В настоящем изобретении в основном предлагаются соединения, проявляющие противораковую активность, и более конкретно соединения, которые ингибируют активность киназы PI3. В настоящем изобретении предлагаются также способы применения соединений для диагностики или обработки in vitro, in situ и in vivo клеток млекопитающих, или для лечения сопутствующих патологических состояний.
В настоящем изобретении предлагаются бензоксазепины и их фармацевтические композиции, которые можно использовать для лечения заболеваний, состояний и/или нарушений, опосредованных киназами PI3.
В одном объекте настоящего изобретения предлагаются соединения формулы I
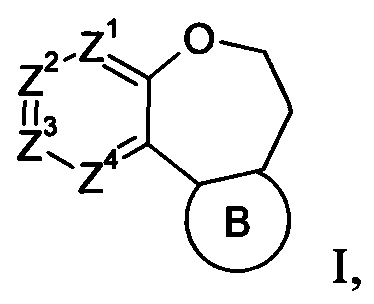
включая стереоизомеры, геометрические изомеры, таутомеры или фармацевтически приемлемые соли указанных соединений, где Z1 обозначает CR1 или N, Z2 обозначает CR2 или N, Z3 обозначает CR3 или N, Z4 обозначает CR4 или N, а В обозначает пиразолил, имидазолил, или триазолил, конденсированный с циклом бензоксазепина. Различные заместители имеют значения, как определено в данном контексте.
Более конкретно, в настоящем изобретении предлагаются соединения формулы I:

стереоизомеры, геометрические изомеры, таутомеры и фармацевтически приемлемые соли указанных соединений, где
Z1 обозначает CR1 или N,
Z2 обозначает CR2 или N,
Z3 обозначает CR3 или N,
Z4 обозначает CR4 или N,
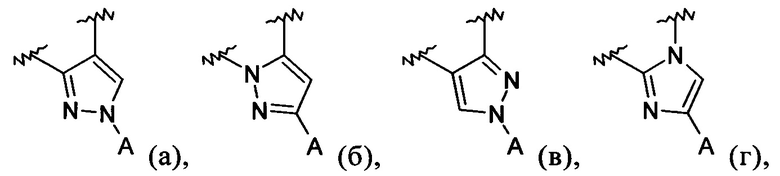
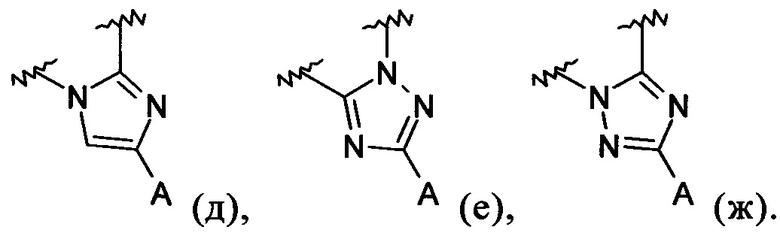
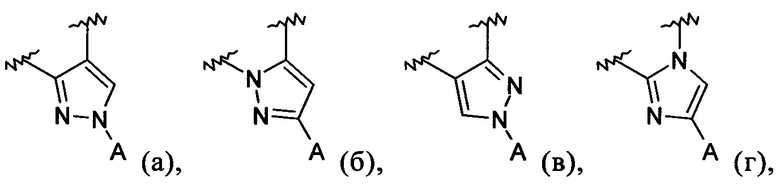
В обозначает пиразолил, имидазолил или триазолил, конденсированный с циклом бензоксазепина, и указанный заместитель выбирают из следующих структур:
R1, R2, R3 и R4 независимо выбирают из Н, F, Cl, Br, I, -CN, -COR10, -CO2R10, -C(=O)N(R10)OR11, -C(=NR10)NR10R11, -C(=O)NR10R11, -NO2, -NR10R11, -NR12C(=O)R10, -NR12C(=O)OR11, -NR12C(=O)NR10R11, -NR12C(=O)(C1-C12алкилен)NR10R11, -NR12(C1-C12алкилен)NR10R11, -NR12(C1-C12алкилен)OR10, -NR12(C1-C12алкилен)C(=O)NR10R11, -OR10,
-S(O)2R10, -C(=O)NR10(C1-C12алкилен)NR10R11,
-C(=O)NR10(C1-C12алкилен)NR10C(=O)OR11,
-C(=O)NR10(C1-C12алкилен)NR10C(=O)R11,-C(=O)NR10(C1-C12алкилен)R10, С1-С12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила, С1-С20гетероарила, -(С3-С12карбоциклил)-(С1-С12алкила), -(С2-С20гетероциклил) (С1-С12алкила), -(С6-С20арил)-(С1-С12алкила), -(С1-С20гетероарил) (С1-С12алкила), -(С1-С12алкилен) (С3-С12карбоциклила), -(С1-С12алкилен) (С2-С20гетероциклила), -(С1-С12алкилен) (С2-С20гетероциклил)(С2-С20гетероциклила), -(С1-С12алкилен)(С2-С20гетероциклил)(С3-С12карбоциклила), -(С1-С12алкилен)(С2-С20гетероциклил)-С(=O)-(С2-С20гетероциклила), -(С1-С12алкилен)(С1-С20гетероарила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С1-С12алкила),
-(С1-С12алилен)(С6-С20арил)(С1-С12алкила),
-(С1-С12алкилен)(С1-С20гетероарил)(С1-С12алкила),
-(С1-С12алкилен)-С(=O)-(С2-С20гетероциклила), -(С1-С12алкилен)С(=O)OR10, -(С1-С12алкилен)-NR10R11, -(С1-С12алкилен)NR12C(=O)R10,
-(С1-С12алкилен)OR10,
-(С1-С12алкилен)-NR10-(С1-С12алкилен)(С1-С20гетероарила),
-(С1-С12алкилен)-NR10-(С1-С12алкилен)(С1-С20гетероциклила),
-(С1-С12алкилен)-NR10-(С1-С12алкилен)-NHC(=O)-(С1-С20гетероарила), -(С1-С12алкилен)(С2-С20гетероциклил)-NR10R11 и
-(С1-С12алкилен)(C2-C20гетероциклил)(C1-C12алкил)-NR10R11,
где алкил, алкенил, алкинил, алкилен, карбоциклил, гетероциклил, арил и гетероарил необязательно замещены одной или более группами, независимо выбранными из F, Cl, Br, I, R10, -SR10, -S(O)2R10, -S(O)2NR10R11, -NR10R11, -NR12C(O)R10, -CO2R10, -C(O)R10, -CONR10R11, оксогруппы и -OR10,
А выбирают из -C(=O)NR5R6, -NR5R6, С6-С20арила, С2-С20гетероциклила и С1-С20гетероарила, где арил, гетероциклил и гетероарил необязательно замещены одной или более группами, независимо выбранными из F, Cl, Br, I, -CN, -COR10, -CO2R10, -C(=O)N(R10)OR11, -C(=NR10)NR10R11, -C(=O)NR10R11, -NO2, -NR10R11, -NR12C(=O)R10, -NR12C(=O)OR11, -NR12C(=O)NR10R11, -NR12C(=O)(С1-С12алкилен)NR10R11, -NR12(С1-С12алкилен)NR10R11, -NR12(C1-C12алкилен)OR10, -NR12(C1-C12алкилен)C(=O)NR10R11, -OR10, -S(O)2R10, -C(=O)NR10(С1-С12алкилен)NR10R11,
-C(=O)NR10(C1-С12алкилен)NR10C(=O)OR11,
-C(=O)NR10(C1-C12алкилен)NR10C(=O)R11, -C(=O)NR10(C1-C12алкилен)R10, С1-С12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила, С1-С20гетероарила,
-(С3-С12карбоциклил)(С1-С12алкила), -(С2-С20гетероциклил)(С1-С12алкила), -(С6-С20арил)(С1-С12алкила), -(С1-С20гетероарил)(С1-С12алкила),
-(С1-С12алкилен)(С3-С12карбоциклила),
-(С1-С12алкилен)(С2-С20гетероциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С2-С20гетероциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С3-С12карбоциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)-С(=O)-(С2-С20гетероциклила),
-(С1-С12алкилен)(С1-С20гетероарила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С1-С12алкила),
-(С1-С12алкилен)(С6-С20арил)(С1-С12алкила),
-(С1-С12алкилен)(С1-С20гетероарил)(С1-С12алкила),
-(С1-С12алкилен)-С(=O)-(С2-С20гетероциклила), -(С1-С12алкилен)С(=O)OR10, -(C1-C12алкилен)-NR10R11,-(C1-C12алкилен)NR12C(=O)R10,
-(С1-С12алкилен)OR10,
-(С1-С12алкилен)-NR10 -(С1-С12алкилен)(С1-С20гетероарила),
-(С1-С12алкилен)-NR10-(С1-С12алкилен)(С1-С20гетероциклила),
-(С1-С12алкилен)-NR10-(С1-C12алкилен)-NHC(=O)-(C1-С20гетероарила),
-(С1-С12алкилен)(С2-С20гетероциклил)-NR10R11 и
-(С1-С12алкилен)(С2-С20гетероциклил)(С1-С12алкил)-NR10R11,
где алкил, алкенил, алкинил, алкилен, карбоциклил, гетероциклил, арил и гетероарил необязательно замещены одной или более группами, независимо выбранными из F, Cl, Br, I, R10, -SR10, -S(O)2R10, -NR10R11, -NR12C(=O)R10, -CO2R10, -C(O)R10, -CONR10R11 и -OR10,
R5 выбирают из Н и С1-С12алкила, необязательно замещенного одной или более группами, независимо выбранными из F, Cl, Br, I, -CN, -CO2H, -CONH2, -CONHCH3, -NH2, -NO2, -N(СН3)2, -NHCOCH3, -NHS(O)2CH3, -ОН, -ОСН3, -ОСН2СН3, -S(O)2NH2 и -S(O)2СН3,
R6 выбирают из С1-С12алкила, С3-С12карбоциклила, С2-С20гетероциклила, С1-С20гетероарила и С6-С20арила, каждый из которых независимо замещен одной или более группами, независимо выбранными из F, Cl, Br, I, -СН3, -CH2OH, -СН2С6Н5, -CN, -CF3, -CO2H, -С(O)СН3, -NH2, -NO2, -N(CH3)2, -NHCOCH3, -NHS(O)2CH3, -ОН, оксогруппы, -ОСН3, -ОСН2СН3, -S(O)2NH2, -S(O)2СН3, -C(=O)NR10(C1-C12алкилен)NR10R11, фенила, пиридинила, тетрагидрофуран-2-ила, 2,3-дигидробензофуран-2-ила, 1-изопропилпирролидин-3-илметила, морфолин-4-ила, пиперидин-1-ила, пиперазинила, пиперазин-4-ил-2-она, пиперазин-4-ил-3-она, пирролидин-1-ила, тиоморфолин-4-ила, S-диоксотиоморфолин-4-ила, -C≡CR13, -CH=CHR13 и -C(=O)NR10R11, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют С2-С20гетероциклил или C1-C20гетероциклил, необязательно замещенный одной или более группами, выбранными из F, Cl, Br, I, СН3, С(СН3)3, -СН2ОН, -CH2CH2OH, -СН2С6Н5, пиридин-2-ила, 6-метилпиридин-2-ила, пиридин-4-ила, пиридин-3-ила, пиримидин-2-ила, пиразин-2-ил, тетрагидрофуранкарбонила, 2-метоксифенила, бензоила, циклопропилметила, (тетрагидрофуран-2-ил)метила, 2,6-диметилморфолин-4-ила, 4-метилпиперазинкарбонила, пирролидин-1-карбонила, циклопропанкарбонила, 2,4-дифторфенила, пиридин-2-илметила, морфолин-4-ила, -CN, -CF3, -CO2H, -CONH2, -CONHCH3, -CON(CH3)2, -COCF3, -СОСН3, -СОСН(СН3)2, -NO2, NHCH3, -N(CH3)2, -N(СН2СН3)2, -NHCOCH3, -NCH3COCH3, -NHS(O)2СН3, -ОН, -ОСН3, -ОСН2СН3, -CH2OCH3, -CH2CH2OCH3, -CH2S(O)2NHCH3, -CH2S(O)2СН2СН3, -S(O)2NHCH3, -S(O)2СН2СН3, -S(O)2NH2, -S(O)2N(СН3)2 и -S(O)2СН3,
R10, R11 и R12 независимо выбирают из Н, С1-С12алкила, -(С1-С12алкилен)-(С2-С20гетероциклила), -(С1-С12алкилен)-(С6-С20арила), -(С1-С12алкилен)-(С3-С12карбоциклила), С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арилаи С1-С20гетероарила, каждый из которых необязательно замещен одной или более группами, независимо выбранными из F, Cl, Br, I, -СН3, -СН2СН3, -СН(СН3)2, -CH2OH, -CH2OCH3, -CH2CH2OH, -С(СН3)2OH, -СН2С(СН3)2OH, -СН2СН(СН3)ОН, -CH2CO2H, -CH2CO2CH3, -CH2NH2, -(СН2)2Н(СН3)2, -СН2С6Н5, -CN, -CF3, -CO2H, -С(O)СН3, -С(O)СН(ОН)СН3, -CO2CH3, -CONH2, -CONHCH3, -CON(CH3)2, -С(СН3)2CONH2, -NH2, -NO2, -N(СН3)2, -N(СН3)С(СН3)2CONH2, -N(СН3)CH2CH2S(O)2СН3, -NHCOCH3, -NHS(O)2СН3, =O (оксогруппы), -ОН, -ОСН3, -ОСН2СН3, -OCH2CH2OH, -ОР(O)(ОН)2, -SCH3, -S(O)2СН3, -S(O)2NH2, -S(O)2N(CH3)2, -CH2S(O)2NHCH3, -CH2S(O)2СН2СН3, -S(O)2NHCH3, -S(O)2СН2СН3, пирролидин-1-ила, 2-оксопирролидин-1-ила, циклопропила, циклопентила, оксетанила, 4-метилпиперазин-1-ила и 4-морфолинила, или
R10 и R11, присоединенные к атому азота, образуют вместе с атомами азота, к которым они присоединены, цикл С2-С20гетероциклила или
С1-С20гетероарила, каждый из которых необязательно замещен одной или более группами, независимо выбранными из F, Cl, Br, I, -СН3, -CH2OH, -СН2С6Н5, -CN, -CF3, -CO2H, -CONH2, -CONHCH3, -NO2, -N(CH3)2, -NHCOCH3, -NHS(O)2CH3, -ОН, оксогруппы, -ОСН3, -ОСН2СН3, -S(O)2NH2, -S(O)2СН3, -СН(СН3)2, -CH2CF3, -CH2CH2OH и -С(СН3)2OH, и
R13 выбирают из Н, F, Cl, Br, I, -СН3, -СН2СН3, -CN, -CF3, -CH2N(СН3)2, -CH2OH, -CO2H, -CONH2, -CON(CH3)2, -NO2 и -S(O)2СН3.
Более конкретно, в настоящем изобретении предлагаются соединения формулы I:
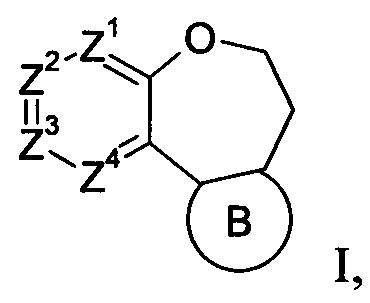
стереоизомеры, геометрические изомеры, таутомеры и фармацевтически приемлемые соли указанных соединений, где
Z1 обозначает CR1 или N,
Z2 обозначает CR2 или N,
Z3 обозначает CR3 или N,
Z4 обозначает CR4 или N,
В обозначает пиразолил, имидазолил или триазолил, конденсированный с циклом бензоксазепина, и указанный заместитель выбирают из следующих структур:

R1, R2, R3 и R4 независимо выбирают из Н, F, Cl, Br, I, -CN, -COR10, -CO2R10, -C(=O)N(R10)OR11, -C(=NR10)NR10R11, -C(=O)NR10R11, -NO2, -NR10R11, -NR12C(=O)R10, -NR12C(=O)OR11, -NR12C(=O)NR10R11, -NR12C(=O)(C1-C12алкилен)NR10R11, -NR12(C1-C12алкилен)NR10R11, -NR12(C1-C12алкилен)OR10, -NR12(C1-C12алкилен)C(=O)NR10R11, -OR10, -S(O)2R10, -C(=O)NR10(C1-C12алкилен)NR10R11,
-C(=O)NR10(C1-C12алкилен)NR10C(=O)OR11,
-C(=O)NR10(C1-C12алкилен)NR10C(=O)R11, -C(=O)NR10(C1-C12алкилен)R10, С1-С12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила, С1-С20гетероарила,
-(С3-С12карбоциклил)(С1-С12алкила), -(С2-С20гетероциклил)(С1-С12алкила), -(С6-С20арил)(С1-С12алкила), -(С1-С20гетероарил)(С1-С12алкила), -(С1-С12алкилен)(С3-С12карбоциклила),
-(С1-С12алкилен)(С2-С20гетероциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С2-С20гетероциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С3-С12карбоциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)С(=O)-(С2-С20гетероциклила),
-(С1-С12алкилен)(С1-С20гетероарила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С1-С12алкила),
-(С1-С12алилен)(С6-С20арил)(С1-С12алкила),
-(С1-С12алкилен)(С1-С20гетероарил)(С1-С12алкила),
-(С1-С12алкилен)-С(=O)-(С2-С20гетероциклила), -(С1-С12алкилен)С(=O)OR10, -(C1-C12алкилен)-NR10R11, -(C1-C12алкилен)NR12C(=O)R10,
-(С1-С12алкилен)OR10,
-(C1-C12алкилен)-NR10-(С1-C12алкилен)(С1-С20гетероарила),
-(C1-C12алкилен)-NR10-(С1-С12алкилен)(С1-С20гетероциклила),
-(С1-C12алкилен)-NR10-(С1-С12алкилен)-NHC(=O)-(С1-С20гетероарила),
-(С1-С12алкилен)(С2-С20гетероциклил)-NR10R11 и
-(С1-С12алкилен)(С2-С20гетероциклил)(С1-С12алкил)-NR10R11,
где алкил, алкенил, алкинил, алкилен, карбоциклил, гетероциклил, арил и гетероарил необязательно замещены одной или более группами, независимо выбранными из F, Cl, Br, I, R10, -SR10, -S(O)2R10, -S(O)2NR10R11, -NR10R11, -NR12C(O)R10, -CO2R10, -C(O)R10, -CONR10R11, оксогруппы и -OR10,
А выбирают из -C(=O)NR5R6, -NR5R6, С6-С20арила, С2-С20гетероциклила и С1-С20гетероарила, где арил, гетероциклил и гетероарил необязательно замещены одной или более группами, независимо выбранными из F, Cl, Br, I, -CN, -COR10, -CO2R10, -C(=O)N(R10)OR11, -C(=NR10)NR10R11, -C(=O)NR10R11, -NO2, -NR10R11, -NR12C(=O)R10, -NR12C(=O)OR11, -NR12C(=O)NR10R11, -NR12C(=O)(C1-C12алкилен)NR10R11, -NR12(C1-C12алкилен)NR10R11, -NR12(C1-C12алкилен)OR10, -NR12(C1-C12алкилен)C(=O)NR10R11, -OR10, -S(O)2R10, -C(=O)NR10(C1-C12алкилен)NR10R11,
-C(=O)NR10(C1-C12алкилен)NR10C(=O)OR11,
-C(=O)NR10(C1-C12алкилен)NR10C(=O)R11,-C(=O)NR10(C1-C12алкилен)R10, С1-С12алкила, С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила, С1-С20гетероарила,
-(С3-С12карбоциклил)(С1-С12алкила), -(С2-С20гетероциклил)(С1-С12алкила), -(С6-С20арил)(С1-С12алкила), -(С1-С20гетероарил)(С1-С12алкила),
-(С1-С12алкилен)(С3-С12карбоциклила),
-(С1-С12алкилен)(С2-С20гетероциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С2-С20гетероциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С3-С12карбоциклила),
-(С1-С12алкилен)(С2-С20гетероциклил)-С(=O)-(С2-С20гетероциклила),
-(С1-С12алкилен)(С1-С20гетероарила),
-(С1-С12алкилен)(С2-С20гетероциклил)(С1-С12алкила),
-(С1-С12алкилен)(С6-С20арил)(С1-С12алкила),
-(С1-С12алкилен)(С1-С20гетероарил)(С1-С12алкила),
-(С1-С12алкилен)-С(=O)-(С2-С20гетероциклила), -(С1-C12алкилен)С(=O)OR10, -(C1-C12алкилен)NR10R11, -(C1-C12алкилен)NR12C(=O)R10,
-(С1-С12алкилен)OR10,
-(С1-С12алкилен)-NR10-(С1-С12алкилен)(С1-С20гетероарила),
-(С1-С12алкилен)-NR10-(С1-С12алкилен)(С1-С20гетероциклила),
-(С1-С12алкилен)-NR10-(С1-С12алкилен)-NHC(=O)-(С1-С20гетероарила),
-(С1-С12алкилен)(С2-С20гетероциклил)-NR10R11 и
-(С1-С12алкилен)(С2-С20гетероциклил)(С1-С12алкил)-NR10R11,
где алкил, алкенил, алкинил, алкилен, карбоциклил, гетероциклил, арил и гетероарил необязательно замещены одной или более группами, независимо выбранными из F, Cl, Br, I, R10, -SR10, -S(O)2R10, -NR10R11, -NR12C(O)R10, -CO2R10, -C(O)R10, -CONR10R11 и -OR10,
R5 выбирают из Н и С1-С12алкила, необязательно замещенного одной или более группами, независимо выбранными из F, Cl, Br, I, -CN, -СО2Н, -CONH2, -CONHCH3, -NH2, -NO2, -N(СН3)2, -NHCOCH3, -NHS(O)2СН3, -ОН, -ОСН3, -ОСН2СН3, -S(O)2NH2 и -S(O)2СН3,
R6 выбирают из С1-С12алкила, С3-С12карбоциклила, С2-С20гетероциклила, С1-С20гетероарила и С6-С20арила, каждый из которых необязательно замещен одной или более группами, независимо выбранными из F, Cl, Br, I, -СН3, -СН2ОН, -СН2С6Н5, -CN, -CF3, -CO2H, -С(O)СН3, -NH2, -NO2, -N(СН3)2, -NHCOCH3, -NHS(O)2CH3, -ОН, оксогруппы, -ОСН3, -ОСН2СН3, -S(O)2NH2, -S(O)2СН3, -C(=O)NR10(C1-C12алкилен)NR10R11, фенила, пиридинила, тетрагидрофуран-2-ила, 2,3-дигидробензофуран-2-ила, 1-изопропилпирролидин-3-илметила, морфолин-4-ила, пиперидин-1-ила, пиперазинила, пиперазин-4-ил-2-она, пиперазин-4-ил-3-она, пирролидин-1-ила, тиоморфолин-4-ила, S-диоксотиоморфолин-4-ила, -C≡CR13, -CH=CHR13 и -C(=O)NR10R11, или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют С2-С20гетероциклил или С1-С20гетероарил, необязательно замещенный одной или более группами, выбранными из F, Cl, Br, I, СН3, С(СН3)3, -CH2OH, -CH2CH2OH, -СН2С6Н5, пиридин-2-ила, 6-метилпиридин-2-ила, пиридин-4-ила, пиридин-3-ила, пиримидин-2-ила, пиразин-2-ил, тетрагидрофуранкарбонила, 2-метоксифенила, бензоила, циклопропилметила, (тетрагидрофуран-2-ил)метила, 2,6-диметилморфолин-4-ила, 4-метилпиперазинкарбонила, пирролидин-1-карбонила, циклопропанкарбонила, 2,4-дифторфенила, пиридин-2-илметила, морфолин-4-ила, -CN, -CF3, -CO2H, -CONH2, -CONHCH3, -CON(CH3)2, -COCF3, -СОСН3, -СОСН(СН3)2, -NO2, NHCH3, -N(CH3)2, -N(CH2CH3)2, -NHCOCH3, -NCH3COCH3, -NHS(O)2CH3, -ОН, -ОСН3, -ОСН2СН3, -CH2OCH3, -CH2CH2OCH3, -CH2S(O)2NHCH3, -CH2S(O)2СН2СН3, -S(O)2NHCH3, -S(O)2СН2СН3, -S(O)2NH2, -S(O)2N(CH3)2 и -S(O)2СН3,
R10, R11 и R12 независимо выбирают из Н, С1-С12алкила, -(С1-С12алкилен)-(С2-С20гетероциклила), -(С1-С12алкилен)-(С6-С20арила), -(С1-С12алкилен)-(С3-С12карбоциклила), С2-С8алкенила, С2-С8алкинила, С3-С12карбоциклила, С2-С20гетероциклила, С6-С20арила и C1-C20гетероарила, каждый из которых необязательно замещен одной или более группами, независимо выбранными из F, Cl, Br, I, -СН3, -СН2СН3, -СН(СН3)2, -CH2OH, -СН2ОСН3, -CH2CH2OH, -С(СН3)2OH, -СН2С(СН3)2OH, -СН2СН(СН3)ОН, -CH2CO2H, -CH2CO2CH3, -CH2NH2, -(СН2)2N(СН3)2, -СН2С6Н5, -CN, -CF3, -CO2H, -С(O)СН3, -С(O)СН(ОН)СН3, -CO2CH3, -CONH2, -CONHCH3, -CON(СН3)2, -С(СН3)2CONH2, -NH2, -NO2, -N(СН3)2, -N(CH3)C(CH3)2CONH2, -N(CH3)CH2CH2S(O)2CH3, -NHCOCH3, -NHS(O)2СН3, =O (остатка оксогруппы), -ОН, -ОСН3, -ОСН2СН3, -OCH2CH2OH, -ОР(O)(ОН)2, -SCH3, -S(O)2СН3, -S(O)2NH2, -S(O)2Н(СН3)2, -CH2S(O)2NHCH3, -CH2S(O)2СН2СН3, -S(O)2NHCH3, -S(O)2СН2СН3, пирролидин-1-ила, 2-оксопирролидин-1-ила, циклопропила, циклопентила, оксетанила, 4-метилпиперазин-1-ила и 4-морфолинила, или
R10 и R11 вместе с атомом азота, к которому они присоединены, образуют цикл С2-С20гетероциклила или С1-С20гетероарила, каждый из которых необязательно замещен одной или более группами, независимо выбранными из F, Cl, Br, I, -СН3, -CH2OH, -СН2С6Н5, -CN, -CF3, -СО2Н, -CONH2, -CONHCH3, -NO2, -N(СН3)2, -NHCOCH3, -NHS(O)2СН3, -ОН, оксогруппы, -ОСН3, -ОСН2СН3, -S(O)2NH2, -S(O)2СН3, -СН(СН3)2, -CH2CF3, -CH2CH2OH и -С(СН3)2OH, и
R13 выбирают из Н, F, Cl, Br, I, -СН3, -СН2СН3, -CN, -CF3, -CH2N(CH3)2, -CH2OH, -CO2H, -CONH2, -CON(CH3)2, -NO2 и -S(O)2СН3.
Следует подробно остановиться на некоторых вариантах осуществления настоящего изобретения, примеры которых проиллюстрированы прилагаемыми структурами и формулами. Хотя в настоящем изобретении описаны перечисленные варианты, следует понимать, что указанные варианты не ограничивают объем настоящего изобретения. Напротив, в объем настоящего изобретения включены все альтернативные варианты, модификации и эквивалентные варианты, как определено в пунктах формулы настоящего изобретения. Специалисту в данной области техники представляется очевидным множество способов и соединений, аналогичных или эквивалентных описанным в данном контексте, которые можно использовать при осуществлении настоящего изобретения на практике. Настоящее изобретение не ограничивается описанными способами и соединениями. В случае, если содержание одной или более включенных в качестве ссылок публикаций, патентов и аналогичных материалов отличается или противоречит содержанию настоящей заявки, включая, но не ограничиваясь только ими, определенные термины, использование терминов, описанные методики и т.п., приоритет отдается настоящей заявке.
Использованный в данном контексте термин «алкил» обозначает насыщенный одновалентный углеводородный радикал с прямой или разветвленной цепью, содержащий от одного до двенадцати атомов углерода (С1-С12), при этом алкильный радикал необязательно независимо замещен одним или более заместителями, описанными ниже. В другом варианте алкильный радикал содержит от одного до восьми атомов углерода (C1-C8), или от одного до шести атомов углерода (C1-С6). Примеры алкильных групп включают, но не ограничиваясь только ими, метил (Me, -СН3), этил (Et, -СН2СН3), 1-пропил (n-Pr, н-пропил, -СН2СН2СН3), 2-пропил (i-Pr, изопропил, -СН(СН3)2), 1-бутил (n-Bu, н-бутил, -СН2СН2СН2СН3), 2-метил-1-пропил (i-Bu, изобутил, -СН2СН(СН3)2), 2-бутил (s-Bu, втор-бутил, -СН(СН3)СН2СН3), 2-метил-2-пропил (t-Bu, трет-бутил, -С(СН3)3), 1-пентил (н-пентил, -СН2СН2СН2СН2СН3), 2-пентил (-СН(СН3)СН2СН2СН3), 3-пентил (-СН(СН2СН3)2), 2-метил-2-бутил (-С(СН3)2СН2СН3), 3-метил-2-бутил (-СН(СН3)СН(СН3)2), 3-метил-1-бутил (-СН2СН2СН(СН3)2), 2-метил-1-бутил (-СН2СН(СН3)СН2СН3), 1-гексил (-СН2СН2СН2СН2СН2СН3), 2-гексил (-СН(СН3)СН2СН2СН2СН3), 3-гексил (-СН(СН2СН3)(СН2СН2СН3)), 2-метил-2-пентил (-С(СН3)2СН2СН2СН3), 3-метил-2-пентил (-СН(СН3)СН(СН3)СН2СН3), 4-метил-2-пентил (-СН(СН3)СН2СН(СН3)2), 3-метил-3-пентил (-С(СН3)(СН2СН3)2), 2-метил-3-пентил (-СН(СН2СН3)СН(СН3)2), 2,3-диметил-2-бутил (-С(СН3)2СН(СН3)2), 3,3-диметил-2-бутил (-СН(СН3)С(СН3)3, 1-гептил, 1-октил и т.п.
Использованный в данном контексте термин «алкилен» обозначает насыщенный двухвалентный углеводородный радикал с прямой или разветвленной цепью, содержащий от одного до двенадцати атомов углерода (С1-С12), при этом алкиленовый радикал необязательно независимо замещен одним или более заместителями, описанными ниже. В другом варианте алкиленовый радикал содержит от одного до восьми атомов углерода (C1-C8), или от одного до шести атомов углерода (C1-С6). Примеры алкиленовых групп включают, но не ограничиваясь только ими, метилен (-СН2-), этилен (-СН2СН2-), пропилен (-СН2СН2СН2-) и т.п.
Термин «алкенил» обозначает одновалентный углеводородный радикал с прямой или разветвленной цепью, содержащий от двух до восьми атомов углерода (С2-C8) и по крайней мере один ненасыщенный участок, т.е. углерод-углеродную двойную связь (sp2 гибридизация), при этом алкенильный радикал необязательно независимо замещен одним или более заместителями, описанными в данном контексте, и включает радикалы в «цис» и «транс» конформациях, или, в другом варианте, в конфигурациях «Е» и «Z». Примеры включают, но не ограничиваясь только ими, этиленил или винил (-СН=СН2), аллил (-СН2СН=СН2) и т.п.
Термин «алкенилен» обозначает двухвалентный углеводородный радикал с прямой или разветвленной цепью, содержащий от двух до восьми атомов углерода (С2-С8) и по крайней мере один ненасыщенный участок, т.е. углерод-углеродную двойную связь (sp2 гибридизация), при этом алкениленовый радикал необязательно замещен и включает радикалы в «цис» и «транс» конформациях, или, в другом варианте, в конфигурациях «Е» и «Z». Примеры включают, но не ограничиваясь только ими, этиленилен или винилен (-СН=СН-), аллил (-СН2СН=СН-) и т.п.
Термин «алкинил» обозначает одновалентный углеводородный радикал с прямой или разветвленной цепью, содержащий от двух до восьми атомов углерода (C2-C8) и по крайней мере один ненасыщенный участок, т.е. углерод-углеродную тройную связь (sp гибридизация), при этом алкинильный радикал необязательно независимо замещен одним или более заместителями, описанными в данном контексте. Примеры включают, но не ограничиваясь только ими, этинил (-С≡СН), пропинил (пропаргил, -СН2С≡СН) и т.п.
Термин «алкинилен» обозначает двухвалентный углеводородный радикал с прямой или разветвленной цепью, содержащий от двух до восьми атомов углерода (С2-C8) и по крайней мере один ненасыщенный участок, т.е. углерод-углеродную тройную связь (sp гибридизация), при этом алкиниленовый радикал необязательно замещен. Примеры включают, но не ограничиваясь только ими, этинилен (-С≡С-), пропинилен (пропаргилен, -СН2С≡С-) и т.п.
Термины «карбоцикл», «карбоциклил», «карбоциклическое кольцо» и «циклоалкил» обозначают одновалентный неароматический насыщенный или частично ненасыщенный цикл, содержащий от 3 до 12 атомов углерода (С3-C12) в виде моноциклического кольца или от 7 до 12 атомов углерода в виде бициклического кольца. Бициклические карбоциклы, содержащие от 7 до 12 атомов, представляют собой, например, бициклические системы [4,5], [5,5], [5,6] или [6,6], а бициклические карбоциклы, содержащие 9 или 10 атомов в цикле, представляют собой бициклические системы [5,6] или [6,6], или мостиковые системы, такие как бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан. Примеры моноциклических карбоциклов включают, но не ограничиваясь только ими, циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил, циклододецил и т.п.
Термин «арил» обозначает одновалентный ароматический углеводородный радикал, содержащий 6-20 атомов углерода (С6-С20), полученный при удалении одного атома водорода от одного атома углерода в составе исходной ароматической циклической системы. Некоторые арильные группы обозначены на приведенных в качестве примеров структурах как «Ar». Арил включает бициклические радикалы, содержащие ароматический цикл, конденсированный с насыщенным, частично ненасыщенным циклом или ароматическим карбоциклом. Примеры арильных групп включают, но не ограничиваясь только ими, радикалы, полученные из бензола (фенил), замещенных бензолов, нафталина, антрацена, бифенила, инденила, инданила, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и т.п. Арильные группы необязательно независимо замещены одним или более заместителями, описанными в данном контексте.
Термин «арилен» обозначает двухвалентный ароматический углеводородный радикал, содержащий 6-20 атомов углерода (С6-С20), полученный при удалении двух атомов водорода от двух атомов углерода в составе исходной ароматической циклической системы. Некоторые ариленовые группы обозначены на приведенных в качестве примеров структурах как «Ar». Арилен включает бициклические радикалы, содержащие ароматический цикл, конденсированный с насыщенным, частично ненасыщенным циклом или ароматическим карбоциклическим кольцом. Примеры ариленовых групп включают, но не ограничиваясь только ими, радикалы, полученные из бензола (фенилен), замещенных бензолов, нафталина, антрацена, бифенилена, инденилена, инданилена, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафтила и т.п. Ариленовые группы необязательно замещены.
Термины «гетероцикл», «гетероциклил» и «гетероциклическое кольцо» используются в данном контексте взаимозаменяемо и обозначают насыщенный или частично ненасыщенный (т.е. содержащий одну или более двойных и/или тройных связей в цикле) карбоциклический радикал, содержащий от 3 до приблизительно 20 атомов в цикле, в котором по крайней мере одним атомом цикла является гетероатом, выбранный из азота, кислорода, фосфора и серы, а остальными атомами цикла являются атомы С, при этом один или более атомов в цикле необязательно независимо замещен одним или более заместителями, описанными ниже. Гетероциклом является моноцикл, содержащий от 3 до 7 атомов в цикле (от 2 до 6 атомов углерода и от 1 до 4 гетероатомов, выбранных из N, О, Р и S), или бицикл, содержащий от 7 до 10 атомов в цикле (от 4 до 9 атомов углерода и от 1 до 6 гетероатомов, выбранных из N, О, Р и S), например, бициклическая система [4,5], [5,5], [5,6] или [6,6]. Гетероциклы описаны в книге Paquette Leo A., «Principles of Modern Heterocyclic Chemistry» (Benjamin W.A., Нью-Йорк (1968)), прежде всего в главах 1, 3, 4, 6, 7 и 9, в сериях монографий «The Chemistry of Heterocyclic Compounds, A series of Monographs» (John Wiley & Sons, Нью-Йорк, издаваемых с 1950 до настоящего времени), прежде всего в тт. 13, 14, 16, 19 и 28, а также в журнале J. Am. Chem. Soc., 82, с. 5566 (1960). Термин «гетероциклил» также включает радикалы, где гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным циклом или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но не ограничиваясь только ими, морфолин-4-ил, пиперидин-1-ил, пиперазинил, пиперазин-4-ил-2-он, пиперазин-4-ил-3-он, пирролидин-1-ил, тиоморфолин-4-ил, 8-диоксотиоморфолин-4-ил, азокан-1-ил, азетидин-1-ил, октагидропиридо[1,2-а]пиразин-2-ил, [1,4]диазепан-1-ил, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидине, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло [3.1.0] гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3Н-индолилхинолизинил и N-пиридилмочевины. В объем указанного определения включены также спиро-структуры. Примерами гетероциклической группы, в которой 2 атома углерода в цикле заменены на остаток оксогруппы (=O), являются пиримидинонил и 1,1-диоксотиоморфолинил. Гетероциклические группы в данном контексте необязательно независимо замещены одним или более заместителями, описанными в данном контексте.
Термин «гетероарил» обозначает одновалентный 5-, 6- или 7-членный ароматический радикал и включает конденсированные циклические системы (в составе которых по крайней мере один цикл является ароматическим), состоящие из 5-20 атомов, содержащие один или более гетероатомов, независимо выбранных из азота, кислорода и серы. Примерами гетероарильных групп являются пиридинил (включая, например, 2-гидроксипиридинил), имидазолил, имидазопиридинил, пиримидинил (включая, например, 4-гидроксипиримидинил), пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксадиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, тетрагидроизохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Гетероарильные группы необязательно независимо замещены одним или более заместителями, описанными в данном контексте.
Гетероциклические или гетероарильные группы связаны через атом углерода или азота, если указанное соединение существует. Например, но не ограничиваясь только ими, гетероциклы или гетероарилы связаны через атом углерода в положении 2, 3, 4, 5 или 6 пиридина, положении 3, 4, 5 или 6 пиридазина, положении 2, 4, 5 или 6 пиримидина, положении 2, 3, 5 или 6 пиразина, положении 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, положении 2, 4 или 5 оксазола, имидазола или тиазола, положении 3, 4 или 5 изоксазола, пиразола или изотиазола, положении 2 или 3 азиридина, положении 2, 3 или 4 азетидина, положении 2, 3, 4, 5, 6, 7 или 8 хинолина или положении 1, 3, 4, 5, 6, 7 или 8 изохинолина.
Например, но не ограничиваясь только ими, гетероциклы или гетероарилы связаны через атом азота в положении 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1H-индазола, положении 2 изоиндола или изоиндолина, в положении 4 морфолина, а также в положении 9 карбазола или β-карболина.
Термин «лечение» обозначает терапевтическое лечение и профилактические или превентивные меры, которые предназначены для предотвращения или замедления (уменьшения) у субъекта нежелательного физиологического изменения или нарушения, например, развития или распространения рака. Согласно настоящему изобретению благоприятные или желательные, проявляющиеся или не проявляющиеся клинические результаты включают, но не ограничиваясь только ими, ослабление симптомов, уменьшение тяжести заболевания, стабилизированное (т.е. без ухудшения) состояние заболевания, отсрочку или замедление развития заболевания, улучшение или ослабление патологического состояния, а также ремиссию (частичную или полную). Термин «лечение» также включает увеличение продолжительности жизни по сравнению с продолжительностью жизни, ожидаемой при отсутствии лечения. Субъекты, нуждающиеся в лечении, включают субъектов, у которых уже установлен диагноз состояния или нарушения, а также субъектов, предрасположенных к развитию состояния или нарушения, или субъектов, которым требуется профилактика развития состояния или нарушения.
Термин «терапевтически эффективное количество» обозначает количество соединения по настоящему изобретению, которое является достаточным для (1) лечения или профилактики конкретного заболевания, состояния или нарушения, (2) снижения интенсивности, улучшения или устранения одного или более симптомов конкретного заболевания, состояния или нарушения, или (3) профилактики или замедления развития одного или более симптомов конкретного заболевания, состояния или нарушения, описанного в данном контексте. В случае рака терапевтически эффективное количество лекарственного средства снижает число раковых клеток, снижает размер опухоли, подавляет (т.е. замедляет до некоторой степени и предпочтительно останавливает) инфильтрацию раковых клеток в периферические органы, подавляет (т.е. замедляет до некоторой степени и предпочтительно останавливает) метастазирование опухоли, подавляет до некоторой степени рост опухоли и/или снижает до некоторой степени интенсивность одного или более симптомов, связанных с раком. В зависимости от степени, с которой лекарственное средство предотвращает рост и/или уничтожает существующие раковые клетки, оно относится к цитостатическим и/или цитотоксическим средствам. Эффективность лечения рака оценивают, например, определяя время до начала прогрессирования заболевания (ВП) и/или скорость ответной реакции (СОР).
Термин «рак» обозначает или описывает физиологическое состояние млекопитающих, которое обычно характеризуется разрегулированным клеточным ростом. Термин «опухоль» обозначает одну или более раковых клеток. Примеры рака включают, но не ограничиваясь только ими, карциному, лимфому, бластому, саркому и лейкоз или лимфолейкоз. Более конкретные примеры указанных видов рака включают плоско-клеточный рак (например, эпителиальный плоско-клеточный рак), рак легких, включая мелкоклеточный рак легких, немелкоклеточный рак легких («НМРЛ»), аденокарциному легких и плоско-клеточный рак легких, перитонеальный рак, гепатоцеллюлярный рак, рак желудка, включая рак желудочно-кишечного тракта, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичников, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак ободочной кишки, рак прямой кишки, колоректальный рак, карциному эндометрия или рак матки, рак слюнных желез, рак почек, рак предстательной железы, рак вульвы, рак щитовидной железы, гепатокарциному, анальную карциному, карциному полового члена, а также рак головы и шеи.
Термин «химиотерапевтический агент» обозначает химическое соединение, пригодное для лечения рака независимо от механизма действия. Классы химиотерапевтических агентов включают, но не ограничиваясь только ими: алкилирующие агенты, антиметаболиты, растительные алкалоиды веретенного яда, цитотоксические/противоопухолевые антибиотики, ингибиторы топоизомеразы, антитела, фотосенсибилизаторы и ингибиторы киназ. Химиотерапевтические агенты включают соединения, используемые для «целевой терапии» и стандартной химиотерапии. Примеры химиотерапевтических агентов включают: эрлотиниб (TARCEVA®, Genentech/OSI Pharm.), доцетаксел (TAXOTERE®, Sanofi-Aventis), 5-ФУ (фторурацил, 5-фторурацил, CAS №51-21-8), гемцитабин (GEMZAR®, Lilly), PD-0325901 (CAS №391210-10-9, Pfizer), цисплатин (цис-диаминдихлорплатину(II), CAS №15663-27-1), карбоплатин (CAS №41575-94-4), паклитаксел (TAXOL®, Bristol-Myers Squibb Oncology, Принстон, N.J.), трастузумаб (HERCEPTIN®, Genentech), темозоломид (4-метил-5-оксо-2,3,4,6,8-пентазабицикло[4.3.0]нона-2,7,9-триен-9-карбоксамид CAS №85622-93-1, TEMODAR®, TEMODAL®, Schering Plough), тамоксифен ((Z)-2-[4-(1,2-дифенилбут-1-енил)фенокси]-N,N-диметилэтанамин, NOLVADEX®, ISTUBAL®, VALODEX®), а также доксорубицин (ADRIAMYCIN®), Akti-1/2, HPPD и рапамицин.
Дополнительные примеры химиотерапевтических агентов включают: оксалиплатин (ELOXATIN®, Sanofi), бортезомиб (VELCADE®, Millennium Pharm.), сутент (SUNITINIB®, SU11248, Pfizer), летрозол (FEMARA®, Novartis), мезилат иматиниба (GLEEVEC®, Novartis), XL-518 (ингибитор Mek, Exelixis, WO 2007/044515), ARRY-886 (ингибитор Mek, AZD6244, Array BioPharma, Astra Zeneca), SF-1126 (ингибитор P13K, Semafore Pharmaceuticals), BEZ-235 (ингибитор P13K, Novartis), XL-147 (ингибитор P13K, Exelixis), PTK787/ZK 222584 (Novartis), фулвестрант (FASLODEX®, AstraZeneca), лейковорин (фолиновая кислота), рапамицин (сиролимус, RAPAMUNE®, Wyeth), лапатиниб (TYKERB®, GSK572016, Glaxo Smith Kline), лонафарниб (SARASAR™, SCH 66336, Sobering Plough), сорафениб (NEXAVAR®, BAY43-9006, Bayer Labs), гефитиниб (IRESSA®, AstraZeneca), иринотекан (CAMPTOSAR®, CPT-11, Pfizer), типифарниб (ZARNESTRA™, Johnson & Johnson), ABRAXANE™ (не содержащий кремофора), композицию наночастиц паклитаксела на основе альбумина (American Pharmaceutical Partners, Schaumberg, Il), вандетаниб (rINN, ZD6474, ZACTIMA®, AstraZeneca), хлорамбуцил, AG1478, AG1571 (SU 5271, Sugen), темсиролимус (TORISEL®, Wyeth), пазопаниб (GlaxoSmithKline), канфосфамид (TELCYTA®, Telik), тиотепа и циклофосфамид (CYTOXAN®, NEOSAR®), алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан, азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа, этиленимины и метиламеламины, включая альтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметиломеламин, ацетогенины (прежде всего буллатацин и буллатацинон), камптотецин (включая синтетический аналог - топотекан), бриостатин, каллистатин, СС-1065 (включая синтетические аналоги - адозелезин, карзелезин и бизелезин), криптофицины (прежде всего криптофицин 1 и криптофицин 8), доластатин, дуокармицин (включая синтетические аналоги - KW-2189 и СВ1-ТМ1), элейтеробин, панкратистатин, саркодиктин, спонгистатин, азотистые иприты, такие как хлорамбуцил, хлорнафазин, хлорфосфамид, эстрамустин, ифосфамид, мехлоретамин, гидрохлорид оксида мехлоретамина, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урамустин, нитрозомочевины, такие как кармустин, хлорзотоцин, фотемустин, ломустин, нимустин и ранимнустин, антибиотики такие, как энедииновые антибиотики (например, калихеамицин, калихеамицин-γ II и калехеамицин-ω II (Angew Chem. Intl. Ed. Engl., 33, cc. 183-186 (1994)), динемицин, динемицин А, бисфосфонаты, такие как клодронат, эсперамицин, а также неокарциностатиновый хромофор и родственные хромопротеиновые энедииновые хромофоры-антибиотики, аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карзинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, морфолино-доксорубицин, цианоморфолино-доксорубицин, 2-пирролино-доксорубицин и дезоксидоксорубицин, эпирубицин, эзорубицин, идарубицин, неморубицин, марцелломицин, митомицины, такие как митомицин С, микофеноловая кислота, ногаламицин, оливомицины, пепломицин, порфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин, антиметаболиты, такие как метотрексат и 5-фторурацил (5-ФУ), аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат, пуриновые аналоги, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин, пиримидиновые аналоги, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин, андрогены, такие как калустерон, пропионат дромостанолона, эпитиостанол, мепитиостан, тестолактон, анти-адреналиновые соединения, такие как аминоглютетимид, митотан, трилостан, средства, восполняющие фолиевую кислоту, такие как фролиновая кислота, ацеглатон, альдофосфамида гликозид, аминолевулиновая кислота, энилурацил, амсакрин, бестрабуцил, бизантрен, эдатраксат, дефофамин, демеколцин, диазиквон, элфорнитин, ацетат эллиптиния, эпотилон, этоглюцид, нитрат галлия, гидроксимочевина, лентинан, лонидаинин, майтанзиноиды, такие как майтанзин и ансамитоцины, митогуазон, митоксантрон, мопиданмол, нитраэрин, пентостатин, фенамет, пирарубицин, лозоксантрон, подофиллиновая кислота, 2-этилгидразид, прокарбазин, полисахаридный комплекс PSK® (JHS Natural Products, Eugene, OR), разоксан, ризоксин, сизофиран, спирогерманий, тенуазоновая кислота, триазиквон, 2,2',2''-трихлортриэтиламин, трихотецены (прежде всего токсин Т-2, верракурин А, роридин А и ангуидин), уретан, виндезин, дакарбазин, манномустин, митобронитол, митолактол, пипоброман, гацитозин, арабинозид («Ara-С»), циклофосфамид, тиотепа, 6-тиогуанин, меркаптопурин, метотрексат, платиновые аналоги, такие как цисплатин и карбоплатин, винбластин, этопозид (VP-16), ифосфамид, митоксантрон, винкристин, винорелбин (NAVELBINE®), новантрон, тенипозид, эдатрексат, дауномицин, аминоптерин, капецитабин (XELODA®, Roche), ибандронат, СРТ-11, ингибитор топоизомеразы RFS 2000, дифторметилорнитин (DMFO), ретиноиды, такие как ретиноевая кислота, а также фармацевтически приемлемые соли, кислоты и производные любого указанного выше соединения.
В определение термина «химиотерапевтический агент» включены также: (1) антигормональные агенты, которые действуют за счет регуляции или подавления действия гормонов на опухоли, такие как антиэстрогены и селективные модуляторы рецептора эстрогена (SERM), включая, например, тамоксифен (включая NOLVADEX®, цитрат тамоксифина), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и FARESTON® (цитрат торемифена), (2) ингибиторы ароматазы, которые ингибируют фермент ароматазу, которая регулирует продуцирование эстрогенов в надпочечниках, такие как, например, 4(5)-имидазолы, аминоглутетимид, MEGASE® (ацетат мегестрола), AROMASIN® (экземестан, Pfizer), форместан, фадрозол, RIVISOR® (ворозол), FEMARA® (летрозол, Novartis) и ARIMIDEX® (анастрозол, AstraZeneca), (3) антиандрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин, а также троксацитабин (аналог 1,3-диоксоланнуклеозидацитозина), (4) ингибиторы протеинкиназ, такие как ингибиторы MEK (WO 2007/044515), (5) ингибиторы липиднкиназ, (6) антисмысловые олигонуклеотиды, прежде всего подавляющие экспрессию генов на сигнальных путях, принимающих участие в аномальной пролиферации клеток, например, PKC-α, Raf и H-Ras, такие как облимерсен (GENASENSE®, Genta Inc.), (7) рибозимы, такие как ингибиторы экспрессии VEGF (например, ANGIOZYME®) и ингибиторы экспрессии HER2, (8) вакцины, такие как вакцины для генной терапии, например, ALLOVECTIN®, LEUVECTIN® и VAXID®, PROLEUKIN® rIL-2, ингибиторы топоизомеразы 1, такие как LURTOTECAN®, ABARELIX® rmRH, (9) антиангиогенные агенты, такие как бевасизумаб (AVASTIN®, Genentech), а также фармацевтически приемлемые соли, кислоты и производные любого указанного выше соединения.
В определение термина «химиотерапевтический агент» включены также терапевтические антитела, такие как алемтузумаб (Campath), бевацизумаб (AVASTIN®, Genentech), цетуксимаб (ERBITUX®, Imclone), панитумумаб (VECTIBIX®, Amgen), ритуксимаб (RITUXAN®, Genentech/Biogen Idec), пертузумаб (OMNITARG™, 2C4, Genentech), трастузумаб (HERCEPTIN®, Genentech), тозитумомаб (Bexxar, Corixia) и конъюгаты антител с лекарственным средством - гемтузумаб озогамицин (MYLOTARG®, Wyeth).
Гуманизированные моноклональные антитела, проявляющие терапевтическую эффективность в качестве химиотерапевтических агентов в комбинации с ингибиторами PI3K по настоящему изобретению, включают: алемтузумаб, аполизумаб, азелизумаб, атлизумаб, бапинейзумаб, бевасизумаб, биватузумаб мертансин, кантузумаб мертансин, цеделизумаб, цертолизумаб пегол, цидфузитузумаб, цидтузумаб, даклизумаб, экулизумаб, эфализумаб, эпратузумаб, эрлизумаб, фелвизумаб, фонтолизумаб, гемтузумаб озогамицин, инотузумаб озогамицин, ипилимумаб, лабетузумаб, линтузумаб, матузумаб, меполизумаб, мотавизумаб, мотовизумаб, натализумаб, нимотузумаб, ноловизумаб, нумавизумаб, окрелизумаб, омализумаб, паливизумаб, пасколизумаб, пекфуситузумаб, пектузумаб, пертузумаб, пекселизумаб, раливизумаб, ранибизумаб, ресливизумаб, реслизумаб, ресивизумаб, ровелизумаб, руплизумаб, сибротузумаб, сиплизумаб, сонтузумаб, такатузумаб тетраксетан, тадоцизумаб, тализумаб, тефибазумаб, тоцилизумаб, торализумаб, трастузумаб, тукотузумаб целмолейкин, тукуситузумаб, умавизумаб, уртоксазумаб и визилизумаб.
Термин «метаболит» обозначает продукт, образующийся при метаболизме конкретного соединения или его соли в организме. Метаболиты соединения можно идентифицировать с использованием стандартных методик, известных в данной области техники, при этом активность метаболитов определяют методом анализа, как описанj в данном контексте. Указанные продукты образуются в результате, например, окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, омыления, ферментативного расщепления и т.п. введенного соединения. Соответственно, настоящее изобретение включает метаболиты соединений по настоящему изобретению, включая соединения, полученные способом, который заключается в том, что соединение по настоящему изобретению контактирует с организмом млекопитающего в течение периода времени, достаточного для образования метаболического продукта указанного соединения.
Термин «листок-вкладыш» обозначает инструкции, которые обычно вкладывают в коммерческие упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях и/или предупреждениях, касающихся применения указанных терапевтических продуктов.
Термин «хиральные» обозначает молекулы, которые пространственно не совместимы со своим зеркальным отображением, в то время как термин «ахиральные» обозначает молекулы, которые пространственно совместимы со своим зеркальным отображением.
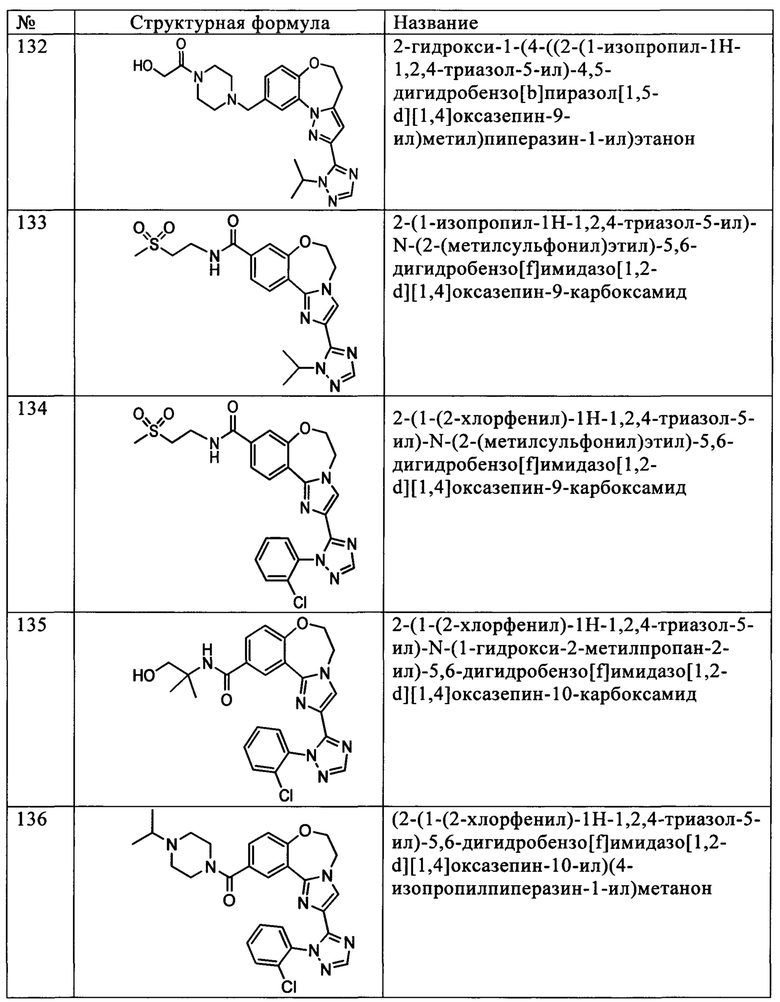
Термин «стереоизомеры» обозначает соединения, которые характеризуются одинаковым химическим строением, но отличаются расположением атомов или групп атомов в пространстве.
Термин «диастереомер» обозначает стереоизомер, содержащий два или более хиральных центров, и молекулы которого не являются зеркальными отображениями друг друга. Диастереомеры отличаются по физическим свойствам, например, температурой плавления, температурой кипения, спектральными свойствами и реакционной способностью. Смеси диастереомеров можно разделить методами высокого разрешения, такими как электрофорез и хроматография.
Термин «энантиомеры» обозначает два стереоизомера соединения, которые пространственно не совместимы с зеркальным отображением друг друга.
Использованные в данном контексте стереохимические определения и условные обозначения в основном соответствуют приведенным в справочнике McGraw-Hill Dictionary of Chemical Terms, Parker S.P., ред., McGraw-Hill Book Company, Нью-Йорк (1984) и в сборнике Eliel Е. и Wilen S., «Stereochemistry of Organic Compounds», John Wiley & Sons, Inc., Нью-Йорк (1994). Соединения по настоящему изобретению могут содержать асимметрические или хиральные центры и в связи с этим могут существовать в различных стереоизомерных формах. В объем настоящего изобретения включены все стереоизомерные формы соединений по настоящему изобретению, включая, но не ограничиваясь только ими, диастереомеры, энантиомеры и атропизомеры, а также их смеси, такие как рацемические смеси. Множество органических соединений существует в оптически активных формах, т.е. характеризуются способностью вращать плоскость плоско-поляризованного света. При описании оптически активного соединения буквы D и L, или R и S, используют для обозначения абсолютной конфигурации молекулы относительно ее хирального центра(ов). Буквы d и l или знаки (+) и (-) используют для обозначения знака вращения плоско-поляризованного света соединением, при этом (-) или 1 обозначает, что соединение является левовращающим. Соединение со знаком (+) или буквой d является правовращающим. Для заданной химической структуры указанные стереоизомеры являются идентичными за исключением того, что они являются зеркальными отображениями друг друга. Конкретный стереоизомер называют также энантиомером, а смесь указанных изомеров в большинстве случаев называют энантиомерной смесью. Смесь энантиомеров 50:50 называется рацемической смесью или рацематом, при этом указанная смесь образуется при отсутствии стереоселективности или стереоспецифичности в ходе химической реакции или процесса. Термины «рацемическая смесь» и «рацемат» обозначают эквимолярную смесь двух энантиомерных соединений, которая не обладает оптической активностью.
Термин «таутомер» или «таутомерная форма» обозначает структурные изомеры с различной энергией, которые превращаются друг в друга при преодолении низкого энергетического барьера. Например, протонные таутомеры (также называемые прототропными таутомерами) характеризуются взаимопревращениями за счет миграции протона, такой как кето-енольная и имин-енаминная изомеризации. Валентные таутомеры характеризуются взаимопревращениями за счет перегруппировки некоторых связывающих электронов.
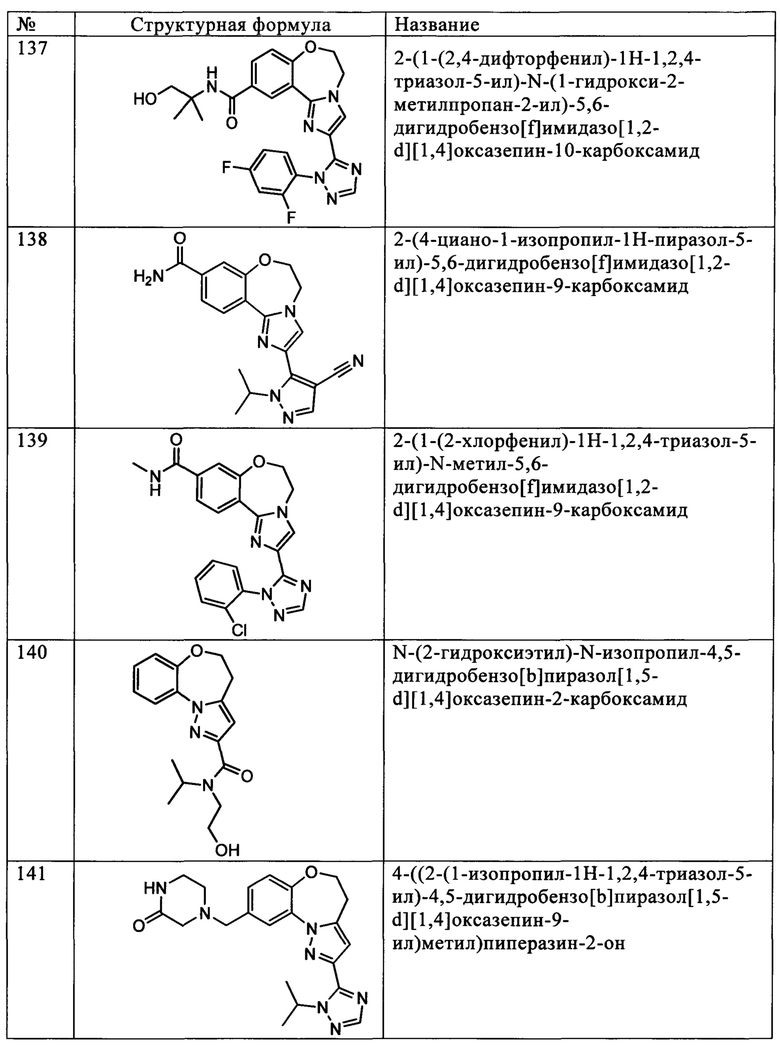
Использованный в данном контексте термин «фармацевтически приемлемая соль» обозначает фармацевтически приемлемые органические или неорганические соли соединения по настоящему изобретению. Примеры солей включают, но не ограничиваясь только ими, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, гидрофосфат, изоникотинат, лактат, салицилат, гидроцитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глютамат, метансульфонат («мезилат»), этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат (т.е. 1,1'-метиленбис(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может включать другой ион, такой как ацетат ион, сукцинат ион или другой противоион. Противоионом является любой органический или неорганический остаток, который стабилизирует заряд исходного соединения. Кроме того, структура фармацевтически приемлемой соли может включать более одного заряженного атома. Если в состав фармацевтически приемлемой соли входит множество заряженных атомов, указанная фармацевтическая соль включает множество противоионов. Следовательно, фармацевтически приемлемая соль может включать один или более заряженных атомов и/или один или более противоионов.
Если соединением по настоящему изобретению является основание, требуемую фармацевтически приемлемую соль получают любым пригодным способом, известным в данной области техники, например, обработкой свободного основания неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, метансульфоновая кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, трифторуксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, α-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как пара-толуолсульфоновая кислота или этансульфоновая кислота, или т.п.
Если соединением по настоящему изобретению является кислота, требуемую фармацевтически приемлемую соль получают любым пригодным способом, например, обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла, или т.п. Типичные примеры пригодных солей включают, но не ограничиваясь только ими, органические соли аминокислот, таких как глицин и аргинин, аммония, первичных, вторичных и третичных аминов, а также циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Термин «фармацевтически приемлемый» обозначает, что соединение или состав совместимы в химическом и/или токсикологическом отношении с другими ингредиентами, входящими в состав композиции, и/или с организмом млекопитающего, который проходит курс лечения указанным соединением или составом.
Термин «сольват» обозначает ассоциацию или комплекс одной или более молекул растворителя и соединения по настоящему изобретению. Примеры растворителей, которые образуют сольваты, включают, но не ограничиваясь только ими, воду, изопропанол, этанол, метанол, диметилсульфоксид (ДМСО), этилацетат, уксусную кислоту и этаноламин.
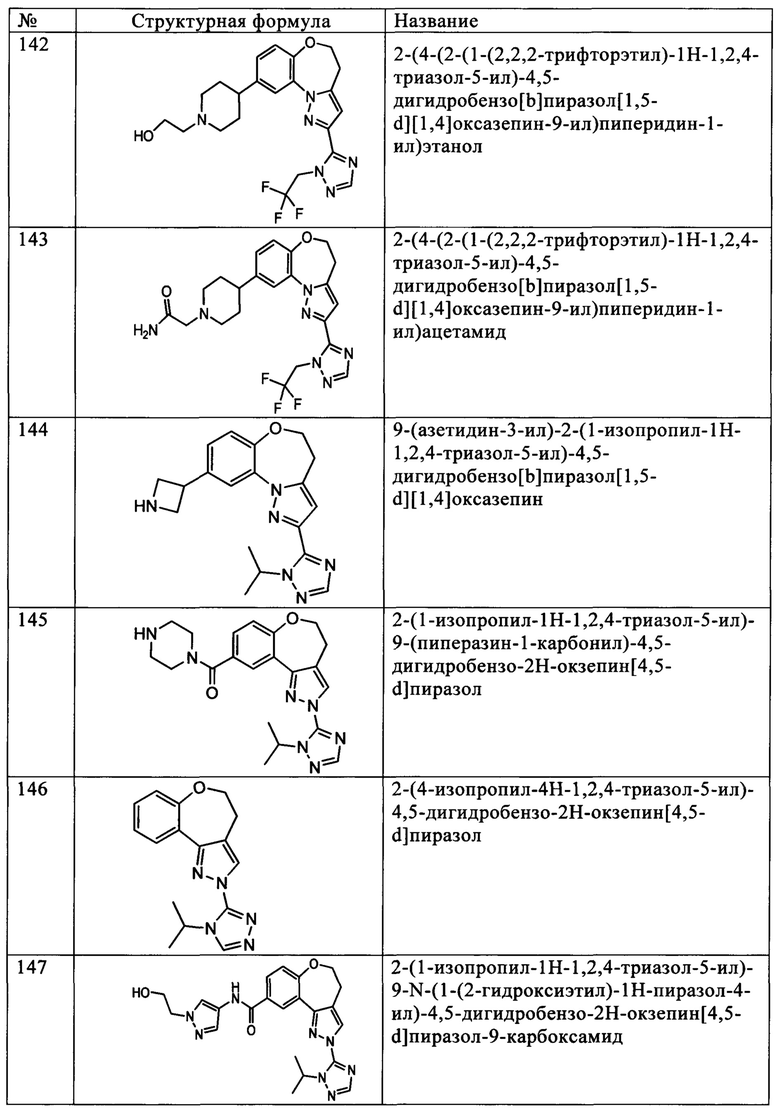
Термины «соединение по настоящему изобретению» и «соединения по настоящему изобретению», а также «соединения формулы I» обозначают соединения формулы I и стереоизомеры, геометрические изомеры, таутомеры, сольваты, метаболиты, а также фармацевтически приемлемые соли и пролекарства указанных соединений.
Любая формула или структура, приведенная в настоящем описании, включая соединения формулы I, также обозначает гидраты, сольваты и полиморфные формы указанных соединений, а также их смеси.
Любая формула или структура, приведенная в настоящем описании, включая соединения формулы I, также обозначает немеченные формы, а также изотопно меченные формы соединений. Изотопно меченные соединения характеризуются структурами, которые представлены формулами, приведенными в настоящем описании, за исключением того, что один или более атомов заменены на атом, характеризующийся выбранной атомной массой или массовым числом. Примеры изотопов, которые можно включать в состав соединений по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, например, но не ограничиваясь только ими, 2Н (дейтерий, D), 3Н (тритий), 11С, 13С, 14С, 15N, 18F, 31Р, 32Р, 35S, 36Cl и 125I. В объем настоящего изобретения включены различные изотопно меченные соединения по настоящему изобретению, например, соединения, в состав которых входят радиоактивные изотопы, такие как 3Н, 13С и 14С. Указанные изотопно меченные соединения можно использовать для исследований метаболизма, исследований кинетики реакций, в методах детектирования или визуализации, таких как позитронно-эмиссионная томография (ПЭТ) или однофотонная эмиссионная компьютерная томография (ОЭКТ), включая анализ распределения лекарственного средства или субстрата в тканях, или для лечения пациентов с использованием лучевой терапии. Терапевтические соединения по настоящему изобретению, меченные или замещенные дейтерием, характеризуются улучшенными свойствами метаболизма лекарственных средств и фармакокинетики (МЛСФК) в отношении распределения, метаболизма и выведения (РМВ). Замещение более тяжелыми изотопами, такими как дейтерий, обеспечивает определенные терапевтические преимущества за счет более высокой метаболической стабильности, например, увеличенного периода полураспада in vivo или снижения необходимой дозировки. В исследованиях методом ПЭТ или ОЭКТ можно использовать соединение, меченное 18F. В связи с этим, изотопно меченные соединения по настоящему изобретению обычно получают по методикам, показанным на схемах, или аналогично тому, как описано в приведенных ниже примерах и способах получения, заменяя реагент без изотопной метки на коммерческий изотопно меченный реагент. Кроме того, замещение более тяжелыми изотопами, прежде всего дейтерием (т.е. 2Н или D) обеспечивает определенные терапевтические преимущества за счет более высокой метаболической стабильности, например, увеличенного периода полураспада in vivo или снижения необходимой дозировки или повышения терапевтического индекса. Следует понимать, что в данном контексте дейтерий рассматривается в качестве заместителя в составе соединения формулы (I). Концентрацию такого более тяжелого изотопа, прежде всего дейтерия, определяют по коэффициенту изотопного обогащения. Любой атом в составе соединений по настоящему изобретению, не обозначенный специально как конкретный изотоп, обозначает любой стабильный изотоп указанного атома. Если не указано иное, положение, конкретно обозначенное как «Н» или «водород», обозначат водород в его природном изотопном составе. Соответственно, любой атом в составе соединений по настоящему изобретению, конкретно обозначенный как дейтерий (D), представляет собой дейтерий.
Бензоксазепины
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4.
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает N, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4.
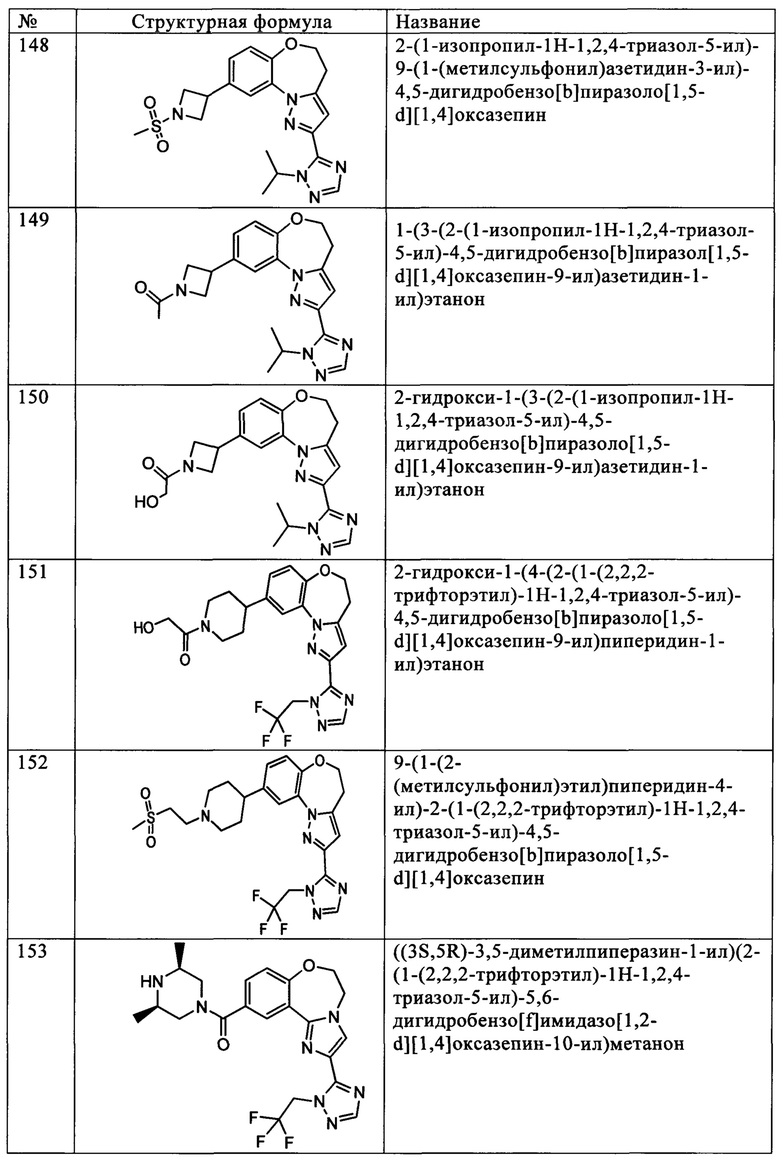
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает N, Z3 обозначает CR3, и Z4 обозначает CR4.
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает N, и Z4 обозначает CR4.
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает N.
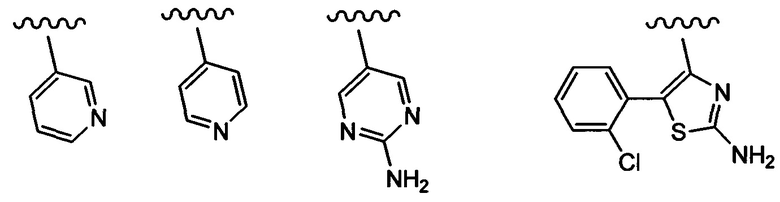
Примеры вариантов соединений формулы I включают соединения, где В обозначает структуру формулы (а).
Примеры вариантов соединений формулы I включают соединения, где В обозначает структуру формулы (б).
Примеры вариантов соединений формулы I включают соединения, где В обозначает структуру формулы (в).
Примеры вариантов соединений формулы I включают соединения, в где В обозначает структуру формулы (г).
Примеры вариантов соединений формулы I включают соединения, где В обозначает структуру формулы (д).
Примеры вариантов соединений формулы I включают соединения, где В обозначает структуру формулы (е).
Примеры вариантов соединений формулы I включают соединения где В обозначает структуру формулы (ж).
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4, а В обозначает структуру формулы (а), (б), (г), (д), (е) или (ж).
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4, а В обозначает структуру формулы (а), (б), (г) или (ж).
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4, а В обозначает структуру формулы (а) или (г).
Примеры вариантов соединений формулы I включают соединения, где А обозначает -C(=O)NR5R6.
Примеры вариантов соединений формулы I включают соединения, где R5 обозначает СН3.
Примеры вариантов соединений формулы I включают соединения, где R6 обозначает фенил, замещенный одной или более группами, независимо выбранными из F, Cl, Br, I, -СН2ОН, -СН2С6Н5, -CN, -CF3, -CO2H, -CONH2, -CONHCH3, -NO2, -N(CH3)2, -NHCOCH3, -NHS(O)2CH3, -ОН, -ОСН3, -ОСН2СН3, -S(O)2NH2, -S(O)2СН3, морфолин-4-ила, пиперидин-1-ила, пиперазинила, пиперазин-4-ил-2-она, пиперазин-4-ил-3-она, пирролидин-1-ила, тиоморфолин-4-ила, S-диоксотиоморфолин-4-ила, -C≡CR13 и -CH=CHR13.
Примеры вариантов соединений формулы I включают соединения, где R5 и R6 вместе с атомом азота, к которому они присоединены, образуют морфолин-4-ил, пиперидин-1-ил, пиперазинил, пиперазин-4-ил-2-он, пиперазин-4-ил-3-он, пирролидин-1-ил, тиоморфолин-4-ил, S-диоксотиоморфолин-4-ил, азокан-1-ил, азетидин-1-ил, октагидропиридо[1,2-а]пиразин-2-ил, [1,4]диазепан-1-ил или индолил.
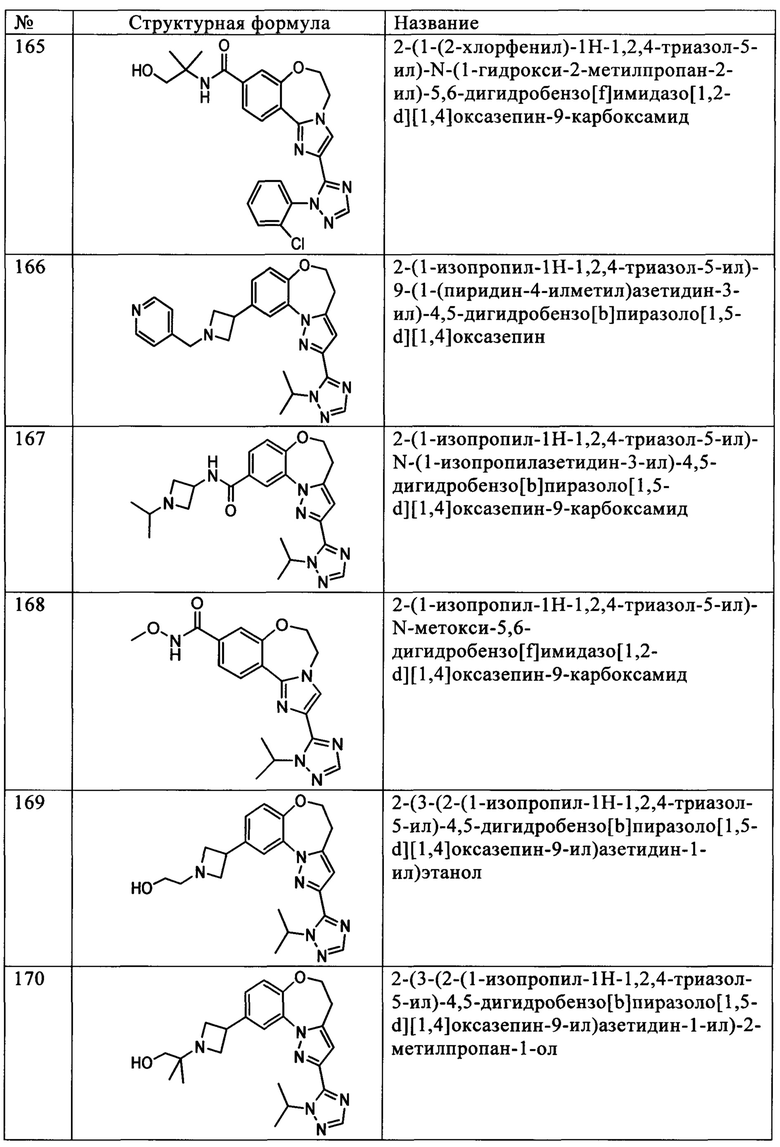
Примеры вариантов соединений формулы I включают соединения, где А обозначает -C(=O)NR5R6, где R5 обозначает СН3, а R6 обозначает фенил, замещенный одной или более группами, независимо выбранными из F, Cl, Br, I, -CH2OH, -СН2С6Н5, -CN, -CF3, -CO2H, -CONH2, -CONHCH3, -NO2, -N(CH3)2, -NHCOCH3, -NHS(O)2CH3, -ОН, -ОСН3, -ОСН2СН3, -S(O)2NH2, -S(O)2СН3, морфолин-4-ила, пиперидин-1-ила, пиперазинила, пиперазин-4-ил-2-она, пиперазин-4-ил-3-она, пирролидин-1-ила, тиоморфолин-4-ила, S-диоксотиоморфолин-4-ила, -C≡CR13 и -CH=CHR13.
Примеры вариантов соединений формулы I включают соединения, где А обозначает -C(=O)NR5R6, где R5 обозначает СН3, а R6 обозначает фенил, замещенный одним или более атомами F.
Примеры вариантов соединений формулы I включают соединения, где А обозначает С2-С20гетероциклил или С1-С20гетероарил, замещенный -СН2ОН, -CH2CO2H, -СН(СН3)CH2OCH3, -СН3, -СН(СН3)2, -СН2СН(СН3)2, -CH2CF3, -С(=O)СН3, -C(=O)NHCH3, -С(=O)N(СН3)2, -CO2H, -CO2CH3, -CH2CO2CH3, -NH2, -NHC(=O)СН3, -ОН, -ОСН3, -S(O)2СН3, 1-метилпиперид-4-илом, 4-метилпиперазин-1-илом, 4-морфолинилом, (4-метилпиперазин-1-ил)карбоксамидом, -СН2(1Н-1,2,4-триазол-5-илом), циклопропилом, циклопропилметилом или циклобутилом.
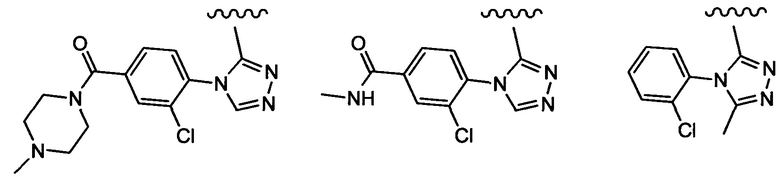
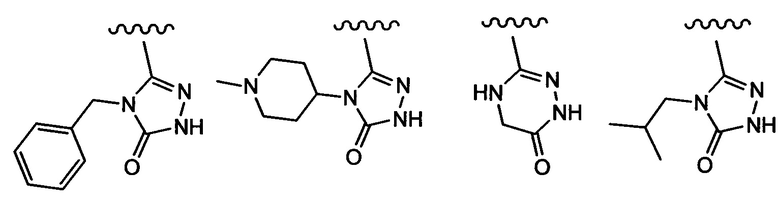
Примеры вариантов соединений формулы I включают соединения, где А обозначает С1-С20гетероарил, выбранный из пиридила, изоксазолила, имидазолила, пиразолила, пирролила, тиазолила, пиридазинила, пиримидинила, пиразинила, оксазолила, оксадиазолила, 1,3,4-оксадиазол-2(3Н)-она, фуранила, тиенила, 1,2,3-триазолила, 1,2,4-триазолила, 1,2,4-триазол-5(4Н)-она, 4,5-дигидро-1,2,4-триазин-6(1Н)-она, тетразолила, пирроло[2,3-b]пиридинила, индазолила, 3,4-дигидрохинолинила и бензо[d]тиазола.
Примеры вариантов соединений формулы I включают соединения, где А выбирают из следующих структур:

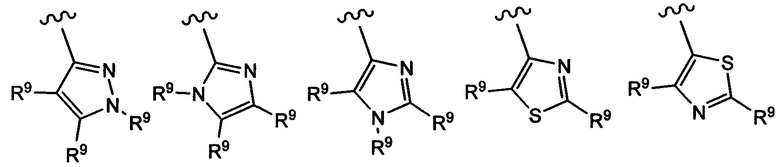
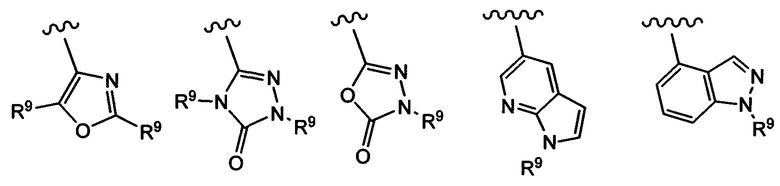
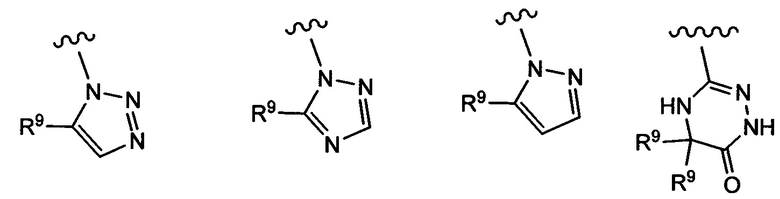
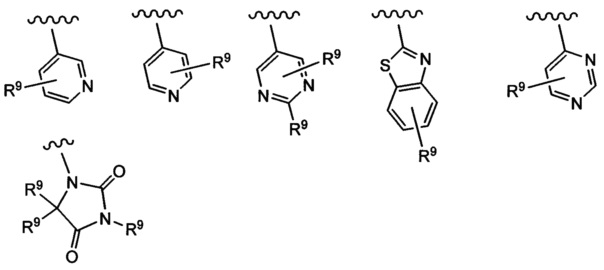
где R9 независимо выбирают из Н, F, Cl, Br, I, -СН3, -СН2СН3, -СН(СН3)2, -С(СН3)3, -СН2СН(СН3)2, -СН2ОН, -CH2CO2H, -СН(СН3)CH2OCH3, -CN, -CF3, -CH2CF3, -CH2NH2, -CH2CH2NH2, -С(=O)СН3, - СН2С(=O)NHCH3, -C(=O)NHCH3, -CO2H, -CH2CO2CH3, -NH2, -ОН, -ОСН3, -SCH3, -S(O)2СН3, циклопропила, циклопропилметила, 1-метилпиперид-4-ила, 4-метилпиперазин-1-ила, 4-морфолинила, морфолин-4-илэтила, бензила и фенила, где бензил и фенил необязательно замещены одной или более группами, выбранными из F, Cl, Br, I, -CH2OH, -CH2CO2H, -CN, -CH2NH2, -СН3, -С(=O)СН3, -C(=O)NHCH3, -CO2H, -CH2CO2CH3, -NH2, -ОСН3, -S(O)2СН3, 1-метилпиперид-4-ила, 4-метилпиперазин-1-ила и 4-морфолинила, и где волнистая линия обозначает место присоединения.
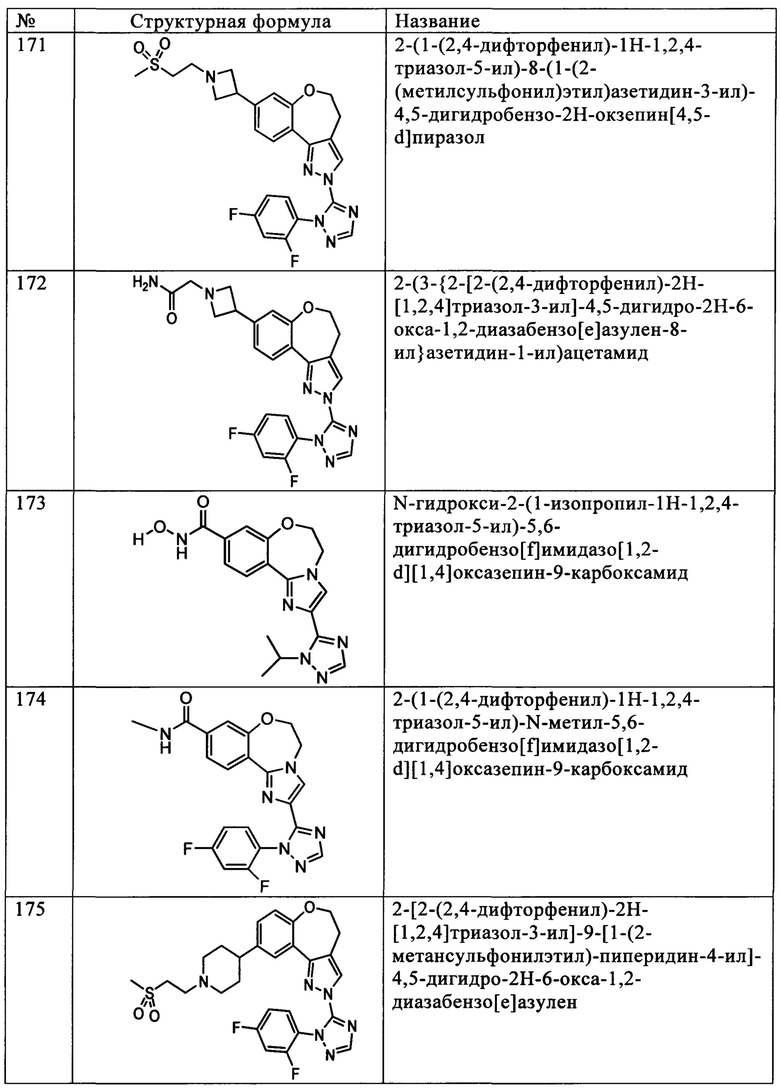
Примеры вариантов соединений формулы I включают соединения, где А обозначает группу формулы (I):
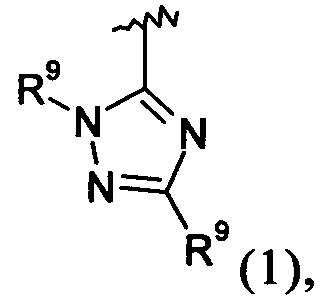
где R9 независимо выбирают из Н, F, Cl, Br, I, -СН3, -СН2СН3, -СН(СН3)2, -С(СН3)3, -СН2СН(СН3)2, -CH2OH, -CH2CO2H, -СН(СН3)CH2OCH3, -CN, -CF3, -CH2CF3, -CH2NH2, -CH2CH2NH2, -С(=O)СН3, - СН2С(=O)NHCH3, -C(=O)NHCH3, -CO2H, -CH2CO2CH3, -NH2, -ОН, -ОСН3, -SCH3, -S(O)2СН3, циклопропила, циклопропилметила, 1-метилпиперид-4-ила, 4-метилпиперазин-1-ила, 4-морфолинила, морфолин-4-илэтила, бензила и фенила, где бензил и фенил необязательно замещены одной или более группами, выбранными из F, Cl, Br, I, -CH2OH, -CH2CO2H, -CN, -CH2NH2, -СН3, -С(=O)СН3, -С(=O)NHCH3, -CO2H, -CH2CO2CH3, -NH2, -ОСН3, -S(O)2СН3, 1-метилпиперид-4-ила, 4-метилпиперазин-1-ила и 4-морфолинила, и где волнистая линия обозначает место присоединения.
Примеры вариантов соединений формулы I включают соединения, где А обозначает группу формулы (1), где R9 независимо выбирают из -СН3, -СН(СН3)2, -NH2 и фенила, где фенил необязательно замещен одной или более группами, выбранными из F и Cl.
Примеры вариантов соединений формулы I включают соединения, где А выбирают из структур:
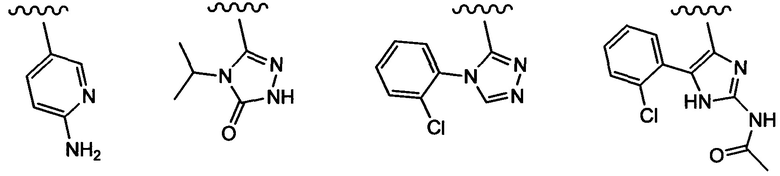
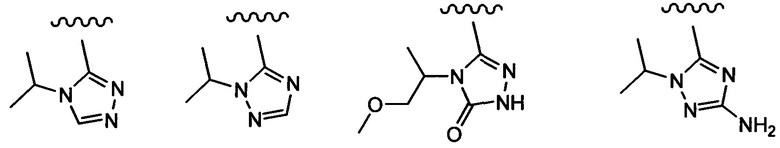
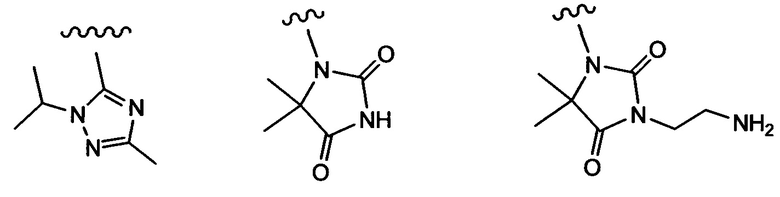
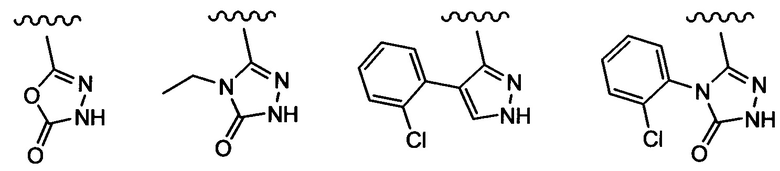
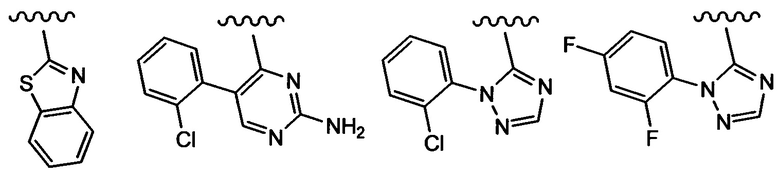



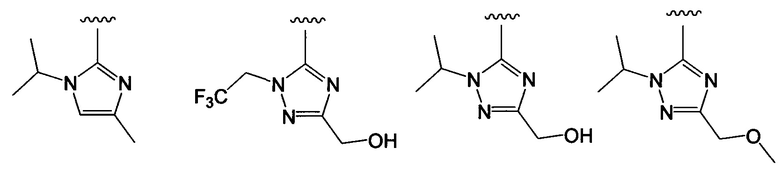


где волнистая линия обозначает участок присоединения.
Примеры вариантов соединений формулы I включают соединения, где А обозначает группу формулы (2), (3), (4), (5) или (6):
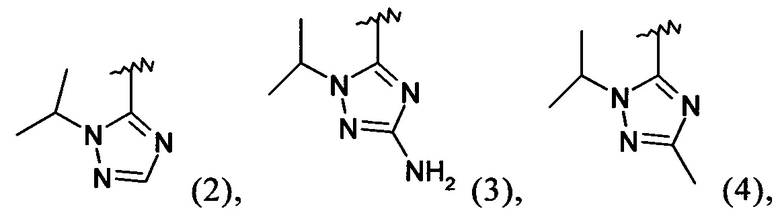
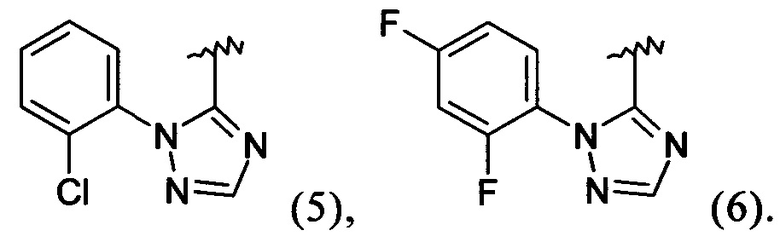
Примеры вариантов соединений формулы I включают соединения, где А обозначает группу формулы (2) или (6).
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4, при этом В обозначает структуру формулы (а) или (г), и А обозначает группу формулы (2) или (6).
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4, при этом В обозначает структуру формулы (а), и А обозначает группу формулы (6).
Примеры вариантов соединений формулы I включают соединения, где Z1 обозначает CR1, Z2 обозначает CR2, Z3 обозначает CR3, и Z4 обозначает CR4, при этом В обозначает структуру формулы (г), и А обозначает группу формулы (6).
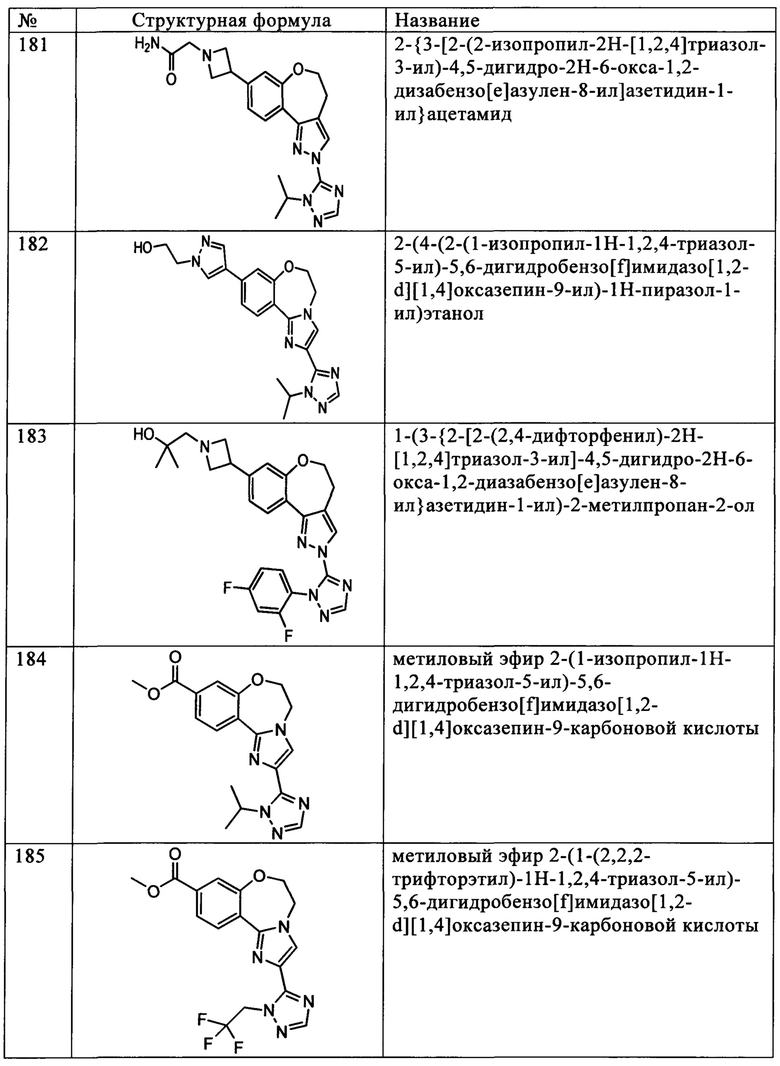
Примеры вариантов соединений формулы I включают соединения формулы Iз:

где R1, R2, R3, а также R4 и А имеют значения, как определено выше для соединений формулы I.
Примеры вариантов соединений формулы Iз включают соединения, где R1, R2, R3 и R4 независимо выбирают из Н, Br, -C(=O)NR10R11,
С2-С20гетероциклила или -(С2-С20гетероциклил)-(С1-С12алкила), где алкил необязательно замещен -S(O)2R10, при этом А выбирают из C1-С20гетероарила, где гетероарил необязательно замещен одним или более -(С6-С20арилом), где арил необязательно замещен одной или более группами, независимо выбранными из F и Cl, a R10 и R11 независимо выбирают из водорода и C1-C12алкила.
Примеры вариантов соединений формулы Iз включают соединения, где R1 обозначает водород, R2 обозначает водород, Br, -C(O)NH2 или
1-(2-метансульфонилэтил)азетидин-3-ил, при этом R3 обозначает водород или пиперидин-4-ил, R4 обозначает водород, и А обозначает 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил или 2-(хлорфенил)-2Н-[1,2,4]триазол-3-ил.
Примеры вариантов соединений формулы I включают соединения формулы Iи:
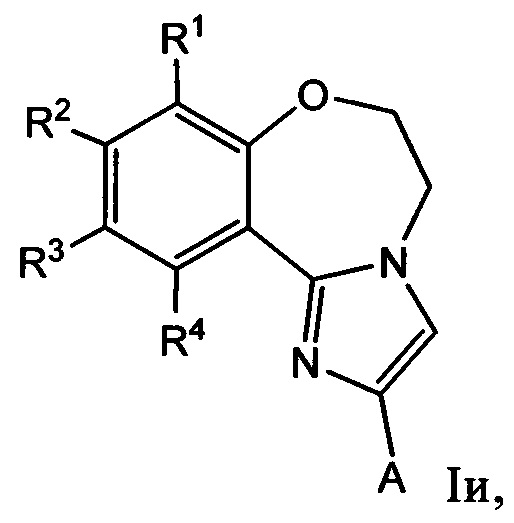
где R1, R2, R3, а также R4 и А имеют значения, как определено выше для соединений формулы I.
Примеры вариантов соединений формулы Iи включают соединения, где R1, R2, R3 и R4 независимо выбирают из Н, F, -C(=O)NR10R11,
-(С2-С20гетероциклил)-(С1-С12алкила), -(С1-С20гетероарил)-(С1-С12алкила), где алкил необязательно замещен одной или более группами, независимо выбранными из -NR10R11, оксогруппы и -OR10, и А обозначает C1-С20гетероарил, где гетероарил необязательно замещен одной или более группами, независимо выбранными из -NR10R11, С1-С12алкила и С6-С20арила, где арил необязательно замещен одним или более атомами Cl, a R10 и R11 независимо выбирают из Н и С1-С12алкила.
Примеры вариантов соединений формулы Iи включают соединения, где
R1 обозначает водород,
R2 обозначает водород, F или группу формулы

R3 обозначает водород или -C(O)NH2,
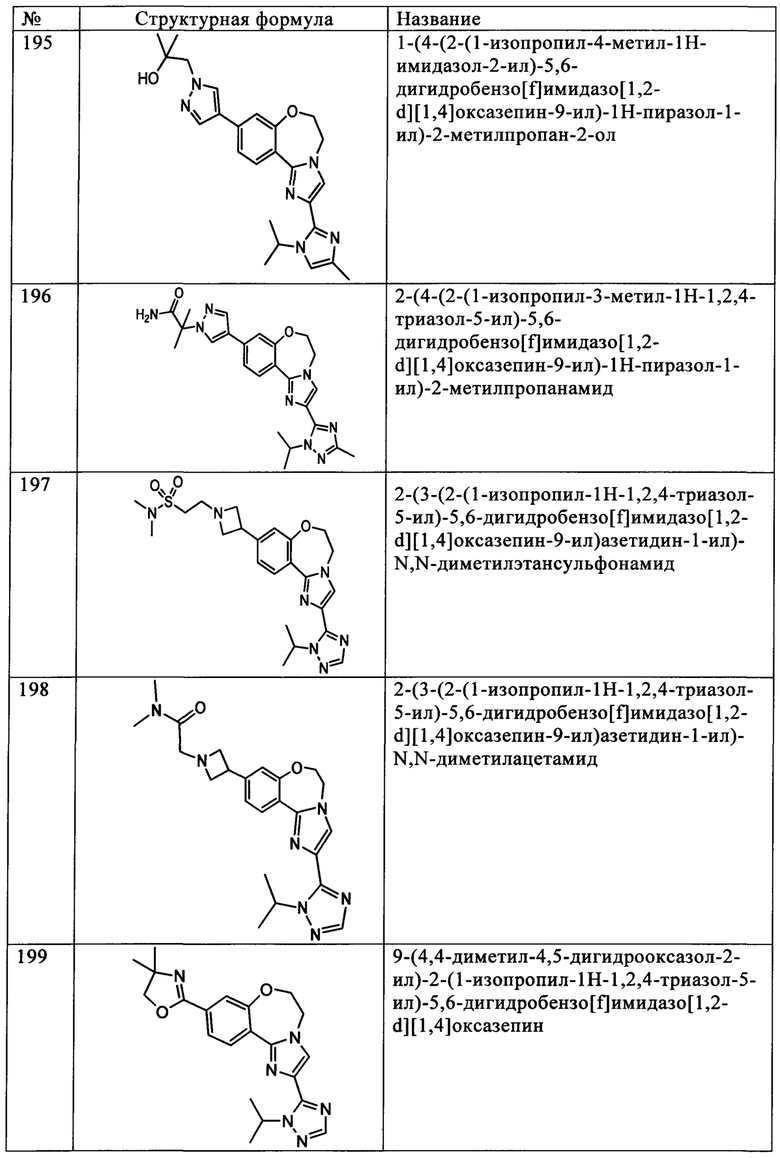
R4 обозначает водород, и
А обозначает 1-изопропил-1Н-[1,2,4]триазол-5-ил, 1-изопропил-3-метил-1Н-[1,2,4]триазол-5-ил, 1-изопропил-3-амино-1Н-[1,2,4]триазол-5-ил или 1-(2-хлорфенил-1Н-[1,2,4]триазол-5-ил.
Примеры вариантов соединений формулы I включают соединения формулы Iк:

где R1, R2, R3, R4 и А имеют значения, как определено выше для соединений формулы I.
Примеры вариантов соединений формулы Iк включают соединения, где R1, R2, R3 и R4 независимо выбирают из Н и
-(С2-С20гетероциклил)-(С1-С12алкила), где гетероциклил необязательно замещен одним или более -OR10, А выбирают из -C(=O)NR5R6 и C1-С20гетероарила, где гетероарил необязательно замещен одним или более С1-С12алкилом, R5 обозначает С1-С12алкил, R6 обозначает С6-С20арил, необязательно замещенный одним или более атомами F, и R10 обозначает Н.
Примеры вариантов соединений формулы Iк включают соединения, где
R1 обозначает водород,
R2 обозначает водород,
R3 обозначает водород или группу формулы
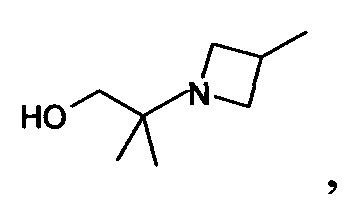
R4 обозначает водород, и
А обозначает группу формулы
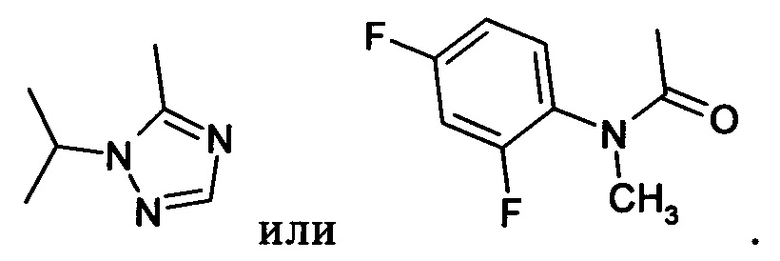
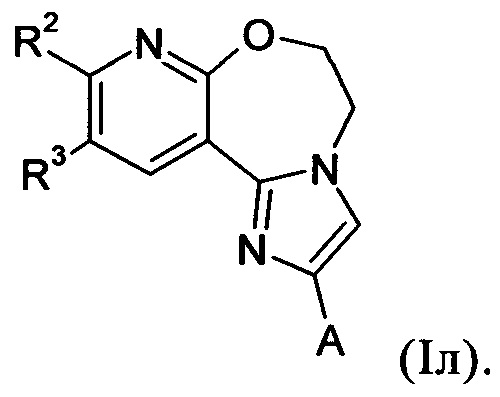
Примеры вариантов соединений формулы I включают соединения формулы Iл:
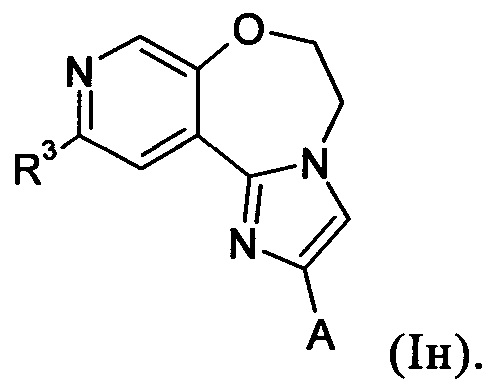
Примеры вариантов соединений формулы I включают соединения формулы Iн:
Примеры вариантов соединений формулы I включают соединения формулы Iо:
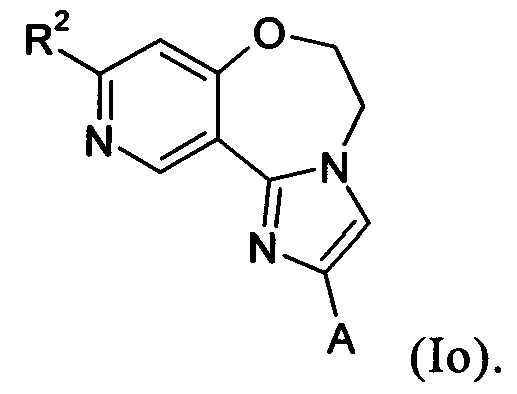
Примеры вариантов соединений формулы I включают соединения формулы Iр:

