Область техники, к которой относится изобретение
Изобретение относится к фармацевтическим композициям в форме частиц или к твердым дозированным формам, включающим сочетание фибрата, а именно: фенофибрата и статина (так же известного как ингибитор HMG-CoA-редуктазы). При создании композиций нет необходимости добавлять воду или водную среду. В результате получают композиции, имеющие очень низкое содержание влаги (менее 2 мас.% воды), что обеспечивает подходящую стабильность при хранении (и фибраты, и статины разлагаются под действием воды). Кроме того, 80% активных веществ (то есть фибрата и статина) присутствует в композиции в растворенном виде, что обеспечивает подходящую биологическую доступность активных ингредиентов при приеме через рот.
Предпосылки создания изобретения
Фибраты представляют собой лекарственные вещества, которые в основном очень плохо и неоднородно абсорбируются после перорального введения. Обычно их рекомендуют принимать с пищей для повышения биологической доступности. Имеется множество улучшенных дозированных форм, содержащих наиболее часто используемый в настоящее время фибрат, фенофибрат, которые были разработаны с целью повышения биологической доступности лекарственного вещества и, в этой связи, его эффективности. Кроме того, в клинических руководствах указывается, что наиболее эффективным способом регуляции холестеринового и липидного обмена является не только терапия с использованием фибрата, но также комбинированная терапия с использованием, например, фенофибрата и статина. Фактически, лечение фенофибратом часто рекомендуют в сочетании с введением статина, поскольку клиницисты предпочитают использовать, например, фенофибрат в связи с его способностью снижать уровень триглицеридов, тогда как статин используется в связи с его положительным эффектом по снижению уровня ЛПНП-Х и по повышению уровня ЛПВП-Х. Однако, в настоящее время такая комбинированная терапия может быть проведена только при использовании двух отдельных продуктов, то есть пациентам нужно принимать, например, одну таблетку фибрата и другую таблетку или капсулу, содержащую статин.
Как отмечалось выше, существует потребность в получении улучшенных композиций фенофибрата, и, соответственно, в этой связи, появилось множество публикаций, относящихся к такого рода композициям (см., например, WO 04/041250). Хотя такие композиции удобнее для введения фибрата при необходимости соответствующего лечения, они не удовлетворяют имеющимся потребностям в композиции, содержащей сочетание фибрата и статина, которая была бы стабильна при хранении и в то же самое время обладала подходящей биологической доступностью относительно обоих активных веществ.
В WO 03/013608 описываются композиции, содержащие фибрат и статин. Однако, в связи с особенностями использованного способа их получения (активные вещества расплавляются вместе, вводятся в желатиновые капсулы, которые далее охлаждают), при этом могут быть получены только капсулы. Кроме того, с фармацевтической точки зрения процесс производства представляется сложным для его масштабирования, в плане требований, предъявляемых при регистрации препарата, из-за таких свойств, как изменения массы вещества, изменения содержания лекарственного компонента и т.п. Несмотря на то, что описываемая композиция может представлять собой твердую композицию, в данной работе не предусмотрены отступления от основного принципа создания препарата иного типа дозированной формы, нежели капсулы.
Соответственно, все еще сохраняется потребность в разработке фармацевтической композиции, которая представляла бы собой единую форму, содержащую фибрат и статин в качестве активных веществ, причем указанная композиция была бы стабильна и обеспечивала возможность проявления соответствующих биофармацевтических свойств входящих в нее активных веществ (например, подходящую биологическую доступность, меньшую зависимость от приема пищи и т.п.) и, кроме того, чтобы производство данной композиции могло без труда подвергаться масштабированию. Кроме того, все еще сохраняется потребность в разработке композиций, содержащих фибрат и статин, так чтобы такие композиции могли подвергаться дальнейшей обработке с получением фармацевтических дозированных форм, позволяющих выбирать конкретный вид дозирования формы. В фармацевтической области такая гибкость может быть достигнута в том случае, когда такая композиция представляет собой форму твердого продукта в виде порошка или в виде частиц. Соответственно, настоящее изобретение относится к такому материалу в форме частиц, который подходит для дальнейшей обработки с получением, например, таблеток.
Дополнительно, все еще сохраняется потребность в композиции, которая обладала бы подходящей биологической доступностью, что позволило бы существенно снизить или преодолеть существующие различия между биологической доступностью лекарственного вещества, характерной для пациентов, принимающих препарат натощак, и биологической доступностью лекарственного вещества (в частности, фенобирата) у пациентов, принимающих препарат после приема пищи, и/или которая позволила бы существенно снизить или преодолеть вариации, отмечаемые при приеме как для отдельного индивидуума, так и между отдельными индивидуумами при современном режиме лечения. Кроме того, также сохраняется потребность в композиции, которая позволяла бы снизить наблюдаемые подобные эффекты.
В целом, известно, что абсорбция и биологическая доступность терапевтически активного вещества может подвергаться воздействию множества факторов, в случае его введения через рот. Такие факторы включают наличие пищи в желудочно-кишечном тракте и, в целом, более длительное время пребывания лекарственного вещества в желудке в присутствии пищи, чем в состоянии натощак. Если биологическая доступность лекарственного вещества подвергается воздействию сверх некоторой определенной точки, в связи с наличием пищи в желудочно-кишечном тракте, считается, что лекарственное вещество оказывает пищевой эффект. Пищевые эффекты важны, поскольку, при их наличии, у пациента, который недавно принимал пищу, возникает риск, связанный с введением лекарственного вещества. Риск определяется возможностью того, что абсорбция в кровоток может быть ухудшена до такой степени, что может стать недостаточной для пациента, который принимает данный препарат для облегчения того состояния, которое требует данного лечения. В случае, например, фенофибрата, ситуация отличается в том смысле, что принимаемая пища, наоборот, повышает абсорбцию. Так что отсутствие приема пищи одновременно с лекарственными веществами может привести к недостаточной абсорбции. Степень абсорбции коммерчески доступного продукта Трикор® (Tricor®), содержащего фенофибрат (от компании Abbott), повышается примерно на 35% в случае приема пищи, в сравнении с условиями приема препарата натощак.
Как указывалось выше, все еще остается потребность в новых фармацевтических композициях, включающих один или несколько фибратов, которые проявляют подходящую биологическую доступность для активного соединения и/или сниженный или полностью отсутствующий пищевой эффект. В данном контексте термин "подходящая биологическая доступность" означает, что введение композиции по настоящему изобретению будет приводить к достижению повышенной биологической доступности, сравнительно с биологической доступностью, достигаемой после введения активного(ых) вещества(в) в виде простой таблетки; или биологическая доступность будет примерно такой же или повышенной в сравнении с биологической доступностью, получаемой после введения коммерчески доступного продукта, содержащего то/те же самое/самые активное(ые) вещество(а), в тех же количествах. В частности, желательно достигать более быстрой и более значительной и/или более полной абсорбции активного соединения, что позволит, в этой связи, снизить вводимые дозировки или уменьшить число ежедневных введений. Далее, фармацевтические композиции по настоящему изобретению также позволяют снизить или устранить потребность в приеме пищи одновременно с приемом дозированных форм (в частности, это релевантно для одного или нескольких активных веществ, содержащихся в композиции по настоящему изобретению, а именно фибрата, такого как фенофибрат), что даст пациентам больше свободы при выборе времени приема лекарственного препарата.
Описание изобретения
Настоящее изобретение относится к фармацевтическим композициям в форме частиц и твердых дозированных форм, предназначенных для лечения состояний, отзывчивых на лечение фибратом и статином.
Как указывалось выше, имеется потребность в разработке фармацевтических композиций, содержащих сочетание фибрата и статина или их фармацевтически приемлемую соль, для перорального введения, которые улучшали бы лечение тех состояний, при которых требуется регуляция липидного метаболизма (например, при атеросклерозе, ишемической болезни сердца, при диабете, ожирении, избыточном весе, метаболическом синдроме и т.п.).
Кроме того, было бы полезно добиться повышенной биологической доступности, особенно фибратного компонента, что, как будет описано ниже, в ряде случаев будет также существенно и для статинового компонента. Фибрат типа фенофибрата обладает очень низкой растворимостью в воде, и это его свойство рассматривается как одна из основных причин низкой биологической доступности фенофибрата. Соответственно, было бы выгодно получить композицию, в которой фибрат находится преимущественно в растворенном состоянии. То же самое относится к статинам, которые также обладают плохой водорастворимостью.
Повышенная биологическая доступность способствует улучшению результатов лечения. При этом возникает также возможность получить такой же самый терапевтический ответ, но при использовании сниженной дозы и/или при меньшем числе введений, при меньшей вариабельности плазменных уровней и без пищевых ограничений. Другой путь достижения улучшенных результатов лечения таких состояний, при которых показано введение фенофибрата, связан с регуляцией высвобождения фенофибрата в желудочно-кишечном тракте, таким образом, чтобы повышенная концентрация фенофибрата в плазме достигалась сразу же или с некоторой временной задержкой относительно времени введения. Дальнейшее терапевтическое усовершенствование может представлять собой разработку композиций с модифицированным или задержанным высвобождением, которые содержат один или несколько фибратов.
В особенности, имеется потребность в разработке твердой композиции в виде частиц, которая может быть в дальнейшем обработана с получением твердых дозированных форм (например, таблеток и т.п.). Такая композиция должна содержать активные лекарственные вещества, например, фибрат и статин, преимущественно в растворенном виде, но в то же самое время композиция должна быть в форме частиц, которые можно далее подвергнуть обработке с получением твердой дозированной формы, такой как таблетки, то есть материала, состоящего из частиц, который содержит активные вещества преимущественно в растворенном виде и который должен иметь приемлемые свойства, такие как, например, приемлемые свойства применительно к текучести, прилипаемости (она должна быть устранена), способности к прессованию и т.п.
В одном аспекте настоящее изобретение относится к материалу в форме частиц, включающему в качестве активного ингредиента один или несколько фибратов и один или несколько статинов, в котором по меньшей мере 80 мас.% от суммарного количества активных веществ растворено в носителе, выбранном из группы, состоящей из гидрофобного, гидрофильного и смешивающегося с водой носителя.
В норме, по меньшей мере 85 мас.%, по меньшей мере 90 мас.%, по меньшей мере 95 мас.% или по меньшей мере 98 мас.% от общего количества активных веществ растворяется в носителе.
Если имеют место варианты, при которых 100% активных веществ растворено в носителе, то активные вещества присутствуют в форме раствора твердого материала в композиции в форме частиц. Наличие раствора твердого материала может быть определено по методу ДСК (DSC), приведенному в описании. Однако может происходить некоторая кристаллизация активных веществ из растворов твердых компонентов при их хранении. Соответственно, настоящее изобретение включает материал в форме частиц, в котором активные вещества присутствуют в форме раствора твердого материала, но настоящее изобретение также включает в свои рамки возможность осаждения активных веществ при хранении.
Как указывалось выше, требуется достаточная текучесть материала в форме частиц в соответствии с настоящим изобретением, с тем чтобы достичь подходящей текучести, открывающей возможность получения разных дозированных форм. Соответственно, материал в форме частиц по настоящему изобретению имеет приемлемую текучесть, определяемую, в соответствии с методикой Европейской Фармакопеи, путем измерения скорости истечения композиции из воронки с диаметром сопла 10,0 мм.
Материал в форме частиц по настоящему изобретению включает два активных вещества, а именно: фибрат и статин, или их фармацевтически приемлемую соль. Ниже приводится описание активных веществ.
Фибраты
Фибраты включают гемифиброзил, фенофибрат, бензафибрат, клофибрат и ципрофибрат. Они используются в качестве средств, регулирующих липидный обмен. Они рассматриваются как пролекарства, которые метаболизируются in vivo до активных метаболитов.
В конкретном варианте осуществления настоящего изобретения фибрат представляет собой фенофибрат или его аналог. Обычно концентрация фибрата в носителе составляет по меньшей мере 10 мас.% относительно общего веса фибрата, статина и носителя. В частности, концентрация фибрата в носителе составляет по меньшей мере 15 мас.%, по меньшей мере 16 мас.%, по меньшей мере 17 мас.% или по меньшей мере 20 мас.%, предпочтительно по меньшей мере 25 мас.%, более предпочтительно по меньшей мере 30 мас.%, и в особенности по меньшей мере 35 мас.% относительно общего веса фибрата, статина и носителя.
Только для целей иллюстрации можно указать конкретный пример фибрата, а именно фенофибрат. Так, например, фенофибрат метаболизируется до фенофибриновой кислоты, которая является активным веществом. Фенофибриновая кислота имеет период полувыведения примерно 20 часов. Определение выявляемых количеств фенофибриновой кислоты в крови пациента может отражать эффективность поглощения фенофибрата.
Химическое название фенофибрата: 1-метилэтиловый сложный эфир 2-[4-(4-хлорбензоил)]-2-метилпропаноевой кислоты, который описывается следующей структурной формулой:
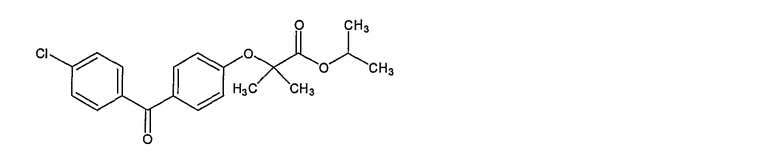
Фенофибрат представляет собой белое твердое вещество, нерастворимое в воде. Точка плавления составляет 79-82°С.
Результаты клинических исследований показали, что повышенные уровни общего холестерина, холестерина типа липопротеина низкой плотности (ЛПНП-Х) и аполипопротеина В (apo B) ассоциируются с развитием атеросклероза у человека. Сниженные уровни холестерина типа липопротеина высокой плотности (ЛПВП-Х) и комплекса, осуществляющего его транспортировку, аполипопротеина А (apo AI и apo AII), ассоциированы с развитием атеросклероза.
Фенофибрат также эффективен при лечении диабета типа II.
Фенофибриновая кислота, то есть активный метаболит фенофибрата, приводит к снижению у пациентов в ходе лечения общего уровня холестерина, ЛПНП-холестерина, аполипопротеина В, уровня суммарных триглицеридов и липопротеина, обогащенного триглицеридами. Кроме того, лечение с использованием фенофибрата приводит к повышению уровня ЛПВП-холестерина и apo AI и apo AII. Фенофибрат действует как мощный регулятор липидного метаболизма, обеспечивающий уникальные клинически выраженные преимущества относительно существующих в настоящее время продуктов из семейства лекарственных веществ на основе фибрата. Фенофибрат приводит к существенному снижению уровня триглицеридов в плазме у пациентов с гипертриглицеридемией и плазменного уровня холестерина и ЛПНП-Х у пациентов с гиперхолестеринемией и со смешанной формой дислипидемии.
Фенофибрат также снижает уровень мочевой кислоты в сыворотке у пациентов с гиперуремией и повышение экскреции мочевой кислоты с мочой у здоровых субъектов.
Фенофибрат также показан в качестве дополнительной терапии в сочетании с диетой, рекомендуемой при лечении взрослых пациентов с гипертриглицеридемией (гиперлипидемия Фредриксона типа IV и V). Улучшение гликемического контроля у пациентов с диабетом, имеющих хиломикронемию натощак, обычно приводит к снижению уровня триглицеридов натощак и удаляет хиломикронемию, что устраняет необходимость фармакологического вмешательства.
Статины
Статины включают симвастатин, аторвастатин, ловастатин, правастатин, розувастатин, питавастатин и флувастатин и их фармацевтически приемлемые соли, такие как соли щелочного металла и щелочно-земельного металла. Статины оказывают положительный эффект при регуляции метаболизма липидов и используются при различных заболеваний, для которых важно контролировать уровень липидов в плазме. В целом, липиды хорошо всасываются (например, флувастатин и симвастатин) после перорального введения, хотя некоторые статины имеют плохую биологическую доступность (ловастатин имеет уровень биологической доступности около 30-40%, правастатин около 30%, симвастатин около 5% и розувастатин около 50%). Некоторые статины водорастворимы (например, правастатин и флувастатин), тогда как другие статины характеризуются плохой водорастворимостью (например, аторвастатин, ловастатин, питавастатин, розувастатин и симвастатин). В целом, статины чувствительны к влаге, то есть считается, что повышенную стабильность имеют композиции, не содержащие влаги или содержащие очень небольшой уровень воды, и то же самое относится к композициям, которые производятся без использования водной среды.
