения вышеуказанных эпотилонов, производных эпотилона и их солей, который таким образом осуществим в промышленных масштабах за счет более короткого пути синтеза, причем указанный способ обеспечивает в среднем высокий общий выход и получение высокоочищенных предшественников и конечных продуктов.
Синтез с целью получения производных эпотилона формулы 9
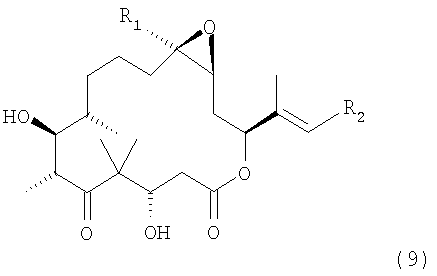
где R1 означает метил, а
R2 означает незамещенный или замещенный арил, незамещенный или замещенный гетероарил или незамещенный или замещенный гетероциклический радикал, конденсированный с бензольным циклом,
и их солей.
Приставка «низш.» означает радикал, содержащий вплоть до и, включая максимум 6, прежде всего вплоть до и включая максимум 4 атома углерода, причем указанный радикал является неразветвленным или разветвленным с одним или более разветвлениями.
Незамещенный или замещенный арил R2 предпочтительно означает ароматический радикал, содержащий от 6 до 14 атомов углерода, прежде всего фенил, нафтил, флуоренил или фенантренил, причем указанный радикал незамещен или замещен одним или более заместителями, предпочтительно до трех, более предпочтительно одним или двумя заместителями, прежде всего выбранными из группы, включающей аминогруппу; (низш.)алканоиламиногруппу, прежде всего ацетиламиногруппу; галоген, прежде всего фтор, хлор или бром; (низш.)алкил, прежде всего метил или также этил или пропил; галоген(низш.)алкил, прежде всего трифторметил; гидрокси; (низш.)алкокси, прежде всего метокси или также этокси; фенил(низш.)алкокси, прежде всего бензилокси; нитро, циано, С8-С12алкокси, прежде всего н-децилокси, карбамоил, (низш.)алкилкарбамоил, такой как N-метил- или N-трет-бутилкарбамоил, (низш.)алканоил, такой как ацетил, фенилокси, галоген(низш.)алкокси, такой как трифторметокси или 1,1,2,2-тетрафторэтилокси, (низш.)алкоксикарбонил, такой как этоксикарбонил, (низш.)алкилмеркапто, такой как метилмеркапто, галоген(низш.)алкилмеркапто, такой как трифторметилмеркапто, гидрокси(низш.)алкил, такой как гидроксиметил или 1-гидроксиметил, (низш.)алкансульфонил, такой как метансульфонил, галоген(низш.)алкансульфонил, такой как трифторметансульфонил, фенилсульфонил, дигидроксибор (-В(ОН)2), 2-метилпиримидин-4-ил, оксазол-5-ил, 2-метил-1,3-диоксолан-2-ил, 1H-пиразол-3-ил, 1-метилпиразол-3-ил и (низш.)алкилендиокси, который связан с двумя соседними атомами углерода, такой как метилендиокси.
Арил R2 прежде всего означает фенил.
Галоген прежде всего означает фтор, хлор, бром или иод, прежде всего фтор или хлор.
Незамещенный или замещенный гетероарил R2 означает, например, радикал выбранный из следующих структур:
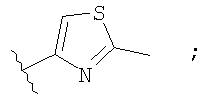
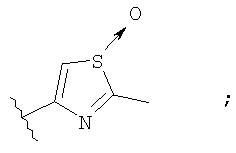
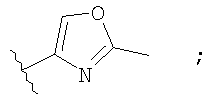
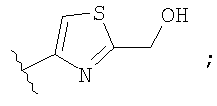
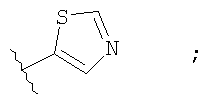
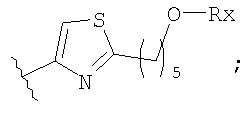
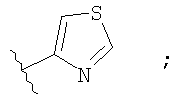

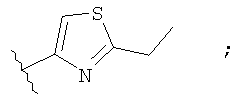
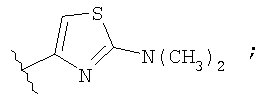
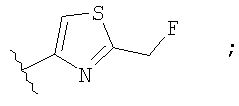
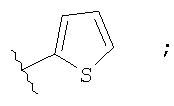
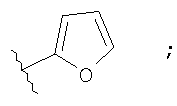
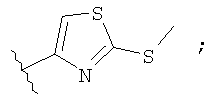
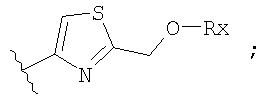
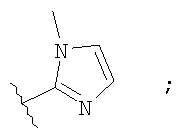
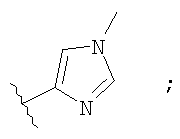
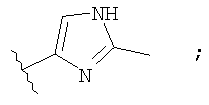
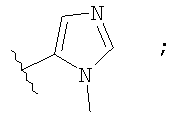
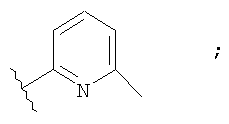
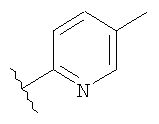 и
и 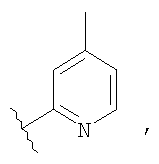
где Rx означает ацил.
Гетероциклический радикал R2, конденсированный с бензольным циклом, означает радикал, выбранный из радикалов формулы
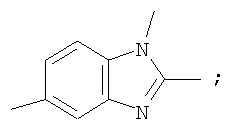
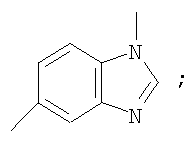
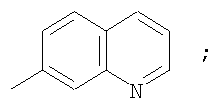

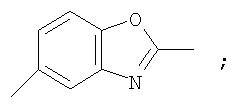
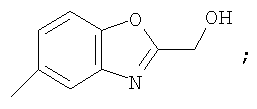
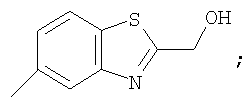
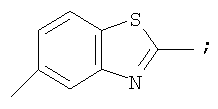
и 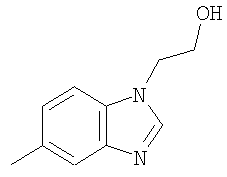
Новый синтез включает следующие стадии:
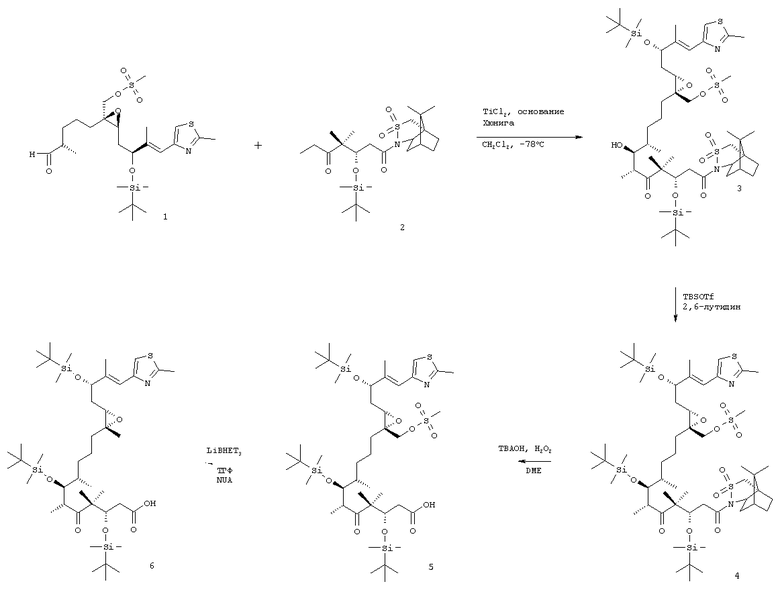
а) взаимодействие соединения формулы 1
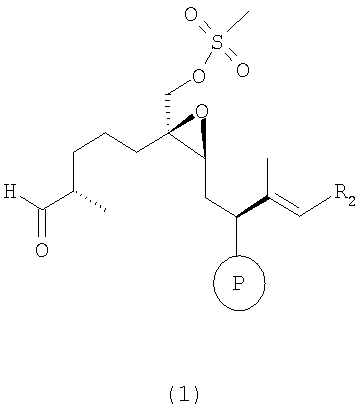
где R2 имеет значения, указанные выше, а мезилгруппа заменена тозилгруппой и т.п., а Р означает ОН-защитную группу, предпочтительно силильную защитную группу, более предпочтительно любую из нижеуказанных силильных групп, и наиболее предпочтительно (низш.)алкилсилильную защитную группу, предпочтительно выбранную из ряда TES, TBDS, TPS,
с сультампроизводным формулы 2, например
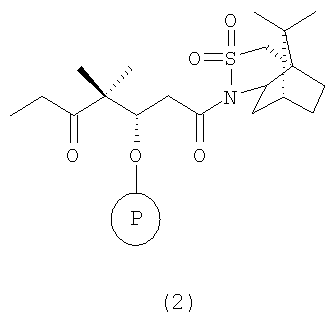
где Р означает ОН-защитную группу, предпочтительно силильную защитную группу, более предпочтительно любую из нижеуказанных силильных групп и наиболее предпочтительно (низш.)алкилсилильную защитную группу, предпочтительно выбранную из ряда TES, TBDS, TPS, с использованием селективной альдольной конденсации в присутствии кислоты Льюиса при добавлении основания в инертном растворителе при пониженной температуре от -50 до -100°С, а затем при повышенной температуре от -20 до +20°С, с образованием соединения формулы 3
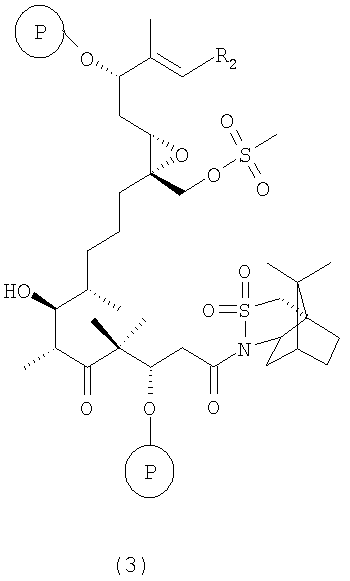
где R2 имеет значения, указанные выше.
Указанное выше соединение формулы 2 можно заменить соединением формулы 2, где Р означает ОН-защитную группу, предпочтительно силильную защитную группу, более предпочтительно любую из нижеуказанных силильных групп и наиболее предпочтительно (низш.)алкилсилильную защитную группу, предпочтительно выбранную из ряда TES, TBDS, TPS.
Соединения формулы 3 являются новыми соединениями и используются в качестве промежуточных продуктов на стадии б) указанного способа, где
б) полученные соединения вышеуказанной формулы 3 взаимодействуют с соединением, образующим простой силильный эфир, при температуре от -70 до +25°С в присутствии 2,6-лутидина с образованием соединения формулы 4
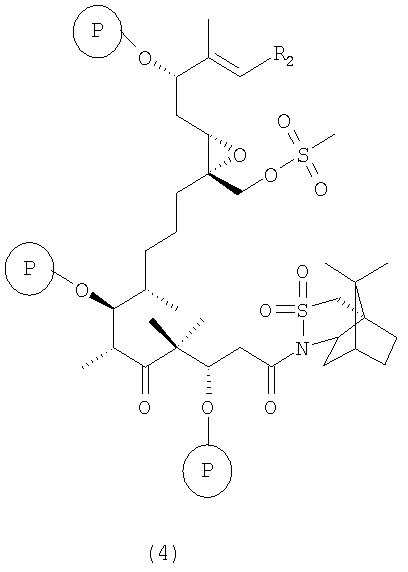
где R2 и Р имеют значения, указанные выше.