Примеры вариантов соединений формулы I включают соединения формулы Iс:
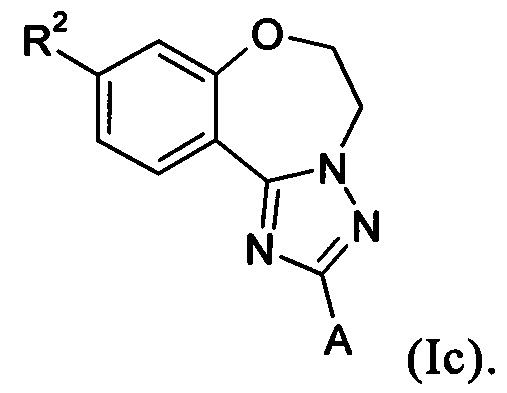
Примеры вариантов соединений формулы I включают соединения формулы Iт:
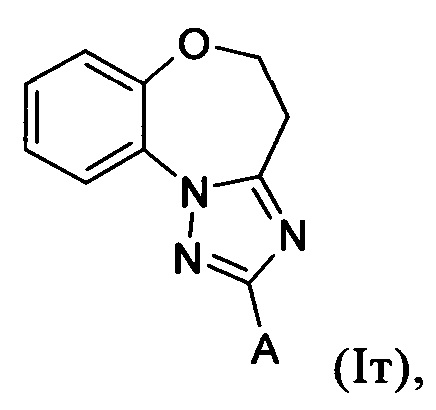
где А имеет значение, как определено выше для соединений формулы I.
Примеры вариантов соединений формулы Iт включают соединения, где А обозначает С1-С20гетероарил, причем гетероарил необязательно замещен одной или более группами, независимо выбранными из С1-С12алкила, например, А обозначает группу формулы
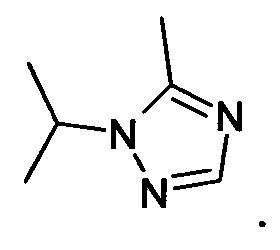
В настоящем изобретении в основном предлагаются бензоксазепины формулы I, проявляющие противораковую активность, и более конкретно ингибирующие активность киназы PI3. Некоторые гиперпролиферативные нарушения характеризуются модуляцией функции киназы PI3, например, при мутациях или сверхэкспресии белков. Соответственно, соединения по настоящему изобретению можно использовать при лечении гиперпролиферативных нарушений, таких как рак. Соединения подавляют рост опухоли у млекопитающих, и их можно использовать для лечения человека с диагнозом рак.
В настоящем изобретении предлагаются также способы применения бензоксазепинов формулы I для диагностики или обработки in vitro, in situ и in vivo клеток млекопитающих и для лечения организмов, или сопутствующих патологических состояний.
В другом объекте настоящего изобретения предлагаются способы ингибирования активности киназы PI3, которые заключаются в том, что киназа PI3 контактирует с эффективным ингибирующим количеством соединения формулы I.
Активность соединения формулы I в отношении киназы PI3 можно определить рядом прямых и косвенных способов детектирования. Для некоторых типичных соединений, описанных в данном контексте, определяли связывающую активность в отношении р110α (альфа) и другой изоформы PI3K (пример 901), а также активность против опухолевых клеток in vitro (пример 902). Связывающая активность в отношении PI3K для некоторых соединений по настоящему изобретению характеризуется величиной IC50 менее 10 нМ. Активность в отношении опухолевых клеток для некоторых соединений по настоящему изобретению характеризуется величиной ЕС50 менее 100 нМ.
В одном объекте настоящего изобретения предлагается способ ингибирования или модуляции активности липидкиназы, причем указанный способ заключается в том, что липидкиназа контактирует с эффективным ингибирующим количеством соединения, как определено в данном контексте. В одном объекте настоящего изобретения предлагается способ ингибирования или модуляции активности PI3K, причем указанный способ заключается в том, что PI3K контактирует с эффективным ингибирующим количеством соединения, как определено в данном контексте. В еще одном объекте настоящего изобретения предлагается способ ингибирования или модуляции активности субъединицы p110α PI3K, причем указанный способ заключается в том, что субъединица p110α PI3K контактирует с эффективным ингибирующим количеством соединения, как определено в данном контексте.
В одном объекте настоящего изобретения предлагается способ ингибирования или модуляции активности липидкиназы, например, активности PI3K у млекопитающего, причем указанный способ заключается в том, что указанному млекопитающему вводят терапевтически эффективное количество соединения, как определено в данном контексте.
Цитотоксическую или цитостатическую активность типичных соединений формулы I определяли, получая линию опухолевых клеток млекопитающего, характеризующуюся пролиферативной активностью, в культуральной клеточной среде, добавляя соединение формулы I, культивируя клетки в течение от приблизительно 6 ч до приблизительно 5 сут и определяя жизнеспособность клеток (пример 902). Для определения жизнеспособности, т.е. пролиферации (IC50), цитотоксичности (ЕС50) и индукции апоптоза (активации каспазы), использовали методы анализа in vitro с использованием клеток.
Эффективность типичных соединений формулы I in vitro определяли методом анализа пролиферации клеток с использованием набора для люминесцентного анализа жизнеспособности клеток CellTiter-Glo® Luminescent Cell Viability Assay, выпускаемого фирмой Promega Corp., Madison, WI (пример 902). Указанный метод гомогенного анализа основан на рекомбинантной экспрессии люциферазы жесткокрылых (Coleoptera) (US 5583024, US 5674713, US 5700670) и определении числа жизнеспособных клеток в культуре по количеству присутствующего АТФ, который является индикатором метаболически активных клеток (Crouch и др., J. Immunol. Meth., 160, cc. 81-88 (1993), US 6602677). Анализ CellTiter-Glo® проводят в 96- или 384-луночном планшете, пригодном для автоматического экспресс-анализа (Cree и др., AntiCancer Drugs, 6, cc. 398-404 (1995)). Методика гомогенного анализа включает добавление одного реагента (реагента CellTiter-Glo®) напрямую в клетки, культивированные в содержащей сыворотку среде. Промывка клеток, удаление среды и многократные стадии добавления с использованием пипетки не требуются. Система обеспечивает определение по крайней мере 15 клеток/лунке в 384-луночном планшете в течение 10 мин после добавления реагента и смешивания.
В ходе гомогенного анализа «добавление-смешивание-измерение» происходит лизис клеток и генерирование люминесцентного сигнала, пропорционального количеству присутствующего АТФ. Количество АТФ прямо пропорционально числу клеток, присутствующих в культуре. При проведении анализа CellTiter-Glo® генерируется люминесцентный сигнал (тлеющий разряд), продуцируемый лициферазной реакцией, который характеризуется полупериодом спада свечения более 5 ч в зависимости от типа клеток и используемой среды. Жизнеспособные клетки регистрируют в относительных единицах люминесценции (ОЕЛ). Субстрат, люциферин жуков, вступает в реакцию окислительного декарбоксилирования с рекомбинантной люциферазой светляков, при этом происходит одновременное превращение АТФ в АМФ и генерация фотонов. Увеличенный полупериод спада свечения устраняет необходимость применения инжекторов для реагентов и обеспечивает широкие возможности для непрерывной или серийной обработки множества планшетов. Для этого метода анализа пролиферации клеток можно использовать различные многолуночные планшеты, например, 96- или 384-луночные планшеты. Данные можно регистрировать с использованием люминометра или полупроводниковой видеокамеры на основе устройства с зарядовой связью. Выходной люминесцентный сигнал представляют в ОЕЛ, регистрируемых с течением времени.
Антипролиферативное действие типичных соединений формулы I в присутствии нескольких линий опухолевых клеток оценивали методом анализа CellTiter-Glo® (пример 902). Для исследуемых соединений определяли значения ЕС50, характеризующие эффективность. Диапазон значений потенциальной активности соединений в присутствии клеток in vitro составлял приблизительно от 100 нМ до приблизительно 10 мкМ. Некоторые исследуемые соединения характеризуются значениями ЕС50 менее 1 мкМ в отношении подавления пролиферации определенных линий опухолевых клеток.
Для некоторых типичных соединений определяли некоторые свойства следующими методами анализа РМВ: проницаемость Сасо-2 (пример 903), клиренс гепатоцитов (пример 904), ингибирование цитохрома Р450 (пример 905), индукция цитохрома Р450 (пример 906), связывание с белками плазмы (пример 907) и блокирование каналов hERG (пример 908).
Испытывали эффективность некоторых типичных соединений при проведении испытаний с увеличением дозы на моделях «голых» мышей Taconic с ксенотрансплантатами опухоли (пример 909). Модельным мышам с клеточной линией рака молочной железы MDA-MB-361.1 вводили некоторые типичные соединения формулы I и носитель (МСТ, отрицательный контроль). Замедление роста опухоли определяли при ежедневном введении соединений в дозе 50 мг/кг и 100 мг/кг перорально в течение 21 дней. В качестве показателя безопасности в ходе всего курса лечения регистрировали массу тела. При лечении модельных мышей MDA-MB-361.1 некоторыми типичными соединениями формулы I при ежедневном введении дозы перорально в течение 21 дней наблюдалась замедление роста опухоли, подавление или регрессия.
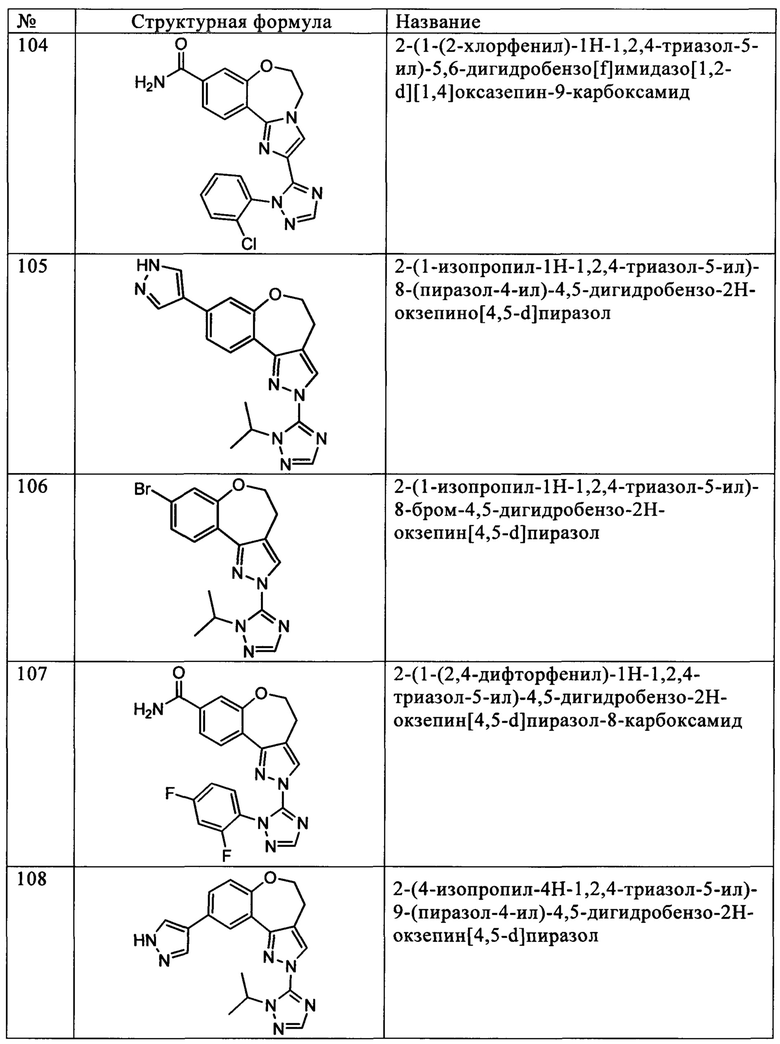
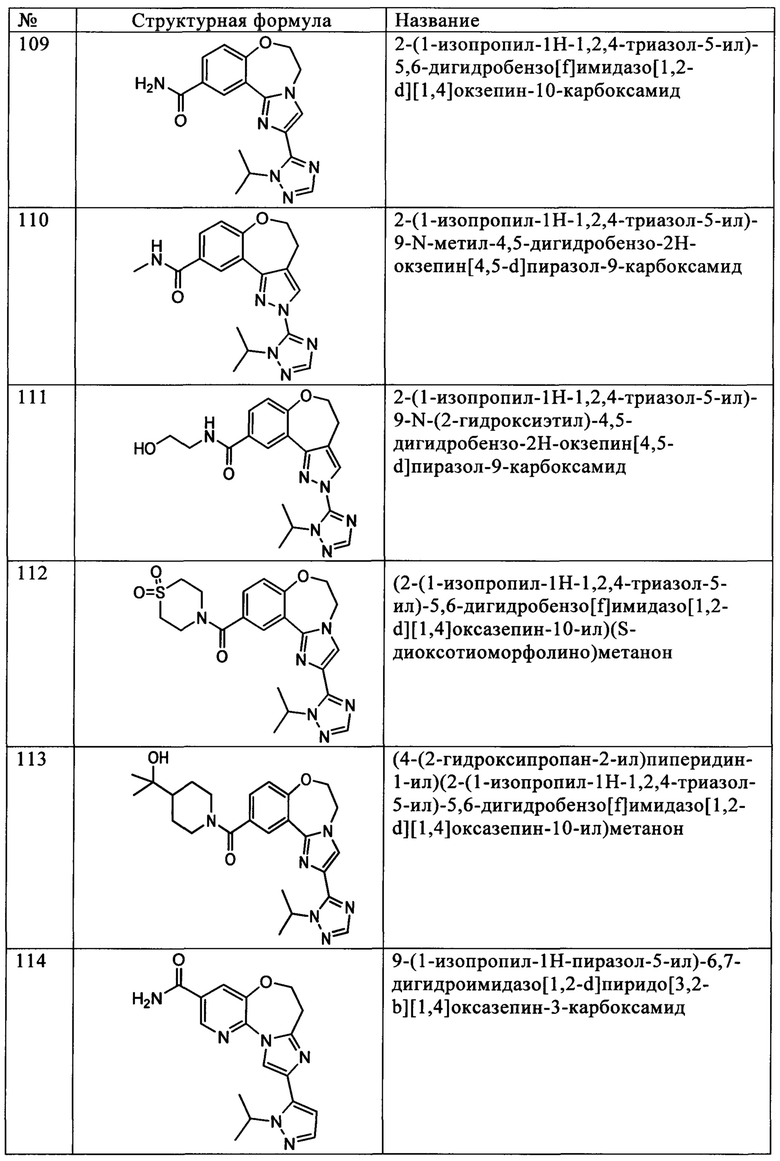
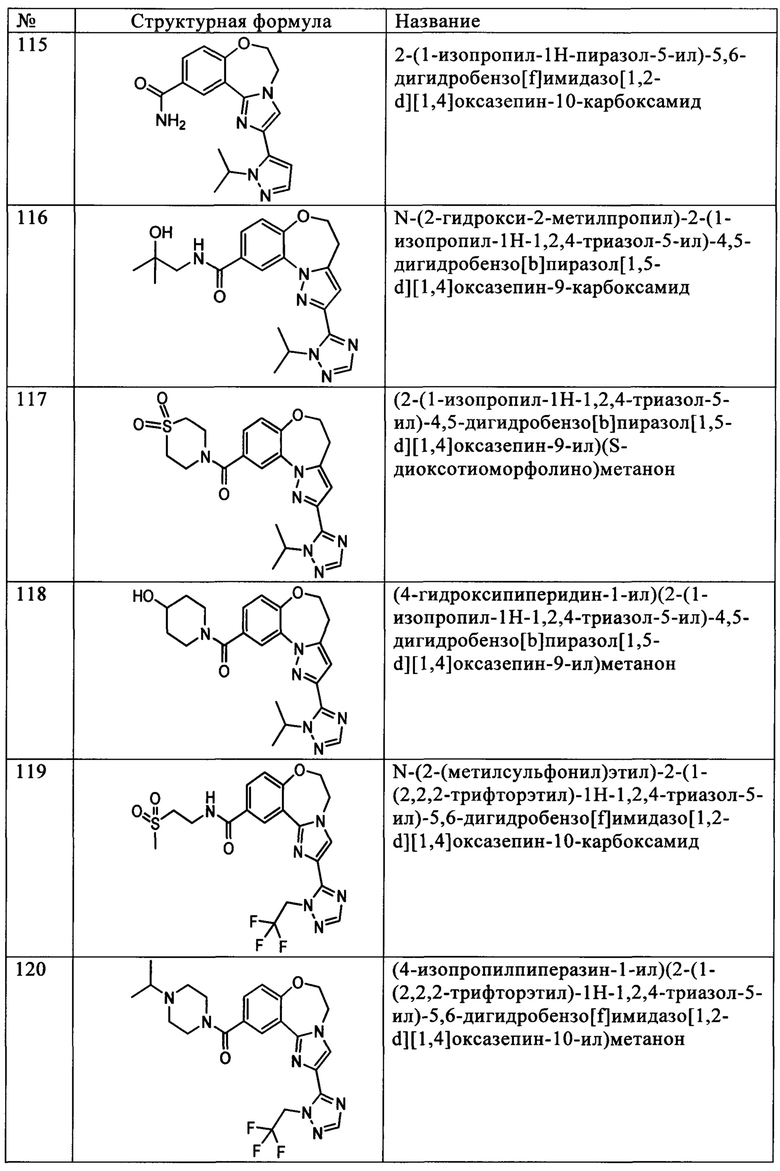
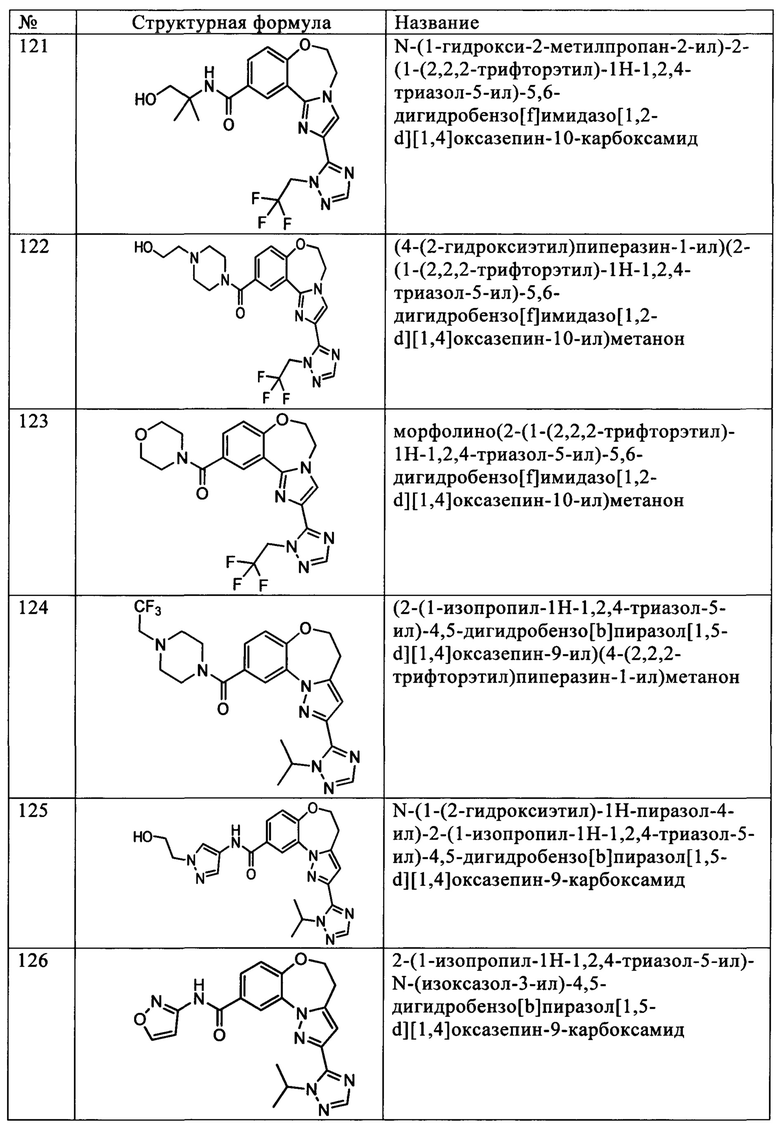
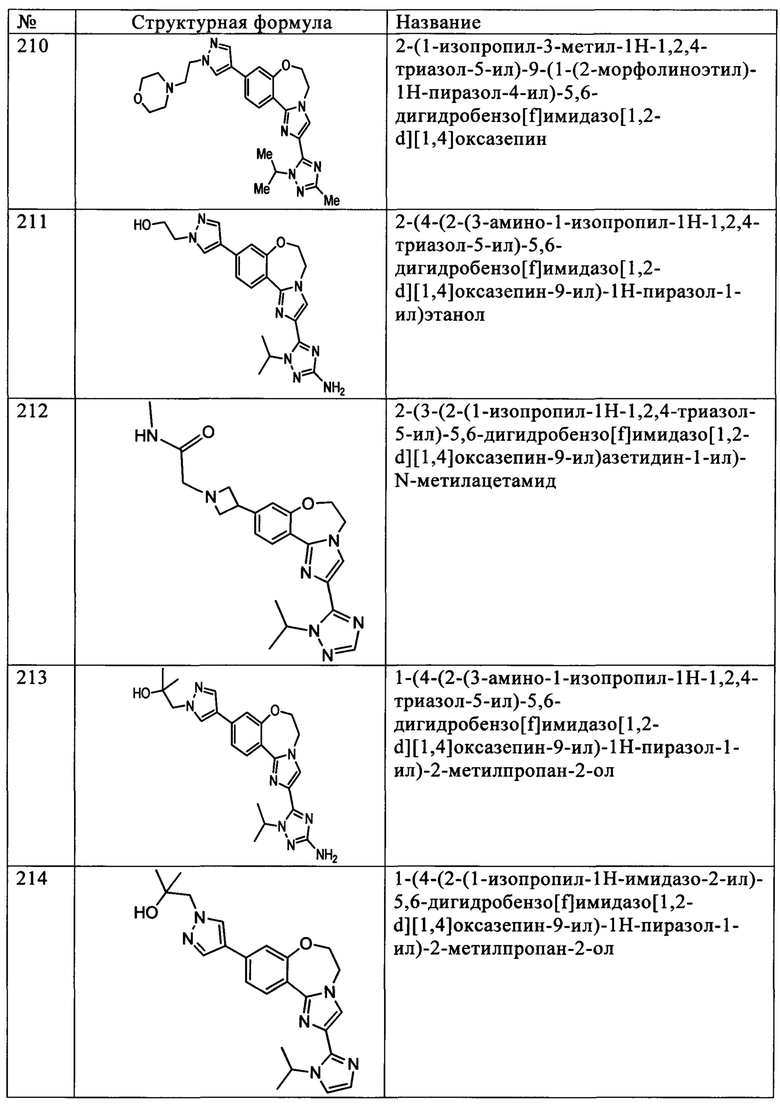
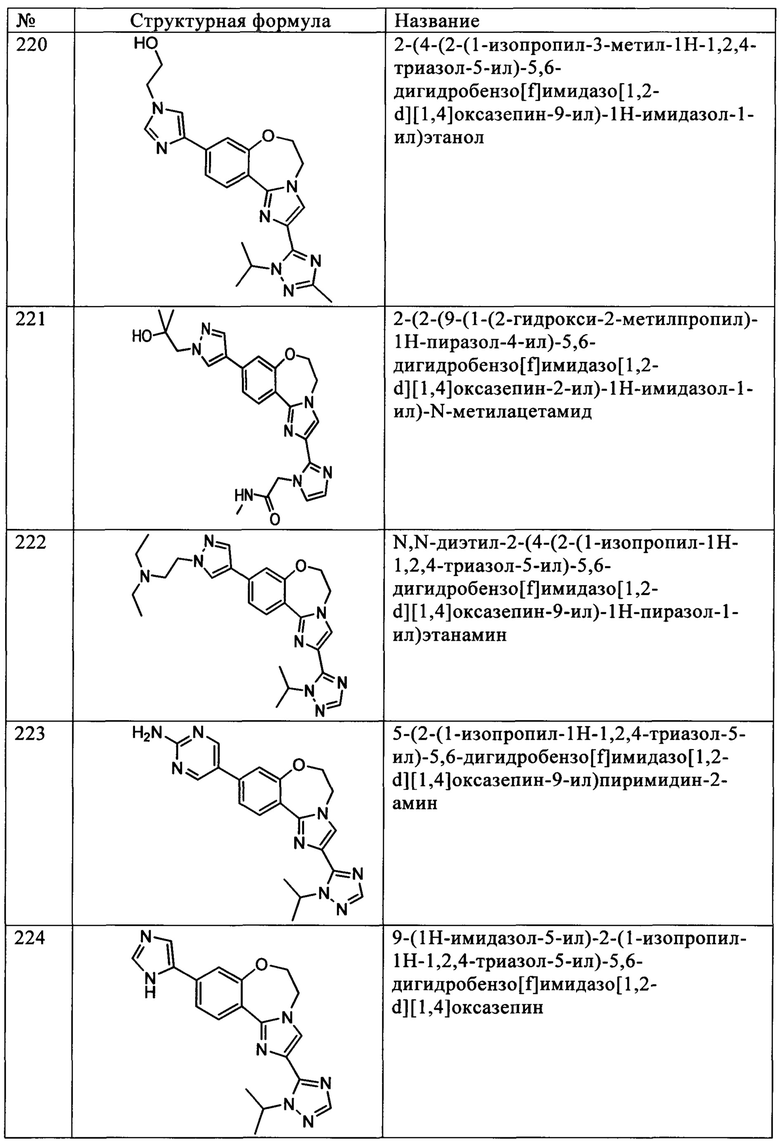
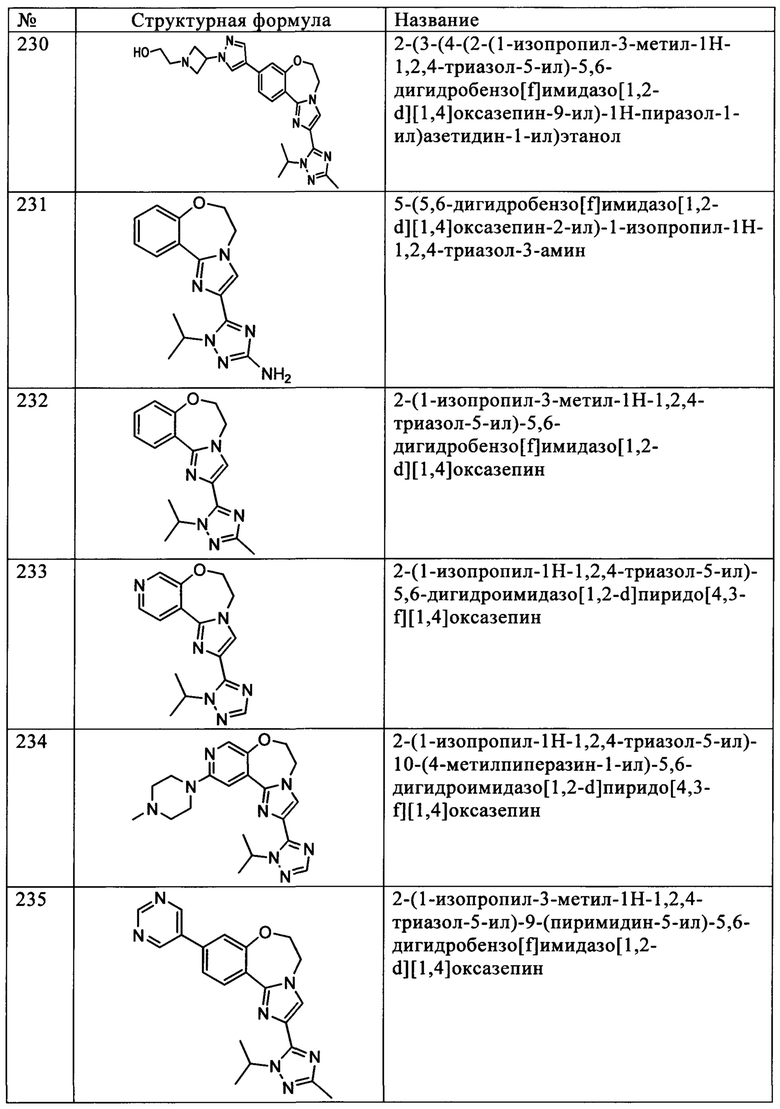
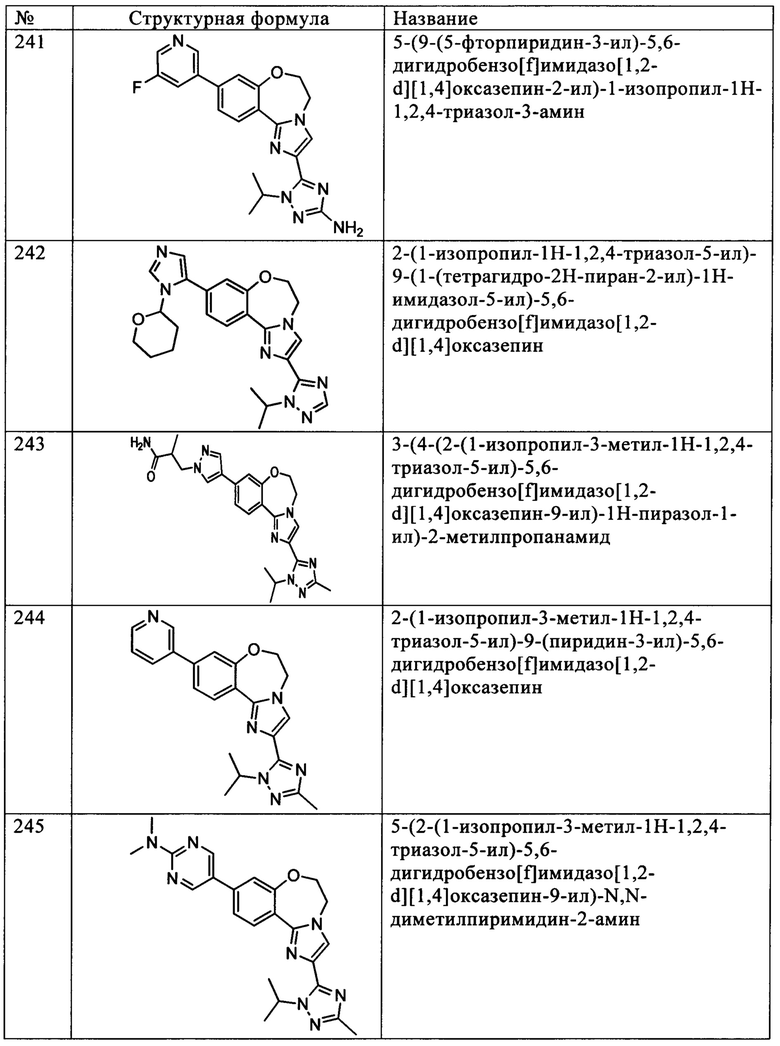
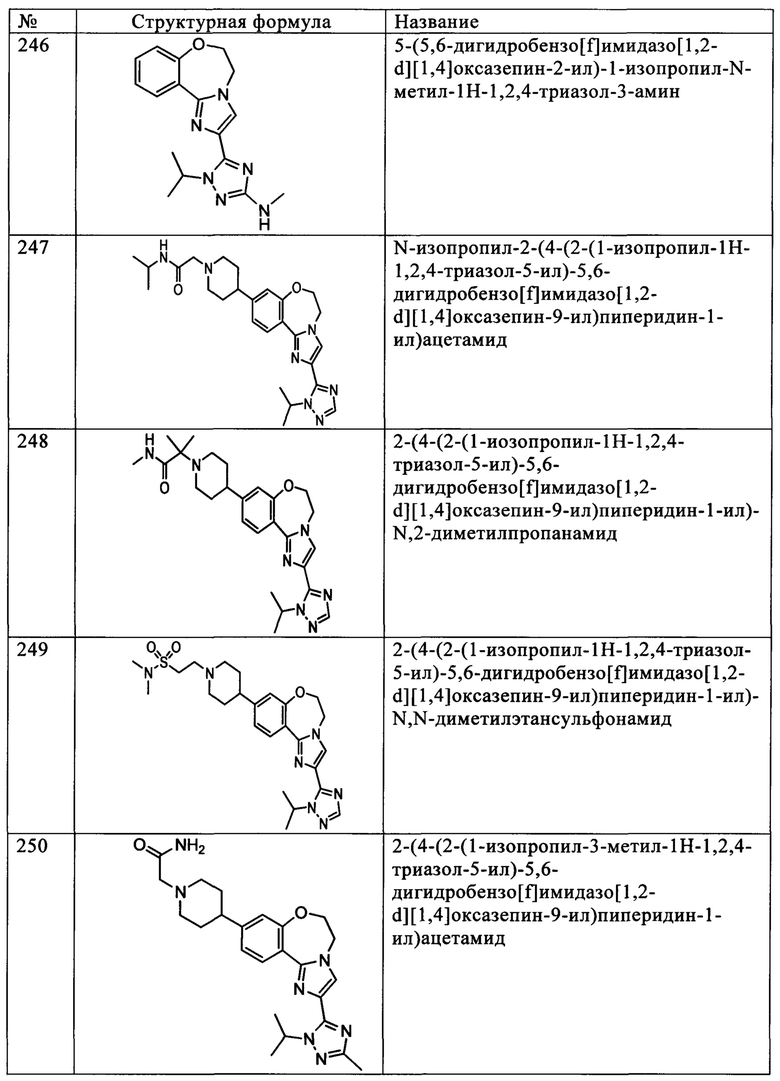
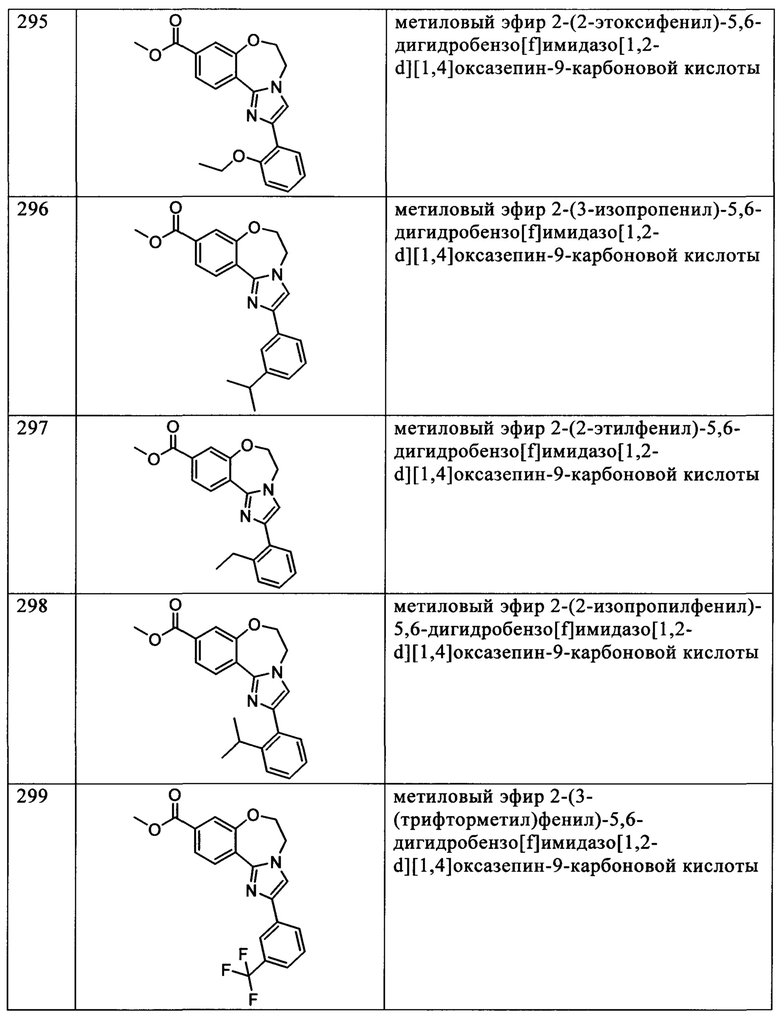
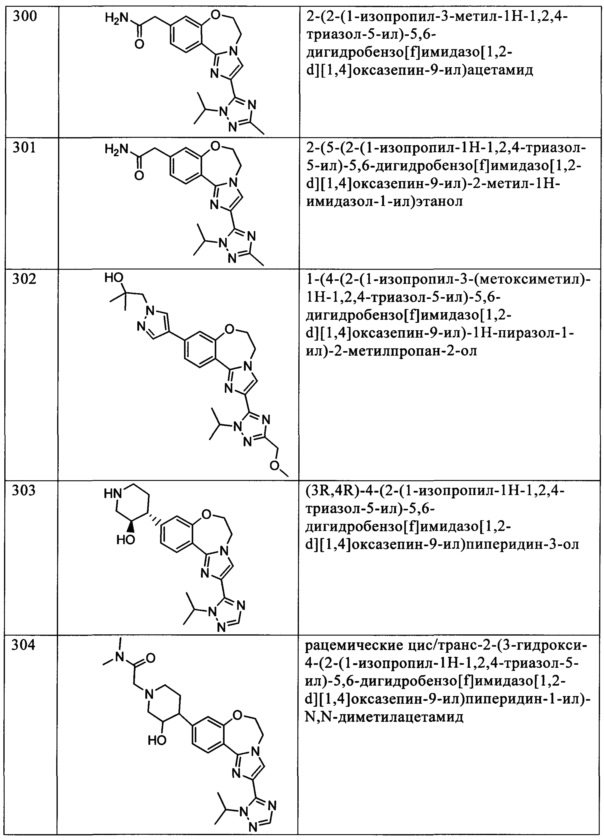
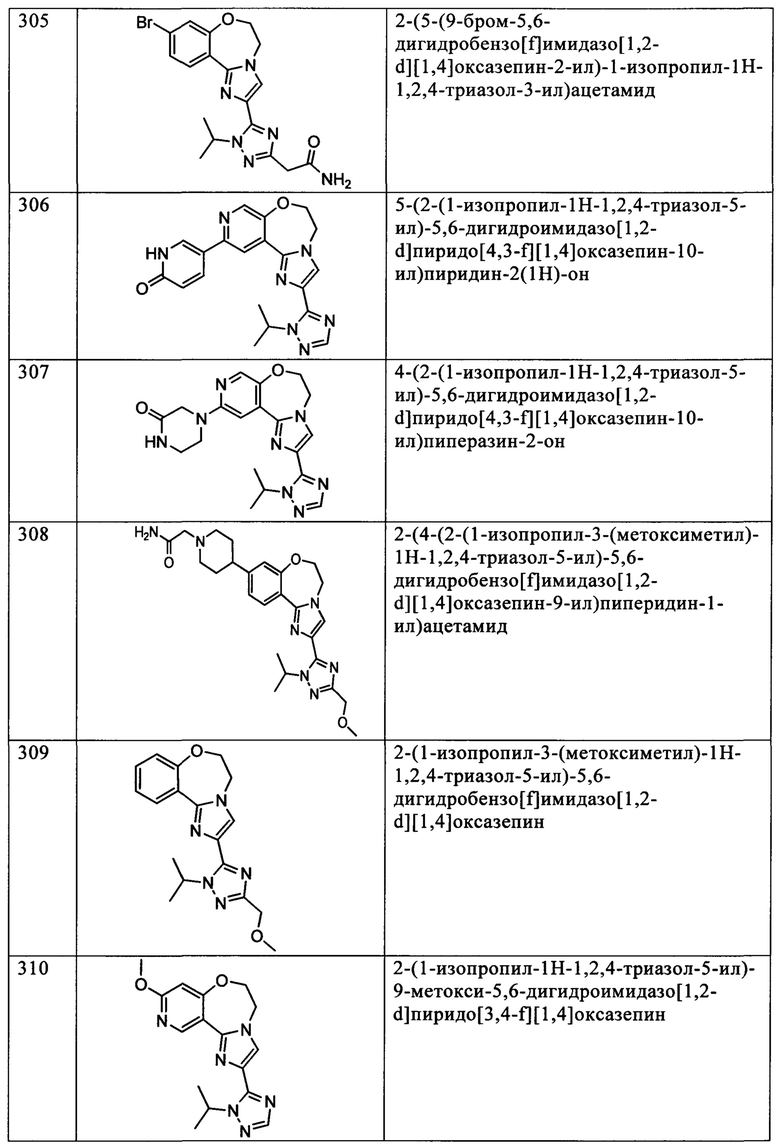
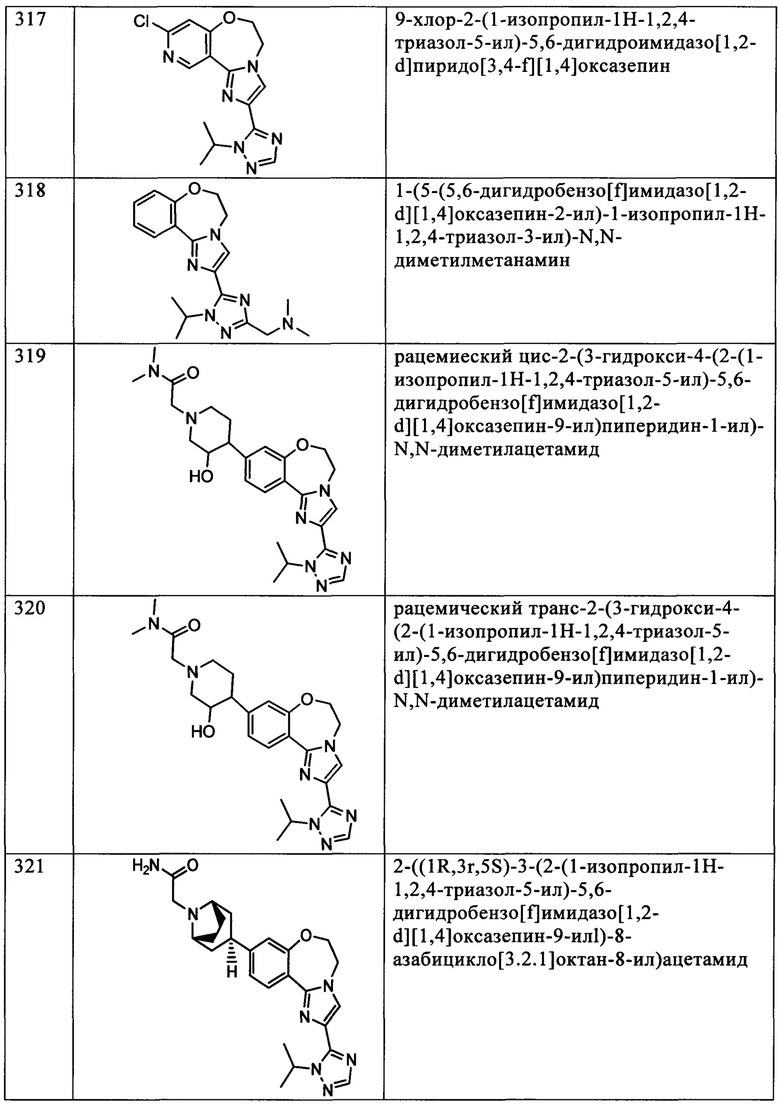
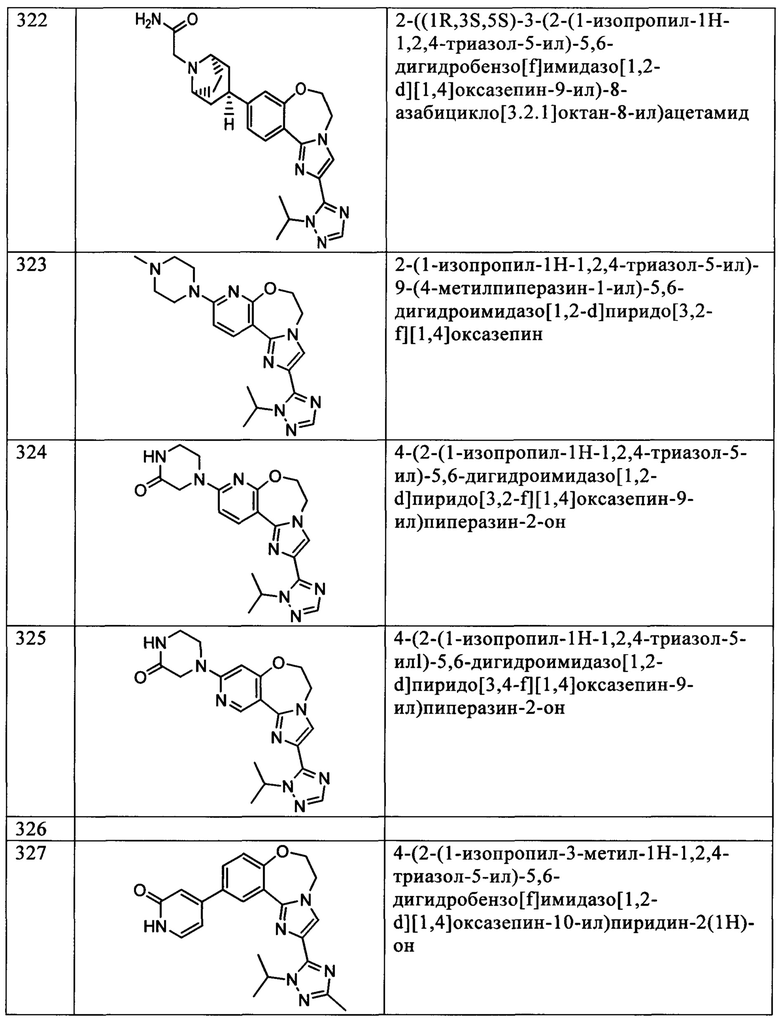
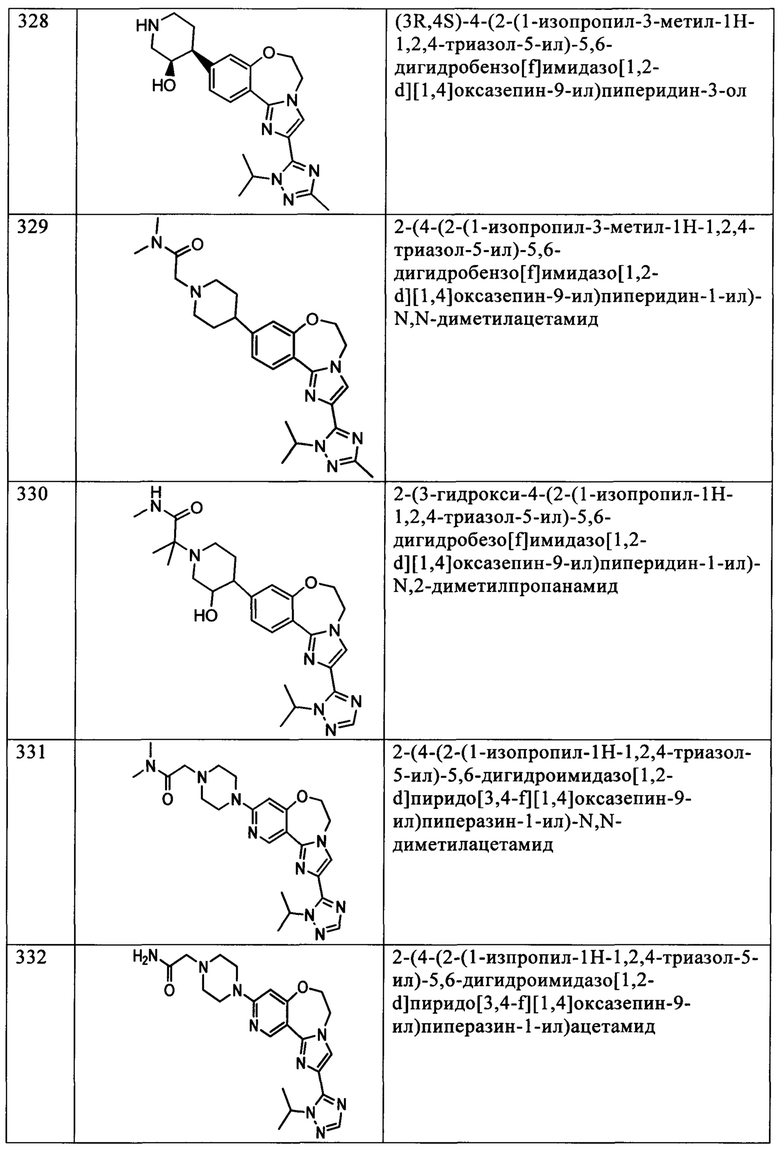
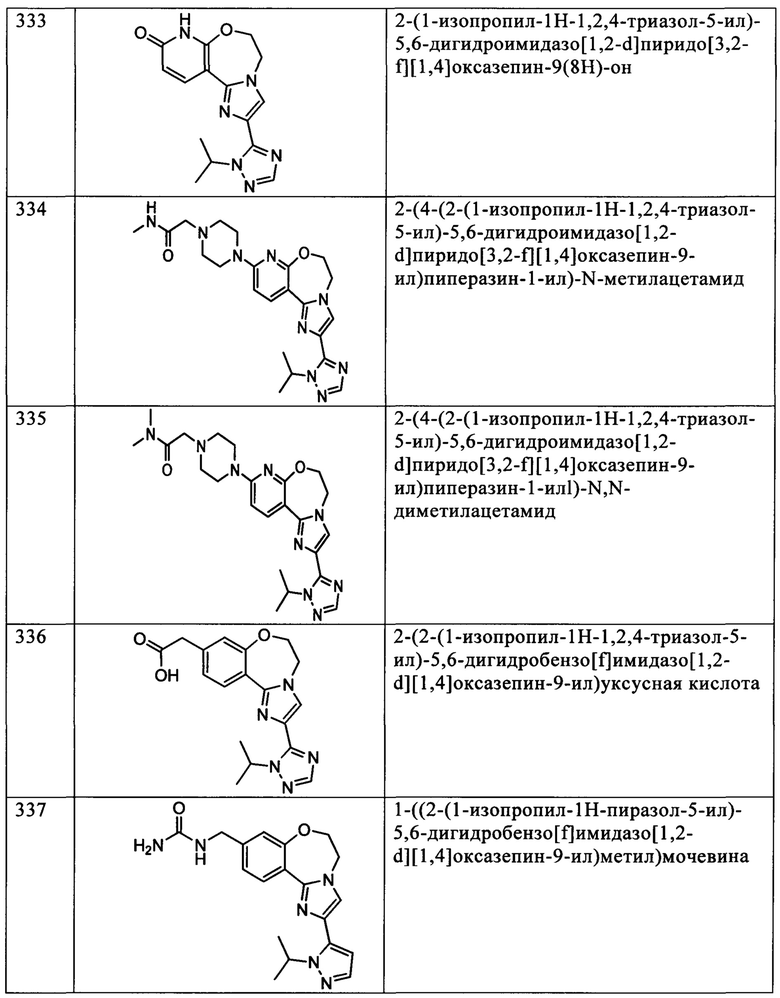
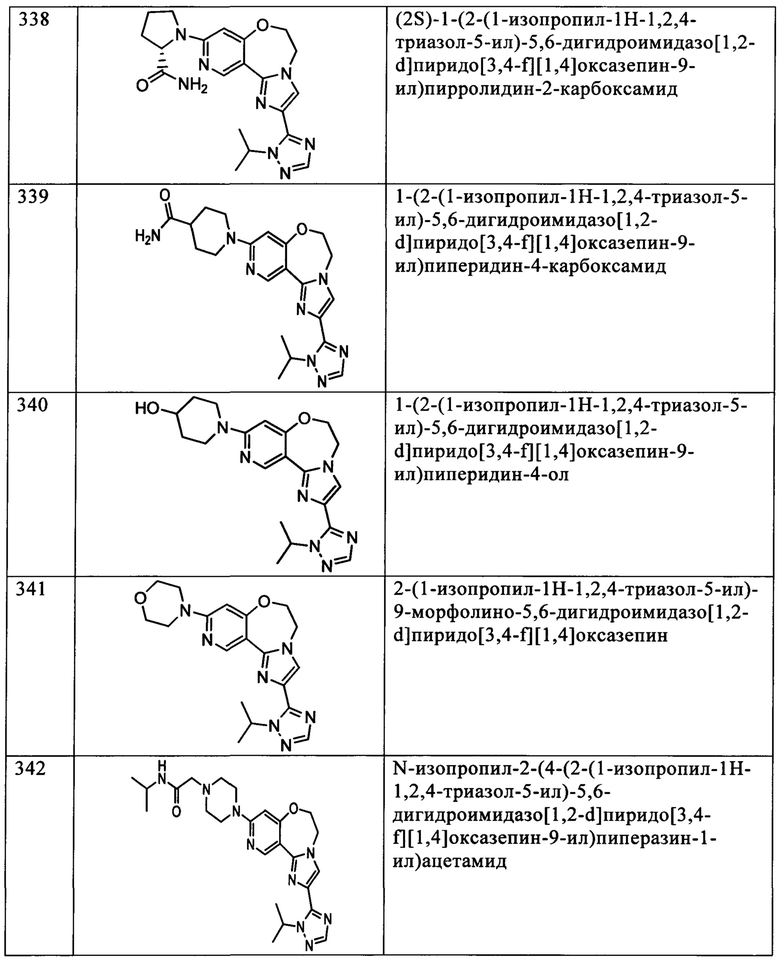
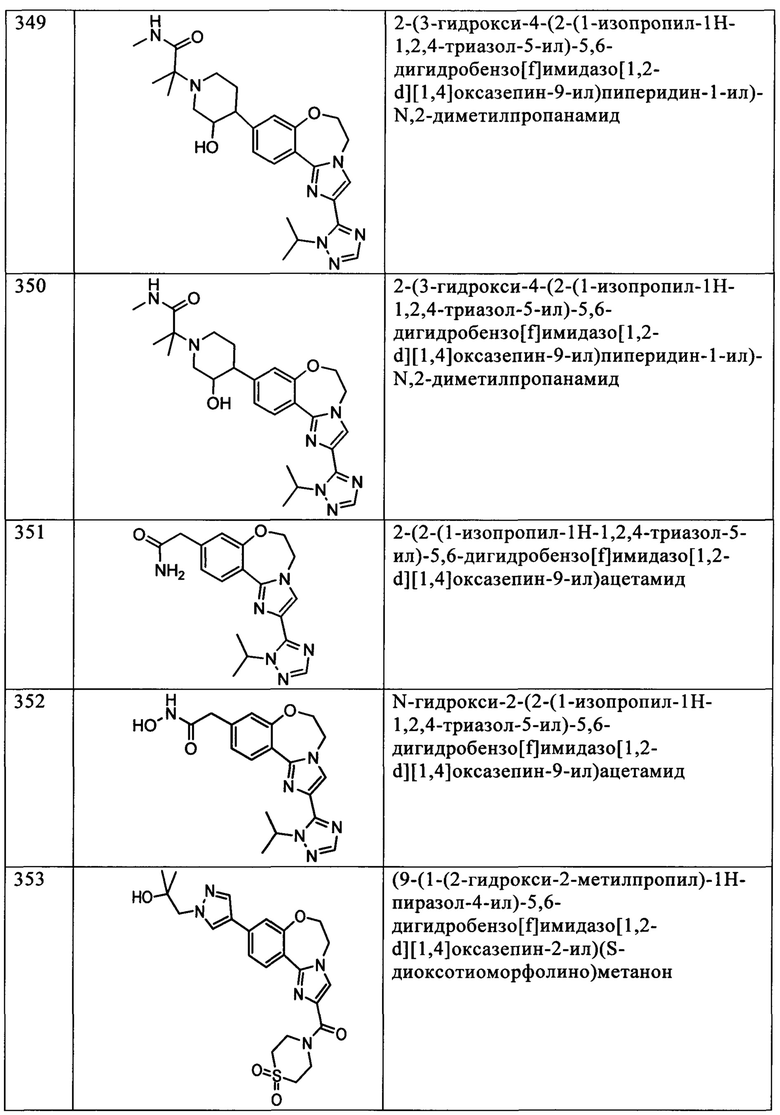
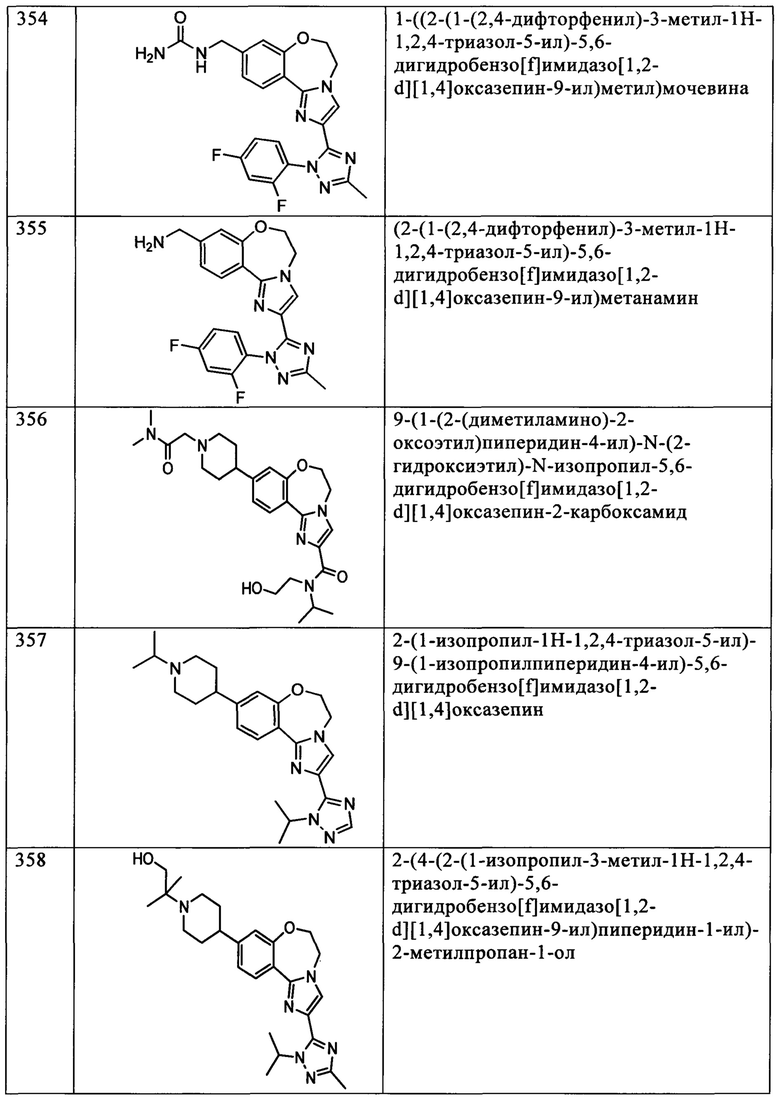
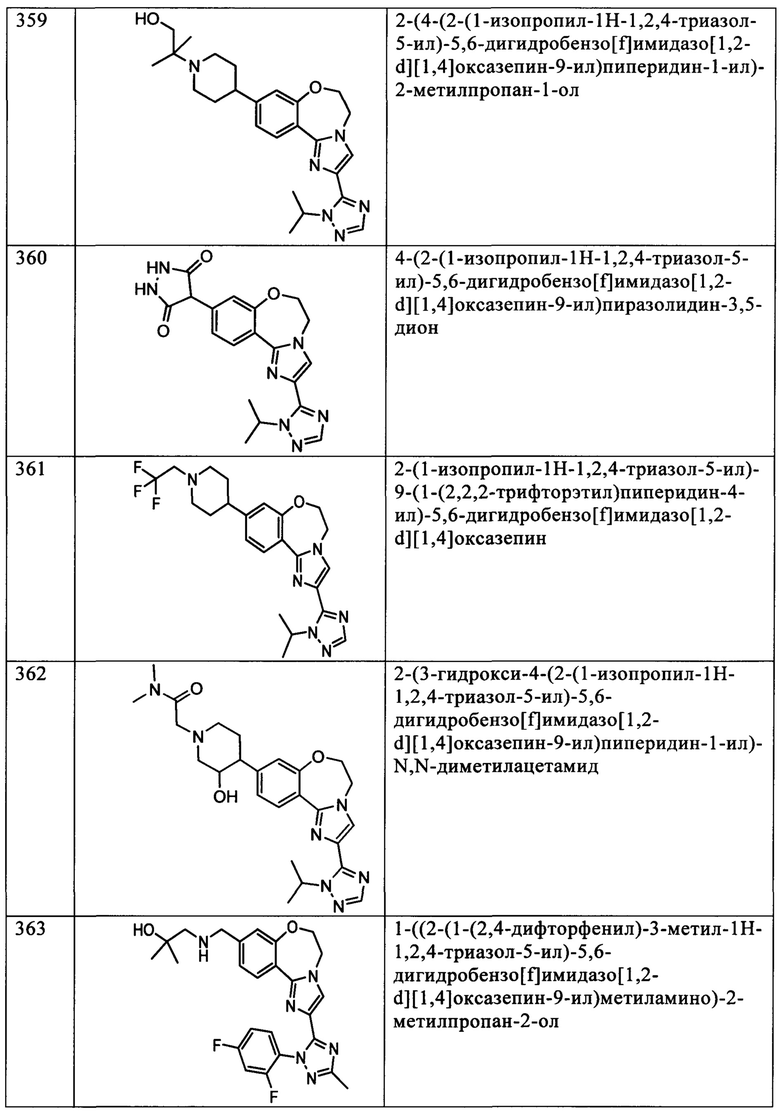
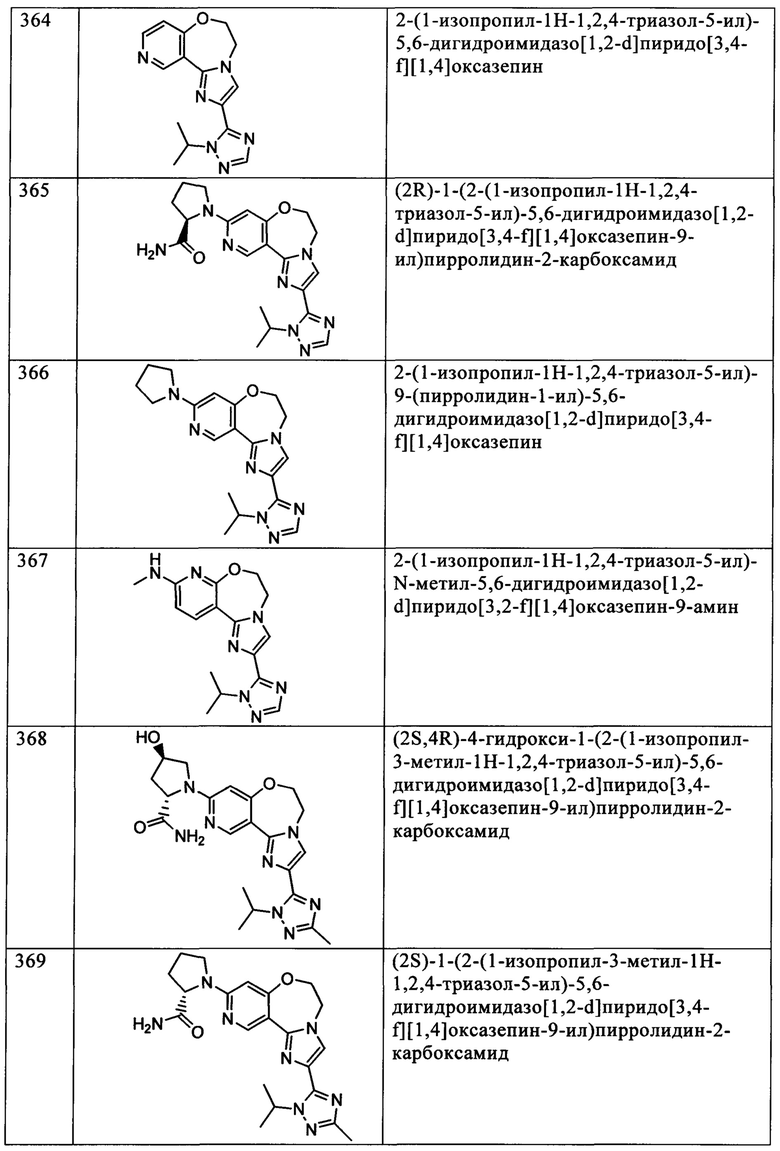
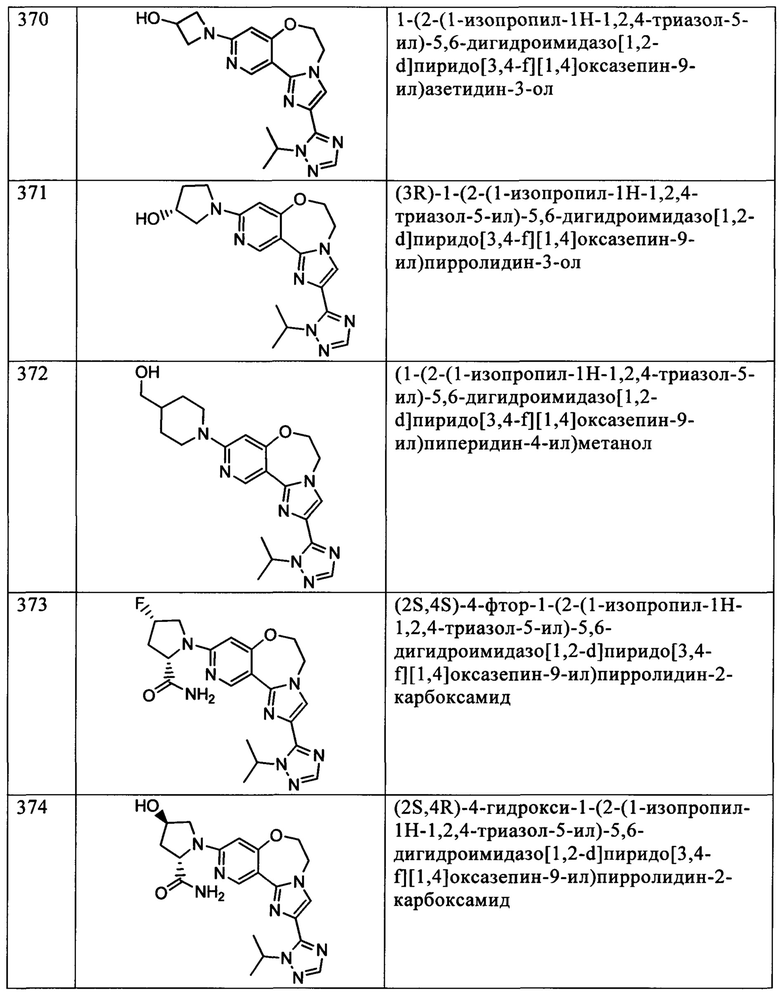
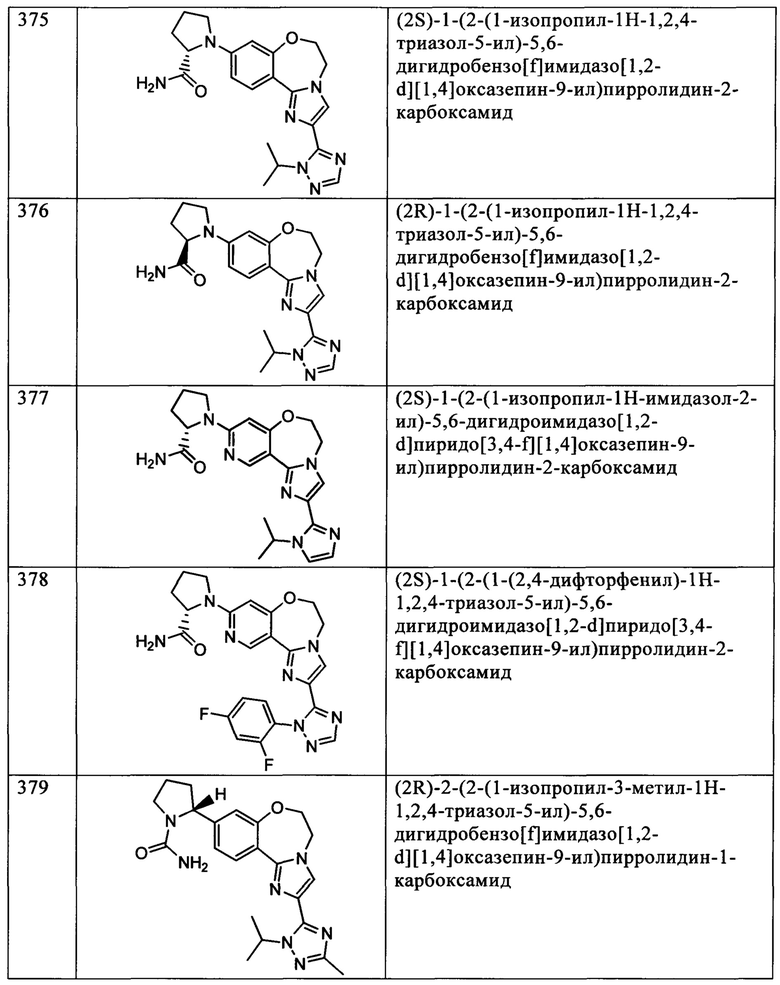
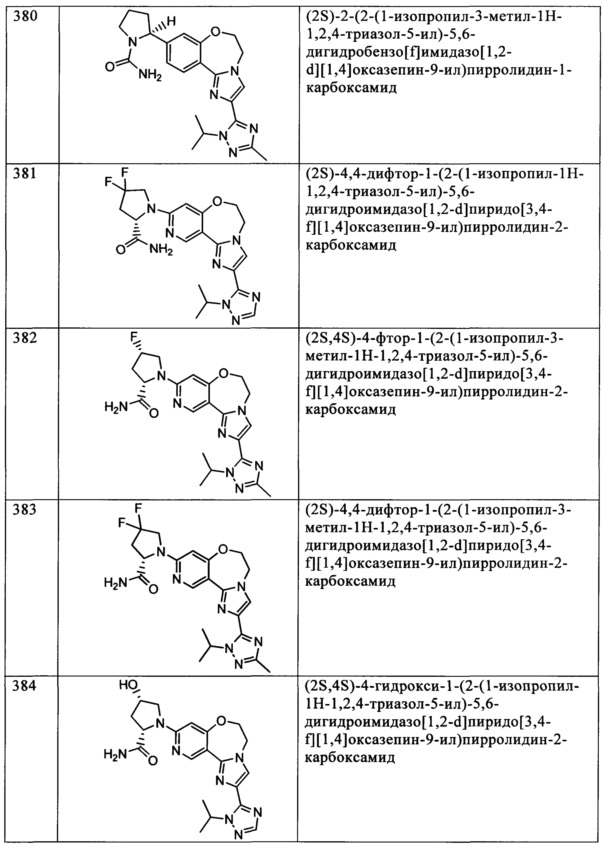
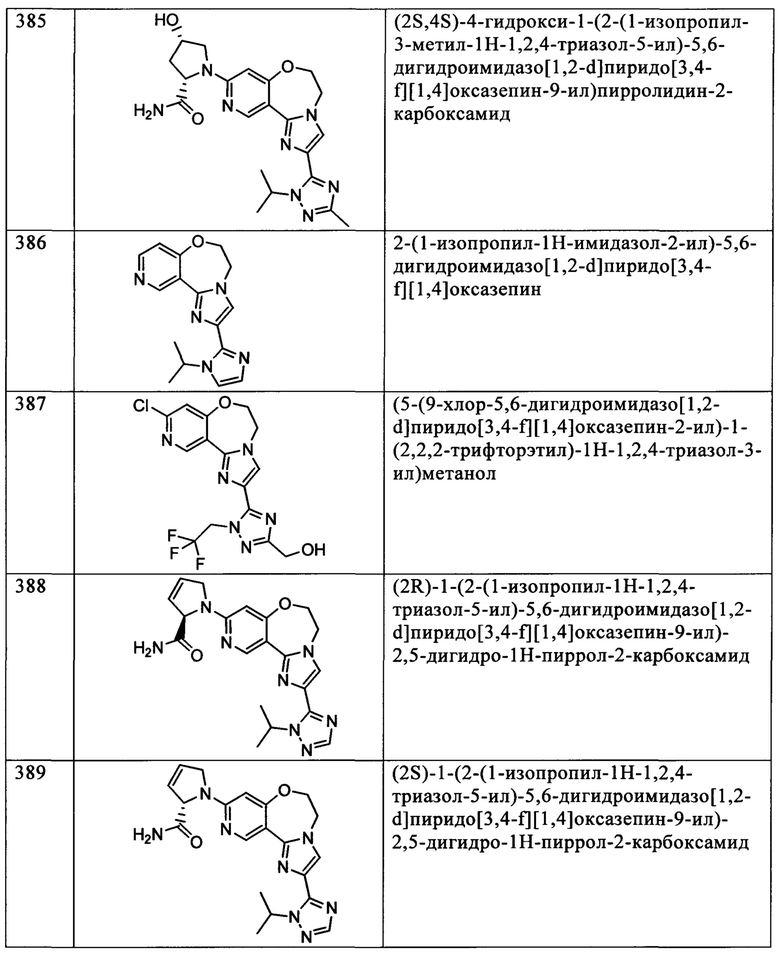
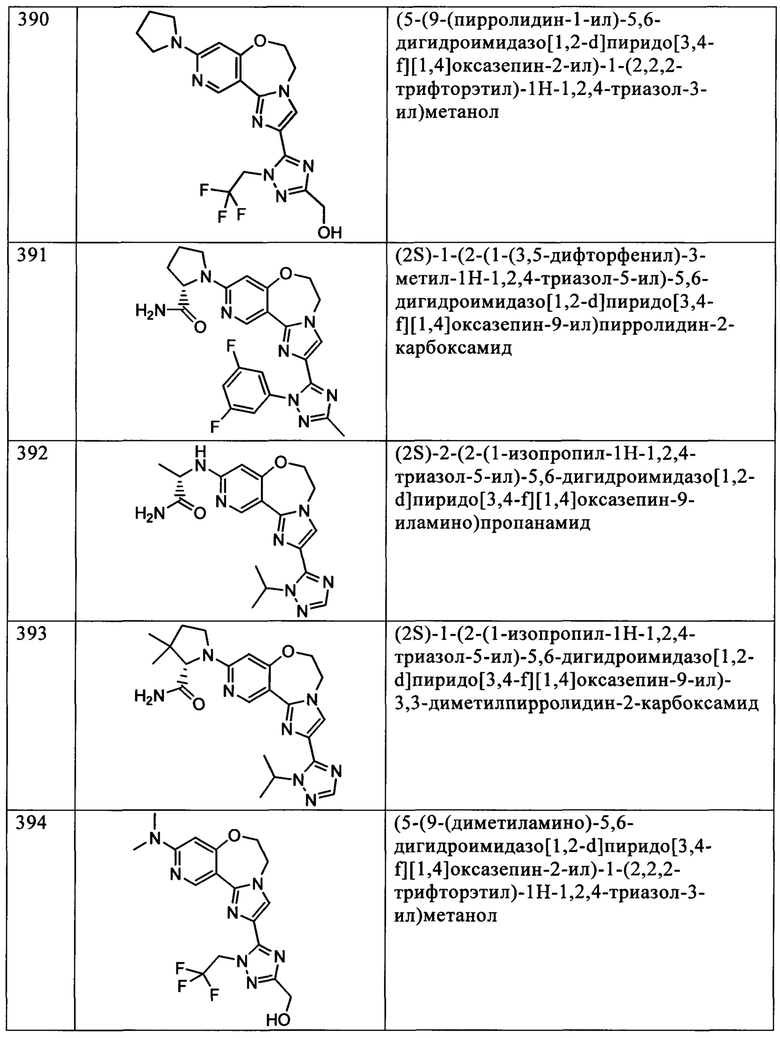
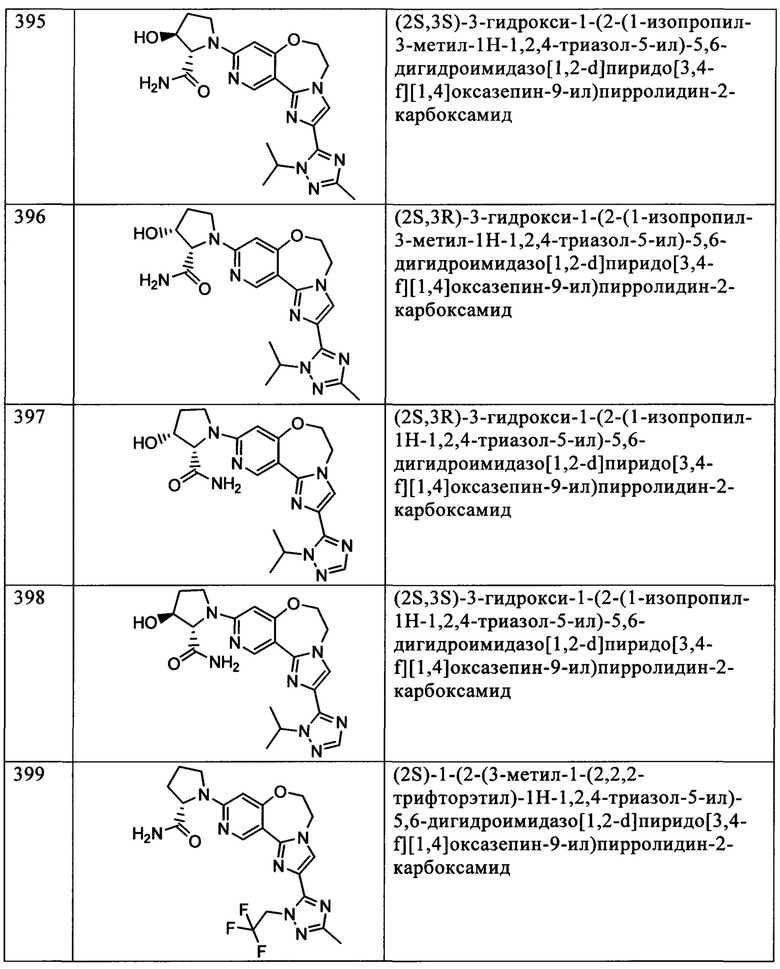
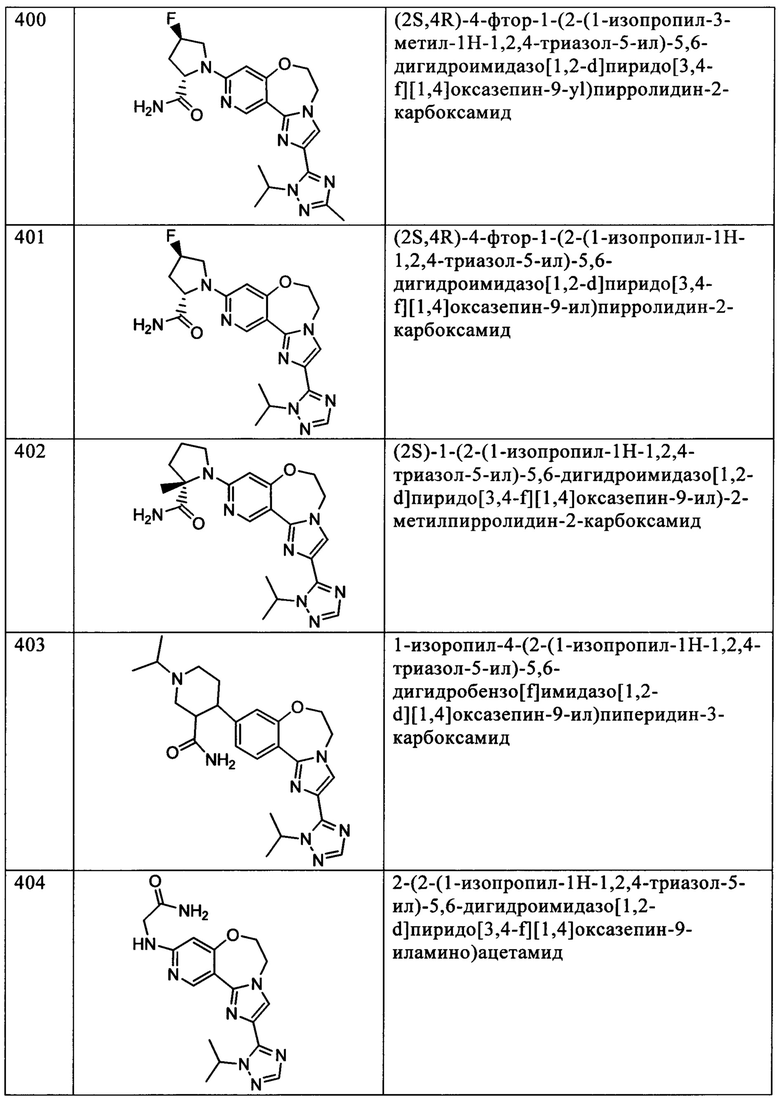
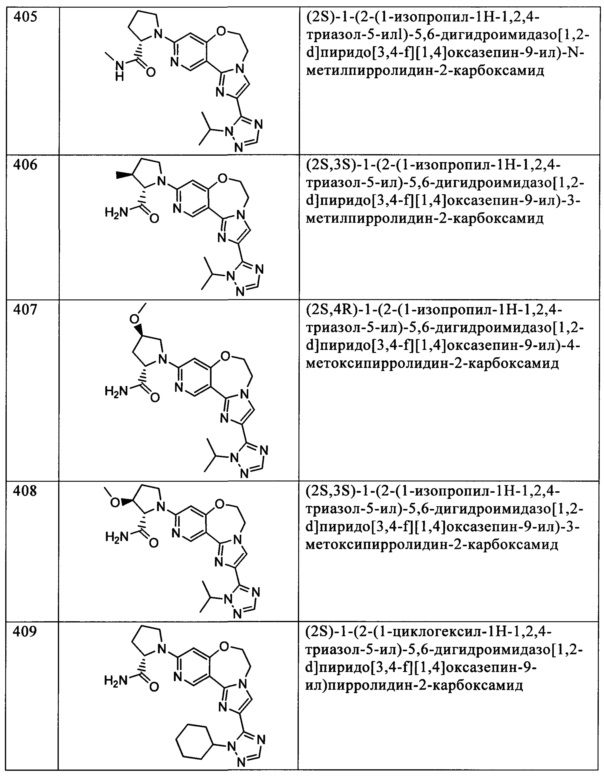
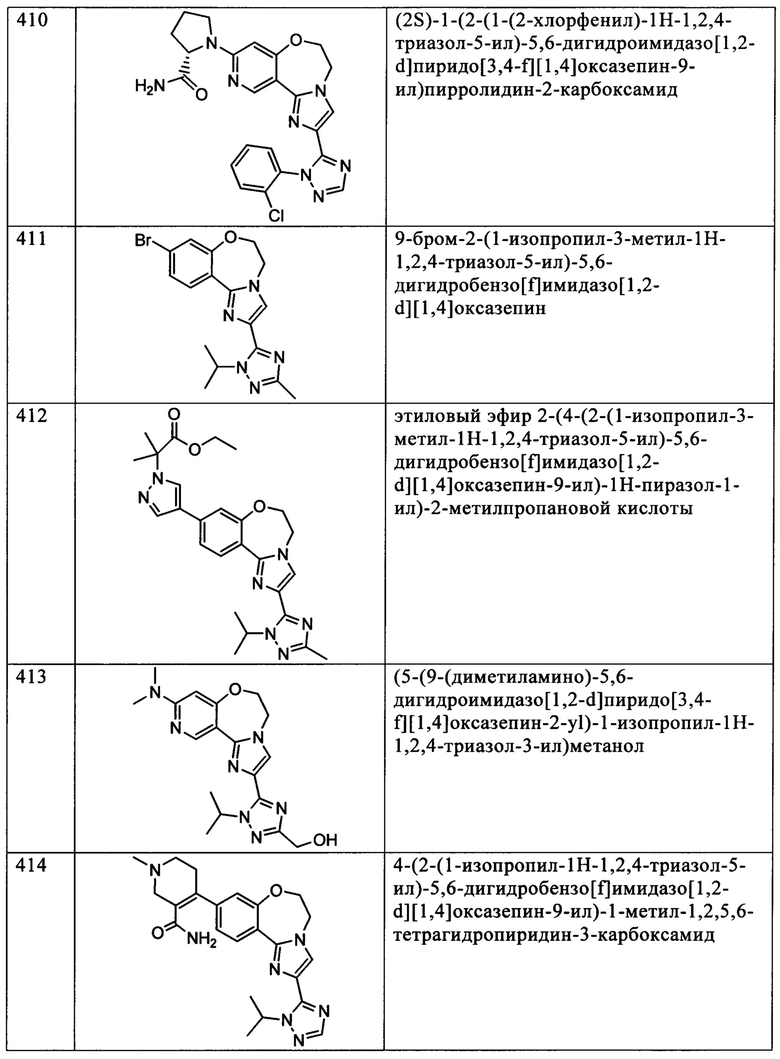
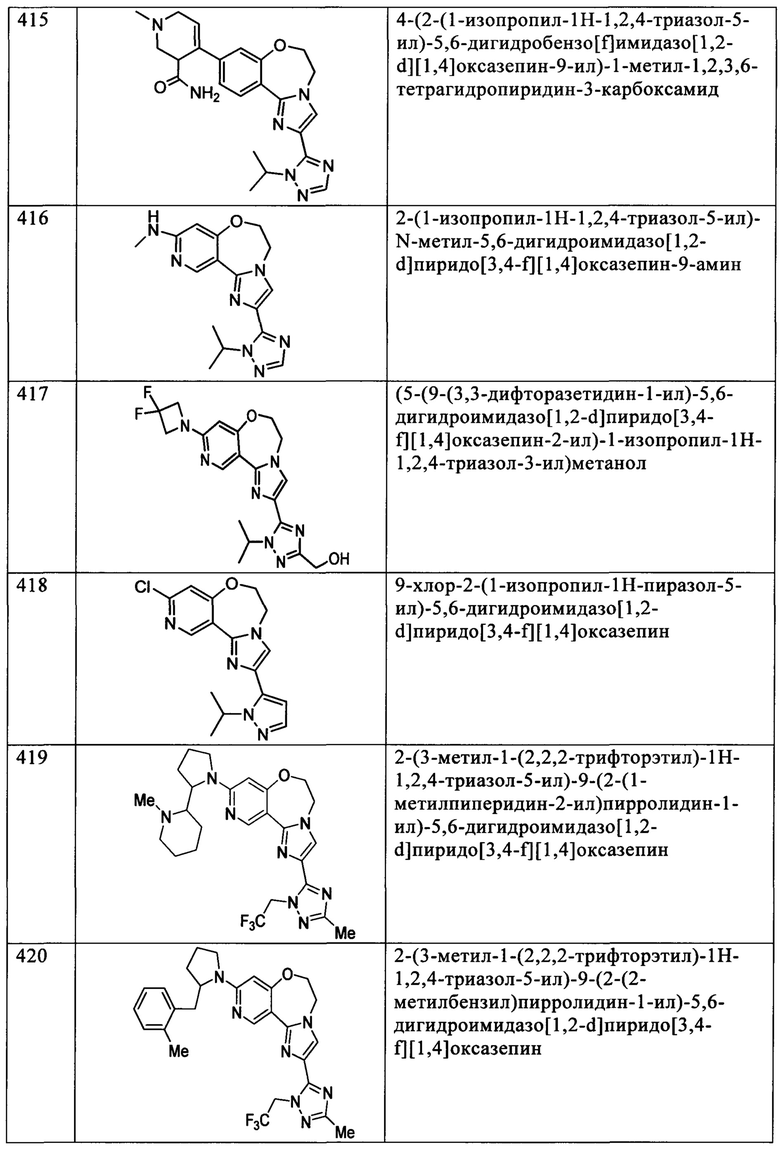
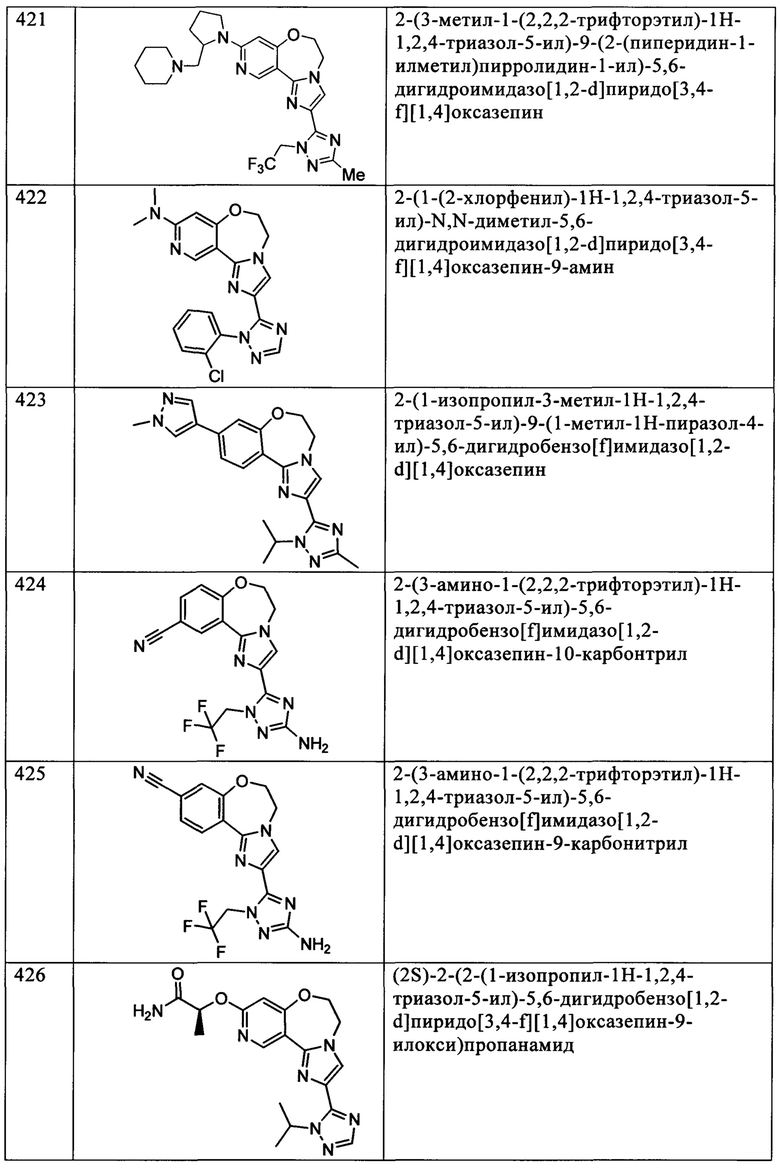
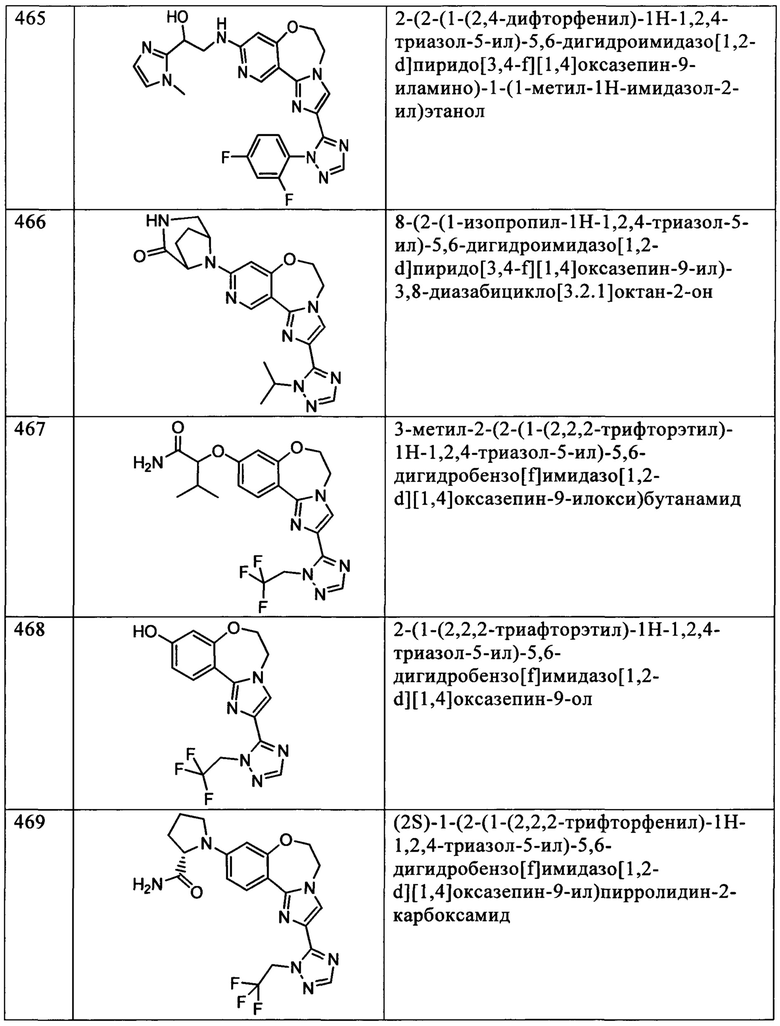
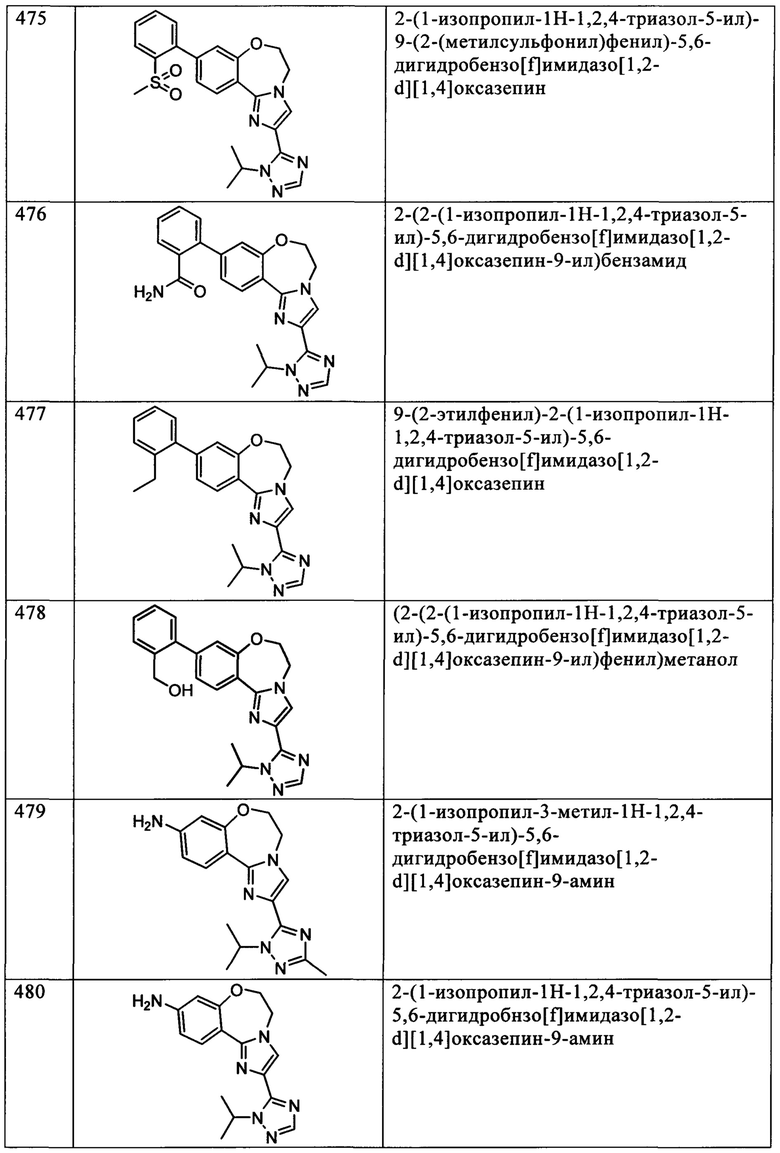
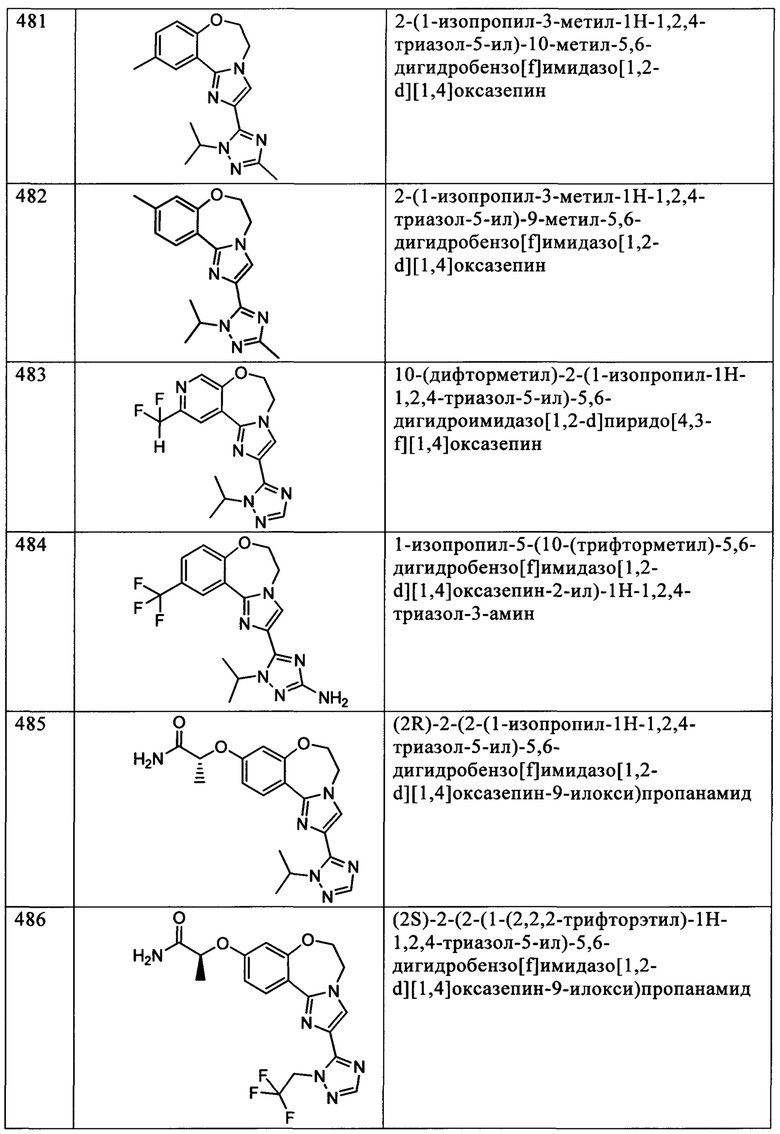
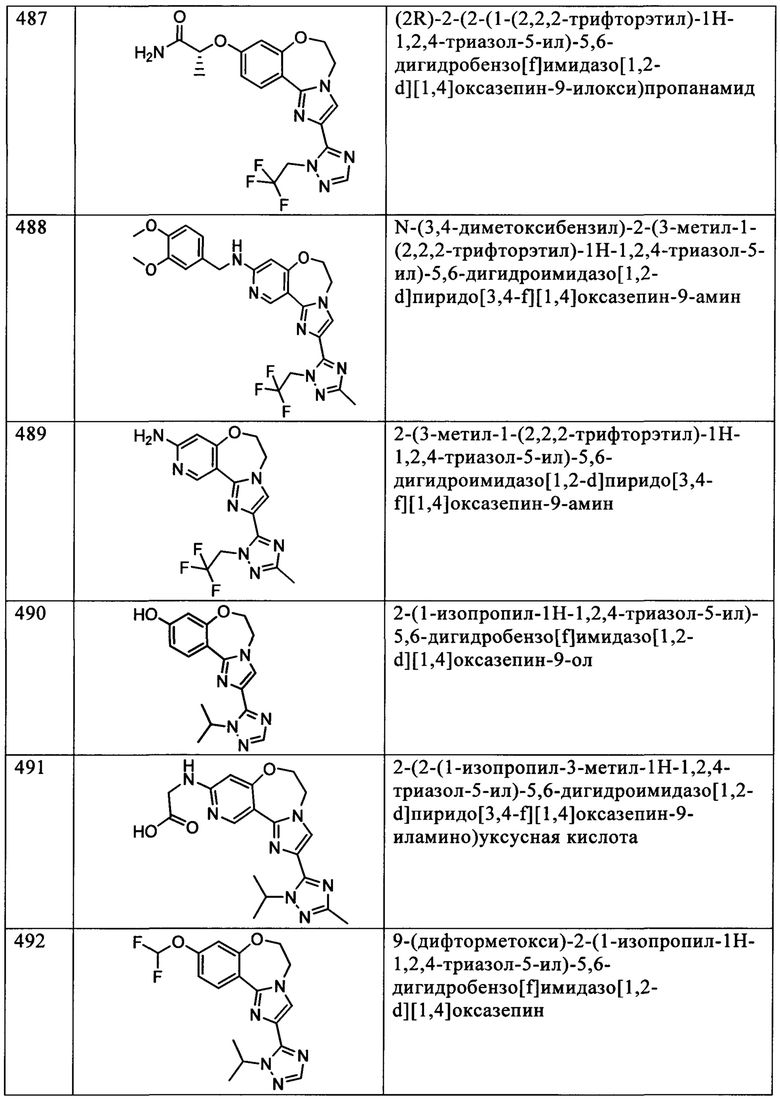
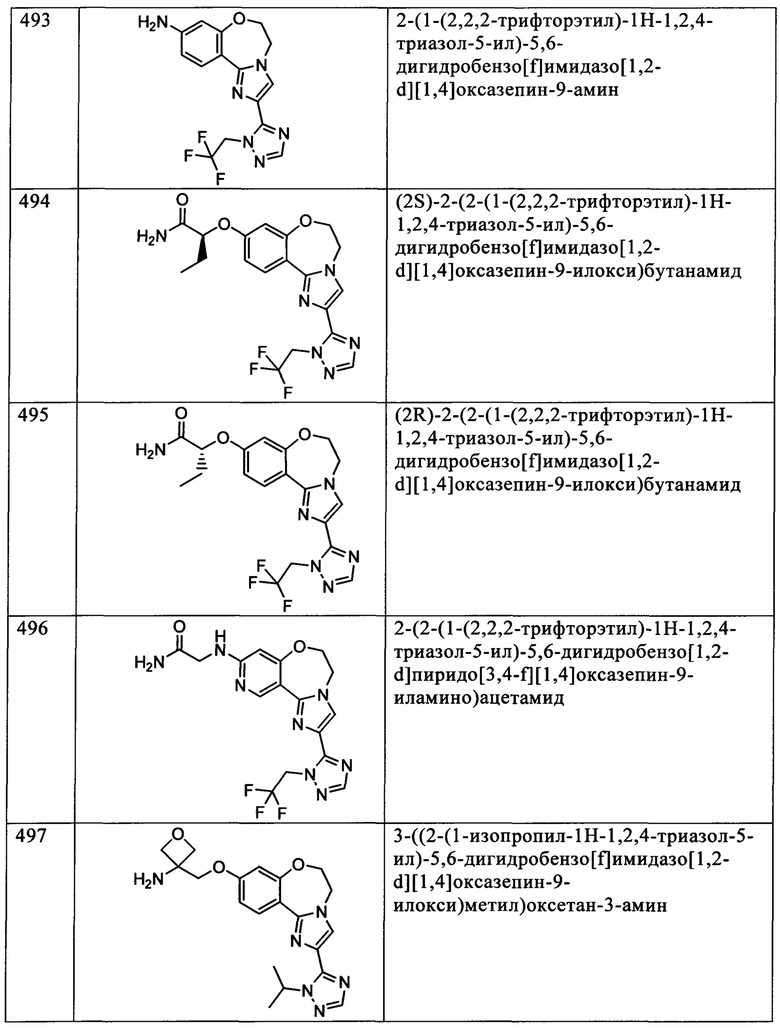
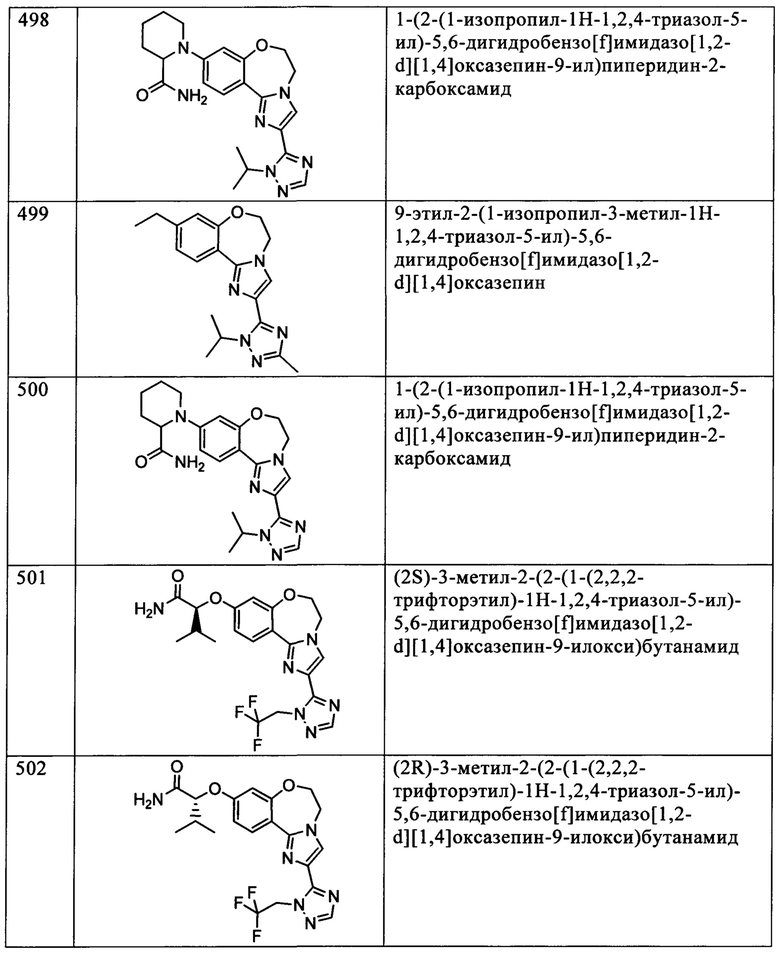
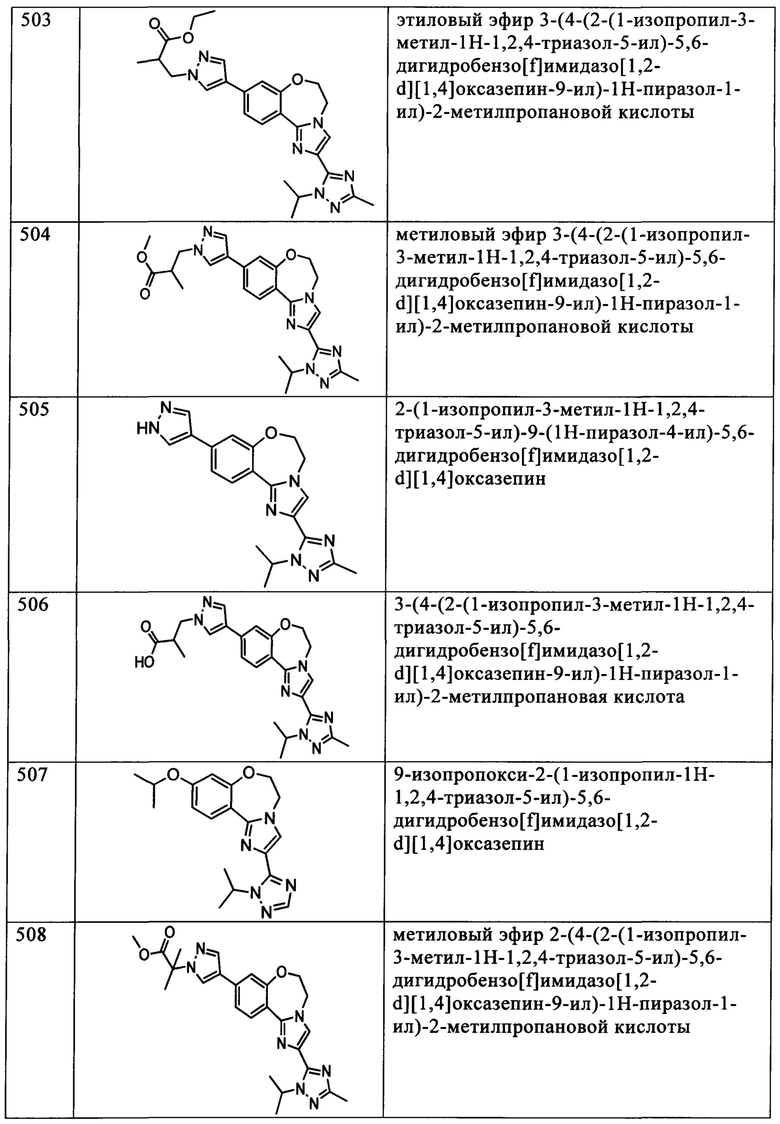
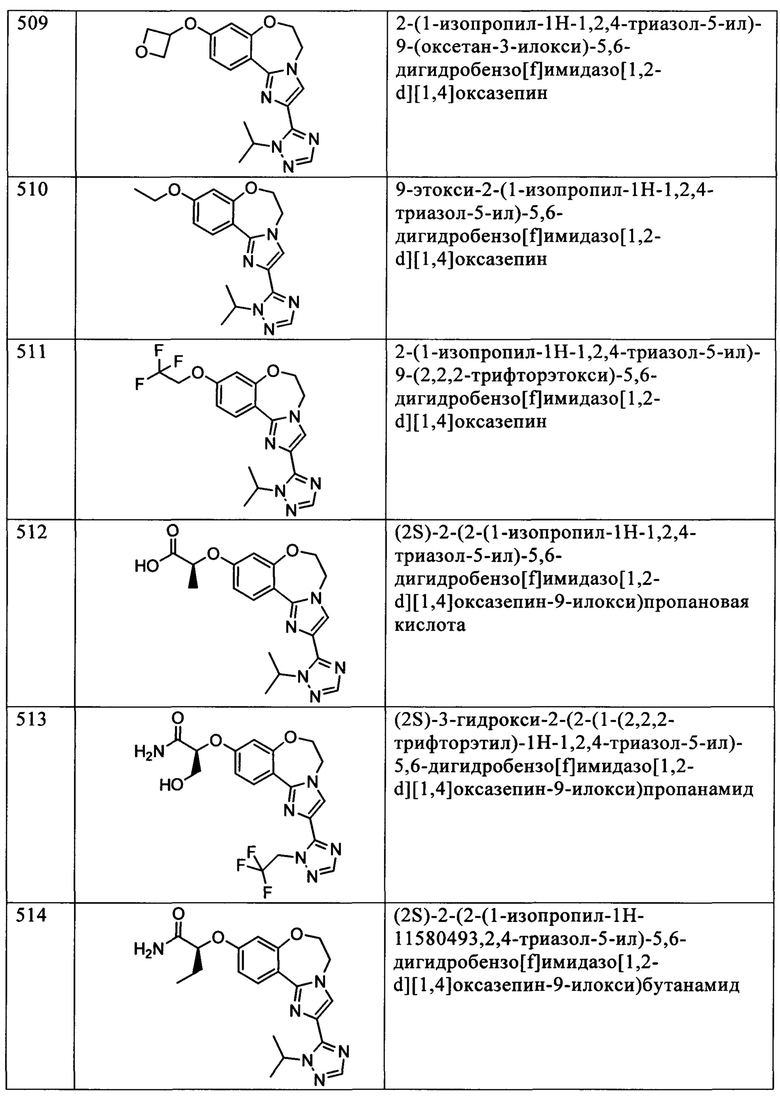
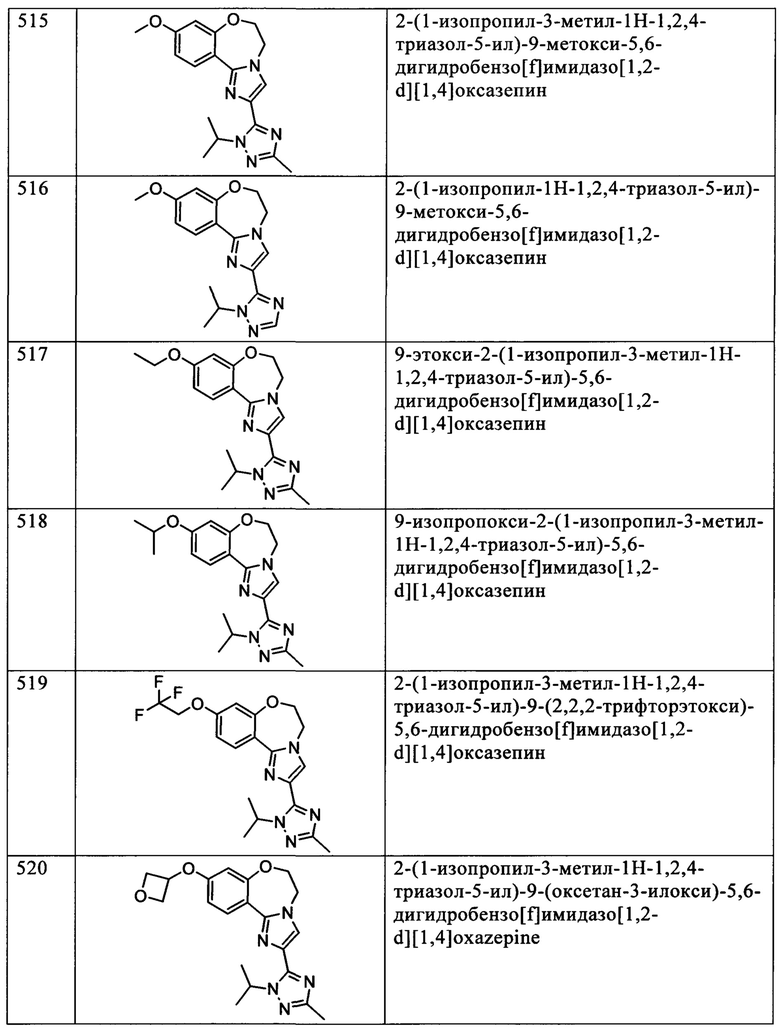
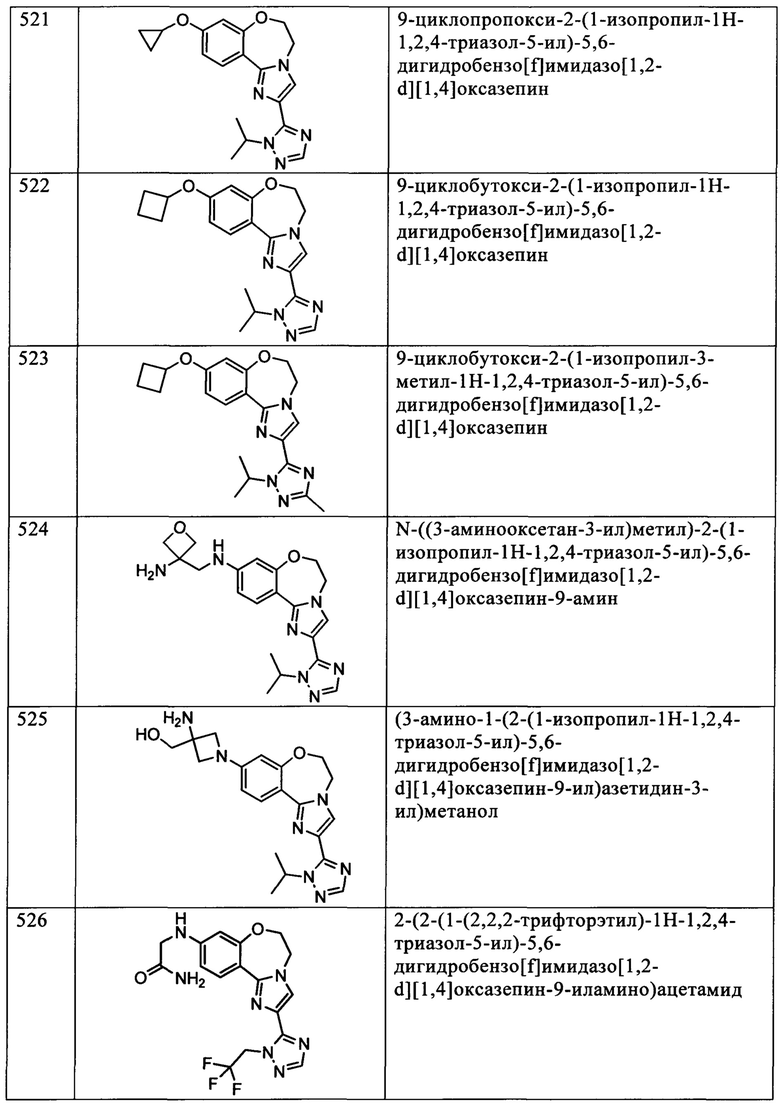
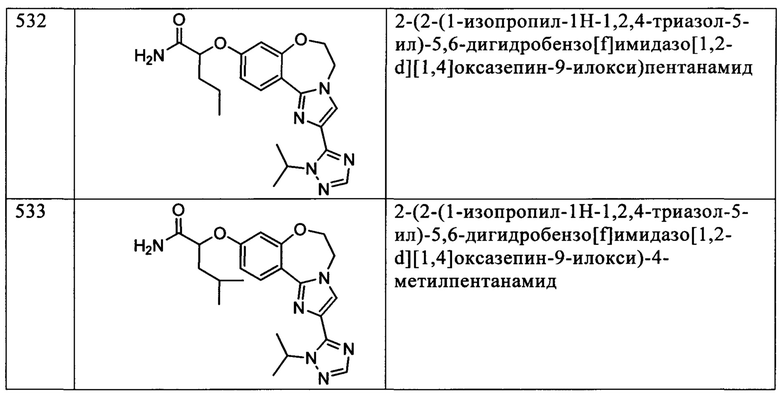
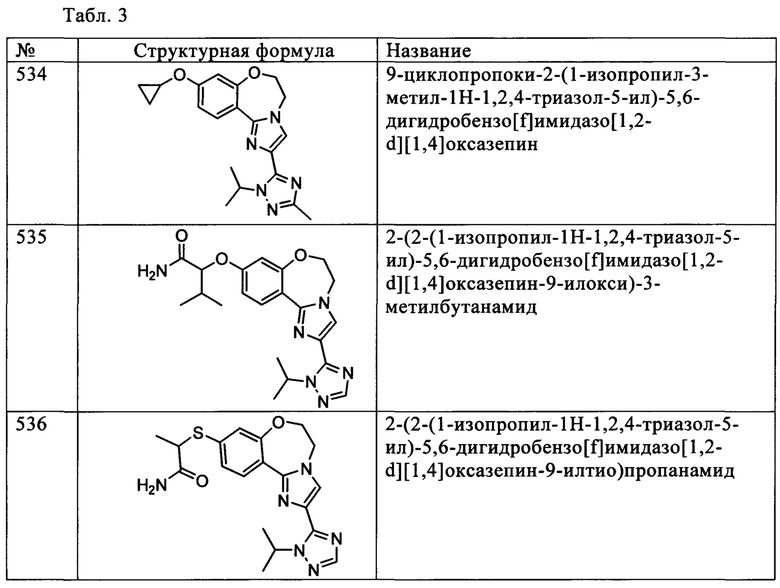
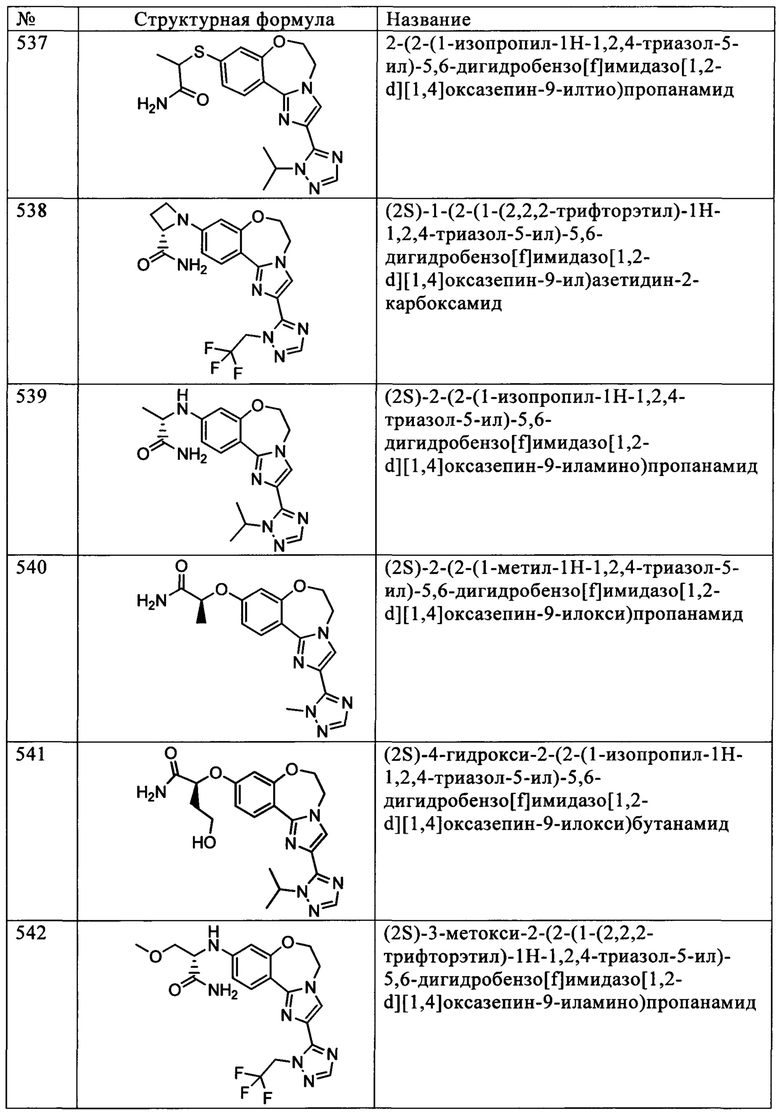
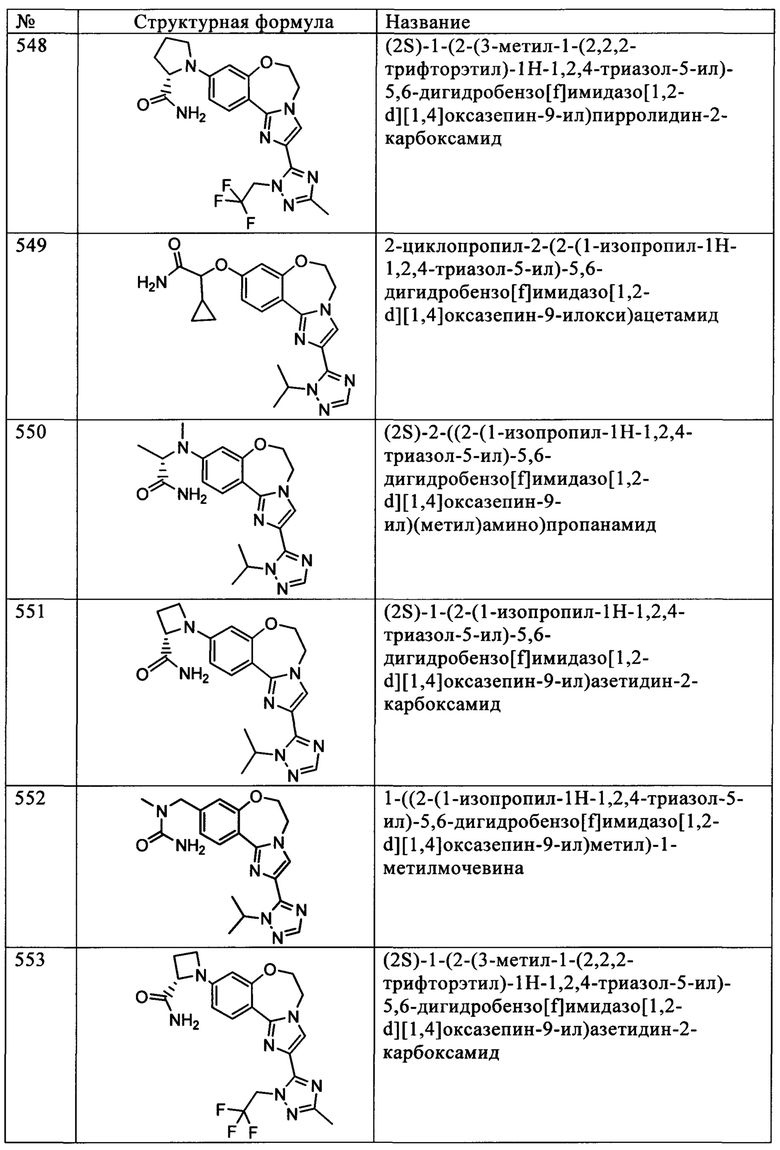
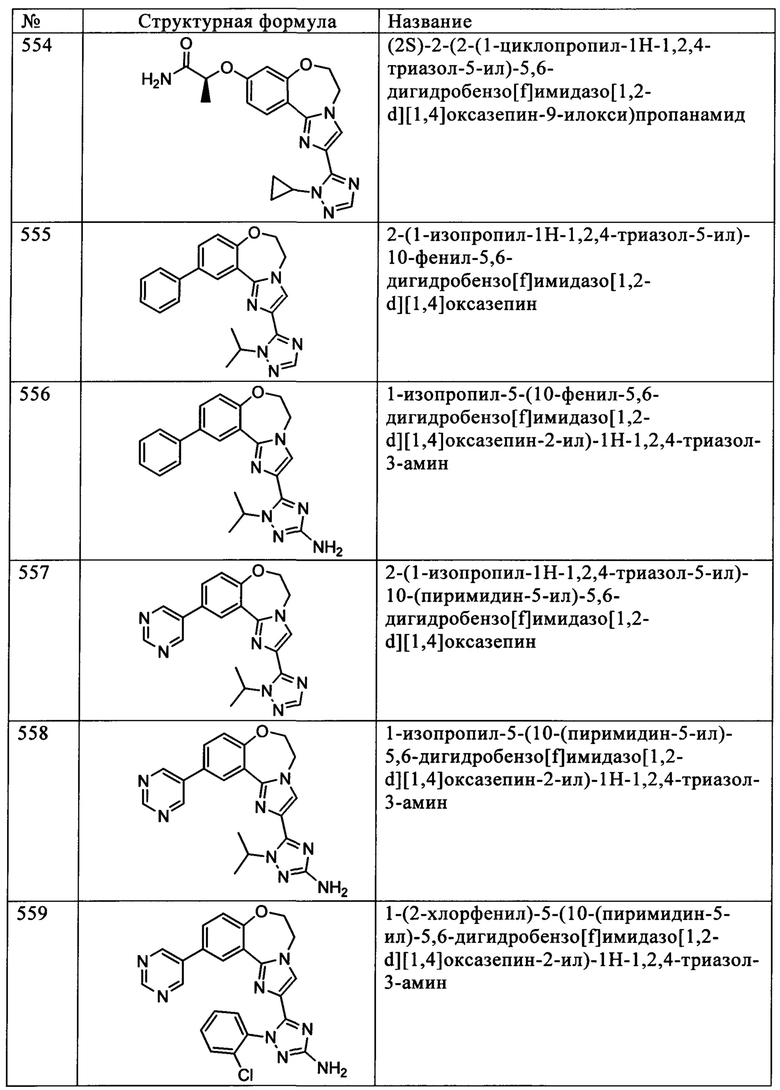
Типичные соединения формулы I (№№101-294 в табл. 1, №№295-533 в табл. 2 и №№534-570 в табл. 3), характеризующиеся следующими структурами и соответствующими названиями (ChemDraw Ultra, версия 9.0.1, CambridgeSoft Corp., Cambridge MA), получали, характеризовали и оценивали их ингибирующую активность в отношении P13Kα (IC50 р110α составляет менее 1 мкМ) и селективность способами по настоящему изобретению. Например,
соединение 101 характеризуется значением IC50, равным 0,77 мкМ, соединение 102 характеризуется значением IC50, равным 0,003 мкМ, соединение 103 характеризуется значением IC50, равным 0,058 мкМ, соединение 154 характеризуется значением IC50, равным 0,00091 мкМ, соединение 170 характеризуется значением IC50, равным 0,022 мкМ, соединение 171 характеризуется значением IC50, равным 0,00066 мкМ, соединение 180 характеризуется значением IC50, равным 0,00018 мкМ, соединение 200 характеризуется значением IC50, равным 0,0020 мкМ, соединение 248 характеризуется значением IC50, равным 0,00037 мкМ, соединение 251 характеризуется значением IC50; равным 0,0014 мкМ, соединение 253 характеризуется значением IC50, равным 0,0044 мкМ, а соединение 280 характеризуется значением IC50; равным 0,20 мкМ.