Концентрация статина в носителе в случае материала в форме частиц или твердой дозированной формы согласно настоящему изобретению составляет по меньшей мере 1 мас.% от общего веса фибрата, статина и носителя. Более конкретно, концентрации статина в носителе составляет по меньшей мере 1,5 мас.%, или по меньшей мере 2,5 мас.%, или по меньшей мере 5 мас.%, или по меньшей мере 7,5 мас.% или по меньшей мере 10 мас.% от общего веса фибрата, статина и носителя.
В конкретном варианте настоящее изобретение относится к материалу в форме частиц, содержащему фенофибрат и симвастатин или их фармацевтически приемлемую соль, материал в форме частиц, содержащий фенофибрат и аторвастатин или их фармацевтически приемлемую соль, материал в форме частиц, содержащий фенофибрат и ловастатин или их фармацевтически приемлемую соль, материал в форме частиц, содержащий фенофибрат и правастатин или их фармацевтически приемлемую соль, материал в форме частиц, содержащий фенофибрат и розувастатин или их фармацевтически приемлемую соль, материал в форме частиц, содержащий фенофибрат и симвастатин или их фармацевтически приемлемую соль.
Ниже приведены примеры конкретных статинов, однако изобретение не ограничивается указанными конкретными примерами.
Аторвастатин
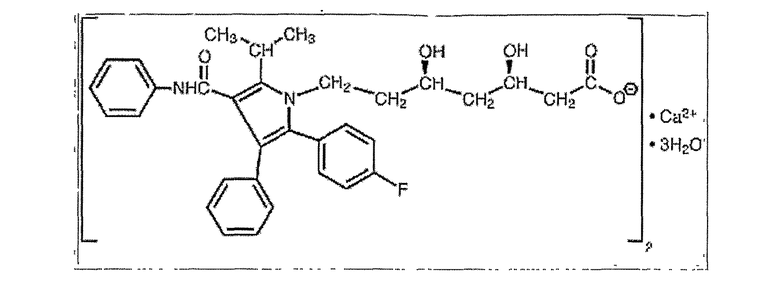
Аторвастатин-кальций представляет собой кристаллический порошок от белого до желтовато-белого цвета, не растворимый в водных растворах с pH 4 и ниже. Аторвастатин-кальций очень слабо растворим в дистиллированной воде, фосфатном буфере с pH 7,4 и ацетонитриле, слабо растворим в этаноле и хорошо растворим в метаноле.
Таблетки Липитор (Lipitor) для перорального введения содержат 10, 20, 40 или 80 мг аторвастатина и следующие неактивные ингредиенты: карбонат кальция, USP; канделильский воск, FCC; натрий-кроскармеллозу, НФ; гидроксипропилцеллюлозу, НФ; моногидрат лактозы, НФ; стеарат магния, НФ; микрокристаллическую целлюлозу, НФ; белый краситель Opadry White YS-1-7040 (гидроксипропилметилцеллюлоза, полиэтиленгликоль, тальк, диоксид титана); полисорбат 80, НФ; эмульсию циметикона.
Аторвастатин представляет собой синтетический агент, снижающий уровень липидов. Аторвастатин представляет собой ингибитор редуктазы 3-гидрокси-3-метилглутарил-коэнзима А (HMG-CoA). Данный фермент катализирует превращение HMG-CoA в мевалонат, что представляет собой раннюю и лимитирующую скорость стадию в биосинтезе холестерина.
Аторвастатин представляет собой селективный конкурентный ингибитор HMG-CoA-редуктазы, фермент, ограничивающий скорость реакции, которая осуществляет превращение 3-гидрокси-3-метилглутарил-коэнзима А в мевалонат, предшественник стеролов, включая холестерин. Холестерин и триглицериды циркулируют в кровотоке в виде части липопротеиновых комплексов. При ультрацентрифугировании такие комплексы разделяются на ЛПВП (липопротеин высокой плотности) и ЛПСП (липопротеин средней плотности), ЛПНП (липопротеин низкой плотности) и ЛПОНП (липопротеин очень низкой плотности). Триглицериды (ТГ) и холестерин в печени включаются в состав ЛПОНП и высвобождаются в плазму для доставки в периферические ткани. ЛПНП формируется из ЛПОНП и катаболизируется преимущественно через рецепторы, имеющие высокую активность к ЛПНП. Клинические исследования и исследования патологических состояний показывают, что повышенные уровни в плазме общего холестерина (общий-Х), ЛПНП-холестерина (ЛПНП-Х) и аполипопротеина В (apo B) способствуют развитию атеросклероза у человека и являются факторами риска развития сердечно-сосудистого заболевания, тогда как повышенные уровни ЛПВП-Х ассоциированы со сниженным риском развития сердечно-сосудистого заболевания.
Аторвастатин быстро абсорбируется при пероральном введении; максимальные концентрации в плазме достигаются в течение 1-2 часов. Уровень абсорбции повышается пропорционально принятой дозе аторвастатина. Абсолютная биологическая доступность аторвастатина (исходный препарат) составляет примерно 14% и системная доступность ингибиторной активности в отношении редуктазы HMG-CoA составляет примерно 30%. Низкая системная доступность связана с пресистемным клиренсом в слизистой желудочно-кишечного тракта и/или пресистемным метаболизмом в печени. Хотя пища снижает скорость и степень абсорбции лекарственного вещества примерно на 25% и 9%, по данным определения Cmax и AUC, отмечается, что снижение ЛПНП-Х аналогично в случае приема аторвастатина с пищей и без пищи. Концентрация аторвастатина в плазме ниже (примерно на 30%, по данным определения Cmax и AUC) после вечернего приема препарата в сравнении с утренним приемом. Однако отмечается, что снижение ЛПНП-Х не зависит от времени дневного приема препарата.
Правастатин
Правастатин-натрий (PRAVACHOL®) является представителем нового класса липид-снижающих соединений, ингибиторов редуктазы HMG-CoA, которые снижают биосинтез холестерина. Данные агенты являются конкурентными ингибиторами редуктазы 3-гидрокси-3-метилглутарил-коэнзима А (HMG-CoA), фермента, катализирующего раннюю, лимитирующую скорость стадию биосинтеза холестерина, а именно: превращение HMG-CoA в мевалонат.
Химически правастатин-натрий представляет собой 1-нафталин-гептаноевую кислоту, 1,2,6,7,8,8а-гексагидро-(бета),(бета),6-тригидрокси-2-метил-8-(2-метил-1-оксобутокси)-мононатриевую соль, [1S-[1(альфа)((бета)S*, (бета)S*),2(альфа),6(альфа),8(бета)(R*),8а(альфа)]]-. Структурная формула показана ниже.
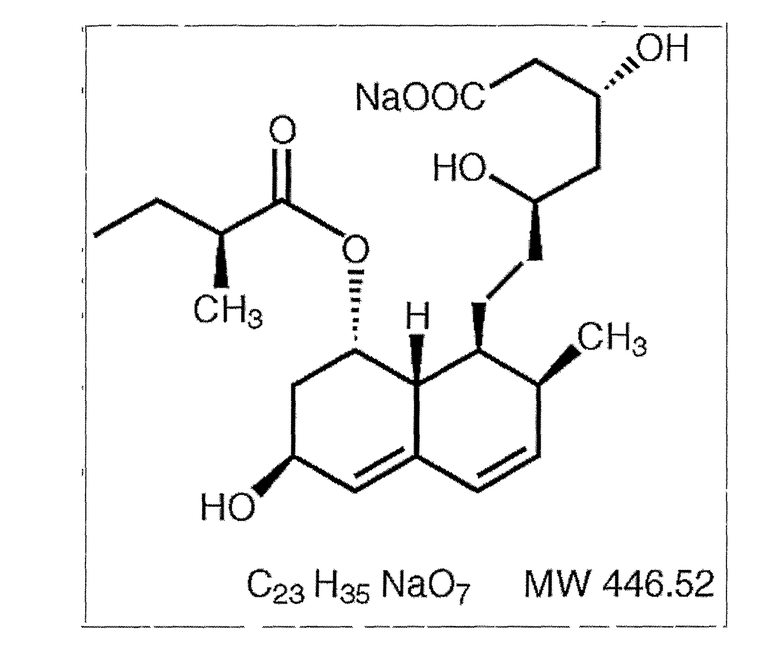
Правастатин-натрий представляет собой тонкоизмельченный или кристаллический порошок без запаха, белого или желтовато-белого цвета. Это относительно полярное гидрофильное соединение с коэффициентом распределения (октанол/вода) 0,59 при pH 7,0. Он растворим в метаноле и воде (>300 мг/мл), слабо растворим в изопропаноле и практически не растворим в ацетоне, ацетонитриле, хлороформе и диэтиловом эфире.
Правахол (PRAVACHOL) доступен для перорального введения в виде таблеток по 10 мг, 20 мг, 40 мг и 80 мг. Неактивные ингредиенты включают: натрий-кроскармеллозу, лактозу, оксид магния, стеарат магния, микрокристаллическую целлюлозу и повидон. Таблетка 10 мг также содержит красный железооксидный пигмент, таблетки по 20 мг и 80 мг содержат также желтый железооксидный пигмент, а таблетка 40 мг содержит зеленую пигментную смесь (Green Lake Blend) (смесь желтого пигмента D&C Yellow No. 10-Aluminum Lake и синего пигмента FD&C Blue No. 1-Aluminum Lake).
Правастатин индуцирует снижение уровня липидов двумя путями. Во-первых, вследствие обратимого ингибирования активности редуктазы HMG-CoA, он оказывает умеренное снижение внутриклеточного пула холестерина. Это приводит к повышению числа рецепторов ЛПНП на клеточных поверхностях и усиливает опосредованный рецепторами метаболизм, а также клиренс циркулирующего ЛПНП. Во-вторых, правастатин ингибирует продукцию ЛПНП за счет ингибирования синтеза ЛПОНП, предшественника ЛПНП.
Клинические и патологические исследования показали, что повышенные уровни в плазме общего холестерина (общего-Х), холестерина типа липопротеина низкой плотности (ЛПНП-Х) и аполипопротеина В (apo B - комплекс, осуществляющий транспортировку ЛПНП через мембрану) способствуют развитию атеросклероза у человека. Аналогично, сниженные уровни ЛПВП-Х и его транспортного комплекса аполипопротеина А ассоциированы с развитием атеросклероза. Эпидемиологические исследования установили, что заболеваемость и смертность от сердечно-сосудистых болезней варьирует, находясь в прямой зависимости от уровня общего-Х и ЛПНП-Х и в обратной зависимости от уровня ЛПВП-Х. Как и ЛПНП, холестерин, обогащенный липопротеинами, обогащенными триглицеридами, включая ЛПНОП, ЛПСП и другие, может также способствовать развитию атеросклероза. Повышенный уровень ТГ в плазме часто встречается в триаде вместе с низким уровнем ЛПВП-Х и малыми частицами ЛПНП, а также в сочетании с нелипидными метаболическими факторами риска ишемической болезни сердца. В качестве такового фактора, общий плазменный уровень ТГ не всегда является независимым фактором риска КБС. Кроме того, независимый эффект повышенного ЛПВП или сниженного ТГ на риск заболеваемости и смертности от ишемической болезни и сердечно-сосудистых заболеваний не определен. Как у здоровых волонтеров, так и у пациентов с гиперхолестеринемией лечение с использованием правахола (PRAVACHOL) (правастатина-натрия) снижает уровень общего-Х, ЛПНП-Х и аполипопротеина В. Правахол (PRAVACHOL) также снижает уровень ЛПОНП-Х и ТГ и приводит к повышению уровня ЛПВП-Х и аполипопротеина А. Эффект правастатина на Lp (а), фибриноген и некоторые другие независимые биохимические маркеры риска развития ишемической болезни сердца неизвестен. Хотя правастатин относительно более гидрофилен, чем другие ингибиторы редуктазы HMG-CoA, влияние относительной гидрофильности, при ее наличии, на эффективность или безопасность не установлено.
Правастатин, как и другие ингибиторы редуктазы HMG-CoA, характеризуется варьирующей биологической доступностью. Коэффициент вариации (КВ) на основании данных, полученных по вариабельности для разных пациентов, составляет от 50% до 60%, по данным AUC. Правастатин в дозе 20 мг вводят натощак взрослым. Среднее геометрическое значение Cmax и AUC составляет от 23,3 до 26,3 нг/мл и от 54,7 до 62,2 нг·час/мл, соответственно.
Розувастатин
Розувастатин является мощным ингибитором редуктазы HMG-CoA (статином). Розувастатин разрешен для лечения первичной гиперхолестеринемии, смешанной дислипидемии, гипертриглицеридемии и гомозиготной семейной гиперхолестеринемии. Он оказывает более сильное снижение уровня холестерина типа липопротеина низкой плотности (ЛПНП), чем аторвастатин, симвастатин и правастатин.
Дозу от 5 до 40 миллиграмм (мг) вводят один раз день перорально, при этом начальная доза составляет от 5 до 20 мг один раз в день. Дозы могут титроваться до 40 мг/день у тех пациентов, для которых не достигается ожидаемого снижения 20 мг/день. Лекарственное средство может вводиться вместе с пищей или без нее в любое время дня. Корректировка дозы предусматривается для пациентов с тяжелым нарушением функции почек и в случае пациентов, получающих в качестве сопутствующего лечения циклоспорин или гемфиброзил.
Пиковые уровни в плазме достигаются в период от 3 до 5 часов после введения пероральных доз и описываются линейными характеристиками в диапазоне от 5 до 80 мг; накопление в равновесном состоянии минимально. Розувастатин, по всей видимости, поступает селективно в печень, в сравнении с не печеночными тканями, что определяется его относительной гидрофильностью. Лекарственное вещество подвергается лишь минимальному метаболизму в печени, и большая часть дозы выделяется с желчью. Биологическая доступность составляет примерно 20%.
Симвастатин
Симвастатин представляет собой агент, снижающий содержание липидов, который получают путем синтеза из продукта ферментации Aspergillus terreus. После перорального приема симвастатин, который является неактивным лактоном, гидролизуется до соответствующей (бета)-гидроксикислоты. Указанный агент представляет собой ингибитор редуктазы 3-гидрокси-3-метилглутарил-коэнзима А (HMG-CoA). Данный фермент катализирует превращение HMG-CoA в мевалонат, что представляет собой раннюю, лимитирующую скорость стадию в процессе биосинтеза холестерина.
Сивмастатин представляет собой бутаноевую кислоту, 2,2-диметил-1,2,3,7,8,8а-гексагидро-3,7-диметил-8-[2-(тетрагидро-4-гидрокси-6-оксо-2Н-пиран-2-ил)-этил]-1-нафталиновый сложный эфир, [1S-[1(альфа),3(альфа),7(бета),8(бета),(2S*,4S*)-8a(бета)]]. Эмпирическая формула симвастатина: C25H38O5, а его молекулярный вес составляет 418,57. Структурная формула представлена ниже.
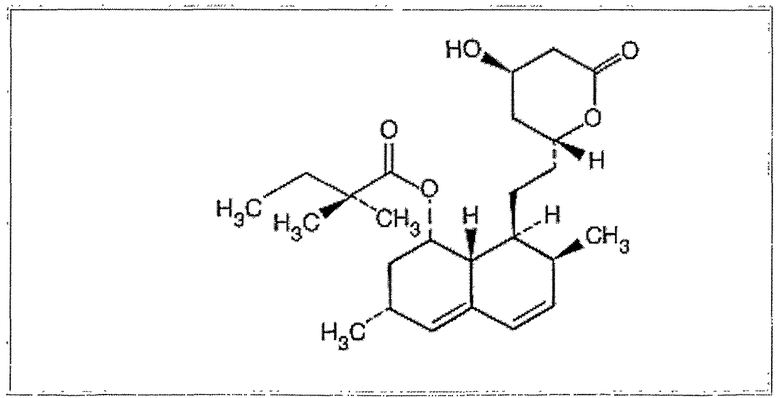
Симвастатин представляет собой кристаллический негигроскопичный порошок от белого до желтовато-белого цвета, который практически нерастворим в воде и свободно растворим в хлороформе, метаноле и этаноле.