Соединения формулы 4 являются новыми соединениями и используются в качестве предшественников на стадии в) указанного способа, где
в) из вышеуказанного соединения формулы 4 после отщепления хиральной вспомогательной группы при обработке ТВАОН/Н2О2 в ДМЭ или LiO2H в ТГФ/МеОН/Н2О получают карбоновую кислоту, т.е. соединение формулы 5
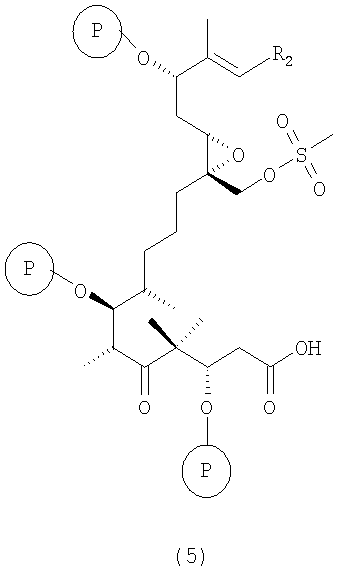
где R2 и Р имеют значения, указанные выше.
Полученные соединения формулы 5 являются новыми соединениями и используются в качестве промежуточных продуктов на стадии г) указанного способа, где
г) соединение формулы 5 взаимодействует с восстанавливающим агентом в инертном растворителе с отщеплением мезил- или тозилгруппы или другой подобной группы с образованием соединения формулы 6
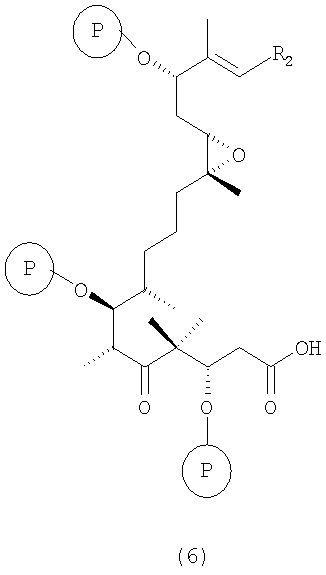
где R2 и Р имеют значения, указанные выше.
Полученные соединения формулы 6 являются новыми соединениями и используются в качестве промежуточных продуктов на стадии д) указанного способа, где
д) тризамещенные трис-силильные простые эфиры вышеуказанной формулы 6 гидролизуют в присутствии десилилирующего реагента или кислоты в инертном растворителе или в их смеси, например TASF или HF•пиридин в ТГФ, при этом получают селективно десилилированное соединение формулы 7
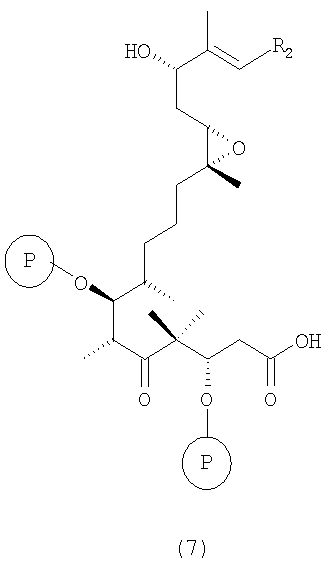
где R2 и Р имеют значения, указанные выше.
Соединения формулы 7 являются новыми соединениями и используются в качестве предшественников на стадии е) указанного способа, где
е) полученные соединения формулы 7 макролактонизируют в условиях, описанных в статье M.Yamaguchi и др., Bull.Chem.Soc. Jpn., 52, 1989 (1979), при этом получают полностью защищенное производное эпотилона В формулы 8
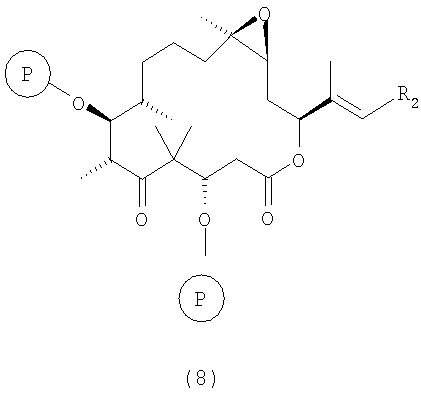
где R2 и Р имеют значения, указанные выше.
Полученные соединения формулы 8 являются новыми соединениями и используются в качестве предшественников на стадии ж) указанного способа, где
ж) полученное соединение формулы 8 обрабатывают HF•пиридином в инертном растворителе при температуре от 0 до 30°С и отщепляют силильные защитные группы, при этом получают производное эпотилона формулы 9
где R1 означает метил, а R2 имеет значения, указанные выше,
и соединения формулы 9 необязательно превращают в соли металлов стандартными методами.
Превращение эпотилона В в соответствующий лактам показано на схеме 21 и описано в примере 3 в WO 99/02514 (стр.31-32 и стр.48-50 соответственно). Превращение соединения формулы 9, которое отличается от эпотилона В, в соответствующий лактам проводят аналогичным способом.
Инертные растворители, указанные на любой из стадий а)-ж), включают, без ограничения перечисленным, метанол, этанол, пропанол, дихлорметан, дихлорэтан, ДМФА, тетрагидрофуран (ТГФ), бензол, толуол, пиридин, этилацетат, ацетон или трет-бутилметиловый эфир (ТВМЕ), гексан или гептан и т.п. и их смеси.
Органические основания, указанные на любой из стадий а)-ж), включают, без ограничения перечисленным, органические амины, такие как незамещенные или гидроксизамещенные моно-, ди- или триалкиламины, прежде всего моно-, ди- или три(низш.)алкиламины, например метиламин, диметиламин, ди-н-пропиламин, триэтиламин, три-н-пропиламин, три-н-бутиламин и диизопропилэтиламин (iPr2NEt), азотсодержащие гетероциклы, этиленимин, пирролидин, пиперидин и морфолин, прежде всего 4-диметиламинопиридин (DMAP), пиридин, 2,6-лутидин, 2,6-ди-трет-бутилпиридин и т.п.
Металлорганические основания включают, например, LDA (диизопропиламинлитий), BuLi, втор-BuLi, KHMDS, LiHMDS или NaHMDS.
Восстанавливающие агенты означают, например, DIBAL-H (диизобутилалюминийгидрид), LiAlH4 (литийалюминийгидрид), литийтриэтилборогидрид и т.п.
Группа Р означает ОН-защитную группу, обычно используемую в органической химии, предназначенную для защиты функциональных гидроксигрупп от нежелательных побочных реакций. Простые силильные эфиры представляют собой, например, стандартные защитные группы, которые обычно используются в органическом синтезе, и предпочтительно защитная силильная группа представляет собой (низш.)алкилсилил, более предпочтительно силильную защитную группу выбирают из ряда TMS (триметилсилил), TES (триэтилсилил), TPS (три-н-пропилсилил), TBDS (трет-бутилдиметилсилил), DEIPS (диэтилизопропилсилил), IPDMS (диметилизопропилсилил), TDS (тексилдиметилсилил), TIPS (триизопропилсилил), ТНР (тетрагидропиранилсилил) или т.п., предпочтительно из ряда TES, TPS или TBDMS, наиболее предпочтительно защитная группа означает TPS. Если соединение содержит более одной защитной группы, то защитные группы выбирают независимо друг от друга, причем они являются идентичными или различными или их можно использовать в любой комбинации, предпочтительно защитные группы идентичны, наиболее предпочтительно защитная группа означает TPS.
Термин «фармацевтически приемлемые соли металлов» означает соли натрия, калия, кальция, магния, алюминия, железа или цинка. Соли получают стандартными методами.
Пригодными кислотами для расщепления бис-триметилсиланилоксигрупп в соединении формулы 4 гидролизом являются слабые органические кислоты, которые не раскрывают эпоксидный цикл в структуре эпотилона, причем указанные кислоты включают разбавленную уксусную кислоту, пропионовую кислоту, гликолевую кислоту, молочную кислоту, фумаровую кислоту, янтарную кислоту, адипиновую кислоту, пимелиновую кислоту, субериновую кислоту, азелаиновую кислоту, яблочную кислоту, винную кислоту, малеиновую кислоту, миндальную кислоту, аминокислоты, такие как глутаминовая кислота или аспарагиновая кислота, прежде всего лимонную кислоту и т.п. Прежде всего следует отметить HF•пиридин в ТГФ или HF•пиридин в пиридине.
Хиральные вспомогательные группы, которые используются при поведении альдольной конденсации на стадии а), означают, например, сультамовую вспомогательную группу или оксазолидиноновые группы, например
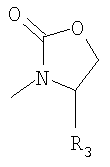 , прежде всего
, прежде всего 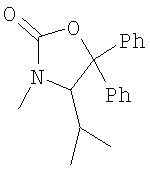
и другие известные хиральные вспомогательные группы.
Стадии а)-ж) в указанной последовательности реакций в способе получения соединений формулы 9 проводят, как более подробно описано ниже:
а) соединения формулы 1 вводят в реакцию с соединением формулы 2, например, в присутствии TiCl4 и основания Хюнига (iPr2NEt) в дихлорметане при температуре -78°С, а затем при температуре 0°С с образованием новых соединений формулы 3;
б) полученное соединение формулы 3 вводят в реакцию с соединением, образующим группу простого силильного эфира, например TPSCl, TBDMSOTf, в присутствии 2,6-лутидина при температуре от -20 до 25°С, прежде всего при температуре -10°С, в дихлорметане в качестве инертного растворителя с образованием соединения формулы 4;
в) в полученном соединении формулы 4 отщепляют вспомогательную сультамовую группу при обработке ТВАОН/Н2О2 в ДМЭ или LiO2Н в ТГФ/МеОН/Н2О с образованием карбоновой кислоты, т.е. соединения формулы 5;
г) полученное соединение формулы 5 обрабатывают с использованием в качестве восстановителя LiBHEt3 в ТГФ в качестве инертного растворителя с отщеплением мезил- или тозилгруппы или аналогичной группы и образованием соединения формулы 6;
д) полученные соединения формулы 6 гидролизуют при обработке десилилирующим реагентом, прежде всего TASF или органической кислотой, прежде всего HF•пиридином в инертном растворителе, например в пиридине или ТГФ, при этом получают соединения формулы 7;
е) полученные соединения формулы 7 подвергают макролактонизации по методике M.Yamaguchi и др., например, при обработке гидроксикислоты реагентом Et3N и 2,4,6-трихлорбензоилхлоридом при пониженной температуре, например при 0°С, а затем реакционную смесь добавляют к раствору 4-DMAP в толуоле и температуру повышают до приблизительно 75°С, при этом получают соединения формулы 8;
ж) полученные защищенные производные эпотилона формулы 8 обрабатывают HF•пиридином в пиридине в качестве инертного растворителя и после отщепления двух силильных защитных групп (TPS, TES, TBDMS) получают производные эпотилона формулы 9, где R1 означает метил, a R2 имеет значения, указанные выше, и соединения формулы 9, где R1 и R2 имеют значения, указанные выше в формуле 9, необязательно превращают в соли металлов стандартными методами.
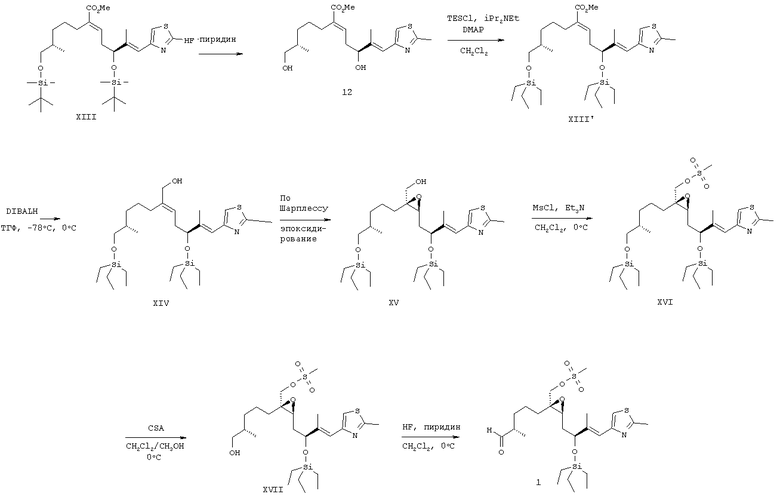
Используемые в указанном способе исходные соединения формулы 1 (где R2 означает незамещенный или замещенный арил, незамещенный или замещенный гетероарил или незамещенный или замещенный гетероциклический радикал, конденсированный с бензольным циклом, арил) используются в качестве ключевых промежуточных соединений для получения производных эпотилона формулы 9, причем соединения формулы 1 являются новыми соединениями и их можно получить по следующей последовательности реакций, включающей стадии а)-ж), как указано ниже.