Соединения формулы I по изобретению можно вводить любым способом, пригодным для лечения состояния, подлежащего лечению. Пригодные способы включают пероральный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутриартериальный, внутрикожный, подоболочечный и эпидуральный), чрескожный, ректальный, назальный, местный (включая подщечный и подъязычный), вагинальный, внутрибрюшинный, внутрилегочный и внутриназальный. В случае иммунодепрессантного лечения соединения можно вводить внутрь очага, включая перфузию или иного типа контактирование трансплантата с ингибитором перед трансплантацией. Следует понимать, что предпочтительный способ введения может изменяться в зависимости, например, от состояния реципиента. Если соединение вводят перорально, его можно перерабатывать в пилюли, капсулы, таблетки и т.п. в смеси с фармацевтически приемлемым носителем или эксципиентом. Если соединение вводят парентерально, его можно перерабатывать в смеси с фармацевтически приемлемым носителем в стандартную лекарственную форму для инъекции, как описано ниже.
Доза соединения формулы I для лечения человека изменяется в интервале от приблизительно 10 мг до приблизительно 1000 мг. Типичная доза соединения составляет от приблизительно 100 мг до приблизительно 300 мг. Дозу можно вводить один раз в сут (1 р/с), два раза в сут (2 р/с), или более часто в зависимости от фармакокинетических и фармакодинамических свойств, включая абсорбцию, распределение, метаболизм и выведение конкретного соединения. Кроме того, на дозировку и режим введения оказывают влияние факторы токсичности. При пероральном введении пилюлю, капсулу или таблетку можно принимать ежедневно или менее часто в течение указанного периода времени. Курс можно повторять в течение ряда циклов лечения.
В другом объекте настоящего изобретения предлагаются способы профилактики или лечения гиперпролиферативного заболевания или нарушения, опосредованного киназами PI3, и указанные способы заключаются в том, что млекопитающему, нуждающемуся в указанном лечении, вводят эффективное количество соединения формулы I. Примеры такого гиперпролиферативного заболевания или нарушения включают, но не ограничиваясь только им, рак.
В еще одном объекте настоящего изобретения предлагаются способы профилактики или лечения гиперпролиферативного нарушения, и указанные способы заключаются в том, что млекопитающему, нуждающемуся в указанном лечении, вводят эффективное количество соединения формулы I, в отдельности или в комбинации с одним или более дополнительных соединений, характеризующихся антигиперпролиферативными свойствами.
В одном объекте настоящего изобретения предлагается способ применения соединения по настоящему изобретению для лечения у млекопитающего гиперпролиферативного заболевания или состояния, опосредованного киназой PI3.
В дополнительном объекте настоящего изобретения предлагается применение соединения по настоящему изобретению для лечения у млекопитающего рака, опосредованного киназой PI3.
Соединения формулы I по настоящему изобретению пригодны для лечения гиперпролиферативных заболеваний, состояний и/или нарушений, включая, но не ограничиваясь только ими, заболевания, характеризующиеся сверхэкспрессией липидкиназ, например, киназы PI3. Соответственно, в объекте настоящего изобретения предлагаются способы лечения или профилактики заболеваний или состояний, которые могут поддаваться лечению или профилактике при ингибировании липидкиназ, включая PI3. В одном варианте способ заключается в том, что млекопитающему, нуждающемуся в указанном лечении, вводят терапевтически эффективное количество соединения формулы I, или стереоизомера, геометрического изомера, таутомера или фармацевтически приемлемой соли указанного соединения. В другом варианте человеку, нуждающемуся в таком лечении, вводят соединение формулы I в смеси с фармацевтически приемлемым носителем, адъювантом или носителем, причем указанное соединение формулы I присутствует в количестве, достаточном для детектируемого ингибирования активности киназы PI3.
Соединения формулы I можно также использовать для лечения гиперпролиферативных заболеваний, характеризующихся сверхэкспрессией протеинкиназ, таких как протеинкиназы, кодируемые генами PIM, генами Pim-1, Pim-2 и Pim-3 (Proviral Insertion, Moloney), которые принимают участие в развитие лимфомы и солидных опухолей (Cuypers и др., Cell, т. 37 (1), cc. 141-150 (1984), Selten и др., EMBO J., т. 4 (7), cc. 1793-1798 (1985), van der Lugt и др., EMBO J., т. 14 (11), cc. 2536-2544 (1995), Mikkers и др., Nature Genetics, т. 32 (1), cc. 153-159 (2002), van Lohuizen и др., Cell, т. 65 (5), cc. 737-752 (1991)).
Рак, который можно лечить способами по настоящему изобретению, включает, но не ограничиваясь только ими, рак молочной железы, яичников, шейки матки, предстательной железы, яичка, мочеполового тракта, пищевода, гортани, глиобластому, нейробластому, рак желудка, кожи, кератоакантому, рак легких, плоско-клеточный рак, крупноклеточный рак, немелкоклеточный рак легких (НМРЛ), мелкоклеточную карциному, аденокарциному легких, рак костной ткани, ободочной кишки, аденому, рак поджелудочной железы, аденокарциному, рак щитовидной железы, фолликулярную карциному, недифференцированную карциному, папиллярную карциному, семиному, меланому, саркому, карциному мочевого пузыря, карциному печени и желчных протоков, карциному почек, миелоидные нарушения, лимфоидные нарушения, волосистоклеточный рак, рак ротовой полости и глотки (пероральный), рак губы, языка, рак полости рта, глотки, тонкой кишки, колоректальный рак, рак толстой кишки, прямой кишки, рак мозга и центральной нервной системы, болезнь Ходжкина и лейкоз.
Соединения формулы I можно использовать для диагностики или обработки in vitro, in situ и in vivo клеток млекопитающих, для лечения организмов или сопутствующих патологических состояний, таких как системное и очаговое воспаление, иммуновоспалительные заболевания, такие как ревматоидный артрит, подавление иммунитета, отторжение трансплантированных органов, аллергия, язвенный колит, болезнь Крона, дерматит, астма, системная красная волчанка, синдром Шегрена, рассеянный склероз, склеродермия/системный склероз, идиопатическая тромбоцитопеническая пурпура (ИТП), васкулит, связанный с антинейтрофильными цитоплазматическими антителами (АНЦА), хроническое обструктивное заболевание легких (ХОЗЛ), псориаз, а также для общего совместного защитного действия.
Соединения формулы I можно использовать для лечения состояний мозга и центральной нервной системы, при лечении которых требуется перенос через гематоэнцефалический барьер. Определенные соединения формулы I характеризуются благоприятными свойствами для доставки в мозг. Мозговые нарушения, которые можно эффективно лечить соединениями формулы I, включают метастатические и первичные опухоли мозга, такие как глиобластома и меланома.
Соединения формулы I можно использовать для лечения офтальмологических нарушений при направленной доставке в глаза. Определенные соединения формулы I характеризуются благоприятными свойствами для доставки в глаза.
В другом объекте настоящего изобретения предлагается соединение по настоящему изобретению, предназначенное для применения при лечении заболеваний или состояний, описанных в данном контексте, у млекопитающего, например, человека, страдающего от указанного заболевания или состояния. Предлагается также применение соединения по настоящему изобретению для получения лекарственного средства, предназначенного для лечения заболеваний и состояний, описанных в данном контексте, у теплокровного животного, такого как млекопитающее, например, человек, страдающего от указанного нарушения.
Чтобы использовать соединения формулы I для терапевтического лечения (включая профилактическое лечение) млекопитающих, включая человека, указанное соединение обычно перерабатывают в фармацевтическую композицию в соответствии со стандартной фармацевтической практикой. В указанном объекте настоящего изобретения предлагается фармацевтическая композиция, включающая соединение по настоящему изобретению в смеси с фармацевтически приемлемым разбавителем или носителем.
В другом объекте настоящего изобретения предлагается фармацевтическая композиция, включающая бензоксазепин формулы I и фармацевтически приемлемый носитель. Фармацевтическая композиция дополнительно включает один или более дополнительных терапевтических агентов.
Типичную композицию получают при смешивании соединения формулы I с носителем, разбавителем или эксципиентом. Пригодные носители, разбавители и эксципиенты известны специалистам в данной области техники и включают соединения, такие как углеводороды, воски, водорастворимые и/или набухающие в воде полимеры, гидрофильные или гидрофобные соединения, желатин, масла, растворители, вода и т.п. Конкретный используемый носитель, разбавитель или эксципиент зависит от способа введения и назначения соединения по настоящему изобретению. Растворители обычно выбирают из растворителей, признанных специалистами в данной области техники безопасными (признанных полностью безвредными, GRAS) для введения млекопитающему. В основном безопасными растворителями являются нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или смешиваются с водой. Пригодные водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, ПЭГ 400, ПЭГ 300) и т.п., а также их смеси. Композиции включают также одно или более буферных веществ, стабилизаторов, ПАВ, смачивающих агентов, смазывающих веществ, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, замутнителей, скользящих веществ, агентов для повышения технологичности, красителей, подсластителей, ароматизаторов, вкусовых добавок и других известных добавок, чтобы обеспечить привлекательный внешний вид лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или для упрощения способа получения фармацевтического продукта (т.е. лекарственного средства).
Композиции можно получить с использованием стандартных методик растворения и смешивания. Например, нерасфасованное лекарственное соединение (т.е. соединение по настоящему изобретению или стабилизированную форму соединения формулы I (например, комплекс с производным циклодекстрина или другим известным комплексообразователем)) растворяют в пригодном растворителе в присутствии одного или более эксципиентов, описанных выше. Соединение по настоящему изобретению обычно перерабатывают в фармацевтическую лекарственную форму, чтобы упростить контролируемую дозировку лекарственного средства и повысить согласие пациента с назначенным курсом лечения.
Фармацевтическую композицию (или состав) определенного назначения упаковывают различными способами в зависимости от способа, используемого для введения лекарственного средства. Как правило, изделие для распространения включает контейнер, который содержит фармацевтическую композицию в соответствующей форме. Пригодные контейнеры известны специалистам в данной области техники и включают тару, такую как флаконы (пластиковые и стеклянные), пакетики, ампулы, пластиковые мешки, металлические пробирки и т.п. Контейнер может содержать также устройство для предотвращения недозволенного доступа к содержимому упаковки. Кроме того, на контейнер наносят этикетку с описанием содержимого контейнера. Этикета содержит также соответствующие предостережения.
Фармацевтические композиции соединений по настоящему изобретению получают для различных способов и типов введения. Например, соединение формулы I, характеризующееся требуемой степенью чистоты, необязательно смешивают с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences, 16ое изд., Osol А., ред. (1980)) в форме лиофилизованного состава, измельченного порошка, или водного раствора. Композицию получают при смешивании при температуре окружающей среды при соответствующем значении рН и при требуемой степени чистоты с физиологически приемлемыми носителями, т.е. носителями, которые являются нетоксичными для реципиентов при используемых дозировках и концентрациях. Значение рН композиции зависит в основном от конкретного применения и концентрации соединения, но может изменяться от приблизительно 3 до приблизительно 8. Пригодным вариантом является композиция в ацетатном буферном растворе при рН 5.
Соединение по настоящему изобретению, предназначенное для применения по настоящему изобретению, предпочтительно является стерильным. Прежде всего, стерильными являются композиции, предназначенные для введения in vivo. Указанную стерилизацию проводят при фильтровании через стерильные фильтрационные мембраны.
Соединение обычно хранят в виде твердой композиции, лиофилизованного состава или в виде водного раствора.
Фармацевтические композиции по настоящему изобретению, включающие соединение формулы I, получают, дозируют и вводят в количествах, при концентрациях, режимами, курсом лечения, в смеси с носителями и с использованием способа введения в соответствии с общепринятой медицинской практикой. Рассматриваемые в связи с этим факторы включают конкретное нарушение, подлежащее лечению, конкретное млекопитающее, нуждающееся в таком лечении, клиническое состояние конкретного пациента, причину нарушения, участок доставки агента, способ введения, режим введения, а также другие факторы, известные практикующим врачам. Термин «фармацевтически эффективное количество» соединения, предназначенного для введения, определяется с учетом указанных факторов и обозначает минимальное количество, необходимое для профилактики, снижения интенсивности симптомов или лечения нарушения, опосредованного факторами свертывания крови. Указанное количество предпочтительно должно быть меньше количества, которое является токсическим для организма-хозяина или повышает предрасположенность организма-хозяина к кровотечению.
В основном предполагается, что исходное фармацевтически эффективное количество соединения формулы I, дозу которого вводят парентерально, составляет величину в диапазоне приблизительно 0,01-100 мг/кг, а именно приблизительно от 0,1 мг/кг до 20 мг/кг массы пациента в сут, при этом типичная исходная доза используемого соединения составляет от 0,3 мг/кг/сут до 15 мг/кг/сут.
Пригодные разбавители, носители, эксципиенты и стабилизаторы являются нетоксичными для реципиентов при используемых дозировках и концентрациях и включают буферные вещества, такие как фосфат, цитрат и соли других органических кислот, антиоксиданты, включая аскорбиновую кислоту и метионин, консерванты (такие как хлорид октадецилдиметилбензиламмония, хлорид гексаметония, хлорид бензалкония, хлорид бензетония, фенол, бутиловый или бензиловый спирт, алкилпарабены, такие как метил- или пропилпарабен, катехол, резорцин, циклогексанол, 3-пентанол и мета-крезол), низкомолекулярные (менее приблизительно 10 остатков) полипептиды, белки, такие как сывороточный альбумин, желатин или иммуноглобулины, гидрофильные полимеры, такие как поливинилпирролидон, аминокислоты, такие как глицин, глютамин, аспарагин, гистидин, аргинин или лизин, моносахариды, дисахариды и другие углеводороды, включая глюкозу, маннозу или декстрины, хелатирующие агенты, такие как ЭДТУ, сахара, такие как сахароза, маннит, трегалоза или сорбит, солеобразующие противоионы, такие как натрий, комплексы металлов (например, цинк-белковые комплексы), и/или неионогенные ПАВ, такие как TWEEN™, PLURONICS™ или полиэтиленгликоль (ПЭГ). Активные фармацевтические ингредиенты включают также в микрокапсулы, полученные, например, по методикам коацервации или межфазной полимеризацией, например, гидроксиметилцеллюлозные или желатиновые микрокапсулы и микрокапсулы из поли(метилметакрилата), соответственно, в коллоидные системы для доставки лекарственных средств (например, в липосомы, альбуминовые микросферы, микроэмульсии, наночастицы и нанокапсулы) или в макроэмульсии. Указанные методики описаны в книге Remington's Pharmaceutical Sciences, 16ое изд., Osol А., ред. (1980).
Можно получить препараты с замедленным высвобождением соединений формулы I. Пригодные примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащие соединение формулы I, при этом матрицы перерабатывают в формованные изделия, например, в пленки или микрокапсулы. Примеры матриц с замедленным высвобождением включают сложные полиэфиры, гидрогели (например, на основе поли(2-гидроксиэтилметакрилата), или поли(винилового спирта)), полилактиды (US 3773919), сополимеры L-глютаминовой кислоты и γ-этил-L-глютамата, недеградируемый этиленвинилацетат, деградируемые сополимеры молочной кислоты и гликолевой кислоты, такие как LUPRON DEPOT™ (инъецируемые микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и лейпролид ацетата), и поли-D-(-)-3-гидроксимасляную кислоту.
Композиции можно использовать в способах введения, подробно описанных в данном контексте. Композиции перерабатывают в стандартную лекарственную форму и получают любыми способами, известными в области фармацевтики. Методики и композиции в основном описаны в книге Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA). Указанные способы включают стадию комбинирования активного ингредиента с носителем, который включает один или более дополнительных ингредиентов. Как правило, композиции получают, равномерно и тщательно смешивая активный ингредиент с жидкими носителями или тонко дисперсными твердыми носителями или с обоими указанными типами носителей, и затем при необходимости перерабатывают в требуемую форму продукта.
Композиции соединения формулы I, пригодные для перорального введения, получают в виде дискретных лекарственных форм, таких как пилюли, капсулы, пакетики или таблетки, каждая из которых содержит предварительно определенное количество соединения формулы I.
Прессованные таблетки получают прессованием в пригодной машине активного ингредиента в свободно текущей форме, такой как порошок или гранулы, необязательно в смеси со связующим, смазывающим веществом, инертным разбавителем, консервантом, ПАВ или диспергирующим агентом. Формованные таблетки получают формованием в пригодной машине смеси порошкообразного активного ингредиента, смоченного инертным жидким разбавителем. На таблетки необязательно наносят покрытие или насечки и необязательно получают в форме с медленным или контролируемым высвобождением активного ингредиента.
Для перорального применения получают таблетки, пастилки, лепешки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, например, желатиновые капсулы, сиропы или эликсиры. Композиции соединений формулы I, предназначенные для перорального применения, получают любым способом, известным в области получения фармацевтических композиций, и указанные композиции включают один или более агентов, включая подсластители, ароматизаторы, красители и консерванты, чтобы обеспечить привлекательный вкус препарата. Приемлемыми являются таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым эксципиентом, который пригоден для получения таблеток. Указанными эксципиентами являются, например, инертные разбавители, такие как карбонат кальция или натрия, лактоза, фосфат кальция или натрия, агенты для грануляции и дезинтегрирующие агенты, такие как маисовый крахмал или альгиновая кислота, связующие агенты, такие как крахмал, желатин или аравийская камедь, а также смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк. Таблетки получают без покрытия или на таблетки наносят покрытие по известным методикам, включая микроинкапсулирование, чтобы замедлить распадаемость и адсорбцию в желудочно-кишечном тракте и таким образом обеспечить замедленное действие в течение более продолжительного периода. Например, можно использовать материал для замедления времени высвобождения активного ингредиента, такое как моностеарат глицерина или дистеарат глицерина в отдельности или в смеси с воском.
Для лечения глаз или других внешних тканей, например, ротовой полости и кожи, композиции используют в виде мази или крема для местного применения, содержащих активный ингредиент(ы) в количестве, например, от 0,075 мас. % до 20 мас. %. Если композиция представлена в виде мази, активные ингредиенты применяют в смеси с парафиновой или смешивающейся с водой основой мази. В другом варианте активные ингредиенты перерабатывают в крем в смеси с основой крема типа масло-в-воде.
При необходимости водная фаза основы крема включает многоатомный спирт, т.е. спирт, содержащий две или более гидроксильные группы, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая ПЭГ 400), а также их смеси. Композиции для местного применения предпочтительно включают соединение, которое повышает абсорбцию или проницаемость активного ингредиента через кожу или другие обрабатываемые области. Примеры указанных агентов, повышающих проницаемость через кожу, включают ДМСО и родственные аналоги.
Масляную фазу эмульсий по настоящему изобретению получают известным способом из известных ингредиентов. Хотя указанная фаза включает только эмульгатор, предпочтительно, чтобы указанная фаза содержала смесь по крайней мере одного эмульгатора с жиром или маслом, или и с обоими, жиром и маслом. Гидрофильный эмульгатор предпочтительно добавляют наряду с липофильным эмульгатором, который действует в качестве стабилизатора. Предпочтительно также включать оба агента, и масло, и жир. Вместе эмульгатор(ы) в присутствии или в отсутствие стабилизатора(ов) составляют так называемый эмульгирующий воск, а воск вместе с маслом и жиром составляют так называемую эмульгирующую основу мази, которая образует маслодиспергируемую фазу композиций крема. Эмульгаторы и стабилизаторы эмульсии, пригодные для применения в композиции по настоящему изобретению, включают Tween® 60, Span® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, моностеарат глицерина и лаурилсульфат натрия.
Водные суспензии соединений формулы I содержат активные соединения в смеси с эксципиентами, пригодными для получения водных суспензий.
Указанные эксципиенты включают суспендирующий агент, такой как карбоксиметилцеллюлоза, кроскармеллоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь, а также диспергирующие или смачивающие агенты, такие как природный фосфатид (например, лецитин), продукт конденсации алкиленоксида с жирной кислотой (например, стеарат полиоксиэтилена), продукт конденсации этиленоксида с длинноцепочечным алифатическим спиртом (например, гептадекаэтиленоксицетанолом), продукт конденсации этиленоксида с частичным сложным эфиром, полученным из жирной кислоты и ангидрида гекситола (например, моноолеат полиоксиэтиленсорбита). Водная суспензия включает также один или более консервантов, таких как этил- или н-пропил-пара-гидроксибензоат, один или более красителей, один или более ароматизаторов, а также один или более подсластителей, таких как сахароза или сахарин.
Фармацевтические композиции соединений формулы I могут представлять собой стерильный инъецируемый препарат, такой как стерильная инъецируемая водная или масляная суспензия. Указанную суспензию получают, как известно в данной области техники, с использованием упомянутых выше пригодных диспергирующих или смачивающих агентов, а также суспендирующих агентов. Стерильным инъецируемым препаратом является также стерильный инъецируемый раствор или стерильная инъецируемая суспензия в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле, или препарат, полученный в виде лиофилизованного порошка. Приемлемыми носителями и растворителями, которые можно использовать, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды можно использовать стандартные стерильные нелетучие масла. Для указанной цели можно использовать любое легкое масло, включая синтетические моно- или диглицериды. Кроме того, при получении инъецируемых препаратов аналогичным образом можно использовать жирные кислоты, такие как олеиновая кислота.
Количество активного ингредиента, которое можно комбинировать с носителем для получения единой лекарственной формы, изменяется в зависимости от организма-хозяина, нуждающегося в лечении, и конкретного способа введения. Например, композиция с замедленным высвобождением, предназначенная для перорального введения человеку, содержит приблизительно от 1 мг до 1000 мг активного ингредиента, смешанного с соответствующим и пригодным количеством носителя, которое изменяется от приблизительно 5 мас. % до приблизительно 95 мас. % в расчете на общую массу композиций. Получают также фармацевтическую композицию, которую можно простым способом отмерять для введения. Например, водный раствор, предназначенный для внутривенного вливания, может содержать приблизительно от 3 мкг до 500 мкг активного ингредиента в мл раствора, чтобы обеспечить вливание пригодного объема при скорости приблизительно 30 мл/ч.
Композиции, пригодные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые содержат антиоксиданты, буферные вещества, бактериостатические агенты и растворенные вещества, которые придают композиции изотоничность в отношении к крови предполагаемого реципиента, а также водные и неводные стерильные суспензии, которые включают суспендирующие агенты и загустители.
Композиции, пригодные для местного применения в глаза, включают также глазные капли, где активный ингредиент растворен или суспендирован в пригодном носителе, прежде всего в водном растворителе для активного ингредиента. Активный ингредиент предпочтительно присутствует в указанных композициях при концентрации приблизительно от 0,5 мас. % до 20 мас. %, например, приблизительно от 0,5 мас. % до 10 мас. %, например, приблизительно 1,5 мас. %.
Композиции, пригодные для местного применения в полости рта, включают лепешки, содержащие активный ингредиент в ароматизированной основе, обычно сахарозе и аравийской камеди или трагаканте, пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин, или сахароза или аравийская камедь, и ополаскиватели для полости рта, содержащие активный ингредиент в пригодном жидком носителе.
Композиции, предназначенные для ректального введения, представляют собой суппозитории в смеси с пригодной основой, содержащей, например, масло какао или салицилат.
Композиции, пригодные для внутрилегочного или назального введения, характеризуются размером частиц, например, в диапазоне от 0,1 мкм до 500 мкм (включая размеры частицы в диапазоне от 0,1 мкм до 500 мкм с шагом, таким как 0,5 мкм, 1 мкм, 30 мкм, 35 мкм и т.п.), при этом указанные композиции вводят быстрой ингаляцией через носовую полость или ингаляцией через полость рта, чтобы обеспечить доставку в альвеолярные мешочки. Пригодные композиции включают водные или масляные растворы активного ингредиента. Композиции, пригодные для введения в виде аэрозоля или сухого порошка, получают стандартными способами и доставляют в смеси с другими терапевтическими агентами, такими как соединения, используемые в настоящее время при лечении или профилактике описанных ниже нарушений.
Композиции, пригодные для вагинального введения, представляют собой вагинальные суппозитории, тампоны, кремы, гели, пасты, пены или композиции спреев, содержащие в дополнение к активному ингредиенту известные в данной области техники соответствующие носители.
Композиции можно упаковывать в стандартные контейнеры с одной дозой или множеством доз, например, в запаянные ампулы и закрытые флаконы, и их можно хранить в лиофилизированном виде, при этом непосредственно перед применением для инъекции требуется только добавление стерильного жидкого носителя, например, воды. Готовые инъекционные растворы и суспензии для немедленного применения получают из стерильных порошков, гранул и таблеток описанных выше типов. Предпочтительными композициями для стандартных лекарственных форм являются композиции, содержащие суточную дозу активного ингредиента или суточную суб-дозу, как указано выше, или ее соответствующую часть.
В настоящем изобретении кроме того предлагаются ветеринарные композиции, включающие по крайней мере один активный ингредиент, как определено выше, в смеси с ветеринарным носителем. Ветеринарными носителями являются соединения, пригодные для введения композиции, и они представляют собой твердые, жидкие или газообразные соединения, которые в других отношениях являются инертными или приемлемыми в ветеринарии и совместимы с активным ингредиентом. Указанные ветеринарные композиции вводят парентерально, перорально или другим требуемым способом.
Соединения формулы I можно использовать в отдельности или в комбинации с другими терапевтическими агентами для лечения заболевания или нарушения, описанного в данном контексте, такого как гиперпролиферативное нарушение (например, рак). В некоторых вариантах соединение формулы I комбинируют в составе фармацевтической комбинированной композиции или для курса лечения, такого как комбинированное лечение, со вторым соединением, которое характеризуется антигиперпролиферативными свойствами, или которое пригодно для лечения гиперпролиферативного нарушения (например, рака). Второе соединение в составе фармацевтической комбинированной композиции или курса лечения предпочтительно характеризуется дополнительной активностью в отношении соединения формулы I, при этом исключено взаимное неблагоприятное влияние указанных соединений. Указанные соединения соответственно присутствуют в комбинации в количествах, которые являются эффективными для указанной цели. В одном варианте, композиция по настоящему изобретению включает соединение формулы I в комбинации с химиотерапевтическим агентом, таким как агент, описанный в данном контексте.
Комбинированное лечение проводят в одновременном или последовательном режиме. При последовательном режиме введения комбинацию вводят два или более раз. Комбинированное введение включает совместное введение с использованием отдельных композиций или одной фармацевтической композиции, а также последовательное введение в любом порядке, при этом предпочтительно оба (или все) активных агента одновременно проявляют биологическую активность в течение некоторого периода времени.
Пригодными дозировками любого из упомянутых выше совместно вводимых агентов являются используемые в настоящее время дозировки и их можно снизить за счет комбинированного действия (синергизма) новых идентифицированных агентов и других химиотерапевтических агентов или курсов лечения.
Комбинированное лечение может обеспечить «синергизм» и оказывать «синергетическое» действие, т.е. лечение, в ходе которого действие, достигаемое при совместном применении активных ингредиентов, превосходит суммарное действие, которое является результатом применения соединений в отдельности. Синергетическое действие достигается, когда активные ингредиенты: (1) получают в виде совместного состава и вводят или доставляют одновременно в виде комбинированной стандартной лекарственной формы, (2) доставляют поочередно или параллельно в виде отдельных композиций, или (3) применяют в ходе других курсов лечения. При доставке в ходе поочередного лечения синергетическое действие достигается, когда соединения вводят или доставляют последовательно, например, различными инъекциями из отдельных шприцев, в виде отдельных пилюль или капсул, или отдельными вливаниями. В основном, в ходе поочередного лечения эффективную дозировку каждого активного ингредиента вводят последовательно, т.е. сериями, в то время как при комбинированном лечении эффективные дозы двух или более активных ингредиентов вводят совместно.
В конкретном варианте противоракового лечения соединение формулы I, или стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемую соль или пролекарство указанного соединения можно комбинировать с другими химиотерапевтическими, гормональными агентами или антителами, такими как описано в данном контексте, а также комбинировать с хирургическим вмешательством и лучевой терапией. В связи с этим, курсы комбинированного лечения по настоящему изобретению включают введение по крайней мере одного соединения формулы I, или стереоизомера, геометрического изомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли или пролекарства указанного соединения и применение по крайней мере одного другого способа лечения рака. Для обеспечения требуемого комбинированного терапевтического действия выбирают количества соединения(й) формулы I и другого фармацевтически активного химиотерапевтического агента(ов), а также относительные сроки введения.
В объем настоящего изобретения включены также описанные в данном контексте метаболические продукты формулы I in vivo. Указанные продукты образуются в результате, например, окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, омыления, ферментативного расщепления и т.п. введенного соединения. Соответственно, настоящее изобретение включает метаболиты соединений формулы I, включая соединения, образующиеся при контактировании соединения по настоящему изобретению с организмом млекопитающего в течение периода времени, достаточного для образования метаболического продукта указанного соединения.
Метаболические продукты, как правило, идентифицируют, получая радиоактивно меченное (например, 14С или 3Н) изотопом соединение по настоящему изобретению, вводя его парентерально в детектируемой дозе (например, приблизительно более 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, выдерживая в течение времени, достаточного для метаболизма (обычно приблизительно от 30 с до 30 ч) и выделяя продукты превращения соединения из мочи, крови или других биологических образцов. Простота выделения указанных продуктов обусловлена присутствием метки (другие соединения выделяют при использовании антител, способных связывать эпитопы, которые сохраняются в метаболите). Структуры метаболитов определяют стандартным способом, например, методами анализа МС, ЖХ/МС или ЯМР. В основном, анализ метаболитов проводят тем же способом, как и стандартные исследования метаболизма лекарственных средств, известные специалистам в данной области техники. Метаболические продукты, при условии, что они иным способом не обнаружены in vivo, можно использовать при диагностических анализах для терапевтической дозировки соединений по настоящему изобретению.
В другом варианте осуществления настоящего изобретения предлагается изделие, или «набор», содержащий соединения, пригодные для лечения описанных выше заболеваний и нарушений. Набор включает контейнер, содержащий соединение формулы I. Набор может дополнительно содержать этикетку или листок-вкладыш, или они прикрепляются к контейнеру. Использованный термин «листок-вкладыш» обозначает инструкции, обычно включаемые в коммерческие упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях, и/или предостережения, касающиеся применения указанных терапевтических продуктов. Пригодные контейнеры включают, например, флаконы, бутыли, шприцы, блистерную упаковку и т.п. Контейнер может быть изготовлен из различных материалов, таких как стекло или пластик. Контейнер может содержать соединение формулы I или II или их композицию, которая является эффективной для лечения состояния, а также может быть снабжен стерильным входным отверстием (например, контейнером является пакет для внутривенного раствора или флакон, снабженный мембраной, прокалываемой иглой для подкожной инъекции). По крайней мере одним активным соединением в составе композиции является соединений формулы I. На этикетке или листке-вкладыше указывается, что композиция предназначена для лечения выбранного состояния, такого как рак. Кроме того, на этикетке или листке-вкладыше указаны показания для пациента, нуждающегося в лечении, такие как гиперпролиферативное нарушение, нейродегенеративное нарушение, гипертрофия сердца, болевое состояние, мигрень или нейротравматическое нарушение или эпизод. В одном варианте на этикетке или листке-вкладыше указывается, что композицию, включающую соединение формулы I, можно использовать для лечения нарушения, являющегося результатом аномального клеточного роста. На этикетке или листке-вкладыше также указывается, что композицию можно использовать для лечения других нарушений. В другом варианте, или дополнительно, готовое изделие дополнительно может включать второй контейнер, содержащий фармацевтически приемлемый буферный раствор, такой как бактериостатическая вода для инъекций (БВДИ), фосфатно-солевой буферный раствор (ФСБ), раствор Рингера и раствор декстрозы. Второй контейнер дополнительно может содержать другие материалы, необходимые с коммерческой и потребительской точки зрения, включая другие буферные растворы, разбавители, фильтры, иглы и шприцы.
Набор дополнительно может содержать инструкции по введению соединения формулы I и, если присутствует, второй фармацевтической композиции. Например, если набор включает первую композицию, содержащую соединение формулы I, и вторую фармацевтическую композицию, набор дополнительно содержит инструкции по одновременному, последовательному или раздельному введению первой и второй фармацевтических композиций пациенту, нуждающемуся в указанном лечении.
В другом варианте наборы пригодны для доставки твердых пероральных форм соединения формулы I или II, таких как таблетки или капсулы. Указанный набор предпочтительно содержит множество стандартных лекарственных форм. Указанные наборы включат упаковку, в которой дозы расположены в порядке их рекомендованного применения. Примером указанного набора является «блистерная упаковка». Блистерные упаковки известны в тароупаковочной промышленности и широко используются для упаковки фармацевтических стандартных лекарственных форм. При необходимости прилагается памятка, например, в форме чисел, букв или других отличительных знаков, или прилагается календарь, на котором отмечены дни курса лечения, когда следует вводить дозы.
Согласно одному варианту набор может включать (а) первый контейнер, содержащий соединение формулы I, и необязательно (б) второй контейнер, содержащий вторую фармацевтическую композицию, при этом вторая фармацевтическая композиция содержит второе соединение с антигиперпролиферативной активностью. В другом варианте, или дополнительно, набор дополнительно включает третий контейнер, содержащий фармацевтически приемлемый буферный раствор, такой как БВДИ, ФСБ, раствор Рингера и раствор декстрозы. Третий контейнер может также включать другие материалы, необходимые с коммерческой и потребительской точки зрения, включая другие буферные растворы, разбавители, фильтры, иглы и шприцы.
В некоторых других вариантах, где набор включает композицию формулы I и второй терапевтический агент, набор может содержать контейнер для размещения отдельных композиций, такой как флакон с отделениями или пакет из фольги с отделениями, однако отдельные композиции можно помещать также в единый контейнер без отделений. Как правило, набор содержит инструкции по введению отдельных компонентов. Форма набора является прежде всего целесообразной, когда отдельные компоненты предпочтительно вводят в виде различных лекарственных форм (например, пероральных и парентеральных), с различными интервалами дозировки, или в случае, если по мнению лечащего врача необходимо титрование индивидуальных компонентов комбинации.
В другом объекте настоящего изобретения предлагаются наборы, включающие соединение формул I, контейнер и необязательно листок-вкладыш или этикетку с описанием лечения.
Бензоксазепины формулы I можно синтезировать способами, которые включают известные или аналогичные методики, прежде всего, как описано в настоящем изобретении. Исходные соединения обычно являются коммерческими продуктами таких фирм, как Aldrich Chemicals (Milwaukee, WI), или их получают с использованием способов, известных специалистам в данной области техники (например, получают способами, в основном описанными в книге Fieser Louis F. и Fieser Mary, Reagents for Organic Synthesis, тт. 1-23, Wiley, Нью-Йорк (1967-2006 изд.), или в книге Beilsteins Handbuch der organischen Chemie, 4, Aufl. ред. Springer-Verlag, Берлин, включая дополнения (или в режиме он-лайн в базе данных Beilstein)).
В некоторых вариантах соединения формулы I можно получать с использованием широко известных методик получения бензоксазепинов (Sekhar и др., Sulfur Letters, 9 (6), cc. 271-277 (1989), Katsura и др., J. Med. Chem., 43, cc. 3315-3321 (2000), Rueeger и др., Biorganic & Med. Chem. Letters, 14, cc. 2451-2457 (2004), Reiter и др., Biorganic & Med. Chem. Letters, 17, cc. 5447-5454 (2007), Banaszak и др., Tetrahedron Letters, 47, cc. 6235-6238 (2006)) и других гетероциклов, которые описаны в книге Comprehensive Heterocyclic Chemistry II, Katritzky и Rees, ред., Elsevier (1997), например, в т. 3, в Liebigs Annalen der Chemie, 9, cc. 1910-1916 (1985), Helvetica Chimica Acta, 41, cc. 1052-1060 (1958), Arzneimittel-Forschung, 40 (12), cc. 1328-1331 (1990).
Соединения формулы I можно получать в отдельности или в виде библиотек соединений, включающих по крайней мере 2, например, от 5 до 1000 соединений, или от 10 до 100 соединений. Библиотеки соединений формулы I можно получать с использованием комбинаторного принципа «расщепление и смешивание» или множеством параллельных синтезов в растворе или методом твердофазного синтеза, известным специалистам в данной области техники. В связи с этим, в другом объекте настоящего изобретения предлагается библиотека соединений, включающая по крайней мере 2 соединения, или их фармацевтически приемлемые соли.
В целях иллюстрации в разделе «Общие методики» приведены общие способы, которые можно использовать для получения соединений формулы I, a также важных промежуточных соединений. В разделах «Схемы» и «Примеры» приведено более подробное описание конкретных стадий реакций. Специалистам в данной области техники представляется очевидным, что для получения соединений по настоящему изобретению можно использовать другие способы синтеза. Хотя на схемах, в основных методиках и примерах указаны определенные исходные соединения и способы, для получения различных производных и/или изменения условий реакций их можно заменять на другие аналогичные исходные соединения и способы. Кроме того, многие соединения, полученные описанными ниже способами, можно затем модифицировать в соответствии с настоящим описанием с использованием стандартных методов синтеза, известных специалистам в данной области техники.
При получении соединений формулы I может потребоваться защита функциональной группы (например, первичного или вторичного амина) в составе промежуточных продуктов. Необходимость такой защиты изменяется в зависимости от природы функциональной группы и условий способов получения. Пригодные аминозащитные группы включают ацетил, трифторацетил, трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz) и 9-фторенилметиленоксикарбонил (Fmoc). Необходимость такой защиты определяет специалист в данной области техники. Общее описание защитных групп и их применения приведено в книге Greene Т.W., Protective Groups in Organic Synthesis, John Wiley & Sons, Нью-Йорк, 3е изд. (1999).
В способах получения соединений по настоящему изобретению продукты реакции предпочтительно отделяют один от другого и/или от исходных соединений. Требуемые продукты, полученные на каждой стадии или серий стадий, отделяют и/или очищают (в данном контексте отделяют) по известным в данной области техники методикам, при этом достигается требуемая степень гомогенности. Обычно указанное отделение включает многофазную экстракцию, кристаллизацию из растворителя или смеси растворителей, перегонку, сублимацию или хроматографию. Хроматография включает любое количество способов, включая, например, обращенно-фазную и нормально-фазную хроматографию, эксклюзионную, ионообменную хроматографию, способы и приборы для жидкостной хроматографии высокого, среднего и низкого давления, хроматографию в аналитическом масштабе, хроматографию с псевдодвижущимся слоем и препаративную тонкослойную или толстослойную хроматографию, а также методики тонкослойной хроматографии в аналитическом масштабе и экспресс-хроматографии.
Другой класс способов отделения включает обработку смеси реагентом, предназначенным для связывания или в другом случае для превращения отделяемого требуемого продукта, непрореагировавших исходных соединений, побочного продукта реакции или т.п. Указанные реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ионообменные среды или т.п. В другом варианте, реагентами являются кислоты в случае свободного основания, основания в случае свободной кислоты, связывающие реагенты, такие как антитела, связывающие белки, селективные хелатирующие агенты, такие как краун-эфиры, реагенты для ионной экстракции жидкость/жидкость или т.п.
Выбор соответствующих способов отделения зависит от природы используемых соединений. Например, температура кипения и молекулярная масса при перегонке и сублимации, присутствие или отсутствие полярных функциональных групп при хроматографии, стабильность соединений в кислотных и щелочных средах при многофазной экстракции и т.п. Специалист в данной области техники может выбрать методики для достижения требуемого отделения.
Диастереомерные смеси можно разделить на индивидуальные диастереомеры на основе их физико-химических различий способами, известными специалистам в данной области техники, такими как хроматография и/или фракционная кристаллизация. Энантиомеры можно разделить за счет превращения энантиомерной смеси в диастереомерную смесь при взаимодействии с соответствующим оптически активным соединением (например, хиральным вспомогательным соединением, таким как хиральный спирт или хлорангидрид кислоты Мошера), разделяя диастереомеры и превращая (например, при проведении гидролиза) индивидуальные диастереомеры в соответствующие чистые энантиомеры. Некоторые соединения по настоящему изобретению также представляют собой атропизомеры (например, замещенные биарилы) и включены в объем настоящего изобретения. Энантиомеры можно также разделить с использованием хиральной колонки ЖХВР.
Индивидуальный стереоизомер, например, энантиомер, в основном не содержащий его стереоизомера, можно получить при разделении рацемической смеси с использованием способа, такого как получение диастереомеров с использованием оптически активных разрешающих агентов (Eliel Е. и Wilen S., «Stereochemistry of Organic Compounds», John Wiley & Sons, Inc., Нью-Йорк (1994), Lochmuller С.H., J. Chromatogr., 113 (3), cc. 283-302 (1975)). Рацемические смеси хиральных соединений по настоящему изобретению можно разделить и выделить любым пригодным способом, включая: (1) получение ионных диастереомерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими способами, (2) получение диастереомерных соединений с реагентами, модифицирующими хиральные производные, разделение диастереомеров и превращение в чистые стереоизомеры, и (3) прямое выделение в значительной степени чистых или обогащенных стереоизомеров в хиральных условиях, см. «Drug Stereochemistry, Analytical Methods and Pharmacology», Wainer Irving W., ред., Marcel Dekker, Inc., Нью-Йорк (1993).
В способе (1) диастереомерные соли получают при взаимодействии энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и т.п. с асимметрическими соединениями, содержащим кислотную функциональную группу, такими как карбоновая кислота и сульфоновая кислота. Разделение диастереомерных солей проводят методами фракционной кристаллизации или ионной хроматографии. Для разделения оптических изомеров аминосоединений добавляют хиральные карбоновые или сульфоновые кислоты, такие как камфорсульфоновая кислота, винная кислота, миндальная кислота или молочная кислота, при этом получают диастереомерные соли.
В другом варианте в способе (2) субстрат, подлежащий разделению, взаимодействует с одним энантиомером хирального соединения, при этом образуется диастереомерная пара (Eliel Е. и Wilen S., «Stereochemistry of Organic Compounds», John Wiley & Sons, Inc., c. 322 (1994)). Диастереомерные соединения получают при взаимодействии асимметрических соединений с энантиомерно чистыми реагентами, модифицирующими хиральные производные, с такими как ментиловые производные, с последующим разделением диастереомеров и гидролизом, при этом получают чистый или обогащенный энантиомер. Способ определения оптической чистоты включает получение хиральных сложных эфиров, таких как ментиловый сложный эфир, например, (-)-ментилхлорформиат в присутствии основания, или сложного эфира Мошера, α-метокси-α-(трифторметил)фенилацетата (Jacob III., J. Org. Chem., 47, с. 4165 (1982)), рацемической смеси и определение присутствия двух атропизомерных энантиомеров или диастереомеров методом 1Н ЯМР. Стабильные диастереомеры атропизомерных соединений можно разделить и выделить нормально- и обращенно-фазной хроматографией с использованием методов разделения атропизомерных нафтилизохинолинов (WO 96/15111). В способе (3) рацемическую смесь двух энантиомеров можно разделить хроматографией с использованием хиральной стационарной фазы («Chiral Liquid Chromatography», Lough W.J., ред., Chapman and Hall, Нью-Йорк (1989), Okamoto, J. Chromatogr., 513, cc. 375-378 (1990)). Обогащенные или очищенные энантиомеры можно различить способами, используемыми для идентификации других хиральных молекул с асимметрическими атомами углерода, такими как оптическое вращение и круговой дихроизм.
Следующие схемы иллюстрируют возможные способы получения соединений по настоящему изобретению.
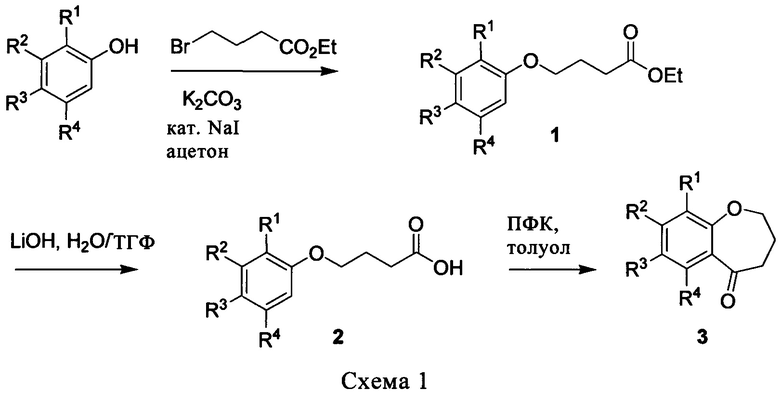
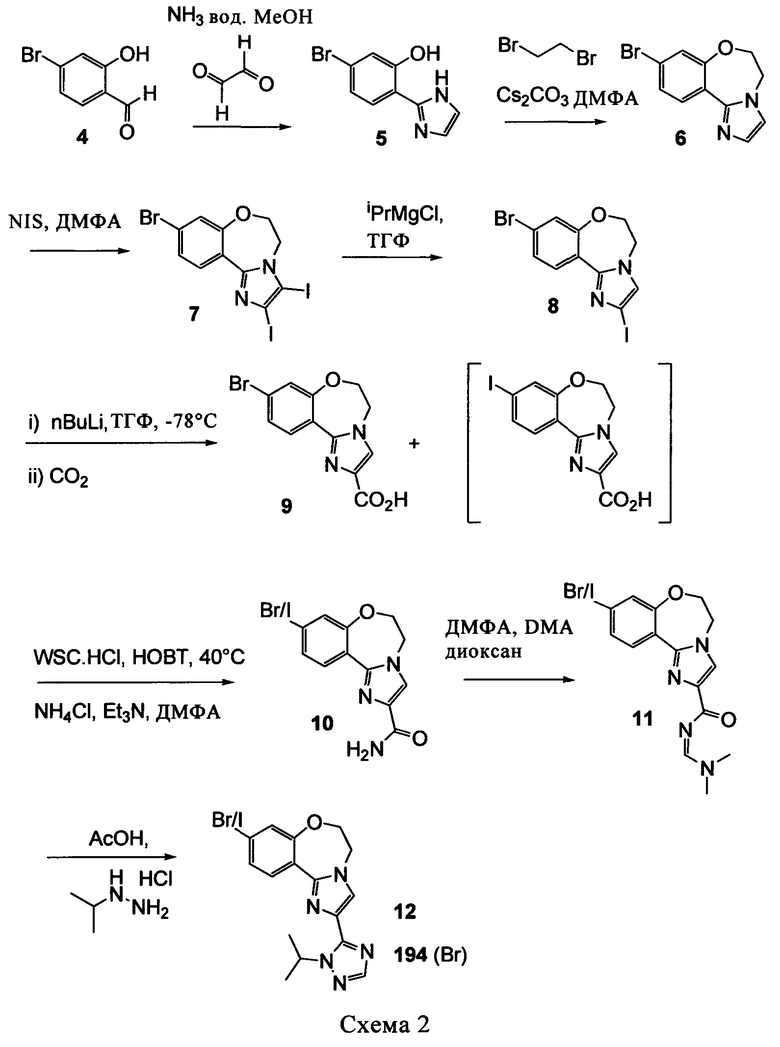
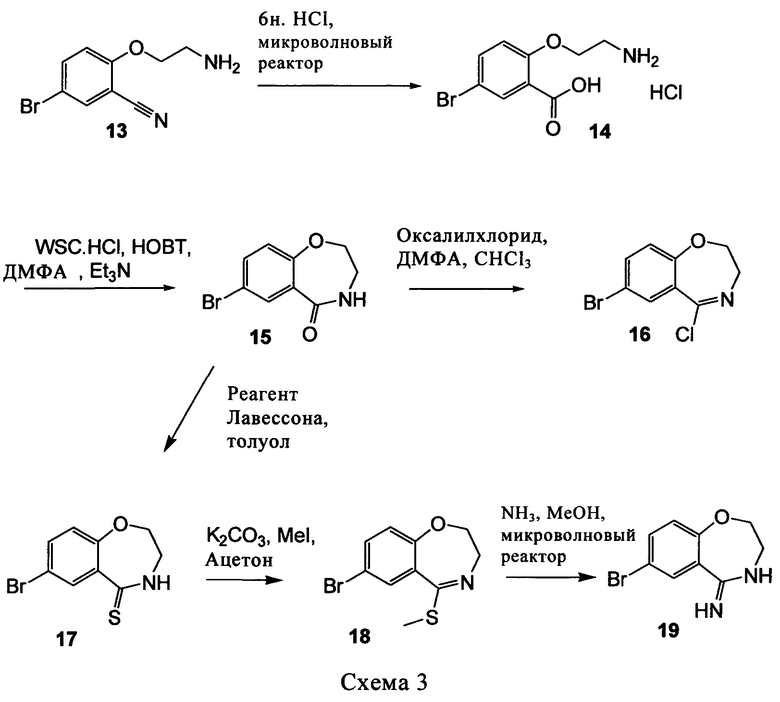
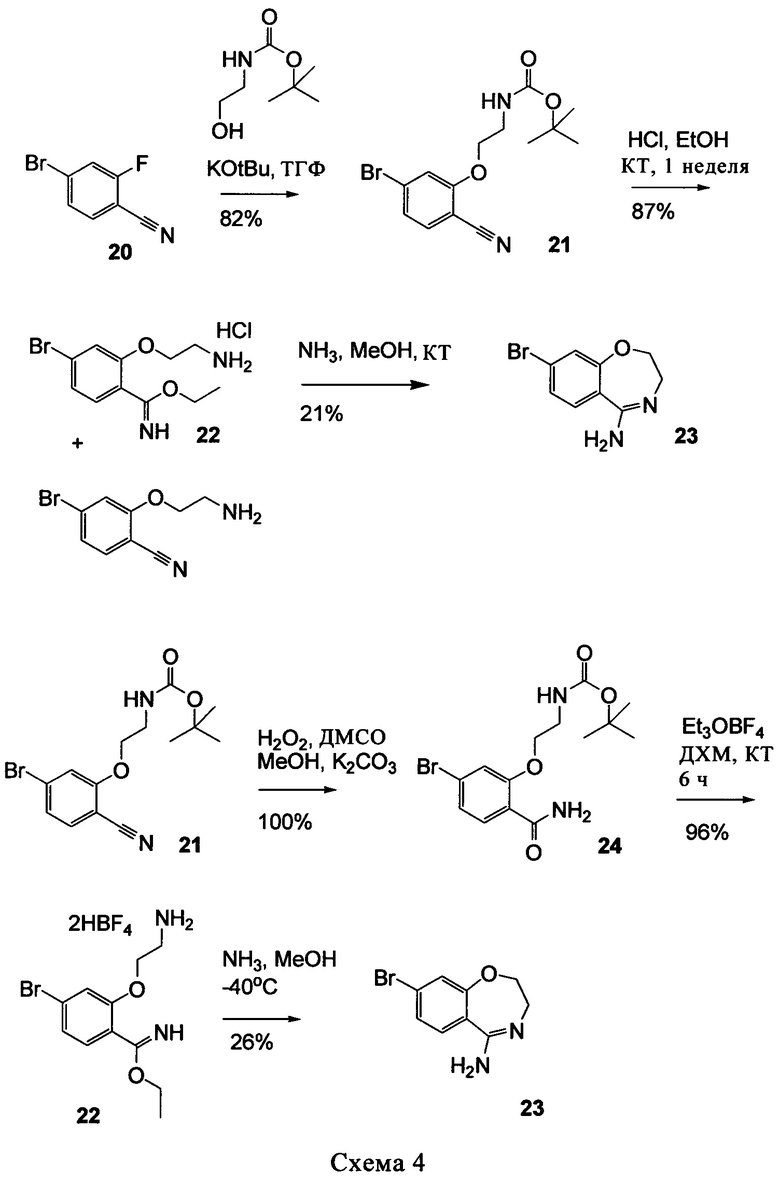
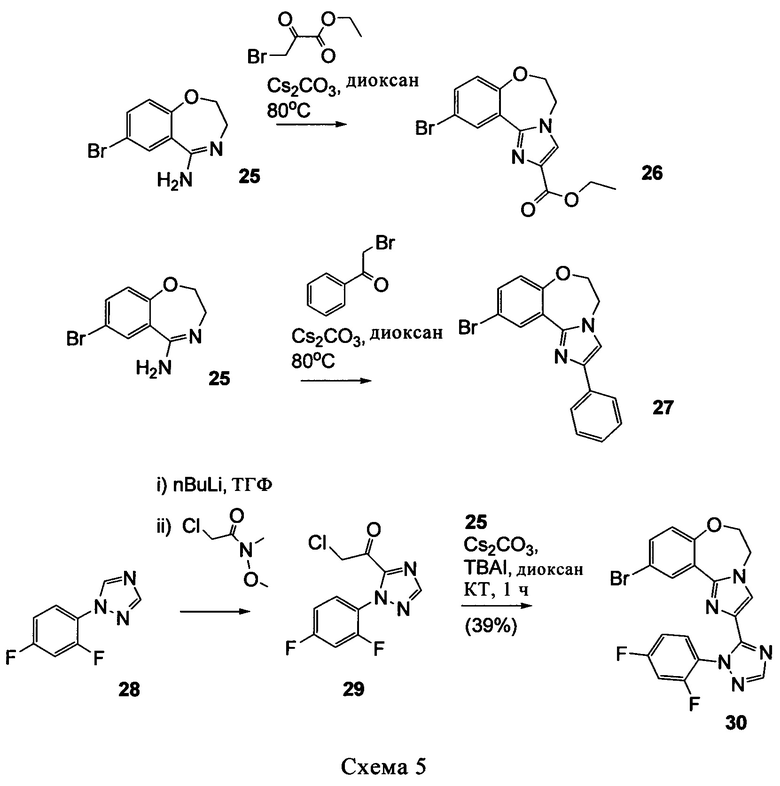
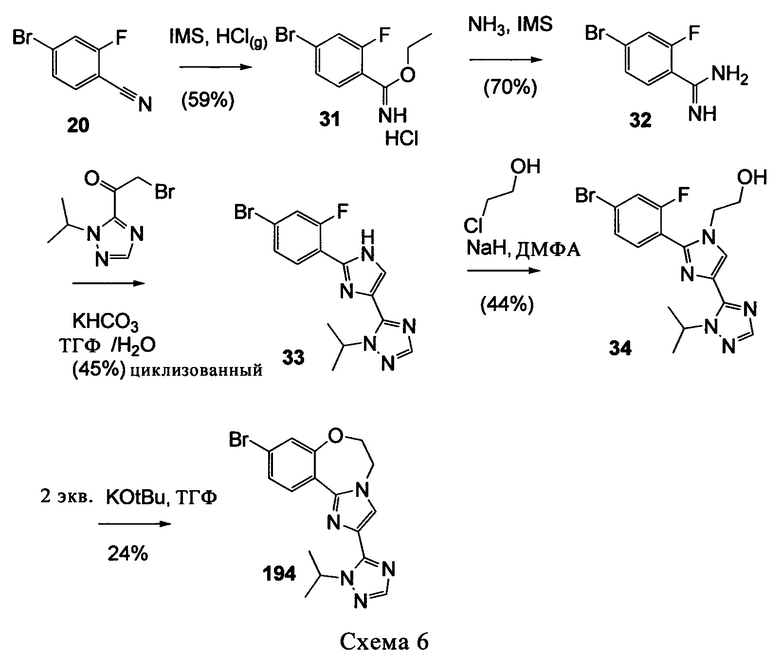

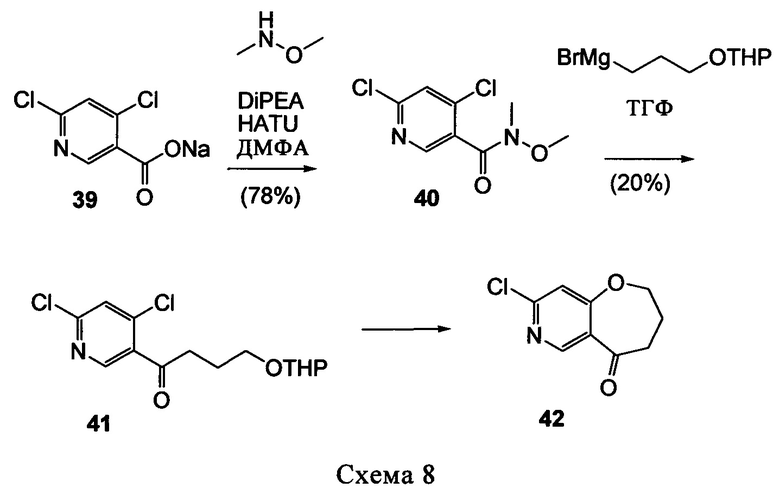
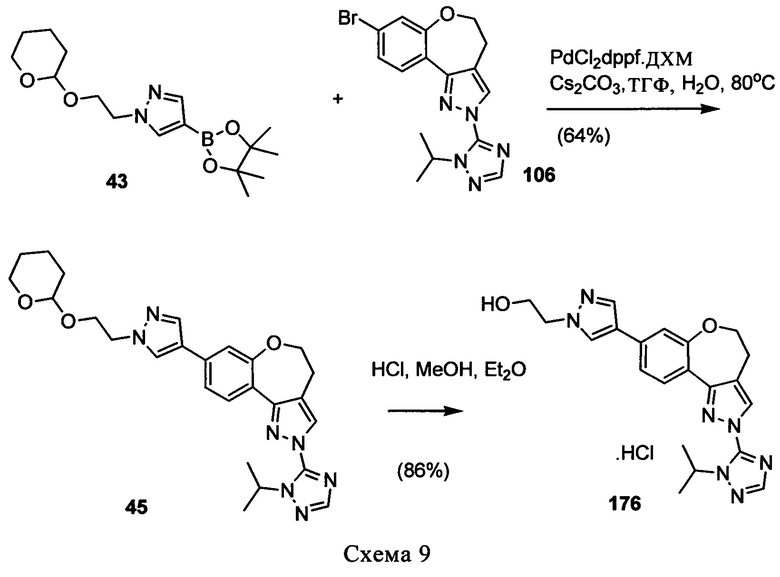
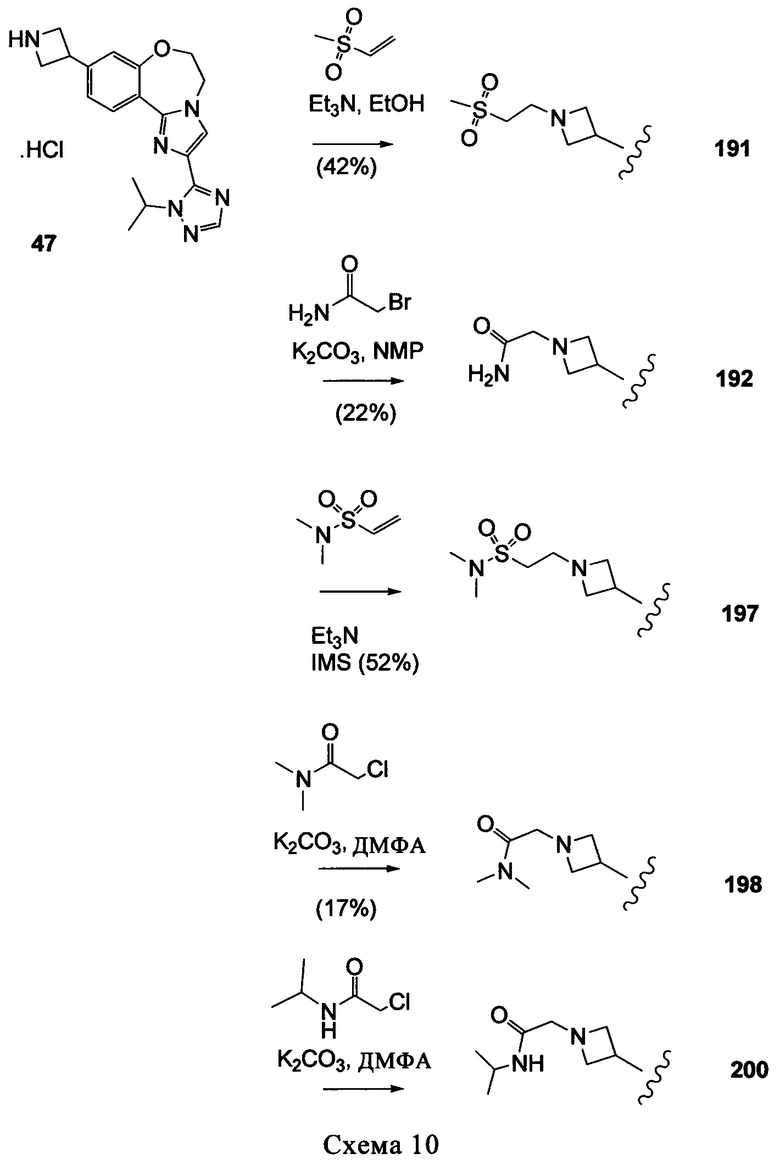
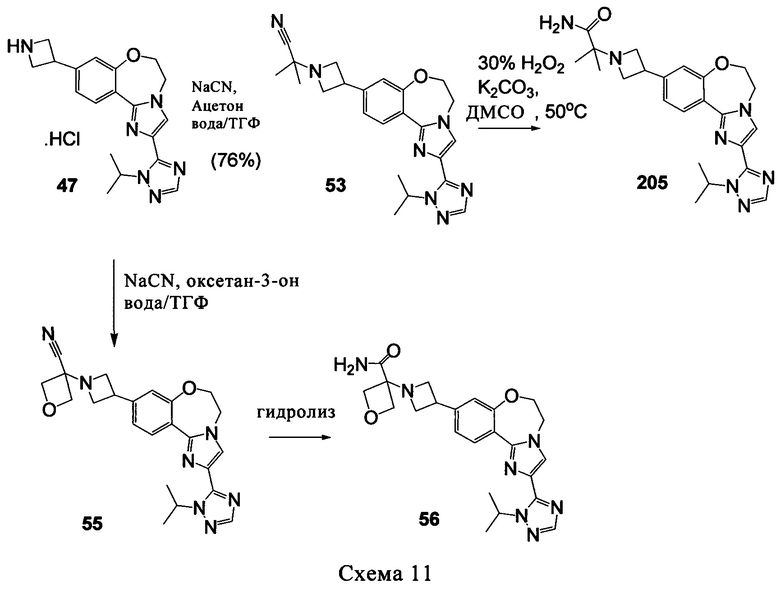


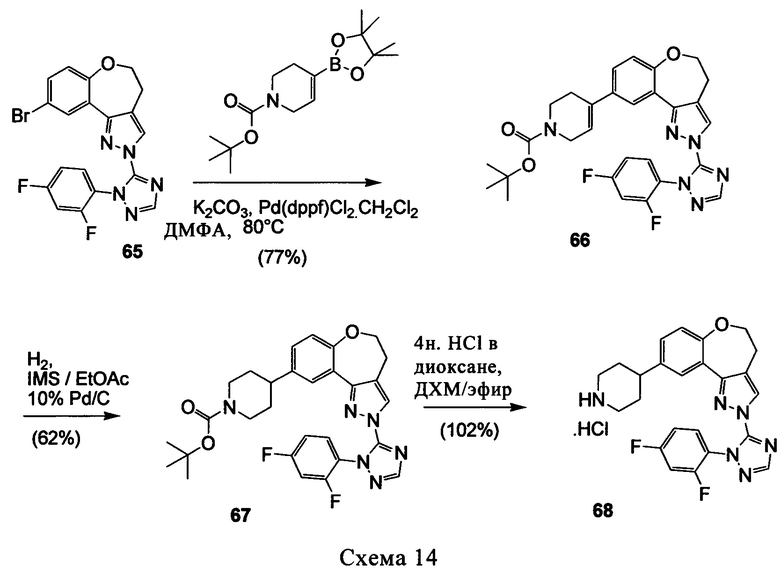
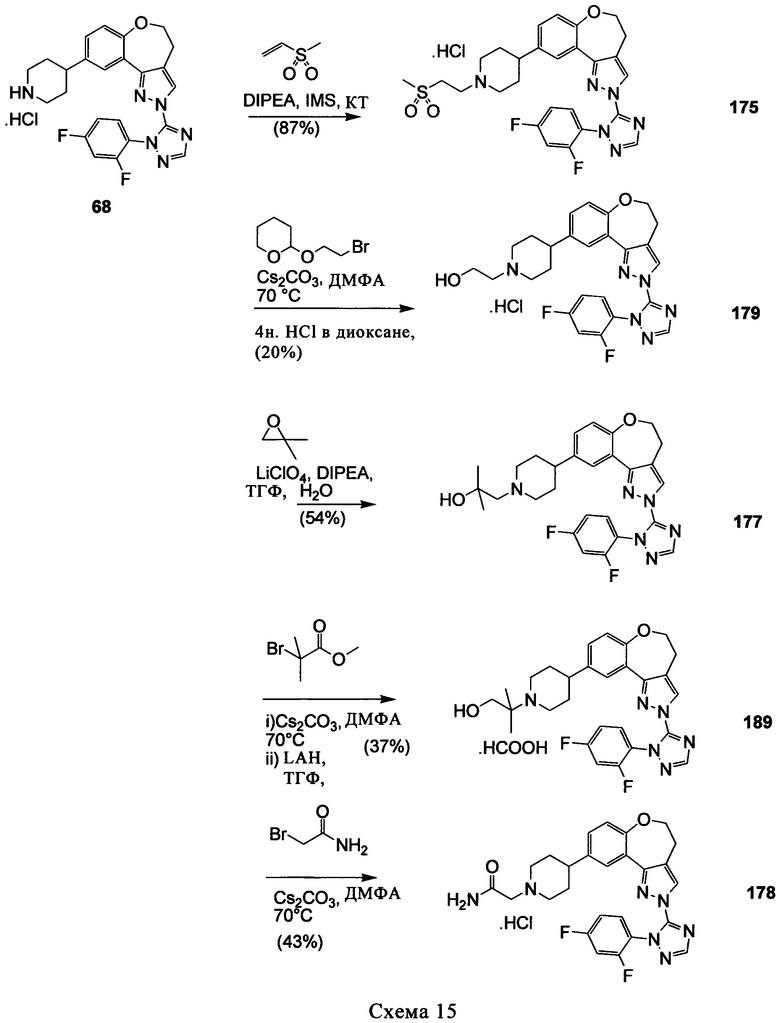
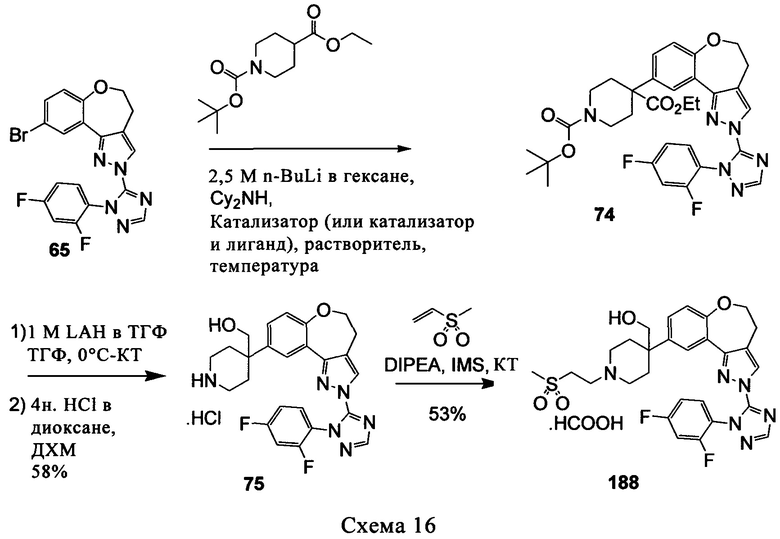
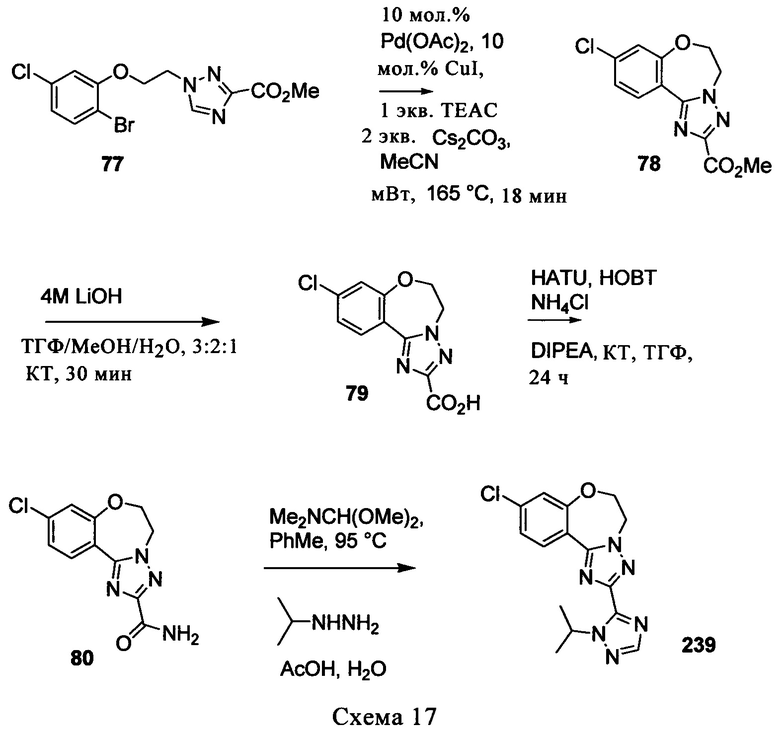
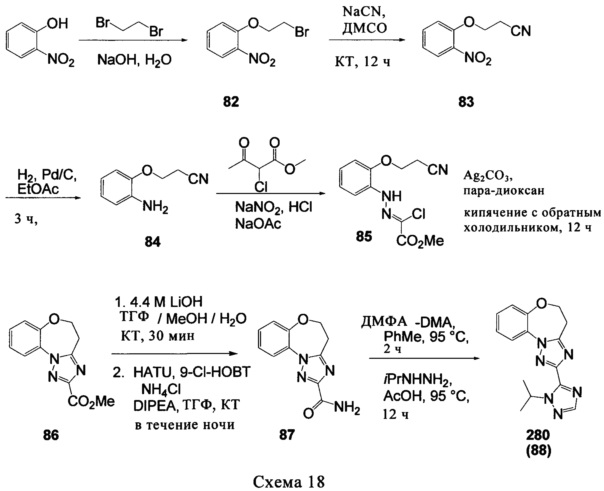

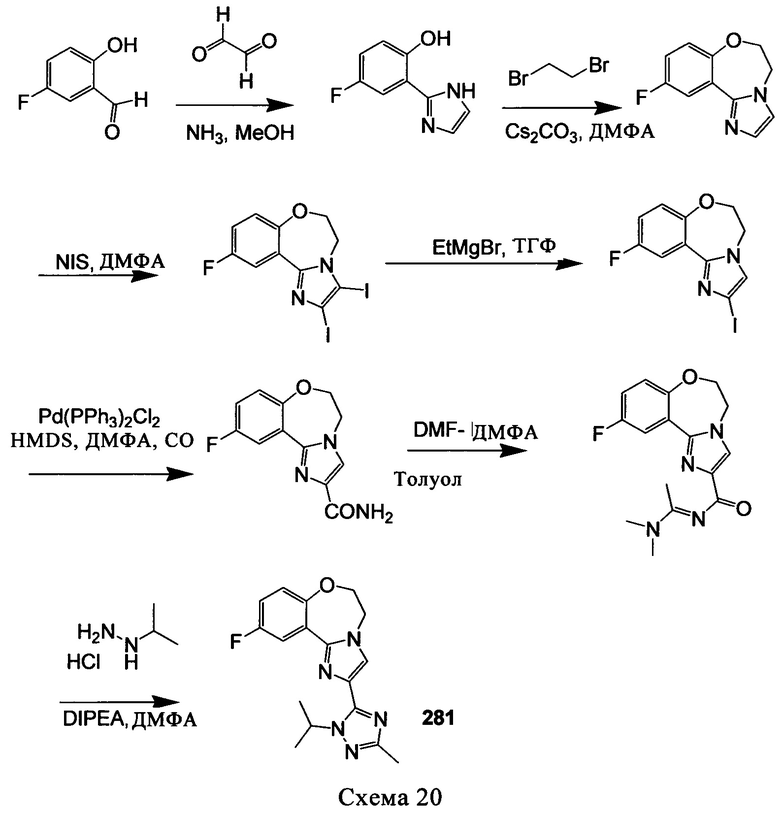
В другом объекте настоящего изобретения предлагаются способы получения, способы выделения и способы очистки соединений формулы I.
В еще одном объекте настоящего изобретения предлагаются новые промежуточные соединения, пригодные для получения соединений формулы I.
Примеры
Химические реакции, описанные в разделе «Примеры», можно использовать для получения ряда других ингибиторов PI3K по настоящему изобретению, при этом подразумевается, что в объем настоящего изобретения включены альтернативные способы получения соединений по настоящему изобретению. Например, соединения по настоящему изобретению, не приведенные в примерах, можно получить очевидными специалистам в данной области техники способами, например, при соответствующем введении защитных групп, использовании других пригодных реагентов, известных в данной области техники, которые отличаются от описанных, и/или при стандартных изменениях условий реакций. Кроме того, для получения других соединений по настоящему изобретению можно использовать другие реакции, описанные в данном контексте или известные в данной области техники.
Использованные реагенты являются коммерческими продуктами таких фирм, как Sigma Aldrich Chemical Company, и были использованы без дополнительной очистки, если не указано иное. Приведенные ниже реакции в основном проводили под давлением азота или аргона или с использованием хлоркальциевой трубки (если не указано иное) в безводных растворителях, при этом реакционные колбы, как правило, были снабжены каучуковой мембранной перегородкой для добавления субстратов и реагентов через шприц. Стеклянные изделия высушивали в сушильном шкафу и/или при нагревании. Колоночную хроматографию проводили в системе Biotage (фирмы Dyax Corporation), снабженной колонкой с силикагелем или картриджем с коллоидным диоксидом кремния SEP PAK® (Waters). 1H ЯМР спектры получали при 400 МГц в дейтерированных растворах CDCl3, d6-ДМСО, CH3OD или d6-ацетона (в част./млн) с использованием хлороформа в качестве внутреннего стандарта (7,25 част./млн). При регистрации мультиплетных сигналов использовали следующие сокращения: s (синглет), d (дублет), t (триплет), m (мультиплет), ушир. (уширенный), дд (дублет дублетов), дт (дублет триплетов). Если указаны константы взаимодействия, они приведены в Гц.
При проведении анализа методом жидкостной хроматографии - масс-спектрометрии (ЖХ-МС) определяли значения времени удерживания (в.у.) и регистрировали ионы определенной массы с использованием различных способов, известных специалистам в области аналитических методов анализа органических соединений.
Названия химических структур приведены в соответствии с торговыми названиями фирмы-производителя, номенклатурой ИЮПАК, программой ChemDraw Ultra, версия 9.0.1, фирмы CambridgeSoft Corp., Cambridge MA, или программой Autonom 2000 Name фирмы MDL Inc. Специалистам в данной области техники представляется очевидным, что существует более одного названия соединения в соответствии с различными условными обозначениями.
Использованы следующие сокращения: ДХМ - дихлорметан или хлористый метилен, ДМФА - N,N-диметилформамид, ДМСО - диметилсульфоксид, EtOAc - этилацетат, HATU - гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урания, IPA - изопропиловый спирт, NIS - N-иодсукцинимид, Pd(PPh3)4 - тетракис(трифенилфосфин)палладий(0), ПФК - полифосфорная кислота, КТ - комнатная температура, ТЭА - триэтиламин, ТФУ - трифторуксусная кислота, ТГФ - тетрагидрофуран, IMS - промышленный денатурированный спирт.
Пример 1
8-Бром-3,4-дигидробензо[b]оксепин-5(2Н)-он 1
Стадия 1
Этиловый эфир 4-(3-бромфенокси)бутановой кислоты
Твердый 3-бромфенол (10,0 г, 58 ммоль) добавляли порциями при перемешивании в суспензию K2CO3 в ацетоне (100 мл) при комнатной температуре. Затем добавляли иодид натрия (NaI, 1,0 г) и этил-4-бромбутират (9,2 мл, 64 ммоль). Реакционную смесь нагревали при 80°С в течение ночи, охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом, при этом получали этиловый эфир 4-(3-бромфенокси)бутановой кислоты 6.
Стадия 2
4-(3-Бромфенокси)бутановая кислота
Этиловый эфир 4-(3-брофенокси)бутановой кислоты 6 заливали 100 мл ТГФ и 50 мл воды и обрабатывали гидроксидом лития LiOH (гидрат, 4,9 г). Смесь нагревали при 50°С в течение 2 сут. Смесь охлаждали до КТ и подкисляли до рН 1 с использованием 2н. HCl. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором и сушили над сульфатом магния, при этом получали неочищенную 4-(3-бромфенокси)бутановую кислоту в виде клейкого твердого вещества. 1Н ЯМР (ДМСО-d6, 500 МГц): 7,24 (m, 1Н), 7,13 (m, 1H), 7,11 (m, 1H), 6,95 (m, 1H), 3,99 (m, 2Н), 2,37 (m, 2Н), 1,94 (m, 2Н).
Стадия 3
При перемешивании к суспензии РРА (приблизительно 60 г) и целита (приблизительно 40 г) в 1000 мл толуола добавляли неочищенную 4-(3-бромфенокси)бутановую кислоту 7 (приблизительно 58 ммоль) одной порцией, остатки смывали 10 мл толуола. Полученную суспензию нагревали при 110°С в течение 5 ч. Толуол декантирповали с использованием слоя целита и оставшуюся эмульсию промывали повторно толуолом и этилацетатом. Элюент концентрировали и очищали проточной хроматографией (4:1 гексан : EtOAc), при этом получали соединение 1 (7 г, выход приблизительно 50%). 1Н ЯМР (ДМСО-d6, 500 МГц) 7,55 (d, J=8,5 Гц, 1Н), 7,37 (d, J=1,5 Гц, 1Н), 7,35 (dd, J=8,5, 1,5 Гц, 1Н), 4,24 (t, J=6,5 Гц, 2Н), 2,79 (t, J=7,0 Гц, 2Н), 2,14 (m, 2H).
Пример 2
(Z)-8-бром-5-хлор-2,3-дигидробензо[b]окзепин-4-карбальдегид 2
Оксихлорид фосфора POCl3 (1,88 мл, 20,8 ммоль) по каплям добавляли в ДМФ (5 мл) при 0°С. Через 30 мин по каплям добавляли раствор соединения 18 (2,0 г, 8,3 ммоль, пример 1) в 8 мл ДМФ. Реакционную смесь охлаждали до КТ при перемешивании в течение 2 ч, затем медленно выливали в ледяную воду при быстром перемешивании. Водную фазу экстрагировали этилацетатом и объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия и концентрировали, при этом получали соединение 2.
Пример 3
7-бром-3,4-дигидробензо[b]окзепин-5(2Н)-он 3
В эмульсию NaH (60% дисперсия в минеральном масле, 1,48 г, 37,1 ммоль) в ТГФ (приблизительно 50 мл) при КТ добавляли 1-(5-бром-2-(2-бромэтокси)фенил)этанон (8,07 г, 25,1 ммоль). Реакционную смесь медленно нагревали с обратным холодильником и перемешивали в течение 20 ч. Растворитель удаляли в вакуме, сконцентрированный остаток наносили на силикагель и очищали хроматографией на колонке (4:1 EtOAc/петролейный эфир). Продукт получали в виде маслообразного вещества желтого цвета после удаления растворителей в виде 4,22 г (выход 70%) соединения 3. 1Н ЯМР (CDCl3): δ 7,87 (d, J=2,6 Гц, 1Н), 7,50 (dd, J=2,6, 8,1 Гц, 1Н), 6,97 (d, J=8,8 Гц, 2Н), 4,24 (t, J=6,6 Гц, 2Н), 2,89 (t, J=7,0 Гц, 2Н), 2,15-2,29 (m, 2Н).
Пример 4
4,7-Дибром-3,4-дигидробензо[b]окзепин-5(2Н)-он 4
К соединению 3 (3 г, 12 ммоль) в этиловом эфире (110 мл) добавляли бром (0,7 мл, 14 ммоль) и перемешивали при КТ в течение ночи. Реакционную смесь концентрировали при пониженном давлении и очищали хроматографией на колонке ISCO (элюент: гексан - 20% гексан в EtOAc в течение 45 мин). После сбора и концентрирования фракций получали соединение 4 (3,53 г, 89%). 1Н ЯМР (500 МГц, CDCl3): δ 7,86 (d, J=2,5, 1H), 7,52 (dt, J=28,5, 14,2, 1H), 6,97 (d, J=8,7, 1H), 4,95 (dd, J=7,6, 6,8, 1H), 4,53-4,36 (m, 1H), 4,17 (ddd, J=12,8, 9,9, 4,4, 1H), 3,04-2,84 (m, 1H), 2,52 (ddt, J=14,7, 7,8, 4,5, 1H)
Пример 5
3-изопропил-1-метил-1Н-1,2,4-триазол-5 (4Н)-он 5
Стадия 1
1-метилгидразинкарбоксамид
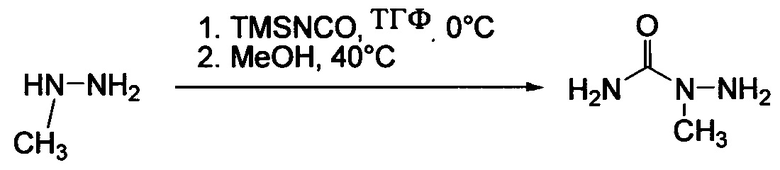
Реакцию между метилгидразином и триметилсилилизоцианатом проводили в ТГФ при 0°С, которую затем останавливали и гидролизовали метанолом, при этом получали 1-метилгидразинкарбоксамид.
Стадиия 2
2-изобутирил-1-метилгидразинкарбоксамид
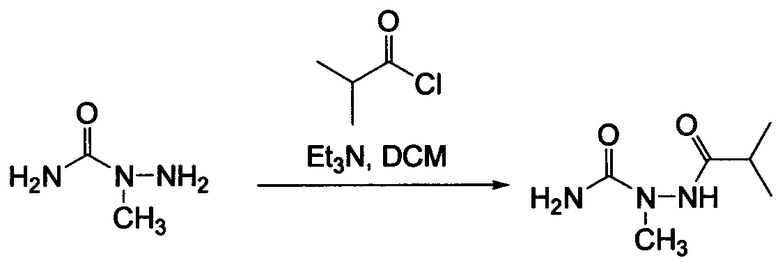
1-Метилгидразинкарбоксамид ацилировали в TEA и ДХМ, при этом получали 2-изобутирил-1-метилгидразинкарбоксамид.
Стадия 3

2-Изобутирил-1-метилгидразинкарбоксамид циклизовали с использованием 10-камфоросульфокислоты при кипячении с обратным холодильником в этилацетате, при этом получали соединение 5.
Пример 6
1,3-диметил-1Н-1,2,4-триазол-5(4Н)-он 6а и 1-изопропил-3-метил-1Н-1,2,4-триазол-5(4Н)-он 6b

Ацетамид и этиловый эфир хлормуравьиной кислоты смешивали при 45°С, при этом получали гидрохлоридэтилацетамидата, который затем вводили в реакцию с этиловым эфиром хлормуравьиной кислоты, диизопропилэтиламина и ДХМ при 0°С, при этом получали этил-N-этоксикарбонилацетамидат, который вводили в реакцию с гидрохлоридом метилгидразина или изопропилгидразина в TEA и толуоле, при этом получали соединения 6а и 6b, соответственно.
Пример 7
4-изопропил-1-(4-метоксибензил)-1Н-имидазол-2(3Н)-он 7