Таблетки Зокор® (ZOCOR®) для перорального введения содержат 5 мг, 10 мг, 20 мг, 40 мг или 80 мг симвастатина и указанные ниже неактивные ингредиенты: целлюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, оксиды железа, лактозу, стеарат магния, крахмал, тальк, диоксид титана и другие ингредиенты. В качестве консерванта добавляют бутилированный гидроксианизол.
Повышенные плазменные уровни общего холестерина (общий-Х) ЛПНП-холестерина и аполипопротеина В (apo B) способствуют развитию атеросклероза у человека и являются факторами риска развития сердечно-сосудистого заболевания, тогда как повышенные уровни холестерина типа липопротеина высокой плотности (ЛПВП-Х) и его транспортного комплекса apo A-I, ассоциированы со сниженным риском развития сердечно-сосудистых заболеваний. Высокие плазменные уровни триглицеридов (ТГ) и холестерина, обогащенного липопротеинами, обогащенными ТГ, включающими липопротеины очень низкой плотности (ЛПОНП), липопротеины средней плотности (ЛПСП) и других, также могут способствовать развитию атеросклероза. Повышенный уровень ТГ в плазме зачастую обнаруживается в триаде вместе с низким уровнем ЛПВП-Х и малыми частицами ЛПНП в сочетании с нелипидными факторами риска ИБС. Не было показано, что общий уровень ТГ является независимым фактором риска для ИБС. Кроме того, независимый эффект повышенного уровня ЛПВП-Х или сниженного уровня ТГ на риск развития ишемической болезни или сердечно-сосудистых заболеваний и смертности не был определен. Было показано, что симвастатин снижает и нормальные и повышенные концентрации ЛПНП-Х. ЛПНП образуется из липопротеина очень низкой плотности (ЛПОНП) и катаболизируется преимущественно с вовлечением рецептора, имеющего высокое сродство к ЛПНП.
Симвастатин подвергается мощной экстракции при его пресистемном поступлении в печень, которая является первичным сайтом действия, с последующим выделением лекарственных эквивалентов с желчью. Вследствие обширной печеночной экстракции симвастатина (оцениваемой на уровне >60% у человека), доступность лекарственного вещества для общего кровотока низка.
Настоящее изобретение относится к материалу в форме частиц и к твердым дозированным формам, предназначенным для усовершенствованного режима лечения состояний, которые реагируют на терапию с использованием фибрата и статина. Кроме того, такие композиции и твердая дозированная форма могут также включать другие активные вещества или использоваться в сочетании с другими активными веществами, такими как, например, другие антилипидемические агенты, антидиабетические вещества, применяемые при диабете типа II, такие как, например, глимепирид, глибенкламид, гликлазид, репаглинид, натеглинид, метформин, пиоглитазон, розиглитазин или акарбоза. К ним относятся также сочетания с ингибиторами абсорбции холестерина, такие как эзетимиб и раскислители холестерина типа колесевелама. Указанные средства могут быть также включены в состав препарата или использоваться в сочетании с лекарственными агентами, которые могут приводить к нежелательному уровню триглицеридов и/или холестерина. Таким образом, композиция по настоящему изобретению может быть включена или использована в сочетании с лекарственными веществами типа, например, изотретионина и ингибитора протеазы ретровирусов, таких как ингибиторы протеазы ВИЧ и другие.
Материал в форме частиц по настоящему изобретению обладает подходящей текучестью, определяемой по методу, описанному в Европейской Фармакопее, в соответствии с которым проводят измерение скорости истечения композиции из воронки с соплом 10,0 мм. Для того чтобы исключить прилипание к производственному оборудованию и аппаратуре, используемой для заполнения, важно, чтобы материал в форме частиц был легко текучим. Эти характеристики также важны в тех случаях, когда желательно обрабатывать материал в форме частиц с получением в итоге других видов композиции, таких как, например, твердые дозированные формы.
Как отмечалось выше, материал в форме частиц по настоящему изобретению содержит носитель. В некоторых вариантах данный носитель является масляным и/или является маслоподобным, и/или он присутствует в относительно высоких количествах. В таких случаях может возникнуть необходимость включить в материал вещество, которое обладает адсорбирующими или абсорбирующими свойствами, так чтобы окончательный материал в форме частиц выглядел как немаслянистый порошок и при хранении из него не высвобождались никакие компоненты носителя, которые могли бы приводить к созданию маслянистой поверхности. Соответственно, материал в форме частиц может содержать один или несколько сорбирующих масло материалов, так что он при тестировании в соответствии с приведенным в настоящем описании способом:
i) имеет значение порога маслянистости 10% или более, при определении по приведенному в настоящем описании тесту оценки порогового значения,
и обладает по меньшей мере одним из следующих свойств:
ii) высвобождает по меньшей мере 30% масла, по данным тестирования в рамках описанного теста на высвобождение и
iii) в форме таблетки характеризуется временем дезинтеграции, равным максимум 1 час, при тестировании по методу Европейской Фармакопеи оценки степени дезинтеграции, так что таблетки содержат примерно 90 мас.% или более маслосорбирующего материала. В некоторых ситуациях было показано, что включение сорбционного материала в композицию является некоторым преимуществом, поскольку при этом, например, достигается высокая концентрация носителя, который обладает масляным или маслоподобным характером. В некоторых случаях, когда носитель имеет точку плавления максимум примерно 25°С, может быть особенно удобно включать сорбционный материал. Соответствующие примеры материалов, подходящих для использования в качестве носителя, а также сорбционных материалов приведены ниже в описании.
В другом варианте настоящее изобретение относится к материалу в форме частиц, включающему один или несколько фибратов и гидрофобный, или гидрофильный, или смешивающийся с водой носитель, в котором концентрация носителя составляет по меньшей мере примерно 10 мас.%. Ниже описаны носители, подходящие для использования в материале в форме частиц по настоящему изобретению.
Гидрофобные, или гидрофильные, или смешивающиеся с водой носители
В контексте настоящего описания термин "гидрофобный, или гидрофильный, или смешивающийся с водой носитель" используется в очень широком смысле, включая масла, воски, полутвердые материалы и материалы, которые обычно используются в качестве растворителей (такие как органические растворители) или сорастворителей, применяемых в фармацевтической промышленности, и, кроме того, данный термин также включает терапевтически и/или профилактически активные вещества, которые при температуре окружающей среды имеют жидкую форму, а также включает эмульсии, такие как, например, микроэмульсии и наноэмульсии, и суспензии. Гидрофобные, или гидрофильные, или смешивающиеся с водой носители обычно являются жидкостями при температуре окружающей среды или при повышенной температуре (по практическим соображениям, максимальная температуре составляет примерно 250°С). Они могут быть гидрофильными, липофильными, гидрофобными и/или амфифильными материалами. В конкретном варианте осуществления настоящего изобретения носитель является неводным материалом.
Гидрофобные, или гидрофильные, или смешивающиеся с водой носители, которые приемлемы для использования в рамках настоящего изобретения, представляют собой вещества или материалы, которые имеют точку плавления по меньшей мере примерно 0°С и максимум примерно 250°С.
В конкретных вариантах осуществления настоящего изобретения гидрофобные, или гидрофильные, или смешивающиеся с водой растворители имеют точку плавления, равную примерно 5°С или более, такую, например, как примерно 10°С или более, примерно 15°С или более, примерно 20°С или более или примерно 25°С или более. Как указывалось выше, обычно требуется добавление маслосорбирующего материала, если используются носители, которые имеют такую низкую точку плавления. Однако специалисты со средним уровнем знаний в данной области хорошо понимают, когда необходимо добавлять такой маслосорбирующий материал.
В других вариантах осуществления настоящего изобретения гидрофобные, или гидрофильные, или смешивающиеся с водой растворители имеют такую точку плавления, которая составляет примерно 25°С, такую, например, как по меньшей мере примерно 30°С, по меньшей мере примерно 35°С или по меньшей мере примерно 40°С. По практическим причинам точка плавления в норме не является слишком высокой, так, масляный или маслоподобный материал обычно имеют точку плавления, равную максимум примерно 300°С, такую, например, как максимум примерно 250°С, максимум примерно 200°С, максимум примерно 150°С или максимум примерно 100°С. Если точка плавления выше, то относительно высокая температура может способствовать, например, окислению или другим видам деградации активного вещества, в тех случаях, когда включается, например, терапевтически и/или профилактически активное вещество.
В контексте настоящего изобретения точку плавления определяют по методу ДСК (DSC) (дифференциальная сканирующая калориметрия). Точку плавления определяют как температуру, при которой линейное повышение кривой ДСК пересекается с температурной осью.
Интересующие гидрофобные, или гидрофильные, или смешивающиеся с водой носители в основном являются веществами, которые используют при производстве фармацевтических препаратов, представляя собой так называемые плавкие связующие вещества или твердые растворители (в виде твердой дозированной формы) или выполняют функцию сорастворителей или ингредиентов фармацевтических средств, используемых для местного применения.
Указанные средства могут иметь гидрофильный, гидрофобный характер или они могут обладать свойствами поверхностно-активного материала. В целом, гидрофильные и/или гидрофобные носители пригодны для использования в процессе производства материала в форме частиц или твердой дозированной формы по настоящему изобретению. В конкретном варианте они могут использоваться в том случае, когда высвобождение активного вещества из фармацевтической композиции отвечает целям мгновенного или модифицированного или немодифицированного высвобождения. Гидрофобные носители обычно используют при производстве фармацевтической композиции с модифицированным высвобождением. Приведенный выше подход упрощен для иллюстрации основных принципов, но имеется множество случаев, когда приемлемы другие сочетания носителей, и другие цели являются релевантными и, в этой связи, приведенные выше примеры никоим образом не ограничивают настоящее изобретение.
В типичном случае подходящий гидрофильный или смешивающийся с водой носитель выбирают из группы, состоящей из полиэфирных гликолей, таких как полиэтиленгликоли, полипропиленгликоли, полиоксиэтилены, полиоксипропилены, полоксамеры и их смеси, или он может быть выбран из группы, состоящей из ксилита, сорбита, тартрата калия-натрия, трибегената сахарозы, глюкозы, рамнозы, лактита, бегеновой кислоты, монометилового эфира гидрохинона, ацетата натрия, этилфумарата, миристиновой кислоты, лимонной кислоты, Gelucire 50/13, других типов Gelucire, таких как, например, Gelucire 44/14 и т.п., Gelucire 50/10, Gelucire 62/05, сложного сахарного эфира 7, сложного сахарного эфира 11, сложного сахарного эфира 15, мальтозы, маннита и их смесей.
Подходящие гидрофобные или смешивающиеся с водой носители выбирают из группы, включающей линейно-цепочечные насыщенные углеводороды, сложные эфиры сорбитана, парафины, жиры и масла, такие как, например, какао-масло, таловый жир, свиной жир, сложные эфиры полиэфиргликоля, высшая жирная кислота, такая как, например, стеариновая кислота, миристиновая кислота, пальмитиновая кислота, высшие спирты, такие как, например, цетанол, стеариловый спирт, низкоплавящиеся воски, такие как, например, глицерилмоностеарат, глицерилмоноолеат, гидрогенизированное таловое масло, миристиловый спирт, стеариловый спирт, замещенные и/или незамещенные моноглицериды, замещенные и/или незамещенные диглицериды, замещенные и/или незамещенные триглицериды, желтый пчелиный воск, белый пчелиный воск, карнаубский воск, касторовый воск, японский воск, ацетилат моноглицеридов, NVP полимеры, ПВП полимеры, акриловые полимеры или их смеси.
В интересующем варианте носитель представляет собой полиэтиленгликоль со средней молекулярной массой в диапазоне примерно от 400 до примерно 35000, такой как, например, от примерно 800 до примерно 35000, от примерно 1000 до примерно 35000, такой как, например, полиэтиленгликоль 1000, полиэтиленгликоль 2000, полиэтиленгликоль 3000, полиэтиленгликоль 4000, полиэтиленгликоль 5000, полиэтиленгликоль 6000, полиэтиленгликоль 7000, полиэтиленгликоль 8000, полиэтиленгликоль 9000, полиэтиленгликоль 10000, полиэтиленгликоль 15000, полиэтиленгликоль 20000 или полиэтиленгликоль 35000. В некоторых ситуациях может быть использован полиэтиленгликоль с молекулярной массой от примерно 35000 до примерно 100000.
В другом варианте носитель представляет собой полиэтиленоксид с молекулярной массой от примерно 2000 до примерно 7000000, такой как от примерно 2000 до примерно 100000, от примерно 5000 до примерно 75000, от примерно 10000 до примерно 60000, от примерно 15000 до примерно 50000, от примерно 20000 до примерно 40000, от примерно 100000 до примерно 7000000, такой, например, как от примерно 100000 до примерно 1000000, от примерно 100000 до примерно 600000, от примерно 100000 до примерно 400000 или от примерно 100000 до примерно 300000.
В другом варианте носитель представляет собой полоксамер, такой как, например, Полоксамер 188 (Poloxamer 188), Полоксамер 237 (Poloxamer 237), Полоксамер 338 (Poloxamer 338) или Полоксамер 407 (Poloxamer 407) или другие блок-сополимеры этиленоксида или полиэтиленоксида, такие как средства серии Плуроник® (Pluronic®) и/или Тетроник® (Tetronic®). Подходящие блок-сополимеры серии Плуроник® включают полимеры с молекулярной массой от примерно 3000 или более, такой, например, как от примерно 4000 до примерно 20000, и/или с вязкостью (Brookfield) от примерно 200 до примерно 4000 сантипуаз, такой, например, как от примерно 250 до примерно 3000 сантипуаз. Подходящие примеры включают Плуроник® (Pluronic®) F38, P65, P68LF, P75, F77, P84, P85, F87, F88, F98, P103, P104, P105, F108, P123, F123, F127, 10R8, 17R8, 25R5, 25R8 и т.п. Подходящие блок-сополимеры серии Тетроник® (Tetronic®) включают полимеры с молекулярной массой от примерно 8000 или более, такой, например, как от примерно 9000 до примерно 35000, и/или с вязкостью (Brookfield) от примерно 500 до примерно 45000 сантипуаз, такой, например, как от примерно 600 до примерно 40000 сантипуаз. Указанное выше значение вязкости определяют при температуре 60°С для веществ, которые имеют пастообразную форму при комнатной температуре, и при 77°С для веществ, которые при комнатной температуре являются твердыми материалами.
В конкретном варианте материал в форме частиц по настоящему изобретению включает в качестве носителя смесь полиэтиленгликоля и полоксамера в пропорции (весовой) от примерно 1:3 до примерно 10:1, предпочтительно от 1:1 до 5:1, более предпочтительно от 3:2 до 4:1, и в особенности от 2:1 до 3:1, в частности, примерно 7:3. В конкретном варианте используемый полоксамер представляет собой полоксамер 188.
В другом варианте в качестве носителя используют полиэтиленгликоль, который имеет среднюю молекулярную массу примерно 6000 (ПЭГ6000).
Носитель может также представлять собой сложный эфир сорбитана, такой как, например, диизостеарат сорбитана, диолеат сорбитана, монолаурат сорбитана, моноизостеарат сорбитана, моноолеат сорбитана, монопальмитат сорбитана, моностеарат сорбитана, сесквиизостеарат сорбитана, сесквиолеат сорбитана, сесквистеарат сорбитана, триизостеарат сорбитана, триолеат сорбитана, тристеарат сорбитана или их смеси.
Носитель может также включать смесь различных носителей, такую как, например, смесь гидрофильного и/или гидрофобного материала.
