Изобретение относится к новым 3-гидрокси-4-пиридиноновым производным и к их применению в образовании хелатных комплексов с ионами железа (III). Точнее, изобретение относится к циклоалкильным производным 3-гидрокси-4-пиридинона.
3-Гидрокси-4-пиридиноны являются бидентатными лигандами, которые образуют хелатный комплекс с ионом Fe(III) в отношении 3:1, и используются для удаления избытка железа из организма человека. Перенасыщение железом может возникать вследствие избыточного потребления железа с пищей, наследственных генетических состояний, таких как гемохроматоз и регулярное переливание крови. Такие переливания применяют для лечения медицинских состояний, таких как талассемия, серповидно-клеточная анемия, идиопатический гемохроматоз и апластическая анемия. Повышенное всасывание железа при трансфузии приводит к перенасыщению железом. При достижении насыщения в организме ферритином и трансферрином, железо откладывается во многих тканях, таких как миокард, печень и эндокринные органы, приводя к токсическим эффектам.
Исследования в области образования хелатных комплексов железа и применимость хелатообразующих соединений были описаны в обзоре (Current Medicinal Chemistry, 2003, 10, 983-985, Tim F. Tam, et al.). Хелатирующие железо соединения могут быть использованы для предотвращения образования окси-радикалов, лечения опухолей, малярии, пост-ишемической реперфузии и нейродегенеративных заболеваний. Соединения, образующие комплексы с железом, такие как десфералТМ (мезилат десферриоксамина) и феррипроксТМ (деферипрон), используют для удаления избытка железа из организма больного с талассемией, поскольку организм человека не располагает эффективными средствами, способствующими выведению железа, накопленного в процессе переливания крови. Десферриоксамин вводят ежедневно путем подкожной инфузии в течение 8-12 часов. В настоящее время деферипрон (1,2-диметил-3-гидрокси-4-пиридинон) является единственным перорально доступным лекарственным средством. Данное средство подвергается метаболизму в печени, и более 85% введенной дозы обнаруживается в моче в виде нехелатообразующего О-глюкуронида (Drug Metab. Dispo. 1992, 20(2), 256-261, S.Singh, et al.). Относительно высокая пероральная доза 75 мг/кг (3,5-4 г в день) требуется для лечения состояний, связанных с перенасыщением железом. Поэтому существует необходимость в создании нового активного гидроксипиридинона для перорального введения с улучшенными по сравнению с деферипроном фармакологическими характеристиками.
Voest et al. (Annals of Internal Medicine 1994, 120, 490-499) суммировал клинический опыт по испытанию соединений, образующих хелатные комплексы с железом, при состояниях, не связанных с перенасыщением железом. Соединения, образующие хелатные комплексы с железом, использовали для достижения антиоксидантных эффектов, антипролиферативных эффектов, противопротозойных эффектов и для хелатирования алюминия, и могут быть использованы для лечения ряда болезненных состояний, например, для лечения ревматоидного артрита, для защиты от кардиотоксических эффектов, вызываемых антрациклиновыми соединениями, для ограничения повреждения миокарда вследствие ишемии-реперфузии, в качестве протипоопухолевых средств и для лечения малярии. Кроме того, исследователь van Asbeck B.S. et al. (J. Clin. Virol. 2001 Feb; 20(3): 141-7) сообщил о том, что соединения, образующие хелатные комплексы с железом, обладают активностями против ВИЧ-инфекции. Следовательно, применение соединений, образующих хелатные комплексы с железом, не ограничивается только лечением состояний, вызванных перенасыщением железом.
Члены класса 3-гидрокси-4-пиридинонов являются известными из-за их способности образовывать хелатные комплексы с железом. Предыдущий уровень техники включает патенты RE 35948, США 6448273, США 6335353 и США 5480894. В патенте США 6335353 эфирные пролекарственные производные 3-гидрокси-4-пиридинонов использовали для усиления выведения железа из печени, однако ни одно из разработанных соединений не получило оценки для человека.
В других подходах выбранные новые соединения были разработаны с целью блокировки фазы II метаболизма деферипрона, т.е. О-глюкуронидации у кислорода, расположенного у С3-атома остова деферипрона. В патенте США 5688815 описаны 1-алкил-3-гидрокси-4-пиридиноны с С2-метильной группой, замещенной фенильным или гетероильным кольцом и гидроксигруппой, и заместителем у N1, которым является низший алкил. В патенте США 6335353 описан 1-алкил-3-гидрокси-4-пиридинон с С2-алкилкарбомоилом, арилкарбамоилом или аралкилкарбамоильной группой, где N1-заместителем является алифатическая углеводородная группа. Использование С2-метилкарбамоильной функциональности в соединении, таком как СР502 (1,6-диметил-3-гидрокси-4(1Н)пиридинон-2-карбокси-(N-метил)амида хлоргидрат; патент США 6335353), приводит к эффективной блокировке О-глюкуронидирования у С3-кислорода. Другие аналоги, описанные в патенте США 6335353, включают в себя СР506 (1,6-диметил-3-гидрокси-4(1Н)пиридинон-2-карбокси-(N-изопропил)амида хлоргидрат), С2-изопропилкарбомоильный аналог и СР508 (1,6-диметил-3-гидрокси-4(1Н)пиридинон-2-карбокси-(N,N-диметил)амида хлоргидрат), диметилкарбамоильный аналог. СР502, СР506 и СР508 являются соединениями, разработанными в уровне техники, и они не получили оценки для человека.
Сущность изобретения
Первый аспект настоящего изобретения относится к производному 3-гидроксипиридин-4-она формулы I и его фармацевтически приемлемой соли,

где:
R1 является X при условии, что R2 является Y;
или
R1 является T при условии, что R2 является W;
или
R1 является X при условии, что R2R5N, вместе взятые, образуют гетероциклическое кольцо, выбранное из пиперидинила, морфолинила, пирролидинила или пиперазинила, где пиперидинильная, морфолинильная, пирролидинильная или пиперазинильная группа является либо незамещенной, либо замещенной одной-тремя С1-С6 алкильными группами;
Х является С3-С6 циклоалкилом;
Y выбирают из группы, состоящей из С3-С6 циклоалкила, С1-С6 алкила и С1-С6 алкила, монозамещенного С3-С6 циклоалкилом;
Т означает С1-С6 алкил;
W означает С3-С6 циклоалкил;
R3 выбирают из группы, состоящей из водорода и С1-С6 алкила;
R4 выбирают из группы, состоящей из водорода и С1-С6 алкила и
R5 выбирают из группы, состоящей из водорода и С1-С6 алкила.
Второй аспект настоящего изобретения относится к применению соединения формулы I в лечении заболевания, связанного с перенасыщением железом.
Третий аспект изобретения относится к фармацевтической композиции, содержащей соединение формулы I.
Одним предпочтительным классом соединений согласно данному изобретению является соединение формулы I, где R1 является X при условии, что R2 является Y, Х является С3-С6 циклоалкилом; Y является С1-С6 алкилом; R3 является водородом; R4 является С1-С6 алкилом и R5 является водородом.
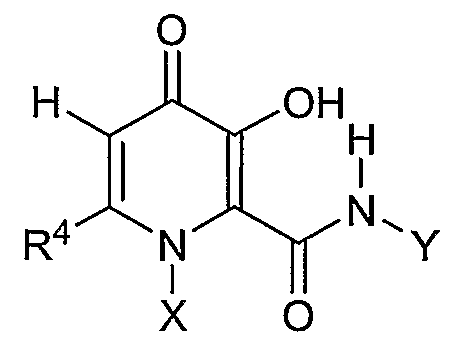
Еще более предпочтительным соединением в этой подгруппе является соединение формулы I, где R4 является метилом, X означает циклопропил и Y означает метил, причем соединение является метиламидом 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты.

Второй предпочтительный класс соединений согласно данному изобретению представлен соединением формулы I, где R1 является X при условии, что R2 является Y, Х является С3-С6 циклоалкилом, Y является С3-С6 циклоалкилом, R3 является водородом, R4 является С1-С6 алкилом и R5 является водородом.
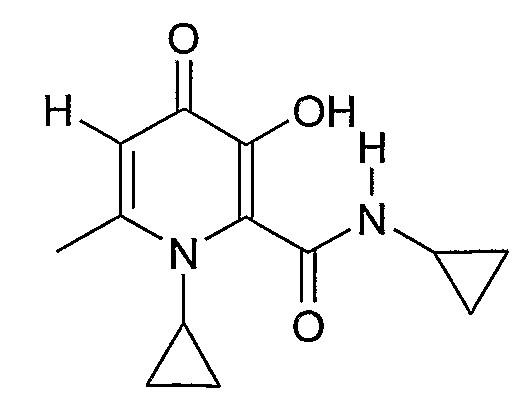
Предпочтительным соединением в данной подгруппе является соединение, где R4 является метилом, X=Y=циклопропил, причем соединением является циклопропиламид 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты.
Третий предпочтительный класс соединений формулы I представлен соединением, где R1 является T при условии, что R2 является W; T означает С1-С6 алкил; W означает С3-С6 циклоалкил, R3 является водородом, R4 является С1-С6 алкилом и R5 является водородом.
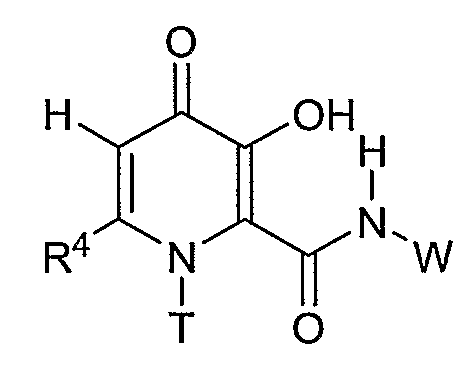
Более предпочтительным соединением в данной подгруппе является соединение, где R4 является метилом, T означает метил и W означает циклопропил, причем соединением является циклопропиламид 3-гидрокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты.

Четвертый предпочтительный класс соединений согласно настоящему изобретению представлен соединением формулы I, где R1 является X при условии, что R2 является Y, X означает С3-С6 циклоалкил; Y означает С1-С6 алкил; R3 является водородом; R4 означает С1-С6 алкил, R5 является метилом.

Еще более предпочтительным соединением в данной подгруппе является соединение формулы I, где R4 является метилом, X является циклопропилом и Y означает метил, и соединением является 1-циклопропил-3-гидрокси-N,N,6-триметил-4-оксо-1,4-дигидропиридин-2-карбоксамид.

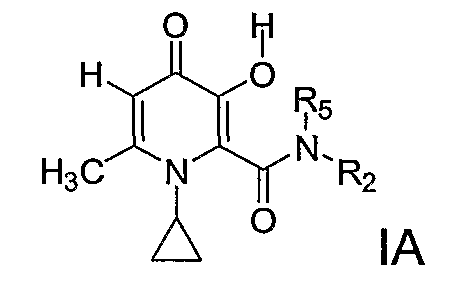
Наиболее предпочтительными соединениями согласно данному изобретению являются соединение IA, соединение формулы I, где R3=H, R4=метил, при условии, что R1=X=циклопропил, R2=Y и Y выбирают из группы С3-С6 циклоалкила; С1-С6 алкила; С1-С6 алкила, монозамещенного С3-С6 циклоалкилом; или R1=X=циклопропил, R2R5N, вместе взятые, образуют гетероциклическое кольцо, выбранное из пиперидинила, морфолинила, пирролидинила или пиперазинила, где пиперидинильная, морфолинильная, пирролидинильная или пиперазинильная группа является либо незамещенной, либо замещенной одной-тремя С1-С6 алкильными группами.
Таким образом, настоящее изобретение относится к циклоалкильному производному 3-гидрокси-4-пиридинона, обладающему улучшенными свойствами по сравнению с соединениями известного уровня техники. Циклоалкильная группа присоединена к N1-атому и/или к атому N С2-амидо группы. До настоящей заявки, соединения с N1-циклоалкильным заместителем или N-циклоалкильным заместителем С2-амидогруппы не были описаны в литературе. Указанные соединения не являются пролекарствами и обладают превосходной избирательностью по отношению к металлу. Они не образуют комплексы с существенными металлами, такими как кальций и магний при рН 7,4 в химических анализах. Величина D7,4 находится в области, установленной для лекарственного средства деферипрона, и соединение является перорально активным в модели крысы с перенасыщением железом. Данные соединения разработаны с подходящими величинами рК фенольной ОН-группы у атома С3 в области от 8,3 до 8,8, величиной pFe3+ выше 20, легко образуют хелатный комплекс с железом при соотношении 1:3, как установлено с помощью графика согласно Job, и имеют величину D7,4>0,1. Исследование монокристаллической структуры хелатного комплекса Fe(III) подтверждает, что соединение формулы I является бидентатным лигандом.
Краткое описание чертежей
На фиг.1 представлен график согласно Job для соединения Аро6622 (циклопропиламид 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты), соединения формулы I.
На фиг.2 представлен график согласно Job для соединения Аро6617 (циклопропиламид 1,6-диметил-3-гидрокси-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты), соединения формулы I.
На фиг.3 представлен график согласно Job для соединения Аро6619 (метиламид 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты), соединения формулы I.
На фиг.4 представлено графическое изображение образования комплекса Fe3+-Аро6619.
На фиг.5 представлено графическое изображение образования комплекса Fe3+-Аро6617.