Конкретные значения R2 и Р указаны при определении формулы 9 и формулы 1 соответственно.
Предлагаемый новый синтез включает следующие стадии:
а) взаимодействие соединения формулы Х
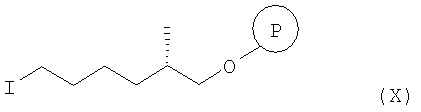
с РРН3 при температуре от 50 до 150°С, более конкретно при температуре 100°С, а затем с KHMDS в инертном растворителе, прежде всего в ТГФ, при 0°С, а затем после охлаждения смеси до температуры от -50 до -100°С, обработку продукта реагентом СН3СО2Cl, более конкретно при температуре -78°С, с образованием соединения формулы XI
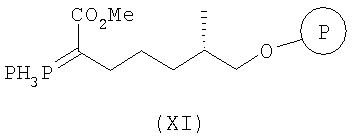
и
б) взаимодействие полученного соединения формулы XI с соединением формулы XII
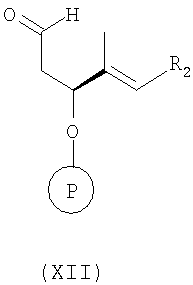
в инертном растворителе, например в толуоле, при температуре от 20 до 60°С, более конкретно при температуре 40°С, с образованием соединения формулы
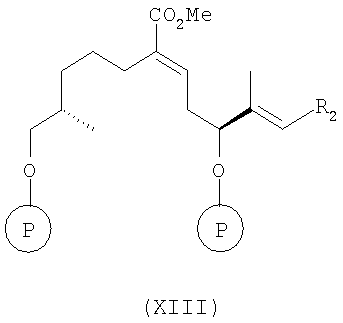
в) восстановление соединения формулы (XIII) восстанавливающим агентом, прежде всего DIBALH, в инертном растворителе, например в толуоле, при температуре от -50 до -100°С, более конкретно при температуре -78°С, а затем при повышенной температуре до 0°С, с образованием соединения формулы XIV
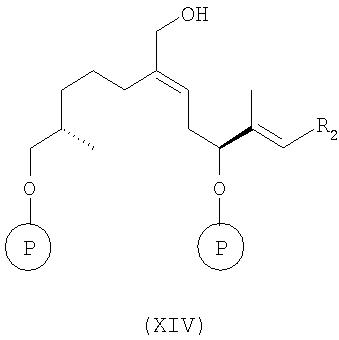
(формула XIV и последующие соединения приводятся в упрощенном виде), где R2 и Р имеют значения, указанные выше, и
г) эпоксидирование соединения формулы XIV в условиях Шарплесса [(+)-диэтил-L-тартрат, Ti(OPr)4, t-BuOOH] с образованием соединения формулы XV
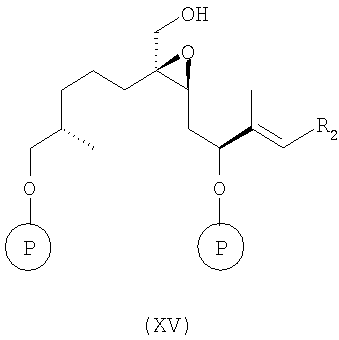
где R2 и Р имеют значения, указанные при определении формулы 9 и формулы 1 соответственно.
Полученное соединение формулы XV является новым соединением и используется в качестве предшественника на следующей стадии д), где
д) в соединение формулы XV вводят мезильную группу при обработке мезилхлоридом (мезилгруппу можно заменить соответствующей тозилгруппой или подобной группой в присутствии триэтиламина (Et3N) в инертном растворителе, например дихлорметане, с образованием соединения формулы XVI
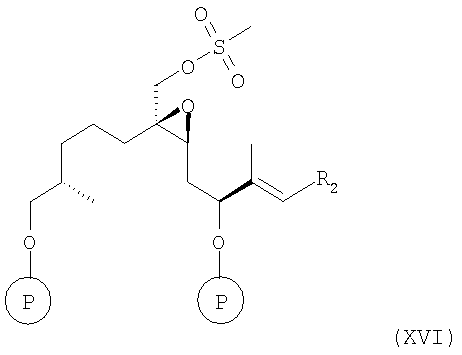
где R2 и Р имеют значения, указанные выше.
Полученное соединение формулы XVI является новым соединением и используется в качестве промежуточного соединения на следующей стадии е), где
е) полученное соединение формулы XVI обрабатывают органической кислотой в инертном растворителе, более конкретно пара-толуолсульфонатом пиридиния или камфорсульфоновой кислотой в абсолютном этаноле, гидролизуют одну защитную группу, и при этом получают соединение формулы XVII
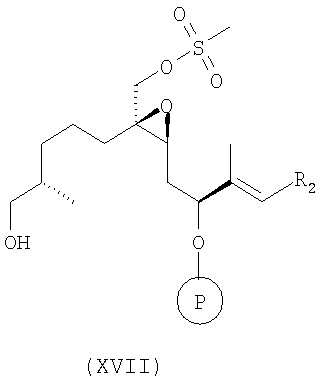
где R2 имеет значения, указанные выше, и
ж) проводят окисление по Сверну, например окисление спиртовой группы в присутствии промотора, оксалилхлорида, и активатора, диметилсульфоксида, получают соль алкоксисульфония и после добавления основания и внутримолекулярной перегруппировки получают кетосоединения формулы 1, которое используют в вышеуказанном способе по изобретению в качестве исходного соединения.
Способ получения производных эпотилона формулы 9 по изобретению обладает множеством преимуществ по сравнению с другими известными и опубликованными методами синтеза.
- Неожиданно установлено, что введение эпоксидной группы на ранних стадиях позволяет исключить стадию эпоксилирования более сложных промежуточных соединений на последующих стадиях синтеза.
- Наблюдается заметная стабилизация мезилата и тозилата эпоксидов. В присутствии указанных функциональных групп можно проводить ряд других стадий.
- Достигаются высокий выход и высокая диастереоселективность прохождения альдольной конденсации ключевых фрагментов, α-разветвленного альдегида в виде енолята титана с этилкетоном.
- Неожиданно установлено, что альдольную конденсацию можно проводить в присутствии мезилата и тозилата эпоксида.
- Альдольная конденсация обеспечивает высокую воспроизводимость синтеза производных эпотилона.
- Неожиданно установлено, что альдольная конденсация позволяет использовать вспомогательную хиральную сультамовую группу в качестве карбоксил-защитной группы и, следовательно, исключить требующие дополнительного времени стадии восстановления и окисления перед проведением заключительной стадии макролактонизации.
- Способ по изобретению представляет собой более короткий путь синтеза по сравнению с другими известными методами синтеза, и, кроме того, при этом достигается общий высокий выход.
Все указанные преимущества подтверждают изобретательский уровень указанной последовательности реакций.
Другим объектом настоящего изобретения являются новые промежуточные соединения, которые получают по описанным реакциям и которые применяются при получении соединений формулы 9.
Производные эпотилона формулы 9, предназначенные для лечения, можно вводить любым известным способом, который выбирают из группы, включающей внутривенный способ, внутриартериальный способ, внутрикожный способ, подкожный способ, пероральный способ, трансбуккальный способ, внутримышечный способ, анальный способ, чрескожный способ, интрадермальный способ, подоболочечный способ и т.п.
Кроме того, можно получать составы с замедленным высвобождением, включающие биодеградируемые микросферы, такие как микросферы полиакриловой кислоты, полигликолевой кислоты или их смесей.
Соединения формулы 9 по изобретению можно использовать отдельно или в комбинации с другими фармацевтически активными соединениями, например с другими химиотерапевтическими агентами, такими как классические цитостатики. В случае комбинаций с другими химиотерапевтическими агентами фиксированные комбинации двух или более компонентов (например, набор компонентов) получают способами, известными специалисту в данной области, а соединение по настоящему изобретению и любой другой химиотерапевтический агент вводят с интервалом, который обеспечивает достижение общего, дополнительного или предпочтительно синергетического действия при лечении опухолевого заболевания.
Предпочтительный способ получения соединений по настоящему изобретению иллюстрируется следующими примерами, не ограничивающими его объем.
Пример 1: соединение X
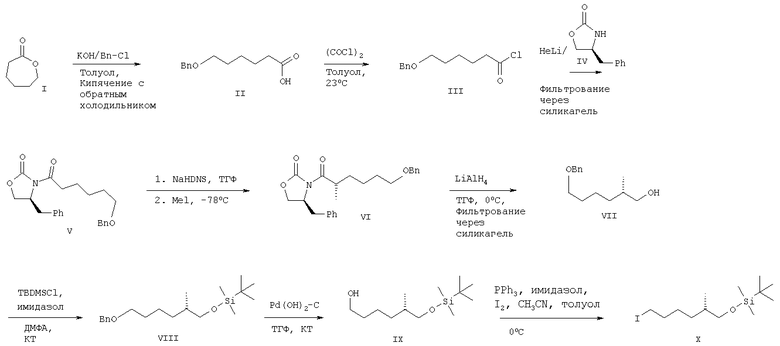
6-Бензилоксигексановая кислота (II) (Can. J.Chem., 70, 1427 (1992))
К раствору 102,2 г (0,90 молей) 6-капролактона в 1,5 л толуола добавляли 250,6 г (4,48 моля) КОН и 113,3 мл (0,98 моля) бензилхлорида и реакционную смесь кипятили с обратным холодильником при перемешивании в течение 20 ч. Реакцию останавливали добавлением 1,7 л воды при комнатной температуре. Водный слой отделяли, раствор в толуоле экстрагировали 200 мл воды. К объединенной водной фазе при 12°С добавляли 300 мл конц. HCl до рН 1,0 и раствор экстрагировали двумя порциями изопропилацетата (1,1 л). Объединенные экстракты промывали 300 мл солевого раствора, сушили над MgSO4 и концентрировали при пониженном давлении, при этом получали 151,2 г (76%) кислоты II в виде масла желтоватого цвета. Rf 0,35 (SiO2, элюент: СН2Cl2/СН3ОН, 95:5).
Хлорангидрид 6-бензилоксигексановой кислоты (III)
Неочищенную кислоту (II) (110 г, 0,50 моля) растворяли в 320 мл толуола и к раствору добавляли 140 мкл ДМФА, а затем 56,7 мл (0,66 моля) оксалилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали в вакууме, при этом получали 126 г хлорангидрида кислоты III, который непосредственно использовали на следующей стадии.
(S)-4-Бензил-3-(6-бензилоксигексаноил)оксазолидин-2-он (V)
Раствор 87,7 г (0,50 моля) (S)-4-бензил-2-оксазолидинона IV растворяли в 1,4 л ТГФ и охлаждали до -78°С. Затем к раствору добавляли гексиллитий (0,45 моля, 178 мл 2,5 М раствора в гексане), а затем медленно добавляли раствор хлорангидрида кислоты III (126 г, 0,50 моля) в 100 мл ТГФ. Смесь перемешивали при -78°С в течение 1 ч, а затем нагревали до 0°С. Реакцию останавливали добавлением насыщенного раствора хлорида аммония (400 мл), слои разделяли и органический слой промывали 100 мл насыщенного раствора хлорида аммония. Слой ТГФ дважды промывали 200 мл 1 М раствора NaOH и 500 мл солевого раствора, сушили над MgSO4 и концентрировали. Неочищенный продукт (162,4 г) очищали экспресс-хроматографией (200 г SiO2, элюент: гептан/ТВМЕ, от 80:20 до 60:20), при этом получали 110,1 г (58% в расчете на две стадии) амида V. Rf 0,41 (SiO2, элюент: гексан/ТВМЕ, 1:1).