Другие приемлемые носители могут представлять собой растворители или полутвердые эксципиенты, такие как, например, пропиленгликоль, полигликозилированные глицериды, включающие Gelucire 44/14, комплекс жирных материалов растительного происхождения, включающий теобромное масло, карнаубский воск, растительные масла, например, миндальное масло, кокосовое масло, кукурузное масло, хлопковое масло, кунжутное масло, соевое масло, оливковое масло, касторовое масло, масло из пальмовых косточек, арахисовое масло, рапсовое масло, масло из виноградных косточек и т.п.; гидрогенизированные растительные масла, такие как, например, гидрогенизированное арахисовое масло, гидрогенизированное масло из пальмовых косточек, гидрогенизированное хлопковое масло, гидрогенизированное соевое масло, гидрогенизированное касторовое масло, гидрогенизированное кокосовое масло; природные жирные материалы животного происхождения, включающие пчелиный воск, ланолин, жирные спирты, включающие цетиловый, стеариловый, лауриловый, миристиновый, пальмитиновый, стеариновый жирные спирты; сложные эфиры, включающие глицеролстеарат, гликольстеарат, этилолеат, пропилмиристат; жидкие полусинтетические глицериды со сшивающими сложноэфирными связями, включающие Мигликол (Miglycol 810/812); амид или алколамиды жирных кислот, включающие этанолстеарамид, диэтаноламид или жирные кислоты кокосового масла, сложные эфиры уксусной кислоты и моно- и ди-глицеридов, сложные эфиры лимонной кислоты и моно- и ди-глицеридов, сложные эфиры молочной кислоты и моно- и ди-глицеридов, моно- и ди-глицериды, полиглицероловые эфиры жирных кислот, полиглицеролполирицинолеат, сложные эфиры пропиленгликоля и жирных кислот, сорбитанмоностеараты, сорбитантристеараты, стеароиллактилаты натрия, стеароиллактилаты кальция, сложные эфиры диацетилвинной кислоты и моно- и ди-глицеридов и т.п.
В норме материал в форме частиц или твердая дозированная форма по настоящему изобретению имеют концентрацию носителя в материале в форме частиц или в твердой дозированной форме примерно 5 мас.% или более, такую, например, как примерно 10 мас.% или более, примерно 15 мас.% или более, примерно 20 мас.% или более, примерно 25 мас.% или более, примерно 30 мас.% или более, примерно 35 мас.% или более, примерно 40 мас.% или более, примерно 45 мас.% или более, примерно 50 мас.% или более, примерно 55 мас.% или более, примерно 60 мас.% или более, примерно 65 мас.% или более, примерно 70 мас.% или более, примерно 75 мас.% или более, примерно 80 мас.% или более, примерно 85 мас.% или более, примерно 90 мас.% или более или примерно 95 мас.% или более.
В конкретных вариантах осуществления настоящего изобретения концентрация носителя в материале в форме частиц или в твердой дозированной форме по настоящему изобретению находится в диапазоне от примерно 20 мас.% до примерно 80 мас.%, таком, например, как от примерно 25 мас.% до примерно 75 мас.%.
Одним из преимуществ указанного подхода является возможность включать относительно большое количество носителя, имея при этом все еще твердый материал. Таким образом, возможно получать твердую композицию с относительно высоким содержанием носителя при использовании маслосорбирующего материала, как указано выше. В фармацевтической области возможность включать относительно большое количество носителя (например, с масляными или маслоподобными характеристиками) в твердую композицию является плюсом, особенно в тех случаях, когда активные вещества не обладают соответствующими свойствами по водорастворимости (например, имеют плохую водорастворимость), стабильности в водной среде (то есть в водной среде происходит их деградация), биологической доступности при пероральном приеме (например, имеют низкую биологическая доступность) и т.п., или в тех ситуациях, когда желательно модифицировать уровень высвобождения активного вещества из композиции, с тем чтобы получить контролируемую, модифицированную, задержанную, пролонгированную и/или пульсовую доставку активного вещества.
Каждый практикующий специалист со средним уровнем знаний в данной области, с использованием общих принципов и рутинных экспериментов, в состоянии выбрать подходящий носитель, который будучи фармацевтически приемлемым, будет также способен к диспергированию, растворению или по меньшей мере частичному растворению активных веществ и который имеет точку плавления в желательном диапазоне. Потенциально возможные носители описаны в документе WO 03/004001, который включен в настоящее описание в качестве ссылки.
В контексте настоящего описания подходящие носителя включают, например, указанные выше носители, а также носители, описанные в WO 03/004001.
Другим преимуществом изобретения является тот факт, что получаемый материал в форме частиц представляет собой свободно текучий порошок, который, в этой связи, легко поддается дальнейшей обработке с получением твердых дозированных форм, таких как таблетки, капсулы или пакетики. Обычно материал в форме частиц имеет свойства, которые приемлемы для изготовления таблеток путем прямого прессования, без внесения больших количеств других добавок. Подходящий тест для определения текучести материала в форме частиц включает способ, описанный в Европейской Фармакопее, в соответствии с которым проводят измерение скорости истечения материала из воронки через сопло (отверстие) с диаметром 10,0 мм.
Фармацевтически приемлемые эксципиенты
В контексте настоящего описания термин "фармацевтически приемлемый эксципиент" употребляют для обозначения любого материала, который является инертным в том смысле, что он по существу не оказывает какого-либо терапевтического и/или профилактического эффекта per se. Такой эксципиент может добавляться с целью придания фармацевтической, косметической и/или пищевой композиции приемлемых технических свойств. Материал в форме частиц или твердая дозированная форма по настоящему изобретению могут содержать один или несколько фармацевтически приемлемых эксципиентов.
Примеры приемлемых для использования в композиции или в твердой дозированной форме по настоящему изобретению эксципиентов включают наполнители, разбавители, дезинтегрирующие средства, связующие вещества, замасливатели и т.п. или их смесь. Поскольку композиция или твердая дозированная форма по настоящему изобретению может использоваться для разных целей, выбор эксципиентов обычно осуществляют с учетом таких разнообразных вариантов использования. Другие фармацевтически приемлемые эксципиенты включают, например, подкислители, подщелачивающие вещества, консерванты, антиоксиданты, буферные средства, хелатирующие агенты, красители, комплексообразователи, эмульгаторы и/или солюбилизирующие средства, вкусовые вещества и отдушки, увлажнители, подсластители, смачивающие вещества и т.п.
Примеры подходящих наполнителей, разбавителей и/или связующих веществ включают лактозу (например, лактозу, полученную путем распылительной сушки, α-лактозу, β-лактозу, Таблетозу® (Tabletose®), разной степени чистоты Фарматозу® (Pharmatose®), Микротозу® (Microtose®) или Фаст-Флок® (Fast-Floc®), микрокристаллическую целлюлозу (разной степени чистоты Авицель® (Avicel®), Элцема® (Elcema®), Вивацель® (Vivacel®), Минг Тай® (Ming Tai®) или Солка-Флок® (Solka-Floc®)), гидроксипропилцеллюлозу, L-гидроксипропилцеллюлозу (низкозамещенную), гидроксипропилметилцеллюлозу (ГПМЦ), (например, Метоцель (Methocel) E, F и K, Метолозу (Metolose) SH of Chin-Etsu, Ltd., такую, например, как продукт с вязкостью 4000 сантипуаз Метоцель (Methocel) Е и Метолоза (Metolose) 60 SH, с вязкостью 4000 сантипуаз Метоцель (Methocel) F и Метолоза (Metolose) 65 SH, с вязкостью 4000, 15000 и 100000 сантипуаз Метоцель (Methocel) К, с вязкостью 4000, 15000, 39000 и 100000 сантипуаз Метолоза (Metolose) 90 SH), метилцеллюлозные полимеры (такие как Метоцель (Methocel) A, Метоцель (Methocel) A4C, Метоцель (Methocel) A15C, Метоцель (Methocel) A4M), гидроксиэтилцеллюлозу, натрий-карбоксиметилцеллюлозу, карбоксиметилен, карбоксиметилгидроксиэтилцеллюлозу и другие производные целлюлозы, сахарозу, агарозу, сорбит, маннит, декстрины, мальтодекстрины, крахмалы или модифицированные крахмалы (включая картофельный крахмал, кукурузный крахмал и рисовый крахмал), фосфат кальция (например, основной фосфат кальция, гидрофосфат кальция, гидрат фосфата дикальция), сульфат кальция, карбонат кальция, альгинат натрия, коллаген и т.п.
Конкретные примеры разбавителей включают, например, карбонат кальция, двуосновный фосфат кальция, трехосновный фосфат кальция, сульфат кальция, микрокристаллическую целлюлозу, порошковую целлюлозу, декстраны, декстрин, декстрозу, фруктозу, каолин, лактозу, маннит, сорбит, крахмал, предварительно желатинизированный крахмал, сахароза, сахар и т.п.
Конкретные примеры средств, способствующих дезинтеграции, включают, например, альгиновую кислоту или альгинаты, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу и другие производные целлюлозы, натрий-кроскармеллозу, кросповидон, калий-полакриллин, натрий-гликолят-крахмал, крахмал, предварительно желатинизированный крахмал, карбоксиметилкрахмал (например, Примогель® (Primogel®) и Эксплотаб® (Explotab®)) и т.п.
Конкретные примеры связующих веществ включают, например, аравийскую камедь, альгиновую кислоту, агар, кальций-каррагенан, натрий-карбоксиметилцеллюлозу, микрокристаллическую целлюлозу, декстрин, этилцеллюлозу, желатин, жидкую глюкозу, гуаровую камедь, гидроксипропилметилцеллюлозу, метилцеллюлозу, пектин, ПЭГ, повидон, предварительно желатинизированный крахмал и т.п.
Средства, способствующие скольжению, и замасливатели также могут быть включены во вторую композицию. Соответствующие примеры включают стеариновую кислоту, стеарат магния, стеарат кальция и другие стеараты металлов, тальк, воски и глицериды, легкое минеральное масло, ПЭГ, глицерилбегенат, коллоидный кремнезем, гидрогенизированные растительные масла, кукурузный крахмал, стеарилфумарат натрия, полиэтиленгликоли, алкилсульфаты, бензоат натрия, ацетат натрия и т.п.
Другие эксципиенты, которые могут быть введены в композицию или в твердую дозированную форму по настоящему изобретению, включают, например, вкусовые вещества, красители, средства, маскирующие вкус, средства, регулирующие pH, буферные вещества, консерванты, стабилизаторы, антиоксиданты, смачивающие вещества, корректоры содержания влаги, поверхностно-активные вещества, средства, способствующие суспендированию, средства, усиливающие абсорбцию, средства, модифицирующие высвобождение и т.п.
Другие добавки, включаемые в композиции или в твердую дозированную форму по настоящему изобретению, могут представлять собой антиоксиданты, такие как, например, аскорбиновая кислота, аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, гипофосфорная кислота, монотиоглицерол, метабисульфит калия, пропилгаллат, сульфоксилат натрий-формальдегида, метабисульфит натрия, тиосульфат натрия, диоксид серы, токоферол, токоферол-ацетат, токоферол-гемисукцинат, TPGS или другие производные токоферолов и т.п. Композиция с носителем может также включать, например, стабилизирующие агенты. Концентрация антиоксиданта и/или стабилизатора в композиции с носителем составляет от примерно 0,1 мас.% до примерно 5 мас.%.
Композиция или твердая дозированная форма по настоящему изобретению может также включать один или несколько поверхностно-активных веществ или веществ, обладающих поверхностно-активными свойствами. Считается, что такие вещества вовлекаются в увлажнение слабо растворимого активного вещества и, таким образом, способствуют улучшению растворимости активного вещества.
Ниже даны примеры поверхностно-активных веществ.
Подходящие для использовании в композиции или в твердой дозированной форме по настоящему изобретению эксципиенты включают поверхностно-активные вещества, такие, как, например, гидрофобные и/или гидрофильные поверхностно-активные вещества, в частности описанные в WO 00/50007 (Lipocine, Inc). Примеры подходящих поверхностно-активных веществ включают:
i) полиоксиэтилированные жирные кислоты, такие как, например, моно- или ди-эфиры жирной кислоты и полиэтиленгликоля или их смеси, такие как моно- или ди-эфиры полиэтиленгликоля и лауриновой кислоты, олеиновой кислоты, стеариновой кислоты, миристиновой кислоты, рицинолевой кислоты, где полиэтиленгликоль может быть выбран из ПЭГ 4, ПЭГ 5, ПЭГ 6, ПЭГ 7, ПЭГ 8, ПЭГ 9, ПЭГ 10, ПЭГ 12, ПЭГ 15, ПЭГ 20, ПЭГ 25, ПЭГ 30, ПЭГ 32, ПЭГ 40, ПЭГ 45, ПЭГ 50, ПЭГ 55, ПЭГ 100, ПЭГ 200, ПЭГ 400, ПЭГ 600, ПЭГ 800, ПЭГ 1000, ПЭГ 2000, ПЭГ 3000, ПЭГ 4000, ПЭГ 5000, ПЭГ 6000, ПЭГ 7000, ПЭГ 8000, ПЭГ 9000, ПЭГ 1000, ПЭГ 10000, ПЭГ 15000, ПЭГ 20000, ПЭГ 35000,
ii) сложные эфиры полиэтиленгликольглицерола и жирной кислоты, например, сложные эфиры указанных выше типов, но в виде глицериловых сложных эфиров индивидуальных жирных кислот;
iii) глицерол, пропиленгликоль, этиленгликоль, ПЭГ или сложные эфиры сорбита, например, с растительными маслами, такими как гидрогенизированное касторовое масло, миндальное масло, масло из пальмовых косточек, касторовое масло, масло из абрикосовых косточек, оливковое масло, арахисовое масло, гидрогенизированное масло из пальмовых косточек и т.п.;
iv) полиглицериловые эфиры жирных кислот, такие как, например, полиглицеролстеарат, полиглицерололеат, полиглицеролрицинолеат, полиглицероллинолеат;
v) сложные эфиры пропиленгликоля и жирной кислоты, такие как, например, монолаурат пропиленгликоля, рицинолеат пропиленгликоля и т.п.;
vi) моно- и ди-глицериды, такие как, например, глицерилмоноолеат, глицерилдиолеат, глицерилмоно- и/или диолеат, глицерилкаприлат, глицерилкапрат и т.п.;
vii) стерол и стероловые производные;
viii) сложные эфиры полиэтиленгликольсорбитана и жирной кислоты (сложные эфиры ПЭГ-сорбитана и жирной кислоты), такие как сложные эфиры на основе ПЭГ с разной молекулярной массой, как указано выше, и различных продуктов серии Твин® (Tween®);
ix) полиэтиленгликольалкиловые эфиры, такие как, например, ПЭГ-олеиловый эфир и ПЭГ-лауриловый эфир;
x) сложные эфиры сахаров, такие как, например, монопальмитат сахарозы и монолаурат сахарозы;
xi) полиэтиленгликольалкилфенолы, такие как Тритон® (Triton®) серии X или N;
xii) полиоксиэтилен-полиоксипропиленовые блок-сополимеры, такие как сополимеры серии Плуроник® (Pluronic®), серии Синпероник® (Synperonic®), серии Эмкаликс® (Emkalyx®), серии Лутрол® (Lutrol®), серии Супроник® (Supronic®) и т.п. Генерическое обозначение данных полимеров - "полоксамеры" и релевантные примеры в контексте настоящего описания включают Полоксамер (Poloxamer) 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403 и 407;
xiii) сложные эфиры сорбитана и жирных кислот, такие как, например, продукты серии Спан® (Span®), продукты серии Ариацель® (Ariacel®), такие как, например, сорбитан-монолаурат, сорбитан-монопальмитат, сорбитан-моноолеат, сорбитан-моностеарат и т.п.;
xiv) сложные эфиры низшего спирта и жирной кислоты, такие как, например, олеат, изопропилмиристат, изопропилпальмитат и т.п.;
xv) ионные поверхностно-активные вещества, включающие катионные, анионные и цвиттерионные поверхностно-активные вещества, такие как, например, соли жирной кислоты, желчные соли, фосфолипиды, сложные эфиры фосфорной кислоты, карбоксилаты, сульфаты и сульфонаты и т.п.