3-метилбутан-2-он бромировали бромом в метаноле, при этом поучали 1-бром-3-метилбутан-2-он, который вводили в реакцию с 4-метоксибензиламином и циклизовали с использованием цианата натрия, при этом получали соединение 7.
Пример 8
Метиловый эфир 6,7-дигидроимидазо[1,2-d]пиридо[3,2-b][1,4]оксазепин-3-карбоновой кислоты 8
Стадия 1
2-Метил-1-тритил-1Н-имидазол
Трифенилметилхлорид (16,0 г, 57,5 ммоль) добавляли порциями в раствор 2-метилимидазола (4,10 г, 50,0 ммоль) и TEA (9,02 мл, 64,7 ммоль) в 20 мл N,N-диметилформамида. Смесь перемешивали в течение 18 ч, смешивали с 300 мл воды и экстрагировали 1000 мл EtOAc. Органический экстракт промывали 1 л воды, солевым раствором, сушили над MgSO4 и концентрировали в вакууме, при этом поучали 50 мл смеси. Остаток собирали, промывали EtOAc и сушили в высоком вакууме в течение 18 ч, при этом получали 15,0 г (92,5%) продукта. 1Н ЯМР (400 МГц, CDCl3): δ 7,34-7,29 (m, 9H), 7,16-7,11 (m, 6H), 6,90 (d, J=1,5, 1H), 6,71 (d, J=1,5, 1H), 1,65 (s, 3H).
Стадия 2
2-(1-Тритил-1Н-имидазол-2-ил)ацетальдегид
1,6 M раствор n-бутиллития в гексане (7,5 мл) по каплям добавляли в раствор 2-метил-1-тритил-1H-имидазола (3,244 г, 10,00 ммоль) в ТГФ (100,0 мл, 1233 ммоль) при t -76°С. Смесь темно-красного цвета перемешивали в течение 45 мин. Затем быстро добавляли этиловый эфир муравьиной кислоты (4,039 мл, 50,00 ммоль) и смесь (цвет которой изменился на желтый) перемешивали в течение 20 мин. Затем добавляли 6 мл 5% водного раствора лимонной кислоты, смесь смешивали с 60 мл водного раствора лимонной кислоты и экстрагировали EtOAc. Органический слой промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме. Полутвердый материал бледно-желтого цвета (2,025 г, 57,5%) использовали на следующей стадии без дополнительной очистки.
Стадия 3
2-(1-тритил-1Н-имидазол-2-ил)этанол
Неочищенный 2-(1-тритил-1Н-имидазол-2-ил)ацетальдегид (2,025 г, 5,75 ммоль) растворяли в смеси МеОН/ТГФ (1:1, 40 мл) и в полученную смесь порциями добавляли NaBH4 (0,435 г, 11,5 ммоль). Смесь перемешивали в течение 18 ч, разбавляли 100 мл воды и 2 раза экстрагировали ДХМ. Объединенные органические экстракты промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали в вакууме, при этом получали 1,915 г (94%) остатка. 1Н ЯМР (500 МГц, CDCl3): δ 7,35-7,31 (m, 9H), 7,12 (dd, J=6,7, 2,7, 6H), 6,93 (d, J=1,0, 1H), 6,74 (d, J=1,0, 1H), 5,04 (ушир., 1H), 3,46 (t, J=5,4, 2H), 2,00 (t, J=5,4, 2H).
Стадия 4
Метиловый эфир 6-иод-5-(2-(1-тритил-1Н-имидазол-2-ил)этокси)никотиновой кислоты
Диизопропиловый эфир азодикарбоновой кислоты (1160 пкл, 5,90 ммоль) по каплям добавляли в смесь 2-(1-тритил-1Н-имидазол-2-ил)этанола (1900 мг, 5,4 ммоль), метилового эфира 5-гидрокси-6-иодникотиновой кислоты (1570 мг, 5,63 ммоль) и трифенилфосфина (1550 мг, 5,90 ммоль) в ТГФ (45,0 мл, 555 ммоль) при 0°С. После перемешивания в течение 3 ч смесь смешивали с водой и экстрагировали EtOAc. Органические слои промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали на колонке с 40 г силикагеля (элюент: 50% EtOAc в ДХМ), при этом получали 1,45 г (44%) метилового эфиар 6-иод-5-(2-(1-тритил-1Н-имидазол-2-ил)этокси)никотиновой кислоты. MC(ESI+): 616,0. 1H ЯМР (400 МГц, CDCl3): δ 8,52 (d, J=1,9, 1H), 7,40-7,28 (m, 10Н), 7,20-7,16 (m, 6H), 6,99 (d, J=1,5, 1H), 6,81 (d, J=1,5, 1H), 3,98-3,91 (m, 5H), 2,46 (t, J=7,3, 2H).
Стадия 5
Метиловый эфир 5-(2-(1Н-имидазол-2-ил)этокси)-6-иодникотиновой кислоты
Триэтилсилан (0,160 мл, 1,00 ммоль) добавляли в раствор 1,45 г (2,36 ммоль) метилового эфиар 6-иод-5-(2-(1-тритил-1Н-имидазол-2-ил)этокси)никотиновой кислоты в ТФУ (30,0 мл, 389 ммоль). Смесь перемешивали в течение 4 ч, концентрировали в вакууме и обрабатывали 50 мл безводного этилового эфира. Твердый материал собирали, промывали несколькими порциями этилового эфира и распределяли между 1 М водным раствором карбоната натрия и EtOAc. Органические слои промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме, при этом получали остаток (0,55 г, 62%) метилового эфира 5-(2-(1Н-имидазол-2-ил)этокси)-6-иодникотиновой кислоты. MC(ESI+): 374,0
Стадия 6
Смесь метилового эфира 5-(2-(1Н-имидазол-2-ил)этокси)-6-иодникотиновой кислоты (373 мг, 1,00 ммоль), оксида меди(1) (14,3 мг, 0,10 ммоль), нингидрина (35,6 мг, 0,20 ммоль) и карбоната калия (290 мг, 2,10 ммоль) в ДМСО (10,0 мл) нагревали при 110°С в течение 2 ч. Смесь выливали в 20 мл воды и экстрагировали EtOAc (3×15 мл). Органические экстракты промывали водой (3×15 мл), солевым раствором, сушили над MgSO4 и концентрировали. Остаток (0,220 г, 90%) использовали на следующей стадии без дополнительной очистки. MC(ESI+): 246,0. 1Н ЯМР (500 МГц, CDCl3): δ 8,77 (d, J=1,9, 1H), 8,10 (s, 1H), 8,04 (d, J=1,9, 1H), 7,08 (s, 1H), 4,47 (t, J=5,1, 2H), 3,97 (s, 3H), 3,46 (t, J=5,1, 2H).
Пример 9
Метиловый эфир
9,10-дииодо-6,7-дигидроимидазо[1,2-d]пиридо[3,2-b][1,4]оксазепин-3-карбоновой кислоты 9
N-Иодсуцинимид (394 мг, 1,75 ммоль) добавляли в раствор метилового эфира 5-(2-(1Н-имидазол-2-ил)этокси)-6-иодникотиновой кислоты (220 мг, 0,90 ммоль) в ДМФ (8,0 мл, 100 ммоль). Смесь перемешивали в течение 6 ч при КТ и 18 ч при 60°С. смесь концентрировали в вакууме и остаток распределяли между EtOAc и 1М водным раствором Na2CO3. Органический слой промывали водой (3×15 мл), солевым раствором, сушили над MgSO4 и концентрировали. Остаток очищали на колонке с 4 г силикагеля (элюент: 40% EtOAc в гептане), при этом получали 130 мг продукта. MC(ESI+): 497,9. 1H ЯМР (500 МГц, CDCl3): δ 9,02 (d, J=1,9, 1H), 8,21 (d, J=1,9, 1H), 4,65 (t, J=6,4, 2H), 4,00 (s, 3H), 3,14 (t, J=6,4, 2H).
Пример 10
Метиловый эфир 10-иод-6,7-дигидроимидазо[1,2-d]пиридо[3,2-b][1,4]оксазепин-3-карбоновой кислоты 10
Бромид этилмагния в этиловом эфире (3,0 М, 0,104 мл) добавляли по каплям в суспензию соединения 9 (130 мг, 0,26 ммоль) в ТГФ (5,0 мл, 62 ммоль) при -15°С. Смесь перемешивали в течение 15-20 мин (завершение реакции подтверждани ЖХМС), выливали в 20 мл насященного водного раствора NH4Cl и экстрагировали EtOAc. Органические экстракты промывали водой (2×20 мл), солевым раствором, сушили над MgSO4 и концентрировали в вакууме, при этом получали 92 мг (94%) продукта. MC(ESI+): 372,0.
Пример 11
Метиловый эфир 9-(1-изопропил-1Н-пиразол-5-ил)-6,7-дигидроимидазо[1,2-d]пиридо[3,2-b][1,4]оксазепин-3-карбоновой кислоты 11
Смесь 1-изопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (117,1 мг, 0,4958 ммоль), соединения 10 (92,0 мг, 0,248 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II), комплекса с ДХМ (1:1, 20,24 мг, 0,02479 ммоль) и 1,0 М ацетата калия в воде (0,49 мл) в 1,2-диметоксиэтане (5,0 мл, 48 ммоль) дегазировали. Реакционную смесь обрабатывали микроволнами при 200 Ватт, 140°С в течение 40 мин. Реакционную смесь фильтровали, промывали ДМЭ, смешивали с водой и экстрагировали EtOAc. Объединенные органические экстракты промывали 1% водным раствором NaOH для удаления фенольных побочных продуктов, затем 5% водным раствором лимонной кислоты, водой, солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали на колонке с 4 г силикагеля (элюент: 60-70% раствор EtOAc в гептане), при этом получали 21 мг продукта. MC(ESI+): 354,2.
Пример 12
9-(1-Изопропил-1Н-пиразол-5-ил)-6,7-дигидроимидазо[1,2-d]пиридо[3,2-b][1,4]оксазепин-3-карбоновая кислота 12
Смесь 21 мг (0,06 ммоль) соединения 11 и 1,0 мл 1Н. водного раствора LiOH в 4 мл метанола и 4 мл ТГФ перемешивали в течение 4 ч. Смесь подкисляли до рН 3 добавлением 1Н. HCl и концентрировали в вакууме. Остаток распределяли между EtOAc и водой, органический слой промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали, при этом получали 17 мг продукта. MC(ESI+): 340,1
Пример 13
Метиловый эфир 2-(1-изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d] [1,4]оксазепин-10-карбоновой кислоты 13
Смесь соединения 26 (370,1 мг, 1,000 ммоль), 1-изопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (354 мг, 1,50 ммоль), соединения 10 (92,0 мг, 0,248 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II), комплекса с ДХМ (1:1, 40,8 мг, 0,0500 ммоль) и 2,0 М ацетата калия в воде (1,0 мл) в ацетонитриле (12 мл, 230 ммоль) дегазировали. Реакционную смесь обрабатывали микроволнами при 200 Ватт, 140°С в течение 30 мин. Реакционную смесь распределяли между водой и EtOAc, фильтровали, органический слой промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали на колонке с 12 г силикагеля (элюент: 35-40% EtOAc в гептане), при этом получали 119 мг (34%). МС: (ESI+): 353,1.
Пример 14
2-(1-Изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновая кислота 14
Соединение 13 гидролизовали, как описано в примере 10, при этом получали соединение 14. MC(ESI+): 339,4.
Пример 15
Метиловый эфир 2-(4-циано-1-изопропил-1Н-празол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 15
Стадия 1
5-Амино-1-изопропил-1Н-пиразол-4-карбонитрил
Метоксид натрия (2,139 г, 39,60 ммоль) добавляли в раствор этоксиметиленмалонитрила (2,198 г, 18,00 ммоль) и гидрохлорида изопропилгидразина (2,212 г, 20,00 ммоль) в этаноле (50 мл, 800 ммоль). Смесь кипятили с обратным холодильником в течение 18 ч. Растворитель удаляли в вакууме, остаток распределяли между EtOAc и водой. Органические слои промывали водой, солевым раствором, сушили над Na2SO4, концентрировали в вакууме и очищали на колонке с 25 г силикагеля (элюент: 25-30% EtOAc в гептане), при этом получали 5-амино-1-изопропил-1Н-пиразол-4-карбонитрила (выход 1,77 г, 65%). MC(ESI+): 151,2. 1H ЯМР (400 МГц, CDCl3): δ 7,51 (d, J=6,4, 1H), 4,23 (ddd, J=19,8, 16,6, 9,8, 3Н), 1,46 (d, J=6,6, 7H).
Стадия 2
5-Иод-1-изопропил-1Н-пиразол-4-карбонитрил
Амилнитрил (13,00 г, 111,0 ммоль) добавляли в суспензию 5-амино-1-изопропил-1Н-пиразол-4-карбонитрил (1,77 г, 11,8 ммоль) в дииодметане (56,0 мл, 695 ммоль) при -10°С в течение 30 мин. Смесь перемешивали в теение 30 мин при КТ и затем нагревали при 100°С в течение 2 ч. Затем смесь охлаждают и концентрируют в высоком вакууме, при этом получабт остаток, коорый распределяют между EtOAc и 5% Na2S2O5. Органический слой промывают водой, 0,1% водным раствором HCl, водой, солевым раствором, сушат и концентрируют в вакууме. Остаток очищают на колонке с силикагелем (элюент: 20-30% EtOAc в гептане). Выход составляет 1,68 г (55%). MC(ESI+): 262,2
Стадия 3
Метиловый эфир 2-(трибутилстаннил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты
Хлорид изопропилмагния в ТГФ (2,0 М, 1,5 мл, 3,00 ммоль) по каплям добавляли в раствор соединения 40 (740 мг, 2,00 ммоль) в ТГФ (12 мл, 150 ммоль) при КТ. Смесь перемешивали в течение 2,5 ч. Хлорид трибутилолова (0,8138 мл, 3,000 ммоль) добавляли в смесь и перемешивали в течение 18 ч. Смесь смешивали с насыщенным водным раствором NH4Cl и экстрагировали этилацетатом. Органический слой промывали солевым раствором, сушили над Na2SO4 и очищали на колонке с 25 г силикагеля (элюент: 15-20% EtOAc в гептане). Выход составлял 160 мг (15%). MC(ESI+): 535,2
Стадия 4
Смесь метилового эфира 2-(трибутилстаннил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (155 мг, 0,291 ммоль), 5-амино-1-изопропил-1Н-пиразол-4-карбонитрила (133 мг, 0,509 ммоль) и Pd(PPh3)4 (16,8 мг, 0,0145 ммоль) в толуоле (6,0 мл, 56 ммоль) нагревали в течение 18 ч. Смесь концентрировали в вакууме, остаоок очищали на колонке с 4 г силикагеля (элюент: 30% EtOAc в гептане). Выход составлял 65 мг (59%). MC(ESI+): 378,2
Пример 16
2-(4-циано-1-изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 16
Соединение 15 гидролизовали, как описано в примере 10, при этом получали соединение 16. MC(ESI+): 364,3
Пример 17
10-Хлор-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин 17
Стадия 1
2-хлор-5-(метоксиметокси)пиридин
60% Дисперсию гидрида натрия в минеральном масле (3:2, гидрид натрия: минеральное масло, 2,32 г) порциями добавляли в раствор 6-хлорпиридин-3-ола (5,00 г, 38,6 ммоль) в смеси ТГФ (10,0 мл, 123 ммоль) и ДМФ (20,0 мл, 258 ммоль). Полученную смесь перемешивали в течение 15 мин и по каплям добавляли хлорметилметиловый эфир (3,66 мл, 48,2 ммоль). Полученную смесь перемешивали в течение 6 ч (окончание реакции определяли ЖХМС), выливали в воду и экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали на колонке с 40 г силикагеля (элюент6 10-40% EtOAc в гептане), при этом получали 6,33 г of 2-хлор-5-(метоксиметокси)пиридина.
Стадия 2
2-Хлор-5-(метоксиметокси)изоникотинальдегид
трет-Бутиллитий в пентане (1,7 М, 19,0 мл) по каплям добавляли в раствор 2-хлор-5-(метоксиметокси)пиридина (4,880 г, 28,11 ммольl) в 100 мл этилового эфира при -76°С. Наблюдали образование некоторого количества осадка. Смесь выдерживали при -76°С в течении 20 мин, затем по каплям добавляли ДМФ (2,938 мл, 37,95 ммоль). Смесь перемешивали в течение 10 мин при -76°С и нагревали до 0°С в течение 1 ч. Затем добавляли 10% водный раствор NH4Cl и смесь экстрагировали EtOAc. Органический раствор промывали водой, солевым раствором и сушили над Na2SO4. После концентрирования в вакууме получали неочищенный 2-хлор-5-(метоксиметокси)изоникотинальдегид в количестве 5,49 г, который использовали на следующией стадии без дополнителной очистки. МС: 202.0, 172,0.
Стадия 3
2-Хлор-4-(1Н-имидазол-2-ил)-5-(метоксиметокси)пиридин
Неочищенный 2-хлор-5-(метоксиметокси)изоникотинальдегид (5,20 г, 25,87 ммоль) растворяли в 60 мл метанола и смешивали с 40% водным раствором этандиала (16,31 г, 112,4 ммоль) и водным раствором аммиака (19,15 г, 337,3 ммоль). Смесь перемешивали в течение 3 ч, концентрировали в вакууме и подкисляли до рН<1 60 мл 1Н. Водного раствора HCl. Водный раствор экстрагировали EtOAc (3×30 мл). Органические экстракты отбрасывали, а водную фазу подщелачивали добавлением насыщенного раствора NaHCO3. Смесь экстрагировали EtOAc (3×30 мл), объединенные органические экстракты промывали водой, солевым раствором, сушили и концентрировали в вакууме. Неочищенный остаток (4,185 г) очищали на колонке с 40 г силикагеля (элюент: 60-70% EtOAc в гептане, при этом получали 2,06 г (33%) продукта. MC(ESI+): 208 (потеря НОМе). 1Н ЯМР (500 МГц, CDCl3): δ 10,56 (s, 1H), 8,35 (s, 1H), 8,23 (s, 1H), 7,30 (s, 1H), 7,22 (s, 1H), 5,43 (s, 2H), 3,54 (d, J=14,0, 3H).
Стадия 4
6-Хлор-4-(1Н-имидазол-2-ил)пиридин-3-ол
Хлористый водород в диоксане (4 М, 40 мл) по каплям добавляли в раствор 2,06 г (8,60 ммоль) of 2-хлор-4-(1Н-имидазол-2-ил)-5-(метоксиметокси)пиридина в ДХМ (40 мл, 600 ммоль). Суспензию перемешивали в течение 2 ч и фильтровали. Твердый продукт промывали ДХМ, эфиром и сушили в вакууме. Выход дигидрохлорида 6-хлор-4-(1Н-имидазол-2-ил)пиридин-3-ола составлял 2,31 г (100%). MC(ESI+): 196,2. 1H ЯМР (400 МГц, ДМСО): δ 13,20 (s, 1H), 8,14 (s, 1H), 7,96 (s, 1H), 7,42 (s, 2H).
Стадия 5
Смесь 2,30 г (8,55 ммоль) дигидрохлорида 6-хлор-4-(1Н-имидазол-2-ил)пиридин-3-ола, 1,2-дибромэтана (1,842 мл, 21,37 ммоль) и карбоната цезия (19,46 г, 59,74 ммоль) в 120 мл ДМФ нагревали в течение 3 ч при 90°С. Смесь фильтровали и концентрировали в высоком вакууме, при этом получали соединение 17 в количестве 1,88 г (99%). MC(ESI+): 222,2. 1H ЯМР (400 МГц, CDCl3): δ 8,37 (s, 1H), 8,17 (s, 1H), 7,24 (d, J=1,0, 1H), 7,10 (d, J=0,9, 1H), 4,51-4,45 (m, 4H).
Пример 18
10-Хлор-2,3-дииод-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин 18
NIS (5,771 г, 25,65 ммоль) добавляли в смесь 1,89 г (8,55 ммоль) соединения 17 в ДМФ (28 мл, 360 ммоль) и смесь нагрвали при 80°С в течение 48 ч. Осадок собирали, промывали ДМФ и этиловым эфиром и сушили на воздухе, а затем в высоком вакууме. Выход соединения составлял 2,85 г (70%). МС: 473,9. 1Н ЯМР (500 МГц, CDCl3): δ 8,33 (s, 1H), 8,19 (s, 1H), 4,53-4,46 (m, 2H), 4,45-4,38 (m, 2H).
Пример 19
10-Хлор-2-иод-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин 19
Хлорид изопропилмагния в ТГФ (2,0 M, 3,311 мл) по каплям добавляли в раствор соединения 18 (2,850 г, 6,020 ммоль) в 110 мл ТГФ при -10°С. Смесь нагрвали до 10°С в течение 45 мин и смешивали с 250 мл холодного 10% NH4Cl. Органический слой промывали солевым раствором и сушили над Na2SO4. После концентрирования в вакууме получали 2,06 г (98,5%). МС: 348,0. 1Н ЯМР (500 МГц, CDCl3): δ 8,33 (d, J=10,1, 1H), 8,18 (s, 1H), 7,18 (s, 1H), 4,46 (q, J=5.8, 4H).
Пример 20
10-Хлор-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-2-карбоксамид 20
Смесь соединения 19 (2056 мг, 5,916 ммоль), хлорида бис(триметилфосфин)палладия(II) (0,00210 мг, 0,300 ммоль) и гексаметилдисилазана (7,488 мл, 35,50 ммоль) в 60 мл ДМФ карбонилировали при давлении 1 атм из баллона с СО. Реакционную смесь нагревали при 70°С в течение 1 ч. Смесь концентрировали в вакууме, остаток распределяли между EtOAc и 1 М водным раствором карбоната натрия. Органические экстракты промывали водой, солевым раствором, сушили над сульфатом магния, концентрировали в вакууме и очищали на колонке с 12 г силикагеля (элюент: 0-5% МеОН в ДХМ), при этом получали 1300 мг (83%) продукта. MC(ESI+): 265,0. 1H ЯМР (500 МГц, ДМСО): δ 8,37 (s, 1H), 8,22 (s, 1H), 7,93 (s, 1H), 7,70 (s, 1H), 7,25 (s, 1H), 4,56 (s, 4H).
Пример 21
10-Хлор-N-((диметиламино)метилен)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-2-карбоксамид 21
Смесь соединения 20 (1,290 г, 4,875 ммоль) и 1,1-диметокси-N,N-диметилметанамина (3,238 мл, 24,37 ммоль) в 70 мл толуола кипятили с обратным холодильником в течение 1 ч. После охлаждения продукт осаждали из реакционной смеси, собирали, промывали этиловым эфиром и сушили на воздухе, при этом получали 0,705 г (85%) продукта. MC(ESI+): 320,1
Пример 22
10-Хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин 22
Смесь 660 мг (2,06 ммоль) соединения 21 и гидрохлорида изопропилгидразина (0,332 г, 3,00 ммоль) в 44 мл уксусной кислоты нагревали при 85°С в течение 3 ч. Затем смесь охлаждали, фильтровали и смешивали с 15 мл воды. Осадок отфильтровывали, промывали водой и сушили в высоком вакууме. Полученное твердое вещество обрабатывали EtOAc, отфильтровывали, промывали EtOAc, этиловым эфиром и сушили на воздухе, при этом получали 0,710 г продукта. МС: 331,2. 1H ЯМР (500 МГц, ДМСО): δ 8,26 (s, 1H), 8,20 (s, 1H),8,11 (s, 1H), 7,96 (s, 1H), 5,76 (dt, J=13,1, 6,6, 1H), 4,62 (q, J=5,6, 4H), 1,50 (d, J=6,6, 6H).
Пример 23
Метиловый эфир 4-гидрокси-3-(1Н-имидазол-2-ил)бензойной кислоты 23
Метиловый эфир 3-формил-4-гидроксибензойной кислоты вводили в реакцию с этаналем и аммиаком, как описано в примере 22, при этом получали соединение 23 с выходом 78%. MC(ESI+): 219,1
Пример 24
Метиловый эфир 5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты 24
Соединение 23 вводили в реакцию с 1,2-дибромэтаном, как описано в примере 17, при этом получали соединение 24 с выходом 76%. MC(ESI+): 245,0. 1Н ЯМР (400 МГц, CDCl3): δ 9,21 (d, J=2,2, 1H), 7,91 (dd, J=8,6, 2,2, 1H), 7,20 (t, J=4,8, 1H), 7,05 (d, J=8,6, 1H), 7,00 (d, J=0,8, 1H), 4,53-4,48 (m, 2H), 4,43-4,39 (m, 2H), 3,91 (d, J=5,9, 3H).
Пример 25
Метиловый эфир 2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты 25
Смесь соединения 24 (2670 мг, 9,29 ммоль) и NIS (5230 мг, 23,2 ммоль) в 100 мл ДМФ нагревали при 80°С в течение 3 ч. Смесь смешивали с 300 мл воды и экстрагировали ДХМ (3×120 мл). Объединенные органические экстракты промывали 5% водным раствором бикарбоната натрия, 2×50 мл 10% водного раствора тиосульфата натрия, водой, солевым раствором, сушили над MgSO4 и концентрировали в акууме до малого объема. Осадок фильтровали, промывали ДХМ и сушили в вакууме, при этом получали 3,86 г (84%) продукта. МС 497,0. 1H ЯМР (500 МГц, CDCl3): δ 9,12 (d, J=2,0, 1H), 7,93 (dd, J=8,6, 2,1, 1H), 7,05 (d, J=8,6, 1H), 4,55-4,46 (m, 2H), 4,38 (dd, J=5,0, 2,9, 2H), 3,92 (s, 3H).
Пример 26
Метиловый эфир 2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты 26
Соединение 25 превращали в соединение 26, как описано в примере 19, при этом получали продукт с выходом 95%. MC(ESI+): 370,9. 1Н ЯМР (400 МГц, CDCl3): δ 9,15 (d, J=2,1, 1H), 7,92 (dd, J=8,6, 2,2, 1H), 7,08 (s, 1H), 7,04 (t, J=7,9, 1H), 4,48 (dd, J=9,5, 5,5, 2H), 4,40 (dd, J=9,4, 5,5, 2H), 3,92 (s, 3H).
Пример 27
Метиловый эфир 2-циано-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты 27
2-Иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксилат (370,1 мг, 1,0 ммоль) и цианид меди (268,6 мг, 3,000 ммоль) смешивали с 8 мл ДМФ. Реакционную смесь подвергали микроволновой обработке при 200 Ватт и 150°С в течение 40 мин. Реакционную смесь распределяли между 25 мл 5% водного раствора аммиака и 25 мл EtOAc. Водный слой дополнительно экстрагировали 3×20 мл EtOAc, объединенные органические слои промывали водой, солевым раствором и сушили над MgSO4, при этом получали 225 мг соединения 27 с выходом 81%. (МС: 270,0).
Пример 28
Метиловый эфир 2-карбамоил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты 28
Соединение 27 (220 мг, 0,82 ммоль) растворяли в 4 мл ДМСО и обрабатывали раствором карбоната калия (136 мг, 0,980 ммоль) в воде (1,60 мл, 88,8 ммоль). После охлаждения до 0°С медленно добавляли пероксид водорода (0,751 мл, 9,80 ммоль). Смесь перемешивали при КТ в течение 2 ч. Смесь разбавляли 20 мл воды и экстрагировали EtOAc (3×20 мл). Органические экстракты промывали 5% раствором тиосульфата натрия, насыщенным раствором NaHCO3, солевым раствором, сушили над сульфатом магния и концентрировали, при этом получали 180 мг (77%) неочищенного соединения 28. MC(ESI+): 288,0.
Пример 29
Метиловый эфир 2-((диметиламино)метиленкарбамоил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты 29
Соединение 28 превращали в соединение 29, как описано в примере 21, с выходом 82%. MC(ESI+): 343,1
Пример 30
Метиловый эфир 2-(1-(2-хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты 30
Соединение 29 вводили в реакцию с гидрохлоридом 2-хлорфенилгидразина, как описано в примере 22, при этом получали соединение 30 с выходом 59%. MC(ESI+): 422,1
Пример 31
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновая кислота 31
Соединение 30 гидролизовали, как описано в примере 12, в соединение 31 с выходом 75%. MC(ESI+): 408,1
Пример 33
9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбальдегид 33
Бромид этилмагния в этиловом эфире (3,0 М, 3,472 мл) по каплям добавляли в раствор 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1173 мг, 3,000 ммоль) в 20 мл ТГФ при -30°С. Смесь перемешивали при указанной температуре в течение 20 мин и нагревали до 15°С. Затем смесь снова охлаждали до -25°С и добавляли ДМФ (929,2 мкл, 12,00 ммоль). Смесь выдерживали в течение 18 ч. Реакцию останавливали насыщенным водным раствором. NH4Cl и экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме, при этом получали 0,92 г. МС: 293,1
Пример 34
9-Бром-2-(4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 34
Раствор аммиака в воде (16,0 М, 0,819 мл) добавляли в раствор соединения 33 (640 мг, 2,2 ммоль) и пирувальдегида (0,787 г, 4,37 ммоль) в метаноле (17 мл, 420 ммоль) и ТГФ (6 мл, 70 ммоль). Через 1 ч в смесь длбавляли аналогичные количества 16,0 М раствора аммиака в воде и пирувальдегида. Смесь перемешивали в течение 2 ч, концентрировали в вакууме и остаток распределяли между EtOAc и водой. Органические экстракты промывали водой, солевым раствором, сушили над MgSO4 и концентрировали. Остаток очищали на колонке с 4 г силикагеля (элюент: градиент EtOAc в ДХМ), при этом получали 0,417 г. МС: 344,9.
Пример 35
9-Бром-2-(1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 35
Этандиаль (0,689 мл, 6,01 ммоль) м 16,0 М раствор аммиака в воде (1,50 мл) добавляли в раствор соединения 33 (550 мг, 1,5 ммоль) в метаноле (30,0 мл, 742 ммоль). Через 1 ч добавляли аналогичные количества этандиаля и аммиака и смесь перемешивали в течение 4 ч. Смесь концентрировали в вакууме и распределяли между 0,5 н. HCl и EtOAc. Органический экстракт отбрасывали, кислый водный раствор осторожно подщелачивали добавлением насыщенного водного раствора. NaHCO3. Смесь экстрагировали EtOAc, органические экстракты промывали водой, солевым раствором, сушили над MgSO4 и концентрировали. Остаток обрабатывали ДХМ, при этом получали осадок, который собирали, промывали охлажденным ДХМ и сушили, при этом получали соединение 35. МС: (ESI+)=331,2
Пример 36
9-Бром-2-(1-изопропил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 36
В раствор соединения 35 (0,237 г, 0,716 ммоль) и карбоната цезия (0,280 г, 0,859 ммоль) в ДМФ (4,74 мл, 61,2 ммоль) добавляли изопропилиодид (0,0859 мл, 0,859 ммоль). Реакционную смесь перемешивали в течение 18 ч при 50°С. Реакцию останавливали водой и дважды экстрагировали EtOAc. Неочищенный продукт очищали, при этом получали соединение 36. МС: (ESI+)=373,1
Пример 37
Метиловый эфир 3-гидрокси-4-(1Н-имидазол-2-ил)бензойной кислоты 37
4-Формил-3-гидроксибензойную кислоту (5 г, 30 ммоль) суспендировали в метаноле (70 мл) и по каплям обрабатывали тионилхлоридом (3,29 мл, 45 ммоль). Смесь кипятили с обратным холодильником в течение ночи. Смесь концентрировали досуха, добавляли 50 мл толуола и снова концентрировали. Остаток перекристаллизовывали из смеси EtOAc-гексан. Получали 4,8 г (85%) метилового эфира 4-формил-3-гидроксибензойной кислоты.
Смесь метилового эфира 4-формил-3-гидроксибензойной кислоты (4,8 г, 27 ммоль), 40% водноо раствора этандиаля (11,6 г, 79,93 ммоль) и 50% водного раствора аммиака (6,8 г, 399 ммоль) в метаноле (50 мл) перемешивали в течение 2 ч или дольше до завершения реакции. Растворитель удаляли на роторном испарителе и остаток распределяли между EtOAc и водой. Смесь фильтровали для удаления остатка. Величину рН доводили до 5-6 осторожным добавлением 1Н. HCl. Водный слой трижды экстрагировали EtOAc. Объединенный органический слой промывали водой, солевым раствором и сушили над MgSO4. Остаток очищали экспресс-хроматографией, при этом получали твердое вещество желтого цвета (4 г, 71%).
Пример 39
Метиловый эфир 5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 38
Смесь соединения 37 (2,2 г, 10 ммоль), 1,2-дибромэтана (3,12 мл, 36 ммоль) и карбоната цезия (13,14 г, 40 ммоль) в ДМФ (100 мл) нагревали при 90°С в течение 12 ч. Смесь фильтровали, надосадочную жидкость концентрировали в вакууме, и остаток распределяли между водой и EtOAc. Суспензию фильтровали и твердый материал являся побочным продуктом. Органический слой промывали водой, солевым раствором и сушили над MgSO4 и концентрировали, при этом получали соединение 38 (2 г, 80%).
Пример 38а
10-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 38а
В расвтор 10-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (9 г, 20 ммоль) в ТГФ (40 мл) добавляли бромид этилмагния в этиловом эфире (22 мл) при -20°С. Реакционную смесь нагревали до КТ и через 1,5 ч завершение реакции подтверждали методом ЖХМС. Реакционную смесь выливали в 10% NH4Cl и экстрагировали EtOAc. Органический слой промывали водой, солевым раствором и сушили над MgSO4 и концентрировали. Неочищенный продукт очищали хроматографией на колонке Isco, при этом получали соединение 38а. ЖХ/МС (ESI+): m/z 265 (М+Н).
Пример 38b
10-(2-фторпиридин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 38b
В смесь соединения 38а (140 мг, 0,53 ммоль) в ДМФ (20 мл) и воды (2 мл) добавляли 2-фторпиридин-3-борную кислоту (89 мг, 0,632 ммоль), ацетат калия (207 мг, 2,11 ммоль) и тетракис(трифенилфосфин)палладия (30 мг, 0,0264 ммоль). Реакционную смесь дегазировали в течение 5 мин и нагрвали при 100°С в течение ночи. Методом ЖХМС наблюдали требуемый пик продукта. Реакционную смесь охлаждали до КТ и фильтровали через тонкий слой целита. Фильтрат промывали водой, солевым раствором, сушили над MgSO4 и концентрировали. Неочищенный остаоок очищали методом препаративной ЖХВР, при этом получали соединение 38b. ЖХ/МС (ESI+): m/z 282 (М+Н)
Пример 38с
3-(5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)пиридин-2(1Н)-он 38с
В раствор соединения 38b (100 мг, 0,4 ммоль) в ДМЭ (4 мл) добавляли 10% водный раствор HCl (4 мл). Реакционную смесь перемешивали и нагревали при 80°С в течение ночи. Реакционную смесь охлаждали до КТ и концентрировали при пониженном давлении. Неочищенный остаоок очищали методом препаративной ЖХВР, при этом получали соединение 38с. ЖХ/МС (ESI+): m/z 280 (М+Н). 1H ЯМР (500 МГц, ДМСО): δ 11,73 (s, 1Н), 8,71 (d, J=2,3, 1H), 7,72-7,50 (m, 1H), 7,47-7,21 (m, 1H), 7,15-6,86 (m, 2H), 6,29 (t, J=6.6, 1H), 4,44 (d, J=6,1, 4H).
Пример 38d
4-(5,6-Дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)пиридин-2(1Н)-он 38d
Соединение 38d получали, как описано в примерах 38а-с. ЖХ/МС (ESI+): m/z 280 (М+Н). 1Н ЯМРR (500 МГц, ДМСО): δ 8,70 (d, J=2,5, 1H), 7,59 (dd, J=8,5, 2,5, 1H), 7,45 (d, J=6,8, 1H), 7,35 (s, 1H), 7,09 (dd, J=16,9, 4,7, 2H), 6,57-6,36 (m, 2H), 4,47 (dd, J=11,6, 5,6, 4H).
Пример 38е
5-(5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)пиридин-2(1Н)-он 38е
Соединение 38е получали, как описано в примерах 38а-с. ЖХ/МС (ESI+): m/z 280 (М+Н). 1H ЯМР (500 МГц, ДМСО): δ 8,48 (s, 1H), 8,03 (s, 1H), 7,94 (s, 1H), 7,83 (d, J=10,8, 1H), 7,77 (d, J=8,7, 1H), 7,21 (d, J=8,7, 2H), 6,46 (d, J=9,8, 1H), 4,65 (dd, J=24,3, 4,8, 4H).
Пример 39
Метиловый эфир 2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 39
Смесь соединения 38 (2 г, 8 ммоль) и NIS (9,2 г, 41 ммоль) в ДМФ нагревали при 80°С в течение ночиt. Смесь разбавляли EtOAc и водой. Вязкую суспензию фильтровали через стеклянный фильтр. Твердый продукт промывали EtOAc, разбавляли ТГФ и сушили над MgSO4. По данным ЖХМС указанный раствор содержал чистый продукт. Раствор коричневого цвета промывали 10% раствором тиосульфата натрия, водой, солевым раствором, сушили над MgSO4 и концентрировали до небольшого объема. Осадок фильтровали и сушили, при этом получали соединение 39 (3,4 г, выход 81%).
Пример 40
Метиловый эфир 2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 40
Свежеприготовленный бромид этилмагния в этиловом эфире (3,0 M, 1,1 мл) по каплям добавляли в суспензию соединения 39 (1,1 г, 2,2 ммоль) в ТГФ при - 15°С. Смесь перемешивали и контролировали с помощью ЖХМС. Через 1 ч в смеси отсутствовал исходный материал и реакционную смесь выливали в насыщенный раствор NH4Cl и экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором, сушили над MgSO4 и концентрировали, при этом 0,7 г (80%) соединения 40.
Пример 41
Метиловый эфир 2-циано-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 41
Соединение 40 (740 мг, 2,3 ммоль) и цианид меди (537 мг, 6,9 ммоль) смешивали в ДМФ (8 мл). Реакционную смесь обрабатывали ультразвуком при 200 Ватт, 150°С в течение 40 мин. Реакционную смесь распределяли между 15% раствром аммиака в воде и EtOAc. Водный слой три раза экстрагировали EtOAc, объединенные экстракты промывали водой, солевым раствором, сушили над MgSO4, при этом получали 0,46 г (выход 74%) соединения 41.
Пример 42
Метиловый эфир 2-карбамоил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 42
Соединение 41 (0,46 г, 1,7 ммоль) перемешивали с карбонатом калия (469 мг, 3,4 ммоль), водой (1,2 мл) и пероксидом водорода (408 мг, 6 ммоль) в ДМСО (7 мл) в течение 4 ч. Смесь разбавляли 70 мл воды и экстрагировали EtOAc. Раствор в EtOAc промывали водой, 5% Na2S2O3, водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме, при этом получали соединение 42 (0,37 г).
Пример 43
9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 43
Стадия 1
5-Бром-2-(1Н-имидазол-2-ил)фенол
Водный раствор 4-бром-2-гидроксибензальдегида (1,0 г, 5 ммоль), 40% водный раствор этандиаля (3,6 г, 24,87 ммоль) и 50% водный раствор аммиака (2,5 г) в метаноле (20 мл) перемешивали в течение 2 ч или долее до завершения реакции. Растворитель удаляли на роторном испарителе и остаток распределяли между EtOAc и водой. Смесь фильтровали для удаления осадка. Величину рН доводили до 5-6 аккуратным добавлением 1Н. HCl. Водный слой трижды экстрагировали EtOAc. Объединенный органический слой промывали водой, солевым раствором и сушили над MgSO4. После очистки хроматографией на колонке ISCO (элюент: 30% EtOAc/ДХМ) получали 0,9 г 5-бром-2-(1Н-имидазол-2-ил)фенола в виде твердого вещества желтого цвета.
Стадия 2
9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
Смесь 5-бром-2-(1Н-имидазол-2-ил)фенола (0,9 g, 4 ммоль), 1,2-дибромэтана (1,3 мл, 15 ммоль) и карбоната цезия (4,9 г, 15 ммоль) в ДМФ (20 мл) нагревали при 90°С в течение 12 ч. Смесь распределяли между водой и EtOAc. Органический слой промывали водой, солевым раствором, сушили над MgSO4 и концентрировали в вакууме, при этом получали 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,8 г).
Стадия 3
9-Бром-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
Смесь 9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (0,8 г, 3 ммоль) и NIS (1,87 г, 8,3 ммоль) в ДМФ перемешивали при КТ в течение 48 ч. Смесь разбавляли EtOAc, промывали 5% раствором бикарбоната натрия, 10% раствором тиосульфата натрия, водой и солевым раствором, и органический слой сушили над MgSO4 и концентрировали, при этом получали твердый остаток. После очистки хроматографией на колонке ISCO (элюент: 30% EtOAc/гептан) получали 9-бром-2,3-дииод-5,6-дигидробензо[f]imidazo[1,2-d][1,4]оксазепин в количестве 1,2 г.
Стадия 4
9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
3,0 М раствор бромида этилмагния в эфире (1,1 мл) по каплям добавляли в суспензию 9-бром-2,3-дииод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,1 г, 2,2 ммоль) в ТГФ при -15°С. Смесь перемешивали и контролировали ЖХ/МС. Через 1 ч исходный материал отсутствовал и реакционную смесь выливали в насыщенный раствор NH4Cl и экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором, сушили над MgSO4 и концентрировали. Неочищенный остаток очищали экспресс-хроматографией на колонке, при этом получали 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин в виде твердого вещества белого цвета (0,7 г).
Стадия 5
Смешивали 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (1,5 г, 3,8 ммоль), хлорид бис(трифенилфосфин) палладия(II) (142 мг, 0,202 ммоль), ДМФ (45 мл) и гексаметилдисилан (4,34 мл, 20,6 ммоль). Расвтор пробулькивали СО и герметизировали, при этом баллон с СО не отсоединяли. Реакционный сосуд нагревали 70°С в течение 2 ч. По данным ЖХ/МС наблюдали завершение реакции. Смесь охлаждали до КТ и выливали в 1н. HCl (30 мл). Смесь перемешивали в течение 5 мин и нейтрализовали добавлением насыщенного водного раствора NaHCO3. Смесь трижды экстрагировали EtOAc, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток обрабатывали изопропиловым спиртом, твердые продукты объединяли после фильтрования и промывания EtOAc, при этом получали 734 мг (выход 62%) соединения 43 в виде твердого вещества темно-коричневого цвета. ЖХ/МС (ESI+): m/z 310 (М+Н). 1Н ЯМР (400 МГц, CDCl3): δ 8,36 (d, J=8,5, 1H), 7,63 (s, 1H), 7,24 (dd, J=7,2, 4,2, 1H), 7,09-6,99 (m, 1H), 4,51-4,36 (m, 4H).
Пример 44
9-Бром-N-формил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 44
9-Бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (10 г, 25,6 ммоль) нагревали в формамиде (200 мл) в присутствии Pd(dppf)Cl2 (0,94 г, 1,28 ммольl) и DMAP (3,13 г, 25,6 ммоль) при пробулькивании СО при 70°С в течение 2,5 ч. Смесь охлаждали до КТ, разбавляли EtOAc и фильтровали. Полученный остаток сушили, при этом получали соединение 44 (6,7 г, 78%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 11,10 (d, J=9,6Hz, 1H), 9,21 (d, J=9,6Hz, 1H), 8,53 (d, J=8,8 Hz, 1H), 8,24 (s, 1H), 7,34-7,28 (m, 2H), 4,53-4,50 (m, 4H). ЖХ/МС: (ESI, m/z)=336 [M+H]+.
Пример 46
[1-Диметиламиноэт-(Е)-илиден]-амид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты 46
В раствор соединения 51 (0,280 г, 0,000909 моль) в толуоле (5 мл) добавляли диметилацетамид диметилацеталя (0,405 мл, 0,00273 моль). Раствор перемешивали при 95°С в течение 4 ч. Толуол удаляли в вакууме, при этом получали соединение 46. MC(ESI+) 377,1/379,1.
Пример 47
трет-Бутиловый эфир [5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-ил]карбаминовой кислоты 47
Стадия 1
Метиловый эфир 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты
8-Бром-2-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен (6,000 г, 0,01534 моль), ацетат палладия (0,1722 г, 0,0007672 моль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (0,8879 г, 0,001534 моль) последовательно добавляли в сухую заполненную азотом колбу. В колбу добавляли дегазированный ТЭА (180 мл, 1,3 моль) и метанол (60 мл), и реакционную смесь тщательно пробулькивали монооксидом углерода в течение приблизительно 3 мин. К колбе присоединяли 2 баллона с монооксидом углерода и реакционную смесь нагревали при 50°С в течение 3 ч. Реакционную смесь пробулькивали азотом, концентрировали в вакууме и в сухом виде наносили на силикегель. Неочищенный продукт очищали экспресс-хроматографией (элюент: 40-100% EtOAc в гексане, затем 5-15% МеОН в ДХМ), при этом получали метиловый эфир 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (4,242 г) в виде твердого веществ светло-коричневого цвета. MS(ESI+) 323,0/325,0
Стадия 2
8-Бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновая кислота
В раствор метилового эфира 8-бром-4,5-дигидроо-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (1,000 г, 0,003095 моль) в ТГФ (7,50 мл) и воды (4,5 мл) добавляли гидроксид лития (0,2964 г, 0,01238 моль). Реакционную смесь перемешивали при 45°С в течение 2 ч. Смесь подкисляли до рН=1 добавлением 2н. HCl. Полученный осадок отфильтровывали, промывали хлодной водой, при этом получали 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновую кислоту (860 мг) в виде твердого вещества грязно-белого цвета. MC(ESI+) 309,0/311,0.
В другом варианте в раствор 8-бром-2-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (10 г, 25,6 ммоль) в ТГФ (120 мл) при -78°С добавляли nBuLi (19,2 мл, 1,6 М в гексане, 30,7 ммоль) со скоростью, обеспечивающей Тмакс<-73°С. В процессе добавление фиолетовое окрашивание ослабевало и наблюдалось образование осадка желто-коричневого цвета. Реакционную смесь перемешивали при -78°С в течение 20 мин. СО2, полученный из сухого льда и пропущенный через осушитель из диоксида кремния, пробулькивали через реакционную смесь в течение 30 мин. Температуру поднимали до -55°С, затем снова опускали до -78°С. В процессе добавления CO2 быстро образовывался плотный осадок. Реакционную смесь перемешивали при -78°С в течение 1 ч. Реакцию останавливали выливанием реакционной смеси в 20 мл воды (предупреждение: возможно образование искр). Смесь нагрвали до КТ. Величину рН доводили приблизительно до рН 8 добавлением насыщенного водного раствора NaHCO3 и водный слой промывали EtOAc. Водные фракции собирали и величину рН доводили приблизительно до рН 4 добавлением АсОН. Полученный осадок собирали фильтрованием, промывали водой и сушили в вакууме, при этом получали 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновую кислоту в виде твердого вещества бежевого цвета (4,38 г, 55%). 1H ЯМР (400 МГц, d6-ДМСО): 8,31 (1Н, d, J=8,5 Гц), 7,98 (1Н, s), 7,32 (1Н, dd, J=8,5, 2,2 Гц), 7,27 (1Н, d, J=2,2 Гц), 4,51-4,47 (4Н, m). ЖХМС: RT=3,67 мин, M+H+=309/311 (40%), M+Na+=323/325 (100%). По данным 1Н ЯМР продукт содержал приблизительно 5% 8-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты.
Стадия 3
трет-Бутиловый эфир{[(Е)-8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбонилимино]метилтиометил}карбаминовой кислоты
В раствор 8-бром-4,5-дигидроокса-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (0,839 г, 0,00271 моль) и оксалилхлорида (2 М раствор в ДХМ, 1,36 мл, 0,002714 моль) в ДХМ (16,70 мл) в атмосфере азота добавляли 1 каплю ДМФ. Расвтор перемешивали при КТ в течение 2 ч. Реакционную смесь концентрировали в вакууме и гидрохлорид повторно растворяли в ДХМ (9,0 мл). Полученный раствор по каплям добавляли в раствор N-трет-бутоксикарбонил-3-метилпсевдотиомочивины (0,5164 г, 0,002714 моль) и ТЭА (1,173 мл, 0,008414 моль) в ДХМ (9,0 мл). Реакционную смесь перемешивали при КТ в течение 1,5 ч. Добавляли ДХМ и воду и смесь трижды экстрагировали ДХМ. Добавляли насыщенный раствор карбоната натрия и смесь экстрагировали хлороформом. Органические слои объединяли и концентрировали. Продукт повтрно растворяли в ДХМ и метаноле и фильтровали. Фильтрат собирали, сушили и в сухом виде наносили на силикагель и очищали экспресс-хроматографией (элюент: 0-15% МеОН в ДХМ), при этом получали трет-бутиловый эфир {[(Е)-8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбонилимино]метилтиометил}карбаминовой кислоты (658 мг) в виде твердого вещества грязно-белого цвета. MC(ESI+) 481,0/483,0.
Стадия 4
В раствор трет-бутилового эфира {[(Е)-8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбонилимино]метилтиометил}карбаминовой кислоты (0,658 г, 0,00137 моль) в ДМФ (7,50 мл) добавляли N,N-диизопропилэтиламин (0,9524 мл, 0,005468 моль), затем гидрохлорид изопропилгидразина (0,2267 г, 0,002050 моль). Реакционную смесь перемешивали при КТ в течение 4 ч. В смесь добавляли воду и ДХМ и смесь трижды экстрагировали ДХМ. Органические слои объединяли, сушили над MgSO4 и концентрировали. Неочищенный остаток очищали экспресс-хроматографией (элюент: 0-10% МеОН в ДХМ), при этом получали соединение 47 (642 мг) в виде клейкого твердого вещества светло-желтого цвета. Материал использовали на следующей стадии без дополнительной очистки. MC(ESI+) 489,1/491,1
Пример 48
8-Бром-2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен 48
В раствор соединения 46 (0,340 г, 0,000901 моль) в уксусной кислоте (3,0 мл, 0,053 моль) добавляли гидрохлорид изопропилгидразина (0,1196 г, 0,001082 моль). Реакионную смесь нагревали при 95°С в течение 3 ч. Уксусную кислоту удаляли в вакууме и продукт наносили на силикагель и очищали экспресс-хроматографией (элюент: 0-10% МеОН в ДХМ), при этом получали соединение 48 (293 мг) в виде твердого вещества оранжевого цвета. MC(ESI+) 388,1/390,1
В другом варианте соединение 48 получали с использованием смеси гидрохлорида 4-бром-2-фторбензамидина (5,67 г, 22,3 ммоль), гидрокарбоната калия (8,95 г, 89,4 ммоль), ТГФ (45 мл) и воды (10 мл), которую нагрвали до кипения и по каплям добавляли раствор соединения 91 (5,5 г, 22,3 ммоль) в тГФ (15 мл). Реакционную смесь кипятили с обратным холодильником в течение 18 ч, затем летучий растворитль удаляли в вакууме. Полученную суспензию фильтровали и и остаток обрабатывали горячим этиловым эфиром, при этом получали 5-[2-(4-бром-2-фторфенил)-1Н-имидазол-4-ил]-1-изопропил-3-метил-1Н-[1,2,4]триазол в виде твердого вещества грязно-белого цвета (6,4 г, 79%). 1Н ЯМР 400 МГЦ (ДМСО-d6): δ 7,97 (1Н, t, J=8,30 Гц), 7,81 (1H, s), 7,76 (1H, dd, J=10,68, 1,92 Гц), 7,58 (1H, dd, J=8,42, 1,93 Гц), 5,79 (1Н, ушир., m), 2,26 (3H, s), 1,44 (6H, d, J=6,60 Гц).
Суспензию 5-[2-(4-бром-2-фторфенил)-1Н-имидазол-4-ил]-1-изопропил-3-метил-1Н-[1,2,4]триазола (2,9 г, 7,96 ммоль) в толуоле (50 мл) обрабатывали этиленкарбонатом (25 мл) и кипятили с обратным холодильником в течение 5 ч. Охлажденную реакционную смесь разбавляли ДХМ и пропускали через слой силикагеля, используя в качестве элюента ДХМ, а затем 20% раствор метанола в ДХМ. Фракции в метаноле объединяли и концентрировали в вакууме, при этом получали твердое вещество светло-коричневого цвета. Твердое вещество обрабатывали диэтиловым эфиром, при этом получали 2-[2-(4-бром-2-фторфенил)-4-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)имидазол-1-ил]этанол в виде твердого вещества белого цвета (2,3 г, 71%). ЖХМС: RT=2,85 мин, [М+Н]+=408/410. 1Н ЯМР 400 МГц (CDCl3): δ 8,16 (1H, s), 7,67-7,20 (3H, m), 5,83 (1H, m), 4,05 (2H, t, J=5,10 Гц), 3,92 (2H, t, J=5,10 Гц), 2,44 (3H, s), 1,50 (6H, d, J=6,65 Гц).
Суспензию 2-[2-(4-бром-2-фторфенил)-4-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)имидазол-1-ил]этанола (2,3 г, 5,6 ммоль) в ДМФ (50 мл) порциями в течение 5 мин обрабатывали гидридом натрия (60% дисперсия, 247 мг, 6,2 ммоль) и смесь перемешивали при КТ в течение 1 ч. Реакцию останавливали медленным добавлением воды (200 мл). Образовавшийся осадок отфильтровывали, промывали водой и при этом получали соединение 48 в виде твердого вещества белого цвета (1,64 г, 53%). ЖХМС: RT=3,43 мин, [М+Н]+=388/390. 1H ЯМР 400 МГц (CDCl3): δ 8,37 (1H, d, J=8,61 Гц), 7,70 (1H, s), 7,26-7,25 (2H, m), 5,87-5,86 (1H, m), 4,50-4,48 (2H, m), 4,46-4,42 (2H, m), 2,42 (3H, s), 1,57 (6H, d, J=6,64 Гц)
Пример 49
9-Бром-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен 49
Стадия 1
7-Бром-3,4-дигидро-2Н-бензо[b]окзепин-5-он
К раствору 5'-бром-2'-гидроксиацетофенона (10 г, 46,5 ммоль) в метилэтилкетоне (100 мл) при перемешивании добавляли K2CO3 (13,5 г, 97,7 ммоль) и 1,2-дибромэтан (20 мл, 232,5 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 16 ч, затем охлаждали до КТ. Реакционную смесь фильтровали и концентрировали в вакууме. Полученный остаток растворяли в смеси диэтиловый эфир/EtOAc (4:1, 500 мл) и образовавшийся осадок удаляли фильтрованием. Фильтрат промывали 2н. NaOH (100 мл) и органическую фракцию сушили над Na2SO4 и концентрировали в вакууме, при этом получали 1-[5-бром-2-(2-бромэтокси)фенил]этанон (8,07 г, 55%), который использовали на следующей стадии без дополнительной очистки.
В суспензию NaH (60% дисперсия в минеральном масле) (1,48 г, 37,1 ммоль) в ТГФ (50 мл) при КТ добавляли [5-бром-2-(2-бромэтокси)фенил]этанон (8,07 г, 25,1 ммоль). Реакционную смесь медленно нагревали до кипения и перемешивали в течение 20 ч. Растворитель удаляли в вакууме и остаток очищали экспресс-хроматографией (SiO3, элюент: 4:1 EtOAc/петролейный эфир), при этом получали 7-бром-3,4-дигидро-2Н-бензо[b]окзепин-5-он в виде маслянистого вещества желтого цвета (4,22 г, 70%). 1H ЯМР (CDCl3): δ 2,15-2,29 (2Н, m), 2,89 (2Н, t, J=7,0 Гц), 4,24 (2Н, t, J=6,6 Гц), 6,97 (1Н, d, J=8,8 Гц), 7,50 (1Н, dd, J=2,6, 8,1 Гц), 7,87 (1Н, d, J=2,6 Гц).
Стадия 2
7-Бром-4-[1-диметиламиномет-(Е)-илиден]-3,4-дигидро-2Н-бензо[b]-окзепин-5-он
7-Бром-3,4-дигидро-2Н-бензо[b]окзепин-5-рн (10,0 г, 41,5 ммоль) в диметилацтальдиметилформамиде (100 мл) нагревали при 110°С в течение 18 ч. Реакционную смесь охлаждали до КТ и добавляли циклогексан (100 мл). Образовавшийся твердый осадок собирали фильтрованием, промывали циклогексаном и сушили в вакууме при 40°С, при этом получали 7-бром-4-[1-диметиламиномет-(Е)-илиден]-3,4-дигидро-2Н-бензо[b]окзепин-5-он в виде кристаллического вещества желтого цвета (8,19 г, 67%). 1Н ЯМР (CDCl3): δ (ч/млн) 7,83 (1Н, d, J=2,59 Гц), 7,74 (1H, s), 7,46 (1H, dd, J=8,51, 2,58 Гц), 6,88 (1H, d, J=8,52 Гц), 4,27-4,19 (2H, m), 3,14 (6H, s), 2,76-2,69 (2H, m).
Стадия 3
В суспензию 8-бром-4-[1-диметиламиномет-(Е)-илиден]-3,4-дигидро-2Н-бензо[b]окзепин-5-она (8,19 г, 27,7 ммоль) в этаноле (100 мл) добавляли порошкообразный дигидрохлорид гидразина (5,81 г, 55,3 ммоль) при КТ и смесь перемешивали в течение 3 ч. Реакционную смесь концентрировали почти досуха в вакууме и добавляли изопропиловый спирт (200 мл) и воду (100 мл). Полученную смесь кипятили с обратным холодильником в течение 3 ч и затем охлаждали до КТ. Смесь концентрировали в вакууме для удаления летучих раствортелей, затем разбавляли 400 мл воды. Полученный твердый осадок собирали фильтрованием, промывали водой и сушили в вакууме при 40°С, при этом получали соединение 49 в виде твердого вещества желтого цвета (7,8 г, 106%). 1Н ЯМР (CDCl3): δ (ч/млн) 8,27 (1H, d, J=2,45 Гц), 7,59 (1H, s), 7,32 (1H, dd, J=8,64, 2,41 Гц), 6,94 (1H, d, J=8,64 Гц), 4,34-4,28 (2H, m), 3,15-3,09 (2H, m).
Пример 50
8-Бромо-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен 50
Стадия 1
8-Бром-4-[1-диметиламиномет-(Е)-илиден]-3,4-дигидро-2Н-бензо[b]-окзепин-5-он
8-Бром-3,4-дигидро-2Н-бензо[b]окзепин-5-он (5,0 г, 20,7 ммоль) в диметилацетальдиметилформамиде (15 мл) нагревали при 110°С в течение 18 ч.
Реакционную смесь охлаждали до КТ и добавляли циклогексан (20 мл). Полученный твердый осадок собирали фильтрованием, промывали водой и сушили в вакууме при 40°С, при этом получали 8-бром-4-[1-диметиламиномет-(Е)-илиден]-3,4-дигидро-2Н-бензо[b]-окзепин-5-он в виде кристаллического вещества желтого цвета (5,32 г, 86%). 1Н ЯМР (CDCl3): δ (ч\млн) 7,73 (1Н, s), 7,61 (1Н, d, J=8,29 Гц), 7,29 (1Н, dd, J=8,29, 1,94 Гц), 7,18 (1Н, d, J=1,91 Гц), 4,28-4,21 (2Н, m), 3,14 (6Н, s), 2,77-2,70 (2Н, m).
Стадия 2
В суспензию 8-бром-4-[1-диметиламиномет-(Е)-илиден]-3,4-дигидро-2Н-бензо[b]окзепин-5-она (5,32 г, 17,9 ммоль) в изопропиловом спирте (50 мл) добавляли порошкообразный дигидрохлорид гидразина (3,77 г, 35,9 ммоль) при КТ и смесь перемешивали в течение 2 ч. Реакционную смесь разбавляли водой (20 мл) и затем нагревали при 100°С в течение 2 ч и затем охлаждали до КТ. Реакционную смесь концентрировали в вакууме для удаления летучего растворителя. Полученную суспензию фильтровали и фильтрат промывали водой и сушили в вакууме при 40°С, при этом получали соединение 50 в виде твердого вещества светло-желтого цвета (4,28 г, 90%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,07 (1Н, d, J=8,52 Гц), 7,64 (1Н, s), 7,30-7,24 (1Н, m), 7,21 (1Н, d, J=2,07 Гц), 4,24 (2Н, dd, J=5,63, 4,50 Гц), 3,00 (2Н, t, J=5,09 Гц).
Пример 51
Амид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты 51
В раствор 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (8,27 г, 26,7 ммоль), EDCI (6,66 г, 34,8 ммоль), HOBt (4,69 г, 34,8 ммоль) и хлорида аммония (4,29 г, 80,2 ммоль) в ДМФ (80 мл) добавляли ТЭА (7,49 мл, 53,5 ммоль) и реакционную смесь перемешивали при 45°С в течение 1,5 ч. Реакционную смесь концентрировали в вакууме и остаток обрабатывали водой (250 мл). Осадок собирали фильтрованием и сушили в вакууме при 45°С в течение 16 ч, при этом получали соединение 51 в виде твердого вещества темно-желтого цвета (7,67 г, 93%). 1Н ЯМР (400 МГц, d6-ДМСО): 8,40 (1Н, d, J=8,7 Гц), 7,80 (1Н, s), 7,42 (1Н, ушир. s), 7,32 (1Н, dd, J=8,7, 2,0 Гц), 7,27 (1Н, d, J=2,1 Гц), 7,15 (1Н, ушир. s), 4,50-4,46 (4H, m). ЖХМС: RT=3,07 мин, М+Н+=308/310. По данным 1Н ЯМР продукт содержал 5% амида 8-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты.
В другом варианте раствор 8-бром-2-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (10,00 г, 0,02558 моль) в ДМФ (250 мл) осторожно дегазировали N2. В раствор добавляли хлорид бис(трифенилфосфин)палладия(II) (0,807 г, 0,00115 моль) и дисилазан (21,58 мл, 0,1023 моль). Раствор продували СО в течение 2 мин и затем герметизировали, при этом балло с СО не отсоединяли. Реакционную смесь нагревали до 70°С в течение 2,5 ч. Добавляли ДХМ и насыщенный раствор NH4Cl и смесь экстрагировали 4 раза ДХМ. Органические фазы объединяли и сушиди над MgSO4 и концентрировали. Добавляли небольшое количество изопропанола и смесь растирали в течение ночи. Смесь фильтровали, при этом получали 5,97 г (выход 76%) соединения 51 в виде порошкообразного продукта коричневого цвета. MS(ESI+) 308,.0/310,0
Пример 52
8-Бром-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен 52
Стадия 1
1-Диметиламиномет-(Z)-илиденамид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты
В суспензию соединения 51 (7,67 г, 24,9 ммоль) в диоксане (150 мл) добавляли ДМФ-ДМА (9,92 мл, 74,7 ммоль) и реакционную смесь нагревали при 100°С в течение 1 ч. В ходе реакции наблюдали растворение твердого вещества и образование раствора коричневого цвета. Реакционную смесь концентрировали в вакууме и твердый остаток обрабатывали диэтиловым эфиром (приблизительно 150 мл). Продукт собирали фильтрованием и сушили в вакууме при 45°С в течение 3 ч, при этом получали 1-диметиламиномет-(Z)-илиденамид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты в виде твердого вещества темно-коричневого цвета (8,52 г, 94%). 1Н ЯМР (400 МГц, d6-ДМСО): 8,56 (1Н, s), 8,34 (1Н, d, J=8,6 гц), 7,96 (1Н, s), 7,32 (1H, dd, J=8,6, 2,0 Гц), 7,26 (1Н, d, J=2,1 гц), 4,51-4,46 (4H, m), 3,16 (3H, s), 3,08 (3H, s). По данным 1Н ЯМР продукт содержит 5% 1-диметиламиномет-(Z)-илиденамида 8-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты.
Стадия 2
В раствор 1-диметиламиномет-(Z)-илиденамида 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты в уксусной кислоте добавляли гидрохлорид изопропилгидразина. Реакционную смесь нагревали при 95°С в течение 3 ч. Уксусную кислоту удаляли в вакууме и продукт в твердом виде наносили на силикагель и очищали экспресс-хроматографией (элюент: 0-10% МеОН в ДХМ), при этом получали 1-диметиламиномет-(Z)-илиденамид 8-бромо-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты.
Пример 53
1-(2-(Тетрагидро-2Н-пиран-2-илокси)этил)-4-(трибутилстаннил)-1Н-имидазол 53а и 1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-5-(трибутилстаннил)-1H-имидазол 53b
Хлрид изопропилмагния (iPrMgCl-LiCl, 4,3 мл 1,3 М) в ТГФ по каплям добавляли в раствор 4-иод-1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол (1,50 г, 4,66 ммоль, смесь региоизомеров) в ТГФ (20 мл, 0,3 моль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч. Затем добавляли хлорид трибутилолова (1,64 мл, 6,05 ммоль) и смесь нагревали до КТ и перемешивали в течение ночи. Реакционную смесь упаривали на роторном испарителе и реакцию останавливали добавлением воды, разбалвляи ДХМ и фильтровали через целит. Водный слой экстрагировали и неочищенные концентрированные органические слои очищали экспресс-хроматографией на колонке (элюент: 50-100% этилацетат в гексане). По данным ЯМР соотношение соединение 53а и 53b составляло 2:1 (в предположении из литературных данных об одинаковых заместителях имидазола). Региоизомеры не разделяли.
Пример 54
1-(4-Бром-1Н-имидазол-1-ил)-2-метилпропан-2-ол и 1-(5-бром-1Н-имидазол-1-ил)-2-метиопропан-2-ол 54
В суспензию 4-бром-1H-имидазола (1,0 г, 6,8 ммоль) и изобутиленоксида (0,665 мл, 7,48 ммоль) в метаноле (0,331 мл, 8,16 ммоль) добавляли карбонат цезия (0,63 г, 1,9 ммоль). Реакционную смесь осторожно нагревали в закрытом сосуде при 110°С в течение 1,5 ч. Реакционную смесь охлаждали до КТ, разбавляли диэтиловым эфиром и 2 раза промывали водой. Органические фазы промывали солевым раствором, сушили над сульфатом магния и концентрировали в вакууме, при этом получали твердое вещество белого цвета, которое очищали экспресс-хроматографией (элюент: 100% EtOAc), при этом получали 2 индивидуальных промежуточных соединения. Основным региоизомером являлся 1-(4-бром-1Н-имидазол-1-ил)-2-метилпропан-2-ол (0,8 г, выход 54%, М+1 220), побочным региоизомером являлся 1-(5-бром-1Н-имидазол-1-ил)-2-метилпропан-2-ол (0,32 г, выход 21%, М+1 220).
Пример 55
N,N-Диэтил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)-1Н-пиразол-1-ил)этанамин 55
В раствор 4,4,5,5-тетраметил-2-(1Н-пиразол-4-ил)-1,3,2-диоксаборолана (250 мг, 1,29 ммоль) и гидрида натрия (61,8 мг, 2,58 ммоль) в ТГФ при 0°С добавляли 2-бром-N,N-диэтилэтанамин (558 мг, 2,58 ммоль). Реакционную смесь нагревают до КТ и за ходом реакции наблюдают с помощью ЖХМС. Через 90 мин реакция практически останавливалась, в реакционную смесь добавляли иодид калия (1,71 г, 10,3 ммоль) и реакционную смесь нагревали в течение ночи при 50°С. Реакционную смесь разбавляли большим объемом EtOAc и воды и распределяли. Органический слой, содержащий продукт, промывали солевым раствором и концентрировали в вакууме, при этом получали вязкий прозрачный маслообразный продукт, который по данным ЖХМС на 100% составлял чистое соединение 55 (340 мг, выход 90%, М+1 294,2)
Пример 56
1-(2-(трет-Бутилдиметилсилилокси)-2-метилпропил)-4-(триметилстаннил)-1H-имидазол 56
Стадия 1
2,4,5-трииод-1Н-имидазол
В смесь 1H-имидазола (50 г, 0,73 моль) в ДМФ (200 мл) порциями добавляли NIS (328 г, 1,46 моль), реакционную смесь перемешивали при КТ в течение 4 ч. Реакционную смесь выливали в насыщенный раствор Na2CO3. фильтровали, остаток промывали водой и сушили, при этом получали 150 г 2,4,5-трииод-1H-имидазола (выход 46%).
Стадия 2
4-иод-1Н-имидазол
2,4,5-Трииод-1Н-имидазол вводиди в реакцию с Na2SO3 в ДМФ (250 мл) и перемешивали при 110°С в течение ночи в атмосфере N2. Реакционную смесь фильтровали, фильтрат концентрировали и выливали в воду, затем экстрагировали EtOAc, органический слой промывали водой, сушили над Na2SO4, концентрировали и очищали на колонке с силикагелем, при этом получали 4-иод-1Н-имидазол (Выход 55%). ЖХ-МС: m/z=195 [М+Н+].
Стадия 3
1-(4-Иод-1Н-имидазол-1-ил)-2-метилпропан-2-ол
Смесь 4-иод-1H-имидазола, 0,5 экв. Cs2CO3 в 2,2-диметилоксиране перемешивали при 120°С в течение 20 мин при микроволновом облучении. Реакционную смесь концентрировали и очищали, при этом получали 1-(4-иод-1Н-имидазол-1-ил)-2-метилпропан-2-ол (выход 71%). ЖХ-МС: m/z=266 [М+Н+]. 1H ЯМР (CDCl3, 400 МГц): δ 7,36 (s, 1Н), 7,06 (s, 1H), 3,84 (s, 2H), 1,22 (s, 6H).
Стадия 4
1-(2-(трет-Бутилдиметилсилилокси)-2-метилпропил)-4-иод-1Н-имидазол
1-(4-Иод-1Н-имидазол-1-ил)-2-метилпропан-2-ол растворяли в ДХМ и по каплям при 0°С добавляли лутидин. Смесь перемешивали при 0°С в течение 30 мин и затем по каплям добавляли трет-бутилдиметилсилилтрифлат (TBSOTf). Смесь нагревали до КТ и оставляли на 1 ч, затем реакцию останавливали добавлением 30% уксусной кислоты, экстрагировали этилацетатом, сушили и концентрировали, при этом получали 1-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-иод-1Н-имидазол (выход 74%). ЖХ-МС: m/z=381 [M+H+].
Стадия 5
В смесь 1-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-иод-1Н-имидазола и ДХМ добавляли бромид этилмагния (1,5 экв.) при -78°С. Температуру смеси медленно поднимало приблизительно на 10°С, а затем снова снижали. В смесь по каплям добавляли хлорид триметилолова (1,6 экв.) при -78°С. Температуру смеси медленно поднимали до КТ. Реакционную смесь выливали в насыщенный раствор NH4Cl и экстрагировали ДХМ. Органическую фазу дважды промывали водой, сушили над безводным Na2SO4 и концентрировали, при этом получали соединение 56 (выход 74%). ЖХ-МС: m/z=419[M+H+]. 1H ЯМР (CDCl3, 400 МГц): δ 7,63 (s, 1H), 7,00 (s, 1H), 3,79 (s, 2H), 1,22-1,19 (s, 6H), 0,86 (s, 9H), 0,27 (s, 6H), 0,02 (s, 6H).
Пример 57
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-9-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 57
Стадия 1
9-Бром-2-(1-изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин
Изопропилиодид (165 мкл, 1,65 ммоль) добавляли в смесь 417 мг (1,21 ммоль) соединения 34 и карбоната цезия (538 мг, 1,65 ммоль) в 3 мл ДМФ. Реакционную смесь перемешивали при КТ в течение 18 ч, смешивали с водой и экстрагировали EtOAc. Органический экстракт промывали водой, солевым раствором и сушили над MgSO4, окнцентрировали и очищали на колонке с 4 г силикагеля (элюент: 4-5% метанол в ДХМ), при этом получали 210 мг 9-бром-2-(1-изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина. МС: 387,1.
Стадия 2
Раствор 9-бром-2-(1-изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,00 г, 0,00258 моль) и ацетата калия (0,758 г, 0,00773 моль) в ДМСО (8,5 мл, 0,12 моль) в круглодонной колбе, снабженной магнитным мешальником, тщательно прокачивали азотом. Биспинасоловый эфир борной кислоты (0,719 г, 0,00283 моль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с ДХМ (1:1) (0,210 г, 0,258 ммоль) добавляли в колбу и реакционную смесь нагревали при 85°С в инертной атмосфере. За ходом реакции следили с помощью ЖХ/МС, которая завершалась через 6 ч. Смесь распределяли между водой и ДХМ и смесь три раза экстрагировали ДХМ. Органические фазы объединяли, сушили над MgSO4 и концентрировали. Продукт наносили на силикагель и очищали экспресс-хроматографией (элюент: 0-10% МеОН в ДХМ, затем 100% EtOAC), при этом получали соединение 57 (488 мг) в виде твердого вещества бежевого цвета. MC(ESI+) 436,2.
Пример 58
9-Бром-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен 58
Соединение 58 получали аналогично тому, как получали 8-бром-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен, из соединения 49 (450 мг, 1,7 ммоль) и 5-хлор-1-изопропил-1Н-[1,2,4]триазола (369 мг, 2,55 ммоль) в виде твердого вещества белого цвета (375 мг, 59%). ЖХМС: RT=5,05 мин, М+Н+=374/376.
Пример 59
9-Бромо-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен 59
Соединение 59 получали аналогично тому, как получали 8-бром-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен, из 5-хлор-1-(2,4-дифторфенил)-1Н-[1,2,4]триазола (1,33 г, 6,16 ммоль) и соединения 49 (1,36 г, 5,13 ммоль), неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: от 0 до 35% EtOAc в циклогексане), при этом получали соединение 59 (1,42 г, 62%). ЖХМС: RT=4,80 мин, M+H+=444/446.
Пример 60
9-Бром-2-[2-(2-хлорфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен 60
Согласно тому, как описано для получения 1-(2,4-дифторфенил)-1Н-[1,2,4]триазола (Пример 103), из гидрохлорида 2,4-дихлорфенилгидразина и формамида получали 1-(2-хлорфенил)-1Н-[1,2,4]триазол в виде твердого вещества грязно-белого цвета. 1Н ЯМР (CDCl3) δ (ч/млн): 8,54 (1Н, s), 8,14 (1H, s), 7,61-7,54 (2H, m), 7,46-7,39 (2H, m).
Согласно тому, как описано для получения 1-(2,4-дифторфенил)-1Н-[1,2,4]триазола (Пример 103), из 1-(2-хлорфенил)-1Н-[1,2,4]триазола и n-бутиллития и гексахлорэтана получали 5-хлор-1-(2-хлорфенил)-1Н-[1,2,4]триазол в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3) δ (ч/млн): 8,05 (1Н, s), 7,61-7,58 (1H, m), 7,55-7,48 (1H, m), 7,46-7,43 (2H, m).
Соединение 60 получали аналогично тому, как описано для получения 8-бром-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена, из 5-хлор-1-(2-хлорфенил)-1Н-[1,2,4]триазола (2,25 г, 10,5 ммоль) и соединения 49 (1,9 г, 7 ммоль), неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 60% ДХМ (+10% EtOAc) в циклогексане), при этом получали соединение 60 (1,3 г, 33%). ЖХМС RT=4,82 мин, M+H+=442/444.
Пример 61
9-Бром-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен 61
Стадия 1
4-Бром-1-бут-3-инилокси-2-нитробензол 61_1
Смесь 4-бром-1-фтор-2-нитробензола (20,0 г, 90 ммоль), 3-бутин-1-ола (7,0 г, 99,8 ммоль) и карбоната калия (13,8 г, 99,8 ммоль) в сухом ДМФ (20 мл) нагревали в присутствии молекулярных сит с диаметром пор  в течение 43 ч. Смесь охлаждали, разбавляли водой до объема приблизительно 500 мл и 3 раза экстрагировали EtOAc. Объединенные органические экстракты промывали водой и солевым раствором, сушили и концентрировали в вакууме. Полученный осадок очищали экспресс-хроматографией (SiO2, элюент: градиент от 5 до 10% EtOAc в циклогексане), при этом получали соединение 61_1 в виде твердого вещества желтого цвета (17,35 г, 71%). По данным ЯМР продукт содержал 19% примесей, которые на данной стадии не удаляли. ЖХМС: RT=4,41 мин, [M+Na]+=292/294.
в течение 43 ч. Смесь охлаждали, разбавляли водой до объема приблизительно 500 мл и 3 раза экстрагировали EtOAc. Объединенные органические экстракты промывали водой и солевым раствором, сушили и концентрировали в вакууме. Полученный осадок очищали экспресс-хроматографией (SiO2, элюент: градиент от 5 до 10% EtOAc в циклогексане), при этом получали соединение 61_1 в виде твердого вещества желтого цвета (17,35 г, 71%). По данным ЯМР продукт содержал 19% примесей, которые на данной стадии не удаляли. ЖХМС: RT=4,41 мин, [M+Na]+=292/294.
Стадия 2
5-Бром-2-бут-3-инилоксифениламин 61_2
4-Бром-1-бут-3-инилокси-2-нитробензол (чистота 82%, 4,22 г, 12,5 ммоль) нагревали в смеси с IMS (40 мл) и ледяной уксусной кислотой (2 мл) при приблизительно 50°С до образования раствора. В раствор добавляли порошкообразное железо (5,05 г, 89,8 ммоль) и гексагидрат хлорида железа (III) (0,56 г, 1,56 ммоль) и смесь кипятили с обратным холодильником в течение 18 ч. Охлажденную смесь фильтровали через целит и тщательно промывали EtOAc. Фильтрат промывали водой, солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 10 до 20% EtOAc в циклогексане), при этом получали соединение 61_2 в виде маслообразного продукта оранжевого цвета (2,68 г, 89%). ЖХМС: RT=4,10 мин, М+H+=240/242.
Стадия 3
Этиловый эфир хлор-(5-бром-2-бут-3-инилоксофенилгидразоно)уксусной кислоты 61_3
Этиловый эфир 2-хлор-3-оксобутановой кислоты (1,94 г, 11,2 ммоль) и ацетат натрия (1,45 г, 17,8 ммоль) перемешивали в смеси с IMS (100 мл), при этом получали прозрачный раствор, который охлаждали до 0°С. В отдельности 5-бром-2-бут-3-инилоксифениламин (2,68 г, 11,2 ммоль) в 6 М хлористоводородной кислоте (6,8 мл) охлаждали до 0°С и по каплям при перемешивании добавляли раствор нитрита натрия (0,77 г, 11,2 ммоль) в воде (11,2 мл) с последующим перемешиванием при температуре менее 5°С. Кислый водный раствор добавляли в раствор IMS, промытом небольшим количеством воды, при температуре менее 5°С. Через 1 ч выдерживания при 0-5°С смесь разбавляли водой и несколь раз экстрагировали EtOAc. Объединенные органические экстракты промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали соединение 61_3 в виде твердого вещества светло-коричневого цвета (3,96 г, 95%). ЖХМС: RT=4,97 мин, [M+Na]+=395/397/399.
Стадия 4
Этиловый эфир 9-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновой кислоты 61_4
Смесь этилового эфира хлор-(5-бром-2-бут-3-инилоксифенилгидразоно)уксусной кислоты (3,28 г, 8,78 ммоль) и ТЭА (12,2 мл, 88 ммоль) в сухом толуоле (900 мл) кипятили с обратным холодильником (120°С) в течение 54 ч. Охлажденную смесь фильтровали, остаток промывали EtOAc и фильтрат концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 10 до 15% EtOAc в циклогексане), при этом получали соединение 61_4 в виде твердого вещества желтого цвета (2,52 г, 85%). ЖХМС: RT=4,52 мин, M+H+=337/339, [M+Na]+=359/361.
Стадия 5
Амид 9-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновой кислоты 61_5
Этиловый эфир 9-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновой кислоты (1,51 г, 4,48 ммоль) в 2М растворе смеси аммиак/метанол (70 мл) нагревали в автоклаве при приблизительно 120°С (внешняя температура) в течение 30 ч, затем охлаждали. Смесь фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 50 до 100% EtOAc в циклогексане, при этом получали соединение 61_5 в виде твердого вещества светло-желтого цвета (1,11 г, 80%). ЖХМС: RT=4,00 мин, M+H+=308/310, [M+Na]+=330/332.
Стадия 6
1-Диметиламинометилиденамид 9-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновой кислоты 61_6
Смесь амида 9-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновой кислоты (1,11 г, 3,60 ммоль) и диметлацетальдиметилформамида (1,44 мл, 10,8 ммоль) в сухом 1,4-диоксане (25 мл) нагревали при 100°С в течение 2 ч и концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром, при этом получали соединение 61_6 в виде твердого вещества желтого цвета (1,27 г, 97%). ЖХМС: RT=3,27 мин, М+Н+=363/365.
Стадия 7
Смесь 1-диметиламинометилиденамида 9-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновой кислоты (1,27 г, 3,5 ммоль), гидрохлорида изопропилгидразина (0,48 г, 4,37 ммоль) и ледяной уксусной кислоты (6 мл) нагревали при 110°С в течение 6,5 ч, затем охлаждали и концентрировали в вакууме. Полученный остаток растворяли в водном растворе бикарбоната натрия и ДХМ и фазы разделяли. Водную фазу несколько раз экстрагировали ДХМ, объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 30 до 70% EtOAc в циклогексане, при этом получали соединение 61 (0,99 г, 76%). ЖХМС: RT=5,07 мин, М+Н+=374/376. 1Н ЯМР (CDCl3) δ (ч/млн): 8,07 (1Н, d, J=2,41 Гц), 7,96 (1H, s), 7,39 (1H, dd, J=8,63, 2,43 Гц), 7,08 (1Н, d, J=8,63 Гц), 6,91 (1H, s), 5,73-5,65 (1H, m), 4,53 (2H, t, J=5,91 гц), 3,18 (2H, t, J=5,91 гц), 1,60 (6H, d, J=6,62 Гц).
Пример 62
9-Бром-2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен 62
Согласно тому, как описано для соединения 61, из 1-диметиламинометилиденамида 9-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновой кислоты и трифторэтилгидразина (70% водный раствор) получали соединение 62 в виде твредого вещества белого цвета. ЖХМС RT=4,49 мин, M+H+=414/416.
Пример 63
Гидрохлорид 8-азетидин-3-yl-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен 63
Стадия 1
трет-Бутиловый эфир 1-иодцинк-3-азетидин-1-карбоновой кислоты 63_1
В закрытую колбу помещали цинковую пыль (276 мг, 4,22 ммоль) и фильтрующий агент Celpure P65 (60 мгg) и смесь нагревали при 300°С в вакууме в течение 10 мин. Колбу продували аргоном и охлаждали до КТ. В смесь добавляли ДМА (2,4 мл), затем по каплям добавляли смесь хлортриметилсилана (TMSCl) и 1,2-дибромэтана (84 мкл, 7:5 об.:об.), что вызывало небольшое повышение температуры реакционной смеси и образование небольшого количества искр. Реакционную смесь выдерживали при КТ в течение и в нее добавляли по каплям трет-бутиловый эфир 3-иодазетидин-1-карбоновой кислоты (0,96 г, 3,38 ммоль) в виде раствора в ДМА (2 мл). Реакционную смесь перемешивали при КТ в течение 1,5 ч, фильтровали, при этом получали соединение 63_1 в виде бесцветного раствора в ДМА.
Стадия 2
трет-Бутиловый эфир 3-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-карбоновой кислоты 63_2
Раствор 8-бром-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена (1 г, 2,25 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) в виде комплекса с ДХМ (183 мг, 0,22 ммоль) и иодида меди(I) (56 мг, 0,29 ммоль) в ДМА (10 мл) дегазировали в вакууме с последуещим пропусканием аргона (3-кратное повторение. В смесь темно-красного цвета добавляли трет-бутиловый эфир 1-иод-цинк-3-азетидин-1-карбоновой кислоты (1,17 г, 3,38 ммоль) в виде раствора в ДМА (4,4 мл) и смесь нагревали при 85°С в течение 2 ч. В ходе реакции смесь окрашивалась в зеленый цвет, затем в светло-оранжевый, и в конце - в черный. Реакционную смесь разбавляли водой (20 мл) и EtOAc (20 мл) и смесь фильтровали через целит. Органическую часть фильтрата отделяли и водный слой экстрагировали EtOAc (2×20 мл). Объединенные органические фракции промывали солевым раствором (100 мл), сушили (MgSO4) и затем концентрировали в вакууме. Полученный остаток очищали экспресс-ххроматографией (SiO2, элюент: градиент от 0 до 100% EtOAc в циклогексане, при этом получали соединение 63_2 в виде маслянистого вещества желтого цвета (1,1 г, 94%). ЖХМС: RT=4,81 мин, M+H+=521 (100%), M+H+-OtBu=465 (60%), M+H+-Boc=421 (20%).
Стадия 3
трет-Бутиловый эфир 3-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-карбоновой кислоты (1,1 г, 2,11 ммоль) растворяли в растворе хлористоводородной кислоты в диоксане (10 мл, 4 н.) и реакцонную смесь перемешивали при КТ в течение 1 ч. Примерно через 5 мин наблдали образование плотного осадка белого цвета. Реакционную смесь концентрировали в вакууме, при этом получали соединение 63 в виде твердого вщества желтого цвета (1,0 г, 100%). ЖХМС: RT=3,00 мин, M+H+=421.
Пример 64
Гидрохлорид 8-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена 64
Стадия 1
трет-Бутиловый эфир 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил]азетидин-1-карбоновой кислоты
трет-Бутиловый эфир 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил]азетидин-1-карбоновой кислоты получали аналогично тому, как описано для получения трет-бутилового эфира 3-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-карбоновой кислоты из 8-бром-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена и трет-бутилового эфира 1-иодцинк-3-азетидин-1-карбоновой кислоты. ЖХМС: RT=4,85 мин, M+H+=451 (40%), M+H+-OtBu=395 (100%), М+Н+-Вос=351 (10%).
Стадия 2
Соединение 64 получали аналогично тому, как описано для соединения 63 из трет-бутилового эфира 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил]азетидин-1-карбоновой кислоты. ЖХМС: RT=2,86 мин, M+H+=351 (20%), M+H+-iPr=308 (100%).
Пример 65
Гидрохлорид 8-азетидин-3-yl-2-(2-iизопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена 65
Стадия 1
трет-Бутиловый эфир 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]азетидин-1-карбоновой кислоты
трет-Бутиловый эфир 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]азетидин-1-карбоновой кислоты получали аналогично тому, как описано для получения трет-бутилового эфира 3-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-карбоновой кислоты, из соединения 52 и трет-бутилового эфира 1-иодцинк-3-азетидин-1-карбоновой кислоты. ЖХМС: RT=4,61 мин. М+H+=451.
Стадия 2
Соединение 65 получали аналгично тому, как описано для соединения 63 из трет-бутилового эфира 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]азетидин-1-карбоновой кислоты. ЖХМС: RT=2,44 мин, М+Н+=351.
Пример 66
Трифторацетат 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена 66
Стадия 1
трет-Бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты 66_1
трет-Бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты получали аналогично тому, как описано для получения трет-бутилового эфира 3-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-карбоновой кислоты, из соединения 52 (3,0 г, 8,0 ммоль) и трет-бутилового эфира 1-иодцинк-4-пиперидин-1-карбоновой кислоты (12 ммоль, полученного аналогично тому, как описано для получения и трет-бутилового эфира 1-иодцинк-3-азетидин-1-карбоновой кислоты), при этом получали соединение 66_1 (1,2 г, 31%). ЖХМС: RT=5,06 мин, М+Н+=479.
Стадия 2
В раствор трет-бутилового эфира 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (1,2 г, 2,51 ммоль) в ДХМ (12 мл) добавляли ТФУ (8 мл) и реакционную смесь перемешивали при КТ в течение 1 ч. Реакционную смесь концентрировали в вакууме, остаток растирали с диэтиловым эфиром, при этом получали соединение 66 в виде твердого вещества серого цвета (1,34 г, 100%). ЖХМС: RT=2,88 мин, М+Н+=379.
В другом варианте гидрохлорид 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена получали при смешивании соединения 52 (2,1 г, 5,4 ммоль), пинаколинового эфира 3,6-дигидро-2Н-пиридин-1-N-Вос-4-борной кислоты (2,59 г, 8,3 ммоль) и карбоната калия (1,92 г, 13,9 ммоль) в ДМФ (13 мл) и продувании аргоном. В смесь добавляли комплекс PdCl2dppf c ДХМ (310 мг, 0,42 ммоль), снова продували аргоном и нагревали при 80°С в течение 18 ч. После охлаждения реакционную смесь фильтровали через целит, промыввали EtOAc, и фильтрат концентрировали в вакууме. Остаток распределяли между EtOAc и водой, органический слой отделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 2% метанола в EtOAc), при этом получали трет-бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (2,56 г, 96%). ЖХМС RT=4,79 мин, [М+Н]+=477. 1H ЯМР 400 МГц (CDCl3) δ: 8,45 (1H, d, J=8,46 Гц), 7,89 (1H, s), 7,73 (1H, s), 7,19 (1H, dd, J=8,37, 1,80 Гц), 7,04 (1H, d, J=1,87 Гц), 6,15 (1H, s), 6,04-5,96 (1H, m), 4,51-4,43 (4H, m), 4,09 (2H, d, J=3,68 Гц), 3,64 (2H, t, J=5,64 Гц), 2,52 (2H, s), 1,59 (6H, d, J=6,63 Гц), 1,49 (9H, s)
трет-Бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты обрабатывали хлористоводородной кислотой, при этом получали гидрохлорид 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена. 1H ЯМР 400 МГц (ДМСО-d6) δ: 9,08 (2H, s), 8,37 (1H, d, J=8,30 Гц), 8,18 (1H, s), 8,07 (1H, s), 7,06 (1H, dd, J=8,35, 1,80 гц), 6,91 (1H, d, J=1,80 Гц), 5,85 (1H, m), 4,53 (4H, m), 3,35 (2H, d, J=12,46 Гц), 2,98 (2H, m), 2,87 (1H, m), 1,93 (4H, m), 1,50 (6H, d, J=6,57 Гц).
Пример 67
Гидрохлорид 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена 67
Стадия 1
трет-Бутиловый эфир 4-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил1-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты 67_1
трет-Бутиловый эфир 4-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты получали аналогичные тому, как описано для получения трет-бутилового эфира 4-{2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты, из соединения 59 (1,55 мг, 1,13 ммоль), при этом получали соединение 67_1 в виде безцветногоклйкого вещества (1,47 г, 77%). ЖХМС RT=5,01 мин, M+H+=547.
Стадия 2
Гидрохлорид 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена получали аналогично тому, как описано для 9-пиперидин-4-ил-2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена, из трет-бутилового эфира 4-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (2,06 г, 3,77 ммоль), при этом получали соединение 67 в виде твердого вещества белого цвета (1,15 г, 62%). ЖХМС RT=3,04 мин, M+H+=449.
Пример 68
Гидрохлорид (4-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-yl]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-4-ил)метанола 68
Стадия 1
1-трет-Бутиловый эфир 4-этиловый эфир 4-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-1,4-дикарбоновой кислоты 68_1
В раствор дициклогексиламина (291 мкл, 1,463 ммоль) в безводном толуоле (3 мл) по каплям при КТ в атмосфере азота добавляли 2,5 М раствор n-бутиллития в гексане (563 мкл, 1,575 ммоль). Затем смесь перемешивали при КТ в течение 10 мин, затем по каплям при КТ добавляли этиловый эфир N-Boc-пиперидин-4-карбоновой кислоты (305 мкл, 1,24 ммоль) и полученную смесь перемешивали в течение 30 мин. Полученную смесь добавляли в смесь соединения 59 (500 мг, 1,13 ммоль), ди(дибензилиденацетон)палладия (35 мг, 0,06 ммоль), тетрафторбората три-трет-бутилфосфония (17,4 мг, 0,06 ммоль) при КТ в атмосфере азота и затем смесь нагревали до 100°С. После нагрвания в течение 17 ч смесь охлаждали до КТ и очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 50% EtOAc в циклогексане), при этом получали соединение 68_1 (200 мг, 29%). ЖХМС RT=4,93 мин, М+Н+=621.
Стадия 2
В раствор 1-трет-бутилового эфира 4-этилового эфира 4-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-1,4-дикарбоновой кислоты (200 мг, 0,323 ммоль) в безводном ТГФ (10 мл) при 0°С в атмосфере азота по каплям добавляли 1М раствор гидрида лития-алюминия в ТГФ (485 мкл, 0,485 ммоль). Смесь нагревали при 0°С в течение 15 мин и нагревали до КТ. Через 60 мин добавляли новую порцию 1М раствора гидрида лития-алюминия в ТГФ (485 мкл, 0,485 ммоль) и продолжали перемешивание. Через 2 ч смесь охлаждали до 0°С и реакцию останавливали осторожным добавлением насыщенного раствора NH4Cl. Смесь экстрагировали ДХМ и органический слой промывали солевым раствором, сушили (Na2SO4) и растворители удаляли в вакууме. Полученный остаток растворяли в ДХМ (10 мл) и обрабатывали 4 н. Раствором HCl в диоксане (2 мл) при КТ. После перемешивания в течение 5 ч растворитель удаляли в вакууме, твердый остаток растирали с диэтиловым эфиром и собирали фильтрованием (97 мг, 58%). ЖХМС RT=2,84 мин, М+H+=479.
Пример 69
2-[2-(2,2,2-Трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбальдегид 69
Стадия 1
Метиловый эфир 2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты
Суспензию соединения 62 (2,18 г, 5,28 ммоль), гексакарбонилмолибдена (696 мг, 2,64 ммоль), транс-ди(му-ацетато)бис[о-(ди-о-толилфосфино)бензил]дипалладия(II) (240 мг, 0,24 ммоль), тетрафторбората три-трет-бутилфосфония (156 мг, 0,52 ммоль) и ДБУ (792 мкл, 5,28 ммоль) в метаноле (15 мл) и диоксане (15 мл) дегазировали, затем нагревали при 150°С в течение 30 мин с использованием микроволнового излучения. Реакционную смесь разбавляли EtOAc (20 мл), фильтровали и фильтрат концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 30 до 60% EtOAc в циклогексане), при этом получали указанное в заголовке соединение (1,02 г, 49%). 1Н ЯМР (CDCl3): δ (ч/млн) 8,69 (1Н, d, J=2,12 Гц), 8,03 (1Н, s), 7,96 (1Н, dd, J=8,48, 2,12 Гц), 7,22 (1Н, d, J=8,50 Гц), 6,94 (1Н, s), 5,57 (2Н, dd, J=16,24, 8,12 Гц), 4,62-4,56 (2Н, m), 3,94 (3Н, s), 3,29-3,23 (2Н, m).
Стадия 2
2-[2-(2,2,2-Трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновая кислота
В раствор метилового эфира 2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты (553 мг, 1,4 ммоль) в диоксане (12,5 мл) и воде (12,5 мл) добавляли гидроксид лития (67 мг, 2,8 ммоль) и реакционную смесь перемешивали при КТ в течение 2 ч. Реакционную смесь концентрировали в вакууме для удаления диоксана и полученный раствор подкисляли до рН добавлением HCl (12Н.). Полученный осадок собирали фильтрованием, промывали водой и сушили в вакууме при 40°С, при этом получали указанное в заголовке соединение (519 мг, 98%). ЖХМС: RT=4,04 мин, М+Н+=380.
Стадия 3
{2-[2-(2,2,2-Трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}метанол
В раствор метилового эфира 2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты (393 мг, 1 ммоль) в ТГФ (10 мл) при -70°С добавляли DIBAL (3 мл, 1 М раствор в толуоле, 3 ммоль) и реакционную смесь пемерешивали при 0°С в течение 1 ч. Реакционную смесь разбавляли метанолом (5 мл), затем насыщенным водным раствором тартрата натрия-калия. Полученную смесь экстрагировали EtOAc (3×20 мл), затем объединенные органические фракции сушили (MgSO4) и концентрировали в вакууме, при этом получали указанное в заголовке соединение (370 мг, 100%).
Стадия 4
В раствор {2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}метанола (370 мг, 1 ммоль) в ДХМ (20 мл) добавляли периодинан Десса-Мартина (467 мг, 1,1 ммоль) и реакционную смесь перемешивали при КТ в течение 30 мин. Реакционнуюю смесь разбавляли ДХМ (20 мл) и раствор промывали раствором гидроксида натрия (1 М, водный). Органический слой отделяли, сушили (MgSO4) и затем концентрировали в вакууме. Остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 90% EtOAc в циклогексане), при этом получали соединение 69 в виде твредого вещества белого цвета (253 г, 70%). ЖХМС: RT=4,10, М+Н+=364.
Пример 70
2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновая кислота 70
Стадия 1
Метиловый эфир 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты
Метиловый эфир 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты получали аналогично тому, как описано для метилового эфира 2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]-триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты, из соединения 61 (0,99 г, 2,65 ммоль). Реакционную смесь растворяли EtOAc (20 мл), фильтровали и фильтрат концентрировали в вакууме.
Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 50 до 100% EtOAc в циклогексане), при этом получали указанное в заголовке соединение (0,32 г, 34%). ЖХМС: RT=4,73, М+Н+=354.
Стадия 2
[2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил]метанол
[2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил]метанол получали аналогично тому, как описнао для получения {2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}метанола, из метилового эфира 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты (0,50 г, 1,42 ммоль), при этом получали указанное в заголовке соединение (360 мг, 78%). ЖХМС: RT=3.81, M+H+=326.
Стадия 3
2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбальдегид
2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбальдегид получали аналогично тому, как описано для получения соединения 69, из [2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил]метанола (360 мг, 1,11 ммоль). Реакционную смесь разбавляли ДХМ (20 мл) и раствор промывали раствором гидроксида натрия (1 М, водный). Органический слой отделяли, сушили (MgSO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: 100% EtOAc), при этом получали указанное в заголовке соединение в виде твердого вещества белого цвета (410 мг, 114%). ЖХМС: RT=4,15, М+Н+=324.
Стадия 4
2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновую кислоту получали аналогично тому, как описано для получения 2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты из метилового эфира 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты (720 мг, 2,04 ммоль). Реакционную смесь концентрировали в ваууме для удаления диоксана и полученный раствор подкисляли до рН 1 добавлением HCl (12Н.). Полученный осадок собирали фильтрованием, промывали водой и сушили в вакумме при 50°С, при этмо получали соединение 70 (584 мг, 84%). ЖХМС: RT=4,61 мин, M+H+=340.
Пример 72
2-(1-(2-Хлорфенил)-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновая кислота 72
Стадия 1
Метиловый эфир 2-(1-(2-хлорфенил)-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,41оксазепин-10-карбоновой кислоты
В раствор 1-(2-хлорфенил)-1Н-имидазола (0,133 г, 0,743 ммоль) в ТГФ (5,43 мл, 66,9 ммоль) при -78°С по каплям добавляли 1,60 М раствор n-бутиллития в гексане (0,464 мл). Реакционную смесь перемешивали при -78°С в течение 1 ч, затем добавляли 0,50 М раствор дихлорида цинка в ТГФ (1,48 мл). Реакционную смесь нагревали до КТ в течение 30 мин, затем в нее добавляли Pd(PPh3)4 (0,0780 г, 0,0675 ммоль), раствор соединения 26 (0,250 г, 0,675 ммоль) в 2 мл ТГФ. Реакционную смесь кипятили с обратным холодильником в течение 2 ч, затем добавляли 2,2 мл 0,50 М раствора дихлорида цинка в ТГФ и смесь кипятили с обратным холодильником в течение 3 ч. Смесь разбавляли EtOAc, затем промывали насыщенным раствором Na2CO3 и солевым раствором. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт метиловый эфир 2-(1-(2-хлорфенил)-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты очищали хроматографией. МС: (ESI+)=421.2
Стадия 2
В раствор метилового эфира 2-(1-(2-хлорфенил)-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты (0,100 г, 0,238 ммоль) в ТГФ (5,56 мл, 68,5 ммоль) и воде (5,56 мл, 308 ммоль) добавляли моногидрат гидроксида лития (0,0399 г, 0,950 ммоль). Реакционную смесь перемешивали при КТ в течение ночи. Реакционную смесь концентрировали. Реакционную смесь подкисляли добавлением 1 М HCl и экстрагировали ДХМ (3Х). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали, при этом получали соединение 72. МС: (ESI+)=407,2.
Пример 74
10-Бром-2-(1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 74
Стадия 1
10-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбальдегид
10-Бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин формилировали, при этом получали 10-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбальдегид с выходом 84%. МС: 293,1.
Стадия 2
10-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбальдегид конденсировали с этандиаделм в присутствии аммиака, при этом получали соединение 74 с выходом 37%. МС: 331,0.
Пример 82
1-(2-Бромэтокси)-2-нитробензол 82
К смеси 2-нитрофенола (25,0 г, 0,180 моль), гидроксида натрия (14,4 г, 359 ммоль) и воды (6,0 мл, 330 ммоль) в колбе объемом 500 мл, снабженной обратным холодильником, при 107°С добавляли 1,2-дибромэтан (61,9 мл, 719 ммоль), и колбу нагревали при 107°С в течение 3 сут (схема 18). Затем продукт 2 раза экстрагировали по 100 мл ДХМ, промывали 2 М раствором NaOH и солевым раствором, сушили над сульфатом натрия и концентрировали. После очистки хроматографией на колонке с силикагелем (элюент: гексан и EtOAc) получали бромид 82 с выходом 63%. 1H ЯМР (500 МГц, CDCl3): δ 7,83 (dd, J=8,4, 1,6 Гц, 1Н), 7,53 (td, J=8,1, 1,6 Гц, 1Н), 7,12-7,01 (m, 2H), 4,45-4,34 (m, 2H), 3,67 (t, J=6,5 Гц, 2H), согласно тому, ка кописано в заявке WO 2002076926.
Пример 83
3-(2-Нитрофенокси)пропаннитрил 83
В раствор цианида натрия (0,398 г, 8,13 ммоль) в ДМСО (29,0 мл, 409 ммоль) при 45°С одной порцией добавляли бромид 82 (2,00 г, 8,13 ммоль) и реакционную смесь перемешивали в течение 4 ч при 70°С (схема 18). Затем реакционную смесь экстрагировали EtOAc, органические слои сушили над сульфатом натрия и концентрировали. После очистки хроматографией на колонке с силикагелем (элюент: гексан и EtOAc) получали нитрил 83 с выходом 43%. 1H ЯМР (400 МГц, CDCl3): δ 7,89 (dd, J=8,1, 1,7 Гц, 1Н), 7,62-7,55 (m, 1H), 7,19-7,13 (m, 1H), 7,11 (dd, J=8,4, 0,8 Гц, 1Н), 4,36 (t, J=6,6 Гц, 2Н), 2,94 (t, J=6,6 Гц, 2Н), согласно тому, как описано в работе Vitale и др., Anales de la Asociacion Quimica Argentina, т. 82(1), с. 19-23 (1994).
Пример 84
3-(2-Аминофенокси)пропаннитрил 84
В колбу объемом 50 мл, снабженную мешальником и содержащую палладий (0,00748 г, 0,0702 ммоль), добавляли EtOAc (11,7 г, 133 ммоль) в атмосфере азота, а затем соединение 83 (0,675 г, 3,51 ммоль, схема 18). Колбу подсоединяли к баллону с водородом и отсоединяли от баллона с азотом. Реакционную смесь интенсивно перемешивали в течение 4 ч, фильтровали через целит и промывали EtOAc. Продукт 84 не требовал дальнейшей очистки и его выход составлял 98%. 1H ЯМР (500 МГц, CDCl3): δ 6,85-6,77 (m, 1Н), 6,74-6,62 (m, 3H), 4,08 (t, J=6,1 Гц, 2Н), 3,94-3,74 (m, 2Н), 2,72 (t, J=6,1 Гц, 2Н). ЖХМС m/z Рассч. для C9H10N2O: 162,07931, найд.: 163,1 [M+1].
Пример 85
(Е/Z)-Метиловый эфир 2-хлор-2-(2-(2-(2-цианоэтокси)фенил)гидразоно)уксусной кислоты 85
К раствору анилина 84 (1,65 г, 10,2 ммоль) в уксусной кислоте (6,80 мл, 120 ммоль) при интенсивном перемешивании добавляли 2 М раствор хлористоводородной кислоты (13,59 мл), и затем нитрит натрия (1,0290 г, 14,914 ммоль) при 0°С (схема 18). Через 20 мин по каплям из шприца добавляли метиловый эфир 2-хлорацетоуксусной кислоты (1,5317 г, 10,173 ммоль) и смесь нагрвали при КТ в течение 5 ч. Органический слой дважды экстрагировали диэтиловым эфиром по 100 мл, сушили над сульфатом натрия и концентрировали. Продукт 85 использовали на следующей стадии без дополнительной очистки. ЖХМС m/z Рассч. для C12H12ClN3O3: 281,05672, найд.: 282,1 [M+1].
Пример 86
Метиловый эфир 4,5-дигидробензо[b][1,2,4]триазол[1,5-d][1,4]оксазепин-2-карбоновой кислоты 86
В колбу объемом 200 мл в атмосфере азота, содержащую хлоргидразон 85 (2,87 г, 10,2 ммоль), добавляли 1,4-диоксан (100 мл) и карбонат серебра (4,22 г, 15,3 ммоль, схема 18). Колбу подсоединеняли к обратному холодильнику и заворачивали в фольгу (для обеспечения отсутствия доступа света). Затем реакционную смесь кипятили с обратным холодильником в течение 4 ч. Реакционную смесь фильтровали, концентрировали и очищали хроматографией на колонке с силикагелем, при этом получали сложный эфир 86 с выходом 7% после двух стадий. 1H ЯМР (500 МГц, CDCl3): δ 8,19 (dd, J=8,2, 1,4 Гц, 1Н), 7,31 (td, J=8,0, 1,6 Гц, 1Н), 7,25-7,20 (m, 1Н), 7,18 (dd, J=8,1, 1,3 Гц, 1Н), 4,50 (t, J=5,7 Гц, 2Н), 4,03 (s, 3H), 3,50 (t, J=5,7 Гц, 2Н). ЖХМС m/z Рассч. для C12H11N3O3: 245,08004, найд.: 246,1 [M+1].
Пример 87
4,5-Дигидробензо[b][1,2,4]триазол[1,5-d][1,4]оксазепин-2-карбоксамид 87
Сложный эфир 86 (0,166 г, 0,677 ммоль) растворяли в смеси 3:2:1 ТГФ : МеОН : H2O (31,2 мл), обрабатывали 4 н. водным раствором гидроксида лития (1,32 мл), и смесь перемешивали в течение 20 мин при 25°С (схема 18). Реакцию останавливали добавлением 1н. водной HCl (20 мл) и раствор три раза экстрагировали по 100 мл EtOAc. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали, при этом получали неочищенную 4,5-дигидробензо[b][1,2,4]триазол[1,5-d][1,4]оксазепин-2-карбоновую кислоту, коорую использовали на следующей стадии без дополнительной очистки. ЖХМС m/z расч. для C12H9N3O3: 231,06439, найд.: 232,1 [M+1].
В раствор неочищенной 4,5-дигидробензо[b][1,2,4]триазол[1,5-d][1,4]оксазепин-2-карбоновой кислоты (0,177 г) в ДМФ (1,55 мл, 20,0 ммоль) добавляли гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,761 г, 2,00 ммоль) и 6-хлор-1-гидроксибензотриазол (0,339 г, 2,00 ммоль, схема 18). Реакционную смесь интенсивно перемешивали и в реакционную смесь добавляли хлорид аммония (0,285 г, 5,34 ммоль). Через 10 мин добавляли N,N-диизопропилэтиламин (0,465 мл, 2,67 ммоль). Через 3 ч реакционную смесь сушили досуха. После очистки препаративной ЖХВР (элюент: ацетонитрил/вода) получали амид 87 (0,0485 г, выход 31% после двух стадий). 1Н ЯМР (500 МГц, CDCl3): δ 8,22 (d, J=8,2 Гц, 1Н), 7,30 (t, J=7,7 Гц, 1H), 7,22 (t, J=7,7 гц, 1Н), 7,18 (d, J=8,0 гц, 1Н), 6,97 (s, 1Н), 5,75 (s, 1Н), 4,49 (t, J=5,7 Гц, 2Н), 3,48 (t, J=4,0 гц, 2Н). МСНР m/z Рас. для C12H10N4O2: 230,08038, найд.: 231,08 [M+1].
Пример 89
трет-Бутиловый эфир 5-(9-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-илкарбоновой кислоты 89
Стадия 1
4-Фтор-2-гидроксибензальдегид (1,918 г, 0,01369 моль), этандиаль (1,884 мл, 0,04107 моль), 14,8 М раствор гидроксида аммония в воде (14 мл, 0.21 моль) и метанол (34 мл, 0,84 моль) смешивали в круглодонной колбе и реакционную смесь перемешивали при КТ. Заверешение реакции регистрировали методом ЖХМС. Смесь концентрировали в вакууме и неочищенное твердое вещество растворяли в 1 М HCl до рН приблизительно 8 с использованием чувствительной к рН бумаги. Продукт экстрагировали EtOAc, промывали солевым раствором, сушили над сульфатом магния и снова концентрировали в вакууме. Остаток очищали экспресс-хроматографией на колонке ISCO (элюент: от 0% до 50% EtOAc в гептане), концентрировали в вакууме, при этом получали 5-фтор-2-(1Н-имидазол-2-ил)фенол (0,92 г, выход 37,7%).
Стадия 2
5-Фтор-2-(1Н-имидазол-2-ил)фенол (0,90 г, 5,0 ммоль) растворяли в ДМФ (40 мл, 500 ммоль). В раствор добавляли карбонат цезия (6,6 г, 20 ммоль), затем добавляли 1,2-дибромэтан (1,7 мл, 20 ммоль) и нагревали при 90°С в течение 3 ч в присутствии колонки для конденсации. Заверешение реакции регистрировали методом ЖХМС. Смесь разбавляли водой и экстрагировали EtOAc. Водный слой подкисляли до рН приблизительно 5 добавлением HCl и экстрагировали EtOAc. Объединенные органические слои концентрировали в вакууме, очищали экспресс-хроматографией на колонке ISCO (элюент: 0-50% EtOAc в гексане) и концентрировали в вакууме, при этом получали 9-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,69 г, выход 67%).
Стадия 3
9-Фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,69 г, 3,4 ммоль), NIS (2,83 г, 12,6 ммоль) и ДМФ в круглодеонной колбе перемешивали в течение 4 сут. Смесь разбавляли EtOAc и распределяли между насыщенным раствором бикарбоната натрия и водой (50/50). Водный слой экстрагировали EtOAc и объединенные органические слои сушили над сульфатом магния, концентрировали в вакууме и очищали экспресс-хроматографией на колонке ISCO (элюент: 0-40% EtOAc в гексане), при этом получали 9-фтор-2,3-диод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (1,25 г, выход 81%).
Стадия 4
9-Фтор-2,3-диод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (1,24 г, 2,74 ммоль) растворяли в ТГФ (25 мл, 310 ммоль) и охлаждали до -78°С на бане сухой лед/ацетон. В смесь добавляли 3,0 М раствор бромида этилмагния в эфире (1,37 мл) и нагревали реакционную смесь до -40°С и перемешивали в течение 4 ч. Завершение реакции контролировали методом ЖХВР. Смесь разбавляли 100 мл насыщенного раствора хлорида аммония и экстрагировали EtOAc. Экстракт сушили над сульфатом магния, концентрировали в вакууме и очищали экспресс-хроматографией на колонке ISCO (элюент: 0-40% EtOAc в гексане), при этом получали 9-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,794 г, выход 88%).
Стадия 5
Круглодонную колбу, содержащую 9-фтор-2-иод-5,6-дигидробензо[f]имидазо[1,2d][1,4]оксазепин (0,794 г, 2.40 ммоль) интенсивно продували азотом. При продолжении прокачивания в колбу добавляли ацетат палладия(II) (27 мг, 0,12 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантан (139 мг, 0,24 ммоль). Затем добавляли прокаченные азотом метанол (10 мл, 200 ммоль) и ТЭА (30 мл, 200 ммоль) и реакционную смесь продували монооксидом углерода в течение 5 мин. Колбу подсоединяли к двум баллонам с монооксидом углерода и реакционную смесь нагревали при 50°С в течение 4,5 ч. Завердение образования метилового эфира подтверждали методом ЖХМС. Реакционную смесь продували азотом и концентрировали в вакууме. Эфир очищали экспресс-хроматографией на колонке ISCO 0 (элюент: 50% EtOAc в гептане) и концентрировали в вакууме. Эфир растворяли в ТГФ (20 мл, 200 ммоль) и добавляли 1 М раствор гидроксида лития (7,22 мл) и реакционную смесь перемешивали в течение 3 сут. Завершение гидролиза контролировали методом ЖХМС. рН смеси доводили до приблизительно 5 добавлением 1 М HCl и рподукт экстрагировали ДХМ и 5% метанолом, при этом получали 9-фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоновую кислоту (0,386 г, выход 64,6%).
Стадия 6
9-Фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоновую кислоту (0,65 г, 2,6 ммоль) суспендировали в ДХМ (15 мл, 230 ммоль) и добавляли 2,0 М раствор оксалилхлорида в ДХМ (2,0 мл) и ДМФ (81 мкл), а затем толуол (15 мл, 140 ммоль) и смесь нагревали на струйной воздушной сушилке до растворения примерно половины твердого вещества. Перемешивали в течение 30 мин и концентрировали в вакууме, при этом получали гидрохлорид. Гидрохлорид растворяли в 20 мл ДХМ и добавляли промежуточное соединение (0,50 г, 2,6 ммоль) и ТЭА (1,1 мл, 7,8 ммоль) в ДХМ (50 мл, 800 ммоль). Реакционную смесь перемешивали в течение 3 ч и по данным ЖХМС реакция была закончена. В смесь добавляли воду и три раза экстрагировали ДХМ. Продукт промывали солевым раствором, сушили над сульфатом магния, концентрировали в вакууме и очищали экспресс-хроматографией на колонке ISCO (элюент: 0-50% EtOAc в гептане), при этом получали промежуточное соединение ацилтиомочевину (0,20 г, выход 18%).
Стадия 7
Промежуточное соединение ацилтиомочевину (200 мг, 0,4 ммоль) растворяли в ДМФ (10 мл, 100 ммоль) и в полученный раствор добавляли N,N-диизопропиламин (0,29 мл, 1,662 ммоль) и гидрохлорид изопропилгидразина (68,92 мг, 0,62 ммоль). Реакционную смесь перемешивали при КТ в течение ночи. Завершение реакции подтверждали методом ЖХМС. Смесь разбавляли водой и 3 раза экстрагрировали ДХМ. Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Продукт очищали экспресс-хроматографией на колонке ISCO (элюент: от 0 до 10% метанол в ДХМ), при этом получали соединение 89 (200 мг, выход 100%).
Пример 90
10-Фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-скарбоксамид 90
Стадия 1
4-Фтор-2-(1Н-имидазол-2-ил)фенол
5-Фтор-2-гидроксибензальдегид (5,0 г, 36 ммоль), этандиаль (4,912 мл, 107 ммоль), 14,8 М раствор гидроксида аммония в воде (40 мл, 600 ммоль) и метанол (90 мл, 2000 ммоль) смешивали в круглодонной колбе и перемешивали при КТ в течение ночи. Завершенность реакции подтверждали методом ЖХМС. Продукт концентрировали в вакууме и в него добавляли 1 М HCL до рН приблизительно 8. Продукт экстрагировали EtOAc, промывали солевым раствором, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (элюент: от 0 до 50% EtOAc в гептане), при этом получали 4-фтор-2-(1Н-имидазол-2-ил)фенол (2,24 г, выход 35%).
Стадия 2
4-Фтор-2-(1Н-имидазол-2-ил)фенол превращали в соединение 90.
Пример 91
2-Бром-1-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)этанон 91
Стадия 1
Гидразид уксусной кислоты (100 г, 1,35 моль) суспендировали в ацетоне (991 мл, 13,5 моль) и циклогексане (1,5 л). Реакционную смесь перемешивали при 55°С в течение 16 ч, при этом твердое вещество растворялось и получали бесцветный раствор. Реакционную смесь концентрировали в вакууме, при этом получали изопропилиденгидразид уксусной кислоты в виде твердого вещества белого цвета (153 г, выход 100%). 1H ЯМР 400 МГц (CDCl3) δ: 8,25 (1Н, ушир. s), 2,26 (3Н, s), 2,00 (3Н, s), 1,83 (3Н, s).
Стадия 2
В раствор изопропилиденгидразида уксусной кислоты (153 г, 1,35 моль) в IMS (1,5 л) добавляли оксид платины (0,66 г) и реакционную смесь перемешивали в атмосфере водорода при КТ до тех пор, пока по данным 1Н ЯМР не наблюдалось полное поглощение изопропилиденгидразида уксусной кислоты (приблизительно 48 ч). Реакционную смесь фильтровали через слой целлита и фильтрат концентрировали в вакууме, при этом получали N'-изопропилгидразид уксусной кислоты в виде бесцветного маслянистого вещества, которое кристаллизовалось при стоянии (154,6 г). 1H ЯМР 400 МГц (CDCl3) δ: 3,12 (1Н, септет, J=6,3 Гц), 1,96 (3Н, s), 1,04 (6H, d, J=6,3 Гц).
Стадия 3
В раствор этилового эфира тиощавелевой кислоты (29,6 г, 0,22 моль) в ДХМ (260 мл) при КТ добавляли тетрафторборат триметилоксония (34,5 г, 0,23 моль) и смесь перемешивали при КТ в течение 2 ч. С тчением времени желтое окрашивание ослабевало и образовывался плотный осадок белого цвета. N'-Изопропилгидразид уксусной кислоты (27,1 г, 0,23 моль) ТЭА (30,9 мл, 0,22 моль) добавляли в виде раствора в ДХМ (75 мл), что приводило к растворению осадка. Реакционную смесь кипятили с обратным холодильником в течение нескольких ч, а затем перемешивали при КТ в течение 10 с. Реакционную смесь промывали водой и водный слой экстрагировали ДХМ (2×50 мл). Объединенные органические экстракты промывали солевым раствором, сушили (MgSO4) и концентрировали в вакууме. Получнный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-100% EtOAc в циклогексане), при этом получали этиловый эфир 2-изопропил-5-метил-2Н-[1,2,4]триазол-3-карбоновой кислоты в виде маслянистого вещества светло-желтого цвета, которое кристаллизовалось при стоянии (15,6 г, выход 32%). 1Н ЯМР 400 МГц (CDCl3) δ: 5,49 (1Н, септ., J=6,7 Гц), 4,45 (2Н, t, J=7,2 Гц), 2,43 (3Н, s), 1,50 (6H, d, J=6,7 Гц), 1,.44 (3Н, t, J=7,2 Гц).
Стадия 4
В раствор этилового эфир а2-изопропил-5-метил-2Н-[1,2,4]триазол-3-карбоновой кислоты (12,09 г, 61,3 ммоль) и дибромэтана (8,63 мл, 122,6 ммоль) в ТГФ (500 мл) при -78°С по каплям добавляли метиллитий (40,9 мл, 122,6 ммоль, 3 М раствор в диэтоксиметане). Реакционную смесь перемешивали при -78°С в течение 15 мин. Затем добавляли уксусную кислоту (3 мл) и реакционную смесь нагревали до КТ. Реакционную смесь разбавляли водой и экстрагировали EtOAc (3×30 мл). Объединенные органические экстракты промывали солевым раствором, сушили (MgSO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-100% EtOAc в циклогексане), при этом получали соединение 91 в виде маслянистого бесцветное вещества, которое кристаллизовалось при стоянии (11,26 г, выход 75%). 1Н ЯМР 400 МГц (CDCl3) δ: 5,41 (1Н, септ., J=6,6 Гц), 4,67 (2Н, s), 2,44 (3H, s), 1.49 (6H, d, J=6,6 Гц).
Пример 92
2-(2-Изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-ол 92
Стадия 1
4-Хлор-5-иодпиридин-2-иламин
В раствор 2-амино-4-хлорпиридина (150 г, 0,78 моль) в ДМФ (1,5 л) добавляли NIS (341 г, 1,52 моль) и реакционную смесь перемешивали при КТ в течение 18 ч, затем концентрировали в вакумме до объема 300 мл. Полученный остаток выливали в 10% водный раствор тиосульфата натрия (1,2 L), перемешивали в течение 15 мин и полученный осадок собирали фильтрованием, промывали водой и сушили при 35°С в вакууме, при этом получали указанное в заголовке соединения в виде твердого вещества светло-коричневого цвета (185 г, выход 62%). 1H ЯМР 400 МГц (CDCl3) δ: 8,33 (1H, s), 6,68 (1H, s), 4,52 (2H, s).
Стадия 2
4-Хлор-5-иод-2-метоксипиридин
В раствор 4-хлор-5-иодпиридин-2-иламина (64,2 г, 0,25 моль) в метаноле (1,1 л) и ТФА (93,7 мл, 1,26 моль) добавляли трет-бутилнитрит (150 мл, 1,26 моль) при температуре менее 3°С. Полученную смесь перемешивали при КТ в течение 1 ч, нагревали до КТ и перемешивали в течение 16 ч. Реакцию останавливали осторожным добавлением воды и концентрировали до ¼ части от исходного объема. Полученный остаток обрабатывали водой (1 л), осадок собирали фильтрованием и сушили в вакумме при 35°С, при этом получали указанное в заголовке соединение (62,3 г, выход 92%), которое содержало 16% примесей. 1Н ЯМР 400 МГц (DMSO-d6) δ: 8,56 (1H, s), 7,20 (1H, s), 3,86 (3H, s).
Стадия 3
4-Хлор-6-метоксиникотиннитрил
Суспензию 4-хлор-5-иод-2-метоксипиридина (30,5 г, 0,11 моль), цианида цинка (II) (7,97 г, 68 ммоль), Pd(PPh3)4 (6,56 г, 5,66 ммоль) и ДМФ (450 мл) дегазировали и нагревали при 120°С в течение 1 ч и концентрировали в вакууме. Полученный остаток обрабатывали водой, экстрагировали ДХМ, объединенные органические фракции сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток перекристаллизовывали из ДХМ и получали указанное в заголовке соединение (10,1 г, выход 54%). Супернатанты концентрировали в вакууме и остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 100% EtOAc в циклогексане), затем перекристаллизовывали из циклогексана и получали указанное в заголовке соединение (5,16 г, выход 28%, общий выход 82%). 1Н ЯМР 400 МГц (CDCl3) δ: 8,45 (1Н, s), 6,90 (1H, s), 4,01 (3H, s).
Стадия 4
Гидрохлорид 4-хлор-6-метоксиникотинамидина
В раствор 4-хлор-6-метоксиникотиннитрила (10,1 г, 59,7 ммоль) в ТГФ (300 мл) при -78°С по каплям добавляли LiHMDS (65,7 мл) и реакционную смесь перемешивали в течение 30 мин, затем нагрвали до КТ и перемешивали в течение 1 ч. Реакцию останавливали добавлением 1N HCl (до рН приблизительноl) и затем экстрагировали 3 раза EtOAc. Водный слой концентрировали в акумме и получали твердое вещество коричневого цвета, которое разгоняли с азеотропом с толуолом, при этом получали указанное в заголовке соединение в виде твердого вещества желто-коричневого цвета. Продукт содержал смесь хлорида аммония и 72 мас. % указанного в заголовке соединения (15,2 г, выход 83%). 1Н ЯМР 400 МГц (DMSO-d6) δ: 9,68 (4H, d, J=15,79 Гц), 8,46 (1H, s), 7,47 (5H, t, J=50,66 Гц), 7,27 (1H, s), 3,95 (3H, s).
Стадия 5
4-Хлор-5-[4-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-1Н-имидазол-2-ил]-2-метоксипиридин
Суспензию гидрохлорида 4-хлор-6-метоксиникотинамидина (18,4 ммоль) и бикарбоната калия (7,37 г, 73,6 ммоль) в ТГФ (42 мл) и воде (8,5 мл) нагревали до кипения и по каплям обрабатывали раствором соединения 91 (4,53 г, 18,4 ммоль) в ТГФ (14 мл). Реакционную смесь кипятили с обратным холодильником в течение 18 ч и летучий растворитель удаляли в вакууме. Полученную суспензию фильтровали, остаток промывали водой, затем сушили, при этом получали указанное в заголовке соединение в виде твердого вещества коричневого цвета (5,91 г, выход 97%). ЖХМС: RT=2,68 мин, [М+Н]+=333/335. 1Н ЯМР 400 МГц (CDCl3) δ: 10,41 (1Н, s), 9,02 (1H, s), 7,81 (1H, s), 6,87 (1H, s), 5,91 (1H, m), 4,00 (3H, s), 2,41 (3H, s), 1.55 (6H, d, J=6,71 Гц).
Стадия 6
2-[2-(4-Хлор-6-метоксипиридин-3-ил)-4-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)имидазол-1-ил]этанол
Суспензию 4-хлор-5-[4-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-1Н-имидазол-2-ил]-2-метоксипиридина (5,9 г, 17,7 ммоль) в толуоле (20 мл) обрабатывали этиленкарбонатом (50 мл) и нагревали при 130°С в течение 2,5 ч. Охлажденную реакционную смесь концентрировали в вакууме, разбавляли ДХМ и пропускали через слой силикагеля (элюент: ДХМ, затем 20% метанол в ДХМ). Фракции в метаноле объединяли и концентрировали в вакууме, полученный остаток перекристаллизовывали из ацетонитрила, при этом получали указанное в заголовке соединение в виде твердого вещества светло-желто-коричневого цвета (2,27 г, выход 34%). ЖХМС: RT=2,53 мин, [М+Н]+=377/379. 1Н ЯМР 400 МГц (CDCl3) δ: 8,25 (1Н, s), 8,05 (1Н, s), 6,92 (1Н, s), 5,82-5.80 (1Н, m), 4,00 (3Н, s), 3,97 (2Н, t, J=4,92 Гц), 3,88 (2Н, t, J=4,92 Гц), 2,38 (3Н, s), 1,48 (6H, d, J=6,63 Гц).
Стадия 7
2-(2-Изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-метокси-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен
Раствор 2-[2-(4-хлор-6-метоксипиридин-3-ил)-4-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)имидазол-1-ил]этанола (2,25 г, 5,97 ммоль) в ДМФ (30 мл) охлаждали до 0°С и обрабатывали гидридом натрия (239 мг, 5,97 ммоль), реакционную смесь перемешивали при 0°С в течение 30 мин, нагревали до КТ и перемешивали в течение 2 ч. Реакционную смесь охлаждали до 0°С и обрабатывали водой (400 мл), осадок отфильтровывали, промывали водой, сушили в вакууме, при этом полвчали указанное в заголовке соединение в виде твердого вещества белого цвета (1,02 г, выход 50%). ЖХМС RT=2,68 мин, [М+Н]+=341. 1Н ЯМР 400 МГц (DMSO-d6) δ: 9,15 (1H, s), 7,87 (1H, s), 6,42 (1H, s), 5,84 (1H, m), 4,57-4,56 (4H, m), 3,89 (3H, s), 2.25 (3H, s), 1,46 (6H, d, J=6,60 Гц).
Стадия 8
Раствор 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-метокси-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена (1,0 г, 2,97 ммоль) в 48% водном растворе HBr (5 мл) и уксусной кислоте (5 мл) нагревали при 80°С в течение 7,5 ч, затем концентрировали в вакууме. Полученный остаток суспендировали в воде (10 мл) и доводили величину рН приблизительно до 6 с использованием 5 н. водного раствора NaOH. Полученный осадок отфильтровывали, промывали водой, сушили в вакууме, при этом получали соединение 92 в виде твердого везщества белого цвета (1,01 г, выход 100%). ЖХМС RT=2,01 мин, [М+Н]+=327. 1Н ЯМР 400 МГц (DMSO-d6) δ: 8,42 (1Н, s), 7,85 (1Н, s), 5,85 (1Н, s), 5,69-5.65 (1Н, m), 4,55-4,54 (2Н, m), 4,50-4,46 (2Н, m), 2,27 (3Н, s), 1,44 (6Н, d, J=6.59 Гц).
Пример 93
2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-ол 93
Стадия 1
4-Хлор-5-[4-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-1Н-имидазол-2-ил]-2-метоксипиридин
Суспензию гидрохлорида 4-хлор-6-метоксиникотинамидина (50,9 ммоль) и бикарбоната калия (20,4 г, 202,5 ммоль) в ТГФ (128 мл) и воде (21 мл) нагревали до кипения и по каплям обрабатывали раствором 2-хлор-1-(2-изопропил-2Н-[1,2,4]триазол-3-ил)этанона (9,55 г, 50,9 ммоль) в ТГФ (25 мл). Реакционную смесь кипятили с обратным холодильником в течение 24 ч и летучий растворитель удаляли в вакууме. Полученный остаток разбавляли водой и экстрагировали EtOAc. Объединенные экстракты сушили (Na2SO4), обрабатывали активированным углем (15 г), фильтровали и концентрировали в вакууме, при этом получали твердое вещество. Твердое вещество растирали с 10% раствором диэтилового эфира в пентане, сушили при 50°С в вакууме, при этом получали указанное в заголовке вещество в виде твердого вещества светло-коричневого цвета (8,74 г, выход 54%). ЖХМС RT=2,86 мин, [М+Н]+=319/321. 1H ЯМР 400 МГц (CDCl3) δ: 9,03 (1Н, s), 7,89 (1Н, s), 7,83 (1Н, s), 7,26 (1Н, s) 6,88 (1Н, s), 4,01 (3Н, s), 1,58 (6Н, d, J=6,63 Гц).
Стадия 2
2-[2-(4-Хлор-6-метоксипиридин-3-ил)-4-(2-изопропил-2Н-[1,2,4]триазол-3-ил)имидазол-1 -ил]этанол
В теплый этиленкарбонат (34 г) добавляли 4-хлор-5-[4-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-1Н-имидазол-2-ил]-2-метоксипиридин (8,74 г, 27,4 ммоль) и смесь нагревали при 130°С в течение 3 ч. Охлажденную реакционную смесь разбавляли ДХМ и наносили на силикагель (150 г). Силикагельпромывали ДХМ, затем 5% метанолом в ДХМ. Фракции в метаноле объединяли и концентрировали в вакууме, при этом получали указанное в заголовке соединение в виде пенообразного материала корчневого цвета (7,52 г, выход 75%). ЖХМС RT=2,65, [М+H]+=363/365. 1H ЯМР 400 МГЦ (CDCl3) δ: 8,27 (1H, s), 8,02 (1H, s), 7,85 (1H, s), 6,93 (1H, s), 5,98-5.82 (1H, m), 4,00 (5H, m), 3,88 (2H, t, J=5,11 Гц), 1,51 (6H, d, J=6,62 Гц).
Стадия 3
2-(2-Изопропил-2Н-[1,2,4]триазолl-3-ил)-8-метокси-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен
Раствор 2-[2-(4-хлор-6-метоксипиридин-3-ил)-4-(2-изопропил-2Н-[1,2,4]триазол-3-ил)имидазол-1-ил]этанола (7,52 г, 20,7 ммоль) в ДМФ (100 мл) охлаждали до 0°С и обрабатывали гидридом натрия (804 мг, 20,1 ммоль), реакционную смесь перемешивали при 0°С в течение 10 мин, нагревали до КТ и перемешивали в течение 72 ч. В смесь добавляли гидрид натрия (150 мг) и перемешивание продолжали до тех пор, пока из смеси не пропадал исходный материал, затем растворитель удаляли в вакууме. Остаток растворяли EtOAc, полученный раствор 3 раза промывали насыщенным солевым раствором, затем сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток растирали со смесью пентан/диэтиловый эфир (5:1), при этом получали указанное в заголовке соединение в виде твердого вещества коричневого цвета (5,38 г, выход 79%). ЖХМС RT=2,86, [М+Н]+=327. 1Н ЯМР 400 МГц (CDCl3) δ: 9,35 (1H, s), 7,87 (1H, s), 7,63 (1H, s), 6,37 (1H, s), 6,03-6.02 (1H, m), 4,54-4,53 (2H, m), 4,53-4,33 (2H, m), 3,99 (3H, s), 1,57 (6H, d, J=6,63 Гц).
Стадия 4
Раствор 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-метокси-4,5-дигидро-6-окса-1,3а,9-тразабензо[е]азулена (1,0 г, 2,97 ммоль) в уксусной кислоте (40 мл) обрабатывали 48% водным раствором HBr (37,7 мл) и нагревали при 80°С в течение 5 ч, затем концентрировали в вакууме. Полученный остаток суспендировали в воде (60 мл) и величину рН доводили до приблизительно 6 с использованием 5н. водного расвтора NaOH. Полученный осадок отфильтровывали, промывали водой и сушили в вакууме. Полученное твердое вещество растирали с ацетоном, при этом получали соединение 93 в твердого вещства бежевого цвета (3,58 г, выход 69%). ЖХМС RT=2,04 мин, [М+Н]+=313. 1Н ЯМР 400 МГц (DMSO-d6) δ: 8,42 (1H, s), 7,90 (1H, s), 7,83 (1H, s), 5,84 (1H, s), 5,78 (1H, m), 4,71-4,30 (4H, m), 1,45 (6H, d, J=6,60 Гц).
Пример 94
Гидрохлорид 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена 94
Стадия 1
2-(2-Изопропил-2Н-[1,2,4]тразол-3-ил)-4,5-дигидро-6-окса-1,3а,9-тразабензо[е]азулен-8-иловый эфир трифторметансульфоновой кислоты