Фиг.6: Эффективность Аро6619 и Аро6617, способствующих выведению железа с мочой в модели крысы с перенасыщением железом (n=6).
Фиг.7: Монокристаллическая структура хелатного комплекса Fe(Apo6617)3.
Фиг.8: Монокристаллическая структура хелатного комплекса Fe(Apo6619)3.
Фиг.9: Картины циклической вольтамперометрии системы Fe-Apo6619 при рН 7,4.
Таблица 1: Химические свойства соединения формулы I.
Таблица 2: Селективность связывания Аро6619 с металлом.
Таблица 3: Эффективность соединений Аро6619 и Аро6617, способствующих выведению железа с фекалиями в модели крысы с перенасыщением железом (n=6). Величины выражены в виде мкг/день/кг.
Таблица 4: Эффективность соединений Аро6619 и Аро6617, способствующих выведению железа с мочой и фекалиями в модели крысы с перенасыщением железом (n=6/группу). Величины выражены в виде мкг/день/кг. Величины выделения с фекалиями даны за 3 дня после введения хелатного соединения и сравнены с базовыми величинами, определенными за 3 дня до введения хелатного соединения. Величины представлены как средние ± 1SD.
Таблица 5: Данные кристаллографии и уточнение структуры Fe(Apo6617)3.
Таблица 6: Длины связей [Å] и углы [°] для Fe(Apo6617)3.
Таблица 7: Данные кристаллографии и уточнение структуры Fe(Apo6619)3.
Таблица 8: Длины связей [Å] и углы [°] для Fe(Apo6617)3.
Подробное описание изобретения
Употребляемые в тексте термины:
Алкил означает разветвленную или неразветвленную насыщенную углеводородную цепь, содержащую, если не указано особо, от одного до шести атомов углерода, включающий, но не ограничивающийся метилом, этилом, пропилом, изопропилом, н-пропилом, бутилом, втор-бутилом, изобутилом, н-пентилом, гексилом.
Термин "циклоалкил", употребляемый в тексте отдельно или как часть другой группы, включает в себя насыщенные циклические углеводородные группы, содержащие 1 кольцо, включающее моноциклический алкил, содержащий от 3 до 6 углеродов, образующих кольцо, который включает в себя циклопропил, циклобутил, циклопентил и циклогексил.
Фармацевтически приемлемые нетоксичные соли относятся к фармацевтически приемлемым солям соединений согласно настоящему изобретению, которые сохраняют биологическую активность родительских соединений и не являются биологически или в других отношениях неподходящими (например, соли являются устойчивыми). Соли двух типов могут быть образованы из соединений согласно настоящему изобретению: (1) соли неорганических и органических оснований соединений формулы I, имеющие фенольную функциональную группу, и (2) аддитивные соли кислоты могут быть образованы у аминной функциональной группы соединений формулы I согласно данному изобретению.
Фармацевтически приемлемые соли неорганических оснований включают натриевые, калиевые, литиевые, аммониевые, кальциевые и магниевые соли. Особенно предпочтительными являются натриевые, кальциевые и магниевые соли. Фармацевтически приемлемые нетоксичные соли органических оснований включают соли первичных, вторичных и третичных аминов, замещенных аминов, включающих природные, замещенные амины, циклические амины и основные ионообменные смолы. Примерами таких солей являются 2-амино-2-гидроксиметилпропан 1,3-диол, изопропиламин, трометамин, глюкозамин, метилглюкамин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминные смолы и тому подобное.
Фармацевтически приемлемые аддитивные соли кислоты образуются с неорганическими и органическими кислотами, такими как галоидводородная кислота, серная кислота, азотная кислота, фосфорная кислота, метансульфокислота и этансульфокислота.
Соединениями согласно данному изобретению являются 2-амидопроизводные 4-оксо-1,4-дигидропиридин-2-карбоксамидных производных, имеющих общую структуру:

Большая часть соединений названа как производное 4-оксо-1,4-дигидропиридин-2-карбоксамида, например:
1-циклопропил-N-гексил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид:

N-циклогексил-1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид:
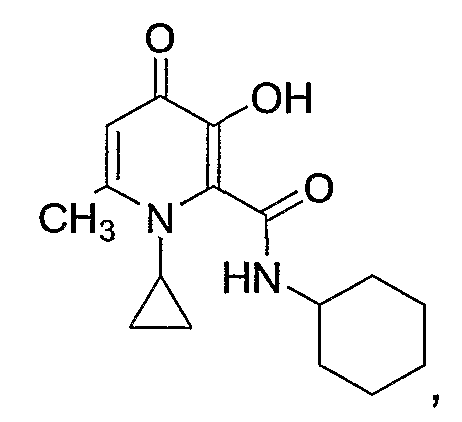
N-(циклогексилметил)-1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидроксипиридин-2-карбоксамид:

В некоторых случаях соединения названы с включением в название "пиридин-4(1Н)-он" в качестве основного скелета. Примерами являются:
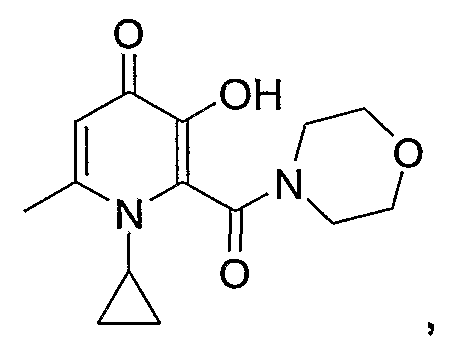
1-циклопропил-3-гидрокси-6-метил-2-(морфолин-4-илкарбонил)пиридин-4-(1Н)-он:
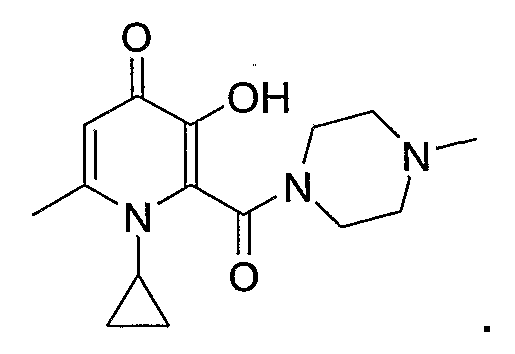
1-циклопропил-3-гидрокси-6-метил-2-[(4-метилпиперазин-1-ил)карбонил]пиридин-4(1Н)-он:
Термин "животные" относится к человеку, а также к другим видам животных, в частности к млекопитающим (например, собаки, кошки, лошади, крупный рогатый скот, свиньи и т.д.), рептилиям, рыбам, насекомым и глистам.
Соединения согласно данному изобретению разработаны с целью улучшения свойств известных аналогов деферипрона. Одним критерием, применяемым для логического обоснования разработки перорально активных хелатных соединений в серии 3-гидрокси-4-пиридинонов, являются соединения, имеющие величины pFe3+ выше, чем у деферипрона (pFe3+=19,7). Используемое в тексте определение pFe3+ представляет собой концентрацию иона железа в растворе, когда общее количество железа равно 10-6 М, и концентрация лиганда составляет 10-5 М, и рН составляет 7,4. Ее рассчитывают, используя экспериментально определенную величину рКа и константы образования комплекса с металлом с помощью программы Hyperquad (Version 2.1, Peter Gans, University of Leeds). Снижение величины рКа фенольной ОН-группы у атома С3 до менее чем 8,8 свидетельствует о том, что более высокая величина pFe3+ достигается при комбинировании с соединениями с подходящей константой комплексообразования β3. Концепция комплексообразования обсуждается подробно ниже.
Ступенчатыми и общими константами комплексообразования для бидентатного лиганда, такого как 3-гидрокси-4-пиридинон, являются следующие:
Константа комплексообразования β3 = K1·K2·K3
Хелатирующее железо лекарственное средство деферипрон (1,2-диметил-3-гидрокси-4-пиридинон) образует хелатный комплекс с железом с константами комплексообразования (log β3) 36,3 и pFe3+ 19,7. Величины рК деферипрона составляют 3,56 и 9,64. Большая часть соединений согласно настоящему изобретению имеет подобные константы комплексообразования (log β3) в области от 34 до 36, величину pFe3+>20 и подходящие величины рКа фенола от 8,3 до 8,8. Следовательно, соединения согласно данному изобретению являются превосходными хелатирующими агентами Fe(III). Величина эффективной проницаемости [Рэфф] через тощую кишку человека соединений согласно данному изобретению теоретически рассчитана, исходя из вычислительных расчетов с помощью программы QMPRPlusTM (из Simulationplus Inc.). Большая часть соединений согласно настоящему изобретению имеет рассчитанную величину Рэфф в области 1±0,3 (см/с × 10-4), что позволяет предположить, что соединения имеют хорошую проницаемость через тощую кишку человека. Химические свойства представленных соединений формулы I перечислены в таблице 1.
Соединения формулы I с циклоалкильными группами у R1 и/или R2 являются комплексонами металла с высокими величинами pFe3+. ВеличиныD7,4 соединений формулы I подобны деферипрону, и последующие исследования на модели крыс с перенасыщением железом показали, что соединения формулы I оказывают эффективное действие в удалении железа in vivo. Подробности исследования эффективности соединений у животных описаны в примерах ниже.
Соединения формулы I не связывают существенные металлы, такие как марганец, кальций и магний. Величины рМ и константы комплексообразования типичного примера соединения формулы I представлены в таблице 2 (и обсуждены подробно в примере 11). Соединение преимущественно связывается с Fe3+ по сравнению с другими двухвалентными и трехвалентными металлами, такими как Cu, Zn и Al.
Соединения формулы I являются новыми циклоалкильными производными 3-гидрокси-4-пиридинонов. Они имеют величины pFe3+ выше 20, подходящие величины D7,4, сравнимые с деферипроном, предпочтение относительно образования хелатного комплекса Fe3+ и фрагмента С2-алкилкарбамоила или С2-циклоалкилкарбамоила, который предназначен блокировать фазу II метаболизма 3-ОН-группы.
Кроме вышесказанного, соединения формулы I связывают Fe3+ в отношении 1:3 при физиологических условиях при рН 7,4. Анализ графика согласно Job подтверждает соотношение 1:3 комплексона к железу (фиг.(1-3) и пример 9).
Графики образования Fe-комплекса в отношении различных значений рН могут быть представлены путем использования программы Hyperquad Stimulation and Speciation (HYSS2 © 2000 Protonic Software) с включением экспериментальных величин рКа (пример 10 и 11) и констант комплексообразования K1, K2 и K3 (пример 14). Фигуры 4 и 5 иллюстрируют график образования комплексов металла с соединениями формулы I при различных значениях рН. В обоих исследованиях типичные примеры соединений формулы I исключительно образуют хелаты FeL3 при рН выше 7,0 (где L является бидентатным лигандом), таким образом, подтверждая отсутствие видов FeL2 + или FeL2+ при физиологическом значении рН. Отсутствие указанных видов свидетельствует о том, что не существует незащищенного железа in vivo при физиологическом значении рН 7,4.
Соединения формулы I, где R1 означает X при условии, что R2 означает Y; или R1 означает T при условии, что R2 является W, получают способом, отраженным на схеме 1.
Схема 1
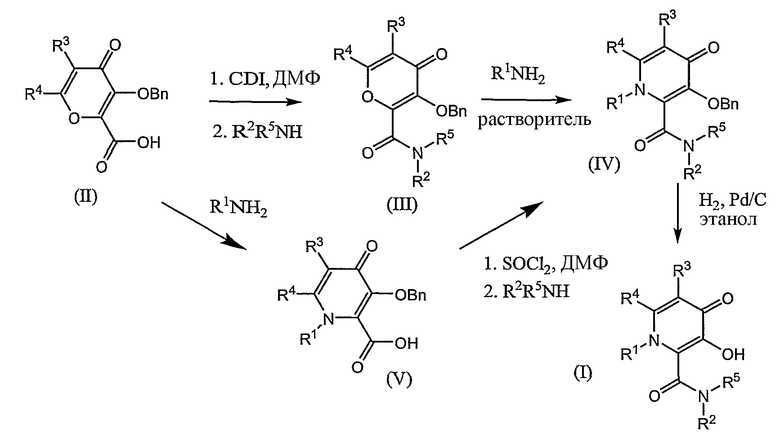
Кислота (II) взаимодействует с 1,1'-карбонилдиимидазолом в инертном растворителе в течение от 2 до 5 часов, предпочтительно 5 часов в инертном растворителе при температурах от 30 до 70°С. После добавления амина R2R5NH, соединение (III) выделяют обычными способами. Раствор соединения (III) и амина R1NH2 в инертном растворителе, таком как спирт, нагревают при температуре от 50 до 80°С, чтобы осуществить введение амина (III) в течение от 3 до 48 часов с получением соединения (IV). Альтернативный способ получения соединения (IV) заключается во взаимодействии соединения формулы (II) с амином R1NH2 в инертном растворителе с получением кислоты формулы (V). Затем соединение (V) подвергают взаимодействию с тионилхлоридом и диметилформамидом с получением соединения формулы (IV). Соединения выделяют традиционными способами, например, колоночной хроматографией и кристаллизацией. Гидрирование соединения (IV) в спирте над катализатором гидрирования приводит к соединению (I), которое выделяют обычными средствами. Предпочтительным катализатором гидрирования является палладий-на-угле или гидроокись палладия-на-угле и Ni Ренея. Получение исходного материала кислоты (II) сообщено в патенте США 6426418. Общая процедура получения кислоты формулы (V) может быть найдена в патенте Канады СА 2379370.