(S)-4-Бензил-3-[(S)-6-бензилокси-2-метилгексаноил]оксазолидин-2-он (VI)
К 372 мл (0,37 моля) NaHMDS (1,0 М раствор в ТГФ) в 380 мл ТГФ при -78°С добавляли раствор амида V (120,1 г, 0,32 моля) в ТГФ (200 мл), а затем добавляли раствор СН3I (78,4 мл, 1,26 моля) в 70 мл ТГФ. Реакционную смесь перемешивали при -78°С в течение 2 ч и реакцию останавливали добавлением насыщенного раствора хлорида аммония (670 мл). Слои разделяли и водную фазу экстрагировали 200 мл ТВМЕ. Объединенные органические слои дважды промывали 250 мл солевого раствора, сушили над MgSO4 и концентрировали в вакууме, при этом получали продукт VI (123,9 г, 87%), который непосредственно использовали на следующей стадии. Rf 0,50 (SiO2, элюент: гептан/ТВМЕ, 1:1).
(S)-6-Бензилокси-2-метилгексан-1-ол (VII)
К суспензии 13,2 г (0,35 моля) LiAlH4 в 380 мл ТГФ добавляли раствор амида VI в 250 мл ТГФ при 0°С. Смесь перемешивали при 0°С в течение 2 ч и реакцию останавливали добавлением 14 мл Н2О, 14 мл 15% раствора NaOH и 25 мл Н2О. Выпавшие в осадок соли алюминия удаляли фильтрованием, фильтрат концентрировали, неочищенный продукт очищали фильтрованием через слой силикагеля (570 г SiO2, элюент: толуол/EtOAc, 3 л 90:10 и 1 л 80:20). Продукт VII получали в виде бесцветного масла (54,5 г, 81% в расчете на две стадии). Rf 0,24 (SiO2, элюент: гептан/ТВМЕ, 1:1). ЖХВР: 87% (колонка: Chiralcel OD, элюент: н-гексан/i-PrOH, 95:5, 1 мл/мин, 30°С), tR 10,07 мин.
[(S)-6-Бензилокси-2-метилгексилокси]-трет-бутилдиметилсилан (VIII)
Спирт VII (54,4 г, 0,25 моля) растворяли в 100 мл ДМФА, добавляли 33,3 г (0,49 моля) имидазола, а затем при 0°С медленно добавляли раствор 55,4 г (0,37 моля) трет-бутилдиметилсилилхлорида в 100 мл ДМФА. Смесь перемешивали при 20°С в течение 2 ч, выливали в 240 мл ледяной 0,1 н. HCl и экстрагировали 300 мл гептана. Органический слой промывали 100 мл 0,1 н. HCl, 200 мл насыщенного раствора NaHCO3, 200 мл солевого раствора, сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт (81,5 г) очищали фильтрованием через слой силикагеля (670 г SiO2, элюент: толуол), при этом получали 64,8 г (79%) бесцветного масла. Rf 0,64 (SiO2, элюент: гептан/ТВМЕ, 1:1).
(S)-6-(трет-Бутилдиметилсиланилокси)-5-метилгексан-1-ол (IX)
Раствор 64,0 г (0,19 моля) бензилового эфира VIII в 500 мл ТГФ гидрировали (3,5 бар) в присутствии 7,0 г 20% Pd(OH)2/C при комнатной температуре в течение 30 мин. Катализатор удаляли фильтрованием и фильтрат концентрировали, при этом получали спирт IX в виде бесцветного масла (46,9 г, 100%). Rf 0,33 (SiO2, элюент: гептан/ТВМЕ, 1:1).
трет-Бутил-[(S)-6-иод-2-метилгексилокси]диметилсилан (X)
К раствору 15 г (60,9 ммоля) спирта IX в 390 мл ацетонитрила/толуола (1:5) при комнатной температуре добавляли трифенилфосфин (23,91 г, 91,16 ммолей) и имидазол (12,45 г, 182,9 ммоля). Смесь охлаждали до 0°С, четырьмя порциями в течение 10 мин добавляли 23,13 г (91,13 ммоля) иода и гетерогенный раствор перемешивали в течение 90 мин. Затем добавляли раствор бисульфита (4%, 300 мл) и 100 мл толуола. Водный слой отделяли и снова экстрагировали толуолом (100 мл). Объединенные слои толуола фильтровали через силикагель и концентрировали в вакууме. К остатку добавляли гептан (225 мл), полученную суспензию перемешивали в течение 10 мин и выдерживали при 4°С в течение 12 ч. Затем смесь фильтровали и фильтрат концентрировали, при этом получали 21,05 г (97%) иодида Х в виде масла светло-желтого цвета. Rf 0,66 (SiO2, элюент: гептан/AcOEt, 7:3).
Пример 2
Альдегид 1, содержащий TBDMS-защитные группы
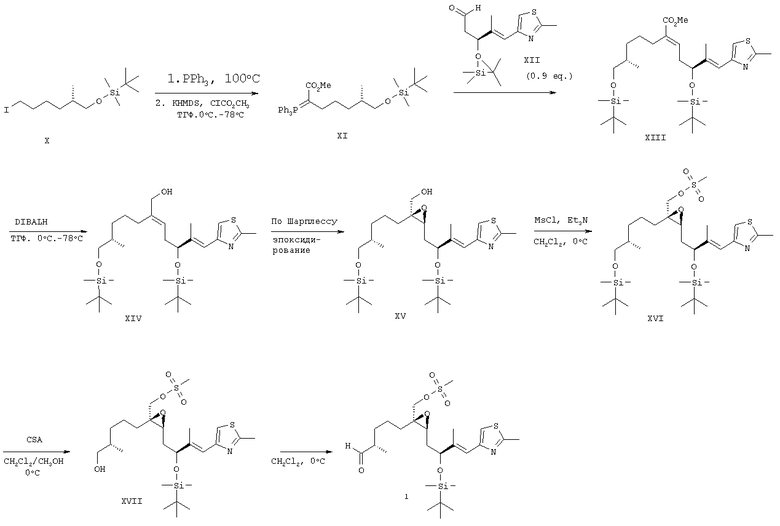
Метиловый эфир (S)-7-(трет-бутилдиметилсиланилокси)-6-метил-2-(трифенилфосфанилиден)гептановой кислоты (XI)
К 8 мл толуола при комнатной температуре в атмосфере аргона добавляли иодид Х (15,46 г, 43,4 ммоля) и трифенилфосфин (12,52 г, 45,75 ммоля) и полученную смесь нагревали при 100°С в течение 3 ч. Затем при пониженном давлении удаляли толуол, к остатку (29,67 г) добавляли ТГФ (400 мл), раствор охлаждали до 0°С, а затем добавляли раствор бис-(триметилсилил)амида калия в толуоле (0,5 М, 174 мл, 86,8 ммоля). Полученную суспензию оранжевого цвета перемешивали при 0°С в течение 1 ч, а затем охлаждали до -75°С. К смеси добавляли метиловый эфир хлормуравьиной кислоты (3,72 мл, 48,2 ммоля), смесь перемешивали при -75°С в течение 1 ч и реакционную смесь желтоватого цвета нагревали до -40°С. К смеси добавляли раствор NaHCO3 (300 мл), а затем добавляли EtOAc (300 мл) и воду (150 мл). Слои разделяли и водную фазу экстрагировали EtOAc (150 мл). Объединенные органические слои промывали насыщенным раствором NaCl (2×200 мл), сушили над MgSO4 и концентрировали в вакууме, при этом получали неочищенный продукт XI (27,02 г) в виде вязкого масла оранжевого цвета, которое непосредственно использовали на следующей стадии.
Метиловый эфир (2Е,6Е)-(S)-5-(трет-бутилдиметилсиланилокси)-2-[(S)-5-(трет-бутилдиметисиланилокси)-4-метилпентил]-6-метил-7-(2-метилтиазол-4-ил)гепта-2,6-диеновой кислоты (XIII)
Раствор илида (XI) (27 г) в толуоле (200 мл) при комнатной температуре в атмосфере аргона добавляли к раствору альдегида (XII) (12,7 г, 39,0 ммоля) в толуоле (80 мл) и полученную смесь перемешивали при 70°С в течение 5 ч. Растворитель удаляли при пониженном давлении, полученный остаток в виде твердого вещества (37,8 г) переносили в 370 мл гептана и последовательно перемешивали при 40°С в течение 30 мин, при комнатной температуре в течение 2,5 ч и, наконец, при 0°С в течение 30 мин. Полученную суспензию фильтровали и остаток на фильтре промывали гептаном (2×60 мл). Фильтраты объединяли и концентрировали в вакууме, при этом получали 24,85 г продукта реакции Виттига XIII в виде масла желтоватого цвета, которое использовали на следующей стадии без дополнительной очистки. Rf 0,59 (SiO2, элюент: гептан/AcOEt, 1:1).
1H-ЯМР (ДМСО-d6, 300 МГц, 300 К): δ 6,90 (s, 1H, С5''-Н), 6,77 (t, J 7,5 Гц, 1Н, С3-Н), 6,47 (s, 1H, С7-Н), 4,20 (dd, J 7,2, 5,4 Гц, 1H, C5-H), 3,69 (s, 3Н, CO2СН3), 3,40 (dd, J 9,9, 5,7 Гц, 1H, C5'-Ha), 3,31 (dd, J 9,9, 6,6 Гц, 1H, С5'-Нb), 2,68 (s, 3H, С2"-СН3), 2,44-2,36 и 2,28-2,21 (два m, 4H, C4-H2 и C1'-H2), 2,00 (s, 3H, С6-СН3), 1,30-1,15 (два m, 4H, C2'-H2, С3'-На и С4'-Н), 1,06-0,99 (m, 1H, С3'-Нb), 0,86 (s, 9Н, SiC(СН3)3), 0,85 (s, 9H, SiC(СН3)3), 0,83 (d, J 6,6, 3Н, С5'-СН3), 0,05 (s, 3H, SiCH3), 0,01 (s, 6H, два SiCH3), -0,01 (s, 3H, SiCH3).
(2Е,6Е)-(S)-5-(трет-Бутилдиметилсиланилокси)-2-[(S)-5-(трет-бутилдиметилсиланилокси)-4-метилпентил]-6-метил-7-(2-метилтиазол-4-ил)гепта-2,6-диен-1-ол (XIV)
К 13,9 г (23 ммоля) аллилового эфира XIII в 500 мл ТГФ при -78°С в течение 10 мин добавляли 47 мл (70 ммолей) DIBALH (1,5 М раствор в толуоле). Реакционную смесь последовательно перемешивали при -78°С в течение 3 ч, нагревали до 0°С в течение 30 мин, а затем перемешивали при 0°С в течение 30 мин и реакцию останавливали добавлением 50 мл 0,1 н. HCl. Слои разделяли и водную фазу промывали ТВМЕ (2×50 мл). Объединенные органические экстракты промывали насыщенным раствором NaHCO3 (2×50 мл) и солевым раствором (50 мл), сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: гептан/EtOAc, 5:1), при этом получали 10,0 г (76%) аллилового спирта XIV в виде масла светло-желтого цвета. Rf 0,16 (SiO2, элюент: гептан/AcOEt, 3:1).