Суспензию соединения 93 (238 мг, 0,76 ммоль) в ДМФ (2,2 мл) обрабатывали гидридом натрия (65% дисперсия в минеральном масле, 34 мг, 0,91 ммоль), реакционную смесь нагревали при 40°С в течение 1,5 ч, затем охлаждали до КТ. В смесь добавляли бензилбис(трифторметан)сульфонамид (327 мг, 0,91 ммоль) и реакционную смесь перемешивали при КТ в течение 48 ч, затем смесь разбавляли EtOAc (60 мл) и промывали солевым раствором (4×20 мл). Полученный раствор сушили (MgSO4), фильтровали и концентрировали в вакууме, при этом получали твердое вещество, которое растирали с диэтиловым эфиром, при этом получали 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-иловый эфир трифторметансульфоновой кислоты (44 мг). Супернатанты, полученные после растирания, концентрировали в вакууме, полученный остаток перекристаллизовывали из метанола, при этом получали указанное в заголовке соединение в виде твердого вещества белого цвета (39 мг, общий выход 25%). ЖХМС RT=3,27 мин, [М+Н]+=445. 1Н ЯМР 400 МГц (DMSO-d6) δ: 9,32 (1H, s), 8,04 (1H, s), 7,93 (1H, s), 7,36 (1H, s), 5,89 (1H, m), 4,74 (2H, m), 4,63 (2H, m), 1,48 (6H, d, J=6,58 Гц).
Стадия 2
В смесь 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-илового эфира трифторметансульфоновой кислоты (83 мг, 0,19 ммоль) и 2 М водного раствора карбоната натрия (600 мкл) в ДМФ (1,2 мл) добавляли бис(дибензилиденацетон)палладия (6 мг, 0,01 ммоль), трифенилфосфин (4 мг, 0,015 ммоль) и трет-бутиловый эфир 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (75 мг, 0,24 ммоль). Реакционную смесь дегазировали и нагревали при 90°С в атмосфере аргона в течение 2 ч, затем концентрировали в вакууме. Полученный остаток распределяли между EtOAc и водой, водный слой экстрагировали EtOAc (х 3) и объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% метанол в EtOAc), при этом получали трет-бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]тразол-3-ил)-4,5-дигидро-6-окса-1,3а,9-тразабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты в виде втердого вещества белого цвета (41 мг, выход 45%). ЖХМС RT=3,24 мин, [М+Н]+=478. 1H ЯМР 400 МГц (CDCl3) δ: 9,65 (1H, s), 7,94 (1H, s), 7,89 (1H, s), 7,00 (1H, s), 6,84 (1H, s), 4,60 (2H, s), 4,50 (2H, s), 4,18 (2H, s), 3,67 (2H, s), 2,62 (2H, s), 1,59 (6H, d, J=6,62 Гц), 1,50 (9Н, s).
Стадия 3
Смесь трет-бтилового эфира 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (89 мг, 0,19 ммольl) в IMS (10 мл) обрабатывали оксидом платины (10 мг), реакционную смесь дегазировали и перемешивали при КТ в атмосфере водорода в течсение 72 ч. В смесь добавляли оксид платины (10 мг) и перемешивание продолжали при КТ в течение 18 ч, затем фильтровали через целит и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 5% метанол в ДХМ), при этом получали трет-бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-ил]пиридин-1-карбоновой кислоты (58 г, выход 64%). ЖХМС RT=2,72, [М+Н]+=480.
Стадия 4
Раствор трет-бутилового эфира 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-ил]пиридин-1-карбоновой кислоты (58 мг, 0,12 ммоль) в ДХМ (0,5 мл) и метаноле (0,3 мл) обрабатывали 4 М HCl в диоксане (0,8 мл) и реакционную смесь перемешивали при КТ в течение 1,5 ч, затем концентрировали в вакууме. Полученный остаоок растирали с диэтиловым эфиром, при этом получали соединение 94 (66 мг, выход 100%). ЖХМС RT=1,68 мин, [М+Н]+=380.
Пример 102
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 102
Оксалилхлорид в ДХМ (2,00 М, 3,0 мл) добавляли в суспензию соединения 31 (112 мг, 0,178 ммоль) в 30 мл ДХМ. В смесь добавляли каталитическое количество ДМФ (1,0 мкл, 0,013 ммоль) и смесь перемешивали в течение 2 ч. Смесь фильтровали, фильтрат концентрировали в ваккуме и остаток сушили в высоком вакууме в течение 1 ч. Остаток растворяли в N,N-диметилацетамиде (3,0 мл, 32 ммоль) и насыщали газообразным аммиаком. Смесь перемешивали в течение 20 мин, концентрировали в вакууме, растворяли в водном растворе метанола и очищали RP ЖХВР. Выход составлял 18,5 мг (26%), МС: 407,1. 1Н ЯМР (400 МГц, ДМСО) δ: 8,26 (d, J=2,2, 1H), 8,20 (s, 1H), 7,80 (s, 1H), 7,72-7,51 (m, 6H), 7,29 (s, 1H), 7,00 (d, J=8,5, 1H).
Пример 103
2-(1-(2,4-Дифторфенил)-1Н-1,2,4-триазол-5-ил)-8-бром-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 103
Раствор 2,4-дифторфенилгидразина (20 г, 0,14 моль) в формамиде (60 мл) нагревали при 120°С в течение 18 ч. Охлажденную реакционную смесь разбавляли насыщенным водным раствором гидроксрбоната натрия и EtOAc, при этом получали эмульсию. Эмульсию фильтровали через целит, водный слой экстрагировали EtOAc. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, полученное твердое вещество очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 100% EtOAc в циклогексане), при этом получали 1-(2,4-дифторфенил)-1Н-[1,2,4]триазол в виде твердого вещества белого цвета (15,7 г, выход 62%). 1Н ЯМР (CDCl3) δ (ч/млн): 8,60 (1H, d, J=2,83 Гц), 8,12 (1H, s), 7,91-7,83 (1H, m), 7,10-7,04 (2H, m).
Раствор 1-(2,4-дифторфенил)-1Н-[1,2,4]триазола (15,7 г, 86,7 ммоль) в ТГФ (300 мл) при -78°С в атмосфере азота по каплям обрабатывали n-бутиллитием (38 мл, 2,5 M, 95,3 ммоль). После перемешивания в течение 1,5 ч в смесь по каплям добавляли раствор гексахлорэтана (22,6 г, 95,3 ммоль) в ТГФ (30 мл). Полученнуюреакционную смесь перемешивали в течение 1,5 ч, нагревали до КТ и реакцию останавливали добавлением воды. Полученную смесь разбавляли водой и экстрагировали EtOAc, объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали маслянистое вещество, которое кристаллизовалось при стоянии. Твердое вещество перекристаллизовывали из циклогексана, при этом получали 5-хлор-1-(2,4-дифторфенил)-1Н-[1,2,4]тразол (12,4 г, выход 66%). 1Н ЯМР (CDCl3) δ (ч/млн): 8,04 (1H, s), 7,51-7,43 (1H, m), 7,10-7,04 (2H, m).
В раствор соединения 50 (2,12 г, 7,99 ммоль) и 5-хлор-1-(2,4-дифторфенил)-1Н-[1,2,4]триазол (2,58 г, 12,0 ммоль) в ТГФ (10 мл) добавляли карбонат цезия (3,9 г, 12,0 ммоль) и реакционную смесь нагревали при 180°С в течение 60 мин с использованием микроволнового излучения. Охлажденную реакционную смесь разбавляли водой и экстрагировали EtOAc, объединенные органические экстракты промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток растирали с горячим циклогексаном, при этом получали соединение 103 (1,62 г, выход 46%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,45 (s, 1H); 8,30 (s, 1H); 7,84-7,77 (m, 1H); 7,66-7,58 (m, 1H); 7,37-7,30 (m, 1H); 7,27 (d, J=8,6 Гц, 1H); 7,22 (d, J=2,0 Гц, 1H); 7,13 (dd, J=8,6, 2,1 Гц, 1H); 4,26 (t, J=5,0 Гц, 2H); 3,06 (t, J=5,0 Гц, 2H).
Пример 104
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-5.6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 104
Согласно тому, как описано для получения соединения 243, получали соединение 014 из 2-(1-(2-хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты, хлорида аммония, HATU, диизопропилэтиламина и ДМФ. Выход составлял 51%. МС: 407,0. 1Н ЯМР (400 МГц, ДМСО) δ: 8,21 (s, 1H), 7,99 (s, 1H), 7,95 (s, 1H), 7,74 (d, J=7,0, 1H), 7,69-7,54 (m, 4H), 7,47 (s, 1H), 7,44-7,36 (m, 2H), 4,48 (d, J=7,6, 4H).
Пример 105
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-8-(пиразол-4-ил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 105
8-Бром-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен (53 мг, 0,14 ммоль), 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразол (36 мг, 0,18 ммоль), трис(дибензилиденацетон) дипалладия (0) (2,2 мг, 1,7 мол. %), 2,4',6'-диизопропил-1,1'-бифенил-2-илдициклогексилфосфин (5,3 мг, 9 мол. %) и K3PO4 (89 мг, 0,42 ммоль) смешивали в реакционном сосуде, воздух откачивали и сосуд заполняли азотом. В смесь добавляли диоксан (1 мл) и воду (0,1 мл) реакционную смесь кипятили с обратным холодильником в течение 18 ч. В смесь добавляли дополнительные количества 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразола (36 мг, 0,18 ммоль), трис(дибензилиденацетона) дипалладия (0) (2,2 мг, 1,7 мол. %), 2,4',6'-диизопропил-1,1'-бифенил-2-илдициклогексилфосфина (5,3 мг, 9 мол. %) и нагревание продолжали в течение еще 18 ч. Реакционную смесь разбавляли EtOAc, декантировали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 10 до 60% EtOAc в циклогексане), при этом получали соединение 105 в виде твердого вещества белого цвета (12 мг, выход 24%). 1H ЯМР (CDCl3, 400 МГц): δ 8,28 (d, J=8,2 Гц, 1Н); 8,08 (s, 1Н); 7,90 (s, 2Н); 7,80 (s, 1Н); 7,30 (dd, J=8,2, 1,8 Гц, 1Н); 7,23 (d, J=1,8 Гц, 1Н); 5,73-5,59 (m, 1Н); 4,42-4,35 (m, 2Н); 3,20-3,13 (m, 2Н); 1,62 (d, J=6,6 Гц, 6Н). Наличие одного заменяемого протона не наблюдали. ЖХМС (способ D*): RT=9,48 мин, M+H+=362.
Пример 106
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-8-бром-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 106
Стадия 1
1-Изопропил-1Н-[1,2,4]тразол
Раствор гидрохлорида изопропилгидразина (60 г, 0,54 ммоль) в формамиде (270 мл) нагревали при 130°С в течение 3 сут. Охлажденный раствор разбавляли насыщенным солевым раствором (700 мл) и экстрагировали EtOAc (4×1 л). Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали маслянистое вещество. Маслянистое вещество перегоняли при пониженном давлении (25 мБар, Ткип. 85-90°С), при этом получали 1-изопропил-1Н-[1,2,4]триазол в виде бесцветного маслянистого вещества (54 г, выход 90%). 1Н ЯМР (CDCl3) δ (ч/млн): 8,10 (1Н, s), 7,95 (1Н, s), 4,63-4,50 (1Н, m), 1,56 (6Н, d, J=6,69 Гц).
Стадия 2
5-Хлор-1-изопропил-1Н-[1,2,4]триазол
Раствор 1-изопропил-1Н-[1,2,4]триазола (19 ммоль) в ТГФ (50 мл) при -78°С в атмосфере азота по каплям обрабатывали n-бутиллитием (11,4 мл, 2,5 M, 28,5 ммоль), при этом получали кремообразную суспнзию желтого цвета. Через 1 ч в суспензию добавляли дополнительное количество n-бутиллития (3,8 мл, 2,5 M, 9,5 ммоль) и смесь перемешивали в течение 1,5 ч. 1,1,2-Трихлортрифторэтан (4,57 мл, 38 ммоль) добавляли по каплям и получали раствор темно-коричневого цвета. Реакционную смесь перемешивали в течение 15 мин, затем реакцию останавливали добавлением насыщенного водного раствора NaHCO3 (20 мл), затем смесь нагревали до КТ. Полученную смесь два раза экстрагировали диэтиловым эфиром, объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали 5-хлор-1-изопропил-1Н-[1,2,4]триазол в виде темного маслянистого вещества (3,9 г), который использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (CDCl3) δ (ч/млн): 7,85 (1Н, s), 4,73-4,63 (1Н, m), 1,54-1,46 (6Н, m).
Охлажденную льдом суспезию гидрида натрия (60% дисперсия, 1,09 г, 38 ммоль) порциями обрабатывали соединением 50 (3,6 г, 13,6 ммоль), при этом получали суспензию темно-красного цвета. В суспензию добавляли 5-хлор-1-изопропил-1Н-[1,2,4]триазол (19 ммоль) и смесь нагревали при 80°С в атмосфере азота в течение 72 ч. После охлаждения реакцию останавливали добавлением воды и 2 раза экстрагировали EtOAc. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 to 15% EtOAc в циклогексане). Соответствующие фракции объединяли и перекристаллизовывали из метанола и смеси циклогексан/EtOAc, при этом получали соединение 106 в виде твердого вещества желто-коричневого цвета (789 мг, выход 16%). 1H ЯМР (CDCl3, 400 МГц): δ 8,15-8,11 (m, 1H); 8,08 (s, 1H); 7,80 (s, 1H); 7,28-7,23 (m, 2H); 5,65-5,53 (m, 1H); 4,38-4,31 (m, 2H); 3,18-3,10 (m, 2H); 1,60 (d, J=6.6 Гц, 6H).
Пример 107
2-(1-(2,4-Дифторфенил)-1H-1,2,4-триазол-5-ил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол-8-карбоксамид 107
В суспензию 8-бромо-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена (80 мг, 0,18 ммоль), гидрохлорида гидроксиламина (25 мг, 0,36 ммоль), гексакарбонилмолибдена (24 мг, 0,09 ммоль), три-трет-бутилфосфинотетрафторбората (5 мг, 10 мол. %), транс-ди-μ-ацетобис[2-(ди-о-толилфосфино)бензил]дипалладия(II) (8,4 мг, 5 мол. %) в диоксане (4 мл) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (27 мкл, 0,18 ммоль) и DIPEA (62 мкл, 0,36 ммоль). Реакционную смесь нагревали при 150°С в течение 30 мин с использованием микроволнового излучения и разбавляли EtOAc. Органический слой промывали водой, насыщенным водным раствором бикарбоната натрия, солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 20 до 90% EtOAc в циклогексане), при этом получали твердое вещество, которое затем перекристаллизовывали из метанола, при этом получали соединение 107 в виде твердого вещества белого цвета (17 мг, выход 23%). 1Н ЯМР (ДМСО-d6, 400 МГц) δ: 8,48 (s, 1H); 8,31 (s, 1H); 7,98 (s, 1H); 7,87-7,79 (m, 1H); 7,69-7,61 (m, 1H); 7,49 (d, J=1,7 Гц, 1H); 7,43 (dd, J=8,3, 1,8 Гц, 1Н); 7,40-7,32 (m, 3H); 4,27 (t, J=5,0 Гц, 2Н); 3,09 (t, J=5,0 Гц, 2Н). ЖХМС (способ D*): RT=8,72 мин, M+H+=409.
Пример 108
2-(4-Изопропил-4Н-1,2,4-триазол-5-ил)-9-(пиразол-4-ил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 108
Суспезию соединения 58 (78 мг, 0,21 ммоль), 4,4,5,5-тетраметил-2-(1Н-пиразол-4-ил)-1,3,2-диоксаборолана (61 мг, 0,31 ммоль), тетракис(трифенилфосфин) палладия (0) (24 мг, 0,021 ммоль) и карбоната натрия (45 мг, 0,42 ммоль) в ацетонитриле (6 мл) и воде (3 мл) в атмосфере азота нагревали при 140°С в течение 25 мин с использованием микроволнового излучения. Реакционную смесь разбавляли EtOAc водой, органический слой отделяли, промывали солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 50% EtOAc в циклогексане), перекристаллизовывали из ацетонитрила и растирали с пентаном, при этом получали соединение 69 в виде твердого вещества грязно-белого цвета (16 мг, выход 20%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,91 (s, 1Н); 8,38 (d, J=2,3 Hz, 1Н); 8,32 (s, 1Н); 8,03 (s, 1Н); 7,97 (s, 2Н); 7,52 (dd, J=8,4, 2,3 Гц, 1Н); 7,05 (d, J=8,4 Гц, 1Н); 5,41-5,26 (m, 1Н); 4,35-4,26 (m, 2Н); 3,17-3,08 (m, 2Н); 1,55 (d, J=6,6 Гц, 6Н). ЖХМС: RT=9,38 мин, М+Н+=362.
Пример 109
2-(1-Изопропилl-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 109
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновую кислоту (0,170 г, 0,000501 моль) раствоярли в ДМФ (7,84 мл, 0,101 моль) и последовательно обрабатывали N,N-диизопропилэтиламином (0,524 мл, 0,00300 моль), хлоридом аммония (0,107 г, 0,00200 моль) гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,228 г, 0,000601 моль). Реакционную смесь перемешивали при КТ в течение ночи. Реакцию останавливали добавлением бикарбоната натрия и экстрагировали EtOAc (3х). Органические слои сушили (Na2SO4) и концентрировали в вакууме, при этом получали соединение 109, которое очищали на обращено-фазной ЖХВР. МС: (ESI+)=339,1. 1H ЯМР (400 МГц, ДМСО): δ 8,93 (d, J=2,2 Гц, 1H), 7,95 (s, 2H), 7,92 (s, 1H), 7,79 (dd, J=8,5, 2,2 Гц, 1H), 7,10 (d, J=8,5 Гцz, 1H), 5,82 (dt, J=13,2, 6,6 Гц, 1H), 4,56 (s, 4H), 1,49 (d, J=6,6 Гц, 6Н).
Пример 110
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-N-метил-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол-9-карбоксамид 110
Соединение 110 получали из соединения 58 и гидрохлорида N,O-диметилгидроксиламина в виде твердого вещества белого цвета (29 мг, выход 31%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,75 (d, J=2,3 Гц, 1Н); 8,44-8,38 (m, 1H); 8,33 (s, 1H); 8,05 (d, J=0,6 Гц, 1Н); 7,73 (dd, J=8,5, 2,3 Гц, 1Н); 7.10 (d, J=8,5 Гц, 1Н); 5,31-5,21 (m, 1H); 4,35 (t, J=5,0 Гц, 2Н); 3,13 (t, J=5,0 Гц, 2Н); 2,78 (d, J=4,5 Гц, 3H); 1,51 (d, J=6,6 Гц, 6Н). ЖХМС (метод D*): RT=8,24 мин, M+H+=353.
Пример 111
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-N-(2-гидроксиэтил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол-9-карбоксамид 111
Соединение 111 получали согласно тому, как описано для получения соединения 107, из соединения 58 и этаноламина в виде твердого вещства белого цвета (23 мг, выход 22%). 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,71 (d, J=2,29 Гц, 1Н); 8,35 (t, J=5,57 Гц, 1Н); 8,29 (s, 1H); 8,00 (d, J=0,60 Гц, 1Н); 7,71 (dd, J=8,51, 2,33 Гц, 1Н); 7,06 (d, J=8,49 Гц, 1Н); 5,29-5,21 (m, 1H); 4,65 (t, J=5,60 Гц, 1Н); 4,30 (t, J=4,98 Гц, 2Н); 3,47 (q, J=5,99 Гц, 2Н); 3,28 (t, J=4,93 Гц, 2Н); 3,09 (t, J=4,98 Гц, 2Н); 1,46 (d, J=6,59 Гц, 6Н). ЖХМС RT=7,41 мин, M+H+=383.
Пример 112
(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)(S-диоксотиоморфолино)метанон 112
Соединение 112 получали аналогично тому, как описано для получения соединения 116, из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты и диоксида тиоморфолина. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,06 (d, J=2,1 Гц, 1Н); 8,04 (d, J=0,6 Гц, 1Н); 7,50 (dd, J=8,3, 2,1 Гц, 1Н); 7,34 (d, J=8,3 Гц, 1Н); 6,92 (s, 1H); 5,67-5,54 (m, 1Н); 4,57 (t, J=5,9 Гц, 2Н); 3,35-3,26 (br m, 8H); 3,24 (t, J=6,0 Гц, 2Н); 1,48 (d, J=6,6 Гц, 6Н). ЖХМС: RT=8,24 мин, M+H+=457.
Пример 113
(4-(2-Гидроксипропан-2-ил)пиперидин-1-ил)(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)метанон 113
Соединение 113 получали аналогично тому, как описано для получения соединения 109, из 2-(1-изопропил-1Н-1,2,4-триазо-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 2-(пиперидин-4-ил)пропан-2-ола. МС: (ESI+)=465,2. 1H ЯМР (400 МГц, ДМСО): δ 8,40 (d, J=1,6 Гц, 1Н), 7,96 (s, 1H), 7,92 (s, 1H), 7,35 (dd, J=8,4, 1,9 Гц, 1Н), 7,11 (d, J=8,4 Гц, 1H), 5,87-5,69 (m, 1H), 4,56 (s, 4H), 4,15 (s, 1H), 1,75 (s, 2H), 1,48 (d, J=6,6 Гц, 7H), 1,19 (dd, J=23,3, 11.1 Гц, 2H), 1,05 (s, 7H).
Пример 114
9-(1-Изопропил-1Н-пиразол-5-ил)-6,7-дигидроимидазо[1,2-d]пиридо[3,2-b][1,4]оксазепин-3-карбоксамид 114
Смесь 17 мг (0,05 ммоль) соединения 12, 30 мг (0,061 ммоль) HATU и 0,022 мл (0,155 ммоль) ТЭА в 2 мл ДМФ перемешивали в течение 10 мин. Через смесь пробулькивали газообразный аммиак в течение 5 мин. Смесь перемешивали в течение 1 ч, концентрировали в вакууме и распределяли между EtOAc и 0,01 н. водным раствором HCl. Органический слой концентрировали и остаток очищали на колонке с 4 г силикагеля (элюент: 7-8% метанол в ДХМ), при этом получали соединение 114 с выходом 2,4 мг. MC(ESI+) 339,2. 1Н ЯМР (400 МГц, СН3ОН+D2O): δ 8,70 (d, J=1,8, 1H), 8,33 (s, 1H), 8,05 (d, J=1,8, 1H), 7,52 (s, 1H), 6,48 (d, J=1,6, 1H), 5,16 (dd, J=13,2, 6.6, 1H), 4,56 (t, J=5,1, 2H), 3,52-3,45 (m, 2H), 1,49 (d, J=6,6, 6H).
Пример 115
2-(1-Изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 115
Соединение 115 получали аналогично тому, как описано в примере 51, из соединения 14 и аммиака. MC(ESI+): 338,1. 1H ЯМР (400 МГц, ДМСО): δ 8,94 (d, J=2,2, 1H), 7,94 (s, 1H), 7,77 (dd, J=8,5, 2,2, 1H), 7,69 (s, 1H), 7,44 (d, J=1,4, 1H), 7,25 (s, 1H), 7,08 (d, J=8,5, 1H), 6,40 (d, J=1,7, 1H), 5,34 (dt, J=13,0, 6,4,1H), 4,52 (dd, J=11,0, 5,6, 4H), 1,44 (d, J=6,6, 6H).
Пример 116
N-(2-Гидрокси-2-метилпропил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-карбоксамид 116
2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновую кислоту (0,2 г, 0,59 ммоль) суспендировали в ДМФ (5 мл) и добавляли DIPEA (0,21 мл, 1,2 ммоль). Полученный раствор обрабатывали HATU (0,23 г, 0,6 ммоль) и НОВТ (81 мг, 0,6 ммоль), затем добавляли 1-амино-2-метилпропан-2-ол (52,5 мг, 0,59 ммоль) и полученную смесь перемешивали при КТ в течение 18 ч. Реакционную смесь концентрировли в вакууме и полученный остаток распределяли между ДХМ и водой. Водный слой 3 раза экстрагировали ДХМ, объединенные органические экстракты промывали водой и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в EtOAc) и перекристаллизовывали из EtOAc, при этом получали соединение 116 (163 мг, выход 67%). 1Н ЯМР (CDCl3, 400 МГц): δ 8,46 (d, J=2,2 Гц, 1Н); 7,94 (s, 1Н); 7,72 (dd, J=8,4, 2,2 Гц, 1Н); 7,22 (d, J=8,4 Гц, 1Н); 6,86 (s, 1Н); 6,66 (s, 1Н); 5,74-5,61 (m, 1Н); 4,57 (t, J=5,7 Гц, 2Н); 3,50 (d, J=5,9 Гц, 2Н); 3,22 (t, J=5,7 Гц, 2Н); 2,18 (s, 1Н); 1,59 (d, J=6,6 Гц, 6Н); 1,31 (s, 6H). ЖХМС:RT=9,53 мин, M+H+=411.
Пример 117
(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)(S-диоксотиоморфолино)метанон 117
Соединение 117 получали из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты и диоксида тиоморфолина. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,06 (d, J=2,1 Гц, 1Н); 8,04 (d, J=0,6 Гц, 1Н); 7,50 (dd, J=8,3, 2,1 Гц, 1Н); 7,34 (d, J=8,3 Гц, 1Н); 6,92 (s, 1Н); 5,67-5,54 (m, 1Н); 4,57 (t, J=5,9 Гц, 2Н); 3,35-3,26 (br m, 8H); 3,24 (t, J=6,0 Гц, 2Н); 1,48 (d, J=6,6 Гц, 6Н). ЖХМС: RT=8,24 мин, M+H+=457.
Пример 118
(4-Гидроксипиперидин-1-ил)(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)метанон 118
Соединение 118 получали аналогично тому, как описано для получения соединения 116, из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты и 4-гидроксипиперидина в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3, 400 МГц): δ 8,02 (d, J=2,1 Гц, 1Н); 7,94 (s, 1Н); 7,35 (dd, J=8,3, 2,1 Гц, 1Н); 7,23 (d, J=8,3 Гц, 1Н); 6,88 (s, 1Н); 5,76-5,62 (m, 1Н); 4,58 (t, J=5,8 Гц, 2Н); 4,19 (ушир. s, 1Н); 4,07-3,98 (m, 1Н); 3,82 (ушир. s, 1Н); 3,37 (ушир. s, 2H); 3,20 (t, J=5,8 Гц, 2H); 1,95 (ушир. s, 2H); 1,70 (d, J=3,9 Гц, 1Н); 1,67-1,54 (d, J=6,6 Гц, 8Н). ЖХМС: RT=8,77 мин, М+Н+=423.
Пример 119
N-(2-(Метилсульфонил)этил)-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 119
Соединение 119 получали аналогично тому, как описано для получения соединения 109, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 2-(метилсульфонил)этанамина. МС: (ESI+)=485,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,93 (d, J=2,1 Гц, 1Н), 8,72 (t, J=5,4 Гц, 1Н), 8,10 (s, 2H), 7,78 (dd, J=8,6, 2,2 Гц, 1Н), 7,14 (d, J=8,5 Гц, 1Н), 5,94 (q, J=8,9 Гц, 2H), 4,58 (s, 4H), 3,69 (dd, J=12,7, 6,5 Гц, 2H), 3,38 (t, J=6,8 Гц, 2H), 3,04 (s, 3H).
Пример 120
(4-Изопропилпиперазин-1-ил)(2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-тразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)метанон 120
Соединение 120 получали аналогично тому, как описано для получения соединения 109, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 1-изопропилпиперазина. МС: (ESI+)=490,2. 1H ЯМР (400 МГц, ДМСО): δ 8,38 (d, J=2,0 Гц, 1Н), 8,10 (d, J=5,4 Гц, 2H), 7,37 (dd, J=8,4, 2,1 Гц, 1Н), 7,12 (d, J=8,4 Гц, 1Н), 5,87 (q, J=8,8 Гц, 2H), 4,57 (s, 4H), 3,49 (s, 4H), 2,68 (dt, J=13,0, 6,6 Гц, 1Н), 2,45 (s, 4H), 0,97 (d, J=6,5 Гц, 6Н).
Пример 121
N-(1-Гидрокси-2-метилпропан-2-ил)-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 121
Соединение 121 получали аналогично тому, как описано для получения соединения 109, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 2-амино-2-метилпропан-1-ола. МС: (ESI+)=451,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,82 (d, J=2,2 Гц, 1Н), 8,09 (d, J=2,9 Гц, 2Н), 7,74 (dd, J=8,6, 2,3 Гц, 1Н), 7,58 (s, 1H), 7,10 (d, J=8,5 Гц, 1Н), 5,94 (q, J=8,9 Гц, 2Н), 4,91 (t, J=5,9 Гц, 1Н), 4,57 (dd, J=10,9, 5,6 Гц, 4Н), 3,51 (d, J=5,9 Гц, 2Н), 1,32 (s, 6H).
Пример 122
(4-(2-Гидроксиэтил)пиперазин-1-ил)(2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)метанон 122
Соединение 122 получали аналогично тому, как описано для получения соединения 109, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 2-(пиперазин-1-ил)этанола. МС: (ESI+)=492,2. 1Н ЯМР (400 МГц, ДМСО): δ 8,39 (d, J=2,0 Гц, 1Н), 8,10 (d, J=5,4 Гц, 2Н), 7,38 (dd, J=8,4, 2,1 Гц, 1Н), 7,13 (d, J=8,4 Гц, 1Н), 5,89 (q, J=8,7 Гц, 2Н), 4,57 (s, 4Н), 4,40 (t, J=5,4 Гц, 1Н), 4,06 (q, J=5,3 Гц, 1H), 3,51 (dd, J=11,6, 6,0 Гц, 4H), 3,17 (d, J=5,2 Гц, 1Н), 2,46-2,39 (m, 5H).
Пример 123
Морфолино(2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)метанон 123
Соединение 123 получали аналогично тому, как описано для получения соединения 109, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и морфолина. МС: (ESI+)=449,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,42 (d, J=2,0 Гц, 1Н), 8,10 (d, J=5,6 Гц, 2Н), 7,40 (dt, J=21,4, 10,7 Гц, 1Н), 7,13 (d, J=8,4 Гц, 1Н), 5,89 (q, J=8,8 Гц, 2Н), 4,57 (s, 4Н), 3,61 (s, 4Н), 3,52 (s, 4Н).
Пример 124
(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)(4-(2,2,2-трифторэтил)пиперазин-1-ил)метанон 124
Соединение 124 получали аналогично тому, как описано для получения соединения 116, из 2-(2-изопроппил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты и дигидрохлорида 1-(2,2,2-трифторэтил)пиперазина в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3, 400 МГц): δ 8,02 (d, J=2,1 Гц, 1Н); 7,95 (s, 1Н); 7,35 (dd, J=8,3, 2,1 Гц, 1Н); 7,23 (d, J=8,3 Гц, 1Н); 6,88 (s, 1Н); 5,73-5,60 (m, 1Н); 4,58 (t, J=5,8 Гц, 2Н); 3,94-3,44 (ушир. m, 4H); 3,20 (t, J=5,8 Гц, 2Н); 3,04 (q, J=9,4 Гц, 2Н); 2,74 (ушир. s, 4H); 1,58 (d, J=6,6 Гц, 6Н). ЖХМС: RT=10,93 мин, M+H+=490.
Пример 125
N-(1-(2-Гидроксиэтил)-1Н-пиразол-4-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-карбоксамид 125
Соединение 125 получали аналогично тому, как описано для получения соединения 126, из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты и 4-амино-1-(2-гидроксиэтил)пиразола в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3, 400 МГц): δ 8,50 (d, J=2,2 Гц, 1Н); 8,38 (s, 1Н); 8,10 (s, 1Н); 7,96 (s, 1Н); 7,78-7,73 (m, 1Н); 7,56 (s, 1Н); 7,22 (d, J=8,4 Гц, 1Н); 6,81 (s, 1H); 5,60-5,47 (m, 1Н); 4,57 (t, J=5,7 Гц, 2H); 4,23 (t, J=4,8 Гц, 2H); 4,01 (t, J=4,8 Гц, 2H); 3,20 (t, J=5,7 Гц, 2H); 1,57 (d, J=6,6 Гц, 6Н). Один заменяемый протон отсутствует. ЖХМС: RT=9,27 мин, М+Н+=449.
Пример 126
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-N-(изоксазол-3-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-карбоксамид 126
В охлажденную льдом суспензию 2-(2-изопропли-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты (0,15 г, 0,44 ммоль) в ДХМ (3,5 мл) добавляли оксалилхлорид (79 мкл, 0,93 ммоль) и ДМФ (25 мкл) и смесь перемешивали при КТ в течение 2 ч. В смесь добавляли 3-аминоизоксазол (185 мг, 2,2 ммоль) и ТЭА (0,12 мл, 0,88 ммоль) и смесь перемешивали при КТ в течение 18 ч, затем в смесь добавляли насыщенный водный раствор гидрокарбоната натрия. Полученную смесь экстрагировали ДХМ, затем 5% МеОН и ДХМ и объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в ДХМ) и растирали с диэтиловым эфиром, при этом получали соединение 126 в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3, 400 МГц): δ 9,49 (s, 1Н); 8,65 (d, J=2,3 Гц, 1Н); 8,30 (d, J=1,8 Гц, 1Н); 7,96 (d, J=0,7 Гц, 1Н); 7,86 (dd, J=8,5, 2,3 Гц, 1Н); 7,30 (d, J=8,5 Гц, 1Н); 7,23 (d, J=1,8 Гц, 1Н); 6,88 (s, 1Н); 5,72-5,59 (m, 1Н); 4,61 (t, J=5,6 Гц, 2Н); 3,25 (t, J=5,6 Гц, 2Н); 1,58 (d, J=6,6 Гц, 6Н). ЖХМС: RT=10,24 мин, М+H+=406.
Пример 127
N-(1Н-Пиразол-4-ил)-2-(1-(2,2,2-трифторэтид)-1Н-1,2,4-тразол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5][1,4]оксазепин-9-карбоксамид 127
Согласно тому, как описано для получения соединения 126, 2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновую кислоту конденсировали с трет-бутиловым эфиром 4-аминопиразол-1-карбоновой кислоты. Промежуточный трет-бутиловый эфир карбоновой кислоты растворяли в ДХМ (5 мл) и обрабатывали ТФУ (2 мл), затем перемешивали при КТ в течение 2 ч. Реакционную смесь концентрировали в вакууме и остаток растирали с водой, при этом получали соединение 127 в виде твердого вещества белого цвета. 1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,53 (s, 1Н); 8,51 (d, J=2,2 Hz, 1Н); 8,23 (s, 1Н); 7,96 (dd, J=8,5, 2,2 Hz, 1Н); 7,84 (s, 2Н); 7,39 (d, J=8,5Hz, 1Н); 7,02 (s, 1Н); 5,77 (q, J=8,7 Hz, 2Н); 4,59 (t, J=5,8 Hz, 2Н). Пики трех протонов перекрывались с пиком воды. ЖХМС: RT=9,11 мин, М+Н+=445.
Пример 128
2-(4-((2-(1-(2,2,2-Трифторэфтил)-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)метил)пиперазин-1-ил)этанол 128
Раствор соединения 69 (175 мг, 0,48 ммоль) и пиперазинэтанола в ДХЭ (15 мл) обрабатывали триацетоксиборгидридом натрия (153 мг, 0,72 ммоль), добавляли каталитическое количество уксусной кислоты и перемешивали при КТ в течение 72 ч. Полученную смесь разбавляли ДХМ, промывали насыщенным водным раствором гидрокарбоната натрия и сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали смолистое вещество, которое очищали экспресс-хрматографией (SiO2, элюент: градиент 0-10% МеОН в ДХМ). Соответствующие фракции объединяли, концентрировали в вакууме, полученный остаток растворяли в диэтиловом эфире и обрабатывали 1М HCl в диэтиловом эфире (2 мл, 2 ммоль). Полученный осадок отфильтровывали, промывали эфиром и сушили в вакууме, при этом получали соединении 128 в виде твердого вещества белого цвета. 1Н ЯМР (ДМСО-d6 + дейтерированная ТФУ, 400 МГц): δ 8,22 (s, 1Н); 8,16-8,12 (m, 1Н); 7,66-7,60 (m, 1Н); 7,39 (d, J=8,3 Гц, 1Н); 7,02 (s, 1Н); 5,75 (q, J=8,7 Гц, 2H); 4,57 (t, J=5,9 Гц, 2H); 4,52 (s, 2H); 3,76 (t, J=5,0 Гц, 2H); 3,61 (ушир. s, 8H); 3,32 (s, 2H); 0,24 (t, J=5,9 Гц, 2H). Один заменяемый протон не наблюдали. ЖХМС: RT=6,56 мин, М+Н+=478.
Пример 129
(4-Гидроксипиперидин-1-ил)(2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)метанон 129
Соединение 129 получали аналогично тому, как описано для получения соединения 109, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 4-гидроксипиперидина. МС: (ESI+)=463,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,39 (d, J=2,0 Гц, 1Н), 8,09 (d, J=4,4 Гц, 2Н), 7,37 (dd, J=8,4, 2,1 Гц, 1Н), 7,12 (d, J=8,4 Гц, 1Н), 5,87 (q, J=8,5 Гц, 2Н), 4,76 (d, J=3,8 Гц, 1Н), 4,57 (s, 4H), 3,76 (d, J=3,6 Гц, 2Н), 1,74 (s, 2Н), 1,39 (s, 2Н).
Пример 130
9-(Пиперидин-4-ил)-2-(1-(2,2,2-трифторметил)-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин 130
Стадия 1
трет-Бутиловый эфир 4-{2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты
Соединение 62 (207 мг, 0,5 ммоль), трет-бутиловый эфир 4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (232 мг, 0,75 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) в ДХМ (10 мол. %) и карбонат калия (138 мг, 1,0 ммоль) помещали в реакционный сосуд, воздух откачивали и сосуд заполняли азотом. Добавляли 1 мл ДМФ, дегазирование повторяли и смесь перемешивали при 80°С в течение 18 ч. Охлажденную реакционную смесь разбавляли EtOAc и промывали водой, затем сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 20 до 60% EtOAc в циклогексане) при этом получали трет-Бутиловый эфир 4-{2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты в виде твердого кристаллического вещества белого цвета (232 мг, выход 90%). ЖХМС: RT=4,87 мин, M+H+=517.
Стадия 2
Раствор трет-бутилового эфира 4-{2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (232 мг, 0,41 ммоль) в IMS (10 мл) обрабатывали суспензией Pd/C (170 мг, 10 мас. % Pd на углероде, 50% воды) в IMS. Смесь дегазировали, воздух откачивали и реакционный сосуд заполняли водородом и перемешивали при КТ в течение 18 ч. Реакционную смесь фильтровали через целит, промывали EtOAc и концентрировали в вакууме. Полученный остаток растворяли в метаноле (5 мл), обрабатывали 1М HCl в диэтиловом эфире (2 мл, 2 ммоль) и смесь перемешивали при КТ в течение 18 ч. Растворитель удаляли в акууме, остаток раствирали с диэтиловым эфиром и при этом получали соединение 130 в виде твердого вещнства светло-желтого цвета (127 мг, Выход 74%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,08-8,97 (ушир. m, 1Н); 8,96-8,81 (ушир. m, 1Н); 8,22 (s, 1Н); 7,69 (s, 1Н); 7,31-7,23 (m, 2Н); 6,99 (s, 1Н); 5,72 (q, J=8,8 Гц, 2Н); 4,51 (t, J=6,1 Гц, 2Н); 3,42-3,32 (m, 2Н); 3,22-3,15 (m, 2Н); 3,08-2,88 (m, 3Н); 2,04-1,95 (m, 2Н); 1,94-1,80 (m, 2Н). ЖХМС: RT=6,75 мин, М+Н+=419,2.
Пример 131
N-((2-(1-(2,2,2-Трифторэтил)-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)метил)пиразин-2-амин 131
Соединение 131 получали согласно тому, как описано для получения 128, но без использования HCl в диэтиловом эфире, из соединения 69 и 2-аминопиразина в виде твердого вещства белого цвета. 1Н ЯМР (CDCl3, 400 МГц): δ 8,03-7,99 (m, 3Н); 7,87 (d, J=2,6 Гц, 1Н); 7,84 (d, J=2,2 Гц, 1Н); 7,31 (dd, J=8,3, 2,2 Гц, 1Н); 7,20 (d, J=8,3 Гц, 1Н); 6,92 (s, 1Н); 5,49 (q, J=8,2 Гц, 2Н); 5,42 (ушир. s, 1Н); 4,66 (d, J=5,4 Гц, 2Н); 4,55 (t, J=6,0 Гц, 2Н); 3,16 (t, J=6,0 Гц, 2Н). ЖХМС: RT=9,93 мин, M+H+=443.
Пример 132
2-Гидрокси-1-(4-((2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)метил)пиперазин-1-ил)этанон 132
Соединение 132 получали согласно тому, как описано для получения 128, но без использования HCl в диэтиловом эфире, из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбальдегида и 2-гидрокси-1-пиперазин-1-илэтанона в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3, 400 МГц): δ 7,95 (s, 1Н); 7,86 (d, J=2,0 Гц, 1Н); 7,28-7,23 (m, 1Н); 7,17 (d, J=8,2 Гц, 1Н); 6,86 (s, 1Н); 5,73-5,60 (m, 1Н); 4,55 (t, J=6,0 Гц, 2Н); 4,15 (s, 2Н); 3,69 (t, J=4,8 Гц, 2Н); 3,64-3,55 (m, 3Н); 3,29 (t, J=4,8 Гц, 2Н); 3,16 (t, J=6,0 Гц, 2Н); 2,55-2,46 (m, 4Н); 1,58 (d, J=6,6 Гц, 6Н). ЖХМС: RT=5,70 мин, M+Н+=452.
Пример 133
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-N(2-(метилсульфонил)этил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 133
Метиловый эфир 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 184 омыляли, при этом получали 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновую кислоту (30 мг, 0,09 ммоль), которую растворяли в ДМФ (0,5 мл) и обрабатывали диизопропилэтиламином (0,077 мл, 0,44 ммоль), 2-(метилсульфонил)этанамином (22 мг, 0,18 ммоль) и затем HATU (67 мг, 0,18 ммоль). Полученную смесь перемешивали при КТ в течение 12 ч. Смесь разбавляли EtOAc и Н2О, водный слой экстрагировали EtOAc (2х) и объединенные органические экстракты промывали соевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали препаративной ЖХВР и получали соединение 133. ЖХ/МС (ESI+): m/z 462 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 8,76 (t, J=5,1, 1Н), 8,48 (d, J=8,4, 1H), 7,99 (s, 1H), 7,92 (s, 1H), 7,59 (d, J=8,4, 1H), 7,53 (s, 1H), 5,86 (dt, J=13,0, 6,6, 1H), 4,56 (d, J=1,9, 4H), 3,69 (dd, J=12,7, 6,5, 2H), 3,39 (t, J=6,7, 2H), 3,05 (d, J=9,0, 3Н), 1,49 (d, J=6,6, 5H).
Пример 134
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-N-(2-(метилсульфонил)этил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 134
Соединение 134 получали из 2-(1-(2-хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (40 мг, 0,1 ммоль) и 2-(метилсульфонил)этанамина. ЖХ/МС (ESI+): m/z 514 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,72 (s, 1H), 8,56 (s, 0Н), 8,20 (s, 1H), 7,94 (s, 1H), 7,73 (d, J=7,0, 1H), 7,71-7,52 (m, 3Н), 7,43 (s, 1H), 7,36 (d, J=8,9, 1H), 4,48 (d, J=6,6, 3Н), 3,73-3,58 (m, 2H), 3,02 (s, 3H).
Пример 135
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-N-(1-гидрокси-2-метилпропан-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 135
Соединение 135 получали аналогично тому, как описано для соединения 109, из соединения 72 и 2-амино-2-метилпропан-1-ола. МС: (ESI+)=479,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,19 (s, 1H), 8,11 (d, J=2,2 Гц, 1H), 7,78 (s, 1H), 7,71-7,66 (m, 1H), 7,63 (dd, J=7,2, 2,1 Гц, 1H), 7,60-7,50 (m, 3H), 7,37 (s, 1H), 6,98 (d, J=8,5 Гц, 1H), 4,99 (t, J=5,9 Гц, 1H), 4,46 (d, J=3,7 Гц, 4Н), 3,54 (d, J=5,9 Гц, 2Н), 1,36 (s, 6Н).
Пример 136
(2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)(4-изопропилпиперазин-1-ил)метанон 136
Соединение 136 получали аналогично тому, как описано для соединения 109, из соединения 72 и 4-изопропилпиперазина. МС: (ESI+)=518,2. 1Н ЯМР (400 МГц, ДМСО): δ 8,20 (s, 1H), 7,84 (s, 1H), 7,70 (s, 2H), 7,64 (d, J=7,4 Гц, 1H), 7,62-7,51 (m, 2H), 7,36 (d, J=8,5 Гц, 1H), 7,06 (d, J=8,5 Гц, 1Н), 4,48 (s, 4Н), 3,57 (s, 2H), 3,04 (s, 2H), 1,29 (d, J=6,5 Гц, 6Н).
Пример 137
2-(1-(2,4-Дифторфенил)-1Н-1,2,4-триазол-5-ил)-N-(1-гидрокси-2-метилпропан-2-ил)-5,6-дигидробензо[f]imidazo[1,2-d][1,4]оксазепин-10-карбоксамид 137
Соединение 137 получали аналогично тому, как описано для соединения 109, из 2-(1-(2,4-дифторфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 2-амино-2-метилпропан-1-ола. МС: (ESI+)=481,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,26 (d, J=2,2 Гц, 1H), 8,21 (s, 1H), 7,94 (s, 1H), 7,73-7,65 (m, 1H), 7,62 (dd, J=8,5, 2,3 Гц, 1H), 7,51-7,42 (m, 1H), 7,39 (s, 1H), 7,25 (t, J=7,7 Гц, 1H), 7,00 (d, J=8,5 Гц, 1H), 4,96 (t, J=6,0 Гц, 1H), 4,49 (d, J=6,1 Гц, 4Н), 3,52 (d, J=5,8 Гц, 2Н), 1,35 (s, 6H).
Пример 138
2-(4-Циано-1-изопропил-1Н-пиразол-5-ил)-5,6-дигидрробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 138
Соединение 138 получали аналогично тому, как описано в примере 51, из соединения 16 и аммиака. Выход составлял 27,8 мг (44%). MC(ESI): 363,1. 1H ЯМР (400 МГц, ДМСО): δ 8,46 (d, J=8,4, 1H), 8,11 (s, 1H), 8,00 (d, J=4,7, 2H), 7,62 (dd, J=8,4,1,5, 1H), 7,57 (d, J=1,4, 1H), 7,44 (s, 1H), 5,46 (dt, J=13,1, 6,5, 1H), 4,59 (dd, J=17,5, 4,8, 4Н), 1,48 (d, J=6,6, 6H).
Пример 139
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-N-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 139
Соединение 139 получали аналогично тому, как описано для соединения 133, из 2-(1-(2-хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (40 мг, 0,1 ммоль) и метиламина (2 М, 0,2 мл) в ТГФ. ЖХ/МС (ESI+): m/z 422 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 8,43 (d, J=4,5, 1H), 8,19 (s, 1H), 7,93 (s, 1H), 7,72 (t, J=10,9, 1H), 7,68-7,51 (m, 4H), 7,47-7,28 (m, 2H), 4,50 (t, J=15,6, 4H), 2,76 (d, J=4,5, 3Н).
Пример 140
N-(2-Гидроксиэтил)-N-изопропил-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-2-карбоксамид 140
Согласно тому, как описано для получения соединения 116, 8-бром-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-2-карбоновую кислоту конденсировали с 2-изопропилэтанолом. Подученное промежуточное соединение (145 мг, 0,37 ммоль) растворяли в IMS (10 мл) и затем добавляли ТЭА (50 мкл, 0,37 ммоль) и 10% Pd/C (20 мг), полученную реакционную смесь перемешивали в атмосфере водорода (1 атм.) в течение 2,75 ч. Реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме. Полученный остаток распределяли между ДХМ и водой, водный слой отделяли и экстрагировали ДХМ. Объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток растворяли в диэтиловом эфире, раствор промывали водой, сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали соединение 140 (79 мг, выход 68%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 7,83 (dd, J=8,0, 1,6 Гц, 1Н); 7,30 (td, J=7,6, 1,8 Гц, 1Н); 7,26-7,16 (m, 2Н); 6,54 (s, 1Н); 4,67-4,53 (m, 1Н); 4,46 (t, J=6,0 Гц, 2Н); 4,35-4,27 (m, 1Н); 3,64-3,56 (m, 2Н); 3,55-3,46 (m, 2Н); 3,13 (t, J=6,0 Гц, 2Н); 0,22 (d, J=6,8 Гц, 6Н). ЖХМС: RT=9,00 мин, М+Н+=316.
Пример 141
4-((2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)метил)пиперидин-2-он 141
Соединение 141 получали согласно тому, как описано для получения 128, но без использования HCl в диэтиловом эфире, из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбальдегида и 2-гидрокси-1-пиперазин-1-илэтанона в виде твердого вещества белого цвета. 1Н ЯМР (MeOD, 400 МГц): δ 8,00 (s, 1Н); 7,92 (d, J=2,1 Гц, 1Н); 7,34 (dd, J=8,3, 2,1 Гц, 1Н); 7,22 (d, J=8,3 Гц, 1Н); 6,88 (s, 1Н); 5,80-5,67 (m, 1Н); 4,54 (t, J=6,1 Гц, 2Н); 3,69 (s, 2Н); 3,36-3,28 (m, 2Н); 3,19 (t, J=6,1 Гц, 2Н); 3,13 (s, 2Н); 2,76-2,70 (m, 2Н); 1,56 (d, J=6,6 Гц, 6Н). Один заменяемый протон не наблюдали. ЖХМС: RT=7,45 мин, М+Н+=408.
Пример 142
2-(4-(2-(1-(2,2,2-Трифторэтил)-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)пиперидин-1-ил)этанол 142
Смесь 9-пиперидин-4-ил-2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена (180 мг, 0,43 ммоль) в ДМФ (1 мл), карбоната калия (89 мг, 0,65 ммоль) и 2-(2-бромэтокси)тетрагидропирана (97 мкл, 0,65 ммоль) нагревали при 50°С в течение 18 ч. Охлажденную реакционную смесь распределяли между EtOAc и водой, водный слой экстрагировали EtOAc и объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН (+2 М NH3) в ДХМ), при этом получали бесцветное маслянистое вещество. Маслянистое вещество растворяли в метаноле и обрабатывали 1 М HCl в метаноле (2 мл, 2 ммоль), реакционную смесь перемешивали при КТ в течение 2 ч и концентрировали в вакууме. Полученный остаток растирали со смесью диэтилового эфира и метанола, при этом получали соединение 142 в виде твердого вещества светло-желтого цвета (115 мг, выход 54%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 9,81 (ушир. s, 1Н); 8,22 (s, 1Н); 7,70 (s, 1Н); 7,32-7,24 (m, 2Н); 6,99 (s, 1Н); 5,72 (q, J=8,7 Гц, 2Н); 5,36 (ушир. s, 1Н); 4,51 (t, J=6,0 Гц, 2Н); 3,80 (t, J=4,9 Гц, 2Н); 3,67-3,57 (m, 2H); 3,22-3,06 (m, 6H); 2,97-2,85 (m, 1Н); 2,10-2,00 (m, 4H). ЖХМС: RT=6,62 мин, M+H+=463,1.
Пример 143
2-(4-(2-(1-(2,2,2-Трифторэтил)-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 143
Смесь 9-пиперидин-4-yl-2-[2-(2,2,2-триафторэфтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена (180 мг, 0,43 ммоль) в ДМФ (1 мл) обрабатывали карбрнатом калия (89 мг, 0,65 ммоль) и бромацетамидом (77 мг, 0,56 ммоль) и затем перемешивали при КТ в течение 18 ч. Реакционную смесь распределяли между EtOAc и водой, водный слой экстрагировали EtOAc и объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром, при этом получали соединение 143 в виде твердого вещества светло-желтого цвета (115 мг, выход 54%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 8,21 (s, 1Н); 7,75 (d, J=2,2 Гц, 1Н); 7,28 (dd, J=8,4, 2,2 Гц, 1Н); 7,20 (d, J=8,3 Гц, 1Н); 7,15 (s, 2H); 6,97 (s, 1Н); 5,73 (q, J=8,8 Гц, 2H); 4,49 (t, J=6,0 Гц, 2H); 3,20 (t, J=6,0 Гц, 2H); 2,96-2,85 (m, 4H); 2,62-2,52 (m, 1Н); 2,24-2,14 (m, 2H); 1,84-1,66 (m, 4H). ЖХМС: RT=6,55 мин, M+H+=476.
Пример 144
9-(Азетидин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин 144
трет-Бутиловый эфир 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил]азетидин-1-карбоновой кислоты (0,47 г, выход 49%) получали из соединения 61 (0,52 г, 1,40 ммоль) и иодида цинка трет-бутилового эфира 3-азетидин-1-карбоновой кислоты (2,0 ммоль). ЖХМС: RT=4,70, М+H+=451.
9-Азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен (0,41 г, 0,76 ммоль) обрабатывали кислотой, при этом получали неочищенный гидрохлорид, который распределяли между EtOAc и насыщенным водным раствором гидрокарбоната натрия и 3 раза экстрагировали ДХМ. Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% 2 М NH3 (в МеОН) в ДХМ), при этом получали соединение 144 (162 мг, выход 31%). 1Н ЯМР (400 МГц, CDCl3): δ 7,97-7,93 (m, 1Н); 7,82 (d, J=2,19 Гц, 1Н); 7,29 (dd, J=8,36, 2,25 Гц, 1Н); 7.19 (d, J=8,32 Гц, 1Н); 6,87 (s, 1Н); 5,74-5,66 (m, 1Н); 4,57-4,50 (m, 2H); 4,12-4,02 (m, 1Н); 4,00 (t, J=7,46 Гц, 2Н); 3,87 (t, J=7,27 Гц, 2Н); 3,16 (t, J=6,02 Гц, 2Н); 1,62 (d, 6Н). ЖХМС: RT=6,01 мин, М+Н+=351.
Пример 145
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(пиперазин-1-карбонил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 145
Смесь N-Boc-пиперазина (101 мг, 0,54 ммоль), 2-(2-изопропилl-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-карбоновой кислоты (180 мг, 0,53 ммоль), EDCI (151 мг, 0,79 ммоль), HOBt (107 мг, 0,795 ммоль) и ТЭА (216 мкл, 1,54 ммоль) в ДМФ (2 мл) перемешивали при КТ в течение 20 ч. Смесь разбавляли этилацетатом и промывали (вода, насыщенный водный раствор бикарбоната натряи и солевой раствор), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в ДХМ), при этмо получали трет-бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-карбонил]пиперазин-1-карбоновой кислоты (258 мг, выход 96%). Промежуточный трет-бутиловый эфир карбюоновой кислоты растворяли в ДХМ (20 мл) и обрабатывали 4 н. HCl в диоксане (4 мл) и перемешивали при КТ в течение 3 ч, затем добавляли диэтиловый эфир (20 мл). Полученный осадок собирали фильтрованием и промывали диэтиловым эфиром, при этом получали соединение 145 в виде твердого ещства белого цвета (209 мг, выход 93%). 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,32 (s, 2H); 8,37 (s, 1Н); 8,31 (d, J=2,20 Гц, 1Н); 8,06 (s, 1Н); 7,44 (dd, J=8,37, 2,21 Гц, 1Н); 7,15 (d, J=8,37 Гц, 1Н); 5,32-5,23 (m, 1Н); 4,37 (t, J=4,98 Гц, 2Н); 3,77 (s, 4H); 3,15 (t, J=6,94 Гц, 6Н); 1,51 (d, J=6,59 Гц, 6Н). ЖХМС: RT=6,02 мин, М+H+=408.
Пример 146
2-(4-Изопропил-4Н-1,2,4-триазол-5-ил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 146
Стадия 1
4-Изопропил-3-метилсульфанил-4Н-[1,2,4]триазол
В раствор 4-изопропил-3-тиосемикарбазида (7,0 г, 52,54 ммоль) в диоксане (50 мл) добавляли ДМФ-ДМА (14,1 мл, 105,08 ммоль) и смесь нагрвали до 100°С. Через 3 ч добавляли дополнительное количество ДМФ-ДМА (52,54 ммоль) и нагревание продолжали. Через 18 ч смесь охлаждали до КТ и растворитель удаляли в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 100% EtOAc в циклогексане), при этом получали 4-изопропил-3-метилсульфанил-4Н-[1,2,4]триазол (4,05 г, выход 49%). ЖХМС: RT=2,55 мин, M+H+=158.
Стадия 2
4-Изопропил-3-метансульфанил-4Н-[1,2,4]триазол
В раствор 4-изопропил-3-метилсульфанил-4Н-[1,2,4]триазола (3,05 г, 19,43 ммоль) в ДХМ (30 мл) добавляли муравьиную кислоту (2,9 мл, 76,36 ммоль) и тетрагидрат молибдата аммония (56 мг, 0,047 ммоль). В смесь при интенсивном перемешивании порциями добавляли раствор пероксида водорода (50 мас. % в Н2О, 8 мл, 116,58 ммоль) во избежания неконтролируемого разогрева. После завершения добавления смесь перемешивали при КТ в течение 18 ч. Смесь охлаждали на ледяной бане, реакцию останавливали осрожным добавлением насыщенного раствора сульфита натрия и экстрагировали ДХМ (3×100 мл). Органические экстракты объединяли и сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали 4-изопропил-3-метансульфанил-4Н-[1,2,4]тразол (3,29 г, выход 90%). ЖХМС: RT=2,02 мин, M+H+=190.
Стадия 3
9-Бром-2-(4-изопропил-4Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен
В сосуд для микроволнового излучения помещали соединение 49 (200 мг, 0,755 ммоль), 4-изопропил-3-метансульфанил-4Н-[1,2,4]триазол (143 мг, 0,755 ммоль), карбонат цезия (246 мг, 0,755 ммоль) и ТГФ (2 мл). Реакционную смесь нагрвали при 150°С в течение 2 ч, затем экстрагировали EtOAc (20 мл), промывали водой (20 мл), сушили (Na2SO4), фильтровали и концентрировли в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 100% EtOAc вциклогексане), при этом получали 9-бром-2-(4-изопропил-4Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен (122 мг, выход 43%). ЖХМС: RT=4,34 мин, М+Н+=374/376.
Стадия 4
2-(4-Изопропил-4Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен
В дегазированный раствор 9-бром-2-(4-изопропил-4Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена (122 мг, 0,326 ммоль) в IMS (10 мл) добавляли ТЭА (51 мкл, 0,359 ммоль) и 10% Pd/C (15 мг). Смесь перемешивали в атмосфере водорода (1 атм.) в течение 60 мин, фильтровали и фильтрат концентрировали в вакууме. Полученный остаток растворяли в ДХМ (20 мл) и промывали водой (20 мл), органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток лиофилизовали из ацетонитрила и воды, при этом получали соединение 146 (54 мг, выход 56%). ЖХМС: RT=10,29 мин, M+H+=296.
В другом варианте дегазированный раствор 8-бром-2-(4-изопропил-4Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена (122 мг, 0,326 ммоль) в IMS (10 мл) добавляли 10% Pd/C (15 мг), реакционную смесь перемешивали в атмосфере водорода в течение 1 ч, реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Полученный остаток растворяли в ДХМ, раствор промывали водой, органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток растворяли в ацетонитриле и воде и раствор лиофилизовали, при этом получали 2-(4-изопропил-4Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен в виде твердого вещества белого цвета (54 мг, выход 56%). 1Н ЯМР (400 МГц, CDCl3): δ 8,27-8,23 (m, 2H); 8,12 (t, J=1,02 Гц, 1Н); 7,30-7,24 (m, 1Н); 7,14-7,06 (m, 2H); 5,38-5,29 (m, 1Н); 4,36 (t, J=5,10 Гц, 2H); 3,16 (td, J=5,10, 1,06 Гц, 2Н); 1,63-1,54 (d, J=6,59 Гц, 6Н). ЖХМС: RT=10,29 мин, M+H+=296.
Пример 147
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-N-(1-(2-гидроксиэтил)-1Н-пиразол-4-ил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол-9-карбоксамид 147
Соединение 147 получали аналогично тому, как описано для получения соединения 116, из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-карбоновой кислоты и 4-амино-1-(2-гидроксиэтил)пиразола в виде твердого вещества белого цвета. 1Н ЯМР (400 MHz, ДМСО-d6): δ 10,45 (s, 1Н); 8,85 (d, J=2,32 Гц, 1Н); 8,36 (s, 1Н); 8,08-8,05 (m, 2Н); 7,87 (dd, J=8,53, 2,35 Гц, 1Н); 7,58 (d, J=0,69 Гц, 1Н); 7,18 (d, J=8,50 Гц, 1Н); 5,36-5,28 (m, 1Н); 4,88 (t, J=5,31 Гц, 1Н); 4,39 (t, J=4,97 Гц, 2Н); 4,13 (t, J=5,61 Гц, 2Н); 3,73 (q, J=5,51 Гц, 2Н); 3,16 (t, J=4,96 Гц, 2Н); 1,53 (d, J=6,59 Гц, 6Н). ЖХМС: RT=9,63 мин, М+H+=449.
Пример 148
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(метилсульфонил)азетидин-3-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,41оксазепин 148
Охлажденный льдом раствор 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена (0,16 г, 0,46 ммоль) в ДХМ (5 мл) обрабатывали ТЭА (0,14 мл, 1,0 ммоль) и метансульфонилхлоридом (40 мкл, 0,51 ммоль) и смесь перемешивали при КТ в течение 2 ч. Реакционную смесь промывали водой и водный слой экстрагировали ДХМ, орагнические экстракты объединяли и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 50 до 100% EtOAc в циклогексане), при этом получали соединение 148 в виде твердого вещества белого цвета (126 мг, выход 64%). 1Н ЯМР (400 МГц, CDCl3): δ 7,95 (d, J=2,49 Гц, 2Н); 7,31 (dd, J=8,35, 2,29 Гц, 1Н); 7,21 (d, J=8,32 Гц, 1Н); 6,89 (s, 1Н); 5,75-5,66 (m, 1Н); 4,56 (t, J=5,89 Гц, 2Н); 4,32 (t, J=8,24 Гц, 2Н); 4,12 (t, J=7,34 Гц, 2Н); 3,92-3,82 (m, 1Н); 3,20 (t, J=5,89 Гц, 2Н); 2,93 (s, 3Н); 1,62 (d, 6Н). ЖХМС: RT=9,68 мин, M+H+=429.
Пример 149
1-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)азетидин-1-ил)этанон 149
Соединение 149 получали аналогично тому, как описано для соединения 148, из 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена и уксусного ангидрида в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3): δ 7,93 (s, 1Н); 7,85 (d, J=2,17 Гц, 1Н); 7,27-7,20 (m, 1Н); 7,19 (d, J=8,32 Гц, 1Н); 6,85 (s, 1Н); 5,68-5,59 (m, 1Н); 4,59-4,50 (m, 3Н); 4,44 (t, J=9,38 Гц, 1Н); 4,18-4,11 (m, 1Н); 4,13-4,05 (m, 1Н); 3,89-3,80 (m, 1Н); 3,16 (t, J=5,93 Гц, 2Н); 1,91 (s, 3Н); 1,57 (d, J=6,62 Гц, 6Н). ЖХМС: RT=9,50 мин, M+H+=393.
Пример 150
2-Гидрокси-1-(3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)азетидин-1-ил)этанон 150
Соединение 150 получали аналогично тому, как описано для соединения 116, из 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена и гликолевой кислоты в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3): δ 7,95 (s, 1Н); 7,87 (d, J=2,09 Гц, 1Н); 7,28-7,21 (m, 1Н); 7,24-7,19 (m, 1Н); 6,87 (s, 1Н); 5,68-5,59 (m, 1Н); 4,60-4,48 (m, 4Н); 4,20 (dd, J=9,89, 6,11 Гц, 1Н); 4,16-4,07 (m, 1Н); 4,05 (d, J=3,28 Гц, 2Н); 4,03-3,96 (m, 1Н); 3,18 (t, J=5,91 Гц, 2Н); 1,58 (d, J=6,62 Гц, 6Н). Один заменяемый протон не наблюдали. ЖХМС: RT=8,95 мин, М+Н+=409.
Пример 151
2-Гидрокси-1-(4-(2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)пиперидин-1-ил)этанон 151
Соединение 151 получали аналогично тому, как описано для соединения 116, из 9-пиперидин-4-ил-2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена и гликолевой кислоты в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,21 (s, 1Н); 7,73 (d, J=2,16 Гц, 1Н); 7,29 (dd, J=8,37, 2,17 Гц, 1Н); 7,21 (d, J=8,32 Гц, 1Н); 6,97 (s, 1Н); 5,72 (q, J=8,78 Гц, 2Н); 4,55-4,43 (m, 4Н); 4,13-4,05 (m, 2Н); 3,79 (d, J=13,44 Гц, 1Н); 3,23-3,15 (m, 3Н); 3,09 (t, J=12,91 Гц, 1Н); 2,91-2,82 (m, 1Н); 2,77-2,65 (m, 1Н); 1,86 (d, J=12,87 Гц, 2Н). ЖХМС: RT=9,76 мин, M+H+=477.
Пример 152
9-(1-(2-(Метилсульфонил)этил)пиперидин-4-ил)-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин 152
Смесь 9-пиперидин-4-ил-2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена (90 мг, 0,22 ммоль), ТЭА (150 мкл) и IMS (0,6 мл) обрабатывали винилсульфоном (48 мл, 0,54 ммоль), затем разбавляли ДХМ и перемешивали при КТ в течение 18 ч. Реакционную смесь концентрировали в вакууме, остаток растирали с диэтиловым эфиром, при этом получали твердое вещество, которое очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 5% МеОН в ДХМ), при этом получали соединение 152 в виде твердого вещества белого цвета (96 мг, выход 85%). 1Н ЯМР (400 Мгц, CDCl3): δ 8,01 (s, 1Н); 7,69 (s, 1Н); 7,19-7,08 (m, 2H); 6,92 (s, 1Н); 5,53 (q, J=8,15 Гц, 2H); 4,54 (t, J=6,01 Гц, 2H); 3,21-3,12 (m, 4H); 3,21-2,92 (m, 5H); 2,93 (t, J=6,33 Гц, 2H); 2,63-2,54 (m, 1Н); 2,21 (t, J=11,57 Гц, 2H); 1,94 (d, J=12,96 Гц, 2H); 1,81-1,66 (m, 2H). ЖХМС: RT=6,57 мин, M+H+=525.
Пример 153
((3S,5R)-3,5-Диметилпиперазин-1-ил)(2-(1-(2,2,2-trifluoroethyl)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)метанон 153
Соединение 153 получали аналогично тому, как описано для соединения 109, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 3S,5R-диметилпиперазина. МС: (ESI+)=476,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,39 (d, J=2,0 Гц, 1Н), 8,10 (d, J=4,9 Гц, 2Н), 7,37 (dd, J=8,4, 2,1 Гц, 1Н), 7,12 (d, J=8,4 Гц, 1Н), 5,88 (s, 2Н), 4,57 (s, 4H), 4,06 (q, J=5,3 гц, 2Н), 3,17 (d, J=5,2 Гц, 2Н), 2,67 (s, 2Н), 0,92 (s, 6H).
Пример 154
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-9-пиперид-4-ил-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 154
Стадия 1
трет-Бутиловый эфир 4-{2-[2-(2-Хлорфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты
трет-Бутиловый эфир 4-{2-[2-(2-Хлорфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты получали аналогично трет-бутиловому эфиру 4-{2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты из соединения 60 (500 мг, 1,13 ммоль) в виде бесцветного смолообразного вещества (490 мг, выход 80%). ЖХМС: RT=5,14 мин, M+Na+=567.
Стадия 2
2-[2-(2-Хлорфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен
2-[2-(2-Хлорфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен получали аналогично 9-пиперидин-4-ил-2-[2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулену из трет-бутилового эфира 4-{2-[2-(2-хлорфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (490 мг, 0,9 ммоль). Неочищенную соль распределяли между ДХМ и насыщенным водным раствором гидрокарбоната натрия и водный слой эксрагировали ДХМ. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали обращенно-фазной ЖХВР (колонка Gemini C18, элюент: градиент МеОН в Н2О+0,1% НСО2Н), при этом получали соединение 154 (80 мг, выход 18%) в виде соли муравьиной кислоты. 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,44 (s, 1Н); 8,39 (s, 1Н); 8,27 (s, 1Н); 7,81-7,68 (m, 3H); 7,64 (td, J=7,60, 1,51 Гц, 1Н); 7,09-7,03 (m, 2H); 6,90 (d, J=8,28 Гц, 1Н); 4,19 (t, J=5,06 Гц, 2H); 3,32 (d, J=12,42 гц, 2H); 3,04 (t, J=5,01 Гц, 2H); 2,89 (dd, J=13,45, 11,09 Гц, 2Н); 2,54-2,56 (m, 1Н); 1,69 (d, J=13,20 Гц, 2Н); 1,58-1,45 (m, 2Н). ЖХМС: RT=7,99 мин, М+H+=447.
Пример 155
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)азетидин-1-ил)ацетамид 155
Соединение 155 получали аналогично тому, как описано для соединения 143, из 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]и бромацетамида. Неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в DCM, при этом получали твредое вещество белого цвета. 1Н ЯМР (400 МГц, CDCl3): δ 7,95 (s, 1Н); 7,83 (d, J=2,18 Гц, 1Н); 7,24 (d, J=2,21 Гц, 1Н); 7,18 (d, J=8,31 Гц, 1Н); 6,86 (s, 1Н); 5,71-5,63 (m, 1Н); 5,45 (s, 1Н); 4,54 (t, J=5,98 Гц, 2Н); 3,87 (t, J=7,28 Гц, 2Н); 3,81-3,72 (m, 1Н); 3,38 (t, J=6,84 Гц, 2Н); 3,22 (s, 2Н); 3,16 (t, J=5,99 Гц, 2Н); 1,59 (d, J=6,62 Гц, 6Н). Один заменяемый протон не наблюдали. ЖХМС: RT=5,84 мин, М+Н+=408.
Пример 156
N-(Азетидин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-карбоксамид 156
Указанный в заголовке продукт получали аналогично тому, как описано для соединения 126 из 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты и 1,1-диметилэтилового эфира 3-аминоазетидин-1-карбоновой кислоты, неочищенный продукт суспендировали в ДХМ и обрабатывали карбонатной смолой МР и перемешивали в течение 1.5 ч. Смесь фильтровали, фильтрат концентрировали в вакууме, полученный остаток растирали с диэтиловым эфиром и при этом получали соединение 156 в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,11 (d, J=6,84 Гц, 1Н); 8,46 (d, J=2,18 Гц, 1Н); 8,05 (t, J=0,64 Гц, 1Н); 7,90-7,85 (m, 1Н); 7,37-7,30 (m, 1Н); 6,92 (s, 1Н); 5,62-5,53 (m, 1Н); 4,82-4,73 (m, 1Н); 4,57 (t, J=5,76 Гц, 2Н); 3,95-3,81 (m, 4Н); 3,25 (t, J=5,78 Гц, 2Н); 1,50 (d, J=6,58 Гц, 6Н). ЖХМС: RT=6,45 мин, M+H+=394.
Пример 157
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(метилсульфонил)этил)азетидин-3-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин 157
Соединение 157 получали аналогично тому, как описано для соединения 152, из 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена и винилсульфона, при этом получали соединение 157 в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,01 (d, J=0,64 Гц, 1Н); 7,84 (d, J=2,19 Гц, 1Н); 7,36 (dd, J=8,32, 2,21 Гц, 1Н); 7,21 (d, J=8,30 Гц, 1Н); 6,87 (s, 1Н); 5,60-5,51 (m, 1Н); 4,48 (t, J=6,03 Гц, 2Н); 3,67 (s, 3Н); 3,20-3,10 (m, 6Н); 3,03 (s, 3Н); 2,83 (t, J=6,83 Гц, 2Н); 1,48 (d, J=6,58 Гц, 6Н). ЖХМС: RT=5,99 мин, M+H+=457.
Пример 158
N-Метил-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробезно[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 158
Соединение 158 получали аналогично тому, как описано для соединения 133, из 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (60 мг, 0,1 ммоль) и 2 М раствора метиламина (0,26 мл) в ТГФ. ЖХ/МС (ESI+): m/z 393 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,49 (d, J=4,2, 1H), 8,42 (s, 1H), 8,11 (d, J=9,8, 1H), 7,60 (dd, J=8,4, 1,6, 1H), 7,52 (d, J=1,4, 1H), 5,90 (q, J=8,8, 2H), 4,57 (dd, J=11,5, 5,4, 4H), 2,79 (d, J=4,5, 3Н).
Пример 159
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-N-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 159
Соединение 159 получали аналогично тому, как описано для соединения 133, из 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (100 мг, 0,3 ммоль) и 2 М раствора метиламина (0,59 мл) в ТГФ. ЖХМС (ESI+): m/z 353 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,54-8,39 (m, 1H), 7,93 (dd, J=18,0, 11,2, 1H), 7,67-7,46 (m, -1H), 6,47 (d, J=33,7, -2H), 5,94-5,78 (m, -2H), 4,68-4,47 (m, -4H), 3,52-3,55 (s, -3H), 2,90-2,68 (m, 1H), 1,47 (t, J=13,6, 6H).
Соединение 160
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-10-(1Н-пиразол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 160
Соединение 160 получали аналогично тому, как описано для соединения 187, из 10-бром-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина 187 и трет-бутилового эфира 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-карбоновой кислоты. МС: 362,3. 1H ЯМР (400 МГц, ДМСО): δ 8,57 (t, J=4,0, 1H), 7,99-7,94 (m, 4H), 7,56 (dt, J=11,2, 5,6, 1H), 7,06 (t, J=8,6, 1H), 5,81 (p, J=6,6, 1H), 4,53 (d, J=9,5, 4H), 1,53 (d, J=6,6, 7H).
Пример 161
2-(1-(2-Хлорфенил)-1Н-имидазол-2-ил)-N-(1-гидрокси-2-метилпропан-2-ил)-5,6-дигидробезно[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 161
Соединение 161 получали аналогично тому, как описано для соединения 109, из соединения 72 и 2-амино-2-метилпропан-1-ола. МС: (ESI+)=478,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,12 (d, J=2,1 Гц, 1H), 7,63 (dd, J=6,0, 3,4 Гц, 1H), 7,53 (dd, J=6,1, 3,2 Гц, 2Н), 7,50-7,44 (m, 3Н), 7,36 (s, 1H), 7,28 (d, J=1,1 Гц, 1H), 7,12 (s, 1H), 6,96 (d, J=8,4 Гц, 1H), 5,00 (s, 1H), 4,41 (s, 4H), 3,55 (d, J=5,1 Гц, 2Н), 1,36 (s, 6Н).
Пример 162
(2-(1-(2,4-Дифторфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)(4-(2-гидроксиэтил)пиперазин-1-ил)метанон 162
Соединение 162 получали аналогично тому, как описано для соединения 109, из 2-(1-(2,4-дифторфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 2-(пиперазин-1-ил)этанола. МС: (ESI+)=522,2.
Пример 163
N-(1-Гидрокси-2-метилпропан-2-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4] оксазепин-9-карбоксамид 163
Соединение 163 получали аналогично тому, как описано для соединения 133, из 2-(1-шзопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (20 мг, 0,06 ммоль) и 2-амино-2-метил-1-пропанола (11 мг, 0,12 ммоль) в ТГФ. ЖХ/МС (ESI+): m/z 411 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,45 (d, J=8,4, 1H), 7,95 (d, J=22,3, 2H), 7,64-7,45 (m, 3Н), 5,86 (dt, J=13,1, 6,5, 1H), 4,88 (t, J=6,0, 1H), 4,63-4,44 (m, 4H), 3,58-3,45 (m, 3Н), 1,49 (d, J=6,6, 6H), 1,32 (s, 6H).
Соединение 164
N-(1-Гидрокси-2-метилпропан-2-ил)-2-(1-(S-диоксотетрагидротиофен-3-ил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоксамид 164
Соединение 164 получали аналогично тому, как описано для соединения 109, из 2-(1-(S-диоксотетрагидротиофен-3-ил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбоновой кислоты и 2-амино-2-метилпропан-1-ола. МС: (ESI+)=487,1.
Пример 165
2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-N-(1-гидрокси-2-метилпропан-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 165
Соединение 165 получали аналогично тому, как описано для соединения 133, из 2-(1-(2-хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (20 мг, 0,06 ммоль) и 2-амино-2-метил-1-пропанола (11 мг, 0,12 ммоль) в ТГФ. ЖХ/МС (ESI+): m/z 480 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 8,19 (s, 1H), 7,93 (s, 1H), 7,78-7,48 (m, 6H), 7,43-7,26 (m, 2H), 4,85 (t, J=6,1, 1H), 4,47 (d, J=9,0, 4H), 3,49 (d, J=6,0, 2H), 1,29 (s, 6H).
Пример 166
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(пиридин-4-илметил)азетидин-3-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин 166
Соединение 166 в виде твердого вещества белого цвета получали аналогично тому, как описано для соединения 128, из 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена и пиридин-4-карбальдегида, the неочищенный продукт очищают экспресс-хроматографией (SiO2, элюент: градиент от 0 до 20% МеОН в EtOAc) и растиранием с циклогексаном. 1Н NMR (400 MHz, CDCl3): δ 8,55 (d, J=5,20 Гц, 2Н); 7,95 (s, 1Н); 7,86 (d, J=2,20 Гцг, 1Н); 7,30-7,24 (m, 3Н); 7,21-7,12 (m, 1Н); 6,88-6,83 (m, 1Н); 5,73-5,65 (m, 1Н); 4,54 (t, J=5,99 Гц, 2Н); 3,89-3,74 (m, 3H); 3,71 (s, 2H); 3,30 (s, 2H); 3,16 (t, J=5,99 Гц, 2Н); 1,58 (d, J=6,62 Гц, 6Н). ЖХМС (способ F): RT=6,07 мин, М+Н+=442.
Пример 167
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-N-(1-изопропилазетидин-3-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-карбоксамид 167
Раствор азетидин-3-иламида 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-карбоновой кислоты (0,15 г, 0,38 ммоль), ацетона (84 мкл, 1,14 ммоль), уксусной кислоты (22 мл), метанола (1 мл) и ДХМ (1 мл) перемешивали при КТ втечение 1 ч. Затем добавляли триацетоксиборгидрид натрия (0,2 г, 0,95 ммоль) т полученную смесь перемешивали при КТ в течение 72 ч. Затем добавляли ацетон (84 мклL) и молекуялрные сита с диаметром пор , перемешивание продолжали в течение 1 ч и добавляли триацетоксиборгидрид натрия (0,2 г, 0,95 ммоль). Реакционную смесь перемешивали при КТ в течение 18 ч, затем добавляли насыщенный водный раствор гидрокарбоната натрия. Полученную смесь экстрагировали 10% МеОН в ДХМ. Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в ДХМ), затем растирали с диэтиловым эфиром и перекристаллизовали из EtOAc, при этом получали соединение 167 в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, CDCl3): δ 8,47 (d, J=2,22 Гц, 1Н); 7,95 (s, 1Н); 7,71 (dd, J=8,44, 2,24 Гц, 1Н); 7,23 (d, J=8,43 Гц, 1Н); 6,87 (s, 1Н); 5,72-5,64 (m, 1Н); 4,74-4,67 (m, 1Н); 4,58 (t, J=5,67 Гц, 2Н); 3,68 (t, J=7,42 Гц, 2Н); 3,22 (t, J=5,69 Гц, 2Н); 3,12 (s, 2H); 2,38 (t, J=7,02 Гц, 1Н); 1,61 (d, J=6,61 Гц, 6Н); 0,98 (d, J=6,21 Гц, 6Н). ЖХМС (способ F): RT=6,82 мин, M+H+=436.
Пример 168
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-N-метокси-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 168
Соединение 168 получали аналогично тому, как описано для соединения 133, из 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (20 мг, 0,06 ммоль) и гидрохлорида метоксиамина (9,8 мг, 0,12 ммоль) в ТГФ. ЖХ/МС (ESI+): m/z 369 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,45 (t, J=16,8, 1H), 7,95 (d, J=26,9, 2H), 7,49 (dd, J=20,1, 18,6, 2H), 5,86 (dt, J=13,1, 6,4, 1H), 4,56 (d, J=2,8, 4H), 3,72 (s, 2H), 1,61-1,32 (m, 5H).
Пример 169
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)азетидин-1-ил)этанол 169
Соединение 169 получали аналогично тому, как описано для соединения 142, из 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена и 2-(2-бромэтокси)тетрагидропирана в виде твердого вещества белого цвета. 1Н ЯМР (400 Мгц, ДМСО-d6): δ 8,03 (s, 1Н); 7,88 (d, J=2,22 Гц, 1Н); 7,42 (dd, J=8,35, 2,22 Гц, 1Н); 7,26 (d, J=8,32 Гц, 1Н); 6,89 (s, 1Н); 5,62-5,54 (m, 1Н); 4,83 (s, 1Н); 4,50 (t, J=6,03 Гц, 2Н); 4,06 (s, 2Н); 3,94 (t, J=8,52 Гц, 1Н); 3,70 (s, 2Н); 3,51 (s, 3Н); 3,18 (t, J=6,05 Гц, 2Н); 2,94 (s, 2Н); 1,49 (d, J=6,58 Гц, 6Н). ЖХМС: (способ F): RT=6,01 мин, M+H+=395.
Пример 170
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b]пиразоло[1,5-d][1,4]оксазепин-9-ил)азетидин-1-ил)-2-метилпропан-1-ол 170
Смесь 9-азетидин-3-ил-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулена (200 мг, 0,55 ммоль), 2-бром-2-метилового эфира пропионовой кислоты (71 мкл, 0,55 ммоль), ТЭА (75 мкл, 0,55 ммоль) в ДМФ нагревали при 55°С в течение 30 ч, затем концентрировали в вакууме. Остаток распределяли между 10% МеОН в ДХМ и водой, водный слой экстрагировали 10% МеОН в ДХМ и объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в ДХМ), при этом получали метиловый эфир 2-{3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил]азетидин-1-ил}-2-метилпропионовой кислоты (56 мг, выход 23%). В раствор метилового эфира 2-{3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,10b-диазабензо[е]азулен-9-ил]азетидин-1-ил}-2-метилпропионовой кислоты (56 мг, 0,12 ммоль) в ТГФ (1,5 мл) при -78°С добавляли DIBAL (1,5 М раствор в толуоле, 0,24 мл, 0,36 ммоль) и смесь нагревали при КТ в течение 18 ч. Смесь охлаждали до 0°С, по каплям добавляли DIBAL (0,12 мл, 0,18 ммоль) и перемешивание продолжали в течение 45 мин. Реакцию останавливали добавлением МеОН (0,5 мл) и насыщенным водным раствором соли Рошаля (0,5 мл), разбавляли EtOAc, фильтровали через целит и промывали EtOAc. Фильтрат концентрировали в вакууме и остаток очищали экспресс-хроматографией (SiO2, элюент: градеинт от 0 до 5% МеОН (+2 М NH3) в ДХМ), при этом получали соединение 170 в виде твердого вещества белого цвета (27 мг, выход 53%). 1Н ЯМР (400 МГц, CDCl3): δ 7,95 (s, 1Н); 7,85 (d, J=2,18 Гц, 1Н); 7,23 (d, J=2,23 Гц, 1Н); 7,17 (d, J=8,29 Гц, 1Н); 6,87 (s, 1Н); 5,74-5,66 (m, 1Н); 4,55 (t, J=6,02 Гц, 2Н); 3,71 (s, 3H); 3,43 (s, 2H); 3,28 (s, 2H); 3,16 (t, J=6,03 Гц, 2Н); 1,59 (d, J=6,62 Гц, 6Н); 1,04 (s, 6Н). Один заменяемый протон не наблюдали. ЖХМС: (способ F): RT=6,43 мин, М+Н+=423.
Пример 171
2-(1-(2,4-Дифторфенил)-1Н-1,2,4-триазол-5-ил)-8-(1-(2-(метилсульфонил)этил)азетидин-3-ил)-4,5-дигидробензо-2Н-окзепино[4,5-d]пиразол 171
Согласно тому, как описано для получения соединения 152, проводили реакцию между соединением 63 и винилсульфоном. Неочищенный продукт очищали обращенно-фазной ЖХВР (колонка Gemini С6-фенил, элюент: градиент от 40 до 90% метанол воде + 0,1% НСО2Н), при этом получали соединение 171 в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,42 (s, 1Н); 8,29 (s, 1Н); 8,14 (s, 1Н); 7,81 (td, J=8,75, 5,92 Гц, 1Н); 7,64 (ddd, J=10,33, 9,02, 2,80 Гц, 1Н); 7,38-7,32 (m, 1Н); 7,29 (d, J=8,17 Гц, 1Н); 6,96 (d, J=1,71 Гц, 1Н); 6,91 (dd, J=8,25, 1,80 Гц, 1Н); 4,23 (t, J=5,01 Гц, 2H); 3,66 (t, J=7,11 Гц, 2H); 3,76-3,41 (m, 1Н); 3,16 (t, J=7,36 Гц, 5H); 3,05 (s, 4H); 2,88 (t, J=6,91 Гц, 2Н). ЖХМС (способ F): RT=7,25 мин, M+H+=527.
Пример 172
2-(3-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-ил)ацетамид 172
Соединение 172 в виде твердого вещества белого цвета получали аналогично тому, как описано для соединения 143, из соединения 63 и бромацетамида, неочищенный продукт очищали экспресс-хроматографии (SiO2, элюент: градиент от 0 до 10% МеОН в ДХМ). 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,42 (s, 1Н); 8,28 (s, 1Н); 7,81 (td, J=8,75, 5,92 Гц, 1Н); 7,63 (ddd, J=10,33, 9,01, 2,79 Гц, 1Н); 7,39-7,27 (m, 2Н); 7,11 (s, 1Н); 7,03 (s, 1Н); 6,96-6,89 (m, 2Н); 4,23 (t, J=5,01 Гц, 2Н); 3,66 (t, J=7,02 Гц, 2Н); 3,62-3,52 (m, 1Н); 3,16 (t, J=6,65 Гц, 2Н); 3,10-2,98 (m, 4Н). ЖХМС: (способ F): RT=7,00 мин, М+H+=478.
Пример 173
N-Гидрокси-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 173
Соединение 173 получали аналогично тому, как описано для соединения 133, из 2-(1-изопропил- 1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (20 мг, 0,06 ммоль) и гидрохлорида гидроксиаммония (8 мг, 0,1 ммоль) в ТГФ. ЖХ/МС (ESI+): m/z 355 (М+Н).
Пример 174
2-(1-(2,4-Дифторфенил)-1Н-1,2,4-триазол-5-ил)-N-метил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 174
Соединение 174 получали аналогично тому, как описано для соединения 133, из 2-(1-(2,4-дифторфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты (30 мг, 0,07 ммоль) и 2 М раствора метиламина (0,06 мл) в ТГФ. ЖХ/МС (ESI+): m/z 422 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,52 (t, J=9,7, 1H), 8,23 (s, 1H), 8,03 (s, 1H), 7,80-7,67 (m, 2H), 7,67-7,55 (m, 1H), 7,47-7,38 (m, 2H), 7,33 (t, J=8,4, 1H), 4,50 (d, J=7,7, 3Н), 2,74 (t, J=17,2, 3Н).
Пример 175
2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-[1-(2-метансульфонилэтил)пиперидин-4-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен 175
Согласно тому, как описано для получения соединения 152, проводили реакцию между гидрохлоридом 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азуленаи винилсульфона. Неочищенный продукт растворяли в ДХМ и обрабатывали 4 н. HCl. После перемешивания в течение 10 мин добавляли диэтиловый эфир и твердый осадок собирали фильтрованием, при этом получали соединение 175 в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, ДМСО-d6): δ 10,63 (s, 1Н); 8,45 (s, 1Н); 8,30 (s, 1Н); 7,85 (td, J=8,75, 5,90 Гц, 1Н); 7,72 (t, J=9,62 Гц, 1Н); 7,42 (t, J=8,57 Гц, 1Н); 7,22 (d, J=2,33 Гц, 1Н); 7,11 (dd, J=8,45, 2,37 Гц, 1Н); 6,97 (d, J=8,34 Гц, 1Н); 4,22 (t, J=5,02 Гц, 2Н); 3,82 (t, J=7,50 Гц, 2Н); 3,76-3,63 (m, 2Н); 3,59 (d, J=9,30 Гц, 2Н); 3,16 (s, 3Н); 3,14 (m, 2Н); 3,06 (t, J=5,04 Гц, 2Н); 2,66 (s, 1Н); 1,96-1,76 (m, 4Н). ЖХМС: (способ F): RT=7,54 мин, M+H+=555.
Пример 176
2-{4-[2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}пиразол-1-ил}этанол 176
8-Бром-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен (160 мг, 0,43 ммоль), 1-[2-(тетрагидропиран-2-илокси)этил]-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1Н-пиразол (165 мг, 0,51 ммоль), карбонат цезия (279 мг, 0,85 ммоль), дихлорид 1,1'-бис(дифенилфосфино)ферроценпалладия(II) в комплексе с ДХМ (17 мг, 5 мол. %) смешивали в реакционном сосуде, воздух откачивали и сосуд заполняли азтом. В сосуд добавляли ТГФ (5 мл) и воду (1 мл) и реакционную смесь нагревали при 85°С в течение 4 ч. Реакционную смесь разбавляли EtOAc и водой, органический слой отделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 30% EtOAc в циклогексане), при этом получали 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-{1-[2-(тетрагидропиран-2-илокси)этил]-1Н-пиразол-4-ил}-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен (134 мг, выход 64%).
2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-8-{1-[2-(тетрагидропиран-2-илокси)этил]-1Н-пиразол-4-ил}-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен (134 мг, 0,27 ммоль) растворяли в диэтиловом эфире (5 мл) и обрабатывали 1 М HCl в диэтиловом эфире (1 мл), затем метанолом (5 мл). Через 30 мин реакционную смесь концентрировали досуха, полученный остаток растирали с диэтиловым эфиром, при этом получали соединение 176 в виде твердого вещества светло-кремого цвета (105 мг, выход 86%). 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,32 (s, 1Н); 8,23 (s, 1Н); 8,19 (d, J=8,23 Гц, 1Н); 8,03 (s, 1Н); 7,96 (s, 1Н); 7,39 (dd, J=8,24, 1,74 Гц, 1Н); 7,29 (d, J=1,68 Гц, 1Н); 5,46-5,36 (m, 1Н); 4,34 (t, 2Н); 4,17 (t, J=5,61 гц, 2Н); 3,78 (t, J=5,58 Гц, 2Н); 3,13 (t, J=4,91 Гц, 2Н); 1,52 (d, J=6,57 Гц, 6Н). Один заменяемый протон не наблюдали. ЖХМС (способ F): RT=10,68 мин, M+H+=406.
Пример 177
1-(4-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-1-ил)-2-метилпропан-2-ол 177
Смесь гидрохлорида 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена (100 мгg, 0,21 ммоль), перхлората лития (22 мг, 0,21 ммоль) и DIPEA (72 мкл, 0,41 ммоль) в ТГФ (3 мл) обрабатывали 2,2-диметилоксираном (183 мкл, 2,06 ммоль) и водой (150 мкл). Реакционную смесь перемешивали при КТ в течение 18 ч и концентрировали в вакууме. Полученный остаток распределяли между ДХМ и водой, органический слой отделяли и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 40 до 100% EtOAc в циклогексане), растирали со смесью МеОН и воды, при этом получали соединение 177 в виде твердого вещества белого цвета (58 мг, выход 54%). 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,44 (s, 1Н); 8,28 (s, 1Н); 7,85 (td, J=8,76, 5,90 Гц, 1Н); 7,61 (ddd, J=10,27, 8,88, 2,78 Гц, 1Н); 7,36-7,30 (m, 1Н); 7,22 (d, J=2,31 Гц, 1Н); 7,08 (dd, J=8,38, 2,35 Гц, 1Н); 6,89 (d, J=8,34 Гц, 1Н); 4,21 (t, J=5,02 Гц, 2H); 4,06 (s, 1Н); 3,12-3,02 (m, 4H); 2,31-2,19 (m, 5H); 1,60-1,48 (m, 4H); 1,15 (s, 6Н). ЖХМС (способ F): RT=7,75 мин, М+Н+=521.
Пример 178
2-(4-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-1-ил)ацетамид 178
Согласно тому, как описано для получения соединения 143, проводили реакцию между гидрохлоридом 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азуленаи бромацетамидом. Неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: градиент от 50 до 100% EtOAc в циклогексане). Очищенные фракции объединяли, концентрировали в вакууме, остаток растворяли в ДХМ, обрабатывали 4 M HCl в диоксане и диэтиловым эфиром. Полученный осадок собирали фильтрованием, при этом получали соединение 178 в виде твердого вещества белого цвета. 1Н ЯМР (400 Мгц, ДМСО-d6): δ 9,86 (s, 1Н); 8,48-8,43 (m, 1Н); 8,30 (s, 1Н); 8,05 (s, 1Н); 7,85 (td, J=8,75, 5,93 Гц, 1Н); 7,74-7,63 (m, 2H); 7,41-7,34 (m, 1Н); 7,22 (d, J=2,30 Гц, 1Н); 7,10 (dd, J=8,45, 2,35 Гц, 1Н); 6,97 (d, J=8,39 Гц, 1Н); 4,22 (t, J=5,00 Гц, 2H); 3,98 (d, J=4,37 Гц, 2H); 3,60 (t, J=12,26 Гц, 2H); 3,24-3,13 (m, 2H); 3,06 (t, J=4,95 Гц, 2H); 2,69-2,61 (m, 1Н); 1,87 (s, 4H). ЖХМС (способ F): RT=7,46 мин, M+H+=506.
Пример 179
2-(4-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-1-ил)этанол 179
Соединение 179 в виде твердого вещества белого цвета получали аналогично тому, как описано для соединения 142, из гидрохлорида 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена и 2-(2-бромэтокси)тетрагидропираном. 1Н ЯМР (400 МГц, ДМСО-d6): δ 9,86 (s, 1Н); 8,44 (s, 1Н); 8,29 (s, 1Н); 7,84 (td, J=8,75, 5,94 Гц, 1Н); 7,72 (td, J=9,64, 2,83 Гц, 1Н); 7,43-7,37 (m, 1Н); 7,21 (d, J=2,28 Гц, 1Н); 7,09 (dd, J=8,45, 2,36 Гц, 1Н); 6,96 (d, J=8,40 Гц, 1Н); 5,37 (t, J=4,89 Гц, 1Н); 4,21 (t, J=5,01 Гц, 2H); 3,83 (d, J=5,48 Гц, 2H); 3,65 (d, J=12,04 Гц, 2H); 3,23 (m, 2H); 3,10 (m, 4H); 2,69-2,60 (m, 1Н); 2,01-1.80 (m, 4H). ЖХМС (способ F): RT=7,55 мин, М+Н+=493.
Пример 180
1-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропан-2-ол 180
Согласно тому, как описано для получения соединения 158, раствор соединения 52 (7,84 г, 20,95 ммоль), 2-метил-1-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]пропан-2-ола (11,15 г, 41,90 ммоль) и карбоната цезия (20,47 г, 62,8 ммоль) в диоксане (380 мл) и воде (38 мл) дегазировали откачиванием воздуха/продуванием аргона (х3). В реакционную смесь добавляли комплекс дихлорида 1,1'-бис(дифенилфосфино)ферроценпалладия(II) и ДХМ (1,71 г, 2,09 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 2 ч. Реакционную смесь разбавляли водой (250 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические фракции промывали солевым раствором (100 мл), сушили (MgSO4), и концентрировали в вакууме, при этом получали эмульсию темно-коричневого цвета. Эмульсию растирали с горячим ИПС (приблизительно 50 мл), охлаждали до КТ и фильтровали. Твердое вещество промывали охлажденным ИПС (приблизительно 30 мл), сушили в вакууме, при этом получали соединение 180 в виде твердого вещества грязно-белого цвета (6,6 г, выход 73%). ЖХ/МС (ESI+): m/z 434 (М+Н). 1H NMR (400 MHz, DMSO): δ 8,37 (d, J=8,4, 1H), 8,16 (s, 1H), 7,95 (s, 1H), 7,91 (s, 2H), 7,39 (dd, J=8,4, 1.7, 1H), 7,28 (d, J=1,7, 1H), 5,90 (dt, J=13,0, 6,5, 1H), 4,72 (s, 1H), 4,52 (q, J=6,2, 4H), 4,04 (s, 2H), 1,49 (d, J=6,6, 6H), 1,10 (s, 6H).
Пример 181
2-{3-[2-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил]азетидин-1-ил}ацетамид 181
Соединение 181 в виде твердого вещества белого цвета получали аналогично тому, как описано для соединения 143, из соединения 64 и бромацетамида, неочищенный продукт очищали обращенно-фазной ЖХВР (колонка Gemini C18, элюент: градиент от 10 до 90% МеОН в воде + 0.1% НСО2Н). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 8,33 (s, 1Н); 8,21-8,14 (m, 2H); 8,04 (s, 1Н); 7,21 (dd, J=8,42, 1,92 Гц, 2H); 7,12-7,02 (m, 2H); 5,44-5,36 (m, 1Н); 4,32 (t, J=5,05 Гц, 2H); 3,80-3,61 (m, 3H); 3,30 (t, J=7,44 Гц, 2H); 3,16-3,09 (m, 4H); 1,52 (d, J=6,59 Гц, 6H). ЖХМС (способ F): RT=6,49 мин, M+H+=408.
Пример 182
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)этанол 182
В сосуд для микроволного облучения объемом 10 мл помещали соединение 194 (0,210 г, 0,56 ммоль) и ацетат калия (0,17 г, 1,68 ммоль), MeCN (1 мл) и воду (2 мл). Смесь интенсивно продували N2. В смесь добавляли раствор 1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (0,271 г, 0,84 ммоль) в MeCN (1 мл), тетракис(трифенилфосфин)палладий (65 мг, 0,056 ммоль) и сосуд немедленно запечатывали. Смесь подвергали микроволновому облучению при 150°С в течение 20 мин. Завершение реакции наблюдали методом ЖХМС (наблюдали небольшое количество продукта, не содержащего группу тетрагидропиран). Реакционную смесь разбавляли EtOAc и водой и 3 раза экстрагировали EtOAc. Органические фазы объединяли, сушили над MgSO4 и концентрировали. Остаток очищали хроматграфией на колонке ISCO (элюент: 10% MeOH/EtOAc), при этом получали 170 мг, 0,35 ммоль (выход 62%) твердого пенящегося вещества белого цвета, которое немедленно растворяли в ДХМ (2 мл) и обрабатывали 4 М хлористым водородом в 1,4-диоксане (0,35 мл). В процессе обработки наблюдали образование осадка белого цвета. Реакционную смесь перемешивали при КТ в течение 1 ч. Реакционную смесь концентрировали досуха и растворяли в смеси ДМФ/Н2О. Смесь очищали обращенно-фазной ЖХВр, при этом получали 105 мг (выход 74%) соединения 182 в виде частично кристаллического твердого вещества белого цвета. ЖХ/МС (ESI+): m/z 406 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 8,37 (d, J=8,4, 1H), 8,22 (s, 1H), 7,95 (s, 1H), 7,91 (s, 2H), 7,38 (dd, J=8,4, 1,8, 1H), 7,27 (d, J=1,7, 1H), 5,91 (dq, J=13,3, 6,7, 1H), 4,91 (t, J=5,3, 1H), 4,58-4,44 (m, 4H), 4,16 (t, J=5,6, 2H), 3,77 (q, J=5,4, 2H), 1,49 (d, J=6,6, 6H).
Пример 183
1-(3-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-ил)-2-метилпропан-2-ол 183
Соединение 183 в виде твердого вещества белого цвета получали аналогично тому, как описано для соединения 117, из соединения 63 и 2,2-диметилоксирана. 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,45 (s, 1Н); 8,31 (s, 1Н); 8,17 (s, 1Н); 7,84 (td, J=8,75, 5,92 Гц, 1Н); 7,66 (ddd, J=10,33, 9,02, 2,79 Гц, 1Н); 7,40-7,34 (m, 1Н); 7,31 (d, J=8,17 Гц, 1Н); 7,00 (d, J=1,69 Гц, 1Н); 6,93 (dd, J=8,25, 1,78 Гц, 1Н); 4,26 (t, J=5,06 Гц, 2H); 3,79 (t, J=7,52 Гц, 2H); 3,64 (t, J=7,75 Гц, 1Н); 3,35 (t, 2H); 3,08 (t, J=5,05 Гц, 2H); 2,35 (t, 2H); 1,10 (s, 6H). ЖХМС (способ F): RT=7,45 мин, M+H+=493.
Пример 184
Метиловый эфир 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 184
В смесь соединения 40 (1,0 г, 2,7 ммоль), 1-изопропил-1Н-1,2,4-триазола (0,30 г, 2,7 ммоль), CuI (1,5 г, 8,1 ммоль), Pd(OAc)2 (0,061 г, 0,27 ммоль) и карбоната цезия (2,2 г, 6,8 ммоль) добавляли ДМФ (26 мл). Реакционную смесь перемешивали и нагревали при 100°С в течение 24 ч в закрытом сосуде. Реакционную смесь охлаждали до КТ, выливали в смесь гидроксида аммония и воды (1:2) и EtOAc и фильтровали через слой силикагеля. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали гидроксидом аммония (1:2), водой, солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный материал очищали на колонке с силикагелем (элюент: EtOAc), при этом получали соединение 184 (0,270 г, выход 28%). 1H ЯМР (400 МГц, ДМСО): δ 8,54 (d, J=8,4, 1H), 7,97 (d, J=35,2, 2H), 7,70 (dd, J=8,4, 1,7, 1H), 7,57 (d, J=1,7, 1H), 5,99-5,70 (m, 1H), 4,57 (q, J=5,9, 4H), 3,87 (s, 3Н), 1,49 (d, J=6,6, 6H). MC (ESI(+)): m/z 354,1 (M+H).
В другом варианте соединение 42 (370 мг, 1,3 ммоль) в диметоксиэтане (3 мл) обрабатывали 1,1-диметокси-N,N-диметилметанамидом (1 мл, 7,5 ммоль) и нагревали при 90°С в течение 0,5 ч. По данным ЖХ/МС наблюдали образование ребуемого продукта. После охлаждения реакционную смесь концентрировали, получали неочищенный ациламидин, который суспендировали в уксусной кислоте (2,3 мл), обрабатывали гидрохлоридом изопропилгидразина (0,29 г, 2,5 ммоль). Смесь нагревали при 75°С в течение 30 мин, охлаждали до КТ и концентрировали. После очистки на колонке ISCO (элюент: 100% EtOAc) получали соединение 184 (0,32 г, выход 70%). 1Н ЯМР (400 МГц, ДМСО): δ 8,54 (d, J=8,4, 1H), 8,02 (s, 1H), 7,93 (s, 1H), 7,70 (dd, J=8,4, 1,7, 1H), 7,57 (d, J=1,7, 1H), 5,85 (dq, J=13,3, 6,6, 1H), 4,57 (q, J=6,0, 4H), 3,87 (s, 3Н), 1,49 (d, J=6,6, 6H).
Пример 185
Метиловый эфир 2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 185
Согласно тому, как описано для получения соединения 184, проводили реакцию между соединением 40 и (1-(2,2,2-трифторэтил)-1Н-1,2,4-триазолом). Продукт высаживали из EtOAc и собирали фильтрованием, при этом получали соединение 185 (393 мг, выход 40%). 1H ЯМР (400 МГц, ДМСО): δ 8,51 (d, J=8,4, 1H), 8,15 (s, 1H), 8,10 (s, 1H), 7,71 (dd, J=8,4, 1,7, 1H), 7,58 (d, J=1,6, 1H), 5,89 (q, J=8,8, 2H), 4,64-4,49 (m, 4H), 3,87 (s, 3H). MC (ESI(+)): m/z 394,0 (M+H).
В другом варианте, согласно тому, как описано для получения соединения 184, проводили реакцию между соединением 42 и 1,1-диметокси-N,N-диметилметанамином с последующей обработкой гидрохлоридом трифторэтилгидразина в уксусной кислоте при этом получали соединение 185 (выход 65%). 1Н ЯМР (400 МГц, ДМСО): δ 8,51 (d, J=8,4, 1H), 8,15 (s, 1H), 8,10 (s, 1H), 7,71 (dd, J=8,4, 1,7, 1H), 7,58 (d, J=1,6, 1H), 5,89 (q, J=8,8, 2H), 4,65-4,45 (m, 5H), 3,87 (s, 3H), 3,58-3,37 (m, 7H).
Пример 186
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)этанол 186
Согласно тому, как описано для соединения 182, проводили реакцию Сузуки между 9-бром-2-(1-изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепином и 1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-4-(4,4,5,5-тераметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, при этом получали соединение 186 в виде твердого кристаллического вещества белого цвета с выходом 72% (после удаления группы тетрагидропиран в кислотных условиях). ЖХ/МС (ESI+): m/z 420 (M+H). 1Н ЯМР (400 МГц, ДМСО): δ 8,36 (d, J=8,4, 1H), 8,22 (s, 1H), 7,95 (s, 1H), 7,87 (s, 1H), 7,38 (dd, J=8,4, 1,7, 1H), 7,27 (d, J=1,7, 1H), 5,83 (dt, J=13,2, 6,6, 1H), 4,91 (t, J=5,3, 1H), 4,51 (s, 4H), 4,16 (t, J=5,6, 2H), 3,77 (q, J=5,6, 2H), 1,48 (t, J=9,0, 6H).
Пример 187
10-Бром-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 187
В смесь 10-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (1,5 г, 3,8 ммоль), 1-изопропил-1Н-1,2,4-триазола (0,40 г, 3,0 ммоль), CuI (1,8 г, 9,5 ммоль), Pd(OAc)2 (0,071 г, 0,32 ммоль) и карбоната цезия (2,6 г, 7,9 ммоль) добавляли ДМФ (20 мл). Реакционную смесь перемешивали при 100°С в течение 24 ч в закрытом сосуде. Реакционную смесь охлаждали до КТ, разбавляли EtOAc и фильтровали через целит. Фильтрат концентрировали при пониженном давлении. В неочищенный остаток добавляли EtOAc и твердое вещество собирали фильтровалнием. Фильтрат концентрировали, неочищенный материал растворяли в ДМФ и очищали обращенно-фазной ЖХВР, при этом получали соединение 187 (64 мг, выход 5%). 1Н ЯМР (400 МГц, ДМСО): δ 8,43 (dd, J=47,5, 31,0, 1H), 7,97 (s, 1H), 7,92 (s, 1H), 7,60-7,39 (m, 1H), 7,04 (d, J=8,7, 1H), 5,74 (dt, J=13,2, 6,6, 1H), 4,76-4,33 (m, 4H), 1,49 (d, J=6,6, 6H). MC (ESI(+)): m/z 374,0 (M+H).
Пример 188
[4-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}-1-(2-метансульфонилэтил)пиперидин-4-ил]метанол 188
В смесь соединения 68 (97 мг, 0,1873 ммоль) с IMS (3 мл) при перемешивании добавляли DIPEA (165 мкл, 0,94 ммоль) и винилсульфон (18 пл, 0,206 ммоль) при КТ. Через 3 ч растворитель удаляли в вакууме и остаток очищали ЖХВР (колонка Gemini С6-фенил, элюент: градиент от 10 до 60%, скорость изменения температуры 20 мин), при этом получали соединение 188 (63 мг, выход 53%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,42-8,39 (1Н, m), 8,23 (1Н, s), 8,19 (1Н, s), 7,80 (1Н, td, J=8,77, 5,87 Гц), 7,53 (1Н, ddd, J=10,34, 8,85, 2,77 Гц), 7,36 (1Н, d, J=2,40 Гц), 7,33-7,27 (1Н, m), 7,12 (1Н, dd, J=8,59, 2,48 Гц), 6,85 (1Н, dd, J=8,55, 4,83 Гц), 4,17 (2Н, t, J=5,04 Гц), 3,22-3,14 (6Н, m), 3,02 (2Н, t, J=5,09 Гц), 2,98 (3Н, s), 2,02 (2Н, s), 1,72 (4Н, s). Два протона перекрывались с пиком воды. ЖХМС (способ F): RT=6,57 мин, M+H+=585.
Пример 189
2-(4-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил1-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-1-ил)-2-метилпропан-1-ол 189
Гидрохлорид 2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-9-пиперидин-4-ил-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулена (250 мг, 0,52 ммоль) растворяли в ДМФ (3 мл) и обрабатывали карбонатом цезия (336 мг, 1,03 ммоль) и 2-бром-2-метиловым эфиром пропионовой кислоты (333 мкл, 2,58 ммоль), затем полученную смесь нагревали при 80°С в течение 20 ч. Охлажденную реакционную смесь разбавляли EtOAc и промывали водой и солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 20 до 70% EtOAc в циклогексане), при этом получали метиловый эфир 2-(4-{2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-9-ил}пиперидин-1-ил)-2-метилпропионовой кислоты. Промежуточный метиловый эфир 2-метилпропионовой кислоты (195 мг, 0,355 ммоль) растворяли в ТГФ (5 мл) и раствор охлаждали до 0°С. По каплям добавляли гидрид лития-алюминия (0,533 мл, 1 М раствор в ТГФ) и реакционную смесь нагревали при 0°С в течение 15 мин и при КТ в течение 90 мин. Реакционную смесь охлаждали до 0°С и добавляли воду, смесь экстрагировали EtOAc и органический экстракт промывали солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали обращенно-фазной ЖХВР (колонка Gemini С6-фенил, элюент: градиент от 30 до 60% метанола в воде + 0.1% НСО2Н), при этом получали соединение 189 в виде твердого вещества белого цвета (123 мг, выход 70%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,40 (1Н, s), 8,24 (1Н, s), 8,17 (1Н, s), 7,79 (1Н, td, J=8,76, 5,90 Гц), 7,64-7,57 (1Н, m), 7,34-7,28 (1Н, m), 7,18 (1Н, d, J=2,29 Гц), 7,03 (1Н, dd, J=8,38, 2,33 Гц), 6,84 (1Н, d, J=8,33 Гц), 4,16 (2H, t, J=5,02 Гц), 3,35 (2H, s), 3,14 (3H, d, J=11,68 гц), 3,00 (2H, t, J=5,05 Гц), 2,38-2,23 (3H, m), 1,63 (2H, d, J=12,48 Гц), 1,53-1,39 (2H, m), 1,03 (6H, s). ЖХМС (способ F): RT=7,89 мин, М+H+=521.
Пример 190
1-(4-(2-(1-Изопропил-3-метилl-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропан-2-ол 190
Согласно тому, как описано для соединения 182, проводили реакцию Сузуки между 9-бром-2-(1-изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепином и 2-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропан-2-олом, при этом получали соединение 190. ЖХ/МС (ESI+): m/z 448 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 8,36 (d, J=8,4, 1H), 8,16 (s, 1H), 7,94 (s, 1H), 7,87 (s, 1H), 7,39 (dd, J=8,4, 1,8, 1H), 7,27 (d, J=1,7, 1H), 5,90-5,70 (m, 1H), 4,72 (s, 1H), 4,51 (s, 4H), 4,04 (s, 2H), 2,25 (s, 3Н), 1,47 (d, J=6,6, 6H), 1,10 (s, 6H).
Пример 191
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(метилсульфонил)этил)азетидин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 191
Согласно тому, как описано для соединения 152, проводили реакцию между соединением 152 и винилсульфоном, при этом получали соединение 191 в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3): δ (ч/млн) 8,44 (1Н, d, J=8,28 Гц), 7,84 (1Н, s), 7,60 (1Н, s), 7,05 (1Н, dd, J=8,32, 1,83 Гц), 6,94 (1Н, d, J=1,78 Гц), 5,99-5,89 (1Н, m), 4,48-4,39 (4Н, m), 3,78-3,70 (2Н, m), 3,71-3,62 (1Н, m), 3,25-3,18 (2Н, m), 3,04 (3Н, s), 3,02-2,95 (4Н, m), 1,56 (6Н, d, J=6,64 Гц). ЖХМС (способ F): RT=5,58 мин, M+H+=457.
Пример 192
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-1-ил)ацетамид 192
Согласно тому, как описано для соединения 143, проводили реакцию между соединением 65 и бромацетамидом. Неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в ДХМ) и растирали с диэтиловым эфиром, при этом получали соединени 192 в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3): δ (ч/млн): 8,46 (1Н, d, J=8,28 Гц), 7,84 (1Н, s), 7,61 (1Н, s), 7,07 (1Н, dd, J=8,32, 1,84 Гц), 6,95 (1Н, d, J=1,79 Гц), 6,89 (1Н, s), 5,99-5,90 (1Н, m), 5,44 (1Н, s), 4,48-4,45 (2Н, m), 4,43-4,40 (2Н, m), 3,86-3,78 (2Н, m), 3,73-3,63 (1Н, m), 3,38-3,31 (2Н, m), 3,20 (2Н, s), 1,56 (6Н, d, J=6,64 Гц). ЖХМС (способ F): RT=5,45 мин, M+H+=408.
Пример 193
(1-Аминоциклопропил)(3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-1-ил)метанон 193
Согласно тому, как описано для соединения 127, проводили реакцию между соединением 65 и 1-трет-бутоксикарбониламиноциклопропанкарбоновой кислотой. Неочищенный продукт очищали обращенно-фазной ЖХВР (колонка Gemini C18, элюент: градиент от 20 до 95% МеОН в Н2О + 0.1% НСО2Н), при этом получали соединение 193 в виде твердого вещества белого цвета. 1Н ЯМР (DMSO-d6): δ (ч/млн) 8,36 (1Н, d, J=8,28 Гц), 8,09 (1Н, s), 7,88-7,86 (2Н, m), 7,14 (1Н, dd, J=8,35, 1,82 Гц), 7,01 (1Н, d, J=1,78 Гц), 5,88-5,80 (1Н, m), 4,50-4,44 (4H, m), 3,83-3,71 (2H, m), 1,44 (6H, d, J=6,60 Гц), 1,05 (2H, d, J=4,13 Гц), 0,67 (2H, d, J=4,01 Гц). Два заменяемых протона не видны. Пики 4 протонов перекрываются с пиком воды. ЖХМС (способ F): RT=6,73 мин, М+Н+=434.
Пример 194
9-Бром-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 194
Соединение 43 (4,93 г, 16,0 ммоль) смешивали с 1,1-диметокси-N,N-диметилметанамином (25 мл, 0,18 моль) и 1,2-диметоксиэтаном (66,5 мл, 0,640 моль). Гетерогенную смесь очень интенсивно перемешивали при 65°С в течение 1 ч. По данным ЖХ/МС наблюдали полное исчерпание исходного материала в конце указанного периода. Реакционную смесь концентрировали в вакууме и использовали для проведения следующей реакции без дополнительной очистки. Неочищенный продукт (5,8 г, 16,0 ммоль) суспендировали в ледяной уксусной кислоте (53,2 мл) и добавляли гидрохлорид изопропилгидразина (4,36 г, 39,4 ммоль). Смесь нагревали при 100°С в течение 2 ч. Реакционную смесь охлаждали до КТ и растворитель удаляли в вакууме. Полученный остаток в сухом виде наносили на силикагель и очищали хроматографией на колонке ISCO (масса колонки 120 г, элюент: 100% EtOAc). После двух стадий получали 2,3 г (выход 39% yield) соединения 194. ЖХ/МС (ESI+): m/z 376 (М+Н, + изотоп галогена). 1Н ЯМР (400 МГц, ДМСО): δ 8,34 (d, J=8,6, 1H), 7,95 (s, 1H), 7,91 (s, 1H), 7,36 (dd, J=8,7, 2,0, 1H), 7,30 (d, J=2,0, 1H), 5,85 (dt, J=13,3, 6,6, 1H), 4,55 (d, J=15,5, 4H), 1,48 (d, J=6,6, 6H).
В другом варианте в суспензию 1-диметиламиномет-(Z)-илиденамида 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (8,52 г, 23,5 ммоль) в уксусной кислоте (50 мл) добавляли гидрохлорид изопропилгидразина (3,3 7 г, 30,5 ммоль) и реакционную смесь нагревали при 100°С в течение 1 ч. Реакционную смесь охлаждали до КТ и выливали в воду (500 мл), при этом получали продукт в виде твердого осадка грязно-белого цвета. Продукт собирали фильтрованием, промывали водой (приблизительно 200 мл) и сушили в вакууме при 45°С в течение 16 ч, при этом получали соединение 194 в виде твердого вещества грязно-белого цвета (7,88 г, выход 86%). 1Н ЯМР (400 МГц, d6-ДМСО): 8,43 (1Н, d, J=8,6 Гц), 7,97 (1H, s), 7,92 (1H, d, J=0,6 Гц), 7,36 (1H, dd, J=8,6, 2,0 Гц), 7,30 (1H, d, J=2,0 Гц), 5,86 (1H, септ., J=6,6 Гц), 4,56-4,52 (4Н, m), 1,48 (6H, d, J=6,6 Гц). ЖХМС: RT=4,69 мин, M+H+=374/376. По данным 1Н ЯМР продукт содержал приблизительно 5% 8-иод-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена.
Еще в одном варианте:
Стадия 1
Гидрохлорид этилового эфира 4-бром-2-фторбензимидиновой кислоты
Суспензию 4-бром-2-фторбензонитрила (25,0 г, 125 ммоль) в IMS (88 мл) охлаждали до 0-5°С и по каплям обрабатывали ацетихлоридом (71 мл, 1 моль) при температуре менее 10°С. Реакционный сосуд закрывали и смесь перемешивали при КТ в течение 18 ч, затем концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром, при этом получали гидрохлорид этилового эфира 4-бром-2-фторбензимидиновой кислоты в виде твердого вещества белого цвета (20,3 г, выход 57%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 7,93-7,88 (1Н, m), 7,85-7,76 (1Н, m), 7,72-7,64 (1Н, m), 4,60 (2Н, q, J=7,02 Hz), 1,47-1,38 (3Н, m).
Стадия 2
Гидрохлорид 4-бром-2-фторбензамидина
Смесь гидрохлорида этилового эфира 4-бром-2-фторбензимидиновой кислоты (20,3 г, 72 ммоль) в IMS (250 мл) при 0-5°С насыщали газообразным NH3, колбу закрывали, нагревали до КТ и перемешивали в течение 18 ч. Растворитель удаляли в вакууме, остаток растирали с диэтиловым эфиром, при этом получали гидрохлорид 4-бром-2-фторбензамидина в виде твердого вещества белого цвета (18,1 г, выход 100%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 9,26 (4Н, s), 7,92-7,87 (1Н, m), 7,71-7,62 (2Н, m).
Стадия 3
1-(2-Изопропил-2Н-[1,2,4]триазол-3-ил)этанон
В раствор 1-изопропил-1Н-[1,2,4]триазола (33 г, 300 ммоль) в ТГФ при -10°С по каплям добавляли n-бутиллитий (145 мл, 2,5 M, 360 ммоль) в течение 45 мин и затем смесь перемешивали при 0°С в течение 30 мин. В смесь добавляли DMA (35 мл), смесь нагревали до КТ и перемешивали в течение 1 ч. Полученную суспензию обрабатывали насыщенным водным раствором хлорида аммония (300 мл). Водную фазу экстрагировали EtOAc, объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали 1-(2-изопропил-2Н-[1,2,4]триазол-3-ил)этанон в виде маслянистого вещества светло-оранжевого цвета (40,1 г, выход 87%). 1Н ЯМР (CDCl3) δ (ч/млн): 7,93 (1Н, s), 5,58-5,46 (1Н, m), 2,72 (3H, d, J=0,78 Гц), 1,49 (6H, dd, J=6,61, 0,78 Гц).
Стадия 4
2-Бром-1-(2-изопропил-2Н-[1,2,4] триазол-3-ил)этанон
В раствор 1-(2-изопропил-2Н-[1,2,4]триазол-3-ил)этанона (10 г, 65,3 ммоль) в уксусной кислоте (1 мл) и ТГФ (100 мл) добавляли раствор РТТ (трибромид фенилтриметиламмония, 24,5 г, 65,3 ммоль) в ТГФ (100 мл) в течение 20 мин. Реакционную смесь нагревали до 75°С и охлаждали до КТ. Полученную смесь концентрировали в вакууме и продукт распределяли между EtOAc и насыщенный водный раствор бикарбоната натрия. Органический слой отделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали остаток, который очищали экспресс-хроматографией (SiO2, элюент: градиент от О до 20% EtOAc в циклогесане), при этом получали 2-бром-1-(2-изопропил-2Н-[1,2,4]триазол-3-ил)этанон в виде маслянистого вещества (5,4 г, выход 36%). 1Н ЯМР (CDCl3): δ (ч/млн) 7,98 (1Н, s), 5,53-5,42 (1Н, m), 4,69 (2H, s), 1,52 (6H, d, J=6,63 Гц).
Стадия 5
5-[2-(4-Бром-2-фторфенил)-1Н-имидазол-4-илl]-1-изопропил-1Н-[1,2,4]триазол
В смесь гидрохлорида 4-бром-2-фторбензамидина (9,84 г, 38,8 ммоль), гидрокарбоната калия (15,6 г, 154,8 ммоль), ТГФ (98 мл) и воды (16 мл) при интенсивном перемешивании и кипячении собратным холодильником добавляли раствор 2-бром-1-(2-изопропил-2Н-[1,2,4]триазол-3-ил)этанона (9,0 г, 38,8 ммоль) в ТГФ (19 мл) в течение 15 мин. Полученную смесь перемешивали в течение 18 ч при кипячении с обратным холодильником, затем концентрировали в вакууме. Полученный остаток обрабатывали водой, полученный осадок собирали фильтрованием, промывали (вода, смесь 1:1 диэтиловый эфир:циклогексае, диэтиловый эфир), при этом получлаи 5-[2-(4-бром-2-фторфенил)-1Н-имидазол-4-ил]-1-изопропил-1Н-[1,2,4]триазол в виде твердого вещества коричневого цвета (10,1 г, выход 74%). 1Н ЯМР (CDCl3): δ (ч/млн) 8,21-8,14 (1Н, m), 7,90 (1Н, s), 7,80 (1Н, s), 7,47-7,38 (2H, m), 7,26 (1Н, s), 5,91 (1Н, ушир. s), 1,59 (6H, d, J=6,63 Гц).
Раствор 5-[2-(4-бром-2-фторфенил)-1Н-имидазол-4-ил]-1-изопропил-1Н-[1,2,4]триазола (10,0 г, 28,6 ммоль) в ДМФ (100 мл) обрабатывали этиленкарбонатом (5,3 г, 60,1 ммоль) и карбонатом цезия (13,9 г, 42,5 ммоль) и нагревали при 100°С в течение 72 ч. Затем добавляли карбонат цезия (9,0 г, 27,5 ммоль) и воду (0,5 mL), перемешивание продолжали в течение 24 ч и реакционную смесь концентрировали в вакууме. Полученный остаток распределяли между ДХМ и водой, органический слой отделяли, промывали водой и солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, 1% МеОН в ДХМ), при этом получали соединение 194 в виде твердого вещещства грязно-белого цвета (5,78 г, выход 58%). 1Н ЯМР (CDCl3): δ (ч/млн) 8,04 (1Н, s), 7,83 (1Н, s), 7,50-7,38 (3H, m), 5,93-5,84 (1Н, m), 4,07-4,02 (2H, m), 3,93-3,88 (2H, m), 1,53-1,46 (6H, m).
Пример 195
1-(4-(2-(1-Изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропан-2-ол 195
Как оисано в примере 182, проводили реакцию между 9-бром-2-(1-изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепином и 2-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропан-2-олом, при этом получали соединение 195 с выходом 22%. MC(ESI+): 447,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,34 (d, J=8,4, 1H), 8,14 (s, 1H), 7,93 (s, 1H), 7,63 (s, 1H), 7,36 (dd, J=8,4, 1,7, 1H), 7,25 (d, J=1,7, 1H), 7,00 (d, J - 0,6, 1H), 5,68-5,57 (m, 1H), 4,72 (s, 1H), 4,48 (s, 4H), 4,03 (s, 2H), 2,10 (s, 3Н), 2,07 (s, 1H), 1,42 (d, J=6,7, 6H), 1,09 (s, 6H).
Пример 196
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропанамид 196
Соединение 196 в виде твердого кристаллического вещества белого цвета получали аналогично тому, как описано для получения соединения 215 с выходом 72% из 9-бром-2-(1-изопропил-4-метил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина. ЖХ/МС (ESI+): m/z 461 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 8,40 (s, 1H), 8,36 (d, J=8,4, 1H), 8,01 (s, 1H), 7,87 (s, 1H), 7,45 (dd, J=8,4, 1,8, 1H), 7,35 (d, J=1,7, 1H), 7,17 (s, 1H), 6,81 (s, 1H), 5,82 (dt, J=13,3, 6,6, 1H), 4,52 (s, 4H), 2,25 (s, 3H), 1,74 (s, 6H), 1,47 (d, J=6,6, 6H).
Пример 197
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-1-ил)-N,N-диметилэтансульфонамид 197
Согласно тому, как описано для соединения 152, проводили реакцию между соединением 65 и и N,N-диметилэтансульфонамидом, при этом получали соединение 197 в иде твердого вещества белого цвета. 1Н ЯМР (CDCl3): δ (ч/млн) 8,44 (1Н, d, J=8,28 Гц), 7,84 (1Н, d, J=0,67 Гц), 7,61 (1Н, s), 7,05 (1Н, dd, J=8,32, 1,83 Гц), 6,95 (1Н, d, J=1,78 Гц), 6,00-5,90 (1Н, m), 4,48-4,39 (4Н, m), 3,80-3,72 (2Н, m), 3,72-3,64 (1Н, m), 3,27-3,19 (2Н, m), 3,02-2,89 (4Н, m), 2,87 (6Н, s), 1,56 (6Н, d, J=6,63 Гц). ЖХМС (способ F): RT=6,35 мин, M+H+=486.
Пример 198
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-1-ил)-N,N-диметилацетамид 198
Согласно тому, как описано для соединения 143, проводили реакцию между соединением 65 и 2-хлор-N,N-диметилацетамидом, неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 6% МеОН в ДХМ), при этом получали соединение 198 в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3): δ (ч/млн) 8,44 (1Н, d, J=8,28 Гц), 7,84 (1Н, d, J=0,71 Гц), 7,60 (1Н, s), 7,10 (1Н, dd, J=8,33, 1,81 Гц), 6,97 (1Н, d, J=1,72 Гц), 6,01-5,91 (1Н, m), 4,48-4,39 (4H, m), 3,99-3,90 (2H, m), 3,86-3,77 (1Н, m), 3,46-3,38 (4H, m), 3,00 (3H, s), 2,93 (3H, s), 1,56 (6H, d, J=6,63 Гц). ЖХМС (способ F): RT=5,87 мин, М+H+=436.
Пример 199
9-(4,4-Диметил-4,5-дигидрооксазол-2-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 199
2-Амино-2-метил-1-пропанол (0,20 г, 2,3 ммоль) растворяли в ТГФ (2,2 мл) и добавляли NaH (60% в минеральном масле, 0,0942 г). Полученную дисперсию перемешивали в течение 1 ч при КТ. В смесь добавляли метиловый эфир 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 184 (0,40 г, 1,1 ммоль) в смеси ТГФ/ДМФ (1:1, 10 мл). Полученную реакционную смесь в течение ночи. Реакцию останавливали добавлением воды и разбавляли EtOAc. Смесь экстрагировали, сушили над MgSO4, фильтровали и концентрировали. Остаток растворяли в ДХМ (10 мл, 200 ммоль), охлаждали до 0°С и по каплям обрабатывали тионилхлоридом (0,314 мл, 4,30 ммоль). Затем смесь нагревали до КТ и перемешивали в течение 3 ч. После концентрировали в вакууме и очистки обращенно-фазной ЖХВР получали соединение 199 (209 мг, выход 48%). ЖХ/МС (ESI+): m/z 393 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 8,47 (d, J=8,4, 1H), 7,99 (s, 1H), 7,92 (d, J=3,5, 1H), 7,59 (dd, J=8,4, 1,6, 1H), 7,45 (d, J=1,6, 1H), 5,85 (dt, J=13,2, 6,6, 1H), 4,55 (dd, J=10,6, 6,4, 4H), 4,12 (s, 2H), 1,49 (d, J=6,6, 6H), 1,30 (s, 6H).
Пример 200
N-Изопропил-2-(3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-1-ил)ацетмид 200
Согласно тому, как описано для соединения 143, проводили реакцию между соединением 65 и 2-хлор-N-изопропилацетамидом. Неочищнный продукт очищали обращено-фазной ЖХВР (колонка Gemini С18, элюент: градиент от 0 до 70% МеОН в Н2О + 0.1% НСО2Н), при этом получали соединение 200 в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3): δ (ч/млн) 8,46 (1Н, d, J=8,28 Гц), 7,85 (1Н, s), 7,63 (1Н, s), 7,05 (1Н, dd, J=8,32, 1,82 Гц), 6,94-6,92 (1Н, m), 6,00-5,90 (1Н, m), 4,49-4,40 (4H, m), 4,12-4,02 (1Н, m), 3,88 (2H, t, J=7,51 Гц), 3,82-3,71 (1Н, m), 3,41 (2H, t, J=7,23 Гц), 3,23 (2H, s), 1,56 (6H, d, J=6,63 Гц), 1,16 (6H, d, J=6,57 Гц). Один заменяемый протон не наблюдали. ЖХМС (способ F): RT=6,8 мин, М+Н+=450.
Пример 201
2-(3-{2-[2-(2,4-Дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-2Н-6-окса-1,2-диазабензо[е]азулен-8-ил}азетидин-1-ил)этанол 201
Согласно тому, как описано для соединения 142, проводили реакцию между соединением 63 и 2-(2-бромэтокси)тетрагидропираном, неочищенный продукт очищали обращено-фазной ЖХВР (колонка Gemini C18, элюент: градиент от 10 до 90% МеОН в воде + 0.1% НСО2Н), при этом получали соединение 201 в виде твердого вещества белого цвета. 1Н ЯМР (400 МГц, DMSO-d6): δ 8,41 (s, 1Н); 8,28 (s, 1Н); 8,16 (s, 1Н); 7,80 (td, J=8,75, 5,92 Гц, 1Н); 7,62 (ddd, J=10,34, 9,02, 2,81 Гц, 1Н); 7,37-7,31 (m, 1Н); 7,28 (d, J=8,17 Гц, 1Н); 6,95 (d, J=1,73 Гц, 1Н); 6,89 (dd, J=8,25, 1,81 Гц, 1Н); 4,22 (t, J=5,03 Гц, 2H); 3,69 (t, J=7,37 Гц, 2H); 3,60 (dt, J=15,08, 7,40 Гц, 1Н); 3,39 (t, J=6,62 Гц, 1Н); 3,23 (t, J=7,74 Гц, 2H); 3,14 (s, 2H); 3,05 (t, J=5,14 Гц, 2H); 2,61 (t, J=5,95 Гц, 2H). ЖХМС (способ F): RT=7,11 мин, М+H+=465.
Пример 202
1-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-имидазол-1-ил)-2-метилпропан-2-ол 202
В сосуд, пригодный для микроволнового облучения и содержащий соединение 194 (402 мг, 1,07 ммоль) добавляли ацетат калия (316 мг, 3,22 ммоль) и ДМСО (8 мл, 100 ммоль). Реакционную смесь интенсивно продували азотом и в нее добавляли биспинасоловый эфир (310 мг, 3,22 ммоль), затем комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия и ДХМ (1:1, 87,7 мг, 0,107 ммоль) и сосуд герметично закрывали. Сосуд нагревали на масляной бане в течение 24 ч. Завершение реакции подтверждали методом ЖХМС. Смесь фильтровали через целит с исользованием смеси 8/2 ДХМ/метанол и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (элюент: от 0 до 5% метано/ДХМ), концентрировали в вакууме и при этом получали 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (117 мг, выход 26%).
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (113 мг, 0,268 ммоль), соединение 54 (88,14 мг, 0,40 ммоль), хлорид 1,1'-бис(дифенилфосфино)ферроценпалладия(II) (21,90 мг, 0,027 ммоль), 1,2-диметоксиэтан (3,0 мл, 29 ммоль), и 1 М раствор карбоната цезия в воде (0,54 мл, 0,5 ммоль) смешивали в сосуде, пригодном для микроволнового облучения, и подвергали микроволновому облучению при 140°С в течение 15 мин. Завершение реакции подтверждали методом ЖХМС. Смесь фильтровали через бумажный фильтр и через слой силикагеля. Смесь концентрировали в вакууме, очизали ЖХВР и получали соединение 202 (13,7 мг, выход 12%).
В другом варианте в смесь соединения 224 (300 мг, 0,75 ммоль) и Cs2CO3 (733 мг, 2,25 ммоль) в ДМФ (15 мл) в атмосфере азота добавляли 2,2-диметилоксиран (2 мл, 22,4 ммоль). Реакционную смесь нагревали при 80°С в течение 8 ч. Смесь охлаждали до КТ, полученную смесь выливали в воду и эксрагировали EtOAc. Органические экстракты сушили над сульфатом магния и очищали препаративной ТСХ (ДХМ/МеОН=10: 1), при этом получали соединение 202 в виде твердого вещества белого цвета (75,3 мг, выход 23%). Н ЯМР (ДМСО-d6, 400 МГц): δ 8,37 (d, J=8,4 Гц, 1Н), 7,92 (s, 2H), 7,67 (s, 1H), 7,64 (s 1H), 7,53 (dd, J1=1,6 Гц, J2=8,4 Гц, 1H), 7,42 (d, J=1,6 Гц, 1H), 5,94-5,88 (m, 1H), 4,79 (s, 1H), 4,54-4,50 (m, 4H), 3,89 (s, 2H), 1,49 (d, J=6,4 Гц, 6Н), 1,09 (s, 6Н). МС: (ESI, m/z)=434 [M+H]+.
Пример 203
3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оказепин-10-ил)пиридин-2(1Н)-он 203
Смешивали 10-бром-2-(1-изопроппил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробнзо[f]имидазо[1,2-d][1,4]оксазепин 187 (0,057 г, 0,15 ммоль), 2-фторпиридин-3-илборную кислоту (0,026 г, 0,183 ммоль), ацетат калия (0,059 г, 0,609 ммоль) и Pd(PPh3)4 (8,8 мг, 0,007 ммоль), ДМФ (6 мл) и воду (0,6 мл). Через реакционную смесь в течение 5 мин продували азот. Реакционную смесь перемешивали при 105°С в течение 24 ч, затем охлаждали, разбавляли EtOAc и фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении, затем разбавляли EtOAc. Раствор последовательно промывали водой, солевым раствором, сушили над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт растворяли в ДМФ и очищали обращено-фазной ЖХВР, при этом получали 10-(2-фторпиридин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (47 мг, выход 80%). МС (ESI(+)): m/z 391,1 (М+Н).
В раствор 10-(2-фторпиридин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (0,047 г, 0,12 ммоль) в ДМЭ (2 мл) добавляли 10% водный раствор HCl (2 мл). Реакционную смесь перемешивали при 80°С в течение 18 ч, охлаждали и концентрировали при пониженном давлении. Неочищенный продукт растворяли в ДМФ и очищали обращено-фазной ЖХВР, при этом получали соединение 203 (25 мг, выход 55%). 1H ЯМР (400 МГц, ДМСО): δ 11,82 (s, 1Н), 8,88 (d, J=2,3, 1H), 7,92 (d, J=6,7, 2H), 7,67 (ddd, J=9,0, 7,7, 2,2, 2H), 7,38 (d, J=4,8, 1H), 7,07 (d, J=8,6, 1H), 6,31 (t, J=6,7, 1H), 5,81 (dt, J=13,2, 6,6, 1H), 4,54 (q, J=5,8, 4H), 1,48 (d, J=6,6, 6H). МС (ESI(+)): m/z 389,1 (М+Н).
Пример 204
9-(1-(2-(3-Фторазетидин-1-ил)этилсульфонил)азетидин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 204
Соединение 65 (200 мг, 0,517 ммоль) перемешивали с ДХМ (2 мл) и ТЭА (145 мкл, 1,04 ммоль) в течение 1 ч, затем добавляли 2-хлорэтансульфонилхлорид (84 мг, 0,52 ммоль). После перемешивания в течение 1 ч добавляли еще ТЭА (73 мкл, 0,52 ммоль) и смесь перемешивали в течение 18 ч, затем ее разбавляли ДХМ и промывали водой и солевым раствором. Полученный раствор концентрировали в вакууме, при этом получали маслянистое вещество, которое использовали на следующей стадии без дополнительной очистки. Порцию маслянистого вещества коричневого цвета (81 мг, 0,18 ммоль) перемешивали в 3 мл IMS при КТ с гидрохлоридом 3-фторазетидина (22 мг, 0,22 ммоль) и ТЭА (56 мкл, 0,4 ммоль) в течение 18 ч, затем концентрировали в вакууме. Полученный остаток растворяли в ДХМ, промывали водой, солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученное маслянистое вещество светло-коричневого цвета очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 2% МеОН в ДХМ), при этом получали соединение 204 в виде твердого вещества белого цвета (37 мг, выход 40%). 1Н ЯМР (CDCl3: δ (ч/млн) 8,49 (1Н, d, J=8,30 Гц), 7,84 (1Н, d, J=0,64 Гц), 7,62 (1Н, s), 7,12 (1Н, dd, J=8,34, 1,88 Гц), 7,00 (1Н, d, J=1,83 Гц), 5,99-5,91 (1Н, m), 5,19-5,13 (0.5Н, m), 5,05-4,99 (0.5Н, m), 4,49-4,46 (2Н, m), 4,45-4,41 (2Н, m), 4,26 (2Н, t, J=8,24 Гц), 4,06 (2Н, t, J=7,28 Гц), 3,80-3,63 (3Н, m), 3,27-3,22 (1Н, m), 3,21-3,16 (1Н, m), 3,06-3,00 (2Н, m), 2,97-2,90 (2Н, m), 1,60-1,54 (6Н, m). ЖХМС: RT=2,94 мин, М+Н+=516.
Пример 205
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2,4][1,4]оксазепин-9-ил)азетидин-1-ил)-2-матилпроапнамид 205
Суспензию соединения 65 (0,23 г, 0,6 ммоль) в воде (2,5 мл) обрабатывали цианидом натрия (49,5 мг, 0,6 ммоль) и ацетном (60 мг, 0,91 ммоль) в воде (0,25 мл) и смесь перемешивали при КТ в течение 18 ч. Смесь 4 раза экстрагировали ДХМ и объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали 2-{3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]азетидин-1-ил}-2-метилпропионитрил (0,19 г, выход 76%). ЖХМС: RT=3,76 мин, М+Н+=418.
2-{3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]азетидин-1-ил}-2-метилпропионитрил (0,17 г, 0,41 ммоль) растворяли в конц. H2SO4, (2 мл) и смесь выдерживали при КТ в течение 3,25 ч и затем добавляли лед. Полученный раствор подщелачивали добавлением Na2CO3, затем добавляли воду и смесь экстрагировали 10% МеОН в ДХМ. Объединенные экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% МеОН в ДХМ), при этом получали соединение 205 в виде твердого вещества белого цвета (97 мг, выход 54%). 1Н ЯМР (CDCl3): δ (ч/млн) 8,46 (1Н, d, J=8,29 Гц), 7,84 (1Н, s), 7,61 (1Н, s), 7,13 (1Н, s), 7,08 (1Н, d, J=8,39 Гц), 6,95 (1Н, s), 6,01-5,91 (1Н, m), 5,27 (1Н, s), 4,50-4,40 (4H, m), 3,62 (3H, s), 3,33 (2H, s), 1,56 (6H, d, J=6,63 Гц), 1,23 (6H, s). ЖХМСS: RT=2,53 мин, М+H+=436.
Пример 206
2-(4-(2-(1-(2,2,2-Трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)этанол 206
Согласно тому, как описано для получения соединения 184, проводили реакцию между 9-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепином и 1-(2,2,2-трифторэтил)-1Н-1,2,4-триазолом и получали 9-бром-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,109 г, выход 10%). 1H ЯМР (400 MHz, ДМСО): δ 8,28 (t, J=21,9, 1H), 8,11 (t, J=7,9, 2H), 7,51-7,35 (m, 1H), 7,32 (d, J=2,0, 1H), 5,88 (q, J=8,8, 2H), 4,76-4,29 (m, 4H). МС (ESI(+)): m/z 413,9 (M+H).
Согласно тому, как описано для получения соединения 182, из 9-бром-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина и 1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола получали соединение 206 (0,056 г, выход 48%). 1Н ЯМР (400 МГц, ДМСО): δ 8,33 (d, J=8,4, 1H), 8,24 (s, 1H), 8,07 (d, J=11,7, 2H), 7,95 (d, J=8,9, 1H), 7,41 (dd, J=8,4, 1,7, 1H), 7,28 (d, J=1,7, 1H), 5,91 (q, J=8,8, 2H), 4,91 (t, J=5,3, 1H), 4,54 (dd, J=10,8, 5,6, 4H), 4,16 (t, J=5,6, 2H), 3,87-3,69 (m, 2H). МС (ESI(+)): m/z 446,1 (M+H).
Пример 207
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-имидазол-1-ил)этанол 207
Pd(PPh3)4 (84,0 мг, 0,0727 ммоль) добавляли в дегазированный раствор соединения 194 (272 мг, 0,727 ммоль) и региоизомеров соединений 53а и 53b (600 мг, 1 ммоль) в ацетонитриле (5 мл, 100 ммоль). Реакционную смесь нагревали с использованием микроволнового излучателя при 140°С в течение 30 мин и завершение реакции подтверждали методом ЖХМС. Смесь концентрировали в вакууме и очищали экспресс-хроматографией (элюент: градиент от 0 до 100% метанол/ДХМ). Продукт концентрировали в вакууме и получали 270 мг региоизомеров 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин и 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-4][1,4]оксазепин. Неразделенные соединения растворяли в 4 н. HCl в диоксане (10 мл) и раствор перемешивали в течение 30 мин при КТ. Полное удаление защитной группы подтверждали методом ЖХМС, при этом получали конечные соединения, которые очищали методом SFC, при этмо разделяли региоизомер 207 (159,8 мг, выход 54%, М+1 406,1).
В другом варианте в смесь соединения 224 (300 мг, 0,75 ммоль) и Cs2CO3 (733 мг, 2,25 ммоль) в ДМФ (15 мл) в атмосфере азота добавляли 2-(2-бромэтокси)тетрагидропиран (0,68 мл, 4,52 ммоль). Реакционную смесь нагревали при 80°С в течение 5 ч. После охлаждения до КТ полученную смесь выливали в воду и экстрагировали EtOAc. Органические экстракты сушили над сульфатом натрия и очищали препаративной ТСХ (ДХМ/МеОН=10:1), при этом получали 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин в виде маслянистого вещества желтого цвета (250 мг, выход 68%). ЖХМС: (ESI, m/z)=490 [M+H]+.