Соединения формулы (I) проверяли на крысах с перенасыщением железом. Данные экскреции железа с фекалиями и экскреции железа с мочой для типичных примеров соединений Аро6617 и Аро6619 приведены в таблицах 3 и 4 и фиг.6 соответственно. Оба соединения проявили значительную экскрецию железа с фекалиями по сравнению с контролем при пероральной дозе 113 и 450 мкмоль/кг. Кроме того, Аро6619 и Аро6617 способствуют экскреции железа с мочой в значительно большей степени, чем деферипрон при дозе 450 мкмоль/кг. Оба соединения рассматривают как более эффективные, чем деферипрон, в мобилизации железа у крыс с перенасыщением железом.
Хелатные комплексы железа и соединений формулы I были синтезированы и выделены (пример 16). Монокристаллические структуры Fe(Apo6617)3 и Fe(Apo6619)3 определенно подтверждают, что указанные бидентатные соединения взаимодействуют с Fe(III) с получением трисбидентатного хелатного комплекса 1:3 (таблицы от 5 до 8, фиг.7-8).
Другой критерий в разработке соединений формулы (I) касается регулирования окислительно-восстановительного потенциала системы Fe-комплексон при рН 7,4 до отрицательной величины ниже -320 мВ (против NHE), чтобы предотвратить какие-либо реакции с формами кислорода. Железо существует во множественных состояниях, включающих Fe2+ и Fe3+. Пара железо(II)/железо(III) может действовать в качестве пары восстановителя и окислителя. Согласно Crumbliss (http://www.medicine.uiowa.edu/FRRB/VirtualSchool/Crumbliss-Fe.pdf) и Pierre (BioMetals, 12, 195-199, 1999) избирательное хелатирование железа путем регулирования окислительно-восстановительного потенциала является способом предотвращения участия железа в каталитическом цикле, в результате которого образуются токсические гидроксильные радикалы и/или реактивные формы кислорода (ROS) (например, посредством реакции Fenton или цикла Haber Weiss). Fe(III)-трисхелатная система с окислительно-восстановительным потенциалом ниже -320 мВ (против NHE или -540 мВ против Ag/AgCl) при рН 7,4 не должна восстанавливаться какими-либо биологическими восстановителями, такими как NADPH/NADH, поэтому она не должна принимать участие в цикле Haber Weiss с образованием ROS (реактивные формы кислорода). В организме млекопитающего железо связано с различными белками, такими как трансферрин в крови человека, таким образом, оно остается в форме, которая не может взаимодействовать с какими-либо молекулами кислорода. Величина Е1/2 Fe-трансферрин составляет -500 мВ (против NHE или -720 мВ против Ag/AgCl).
Окислительно-восстановительный потенциал комплексов железа может быть измерен с помощью циклической вольтамперометрии (CV). Использование CV для измерения окислительно-восстановительных потенциалов хелатных комплексов железа с деферипроном, деферриоксамином и Аро6619 (типичным примером соединения согласно данному изобретению), как с комплексонами, соответственно, иллюстрировано в примере 17 ниже. Хелатные комплексы железа, такие как Fe-десферриоксамин (DFO) и Fe-(деферипрон)3, имеют величины окислительно-восстановительных потенциалов Е1/2 при -698 мВ (против Ag/AgCl) и -834 мВ (против Ag/AgCl) при рН 7,4 соответственно. Хелатные комплексы соединений формулы I, такие как Fe(Apo6619)3, имеют величину Е1/2 -691 мВ (против Ag/AgCl), подобную величине для десферриоксамина. Графическое представление циклической вольтамперометрии Fe-DFO, Fe(деферипрон)3 и Fe(Apo6619)3 можно увидеть на фиг.9. Одно преимущество комплексонов согласно данному изобретению заключается в том, что окислительно-восстановительные потенциалы их хелатных комплексов с железом находятся в крайней отрицательной области при физиологическом рН 7,4, поэтому их хелатные комплексы с железом не будут участвовать в окислительно-восстановительном цикле с образованием активных форм кислорода при физиологическом рН. При сочетании с другими новыми свойствами, как описано в данном изобретении, соединения формулы I представляют собой эффективные средства для удаления железа посредством механизма хелатирования.
Для лечения заболеваний, связанных с перенасыщением железом, таких как талассемия, серповидно-клеточная болезнь, гемохроматоз, и лечения больных с токсической концентрацией железа, соединения согласно изобретению могут быть введены перорально, местно или парентерально в стандартных лекарственных формах, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители.
Для лечения состояний, не связанных с перенасыщением железом, таких как ВИЧ-инфекция, защитный эффект от интоксикации сердца антрациклинами, опухоли и малярия, соединения согласно настоящему изобретению могут быть введены перорально, местно или парентерально в стандартных лекарственных формах, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители.
Употребляемый в тексте термин парентеральный включает способы подкожной инъекции или инфузии. Помимо лечения теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки и т.д., соединения согласно изобретению являются эффективными и в лечении человека.
Для использования в фармацевтических композициях подходящие нетоксичные твердые носители, включающие, например, маннит фармацевтических категорий, лактозу, крахмал, стеарат магния, сахарин натрия, тальк, целлюлозу, глюкозу, сахарозу, карбонат магния и тому подобное, могут быть использованы. Активное соединение, определенное выше, может быть приготовлено в виде жидкости. Фармацевтические вводимые композиции могут быть получены, например, путем растворения, диспергирования и т.д., активного соединения, определенного выше, и возможных фармацевтических адъювантов в носителе, таком как, например, вода, физиологический раствор, водная декстроза, глицерин, этанол и тому подобное, с образованием раствора или суспензии. Если желательно, фармацевтическая композиция, которая должна быть введена, также может содержать небольшое количество нетоксичных вспомогательных субстанций, таких как смачивающие и эмульгирующие средства и тому подобное, например ацетат натрия, сорбитанмонолаурат, триэтаноламина натрий ацетат, триэтаноламина олеат и т.д. Способы получения таких дозированных форм известны или будут очевидны специалистам в данной области: например, смотри Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 15th Edition, 1975, ch. 83, p. 1436-1460, и ch. 89, p. 1576-1607. Композиция препарата, которая должна быть введена, так или иначе, будет содержать эффективное количество активного соединения(й) для ослабления симптомов у субъекта, подвергаемого лечению.
Фармацевтические композиции, содержащие активный ингредиент, могут находиться в форме, пригодной для перорального использования, например, в форме таблеток, пилюль, лепешек, водных или масляных суспензий, дисперсных порошков или гранул, эмульсий, твердых и мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального использования, могут быть приготовлены любым способом, известным в данной области для производства фармацевтических композиций, и такие композиции содержат одно или более средств из группы, состоящей из подсластителей, ароматизаторов, красителей и консервантов для получения фармацевтически элегантных и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые пригодны для промышленного производства таблеток. Наполнителями могут быть, например, инертные разбавители, такие как фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие средства, например кукурузный крахмал или альгиновая кислота; связывающие средства, например крахмал, желатин или гуммиарабик, и смазывающие средства, например стеарат магния, стеариновая кислота или тальк. Таблетки могут быть покрыты оболочками известными способами, чтобы задержать дезинтеграцию и всасывание в желудочно-кишечном тракте и, тем самым, обеспечить замедленное действие в течение длительного периода времени. В качестве смягчающего средства, эмульгатора или увлажнителя может быть использован моностеарат или глицерилдистеарат.
Препараты для перорального использования могут быть представлены в форме твердых желатиновых капсул, где активные ингредиенты смешивают с инертным твердым разбавителем, например фосфатом кальция или каолином, или в форме мягких желатиновых капсул, где активный ингредиент смешивают с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии могут содержать активные материалы в смеси с наполнителем, подходящим для промышленного производства водных суспензий. Такими наполнителями являются суспендирующие средства, например карбоксиметилцеллюлоза натрия, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, камедь и аравийская камедь; диспергирующими или смачивающими средствами могут быть природный фосфат, например лецитин, или продукты конденсации простых циклических эфиров с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации окиси этилена с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации окиси этилена с частичными сложными эфирами жирных кислот и ангидридов гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии также могут содержать один или более консервантов, например этиловый или н-пропиловый эфир п-гидроксибензойной кислоты, один или более красителей, таких как сахароза или сахарин.
Масляные суспензии могут быть приготовлены путем суспендирования активного ингредиента в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Подсластители, такие как перечисленные выше, и ароматизаторы могут быть добавлены для получения приятного на вкус перорального препарата. Указанные композиции могут быть защищены путем добавления антиоксиданта, такого как аскорбиновая кислота.
Дисперсные порошки и гранулы, подходящие для приготовления водной суспензии путем добавления воды, способствуют образованию смеси активного ингредиента с диспергирующим или смачивающим средством, суспендирующим средством и одним или более консервантами. Подходящими диспергирующими или смачивающими средствами и суспендирующими средствами являются такие, которые уже были упомянуты выше. Могут быть представлены дополнительные реципиенты, например подсластители, ароматизаторы и красители.
Фармацевтическая композиция согласно изобретению может быть представлена в виде эмульсий масло-в-воде. Масляной фазой может быть растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смеси. Подходящими эмульгирующими средствами могут быть природные фосфаты, сложные эфиры жирных кислот и ангидридов гексита, например сорбитанмоноолеат, и продукты конденсации указанных частичных сложных эфиров с окисью этилена, например полиоксиэтиленсорбитанмоноолеат. Эмульсия также может содержать подсластители и ароматизаторы.
Сиропы и эликсиры могут быть приготовлены с подсластителями, например глицерином, пропиленгликолем, сорбитом или сахарозой. Такие препараты также могут содержать успокоительное средство, консерванты и ароматизаторы и красители. Фармацевтические композиции могут быть приготовлены известными в данной области способами, используя такие подходящие диспергирующие и смачивающие средства и суспендирующие средства, которые были упомянуты выше. Стерильным инъецируемым препаратом также может быть стерильный инъецируемый раствор или суспензия в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, например раствор в 1,3-бутандиоле. Среди приемлемых носителей и разбавителей, которые могут быть использованы, являются вода, растворы Рингера и изотонический раствор хлорида натрия. Кроме того, обычно используют жирные масла, включающие синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, находят применение в препарате или инъецируемых формах.
Соединения формулы (I), или их соответствующая фармацевтически приемлемая соль и/или фармацевтически приемлемый сольват, также могут быть введены в форме препарата для местного нанесения в комбинации с подходящими для местного применения наполнителями. Примерами препаратов для местного применения являются мази, кремы или лосьоны, пропитанные повязки, гели, карандаши-гели, спреи и аэрозоли. Препараты могут содержать соответствующие обычные добавки, такие как консерванты, растворители, чтобы содействовать проникновению лекарственного средства, и мягчительные средства в мазях и кремах. Они также могут содержать совместимые обычные носители, такие как основы для кремов и мазей и этанол или олеиловый спирт для лосьонов. Препараты для местного применения содержат количество активных ингредиентов, способствующих ослаблению симптомов у субъекта, подвергаемого лечению. Соответственно, для соединения формулы (I) или его соответствующей фармацевтически приемлемой соли компромиссным будет содержание приблизительно от 0,5 до 10% масс. препарата. Подходящие препараты в форме кремов, лосьонов, гелей, карандашей, мазей, спреев или аэрозолей, в состав которых могут быть включены соединения формулы (I) или их соответствующая фармацевтически приемлемая соль, являются обычными препаратами, хорошо известными в данной области, описанными в стандартных учебниках, например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 15th Edition, 1975, ch. 83, p. 1436-1460 и ch. 89, p. 1576-1607.
Парентеральное введение обычно характеризуется инъекцией, либо подкожным, внутримышечным, либо внутривенным введением. Инъецируемые препараты могут быть приготовлены в обычном виде, либо как жидкие растворы или суспензии в жидкости до инъекции, либо как эмульсии. Подходящими наполнителями являются, например, вода, физиологический раствор, декстроза, глицерин, этанол или тому подобное. Кроме того, если желательно, фармацевтические композиции, которые должны вводиться, также могут содержать незначительные количества нетоксического вспомогательного вещества, такого как смачивающее и эмульгирующее средство, создающие рН среды реагенты и тому подобные, такие как, например, ацетат натрия, сорбитанмонолаурат, триэтаноламина олеат и т.д.
Количество активного ингредиента, который может быть комбинирован с материалами-носителями с получением однократной дозированной формы, будет изменяться в зависимости от подвергаемого лечению хозяина, и отдельная вводимая человеку дозированная форма может содержать от 0,5 мг до 5 г активного средства, комбинированного с соответствующим количеством материала-носителя, которое может изменяться приблизительно от 5 до 95% от общей композиции. Стандартные лекарственные формы должны обычно содержать приблизительно от 1 мг до 500 мг активного ингредиента.
Однако следует понимать, что специфическая доза для любого отдельного больного будет зависеть от ряда факторов, включающих активность используемого специфического соединения, возраст, вес тела, общее состояние здоровья, пол, питание, время введения, комбинацию лекарственных средств и тяжесть отдельного заболевания, подвергаемого лечению.