{(2S,3S)-3-[(Е)-(S)-2-(трет-Бутилдиметилсиланилокси)-3-метил-4-(2-метилтиазол-4-ил)бут-3-енил]-2-[(S)-5-(трет-бутилдиметилсиланилокси)-4-метилпентил]оксиранил) метанол (XV)
Аллиловый спирт (XIV) (3,00 г, 5,28 ммоля) растворяли в 51 мл СН2Cl2, при -30°С добавляли 0,59 М раствор (+)-диэтил-L-тартрата (4,48 мл, 2,64 ммоля) в СН2Cl2, а затем добавляли 0,34 М раствор изопропоксида титана (IV) (6,21 мл, 2,11 ммоля) в СН2Cl2. Смесь перемешивали при -30°С в течение 30 мин, а затем в течение 5 мин добавляли 2,11 мл (16,6 ммоля) трет-бутилгидропероксида (5,5 М раствор в декане) и реакционную смесь перемешивали при -30°С в течение 24 ч. Затем к смеси добавляли раствор NaHSO3 (4%, 50 мл), слои разделяли и водный слой экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали 50 мл солевого раствора, сушили над MgSO4 и концентрировали в вакууме, при этом получали 3,11 г неочищенного эпоксиспирта XV в виде масла светло-желтого цвета, которое использовали на следующей стадии без дополнительной очистки. Rf 0,22 (SiO2, элюент: гептан/AcOEt, 2:1). МС (ES+): m/z (%) 606 (100, [М+Na]+), 584 (13, [М+Н]+).
(2S,3S)-3-[(Е)-(S)-2-(трет-Бутилдиметилсиланилокси)-3-метил-4-(2-метилтиазол-4-ил)бут-3-енил]-2-[(S)-5-(трет-бутилдиметилсиланилокси)-4-метилпентил]оксиранилметиловый эфир метансульфоновой кислоты (XVI)
К раствору неочищенного эпоксиспирта (XV) (3,11 г, 5,3 ммоля) в СН2Cl2 (40 мл) при 0°С добавляли 2,74 мл (16,0 ммоля) N-этилдиизопропиламина и полученную смесь перемешивали при 0°С в течение 15 мин. Затем к смеси добавляли по каплям метансульфонилхлорид (0,75 мл, 8,0 ммоля) и смесь перемешивали при 0°С в течение 1 ч. Реакцию останавливали добавлением 40 мл H2O и 40 мл ТВМЕ. Слои разделяли и водный слой экстрагировали ТВМЕ (40 мл). Органические экстракты промывали 0,1 н. HCl (40 мл), насыщенным раствором NaHCO3 (2×40 мл), солевым раствором (40 мл) и сушили над MgSO4. После концентрирования в вакууме получали 3,79 г неочищенного мезилата XVI, который использовали на следующей стадии без дополнительной очистки. Rf 0,27 (SiO2, элюент: гептан/AcOEt, 2:1). МС-ВР: m/z 684,3222 ([М+Na]+, C31H59NO6S2Si2, рассч. 684,3220).
(2S,3S)-3-[(Е)-(S)-2-(трет-Бутилдиметилсиланилокси)-3-метил-4-(2-метилтиазол-4-ил)бут-3-енил]-2-((S)-5-гидрокси-4-метилпентил)оксиранилметиловый эфир метансульфоновой кислоты (XVII)
Неочищенный эфир TBDMS XVI (3,79 г, 5,7 ммоля) растворяли в СН2Cl2/СН3ОН (140 мл, 1:1 об./об.), при 0°С добавляли 1,33 г (5,73 ммоля) 10-камфорсульфоновой кислоты и реакционную смесь перемешивали при 0°С в течение 1 ч. После завершения реакции удаления защитной группы добавляли 20 мл насыщенного раствора NaHCO3 и 20 мл ТВМЕ. Слои разделяли и водный слой экстрагировали ТВМЕ (2×20 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (2×20 мл) и солевым раствором (20 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт XVII (3,11 г) получали в виде масла желтоватого цвета и использовали на следующей стадии без дополнительной очистки. Rf 0,28 (SiO2, элюент: гептан/AcOEt, 1:1).
(2S,3S)-3-[(Е)-(S)-2-(трет-Бутилдиметилсиланилокси)-3 -метил-4-(2-метилтиазол-4-ил)бут-3-енил]-2-((S)-4-метил-5-оксопентил)оксиранилметиловый эфир метансульфоновой кислоты (1)
К раствору спирта XVII (3,11 г, 5,7 ммоля) в CH2Cl2 (15 мл) при 0°С последовательно в течение 5 мин добавляли триэтиламин (10 мл), ДМСО (6 мл) и раствор SO3·пиридина (3,61 г, 22,7 ммоля) в ДМСО (40 мл). Смесь перемешивали при 0°С в течение 1 ч, реакцию останавливали добавлением раствора NaHSO4 (10%, 20 мл) и смесь разбавляли ТВМЕ (20 мл). Слои разделяли и водный слой экстрагировали ТВМЕ (2×20 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (2×20 мл) и солевым раствором (20 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Остаток очищали экспресс-хроматографией (SiO2, элюент: гептан/AcOEt, 3:1). Альдегид 1 получали в виде масла светло-желтого цвета (2,20 г, выход 76% в расчете на четыре стадии и на продукт XIV). Rf 0,39 (SiO2, элюент: гептан/AcOEt, 1:1).
Пример 3
Альдегид XVI' содержащий TBDMS-защитные группы
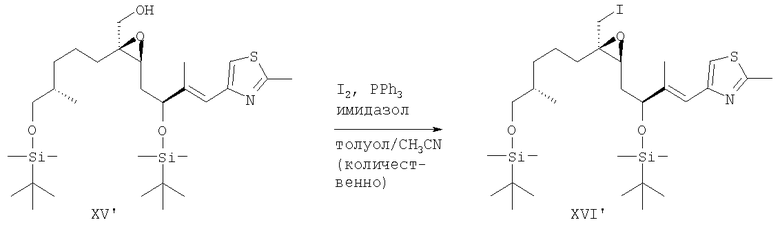
4-[(Е)-(S)-3-(трет-Бутилдиметилсиланилокси)-4-{(2S,3R)-3-[(S)-5-(трет-бутилдиметилсиланилокси)-4-метилпентил]-3-иодметилоксиранил}-2-метилбут-1-енил]-2-метилтиазол (XVI')
К раствору эпоксиспирта XV' (50 мг, 86 мкмолей), трифенилфосфина (34 мг, 0,13 ммоля) и имидазола (18 мг, 0,26 ммоля) в толуоле/ацетонитриле (2,4 мл, 5:1, об./об.) при комнатной температуре в атмосфере аргона одной порцией добавляли иод (33 мг, 0,13 ммоля) и смесь перемешивали в течение 30 мин. После завершения реакции (по результатам ТСХ) реакционную смесь выливали в насыщенный раствор NaHSO4 (5 мл), слои разделяли и водный слой экстрагировали толуолом (2×2 мл). Объединенные органические слои промывали 1 н. HCl (5 мл), насыщенным раствором NaHCO3 (5 мл) и солевым раствором (5 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток суспендировали в гептане и нерастворимый трифенилфосфиноксид отделяли фильтрованием, при этом получали неочищенный иодид XVI' в виде масла светло-желтого цвета (65 мг), которое использовали без дополнительной очистки.
Пример 4
Соединение 2
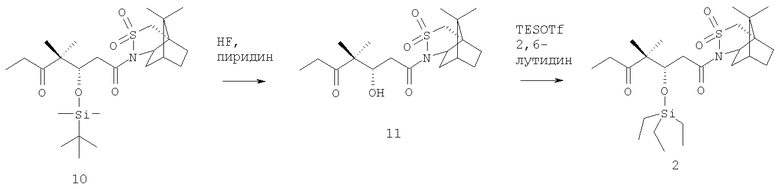
1-(10,10)-Диметил-3,3-диоксо-6-тиа-4-азатрицикло[5.2.1.0]дец-4-ил)-3-гидрокси-4,4-диметилгептан-1,5-дион (11)
Эфир TBDMS (10) (9,21 г, 18,43 ммоля) смешивали в тефлоновом сосуде в атмосфере аргона с комплексом HF·пиридин (26,3 мл, 921 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивали в течение 2 ч, а затем реакцию останавливали добавлением раствора NaHCO3 (78 г в 500 мл воды) и 200 мл ТВМЕ. Слои разделяли, органический слой промывали солевым раствором (2×200 мл), сушили над MgSO4 и концентрировали в вакууме, при этом получали 6,80 г неочищенного продукта, который использовали без дополнительной очистки.
1-(10,10-Диметил-3,3-диоксо-6-тиа-4-азатрицикло[5.2.1.0]дец-4-ил)-4,4-диметил-3-триэтилсиланилоксигептан-1,5-дион (2)
К раствору неочищенного спирта (11) (6,80 г, 17,6 ммоля) в СН2Cl2 (90 мл) при 0°С добавляли 2,6-лутидин (6,14 мл, 52,9 ммоля), а затем триэтилсилилтрифторметансульфонат (7,98 мл, 35,3 ммоля) и полученный раствор перемешивали при 0°С в течение 30 мин. Реакцию останавливали осторожным добавлением 140 мл 1 н. HCl и 150 мл ТВМЕ. Слои разделяли и органический слой последовательно промывали 100 мл насыщенного раствора NaHCO3 и 100 мл солевого раствора, сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали хроматографией (SiO2, элюент: гексан/AcOEt, 4:6), при этом получали 8,3 г очищенного TES-эфира 2 (выход 90% в расчете на две стадии). Rf 0,62 (SiO2, элюент: гептан/AcOEt, 1:1).
1H-ЯМР (ДМСО-d6, 400 МГц, 300 К): δ 4,55 (t, J 5,2 Гц, 1Н, С3-Н), 3,86 (d, J 14,3 Гц, 1Н, C8'-Ha), 3,83 (t, J 6,1 Гц, 1Н, С1'-Н), 3,66 (d, J 14,2 Гц, 1Н, C8'-Hb), 2,84 (dd, J 17,4, 4,6 Гц, 1Н, С2-На), 2,54 (m, 3H, С2-Нb и С7-Н2, частично перекрывается с ДМСО), 2,00-1,92 и 1,87-1,73 (2 m, 6Н, С2'-Н2, С3'-Н и С5'-Н2), 1,48-1,20 и 1,34-1,24 (2 m, 2H, С4'-Н2), 1,10 (s, 3H, С4-СН3), 1,07 (s, 3H, С4-СН3), 0,98 (s, 3H, С9'-СН3), 0,95 (s, 3H, С9'-СН3), 0,94-0,87 (m, 12H, Si(СН2СН3)3 и С7-Н3), 0,60-0,45 (m, 6H, Si(СН2СН3)3).
13С-ЯМР (ДМСО-d6, 100 МГц, 300 К): δ 215,3, 169,7, 73,1, 65,1, 52,9, 52,7, 49,2, 48,1, 45,1, 40, 38,8, 32,8, 32,1, 26,7, 21,5, 20,9, 21,0, 20,4, 8,5, 7,6, 7,5, 5,4.
ИК (KBr): νmax 2958s, 2913m, 2878s, 1702s, 1391m, 1333s, 1312m, 1267m, 1237m, 1219m, 1166m, 1136s, 1087s, 743m, 539m см-1.
MC (ES+): m/z (%) 769 (3, [3М+Са]2+), 538 (6, [М+К]+), 522 (100, [М+Na]+), 519 (17, [2М+Са]2+), 500 (5, [М+Н]+).
Пример 5.