В раствор 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (250 мг, 0,51 ммоль) в EtOH (15 мл) добавляли раствор хлористого водорода в диоксане (1,28 мл, 5,1 ммоль). Смесь кипятили с обратным холодильником, охлаждали до КТ и концентрировали. Полученный осадок промывали EtOAc и получали соединение 207 в виде твердого вещества желтого цвета (115,2 мг, выход 56%). 1H ЯМР (метан-d4, 400 МГц): δ 9,12 (d, J=1,2 Гц, 1Н), 8,78 (s, 1H), 8,65 (d, J=8,4 Гц, 1H), 8,28 (s, 1H), 8,17 (d, J=1,2 Гц, 1H), 7,58-7,53 (m, 2H), 5,85-5,78 (m, 1H), 4,71-4,63 (m, 4H), 4,41 (t, J=5,2 Гц, 2H), 3,96 (t, J=5,2 Гц, 2H), 1,66 (d, J=6,8 Гц, 6Н). MC: (ESI, m/z)=406 [М+Н]+.
Пример 208
2-(5-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-имидазол-1-ил)этанол 208
Pd(PPh3)4 (84,0 мг, 0,0727 ммоль) добавляли в дегазированный раствор соединения 194 (272 мг, 0,727 ммоль) и региоизомеров соединений 53а и 53b (600 мг, 1 ммоль) в ацетонитриле (5 мл, 100 ммоль). Реакционную смесь нагревали с использованием микроволнового излучателя при 140°С в течение 30 мин и завершение реакции подтверждали методом ЖХМС. Смесь концентрировали в вакууме и очищали экспресс-хроматографией (элюент: градиент от 0 до 100% метанол/ДХМ). Продукт концентрировали в вакууме и получали 270 мг региоизомеров 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин и 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин. Неразделенные соединения растворяли в 4 н. HCl в диоксане (10 мл) и раствор перемешивали в течение 30 мин при КТ. Полное удаление защитной группы подтверждали методом ЖХМС, при этом получали конечные соединения, которые очищали методом SFC, при этмо разделяли региоизомер 208 (27 мг, выход 9%, М+1 406.1).
Пример 209
2-(1-(2-Морфолиноэтил)-1Н-имидазол-2-ил)-10-(1Н-пиразол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 209
Соединение 35 алкилировали с 4-(2-хлорэтил)морфолином, при этом получали 9-бром-2-[1-(2-морфолиноэтил)-1Н-имидазол-2-ил]-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (выход: 51%, МС: 444,2), который в условиях конценсации Сузуки конденсировали с трет-бутиловым эфиром 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-при этом получали соединение 209 с выходом 24%. МС: 432,1. 1Н ЯМР (400 МГц, ДМСО): δ 12,95 (s, 1Н), 8,56 (d, J=2,3, 1H), 8,09 (s, 1H), 7,87 (s, 1H), 7,72 (s, 1H), 7,51 (dd, J=8,4, 2,3, 1H), 7,21 (d, J=1,0, 1H), 7,04 (d, J=8,4, 1H), 6,89 (d, J=1,0, 1H), 4,72 (t, J=7,1, 2H), 4,50 (q, J=5,6, 4H), 3,48-3,40 (m, 4H), 2,73 (t, J=7,1, 2H), 2,46-2,36 (m, 4H).
Пример 210
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-морфолиноэтил)-1Н-пиразол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 210
Согласно основной методике С проводили реакцию соединения 48 с 4-(2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)этил)морфолином. Реакционную смесь очищали обращено-фазной ЖХВР и получали соединение 210. ЖХМС: 489,2.
Пример 211
2-(4-(2-(3-Амино-1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)этанол 211
В сосуд, пригодный для микроволнового облучения, добавляли 5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-иламин (0,180 г, 0,000462 моль) и карбонат калия (0,1917 г, 0,001387 моль) в ацетонитриле (2,0 мл, 0,038 моль) и воде (2,0 мл, 0,11 моль). Реакционную смесь интенсивно дегазировали и в течение 5 мин продували азотом. В смесь добавляли Pd(PPh3)4 (0,05344 г, 0,00004624 моль) и 2-[4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]этиловый эфир уксусной кислоты (0,1554 г, 0,0005549 моль) и сосуд немедленно запечатывали. Смесь нагревали при 140°С в течение 20 мин с использованием микроволнового облучения. Реакционную смесь разбавляли ДХМ и фильтровали через слой целита. В смесь добавляли насыщенный раствор NH4Cl и смесь 3 раза экстрагировали ДХМ. Органические слои объединяли, сушили над MgSO4 и концентрировали. Неочищенный продукт очищали обращено-фазной ЖХВР, при этом получали соединение 211 (34,6 мг) в виде бесцветного маслянистого вещества. MC(ESI+) 421,1. 1H ЯМР (400 МГц, ДМСО): δ 8,35 (d, J=8,4, 1H), 8,22 (s, 1H), 7,95 (s, 1H), 7,73 (s, 1H), 7,38 (dd, J=8,4, 1,8, 1H), 7,27 (d, J=1,7, 1H), 5,84-5,69 (m, 1H), 5,19 (s, 2H), 4,93 (t, J=5,3, 1H), 4,50 (s, 4H), 4,16 (t, J=5,6, 2H), 3,77 (q, J=5,6, 2H), 1,42 (d, J=6,6, 6H).
Пример 212
2-(3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)азетидин-1-ил)-N-метилацетамид 212
Раствор соединения 65 (70 мг, 0,2 ммоль) в НМП (2 мл) обрабатывали фосфатом натрия (85 мгg, 0,6 ммоль) и N-метил-2-хлорацетамидом (24 мг, 0,22 ммоль) в НМП (0,2 мл) и смесь перемешивали при КТ в течение 18 ч. Смесь очищали на колонке Isolute® SCX-2 (элюент: МеОН, затем 2 М NH3 в МеОН). Соответствующие фракции объединяли и концентрировали в вакууме, полученный остаток очищали экспресс-хроматографией (SiO2, эолюент: градиент от 0 до 10% МеОН в ДХМ), при этом получали соединение 212 (24 мг, выход 29%). 1Н ЯМР (CDCl3): δ (ч/млн) 8,50 (1Н, d, J=8,29 Гц), 7,89 (1Н, s), 7,68 (1Н, s), 7,22 (1Н, s), 7,10 (1Н, dd, J=8,33, 1,81 Гц), 6,99-6,96 (1Н, m), 6,03-5,94 (1Н, m), 4,53-4,49 (2H, m), 4,48-4,44 (2H, m), 3,96 (2H, t, J=7,77 Гц), 3,81 (1Н, t, J=7,74 Гц), 3,54 (2H, t, J=7,43 Гц), 3,35 (2H, s), 2,86 (3H, d, J=4,93 Гц), 1,60 (6H, d, J=6,63 Гц). ЖХМС: RT=2,53 мин, M+H+=422.
Пример 213
1-(4-(2-(3-Амино-1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропан-2-ол 213
В сосуд, пригодный для микроволнового облучения, добавляли 5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-иламин (0,180 г, 0,000462 моль) и ацетат калия (0,1362 г, 0,001387 моль) в ацетонитриле (2,0 мл, 0,038 моль) и воде (2,0 мл, 0,11 моль). Реакционную смесь интенсивно дегазировали и в течение 5 мин продували азотом. В смесь добавляли Pd(PPh3)4 (0,05344 г, 0,00004624 моль) и 2-метил-1-[4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]пропан-2-ол (0,1477 г, 0,0005549 моль) и сосуд немедленно запечатывали. Смесь нагревали при 140°С в течение 20 мин с использованием микроволнового облучения. Реакционную смесь разбавляли ДХМ и фильтровали через слой целита. В смесь добавляли насыщенный раствор NH4Cl и смесь 3 раза экстрагировали ДХМ. Органические слои объединяли, сушили над MgSO4 и концентрировали. Неочищенный продукт очищали обращено-фазной ЖХВР, при этом получали соединение 213 (68,2 мг) в виде бесцветного твердого вещества. MC(ESI+) 449,2. 1H ЯМР (400 МГц, ДМСО): δ 8,35 (d, J=8,4, 1H), 8,15 (s, 1H), 7,95 (s, 1H), 7,73 (s, 1H), 7,38 (dd, J=8,4, 1,8, 1H), 7,27 (d, J=1,7, 1H), 5,82-5,68 (m, 1H), 5,19 (s, 2H), 4,50 (s, 4H), 4,04 (s, 2H), 1,42 (d, J=6,6, 6H), 1,09 (s, 6H).
Пример 214
1-(4-(2-(1-Изопропил-1Н-имидазол-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропан-2-ол 214
Соединение 36 (157 мг, 0,421 ммоль), 2-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропан-2-ол (139,9 мг, 0,5258 ммоль), и Pd(PPh3)4 (68,05 мг, 0,05889 ммоль) растворяли в ацетонитриле (2,66 мл, 50,9 ммоль) и смешивали с растворенным 2,00 М раствором карбоната калия в воде (0,421 мл). Реакционную смесь дегазировали Реакционную смесь обрабатывали микроволновым облучением при 150 Ватт, 140°С в течение 30 мин. Реакционную смесь охлаждали до КТ и экстрагировали EtOAc, при этом получали неочищенный продукт, который очищали обращенно-фазной ЖХВР, при этом получали соединени 214. МС: (ESI+)=433,2. 1H ЯМР (400 МГц, ДМСО): δ 8,36 (d, J=8,4 Гц, 1H), 8,15 (s, 1H), 7,94 (s, 1H), 7,68 (s, 1H), 7,42-7,31 (m, 2H), 7,26 (d, J=1,7 Гц, 1H), 6,94 (s, 1H), 5,66 (dt, J=13,5, 6,7 Гц, 1H), 4,74 (s, 1H), 4,50 (s, 4H), 4,03 (s, 2H), 1,46 (d, J=6,7 Гц, 6H), 1,09 (s, 6H).
Пример 215
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропанамид 215
Согласно тому, как описано для получения соединения 182, из соединения 194 и 2-метил-2-(4-(4,4,5,5-тераметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропанамида получали промежуточное соединение метиловый эфир 2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановой кислоты (и соответствующую кислоту) с выходом 62%. ЖХ/МС (ESI+): m/z 388 (М+Н).
Смесь, содержащую указанный эфир и соответствующую кислоту (100 мг, 0,22 ммоль) обрабатывали 1 М раствором гидроксида лития в воде (2 мл) и метанолом (0,37 мл). Реакционную смесь перемешивали при КТ в течение 12 ч. Смесь подкисляли добавлением 10% водного раствора лимонной кислоты до рН=5 и 2 раза экстрагировали EtOAc. Объединенные органические слои промывали солевым раствором, сушили и концентрировали. Полученную карбоновую кислоту использовали на следующих стадиях без дополнительной очистки. 1H ЯМР (400 МГц, ДМСО): δ 8,44 (s, 1H), 8,39 (s, 0Н), 8,37 (s, 1H), 7,98 (s, 1H), 7,92 (s, 2H), 7,45 (dd, J=8,4, 1,8, 1H), 7,36 (d, J=1,7, 1H), 5,90 (dt, J=13,2, 6,6, 1H), 4,53 (q, J=6,0, 4H), 1,72 (d, J=42,8, 6H), 1,50 (d, J=6,6, 6H).
Карбоновую кислоту, полученную на предыдущей стадии (100 мг, 0,22 ммоль) растворяли в ДМФ (1 мл) и обрабатывали последовательно N,N-диизопропилэтиламином (0,3 мл, 2,0 ммоль), хлоридом аммония (50 мг, 0,9 ммоль) и гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (200 мг, 0,6 ммоль). Полученную смесь перемешивали при КТ в течение ночи. Добавляли насыщенный раствор бикарбоната натрия и экстрагировали EtOAc. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали. Остаток очищали обращенно-фазной ЖХВР, при этом получали 53 мг (выход 54%) соединения 215. ЖХ/МС (ESI+): m/z 447 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,41 (s, 1H), 8,39 (s, 0Н), 8,37 (s, 1H), 8,02 (s, 1H), 7,46 (dd, J=8,4, 1,7, 1H), 7,35 (t, J=7,2, 1H), 7,20 (s, 1H), 6,85 (s, 1H), 5,90 (гепт., J=6,6, 1H), 4,53 (q, J=5,9, 4H), 1,74 (s, 6H), 1,50 (d, J=6,6, 6H).
Пример 216
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановая кислота 216
Из соединения 194 и этилового эфира 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропановой кислоты в условиях реакции Сузуки (Pd(dppf)Cl2, Cs2CO3) получали этиловый эфир 2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановой кислоты. ЖХ/МС (ESI+): m/z 476 (М+Н).
Этиловый эфир 2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановой кислоты обрабатывали раствором гидроксида лития в воде, при этом получали соединение 216. ЖХ/МС (ESI+): m/z 448 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,44 (s, 1Н), 8,38 (d, J=8,4, 1H), 7,98 (s, 1H), 7,92 (s, 2H), 7,45 (dd, J=8,4, 1,8, 1H), 7,36 (d, J=1,7, 1H), 5,90 (dt, J=13,2, 6,6, 1H), 4,53 (q, J=6,0, 4H), 1,77 (s, 6H), 1,50 (d, J=6,6, 6H).
Пример 217
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(1Н-пиразол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 217
Согласно тому, как описано для получения соединения 182, из соединения 194 и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола получали соединение 217 с выходом 78%. ЖХМС (ESI+): m/z 362 (М+Н). 1H ЯМР (400 МГц, ДМСО): δ 13,02 (s, 1H), 8,37 (d, J=8,4, 1H), 8,29 (s, 1H), 8,00 (s, 1H), 7,54-7,38 (m, 1H), 7,30 (t, J=12,5, 1H), 5,91 (dt, J=13,2, 6,6, 1H), 4,57-4,46 (m, 4H), 1,50 (d, J=6,6, 6H).
Пример 218
3-(2-(1-(2,2,2-Трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)пиридин-2(1Н)-он 218
Стадия 1
Согласно тому, как описано для соединения 187, из 10-бром-2-иод-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина и трифторэтилтриазола (1-(2,2,2-трифторэтил)-1Н-1,2,4-триазола) получали неочищенный продукт. Неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: EtOAc), концентрировали при пониженном давлении, растворяли в ДМФ, очищали обращенно-фазной ЖХВР и получали 10-bromo-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,027 г, выход 2%). 1H ЯМР (400 МГц, ДМСО): δ 8,48 (d, J=2,6, 1H), 8,10 (d, J=5,5, 2H), 7,49 (dd, J=8,7, 2,6, 1H), 7,04 (t, J=7,5, 1H), 5,86 (q, J=8,8, 2H), 4,54 (dt, J=7,4, 3,7, 4H). MC (ESI(+)): m/z 413,9 (М+Н).
Стадия 2
Согласно тому, как описано для соединения 203, из 10-бром-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина и 2-фторпиридин-3-илборной кислоты получали 10-(2-фторпиридин-3-ил)-2-(1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]|оксазепин (0,108 г, выход 55%), 1Н ЯМР (400 МГц, ДМСО): δ 8,71 (t, J=2,0, 1H), 8,57-7,83 (m, 4H), 7,80-7,39 (m, 2H), 7,22 (d, J=8,5, 1H), 5,88 (q, J=8,8, 2H), 4,79-4,38 (m, 4H). MC (ESI(+)): m/z 431,1 (M+H), который гидролизовали в присутствии HCl, при этом получали соединение 218 (0,072 г, выход 72%). 1Н ЯМР (400 МГц, ДМСО): δ 11,85 (s, 1H), 8,78 (d, J=2,3, 1H), 8,09 (d, J=2,8, 2H), 7,69 (ddd, J=8,9, 7,7, 2,2, 2H), 7,36 (t, J=23,8, 1H), 7,08 (d, J=8,6, 1H), 6,30 (t, J=6,7, 1H), 5,91 (q, J=8,8, 2H), 4,56 (dd, J=13,5, 5,5, 4H). MC (ESI(+)): m/z 429,1 (M+H).
Пример 219
5-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиридин-2-амин 219
Согласно тому, как описано для получения соединения 182, из соединения 194 и 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина получали соединение 219 с выходом 62%. ЖХ/МС (ESI+): m/z 388 (M+H). 1Н ЯМР (400 МГц, ДМСО): δ 8,42 (d, J=8,4, 1H), 8,33 (d, J=2,4, 1H), 7,93 (d, J=6,5, 2H), 7,77 (dd, J=8,7, 2,5, 1H), 7,41 (dd, J=8,5, 1,9, 1H), 7,26 (d, J=1,8, 1H), 6,53 (d, J=8,6, 1H), 6,16 (s, 2H), 5,92 (dt, J=13,2, 6,6, 1H), 4,60-4,44 (m, 4H), 1,50 (d, J=6,6, 6H).
Пример 220
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-имидазол-1-ил)этанол 22
1-[2-(Тетрагидропиран-2-илокси)этил]-4-трибутилстаннанил-1Н-имидазол (0,690 г, 0,00142 моль) добавляли в раствор соединения 48 (0,293 г, 0,000755 моль) в ацетонитриле (4,5 мл, 0,086 моль). Смесь интенсивно дегазировали током азота и добавляли Pd(PPh3)4 (0,0872 г, 0,0000755 моль). Сосуд закрывали и нагревали с использованием микроволнового облучения при 140°С в течение 30 мин. Добавляли ДХМ и воду и смесь фильтровали через слой целита. Водную фазу 3 раза экстрагировали ДХМ. Органические фазы объединяли, сушили над MgSO4 и концентрировали.
Неочищенный продукт растворяли в ДХМ (8,0 мл, 0,12 моль). По каплям добавляли раствор хлористого водорода (4 н. В диоксане, 0,47 мл, 0,00189 моль) и реакционную смесь перемешивали при КТ в течение 1 ч. Реакционную смесь концентрировали в вакууме. В остаток добавляли ДХМ и насыщенный раствор карбоната натрия, при этом наблюдали выделение проукта в осадок в водной фазе. Водную фазу фильтровали и твердый продукт очищали обращенно-фазной ЖХВР, при этом получали соединение 220 (42 мг) в виде бесцветного маслянистого вещества. MS(ESI+) 420,2.
Пример 221
2-(2-(9-(1-(2-Гидрокси-2-метилпропил)-1Н-пиразол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1Н-имидазол-1-ил)-N-метилацетамид 221
Согласно тому, как описано для соединения 214, из 2-(2-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1Н-имидазол-1-ил)-N-метилацетамида и 2-метил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропан-2-ола в условиях реакции Сузуки получали соединение 221. МС: (ESI+)=462,2. 1H ЯМР (400 МГц, ДМСО): δ 8,38 (d, J=8,4 Гц, 1Н), 8,18 (s, 1Н), 8,06-7,99 (m, 1Н), 7,96 (s, 1Н), 7,69 (s, 1Н), 7,33 (dd, J=8,4, 1,8 Гц, 1H), 7,26 (d, J=1,7 Гц, 1Н), 7,11 (d, J=1,1 Гц, 1Н), 6,90 (d, J=1,1 Гц, 1Н), 5,18 (s, 2H), 4,76 (s, 1Н), 4,49 (s, 4H), 4,04 (s, 2H), 2,65 (d, J=4,6 Гц, 3Н), 1,10 (s, 6H).
Пример 222
N,N-Диэтил-2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)этанамин 222
В сосуд, пригодный для микроволнового облучения, объемом 5 мл добавляли соединение 194 (347 мг, 0,928 ммоль), соединение 55 (340 мг, 1,16 ммоль), 2М раствор карбоната калия в воде (0,9 мл, 2 ммоль) и ацетонитриле (1,52 г, 37,1 ммоль!) и хлорид 1,1-бис(дифенилфосфино)ферроценпалладия(II) (45,4 мг, 0,056 ммоль) и сосуд закрывали. Реакционную смесь нагревали с использованием микроволнового облучения при 140°С в течение 10 мин. Охлажденную реакционную смесь разбавляли EtOAc и водой и распределяли. Органический слой промывали над солевым раствором, сушили над сульфатом натрия, концентрировали в вакууме и очищали ЖХВР, при этом получали соединени 222 (140 мг, выход 33%, М+1 461,6).
Пример 223
5-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-4][1,4]оксазепин-9-ил)пиримидин-2-амин 223
Согласно тому, как описано для получения соединения 182, из соединения 194 и 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-амина получали соединение 223 с выходом 73%. ЖХ/МС (ESI+): m/z 389 (М+Н). 1Н ЯМР (400 МГц, ДМСО): δ 8,66 (s, 1Н), 8,45 (d, J=8,4, 1H), 7,93 (d, J=9,7, 2H), 7,46 (dd, J=8,5, 1,9, 1H), 7,35 (d, J=1,8, 1H), 6,86 (s, 1H), 5,91 (гепт., J=6,4, 1H), 4,61-4,44 (m, 4H), 1,50 (d, J=6,6, 6H).
Пример 224
9-(1Н-Имидазол-5-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 224
В раствор соединения 242 (120 мг, 0,27 ммоль) в диоксане (4 мл) добавляли раствор гидрохлорида в диоксане (0,34 мл, 1,35 ммоль). Смесь нагревали при 60°С в течение 2 ч, охлаждали до КТ и концентрировали. В смесь добавляли насыщенный раствор бикарбоната натрия и экстрагировали EtOAc. Органические слои сушили над сульфатом натрия, концентрировали и остаток очищали препаративной ТСХ (элюент: ДХМ/МеОН=8:1), при этом получали соединение 224 в виде твердого вещества желтого цвета (36 мг, выход 37%). 1Н ЯМР (ДМСО-d6, 400 МГц): δ 12,29 (s, 1H), 8,37 (d, J=8,4 Гц, 1H), 7,91 (s, 2H), 7,75 (s, 1H), 7,69 (s, 1H), 7,55 (d, J=8,0 Гц, 1H), 7,45 (s, 1H), 5,94-5,88 (m, 1H), 4,53 (ушир. s, 4H), 1,49 (d, J=6,4 Гц, 6H). MC: (ESI, m/z)=362 [M+H]+.
Пример 225
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(2-(метилсульфонил)этил)пиперидин-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 225
Согласно тому, как описано для соединения 152, соединение 225 получали в виде твердого вещества белого цвета из соединения 66 и винилсульфона. 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,27 (1Н, d, J=8,29 Гц), 7,86-7,84 (2Н, m), 7,00 (1Н, dd, J=8,37, 1,76 Гц), 6,85 (1Н, d, J=1,70 Гц), 5,88-5,78 (1Н, m), 4,44 (4Н, q, J=5,99 Гц), 3,26 (3Н, m), 3,00 (3Н, s), 2,95 (2Н, d, J=11,00 Гц), 2,68 (2Н, t, J=6,79 Гц), 2,02 (2H, t, J=11,39 Гц), 1,72 (2H, d, J=12,62 гц), 1,57 (2H, qd, J=12,27, 3,58 Гц), 1,42 (6H, d, J=6,60 гц). ЖХМС: RT=2,68 мин, M+H+=485.
Пример 226
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 226
Согласно тому, как описано для соединения 143, соединение 226 получали в виде твердого вещества белого цвета из соединения 66 и 2-бромацетамида. 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,27 (1Н, d, J=8,27 Гц), 7,84 (2H, d, J=2,70 Гц), 7,15 (1Н, s), 7,06 (1Н, s), 7,01 (1Н, d, J=8,43 Гц), 6,87 (1Н, s), 5,87-5,78 (1Н, m), 4,44 (4H, d, J=6,92 Гц), 2,90-2,78 (4H, m), 2,15-2,06 (2H, m), 1,68 (5H, s), 1,42 (6H, d, J=6,59 Гц). ЖХМС (способ F): RT=2,57 мин, М+Н+=436.
Пример 227
2-Гидрокси-1-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-2-метилпропан-1-он 227
Раствор соединения 66 (350 мг, 0,71 ммоль), 2-гидроксиизобутановой кислоты (111 мг, 1,07 ммоль), EDCI (327 мг, 1,7 ммоль), HOBt (230 мг, 1,7 ммоль) и DIPEA (0,36 мл, 2,13 ммоль) пемермешивали при КТ в течение 5 ч и затем в раствор добавляли насыщенный водный раствор бикарбоната натрия. Полученную смесь экстрагировали ДХМ (2×30 мл), объединенные органические экстракты промывали солевым раствором и сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 5% МеОН в ДХМ), остаток лиофилизировали, при этом получали соединение 227 в виде твредого вещества белого цвета (141 мг, выход 43%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,31 (1Н, d, J=8,29 Гц), 7,82 (1Н, s), 7,78 (1Н, s), 7,03 (1Н, d, J=8,38 Гц), 6,90 (1Н, s), 5,85-5,77 (1Н, m), 4,69 (2H, d, J=13,25 Гц), 4,48 (4H, t, J=7,99 гц), 2,91-2,74 (4H, m), 1,85 (2H, d, J=13,04 гц), 1,63-1,49 (2H, m), 1,49 (6H, d, J=6,64 Гц), 1,37 (6H, s). ЖХМС: RT=3,92 мин, M+H+=465.
Пример 228
(2S)-2-Гидрокси-1-(3-(4-(2-(1-изопропил-3-метил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)азетидин-1-ил)пропан-1-оне 228
Из соединения 48 и трет-бутилового эфира 3-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-ил] азетидин-1-карбновой кислоты в условиях реакции Сузуки получали трет-бутиловый эфир 3-{4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиразол-1-ил}азетидин-1-карбоновой кислоты. MC(ESI+) 531,2.
Из трет-бутиловый эфир 3-{4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиразол-1-ил}азетидин-1-карбоновой кислоты и кислоты получали 8-(1-азетидин-3-ил-1Н-пиразол-4-ил)-2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен. MC(ESI+) 431,2.
Из 8-(1-азетидин-3-ил-1Н-пиразол-4-ил)-2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азуленаи DIPEA, HATU и L-молочной кислоты получали соединение 228. MC(ESI+) 503.2. 1Н ЯМР (400 Мгц, ДМСО): δ 8,45 (d, J=1,7, 1H), 8,37 (d, J=8,4, 1H), 8,12 (s, 1H), 7,89 (s, 1H), 7,42 (dd, J=8,4, 1,7, 1H), 7,32 (d, J=1,7, 1H), 5,92-5,74 (m, 1H), 5,34-5,25 (m, 1H), 5,21 (t, J=5,5, 1H), 4,84-4,66 (m, 1H), 4,60-4,53 (m, 1H), 4,52 (s, 4H), 4,40-4,31 (m, 1H), 4,26-4,09 (m, 2H), 2,25 (s, 3H), 1,47 (d, J=6,6, 6H), 1,22 (d, J=6,7, 3H).
Пример 229
2-(4-(2-(3-Амино-1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-имидазол-1-ил)этанол 229
Соединение 229 получали из 5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-иламина и соеднения 53а после удаления группы тригидропиран с использованием водного раствора HCl и очистки обращенно-фазной ЖХВР (49 мг). ЖХМС: 421,2.
Пример 130
2-(3-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)азетидин-1-ил)этанол 230
В раствор 8-(1-азетидин-3-ил-1Н-пиразол-4-ил)-2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена в ДХМ добавлячли (трет-бутилдиметилсиланокси)ацетальдегид, уксусную кислоту и триацетоксиборгидрид натрия. Реакционную смесь перемешивали при КТ в течение приблизительно 5 ч и реакцию останавливали добавлением 1 н. раствора NaOH. Добавляли ДХМ и смесь 3 раза экстрагировали ДХМ. Органически фазы объединяли, сушили над MgSO4 и концентрировали. Смесь очищали экспресс-хроматографией (элюент: 0-10% МеОН в ДХМ), при этом получали 8-(1-{1-[2-(трет-бутилдиметилсиланилокси)этил]азетидин-3-ил}-1Н-пиразол-4-ил)-2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен, MC(ESI+) 589,3, который обрабатывали кислотой и получали соединение 230. 1H ЯМР (400 МГц, ДМСО): δ 8,45 (s, 1H), 8,36 (d, J=8,4, 1H), 8,03 (s, 1H), 7,88 (s, 1H), 7,41 (d, J=8,3, 1H), 7,32 (d, J=1,6, 1H), 5,90-5,78 (m, 1H), 4,97 (quin, J=6,9, 1H), 4,52 (s, 4H), 4,47 (t, J=5,4, 1H), 3,73 (t, J=7,6, 2H), 3,47-3,37 (m, 4H), 2,58 (t, J=7,7, 2H), 2,25 (s, 3Н), 1,47 (d, J=6,6, 6H).
Пример 231
5-(5,6-Дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-амин 231
5-(8-Бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-иламин гидрировали в присутствии 10% Pd на углероде, продукт очищали обращенно-фазной ЖХВР и получали соединение 231. ЖХМС: 311,2.
Пример 232
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 232
В круглодонную колбу объемом 25 мл помещали 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 411 (1,0 г, 2,6 ммоль), бисборонат биспинасола (0,719 г, 2,83 ммоль), ацетат калия (0,76 г, 7,7 ммоль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с ДХМ (1:1) (0,21 г, 0,26 ммоль) в амтосфере азота. Смесь разбавляли диметилсульфоксидом (8,5 мл) и нагревали при 85°С в течение 12 ч. Реакционную смесь охлаждали до КТ и разбавляли водой и ДХМ. Фазы распределяли и водный слой 3 раза экстрагировали ДХМ. Органические слои объединяли, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали экспресс-хроматграфией, при этом поучали соединение 232 в виде дегалогенированного побочного продукта (66 мг, выход 7%). МС (ESI+) m/z 310,2 (М+Н+), рассч. 310,4. 1H ЯМР (500 МГц, ДМСО): δ 8.41 (dd, J=8,0, 1,6 Гц, 1Н), 7,89 (s, 1H), 7,33 (dd, J=11,0, 4,3 Гц, 1Н), 7,16 (t, J=7,6 Гц, 1Н), 7,07 (d, J=8,1 Гц, 1Н), 5,83 (dt, J=13,0, 6,6 Гц, 1Н), 4,51 (q, J=5,6 Гц, 4H), 2,26 (s, 3Н), 1,46 (d, J=6,6 Гц, 6Н).
Пример 133
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин 233
Смесь соединения 22 (82,7 мг, 0,250 ммоль), палладий на углероде 10% (0,1:0,9, палладий: черный уголь, 83 мг) и ТЭА (0,104 мл, 0,750 ммоль) в 5,0 мл этанола и 5,0 мл ТГФ (5,0 мл, 62 ммоль) гидрировали при давлении 1 атм в течение 3 ч. Катализатор отфильтровывали, фильтрат концентрировали остаток очищали обращенно-фазной ЖХВР (элюент: градиент ацетонитрила), при этом получали соединение 233 с выходом 32 мг (43%). MC(ESI+): 297,2. 1Н ЯМР (500 МГц, ДМСО): δ 8,40 (s, 1Н), 8,30 (d, J=5,2, 1Н), 8,25 (d, J=5,2, 1Н), 8,08 (s, 1Н), 7,94 (s, 1Н), 5,87 (dt, J=13,0, 6,6, 1Н), 4,60 (dd, J=13,1, 5,5, 4H), 1,49 (d, J=6,6, 6Н).
Пример 234
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-10-(4-метилпиперазин-1-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин 234
Смесь соединения 22 (132 мг, 0,400 ммоль), 1-метилпиперазина (88,7 мкл, 0,800 ммоль), ацетата палладия (44,9 мг, 0,200 ммоль), 2,8,9-триизобутил-2,5,8,9-тетрааза-1-фосфабицикло[3.3.3]умдекана (71,0 мкл, 0,200 ммоль) и трет-бутоксида натрия (38,4 mg, 0,400 ммоль) в 1,4-диоксане (8,0 мл) дегазировали. Реакционную смесь подвергали микроволновому облучению при 200 Ватт и 120°С в течение 30 мин. Смесь фильтровали и фильтрат концентрировали в вакууме. Остаток распределяли между водой и EtOAc. Органические экстракты промывали водой, солевым раствором и сушили над MgSO4 и концентрировали в вакууме. Остаток очищали на колонке с 4 г силикагеля (элюент: градиент 5-10% метанола и 1% аммиак в ДХМ), при этом получали соединение 234 с выходом 50 мг (30%). МС: 395,2. 1Н ЯМР (500 МГц, ДМСО): δ 8,04 (s, 1H), 8,03 (s, 1H), 7,94 (s, 1H), 7,63 (s, 1H), 5,69 (dt, J=13,3, 6,6, 1H), 4,61-4,53 (m, 2H), 4,49-4,41 (m, 2H), 3,41 (s, 4H), 2,45 (s, 4H), 2,23 (s, 3Н), 1,50 (d, J=6,6, 6H).
Пример 235
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-9-(пиримидин-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 235
В условиях реакции Сузуки из соединения 48 и 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиримидина получали соединение 235. MC(ESI+) 388,2. 1Н ЯМР (500 МГц, ДМСО): δ 9,24-9,20 (m, 3Н), 8,54 (d, J=8,4, 1H), 7,96 (s, 1H), 7,65 (dd, J=8,4, 1,4, 1H), 7,58 (d, J=1,2, 1H), 5,96-5,71 (m, 1H), 4,57 (s, 4H), 2,26 (s, 3Н), 1,48 (d, J=6,6, 6H).
Пример 236
9-(5-Фторпиридин-3-yl)-2-(1-зопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 236
В условиях реакции Сузуки из соединения 48 и 3-фтор-5-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиридина получали соединение 236. MC(ESI+) 405,2. 1Н ЯМР (500 МГц, ДМСО): δ 8,89 (s, 1H), 8,61 (d, J=2,6, 1H), 8,52 (d, J=8,4, 1H), 8,19-8,14 (m, 1H), 7,96 (s, 1H), 7,63 (dd, J=8,3, 1,3, 1H), 7,54 (d, J=1,4, 1H), 5,89-5,80 (m, 1H), 4,57 (s, 4H), 2,26 (s, 3Н), 1,48 (d, J=6,6, 6H).
Пример 237
2-(3-Амино-1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбонитрил 237
Соединение 237 получали из 5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-иламина и цианида цинка, неочищенный продукт очищали обращенно-фазной ЖХВР. ЖХМС: 336.
Пример 238
N-(5-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиридин-2-ил)ацетамид 238
В условиях реакции Сузуки из соединения 48 и N-[5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-пиридин-2-ил]ацетамида получали соединение 238. MC(ESI+) 444,2. 1H ЯМР (500 МГц, ДМСО): δ 10,63 (s, 1H), 8,72 (s, 1H), 8,48 (d, J=8,4, 1H), 8,17 (s, 2H), 7,93 (s, 1H), 7,55 (dd, J=8,4, 1,4, 1H), 7,43 (d, J=1,4, 1H), 5,89-5,80 (m, 1H), 4,55 (s, 4H), 2,26 (s, 3Н), 2,12 (s, 3Н), 1,47 (d, J=6,6, 6H).
Пример 239
9-Хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f][1,2,4]триазоло[1,5-f][1,4]оксазепин 239
Стадия 1
Метиловый эфир 9-хлор-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-2-карбоновой кислоты
В метиловый эфир 1-(2-(2-бром-5-хлорфенокси)этил)-1Н-1,2,4-триазол-3-карбоновой кислоты в ацетонитриле (10,00 мл, 191,5 ммоль) в сосуде, пригодном для микроволнового облучения и снабженном мешальником, добавляли карбонат цезия (0,9036 г, 2,773 ммоль). Смесь дегазировали продуванием азота через шприц. В сосуд добавляли хлорид тетраэтиламмония (0,2298 г, 1,387 ммоль), ацетат палладия (0,1556 г, 0,6933 ммоль) и иодид меди(1) (0,02641 г, 0,1387 ммоль) и сосуд закрывали. Затем сосуд перемешивали при 165°С в течение 18 мин с использованием микроволновго облучения. Реакционную смесь охлаждали до КТ, фильтровали через целит, концентрировали и очищали хроматографией на силикагеле, при этом получали метиловый эфир 9-хлор-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-2-карбоновой кислоты с выходом 15%. 1H ЯМР (400 МГц, CDCl3: δ 8,59 (d, J=2,6 Гц, 1H), 7,34 (dd, J=8,8, 2,7 Гц, 1H), 7,04 (d, J=8,8 Hz, 1H), 4,75 (m, 2H), 4,53 (m, 2H), 4,05 (s, 3Н). MCHP m/z рассч. для C12H10ClN3O3: 279,04107, найд.: 280,0 [M+1].
Стадия 2
Метиловый эфир 9-хлор-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-2-карбоновой кислоты (0,144 г, 0,515 ммоль) растворяли в смеси 3:2:1 ТГФ : МеОН : H2O (5,0 мл) и обрабатывали 4 н. водным раствором LiOH (0,644 мл). Смесь перемешивали в течение 30 мин при 25°С. Реакцию останавливали добавлением 4 н. водного раствора HCl (10 мл) и раствор экстрагировали (3×20 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали в вакууме, при этом получали 9-хлор-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-2-карбоновую кислоту (выход 89%).
Стадия 3
В раствор 9-хлор-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-2-карбоновой кислоты (0,137 г, 0,515 ммоль) в ДМФ (1,20 мл, 15,4 ммоль) добавляли гексафторфосфат N,N,N',N'-тетраметил-0-(7-азабензотриазол-1-ил)урония (0,587 г, 1,54 ммоль) и 6-хлор-1-гидроксибензотриазол (0,262 г, 1,54 ммоль). Смесь интенсивно перемешивали и в нее добавляли NH4Cl (0,220 г, 4,12 ммоль). Через 10 мин добавляли N,N-диизопропилэтиламин (0,359 мл, 2,06 ммоль). Реакционую смесь перемешивали при КТ в течение 3 ч. Затем реакционную смесь концентрировали досуха и промывали водой. Неочищенный продукт очищали на слое силикагеля (элюент: ДХМ/МеОН) и обращенно-фазной ЖХВР, при этом получали 9-хлор-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-2-карбоксамид (выход 5,3%).
Стадия 4
В раствор 9-хлор-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин-2-карбоксамида (0,0053 г, 0,020 ммоль) в толуоле (0,160 мл, 1,50 ммоль) добавляли 1,1-диметокси-N,N-диметилметанамин (0,0150 мл, 0,113 ммоль) и смесь нагревали в закрытом сосуде при 102°С в течение 2 ч при перемешивании. Затем реакционную смесь охлаждали и концентрировали досуха, в остаток добавляли гидрохлорид изопропилгидразина (0,00376 г, 0,0340 ммоль) и уксусную кислоту (0,0938 г, 1,56 ммполь), реакционный сосуд закрывали и смесь нагревали при 102°С в течение ночи при перемешивании. Затем реакционную смесь концентрировали досуха и смешивали с ДХМ, смесь очищали обращенно-фазной ЖХВР, при этом получали 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f][1,2,4]триазоло[1,5-d][1,4]оксазепин с выходом 98%. 1Н ЯМР (500 МГц, CDCl3: δ 8,56 (d, J=2,6 Гц, 1H), 8,02 (s, 1H), 7,35 (dd, J=8,8, 2,6 Гц, 1H), 7,06 (d, J=8,8 Гц, 1H), 5,84-5,66 (m, 1H), 4,84-4,70 (m, 2H), 4,62- 4,47 (m, 2H), 1,61 (s, 6H). ЖХМС m/z рассч. для C15H15ClN6O: 330,09959, найд.: 331,1 [M+1].
Пример 241
5-(9-(5-Фторпиридин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-амин 241
Проводили реакцию Сузуки между 5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-иламином и 3-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридином. Неочищенный продукт очищали обращенно-фазной ЖХВР, при этом получлаи соединение 241. ЖХМС: 406,2.
Пример 242
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-имидазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 242
1-(Тетрагидро-2Н-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-имидазол получали при гидрировании 2-хлор-1-(тетрагидро-2Н-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-имидазола в присутствии палладия.
В смесь соединения 194 (250 мг, 0,67 ммоль) и 1-(Тетрагидро-2Н-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-имидазола (204 мг, 0,73 ммоль) в сухом ДМФ (2,5 мл) в атмосфере азота добавляли CsF (254 мг, 1,68 ммоль), CuI (13 мг, 0,067 ммоль) и Pd(PPh3)4 (39 мг, 0,034 ммоль). Реакционную смесь нагревали при 130°С в течение 40 мин с использованием микроволного облучения. Смесь охлаждали до КТ, выливали в воду и экстрагировали EtOAc. Органический экстракты сушили над суьфатом натрия и очищали препаративной ТСХ (элюент: 100% EtOAc), при этом получали соединение 242 в виде твердого вещества желтого цвета (49,3 мг, выход 17%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,49 (d, J=8,4 Гц, 1Н), 8,11 (s, 1H), 7,97 (s, 1H), 7,92 (s 1H), 7,28 (dd, J1=1,6 Гц, J2=8,4 Гц, 1H), 7,18 (d, J=6,8 Гц, 2H), 5,92-5,89 (m, 1H), 5,19 (d, J=9,2 Гц, 1H), 4,55-4,52 (m, 4H), 4,04 (d, J=11,2 Гц, 1H), 3,60-3,56 (m, 1H), 2,29-1,90 (m, 3H), 1,58-1,48 (m, 9H). MC: (ESI, m/z)=446 [M+H]+.
Пример 3
3-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропанамид 243
Проводили реакцию Сузуки между 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепином 411 и этиловым эфиром 2-метил-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом-1-ил)пропановой кислоты (Pd(dppf)Cl2, Cs2CO3), при этом получали этиловый эфир 3-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановую кислоту, ЖХМС (ESI+): m/z 490 (М+Н). Полученный продукт гидролизовали в присутствии гидроксида лития в воде, соответствующую кислоту обрабатывали хлоридом аммония, HATU, диизопропилэтиламином и ДМФ, при этом получали соединение 243. ЖХ/МС (ESI+): m/z 461 (М+Н). 1Н ЯМР (500 МГц, ДМСО): δ 8,35 (d, J=8,4, 1H), 8,15 (s, 1H), 7,95 (s, 1H), 7,86 (s, 1H), 7,36 (dd, J=8,4, 1,7, 2H), 7,25 (d, J=1,7, 1H), 6,83 (s, 1H), 5,82 (dt, J=13,2, 6,6, 1H), 4,51 (s, 4H), 4,30 (dd, J=13,5, 7,6, 1H), 4,02 (dd, J=13,5, 7,0, 1H), 2.91 (dd, J=14,3, 7,1, 1H), 2,25 (s, 3H), 1,47 (d, J=6,6, 5H), 1,01 (d, J=7,0, 3H).
Пример 244
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-9-(пиридин-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 244
Из соединения 48 и 3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-пиридина получали соединение 244. MC(ESI+) 387,2.
Пример 245
5-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-N,N-диметилпиперидин-2-амин 245
Из соединения 48 и диметил[5-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиримидин-2-ил]амина получали соединение 245. MC(ESI+) 431,2.
Пример 246
5-(5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-N-метил-1Н-1,2,4-триазол-3-амин 246
5-(9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-N-метил-1Н-1,2,4-триазол-3-амин гидрировали в присутствии 10% Pd на углероде, продукт очищали обращенно-фазной ЖХВР, при этом получали соединение 246. ЖХМС: 325,2.
Пример 247
N-Изопропил-2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[Лимидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 247
В суспензию соединения 66 (350 мг, 0,71 ммоль) в ТГФ добавляли карбонат калия (245 мг, 1,78 ммоль) N-изопропил-2-хлорацетамид (106 мг, 0,78 ммоль) и реакционную смесь перемешивали при КТ в течение 18 ч, затем нагревали при 50°С в течение 2 ч. Полученную смесь концентрировали в вакууме и остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 5% МеОН в ДХМ), при этом получали соединение 247 в виде твердого вещества белого цвета (180 мг, выход 53%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,28 (1Н, d, J=8,28 Гц), 7,86 (1Н, d, J=0,63 гц), 7,85 (1Н, s), 7,40 (1Н, d, J=8,18 гц), 7,03 (1Н, dd, J=8,36, 1,77 Гц), 6,89 (1Н, d, J=1,72 Гц), 5,88-5,79 (1Н, m), 4,48-4,41 (4Н, m), 3,89-3,83 (1Н, m), 2,89-2,79 (4Н, m), 2,13 (2Н, td, J=10,70, 4,11 Гц), 1,74-1,64 (4Н, m), 1,43 (6Н, d, J=6,60 гц), 1,04 (6Н, d, J=6,59 гц). Пик от 1 протона перекрывался с пиком растворителя. ЖХМС: RT=2,94 мин, М+Н+=478.
Пример 248
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N,2-диметилпропанамид 248
Двухфазную смесь 50% водного гидроксида натрия (2 мл) в ДХМ (2,5 мл) обрабатывали 2-бром-2,N-диметилпропионамидом (121 мг, 0,67 ммоль), бромидом тетрабутиламмония (118 мг, 0,37 ммоль) и раствором соединения 66 (300 мг, 0,61 ммоль) в ДХМ (1 мл). Полученную смесь перемешивали при КТ в течение 18 ч и затем добавляли бромид тетрабутиламмония (118 мг, 0,37 ммоль) и перемешивали при КТ в течение 18 ч. Реакционную смесь разбавляли ДХМ и промывали водой. Объединенные органические экстракты промывали ДХМ, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 5% МеОН в ДХМ), растирали с диэтиловым эфиром и получали соединение 248 в виде твердого вещества кремового цвета (120 мг, выход 41%). 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,27 (1Н, d, J=8,26 Гц), 7,86 (1Н, d, J=0,63 гц), 7,85 (1Н, s), 7,64 (1Н, m), 7,02 (1Н, dd, J=8,34, 1,75 гц), 6,91 (1Н, d, J=1,70 гц), 5,88-5,78 (1Н, m), 4,48-4,42 (4H, m), 2,74 (2H, d, J=10,86 Гц), 2,59 (3H, d, J=4,75 Гц), 2,17-2,08 (2H, m), 1,75-1,69 (4H, m), 1,43 (6H, d, J=6,60 Гц), 1,05 (6H, s). Пик 1 протона перекрывался с пиком растворителя. ЖХМС: RT=2,78 мин, М+Н+=478.
Пример 249
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N,N-диметилэтансульфонамид 249
Из соединения 66 и N,N-диметилэтансульфонамида получали соединение 249 в виде твердого вещества белого цвета. 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,28 (1Н, d, J=8,29 Гц), 7,86 (1Н, d, J=0,63 гц), 7,85 (1Н, s), 7,01 (1Н, dd, J=8,37, 1,77 Гц), 6,85 (1Н, d, J=1,72 Гц), 5,88-5,80 (1Н, m), 4,48-4,42 (4H, m), 3,23-3,15 (2H, m), 2,95 (2H, d, J=11,09 Гц), 2,74 (6H, s), 2,68-2,60 (2H, m), 2,09-1,98 (2H, m), 1,73 (2H, d, J=12,61 Гц), 1,64-1,55 (2H, m), 1,43 (6H, d, J=6,61 Гц). Пик 1 протона перекрывался с пиком растворителя. ЖХМС: RT=2,90 М+Н+=513.
Пример 250
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 250
трет-Бутиловый эфир 4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты получали из соединения 52 (500 мг, 1,12 ммоль) и иодида цинка трет-бутилового эфира 4-пиперидин-1-карбоновой кислоты (1,68 ммоль) (569 мг, выход 100%). ЖХМС: RT=4,79 мин, М+H+=493.
В раствор of трет-бутилового эфира 4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (569 мг, 1,16 ммоль) в диоксане (15 мл) и метаноле (5 мл) добавляли 4 М раствор HCl в диоксане (15 мл) и реакционную смесь перемешивали при КТ в течение 20 ч и затем концентрировали в вакууме. Полученный остаток растировали со смесью диэтилового эфира и метанола, при этом получали гидрохлорид 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена в виде твердого вещества желто-коричневого цвета (212 мг, выход 43%). ЖХМС: RT=2,34/2,66 мин, M+H+=393.
Смесь гидрохлорида 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (120 мг, 0,28 ммоль) в ДМФ (1,5 мл) обрабатывали карбонатом калия (97 мг, 0,7 ммоль) и бромацетамидом (43 мг, 0,336 ммоль) и перемешивали при КТ в течение 18 ч. Реакционную смесь разбавляли смесью EtOAc/метанол, промывали водой, органический слой сушили (Na2SO4), фильтровали и концентрировали в ваккуме. Полученный твердый остаток перекристаллизовывали из метанола, при этом получали соединение 250 в виде твердого вещества белого цвета (51 мг, выход 41%). 1H ЯМР (ДМСО-d6): δ (ч/млн) 8,27 (1Н, d, J=8,28 Гц), 7,81 (1Н, s), 7,16 (1Н, s), 7,07 (1Н, s), 7,01 (1Н, dd, J=8,36, 1,78 Гц), 6,87 (1Н, d, J=1,72 Гц), 5,81-5,73 (1Н, m), 4,45-4,41 (4H, m), 2,89-2,82 (2H, m), 2,83 (2H, s), 2,20 (3H, s), 2,16-2,07 (2H, m), 1,72-1,66 (4H, m), 1,40 (6H, d, J=6,61 Гц). Пик 1 протона перекрывался с пиком растворителя. ЖХМС: RT=2,97 [М+Н]+=450.
Пример 251
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановая кислота 251
В раствор 4,4,5,5-тетраметил-2-(1Н-пиразол-4-ил)-1,3,2-диоксаборолана (5 г, 0,03 моль) и карбоната цезия (10 г, 0,03 моль) в ДМФ (50 мл) добавляли этиловый эфир 2-бромизобутановой кислоты (4,2 мл, 0,03 моль). Реакционную смесь перемешивали при 110°С в течение ночи. Реакционню смесь охлаждали до КТ, разбавляли Н2О, водный слой экстрагировали EtOAc (2x) и объединенные органические слои промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищанный продукт представлял собой смесь двух изомеров, которые затем разделяли при растирании с гексаном и при этом получали требуемый этиловый эфир 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропановой кислоты. ЖХ/МС (ESI+): m/z 309 (М+Н). 1H ЯМР (400 МГц, CDCl3): δ 7,89 (d, J=4,1, 1H), 7,84 (s, 1H), 4,22-4,08 (m, 2H), 1,85 (d, J=7,6, 6H), 1,36-1,31 (m, 12H), 1,20 (td, J=7,1, 2,8, 3H).
Из 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина 411 и этилового эфира 2-метил-2-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-ил)пропановой кислоты в условиях реакции Сузуки получали этиловый эфир 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановой кислоты. ЖХ/МС (ESI+): m/z 490 (М+Н).
Этиловый эфир 2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропановой кислоты (750 мг, 0,0015 моль) обрабатывали 1 М раствором LiOH/Н2О (1,6 мл) в МеОН (10 мл). Реакционную смесь перемешивали при КТ в течение 2 ч. Смесь подкисляли добалением 10% водным раствором лимонной кислоты до рН=5, 2 раза экстрагировали EtOAc, сушили над MgSO4 и концентрировали в вакууме. Неочищенный остаток очищали препаративной ЖХВР и получали соединение 251. ЖХ/МС (ESI+): m/z 462 (М+Н). 1Н ЯМР (500 МГц, ДМСО): δ 8,44 (s, 1H), 8,36 (d, J=8,4, 1H), 7,97 (s, 1H), 7,86 (s, 1H), 7,44 (dd, J=8,4, 1,7, 1H), 7,35 (d, J=1,7, 1H), 5,82 (dt, J=13,1, 6,6, 1H), 4,52 (s, 3H), 2,25 (s, 2H), 1,78 (s, 4H), 1,45 (t, J=13,9, 4H).
Пример 252
1-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-имидазол-1-ил)-2-метилпропан-2-ол 252
Из 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена и 1-(4-бромимидазол-1-ил)-2-метилпропан-2-ола получали соединение 252. MC(ESI+) 448,3
Пример 253
5-(9-Фтор-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-амин 253
Соединение 89 (0,200 мг, 0,47 ммоль) растворяли в 1,2-дихлорэтане (3 мл, 40 ммоль) и добавляли ТФУ (3 мл, 40 ммоль). Реакцинную смесь перемешивали при КТ в течение 1.5 ч. Завершение реакции подтверждали методом ЖХМС и реакционную смесь концентрировали в вакууме. Неочищенный осадок очищали ЖХВР и получали соединение 253 (18,3 мг, выход 12%).
Пример 254
2-(4-(2-(3-Метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)этанол 254
Из 8-бром-2-(5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена и 1-[2-(тетрагидропиран-2-илокси)этил]-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1Н-пиразол при микроволновом облучении получали соединение 254. MC(ESI+) 378,2.
Пример 255
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-метил-1Н-имидазол-1-ил)этанол 255
Соединение 256 (90 мг, 0,18 ммоль) растворяли в 3 мл метанола и по каплям добавляли смесь HCl-метанол (3 мл, 4 М) при 0°С. Смесь медленно нагревали до КТ и пермешивали при КТ в течение 2 ч, затем концентрировали. Остаток промывали EtOAc и получали 65 мг гидрохлорида соединения 255 с выходом 82%. 1H ЯМР (CDCl3, 400 МГц): δ 8,65-8,61 (m, 2Н), 8,17 (s, 1Н), 8,00 (s, 1Н), 7,52-7,48 (m, 2Н), 5,81-5,78 (m, 1Н), 4,68-4,60 (m, 4Н), 4,31-4,29 (m, 2Н), 3,95-3,92 (m, 2Н), 2,74 (s, 3Н), 1,65 (d, J=1,2 Гц, 6Н). ЖХМС: m/z=404 [M+H+].
Пример 256
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(2-метил-1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 256
4-Иод-2-метил-1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазол получали из of 4-Иод-2-метил-1H-имидазола, Cs2CO3 и 2-(2-бромэтокси)тетрагидропирана в ДМФ. В смесь 4-иод-2-метил-1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-1Н-имидазола (500 мг, 1,9 ммоль) в ДХМ (10 мл) добавляли бромид этилмагния (0,7 мл, 3 М, 2,2 ммоль) при -78°С. Температуру смеси медленно поднимали до 10°С и затем снова снижали. В смесь по каплям добавляли хлорид триметилолова (2,2 мл, 1 М, 2,2 ммоль) при -78°С. Температуру реакционной смеси медленно поднимали до КТ. Реакционную смесь выливали в насыщенный раствор NH4Cl, экстрагировали ДХМ. Органический слой 2 раза промывали водой, сшили над безводным Na2SO4, концентрировали, при этом получали 0,44 г 2-метил-1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-4-(триметилстаннил)-1Н-имидазола (выход 63%). 1Н ЯМР (CDCl3, 400 МГц): δ 7,04 (s, 1Н, ArH), 3,71(s, 2Н), 2,43 (s, 3Н), 1,24-1,20 (m, 6Н), 0,88 (s, 9 Н), 0,29 (s, 6Н), 0,06 (s, 9 Н). ЖХ-МС: m/z=375 [M+H+].
Смесь соединения 194 (300 мг, 0,8 ммоль), Pd(PPh3)4 (93 мг, 0,08 ммоль), 2-метил-1-(2-(тетрагидро-2Н-пиран-2-илокси)этил)-4-(триметилстаннил)-1Н-имидазола (600 мг, 1,6 ммоль) в диоксане (2 мл) продували N2 в течение 2 мин и перемешивали при 120°С в течении 30 мин с использованием микроволнового облучения. Смесь фильтровали, концентрировали, очищали препаративной ТСХ (элюентЕtOАс) и получали 130 мг соединения 256 (выход 32%). 1Н ЯМР (CDCl3, 400 МГц): δ 8,44-8,42 (m, 1Н, ArH), 7,81 (s, 1Н), 7,58 (s, 1Н), 7,48-7,46 (m, 1Н), 7,42 (d, J=1,6 Hz, 1H), 7,19 (s, 1Н), 4,51-4,95 (m, 1Н), 4,45-4,44 (t, 1H), 4,39-4,38 (m, 2H), 4,38-4,37 (m, 2H), 4,07-4,04 (m, 2H), 4,00-4,97 (m, 1H), 3,63-3,54 (m, 2H), 3,40-3,36 (m, 1Н), 2,49 (s, 3H), 1,76-1,60 (m, 4H), 1,48-1,47 (m, 2Н) ЖХ-МС: m/z=504 [М+H+].
Пример 257
5-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиримидин-2-амин 257
Из соединения 48 и 5-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиримидин-2-иламина получали соединение 257. MC(ESI+) 403,2.
Пример 258
5-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)пиридин-2(1Н)-он 258
Из 10-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина и 6-фторпиридин-3-илборной кислоты получали 10-(6-фторпиридин-3-ил)-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,121 г, выход 39%). 1Н ЯМР (500 МГц, ДМСО): δ 8,67 (d, J=2,4, 1H), 8,52 (d, J=2,6, 1H), 8,25 (td, J=8,2, 2,7, 1H), 7,92 (s, 1H), 7,68 (dd, J=8,5, 2,5, 1H), 7,31 (dd, J=8,4, 2,8, 1H), 7,19 (d, J=8,5, 1H), 5,69 (dt, J=13,3, 6,6, 1H), 4,56 (s, 4H), 2,26 (s, 3H), 1,47 (d, J=6,6, 6H). MC (ESI(+)): m/z 405,2 (M+H).
10-(6-Фторпиридин-3-ил)-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин гидролизовали в присутствии HCl и получали соединение 258 (0,028 г, выход 25%). 1H ЯМР (500 МГц, ДМСО) δ 11,76 (s, 1H), 8,50 (d, J=2,4, 1H), 7,90 (s, 1H), 7,78 (dd, J=9,5, 2,8, 1H), 7,64 (s, 1H), 7,52 (dd, J=8,5, 2,5, 1H), 7,10 (d, J=8,5, 1H), 6,47 (d, J=9,5, 1H), 5,70 (dt, J=13,2, 6,5, 1H), 4,52 (q, J=5,8, 4H), 2,26 (s, 3H), 1,48 (d, J=6,6, 6H). MC (ESI(+)): m/z 403,2 (M+H).
Пример 261
N-(Азетидин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-10-амин 261
Раствор соединения 22 (90,0 мг, 0,272 ммоль), трет-бутилового эфира 3-аминоазетидин-1-карбоновой кислоты (46,9 мг, 0,272 ммоль), ацетата палладия (6,11 мг, 0,0272 ммоль), XPhos (13,0 мг, 0,0272 ммоль) и трет-бутоксида натрия (52,3 мг, 0,544 ммоль) в 1,4-диоксане (1,50 мл, 19,2 ммоль) нагревали при микроволновом облучении при 115°С в течение 20 мин. Реакционную смесь фильтровали через слой целита и промывали EtOAc. Фильтрат промывали водой и солевым раствором. Органически слой сушили над Na2SO4, концентрировали и получали неочищенный трет-бутиловый 3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-10-иламино)азетидин-1-карбоновой кислоты, который растворяли в ДХМ (10,0 мл). В раствор добавляли ТФУ (0,419 мл, 5,44 ммоль) и реакционную смесь перемешивали в течение 3 ч. Реакционную смесь концентрировали и очищали обращенно-фазной ЖХВР, при этом получали соединение 261. МС: (ESI+)=367,2. 1Н ЯМР (500 МГц, ДМСО): δ 8,29 (s, 1Н), 8,23 (s, 1H), 7,98 (s, 1H), 7,79 (s, 1H), 5,74 (dt, J=13,1, 6,5 Гц, 2Н), 4,73-4,60 (m, 3H), 4,58-4,51 (m, 2H), 4,43 (dd, J=12,7, 6,9 Гц, 2H), 4,24 (dt, J=15,9, 5,6 Гц, 1H), 2,77 (m, J=34,8, 13,1, 5,3 Гц, 2Н), 1,50 (d, J=6,6, 2,9 Гц, 6Н).
Пример 262
3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-10-иламино)пропан-1,2-диол 262
Раствор соединения 22 (90,0 мг, 0,272 ммоль), (2,2-диметил-1,3-диоксолан-4-ил)метанамина (0,0353 мл, 0,272 ммоль), ацетата палладия (6,11 мг, 0,0272 ммоль), XPhos (13,0 мг, 0,0272 ммоль) и трет-бутоксида натрия (52,3 мг, 0,544 ммоль) в 1,4-диоксане (1,50 мл, 19,2 ммоль) нагревали при микроволновом облучении при 115°С в течение 20 мин. Реакционную смесь фильтровали через слой целита и промывали EtOAc. Фильтрат промывали водой и солевым раствором. Органический слой сушили над Na2SO4, концентрировали и получали промежуточное соединение N-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-10-амин, в кторый добавляли 4,00 М раствор хлористого водорода в диоксане (5,00 мл). Реакционную смесь перемешивали в течение 3 ч. Реакционную смесь концентрировали и очищали ЖХВР, при этом получали соединение 262. 1Н ЯМР (500 МГц, ДМСО): δ 7,99 (s, 1H), 7,92 (s, 1H), 7,85 (s, 1H), 7,44 (s, 1H), 6,36 (s, 1H), 5,86 (dt, J=13,2, 6,6 гц, 1H), 4,78 (d, J=4,8 Гц, 1H), 4,61-4,46 (m, 3H), 4,46-4,33 (m, 2Н), 3,64 (dd, J=11,4, 5,3 Гц, 1H), 3,47-3,34 (m, 3H), 3,21-3,10 (m, 1H), 1,49 (d, J=6,6 Гц, 6Н).
Пример 263
3-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)пиридин-2(1Н)он 263
Аналогично тому, как описано для соединения 203. получали 10-(2-фторпиридин-3-ил)-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин с выходом 0,756 г, 62%. 1Н ЯМР (500 МГц, ДМСО): δ 8,73 (s, 1H), 8,24 (d, J=4,7, 1H), 8,20-8,10 (m, 1H), 7,91 (s, 1H), 7,60 (d, J=9,4, 1H), 7,55-7,45 (m, 1H), 7,20 (d, J=8,5, 1H), 5,70 (dt, J=13,2, 6,6, 1Н), 4,57 (s, 4H), 2,26 (s, 3H), 1,45 (d, J=6,6, 6H). MC (ESI(+)): m/z 405,2 (M+H).
10-(2-Фторпиридин-3-ил)-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин гидролизовали в присутствии HCl и получали соединение 263 (0,412 г, выход 70%). 1Н ЯМР (500 МГц, ДМСО): δ 11,80 (s, 1Н), 8,86 (d, J=2,3, 1Н), 7,88 (s, 1Н), 7,66 (ddd, J=9,0, 7,7, 2,2, 2H), 7,37 (d, J=5,0, 1Н), 7,06 (d, J=8,5, 1Н), 6,31 (t, J=6,6, 1Н), 5,73 (dt, J=13,2, 6,6, 1Н), 4,61-4,47 (m, 4H), 2,25 (s, 3H), 1,45 (d, J=6,6, 6H). MC (ESI(+)): m/z 403,2 (M+H).
Пример 265
1-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-2-метил-1Н-имидазол-1-ил)-2-метилпропан-2-ол 265
1-(2-(трет-Бутилдиметилсилилокси)-2-метилпропил)-4-иодо-2-метил-1Н-имидазол получали из 1-(4-иод-2-метил-1Н-имидазол-1-ил)-2-метилпропан-2-ол, лутидина, трет-бутилдиметилсилилтриметилсульфоната (TBSOTf) в ДХМ.
В смесь 1-(2-(трет-Бутилдиметилсилилокси)-2-метилпропил)-4-иодо-2-метил-1Н-имидазола (1,5 г, 3,8 ммоль) в ДХМ (15 мл) добавляли бромид этилмагния (1,9 мл, 3 M, 5,7 ммоль) при -78°С. Реакционную смесь медленно нагревали до температуры приблизительно 10°С и затем снова охлаждали. По каплям добавляли хлорид триметилолова (6,5 мл, 1 M, 6,5 ммоль) при -78°С. затем температуру медленно поднимали до КТ. Реакционную смесь выливали в насыщенный раствор NH4Cl и экстрагировали ДХМ. Органическую фазу 2 раза промывали водой, сушили над безводным Na2SO4, концентрировали и получали 0,44 г 1-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-2-метил-4-(триметилстаннил)-1Н-имидазол с выходом 63%. 1Н ЯМР (CDCl3, 400 МГц): δ 7,04 (s, 1Н, ArH), 3,74 (s, 2Н), 2,43 (s, 3Н), 1,24-1,20 (m, 6Н), 0,88 (s, 9 Н), 0,29 (s, 6Н), 0,03 (s, 8Н). ЖХ-МС: m/z=375 [М+Н+].
В смесь соединения 194 (300 мг, 0,8 ммоль), Pd(PPh3)4 (93 мг, 0,08 ммоль), 1-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-2-метил-4-(триметилстаннил)-1Н-имидазола (690 мг, 1,6 ммоль) в диоксане (2 мл) продували N2 в течение 2 мин и перемешивали при 120°С в течение 35 мин под действием микроволнового облучения. Смесь фильтровали, концентрировали и и очищали препаративной ТСХ (элюент: EtOAc), при этом получали 160 мг 9-(1-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-2-метил-1Н-имидазол-4-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин с выходом 46%. ЖХ-МС: m/z=548 [М+Н+].
9-(1-(2-(трет-Бутилдиметилсилилокси)-2-метилпропил)-2-метил-1Н-имидазол-4-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (110 мг, 0,196 ммоль) растворяли в 5 мл ТГФ, добавляли фторид тетрабутиламмония (102 мг, 0,392 ммоль) при 0°С. Температуру медленно повышали до КТ и при КТ смесь перемешивали в течение ночи. Смесь концентрировали, остаток распределяли между EtOAc и водой, при этом получали 30 мг соединения 265 с выходом 36%. 1Н ЯМР (CDCl3, 400 МГц): δ 8,48 (d, J=8,0 Гц, 1Н), 7,87 (s, 1Н), 7,64 (s, 1Н), 7,51-7,47 (m, 2Н), 7,26 (s, 1Н), 6,03-5,99 (m, 1Н), 4,51-4,43 (m, 4Н), 3,85 (s, 1Н), 2,47 (s, 3Н), 1,65-1,56 (m, 6Н), 1,28-1,26 (m, 6Н). ЖХ-МС: m/z=433 [M+H+].
Пример 269
3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f] [1,4]оксазепин-10-ил)пиридин-2(1Н)он 269
В раствор 10-(2-фторпиридин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепина (0,087 г, 0,22 ммоль) в 1,2-диметоксиэтане (3,00 мл, 28,9 ммоль) добавляли 10% водный раствор HCl (3 мл). Реакционную смесь перемешивали при 80°С в течение ночи. Реакционную смесь охлаждали до КТ и концентрировали при пониженном давлении, при этом получалисоединение 269, которое анализировали методом обращенно-фазной ЖХВР. МС: (ESI+)=390,2. 1H ЯМР (500 МГц, ДМСО): δ 9,76 (s, 1H), 8,45-8,39 (m, 2Н), 8,04 (s, 1H), 7,93 (s, 1H), 7,47 (dd, J=6,1, 2,1 Гц, 1H), 6,37 (t, J=6,7 Гц, 1H), 5,91 (dt, J=13,3, 6,7 Гц, 2Н), 5,91 (dt, J=13,3, 6,7 Гц, 1H), 4,61 (dd, J=13,1, 5,5 Гц, 4Н), 1,52 (d, J=6,6 Гц, 6Н).
Пример 270
2-(5-(9-Циклопропил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-3-метил-1Н-1,2,4-триазол-1-ил)пропан-1-ол 270
В сосуд, пригодный для микроволнового облучения, помещали раствор 2-[5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-3-метил-[1,2,4]триазол-1-ил]пропан-1-ола (0,140 г, 0,000346 моль) и карбоната калия (0,220 г, 0,00104 моль) в ТГФ (2,0 мл) и воде (2,0 мл). Смесь интенсивно продували N2. В смесь добавляли 2-циклопропил-4,4,5,5-тетраметил-1,3,2-диоксоборолан (0,189 мл, 0,00104 моль) и РВ(PPh3)4 (0,0400 г, 0,0000346 моль) и сосуд немедленно закрывали. Реакционную смесь нагревали с использованием микроволнового облучения при 120°С в течение 20 мин. Реакционную смесь разбавляли ДХМ и фильтровали через слой целита. В смесь добавляли насыщенный раствор NH4Cl и 3 раза экстрагировали ДХМ. Органические слои объединяли, сушили над MgSO4 и концентрировали. Неочищенный остаток очищали обращенно-фазной ЖХВР и получали 35 мг соединения 270 в виде твердого вещества белого цвета. MC(ESI+) 366,2. 1Н ЯМР (500 МГц, ДМСО): δ 8,26 (d, J=8,3 Гц, 1Н), 7,82 (s, 1H), 6,86 (dd, J=8,4, 1,7 Гц, 1Н), 6,75 (d, J=1,7 Гц, 1Н), 5,73-5,57 (m, 1Н), 4,85 (t, J=5,4 Гц, 1Н), 4,50-4,43 (m, 4H), 3,76 (ddd, J=10,7, 7,5, 6,0 Гц, 1Н), 3,68-3,62 (m, 1Н), 2,24 (s, 3H), 1,97-1,85 (m, 1H), 1,39 (d, J=6,7 Гц, 3H), 1,02-0,93 (m, 2H), 0,75-0,67 (m, 2H).
Пример 271
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(2-метил-1Н-имидазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 271
Согласно тому, как описано для соединения 265, из смеси соединения 194 и смеси региоизомеров 2-метил-1-((2-(триметилсилил)этокси)метил)-4-(триметилстаннил)-1H-имидазола и 2-метил-1-((2-(триметилсилил)этокси)метил)-5-(триметилстаннил)-1Н-имидазола получали 9-(1-((2-(трет-бутилдиметилсилил)этокси)метил)-2-метил-1Н-имидазол-4-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин, который растворяли (360 мг, 0,71 ммоль) в этаноле (3 мл). В смесь по каплям добавляли раствор HCl в метаноле (3 мл, 4 M) при 0°С. Через 30 мин смесь нагрвали до 70°С и перемешивали в течение ночи. Смесь концентрировали, остаоок подщелачивали добавлением ТЭА, затем очищали препаративной ТСХ и получали соединение 271 с выходом 91%. 1Н ЯМР (MeOD, 400 МГц): δ 8,72 (s, 1Н), 8,60 (d, J=4,4 Гц, 1H), 8,19 (s, 1Н), 7,87 (s, 1Н), 7,51 (d, J=2,0 Гц, 1Н), 7,46 (s, 1Н), 5,72-5,72 (m, 1Н), 4,65-4,64 (m, 2Н), 4,60-4,59 (m, 2H), 2,68 (s, 3H), 1,63-1,62 (d, J=6,8 Гц, 6Н). ЖХ-МС: m/z=376 [M+H+].
Пример 272
1-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1-метил-1Н-имидазол-2-ил)-2-метилпропан-2-ол 272
В смесь соединения 194 (500 мг, 1,336 ммоль) и 4,4,4',4',5,5,5',5'-оксаметил-2,2'-би(1,3,2-диоксаборолана) (509 мг, 2,004 ммоль) в сухом ДМФ (4 мл) в атмосфере азота добавляли KOAc (393 мг, 4,01 ммоль) и Pd(dppf)Cl2 (50 мг, 0,067 ммоль). Реакционную смесь нагревали при 120°С в течение 20 мин под действием микроволнового облучения. Смесь охлаждали до КТ, концентрировали, неочищенный продукт очищали хроматографией на колонке (элюент: гексан/EtOAc, от 3:1 до приблизительно 1:2) и при этом получали 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин в виде твердого вещества желтого цвета (275 мг, выход 49%). ЖХМС: (ESI, m/z)=422 [М+Н]+.
В смесь 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-9-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина (200 мг, 0,48 ммоль) и 1-(4-бром-1-метил-1Н-имидазо-2-ил)-2-метилпропан-2-ола (166 мг, 0,71 ммоль) в сухом ДМФ (2 мл) в атмосфере азота добавляли CsF (180 мг, 1,19 ммоль), CuI (9 мг, 0,048 ммоль) и Pd(PPh3)4 (27 мг, 0,024 ммоль). Реакционную смесь нагревали при 130°С в течение 40 мин под действием микроволнового облучения. Смесь охлаждали до КТ, выливали в воду и экстрагировали EtOAc. Органические слои сушили над сульфатом натрия, очищали препаративной ЖХВР и получали соединение 272 в виде твердого вещества белого цвета (34,9 мг, выход 16%). 1H ЯМР (ДМСО-d6, 400 МГц): δ 8,51 (s, 1Н), 7,90 (s, 1H), 7,86 (s, 1H), 7,23-7,11 (m, 3H), 5,93-5,89 (m, 1H), 4,54-4,52 (m, 4H), 3,74-3,66 (m, 5H), 1,46 (dd, J=6,4 Гц, 6H), 1,24 (ушир. s, 6H). MC: (ESI, m/z)=448 [M+H]+.
Пример 274
N-трет-Бутил2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 274
В суспензию соединения 66 (300 мг, 0,61 ммоль) в ТГФ (5 мл) добавляли карбонат калия (295 мг, 2,13 ммоль) и N-трет-бутил-2-хлорацетамид (100 мг, 0,67 ммоль). Смесь перемешивали в течение 5 сут, разбавляли ДХМ и промывали водой (2×30 мл). Органическую фазу сушили (MgSO4) и концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром и получали соединение 274 в виде твердого вещества кремового цвета (169 мг, 0,34 ммоль, выход 56%). ЖХМС: RT=3,12 мин, [М+Н]+=492. 1Н ЯМР (DMSO-d6): δ (ч/млн) 8,29 (1Н, d, J=8,29 Гц), 7,86 (1Н, d, J=0,63 Гц), 7,85 (1Н, s), 7,15 (1Н, s), 7,03 (1Н, dd, J=8,36, 1,77 Гц), 6,89 (1Н, d, J=1,71 Гц), 5,90-5,80 (1Н, m), 4,48-4,41 (4Н, m), 2,85 (2Н, d, J=11,24 Гц), 2,81 (2Н, s), 2,20-2,09 (2Н, m), 1,78-1,67 (2Н, m), 1,70-1,58 (2Н, m), 1,44 (6Н, d, J=6,60 Гц), 1,25 (9Н, s). Пик одного протона перекрывается с пиком растворителя.
Пример 275
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[11имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N-метилацетамид 275
Суспензию соединения 48 (1,2 г, 3,09 ммоль), трет-бутилового эфира 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (1,8 г, 5,82 ммоль), комплекса PdCl2dppf c ДХМ (339 мг, 0,46 ммоль) и карбоната калия (1,9 г, 13,9 ммоль) в ДМФ (10 мл) дегазировали и нагревали при 90°С в атмосфере азота в течение 1,5 ч. Охлажденную реакционную смесь разбавляли EtOAc и водой, водные экстракты экстрагировали EtOAc и объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 100% EtOAc в циклогексане, 1% ТЭА в составе конечного элюента), при этом получали трет-бутиловый эфир 4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты в виде пенящегося вещества желто-коричневого цвета (1,5 г, выход 99%). ЖХМС: RT=3,77 мин, [М+Н]+=491.
Раствор трет-бутилового эфира 4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (1,5 г, 3,06 ммоль) в IMS (15 мл) дегазировали и обрабатывали палладием на угле (10% палладий, 50% в воде, 450 мг) и реакционную смесь перемешивали при КТ в атмосфере азота в течение 18 ч, затем при 40°С в течение 8 ч и затем при КТ в течение 18 ч. Затем добавляли палладий на угле (10% палладий, 50% в воде, 450 мг) и перемещивание продолжали при 40°С в течение 8 ч. Затем фильтровали через слой целита и удаляли растворитель удаляли в вакууме. Полученный остаток растворяли в метаноле (5 мл) и раствор обрабатывали 3 М HCl в метаноле (10 мл), перемешивали при КТ в течение 1,5 ч, затем концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром и получали гидрохлорид 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена в виде твердого вещества желтого цвета (1,26 г, выход 79%). 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,03 (2Н, s), 8,37 (1Н, d, J=8,31 гц), 8,17 (1Н, s), 7,06 (1Н, dd, J=8,37, 1,7 Гц), 6,92 (1Н, d, J=1,7 Гц), 5,78 (1Н, m), 4,54 (4Н, d, J=17,19 Гц), 3,35 (2Н, d, J=12,43 гц), 2,98 (2Н, m), 2,87 (1Н, m), 2,38 (2Н, s), 2,08-1,78 (4Н, m), 1,49 (6Н, d, J=6,57 Гц).
В суспензию гидрохлорида 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (150 мг, 0,35 ммоль) добавляли ТЭА (107 мкл, 0,77 ммоль) и 2-хлор-N-метилацетамид (41 мг, 0,38 ммоль). Полученную смесь перемешивали в течение ночи, затем добавляли иодид тетрабутиламмония (13 мг, 0,04 ммоль) и реакционную смесь перемешивали в течение 24 ч. Смесь промывали водой и экстрагировали 10% МеОН в ДХМ (х 5). Объединенные органические слои сушили (MgSO4), концентрировали в вакууме и очищали экспресс-хроматографией на колонке (SiO2, элюент: градиент 0-10% МеОН в ДХМ) 2 раза, при этом получали соединение 275 (23 мг, 0,05 ммоль, выход 14%). ЖХМС: RT=2,63 мин, [М+Н]+=464. 1Н ЯМР (CDCl3): δ (ч/млн) 8,42 (1Н, d, J=8,30 Гц), 7,59 (1Н, s), 7,00 (1Н, dd, J=8,35, 1,83 Гц), 6,88 (1Н, d, J=1,79 Гц), 5,91-5,81 (1Н, m), 4,48-4,44 (2H, m), 4,42-4,36 (2H, m), 3,13-2,91 (4H, m), 2,85 (3H, d, J=4,98 Гц), 2,56-2,48 (1Н, m), 2,39 (3H, s), 2,33 (2H, s), 1,87 (2H, s), 1,78 (2H, s), 1,54 (6H, d, J=6,65 Гц). Пик NH не наблюдали.
В другом варианте в суспензию соединения 66 (300 мг, 0,61 ммоль) в ТГФ (5 мл) добавляли ТЭА (291 мкл, 2,10 ммоль) м 2-бром-N-метилацетамид (102 мг, 0,67 ммоль). Смесь перемешивали в течение 2 ч, затем добавляли карбонат калия (169 мг, 1,22 ммоль) и перемешивали в течение 2 ч. Реакционную смесь разбавляли ДХМ и промывали водой (2×20 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром и получали соединение 275 в виде твердого вещества кремового цвета (156 мг, 0,35 ммоль, выход 57%). ЖХМС: RT=2,62 мин, [М+Н]+=450. 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,29 (1Н, d, J=8,27 Гц), 7,86 (1Н, s), 7,85 (1Н, s), 7,66 (1Н, s), 7,02 (1Н, dd, J=8,33, 1,76 Гц), 6,88 (1Н, d, J=1,71 Гц), 5,89-5,79 (1Н, m), 4,49-4,42 (4H, m), 2,88 (2H, s), 2,84 (2H, d, J=10,90 Гц), 2,59 (3H, d, J=4,73 Гц), 2,13 (2H, s), 1,77-1,67 (4H, m), 1,44 (6H, d, J=6,60 гц). Пик одного протона перекрывался с пиком растворителя.
Пример 276
N-Этил-2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 276
В суспензию гидрохлорида 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (150 мг, 0,35 ммоль) в ДХМ (2 мо) добавляли ТЭА (107 мкл, 0,77 ммоль). Полученную смесь перемешивали в ечение 10 мин, затем добавляли 2-хлор-N-этилацетамид (46 мг, 0,38 ммоль) и иодид тетрабутиламмония (13 мг, 0,04 ммоль). Реакционную смесь перемешивали при КТ в течение 4 сут, затем смесь промывали насыщенным водным раствором гидрокарбоната натрия. Водный слой экстрагировали 10% МеОН в ДХМ (х 3) и объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-10% МеОН в ДХМ), при этом получлаи соединение 276 (91 мг, 0,19 ммоль, выход 55%). ЖХМС: RT=2,75 мин, [M+H]+=478. 1Н ЯМР (CDCl3): δ (ч/млн) 8,43 (1Н, d, J=8,29 Гц), 7,59 (1Н, s), 7,16 (1Н, s), 7,00 (1Н, dd, J=8,34, 1,81 Гц), 6,88 (1Н, d, J=1,76 Гц), 5,91-5,80 (1Н, m), 4,48-4,44 (2H, m), 4,42-4,37 (2H, m), 3,37-3,26 (2H, m), 3,06-2,88 (4H, m), 2,57-2,47 (1Н, m), 2,38 (3H, s), 2,30 (2H, s), 1,87 (2H, d, J=12,74 Гц), 1,76 (2H, s), 1,54 (6H, d, J=6,65 Гц), 1,15 (3H, t, J=7,26 Гц).
Пример 277
N-Изопропил-2-(4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 277
В суспензию гидрохлорида 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (150 мг, 0,35 ммоль) в ДХМ (2 мл) добавляли ТЭА (107 мкл, 0,77 ммоль). Полученную смесь перемешивали в течение 10 мин, затем добавляли 2-хлор-N-изопропилацетамид (52 мг, 0,38 ммоль) и иодид тетрабутиламмония (13 мг, 0,04 ммоль). Реакционную смесь перемешивали при КТ в течение 4 сут, затем промывали насыщенным водным раствором гидрокарбоната натрия. Водную фазу экстрагировали 10% МеОН в ДХМ (х 3) и объединенные органические слои сушили (Na2SO4), затем концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией на колонке (SiO2, элюент: градиент 0-10% МеОН в ДХМ), при этом получали соединение 277 (88 мг, 0,18 ммоль, выход 51%). ЖХМС: RT=2,85 мин, [М+Н]+=492. 1Н ЯМР (CDCl3: δ (ч/млн) 8,43 (1Н, d, J=8,29 Гц), 7,59 (1Н, s), 7,01 (1Н, dd, J=8,34, 1,82 Гц), 6,97 (1Н, s), 6,89 (1Н, d, J=1,76 Гц), 5,92-5,82 (1Н, m), 4,48-4,44 (2H, m), 4,42-4,38 (2H, m), 4,13-4,02 (1Н, m), 3,10-2,85 (4H, m), 2,58-2,46 (1Н, m), 2,38 (3H, s), 2,35-2,20 (2H, m), 1,94-1,84 (2H, s), 1,81-1,68 (2H, m), 1,54 (6H, d, J=6,64 Гц), 1,16 (6H, d, J=6,55 Гц).
Пример 278
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N,N-диметилацетамид 278
В суспензию соединения 66 (300 мг, 0,61 ммоль) в ТГФ (5 млL) добавляли карбонат калия (295 мг, 2,13 моль) и 2-хлор-N,N-диметилацетамид (82 мг, 0,67 ммоль). Реакционную смесь перемешивали в течение 4 ч, разбавляли ДХМ и смесь промывали водой (2×30 мл), сушили (MgSO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией на колонке (SiO2, элюент: градиент 0-5% МеОН в ДХМ). Полученный материал растирали с диэтиловым эфиром и петролейным эфиром, при этом получали соединение 278 в виде твердого вещества белого цвета (85 мг, 0,17 ммоль, выход 29%). ЖХМС: RT=2,72 мин, [M+H]+=464. 1Н ЯМР (DMSO-d6): δ (ч/млн) 8,28 (1Н, d, J=8,29 Гц), 7,86 (1Н, d, J=0,64 Гц), 7,85 (1Н, s), 7,01 (1Н, dd, J=8,35, 1,78 Гц), 6,86 (1Н, d, J=1,73 Гц), 5,89-5,79 (1Н, m), 4,48-4,41 (4Н, m), 3,10 (2Н, s), 3,00 (3Н, s), 2,89 (2Н, d, J=10,79 Гц), 2,77 (3Н, s), 2,11 (2Н, dd, J=12,35, 10.20 Гц), 1,71 (2Н, d, J=12,52 Гц), 1.61 (2Н, td, J=12,15, 3,73 Гц), 1,44 (6Н, d, J=6,60 Гц). Пик одного протона перекрывался с пиком растворителя.
В другом варианте раствор соединения 94 (66 мг, 0,12 ммоль) в ДХМ (1 мл), метанол (1 мл) и ТЭА (0,07 мл) обрабтывали N,N-диметил-2-хлорацетамидом (17 мг, 0,14 ммоль) и TBAI (5 мг) и затем перемешивали при КТ в течении 48 ч и при 30°С в течение 18 ч. Реакционную смесь концентрировали в вакууме и остаток распределяли между ДХМ и водой. Водный слой экстрагировали 3 раза ДХМ, 10% метанолом в ДХМ, объединенные органические экстракты сушили с использованием картриджа для фазового разделения и затем концентрировали в вакууме. Полученный остаток очищали экспрес-хроматографией (SiO2, элюент: градиент от 0 до 10% метанол в ДХМ), при этом получали соединение 278 в виде твердого вещества белого цвета (34 мг, выход 61%). ЖХМС: RT=2,26 мин, [М+Н]+ 465. 1Н ЯМР 400 МГц (ДМСО-d6) δ: 9,43 (1Н, s), 7,96 (1Н, s), 7,92 (1Н, s), 6,91 (1Н, s), 5,91-5,90 (1Н, m), 4,60-4,58 (4Н, m), 3,15 (2Н, s), 3,06 (3Н, s), 2,93 (2Н, d, J=11,06 Гц), 2,82 (3Н, s), 2,63 (1Н, m), 2,15-2,14 (2Н, m), 1,82-1,69 (4Н, m), 1,50 (6Н, d, J=6,60 Гц).
Пример 280
2-(1-Изопропилl-1Н-1,2,4-триазол-5-ил)-4,5-дигидробензо[b][1,2,4]триазоло[1,5-d][1,4]оксазепин 88 (соединение 280 в табл. 1)
В дисперсию амида 87 (0,0485 г, 0,211 ммоль) в толуоле (1,68 мл, 15,8 ммоль) добавляли 1,1-диметокси-N,N-диметилметанамин (0,158 мл, 1,19 ммоль) и смесь пемерешивали в закрытой колбе при 102°С в течение 2 ч (схема 18). Затем реакционную смесь охлаждали и концентрировали досуха, добавляли гидрохлорид изопропилгидразина (0,0396 г, 0,358 ммоль) и уксусную кислоту (0,934 мл, 16,4 ммоль), реакционную смесь закрывали и перемешивали при 102°С в течение ночи. Реакционную смесь концентрировали досуха и смешивали с ДМФ, полученный остаток очищали препаративной ЖХВР (элюент: ацетонитрил/вода), при этом получали триазол 88 с выходом 42%. 1Н ЯМР (500 МГц, CDCl3): δ 8,23 (d, J=8,2 Гц, 1H), 8,02 (s, 1H), 7,30 (dd, J=15,6, 7,8 Гц, 2Н), 7,23 (d, J=7,4 Гц, 1H), 7,20 (d, J=7,9 Гц, 1H), 5,77-5,69 (m, 1H), 4,51 (t, J=5,6 Гц, 2Н), 3,56 (t, J=5,6 Гц, 2Н), 1,60 (d, J=6,6 Гц, 6Н). ЖХМС m/z рассч. для C12H16N6O: 296,13856, найд.: 297,1 [M+1].
Пример 281
10-Фтор-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 281
Соединение 90 (0,2 г, 0,81 ммоль), диметилацетамид-диметилацеталь (0,36 мл, 2,4 ммоль) и толуол (10 мл, 90 ммоль) смешивали в круголодоной колбе, снабженной обратным холодильником. Смесь нагревали при 95°С в течение более 24 ч и концентрировали в вакууме. Остаток растворяли в уксусной кислоте, добавляли гидрохлорид изопропилгидразина (0.11 г, 0,97 ммоль) и нагрвали при 95°С с обратным холодильником в течение 4 ч. Заврешение реакции подтверждали методом ЖХМС. Смесь конценсировали в вакууме и очищали ЖХМС, при этом получали соединение 281 (67,7 мг, выход 26%, M+1 328,1).
Пример 282
9-(1,2-Диметил-1Н-имидазол-4-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 282
В круглодонную колбу объемом 100 мл, содержащую NaH (12 мг, 0,5 ммоль), по каплям добавляли ДМФ (6 мл) и соединение 271 (100 мг, 0,25 ммоль). После перемешивания в течение 1 ч по каплям добавляли иодметан (48 мг, 0,33 ммоль) при 0°С в ТГФ. Смесь медленно нагревали до КТ и перемешивали в течение 2 ч. Реакционную смесь выливали в воду, экстрагировали EtOAc, органическую фазу сушили над безводным Na2SO4, концентрировали, затем очищали препаративной ТСХ (элюент: EtOAc), при этом получали 50 мг (выход 52%) соединения 282. 1H ЯМР (CDCl3, 400 МГц): δ 8,48 (d, J=8,0 Гц, 1Н), 7,87 (s, 1Н), 7,64 (s, 1Н), 7,48-7,44 (m, 2Н), 7,14 (s, 1Н), 6,04-5,98 (m, 1Н), 4,51-4,43 (m, 4Н), 3,62 (s, 3H), 2,45(s, 3Н),1,60 (d, J=6,8 Гц, 6Н), и 15 мг (выход 16%) региоизомера 9-(1,2-диметил-1Н-имидазол-5-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина 291. 1Н ЯМР (CDCl3, 400 МГц): δ 8,57 (d, J=8,0 Гц, 1Н), 7,87 (s, 1Н), 7,87 (s, 1Н), 7,69 (s, 1Н), 7,16 (d, J=6,8 Гц, 1Н), 7,06-7,04 (m, 2H), 6,02-5,96 (m, 1Н), 4,54-4,52 (m, 2Н), 4,48-4,47 (m, 2Н), 3,59 (s, 3H), 2,46(s, 3Н),1,60 (d, J=6,8 Гц, 6Н). ЖХ-МС: m/z=389 [М+Н+].
Пример 285
2-(4-(2-(1,3-Диметил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)этанол 285
Из соединения 51 и диметилацетамида-диметилацеталя при растворении в уксусной кислоте и обработке гидрохлоридом метилгидразина получали 8-бром-2-(2,5-диметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен. MC(ESI+) 360,0/362,0.
В сосуд, пригодный для микроволнового облучения, добавляли 8-бром-2-(2,5-диметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен (0,150 г, 0,000416 моль) и ацетат калия (0,123 г, 0,00125 моль) в ацетонитриле (2,0 мл) и воде (2,0 мл). Раствор интенсивно продували N2. В раствор добавляли 1-[2-(тетрагидропиран-2-илокси)этил]-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-1Н-пиразол (0,148 г, 0,000458 моль) и Pd(PPh3)4 (0,0481 г, 0,0000416 моль) и сосуд немедленно закрывали. Реакционную смесь нагревали с использованием микроволнового облучения при 140°С в течение 20 мин. Смесь распредеяли между насыщенным NH4Cl и ДХМ и 3 раза экстрагировали ДХМ. Органические фазы объединяли, сушили над MgSO4 и концентрировали. Неочищенный продукт растворяли в ДХМ (5,0 мл) и по каплям добавляли раствор хлористого водорода (0,00125 моль, 4 н. в диоксане, 0,31 мл). Реакционную смесь перемешивали при КТ в течение 1 ч. Смесь концентрировали, распределяли между насыщеным раствором бикарбоната натрия и ДХМ и 3 раза экстрагировали ДХМ. Наблюдали переход большей части продукта в осадок в водной фазе. Смесь фильтровали, очищали обращенно-фазной ЖХВР и перекристаллизовывали из смеси EtOH/MeOH, при этом получали 24 мг соединения 285 в виде твердого вещества белого цвета. MCS(ESI+) 392,2. 1H ЯМР (500 МГц, ДМСО): δ 8,40 (d, J=8,4 Hz, 1H), 8,22 (s, 1H), 7,94 (s, 1H), 7,88 (s, 1H), 7,36 (dd, J=8,4, 1,7 Hz, 1H), 7,27 (d, J=1,7 Hz, 1Н), 4,90 (t, J=5,3 Hz, 1H), 4,54-4,48 (m, 4H), 4,21 (s, 3H), 4,16 (t, J=5,7 Hz, 2H), 3,77 (q, J=5,6 Hz, 2H), 2,24 (s, 3H).
Пример 286
2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-9-(1-(2-метоксиэтил)пиперидин-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 286
В раствор соединения 66 (300 мг, 0,61 ммоль) в ДМФ (3,5 мл) добавляли карбонат калия (290 мг, 2,10 ммоль) и 1-бром-2-метоксиэтан (93 мг, 0,67 ммоль). Полученную реакционную смесь перемешивали при 60°С в течение 4 ч, охлаждали до КТ и разбавляли ДХМ. Смесь последовательно промывали насыщенным водным раствором гидрокарбоната натрия, водой и солевым раствором, затем сушили (MgSO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией на колонке (SiO2, элюент: градиент 0-8% МеОН в ДХМ) и растирали с петролейным эфиром, при этом получали соединение 286 в виде твердого вещества белого цвета (127 мг, 0,29 ммоль, выход 48%). ЖХМС: RT=3,02 мин, [М+Н]+=437. 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,28 (1H, d, J=8,28 Гц), 7,86 (1H, d, J=0,62 Гц), 7,85 (1H, s), 7,01 (1H, dd, J=8,36, 1,77 Гц), 6,85 (1H, d, J=1,72 Гц), 5,90-5,80 (1H, m), 4,45 (4H, m), 3,40 (2H, t, J=5,89 Гц), 3,20 (3H, s), 2,93 (2H, d, J=11,11 Гц), 2,01 (2H, dd, J=12,41, 10,21 Гц), 1,70 (2H, d, J=12,52 Гц), 1,58 (2H, ddd, J=24,42, 12,21, 3,61 Гц), 1,44 (6H, d, J=6,60 Гц). Пики 3 проонов перекрываются с пиком растворителя.
Пример 287
2-(4-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-2-метилпропанамид 287
В колбу, содержащую 2-{4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-ил}-2-метилпропионитрил (312 мг, 0,70 ммоль) добавляли концентрированную серную кислоту (3,5 мл). Полученную смесь перемешивали в течение 3,5 ч при КТ, выливали в лед и подщелачивали добавлением карбоната натрия. Водную смесь экстрагировали 10% МеОН в ДХМ (х 5). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией на колонке (SiO2, элюент: градиент 0-10% МеОН в ДХМ), растирали с диэтиловым эфиром (х 3) и получали соединение 287 (196 мг, 0,42 ммоль, выход 60%). ЖХМС: RT=2,67 мин, [М+Н]+=464. 1Н ЯМР (ДМСО-d6): δ (ч/млн) 8,27 (1Н, d, J=8,27 Гц), 7,86 (1H, d, J=0,63 Гц), 7,85 (1H, s), 7,17 (1H, d, J=3,55 Гц), 7,03 (1H, dd, J=8,34, 1,74 Гц), 6,91-6,89 (2H, m), 5,89-5,79 (1H, m), 4,48-4,41 (4H, m), 2,80 (2H, d, J=10,82 Гц), 2,17-2,09 (2H, m), 1,77-1,60 (4H, m), 1,44 (6H, d, J=6,60 Гц), 1,05 (6H, s). Пик 1 протона перекрывался с пиком растворителя.
Пример 288
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)этанол 288
В суспензию гидрохлорида 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (100 мг, 0,23 ммоль) в ДМФ (2 мл) добавляли фосфат натрия (107 мг, 0,75 ммоль), ТЭА (2 капли) и 2-(2-бромэтокси)тетрагидропиран (38 мкл, 0,25 ммоль). Полученную смесь перемешивали в течение ночи при КТ и затем добавляли ТЭА (100 мкл). Реакционную смесь перемешивали при 50°С в течение 5 ч и выдерживали при КТ в течение 2 сут. Реакционную смесь перемешивали при 50°С в течение ночи и затем добавляли 2-(2-бромэтокси)тетрагидропиран (38 мкл, 0,25 ммоль) и иодид калия (10 мг, 0,06 ммоль) и перемешивали в течение ночи при 55°С. Раствор охлаждали и наносили на картридж SCX-2, промывали МеОН (элюент: 2 М NH3 в МеОН). Полученный остаток очищали экспресс-хроматографией на колонке (SiO2, элюент: градиент 0-10% МеОН в ДХМ), при этом получали соединение 288 (31 мг, 0,07 ммоль, выход 31%). ЖХМС: RT=2,59 мин, [М+Н]+ 437. 1Н ЯМР (CDCl3): δ (ч/млн) 8,42 (1Н, d, J=8,30 Гц), 7,58 (1Н, s), 7,00 (1H, dd, J=8,34, 1,83 Гц), 6,88 (1H, d, J=1,78 Гц), 5,93-5,83 (1H, m), 4,46-4,43 (2H, m), 4,41-4,37 (2H, m), 3,70 (2H, t, J=5,19 Гц), 3,17 (2H, d, J=11,24 Гц), 2,68 (2H, t, J=5,17 Гц), 2,62-2,50 (1H, m), 2,38 (3H, s), 2,36-2,27 (2H, m), 2,05-1,85 (4H, m), 1,54 (6H, d, J=6,65 Гц). Пик ОН группы не наблюдали.
Пример 289
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-2-метилпропанамид 289
В колбу, содержащую 2-{4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-ил}-2-метилпропинитрил (140 мг, 0,31 ммоль), добавляли концентрированную серную кислоту (1,75 мл). Смесь перемешивали при КТ в течение 3,5 ч, разбавляли льдом и нейтрализовалидобавлением карбоната натрия. Водную смесь экстрагировали 10% МеОН в ДХМ (х 5), орагническую фазу сушили (MgSO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией на колонке (SiO2, элюент: градиент 0-10% МеОН в ДХМ). Продукт растирали с диэтиловым эфиром и очищали обращенно-фазной ЖХВР (С18, элюент: градиент 20-70% МеОН/0,1% муравьиная кислота в воде/0,1% муравьиная кислота). Полученный остаток помещали в МеОН (1,2 мл) и обрабатывали 0,2 М HCl в диэтиловом эфире. Раствор концентрировали в вакууме и получали соединение 289 (14 мг, 0,03 ммоль, выход 9%). ЖХМС: RT=2,65 мин, [М+Н]+=478. 1Н ЯМР (ДМСО-d6): δ (ч/млн) 9,50 (1Н, m), 8,33 (1Н, d, J=8,29 Гц), 7,97 (1Н, s), 7,93 (1Н, s), 7,86 (1Н, s), 7,02 (1Н, dd, J=8,36, 1,75 Гц), 6,87 (1Н, d, J=1,71 Гц), 5,78-5,71 (1Н, m), 4,50-4,44 (4Н, m), 3,19-3,04 (2Н, m), 2,91-2,80 (1Н, m), 2,25 (3Н, s), 2,12 (2Н, d, J=13,49 Гц), 1,98 (2Н, d, J=13,54 Гц), 1,51 (6Н, s), 1,43 (6Н, d, J=6,59 Гц). Пики 2 протонов перекрывались с пиками растворителя.
Пример 292
1-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-2-метилпропан-2-ол 292
При перемешивании в суспензию трифторацетата 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (300 мг, 0,61 ммоль) в ТГФ (8 мл) добавляли перхлорат лития (65 мг, 0,61 ммоль) и DIPEA (0,21 мл, 1,21 ммоль) и 1,2-эпокси-2-метилпропан (0,54 мл, 0,81 ммоль) и смесь перемешивали при КТ в течение 5 ч, затем добавляли воду (2,5 мл). Смесь перемешивали в течение 18 ч, затем добавляли DIPEA (0,16 мл, 0,92 ммоль) и нагревали при 45°С в течение 5 ч и при КТ в течение 72 ч. Реакционную смесь разбавляли ДХМ, промывали, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, предварительно обрабатывали 25% ТЭА в ДХМ, элюент: градиент от 0 до 5% метанол в ДХМ), лиофилизовали из смеси метанол/вода и растирали с петролейным эфиром, при этом получали соединение 292 в виде твердого вещества кремового цвета (92 мг, выход 34%). ЖХМС: RT=2,76 мин, [М+Н]+=451. 1Н ЯМР 400МГц (ДМСО-d6): δ 8,32 (1Н, d, J=8,28 Гц), 7,90 (2Н, d, J=2,80 Гц), 7,05 (1Н, dd, J=8,35, 1,77 Гц), 6,90 (1Н, d, J=1,70 Гц), 7,27-4,48 (1Н, m), 4,49 (4Н, q, J=5,87 Hz), 4,03 (1Н, s), 3,04 (2Н, d, J=10,80 Hz), 2,46 (1Н, s), 2,23 (4Н, s), 1,70 (4Н, s), 1,48 (6Н, d, J=6,59 Hz), 1.10 (6H, s).
Пример 293
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)этанол 293
При перемешивании в суспензию трифторацетата 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиридин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (300 мг, 0,61 ммоль) в ДМФ (3,5 мл) добавляли карбонат калия (290 мг, 2,1 ммоль) и 2-(2-бромэтокси)тетрагидро-2Н-пиран и смесь нагревали при 60°С в течение 18 ч, затем разбавляли ДХМ. Полученный раствор промывали насыщенным водным раствором гидрокарбоната натрия, водой и солевым раствором, затем сушили (MgSO4), фильтровали и концентрировли в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 от 8% метанол в ДХМ), при этом получали 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-{1-[2-(тетрагидропиран-2-илокси)этил]пиперидин-4-ил}-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен в виде твердого вещества кремового цвета (147 мг, выход 48%). 1H ЯМР 400 МГц (CDCl3): δ 8,44 (1Н, d, J=8,29 Гц), 7,87 (1Н, s), 7,63 (1Н, s), 7,04 (1Н, dd, J=8,33, 1,81 Гц), 6,92 (1Н, d, J=1,75 Гц), 6,01-6,00 (1Н, m), 4,63 (1Н, t, J=3,54 Гц), 4,46-4,45 (4Н, m), 3,92-3,91 (2Н, m), 3,65 (1Н, m), 3,57-3,49 (1Н, m), 3,15 (2Н, m), 2,73 (2Н, m), 2,53 (1Н, m), 2,25 (2Н, m), 1,87 (5Н, m), 1,73-1,69 (3Н, m), 1,60 (8Н, m).
Раствор 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-{1-[2-(тетрагидропиран-2-илокси)этил]пиперидин-4-ил}-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (371 мг, 0,73 ммоль) в метаноле (3,5 мл) обрабатывали 4 н. HCl в диоксане (3,5 мл) и смесь перемешивали в течение 45 мин и затем концентрировали в вакууме. Реакционную смесь помещали в условия, описанные выше, перемешивали при КТ в течение 1 ч и концентрировали в вакууме. Полученный остаток распределяли между ДХМ и насыщенным водным раствором гидрокарбоната натрия, водный слой 2 раза экстрагировали гексаном и объединенные органические экстракты промывали солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, колонку предварительно обрабатывали 1% ТЭА в ДХМ, элюент: градиент от 0 до 7% метанол в ДХМ), при этом получали соединение 293 в виде пенящегося вещества желтого цвета (94 мг, выход 30%). ЖХМС: RT=2,61 мин, [М+Н]+=423. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,32 (1Н, d, J=8,28 Гц), 7,89 (2Н, d, J=1,96 Гц), 7,05 (1Н, dd, J=8,31, 1,76 Гц), 6,89 (1Н, d, J=1,71 Гц), 5,88-5,87 (1Н, m), 4,49 (4Н, q, J=5,91 Гц), 3,53 (2Н, t, J=7,21 Гц), 3,03 (2Н, d, J=11,27 Гц), 2,52 (3Н, m), 2,16 (2Н, t, J=11,38 Гц), 1,76 (2Н, d, J=12,61 Гц), 1,67 (2Н, d, J=12,75 Гц), 1,47 (6Н, d, J=6,60 Гц).
Пример 294
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(тетрагидро-2Н-пиран-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 294
Смесь 3,6-дигидро-2Н-пиран-4-илового эфира трифторметансульфокислоты (125 мг, 0,54 ммоль), 8-(5,5-диметил[1,3,2]диоксаборинан-2-ил)-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (200 мг, 0,491 ммоль), комплекса PdCl2dppf c ДХМ (41 мг, 0,05 ммоль, 10 мол. %), карбоната цезия (400 мг, 1,23 ммоль), DME (2 мл) и воды (0,2 мл) нагревали при 80°С в течение 90 мин. Охлажденную реакционную смесь разбавляли ДХМ, фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматграфией (SiO2, элюент: градиент от 0 до 100% EtOAc в циклогексане), при этом полчали 8-(3,6-дигидро-2Н-пиран-4-ил)-2-(2-изопропил-2Н-[1,2,4]тразол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен (73 мг, выход 39%). ЖХМС: RT=4,36, [М+Н]+=378.
Смесь 8-(3,6-дигидро-2Н-пиран-4-ил)-2-(2-изопропил-2Н-[1,2,4]тразол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (73 мг, 0,19 ммоль), 20% гидроксида палладия на угле (50 мг) и EtOAc (10 mL) дегазировали и перемешивали при КТ в течение 72 ч в атмосфере водорода. Реакционную смесь фильтровали, фильтрат концентрировали в вакууме, остаток растирали с циклогексаном и при этом получали соединение 294 в виде твердого вещества белого цвета (51 мг, выход 71%). ЖХМС: RT=4,30 мин, [М+Н]+=380. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,34 (1Н, d, J=8,29 Гц), 7,91 (2Н, d, J=1,36 Гц), 7,08 (1Н, dd, J=8,34, 1,79 Гц), 6,92 (1Н, d, J=1,73 Гц), 5,90-5,89 (1Н, m), 4,50 (4Н, q, J=5,58 Гц), 3,96-3,95 (2Н, m), 3,44 (2Н, td, J=11,21, 3,01 Гц), 2,77-2,76 (1Н, m), 1,73-1,66 (4Н, m), 1,49 (6Н, d, J=6,60 Гц).
Пример 295
Метиловый эфир 2-(2-этоксифенил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 295
Соединение 40 (80 мг, 1 экв.), 2-этоксифенилборную кислоту (66 мг, 1,75 экв.) и тетракис(трифенилфосфин)палладий (10 мг, 0,05 экв.) в 1,0 М водном растворе карбоната натряи (1,0 мл) и ацетонитриле (1,0 мл) нагревали при 140°С в течение 10 мин в закрытом микроволновом реакторе. Смесь неочищенного продукта концентрировали, очищали обращенно-фазной ЖХВР и получали соединение 295 (9 мг). ESI-MC 365,1 (М)+.
Метиловый эфир 2-(3-изопропилфенил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 296
Соединение 40 (80 мг, 1 экв.), 3-изопропилфенилборную кислоту (65 мг, 1,75 экв.) и тетракис(трифенилфосфин)палладий (10 мг, 0,05 экв.) в 1,0 М водном растворе карбоната натряи (1,0 мл) и ацетонитриле (1,0 мл) нагревали при 140°С в течение 10 мин в закрытом микроволновом реакторе. Смесь неочищенного продукта концентрировали, очищали обращенно-фазной ЖХВР и получали соединение 296 (4 мг). ESI-MC 363,1 (М)+.
Метиловый эфир 2-(2-этилфенил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 297
Соединение 40 (80 мг, 1 экв.), 2-этилфенилборную кислоту (60 мг, 1,75 экв.) и тетракис(трифенилфосфин)палладий (10 мг, 0,05 экв.) в 1,0 М водном растворе карбоната натряи (1,0 мл) и ацетонитриле (1,0 мл) нагревали при 140°С в течение 10 мин в закрытом микроволновом реакторе. Смесь неочищенного продукта концентрировали, очищали обращенно-фазной ЖХВР и получали соединение 297 (11 мг). ESI-MC 349,1 (M)+.
Метиловый эфир 2-(2-изопропилфенил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 298
Соединение 40 (80 мг, 1 экв.), 2-изопропилфенилборную кислоту (65 мг, 1,75 экв.) и тетракис(трифенилфосфин)палладий (10 мг, 0,05 экв.) в 1,0 М водном растворе карбоната натряи (1,0 мл) и ацетонитриле (1,0 мл) нагревали при 140°С в течение 10 мин в закрытом микроволновом реакторе. Смесь неочищенного продукта концентрировали, очищали обращенно-фазной ЖХВР и получали соединение 298 (23 мг). ESI-MC 363,1 (М)+.
Метиловый эфир 2-(3-(трифторметил)фенил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновой кислоты 299
Соединение 40 (80 мг, 1 экв.), 3-(трифторметил)фенилборную кислоту (76 мг, 1,75 экв.) и тетракис(трифенилфосфин)палладий (10 мг, 0,05 экв.) в 1,0 М водном растворе карбоната натряи (1,0 мл) и ацетонитриле (1,0 мл) нагревали при 140°С в течение 10 мин в закрытом микроволновом реакторе. Смесь неочищенного продукта концентрировали, очищали обращенно-фазной ЖХВР и получали соединение 299 (34 мг). ESI-MC 389,1 (М)+.
Пример 300
2-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)ацетамид 300
Смесь 194 мг (0,500 ммоль) 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина 411, 0,436 мл (2,00 ммоль) 1-(трет-бутилдиметилсилилокси)-1-метоксиэтена, 19,6 мг (0,025 ммоль) дихлоробис(три-о-толилфосфин)палладия(II) (19,6 мг, 0,0250 ммоль) и 309 мг (1,00 ммоль) фторида трибутилолова (309 мг, 1,00 ммоль) в 3,0 мл ТГФ дегазировали и затем нагревали в течение 18 ч при 80°С. Смесь фильтровали через слой целита, фильтрат смешивали с 10 мл воды, смесь подкисляли до рН 2 и экстрагировали EtOAc. Органические фазы объединяли, сушили над MgSO4 и концентрировали. Остаток очищали экспресс-хроматографией (элюент: градиент 0-5% метанол в ДХМ), при этом получали 98 мг метилового эфира 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусной кислоты (выход 51%). M/z 382,2. рассч. 381.18.
Смесь 98 мг (0,257 ммоль) метилового эфира 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусной кислоты и 2,0 мл 1,0 М водного гидроксида лития в 6 мл смеси метанол/ТГФ (1:1), смесь перемешивали при 50°С в течение 3 ч. Смесь концентрировали и подкисляли до рН 3 аккуратным добавлением 1 н. водного раствора хлористого водорода. Осадок собирали, сушили в высоком вакууме в течение 18 ч и при этом получали 2-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусную кислоту с выходом 58 mg. M/z 368,2, расч. 367,16.
Смесь 58 мг (0,158 ммоль) of 2-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусной кислоты, 76 мг (0,20 ммоль) гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония, 16 мг (0,30 ммоль) хлорида аммония и 28 мкл (0,200 ммоль) ТЭА в 3 мл N,N-диметилформамида перемешивали в течение 40 мин. Смесь концентрировали в вакууме и растирали с 10 мл воды. Твердый продукт собирали и очищали обращенно-фазной ЖХВР (элюент: градиент ацетонитрила), при этом получали соединение 300 с выходом 8,1 мг. M/z 367,2, расч. 366,18. 1H ЯМР (500 МГц, ДМСО): δ 8,30 (d, J=8,2, 1H), 7,86 (s, 1H), 7,44 (s, 1H), 7,04 (d, J=8,3, 1H), 6,96 (s, 1H), 6,88 (s, 1H), 5,80 (dt, J=13,1, 6,5, 1H), 4,49 (q, J=6,2, 4H), 3,38 (s, 2H), 2,25 (s, 3Н), 1,45 (d, J=6,6, 6H).
Пример 302
1-(4-(2-(1-Изопропил-3-(метоксиметил)-1Н-1,2,4-тразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1Н-пиразол-1-ил)-2-метилпропан-2-ол 302
Стадия 1
В смесь 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (1,92 г, 6,2 ммоль) и трет-бутилового эфира N'-изопропилгидразинкарбоновой кислоты (1,30 г, 7,5 ммоль) в ДМФ (20 мл) при 0°С добавляли DIPEA (2,70 мл, 15,5 ммоль) и HATU (3,54 г, 9,3 ммоль). Реакционную смесь перемешивали в течение 7 ч при КТ, разбавляли ДМФ (40 мл) и перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме и полученный остаток распределяли между ДХМ и водой. Водную фазу экстрагировали ДХМ (х2), объединенные органические фазы промывали последовательно 10% раствором лимонной кислоты, насыщенным раствором бикарбоната натрия. Солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-90% EtOAc в циклогексане), при этом получали трет-бутиловый эфир N'-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбонил)-N'-изопропилгидразинкарбоновой кислоты в виде твердого вещества белого цвета (3,02 г, выход количественный). ЖХМС: RT=4,76 мин, [М+Н]+=465/467. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,62 (1Н, s), 8,40 (1H, d, J=8,59 Гц), 7,69 (1H, s), 7,23-7,22 (2H, m), 4,81 (1H, s), 4,44 (4H, s), 1,32 (9H, s), 1,13 (6H, d, J=6,64 Гц).
Стадия 2
Раствор трет-бутилового эфира N'-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбонил)-N'-изопропилгидразинкарбоновой кислоты (2,71 г, 5,83 ммоль) в метаноле (52 мл) обрабатывали 4 н. HCl в диоксане (5,83 мл, 23,3 ммоль). Реакционную смесь пемершивали при 50°С в течение ночи, концентрировали в вакууме, полученный остаток растирали с диэтиловым эфиром и при этом получали дигидрохлорид N-изопропилгидразина 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты в виде пенящегося вещества светло-желтого цвета (2,69 г, выход количественный). ЖХМС: RT=4,17 мин, [М+Н]+=365/367.
Стадия 3
Суспензию дигидрохлорид N-изопропилгидразина 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (2,69 г, 6,1 ммоль) в ДХМ (61 мл) обрабатывали ТЭА (3,84 мл, 27,6 ммоль). Полученный раствор охлаждали до 0°С, по каплям добавляли метоксиацетилхлорид (1,12 мл, 12,3 ммоль) и реакционную смесь перемешивали при 0°С в течение 1,75 ч. Реакцию останавливали добавлением насыщенного растора бикарбоната натрия и фазы разделяли. Водную фазу экстрагировали ДХМ (х2) и объединенные органические фазы последовательно промывали 10% раствором лимонной кислоты, насыщенным раствором бикарбоната натрия и солевым раствором. Органический раствор сушили (Na2SO4), концентрировали в вакууме и растирали с диэтиловым эфиром, при этом N-изопропил-N'-(2-метоксиацетил)гидразид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты в виде твердого вещества грязно-белого цвета (2,29 г, 5,24 ммоль, выход 86%). ЖХМС: RT=4,29 мин, [М+Н]+=437/439. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,61 (1Н, s), 8,29 (1H, d, J=8,63 Гц), 7,71 (1H, s), 7,27 (1H, dd, J=8,64, 2,06 Гц), 7,21 (1H, d, J=2,06 гц), 4,84 (1H, t, J=6,91 Гц), 4,45 (4H, s), 3,92 (2H, s), 3,33 (3H, s), 1,15 (6H, d, J=6,66 Гц).
Стадия 4
N-Изопропил-N'-(2-метоксиацетил)гидразид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (1,00 г, 2,29 ммоль) суспендировали в оксихлориде фосфора (V) (23 мл) и перемешивали при 100°С в течение 18 ч. Реакционную смесь концентрировали в вакууме и остаток перегоняли с азеотропом с толуолом (х3), при этом получали твердое вещество коричневого цвета. В колбу, содержащую твердое вещество коричневого цвета, добавляли уксусную кислоту (23 мл) и хлорид аммония (1,76 г, 22,9 ммоль), полученную смесь перемешивали при 125°С в течение 2,5 ч, добавляли хлорид аммония (0,88 г, 11,4 ммоль) и смесь перемешивали при 125°С в течение 1 ч, затем концентрировали в вакууме. Полученный остаток обрабатывали водой и экстрагировали ДХМ (х3). Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия, солевыи раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученное твердое вещество растирали с диэтиловым эфиром и получали 8-бром-2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен в виде твердого вещества светло-коричневого цвета (0,73 г, 1,74 ммоль, выход 76%). ЖХМС: RT=4,95 мин, [М+Н]+=418/420. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,29 (1H, d, J=8,65 Гц), 7,93 (1H, s), 7,31 (1H, dd, J=8,66, 2,05 Гц), 7,25 (1H, d, J=2,05 Hz), 5,79-5,78 (1H, m), 4,49 (4H, s), 4,33 (2H, s), 3,27 (3H, s), 1,43 (6H, d, J=6,60 гц).
Стадия 5
Смесь 8-бром-2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (100 мгg, 0,24 ммоль), 2-метил-1-[4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]пропан-2-ола (127 мг, 0,48 ммоль), комплекса PdCl2(dppf) с ДХМ (9,8 мг, 0,012 ммоль), карбоната цезия (234 мг, 0,72 ммоль), DME (1,6 мл), воды (0,27 мл) и IMS (0,5 мл) дегазировали и нагревали при 140°С в течение 20 мин под действием микроволнового облучения. Реакционную смесь распределяли между ДХМ и водой, водный слой экстрагировали ДХМ и объединенные органические экстракты промывали солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали обращено-фазной ЖХВР (колонка С18, элюент: градиент от 5 до 95% метанол в воде + 0,1% НСО2Н), при этом получали соединение 302 в виде твердого вещества белого цвета (40 мг, выход 35%). ЖХМС: RT=3,77 мин, [М+Н]+ 478. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,37 (1Н, d, J=8,38 Гц), 8,17 (1H, s), 7,94 (2H, d, J=7,28 Гц), 7,40 (1H, dd, J=8,37, 1,80 Гц), 7,28 (1H, d, J=1,77 Гц), 5,89-5,88 (1H, m), 4,74 (1H, s), 4,53 (4H, m), 4,38 (2H, s), 4,04 (2H, s), 3,32 (3H, s), 1,49 (6H, d, J=6,60 Гц), 1,10 (6H, s).
Пример 303
(3R,4R)-4-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-3-ол 303
трет-Бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (1,05 г, 2,21 ммоль) частично растворяли в сухом диэтиленгликольдиметиловом эфире (25 мл) и в раствор по каплям добавляли раствор комплекса боран/ТГФ (1 М в ТГФ, 13,2 мл, 13,2 ммоль). После кратковременного перемешивания при КТ смесь выдерживали в течение 16 ч. Смесь охлаждали во льду и воде (2 мл) и по каплям последовательно добавляли 2 М раствор гидроксида натрияе (6,5 мл) и 35% раствор пероксида водорода (1,7 мл, 16,24 ммоль). Смесь нагревали при 50°С в течение 6 ч, охлаждали, разбавляли водой (приблизительно 45 мл) и 3 раза экстрагировали EtOAc. Объединенные органические экстракты сушили (Na2SO4) и концентрировали. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-8.5% метанол в ДХМ), при этом получали указанное в заголовке соединение (0,78 г, выход 71%), содержащее приблизительно 20% изомера 4-гидроксипиперидина. Продукт 2 раза перекристаллизовывали из смеси EtOAc/метанол, при этом получали рацемический транс-изомер трет-бутилового эфира 3-гидрокси-4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты, содержащий менее 5% цис-изомера (0,38 г). ЖХМС: RT=4,44, [М+Н]+=495. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,39 (1Н, d, J=8,25 Гц), 7,89 (1H, s), 7,47 (1H, d, J=2,62 Гц), 7,03 (1H, dd, J=8,29, 1.80 Гц), 6,77 (1H, s), 6,07-5,99 (1H, m), 4,37-4,37 (6H, m), 4,19-4,18 (1H, m), 3,73-3,73 (1H, m), 2,66-2,66 (3H, m), 1,81-1,81 (1H, m), 1,61 (6H, d, J=6,01 Гц), 1,50 (9H, s).
В раствор рацемического транс-изомера трет-бутилового эфира 3-гидрокси-4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (180 мг, 0,36 ммоль) в ДХМ (1 мл) и метаноле (0,6 мл) медленно добавляли 4 М раствор HCl в диоксане (1,6 мл), реакционную смесь перемешивали при КТ в течение 2,5 ч и концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром и получали 4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-3-ол, который делили на энантиомеры и при этом получали (3R,4R) энантиомер соединения 303 в виде твредого вещества белого цвета (172 мг, выход количественный). ЖХМС: RT=2,41 мин, [М+Н]+ 395. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,30 (1Н, ушир. s), 9,10 (1Н, ушир. s), 8,36 (1H, d, J=8,28 Гц), 8,10 (1Н, ушир. s), 8,06 (1H, s), 7,04 (1H, d, J=8,38 Гц), 6,91 (1H, s), 5,87 (1Н, m), 4,53 (4H, d, J=8,12 Гц), 3,90 (1Н, br, m), 3,25 (2H, m), 2,78 (1Н, m), 2,52 (2H, m), 1,80 (2H, m), 1,50 (1H, d, J=6,58 Гц).
Пример 305
2-(5-(9-Бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-ил)ацетамид 305
Согласно тому, как описано в примере 420, проводили реакцию между 8-бром-2-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азуленом, гидрохлоридом 2-карбамидоилацетамида и гидрохлоридом изопропилгидразина. Неочищенный продукт очищали обращено-фазной ЖХВР и получали соединение 305 (29 мг). ЖХМС: 433,0. 1Н ЯМР (500 МГц, ДМСО): δ 8,33 (d, J=8,6 Гц, 1Н), 7,95 (s, 1Н), 7,39 (s, 1H), 7,35 (dd, J=8,7, 2,0 Гц, 1Н), 7,29 (d, J=2,0 Гц, 1Н), 6,97 (s, 1H), 5,79 (dt, J=13,2, 6,6 Гц, 1H), 4,53 (s, 4H), 3,45 (s, 2H), 1,47 (d, J=6,6 Гц, 6Н).
Пример 306
5-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-10-ил)пиридин-2(1Н)-он 306
Соединение 22 (85,0 мг, 0,257 ммоль) растворяли в ацетонитриле (2 мл, 50 ммоль) и воде (2 мл, 100 ммоль), содержащей растворенный ацетат калия (85,5 мг, 0,871 ммоль). Раствор дегазировали пробулькиванием азота в течение 5 мин. Затем добавляли 2-фторпиридин-5-борную кислоту (47,1 мг, 0,334 ммоль) и Pd(PPh3)4 (40 мг, 0,035 ммоль). Реакционную смесь подвергали микроволновому облучению при 145°С в течение 35 мин. Смесь охлаждали до КТ и экстрагировали EtOAc. Объединенные органические экстракты концентрировали и получали промежуточный 10-(6-фторпиридин-3-ил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин, который растворяли в 1,2-диметоксиэтане (3,00 мл, 28,9 ммоль). В раствор добавляли 10% водный раствор HCl (3 мл). Реакционную смесь перемешивали при 80°С в течение ночи. Реакционную смесь охлаждали до КТ, концентрировали в вакууме при пониженном давлении и получали соединение 306, которое анализировали обращено-фазной ЖХВР. МС: (ESI+)=390,1.
Пример 307
4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[4,3-f][1,4]оксазепин-10-ил)пиперидин-2-он 307
Раствор соединения 22 (80,0 мг, 0,242 ммоль), пиперазин-2-она (48,4 мг, 0,484 ммоль), XPhos (23,0 мг, 0,0484 ммольl) и трет-бутоксида натрия (46,5 мг, 0,484 ммоль) нагревали с использованием микроволнового облучения при 125°С в течение 30 мин. Реакционную смесь фильтровали через слой целита и промывали EtOAc. Фильтрат промывали водой и солевым раствором. Органический слой сушили над Na2SO4, концентрировали и получали соединение 307, которое анализировали методом обращенно-фазной ЖХМС. МС: (ESI+)=395,2. 1H ЯМР (500 МГц, ДМСО): δ 8,06 (s, 1H), 8,03 (d, J=5,7 Гц, 2H), 7,93 (s, 1H), 7,59 (s, 1H), 5,72 (dt, J=13,1, 6,6 гц, 1H), 4,62-4,52 (m, 2H), 4,51-4,39 (m, 2H), 3,94 (s, 2H), 3,73-3,62 (m, 2H), 3,37-3,30 (m, 2H), 1,51 (d, J=6,6 Гц, 6Н).
Пример 308
2-(4-(2-(1-Изопропил-3-(метоксиметил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)ацетамид 308
Стадия 1
8-Бром-2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен (250 мг, 0,60 ммоль), пинасоловый эфир 3,6-дигидро-2Н-пиридин-1-N-Вос-4-борной кислоты (370 мг, 1,20 ммоль), карбонат цезия (585 мг, 1,79 ммоль) и дихлорид 1,1'-бис(дифенилфосфино)ферроценпалладия (II) в комплексе с ДХМ (24 мг, 0,03 ммоль) суспендировали в ДМЭ (4,0 мл), IMS (1,3 мл) и воде (0,68 мл) и реакционную смесь продували аргоном. Реакционную смесь в закрытой пробирке нагревали с использованием микроволнового облучения при 140°С в течение 20 мин. Реационную смесь промывали водой, экстрагировали ДХМ (2415 мл) и объединенные органические экстракты промывали солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: 5% метанол в ДХМ), при этом получали трет-бутиловый эфир 4-[2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты в виде пенящегося вещества коричневого цвета (224 мг, выход 72%). ЖХМС: RT=5,08 мин, [М+Н]+=521.
Стадия 2
В раствор трет-бутиловый эфир 4-[2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (224 мг, 0,43 ммоль) в IMS (3 мл) добавляли каталитическое количество палладия на угле (10 мас. %) и реакционную смесь перемешивали в атмосфере водорода при 50°С в течение 16 ч. Реакционную смесь фильтровали и твердый продукт промывали IMS (10 мл). Фильтрат концентрировали в вакууме и полученный остаток очищали экспресс-хроматографией (SiO2, элюент: 2% МеОН в ДХМ), при этом получали трет-бутиловый эфир 4-[2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты в виде маслянистого вещества желтого цвета (162 мг, выход 72%). ЖХМС: RT=5,05 мин, [М+Н]+=523.
Стадия 3
В раствор трет-бутилового эфира 4-[2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (158 мг, 0,30 ммоль) в ДХМ (1,5 мл) добавляли ТФУ (1,5 мл, 20,2 ммоль) и реакционную смесь перемешивали при КТ в течение 30 мин. Реакционную смесь концентрировали в вакууме и остаток разгоняли с азеотропом с эфиром. Полученное маслянистое вещество растирали с диэтиловым эфиром, при этом получлаи трифторацетат 2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена в виде твердого вещества, которое собирали фильтрованием (103 мг, выход 64%). ЖХМС: RT=3,03 мин, [М+Н]+=423. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,56 (1Н, s), 8,33 (1H, d, J=8,33 Гц), 7,90 (1H, s), 7,00 (1H, dd, J=8,38, 1,78 Гц), 6,85 (1H, d, J=1,73 Гц), 5,81-5,80 (1H, m), 4,46 (4H, d, J=2,51 Гц), 4,33 (2H, s), 3,34 (2H, d, J=12,58 Hz), 3,27 (3H, s), 2,98-2,95 (2H, m), 2,82 (1H, t, J=11,91 Гц), 1,93 (2H, d, J=13,66 Гц), 1,75 (2H, t, J=12,95 Гц), 1,44 (6H, d, J=6,60 Гц).
Стадия 4
Суспензию трифторацетат 2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (99 мг, 0,18 ммоль) в ТГФ (1,8 мл) обрабатывали 2-бромацетамидом (109 мг, 0,2 ммоль) и карбонатом калия (56 мг, 0,41 ммоль), реакционную смесь перемешивали при КТ и разбавляли ДХМ и водой. Водный слой экстрагировали ДХМ и объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия, солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток растворяли в метаноле, раствор охлаждали до 0°С и обрабатывали водой, при этом получали осадок, который отфильтровывали, промывали ледяной смесью метанол/воды и получали твердое вещество белого цвета. Твердое вещетво разгоняли с азеотропом с метанолом, затем с диэтиловым эфиром, при этом получали соединение 308 в виде твердого вещества белого цвета (38 мг, выход 43%). ЖХМС: RT=2,66 мин, [М+Н]+=480. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,39 (1H, d, J=8,32 Гц), 8,16 (1H, s), 7,09-7,08 (1H, m), 6,97 (1H, s), 5,76-5,74 (1H, m), 4,58 (4H, d, J=14,42 гц), 4,51 (2H, s), 3,96 (2H, s), 3,60 (2H, d, J=11,71 гц), 3,37 (3H, s), 3,20 (2H, m), 2,86 (1H, m), 2,03 (4H, m), 1,53 (6H, d, J=6.57 Гц).
Пример 309
2-(1-Изопропил-3-(метоксиметил)-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 309
Стадия 1
Смесь гидрохлорида 9-бром-2-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (2,00 г, 5,1 ммоль), 2-гидрохлорида метоксиацетамида (0,76 г, 6,1 ммоль) и ТЭА (5,00 мл, 35,9 ммоль) в ДМФ (38 мл) вакуумировали и заполняли азотом (х3). Реакционную смесь обрабатывали реагентом Xantphos (0,15 г, 0,26 ммоль) и ацетатом палладия (II) (57 мг, 0,26 ммоль), продували газообразным монооксидом углерода и реакционную смесь перемешивали при 60°С в течение 3,5 ч. Реакционную смесь охлаждали до КТ, продували азотом и обрабатывали гидрохлоридом изопропилгидразина (1,70 г, 15,0 ммоль) и уксусной кислотой (19 мл). После пермешивания при 60°С в течение 1,5 ч реакционную смесь разбавляли EtOAc (350 мл). Раствор промывали 1 н. NaOH (2×50 мл), солевым раствором (50 мл), затем сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-3% метанол в ДХМ), затем растирали с диэтиловым эфиром и получали 9-бром-2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен в виде твердого вещества светло-розового цвета (0,85 г, 2,0 ммоль, выход 40%). ЖХМС: RT=4,95 мин, [М+Н]+=418/420. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,44 (1H, d, J=2,57 Гц), 7,94 (1H, s), 7,43 (1H, dd, J=8,74, 2,58 Гц), 6,99 (1H, d, J=8,74 Гц), 5,72-5,63 (1H, m), 4,49 (4H, d, J=3,06 Гц), 4,33 (2H, s), 3,27 (3H, s), 1,44 (6H, d, J=6,60 Гц).
Стадия 2
В суспензию 9-бром-2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (0,85 г, 2,0 ммоль) в IMS (20 мл) добавляли ДХМ (6 мл). Смесь дегазировали продуванием азота и обрабатывали палладием на угле (10% палладий, 50% воды, 350 мг), из сосуда откачивали воздух, заполняли азотом, перемешивали при КТ в течение 18 ч, добавляли катализатор и реакционную смесь перемешивали в течение 72 ч. Реакционную смесь фильтровалиЮ концентрировали в вакууме, при этом получали соединение 309 в виде твердого вещества светло-желтого цвета (0,73 г, выход количественный). ЖХМС: RT=4,55 мин, [М+Н]+=340. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,37 (2Н, dd, J=9,88, 1,75 Гц), 7,30 (1H, ddd, J=7,93, 7,18, 1,70 Гц), 7,12-7,12 (1H, m), 7,03 (1H, dd, J=8,19, 1,22 Гц), 5,83-5,74 (1H, m), 4,48-4,47 (4H, m), 4,36 (2H, s), 3,28 (3H, s), 1,45 (6H, d, J=6,60 Гц).
Пример 313
9-Бром-2-(3-циклопропил-1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 313
Проводили реакцию между 8-бром-2-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азуленом, гидрохлоридом циклопропанкарбоксамидином и гидрохлоридом изопропилгидразина согласно тому, как опснао в примере 420. Неочищенный продукт очищали обращенно-фазной ЖХВР и получали соединение 313 (75 мг). ЖХМС: 414,0. 1H ЯМР (400 МГц, ДМСО): δ 8,30 (m, H), 7,90 (s, 1H), 7,35 (dd, J=8,7, 2,0 Гц, 1Н), 7,29 (d, J=2,0 Гц, 1Н), 5,75 (dt, J=13,2, 6,6 Гц, 1Н), 4,51 (m, 4H), 2,02-1,91 (m, 1Н), 1,45 (d, J=6,6 Гц, 6Н), 0,93-0,85 (m, 2H), 0,84-0,77 (m, 12H).
Пример 314
9-(1-Этилпиперидин-4-ил)-2-(1-изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 314
При перемешивании в суспензию трифторацетата 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (189 мг, 0,37 ммоль) в ДХЭ (3 мл) добавляли уксусную кислоту (3 капли, каталитическое количество) ацетальдегида (0,023 мл, 0,41 ммоль) и триацетоксиборгидрид натрия (94 мг, 0,44 ммоль). После перемешивания в течение 2 ч при КТ добавляли ДХМ, смесь промывали насыщенным водным раствором гидрокарбоната натрия, водой и солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 д 20% метанола в воде), затем лиофилизовали из смеси метанол/вода и растирали с петролейным эфиром, приэтом получали соединение 314 в виде твердого вещества коричневого цвета (18 мг, выход 12%). ЖХМС: RT=2,73 мин, [М+Н]+=407. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,32 (1Н, d, J=8,29 гц), 7,90 (2Н, d, J=2,38 Гц), 7,06 (1Н, d, J=8,41 Гц), 6,90 (1Н, s), 5,90-5,89 (1Н, m), 4,49 (4Н, d, J=7,47 Гц), 2,97 (2Н, d, J=10,98 Гц), 2,34 (2Н, q, J=7,20 Гц), 1,95 (2Н, t, J=11,47 Гц), 1,76 (2Н, d, J=12,54 Гц), 1,72-1,54 (3Н, m), 1,48 (6Н, d, J=6,59 Гц), 1,02 (3Н, t, J=7,20 Гц).
Пример 315
(5-(5,6-Дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-ил)метанол 315
Раствор 8-бром-2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена 309 (280 мг, 0,67 ммоль) в 48% водном растворе HBr (4,19 мл) нагревали при 100°С в течение 4 ч и охлаждали до КТ. Растворнейтрализовали добавлением 1 М водного раствора карбоната натрия и экстрагировали ДХМ, органический слой промывали водой и сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали [5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-ил]метанол в виде твредого вещества кремого цвета (162 мг, выход 60%). ЖХМС: RT=4,74 мин, [М+Н]+=404. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,30 (1Н, d, J=8,65 Гц), 7,91 (1Н, s), 7,33 (1Н, dd, J=8,65, 2,06 гц), 7,27 (1Н, d, J=2,03 Гц), 5,82-5,75 (1Н, m), 5,18 (1Н, t, J=6,03 гц), 4,51 (4Н, s), 4,40 (2Н, d, J=5,99 Гц), 1,44 (6Н, d, J=6,60 Гц).
Раствор [5-(8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-ил]метанола (67 мг, 0,17 ммоль) в IMS дегазировали, обрабатывали Pd/C (10 мас. %, 120 мг) и перемешивали при КТ в атмосфере водорода в течение 18 ч. Реакционную смесь фильтровали через слой целита, промывали ДХМ и фильтрат концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 7% метанола в ДХМ), при этом получали соединение 315 в виде твердого вещества белого цвета (13 мг, выход 24%). ЖХМС: RT=3,79 мин, [М+Н]+=326. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,42 (1Н, dd, J=8,03, 1,74 Гц), 7,91 (1Н, s), 7,33 (1Н, ddd, J=8,16, 7,12, 1,77 Гц), 7,16-7,16 (1Н, m), 7,06 (1Н, dd, J=8,18, 1,21 Гц), 5,86 (1H, t, J=6,61 Гц), 5,21 (1H, t, J=6,03 Гц), 4,52 (4H, q, J=5,81 Гц), 4,42 (2H, d, J=6,00 Гц), 1,48 (6H, d, J=6,61 Гц).
Пример 316
3-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропанамид 316
Смесь 0,194 г (0,500 ммоль) соединения 194, 0,180 мл (2,00 ммоль) метилакрилата, 22,4 мг (0,0999 ммоль) ацетата палладия, 122 мг (0,400 ммоль) триi-о-толилфосфина и 0,278 мл, (2,00 ммоль) ТЭА в 4,0 мл ДМФ нагревали при 100°С в течение 6 ч. Смесь концентрировали в вакууме и распределяли между EtOAc и водой. Органические экстракты промывали разбавленным водным раствоом HCl, водой, солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали на колонке с 4 г силикагеля (Элюент: градиент гептан-EtOAc), при этом получали (Е)-метиловый эфир 3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)акриловой кислоты с выходом 0,11 г. M/z 380,2, рассч. 379,16.
Раствор 0,11 г (0,28 ммоль) (Е)-метилового эфира 3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)акриловой кислоты в 5 мл смеси ТГФ/метанол гидрировали над 100 мг 10% Pd-C в течение 4 ч. Смесь фильтровали через целит, фильтрат концентрировали в вакууме и получали метиловый эфир 3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропановой кислоты с выходом 96 мг. M/z 382,2, рассч. 381,18, который обрабатывали гидроксидом лития и получали 3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропановую кислоту. M/z 368,2, расч. 367,16.
Согласно мому, как описано в примере 300, из метилового эфира 3-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропановой кислоты получали соединение 316. M/z 367,1, расч. 366,18. 1Н ЯМР (400 МГц, ДМСО): δ 8,29 (d, J=8,2, 1H), 7,85 (s, 1H), 7,28 (s, 1H), 7,00 (d, J=8,3, 1H), 6,89 (s, 1H), 6,75 (s, 1H), 5,81 (dt, J=13,1, 6,4, 1H), 4,48 (s, 4H), 2,80 (t, J=7,5, 2H), 2,36 (dd, J=16,8, 9,2, 2H), 2,25 (s, 3H), 1,45 (d, J=6,5, 6H).
Пример 317
9-Хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин (264) 317
Смесь [9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-карбоксамида (0,520 г, 1,96 ммоль) и 1,1-диметокси-N,N-диметилметанамина (1,305 мл, 9,824 ммоль) в толуоле (28,2 мл, 265 ммоль) кипятили с обратным холодильником в течение 1 ч. ЖХМС: основной пик m/z 320,1. После охлаждение промежуточное соединение концентрировали. Смесь промежуточного соединения и гидрохлорида изопропилгидразина (0,4345 г, 3,929 ммоль) в уксусной кислоте (18 мл, 320 ммоль) нагревали при 85°С в течение 3 ч. Смесь охлаждали и нерастворимые примеси отфильтровывали. Надосадочную жидкость концентрировали в вакууме. Остаток разбавляли EtOAc и промывали водой и солевым раствором. Органические слои сушили над Na2SO4, фильтровали и концентрировали, при этом получали соединение 317. МС: (ESI+)=331,0. 1H ЯМР (400 МГц, ДМСО): δ 9,29 (s, 1H), 7,97 (d, J=24,4 Гц, 1H), 7,94 (s, 1H), 7,31-7,19 (m, 1H), 5,85 (dq, J=13,0, 6,4 Гц, 1H), 4,66 (dd, J=5,2, 2,5 Гц, 2Н), 4,63-4,54 (m, 2H), 1,48 (d, J=6,6 Гц, 6Н).
Пример 138
1-(5-(5,6-Дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-ил)-N,N-диметилметанамин 318
Суспенизию (5-(5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-ил)метанола 315 (166 мг, 0,51 ммоль) в ДХМ (5 мл) обрабатывали периодинаном Десса-Мартина (DMP, 238 мг, 0,56 ммоль) и полученный раствор перемешивали при КТ в течение 2 ч в атмосфере азота. Реакцию останавливали добавлением тиосульфата натрия (620 мг в 1 мл воды), добавляли воду и 2 раза экстрагировали ДХМ. Объединенные органические экстракты промывали водой и сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали 5-(4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-карбальдегид в виде твердого вещества кремового цвета (170 мг, выход количественный). ЖХМС: RT=4,30 мин, [М+Н]+ 324. 1Н ЯМР 400 МГц (ДМСО-d6): δ: 9,91 (1Н, s), 8,43 (1Н, dd, J=8,06, 1,78 Гц), 8,11 (1Н, s), 7,34-7,34 (1Н, m), 7,17-7,17 (1Н, m), 7,07 (1H, dd, J=8,17, 1,23 Гц), 6,03 (1H, m), 4,57-4,48 (4H, m), 1,55 (6H, d, J=6,60 Гц).
Смесь 5-(4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-карбальдегида (85 мг, 0,26 ммоль), уксусной кислоты (катлитическое количество, 2 капли), молекулярны сит с диаметром пор  и гидрохлорида диметиламина (24 мг, 0,29 ммоль) в ТГФ (5 мл) перемешивали при КТ в течение 10 мин и затем в смесь добавляли триацетоксиборгидрид натрия (66 мг, 0,31 ммоль). После перемешивания при КТ в течение 18 ч реакционную смесь разбавляли ДХМ, органический слой промывали насыщенным водным раствором бикарбоната натрия, водой и солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали обращено-фазной ЖХВР (колонка С18, элюент: градиент от 5 до 98% метанол в воде + 0.1% НСО2Н), при этом получали соединение 318 в виде твердого вещества белого цвета (13 мг, выход 14%). ЖХМС: RT=3,18 мин, [М+Н]+ 353. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,45 (1H, dd, J=8,02, 1,76 Гц), 7,97 (1H, s), 7,39-7,32 (1H, m), 7,19-7,19 (1H, m), 7,09 (1H, dd, J=8,15, 1,25 Гц), 5,89 (1H, m), 4,54 (4H, d, J=2,46 Гц), 3,47 (2H, s), 2,24 (6H, s), 1,51 (6H, d, J=6,59 Гц).