Соединения согласно настоящему изобретению отличаются от соединений, описанных в патентах США 6448273, США 6335353, RE 35948 и США 5688815. Первые три патента описывают 3-гидрокси-4-пиридиноны, имеющие алифатическую углеводородную группу у N1. Патент США 5688815 также описывает 3-гидрокси-4-пиридиноны с замещенной или незамещенной низшей алкильной группой у N1. Согласно общепринятому учебному руководству по химии, Organic Chemistry by James B. Hendrickson, Donald J. Cram, George S. Hammond, third edition, 1970, McGraw Hill, p. 72, алифатические углеводороды состоят из цепей атомов углерода, не организованных в кольца. Вещества, принадлежащие к этой группе, иногда рассматривают как соединения с открытой цепью. Примерами алифатической углеводородной группы являются линейные или разветвленные алкилы, такие как метил, этил, пропил, изопропил, изобутил, бутил и трет-бутил. Соединения согласно данному изобретению состоят из 3-гидрокси-4-пиридинонов с (а) N1-циклоалкильным заместителем и С2-циклоалкилкарбамоильным заместителем; или (b) N1-циклоалкильным заместителем и С2-циклоалкилкарбамоильным заместителем; или (с) N1-алкильным заместителем с С2-циклоалкилкарбамоильным заместителем. Они представляют собой соединения с ациклическими углеводородными заместителями. В ациклических углеводородах углеродные цепи образуют кольца. Примерами ациклических углеводородных групп являются циклоалкильные производные, такие как циклопропил, циклобутил, циклопентил и циклогексил. Четыре патента, США 6448273, США 6335353, RE 35948 и США 5688815, не охватывают циклоалкильные производные 3-гидрокси-4-пиридинонов. Настоящее изобретение охватывает 3-гидрокси-4-пиридиноны с N1-циклоалкильной группой с алкилкарбамоильной группой у атома С2 или циклоалкилкарбамоильной группой у атома С2. Изобретение также охватывает 3-гидрокси-4-пиридиноны с циклоалкилкарбамоильной группой у атома С2 с N1-алкильной группой.
Далее, изобретение описывают и иллюстрируют в следующих отдельных примерах.
Специфическое описание наиболее предпочтительных вариантов осуществления изобретения
ПРИМЕР 1: Получение циклогексиламида 3-бензилокси-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты
1,1'-Карбонилдиимидазол (1,99 г, 12,30 ммоль) добавляли к раствору 3-(бензилокси)-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты (2,0 г, 7,69 ммоль) в диметилформамиде (ДМФ, 18 мл) при комнатной температуре. Образовавшийся раствор нагревали при 40°-50°С в течение 3 часов. Получают бледно-желтый раствор. Затем добавляли циклогексиламин (1,23 мл, 10,76 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. ДМФ удаляли при пониженном давлении с получением в качестве сырого продукта масла бледно-желтого цвета, который очищали колоночной флэш-хроматографией (градиент элюции: от 1:1 смесь этилацетат/гексан до 10% метанола в этилацетате) с получением указанного в заголовке соединения (1,60 г, выход 61%) в виде твердого вещества белого цвета.
Т.пл. 118-120°С; 1H-ЯМР (CDCl3, 400 МГц) δ 0,91 (м, 2H, циклогексил-H), 1,29 (м, 2H, циклогексил-H), 1,58 (м, 3H, циклогексил-H), 1,79 (м, 2H, циклогексил-H), 2,37 (с, 3H, CH3), 3,79 (м, 1H, CH), 5,40 (с, 2H, CH2), 6,28 (с, 1H, CH), 7,41 (м, 5H, ArH), 7,67 (ушир., 1H, NH); МС (m/z) 342 (M++1).
Подобным способом, описанным выше, путем замены циклогексиламина другим амином были получены следующие соединения:
Циклопропиламид 3-бензилокси-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты
Т.пл. 79-80°С; 1H-ЯМР (CDCl3, 400 МГц) δ 0,21 (м, 2H, циклопропил-H), 0,70 (м, 2H, циклопропил-H), 2,35 (с, 3H, CH3), 2,71 (м, 1H, CH), 5,35 (с, 2H, CH2), 6,27 (с, 1H, CH), 7,39 (м, 5H, ArH), 7,70 (с, 1H, NH); 13C(CDCl3) δ 6,35, 7,21, 19,87, 22,61, 22,70, 75,56, 115,37, 128,94(2C), 129,17(2C), 129,25, 135,49, 146,14, 146,39, 160,22, 165,74, 176,17; МС (m/z) 300 (M++1).
Метиламид 3-бензилокси-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты
Т.пл. 137-140°С; 1H-ЯМР (CDCl3, 400 МГц) δ 2,38 (с, 3H, CH3), 2,78 (д, 3H, NCH3), 5,39 (с, 2H, CH2), 6,30 (с, 1H, CH), 7,40 (м, 5H, ArH), 7,62 (ушир., 1H, NH); МС (m/z) 300 (M++1).
ПРИМЕР 2: Получение циклогексиламида 3-бензилокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
К раствору циклогексиламида 3-бензилокси-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты (1,40 г, 4,1 ммоль) в 5 мл метанола добавляли раствор метиламина (9 мл 2 М раствора в метаноле, 16 ммоль). Полученный раствор перемешивали при температуре от 70 до 75°С в течение ночи под давлением в запаянной трубке. Растворитель удаляли при пониженном давлении с получением твердого вещества бледно-желтого цвета как сырого продукта. Материал очищали колоночной хроматографией (градиент элюции: от 100% этилацетата до 25% метанола в этилацетате) с получением указанного в заголовке соединения в виде твердого вещества белого цвета (1,20 г, 83,0%).
Т.пл. 258-260°С; 1H-ЯМР (CDCl3, 400 МГц) δ 1,26-1,45 (м, 6H, циклогексил-H), 1,79 (м, 2H, циклогексил-H), 1,95 (м, 2H, циклогексил-H), 2,41 (с, 3H, CH3), 3,82 (с, 3H, NCH3), 3,95 (м, 1H, CH), 5,13 (с, 2H, CH2), 7,19 (с, 1H, CH), 7,36 (м, 3H, ArH), 7,43(м, 2H, ArH), 8,50 (ушир., 1H, NH); МС (m/z) 355 (M++1).
Подобным способом, путем замены циклогексиламида 3-бензилокси-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты другим амидным производным 3-бензилокси-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты, были получены следующие соединения:
Циклопропиламид 3-бензилокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
Т.пл. 187-189°С; 1H-ЯМР (CDCl3, 400 МГц) δ 0,52 (м, 2H, циклопропил-H), 0,74 (м, 2H, циклопропил-H), 2,18 (с, 3H, CH3), 2,78 (м, 1H, CH), 3,50 (с, 3H, NCH3), 5,08 (с, 2H, CH2), 6,12 (с, 1H, CH), 7,33 (м, 3H, ArH), 7,39 (м, 2H, ArH), 7,91 (ушир. 1H, NH); МС (m/z) 313 (M++1).
Метиламид 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
Т.пл. 132-135°С; 1H-ЯМР (CDCl3, 400 МГц) δ 1,05 (м, 4H, циклопропил-H), 2,38 (с, 3H, CH3), 2,70 (д, J=1,8 Гц, 3H, NCH3), 3,35 (м, 1H, CH), 5,07 (с, 2H, CH2), 6,14 (с, 1H, CH), 7,15 (ушир., 1H), 7,35 (м, 5H, ArH); 13C(CDCl3) δ 9,48, 20,30, 25,86, 34,15, 74,01, 118,16, 127,79, 128,06(2C), 128,22(2C), 137,35, 142,05, 143,98, 149,91, 162,01, 173,89; МС (m/z) 313 (M++1).
Циклопропиламид 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
Т.пл. 164-167°С; 1H-ЯМР (CDCl3, 400 МГц) δ 0,54 (м, 2H, циклопропил-H), 0,76 (м, 2H, циклопропил-H), 1,08-1,11 (м, 4H, циклопропил-H), 2,35 (с, 3H, CH3), 2,75 (м, 1H, CH), 3,37 (м, 1H, CH), 5,05 (с, 2H, CH2), 6,13 (с, 1H, CH), 7,33 (м, 5H, ArH), 7,89 (ушир., с, 1H, NH); МС (m/z) 339 (M++1).
ПРИМЕР 3: Циклогексиламид 3-гидрокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты [Аро6621]
Pd(OH)2 на угле (0,18 г, 10% масс. сухой основы) добавляли к раствору циклогексиламида 3-бензилокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты (1,0 г, 2,82 ммоль) в этаноле (50 мл) в атмосфере азота. Смесь подвергали гидрированию при давлении 50 фунтов/кв. дюйм в течение 4 часов. Pd(OH)2 удаляли фильтрованием через слой целита, массу целита затем промывали этанолом (3×10 мл). Этанольный фильтрат упаривали с получением твердого вещества не совсем белого цвета (0,57 г, 77%). Дальнейшая очистка путем перекристаллизации из метанола (15 мл) привела к указанному в заголовке соединению в виде твердого вещества белого цвета (0,18 г).
Т.пл. 280-285°С (с разложением); 1H-ЯМР (CD3OD 400 МГц) δ 1,30-1,43 (м, 5H, циклогексил-H), 1,70 (м, 1H, циклогексил-H), 1,80 (м, 2H, циклогексил-H), 2,00 (м, 2H, циклогексил-H), 2,41 (с, 3H, CH3), 3,63 (с, 3H, CH3), 3,90 (м, 1H, CH), 6,38 (с, 1H, CH); МС (m/z) 265 (M++1).
Подобным способом, путем замены циклогексиламида 3-бензилокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты другими циклоалкиламидами 3-бензилокси-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты, были получены следующие соединения:
Циклопропиламид 3-гидрокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты [Аро6617]
Т.пл. 260-262°С; 1H-ЯМР (MeOD-d4, 400 МГц) δ 0,66 (м, 2H, циклопропил-H), 0,85 (м, 2H, циклопропил-H), 2,41 (с, 3H, CH3), 2,95 (м, 1H, CH), 3,63 (м, 1H, NCH3), 6,38 (с, 1H, CH); МС (m/z) 223 (M++1).
Метиламид 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты [Аро6619]
Т.пл. 258-260°С (с разложением); 1H-ЯМР (MeOD-d4, 400 МГц) δ 1,05 (м, 2H, циклопропил-H), 1,19 (м, 2H, циклопропил-H), 2,54 (с, 3H, CH3), 2,97 (с, 1H, NCH3), 3,46 (м, 1H, CH), 6,33 (с, 1H, CH); МС (m/z) 223 (M++1).
ПРИМЕР 4: Получение 3-(бензилокси)-N-циклобутил-6-метил-4-оксо-4Н-пиран-2-карбоксамида
Смесь 3-(бензилокси)-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты (2,5 г, 9,6 ммоль, 1,0 эквивалент), 1,1'-карбонилдиимидазола (2,49 г, 15,37 ммоль, 1,6 эквивалента) в ДМФ (20 мл) перемешивали при 50°С в течение 5 ч. Смесь охлаждали до комнатной температуры. Хлоргидрат циклобутиламина (1,24 г, 11,52 ммоль, 1,2 эквивалента) и Et3N (1,74 мл, 12,48 ммоль, 1,3 эквивалента) добавляли, и смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении. Очистка хроматографией (1:1 смесь гексаны/EtOAc, затем EtOAc) привела к указанному в заголовке соединению (2,76 г, 91,56%) в виде твердого вещества желтого цвета.
Т.пл 69,3-71,0°С; 1H-ЯМР (CDCl3, 400 МГц) δ 1,51-1,72 (м, 4H, циклобутил H), 2,19-2,28 (м, 2H, циклобутил H), 2,37 (с, 3H, CH3), 4,39-4,41 (м, 1H, CH), 5,41 (с, 2H, OCH2Ph), 6,30 (с, 1H, CH), 7,39-7,49 (м, 5H, ArH), 7,86 (ушир., 1H, NH), и МС (m/z) 314 (M++1), 217, 91.
Подобным способом было получено следующее соединение:
3-(бензилокси)-N-циклопентил-6-метил-4-оксо-4Н-пиран-2-карбоксамид
Т.пл. 108,0-108,5°С; 1H-ЯМР (CDCl3, 400 МГц) δ 1,11-1,16 (м, 2H, циклопентил H), 1,50-1,55 (м, 4H, циклопентил H), 1,87-1,92 (м, 2H, циклопентил H), 2,38 (с, 3H, CH3), 4,17-4,22 (м, 1H, CH), 5,41 (с, 2H, CH2), 6,30 (с, 1H, CH), 7,38-7,43 (м, 5H, ArH), 7,72 (ушир., 1H, NH), МС (m/z) 328 (M++1), 217, 91.
ПРИМЕР 5: Получение 3-(бензилокси)-N-циклобутил-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоксамида
К раствору соединения из примера 4 (2,616 г, 8,35 ммоль, 1,0 эквивалент) в метаноле (10 мл) быстро добавляли метиламин (2 М в метаноле, 20 мл, 40 ммоль, 4,79 эквивалента). Запаянную трубку перемешивали в течение ночи при 70-75°С. Полученный раствор коричневого цвета упаривали досуха и очищали хроматографией (EtOAc, затем 1:4 смесь MeOH/EtOAc) с получением указанного в заголовке соединения (1,70 г, 62,24%) в виде твердого вещества белого цвета.