Соединение 6, содержащее TBDMS-защитные группы
Альдольный продукт (3)
Раствор соединения 2 (750 мг, 1,50 ммолей) в СН2Cl2 (5,6 мл) при -78°С в атмосфере аргона последовательно обрабатывали 0,5 М раствором TiCl4 в СН2Cl2 (3,00 мл, 1,50 ммоля) и N-этилдиизопропиламином (2,57 мкл, 1,50 ммоля), при этом раствор немедленно приобретал интенсивный темно-красный цвет. Смесь перемешивали при -78°С в течение 10 мин, затем добавляли по каплям раствор соединения 1 (900 мг, 1,65 ммоля) в СН2Cl2 (1,9 мл) и раствор перемешивали при -78°С в течение 1 ч. Затем смесь нагревали до 0°С и перемешивали в течение еще 15 мин до завершения реакции (по результатам ТСХ). Реакцию останавливали при 0°С добавлением фосфатного буферного раствора (рН 7, 4 мл) и разбавляли ТВМЕ (5 мл). Водный слой экстрагировали ТВМЕ (2×5 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (2×4 мл) и солевым раствором (4 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (50 г SiO2, элюент: гептан/AcOEt, 2:1), при этом получали альдольный продукт 3 (1200 мг, выход 70% в расчете на продукт 1) в виде масла светло-желтого цвета. Rf 0,42 (SiO2, элюент: гептан/AcOEt, 1:2).
МС (ES+): m/z (%) 1084 (5, [М+К]+), 1067 (100, [M+Na]+), 1045 (26, [М+Н]+).
Эфир TBDMS (4)
Раствор соединения 3 (1,20 г, 1,15 ммоля) в СН2Cl2 (15 мл) при 0°С в атмосфере аргона последовательно обрабатывали 2,6-лутидином (0,87 мл, 7,49 ммоля) и трет-бутилдиметилсилилтрифторметансульфонатом (1,32 мл, 5,74 ммоля). Смесь перемешивали при 0°С в течение 14 ч, а затем добавляли 0,1 н. HCl (5 мл) и СН2Cl2 (5 мл). Слои разделяли и водный слой экстрагировали СН2Cl2 (2×5 мл). Объединенные органические экстракты последовательно промывали насыщенным раствором NaHCO3 (2×5 мл) и солевым раствором (5 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (50 г SiO2, элюент: гептан/AcOEt, 2:1), при этом получали соединение 16 (893 мг, 67%) в виде масла светло-желтого цвета. Rf 0,70 (SiO2, элюент: гептан/AcOEt, 1:2).
МС (ES+): m/z (%) 1197 (12, [М+К]+), 1081 (100, [M+Na]+), 1059 (31, [М+Н]+), 599 (75, [М+Са]2+).
Карбоновая кислота (5)
Раствор соединения 4 (318 мг, 0,274 ммоля) в 1,2-диметоксиэтане (4,8 мл) при 0°С последовательно обрабатывали ТВАОН (730 мкл, 2,76 ммоля) и 30% Н2О2 (280 мкл, 2,74 ммоля) и полученную смесь перемешивали при 0°С в течение 5 ч. Затем к смеси добавляли насыщенный раствор NH4Cl (2 мл) и ТВМЕ (2 мл). Слои разделяли и водный слой экстрагировали ТВМЕ (2×2 мл). Органические экстракты последовательно промывали насыщенным раствором NaHCO3 (2×2 мл) и солевым раствором (2 мл), объединяли, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (15 г SiO2, элюент: гептан/AcOEt, 1:1, содержащий 1% АсОН), при этом получали соединение 5 (103 мг, 39%) в виде масла светло-желтого цвета. Rf 0,41 (SiO2, элюент: СН2Cl2/СН3CN/гексан, 3:3:4). МС (ES+): m/z (%) 984 (100, [M+Na]+), 962 (12, [M+H]+).
Метилэпоксид (6)
К раствору соединения 5 (78 мг, 0,081 ммоля) в ТГФ (1,6 мл) при комнатной температуре добавляли по каплям 1 М раствор LiBHEt3 в ТГФ (970 мкл, 0,97 ммоля) и полученную смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию останавливали добавлением насыщенного раствора NH4Cl (2 мл) и ТВМЕ (2 мл). Слои разделяли и водный слой экстрагировали ТВМЕ (2×2 мл). Объединенные органические экстракты последовательно промывали насыщенным раствором NaHCO3 (2×2 мл) и солевым раствором (2 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (6 г SiO2, элюент: гептан/AcOEt, 1:1, содержащий 1% АсОН), при этом получали соединение 6 (60 мг, 85%) в виде масла светло-желтого цвета. Rf 0,56 (SiO2, элюент: гептан/AcOEt, 1:1, содержащий 1% АсОН). МС (ES+): m/z (%) 890 (100, [М+Na]+), 868 (50, [М+Н]+).
Пример 6
Альдегид 1, содержащий одну TES- и одну TBDMS-защитную группу
Метиловый эфир (2E,6E)-(S)-5-гидрокси-2-[(S)-5'-гидрокси-4'-метилпентил)-6-метил-7-(2"-метилтиазол-4"-ил]гепта-2,6-диеновой кислоты (12)
В тефлоновый сосуд, содержащий 24 мл комплекса HF·пиридин (839 молей), при -10°С добавляли аллиловый эфир (XIII) (10,0 г, 16,8 ммоля) и полученную смесь перемешивали при -10°С в течение 10 мин, после чего реакцию немедленно останавливали поскольку результаты ТСХ указывали на полное потребление исходного материала. Смесь выливали в перемешиваемую суспензию NaHCO3 (70 г) в воде (100 мл) и ТВМЕ (100 мл). Слои разделяли и водную фазу экстрагировали ТВМЕ (3×80 мл). Органические экстракты объединяли и промывали 150 мл 0,1 н. HCl, сушили над MgSO4 и концентрировали в вакууме, при этом получали неочищенный диол (2) (6,34 г), который непосредственно использовали на следующей стадии. Rf 0,06 (SiO2, элюент: гептан/AcOEt, 1:1).
1H-ЯМР (ДМСО-d6, 300 МГц, 300 К): δ 6,90 (s, 1H, C5''-H), 6,75 (t, 1H, J 7,5 Гц, С3-Н), 6,51 (s, 1H, С7-Н), 4,23 (t, J 6,3 Гц, 1H, С3-Н), 3,65 (s, 3Н, СО2СН3), 3,41 (dd, J 10,5, 6,0 Гц, 1H, С5'-На), 3,34 (dd, J 10,5, 6,3 Гц, 1H, С5'-Нb), 2,64 (s, 3H, С2"-СН3), 2,46 (t, J 6,9 Гц, 2Н, С4-Н2), 2,25 (t, J 7,2 Гц, 2Н, С1'-H2), 2,00 (s, 3Н, С6-СН3), 1,70-1,55 и 1,53-1,25 (2 ушир. m, 6Н, С2'-Н2, С3'-Нa, С4-Н, C5-OH и C5'-ОН), 1,15-1,00 (m, 1 Н, С3'-Нb), 0,85 (d, J 6,6 Гц, 3Н, С4'-СН3).
Метиловый эфир (2Е,6Е)-(S)-6-метил-7-(2'-метилтиазол-4'-ил)-2-[(S)-4"-метил-5"-триэтилсиланилоксипентил]-5-триэтилсиланилоксигепта-2,6-диеновой кислоты (XIII')
Раствор неочищенного диола (12) (6,33 г, 14 ммолей) в СН2Cl2 (150 мл) при 0°С последовательно обрабатывали N-этилдиизопропиламином (23,6 мл, 138 ммолей), 4-(N,N-диметиламино)пиридином (70 мг, 0,57 ммоля) и триэтилхлорсиланом (10,9 мл, 86,1 ммоля), и полученный раствор перемешивали при комнатной температуре в течение ночи. Реакцию останавливали добавлением насыщенного раствора NaHCO3 (150 мл) и водный слой экстрагировали СН2Cl2 (3×200 мл). Органические экстракты объединяли, промывали 200 мл солевого раствора, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали хроматографией (SiO2, элюент: градиент гексан/AcOEt от 20 до 30%), при этом получали 8,6 г (86% в расчете на две стадии) TES-защищенного диола (XIII'). Rf 0,63 (SiO2, элюент: гептан/AcOEt, 1:1).
1Н-ЯМР (ДМСО-d6, 400 МГц, 300 К): δ 7,26 (s, δ, С5'-Н), 6,73 (t, J 7,3 Гц, 1Н, С3-Н), 6,48 (s, 1H, C7-H), 4,30 (t, J 6,7 Гц, 1Н, С5-Н), 3,65 (s, 3Н, CO2СН3), 3,41 (dd, J 9,8, 5,9 Гц, 1H, С5"-На), 3,36 (dd, J 9,8, 6,3 Гц, 1H, С5"-Нb), 2,66 (s, 3H, С2'-СН3), 2,45 (t, J 6,7 Гц, 2Н, С4-Н2), 2,30-2,20 (ушир. m, 2H, C1"-H2), 2,04 (s, 3H, С6-СН3), 1,59-1,47 (ушир. m, 1H, С4"-Н), 1,45-1,23 (ушир. m, 3Н, С2"-Н2 и С3"-На), 1,01-1,00 (ушир. m, 1H, С3"-Нb), 0,98-0,88 (m, 18H, два Si(СН2СН3)3), 0,83 (d, J 6,9 Гц, 3Н, С4"-СН3), 0,62-0,52 (m, 12H, два Si(СН2СН3)3).
ИК (пленка) νmax 2953s, 2934s, 2913s, 2875s, 1713s, 1644w, 1505w, 1459m, 1437m, 1414w, 1376w, 1285m, 1269m, 1241m, 1185m, 1120m, 1072m, 1006m, 744m, 723s см-1.
MC (ES+): m/z (%) 618 (100, [M+Na]+), 596 (72, [М+Н]+).
(2E,6E)-(S)-6-Метил-7-(2'-метилтиазол-4'-ил)-2-[(S)-4"-метил-5"-триэтилсиланилоксипентил]-5-триэтилсиланилоксигепта-2,6-диен-1-ол (XIV)
К раствору аллилового эфира (XIII') (45,54 г, 76,4 ммоля) в ТГФ (250 мл) при -78°С добавляли 1,5 М раствор DIBAL-H в толуоле (153 мл, 229 ммоля) и полученную смесь перемешивали при -78°С в течение 3 ч. Реакцию останавливали медленным добавлением 115 мл 2 н. HCl и разбавляли 100 мл ТВМЕ. Слои разделяли и водную фазу экстрагировали ТВМЕ (2×100 мл). Объединенные органические экстракты промывали 100 мл раствора NaHCO3, 100 мл солевого раствора, сушили над MgSO4 и концентрировали в вакууме, при этом получали 51,53 г неочищенного аллилового спирта, который очищали хроматографией (SiO2, элюент: градиент гексан/AcOEt, от 20 до 40%), при этом получали 32,5 г (75%) очищенного аллилового спирта (XIV). Rf 0,47 (SiO2, элюент: гептан/AcOEt, 1:1).
1H-ЯМР (ДМСО-d6, 500 МГц, 300 К): δ 7,29 (s, 1H, С5'-Н), 6,38 (s, 1H, C7-H), 5,29 (t, J 7,1 Гц, 1H, С3-Н), 4,60 (t, J 5,4 Гц, 1H, С1-ОН), 4,10 (t, J 6,4 Гц, 1H, С5-Н), 3,79 (d, J 5,1 Гц, 2H, C1-H2), 3,40 (dd, J 9,8, 5,9 Гц, 1H, C5"-Ha), 3,33 (dd, J 9,8, 6,5 Гц, 1H, С5"-Нb), 2,64 (s, 3Н, С2'-СН3), 2,25 (t, J 6,8 Гц, 2H, С4-Н2), 1,99 (s, 3H, С6-СН3), 2,00-1,90 (ушир. m, 2Н, C1"-H2), 1,58-1,46 (m, 1H, C4"-H), 1,43-1,22 (m, 3Н, С3"-Ha и С2"-Н2), 1,08-0,97 (m, 1H, С3"-Нb), 0,95-0,85 (m, 18H, два Si(СН2СН3)3), 0,82 (d, J 6,7 Гц, 3Н, С4'-СН3), 0,58-0,50 (m, 12Н, два Si(СН2СН3)3).
13С-ЯМР (ДМСО-d6, 125 МГц, 300 К): δ 164,1, 152,5, 141,1, 140,7, 119,9, 118,4, 116,6, 78,0, 67,3, 64,6, 35,2, 34,4, 32,9, 28,0, 25,4, 18,9, 16,6, 13,7, 6,74, 6,72, 4,3, 4,0.