В том случае, когда в композиции или в твердой дозированной форме по настоящему изобретению присутствует поверхностно-активное вещество или смесь поверхностно-активных веществ, концентрация указанного(ых) поверхностно-активного(ых) вещества/веществ находится обычно в диапазоне от примерно 0,1 до 80 мас.% и составляет, например, от примерно 0,1 до 20 мас.%, от примерно 0,1 до примерно 15 мас.%, от примерно 0,5 до примерно 10 мас.%, или, альтернативно, от примерно 0,10 до 80 мас.% и может представлять, например, от примерно 10 до примерно 70 мас.%, от примерно 20 до примерно 60 мас.%, или от примерно 30 до примерно 50 мас.%.
В конкретном аспекте настоящего изобретения по меньшей мере один или несколько фармацевтически приемлемых эксципиентов выбирают из группы, состоящей из кремниевой кислоты, ее производного или ее соли, включая силикаты, диоксид кремния или их полимеры; алюмосиликата магния и/или алюминометасиликата магния, бентонита, каолина, трисиликата магния, монтмориллонита и/или сапонита.
Сорбционные материалы
Материалы, такие как были указаны выше в последнем параграфе, могут использоваться в качестве сорбционного материала для масляных или маслоподобных материалов в фармацевтических, косметических и/или пищевых композициях. В конкретном варианте данный материал используют в качестве сорбционного материала для масел или маслоподобных материалов в фармацевтических препаратах. Данный материал обладает способностью функционировать в качестве сорбционного материала для масел или маслоподобных материалов, который также обозначается как "масло-сорбирующий материал". Как указывалось выше, носитель, подходящий для использования в материале в форме частиц или в твердой дозированной форме по настоящему изобретению, имеет масляный или маслоподобный характер, и в этой связи, в такую композицию может быть выгодно включать сорбционный материал.
Кроме того, в рамках настоящего описания термин "сорбция" используют для обозначения понятия "абсорбция", а также "адсорбция". Следует понимать, что, в случае использования данного термина, он охватывает все явления, включающие как абсорбцию, так и адсорбцию. Термины "сорбционный материал" и "маслосорбирующий материал" имеют одно и то же значение.
Сорбционный материал, подходящий для использования согласно настоящему изобретению, представляет собой твердый фармацевтически приемлемый материал, который при тестировании по приведенному в настоящем описании методу:
i) имеет пороговое значение 10% или более при испытании в соответствии с тестом по определению порогового значения,
и характеризуется по меньшей мере одним из указанных свойств:
ii) высвобождает по меньшей мере 30% масла, при определении по методу высвобождения, приведенному в настоящем описании, и
iii) в форме таблетки характеризуется временем дезинтеграции, равным максимум 1 час, при испытании по методу Европейской Фармакопеи тестирования дезинтеграции, так что данная таблетка содержит примерно 90 мас.% или более фармацевтически приемлемого материала.
Материал особенно полезен в качестве сорбционного материала для масляных или маслоподобных материалов в фармацевтических, косметических и/или пищевых композициях, особенно в фармацевтических препаратах.
Как следует из вышесказанного, важно, чтобы маслосорбирующий материал удовлетворял критериям по меньшей мере двух тестов. Один из этих тестов является обязательным, то есть препарат должен обязательно удовлетворять тесту на пороговое значение. Данный тест показывает, какое количество масла или маслоподобного материала данный сорбирующий материал может абсорбировать, при сохранении подходящих свойств текучести. Важно, что сорбирующий материал, подходящий для использования согласно настоящему изобретению (при наличии или в отсутствие абсорбции масла), имеет подходящую текучесть, так что он может легко смешиваться с другими эксципиентами и/или подвергаться дальнейшей обработке с получением композиций, без серьезных проблем, связанных, например, с прилипаемостью к используемой аппаратуре. Тест, описанный в экспериментальном разделе настоящего описания и в руководстве, описывает методику проведения данного испытания. Тест на определение порогового значения включает измерение текучести твердого материала, содержащего разные количества масла.
Из вышесказанного видно, что пороговое значение обычно должно превышать 10% и часто маслосорбирующий материал имеет пороговое значение, составляющее по меньшей мере примерно 15%, такое, например, как по меньшей мере примерно 20%, по меньшей мере примерно 25%, по меньшей мере примерно 30%, по меньшей мере примерно 35%, по меньшей мере примерно 40%, по меньшей мере примерно 45%.
Особенно приемлемым материалом для использования согласно настоящему изобретению является Аэроперл 300 (Aeroperl 300), который характеризуется очень высоким пороговым значением для масла, составляющим примерно 60%. Соответственно, материалы, которые имеют пороговое значение для масла по меньшей мере примерно 50%, такие, например, как по меньшей мере примерно 55% или по меньшей мере примерно 60%, используют в конкретных вариантах согласно настоящему изобретению.
Кроме того, маслосорбирующий материал, применяемый согласно настоящему изобретению, должен удовлетворять по меньше мере одному из тестов, а именно: тесту на высвобождение и/или тесту на дезинтеграцию.
Тест на высвобождение определяет меру способности сорбционного материала влиять на высвобождение масла, которое абсорбировалось в материал при контакте с водой. Указанная способность очень важна, особенно в тех случаях, когда активное вещество содержится в масле или в маслоподобном материале. Если маслосорбирующий материал не способен высвобождать масло из материала, то имеется большой риск того, что активное вещество будет лишь в незначительной мере высвобождаться из материала. Соответственно, в таких ситуациях могут возникать проблемы, связанные с биологической доступностью, например, с плохой абсорбцией и т.п.
В соответствии с требованиями теста на высвобождение, твердый фармацевтически приемлемый материал при тестировании, описанном выше:
ii) высвобождает по меньшей мере примерно 30%, например, по меньшей мере примерно 35%, по меньшей мере примерно 40%, по меньшей мере примерно 45%, по меньшей мере примерно 50%, по меньшей мере примерно 55% или по меньшей мере 60% масла. Как следует из приведенных примеров, подходящий маслосорбционный материал типа Аэроперла 300 (Aeroperl 300) имеет более высокий уровень высвобождения. В этой связи, в конкретном варианте осуществления настоящего изобретения твердый фармацевтически приемлемый материал при тестировании по описанному выше методу:
ii) высвобождает по меньшей мере примерно 65%, например, по меньшей мере примерно 70%, по меньшей мере примерно 75% или по меньшей мере примерно 80% масла.
Вторым тестом из числа тех тестов, среди которых по меньшей мере одному должен соответствовать маслосорбирующий материал по настоящему изобретению, является тест на дезинтеграцию. Этот тест проводится на твердом материале в форме частиц, с использованием таблетки, изготовленной из твердого материала. Требование относительно дезинтеграции является важным, поскольку дает гарантии того, что твердый материал, при его включении в твердые дозированные формы, не будет привносить нежелательных свойств данной дозированной форме, то есть не приведет к нежелательным показателям растворимости и биологической доступности активного вещества, содержащегося в дозированной форме. Для некоторых материалов, подходящих для использования в настоящем изобретении, возможно получать прессованием таблетки, содержащие 100 мас.% самого твердого материала. Если это имеет место, то тест выполняется на таких таблетках. Однако имеются ситуации, в которых весьма трудно получать таблетки из одного только твердого материала. В таких случаях можно добавлять фармацевтически приемлемые эксципиенты, обычно используемые при изготовлении прессованных таблеток, в концентрации до 10 мас.% или менее. Примеры подходящих фармацевтически приемлемых эксципиентов включают наполнители, разбавители, связующие вещества и замасливатели. Однако включения эксципиентов, классифицируемых обычно как дезинтегрирующие средства, следует избегать.
Соответственно, твердый фармацевтически приемлемый материал для использования в рамках настоящего изобретения, при тестировании, указанном выше:
iii) в форме таблетки должен иметь время дезинтеграции максимум 1 час, при испытании по тесту Европейской Фармакопеи на дизинтеграцию, при этом таблетка содержит примерно 90 мас.% или более, например примерно 92,5 мас.% или более, примерно 95 мас.% или более, примерно 97,5 мас.% или более или примерно 100 мас.% фармацевтически приемлемого материала.
В еще одном материале твердый фармацевтически приемлемый материал, при тестировании по описанному способу:
iii) в форме таблетки характеризуется значением времени дезинтеграции максимум примерно 50 минут, таким, например, как максимум примерно 40 минут, максимум примерно 30 минут, или максимум примерно 20 минут, максимум примерно 10 минут или максимум примерно 5 минут, при испытании в соответствии с тестом Европейской Фармакопеи на дезинтеграцию, при этом таблетка содержит примерно 90 мас.% или более, например, примерно 92,5 мас.% или более, примерно 95 мас.% или более, примерно 97,5 мас.% или более или примерно 100 мас.% фармацевтически приемлемого материала.
В конкретном варианте твердый материал, используемый в качестве сорбционного материала, удовлетворяет всем трем тестам. Таким образом, твердый фармацевтически приемлемый материал, при испытании в соответствии с указанным тестом:
i) характеризуется пороговым значением, равным по меньшей мере примерно 10%, таким, например, как по меньшей мере примерно 15%, по меньшей мере примерно 20%, по меньшей мере примерно 25%, по меньшей мере примерно 30%, по меньшей мере примерно 35%, по меньшей мере примерно 40%, по меньшей мере примерно 45%, по меньшей мере примерно 50%, по меньшей мере примерно 55% или по меньшей мере примерно 60%;
ii) высвобождает по меньшей мере примерно 30%, например, по меньшей мере примерно 35%, по меньшей мере примерно 40%, по меньшей мере примерно 45%, по меньшей мере примерно 50%, по меньшей мере примерно 55%, по меньшей мере примерно 60%, по меньшей мере примерно 65%, по меньшей мере примерно 70%, по меньшей мере примерно 75% или по меньшей мере примерно 80% масла, и
iii) в форме таблетки характеризуется значением времени дезинтеграции максимум 1 час, таким, например, как максимум примерно 50 минут, максимум примерно 40 минут, максимум примерно 30 минут, максимум примерно 20 минут, максимум примерно 10 минут или максимум примерно 5 минут, при испытании в соответствии с тестом Европейской Фармакопеи на дезинтеграцию, при этом таблетка содержит примерно 90 мас.% или более, например, примерно 92,5 мас.% или более, примерно 95 мас.% или более, примерно 97,5 мас.% или более или примерно 100 мас.% фармацевтически приемлемого материала.
Другие конкретные варианты осуществления настоящего изобретения описаны ниже, где твердый фармацевтически приемлемый материал, использованный в качестве сорбционного материала в композиции по настоящему изобретению, при указанном испытании:
i) имеет пороговое значение, равное по меньшей мере 55%;
твердый фармацевтически приемлемый материал, при испытании в соответствии с указанным в описании методом:
ii) высвобождает по меньшей мере примерно 75% масла и/или
твердый фармацевтически приемлемый материал, при испытании в соответствии с указанным в описании тестом:
iii) в виде таблетки характеризуется значением времени дезинтеграции максимум примерно 10 минут, при испытании по тесту Европейской Фармакопеи на дезинтеграцию, при этом таблетка содержит примерно 97,5 мас.% фармацевтически приемлемого материала.
Твердый фармацевтически приемлемый материал, используемый в качестве сорбционного материала в композиции по настоящему изобретению, обычно представляет собой материал в форме частиц в форме, например, порошков, частиц, гранул, гранулята и т.п.
Такой материал в форме частиц, который пригоден для использования в качестве маслосорбирующего материала, обычно имеет объемную плотность, равную примерно 0,15 г/см3 или более, такую, например, как по меньшей мере примерно 0,20 г/см3 или 0,25 г/см3.
Кроме того, маслосорбирующий материал обычно имеет показатель, характеризующий абсорбцию масла, равный примерно по меньшей мере 100 г масла/100 г, такой, например, как по меньшей мере примерно 150 г масла/100 г, по меньшей мере примерно 200 г масла/100 г, по меньшей мере примерно 250 г масла/100 г, по меньшей мере примерно 300 г масла/100 г, по меньшей мере примерно 400 г масла/100 г фармацевтически приемлемого материала. Показатель абсорбции масла определяют в соответствии с процедурой, описанной в экспериментальном разделе настоящей заявки.
Авторы настоящего изобретения показали, что общей характеристикой некоторых материалов, пригодных для использования в качестве маслосорбирующего материала, является то, что они обладают относительно большой площадью поверхности. Соответственно, фармацевтически приемлемый материал, используемый в качестве маслосорбирующего материала по настоящему изобретению, может иметь площадь поверхности BET, равную по меньшей мере 5 м2/г, такую, например, как по меньшей мере примерно 25 м2/г, по меньшей мере примерно 50 м2/г, по меньшей мере примерно 100 м2/г, по меньшей мере примерно 150 м2/г, по меньшей мере примерно 200 м2/г, по меньшей мере примерно 250 м2/г или по меньшей мере примерно 275 м2/г.
Как указывалось выше, одной из характерных особенностей фармацевтически приемлемого материала, используемого в качестве маслосорбирующего материала по настоящему изобретению, является то, что он позволяет удержать хорошую текучесть, даже если вносится вместе с масляным или маслоподобным материалом. Таким образом, текучесть фармацевтически приемлемого материала, содержащего 25 мас.% или более, например, 30 мас.% или более, 40 мас.% или более, 45 мас.% или более, 50 мас.% или более, 55 мас.% или более, 60 мас.% или более, 65 мас.% или более или примерно 70 мас.% или более viscoleo, обычно удовлетворяет требованиям Европейской Фармакопеи.
Следует отметить, что маслосорбирующий материал может включать кремниевую кислоту или ее производное или ее соль, такие, например, как диоксид кремния, или его полимер, в качестве фармацевтически приемлемого эксципиента. При этом, в зависимости от качества, диоксид кремния может использоваться как замасливатель или как маслосорбирующий материал. Количества его, соответствующие последней функции, представляются особенно важными.
В конкретном варианте осуществления изобретения композиция или твердая дозированная форма по настоящему изобретению включает фармацевтически приемлемый эксципиент, который представляет собой диоксид кремния, продукт со свойствами, соответствующими таковым Аэроперла 300 (Aeroperl® 300) (доступен от Degussa, Frankfurt, Germany).
Использование маслосорбирующего материала в композициях или дозированных формах по настоящему изобретению очень полезно для получения фармацевтической, косметической и/или пищевой композиции, где указанная композиция включает масло или маслоподобный материал. Одним из преимуществ является тот факт, что появляется возможность включать относительно большие количества масла и маслоподобного материала и сохранять при этом материал в виде твердой формы. Таким образом, можно получать твердые композиции с относительно высоким содержанием масла и маслоподобного материала, используя маслосорбирующий материал по настоящему изобретению. В фармацевтической области такое свойство рассматривается как благоприятное, поскольку позволяет включать относительно большое количество масла или маслоподобного материала в твердую композицию, особенно в тех случаях, когда активное вещество не обладает подходящими свойствами по водорастворимости (например, имеет плохую растворимость в воде), по стабильности в водной среде (например, происходит дезинтеграция в водной среде), по биологической доступности при пероральном введении (например, обладает низкой биологической доступностью) и т.п., или в тех случаях, когда желательно модифицировать уровень высвобождения активного вещества из композиции для получения контролируемой, задержанной, пролонгированной и/или пульсовой доставки активного вещества. Таким образом, конкретный вариант включает его использование при получении фармацевтических композиций.
Маслосорбирующий материал, применяемый при обработке твердых композиций, обычно абсорбирует примерно 5 мас.% или более, например, примерно 10 мас.% или более, примерно 15 мас.% или более, примерно 20 мас.% или более, примерно 25 мас.% или более, примерно 30 мас.% или более, примерно 35 мас.% или более, примерно 40 мас.% или более, примерно 45 мас.% или более, примерно 50 мас.% или более, примерно 55 мас.% или более, примерно 60 мас.% или более, примерно 65 мас.% или более, примерно 70 мас.% или более, примерно 75 мас.% или более, примерно 80 мас.% или более, примерно 85 мас.% или более, примерно 90 мас.% или более или примерно 95 мас.% или более масла или масляного материала во все еще твердом материале.