Т.пл. 221,3-222,4°С; 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,65-1,69 (м, 2H, циклобутил H), 1,90-1,95 (м, 2H, циклобутил H), 2,14-2,21 (м, 2H, циклобутил H), 2,31 (с, 3H, CH3), 3,42 (с, 3H, NCH3), 4,34-4,30 (м, 1H, CH), 5,05 (с, 2H, OCH2Ph), 6,22 (с, 1H, CH), 7,39-7,30 (м, 5H, ArH), 9,08-9,06 (д, 1H, J=7,08 Гц, NH); МС (m/z) 327(M++1), 230, 166, 91.
Подобным способом было получено следующее соединение:
3-(бензилокси)-N-циклопентил-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоксамид
Т.пл. 233,6-234,4°С; 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,43-1,52 (м, 4H, циклопентил H), 1,54-1,60 (м, 2H, циклопентил H), 1,78-1,83 (м, 2H, циклопентил H), 2,30 (с, 3H, CH3), 3,43 (с, 3H, NCH3), 4,13-4,18 (м, 1H, CH), 5,04 (с, 2H, OCH2Ph), 6,22 (с, 1H, CH), 7,30-7,41 (м, 5H, ArH), 8,80-8,82 (д, J=6,95 Гц, 1H, NH); МС (m/z) 341 (M++1), 230, 166, 91.
ПРИМЕР 6: Получение N-циклобутил-3-гидрокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоксамида [Аро6622]
Смесь N-циклобутил-3-бензилокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоксамида (1,528 г, 4,68 ммоль, 1,0 эквивалент), 10% Pd на активированном угле (200 мг, влажный) и этанола (200 мл) перемешивали при давлении Н2 50 фунтов/кв. дюйм при комнатной температуре в течение 2,5 ч. Катализатор фильтровали через целит, и фильтрат упаривали с получением твердого вещества, которое перекристаллизовывали из МеОН с получением указанного в заголовке соединения (0,57 г, 51,5%) в виде твердого вещества белого цвета.
Т.пл. 277,3°С (с разложением); 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,68-1,70 (м, 2H, циклобутил H), 1,95-2,01 (м, 2H, циклобутил H), 2,20-2,26 (м, 2H, циклобутил H), 2,29 (с, 3H, CH3), 3,41 (с, 3H, NCH3), 4,31-4,35 (м, 1H, CH), 6,13 (с, 1H, CH), 8,98 (ушир., 1H, NH); МС (m/z) 237(M++1), 185, 166, 123.
Подобным способом были получены следующие соединения:
N-циклопентил-3-гидрокси-1,6-диметил-4-оксо-1,4-дигидропиридин-2-карбоксамид [Аро6620]
Т.пл. 289,3°С (с разложением); 1H-ЯМР (ДМСО-d6, 400 МГц) δ 1,49-1,55 (м, 4H, циклопентил H), 1,61-1,68 (м, 2H, циклопентил H), 1,83-1,87 (м, 2H, цикорпентил H), 2,29 (с, 3H, CH3), 3,42 (с, 3H, NCH3), 4,14-4,18 (м, 1H, CH), 6,12 (с, 1H), 8,71-8,73 (д, J=7,05 Гц, 1H, NH); МС (m/z) 251 (M++1), 166.
Циклопропиламид 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты [Аро6618]
Т.пл. 241-243°С; 1H-ЯМР (ДМСО-d6, 400 МГц) δ 0,53 (м, 2H, циклопропил-H), 0,71 (m, 2H, циклопропил-H), 0,94-1,00 (м, 4H, циклопропил-H), 2,42 (с, 3H, CH3), 2,79 (м, 1H, CH), 3,30 (м, 1H, CH), 6,08 (с, 1H, CH), 8,54 (ушир., с, 1H, NH); МС (m/z) 249 (M++1).
ПРИМЕР 7: Получение 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
К суспензии 3-(бензилокси)-6-метил-4-оксо-4Н-пиран-2-карбоновой кислоты (70 г, 0,27 моль) в МеОН (350 мл) в 3-горлой колбе (круглодонная колба), снабженной мешалкой с приводом, добавляли циклопропиламин (120 мл, 1,72 моль). В результате образовался прозрачный бледно-желтый раствор. Реакционную смесь кипятили с обратным холодильником в течение приблизительно 19 ч. Летучие вещества удаляли в вакууме, и остаток растворяли в воде (700 мл) при перемешивании. Водную смесь фильтровали через рыхлый слой целита®. Фильтрат помещали в 3-горлую колбу RBF, снабженную мешалкой с приводом, и охлаждали на бане со льдом. Концентрированную HCl добавляли до тех пор, пока рН фильтрата не достигал приблизительно рН 1-2 и не осаждалась большая масса твердого вещества цвета "апельсина". К суспензии добавляли ацетон (200 мл). Затем, твердое вещество собирали фильтрацией с помощью вакуум-фильтра, тщательно промывали ацетоном и сушили на воздухе. Указанное в заголовке соединение было получено в виде твердого вещества не совсем белого цвета (71,0 г, 88%).
Т.пл. 139,0-139,5°С; 1H-ЯМР (300 МГц, ДМСО-D6) δ (м.д.): 0,98-1,15 (м, 4H, 2 c-CH2), [2,37 (с) + 2,40 (с), ротамеры, отношение 3/2, 3H, CH3)], 3,30-3,50 (м, 1H, C=CH), 5,00-5,05 (м, 2H, CH2Ph), 6,20-6,25 (м, 1H, C=CH), 7,28-7,50 (м, 5H, Ph); МС (m/z): 300,2 (M++1), 256,2, 192,2, 164,4, 91,0 (100%); рассчитано для C17H17NO4: C, 68,21; H, 5,72; N, 4,68%. Найдено: C, 67,76; H, 5,76; N, 4,61%.
ПРИМЕР 8: Синтез метиламида 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
К холодной суспензии (баня со смесью льда с поваренной солью, внутренняя температура = -5°С) 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты (30,0 г, 0,10 моль), CH2Cl2 (150 мл) и ДМФ (7,8 мл, 0,10 моль) в 3-горлой-RBF (круглодонная колба), снабженной мешалкой с приводом, добавляли тионилхлорид (9,5 мл, 0,13 моль) по каплям в течение периода 5 минут. После добавления тионилхлорида, реакционная смесь все еще представляла собой суспензию. Баню со смесью льда с поваренной солью отставляли. Реакционную смесь оставляли нагревать до комнатной температуры. Аликвоты удаляли, и реакцию гасили 2 М раствором метиламина в ТГФ. Затем образовавшуюся смесь анализировали посредством ВЭЖХ. Таким образом, проверка с помощью ВЭЖХ показала, что приблизительно 96% исходного материала было израсходовано после перемешивания реакционной смеси при комнатной температуре в течение 3 ч. (ВЭЖХ, подвижная фаза: 0,035% HClO4/CH3CN, 80/20, колонка: симметрия C18 WAT046980, скорость потока: 1 мл/мин, длина волны для проверки: 260 нм, RT 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты = 2,46 мин, RT метиламида 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты = 5,40 мин).
В другую 1-литровую 3-горлую-RBF, снабженную мешалкой с приводом, помещали дихлорметан (240 мл) и триэтиламин (36 мл, 0,26 моль) (баня со смесью льда с поваренной солью, внутренняя температура = -10°С). К холодному раствору добавляли 2 М метиламин в тетрагидрофуране (73 мл, 0,146 моль). Хлорангидрид, синтезированный in situ выше, переносили в дополнительную воронку и медленно добавляли к раствору амина в течение периода 30 минут. Экзотермическая реакция была отмечена, но температура внутри держалась ниже -5°С. Реакция завершалась через 10 мин, как показано с помощью ТСХ (CH2Cl2/MeOH, отношение 9/1, об./об.). Реакционную смесь гасили водой (100 мл), и смесь перемешивали в течение 5 мин. Органическую фазу собирали и промывали более двух раз водой с последующим промыванием разбавленным раствором NaOH (0,05 М, 3×100 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением твердого вещества коричневого цвета. Твердое вещество суспендировали в 150 мл смеси этанола и этилацетата (отношение 2/8, об./об.), и взвесь перемешивали в течение 2 ч. Твердое вещество собирали фильтрацией с помощью вакуум-фильтра, промывали этилацетатом (50 мл) и затем сушили на воздухе. Таким образом, указанное в заголовке соединение было получено в виде твердого вещества бледно-розового цвета со слегка коричневатым оттенком (14 г, 45%). Затем, материал очищали колоночной хроматографией (5% MeOH:CH2Cl2).
Т.пл. 132-135°С; 1H-ЯМР (CDCl3, 400 МГц) δ 1,05 (м, 4H, циклопропил-H), 2,38 (с, 3H, CH3), 2,70 (д, J=1,8 Гц, 3H, NCH3), 3,35 (м, 1H, CH), 5,07 (с, 2H, CH2), 6,14 (с, 1H, CH), 7,15 (ушир., 1H), 7,35 (м, 5H, ArH); 13C (CDCl3) δ 9,48, 20,30, 25,86, 34,15, 74,01, 118,16, 127,79, 128,06(2C), 128,22(2C), 137,35, 142,05, 143,98, 149,91, 162,01, 173,89; МС (m/z): 313 (M++1).
Подобным способом были получены следующие соединения:
3-(бензилокси)-N-(циклогексилметил)-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид
1H-ЯМР (CD3OD, 400 МГц) δ 0,90-0,96 (м, 3H), 1,13-1,23 (м, 3H), 1,45-1,54 (м, 1H), 1,64 (ушир. м, 4H), 1,73-1,76 (ушир. м, 4H), 2,56 (с, 3H, CH3), 3,12-3,13 (д, J=6,8 Гц, 2H), 3,36-3,40 (м, 1H, CH), 5,09 (с, 2H), 6,43 (с, 1H), 7,31-7,37 (м, 3H), 7,43-7,45 (м, 2H); МС (m/z): 395 (M++1).
3-(бензилокси)-1-циклопропил-6-метил-2-(морфолин-4-илкарбонил)пиридин-4(1Н)-он
1H-ЯМР (CDCl3, 400 МГц) δ 0,87-0,94 (ушир. м, 1H), 1,09-1,13 (м, 1H), 1,25-1,30 (м, 2H), 2,56 (с, 3H, CH3), 3,30-3,42 (м, 2H), 3,45-3,69 (м, 6H), 3,84-3,90 (м, 1H, CH), 4,74-4,77 (д, J=10,4 Гц, 1H), 5,54-5,56 (д, J=10,6 Гц, 1H), 6,80 (ушир. с, 1H, NH), 7,36-7,41 (м, 5H, ArH); МС (m/z): 369 (M++1).
3-(бензилокси)-1-циклопропил-6-метил-N-(3-метилбутил)-4-оксо-1,4-дигидропиридин-2-карбоксамид
1H-ЯМР (CDCl3, 400 МГц) δ 0,86-0,88 (д, J=6,4 Гц, 6H, 2CH3), 1,04-1,09 (м, 4H), 1,27-1,37 (м, 2H), 1,55-1,60 (м, 1H, CH), 2,37 (с, 3H, CH3), 3,20-3,25 (м, 2H, CH2), 3,34-3,37 (м, 1H, CH), 5,09 (с, 2H, CH2), 6,10 (с, 1H), 7,30-7,38 (м, 5H, ArH), 7,23-2,28 (ушир. т, 1H, NH).
3-(бензилокси)-N-циклогексил-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид
1H-ЯМР (CDCl3, 400 МГц) δ 1,15-1,30 (м, 3H), 1,31 (ушир. м, 1H), 1,34 (ушир. м, 5H), 1,66-1,70 (м, 1H), 2,78 (с, 3H, CH3), 3,30-3,34 (м, 1H), 3,42-3,51 (м, 2H), 3,67-3,69 (м, 1H), 3,80-3,83 (м, 1H), 4,82-4,85 (д, J=10,3 Гц, 1H), 5,37-5,40 (д, J=10,5 Гц, 1H), 7,34 (ушир. м, 5H, ArH), 7,86 (с, 1H).
3-(бензилокси)-1-циклопропил-N-гексил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид
1H-ЯМР (CDCl3, 400 МГц) δ 0,89-0,92 (т, J=6,6 Гц, 3H, CH3), 1,25-1,32 (м, 6H), 1,40-1,47 (м, 4H), 1,64-1,70 (м, 2H, CH2), 2,54 (с, 3H, CH3), 3,43-3,48 (м, 2H, CH2), 3,91-3,93 (м, 1H, CH), 5,10 (с, 2H, CH2), 7,37-7,46 (м, 6H, ArH и C=CH), 9,24 (ушир. т, 1H, NH); МС (m/z): 383 (M++1).
3-(бензилокси)-1-циклопропил-6-метил-2-[(4-метилпиперазин-1-ил)карбонил]пиридин-4(1Н)-он
1H-ЯМР (CDCl3, 400 МГц) δ 0,85-0,88 (м, 1H), 1,06-1,29 (м, 4H), 1,40-1,45 (ушир. м, 2H), 1,50-1,58 (ушир. м, 4H), 2,51 (с, 3H, CH3), 3,12-3,17 (м, 1H), 3,35-3,48 (м, 3H), 3,75-3,78 (м, 1H, CH), 4,76-4,78 (д, J=10,6 Гц, 1H), 5,53-5,56 (д, J=10,7 Гц, 1H), 6,68 (ушир. с, 1H, NH), 7,30-7,43 (м, 5H, ArH); МС (m/z): 382 (M++1).