ИК (пленка): νmax 3341s (ушир.), 2952s, 2932s, 2874s, 1508m, 1458m, 1414m, 1378w, 1238w, 1190m, 1071s, 1016s, 883w, 829w, 743m, 728m см-1.
MC (ES+): m/z (%) 590 (28, [M+Na]+), 568 (100, [М+Н]+).
{(2S,3S)-3-[(E)-(S)-3'-Метил-4'-(2"-метилтиазол-4"-ил)-2-триэтилсиланилоксибут-3-енил]-2-[(S)-4'"-метил-5'"-триэтилсиланилоксипентил]оксиранил)метанол (XV)
5,00 г (8,80 ммоля) аллилового спирта (4) растворяли в 100 мл СН2Cl2 в высушенной колбе в атмосфере аргона, добавляли молекулярные сита 4Å (4 г), смесь охлаждали до -30°С и добавляли (+)-диэтил-L-тартрат (7,46 мл, 4,40 ммоля) и изопропоксид титана (IV) (10,35 мл, 3,52 ммоля). Полученную смесь перемешивали при -30°С в течение 1 ч, при этом раствор постепенно приобретал желто-зеленую окраску. Затем добавляли 3,52 мл трет-бутилгидропероксида (19,36 ммоля), реакционную смесь выдерживали при -25°С в течение ночи. Реакцию останавливали, добавив 100 мл NaHSO3, и смесь перемешивали. Фильтрат экстрагировали ТВМЕ (2×80 мл). Объединенные органические слои промывали 80 мл солевого раствора, сушили над MgSO4 и концентрировали в вакууме, при этом получали 7,24 г неочищенного продукта. После очистки хроматографией (элюент: гексан/AcOEt, 1:1) получали 5,2 г (100%) эпоксиспирта (XV). Rf 0,38 (SiO2, элюент: гептан/AcOEt, 1:1).
1Н-ЯМР (ДМСО-d6, 400 МГц, 300 К): δ 7,28 (s, 1H, C5"-H), 6,44 (s, 1H, С4 -Н), 4,30 (dd, J 8,2, 4,1 Гц, 1H, С2'-Н), 3,46 (d, J 12,2 Гц, 1H, C1-Ha), 3,39 (dd, J 10,2, 6,0 Гц, 1H, C5'"-Ha), 3,34 (dd, J 10,2, 6,2 Гц, 1H, С5"-Нb), 3,27 (d, J 12,2 Гц, 1H, C1-Нb), 2,90 (dd, J 7,3, 4,6 Гц, 1H, С3-Н), 2,63 (s, 3Н, С2"-СН3), 1,98 (s, 3Н, С3'-СН3), 1,81 (ddd, J 12,3, 8,2, 4,6 Гц, 1H, C1'-Ha), 1,68-1,20 (4 m, 7H, C1'-Нb, C1"'-Н2, С2'"-Н2, С3"'-На, С4"'-Н), 1,08-0,98 (m, 1H, С3"'-Нb), 0,93-0,83 (m, 18H, два Si(СН2СН3)3), 0,82 (d, J 6,7 Гц, 3Н, С4"'-СН3), 0,59-0,50 (m, 12H, два Si(СН2СН3)3).
13С-ЯМР (ДМСО-d6, 125 МГц, 300 К): δ 164,7, 152,6, 141,2, 118,7, 117,1, 76,3, 67,6, 63,6, 63,6, 60,2, 57,5, 35,5, 35,4, 33,3, 28,7, 22,3, 19,1, 16,8, 14,4, 13,9, 7,1, 7,0, 4,6, 4,3.
ИК (пленка) νmax 3403s (ушир.), 2954s, 2876s, 1507m, 1459m, 1414m, 1377m, 1239m, 1185m, 1084s, 1007s, 976w, 802w, 744s, 676w см-1.
MC (ES): m/z (%) 606 (52, [M+Na]+), 584 (100, [М+Н]+).
(2S,3S)-3-[(Е)-(S)-3'-Метил-4'-(2"-метилтиазол-4"-ил)-2-триэтилсиланилоксибут-3-енил]-2-[(S)-4"'-метил-5"'-триэтилсиланилоксипентил]оксиранилметиловый эфир метансульфоновой кислоты (XVI)
К раствору эпоксиспирта (XV) (11,71 г, 20,0 ммолей) в СН2Cl2 (148 мл) при 0°С добавляли 10,3 мл (60,1 ммоля) N-этилдиизопропиламина и полученную смесь перемешивали при 0°С в течение 15 мин. Затем добавляли метансульфонилхлорид (2,33 мл, 30,1 ммоля) и смесь перемешивали при 0°С в течение 1 ч. Реакцию останавливали добавлением 50 мл Н2O и 50 мл ТВМЕ. Слои разделяли и водный слой промывали ТВМЕ (50 мл). Объединенные органические экстракты промывали 0,1 н. HCl (40 мл), насыщенным раствором NaHCO3 (2×40 мл), солевым раствором (50 мл) и сушили над MgSO4. После концентрирования в вакууме получали 13,7 г эпоксимезилата (XVI), который использовали без дополнительной очистки. Rf 0,55 (SiO2, элюент: гептан/AcOEt, 1:1).
1Н-ЯМР (ДМСО-d6, 400 МГц, 300 К): δ 7,34 (s, 1H, С5"-Н), 6,49 (s, 1H, C4'-H), 4,38 (d, J 11,4 Гц, 1H, C1-Ha), 4,34 (dd, J 8,2, 3,7 Гц, 1H, С2'-Н), 4,07 (d, J 11,4 Гц, 1H, C1-Нb), 3,43 (dd, J 9,8, 5,9 Гц, 1H, C5"'-Ha), 3,37 (dd, J 9,8, 6,3 Гц, 1H, С5"'-Нb), 3,33 (s, 3Н, SO2СН3), 3,06 (dd, J 7,0, 4,6 Гц, 1H, С3-Н), 2,66 (s, 3H, С2"-СН3), 2,03 (s, 3Н, С3'-СН3), 1,89 (ddd, J 15,2, 7,0, 3,7 Гц, 1H, C1'-Ha), 1,77-1,20 (5 m, 7H, C1'-Нb, C1"'-H2, С2"'-Н2, С3"'-На, С4"'-Н), 1,12-1,03 (m, 1H, С3"'-Нb), 0,97-0,88 (m, 18H, два Si(СН2СН3)3), 0,85 (d, J 6,6 Гц, 3Н, С4"'-СН3), 0,63-0,50 (m, 12H, два Si(СН2СН3)3).
13С-ЯМР (ДМСО-d6, 125 МГц, 300 К): δ 164,2, 152,3, 140,5, 118,6, 117,0, 75,8, 72,1, 67,2, 60,4, 57,8, 36,6, 35,1, 34,6, 32,8, 28,1, 21,7, 18,9, 16,5, 13,6, 6,8, 6,7, 4,3, 4,0.
MC (ES+): m/z (%) 684 (100, [M+Na]+), 662 (54, [М+Н]+).
(2S,3S)-2-[(S)-5'-Гидрокси-4'-метилпентил]-3-[(E)-(S)-3"-метил-4"-(2"'-метилтиазол-4'"-ил)-2-триэтилсиланилоксибут-3-енил]оксиранилметиловый эфир метансульфоновой кислоты (XVII)
Раствор бис-TES эфира (XVI) (4,89 г, 7,38 ммоля) в ТГФ/АсОН/Н2О (10:2:1 об./об./об., 115 мл) нагревали при 50°С в течение 15 ч. Реакцию останавливали добавлением 460 мл насыщенного раствора NaHCO3 и смесь экстрагировали ТВМЕ (2×250 мл). Органические слои объединяли, промывали солевым раствором, сушили над MgSO4 и концентрировали в вакууме, при этом получали 4,73 г неочищенного продукта. Продукт очищали экспресс-хроматографией (SiO2, элюент: градиент AcOEt/гексан от 30 до 60%), при этом получали 3,10 г спирта (XVII) (79% в расчете на две стадии и на продукт XV). Rf 0,15 (SiO2, элюент: гептан/AcOEt, 1:1).
1H-ЯМР (ДМСО-d6, 400 МГц, 300 К): δ 7,42 (s, 1H, С5"'-Н), 6,50 (s, 1H, C4"-H), 4,38 (d, J 11,3 Гц, 1Н, C1-Ha), 4,36 (dd, J 8,3, 4,6 Гц, 1Н, С2"-Н, частично перекрывается C1-Ha), 4,07 (d, J 11,8 Гц, 1H, C1-Ha), 3,30-3,23 (m, 1H, С5'-На), 3,20 (s, 3Н, SO2СН3), 3,22-3,14 (m, 1H, C5'-Нb, частично перекрывается SO2СН3), 3,06 (dd, J 7,1, 4,4 Гц, 1H, С3-Н), 2,67 (s, 3Н, С2"'-СН3), 2,04 (s, 3Н, С3"-СН3), 1,90 (ddd, J 14,2, 8,3, 4,4 Гц, 1H, C1"-Ha), 1,70-1,58 (m, 1H, С1"-Нb), 1,54-1,32 (m, 6Н, C1'-H2, C2'-H2, С3'-На, С4'-Н), 1,10-0,98 (m, 1H, С3'-Нb), 0,93 (t, J 8,0 Гц, 9Н, Si(СН2СН3)3), 0,84 (d, J 6,6 Гц, 3Н, С4'-СН3), 0,60 (q, J 8,0 Гц, 6Н, Si(СН2СН3)3).
13С-ЯМР (ДМСО-d6, 100 МГц, 300 К): δ 165,1, 153,2, 141,5, 119,4, 117,9,76,6, 73,3, 67,0, 61,2, 58,6, 37,6, 36,0, 35,5, 33,9, 29,0, 22,6, 19,7, 17,5, 14,9, 14,5, 7,63, 7,58, 5,1.
ИК (пленка): νmax 3410s (ушир.), 2955s, 2876s, 1655w, 1505w, 1459m, 1414m, 1359s, 1270w, 1239w, 1177s, 1081m, 1006m, 958m, 833m, 745m, 529m см-1.
MC (ES+): m/z (%) 586 (5, [М+К]+), 570 (100, [M+Na]+), 548 (8, [М+Н]+).
(2S,3S)-3-[(Е)-(S)-3'-Метил-4'-(2"-метилтиазол-4"-ил)-2-триэтилсиланилоксибут-3-енил]-2-[(S)-4"'-метил-5'"-оксопентил]оксиранилметиловый эфир метансульфоновой кислоты (1)
К раствору спирта (XVII) (2,00 г, 3,65 ммоля) в СН2Cl2 (10 мл) при 0°С последовательно добавляли триэтиламин (3,86 мл, 27,7 ммоля), ДМСО (6,48 мл, 91,3 ммоля) и комплекс пиридиний·SO3 (2,32 г, 14,6 ммоля) в ДМСО (26 мл) и полученный раствор перемешивали при 0°С в течение 1 ч. Реакцию останавливали добавлением раствора NaHSO4 (40%, 120 мл) и ТВМЕ (150 мл). Слои разделяли и органический слой промывали насыщенным раствором NaHCO3 (2×100 мл). Водные слои повторно экстрагировали ТВМЕ (2×100 мл). Органические экстракты объединяли, промывали солевым раствором (100 мл), сушили над MgSO4 и концентрировали в вакууме, при этом получали 1,94 г неочищенного продукта. После очистки экспресс-хроматографией (SiO2, элюент: градиент AcOEt/гексан, от 50 до 70%) получали 1,63 г (82%) очищенного альдегида (1). Rf 0,30 (SiO2, элюент: гептан/AcOEt, 1:1).