и гидрохлорида диметиламина (24 мг, 0,29 ммоль) в ТГФ (5 мл) перемешивали при КТ в течение 10 мин и затем в смесь добавляли триацетоксиборгидрид натрия (66 мг, 0,31 ммоль). После перемешивания при КТ в течение 18 ч реакционную смесь разбавляли ДХМ, органический слой промывали насыщенным водным раствором бикарбоната натрия, водой и солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали обращено-фазной ЖХВР (колонка С18, элюент: градиент от 5 до 98% метанол в воде + 0.1% НСО2Н), при этом получали соединение 318 в виде твердого вещества белого цвета (13 мг, выход 14%). ЖХМС: RT=3,18 мин, [М+Н]+ 353. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,45 (1H, dd, J=8,02, 1,76 Гц), 7,97 (1H, s), 7,39-7,32 (1H, m), 7,19-7,19 (1H, m), 7,09 (1H, dd, J=8,15, 1,25 Гц), 5,89 (1H, m), 4,54 (4H, d, J=2,46 Гц), 3,47 (2H, s), 2,24 (6H, s), 1,51 (6H, d, J=6,59 Гц).
Пример 319
Рацемический цис-изомер 2-(3-гидрокси-4-(2-(1-изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N,N-диметилацетамид 319
Стадия 1
Рацемический транс изомер трет-бутиловый эфир 3-гидрокси-4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты, описанный в примере 328 (содержащий приблизительно 24% 4-гидроксипиперидинового изомера, 0,32 г, 0,65 ммоль) растворяли в ДХМ (15 мл) и охлаждали на ледяной бане. По каплям добавляли периодинан Десс-Мартина (0,3 М в ДХМ, 4,33 мл, 1,3 ммоль) и смесь перемешивали при 0-10°С в течение 7 ч и выдерживали в холодильнике в течение 16 ч. Затем добавляли водные растворы бисульфата натрия и бикарбоната натрия (по 10 мл) и смесь перемешивали при КТ в течение 15 мин. Фазы разделяли и водную фазу 2 раза экстрагировали ДХМ. Объединенные органические экстракты промывали солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-5% метанол в ДХМ), растирали с эфиром и получали трет-бутиловый эфир 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3-оксопиперидин-1-карбоновой кислоты (86 мг). После растирания надосадочной жидкости получали дополнительное количество продукта (59 мг). Общий выход составлял 145 мг (45%). ЖХМС: RT=3,53, [М+Н]+=493, [М+Н+МеОН]+=525.
Стадия 2
Раствор трет-бутилового эфира 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазаюензо[е]азулен-8-ил]-3-оксопиперидин-1-карбоновой кислоты (145 мг, 0,29 ммоль) в сухом ТГФ (10 мл) охлаждали до -78°С и по каплям добавляли раствор три-втор-бутилборгидрида лития в ТГФ (L-Selectride®, 1 M, 0,30 мл, 0,30 ммоль). Смесь перемешивали при -78°С в течение 1 ч, затем по каплям добавляли водный раствор бикарбоната натрия. После нагревания до КТ смесь экстрагировали 3 раза EtOAc. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-5% метанол в ДХМ), при этом получали рацемический цис-изомер трет-бутиловый эфир 3-гидрокси-4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (92 мг, выход 64%). ЖХМС: RT=3,39, [М+Н]+=495.
Стадия 3
В раствор рацемического цис-изомера трет-бутилового эфира 3-гидрокси-4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (92 мг, 0,186 ммоль) в ДХМ (0,5 мл) и метанола (0,3 мл) медленно добавляли раствор хлористого водорода в диоксане (4 M, 0,8 мл). ТСмесь перемешивали при КТ в течение 2 ч 20 мин, затем концентрировали в вакууме. Полученный остаток 2 раза растирали с эфиром и сушили в вакууме, при этом получали рацемический цис-изомер гидрохлорид 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-пиперидин-3-ола (86 мг, выход 108%). ЖХМС: RT=1,93, [М+Н]+=395.
Стадия 4
Из рацемического цис-изомера гидрохлорида 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-пиперидин-3-ола (86 мг, 0,19 ммоль) и N,N-диметил-2-хлорацетамида (26 мг, 0,21 ммоль) получали соединение 319 (51 мг, выход 57%). ЖХМС: RT=2,58 мин, [М+Н]+=480. 1Н ЯМР 400 МГц (ДМСО-d6) δ: 8,29 (1Н, d, J=8,29 Гц), 7,90-7,90 (2Н, m), 7,08 (1Н, dd, J=8,37, 1,72 Гц), 6,98 (1Н, d, J=1,65 Гц), 5,89-5,88 (1Н, m), 4,56-4,41 (4Н, m), 4,08 (1Н, ушир.), 3,79 (1Н, ушир.), 3,17 (2Н, m), 3,06 (3Н, s), 2,88 (2Н, m), 2,82 (3Н, s), 2,62 (1Н, m), 2,39 (1Н, m), 2,22 (2Н, m), 1,53 (1Н, m), 1,48 (6Н, d, J=6,61 Гц).
Пример 320
Рацемический транс-изомер 2-(3-гидрокси-4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N,N-диметилацетамид 320
Из транс-рацемического изомера гидрохлорида 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-3-ола (0,15 г, 0,35 ммоль) и N,N-диметил-2-хлорацетамида (46 мг, 0,38 ммоль), при этом получали соединение 320 (135 мг, выход 80%). ЖХМС: RT=2,50 мин, [М+Н]+=480. 1Н ЯМР 400 МГц (CDCl3): δ 8,40 (1Н, d, J=8,26 Гц), 7,86 (1Н, s), 7,43 (1Н, s), 7,08-7,03 (1Н, m), 6,82 (1Н, d, J=1,72 Гц), 6,02 (1Н, m), 4,38 (4Н, m), 4,10 (1Н, ушир. m), 3,38 (2Н, m), 3,31 (2Н, m), 3,09 (3Н, s), 3,00 (1Н, m), 2,98 (3Н, s), 2,47 (2Н, m), 1,86 (2Н, m), 1,60 (6Н, dd, J=6,59, 2,76 Гц).
Пример 321
2-((1R,3r,5S)-3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-8-азабицикло[3.2.1]оксан-8-ил)ацетамид 321
Стадия 1
В сосуд помещали соединение 52 (1,00 г, 2,67 ммоль, бис(неопентилгликолят)дибор (905 мг, 4,01 ммоль), ацетат калия (918 мг, 9,35 ммоль) и диоксан (12 мл), сосуд закрывали и дегазировали продуванием азта в течение 10 мин. В реакционную смесь добавляли комплекс PdCl2dppf c ДХМ (109 мг, 0,13 ммоль, 5 мол. %) и реакционную смесь продували азотом, а затем перемешивали при 90°С в течение 65 ч. Реакционную смесь охлаждали до КТ, разбавляли ДХМ (200 мл) и обрабатывали активированным углем. Смесь фильтровали и фильтрат промывали водой, сушили (Na2SO4) и концентрировали в вакууме, при этом получали 8-(5,5-диметил-[1,3,2]диоксаборинан-2-ил)-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен (1,00 г, 2,46 ммоль, выход 92%). ЖХМС: RT=3,79 мин, [М-НСС(СН3)2СН+Н]+=340.
Стадия 2
В раствор трет-бутилового эфира 3-оксо-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (500 мг, 2,22 ммоль) в ТГФ (5 мл) при -78°С по каплям добавляли 1 М раствор LiHMDS в ТГФ (2,44 мл, 2,44 ммоль). Полученную смесь перемешивали при -78°С в течение 1 ч и по каплям добавляли раствор N-фенилбис(трифторметансульфонимида) (872 мг, 2,44 ммоль) в ТГФ (5 мл). Реакционную смесь перемешивали при -78°С в течение 4 ч и реакцию останавливали добавлением насыщенного раствора бирабоната натрия. Смесь экстрагировали EtOAc, сушили (MgSO4) и концентрировали в вакууме, остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-40% EtOAc в циклогексане). Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-100% ДХМ в циклогексане), при этом получали трет-бутиловый эфир 3-трифторметансульфонилокси-8-азабицикло[3.2.1]окс-2-ен-8-карбоновой кислоты (402 мг, 0,56 ммоль, выход 25%). 1Н ЯМР 400 МГц (CDCl3): δ 6,11 (1H, s), 4,48 (2H, m), 3,02 (1H, m), 2,26 (1H, m), 2,13 (1H, m), 2,09 (1H, m), 2,04 (1H, m), 2,03 (1H, s), 1,48 (9H, s).
Стадия 3
В сосуд помещали 8-(5,5-диметил-[1,3,2]диоксаборинан-2-ил)-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен (237 мг, 0,56 ммоль), комплекс PdCl2dppf c ДХМ (46 мг, 0,06 ммоль, 10 мол. %) и карбонат цезия (456 мг, 1,40 ммоль), из сосуда откачивали воздух и заполняли азотом. В полученную смесь добавляли трет-бутиловый эфир 3-трифторметансульфонилокси-8-азабицикло[3.2.1]окс-2-ен-8-карбоновой кислоты (402 мг, 0,56 ммоль) в DME (2 мл) и воду (0,2 мл). Смесь ваккумировали, заполняли азотом и перемешивали при 110°С в течение 1,5 ч. Реакционную смесь распределяли между EtOAc и водой, оргнаические слои отделяли, сушили (MgSO4) и концентрировали в вакууме. Остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-100% EtOAc в циклогексане), при этом получали трет-бутиловый эфир 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-8-азабицикло[3.2.1]окт-2-ен-карбоновой кислоты в виде бесцветного маслянистого вещества (177 мг, 0,35 ммоль, выход 63%). ЖХМС: RT=4,84 мин, [М+Н]+=503.
Стадия 4
В дегазированный раствор трет-бутилового эфира 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-8-азабицикло[3.2.1]окт-2-ен-карбоновой кислоты (176 мг, 0,35 ммоль) в уксусной кислоте (7 мл) добавляли гидроксид палладия на угле (20% палладия, 50% воды, 62 мг). Сосуд вакуумировали и заполняли газообразным водородом (х3), полученную смесь перемешивали при КТ в течение 100 ч. Реакционную смесь фильтровали через слой целита, промывали EtOAc, фильтрат концентрировали в вакууме, при этом поучали смесь эндо- и экзо-изомеров трет-бутилового эфира 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты в виде смолистого вещества черного цвета (177 мг, 0,35 ммоль, выход количественный). ЖХМС: RT=4,83 мин, [М+Н]+ 505.
Стадия 5
В раствор трет-бутилового эфира 3-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-8-азабицикло[3.2.1]октан-8-карбоновой кислоты (177 мг, 0,35 ммоль) в диоксане (10 мл) добавляли 4М раствор HCl в диоксане (5 мл) и метанол (5 мл). Полученную смесь перемешивали при КТ в течение 18 ч и концентрировали в вакууме. Полученный остаток смешивали с метанолом и наносили на картридж SCX-2 (элюент: метанол, затем 2 М раствор NH3 в метаноле). Основные фракции концентрировали в вакууме, при этом получали смесь изомеров эндо/экзо 8-(8-азабицикло[3.2.1]окт-3-ил)-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена в виде стеклообразного вещества коричневого цвета (91 мг, 0,22 ммоль, выход 64%). ЖХМС: RT=2,77 мин и 2,91 мин, [М+Н]+=405.
Стадия 6
Раствор смеси изомеров эндо/экзо 8-(8-азабицикло[3.2.1]окт-3-ил)-2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (91 мг, 0,22 ммоль) в ДХМ (1 мл) обрабатывали ТЭА (38 мкл, 0,27 ммоль), бромацетамидом (37 мг, 0,27 ммоль) и реакционную смесь перемешивали при КТ в течение 22 ч, затем концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 7% 2 М NH3 (метанол) в ДХМ) и смесь эндо- и экзо-изомеров разделяли обращено-фазной ЖХВР (колонка С6-фенил, элюент: градиент от 5 до 25% ацетонитрил в воде + 0,1% НСО2Н в течение 20 мин), при этом получали указанные в заголовке соединения в виде твердых веществ белого цвета (3 мг изомера с большей скоростью элюирования и 13 мг изомера с меньшей скоростью элюирования). Изомер с большей скоростью элюирования принимали з соединение 321: ЖХМС: RT=2,64 мин, [М+Н]+=462. 1H ЯМР 400 МГц (CDCl3): δ 8,45 (1H, d, J=8,40 Гц), 8,10 (1Н, ушир.), 7,97 (1H, ушир.), 7,89 (1H, s), 7,67 (1H, s), 7,14 (1H, d, J=8,52 Гц), 7,02 (1H, s), 5,99 (1H, m), 5,81 (1H, br), 4,49-4,47 (4H, m), 3,49 (2H, s), 3,19 (3H, m), 2,66-2,56 (2H, m), 2,04-1,96 (4H, m), 1,66 (2H, m), 1,59 (6Н, d, J=6,63 Гц).
Пример 322
2-((1R,3s,5S)-3-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-8-азабицикло[3.2.1]октан-8-ил)ацетамид 322
Согласно тому, как описано в примере 321, изомер с меньшей скоростью элюирования принимали за соединение 322. ЖХМС: RT=2,70 мин, [М+Н]+=462. 1H ЯМР 400 МГц (CDCl3): δ 8,45 (1H, d, J=8,31 Гц), 8,02 (1H, br),7,88 (1H, d, J=0,67 Гц), 7,74 (1H, br), 7,66 (1H, s), 7,06 (1H, dd, J=8,35, 1,82 Гц), 6,93 (1H, d, J=1,77 Гц), 5,99-5,98 (1H, m), 5,75 (1H, ушир.), 4,47-4,46 (4H, m), 3,42 (2H, m), 3,16 (2H, s), 2,92-2,90 (1H, m), 2,10-2,08 (2H, m), 2,00 (2H, t, J=12,82 Гц), 1,82-1,81 (4H, m), 1,59 (6H, d, J=6,63 Гц).
Пример 323
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(4-метилпиперазин-1-ил)-5,6-дигидроимидазо[1,2-d]иридо[3,2-f][1,4]оксазепин 323
Раствор 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепина (66,0 мг, 0,200 ммоль), 1-метилпиперазина, (88,5 мкл, 0,798 ммоль) и ТЭА (167 мкл, 1,20 ммоль) в N,N-диметилацетамиде (3,00 мл, 32,3 ммоль) нагревали с использованием микроволнового облучения при 160°С в течение 20 мин. Реакционную смесь фильтровали через слой целлита и промывали EtOAc. Фильтрат промывали водой и солевым рствором. Органический слой сушили над Na2SO4, концентрировали. Неочищенный продукт очищали обращено-фазной ЖХВР и получали соединение 323. МС: (ESI+)=395,2. 1H ЯМР (400 МГц, ДМСО): δ 8,50 (d, J=8,7 Гц, 1H), 7,88 (s, 1H), 7,80 (s, 1H), 6,72 (d, J=8,8 Гц, 1Н), 5,88 (dt, J=13,2, 6,7 Гц, 1H), 4,49 (m, 4H), 3,61-3,48 (m, 4H), 2,42-2,34 (m, 4H), 2,21 (s, 3H), 1,47 (d, J=6,6 Гц, 6H).
Пример 324
4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)пиперазин-2-он 324
Согласно тому, как описано в примере 323, из 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепина и пиперазин-2-она получали соединение 324. МС: (ESI+)=395,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,55 (d, J=8,7 Гц, 1H), 8,10 (s, 1H), 7,88 (s, 1H), 7,81 (s, 1H), 6,70 (d, J=8,8 Гц, 1H), 5,88 (dt, J=13,2, 6,6 Гц, 1H), 4,60-4,40 (m, 4H), 4,06 (s, J=8,0 Гц, 2Н), 3,84-3,68 (m, 2H), 1,47 (d, J=6,6 Гц, 6Н).
Пример 325
4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f|[1,4]оксазепин-9-ил)пиперазин-2-он 325
Раствор 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина 264 (55,0 мг, 0,166 ммоль), пиперазин-2-она (0,110 г, 1,10 ммоль) и ТЭА (0,275 мл, 1,97 ммоль) в N-метилпирролидоне (3,00 мл, 31,1 ммоль) нагревали при 150°С в течение 2 сут. Реакционную смесь фильтровали через слой целлита и промывали EtOAc. Филььтрат промывали водой и солевым раствором. Органический слой сушили над Na2SO4, концентрировали и получали соединение 325, которое анализировали методом обращено-фазной ЖХВР. МС: (ESI+)=395,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,55 (d, J=8,7 Гц, 1Н), 8,10 (s, 1H), 7,88 (s, 1H), 7,81 (s, 1H), 6,70 (d, J=8,8 Гц, 1H), 5,88 (dt, J=13,2, 6,6 Гц, 1H), 4,63-4,30 (m, 4H), 4,06 (d, J=8,0 Гц, 2H), 3,81-3,63 (m, 2H), 1,47 (d, J=6,6 Гц, 6Н).
Пример 327
4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-ил)пиридин-2(1Н)он 327
Согласно тому, как описано для получения соединения 203, но с использованием вместо 2-фторпиридин-3-илборной кислоты 2-фторпиридин-4-илборной кислоты получали 10-(2-фторпиридин-4-ил)-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин (0,242 г, выход 24%, МС (ESI(+)): m/z 404,9 (М+Н), который обрабатывали 10% водным раствором HCl и получали соединение 327 (0,141 г, выход 59%). 1Н ЯМР (400 МГц, ДМСО): δ 11,53 (s, 1H), 8,72 (d, J=2,2, 1H), 7,93 (s, 1H), 7,67 (dd, J=8,5, 2,3, 1H), 7,49 (d, J=6,9, 1H), 7,15 (d, J=8,6, 1H), 6,55 (s, 1H), 6,48 (d, J=6,2, 1H), 5,70 (dt, J=13,2, 6,7, 1H), 4,56 (s, 4H), 2,26 (s, 3H), 1,49 (d, J=6,6, 6Н). МС (ESI(+)): m/z 403,1 (М+Н).
Пример 328
(3R,4S)-4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-3-ол 328
Стадия 1
трет-Бутиловый эфир 4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]-3,б-дигидро-2Н-пиридин-1-карбоновой кислоты (0,60 г, 1,22 ммоль) частично растворяли в сухом диэтиленгликольдиметиловом эфире (14 мл) и по каплям добавляли раствор комплекса боран/ТГФ (1 М в ТГФ, 7,29 мл, 7,29 ммоль). После кратковременного перемешивания при КТ смесь выдерживали в течение 16 ч. Смесь охлаждали во льду и воде (1,1 мл) и последовательно по каплям добавляли 2 М растворе гидроксида натрия (3,6 мл) и 35% растворе пероксида водорода (0,94 мл, 8,98 ммоль). Смесь перемишивали при 50°С в течение 8 ч, охлаждали, разбавляли водой (приблизительно 30 мл) и экстрагировали EtOAc. Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экпрессс-хроматографией (SiO2, элюент: градиент 0-10% метанол в ДХМ), при этом получали рацемический транс-изомер трет-бутиловый эфир 3-гидрокси-4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (0,46 г, выход 74%), содержащий приблизительно 20% изомера 4-гидроксипиперидина. ЖХМС: RT=4.48, [М+Н]+=509.
Стадия 2
Рацемический транс-изомер трет-бутиловый эфир 3-гидрокси-4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (0,215 г, 0,42 ммоль) суспендировали в ТГФ (10 мл) и добавляли трифенилфосфин (0,22 г, 0,85 ммоль) и хлоруксусную кислоту (82 мг, 0,85 ммоль). По каплям добавляли диэтиловый эфир азодикарбоновой кислоты (0,133 мл, 0,85 ммоль) и смесь перемешивали при КТ в течение 24 ч. Смесь концентрировали и полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-10% метанол в ДХМ). Полученный неочищенный материал растворяли в сухом ДХМ (5 мл) и добавляли трифенилфосфин, хлоруксусную кислоту и диэтиловый эфир азодикарбоновой кислоты (количества указаны выше). Смесь перемешивали при КТ в течение 16 ч, концентрировали в вакууме, неочищенный продукт растирали с эфиром и маточный раствор концентрировали. Полученный остаток очищали экспресс-хромтаографией (SiO2, элюент: градиент 0-5% метанол в ДХМ), при этом получали неочищенный рацемический цис-изомер трет-бутиловый эфир 3-(2-хлорацетокси)-4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (0,297 г), который использовали на следующей стадии без дополнительной очистки. ЖХМС: RT=4,61, [М+Н]+=585/587.
Стадия 3
Раствор неочищенного рацемического цис-изомера трет-бутилового эфира 3-(2-хлорацетокси)-4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (0,297 г) в диоксане (5 мл) перемешивали с водным раствором гидроксида натрия (1 М, 4,2 мл) при КТ в течение 16 ч, нагревали при 50°С в течение приблизительно 24 ч. Охлажденную смесь 3 раза эстрагировали EtOAc, объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент 0-5% метанол в ДХМ), при этом получали рацемический цис-изомер трет-бутилового эфира 3-гидрокси-4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (17 мг). ЖХМС: RT=4,50, [М+Н]+ 509.
В раствор рацемического цис-изомера трет-бутилового эфира 3-гидрокси-4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-карбоновой кислоты (17 мгg, 0,36 ммоль) в ДХМ (1 мл) и метанола (0,6 мл) медленно добавляли 4 М раствор HCl в диоксане (1,5 мл) и реакционную смесь перемешивали при КТ в течение 2,5 ч, затем концентрировали в вакууме. Полученный остаток растирали с диэтиловым эфиром, при этом получали рацемический цис-изомер 4-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-3-ол в виде твердого вещества белого цвета, который представлял собой (3R,4S) энантиомер 328 (10 мг, выход 67%). МС: RT 2,47, [М+Н]+=409. 1Н ЯМР (ДМСО-d6): δ 8,92 (1Н, ушир. d), 8,33 (1H, d, J=8,30 Гц), 7,97 (1H, s), 7,08 (1H, d, J=8,36 Гц), 6,96 (1H, d, J=1,66 гц), 5,79 (1H, t, J=6,59 Гц), 4,50 (4H, d, J=7,99 Гц), 4,06 (1H, s), 3,27 (2H, m), 3,17 (2H, m), 2,97 (2H, d, J=14,58 Гц), 2,32-2,27 (1Н, m), 2,29 (3H, s), 1,74 (1H, d, J=13,43 Гц), 1,46 (6H, d, J=6,59 Гц).
Пример 329
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N,N-диметилацетамид 329
Суспензию гидрохлорида 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (310 мг, 0,72 ммоль) в ДХМ (6 мл) и ТЭА (0,3 мл, 2,16 ммоль) озвучавали и перемешивали, затем добавляли N,N-диметл-2-хлорацетамид (98 мг, 0,8 ммоль) и TBAI (28 мг, 0,072 ммоль) и полученную смесь перемешивали пр КТ в течение 72 ч, затем концентрировли в вакууме. Полученный остаток распределяли между водой и ДХМ, и водный слой 5 раз экстрагировали ДХМ, объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% метанол в ДХМ), затем растирали с диэтиловым эфиром, при этом получали соединение 329 в иде твердого вещества белого цвета (129 мг, выход 38%). ЖХМС: RT=2,71 мин, [М+Н]+=478. 1Н ЯМР 400 МГц (CDCl3): δ 8,44 (1H, d, J=8,30 Гц), 7,61 (1H, s), 7,04 (1H, d, J=8,43 гц), 6,91 (1H, s), 5,91 (1H, t, J=6,63 Гц), 4,45 (4H, d, J=14,92 Гц), 3,39 (2H, m), 3,11 (2H, m), 3,10 (3H, s), 2,99 (1H, m), 2,98 (3H, s), 2,54 (2H, m), 2,41 (3H, s), 1,90 (4H, s), 1,57 (6H, d, J=6,65 Гц).
Пример 330
2-(3-Гидрокси-4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-N,2-диметилпропанамид 330
Смесь рацемического тран-изомера гидрохлорида 4-[2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-3-ола (199 мг, 0,46 ммоль), 2-бром-2-метил-N-метилпропионамида (83 мг, 0,46 ммоль), NaOH (2 мл, 50% водный раствор), ТВАВ (16 мг, 0,05 ммоль) и ДХМ (2.5 mL) интенсивно перемешивали при КТ в течение 7,5 ч. Фазы разделялии водный слой 3 раза экстрагировали 10% метанолом в ДХМ, объединенные органические экстракты промывали солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 от 10% метанол в ДХМ), при этом получали соединение 330 (84 мг, выход 37%). ЖХМС: RT=2,54 мин, [М+Н]+=494. 1Н ЯМР 400 МГц (CDCl3): δ 8,44 (1H, d, J=8,23 Гц), 7,87 (1H, s), 7,48 (1H, s), 7,17 (1H, ушир.), 7,08 (1H, m), 6,86 (1H, s), 6,03-6,01 (1H, m), 4,77-4,07 (4H, m), 3,82 (1H, ушир.), 3,17 (1H, m), 2,86 (3H, d, J=4,98 Гц), 2,85 (2Н, m), 2,46 (1H, m), 2,27 (1H, m), 2,18 (1H, m), 1,90 (1H, m), 1,70 (1H, m), 1,61 (6H, m), 1,27 (6H, d, J=10,69 Гц).
Пример 331
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперазин-1-ил)-N,N-диметлацетамид 331
Раствор 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина 264 (80,0 мг, 0,242 ммоль), этилового эфира 2-(пиперазин-1-ил)уксусной кислоты (0,275 г, 1,60 ммоль) и ТЭА (0,400 мл, 2,87 ммоль) в N-метилпирролидиноне (4,36 мл, 45,2 ммоль) нагревали при 150°С в течение 2 сут. Реакционную смесь фильтровали через слой целита и промывали EtOAc. Фильтрат промывали водой и солевым раствором. Органический слой сушили над Na2SO4, концентрировали. Враствор неочищенного промежуточного этилового эфира в ТГФ (3,00 мл, 37,0 ммоль) и воде (3,00 мл, 166 ммоль) добавляли гидрат гидроксида лития (0,0406 г, 0,967 ммоль). Реакционную смесь перемешивали при КТ. Реакцию останавливали добавлением воды и промывали EtOAc. Водный слой концентрировали и получали 2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-а]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперазин-1-ил)уксусную кислоту, которую использовали на следующей стадии. МС: (ESI+)=439,2.
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперазин-1-ил)уксусную кислоту (0,050 г, 0,00011 моль) растворяли в ДМФ (1,79 мл, 0,0231 моль) и обрабатывали последовательно N,N-диизопропилэтиламином (0,119 мл, 0,000686 моль), гидрохлоридом диметиламина (0,0373 г, 0,000457 моль) и гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,0521 г, 0,000137 моль). Смесь перемешивали при КТ в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия и экстрагировали EtOAc. Сухие органические слои сушили над сульфатом натрия, концентрировали и получали соединение 331, которое анализировали обращенно-фазной ЖХВР. МС: (ESI+)=466,2. 1Н ЯМР (400 МГц, ДМСО): δ 9,10 (s, 1Н), 7,89 (s, 1H), 7,84 (s, 1H), 6,34 (s, 1H), 6,01-5,79 (m, 1H), 4,50 (d, J=10,0 Гц, 4H), 3,53 (s, 4H), 3,18 (s, 2H), 3,03 (s, 3Н), 2,82 (s, 3Н), 1,47 (d, J=6,5 Гц, 6Н).
Пример 232
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперазин-1-ил)ацетамид 332
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперазин-1-ил)уксусную кислоту (0,050 г, 0,00011 моль) растворяли в ДМФ (1,79 мл, 0,0231 моль) и обрабатывали последовательно N,N-диизопропилэтиламином (0,119 мл, 0,000686 моль), хлоридом аммония (0,0244 г, 0,000457 моль) и гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,0521 г, 0,000137 моль). Смесь перемешивали при КТ в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия и экстрагировали EtOAc. Сухие органические слои сушили над сульфатом натрия, концентрировали и получали соединение 332, которое анализировали обращенно-фазной ЖХВР. МС: (ESI+)=466,2. 1Н ЯМР (400 МГц, ДМСО): δ 9,11 (s, 1Н), 7,89 (s, 1H), 7,85 (s, 1H), 7,20 (s, 2H). 6,35 (s, 1H), 5.93 (dt, J=13,7, 7,0 Гц, 1H), 4,50 (d, J=10,4 Гц, 4Н), 3,57 (s, 5H), 2,91 (s, 2H), 1,47 (d, J=6,5 Гц, 6Н).
Пример 333
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9(8Н)-он 333
Раствор 9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-2-карбоксамида (55,0 мг, 0,166 ммоль) в серной кислоте (0,70 мл, 13 ммоль) и воде (0,70 мл, 39 ммоль) нагревали при 125°С в течение 2 ч. Реакционную смесь разбавляли водой, нейтрализовали 1 М NaOH и экстрагировали EtOAc. Органический слой сушили над Na2SO4, концентрировали, очищали обращенно-фазной ЖХВР и получали соединение 333. МС: (ESI+)=313,0.
Пример 334
2-(4-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)пиперазин-1-ил)-N-метилацетамид 334
Раствор 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепина (150,0 мг, 0,4535 ммоль), этилового эфира 2-(пиперазин-1-ил)уксусной кислоты (0,275 г, 1,60 ммоль) и ТЭА (0,400 мл, 2,87 ммоль) в N-метилпирролидиное (4,36 мл, 45,2 ммоль) нагревали при 150°С в течение 2 сут. Реакционную смесь разбавляли EtOAc, промывали водой и солевым раствором. Органический слой сушили над Na2SO4, концентрировали и получали промежуточный этиловый эфир, который растворяли в ТГФ (8,00 мл, 98,6 ммоль) и воде (8,00 мл, 444 ммоль). В раствор добавляли гидрат гидроксида лития (0,07612 г, 1,814 ммоль). Смесь перемешивали при КТ, реакцию останавливали добавлением воды и промывали EtOAc. Водный слой концентрировали и получали 2-(4-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)пиперазин-1-ил)уксусную кислоту. MS: (ESI+)=439.4
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)пиперазин-1-ил)уксусную кислоту (0,050 г, 0,00011 моль) растворяли в ДМФ (1,79 мл, 0,0231 моль) и обрабатывали последовательно N,N-диизопропилэтиламином (0,119 мл, 0,000686 моль), 2,00 М раствором метиламина в ТГФ (0,119 мл, 0,000686 моль, 0,228 мл) и гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,0521 г, 0,000137 моль). Смесь перемешивали при КТ в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия и экстрагировали EtOAc. Сухие органические слои сушили над сульфатом натрия, концентрировали и получали соединение 334, которое анализировали обращенно-фазной ЖХВР. МС: (ESI+)=452,2.
Пример 335
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)пиперазин-1-ил)-N,N-диметилацетамид 335
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперазин-1-ил)уксусную кислоту (0,050 г, 0,00011 моль), описанную в примере 334, растворяли в ДМФ (1,79 мл, 0,0231 моль) и обрабатывали последовательно N,N-диизопропилэтиламином (0,119 мл, 0,000686 моль), гидрохлоридом диметиламина (0,0373 г, 0,000457 моль) и гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,0521 г, 0,000137 моль). Смесь перемешивали при КТ в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия и экстрагировали EtOAc. Сухие органические слои сушили над сульфатом натрия, концентрировали и получали соединение 335, которое анализировали обращенно-фазной ЖХВР. МС: (ESI+)=466,3.
Пример 337
1-((2-(1-Изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)метил)мочевина) 337
Соединение 40 (0,6 г, 2,0 ммоль), полученное, как описано в примере 40, 1-изопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (0,477 ммоль, 2,02 ммоль) и хлорид бис(трифенилфосфин)палладия(II) (0,059 г, 0,084 ммоль) смесшивали в сосуде для микроволного облучения объемом 35 мл. В сосуд последовательно добавляли карбонат калия (1,0 М водный раствор, 5 мл) и ацетонитрил (5 мл). Реакционный сосуд подвергали микроволновому облучению при 140°С в течение 20 мин. Смесь разбавляли EtOAc и продукт выделяли кислотно-основной экстракцией, при этом получали 0,3 г (выход 50%) 2-(1-изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновую кислоту.
Изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоновую кислоту (0,3 г, 0,9 ммоль) растворяли в ТГФ (3 мл) и в раствор добавляли хлорид аммония (0,19 г, 3,6 ммоль), N,N-диизопропилэтиламин (0,31 мл, 1,8 ммоль) и HATU (0,37 г, 0,98 ммоль) и полученную смесь перемешивали при КТ в течение 2 ч. Завершение реакции наблюдали методом ЖХМС. Реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия и 2 раза экстрагрировали EtOAc. Объединенные оганические слои промывали солевым раствором и сушили над Na2SO4. Жидкую фазу фильтровали и концентрировали досуха. Полученный неочищенный продукт использовали на следующей стадии без дополнительной очистки. В результате получали 0,3 г (выход количественный) 2-(1-изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбоксамид 346. МС (ESI+) m/z 338,1 (М+Н+), рассч. 338,4.
Тетрагидроалюминат лития (0,047 г, 1,3 ммоль) суспендировали в ТГФ (8 мл) и охлаждали до 0°С. В суспензию добавляли раствор соединения 346 (0,3 г, 0,9 ммоль) в ТГФ (2 мл) и реакционную смесь перемешивали на холоде в течение 10 мин. Колбу постепенно нагревали до КТ и перемешивали в течение 16 ч. Реакцию останавливали путем выливания смеси в насыщенный раствор соли Рошеля в диэтиловом эфире (1:1). Эмульсию перемешивали очень интенсивно до фазового разделения (приблизительно в течение 2 ч). Фазы разделяли и водный слой много раз экстрагировали EtOAc. Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали, при этом получали 0,15 г (0,46 ммоль) (2-(1-изопропил-1Н-пиразол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)метанамин 347 (МС (ESI+) m/z 323,1 (М+Н+), рассч. 323,4), который растворяли в ледяной уксусной кислоте (0,8 млL) и воде (5 мл). В раствор по каплям добавляли раствор цианат калия (0,114 г, 1,41 ммоль) в воде. Для облегчения растворения добавляли ДМФ (3 мл). Полученную реакционную смесь перемешивали при КТ в течение 12 ч. Затем реакционную смесь нагревали при 50°С в течение 3 ч. Смесь охлаждали до КТ, фильтровали и получали 0,02 г (выход 10%) соединения 337. МС (ESI+) m/z 367,1 (М+Н+), рассч. 367,4. 1Н ЯМР (400 МГц, ДМСО): δ 8,53 (s, 5H), 8,33 (d, J=8,2 Гц, 1H), 7,61 (d, J=3,7 Гц, 1H), 7,43 (s, 1H), 7,01 (d, J=8,7 Гц, 1H), 6,91 (s, 1H), 6,63 (s, 1H), 6,38 (s, 1H), 5,62 (s, 2H), 5,42 (s, 7H), 4,47 (s, 3H), 4,17 (d, J=5,8 Гц, 2H), 1,44 (d, J=6,4 Гц, 6Н).
Пример 338
(2S)-1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 338
Согласно тому, как описано в примере 339, соединение 93 и L-пролинамид вводили в реакцию и неочищенный продукт очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 8% метанол в ДХМ), затем перекристаллизовывали из метанола и получали соединение 338 в виде твердого вещества белого цвета (115 мг, выход 44%). ЖХМС: RT=2,48 мин, [М+Н]+ 409. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,06 (1Н, s), 7,88 (1H, s), 7,83 (1H, s), 7,33 (1H, ушир.), 6,92 (1Н, ушир.), 5,97-5,96 (1H, m), 5,94 (1H, s), 4,53-4,45 (4H, m), 4,30 (1H, d, J=8,51 Гц), 3,59 (1H, s), 3,37 (1H, d, J=9,93 Гц), 2,18 (1H, m), 1,95 (3H, m), 1,47 (6H, dd, J=6,59, 3,43 Гц).
Пример 339
1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперидин-4-карбоксамид 339
Соединение 93 обрабатывали гидридом натрия (1,2 экв.) и реакционную смесь перемешивали при КТ или при 40°С в течение интервалаот 15 мин до 1,25 ч, затем добавляли бензилбис(трифторметан)сульфонамид (1,2 экв.). Перемешивание продолжали при КТ до полного исчерпания пиридона (по данным ТСХ или ЖХМС), затем добавляли амид пиперидин-4-карбоновой кислоты (от 1 до 2,5 экв.) и реакционную смеь нагревали при температуре от 70 до 100°С до полного завершения реакции. Неочищенные продукты выделяли удалением растворителя в вакууме, переводили в осадок добавлением воды и экстракцией EtOAc или ДХМ, или с использованием картриджа Isolute SCX-2, и неочищенный продукт перекристаллизовывали из метанола, при этом получали соединение 339 в виде твердого вещества белого цвета (161 мг, выход 60%). ЖХМС: RT=2,41 мин, [М+Н]+ 423. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,10 (1Н, s), 7,88 (1Н, d, J=0,65 Гц), 7,83 (1Н, s), 7,27 (1Н, s), 6,76 (1Н, s), 6,34 (1Н, s), 5,93-5,92 (1Н, m), 4,51-4,49 (4Н, m), 4,31 (2Н, d, J=13,19 Гц), 2,94-2,81 (2Н, m), 2,38-2,35 (1Н, m), 1,73 (2Н, m), 1,53 (2Н, dd, J=12,25, 3,87 Гц), 1,47 (6Н, d, J=6,60 Гц).
Пример 340
1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперидин-4-ол 340
Согласно тому, как описано в примере 339, из соединения 93 и 4-гидроксипиперидина получали неочищенный продукт, который перекристаллизовывали из метанола и получали соединение 340 в виде твердого вещества белого цвета (106 мг, выход 42%). ЖХМС: RT=2,48 мин, [М+Н]+=396. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,09 (1Н, s), 7,88 (1Н, d, J=0,64 Гц), 7,83 (1Н, s), 6,33 (1Н, s), 5,97-5,88 (1Н, m), 4,69 (1Н, d, J=4,26 Гц), 4,49-4,48 (4Н, m), 4,03-3,99 (2Н, m), 3,75-3,67 (1Н, m), 3,14-3,13 (2Н, m), 1,80-1,71 (2Н, m), 1,47 (6Н, d, J=6,60 Гц), 1,34-1,33 (2Н, m).
Пример 341
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-морофлино-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин 341
Согласно тому, как описано в примере 339, из соединения 93 и морфолина получали неочищенный продукт, который перекристализовывали из метанола и получали соединение 341 в виде твердого вещества белого цвета (107 мг, выход 44%). ЖХМС: RT=3,12 мин, [М+Н]+ 382. 1Н ЯМР 400 МНц (ДМСО-d6): δ 9,14 (1Н, s), 7,90 (1Н, d, J=0,63 Гц), 7,86 (1Н, s), 6,36 (1Н, s), 5,93-5,92 (1Н, m), 4,57-4,48 (4H, m), 3,70 (4H, t, J=4,74 Гц), 3,50 (4H, t, J=4,74 Гц), 1,49 (6H, d, J=6,60 Гц).
Пример 342
N-Изопропил-2-(4-(2-(1-изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперазин-1-ил)ацетамид 342
Соединение 342 получали, как описнао в примере 341. МС: (ESI+)=480,2.
Пример 343
1-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)азетидин-3-карбоксамид 343
Раствор 9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-2-карбоксамида (45,0 мг, 0,136 ммоль), азетидин-3-карбоновой кислоты (30,0 мг, 0,297 ммоль) и ТЭА (0,300 мл, 2,15 ммоль) в изопропиловом спирте (1,00 мл, 13,1 ммоль) нагревали при 150°С в течение 2 сут. Реакционную смесь разбавляли водой и промывали EtOAc. Водный слой подкисляли и экстрагировали EtOAc. Органический слой сушили над Na2SO4, концентрировали и получали промежуточную карбоновую ксилоту 1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-ил)азетидин-3-карбоновую кислоту (МС: (ESI+) 396,1), которую растворяли (0,060 г, 0,00015 моль) в ДМФ (2,37 мл, 0,0306 моль) и последовательно обрабатывали N,N-диизопропилэтиламином (0,158 мл, 0,000910 моль), хлоридом аммония (0,0325 г, 0,000607 моль) и гексафторфосфатом N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (0,0692 г, 0,000182 моль). Смесь перемешивали при КТ в течение 2 ч. В смесь добавляли насыщенный раствор бикарбоната натрия и экстрагировали EtOAc. Органический экстракт сушили над сульфатом натрия, концентрировали и получали соединение 343, которое анализировали обращенно-фазной ЖХВР. МС: (ESI+)=395,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,50 (d, J=8,5 Гц, 1Н), 7,88 (s, 1H), 7,80 (s, 1Н), 7,50 (s, 1Н), 7,04 (s, 1Н), 6,28 (d, J=8,5 Гц, 1Н), 5,96-5,76 (m, 1Н), 4,48 (d, J=10,2 Гц, 4Н), 4,09 (t, J=8,3 Гц, 2Н), 3,98 (t, J=6,9 Гц, 2Н), 3,44 (dd, J=14,5, 7,4 Гц, 1Н), 1,46 (d, J=6,5 Гц, 6Н).
Пример 345
2-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропанамид 345
Дегазированную смесь 187 мг (0,500 ммоль) соединения 194, 320,6 мг (2,000 ммоль) (1-триметилсилилокси)-1-метоксипроп-1-ена, 19,4 мг (0,025 ммоль) димера бром(три-трет-бутилфосфин)палладия и 154,5 мг (0,500 ммоль) трибутилфосфина в 4,0 мл 1,4-диоксана нагревали в течение 18 ч при 105°С. Затем смесь насыщенного и ненасыщенного эфиров отделяли от продукта дебромирования хроматографией на колонке (элюент: градиент 1-4% метанол в ДХМ). 98 мг указанной смеси и 100 мг 10% палладия на угле в ТГФ гидрировали при 1 атм в течение 3 ч. Смесь фильтровали, фильтрат концентрировали в вакууме и получали 80 мг чистого метилового эфира 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропионовой кислоты. M/z 382,1, рассч. 381,18.
Из метилового эфира 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропионовой кислоты и гидроксида лития получали 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пропановую кислоту. M/z 368,2, расч. 367,16, из которой получали соединение 345. M/z 367,1, расч. 366,18. 1H ЯМР (400 МГц, ДМСО): δ 8,32 (d, J=8,3, 1H), 7,90 (s, 2H), 7,41 (s, 1H), 7,10 (d, J=8,5, 1H), 7,00 (s, 1H), 6,87 (s, 1H), 5,88 (dt, J=12,5, 6,2, 1H), 4,50 (d, J=4,7, 4H), 3,57 (q, J=6,9, 1H), 1,48 (d, J=6,5, 6H), 1,31 (d, J=6,9, 3Н).
Пример 348
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-9-(окзетан-3-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 348
Получение борной кислоты: Раствор 2-(2-Изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (пример 57, 0,495 г, 0,00114 моль) и периодата натрия (0,730 г, 0,00341 моль) в смеси 4:1 ТГФ (7,38 мл) и воды (1,84 мл) перемешивали при КТ в течение 30 мин. В реакционную смесь добавляли хлористый водород (0,000796 моль, 1 н., 0,8 мл) и реакционную смесь перемешивали при КТ в течение ночи. Смесь разбавляли водой и 3 раза экстрагировали ДХМ. Органические слои объединяли и сушили над MgSO4 и концентрировали. Неочищенный материал использовали на следующей стадии без дополнительной очистки.
В сосуд для микроволнового облучения СЕМ добавляли неочищенную борную кислоту (0,154 г, 0,437 ммоль), иодид никеля(II) (0,0186 г, 0,0596 ммоль), гидпрохлорид транс-2-аминогексанола (0,00904 г, 0,0596 ммоль) и гексаметилдисилазан натрия (0,477 ммоль, 2 М в ТГФ, 0,24 мл) в дегазированном изопропиловом спирте (0,91 млL) и ДМСО (1,5 мл). Смесь непрерывно продували азотом. Затем добавляли 3-иодокзетан (0,0731 г, 0,398 ммоль) в изопропиловом спирте (0,21 мл) и сосуд немедленно закрывали. Реакционную смесь нагревали при 85°С с использованием микроволнового облучения в течение 25 мин. Методом ЖХМС подтверждали приблизительно 50%-ное превращение продукта. Смесь разбавляли ДХМ и фильтровали через целит. В смесь добавляли воду и 3 раза экстрагировали ДХМ. Неочищенный продукт в твердом виде наносили на силикагель и очищали экспресс-хроматографией (элюент: 50% EtOAc в гексане) и повторно очищали обращенно-фазной ЖХВР, при этом получали 25,8 мг соединения 348 в виде твердого вещества белого цвета. MS(ESI+) 366,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,40 (d, J=8,2 Гц, 1Н), 7,89 (s, 1H), 7,22 (d, J=8,3 Гц, 1H), 7,06 (s, 1H), 5,93-5,73 (m, 1H), 4,94 (t, J=7,0 Гц, 2H), 4,63 (t, J=6,2 гц, 2Н), 4,55-4,46 (m, 4Н), 4,33-4,18 (m, 1H), 2,25 (s, 3H), 1.46 (d, J=6,4 Гц, 6Н).
Пример 352
N-Гидрокси-2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)ацетамид 352
Согласно тому, как описано в примере 316, из 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусной кислоты 336 и гидроксиламина получали соединение 352. M/z 369,1, расч. 368,16. 1H ЯМР (400 МГц, ДМСО): δ 10,69 (s, 1H), 8,88 (s, 1H), 8,32 (d, J=8,2, 1H), 7,92 (d, J=2,9, 2H), 7,04 (d, J=8,2, 1H), 6,97 (s, 1H), 5,95-5,81 (m, 1H), 4,50 (d, J=5,7, 4Н), 3,29 (s, 2H), 1,48 (d, J=6,6, 6Н).
Пример 354
1-((2-(1-(2,4-Дифторфенил)-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)метил)мочевина 354
Из С-{2-[2-(2,4-дифторфенил)-5-метил-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил}метиламина в смеси уксусной кислоты и воды и и цианата калия в воде получали соединение 354. MC(ESI+) 452,1. 1Н ЯМР (400 МГц, ДМСО): δ 7,89 (s, 1Н), 7,68 (td, J=8,7, 6,2 Гц, 1Н), 7,62-7,54 (m, 2H), 7,32-7,25 (m, 1Н), 6,87-6,80 (m, 2H), 6,44 (t, J=6,1 Гц, 1Н), 5,57 (ушир., 2H), 4,49-4,38 (m, 4H), 4,12 (d, J=6,1 Гц, 2H), 2,35 (s, 3Н).
Пример 355
(2-(1-(2,4-Дифторфенил)-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)метанамин 355
В раствор соединения 51 (5,00 г, 0,0162 моль) в толуоле (85 мл) добавляли диметилацетамид-диметилацеталь (7,23 мл, 0,0487 моль). Реакционную смесь перемешивали при 95°С в течение 4 ч. Толуол удаляли в вакууме и неочищинное соединение 46 использовали на следующей стадии без дополнительной очистки. MC(ESI+) 377,1/379,1.
Соединение 46 (0,0162 моль) растворяли в уксусной кислоте (50 мл). В раствор добавляли гидрохлорид 2,4-дифторфенилгидразина (3,52 г, 0,0195 моль) и реакционную смесь перемешивали при 95°С в течение ночи. Уксусную кислоту удаляли в вакууме. Неочищенный продукт в твердом виде наносили на силикагель и очищали экспресс-хроматографией (элюент: градиент 4-10% метанол в ДХМ), при этом получали 3,662 г 8-бром-2-[2-(2,4-дифторфенил)-5-метил-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена в виде твердого вещества оранжевого цвета. MC(ESI+) 458,0/460,0.
Из 8-бром-2-[2-(2,4-дифторфенил)-5-метил-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена, цианида цинка и Pd(PPh3)4 в ДМФ под действием микроволнового облучения при 60 Ватт в течение 30 мин (Тмакс=175°С) получали 2-[2-(2,4-дифторфенил)-5-метил-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-карбонитрил. MC(ESI+) 405,1.
В раствор 2-[2-(2,4-дифторфенил)-5-метил-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-карбонитрила в ТГФ по каплям добавляли раствор тетрагидроалюмината лития (1 М в ТГФ) при 0°С. Реакционную смесь перемешивали в течение 2 ч и реакцию останавливали добавлением Na2SO4 до прекращения выделения Н2. В смесь добавляли MgSO4 и смесь разбавляли большим количеством ДХМ, фильтровали через целит и концентрировали в вакууме. Неочищенный продукт очищали экспрес-хроматографией (элюент: градиент 1-15% МеОН в ДХМ, содержащем Et3N), приэтом получали соединение 355. MC(ESI+) 409,1. 1Н ЯМР (400 МГц, ДМСО): δ 7,89 (s, 1Н), 7,68 (td, J=8,7, 6,2 гц, 1Н), 7,64-7,52 (m, 2H), 7,32-7,25 (m, 1H), 6,95 (s, 1Н), 6,91 (dd, J=8,2, 1,2 Гц, 1Н), 4,48-4,38 (m, 4H), 3,66 (s, 2H), 2,35 (s, 3Н).
Пример 356
9-(1-(2-(Диметил амино)-2-оксоэтил)пиперидин-4-ил)-N-(2-гидроксиэтил)-N-изопропил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-карбоксамид 356
Стадия 1
Закрытую колбу, содержащую 8-бром-2-иод-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен (500 мг, 1,28 ммоль), хлорид палладия(II) (6 мг, 0,03 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантен (15 мг, 0,03 ммоль) продували СО. В колбу добавляли 2-изопропиламиноэтанол (172 мг, 1,67 ммоль) и ТЭА (0,53 мл, 3,8 ммоль) в виде раствора в толуоле (2,5 мл) и реакционную смесь нагрвали при 100°С в течение 3,5 ч. Реакционную смесь промывали водой и экстрагировали EtOAc (2×30 мл). Объединенные органические экстракты промывали солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хромаографией (SiO2, элюент: градиент 2-3% метанол в ДХМ), при этом получали (2-гидроксиэтил)изопропиламид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (123 мг, выход 23%). ЖХМС: RT=3,11 мин, [М+Н]+=394/396.
Стадия 2
(2-гидроксиэтил)изопропиламид 8-бром-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (114 мг, 0,28 ммоль), пинаколиновыйэфир 3,6-дигидро-2Н-пиридин-1-N-Вос-4-борной кислоты (129 мг, 0,62 ммоль), карбонат калия (96 мг, 0,69 ммоль) и комплекс PdCl2dppf с ДХМ (20 мг, 0,02 ммоль) суспендировали в ДМФ (1,5 мл) и реакционную смесь продували аргоном. Реакционную смесь нагревали при 100°С в течение 3 ч. Реакционную смесь промывали водой, экстрагировали ДХМ (2×15 мл) и объединенные органические экстракты промывали солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Полученный остаток очищали экспресс-хромтаографией (SiO2, элюент: градиент 0-2% МеОН в ДХМ), при этом получали трет-бутиловый эфир 4-{2-[(2-гидроксиэтил)изопропилкарбамоил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (115 мг, выход 80%). ЖХМС: RT=3,48 мин, [М+Н]+=497.
Стадия 3
Гидрохлорид (2-гидроксиэтил)изопропиламида 8-пперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты
В раствор трет-бутилового эфира 4-{2-[(2-гидроксиэтил)изопропилкарбамоил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил}-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (112 мг, 0,23 ммоль) в IMS (3 мл) добавляли хлористоводородную кислоту (2 мл, 2 М, 4,0 ммоль) и палладий на угле (20 мг, 10 мас. %). Реакционную смесь перемешивали в атмосфере водорода при 50°С в течение 4,5 ч. Реакционную смесь фильтровали и твердый продукт промывали IMS (10 мл). Фильтрат концентрировали в вакууме, перегоняли с азеотропом с ацетонитрилом и получали гидрохлорид (2-гидроксиэтил)изопропиламида 8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты в виде густого маслянистого вещества (110 мг). ЖХМС: RT=0,32 мин, [М+Н]+=399.
В смесь гидрохлорида (2-гидроксиэтил)изопропиламида 8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-2-карбоновой кислоты (127 мг, 0,23 ммоль) в ДМФ (2 мл) при перемешивании добавляли карбонат калия (127 мг, 0,92 ммоль), N,N-диметил-2-хлорацетамид (36 мг, 0,3 ммольl) и KI (каталитическое количество) и перемешивание продолжали при КТ в течение 72 ч, затем концентрировали в вакууме. Полученный остаток разбавляли EtOAc, промывали водой, солевым раствором, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток наносили на картридж Isolute SCX-2 (Элюент: ДХМ/метанол, затем 2 М раствор NH3 в метаноле). Основные фракции объединяли и концентрировали в вакууме, остаток очищали обращенно-фазной ЖХВР (колонка С18, элюент: градиент от 5 до 95% CH3CN в воде + 0.1% НСО2Н), при этом получали соединение 356 в виде бесцветного стеклообразного вещества (22 мг, выход 20%). ЖХМС: RT=1,90 мин, [М+Н]+=484. 1H ЯМР 400 МГц (CDCl3): δ 8,42 (2H, s), 8,20 (1H, br, s), 8,34 (1H, d, J=8,32 Гц), 7,81 (1H, s), 7,04 (1H, dd, J=8,40, 1,77 Гц), 6,90 (1H, d, J=1,69 Гц), 4,63 (2H, m), 4,45-4,44 (4H, m), 3,38 (3H, m), 3,32 (2H, m), 3,16 (2H, d, J=11,21 Гц), 3,11 (3H, s), 2,99 (3H, s), 2,56 (1H, m), 2,48 (2H, m), 1,87 (4H, m), 1,42 (6H, d, J=6,46 Гц).
Пример 357
2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-9-(1-изопропилпиперидин-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 357
В суспензию трифторацетата 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (250 мгg, 0,51 ммоль) в ДХЭ (5 мл) добавляли ацетон (0,06 мл, 0,77 ммоль) и молекулярные сита с диаметром пор , реакционную смесь перемешивали в течение 10 мин в атмосфере аргона, затем добавляли триацетоксиборгидрид натрия (216 мг, 1,02 ммоль) и перемешивали при КТ в течение 18 ч. Затем в смесь добавляли ацетон (0,06 мл) и триацетоксиборгидрид натрия (216 мг) и перемешивание продолжали в течение 18 ч. Реакционную смесь разбавляли ДХМ и насыщенным водным раствором гидрокарбоната натрия, органический слой промывали водой, солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный твердый материал очищали обращенно-фазной ЖХВР (элюент: градиент от 20 до 70% метанол в воде + 0.1% НСО2Н), при этом получали соединение 357 в виде твердого вещества желтого цвета (17 мг, выход 8%). ЖХМС: RT=2,81 мин, [М+Н]+=421. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,37-8,26 (1H, m), 7,90-7,89 (2H, m), 7,05 (1H, dd, J=8,33, 1,81 гц), 6,89 (1H, d, J=1,73 гц), 5,89-5,88 (1H, m), 4,49-4,48 (4H, m), 2,91 (2H, d, J=10,95 Гц), 2,76-2,75 (1H, m), 2,46 (1H, s), 2,26 (2H, t, J=11,38 гц), 1,78 (2H, d, J=12,43 Гц), 1,64 (2H, td, J=12,20, 3,68 гц), 1,48 (6H, d, J=6,60 гц), 1,01 (6H, d, J=6,57 Гц).
Пример 358
2-(4-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-2-метилпропан-1-ол 358
Смесь гидрохлорида 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (100 мг, 0,255 ммоль), этилового эфира 2-бром-2-метилпропионовой кислоты (45 мкл, 0,31 ммоль), карбоната цезия (187 мг, 0,57 ммоль) и ДМФ (0,5 мл) нагревали при 70°С в течение 3 ч, перемешивали при КТ в течение 18 ч, затем снова нагрвали при 70°С в течение 4 ч и концентрировали в ваууме. Полученный остаток очищали экспресс-хроматграфией (SiO2, элюент: градиент от 0 до 5% метанол в ДХМ), при этом получали этиловый эфир 2-{4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-ил}-2-метилпропионовой кислоты (47 мг, выход 36%). ЖХМС RT=2,24, [М+Н]+=507.
Раствор этилового эфира 2-{4-[2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил]пиперидин-1-ил}-2-метилпропионовой кислоты (47 мг, 0,092 ммоль) в безводном ТГФ (3 мл) охлаждали до 0°С и обрабатывали гидридом лития-алюминия (1 М раствор в ТГФ, 0,3 мл, 0,3 ммоль), смесь перемешивали и нагревали до КТ, затем реакцию останавливали добавлением насыщеннго водного раствора гидрокарбоната натрия. Полученную смесь 2 раза экстрагировали EtOAc, объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали обращенно-фазной ЖХВР (колонка С18, элюент: градиент от 0 до 60% метанол в воде + 0,1% муравьиная кислота), при этом получали соединение 358 в виде твердого вещества белого цвета (20 мг, выход 47%). ЖХМС: RT=2,74 мин, [М+Н]+=465. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,33 (1Н, d, J=8,28 Гц), 8,26 (1H, s), 7,87 (1H, s), 7,05 (1H, dd, J=8,35, 1,77 Гц), 6,90 (1H, d, J=1,71 Гц), 5,82-5,81 (1H, m), 4,49 (4H, m), 3,38 (2H, s), 3,21 (2H, d, J=11,33 Гц), 2,58 (1H, t, J=11,87 Гц), 2,47 (2H, d, J=11,47 Гц), 2,26 (3H, s), 1,85 (2H, d, J=12,54 Гц), 1,72 (2H, m), 1,46 (6H, d, J=6,60 гц), 1,07 (6H, s).
Пример 359
2-(4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиперидин-1-ил)-2-метилпропан-1-ол 359
Согласно тому, как описано в примере 358, из трифторацетата 2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (103 мг) получали соединение 359 в виде твердого веществ абелого цвета (6,6 мг, общий выход 2%). ЖХМС: RT=2,73 мин, [М+Н]+=451. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,33 (1Н, d, J=8,28 Гц), 7,91 (2H, d, J=2,06 Гц), 7,06 (1H, dd, J=8,35, 1,76 Гц), 6,90 (1H, d, J=1,71 Гц), 5,90 (1H, t, J=6,60 Гц), 4,50 (4H, dd, J=11,70, 5,85 Гц), 3,34 (2Н, ушир. m), 3,13 (2H, d, J=11,26 Гц), 2,55 (1Н, m), 2,35 (2H, t, J=11,38 Гц), 1,81 (2H, d, J=12,50 Гц), 1,64 (2H, d, J=12,44 Гц), 1,49 (6H, d, J=6,60 Гц), 1,02 (6H, s).
Пример 360
4-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пиразолидин-3,5-дион 360
Смесь соединения 194 (1410 мг, 3,77 ммоль), диэтилового эфира малоновой кислоты (2,00 мл, 13,2 ммоль), ацетата палладия(II) (42,3 мг, 0,188 ммоль), 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенила (148 мг, 0,377 ммоль) и фосфата калия (2,80 г, 13,2 ммоль) в 6,0 мл 1,4-диоксана дегазировали и нагревали в течение 24 ч при 100°С. Смесь концентрировали в вакууме и остаток очищали на колонке с 24 г силикагеля (элюент: 1% МеОН в EtOAc), при этом получали диэтиловый эфир 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)малоновой кислоты с выходом 974 мг. M/z 454,3, расч. 453,20.
Смесь 181 мг (0,40 ммоль) диэтиловый эфир 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)малоновой кислоты и 0,314 мл (10,0 ммоль) гидразина в 4,0 мл этанола нагревали при 75°С в течение 18 ч. Смесь концентрировали, остаток растирали с уксусной кислотой. Остаток отфильтровывали, промывали уксусной кислотой и этиловым эфиром, затем перекристаллизовывали из смеси этиловый эфир/этанол, при этом получали соединение 360 с выходом 59 мг (37,5%). M/z 394,1, расч. 393,15. 1Н ЯМР (400 МГц, ДМСО): δ 10,20 (s, 2H), 8,29 (d, J=8,6, 1Н), 7,89 (d, J=12,9, 2Н), 7,70 (d, J=8,5, 1H), 7,63 (s, 1H), 5,93 (dt, J=13,1, 6,7, 1H), 4,48 (d, J=7,2, 4H), 1,49 (d, J=6,6, 6H).
Пример 361
2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-9-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 361
Суспензию трифторацетата2-(2-изопропил-2Н-[1,2,4]триазол-3-ил)-8-пиперидин-4-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (250 мг, 0,51 ммоль) в ТГФ (5 мл) обрабатывали ТЭА и смесь продували аргоном. В смесь добавляли 2,2,2-трифторэтилтрифторметансульфонат (0,15 мл, 1,02 ммоль) и перемешивали при КТ в течение 72 ч. Реакционную смесь распределяли между ДХМ и водой, органический слой промывалинасыщенным водным раствором бикарбоната натрия, солевым раствором и сушили (MgSO4), фильтровали и концентрировали в вакууме. Полученный остаток растворяли в 1,25 М растворе HCl в метаноле, затем концентрировали в вакууме, при этом получали твердое вещество, которое растирали с ИПС и диэтиловым эфиром. Полученное твердое вещество очищали обращенно-фазной ЖХВР (колонка С18, элюент: градиент от 10 до 98% метанол в воде + 0,1% HCO2H), при этом получали соединение 361 в виде твердого вещества белого цвета (49 мг, выход 23%). ЖХМС: RT=4,59 минn, [М+Н]+=461. 1Н ЯМР 400 МГц (ДМСО-d6): δ 8,33 (1H, d, J=8,29 Гц), 7,91-7,91 (2H, m), 7,08 (1H, dd, J=8,36, 1,79 Гц), 6,92 (1H, m), 5,91-5,90 (1H, m), 4,51 (4H, q, J=5,90 Гц), 3,21 (2H, q, J=10,30 Гц), 3,03 (2H, d, J=11,25 Гц), 2,45 (3H, m), 1,76 (2H, m), 1,69-1,67 (2H, m), 1,49 (6H, d, J=6,60 Гц).
Пример 363
1-((2-(1-(2,4-Дифторфенил)-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)метиламино)-2-метилпропан-2-ол 363
В суспензию С-{2-[2-(2,4-дифторфенил)-5-метил-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулен-8-ил}метиламина (0,150 г, 0,000367 моль) и карбоната цезия (0,0335 г, 0,000103 моль) в метаноле (0,5 мл) добавляли бутиленоксид (0,0359 мл, 0,000404 моль). Колбу закрывали и нагревали при 70°С в течение 2 ч. Смесь разбавляли диэтиловым эфиром и добавляли воду. Смесь 3 раза экстрагировали диэтиловым эфиром. Органические фазы объединяли, сушили над MgSO4 и концентрировали, при этом получали 11,2 мг соединения 363 в виде твердого вещества белого цвета (выход 6,4%). MC(ESI+) 481,2. 1Н ЯМР (400 МГц, ДМСО): δ 7,89 (s, 1H), 7,68 (td, J=8,6, 6,0 Гц, 1Н), 7,63-7,51 (m, 2H), 7,29 (td, J=8,2, 1,5 Гц, 1H), 6,94 (s, 1H), 6,90 (dd, J=8,3, 1,3 Гц, 1H), 4,47-4,38 (m, 4H), 4,20 (s, 1H), 3,67 (s, 2H), 2,35 (s, 3Н), 2,33 (s, 2H), 1,08 (s, 6H).
Пример 364
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин 364
В раствор 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина 264 (35,0 мг, 0,106 ммоль) в метаноле (1,91 мл, 47,1 ммоль) добавляли ТЭА (0,0147 мл, 0,106 ммоль) палладий (0,0113 г, 0,0106 ммоль). Реакционную смесь перемешивали при КТ в атмосфере водорода. Реакционную смесь фильтровали через целит. Раствор концентрировали и разбавляли EtOAc, промывали Н2О. Органический слой сушили над Na2SO4, концентрировали. Неочищенный продукт очищали на колонке isco и получали соединение 364. МС: (ESI+)=297,1. 1Н ЯМР (400 МГц, ДМСО): δ 9,49 (s, 1H), 8,36 (d, J=5,7 Гц, 1H), 7,99 (s, 1H), 7,92 (s, 1H), 7,05 (d, J=5,7 Гц, 1H), 5,90 (dt, J=13,2, 6,5 Гц, 1H), 4,59 (dt, J=26,2, 13,1 Гц, 4H), 1,50 (d, J=6,6 Гц, 6H).
Пример 365
(2R)-1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-fl[1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 365
Раствор 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина 264 (25,0 мг, 0,0756 ммоль), R-пролинамида (0,0570 г, 0,499 ммоль) и ТЭА (0,125 мл, 0,897 ммоль) в N-метилпирролидоне (1,36 мл, 14,1 ммоль) нагревали при 150°С в течение 2 сут. Реакционную смесь фильтровали через целит и промывали EtOAc. Фильтрат промывали водой и солевым раствором. Органический слой сушили над Na2SO4, концентрировали и получали соединение 365, которое анализировали методом обращенно-фазной ЖХВР. МС: (ESI+) 409,2.
Пример 366
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(пирролидин-1-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин 366
Соединение 366 получали по методике, описанной в примере 365. МС: (ESI+)=366,2. 1Н ЯМР (400 МГц, ДМСО): δ 9,08 (s, 1Н), 7,88 (s, 1H), 7,82 (s, 1H), 5,94 (dd, J=13,6, 6,9 Гц, 1H), 4,61-4,32 (m, 4H), 3,40 (d, J=6,3 Гц, 4Н), 1,95 (t, J=6,5 Гц, 4H), 1,48 (d, J=6,6 Гц, 6Н).
Пример 367
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-N-метил-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин-9-амин 367
Смесь 1,936 г (11,00 ммоль) 2,6-дихлорникотинальдегида, 6,384 г (44,00 ммоль) водного раствора этандиаля и водного раствора аммиака (4,996 г, 88,00 ммоль) в 60 мл метанола перемешивали в течение 3 ч. Смесь концентрировали в вакууме и подкисляли до рН<1 добавлением 200 мл 0,5 н. водного растора HCl. Водный раствор экстрагировали EtOAc (3×30 мл). Органические экстракты отбрасывали, а водные экстракты подщелачивали добавлением насыщенного раствора NaHCO3. Смесь экстрагировали EtOAc (3×30 мл), объединенные органические экстракты промывали водой, солевым раствором и концентрировали в и получали 2,6-дихлор-3-(1Н-имидазол-2-ил)пиридин (0,85 г неочищенного продукта, выход 36%). M/z 214,0, расч. 212,99. 1Н ЯМР (500 МГц, ДМСО): δ 12,51 (s, 1H), 8,31 (d, J=8,1, 1H), 7,74-7,63 (m, 1H), 7,26 (s, 2H).
Смесь 0,856 г (4,00 ммоль) 2,6-дихлор-3-(1Н-имидазол-2-ил)пиридина, 704 мг (8,00 ммоль) этиленкарбоната и 2930 мг (9,00 ммоль) карбоната цезия нагревали в 25,0 мл ДМФ в течение 13 ч при 90°С. Смесь фильтровали, фильтрат концентрировали в высоком вакууме, остаток очищали на колонке с 10 г силикагеля (элюент: EtOAc в гептане), при этом получали 9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин с выходом 0,359 г (41%). M/z 222,0, расч. 221,04. 1H ЯМР (500 МГц, CDCl3): δ 8.84 (d, J=8,2, 1H), 7,18 (s, 1H), 7,15 (d, J=8,2, 1H), 7,02 (s, 1H), 4,63-4,57 (m, 2H), 4,47-4,41 (m, 2H).
Смесь 0,359 г (1,62 ммоль) 9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепина и 0,913 г (4,06 ммоль) N-иодсукцинимида в 20,0 мл ДМФ нагревали при 80°С в течение 60 ч. Смесь концентрировали в вакууме, остаток распределяли между EtOAc (40 мл) и 0,1 М водным раствором Na2CO3. Органические экстракты промывали 5% водным раствором Na2S2O5, водой, солевым раствором, сушили над Na2SO4 и концентрировали в вакууме, при этом получали 9-хлор-2,3-дииод-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин с выходом 0,644 (84%). M/z 473,9, расч. 472,83. 1Н ЯМР (500 МГц, CDCl3): δ 8,85 (d, J=8,2, 1H), 7,19 (s, 1H), 7,16 (d, J=8,2, 1H), 7,03 (s, 1H), 4,60 (dd, J=9,9, 5,8, 2H), 4,47-4,42 (m, 2H).
2,0 M раствор хлорида изопропилмагния в ТГФ (0,782 мл) по каплям добавляли в раствор 0,644 г (1,36 ммоль) 9-хлор-2,3-дииод-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепина в 12 мл ТГФ при -10°С. Смесь нагревали до 15°С. Реакцию останавливали добавлением 20 мл насыщенного водного раствора NH4Cl и экстрагировали EtOAc. Органические экстракты промывали водой, солевым раствором, сушили над Na2SO4 и концентрировали, при этом получали 9-хлор-2-иод-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин с выходом 448 мг (98%). M/z 348,2, расч. М 346,93. 1H ЯМР (400 МГц, CDCl3): δ 8,83 (d, J=8,2, 1H), 7,16 (dd, J=14,0, 5,9, 1H), 7,10 (s, 1H), 4,61-4,54 (m, 2H), 4,41 (dd, J=5,0, 2,9, 2H).
Согласно методикам, описанным в разделе Примеры, включая примеры 20-22, из 9-хлор-2-иод-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепина получали 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепин. Смесь 110 мг (0,33 ммоль) 9-хлор-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,2-f][1,4]оксазепина, 87 мг (1,3 ммоль) хлорида метиламмония и 0,23 мл (1,3 ммоль) N,N-диизопропилэтиламина в 3,0 мл N-метилпирролидинона облучали микроволновым облучением в течение 90 мин при 170°С. НМР удаляли в высоком вакууме, остаток подщелачивали добавлением 1 М раствора Na2CO3 и распределяли между EtOAc и водой. Органические экстракты промывали 5% водным раствором лимонной кислоты, водой, солевым раствором, сушили над MgSO4 и концентрировали. Остаток очищали обращенно-фазной ЖХВР (элюент: градиент ацетонитрила), при этом получали соединение 367 с выходом 12 мг. M/z 326,3, расч. 325,17. 1Н ЯМР (400 МГц, ДМСО): δ 8,37 (d, J=8,6, 1H), 7,88 (s, 1H), 7,77 (s, 1H), 6,96 (s, 1H), 6,34 (d, J=8,6, 1Н), 5,88 (dt, J=13,0, 6,5, 1H), 4,46 (d, J=9,2, 4H), 2,78 (d, J=2,9, 3Н), 1,46 (d, J=6,6, 6H).
Пример 368
(2S,4R)-4-Гидрокси-1-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 368
Согласно тому, как описано в примере 339, из соединения 92 и 4-транс-гидрокси-L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS и получали соединение 368 в виде твердого вещества белого цвета (86 мг, выход 53%). ЖХМС: RT=2,19 мин, [М+Н]+ 439. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,02 (1Н, s), 7,77 (1H, s), 7,40 (1Н, ушир.), 6,90 (1H, ушир.), 5,90 (1H, s), 5,87-5,85 (1H, m), 5,05 (1H, d, J=3,93 Гц), 4,52-4,41 (4H, m), 4,39 (1H, m), 4,31 (1H, m), 3,66 (1H, dd, J=10,60, 4,98 гц), 2,22 (3H, s), 2,16-2,10 (1H, m), 2,00-1,99 (1H, m), 1,43 (6H, dd, J=6,59, 2,86 Гц).
Пример 369
(2S)-1-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 369
Согласно тому, как описано в примере 339, из соединения 92 и L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS и получали соединение 369 в виде твердого вещества белого цвета (130 мг, выход 67%). ЖХМС: RT=2,48 мин, [М+Н]+ 423. 1H ЯМР 400 МГц (ДМСО-d6): δ 9,06 (1H, s), 7,80 (1H, s), 7,35 (1Н, ушир.), 6,94 (1Н, ушир.), 5,96 (1H, s), 5,90-5,88 (1H, m), 4,50 (4H, d, J=17,28 Гц), 4,32 (1H, m), 3,61 (1H, m), 3,45 (1H, m), 2,26 (2H, s), 2,24-2,15 (1H, m), 1,98-1,97 (3H, m), 1,47 (6H, dd, J=6,59, 3,27 Гц).
Пример 370
1-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)азетидин-3-ол 370
Согласно тому, как описано в примере 339, из соединения 93, гидрохлорида 3-гидроксиазетидина DIPEA (2,2 экв.) получали неочищенный продукт, который очищали экспресс-хроматграфией (SiO2, элюент: градиент от 0 до 8% метанол в ДХМ), перекристаллизовывали из IMS и получали соединение 370 виде кристаллического вещества светло-зеленого цвета (16 мг, выход 14%). ЖХМС: RT=2,26 мин, [М+Н]+ 368. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,09 (1Н, s), 7,90 (1H, d, J=0,63 Гц), 7,84 (1Н, s), 5,95-5,94 (1Н, m), 5,91 (1Н, s), 5,70 (1Н, d, J=6,40 Гц), 4,59 (1H, s), 4,55-4,47 (4H, m), 4,18 (2H, t, J=7,66 Гц), 3,70 (2H, dd, J=8,85, 4,66 Гц), 1,48 (6H, d, J=6,60 Гц).
Пример 371
(3R)-1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролид-3-ол 371
Согласно тому, как описано в примере 339, из соединения 93, гидрохлорида (R)-пролинола и DIPEA (2,2 экв.) получали неочищенный продукт, который перекристаллизовывали из метанола и получали соединение 371 виде кристаллического вещества светло-зеленого цвета (46 мг, выход 34%). ЖХМС: RT=2,26 мин, [М+Н]+=382. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,09 (1Н, s), 7,88 (1Н, d, J=0,63 Гц), 7,82 (1H, s), 5,97-5,96 (1H, m), 5,94 (1H, s), 4,97 (1H, d, J=3,62 Гц), 4,49-4,48 (4H, m), 4,39 (1H, s), 3,53-3,42 (3H, m), 2,02-2,00 (1H, m), 1,90-1,87 (1H, m), 1,48 (6H, d, J=6,60 Гц).
Пример 372
(1-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пиперидин-4-ил)метанол 372
Согласно тому, как описано в примере 339, из соединения 93 и 4-пиперидинметанола получали неочищенный продукт, который очищали экспресс-хроматграфией (SiO2, элюент: градиент от 0 до 8% метанол в ДХМ), перекристаллизовывали из метанола и получали соединение 372 виде твердого вещества белого цвета (69 мг, выход 48%). ЖХМС: RT=2,57 мин, [М+Н]+ 410. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,10 (1Н, s), 7,90 (1H, s), 7,84 (1H, s), 6,32 (1H, s), 5,99-5,90 (1H, m), 4,54-4,44 (5 H, m), 4,34 (2H, d, J=13,11 Гц), 3,28 (2H, t, J=5,64 Гц), 2,84 (2H, t, J=12,55 Гц), 1,71 (2H, d, J=13,74 Гц), 1,65 (1H, m), 1,49 (6H, d, J=6,60 Гц), 1,13 (2H, t, J=12,33 Гц).
Пример 373
(2S,4S)-4-Фтор-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 373
Согласно тому, как описано в примере 339, из соединения 93, гидрохлорида 4-цис-фтор-(L)-пролинола и DIPEA (2,2 экв.) получали неочищенный продукт, который перекристаллизовывали из IMS и получали соединение 373 виде твердого вещества белого цвета (56 мг, выход 22%). ЖХМС: RT=2,59 мин, [М+Н]+=427. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,05 (1Н, s), 7,84 (1Н, d, J=0,63 гц), 7,80 (1Н, s), 7,11 (1Н, s), 6,92 (1Н, s), 5,93-5,91 (2Н, m), 5,42 (1Н, s), 5,29 (1Н, s), 4,46-4,44 (5Н, m), 3,83-3,54 (2Н, m), 2,28 (1Н, dd, J=20,28, 14,80 Гц), 1,43 (6Н, dd, J=6,59, 3,60 Гц).
Пример 374
(2S,4R)-4-Гидрокси-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридоо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 374
Согласно тому, как описано в примере 339, из соединения 93 и 4-транс-гидрокси-L-пролинамида получали неочищенный продукт, который очищали экспресс-хроматграфией (SiO2, элюент: градиент от 0 до 10% метанол в ДХМ), при этом поулчали соединение 374 виде твердого вещества белого цвета (56 мг, выход 27%). ЖХМС: RT=2,16 мин, [М+Н]+ 425. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,05 (1Н, s), 7,88 (1Н, s), 7,83 (1Н, s), 7,42 (1Н, ушир.), 6,92 (1Н, ушир.), 5,95 (1Н, m), 5,92 (1Н, s), 5,07 (1Н, d, J=3,93 Гц), 4,50-4,49 (4Н, m), 4,40 (1Н, m), 4,32 (1Н, m), 3,67 (1Н, t, J=5,33 Гц), 2,16 (1Н, m), 2,04 (1Н, m), 1,47 (6Н, dd, J=6,59, 3,01 Гц).
Пример 375
(2S)-1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 375
Дегазированную смесь 374 мг (1,00 ммоль) соединения 194, 342,5 мг (2,000 ммоль) трет-бутилового эфира L-пролина, 26 мг (0,050 ммоль) бис(три-трет-бутилфосфин)палладия и 192 мг (2,00 ммоль) трет-бутоксида натрия в толуоле (10,0 мл, 93,9 ммоль) нагревали при 95°С в течение 24 ч. В смесь добавляли аналогичные количества трет-бутилового эфира L-пролина, трет-бутоксида натрия и катализатора и смесь нагревали в течение 6 ч при 115°С до полного исчерпания бромида в реакционной смеси. Смесь концентрировали в вакууме, остаток распределяли между EtOAc и 5% водным раствором лимонной кислоты. Органические экстракты промывали водой, насыщенным раствором NaHCO3, водой, солевым раствором, сушили над MgSO4 и очищали на колонке с 12 г силикагеля (элюент: 4% метанол в ДХМ), при этом получали (S)-трет-бутиловый эфир 1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензоо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-крбоновой кислоты с выходом 153 мг (чистота 55-60%, продукт загрязнен побочным продуктом дебромированияч). M/z 465,2, расч. 464,25.
3 мл ТФУ добавляли в смесь 153 мг (S)-трет-бутилового эфира 1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоновой кислоты и 0,2 мл триэтилсилана в 5 мл ДХМ. Реакционную смесь перемешивали в течение 3 ч. Смесь концентрировали и остаток распределяли между 1 М водным раствором Na2CO3 и этиловым эфиром. Водный слой экстрагировали более 2 раз EtOAc. Органические слои отбрасывали, водный расвтор нейтрализовали до рН 6. Остаток собирали, промывали водой, сушили в высоком вакууме в течение 36 ч и получали (S)-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоновую кислоту с выходом 55 мг. M/z 409,3, расч. 408,19.
Смесь 55 мг (0,135 ммоль) (S)-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоновой кислоты, гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (57,0 мг, 0,150 ммоль), N,N-диизорпопилэтиламина (52,2 пл, 0,300 ммоль) и хлорида аммония (8,02 мг, 0,150 ммоль) в N,N-диметилацетамиде (3,0 мл, 32 ммоль) перемешивали в течение 1 ч. Смесь концентрировали в вакууме, остаток растирали с диэтиловым эфиром, 0,01 н. водным раствором HCL, водой, сушили в вакууме и очищали обращенно-фазной ЖХВР и хиральным способом очистки, при этом получали соединение 375 с выходом 12 мг. M/z 408,2, расч. 407,21. 1Н ЯМР (400 МГц, ДМСО): δ 8,19 (d, J=8,9, 1H), 7,87 (s, 1H), 7,77 (s, 1H), 7,40 (s, 1H), 7,04 (s, 1H), 6,36 (dd, J=9,0, 2,4, 1H), 6,07 (d, J=2,4, 1H), 5,91 (dq, J=13,3, 6,5, 1H), 4,44 (d, J=6,5, 4H), 4,01-3,93 (m, 1H), 3,57 (t, J=7,0, 1H), 3,24 (d, J=9,0, 1H), 2,23 (dd, J=12,1, 6,9, 1H), 1,98 (dd, J=14,8, 11,3, 3Н), 1,47(dd, J=6,6, 3,3, 6H).
Пример 376
(2R)-1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 376
Согласно тому, как описано в примере 316, из соединения 194 и 1-(трет-бутилдиметилсилилокси)-1-метоксиэтена получали метиловый эфир 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусной кислоты. M/z 368,2, расч. 367,16.
Из метилового эфир 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусной кислоты и гидроксида лития получали 2-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)уксусную кислоту 336. M/z 354,1, рассч. 353,15.
Из соединения 336 и аммиака получали соединение 376. M/z 353,1, расч. 352,16. 1Н ЯМР (400 МГц, ДМСО): δ 8,32 (d, J=8,3, 1H), 7,91 (s, 2H), 7,48 (s, 1H), 7,04 (d, J=8,2, 1H), 6,97 (s, 1H), 6,92 (s, 1H), 5,95-5,81 (m, 1H), 4,50 (d, J=5,8, 4H), 3,38 (s, 2H), 1,48 (d, J=6,5, 6H).
Пример 377
(2S)-1-(2-(1-Изопропил-1Н-имидазол-2-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 377
Бромид этилмагния (3,0 M в Et2O, 100 ммоль, 33,3 мл) по каплям добавляли в раствор 9-хлор-2-иод-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина (10,0 г, 28,8 ммоль) в ТГФ (173 мл) при -20°С. Смесь выдерживали при указанной температуре в течение 20 мин и нагревали до комнатной температуры в течение 1 ч. Затем реакционную смесь охлаждали до -20°С и в смесь добавляли ДМФ (8,9 мл, 115 ммоль). Перемешивание продолжали в течение 16 ч и реакцию останавливали добавлением насыщенного водного раствора хлорида аммония (220 мл) и разбавляли EtOAc (250 мл). Фазы разделяли и водный слой 2 раза экстрагировали EtOAc. Объединенные органические фазы промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме, при этом получали 7,08 г (выход 98%) 9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-карбальдегид в виде твердого вещества желтого цвета (степенб чистоты более 95% по данным аналитической ЖХВР). МС (ESI+): m/z 249,8 (М+Н+), расч. 249,65.
В круглодонной колбе объемом 100 мл смешивали 9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-карбальдегид (1,1 г, 4,4 ммоль), 40% водный раствор этандиаля (2,1 мл, 18,0 ммоль), гидроксид аммония (2,55 мл, 65,5 ммоль) и метанол (10,5 мл). Полученную реакционную смесь перемешивали при КТ в течение 6 ч. Затем смесь упаривали досуха и маслянистый остаток очищали экспресс-хроматографией на колонке (элюент: медленный градиент 0-100% EtOAc в ДХМ), при этом получали 1,13 г (выход 86%) 9-хлор-2-(1Н-имидазол-2-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин.
В раствор 9-хлор-2-(1Н-имидазол-2-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина (0,402 г, 1,4 ммоль) в ДМФ (9,6 мл) добавляли Cs2CO3 (0,6 г, 2,0 ммоль) и изопропилиодид (0,2 мл, 2 ммоль). Реакционную смесь нагревали при 50°С в течение 20 ч. Смесь постепенно охлаждали до КТ и разбавляли водой и EtOAc. Смесь 2 раза экстрагировали EtOAc, сушили над MgSO4, фильтровали и концентрировали. Полученный остаток очищали экспресс-хроматографией на колонке (элюент: 0-10% МеОН в ДХМ), при этом получали 0,22 г (выход 48%) 9-хлор-2-(1-изопропил-1Н-имидазол-2-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин, который смешивали с L-пролинамидом (0,152 г, 1,33 ммоль) в N,N-диметилацетамиде (6,7 мл) в сосуде, пригодном для высокого давления, который немедленно закрывали. Смесь нагревали при 150°С в течение 40 ч, добавляли L-пролинамид (0,152 г, 1,33 ммоль) и смесь продолжали нагревать в течение 12 ч. В конце указанного периода наблюдали только 50%-ное превращение исходного вещества, нагревание прекращали и материал очищали обращенно-фазной ЖХВР (элюент: 0,1% NH4OH в ацетонитриле), при этом получали 19,8 мг (выход 8%) соединения 377. МС (ESI+) m/z 408,2 (М+Н+), расч. 408,5. 1H ЯМР (400 МГц, ДМСО): δ 9,03 (s, 1Н), 7,58 (s, 1H), 7,43-7,26 (m, 2H), 6,89 (d, J=1,0 Гц, 2Н), 5,94 (s, 1H), 5,72 (dt, J=13,6, 6,8 Гц, 1H), 4,56-4,38 (m, 4H), 4,29 (d, J=8,0 Гц, 1Н), 3,59 (s, 1Н), 2,17 (d, J=8,9 Гц, 1Н), 1,98 (dd, J=31,1, 24,1 Гц, 4Н), 1,44 (d, J=4,2 Гц, 6Н).
Пример 378
(2S)-1-(2-(1-(2,4-Дифторфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 378
В раствор амида 8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-карбоновой кислоты (2,40 г, 0,00907 моль) в толуоле (40 мл) добавляли 1,1-диметокси-N,N-диметилметанамин (4,82 мл, 0,0363 моль). Колбу закрывали и нагревали при 95°С в течение 8 ч. Завершение реакции наблюдали методом ТСХ. Растворитель удаляли в вакууме и получали 1 -диметиламинометилиденамид 8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-карбоновой кислоты, который использовали неочищенным. MC(ESI+) 320,1.
1-Диметиламинометилиденамид 8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-карбоновой кислоты (0,00907 моль) растворяли в уксусной кислоте (36 мл). В раствор добавляли гидрохлорид 2,4-дифторфенилгидразина (1,965 г, 0,01088 моль) и реакционную смесь перемешивали при 95°С в течение 2,5 ч. АсОН удаляли в вакууме. Неочищенный рподукт растирали с изопропиловым спиртом и получали 2,828 г (выход после 2 стадий 78%) 8-хлор-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена в виде порошкообразного вещества светло-коричневого цвета, чистоту которого подтверждали методом 1Н ЯМР. MC(ESI+) 401,1.
Раствор 8-хлор-2-[2-(2,4-дифторфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена (0,200 г, 0,499 ммоль), L-пролинамида (0,171 г, 0,00150 моль) и ТЭА (0,417 мл, 0,00299 моль) в N-метилпирролидиноне (5 мл) в атмосфере азота нагревали при 150°С в течение ночи. Смесь разбавляли ДХМ. В смесь добавляли NH4Cl и смесь 3 раза экстрагировали ДХМ. Органические фазы объединяли, сушили над Na2SO4 и концентрировали. Неочищенный остаток очищали обращенно-фазной ЖХВР и получали 94 мг (выход 39%) соединения 378 в виде твердого вещества светло-розового цвета. MC(ESI+) 479,2. 1Н ЯМР (400 МГц, ДМСО): δ 8,35 (s, 1Н), 8,18 (s, 1Н), 7,86 (s, 1Н), 7,69 (td, J=8,7, 6,0 Гц, 1Н), 7,62-7,53 (m, 1Н), 7,35-7,26 (m, 2Н), 6,92 (ушир., 1Н), 5,84 (s, 1H), 4,47-4,36 (m, 4H), 4,24 (d, J=7,4 Гц, 1Н), 3,62-3,53 (m, 1Н), 3,42-3,32 (m, 1Н), 2,24-2,08 (m, 1Н), 2,02-1,83 (m, 3H).
Пример 376
(2R)-2-(2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-1-карбоксамид
379 и (2S)-2-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)пирролидин-1-карбоксамид
380
В раствор N-(трет-бутоксикарбонил)пирролидина (1,02 мл, 0,00584 моль) и N,N,N',N'-тетраметилэтилендиамина (0,881 мл, 0,00584 моль) в безводном 2-метокси-2-метилпропане (12 мл) при -78°С добавляли втор-бутиллитий (0,00584 моль, 1,4 М в циклогексане, 4,17 мл). Реакционную смесь перемешивали в течение 3 ч при -78°С. В смесь по каплям при интенсивном перемешивании добавляли раствор дихлорида цинка (0,00350 моль, 0,5 М в ТГФ, 7,0 мл) и перемешивали при -78°С в течение 30 мин. Реакционную смесь нагревали до КТ и перемешивали в течение 30 мин. В колбу, заолненную азотом и содержащую соединение 48 (0,900 г, 0,00232 моль), ацетат палладия (0,0260 г, 0,000116 моль) и тетрафторборат три-трет-бутилфосфония (0,0420 г, 0,000145 моль) добавляли раствор хлорида цинка в пирролидине (1,25 экв., 0,243 М, 11,9 мл). Реакционную смесь нагревали при 90°С в течение ночи. Методом ЖХ/МС наблюдали неполное превращение продукта. Смесь разбавляли ДХМ и фильтровали через целит. В смесь добавляли насыщенный раствор NH4Cl и смесь 3 раза экстрагировали ДХМ. Органические слои собирали, сушили над Na2SO4 и концентрировали. Неочищенный остаток растворяли в ДХМ (4,5 мл). В раствор добавляли ТФУ (5,5 мл) и реакционную смесь перемешивали при КТ в течение 30 мин, затем концентрировали. Неочищенный остаток очищали обращенно-фазной ЖХВР, энантиомеры разделяли с помощью хирального SFC и получали по 10 мг каждого из энантиомеров 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пирролидин-2-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена в виде твердых веществ белого цвета. MC(ESI+) 379,2.
В раствор 2-(2-изопропил-5-метил-2Н-[1,2,4]триазол-3-ил)-8-пирролидин-2-ил-4,5-дигидро-6-окса-1,3а-диазабензо[е]азулена (0,009 г, 0,00002 моль) в уксусной кислоте (4,06 мкл, 0,0000713 моль) и воде (0,13 мл) по каплям добавляли раствор цианата калия (0,00579 г, 0,0000713 моль) в воде (0,13 мл). Для растворения реагентов добавляли ДМФ (0,13 мл, 0,0017 моль). Реакционную смесь перемешивали при 50°С в течение ночи, охлаждали, фильтровали и промывали холодной водой. Неочищенный остаток очищали переосаждением из смеси МеОН/вода и получали по 10 мг каждого из энантиомеров 379 и 380. MC(ESI+) 422,2. 1H ЯМР (400 МГц, ДМСО): δ 8,32 (d, J=8,3 Гц, 1Н), 7,85 (s, 1H), 6,95 (dd, J=8,3, 1,6 Гц, 1Н), 6,79 (d, J=1,3 Гц, 1Н), 5,90-5,76 (m, 1Н), 5,69 (ушир., 2Н), 4,89 (dd, J=7,9, 1,3 Гц, 1Н), 4,53-4,44 (m, 4H), 3,58-3,49 (m, 1Н), 3,42-3,34 (m, 1Н), 2,25 (s, 3Н), 2,25-2,17 (m, 1Н), 1,92-1,65 (m, 3H), 1,45 (dd, J=6,6, 3,9 Гц, 6Н).
Пример 381
(2S)-4,4-Дифтор-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 381
Согласно тому, как описано в примере 339, из соединения 93, гидрохлорида 4,4-дифтор-L-пролинамида и DIPEA (2,2 эквq.) получали неочищенный продукт, который очищали обращенно-фазной ЖХВР (колонка С18, элюент: градиент от 5 до 95% ацетонитрил в воде + 0,1% НСО2Н), перекристаллизовывали из IMS и получали соединение 381 в виде твердого вещества белого цвета (24 мгg, пример 11%). ЖХМС: RT=3,21 мин, [М+Н]+=445. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,11 (1Н, s), 7,90 (1Н, d, J=0,64 Гц), 7,87 (1Н, s), 7,53 (1Н, ушир.), 7,14 (1Н, ушир.), 6,08 (1Н, s), 5,99-5,91 (1Н, m), 4,61 (1Н, dd, J=9,53, 4,24 Гц), 4,58-4,49 (4Н, m), 4,00-3,98 (2Н, m), 2,91-2,90 (1Н, m), 2,45 (1Н, dd, J=13,51, 4,18 Гц), 1,49 (6Н, dd, J=6,59, 3,35 Гц).
Пример 382
(2S,4S)-4-Фтор-1-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 382
Согласно тому, как описано в примере 339, из соединения 93, гидрохлорида 4-цис-фтор-L-пролинамида и ТЭА (2,5 экв.) получали неочищенный продукт, который перекристаллизовывали из смеси IMS/ДХМ, при этом получали соединение 382 в виде твердого вещества белого цвета (68 мг, выход 42%). ЖХМС: RT=2,58 мин, [М+Н]+ 441. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,04 (1Н, s), 7,76 (1H, s), 7,10 (1H, s), 6,91 (1H, s), 5,95 (1H, s), 5,87-5,80 (1H, m), 5,42 (1H, s), 5,29 (1H, s), 4,49-4,36 (5 H, m), 3,74-3,73 (2H, m), 2,46-2,45 (1H, m), 2,20 (3H, s), 1,40 (6H, dd, J=6,59, 3,37 Гц).
Пример 383
(2S)-4,4-Дифтор-1-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 383
Согласно тому, как описано в примере 339, из соединения 93, гидрохлорида 4,4-дифтор-L-пролинамида и ТЭА (2,5 экв.) получали неочищенный продукт, который очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 5% метанол в ДХМ), при этом получали соединение 383 в виде твердого соединения белого цвета (29 мг, выход 17%). ЖХМС: RT=3,14 мин, [М+Н]+=459. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,09 (1H, s), 7,85 (1H, d, J=0,64 Гц), 7,55 (1H, ушир.), 7,15 (1H, ушир.), 6,05 (1H, s), 5,93-5,81 (1H, m), 4,61 (1H, dd, J=9,53, 4,24 Гц), 4,58-4,49 (4H, m), 4,00-3,98 (2H, m), 2,91-2,90 (1H, m), 2,45 (1H, dd, J=13,51, 4,18 Гц), 2,25 (3Н, s), 1,49 (6H, dd, J=6,59, 3,35 Гц).
Пример 384
(2S,4S)-4-Гидрокси-1-(2-(1-изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 384
Согласно тому, как описано в примере 339, из соединения 93 и 4-цис-гидрокси-L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS, при этом получали соединение 384 в виде твердого вещества белого цвета (75 мг, выход 28%). ЖХМС: RT=2,32 мин, [М+Н]+ 425. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,08 (1H, s), 7,90 (1H, d, J=0,63 Гц), 7,85 (1H, s), 7,47 (1H, ушир.), 7,13 (1H, ушир.), 6,00 (1H, s), 5,95-5,94 (1H, m), 5,28 (1H, d, J=6,27 ушир.), 4,51 (4H, d, J=11,64 Гц), 4,32 (2H, m), 3,60 (1H, m), 3,50 (1H, m), 2,42 (1H, t, J=4,54 Гц), 1,94 (1H, d, J=13,24 Гц), 1,49 (6H, dd, J=6,59, 2,75 Гц).
Пример 385
(2S,4S)-4-Гидрокси-1-(2-(1-изопропил-3-methyl-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 385
Согласно тому, как описано в примере 339, из соединения 93 и 4-цис-гидрокси-L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS, при этом получали соединение 385 в виде твердого вещества белого цвета (74 мг, выход 46%). ЖХМС: RT=2,34 мин, [М+Н]+ 439. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,05 (1Н, s), 7,79 (1H, s), 7,45 (1H, ушир.), 7,11 (1Н, ушир.), 5,98 (1H, s), 5,90-5,84 (1H, m), 5,27 (1H, d, J=6,27 Гц), 4,48 (4H, m), 4,31-4,28 (2H, m), 3,59 (1H, dd, J=10,61, 4,77 Гц), 3,48 (1H, d, J=10,45 Гц), 2,40-2,38 (1H, m), 2,24 (3H, s), 1,93 (1H, d, J=13,06 Гц), 1,45 (6H, dd, J=6,59, 2,55 Гц).
Пример 386
2-(1-Изопропил-1Н-имидазол-2-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин 386
9-Хлор-2-(1-изопропил-1Н-имидазол-2-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин (0,044 г, 0,13 ммоль), полученный согласно тому, как описано в примере 377, смешивали с этанолом (5 мл) и добавляли 20% гидроксид палладия на угле (19 мг, 0,03 ммоль) и ледяную уксусную кислоту (0,2 мл). Смесь вакуумировали и заполняли водородом. Данный процесс повторяли 2 раза. Затем реакционную смесь перемешивали в атмосфере водорода при КТ в течение 2 ч. Смесь фильтровали через слой целита и концентрировали в вакууме. Остаток очищали обращенно-фазной ЖХВР (элюент: 0,1% NH4OH в ацетонитриле), при этомполучали 13 мг (выход 39%) соединения 386. МС (ESI+) m/z 296,1 (М+Н+), расч. 295,3. 1H ЯМР (400 МГц, ДМСО): δ 9,47 (s, 1Н), 8,33 (d, J=5,6 Гц, 1H), 7,74 (s, 1H), 7,33 (d, J=1,1 Гц, 1H), 7,03 (d, J=5,6 Гц, 1H), 6,92 (d, J=0,9 Гц, 1H), 5,66 (dq, J=13,3, 6,6 Гц, 1H), 4,57 (td, J=7,9, 3,5 Гц, 4Н), 1,38 (d, J=6,6 Гц, 6Н).
Пример 387
(5-(9-Хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f]|[1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-3-ил)метанол 387
Суспензию 8-хлор-2-[5-метоксиметил-2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена (500 мг, 1,20 ммоль) в 48% водном растворе HBr (2,1 мл) закрывали и нагревали при 100°С в течение 3 ч при осторожном мониторинге методом ЖХМС. Смесь охлаждали до КТ и выливали в холодный 10% раствор КОН. Твердое вещество удаляли фильтрованием (чистота приблизительно 90% по данным ЖХМС и 1Н ЯМР). Твердое вещество использовали на следующих стадиях без дополнительной очистки. Небольшое количество твердого вещества очищали ЖХВР и получали 17 мг соединения 387. ЖХМС: 401,0. 1Н ЯМР (400 МГц, ДМСО): δ 9,27 (s, 1Н), 8,11 (s, 1H), 7,24 (s, 1H), 5,81 (q, J=8,9 Гц, 2Н), 5,34 (t, J=6,0 Гц, 1H), 4,71-4,57 (m, 4H), 4,46 (d, J=6,0 Гц, 2Н).
Соединеие 388
(2R)-1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)-2,5-дигидро-1Н-пиррол-2-карбоксамид 388
Раствор 2,64 г (8,00 ммоль) 9-хлор-2-(1-зопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина 264 в 250 мл ледяной уксусной кислоты помещали в стеклянный сосуд объемом 350 мл и пригодный для высокого давления. Сосуд закрывали и смесь нагревали в течение 64 ч при 155°С. Смесь охлаждали до КТ, при этом наблюдали образование осадка белого цвета. Смесь концентрировали в вакууме до объема 50 мл, твердое вещество отфильтровывали, промывали уксусной кислотой, этиловым эфиром и сушили на врздухе, при этом получали 1,68 г продукта. Продукт перемешивали вместе с 15 мл насыщенногораствора NaHCO3 в течение 1 ч, отфильтровывали, промывали 2 раза по 10 мл воды, растворяли в смеси ТГФ/вода (10:1) и концентрировали. Остаток сушили в высоком вакууме в течение 18 ч и получали 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9(10Н)-он с выходом 1,12 г (45%). M/z 313,1, расч. 312,13. 1H ЯМР (400 МГц, ДМСО): δ 11,79 (s, 1H), 8,41 (s, 1H), 7,89 (s, 1H), 7,82 (s, 1H), 5,84 (s, 1H), 5,78 (dt, J=13,2, 6,6, 1H), 4,57-4,51 (m, 2Н), 4,50-4,45 (m, 2Н), 1,45 (d, J=6,6, 6H).
Гидрид натрия (192 мг, 4,80 ммоль, 60% суспензия в минеральном масле) добавляли в суспензию 600 мг (1,92 ммоль) 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9(10Н)-она в 5,0 мл диметилформамида и смесь нагревали при 45°С в течение 1 ч. В смесь добавляли N-фенилбис(трифторметансульфонимид) (1373 мг, 3,842 ммоль) и реакционную смесь нагревали при 45°С в течение 20 ч. Смесь распределяли между EtOAc и 5% лимонной кислотой, органические экстракты промывали водой (×2), солевым раствором, сушили над MgSO4 и концентрировали. Остаток очищали на колонке с 12 г силикагеля (элюент: 4% МеОН в ДХМ), при этом получали 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-илтрифторметансульфонат с выходом 560 мг (66%). M/z 445,2, расч. 444,08.
Смесь 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-илтрифторметансульфоната, (S)-2,5-дигидро-1Н-пиррол-2-карбоксамида (84,1 мг, 0,750 ммоль) и N,N-диизопропилэтиламина (61,0 мкл, 0,350 ммоль) в N,N-диметилацетамиде (3,0 мл, 32 ммоль) нагревали в течение 18 ч при 80°С. Смесь концентрировали в высоком вакууме, остаток растирали с 5% водным раствором лимонной кислоты. Осадок отфильтровывали, промывали водным раствором лимонной кислоты, водой, сушили в вакууме и очищали обращенно-фазной ЖХВР (элюент: градиент ацетонитрила), при этом получали соединение 388 с выходом 28 мг. M/z 407,2, расч. 406,19. 1Н ЯМР (400 МГц, ДМСО): δ 9,08 (s, 1Н), 7,88 (s, 1H), 7,84 (s, 1H), 7,38 (s, 1H), 6,98 (s, 1H), 6,13 (dd, J=6,2, 1,9, 1H), 6,02-5,88 (m, 3H), 4,97 (s, 1H), 4,52 (dd, J=9,3, 7,1, 4H), 4,39-4,18 (m, 2H), 1,48 (dd, J=6,6, 2,5, 6H).
Пример 390
(5-(9-(Пирролидин-1-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-3-ил)метанол 390
Из (5-(9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-3-ил)метанола 387 и пирролидина получали соединение 390 (13 мг) в виде бесцветного твердого вещества. ЖХМС: 436,1. 1H ЯМР (400 МГц, ДМСО): δ 9,05 (s, 1H), 7,93 (s, 1H), 5,95 (s, 1H), 5,84 (q, J=8,9 Гц, 2H), 5,32 (t, J=6,1 Гц, 1H), 4,50 (m, 4H), 4,45 (d, J=6,0 гц, 2H), 3,41 (m, 4H), 1,95 (m, 4H).
Пример 391
(2S)-1-(2-(1-(3,5-Дифторфенил)-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 391
Из амида (S)-1-(2-иод-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-8-ил)-пирролидин-2-карбоновой кислоты, гидрохлорида ацетамида и гидрохлорида 3,5-дифторфенилгидразина, согласно тому, как описано в примере 420, получли неочищенный продукт, который очищали обращенно-фазной ЖХВР, при этом получали соединение 391 (13 мг, выход 11%). ЖХМС: 493,2. 1Н ЯМР (400 МГц, ДМСО): δ 8,58 (s, 1Н), 7,82 (s, 1H), 7,45 (m, 3H), 7,32 (s, 1H), 6,91 (s, 1H), 5,87 (s, 1H), 4,45 (m, 4H), 4,25 (d, J=7,8 Гц, 1H), 3,59 (m, 1H), 3,42-3,31 (m, 1H), 2,34 (s, 3H), 2,22-2,09 (m, 1H), 2,00-1,86 (m, 3H).
Пример 392
(2S)-2-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-иламино)пропанамид 392
Раствор, содержащий 9-хлор-2-(1-изоропил-1Н-1,2,4-триазол-5-ил1)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепина 264 (0,1 г, 0,3 ммоль), полученный согласно тому, как описано в примере 377, и гидрохлорид L-аланинамида (0,15 г, 1,21 ммоль) в N,N-диметилацетамиде (1 мл) нагревали при 90°С в течение 16 ч. Неочищенную реакционную смесь очищали обращенно-фазной ЖХВР и получали 8 мг (выход 7%) соединения 392. МС (ESI+) m/z 383,1 (М+Н+), расч. 383,2. 1H ЯМР (400 МГц, ДМСО): δ 8,99 (s, 1H), 7,88 (s, 1H), 7,81 (s, 1H), 7,30 (s, 1H), 6,90 (s, 1H), 6,81 (d, J=7,4 Гц, 1H), 6,12 (s, 1H), 5,92 (dt, J=13,1, 6,5 Гц, 1H), 4,47 (q, J=5,8 Гц, 4H), 4,37-4,21 (m, 1H), 1,47 (dd, J=6,6 Гц, 6Н), 1,25(d, J=7,0 Гц, 3H).
Пример 393
(2S)-1-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f]оксазепин-9-ил)-3,3-диметилпирролидин-2-карбоксамид 393
Согласно тому, как описао в примере 388, из 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-илтрифторметансульфоната и (S)-3,3-диметилпирролидин-2-карбоксамида получали соединение 393. M/z 437,2, расч. 436,23. 1Н ЯМР (400 МГц, ДМСО): δ 9,05 (s, 1H), 7,88 (s, 1H), 7,82 (s, 1H), 7,40 (s, 1H), 6,97 (s, 1H), 5,95(dd, J=14,5, 7,9, 2Н), 4,49 (dd, J=14,6, 5,6, 4H), 3,90 (s, 1H), 3,63 (t, J=8,5, 1H), 3,43 (d, J=7,8,1H), 1,96 (dd, J=21,1, 9,3, 1H), 1,68 (dd, J=11,9, 5,8, 1H), 1,48 (dd, J=6,6, 3,6, 6H), 1,08 (d, J=6,9, 6H).
Пример 394
(5-(9-(Диметиламино)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-3-yl)метанол 394
Из (5-(9-хлор-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-3-ил)метанола 387 и гидрохлорида диметиламина получали соединение 394 (11 мг) в виде бесцветного твердого вещества. ЖХМС: 410,1. 1H ЯМР (400 МГц, ДМСО): δ 9,06 (s, 1H), 7,94 (s, 1H), 6,13 (s, 1H), 5,84 (q, J=8,9 Гц, 2Н), 5,32 (t, J=6,1 Гц, 1H), 4,50 (m, 4H), 4,45 (d, J=6,0 Гц, 2Н), 3,05 (s, 6H).
Пример 395
(2S,3S)-3-Гидрокси-1-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 395
Согласно тому, как описано в примере 339, из соединения 92 и 3-транс-гидрокси-L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS и получали соединение 395 в виде твердого вещества белого цвета (63 мг, выход 39%). ЖХМС: RT=2,23 мин, [М+Н]+ 439. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,07 (1Н, s), 7,80 (1Н, s), 7,38 (1Н, ушир.), 7,02 (1Н, ушир.), 5,97 (1Н, s), 5,91-5,90 (1Н, m), 5,27 (1Н, d, J=3,77 Гц), 4,50 (5Н, m), 4,35 (1Н, t, J=5,08 гц), 4,27 (2Н, m), 3,65 (1Н, m), 2,25 (3Н, s), 2,07 (1Н, m), 1,89 (1Н, m), 1,46 (6Н, dd, J=6,60, 3,78 Гц).
Пример 396
(2S,3R)-3-Гидрокси-1-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 396
Согласно тому, как описано в примере 339, из соединения 92 и 3-цис-гидрокси-L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS и получали соединение 396 в виде твердого вещества белого цвета (37 мг, выход 23%). ЖХМС: RT=2,29 мин, [М+Н]+ 439. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,04 (1Н, s), 7,78 (1Н, s), 7,17 (1Н, ушир.), 6,91 (1Н, ушир.), 5,91 (1H, s), 5,88 (1H, m), 5,22 (1H, d, J=4,70 Гц), 4,47 (4H, m), 4,24 (1H, m), 3,64 (1H, m), 3,37 (1H, m), 2,24 (3H, s), 2,07 (2H, m), 2,02 (1H, m), 1,44 (6H, dd, J=6,59, 3,11 Гц).
Пример 397
(2S,3R)-3-Гидрокси-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 397
Согласно тому, как описано в примере 339, из соединения 93 и 3-цис-гидрокси-L-пролинамида получали неочищенный продукт, который очищали обращенно-фазной ЖХВР (колонка С18, элюент: градиент от 15 до 55% метанол в воде + 0,1% НСО2Н, при этом получали соединение 397 в виде порошкообразного вещества белого цвета (11 мг, выход 6%). ЖХМС: RT=2,28 мин, [М+Н]+ 425. 1H ЯМР 400 МГц (ДМСО-d6): δ 9,08 (1H, s), 7,91 (1H, d, J=0,63 Гц), 7,85 (1H, s), 7,20 (1Н, ушир.), 6,94 (1Н, ушир.), 6,00-5,98 (1H, m), 5,94 (1H, s), 5,25 (1H, d, J=4,81 Гц), 4,51-4,50 (4H, m), 4,47 (1H, m), 4,27 (1H, m), 3,68 (1H, m), 3,41 (1H, m), 2,09-2,08 (1H, m), 2,04-2,01 (1H, m), 1,50 (6H, dd, J=6,59, 3,26 Гц).
Пример 398
(2S,3S)-3-Гидрокси-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 398
Согласно тому, как описано в примере 339, из соединения 93 и 3-транс-гидрокси-L-пролинамида получали неочищенный продукт, который очищали обращенно-фазной ЖХВР (колонка С18, элюент: градиент от 15 до 55% метанол в воде + 0,1% НСО2Н, при этом получали соединение 398 в виде порошкообразного вещества белого цвета (63 мг, выход 39%). ЖХМС: RT=2,20 мин, [М+Н]+ 425. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,09 (1Н, s), 7,91 (1H, d, J=0,63 Гц), 7,86 (1H, s), 7,40 (1H, ушир.), 7,04 (1H, ушир.), 5,99 (1H, m), 5,89 (1Н, s), 5,29 (1H, d, J=3,76 Гц), 4,53 (4H, m), 4,29 (1H, m), 4,21 (1H, ушир.), 3,66 (1H, d, J=m) 3,50 (1H, m), 2,13-2,02 (1H, m), 1,91 (1H, dd, J=12,83, 6,39 ушир.), 1,50 (6H, dd, J=6,59, 3,95 ушир.).
Пример 399
(2S)-1-(2-(3-Метил-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 399
Согласно тому, как описано в примере 378, из 1-диметиламинометилиденамида 8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-карбоновой кислоты и трифторэтилгидразина получали 8-хлор-2-[5-метил-2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен (MC(ESI+) 385,1/387,1), из которого и L-пролинамида полчали соединение 399. MC(ESI+) 463,2. 1H ЯМР (400 МГц, ДМСО): δ 9,02 (s, 1H), 7,92 (s, 1H), 7,33 (s, 1H), 6,91 (s, 1H), 5,94 (s, 1H), 5,86-5,72 (m, 2H), 4,55-4,54 (m, 4H), 4,35-4,26 (m, 1H), 3,65-3,56 (m, 1H), 3,43-3,35 (m, 1H), 2,28 (s, 3H), 2,22-2,10 (m, 1H), 2,04-1,90 (m, 3H).
Пример 400
(2S,4R)-4-Фтор-1-(2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 400
Согласно тому, как описано в примере 339, из соединения 92 и 4-транс-фтор-L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS и получали соединение 400 в виде твердого вещества белого цвета (53 мг, выход 33%). ЖХМС: RT=2,77 мин, [М+Н]+ 441. 1Н ЯМР 400 МГц (ДМСО-d6): δ 9,01 (1Н, s), 7,75 (1Н, s), 7,51 (1Н, s), 7,00 (1Н, s), 5,96 (1Н, s), 6,00-5,66 (1Н, m), 5,46 (1Н, s), 5,33 (1Н, s), 4,45-4,44 (4Н, m), 4,34 (1Н, t, J=8,00 Гц), 3,86 (1Н, s), 3,71 (1Н, ddd, J=36,66, 12,73, 3,36 Гц), 2,53-2,52 (1Н, m), 2,20 (3Н, s), 1,40 (6Н, dd, J=6,59, 2,53 Гц).
Пример 401
(2S,4R)-4-фтор-1-(2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 401
Согласно тому, как описано в примере 339, из соединения 92 и 4-транс-фтор-L-пролинамида получали неочищенный продукт, который перекристаллизовывали из IMS и получали твердое вещество. Маточные жидкости концентрировали в вакууме и остаток очищали экспресс-хроматографией (SiO2, элюент: градиент от 0 до 10% метанол в ДХМ), чистые фракции объединяли, при этом получали соединение 401 в виде твердого вещества белого цвета (45 мг, выход 28%). ЖХМС: RT=2,80 мин, [М+Н]+=427. 1H ЯМР 400 МГц (ДМСО-d6): δ 9,02 (1Н, s), 7,84 (1H, d, J=0,63 Гц), 7,80 (1Н, s), 7,51 (1H, s), 7,00 (1H, s), 5,97 (1H, s), 5,90-5,89 (1H, m), 5,46 (1H, s), 5,33 (1H, s), 4,50-4,43 (4H, m), 4,34 (1H, t, J=8,09 Гц), 3,73-3,72 (1H, m), 2,53-2,52 (1H, m), 2,13-2,12 (1H, m), 1,43 (6H, dd, J=6,59, 2,68 Гц).
Пример 405
(2S)-1-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)-N-метилпирролидин-2-карбоксамид 405
Согласно тому, как описано в примере 388, из 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-илтрифторметансульфоната и (S)-N-метилпирролидин-2-карбоксамида получали соединение 405. M/z 423,2, расч. 422,22. 1Н ЯМР (400 МГц, ДМСО): δ 9,06 (s, 1Н), 7,88 (s, 1H), 7,83 (s, 1H), 7,74 (d, J=4,3, 1H), 5,95 (p, J=6,7, 2H), 4,58 - 4,41 (m, 4H), 4,35 (d, J=8,5, 1H), 3,67-3,57 (m, 1H), 3,41-3,33 (m, 1H), 2,57 (d, J=4,6, 3H), 2,15 (dd, J=10,8, 7,7, 1H), 1,94 (d, J=6,6, 3H), 1,47 (dd, J=6,6, 2,4, 6H).
Пример 406
(2S,3S)-1-(2-(1-Изопропил-1H-1,2,4-триазол-5-ил)-5,6-дигидроимидазо [1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)-3-метилпирролидин-2-карбоксамид 406
Согласно тому, как описано в примере 388, из 2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-илтрифторметансульфоната и (2S,3S)-3-метилпирролидин-2-карбоксамида получали соединение 406. M/z 432,2, расч. 422,22. 1H ЯМР (400 МГц, ДМСО): δ 9,05 (s, 1H), 7,88 (s, 1H), 7,82 (s, 1H), 7,36 (s, 1H), 6,91 (s, 1H), 6,57 (s, 1H), 6,01-5,88 (m, 2H), 4,49 (d, J=8,8, 4H), 3,88 (s, 1H), 3,55 (t, J=6,7, 2H), 2,32 (dd, J=10,3, 6,4, 1H), 2,18-2,09 (m, 1H), 1,59 (dd, J=12,0, 5,7, 1H), 1,48 (dd, J=6,6, 2,9, 6H), 1,10 (d, J=6,9, 3H).
Пример 409
(2S)-1-(2-(1-Циклогексил-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 409
Согласно тому, как описано в примере 378, из 1-диметиламинометилиденамида 8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-карбоновой кислоты и гидрохлорида циклогексилгибразина получали 8-хлор-2-(2-циклогексил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен (MC(ESI+) 371,2/373,2), из которого и L-пролинамида получали соединение 409. MC(ESI+) 449,2.
Пример 410
(2S)-1-(2-(1-(2-Хлорфенил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-9-ил)пирролидин-2-карбоксамид 410
Согласно тому, как описано в примере 378, из 1-диметиламинометилиденамида 8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-карбоновой кислоты и гидрохлорида 2-хлорфенилгидразина получали 8-хлор-2-[2-(2-хлорфенил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен (MC(ESI+) 399,1/401,1), из которого и L-пролинамида получали соединение 410. MC(ESI+) 477,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,26 (s, 1Н), 8,15 (s, 1H), 7,79 (s, 1H), 7,72 (dd, J=8,0, 1,1 Гц, 1H), 7,66-7,52 (m, 3H), 7,29 (ушир., 1H), 6,91 (ушир., 1H), 5,83 (s, 1H), 4,45-4,38 (m, 4H), 4,23 (d, J=7,7 Гц, 1H), 3,61-3,53 (s, 1H), 3,40-3,32 (m, 1H), 2,23-2,06 (m, 1H), 2,02-1,84 (m, 3H).
Пример 413
(5-(9-(Диметиламино)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-ил)метанол 413
Согласно тому, как описано в примере 420, из гидрохлорида 8-хлор-2-иод-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена, гидрохлорида 2-метоксиацетамидина игидрохлорида изопропилгидразина получали 8-хлор-2-(2-изопропил-5-метоксиметил-2Н-[1,2,4]триазол-3-ил)-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен, который собирали фильтрованием (1,42 г, выход 66%).
Из 8-хлор-2-[5-метоксиметил-2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена и 48% водным раствором HBr получали [5-(8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-ил]метанол после очистки FCC (CH2Cl2/MeOH), выход 29%.
Из [5-(8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-ил]метанола и гидрохлорида димтиламина получали соединение 413 (10 мг) в виде бесцветного твердого вещества. ЖХМС: 370,2. 1H ЯМР (400 МГц, ДМСО): δ 9,09 (s, 1Н), 7,80 (s, 1H), 6,13 (s, 1H), 5,98-5,85 (m, 1H), 5,18 (t, J=6,0 Гц, 1H), 4,56-4,44 (m, 4H), 4,41 (d, J=6,0 Гц, 2Н), 3,05 (s, 6H), 1,47 (d, J=6,6 Гц, 6Н).
Пример 417
(5-(9-(3,3-Дифторазетидин-1-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-ил)метанол 417
Из [5-(8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-yl]метанола и гидрохлорида 3,3-дифторазетидина получали соединение 417 (23 мг) в виде бесцветного твердого вещества. ЖХМС: 418,1. 1Н ЯМР (400 МГц, ДМСО): δ 9,14 (s, 1H), 7,85 (s, 1H), 6,16 (s, 1H), 5,87 (dt, J=13,2, 6,6 Гц, 1H), 5,18 (t, J=6,0 Гц, 1H), 4,60-4,47 (m, 4H), 4,41 (перекрывающийся m, 6H), 1,47 (d, J=6,6 Гц, 6H).
Пример 420
2-(3-Метил-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-9-(2-(2-метилбензил)пирролидин-1-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин 420
Раствор 8-хлор-2-иод-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулена (2,00 г, 0,58 ммоль) и гидрохлорида ацетамидина (0,653 г, 0,69 ммоль) растворяли в ДМФ (20 мл) и Et3N (4,0 мл) и в атмосфере азота обрабатывали 4,5-бис(дифенилфосфино)-9,9-диметлксантеном (реагент Xantphos, 0,166 г, 0,029 ммоль) и Pd(OAc)2 (0,0646 г, 0,029 ммоль). К колбе подсоединяли баллон с монооксидом углерода и реакционную смесь нагревали при 65°С 2 в течение 2 ч. После охлаждения до КТ в смесь добавляли раствор трифторэтилгидразина (70 мас. % в Н2О, 1,12 г, 0,69 ммоль) в уксусной кислоте (5 мл). После нагревания смеси в течение 3 ч при 65°С смесь охлаждали и разбавляли водой. 8-Хлор-2-[5-метил-2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен собирали фильтрованием (после промывания водой и гексаном и высушивания в вакууме получали 1,74 г, выход 78%). 1Н ЯМР (400 МГц, ДМСО): δ 9,26 (s, 1Н), 8,10 (s, 1H), 7,24 (s, 1H), 5,76 (q, J=8,8 Гц, 2Н), 4,71-4,53 (m, 4H), 2,29 (s, 3H).
В сосуд, объемом 10 мл и устойчивый к микроволновому облучению, помещали 8-хлор-2-[5-метил-2-(2,2,2-трифторэтил)-2Н-[1,2,4]триазол-3-ил]-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен (85 мг, 0,22 и) и 2-(2-метилбензил)-пирролидин (231 мг, 6 экв.). Затем в сосуд добавляли 0,5 мл NMП и 0,5 мл ТЭА. Сосуд закрывали и нагревали при 160°С в течение 24 ч. После охлаждения до КТ расвор очищали обращенно-фазной ЖХВР (0,1% NH4OH/ацетонитрил). Выделенное твердое вещество очищали хиральной СФХ (колонка AD, изократический элюент: 35% МеОН), при этом получали 27 мг одного энантиомера и и 29 мг соединения 420 (общий выход 48%). ЖХМС: 524,2. 1Н ЯМР (400 МГц, ДМСО): δ 9,08 (s, 1H), 9,08 (s, 1H), 7,92 (s, 1H), 7,27-7,06 (m, 4H), 5,99 (s, 1H), 5,80 (q, J=8,9 Гц, 2Н), 4,50 (m, 4H), 4,38 (ушир. s, 1H), 3,49 (t, J=8,6 Гц, 1H), 3,15 (d, J=10,0 Гц, 1H), 2,60-2,53 (m, 1H), 2,42 (s, 3H), 2,28 (s, 3H), 2,10-1,89 (m, 2Н), 1,81-1,71 (m, 2H).
Пример 421
2-(3-Метил-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-9-(2-(пиперидин-1-илметил)пиролидин-1-ил)-5,6-дигидроимидазо[1,2-d]пиридо[3,4-f][1,4]оксазепин 421
Из [5-(8-хлор-4,5-дигидро-6-окса-1,3а,9-триазабензо[е]азулен-2-ил)-1-изопропил-1Н-[1,2,4]триазол-3-ил]метанола и 1-пирролидин-2-илметилпиперидина после очистки продукта обращенно-фазной ЖХВР получали соединение 421 (67 мг). ЖХМС: 542,2.
Пример 423
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-9-(1-метил-1Н-пиразол-4-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 423
В сосуд, пргодный для микроволнового облучения, помещали 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина 411 (0,239 мг, 0,617 ммоль) в смеси 3,0 мл ацетонитрила/1,0 мл воды. В суспензию добавляли ацетат калия (182 мг, 1,85 ммоль) и пинаколиновый эфир 1-метилпиразолборной кислоты (154 мг, 740 ммоль). Реакционную суспензию дегазировали пробулькиванием азота с использованием шприца при перемешивании. Через несколько мин шприц отсоединяли, добавляли Pd(PPh3)4 (57 мг, 56 ммоль) и реакционный сосуд быстро закрыт.Закрытый сосуд нагревали с использованием микроволнового облучения при 150°С в течение 30 мин. Охлажденную реакционную смесь разбавляли EtOAc и органический раствор промывали водой, солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Неочищенный остаток очищали препаративной обращенно-фазной ЖХВР и получали 132 мг соединения 423 (теоретический выход 55%). 1H ЯМР (400 МГц, ДМСО): δ 8,36 (d, J=8,4 Hz, 1H), 8,20 (s, 1H), 7,90 (d, J=23,3 Гц, 2H), 7,36 (dd, J=8,4, 1,6 Гц, 1H), 7,25 (d, J=1,6 Гц, 1H), 5,83 (dt, J=13,3, 6,6 Гц, 1H), 4,51 (s, 4H), 3,87 (s, 3Н), 2,25 (s, 3Н), 1,47 (d, J=6,6 Гц, 6Н).
Пример 424
2-(3-Амино-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-10-карбонитрил 424
Согласно тому, как описнао в примере 425, из 2-(10-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-(2,2,2-триафторэтил)-1Н-имидазол-4-амина (150 мг, 0,350 ммоль) получали 31 мг соединения 424 (теоретический выход 24%). 1H ЯМР (400 МГц, ДМСО): δ 8,72 (d, J=2,1 Гц, 1H), 7,94 (s, 1H), 7,76 (dd, J=8,5, 2,1 Гц, 1H), 7,24 (d, J=8,6 Гц, 1H), 5,58 (q, J=8,8 Гц, 2H), 5,48 (d, J=12,2 Гц, 2H), 4,60 (d, J=9,0 Гц, 4H).
Пример 425
2-(3-Амино-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбонитрил 425
В сосуд, устойчивый к микроволновому облучению, помещали раствор 2-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-имидазо-4-амина (0,200 мг, 0,466 ммоль) в 1,0 мл ДМФ. В указанный раствор добавляли цианид цинка (160 мг, 1,4 ммоль). Реакционную смесь дегазировали пробулькиванием азота в смесь при перемешивании с использованием шприца. Через несколько минут шприц отсоединяли, добавляли бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий(II) (66 мг, 0,093 ммоль) и реакционный сосуд быстро закрывали. Закрытый сосуд нагревали при микроволновом облучении при 125°С в течение 30 мин. Охлажденную реационную смесь разбавляли EtOAc и органический раствор промывали водой, солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Неочищенный остаток очищали препаративной обращенно-фазной ЖХВР и получали 81 мг соединения 425 (теоретический выход 38%). 1Н ЯМР (400 МГц, ДМСО): δ 8,51 (dd, J=11,8, 5,7 Гц, 1Н), 8,07 (d, J=75,8 Гц, 1Н), 7,78 - 7,40 (m, 2H), 5,58 (q, J=8,9 Гц, 2Н), 5,50 (s, 2H).
Пример 431
5-(9-Циклопропил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-амин 431
В сосуд, устойчивый к микроволновому облучению, помещали 5-(9-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-изопропил-1Н-1,2,4-триазол-3-амин (100 мг, 0,200 ммоль) в 0,500 мл ТГФ и 0,300 мл воды. В полученный раствор добавляли фосфат калия (164 мг, 0,770 моль) и пинаколновый эфир циклопропилборной кислоты (308 мг, 1,48 моль). Реакционную смесь дегазировали пробулькиванием азота в смесь при перемешивании с использованием шприца. Через несколько минут шприц отсоединяли, добавляли бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий(II) (20 мг, 0,026 ммоль) и реакционный сосуд быстро закрывали. Закрытый сосуд нагревали при микроволновом облучении при 125°С в течение 30 мин. Охлажденную реационную смесь разбавляли EtOAc и органический раствор промывали водой, солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Неочищенный остаток очищали препаративной обращенно-фазной ЖХВР и получали 16,7 мг соединения 431 (теоретический выход 14%). 1Н ЯМР (400 МГц, ДМСО): δ 8,24 (d, J=8,3 Гц, 1Н), 7,69 (s, 1Н), 6,85 (d, J=8,4 Гц, 1Н), 6,74 (s, 1Н), 5,73 (dt, J=13,3, 6,8 Гц, 1Н), 5,15 (s, 2H), 4,46 (d, J=2,3 Гц, 4Н), 2,04-1,86 (m, 1Н), 1,40 (d, J=6,6 Гц, 6Н), 1,11-0,88 (m, 2H), 0,72 (q, J=4,9 Гц, 2H).
Пример 442
5-(10-Циклопропил-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-3-амин 442
Согласно тому, как описано в примере 431, из 5-(10-бром-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-2-ил)-1-(2,2,2-трифторэтил)-1Н-1,2,4-триазол-3-амина (150 мг, 0,350 ммоль) получали 8,4 мг соединения 442 (теоретический выход 6,4%). 1Н ЯМР (400 МГц, ДМСО): δ 8,72 (d, J=2,1 Гц, 1Н), 7,94 (s, 1H), 7,76 (dd, J=8,5, 2,1 Гц, 1Н), 7,24 (d, J=8,6 Гц, 1Н), 5,73 (dt, J=13,3, 6,8 Гц, 1Н), 5,15 (s, 2H), 4,46 (d, J=2,3 Гц, 4Н), 2,04-1,86 (m, 1Н), 1,40 (d, J=6,6 Гц, 6Н), 1,11-0,88 (m, 2H), 0,72 (q, J=4,9 Гц, 2H).
Пример 443
2-(1-Изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-карбонитрил 443
В сосуд, устойчивый к микроволновому облучению, помещали раствор 9-бром-2-(1-изопропил-3-метил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепина 411 (0,300 мг, 0,699 ммоль) в 2,0 мл ДМФ. В указанный раствор добавляли цианид цинка (250 мг, 2,1 ммоль). Реакционную смесь дегазировали пробулькиванием азота в смесь при перемешивании с использованием шприца. Через несколько минут шприц отсоединяли, добавляли бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий(II) (100 мг, 0,140 ммоль) и реакционный сосуд быстро закрывали. Закрытый сосуд нагревали при микроволновом облучении при 125°С в течение 30 мин. Охлажденную реационную смесь разбавляли EtOAc и органический раствор промывали водой, солевым раствором, сушили (Na2SO4) и концентрировали в вакууме. Неочищенный остаток очищали препаративной обращенно-фазной ЖХВР и получали 156 мг соединения 443 (теоретический выход 49%). 1Н ЯМР (400 МГц, ДМСО): δ 8,55 (d, J=8,2 Гц, 1Н), 8,02 (s, 1Н), 7,74-7,46 (m, 2H), 5,79 (dt, J=13,1, 6,6 Гц, 1Н), 4,57 (s, 4Н), 2,29 (d, J=28,5 Гц, 3Н), 1,46 (d, J=6,6 Гц, 6Н).
Пример 475
2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-9-(2-(метилсульфонил)фенил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 475
Раствор соединения 52 (0,050 г, 0,13 ммоль) и измельченного фосфата калия (0,0851 г, 0,401 ммоль) в ДМФ (0,5 мл) интенсивно дегазировали продуванием азота. В раствор добавляли 2-метилсульфонилфенилборную кислоту (0,053 г, 0,27 ммоль), ацетат палладия (0,0015 г, 0,0067 ммоль) и реагент S-Phos (0,00686 г, 0,0167 ммоль) и смесь перемешивали при микроволновом облучении в течение 30 мин при 180°С. Реакционную смесь разбавляли ДХМ и фильтровали через целит. В смесь добавляли насыщенный раствор NH4Cl и смесь 3 раза экстрагировали ДХМ. Органические слои объединяли, сушили над Na2SO4 и концентрировали. Несчитанный остаток очищали обращенно-фазной ЖХВР и получали 8,1 мг соединения 475 в виде твердого вещества белого цвета. MC(ESI+) 450,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,47 (d, J=8,3 Гц, 1Н), 8,11 (dd, J=8,0, 1,1 Гц, 1Н), 7,98 (s, 1Н), 7,92 (s, 1Н), 7,79 (td, J=7,5, 1,3 Гц, 1Н), 7,70 (td, J=7,8, 1,3 Гц, 1Н), 7,45 (dd, J=7,5, 1,1 Гц, 1Н), 7,21 (dd, J=8,3, 1,8 Гц, 1Н), 7,12 (d, J=1,7 Гц, 1Н), 5,93 (hept, J=6,2 Гц, 1Н), 4,57 (q, J=5,7 Гц, 4Н), 2,93 (s, 3Н), 1,50 (d, J=6,6 Гц, 6H).
Пример 476
2-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)бензамид 476
Согласно тому, как описано в примере 475, из соединения 52 и (2-аминокарбонилфенил)борной кислоты получали соединение 476. MC(ESI+) 415,2. 1Н ЯМР (400 МГц, ДМСО): δ 8,42 (d, J=8,3 Гц, 1Н), 7,95 (s, 1Н), 7,91 (s, 1Н), 7,70 (s, 1Н), 7,54-7,39 (m, 4Н), 7,34 (s, 1Н), 7,22 (dd, J=8,3, 1,7 Гц, 1Н), 7,12 (d, J=1,7 Гц, 1Н), 5,93 (гепт., J=6,2 Гц, 1Н), 4,55 (q, J=5,7 Гц, 4Н), 1,50 (d, J=6,6 Гц, 6Н).
Пример 477
9-(2-Этилфенил)-2-(1-изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин 477
Согласно тому, как описано в примере 475, из соединения 52 и 2-этилфенилборной кислоты получали соединение 477. MC(ESI+) 400,2. 1Н ЯМР (400 МГц, ДМСО): δ 8,47 (d, J=8,2 Гц, 1Н), 7,95 (s, 1Н), 7,92 (s, 1Н), 7,38-7,31 (m, 2H), 7,30-7,23 (m, 1Н), 7,20 (d, J=7,3 Гц, 1Н), 7,12 (dd, J=8,2, 1,7 Гц, 1Н), 6,97 (d, J=1,6 Гц, 1Н), 5,93 (гепт., J=6,2 Гц, 1Н), 4,65-4,41 (m, 4Н), 2,61 (q, J=7,6 Гц, 2H), 1,50 (d, J=6,6 Гц, 6Н), 1,07 (t, J=7,5 Гц, 3Н).
Пример 478
(2-(2-(1-Изопропил-1Н-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)фенил)метанол 478
Согласно тому, как описано в примере 475, из соединения 52 и 2-(гидроксиметил)фенилборной кислоты получали соединение 478. MC(ESI+) 402,1. 1Н ЯМР (400 МГц, ДМСО): δ 8,46 (d, J=8,3 Гц, 1Н), 7,96 (s, 1Н), 7,92 (s, 1Н), 7,58 (d, J=7,7 Гц, 1H), 7,41 (td, J=7,4, 1,1 Гц, 1Н), 7,38-7,32 (m, 1H), 7,28 (d, J=7,4 Гц, 1H), 7,20 (dd, J=8,3, 1,6 Гц, 1H), 7,09 (d, J=1,5 Гц, 1H), 6,02-5,83 (m, 1H), 5,16 (t, J=5,3 Гц, 1H), 4,56 (q, J=6,1 Гц, 4Н), 4,45 (d, J=5,3 Гц, 2Н), 1,50 (d, J=6,6 Гц, 6H).
Пример 901
Анализ связывания р110α PI3K
Анализ связывания: эксперименты по первичной поляризации проводили в системе Analyst НТ 96-384 (фирмы Molecular Devices Corp, Саннивейл, Калифорния). Для измерения аффинности поляризации флуоресценции получали трехкратные серийные разведения p110α PI3K (фирмы Upstate Cell Signaling Solutions, Шарлотсвилль, Вирджиния), начальная концентрация составляла 20 мкг/мл в поляризационном буферном растворе (10 мМ Трис, рН 7,5, 50 мМ NaCl, 4 мМ MgCl2, 0,05% Chaps и 1 мМ DTT) в 10 мМ PIP2 (фирмы Echelon-Inc., Солт-Лейк-Сити, Юта). После выдерживания в течение 30 мин при КТ реакции останавливали при добавлении GRP-1 и зонда PIP3-TAMRA (фирмы Echelon-Inc., Солт-Лейк-Сити, Юта) до конечных концентраций 100 нМ и 5 нМ, соответственно. Образцы считывали с использованием стандартных фильтров для флуорофора родамина (λ возбуждения 530 нм, λ испускания 590 нм) в 384-луночных черных планшетах с малым объемом лунок Proxiplate (фирмы Perkin Elmer, Уэллсли, Массачусетс). Строили графики зависимости величины поляризации флуоресценции от концентрации белка, и рассчитывали значения ЕС50 по 4-х параметрическому уравнению с использованием программного обеспечения KaleidaGraph (Synergy software, Рединг, Пенсильвания). В указанном эксперименте также определяли концентрацию белка для использования в последующих экспериментах по конкурентному ингибированию.
Величину ингибирования IC50 определяли при добавлении р110α PI3K (конечная концентрация 0,04 мг/мл) в смеси с PIP2 (конечная концентрация 10 мМ) в лунки, содержащие трехкратные серийные разведения антагонистов в конечной концентрации 25 мМ АТФ (фирмы Cell Signaling Technology, Inc., Данверс, Массачусетс) в поляризационном буферном растворе. После выдерживания в течение 30 мин при КТ реакции останавливали при добавлении GRP-1 и зонда PIP3-TAMRA (фирмы Echelon-Inc., Солт-Лейк-Сити, Юта) в конечных концентрациях 100 нМ и 5 нМ, соответственно. Образцы считывали с использованием стандартных фильтров для флуорофора родамина (λ, поглощения 530 нм, λ испускания 590 нм) в 384-луночных черных планшетах с малым объемом лунок Proxiplate (фирмы Perkin Elmer, Уэллсли, Массачусетс). Строили график зависимости величины поляризации флуоресценции от концентрации антагониста и рассчитывали значения IC50 по 4-х параметрическому уравнению с использованием программного обеспечения Assay Explorer (фирмы MDL, Сан-Рамон, Калифорния).
В другом варианте ингибирование PI3K определяли радиометрическим анализом при использовании очищенного рекомбинантного фермента и АТФ при концентрации 1 мкМ. Готовили серийные разведения соединения формулы I в 100% ДМСО. Реакционную киназную смесь выдерживали в течение 1 ч при КТ и реакцию останавливали при добавлении ФСБ. Затем определяли значения IC50 с использованием сигмоидальной кривой зависимости ответной реакции от дозы (различный наклон).
Пример 902
Анализ пролиферации клеток in vitro
Эффективность соединений формулы I оценивали с использованием анализа пролиферации клеток согласно следующему протоколу (Promega Corp. Technical Bulletin TB288, Mendoza и др.. Cancer Res., 62, cc. 5485-5488 (2002)).
1. Аликвотную часть (100 мкл) клеточной культуры, содержащую приблизительно 104 клеток (РС3, Detroit562 или MDAMB361.1) в среде, помещали в каждую лунку 384-луночного планшета с непрозрачными стенками.
2. В контрольные лунки добавляли среду и не добавляли клетки.
3. Исследуемое соединение добавляли в лунки и выдерживали в течение 3-5 сут.
4. Планшеты уравновешивали при КТ в течение приблизительно 30 мин.
5. В каждую лунку добавляли реагент CellTiter-Glo в объеме, равном объему клеточной культуральной среды в лунке.
6. Содержимое перемешивали в течение 2 мин на роторной качалке для индуцирования лизиса клеток.
7. Планшет выдерживали при КТ в течение 10 мин для стабилизации сигнала люминесценции.
8. Регистрировали значения люминесценции и вводил в графу ОЕЛ (относительные единицы люминесценции).
В другом варианте клетки высевали при оптимальной плотности в лунки 96-луночного планшета и выдерживали в течение 4 сут в присутствии исследуемого соединения. Затем в среду для анализа добавляли Alamar Blue™, клетки выдерживали в течение 6 ч и планшет считывали при длине волны возбуждения 544 нм, длине волны испускания 590 нм. Значения ЕС50 рассчитывали с использованием сигмоидальной зависимости ответной реакции от дозы. Термин «ЕС50» обозначает полумаксимальную эффективную концентрацию, т.е. концентрацию, при которой уровень ответной реакции составляет половину расстояния между базовой линией и максимально возможной величиной ответной реакции через некоторый промежуток времени. Указанная величина обычно используется в качестве меры эффективности лекарственного средства.
Антипролиферативный эффект типичных соединений формулы I оценивали с использованием реагента CellTiter-Glo® для различных линий опухолевых клеток, как показано ниже.
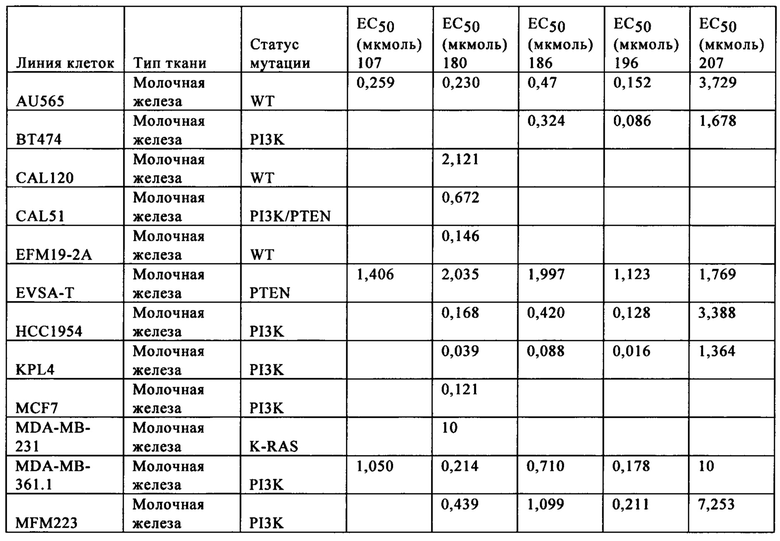