Способ получения материала в форме частиц или твердой дозированной формы по настоящему изобретению
В другом аспекте настоящее изобретение относится к способу получения фармацевтических композиций по настоящему изобретению. В целом, может быть использован любой подходящий способ, имеющийся в фармацевтической области.
Прежде всего, авторы настоящего изобретения разработали способ получения композиции в форме частиц, который особенно подходит для включения активных веществ, слабо растворимых в воде, в случае которых могут возникать проблемы с уровнем биологической доступности, определяемым плохой растворимостью в воде. Фармацевтические композиции могут быть изготовлены любым удобным способом, таким как гранулирование, смешивание, распылительная сушка и т.п. Особенно полезным способом является способ, описанный в WO 03/004001, который включен в настоящее описание в качестве ссылки. В нем описывается способ получения материала в форме частиц с использованием контролируемого способа агломерации, то есть способа, который позволяет контролировать рост размера частиц. Данный способ включает распыление первой композиции, включающей, например, активное вещество и носитель, которые были ранее расплавлены, на вторую твердую среду с носителем. В норме расплавленный носитель имеет точку плавления примерно 5°С, однако ниже, чем точка плавления активного вещества. Точка плавления носителя может находиться в диапазоне от 10°С до 150°С, таком, например, как в диапазоне от 30°С до 100°С или в диапазоне от 40°С до 50°С, что является наиболее предпочтительным.
Соответственно, в другом аспекте настоящее изобретение относится к способу изготовления материала в форме частиц или твердой дозированной формы по настоящему изобретению, который включает стадии:
i) применение носителя в жидкой форме, если это релевантно;
ii) поддержание жидкого носителя при температуре ниже точки плавления фибрата и/или статина;
iii) растворение желательного количества фибрата и статина в носителе;
iv) распыление полученного раствора на твердый носитель, имеющий температуру ниже точки плавления носителя;
v) механическая обработка полученной композиции с получением частиц, например, материала в форме частиц, и
vi) необязательно проведение традиционных методик, позволяющих получать на основе материала в форме частиц твердую дозированную форму.
Преимуществом использования контролируемого метода агломерации, описанного в WO 03/004001, является то, что он позволяет использовать относительно большое количество расплава относительно материала в форме частиц, без соответствующего нежелательного роста размера частиц. Соответственно, в одном варианте осуществления настоящего изобретения материал в форме частиц в фармацевтической композиции имеет среднее геометрическое значение диаметра dgw ≥ 10 мкм, такое, например, как ≥ 20 мкм, от примерно 20 до примерно 2000, от примерно 30 до примерно 2000, от примерно 50 до примерно 2000, от примерно 60 до примерно 2000, от примерно 75 до примерно 2000, например, от примерно 100 до примерно 1500 мкм, от примерно 100 до примерно 1000 мкм или от примерно 100 до примерно 700 мкм, или максимум примерно 400 мкм или максимум 300 мкм, составляя, например, от примерно 50 до примерно 400 мкм, например, от примерно 50 до примерно 350 мкм, от примерно 50 до примерно 300 мкм, от примерно 50 до примерно 250 мкм, от примерно 100 до примерно 300 мкм.
Описание твердой дисперсии и раствора твердого вещества, основанных на органических растворителях
В своем существенном варианте настоящее изобретение относится к варианту, в соответствии с которым по меньшей мере часть фибрата и статина присутствует в композиции в виде твердой дисперсии, включающей молекулярную дисперсию и раствор твердого вещества. В норме 10% или более, например, 20% или более, 30% или более, 40% или более, 50% или более, 60% или более, 70% или более, 80% или более, 90% или более, например, примерно 95% или более или примерно 100 мас.% фибрата или статина присутствует в носителе в форме твердой дисперсии (при условии, что по меньшей мере 80 мас.% от общего количества фибрата и статина растворено в носителе).
Твердая дисперсия может быть получена разными путями, например, с помощью органических растворителей, путем диспегирования или растворения активного вещества в другой подходящей среде (например, в носителе, который находится в жидкой форме при комнатной температуре или при повышенных температурах).
Твердые дисперсии (по методу, включающему использование растворителя) получают путем растворения физической смеси активного вещества (например, лекарственного вещества) и носителя в обычно используемом органическом растворителе, с последующим выпариванием растворителя. Носитель часто представляет собой лиофильный полимер. Подходящие органические растворители включают фармацевтически приемлемый растворитель, в котором растворяют активное вещество, такой как метанол, этанол, метиленхлорид, хлороформ, этилацетат, ацетон и их смеси.
Подходящие водорастворимые носители включают полимеры, такие как полиэтиленгликоль, полоксамеры, полиоксиэтиленстеараты, поли-ε-капролактон, поливинилпирролидон (ПВП), сополимер поливинилпирролидона-поливинилацетата: ПВП-ПВА (Kollidon VA64), полиметакриловые полимеры (Eudragit RS, Eudragit RL, Eudragit NE, Eudragit E) и поливиниловый спирт (ПВС), гидроксипропилцеллюлозу (ГПЦ), гидроксипропилметилцеллюлозу (ГПМЦ), метилцеллюлозу и поли(этиленоксид) (ПЭО).
Полимеры, содержащие кислотные функциональные группы, могут быть пригодны для твердых дисперсий, которые высвобождают активное вещество в предпочтительном диапазоне pH, создавая условия для приемлемой абсорбции в кишечнике. Такие полимеры могут представлять собой один или несколько полимеров, выбранных из группы, включающей фталат гидроксипропилметилцеллюлозы (ГПМЦ), поливиниловый ацетатфталат (ПВАФ), ацетат-сукцинат гидроксипропилметилцеллюлозы (ГПМЦ-АС), альгинат, карбомер, карбоксиметилцеллюлозу, сополимер метакриловой кислоты (Eudragit L, Eudragit S), шеллак, ацетат-фталат целлюлозы (CAP), крахмал-гликолят, полакрилин, ацетат-фталат метилцеллюлозы, ацетат-фталат гидроксипропилцеллюлозы, ацетат-терефталат целлюлозы, ацетат-изофталат целлюлозы и ацетат-тримеллитат целлюлозы.
Что касается количества активных веществ и носителя в дисперсии, то весовое соотношение активных веществ к полимеру может находиться в диапазоне от примерно 3:1 до примерно 1:20. Однако может быть использован более узкий диапазон от примерно 3:1 до примерно 1:5, такой, например, как от 1:1 до примерно 1:3 или аналогичный.
Твердую дисперсию получают предпочтительно по методикам распылительной сушки, контролируемой агломерации, сушки при замораживании или путем нанесения покрытия на частицы носителя, или по любому другому способу, включающему удаление растворителя. Высушенный продукт содержит активное вещество в форме твердой дисперсии, включающей молекулярную дисперсию и раствор твердого вещества.
Фармацевтические композиции, включающие активное вещество, по меньшей мере частично, в форме твердой дисперсии или раствора, могут быть в принципе получены с использованием любой известной в данной области подходящей процедуры изготовления фармацевтических композиций.
Кроме метода, основанного на использовании органического растворителя, твердые дисперсии или растворы твердого вещества одного или нескольких фибратов могут быть получены путем диспергирования и/или растворения активного соединения в композиции с носителем, используемом в рамках методики контролируемой агломерации. Могут быть добавлены стабилизаторы и т.п. для достижения стабильности, твердой дисперсии/раствора.
Твердые дозированные формы
Фармацевтическая композиция по настоящему изобретению имеет форму частиц и может быть использована как таковая. Однако, во многих случаях удобнее представлять композицию в виде гранул, шариков, микросфер, наночастиц и т.п., или в виде твердых дозированных форм, включающих таблетки, капсулы, пакетики и т.п. Твердая дозированная форма по настоящему изобретению может представлять собой единичную дозированную форму или может иметь дозированную форму типа полидепо, включающую множество отдельных стандартных единиц, таких как, например, различные гранулированные формы, шарики и/или гранулы.
В норме фармацевтическая композиция или твердая дозированная форма по настоящему изобретению предназначены для введения пероральным, трансбуккальным или сублингвальным способом.
Настоящее изобретение относится к указанной выше форме представления препарата. В рамках настоящего изобретения рассматривается композиция/твердая дозированная форма, которая предназначена для высвобождения активного вещества в быстром режиме, в режиме задержанного высвобождения или в режиме модифицированного высвобождения.
Твердая дозированная форма по настоящему изобретению включает фармацевтическую композицию в форме частиц, как было описано выше. В норме концентрация фармацевтической композиции в форме частиц находится в диапазоне от примерно 5 до 100 мас.%, таком, например, как в диапазоне от примерно 10 мас.% до примерно 90 мас.%, от примерно 15 мас.% до примерно 85 мас.%, от примерно 20 мас.% до примерно 80 мас.%, от примерно 25 мас.% до примерно 80 мас.%, от примерно 30 мас.% до примерно 80 мас.%, от примерно 35 мас.% до примерно 80 мас.%, от примерно 40 мас.% до примерно 75 мас.%, от примерно 45 мас.% до примерно 75 мас.% или от примерно 50 мас.% до примерно 70 мас.% от дозированной формы. В варианте настоящего изобретения концентрация фармацевтической композиции в форме частиц составляет 50 мас.% или более от дозированной формы.
Твердую дозированную форму по настоящему изобретению получают путем обработки материала в форме частиц по настоящему изобретению с использованием методик, известных специалистам в данной области. В норме, такая методика включает добавление также одного или нескольких фармацевтически приемлемых эксципиентов из числа указанных выше.
Композиция или твердая дозированная форма по настоящему изобретению может также содержать покрытие из пленочного покрытия, энтеросолюбильного покрытия, покрытия, способствующего модификации высвобождения, защитного покрытия, антиадгезивного покрытия и т.п.
Твердая дозированная форма по настоящему изобретению может также содержать покрытие, наносимое с целью достижения подходящих свойств, например, в отношении высвобождения активного вещества. Покрытие может быть нанесено на стандартную дозированную форму (например, на таблетки, капсулы) или может быть нанесено на дозированную форму типа полидепо или на ее отдельные стандартные единицы.
Подходящие для нанесения покрытия материалы включают, например, метилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, акриловые полимеры, этилцеллюлозу, ацетат-фталат целлюлозы, поливинилацетат-фталат, фталат гидроксипропилметилцеллюлозы, поливиниловый спирт, натрий-карбоксиметилцеллюлозу, ацетат целлюлозы, ацетат-фталат целлюлозы, желатин, сополимер метакриловой кислоты, полиэтиленгликоль, шеллак, сахарозу, диоксид титана, карнаубский воск, микрокристаллический воск и зеин.
В материал, используемый для нанесения покрытия, могут быть добавлены пластификаторы и другие ингредиенты. При этом к материалу, используемому для нанесения покрытия, могут быть добавлены одинаковые или разные активные вещества.
Материал в форме частиц или твердая дозированная форма по настоящему изобретению могут быть предназначены для высвобождения фибрата и статина в любом подходящем режиме. В конкретном варианте композиция (например, материала в форме частиц или твердая дозированная форма) может повышать биологическую доступность фибрата и/или статина после перорального приема. Активные вещества могут высвобождаться относительно быстро, приводя к более раннему началу действия, при этом активное вещество может высвобождаться в таком режиме, который описывается нулевой кинетикой или кинетикой первого порядка, или оно может высвобождаться контролируемым или модифицированным образом, так что достигается заданный характер высвобождения. Простые композиции также входят в область настоящего изобретения.
В конкретном варианте твердая дозированная форма по настоящему изобретению ассоциирована с повышением биологической доступности фибрата и/или статина относительно соответствующих показателей для существующих в настоящее время коммерческих дозированных форм фибрата и/или статина для введения млекопитающим, при наличии в этом потребности.
Что касается твердой дозированной формы фенофибрата по настоящему изобретению, то может быть достигнута величина AUC относительно коммерчески доступных таблеток препарата Трикор® (Tricor®), равная по меньшей мере примерно 1,1 или по меньшей мере примерно 1,2, или по меньшей мере примерно 1,3, или по меньшей мере примерно 1,4, или по меньшей мере примерно 1,5, или по меньшей мере примерно 1,75 или более, или по меньшей мере примерно 2,0, или по меньшей мере примерно 2,5, или по меньшей мере примерно 3,0, при этом значение AUC определяют в аналогичных условиях. Кроме того, твердые дозированные формы получают со значением Cmax относительно указанного значения для коммерчески доступного препарата таблеток Трикор® (Tricor®), равным примерно 1,1, или по меньшей мере примерно 1,2, или по меньшей мере примерно 1,3, или по меньшей мере примерно 1,4, или по меньшей мере примерно 1,5, или по меньшей мере примерно 1,6 или более, или по меньшей мере примерно 2,0, или по меньшей мере примерно 2,5, или по меньшей мере примерно 3,0, где значения Cmax определяют в аналогичных условиях.
Другие аспекты изобретения
В одном варианте настоящее изобретение относится к фармацевтической композиции в форме частиц, которая включает фибрат и статин, где указанная композиции при пероральном введении млекопитающим, при наличии в этом потребности, демонстрирует значение AUC/AUCControl, равное по меньшей мере 1,0, где значение AUCControl определяют с использованием коммерчески доступного продукта, содержащего то же самое количество фибрата и/или статина, а значения AUC определяют в аналогичных условиях.
Абсолютное значение биологической доступности для инъецируемых композиций отсутствует, например, для фенофибрата (скорое всего, в связи с проблемами растворимости в водных средах). Коммерчески доступная композиция, содержащая фенофибрат, включает поверхностно-активные вещества и/или, например, липофильную среду. Поверхностно-активные вещества могут улучшать биологическую доступность и, в этой связи, биологическая доступность такой композиции может быть уже достаточно хорошей. Однако, все еще имеется потребность в разработке методики создания гибкой композиции, которая позволяла бы изготавливать множество дозированных форм. Соответственно, потребность в таких улучшенных и/или более гибких композициях может быть связана с достижением той же или лучшей биологической доступности, чем уже было достигнуто при использовании имеющихся коммерчески доступных продуктов.
Соответственно, в других вариантах осуществления изобретения значение AUC/AUCControl составляет по меньшей мере примерно 1,1, например, по меньшей мере примерно 1,2, по меньшей мере примерно 1,3, по меньшей мере примерно 1,4, по меньшей мере примерно 1,5, по меньшей мере примерно 1,75 или более, примерно 1,8 или более, примерно 1,9 или более, примерно 2,0 или более, примерно 2,5 или более, примерно 2,75 или более, примерно 3,0 или более, примерно 3,25 или более, примерно 3,5 или более, примерно 3,75 или более, примерно 4,0 или более, примерно 4,25 или более, примерно 4,5 или более, примерно 4,75 или более или примерно 5,0 или более, где значения AUC определяют в аналогичных условиях.
При сравнении результатов тестирования, указанных выше, коммерчески доступный продукт включает Трикор® (Tricor®) в виде таблеток или Трикор® (Tricor®) в виде капсул, а фибрат представлен фенофибратом. В том случае, когда фибрат представлен гемфиброзилом, соответствующим коммерчески доступным продуктом является Лопид® (Lopid®), в том случае, когда фибрат представлен безафибратом, соответствующим коммерчески доступным продуктом является Безалип® (Bezalip®), в том случае, когда фибрат представлен клофибратом, соответствующим коммерчески доступным продуктом является Атромид® (Atromid®), и, наконец, когда фибрат представлен ципрофибратом, соответствующим коммерчески доступным продуктом является Липанон® (Lipanon®). Рекомендованной дозировкой Трикора® является 54-160 мг/день при приеме вместе с пищей. Таблетки Трикор® предлагаются с активностью 54 и 160 мг, а капсулы Трикор® предлагаются с активностью 67 и 200 мг. Таблетки обладают более высокой биологической доступностью, чем капсулы. Другими торговыми наименованиями препарата являются Липантил® (Lipanthyl® или Lipanthil®) или Каталип® (Catalip®).
Контрольные композиции для отдельных статинов были указаны выше в разделе, относящемся к обсуждению отдельных статинов.