3-(бензилокси)-1-циклопропил-N,N,6-триметил-4-оксо-1,4-дигидропиридин-2-карбоксамид
1H-ЯМР (CDCl3, 400 МГц) δ 1,16-1,20 (м, 2H), 1,27-1,33 (м, 1H), 1,87-1,95 (м, 1H), 2,78 (с, 3H, CH3), 3,05 (с, 3H, CH3), 3,08 (с, 3H, CH3), 3,62-3,68 (м, 1H, CH), 4,86-4,90 (д, J=10,8 Гц, 1H), 5,33-5,38 (д, J=10,8 Гц, 1H), 7,29-7,33 (м, 5H, ArH), 7,77 (с, 1H, NH); МС (m/z): 327 (M++1).
ПРИМЕР 9: А. Получение метиламида 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
Процедура I:
Стадия а. Синтез метиламида 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты.
К суспензии метиламида 3-бензилокси-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты (10,0 г, 0,032 моль) в метаноле (40 мл) и воде (2,6 мл) при температуре, достигаемой в бане со льдом, добавляли концентрированную HCl (3,9 мл) по каплям. Образовавшийся прозрачный раствор коричневого цвета перемешивали при комнатной температуре в течение приблизительно 5 мин, затем в раствор впускали газообразный азот в течение приблизительно 5 мин. Затем добавляли Pd-C (10% влажный, 5% масс./масс., 0,5 г), и реакционный сосуд продували дважды водородом. Смесь подвергали гидрированию в реакторе Парра при давлении водорода 50 фунтов/кв. дюйм при комнатной температуре, и развитие реакции контролировали посредством ВЭЖХ в течение 3 ч. Реакция завершалась через 3 ч.
Избыток водорода удаляли, и газообразный азот впускали в раствор в течение приблизительно 5 мин. Реакционную смесь фильтровали через предварительно обработанный целит (предварительно промытый 0,1 N стандартным раствором метиламида 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты в метаноле), и отжатый осадок на фильтре промывали 6×10 мл метанола. Объем фильтрата снижали приблизительно до 30 мл при пониженном давлении. Остаток охлаждали на льду, и некоторое количество твердого вещества начинало осаждаться. Затем, добавляли 2N раствор NaOH (25 мл) до тех пор, пока рН раствора не достигал приблизительно рН 5, и смесь перемешивали в течение приблизительно 10 мин. Метил-трет-бутиловый эфир (МТВЕ, 30 мл) добавляли, и образовавшуюся смесь перемешивали при температуре, достигаемой в бане со льдом, в течение 30 мин. Твердое вещество собирали фильтрацией с помощью вакуум-фильтра, тщательно промывали дважды 5 мл смеси EtOH/MTBE (отношение 1/2). Для контролирования реакции, которое осуществляли с помощью системы Hewlett Packard series 1100 HPLC, условия ВЭЖХ были следующие: симметрия С18 колонка (WAT046980), градиент 0,035% HClO4/CH3CN, мин % CH3CN: 0-10; 6-10; 7-20 и 15-20, λ при 210, 260 и 285 нм; время удерживания метиламида 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты составляет 2,099 мин.
Стадия b. Очистка метиламида 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты
Суспензию сырого продукта, полученного, как описано на стадии а, в 1/1 смеси EtOH/дистиллированная вода (всего 14 мл) перемешивали при температуре, достигаемой в бане со льдом, в течение 1 ч. Твердое вещество собирали фильтрацией с помощью вакуум-фильтра и тщательно промывали 2 раза посредством 5 мл предварительно охлажденной смеси 1/1 EtOH/дистиллированная вода. Указанное в заголовке соединение, твердое вещество слегка розоватого цвета, сушили до постоянного веса при 40°С в вакууме в течение 16 ч. Полученный продукт дал отрицательный результат при проверке нитратом серебра и имел вес 5,3 г (74% общий выход, стадии а и b).
1H-ЯМР (300 МГц, ДМСО-D6) δ (м.д.): 0,94-0,99 (м, 4H, 2 c-CH2), 2,39 (с, 3H, CCH3), 2,76 (д, J=4,4 Гц, 3H, NHCH3), 3,28-3,31 (м, 1H, c-CH), 6,08 (с, 1H, C=CH), 8,44 (ушир. кв, 1H, NHCH3); 13C-ЯМР (75 МГц, ДМСО-D6) δ (м.д.): 9,1, 19,9, 25,8, 33,7, 112,3, 130,1, 143,3, 148,7, 161,8, 170,6; МС/МС (+ve ES): MC (m/z) 223 (M++1), 192,1, 164,2 (M+-CONHCH3, 100%), 150,1, 136,3. Анализ элементного состава: рассчитанный для C11H14N2O3: C, 59,45; H, 6,35; N, 12,60%. Найденный: C, 59,19; H, 6,07; N, 12,53%; ИК-спектр (KBr) см-1: 3300 (NH), 1670, 1653, 1495 (С=С).
В. N-(Циклогексилметил)-1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид:
Смесь 3-(бензилокси)-N-(циклогексилметил)-1-циклопропил-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамида (2,0 г, 4,8 ммоль), Pd/C (10% влажный, 0,45 г) в этаноле (150 мл) подвергали гидрированию в аппарате Парра при давлении водорода 50 фунтов/кв. дюйм в течение 16 ч. Реакционную смесь фильтровали через рыхлый слой целита, и целит тщательно промывали посредством EtOH (25 мл). Упаривание растворителя привело к твердому веществу бледно-розового цвета. Твердое вещество растворяли в горячем метаноле, затем охлаждали до комнатной температуры, и твердый продукт выпадал в осадок. Твердое вещество собирали фильтрацией с помощью вакуум-фильтра. Маточную жидкость концентрировали в вакууме, и твердый остаток вновь растворяли в горячем метаноле и охлаждали до комнатной температуры, при этом продукт выпадал в осадок, который затем собирали. Данную процедуру повторяли еще один раз. Три объединенные фракции твердого вещества белого цвета имели вес 0,95 г (63% выход).
1H-ЯМР (CDCl3, 400 МГц) δ 0,84-0,88 (м, 2H, CH2 для c-Pr), 1,03-1,09 (м, 2H, CH2 для c-Pr)), 1,06-1,31 (м, 5H), 1,65-1,87 (м, 6H), 2,50 (с, 3H, CH3), 3,33-3,36 (м, 2H, CH2N), 3,51 (с, 1H), 3,58-3,61 (м, 1H, CH для c-Pr) 6,27 (с, 1H, C=CH), 6,80 (ушир.т, 1H, NH); МС (m/z): 305 (M++1).
С. Следующие соединения были получены подобным способом:
1-циклопропил-3-гидрокси-6-метил-N-(3-метилбутил)-4-оксо-1,4-дигидропиридин-2-карбоксамид:

Выход: 88%; 1H-ЯМР (CDCl3, 400 МГц) δ 0,85-0,89 (м, 1H), 0,98-1,00 (д, J=6,4 Гц, 6H, 2CH3), 1,15-1,19 (м, 2H), 1,54-1,60 (м, 2H), 1,72-1,77 (м, 1H, CH), 2,50 (с, 3H, CH3), 3,49-3,53 (м, 2H, CH2), 3,57-3,60 (м, 1H, CH), 3,72 (ушир. с, 1H), 6,27 (с, 1H), 7,23 (ушир. т, 1H, NH); МС (m/z): 279 (M++1).
1-циклопропил-N-гексил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид:

Выход: 87%; 1H-ЯМР (CDCl3, 400 МГц) δ 0,90-0,94 (т, J=6,8 Гц, 3H, CH3), 1,27-1,47 (м, 10H), 1,68-1,73 (м, 2H), 2,70 (с, 3H, CH3), 3,47-3,52 (м, 2H, CH2), 3,85-3,88 (м, 1H, CH), 7,05 (с, 1H, C=CH), 8,30 (ушир. т, 1H, NH); МС (m/z): 293 (M++1).
N-циклогексил-1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид:

Выход: 91%; 1H-ЯМР (CDCl3, 400 МГц) δ 0,98-1,05 (м, 1H), 1,21-1,38 (м, 3H), 1,60-1,80 (ушир. м, 7H), 2,71 (с, 3H, CH3), 3,32-3,37 (м, 1H), 3,46-3,50 (м, 1H), 3,55-3,64 (м, 2H), 3,92-3,99 (м, 1H), 6,88 (с, 1H, C=CH); МС (m/z): 277 (M++1).
1-циклопропил-3-гидрокси-N,N,6-триметил-4-оксо-1,4-дигидропиридин-2-карбоксамид:

Выход: 97%; 1H-ЯМР (CD3OD, 300 МГц) δ 0,98-1,10 (м, 1H), 1,15-1,43 (м, 3H), 2,76 (с, 3H, CH3), 3,07 (с, 3H, CH3), 3,16 (с, 3H, CH3), 3,70-3,76 (м, 1H, CH), 7,10 (с, 1H, C=CH); 13C-ЯМР (CD3OD, 75 МГц) δ 9,5, 10,9, 21,3, 35,0, 38,1, 38,8, 114,4, 138,8, 142,9, 154,7, 162,5, 162,8; МС (m/z): 237 (M++1).
1-циклопропил-3-гидрокси-6-метил-2-[(4-метилпиперазин-1-ил)карбонил]пиридин-4(1Н)-он:

Выход: 96%; 1H-ЯМР (CD3OD, 300 МГц) δ 0,89-1,00 (м, 1H), 1,06-1,29 (м, 3H), 1,52-1,85 (ушир. м, 8H), 2,56 (с, 3H, CH3), 3,40-3,60 (м, 3H), 3,88-3,98 (м, 1H, CH), 6,48 (с, 1H, C=CH); 13C-ЯМР (CD3OD, 75 МГц) δ 10,0, 11,0, 21,0, 25,4, 26,4, 27,0, 36,5, 43,8, 49,2, 114,7, 132,9, 144,5, 152,8, 162,4, 170,2.
N,1-дициклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоксамид:

1H-ЯМР (CDCl3, 400 МГц) δ 0,68-0,70 (м, 2H), 0,85-0,95 (м, 4H), 1,15-1,26 (м, 2H), 2,70 (с, 3H, CH3), 2,91-2,98 (м, 1H), 3,50-3,61 (м, 1H), 6,26 (с, 1H, C=CH), 7,10 (ушир. с, 1H, NH); МС (m/z): 249 (M++1).
1-циклопропил-3-гидрокси-6-метил-2-(морфолин-4-илкарбонил)пиридин-4(1Н)-он:
1H-ЯМР (CD3OD, 300 МГц) δ 1,00-1,10 (м, 1H), 1,20-1,45 (м, 3H), 2,73 (с, 3H, CH3), 3,45-3,53 (м, 2H), 3,62-3,86 (м, 6H), 3,90-4,00 (м, 1H), 7,02 (с, 1H, C=CH); 13C-ЯМР (CD3OD, 75 МГц) δ 10,3, 11,1, 21,3, 38,6, 43,6, 48,3, 67,4, 67,7, 114,5, 137,2, 143,3, 154,7, 161,2, 163,7; МС (m/z): 279 (M++1).
ПРИМЕР 10: Определение рКа для Аро6619 потенциометрическим титрованием
Величины рКа лигандов определяли потенциометрическим титрованием, когда концентрация лиганда в воде выше, чем 1×10-2 М могла быть приготовлена. В обычном эксперименте, раствор образца (2,67×10-2 М) приготовляли следующим способом: Аро6619 (92,6 мг) взвешивали в 25-мл лабораторном стакане с последующим добавлением 0,1 М NaCl (15 мл). Смесь подвергали действию ультразвука в течение 10 минут с получением прозрачного бесцветного раствора. Затем газообразный азот пропускали через раствор. К раствору добавляли 1,000 N хлористоводородную кислоту (624 мкл, 1,5 эквивалента), чтобы достичь рН раствора 1,88. Раствор оставляли для уравновешивания при 22°С в течение 60 минут.
Затем раствор титровали против 1,000 N NaOH при 22°С, чтобы достичь рН 11,8. При каждом добавлении основания, раствор оставляли уравновешиваться до установления постоянного показания рН. Объем добавленного основания и показание рН записывали для каждого измерения. К концу эксперимента было сделано 137 измерений.
Данные значений рН против объема основания анализировали с использованием Hyperquad 2000 (Version 2.1, Peter Gans, University of Leeds). Для данной модели: L- + H+ ↔ LH (pKa1) и LH + H+ ↔ LH+ 2 (pKa2), были получены оптимальные величины pKa для Аро6619, которые составили pKa1=8,6 и pKa2=2,5.
ПРИМЕР 11: Определение рКа для Аро6617 спектрофотометрическим титрованием
Величины рКа лигандов могут быть определены спектрофотометрическим титрованием в случаях, когда как конъюгированная кислота, так и основание поглощает в УФ-видимой областях спектра. В обычном эксперименте, раствор образца приготовляли следующим способом: Аро6617 (0,792 мг) взвешивали в 80-мл лабораторном стакане с последующим добавлением 0,1 М NaCl (50 мл). Смесь подвергали действию ультразвука в течение 5 минут с получением прозрачного бесцветного раствора. Газообразный азот пропускали через раствор. К раствору добавляли 1,000 N NaOH (50 мкл), чтобы достичь рН раствора 10,9. Раствор оставляли уравновешиваться при 22°С в течение 1 часа. Sipper-систему использовали для циркуляции раствора образца между лабораторным стаканом и проточной ячейкой.