1H-ЯМР (CDCl3, 500 МГц, 300 К): δ 9,61 (d, J 1,7 Гц, 1H, CHO), 6,95 (s, 1H, C5"-Н), 6,52 (s, 1H, С4'-Н), 4,33 (dd, J 4,4 Гц, 1H, С2'-Н, частично перекрывается C1-На), 4,34 (d, J 11,4 Гц, 1H, C1-Ha), 4,08 (d, J 11,4 Гц, 1H, C1-Нb), 3,11 (dd, J 7,8, 3,9 Гц, 1H, С3-Н), 3,05 (s, 3H, SO2СН3), 2,71 (s, 3H, С2"-СН3), 2,33 (m, 1H, С4"'-Н), 2,02 (s, 3H, С3'-СН3), 1,99-1,32 (6 m, 8H, C1'-Н2, C1"'-H2, С2"'-Н2, С3"'-Н2), 1,11 (d, J 7,1 Гц, 3H, С4"'-СН3), 0,94 (t, J 8,0 Гц, 9Н, Si(СН2СН3)3), 0,61 (q, J 8,0 Гц, 6Н, Si(СН2СН3)3).
13С-ЯМР (CDCl3, 125 МГц, 300 К): δ 204,7, 164,8, 152,8, 141,8, 119,2, 115,8, 76,0, 72,0, 60,7, 59,0, 46,3, 37,8, 35,3, 30,5, 28,3, 22,5, 19,4, 14,1, 13.5, 7,0, 4,9.
ИК (пленка): ν 2956s, 2876s, 2720w, 1724s, 1658w, 1508m, 1459m, 1414w, 1358s, 1240w, 1177s, 1079m, 1006m, 957m, 883w, 828m, 745m, 529m см-1.
MC (ES+): m/z 546 (100, [М+Н]+).
MS (ES+) m/z 546 (100, [М+Н]+).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭПОТИЛОНА, НОВЫЕ ПРОИЗВОДНЫЕ ЭПОТИЛОНА, А ТАКЖЕ НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ РЕАЛИЗАЦИИ СПОСОБА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2404985C2 |
| ПРОИЗВОДНОЕ АЛЬФА-АМИНОГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2265592C2 |
| ПРОИЗВОДНЫЕ АМИНОКИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2005 |
|
RU2379281C2 |
| ФОТОУПРАВЛЯЕМЫЕ ФОТОХРОМНЫЕ ЭЛЕКТРОЛЮМИНЕСЦИРУЮЩИЕ И ЭЛЕКТРОПРОВОДЯЩИЕ ПОЛИМЕРЫ ДЛЯ ФОТОНИКИ | 2004 |
|
RU2345998C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ТИОКОЛХИЦИНА И БАККАТИНА В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2001 |
|
RU2264393C2 |
| ЭНАНТИОСЕЛЕКТИВНЫЙ СИНТЕЗ ПРОИЗВОДНЫХ ГАММА-АМИНО-АЛЬФА, БЕТА-НЕНАСЫЩЕННЫХ КАРБОНОВЫХ КИСЛОТ | 2009 |
|
RU2512499C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГИДАНТОИНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2005 |
|
RU2376301C2 |
| СПОСОБ ОБРАЗОВАНИЯ ПОЛИГЕДРАЛЬНЫХ ОЛИГОМЕРНЫХ СИЛСЕСКВИОКСАНОВ (ВАРИАНТЫ) | 2000 |
|
RU2293745C2 |
Изобретение относится к хорошо осуществимому в промышленных масштабах за счет более короткого пути синтеза способу получения производных эпотилона формулы (9) и их солей с катионами металлов, согласно которому обеспечивается высокая воспроизводимость синтеза высокоочищенных производных эпотилона. Способ обеспечивает проведение альдольной конденсации в присутствии мезилата и тозилата эпоксида с использованием вспомогательной хиральной сультамовой группы в качестве карбоксил-защитной группы, в результате чего требующие дополнительного времени стадии восстановления и окисления перед проведением заключительной стадии макролактонизации исключаются. Введение эпоксидной группы на ранних стадиях способа позволяет также исключить стадию эпоксилирования более сложных промежуточных соединений на последующих стадиях процесса. 7 з.п. ф-лы.
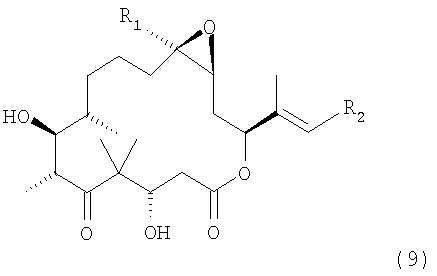
где
R1 означает метил, a R2 означает тиазол, замещенный алкилом, и их солей с катионами металлов, включающий
а) взаимодействие соединения формулы 1
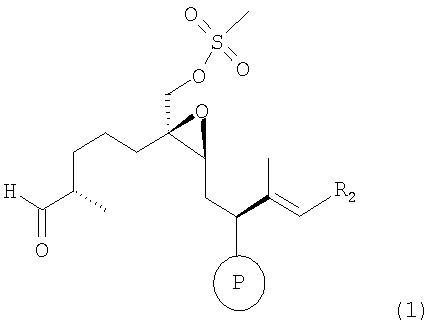
где R2 имеет значения, указанные выше, а мезилгруппа заменена тозилгруппой или подобной группой, а Р означает ОН-защитную группу, с сультам-производным формулы 2, например
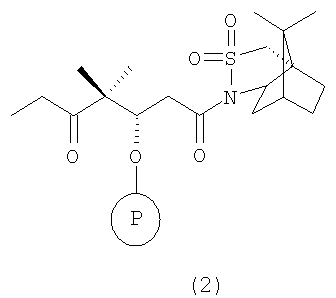
по реакции селективной альдольной конденсации в присутствии кислоты Льюиса и при добавлении основания в инертном растворителе при пониженной температуре от -50 до -100°С, а затем при повышенной температуре от -20 до +20°С, с образованием соединения формулы 3
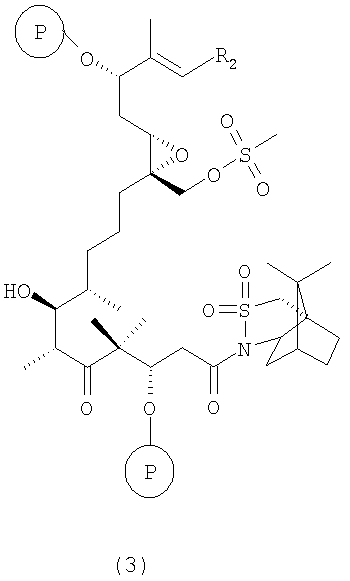
где R2 и Р имеют значения, указанные выше, и
б) взаимодействие полученных соединений вышеуказанной формулы 3 с соединением, образующим простой силиловый эфир, в инертном растворителе при температуре от -70 до 25°С в присутствии 2,6-лутидина с образованием соединения формулы 4
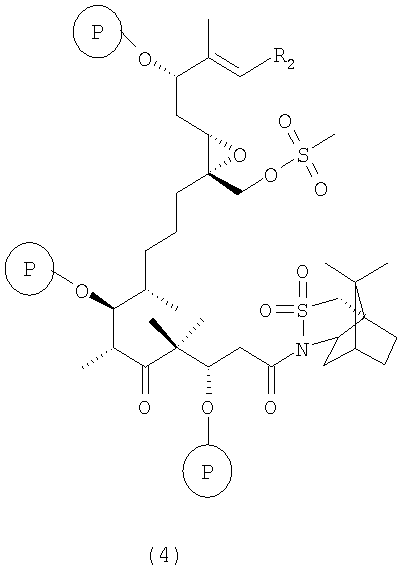
где R2 и Р имеют значения, указанные выше, и
в) превращение вышеуказанного соединения формулы 4 в карбоновую кислоту при отщеплении хиральной вспомогательной группы обработкой ТВАОН/Н2O2 в ДМЭ или LiO2H в ТГФ/МеОН/Н2О с образованием соединения формулы 5
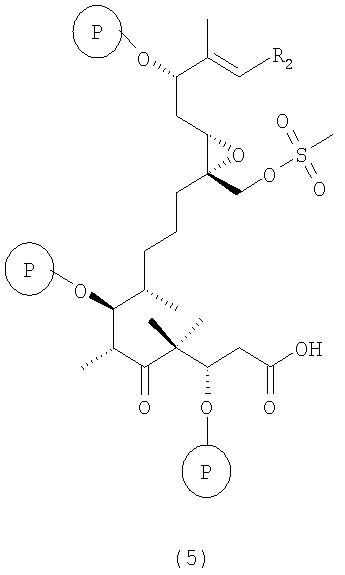
где R2 и Р имеют значения, указанные выше, и
г) взаимодействие соединения формулы 5 с восстанавливающим агентом в инертном растворителе с отщеплением мезил- или тозилгруппы или подобной группы, с образованием соединения формулы 6
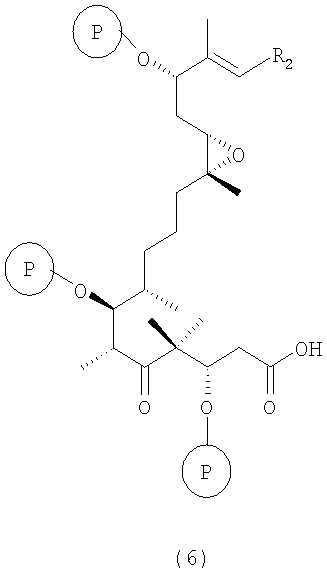
где R2 имеют значения, указанные выше, и
д) гидролиз соединений формулы 6 в присутствии десилилирующего реагента или кислоты в инертном растворителе или в их смеси, например TASF или HF·пиридин в ТГФ, с образованием селективно десилилированного соединения формулы 7
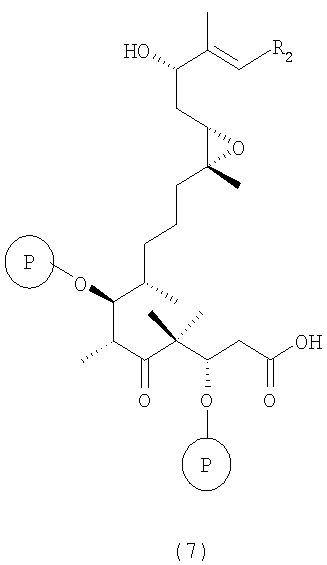
где R2 и Р имеют значения, указанные выше, и
е) макролактонизацию гидроксикислоты формулы 7 посредством обработки реагентом Et3N и 2,4,6-трихлорбензоилхлоридом при пониженной температуре с образованием полностью защищенного производного эпотилона формулы 8
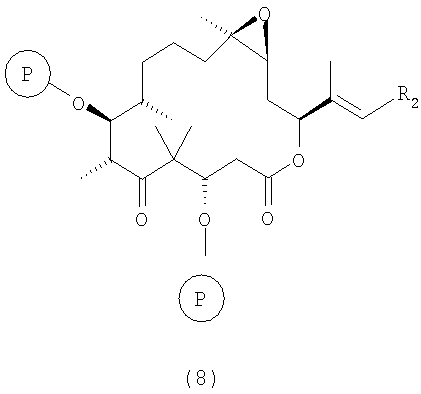
где R2 и Р имеют значения, указанные выше, и
ж) взаимодействие полученного соединения формулы 8 с HF·пиридином в инертном растворителе при температуре от 0 до 30°С и отщепление обеих силильных защитных групп с образованием производных эпотилона формулы 9
где R1 означает метил, a R2 имеет значения, указанные выше, и необязательно получение солей соединений формулы 9, где R1 и R2 имеют значения, указанные при определении формулы 9, с катионами металлов стандартными методами.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| US 6387927 B1, 14.05.2002 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Автоматически действующая центрифуга для побелки сахарных голов | 1939 |
|
SU63962A1 |
| МОДИФИЦИРОВАННЫЕ В БОКОВОЙ ЦЕПИ ЭПОТИЛОНЫ | 1998 |
|
RU2201932C2 |