Пример 903
Проницаемость клеток Сасо-2
Клетки Сасо-2 высевали в планшеты Millipore Multiscreen при плотности 1×105 клеток/см2 и культивировали в течение 20 сут. Затем проводили оценку проницаемости клеток. Соединения наносили на верхнюю поверхность (А) монослоя клеток и измеряли проницаемость в базолатеральный компартмент (В). Исследования проводили в обратном направлении (В-А) для изучения активного транспорта. Рассчитывали коэффициент проницаемости, Рарр, для каждого соединения, указанный коэффициент является мерой скорости диффузии соединения через мембрану. Соединения объединяли в группы с низким (Рарр≤1,0×106 см/с) или высоким (Papp≥10×106 см/с) потенциалом абсорбции по сравнению с контрольными соединениями, для которых установлена величина абсорбции клетками человека.
Для оценки способности соединения активно высвобождаться из клетки оценивали соотношение базолатерального транспорта (В) и транспорта через верхний слой (А) по сравнению с величиной от А до В. Величины В-А/А-В≥1,0 указывают на активное высвобождение соединения из клетки.
Пример 904
Клиренс гепатоцитов
Использовали суспензию криоконсервированных гепатоцитов человека. Образцы выдерживали при концентрации соединения 1 мМ или 3 мкМ при плотности жизнеспособных клеток 0,5×106 клеток/мл. Конечная концентрация ДМСО в среде составляла приблизительно 0,25%. Проводили также контрольный эксперимент в отсутствии клеток для выявления неферментативной деградации. Из смеси отбирали образцы по 50 мкл в двух повторах через 0, 5, 10, 20, 40 и 60 мин (контрольный образец отбирали только через 60 мин) и для остановки реакции в образцы добавляли метанол, содержащий внутренний стандарт (100 мкл). В качестве контрольных соединений использовали толбутамид, 7-гидроксикумарин и тестостерон. Образцы центрифугировали и из супернатантов для каждого промежутка времени отбирали пробы для анализа методом ЖХ-МС/МС. Строили зависимость отношения логарифма площади пиков (площадь пика исходного соединения/площадь пика внутреннего стандарта) от времени и собственный клиренс (CLint) рассчитывали по следующему уравнению: CLint (мкл/мин/миллион клеток)=V×k, где k обозначает константу скорости элиминации, полученную из зависимости градиента концентрации от времени, V обозначает объем, полученный из объема образца после инкубации, измеренный в мкл × 106 × клеток-1.
Пример 905
Ингибирование цитохрома Р450
Соединения формулы I оценивали по их действию на мишени CYP450 (1А2, 2С9, 2С19, 2D6, 3А4) при приблизительно 10 концентрациях в двух повторах, причем максимальная концентрация составляла приблизительно 100 мкМ. В качестве контроля использовали стандартные ингибиторы (фурафиллин, сульфафеназол, транилципромин, квинидин, кетоконазол). Планшеты считывали с использованием прибора BMG LabTechnologies PolarStar в режиме флуоресценции.
Пример 906
Индукция цитохрома Р450
Свежевыделенные гепатоциты человека от одного донора культивировали в течение приблизительно 48 ч, затем добавляли соединение формулы I при трех концентрациях и выдерживали в течение 72 ч. Зонды-субстраты для CYP3A4 и CYP1A2 добавляли за 30 мин и 1 ч до завершения выдерживания. Через 72 ч клетки и среду удаляли и степень метаболизма каждого зонда-субстрата оценивали методом ЖХ-МС/МС. Эксперимент контролировали при использовании индукторов индивидуальных Р450, выдержанных при одной концентрации в трех повторах
Пример 907
Связывание белков плазмы
Растворы соединений формулы I (5 мкм, конечная концентрация ДМСО 0,5%) готовили в буферном растворе и 10% плазме (об./об. в буферном растворе). 96-Луночный планшет для диализа НТ собирали таким образом, чтобы каждая лунка была разделена на две части при помощи полупроницаемой целлюлозной мембраны. С одной стороны мембраны помещали буферный раствор, а с другой стороны раствор плазмы, затем планшет выдерживали при 37°С в течение 2 ч в трех повторах. Затем содержимое лунок удаляли, растворы каждой серии соединений объединяли в две группы (не содержащие и содержащие плазму) и анализировали методом анализа ЖХ-МС/МС с использованием двух типов калибровочных стандартов для не содержащих плазму (6 точек) и содержащих плазму (7 точек) растворов. Рассчитывали долю несвязанного соединения.
Пример 908
Блокирование канала hERG
Оценивали способность соединений формулы I модулировать отток рубидия из клеток HEK-294, стабильно экспрессирующих калиевые каналы hERG, при использовании стандартной методики притока. Клетки готовили в среде, содержащей RbCl, высевали в 96-луночные планшеты и выращивали в течение ночи до образования монослоев. Эксперимент по оттоку инициировали при удалении среды и трижды промывали каждую лунку 100 мкл предварительно выдержанного при КТ буферного раствора (содержащего низкую концентрацию [K+]). После последнего удаления среды в каждую лунку добавляли 50 мкл рабочего (2×) раствора соединения и выдерживали при КТ в течение 10 мин. Затем в каждую лунку добавляли 50 мкл стимулирующего буферного раствора (содержащего высокую концентрацию [K+]), при этом получали конечные концентрации исследуемых соединений. Планшеты с клетками выдерживали при КТ в течение еще 10 мин. Затем 80 мкл супернатанта из каждой лунки переносили в аналогичные лунки 96-луночного планшета и анализировали методом атомной эмиссионной спектроскопии. Соединение анализировали по 10 точкам кривых IC50, n=2, начиная с самой высокой концентрации 100 мкМ.
Пример 909
Ксенографты опухоли in vivo
Животных, пригодных для трансгенных экспериментов, получали из обычных коммерческих источников. Группам «голых» мышей Taconic имплантировали в бок подкожно клетки рака молочной железы MDA-MB-361.1 (мутантный PI3K). Мышам с ксенотрансплантатами каждые сутки в течение 21 дня вводили лекарственное средство или носитель. Объем опухоли регистрировали дважды в неделю в ходе испытаний. Массу мышей также регистрировали дважды в неделю и регулярно обследовали состояние мышей. Объем опухоли регистрировали в двух направлениях (длина и ширина) с использованием штангенциркуля Ultra Cal-IV (модель 54-10-111, Fred V., Fowler Co., Inc., Ньютон, Массачусетс) и данные анализировали с использованием программы Excel, версия 11.2 (фирмы Microsoft Corporation, Редмонд, Вашингтон). Графики подавления роста опухоли строили с использованием программного обеспечения Kaleida Graph, версия 3.6 (Synergy Software, Рединг, Пенсильвания). Объем опухоли рассчитывали по формуле: размер опухоли (мм3) = (длина опухоли × ширина опухоли2) × 0,5.
Массу тела животных измеряли с использованием весов Adventurera Pro AV812 (фирмы Ohaus Corporation, Пайн Брук, Нью-Джерси). Графики строили с использованием программного обеспечения KaleidaGraph, версия 3.6. Изменение массы в % рассчитывали по следующей формуле: изменение массы в % группе = (1-(исходная масса/измеренная масса))×100.
Мышей, у которых объем опухоли превышал 2000 мм3 или потеря массы тела составляла >20% от исходной массы тела умерщвляли в соответствии с правилами испытаний, утвержденными соответствующим учреждением.
Подавление роста опухоли (% INH) после завершения испытаний (EOS) рассчитывали по формуле: % INH=100×(EOS средний объем опухоли у животных, которым вводили носитель, - EOS средний объем опухоли у животных, которым вводили лекарственное средство)/EOS средний объем опухоли у животных, которым вводили носитель.
Частоту опухолей (ЧО) определяли по числу измеряемых опухолей, оставшихся в каждой группе после завершения испытаний. Частичную ответную реакцию (ЧОР) определяли при снижении объема опухоли >50% но <100% по сравнению с исходным объемом при определении в любой день испытаний. Полную ответную реакцию (ПОР) определяли по снижению объема опухоли на 100% по сравнению с исходным объемом опухоли при определении в любой день испытаний. Результаты анализировали и определяли величину р с использованием критерия Даннета и программного обеспечения JMP для статистических рассчетов, версия 5.1.2 (SAS Institute, Кэри, Северная Каролина). Объем индивидуальных опухолей после завершения испытаний и средний объем опухоли ± СО рассчитывали при использовании программного обеспечения JMP для статистических расчетов, версия 5.1.2. Строили графики изменения массы тела на основании среднего изменения в % от исходной массы тела ± СО.
Пример 910
Анализ индукции фосфоAKT
В 6-луночный планшет для тканевых культур высевали клетки при плотности 5×105 клеток/лунка и выдерживали в течение ночи. Клетки обрабатывали соединением формулы I в концентрации EC80. Клетки промывали один раз холодным ФСБ и лизировали в буферном растворе 1X Cell Extraction Buffer, Biosource (Карлсбад, Калифорния) с добавлением ингибиторов протеаз (Roche, Mannheim, Germany), 1 мМ фенилметансульфонилфторида и смеси ингибиторов фосфатаз 1 и 2, Sigma (Сент-Луис, Миссури). Определение концентрации белков проводили с использованием набора для анализа белков Pierce BCA Protein Assay Kit (Рокфорд, Иллинойс). Уровни pAkt (Ser473) и общий уровень Akt определяли с использованием набора Biosource (Карлсбад, Калифорния) и системы Luminex Bio-Plex (фирмы Bio-Rad, Геркулес, Калифорния).
Термины «содержат», «содержащий», «включают», «включающий» и «включает», использованные в данном контексте и в следующей формуле изобретения, обозначают присутствие указанных признаков, целых чисел, компонентов или стадий, но не исключают присутствие или добавление одного или более других признаков, чисел, компонентов, стадий или их групп.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗОКСАЗЕПИНОВЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2649976C2 |
| ИНГИБИТОР, СОДЕРЖАЩИЙ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2019 |
|
RU2800543C2 |
| ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2278865C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2013 |
|
RU2637944C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИПЕРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ АУТОТАКСИНА | 2022 |
|
RU2834850C2 |
Изобретение относится к соединению, имеющему структуру:
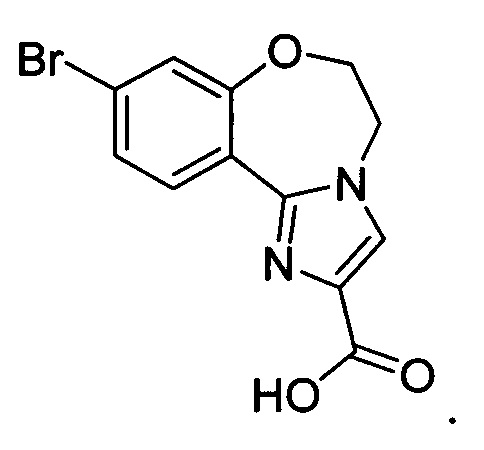
Технический результат: получено новое соединение, которое является промежуточным при синтезе бензоксазепиновых ингибиторов PI3. 3 табл., 910 пр.
1. Соединение, имеющее структуру:
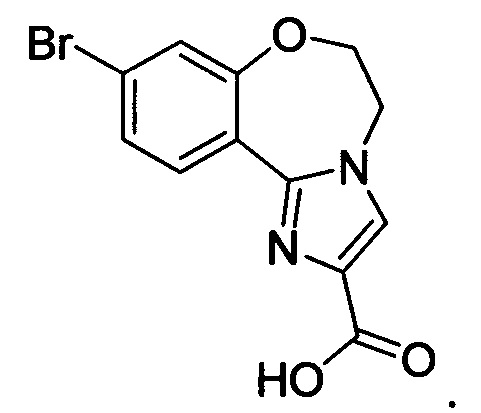
| WO 2007141491 A1, 13.12.2007 | |||
| Wang Q | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ПРОИЗВОДНЫЕ БЕНЗОКСАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СТИМУЛЯТОРОВ РЕЦЕПТОРА АМРА | 2002 |
|
RU2279434C2 |