Раствор образца титровали стандартными растворами хлористо-водородной кислоты при 22°С, чтобы достичь рН 1,40. После каждого добавления кислоты раствор оставляли уравновешиваться до установления постоянного показания рН. Для каждого измерения записывали показание рН и спектр в УФ-видимой областях. Максимальные длины волн для депротонированных видов (L-), нейтральных видов (LH) и протонированных видов (LH2 +) составляли 314 нм, 281 нм и 249 нм соответственно. В области рН>6 после каждого добавления кислоты наблюдали небольшое уменьшение абсорбции при 314 нм и небольшое увеличение абсорбции при 281 нм в каждом спектре, в то время как в области рН<5 после каждого добавления кислоты наблюдали небольшое уменьшение абсорбции при 281 нм и небольшое увеличение абсорбции при 249 нм в каждом спектре. Раствор титровали до тех пор, пока не наблюдали явного изменения в спектрах после нескольких добавлений кислоты. К концу эксперимента было осуществлено 116 измерений.
Полученную серию данных анализировали, используя рНАВ (Peter Gans, University of Leeds). Были получены оптимальные величины pKa для Аро6617, которые составили pKa1=8,6 и pKa2=2,5.
ПРИМЕР 12: Стехиометрия комплексов Fe-Apo6622, определенная методом Job
В обычном эксперименте, растворы комплексов Fe-Apo6622 приготовляли путем смешивания исходного раствора Fe3+ (атомно-абсорбционный стандарт, 1005 мкг/мл в 1% масс. HCl, Aldrich) и исходного раствора Аро6622 (6,98×10-3 М в 0,1 М MOPS-буфере, pH 7,4). Было приготовлено 12 растворов образца. Хотя сумму общей концентрации железа (Собщая железо) и общей концентрации лиганда (Собщая L) в каждом из 12 растворов образца поддерживали постоянной (8,00×10-4 М), молярная доля лиганда, а (а=Собщая L/(Собщая L+Собщая железо), для 12 растворов образца была разной и составляла 0, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,75, 0,8, 0,9 и 1,0, соответственно. Общий объем для каждого из 12 растворов образцов был 5 мл, используя MOPS (0,1 М, рН 7,4) в качестве растворителя. рН 12 растворов подводили путем добавления NaOH до рН 7,4. Растворы образцов перемешивали с помощью вихревого встряхивателя при комнатной температуре в течение 3 часов, и затем помещали в метаболический термостат с перемешиванием типа Dubnoff при 25°С и 90 RPM в течение ночи. Растворы образца центрифугировали при 4000 об/мин в течение 15 минут и затем помещали в термостат при 25°С без перемешивания. Спектр в УФ-видимой областях записывали при 25°С для каждого из 12 растворов.
График согласно Job был построен с величинами абсорбции при 450 нм на оси ординат и "а" на оси абсцисс. Максимальная абсорбция была найдена при а=0,75, которая соответствует отношению железо:лиганд в комплексах, равному 1:3. Результаты построения графика согласно Job показаны на фиг.1.
Подобным образом, были построены графики согласно Job для Fe-Apo6617 и Fe-Apo6619. Они представлены на фиг.2 и фиг.3.
ПРИМЕР 13: Определение коэффициента распределения
Буфер MOPS (50 мМ, рН=7,4) и 1-октанол использовали в качестве водной фазы и органической фазы, соответственно, для определений коэффициента распределения. Буфер MOPS и 1-октанол предварительно насыщали один другим до использования.
В обычном эксперименте органический исходный раствор Аро6618 (циклопропиламид 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты) приготовляли путем взвешивания 0,50 мг соединения в 10-мл мерную колбу и доводили до объема 1-октанолом. Затем, раствор подвергали воздействию ультразвука в течение 60 минут до полного растворения образца. Концентрацию исходного раствора рассчитывали как С°орг.=2,0×10-4 М. Органический стандартный раствор Аро6619 с концентрацией 2,0×10-5 М приготовляли в 10-мл мерной колбе путем 10-кратного разведения исходного раствора 1-октанолом. Раствор образца приготовляли в 10-мл мерной колбе. Исходный раствор образца (3 мл) переносили с помощью пипетки в колбу с последующим добавлением буфера MOPS (3 мл). Затем стандартный раствор и растворы образца перемешивали с помощью вихревого встряхивателя в течение 2 часов. После перемешивания растворы переносили в опытные пробирки и центрифугировали при 4000 об/мин в течение 15 минут. Спектры в УФ-видимой областях для стандартного раствора и органической (верхняя) фазы раствора образца записывали при 22°С. Коэффициент распределения, D7,4, рассчитывали с помощью следующего уравнения:
D7,4=[Aогр./(С°орг.еорг.-Аорг.)]×(Vводн./Vорг.),
где еорг. = коэффициент молярной экстинкции при пиковой длине волны (λмакс.), наблюдаемой в УФ-видимой областях спектра органического стандартного раствора; Аорг. = абсорбция органической фазы в растворе образца при такой же длине волны (λмакс.); С°орг. = концентрация исходного раствора; Vводн. = объем буфера MOPS в растворе образца; Vорг. = объем исходного раствора в растворе образца.
ПРИМЕР 14: Определение констант комплексообразования металла
А. Приборы и реактивы:
Для измерений рН использовали рН-метр (Accumet Research AR15, Fisher) и комбинированный электрод (Accumet Standard-size Glass Combination Electrode, 13-620-285, Fisher). До использования электрод калибровали тремя стандартными буферными растворами (рН 4,00, рН 7,00 и рН 10,00, Fisher). Титрант добавляли вручную путем использования цифровых пипеток (Eppendorf). Спектрофотометр с УФ-видимой областями спектра (Agilent 8453) использовали для измерений абсорбции в УФ-видимой областях спектра.
Sipper-систему (89068D Agilent) использовали всякий раз, когда измеряли рН-зависимую абсорбцию. Встряхиватель (VX-2500 Multi-tube Vortexer, VWR Scientific Products) использовали для приготовления растворов образца, используемых при определении коэффициента распределения и при построении графиков согласно Job.
Исходные растворы металла были поставлены фирмой Aldrich: атомно-абсорбционный стандартный раствор железа (1000 мкг/мл Fe в 1% масс. HCl); атомно-абсорбционный стандартный раствор алюминия (1000 мкг/мл Al в 1% масс. HCl); атомно-абсорбционный стандартный раствор кальция (1000 мкг/мл Ca в 1% масс. HCl); атомно-абсорбционный стандартный раствор меди (1000 мкг/мл Cu в 1% масс. HNO3); атомно-абсорбционный стандартный раствор магния (1000 мкг/мл Mg в 1% масс. HNO3); атомно-абсорбционный стандартный раствор марганца (1000 мкг/мл Mn в 1% масс. HNO3); атомно-абсорбционный стандартный раствор цинка (1000 мкг/мл Zn в 1% масс. HCl). Стандартные растворы гидроокиси натрия и хлористо-водородной кислоты были поставлены фирмой VWR Scientific Products. Реагент MOPS (3-[N-морфолино]пропансульфокислота) был поставлен компанией Sigma-Aldrich.
В. Определение констант ступенчатого образования системы Fe-Apo6619 спектрофотометрическим титрованием. Аро6619 представляет собой метиламид 1-циклопропил-3-гидрокси-6-метил-4-оксо-1,4-дигидропиридин-2-карбоновой кислоты.
Константы ступенчатого образования систем Mn+-лиганд определяли спектрофотометрическим титрованием в случаях, когда комплексы металлов имели значительную абсорбцию в видимой области спектра вследствие переноса заряда от лиганда к металлу. В обычном эксперименте раствор образца приготовляли следующим способом: Аро6619 (10,7 мг) взвешивали в 80-мл лабораторном стакане с последующим добавлением 0,1 М NaCl (50 мл). Смесь подвергали действию ультразвука в течение 10 минут с получением прозрачного бесцветного раствора. Исходный раствор железа (атомно-абсорбционный стандартный раствор, Aldrich, 496 мкл, 8,93Е-06 моль) переносили с помощью пипетки в раствор с последующим добавлением 1,000 N NaOH (137 мкл). Молярное отношение между общим железом и общим Аро6619 составляло 1:5,4. Смесь оставляли для уравновешивания при комнатной температуре в течение ночи. Азот пропускали через раствор. Затем к раствору добавляли 1,000 N раствор хлористо-водородной кислоты (3 мл), чтобы достичь рН 1,5. Раствор оставляли уравновешиваться при 22°С в течение 3 часов.
Sipper-систему использовали для циркуляции раствора образца между лабораторным стаканом и проточной ячейкой.
Раствор образца титровали против стандартных растворов NaOH при 22°С, чтобы достичь рН 6,89. После каждого добавления основания раствор оставляли для уравновешивания до установления постоянного показания рН. Для каждого измерения записывали показания рН и спектр в УФ-видимой областях. Для каждого измерения достаточное количество основания было добавлено так, что наблюдалось небольшое увеличение абсорбции спектра. Раствор титровали до тех пор, пока не наблюдалось явное увеличение абсорбции после нескольких последовательных добавлений основания. К концу эксперимента были сделаны 64 измерения.
Затем, полученную серию данных анализировали, используя рНАВ. Для данной модели: L- + H+ ↔ LH (pKa1), LH + H+ ↔ LH+ 2 (pKa2), Fe3+ + L- ↔ FeL2+ (K1), FeL2+ + L- ↔ FeL2 + (K2), FeL2 + + L- ↔ FeL3 (K3) и β3 = K1K2K3, были получены оптимальные константы ступенчатого образования системы Fe-Apo6619 в виде log K1 = 12,5(1); log K2 = 11,6(1); log K3 = 9,5(1); log β3 = 33,6(2).
С. Определение констант ступенчатого образования системы Al-Apo6619 потенциометрическим титрованием.
Константы ступенчатого образования системы Mn+-лиганд определяли потенциометрическим титрованием в случаях, когда комплексы металлов (≥0,002 М) не выпадали в осадок при титровании. В обычном эксперименте, раствор образца приготовляли следующим способом: Аро6619 (31,91 мг) взвешивали в 25-мл лабораторном стакане с последующим добавлением 0,1 М раствора NaCl (18,9 мл). Смесь подвергали действию ультразвука в течение 10 минут с получением прозрачного бесцветного раствора. Исходный раствор алюминия (атомно-абсорбционный стандартный раствор, Aldrich, 971 мл, 3,59×10-5 моль) переносили в раствор с помощью пипетки с последующим добавлением 1,000 N раствора NaOH (229 мкл) для достижения рН 8,56. Молярное отношение между общим алюминием и общим Аро6619 составляло 1:4. Для металлов М2+, молярное отношение составляло 1:3. Азот пропускали через раствор. Смесь оставляли для уравновешивания при 22°С в течение 2 часов. Затем к раствору добавляли 1,000 N раствор хлористо-водородной кислоты (264 мкл) для достижения рН 2,20. Раствор оставляли для уравновешивания при 22°С в течение 1 часа.
Раствор титровали против 1,000 N NaOH при 22°С для достижения рН 11,0. При каждом добавлении основания раствор оставляли для уравновешивания до установления постоянного показания рН. Объем добавленного основания и показания рН записывали для каждого измерения. В эксперименте было сделано 93 измерения.
Данные серии значений рН против объема основания анализировали с использованием Hyperquad 2000. Для данной модели: L- + H+ ↔ LH (pKa1), LH + H+ ↔ LH+ 2 (pKa2), Al3+ + L- ↔ AlL2+ (K1), AlL2+ + L- ↔ AlL2 + (K2), AlL2 + + L- ↔ AlL3 (K3) и β3 = K1K2K3, были получены оптимальные константы ступенчатого образования системы Al-Apo6619 в виде log K1 = 12,6(2); log K2 = 9,2(1); log K3 = 8,4(1); log β3 = 30,2(2).
Вычисление рМn+
Величины pMn+ определяли в виде- log[M(H2O)m]n+ при физиологических условиях, т.е. рН 7,4, концентрации лиганда 10 мкМ и концентрации металла 1 мкМ. Для того чтобы рассчитать pMn+ для системы MLn, необходимо знать величины βn и рКа (βn являются константами образования системы Mn+ + n L- ↔ MLn; pKa являются константами равновесия для L- + n H+ ↔ LHn (n-1)+). Величины pMn+ могут быть вычислены путем использования программы Hyss (Hyperquad Stimulation and Speciation software: HYSS2 © 2000 Protonic Software).
Данные, полученные при определениях, описанных выше, для соединений формулы I могут быть найдены в таблице 1 и 2.