Другим объектом настоящего изобретения является снижение или устранение пищевого эффекта. Таким образом, в другом аспекте настоящее изобретение относится к фармацевтической композиции (к материалу в форме частиц или твердой дозированной форме), включающей фибрат и статин, так что композиция при пероральном введении млекопитающим, в случае наличия такой потребности, не демонстрирует существенного побочного пищевого эффекта, выявляемого по величине показателя (AUCfed/AUCfasted), которая составляет примерно 0,85 со сниженным пределом 90% доверительности, равным по меньшей мере 0,75, при этом значение AUC определяют для фибрата или статина или их обоих.
В конкретном варианте фармацевтическая композиции по настоящему изобретению имеет значение (AUCfed/AUC fasted), равное примерно 0,9 или более, такое, например, как примерно 0,95 или более, примерно 0,97 или более, примерно 1 или более.
Иными словами, разница между значениями биологической доступности, измеряемыми после перорального введения млекопитающему с пищей и без пищи, соответственно, составляет менее чем 25% и равно, например, менее чем 20%, менее чем 15%, менее чем 10% или менее чем 5%.
В другом аспекте настоящее изобретение относится к фармацевтической композиции в форме частиц, включающей фибрат и статин, так что указанная композиция при пероральном введении млекопитающему, в случае наличия такой потребности, по существу биологически эквивалентна коммерчески доступному продукту, содержащему то же самое количество фибрата и/или то же самое количество статина, при введении в той же самой или в сниженной дозе, что и коммерчески доступный продукт, содержащий такое же количество фибрата и такое же количество статина.
В конкретных вариантах указанная доза составляет примерно 98 мас.%, например, максимум примерно 95 мас.%, максимум примерно 90 мас.%, максимум примерно 85 мас.%, максимум примерно 80 мас.%, максимум примерно 75 мас.%, максимум примерно 70 мас.%, максимум примерно 65 мас.%, максимум примерно 60 мас.%, максимум примерно 55 мас.% или максимум примерно 50 мас.% от дозы фибрата, вводимого в форме коммерчески доступного продукта, содержащего то же самое количество фибрата, и/или от дозы статина, вводимого в форме коммерчески доступного продукта, содержащего те же самое количество статина.
В норме биологическую эквивалентность определяют с помощью по меньшей мере одного из следующих параметров: tmax (время, необходимое для достижения максимальной концентрации в плазме), cmax (максимальная плазменная концентрация), AUC0-t (площадь под кривой от момента времени 0 на момент времени t), AUC0-infinity (площадь под кривой от момента времени 0 до бесконечного времени t), W50 (период времени, в течение которого плазменная концентрация составляет 50% или более от Cmax), W75 (период времени, в течение которого плазменная концентрация составляет 75% или более от Cmax) и/или MRT (среднее время пребывания в организме).
Основная проблема, возникающая при лечении с использованием фибрата, связана с наличием больших вариаций как для отдельного индивидуума, так и между индивидуумами. Так, в еще одном аспекте настоящее изобретение относится к фармацевтической композиции в форме частиц, которая включает фибрат и статин, где данная композиция при пероральном введении млекопитающему, при наличии такой потребности, приводит к снижению вариаций как для одного индивидуума, так и между отдельными индивидуумами, сравнительно с таковыми, наблюдаемыми при введении коммерчески доступного продукта, содержащего то же самое количество фибрата, в тех же условиях и в дозе, которая дает эквивалентный терапевтический эффект.
Удобным способом для определения того, произошла ли абсорбция соответствующего количества фибрата и/или статина, является определение содержания неизмененного фибрата, выделенного с фекалиями. Таким образом, в одном варианте настоящее изобретение относится к твердой фармацевтической композиции, в которой максимум примерно 25 мас.%, например, максимум примерно 20 мас.%, максимум примерно 15 мас.%, максимум примерно 10 мас.% или максимум примерно 5 мас.% фибрата и/или статина, содержащегося в композиции, выделяется с фекалиями после перорального введения.
Фармацевтическая композиция или твердая дозированная форма по настоящему изобретению разработана для высвобождения фибрата соответствующим образом. Конкретные параметры высвобождения, а также конкретные параметры абсорбции описываются ниже.
В конкретных вариантах осуществления настоящего изобретения фибрат и/или статин высвобождаются из композиции в течение примерно 2 часов, например, в течение примерно 1,5 часа или в течение примерно 1 часа после перорального введения и/или примерно 50 мас.% или более фибрата и/или статина высвобождается из композиции в течение 30 минут после перорального введения и/или примерно 50 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 20 минут после перорального введения и/или примерно 60 мас.% или более фибрата высвобождается из композиции в течение примерно 1,5 часа после перорального введения, и/или примерно 60 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 1 часа после перорального введения, и/или примерно 70 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 1,5 часа после перорального введения, и/или примерно 70 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 1 часа после перорального введения, и/или примерно 85 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 45 минут, при испытании in vitro по тесту определении уровня растворения, в соответствии с процедурой Американской Фармакопеи (метод с использованием лопастной мешалки), при использовании воды в качестве растворяющей среды, при скорости 100 об/мин и при температуре примерно 37°С.
В другом варианте примерно 50 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 20 минут, 15 минут или 10 мин и/или примерно 60 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 20 минут или 15 минут, и/или примерно 70 мас.% или более фибрата и/или статина высвобождается из композиции в течение примерно 20 минут или 15 минут, при испытании по тесту определения уровня растворимости in vitro, в соответствии с процедурой Американской Фармакопеи (метод с использованием лопастной мешалки), при использовании воды в качестве растворяющей среды, при скорости 100 об/мин и при температуре примерно 37°С.
В еще одном варианте примерно 50 мас.% или более фибрата и/или статина, содержащихся в композиции, абсорбируется в течение примерно 8 часов, 7 часов, 6 часов, 5 часов и/или примерно 60 мас.% или более фибрата и/или статина, содержащихся в композиции, абсорбируется в течение примерно 8 часов или 7 часов после перорального введения, и/или примерно 60 мас.% или более фибрата, содержащегося в композиции, абсорбируется в течение примерно 7 часов после перорального введения, и/или примерно 70 мас.% или более фибрата, содержащегося в композиции, абсорбируется в течение примерно 8 часов или 7 часов после перорального введения.
Детали и особенности, раскрываемые в рамках данного аспекта изобретения, применимы, в том числе, и к другим аспектам изобретения. Соответственно, свойства, относящиеся к повышению биологической доступности, изменениям в параметрах биологической доступности, к снижению побочного пищевого эффекта, а также к высвобождению одного или более фибратов и т.п., описанных и/или заявленных в настоящем описании для фармацевтической композиции в форме частиц, аналогичны таковым для твердой дозированной формы по настоящему изобретению.
Материалы и методы
Материалы
Фенофибрат (поставляемый компанией Sigma)
Моногидрат лактозы, 200 меш (от компании DMV)
Гранулированный оксид кремния, Aeroperl® 300 (Degussa)
Полиэтиленгликоль 6000, Pluracol® E6000 (от компании BASF)
Полоксамер 188, Pluronic® F-68 (от компании BASF)
Глицерилмоностеарат, Rylo® MD50 (от компании Danisco Cultor), Евр. Фарм.
Avicel PH200 (микрокристаллическая целлюлоза) (от компании FMC)
Стеарат магния
Таблетки, капсулы или гранулы могут содержать энтеросолюбильное покрытие, полученное с использованием разных типов полимеров, таких как ацетат-сукцинат гидроксипропилметилцеллюлозы (Aqoat), ацетат-фталат целлюлозы CAP, фталат гидроксипропилметилцеллюлозы HPMCP или сооплимеры метакриловой кислоты, такие как Eudragit L30D, Eudragit 100/S, Eudragit 100/L.
Композиция таблеток Трикор® (Tricor®)
Таблетки Трикор® (Tricor®) представляют собой фенофибрат-содержащие таблетки, доступные для перорального введения, содержащие 54 мг или 160 мг фенофибрата на таблетку.
Таблетки содержат следующие неактивные ингредиенты: коллоидный диоксид кремния, кросповидон, моногидрат лактозы, лецитин, микрокристаллическую целлюлозу, поливиниловый спирт, повидон, лаурилсульфат натрия, стеарилфумарат натрия, тальк, диоксид титана, ксантановую камедь, краситель.
Оборудование
Оборудование для псевдоожижения в лабораторных условиях: Strea-1.
Устройство для псевдоожиженного слоя представляет собой прототип, состоящий из отдельных единиц для нагревания воздуха, подаваемого для атомайзера, резервуара под давлением и питающей трубы. Гранулят просеивают вручную и смешивают с экстрагранулярными эксципиентами в смесителе Turbula.
Прессование таблеток проводят на прессе с одним штамповочным устройством, Diaf TM20.
Методы
В соответствии со способом настоящего изобретения, лекарственный препарат на основе фенофибрата растворяют в расплавленном(ых) носителе(ях) и наносят на носитель для частиц следующим образом:
Носитель(ли) расплавляют в стакане, помещенном в микроволновую печь. Стакан переносят на нагреваемое плато с контролируемой температурой, к которому подведена магнитная мешалка. Фенофибрат медленно растворяют в расплаве при температуре 75°С при перемешивании на магнитной мешалке. Затем горячий раствор переносят в резервуар под давлением для распыления расплава на носитель в псевдоожиженном слое. Гранулированный продукт выпускают из устройства для псевдоожиженного слоя и просеивают вручную через сито 0,7 мм или 1,0 мм. Просеянный продукт смешивают со стеаратам магния в течение 0,5 минут с использование смесителя Turbula. Если включается супергранулярная фаза, то данную супергранулярную фазу подвергают предварительному смешиванию с гранулятом в течение 3 минут в смесителе Turbula.
Прессование таблеток проводят на прессе с одним штамповочным устройством, Diaf TM20.
Тест для определения порогового значения
Данный тест включает определение текучести по методу, описанному в Европейской Фармакопее, путем измерения скорости течения материала из воронки через сопло с диаметром 10,0 мм.
Продукт Viscoleo (триглицерида со средней длиной цепи MCT; Miglyol 812 N от компании Condea) добавляют к 100 г твердого фармацевтически приемлемого материала, подлежащего тестированию по настоящему изобретению, и перемешивают вручную. Полученную смесь просеивают через сито размером 0,3 мм с получением гомогенной смеси. Затем добавляют масло до того момента, когда поток из 100 г уже не может протекать через данное сопло. Если исследуемый материал обладает крупными объемными параметрами (например, как Aeroperl 300), используют только 50 г смеси при тестировании в данном варианте. Максимальную концентрацию масла, при которой поток материала может быть все еще получен, называют пороговым значением (приводится в виде мас.%).
Тест на высвобождение
Растворимый в жире краситель Sudan II (BDH Gur®), получаемый от компании BDH VWR International, в количестве 14,3 мг растворяют в 50,0 г viscoleo (фракционированные триглицериды со средней длиной цепи).
10 г указанного масла добавляют к 10,0 г твердого фармацевтически приемлемого материала, подлежащего тестированию по настоящему изобретению, и смешивают до того момента, когда масло полностью абсорбируется в твердый материал. Смесь затем просеивают через сито размером 0,3 мм с получением гомогенной смеси.
1,00 г данной смеси переносят в центрифужную пробирку и добавляют 3,0 мл воды. Данную суспензию смешивают с пробой крови в течение 1 часа и затем центрифугируют в течение 10 минут при 5000 об/мин. Верхнюю фазу масла и воды переносят осторожно в стакан и воду выпаривают в печи при температуре 80°С до постоянного веса. Количество высвобожденного продукта из твердого материала вычисляют на основании веса, оставшегося после выпаривания водной фазы.
Тест на дезинтеграцию
Время дезинтеграции определяют по методу, описанному в Европейской Фармакопее.
Тест на растворимость
Данный тест проводят в соответствии с методом Евр. Фарм. 2.9.3 с использованием лопастного смесителя. Количественное определение проводят по методу ВЭЖХ с УФ-детктором.
Определение объемной плотности
Объемную плотность измеряют, внося 100 г исследуемого порошка в градуированный цилиндр на 250 мл. Объемную плотность получают в виде объемной плотности в г/мл после спуска жидкости. Определение проводят по методу Европейской Фармакопеи (кажущийся объем).
Определение маслоабсорбирующего показателя
Показатель маслоабсорбирующей способности определяют при добавлении точных количеств (10 г) viscoleo к точному количеству фармацевтически приемлемого материала (100 г), подлежащего тестированию. Маслоабсорбирующий показатель (выражаемый в виде г viscoleo/100 г материала) определяется в той точке, когда дальнейшее добавление 10 г масла приводит к образованию материала, который уже не обладает соответствующими свойствами текучести, то есть материал не удовлетворяет требованиям Европейской Фармакопеи (тест на текучесть: см., приведенный выше в разделе "Тест на пороговое значение").
Определение площади поверхности BET
Применяемый прибор представляет собой Micromertics Gemini 2375. Данный метод основан на волюметрических методах Американской Фармакопеи, включающих определение множественных точек.
Определение текучести
Текучесть определяют по методу, описанному в Европейской Фармакопее, в соответствии с которым производят измерение скорости течения материала из воронки с диаметром сопла 10,0 мм.
Определение вариаций в весовых значениях
Таблетки, полученные в соответствии с процедурой, описанной в примерах, подвергают тесту для определения весовых вариаций, в соответствии с процедурой Европейской Фармакопеи.
Определение среднего значения твердости
Таблетки, полученные в соответствии с процедурой, описанной в примерах, подвергают тесту для определения среднего значения твердости таблеток с использованием аппарата Schleuniger Model 6D, проводя данный тест в соответствии с общими инструкциями, прилагаемыми к данному аппарату.
Определение показателей для раствора твердого вещества
В соответствии с настоящим изобретением фибрат растворяют в носителе. Для оптимизации процесса проводят тест, включающий дифференциальную сканирующую калориметрию. Тест проводят с использованием композиции в форме частиц, твердой дозированной формы или смеси носителя и фибрата (после того, как, по расчетам, произошло образование раствора твердого вещества). Используют стандартное оборудование для ДСК (DSC), соединенное с персональным компьютером.
Определение среднего геометрического значения диаметра dgw.
Среднее геометрическое значение диаметра определяют при использовании метода дисперсии лазерной дифракции на полученном материале в форме частиц (или на исходном материале) на воздухе. Измерение проводят с использованием оборудования Sympatec Helos под давлением с дисперсией 1 бар, при этом регистрируется распределение эквивалентного сферического диаметра. Указанное распределение сопоставляют со значением log нормального распределения в режиме объем-размер.
В контексте настоящего описания "среднее геометрическое значение диаметра" означает средний диаметр log нормального распределения объем-размер.
Исследование in vivo на собаках Бигль
Исследование in vivo с целью определения биологической доступности композиций по настоящему изобретению относительно биологической доступности коммерчески доступной композиции таблеток с фенофибратом, например, Трикор® (Tricor®), проводят на собаках Бигль.
Экспериментальную работу проводят в Дании с использованием четырех самцов собак Бигль, каждая из которых имеет вес тела 12-18 кг (исходный вес). Исследование проводят в виде открытого, нерандомизированного перекрестного исследования. Для каждого животного ставят собственный контроль. Пероральные дозы фенофибрата вводят в соответствии с приведенными ниже данными.
Собак выдерживают без пищи в течение ночи перед введением дозы (воду дают по потребности) и через 5 часов после введения дозы дают пищу (доступ к воде не ограничивают). Каждой собаке дают дозу определенного количества фенофибрата, без учета веса животного.
Отбирают образцы крови из хвостовой вены в следующих временных точках: перед введением дозы, через 1, 1,5, 2, 3, 4, 6, 8, 12 и 24 часа после введения дозы. Отбирают 4 мл крови, смешивают с ЭДТА и замораживают (-80°С). Образцы крови анализируют с использованием процедуры экстракции и регистрации в режиме on-line LC/MS и результаты приводят в виде мг/мл.
Определенные полные профили концентрации крови для фенофибрата обрабатывают с использованием соответствующего программного обеспечения Pharmacokinetic software WinNonlin® (Pharsight, California; USA) для вычисления фармакокинетических параметров. Все данные корректируют относительно дозы, где это необходимо.
Приведенные ниже примеры даны с целью иллюстрации настоящего изобретения и не рассматриваются как ограничивающие область настоящего изобретения.
Пример 1
Таблетка с мгновенным высвобождением, содержащая фенофибрат и аторвастатин
Фенофибрат и аторвастатин вначале растворяют в полиэтиленгликоле 6000 и Полоксамере 188 (весовое соотношение -70:30) при температуре 70°С. Дисперсию распыляют на 250 г лактозы в псевдоожиженном слое Phast FB-100 с использованием устройства для распыления расплава Phast FS-1.7. Полученный материал в форме частиц просеивают через сито 0,7 мм и смешивают со стеаратом магния в течение 0,5 минут в смесителе Turbula.
Порошковую смесь прессуют в таблетки с диаметром 13 мм с содержанием 160 мг фенофибрата и 20 мг аторвастатина, получая таблетки сложной чашеобразной формы весом 667 мг.
Среднее время дезинтеграции: 20 мин, твердость: 45N
Пример 2
Таблетка с мгновенным высвобождением, содержащая фенофибрат и правастатин
Фенофибрат и правастатин вначале растворяют в полиэтиленгликоле 6000 и Полоксамере 188 (весовое соотношение -70:30) при температуре 70°С. Дисперсию распыляют на 250 г лактозы в псевдоожиженном слое Phast FB-100 с использованием устройства для распыления расплава Phast FS-1.7. Полученный материал в форме частиц просеивают через сито 0,7 мм и смешивают со стеаратом магния в течение 0,5 минут в смесителе Turbula.
Порошковую смесь прессуют в таблетки с диаметром 13 мм с содержанием 160 мг фенофибрата и 40 мг аторвастатина, получая таблетки сложной чашеобразной формы весом 688 мг.
Среднее время дезинтеграции: 25 мин, твердость: 47N
Пример 3
Таблетка с мгновенным высвобождением, содержащая фенофибрат и розувастатин
Фенофибрат и розувастатин вначале растворяют в полиэтиленгликоле 6000 и Полоксамере 188 (весовое соотношение -70:30) при температуре 70°С. Дисперсию распыляют на 250 г лактозы в псевдоожиженном слое Phast FB-100 с использованием устройства для распыления расплава Phast FS-1.7. Полученный материал в форме частиц просеивают через сито 0,7 мм и смешивают со стеаратом магния в течение 0,5 минут в смесителе Turbula.
Порошковую смесь прессуют в таблетки с диаметром 12 мм с содержанием 160 мг фенофибрата и 40 мг розувастатина, получая таблетки сложной чашеобразной формы весом 658 мг.
Среднее время дезинтеграции: 22 мин, твердость: 41N
Пример 4
Таблетка с мгновенным высвобождением, содержащая фенофибрат и симвастатин
Фенофибрат и симвастатин вначале растворяют в полиэтиленгликоле 6000 и Полоксамере 188 (весовое соотношение -70:30) при температуре 70°С. Дисперсию распыляют на 250 г лактозы в псевдоожиженном слое Phast FB-100 с использованием устройства для распыления расплава Phast FS-1.7. Полученный материал в форме частиц просеивают через сито 0,7 мм и смешивают со стеаратом магния в течение 0,5 минут в смесителе Turbula.
Порошковую смесь прессуют в таблетки с диаметром 13 мм с содержанием 160 мг фенофибрата и 40 мг симвастатина, получая таблетки сложной чашеобразной формы весом 688 мг.
Среднее время дезинтеграции: 25 мин, твердость: 39N
Пример 5
Таблетка на основе липофильной матрицы из глицерилмоностеарата
Фенофибрат и розувастатин вначале растворяют в глицерилмоностеарате при температуре 70°С. Раствор распыляют на 200 г лактозы в псевдоожиженном слое Phast FB-100 с использованием устройства для распыления расплава Phast FS-1.7. Полученный материал в форме частиц просеивают через сито 0,7 мм и смешивают со стеаратом магния в течение 0,5 минут в смесителе Turbula.
Порошковую смесь прессуют в таблетки с диаметром 11 мм, имеющие вес 572 мг и сложную чашеобразную форму.
Среднее время дезинтеграции: 42 мин, твердость: 48N
Пример 6
Капсула типа полидепо с модифицированным высвобождением на основе набухающей гидроколлоидной матрицы из гидроксипропилцеллюлозы
Фенофибрат и аторвастатин вначале растворяют в глицерилмоностеарате при температуре 70°С. Раствор распыляют на смесь из 50 г лактозы и 150 г ГПМЦ в псевдоожиженном слое Phast FB-100 с использованием устройства для распыления расплава Phast FS-1.7. Полученный материал в форме частиц просеивают через сито 0,7 мм и вводят в твердые желатиновые капсулы для заполнения (680 мг).
Пример 7
Таблетка с мгновенным высвобождением
Фенофибрат и правастатин вначале растворяют в полиэтиленгликоле 3000 при температуре 70°С. Дисперсию распыляют на 200 г Aeroperl в псевдоожиженном слое Phast FB-100 с использованием устройства для распыления расплава Phast FS-1.7. Полученный материал в форме частиц просеивают через сито 0,7 мм и смешивают со стеаратом магния в течение 0,5 минут в смесителе Turbula.
Порошковую смесь прессуют в таблетки с диаметром 11 мм, с содержанием 160 мг фенофибрата и 40 мг аторвастатина, получая таблетки сложной чашеобразной формы весом 478 мг.
Среднее время дезинтеграции: 29 мин, твердость 51N
Пример 8
Твердая дозированная форма по настоящему изобретению
Приведенную ниже композицию получают по способу, описанному выше в примере 1.
Пример 9
Предпочтительный диапазон количественного содержания фенофибрата и статина в композиции по настоящему изобретению
Композиции, описанные, в частности, в примерах 1-7, могут варьироваться для корректировки содержащегося в них уровня фибрата и статина. Каждый специалист в данной области знает, как откорректировать количество активных веществ и фармацевтически приемлемых эксципиентов без потери целевых объектов изобретения. Ниже приведены подходящие диапазоны содержания фенофибрата и отдельных статинов в композиции по настоящему изобретению.
Пример 10
Стабильность композиции по настоящему изобретению
В случае таких веществ, как фенофибрат и статины, влажность представляет собой существенную опасность для стабильности соединений. Это особенно справедливо, в том случае, когда пытаются объединять два нестабильных соединения в составе одной стандартной таблетки. Даже очень небольшое количество влаги/воды может существенно повысить деградацию при "взаимодействии" лекарственных компонентов. Кроме того, рост кристаллов также представляет собой потенциальную угрозу, связанную с влажностью, содержащейся в комбинированных продуктах.
При устранении воды в процессе получения и тщательном отборе ингредиентов, содержащих низкое количество воды, в целях оптимизации композиции, достигается хорошая стабильность соединения. В среднем, общее содержание воды в готовой композиции составляет менее 0,5 мас.%. Полимерная матрица служит в качестве защитной оболочки от влаги/кислорода для лабильных молекул.
Пример 10
Композиции, используемые в исследовании in vivo на собаках
Композиции по настоящему изобретению исследуют in vivo на собаках. Поскольку фенофибрат представляет собой лекарственное вещество, основной проблемой которого является биологическая доступность, исследования проводят прежде всего для определения, может ли быть достигнута улучшенная биологическая доступность. Соответственно, не приводятся данные относительно статинового компонента.
Таблетки с содержанием 50 мг и 160 мг фенофибрата, имеющие указанный ниже состав, получают по методике, описанной в примере 1:
Пример 11
Тесты по определению растворимости
Композицию А таблеток, полученную в примере 10, подвергают тесту на растворимость, описанному в разделе "Методы", в результате которого были получены следующие результаты:
Пример 12
Тесты на стабильность
Образцы композиции А таблеток, полученных в примере 10, хранят в указанных ниже условиях, соответственно, и через 1 месяц и 3 месяца после начала хранения подвергают испытанию в рамках теста на растворимость (стабильность), описанного в разделе "Методы"; указанный % растворения представляет собой % фенофибрата, растворенного через 45 минут.
Образцы композиции А таблеток хранят в указанных ниже условиях, соответственно, и затем подвергают тесту на определение количества фибрата, получая следующие результаты:
Образцы композиции А таблеток по настоящему изобретению хранят в указанных ниже условиях, соответственно, и затем подвергают тесту для определения степени деградации продукта по процедуре Европейской Фармакопеи (продукты деградации A, B, G и неизвестные, накапливаемые в общем продукте деградации; метод ВЭЖХ), получая следующие результаты:
Пример 13
Исследование in vivo на собаках
Исследование in vivo композиции А с содержанием 160 мг, полученной в примере 10, на собаках Бигль, проводят по процедуре, описанной в разделе "Методы", в сравнении с результатами испытаний таблеток Трикор® (Tricor®), 160 мг (партия No: 098212Е21), с получением следующих результатов:
Концентрация в крови (мг/мл) (среднее значения для 4 собак) после введения композиции:
Относительная биологическая доступность, по данным AUC (композиция А по настоящему изобретению/Tricor®): 306%.
Относительное значение Cmax (композиция А по настоящему изобретению/Tricor®): 356%.
Пример 14
Исследование in vivo на собаках
Второе исследование in vivo композиции А (Пример 10) с содержанием 160 мг было проведено на собаках Бигль, как описано выше в разделе "Методы", в сравнительном аспекте с препаратом Трикор® (Tricor®), 160 мг (партия No: 098212Е21), с получением следующих результатов:
Концентрация в крови (мг/мл) (среднее значения для 4 собак) после введения композиции:
Относительная биологическая доступность, по данным AUC (композиция А по настоящему изобретению/Tricor®): 198%.
Относительное значение Cmax (композиция А по настоящему изобретению/Tricor®): 238%.
Пример 15
Исследование in vivo на собаках
Исследование in vivo композиций B, C и D, 2×50 мг (Пример 10) было проведено на собаках Бигль, как описано выше в разделе "Методы", в сравнительном аспекте с препаратом Липантил®67M (Lipanthyl®67M), 2×67 мг (партия No: 75641), с получением следующих результатов:
Концентрация в крови (мг/мл) (среднее значения для 4 собак) после введения композиции:
Относительная биологическая доступность, по данным AUC (композиция В по настоящему изобретению/Липантил®67M): 532%
Относительное значение Cmax (композиция ВА по настоящему изобретению /Липантил®67M): 548%.
Относительная биологическая доступность, по данным AUC (композиция С по настоящему изобретению/Липантил®67M): 228%.
Относительное значение Cmax (композиция С по настоящему изобретению /Липантил®67M): 161%.
Относительная биологическая доступность, по данным AUC (композиция D по настоящему изобретению/Липантил®67M): 424%.
Относительное значение Cmax (композиция D по настоящему изобретению/Липантил®67M): 329%.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ФЕНОФИБРИНОВУЮ КИСЛОТУ, ЕЕ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМУЮ СОЛЬ ИЛИ ПРОИЗВОДНОЕ | 2003 |
|
RU2356549C2 |
| КОМПОЗИЦИЯ ПРОЛЕКАРСТВА ДЛЯ БОРЬБЫ С ВИРУСОМ ГЕПАТИТА С | 2006 |
|
RU2435592C2 |
| КОМПОЗИЦИЯ, ВКЛЮЧАЮЩАЯ ФЕНОФИБРИНОВУЮ КИСЛОТУ ИЛИ ЕЕ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМУЮ СОЛЬ, И КАПСУЛА, ВКЛЮЧАЮЩАЯ КОМПОЗИЦИЮ | 2008 |
|
RU2500398C2 |
| ЛЕКАРСТВЕННЫЕ КОМБИНАЦИИ, ВКЛЮЧАЮЩИЕ (Е)-7-[-4-(4-ФТОРФЕНИЛ)-6-ИЗОПРОПИЛ-2-[-МЕТИЛ(МЕТИЛСУЛЬФОНИЛ)АМИНО]- ПИРИМИДИН-5-ИЛ](3R,5S)-3,5-ДИГИДРОКСИГЕПТ-6-ЕНОЕВУЮ КИСЛОТУ И ИНГИБИТОР, ИНДУКТОР ИЛИ СУБСТРАТ ИЗОФЕРМЕНТА Р450-3А4 | 2000 |
|
RU2233162C2 |
| НОВЫЕ КОМПОЗИЦИИ ФЕНОФИБРАТА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ЛЕЧЕНИЯ | 2005 |
|
RU2398577C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ДОЗИРОВАННАЯ ФОРМА, СОДЕРЖАЩАЯ ПОЛИМЕРНУЮ КОМПОЗИЦИЮ-НОСИТЕЛЬ | 2009 |
|
RU2519679C9 |
| СТАБИЛИЗАЦИЯ ДОЗИРОВАННОЙ ФОРМЫ НА ОСНОВЕ ТВЕРДОГО ИЛИ ПОЛУЖИДКОГО ЛИПИДА ПУТЕМ ОТВЕРЖДЕНИЯ И ДОБАВЛЕНИЯ ПОВЕРХНОСТНО-АКТИВНОГО ВЕЩЕСТВА (ВЕЩЕСТВ), ОБЛАДАЮЩЕГО НИЗКИМ ЗНАЧЕНИЕМ ПОКАЗАТЕЛЯ ГЛБ | 2019 |
|
RU2833036C2 |
| КОМПОЗИЦИИ ТВЕРДЫХ РАСТВОРОВ И ИХ ПРИМЕНЕНИЕ ПРИ СЕРДЕЧНО-СОСУДИСТОМ ЗАБОЛЕВАНИИ | 2014 |
|
RU2674982C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ И ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2635188C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ГИПЕРЛИПИДИМИИ | 2017 |
|
RU2683641C2 |
Изобретение относится к области лекарственных средств, в частности к материалу в форме частиц для лечения состояний, при которых требуется регуляция липидного метаболизма, включающему в качестве активных веществ один или несколько фибратов и один или несколько статинов или их фармацевтически приемлемую соль, в носителе, выбранном из группы, состоящей из i) смеси полиэтиленгликоля и полоксамера в соотношении от 2:1 до 3:1 и с последующим распылением на лактозу, ii) глицерилмоностеарата с последующим распыленнием на лактозу или на смесь лактозы и ГПМЦ и iii) полиэтиленгликоля, с последующим распыленнием на Aeroperl. Кроме того, изобретение относится к твердым дозированным формам, включающим указанный материал, и способу их получения. Изобретение позволяет установить подходящую биологическую доступность обоих активных ингредиентов при пероральном введении. 3 н. и 53 з.п. ф-лы, 19 табл.
i) от примерно 140 до примерно 170 мг фенофибрата и от примерно 10 до примерно 80 мг аторвастатина или их фармацевтически приемлемой соли, или
ii) от примерно 50 до примерно 100 мг фенофибрата и от примерно 5 до примерно 50 мг аторвастатина или их фармацевтически приемлемой соли, или
iii) фенофибрат или аторвастатин или их фармацевтически приемлемую соль в массовом соотношении фенофибрата и аторвастатина или их фармацевтически приемлемой соли от примерно 1:1 до примерно 20:1, или
iv) от примерно 140 до примерно 170 мг фенофибрата и от примерно 5 до примерно 80 мг симвастатина или их фармацевтически приемлемой соли, или
v) от примерно 50 до примерно 100 мг фенофибрата и от примерно 2,5 до примерно 50 мг симвастатина или их фармацевтически приемлемой соли, или
vi) фенофибрат и симвастатин или их фармацевтически приемлемую соль в массовом соотношении фенофибрата и симвастатина или их фармацевтически приемлемой соли от примерно 1:1 до примерно 40:1.
i) получение носителя в жидкой форме,
ii) поддержание жидкого носителя при температуре ниже точки плавления фибрата и/или статина,
iii) растворение желательного количества фибрата и статина в носителе,
iv) распыление полученного раствора на твердый носитель, имеющий температуру ниже точки плавления носителя,
v) механическая обработка полученных композиций с получением частиц, например материала в форме частиц.
| WO 03013608 А1 20.02.2003 | |||
| WO 03004001 А1 16.01.2003 | |||
| WO 2005034908 А1 21.04.2005. |