ПРИМЕР 15: Оценка соединений формулы I на модели крыс с перенасыщением железом
Эффективности Аро6619 и Аро6617 в усилении экскреции железа с мочой и фекалиями у крыс с перенасыщением железом
Целью данного исследования было определение эффективности Аро6619 и Аро6617 в усилении экскреции железа на модели крыс с перенасыщением железом. Перегрузку железом достигали путем введения комплекса железо-декстран. Перенасыщение железом с помощью комплекса железо-декстран была ранее использована для оценки эффективности комплексона у мышей (Kontoghiorghes G.J., Mol. Pharmacol. 1986, 30(6), 670-3; Bartfay et al., Cardiovasc Res. 1999, 43(4), 892-900), песчанок (Hershko et al., J.Lab. Clin. Med. 2002, 139, 50-58), крыс (Rakba N. Biochem. Pharmacol. 1998, 55(11): 1796-1806) и приматов (Bergeron et al., Blood, 1992, 79(7), 1882-1890). Схема нагрузки железом, использованная в данном изучении, привела к 20-кратному увеличению железа в печени и 3,8-кратному увеличению уровней железа в сердце у самцов крысы. Предварительное изучение на данной модели показало, что указанная модель не связана со значительными отклонениями в увеличении веса животных, потреблении пищи, от параметров клинической химии или гематологических параметров.
Экспериментальный протокол:
Шесть самцов крысы Sprague-Dawley (имеющих вес между 200-250 грамм) получали из Charles River Laboratories, Montreal, Quebec, Canada. Крыс нагружали железом путем введения комплекса железо-декстран внутрибрюшинно при дозе 100 мг/кг, дважды в неделю в течение периода 4 недели, всего 8 инъекций (iron dextran, Sigma). Общий объем инъецируемого комплекса железо-декстран составлял 1 мл/кг. Через восемь недель крыс переносили в метаболические клетки (одна крыса/клетку). Когда животные находились в метаболических клетках, выделения (как моча, так и фекалии) собирали ежедневно в течение, по крайней мере, 3 дней до и 4 дней после введения каждого комплексона. Каждый из двух комплексонов (Аро6619 и Аро6617) вводили непрерывно. Комплексоны вводили в виде однократной дозы 450 мкмоль/кг с помощью желудочного зонда при дозовом объеме 2-4 мл/кг. Животных взвешивали до дозирования, чтобы обеспечить введение точной дозировки. Животных проверяли ежедневно (глаза, кожу и движение) после введения комплексона, чтобы установить, были ли какие-либо заметные признаки отклонений от нормы. Мочу и фекалии хранили при -20°С до анализа для определения концентраций тотального железа.
Питание животных, вода и содержание:
Крыс содержали в условиях регулирования климата и освещения (температура: 19-25°С, относительная влажность 40%, 12-часовой цикл свет/темнота) на протяжении изучения. В течение акклиматизации, нагрузки железом и фаз уравновешивания крыс помещали в стандартные клетки (2 крысы/клетку), кормили стандартной для грызунов пищей и давали регулярно водопроводную воду ad libitum. Крыс переносили в метаболические клетки (одна крыса/клетку, Nalgene, Rochester NY) после нагрузки железом и уравновешивания. За три дня до помещения крыс в метаболические клетки крыс кормили пищей с низким содержанием железа (3 ч/млн железа, Dyets Inc., Bethlehem, PA) и давали воду Millipore ad libitum. Крыс продолжали кормить пищей с низким содержанием железа в течение изучения. Целью кормления животных пищей с низким содержанием железа было снизить уровень фона в фекальных образцах, производимого поступающим с пищей железом.
Приготовление дозированного раствора комплексонов:
Вначале, к 50 мг/мл дозированному раствору комплексонов формулы I (570 мг) добавляли смесь воды Millipore (2 мл) и 6 N HCl (0,4 мл) и доводили до конечного объема (11,4 мл) водой Millipore (9 мл). Конечное значение рН растворов доводили до рН 4 разбавленным раствором гидроокиси натрия. Растворы защищали от света и до каждого введения приготовляли свежие растворы.
Определения железа:
Образцы мочи и фекалий передавали в Trace Elements Laboratory at the London Health Sciences Center, London, Ontario, Canada для анализа общей концентрации железа. Кратко, образцы фекалий приготовляли путем добавления воды, нагревания до 98°С, перемешивания с помощью вихревого встряхивателя и последующей лиофилизации. Затем, образцы смешивали, и типичную субвыборку производили и подвергали перевариванию с помощью кипящей смеси HNO3 и H2O2. Затем образцы фекалий разбавляли до анализа водой 1:100 высшей степени очистки, используя секторное поле высокого разрешения ICP-MS (Finnigan Element 1). Образцы мочи подвергали перевариванию с помощью 0,1% HNO3 и разбавляли 1/10 до изучения на ICP-MS. Соответствующие калибровочные кривые, используя контрольные образцы железа, и пригодные для контроля стандарты, соответствующие стандартам NIST, использовали. Образцы, данные для которых были выше или ниже области определения количества, вновь анализировали. Поскольку общее количество мочи и фекалий, выделяемое в течение заданного периода времени было известно так же, как и вес крыс, общие уровни железа в моче и фекалиях выражали как мкг/день/кг. Статистические сравнения в группах и между группами были сделаны с помощью непарных t-тестов. Величина р <0,05 была принята как достоверная.
Результаты
У крыс не выявили явные признаки нездоровья после введения любого из комплексонов. Все животные продолжали прибавлять в весе нормально после каждого из введенных комплексонов.
Выведение железа с мочой:
Данные по эффективности Аро6619 и Аро6617 в усилении выведения железа с мочой представлены на фиг.6, ниже. Базовый уровень выведения железа с мочой, измеренный в течение 3 дней до введения Аро6619, составлял 6±1 мкг/день/кг.
Выведение железа с мочой повышалось до 240±131 мкг/день/кг в первый день после введения Аро6619 (р=0,007). Впоследствии экскреция снижалась до 16±5 мкг/день/кг на второй день после введения Аро6619, однако даже эти уровни были все еще значительно выше, чем базовые уровни (р=0,004). На третий день, выведение железа с мочой возвращалось до базовых уровней (5±1 мкг/день/кг). Соединение Аро6617 также привело к повышению выведения железа в первый и второй дни после введения (164±55 и 17±13 мкг/день/кг, соответственно). Хотя выведение железа с мочой, производимое посредством Аро6619, было в значительно большей степени, чем выведение, достигаемое посредством Аро6617 (240±131 мкг/день/кг против 164±55 мкг/день/кг, в первый день после введения комплексона), эта разница не достигла статистической значимости вследствие того факта, что одна из шести крыс не выделяла железа с мочой больше с Аро6617, чем с Аро6619.
В целях сравнения деферипрон также изучали при дозе 450 мкмоль/кг на вышеуказанной модели, но в другой группе крыс (n=6). Базовые уровни выводимого с мочой железа составляли 9±3 мкг/день/кг. Уровни железа повышались до 80±32 мкг/день/кг в первый день после введения деферипрона (р=0,06), и уровни возвращались до базового уровня на второй день.
Выведение железа с фекалиями:
Данные по эффективности Аро6619 и Аро6617 в усилении выведения железа с фекалиями представлены в таблице 3. Обе базовые величины, а также величины, достигаемые введением комплексонов, представляют собой сумму выведенного железа в три дня до и после введения комплексона, соответственно. Как соединение Аро6619, так и соединение Аро6617 повышало степень выведения железа с фекалиями при дозе 450 мкмоль/кг, но эта величина достигала статистической значимости только в Аро6617-группе (Аро6619: 4154±1245 мкг/день/кг, р=0,08 против базового уровня; Аро6617 4411±790 мкг/день/кг против базового уровня, р=0,008). В предварительном исследовании на такой же модели введенный шести крысам деферипрон при дозе 450 мкмоль/кг приводил к величинам железа, выведенным с фекалиями, 2157±169 мкг/день/кг на третий день после введения комплексона.
Второе изучение на крысах при дозе 113 мкмоль/кг:
Второе изучение проводили, чтобы подтвердить результаты по эффективности Аро6619 и Аро6617, наблюдаемые в описанном выше исследовании, и также охарактеризовать эффективность указанных соединений при дозах ниже, чем 450 мкмоль/кг. Исследование проводили в двух отдельных группах крыс с перенасыщением железом. Способ нагрузки железом, приготовление дозированных растворов и оценка эффективности у данных крыс были подобны описанному выше исследованию.
Первую группу крыс (n=6) обрабатывали Аро6619 последовательно при дозах 28, 113 и 450 мкмоль/кг. Подобным образом, вторую группу крыс (также n=6) обрабатывали Аро6617 при таких же трех дозах. Суммарно данные экскреции приведены в таблице 4. Подобно результатам предварительного исследования, как соединение Аро6619, так и соединение Аро6617 приводило к повышению уровня выведения железа с мочой при дозе 450 мкмоль/кг (Аро6619: при базовом уровне 11±3 до 335±76 в первый день после введения Аро6619, р=0,0001; Аро6617: при базовом уровне 14±4 до 183±20 в первый день после введения Аро6617, р=0,0003). В противоположность предварительному исследованию, где не наблюдали значительной разницы между эффективностью Аро6619 и Аро6617 в выведении железа с мочой при дозе 450 мкмоль/кг, в данном изучении было очевидно, что соединение Аро6619 оказалось более эффективным, чем Аро6617 (р=0,004) при этой дозе. Подобным образом, при дозе 113 мкмоль/кг (25 мк/кг) как Аро6619, так и Аро6617 повышало уровень выводимого с мочой железа (р<0,005), но соединение Аро6619 было более эффективным, чем Аро6617 (р=0,01). При дозе 28 мкмоль/кг, только соединение Аро6617 оказывало повышение уровня экскреции железа (р=0,01) выше базового уровня. Однако величина повышенной экскреции была небольшой как для Аро6619, так и для Аро6617.
Уровень выведенного с фекалиями железа повышался посредством Аро6619 при дозе 450 мкмоль/кг (р=0,03), и также наблюдали тенденцию к повышению экскреции с Аро6617 (р=0,08). Значительных повышений уровня экскреции железа с фекалиями не наблюдали с любым из комплексонов при дозах ниже, чем 450 мкмоль/кг.
В общем, оба исследования показали, что Аро6619 и Аро6617 приводят к повышению уровня экскреции железа с мочой и фекалиями. Данная экскреция превышает экскрецию, наблюдаемую в гистологических изучениях с деферипроном на такой же модели. Хотя соединение Аро6619 способствует более значительному повышению экскреции железа с мочой в сравнении с Аро6617 при высоких дозах, превосходство Аро6619 над Аро6617 в усилении экскреции железа с фекалиями не было очевидным из этих исследований. По большей части, полученные данные объясняются как высокими, так и высоковариабельными "базовыми" уровнями железа в фекалиях (т.е. низкий сигнал относительно коэффициента шума), что делает индуцируемые комплексоном повышения экскреции железа трудно определяемыми.
ПРИМЕР 16: А. Получение хелатного комплекса Fe(Apo6617)3
Карбонатный буфер рН 9,7 приготовляли путем растворения 0,84 бикарбоната натрия, 1,06 г карбоната натрия в деионизированной воде и разбавления раствора до 50 мл. Аро6617 (1,028 г, 4,62 ммоль) добавляли к карбонатному буферу (25 мл). Гетерогенную смесь перемешивали в течение 15 минут при комнатной температуре с получением прозрачного раствора. Безводный хлорид железа (0,2417 г, 1,49 ммоль) добавляли малыми порциями в течение 5 мин при комнатной температуре с получением темно-красного раствора. Затем колбу герметично закрывали насадкой с перегородкой и перемешивали при комнатной температуре в течение 42 ч. Ацетонитрил (30 мл) добавляли и растворитель упаривали при пониженном давлении с получением сухой массы красного цвета. Твердое вещество растворяли в дихлорметане (90 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Сырой продукт подвергали очистке флэш-хроматографией, используя градиент элюции (смесь дихлорметан/метанол: 95/5, 90/10, 85/15 и 80/20). Получали твердое вещество красного цвета (900 мг). Твердое вещество смешивали со смесью этилацетат/метанол (90/10, 60 мл) и перемешивали при комнатной температуре в течение 1 ч. Нерастворимые частицы фильтровали и фильтрат упаривали при пониженном давлении с получением продукта (800,5 мг). МС (m/z): 742,6 (М++Na), 720,4 (M++1), 634,6, 499,0, 469,6, 360,4, 334,3.
Насыщенный раствор Fe(Apo6617)3 приготовляли путем растворения 0,2 г материала в дихлорметане. Нерастворимые частицы фильтровали. К 1 мл насыщенного раствора материала в дихлорметане в пузырьке добавляли 0,3 мл этилацетата. Пузырек закрывали при комнатной температуре. Кристаллы темно-коричневого цвета отделяли для кристаллографического изучения. Трехмерная монокристаллическая структура представлена на фиг.7. Кристаллографические данные представлены в таблицах 5-6.
В. Приготовление Fe(Apo6619)3
Аро6619 (4,4488 г, 20,0 ммоль) взвешивали в 100-мл 1-горлую круглодонную колбу, снабженную стержневым магнитом для перемешивания. Деионизированную воду (30 мл) добавляли с получением суспензии. К смеси добавляли раствор NaOH (3,336 мл 6,000 N раствора, 20,0 ммоль) при комнатной температуре с получением прозрачного раствора оранжево-красного цвета. FeCl3·6H2O (1,7735 г, BDH, 97-102%, 6,56 ммоль) взвешивали в 30-мл опытную пробирку. Деионизированную воду (4 мл) добавляли в опытную пробирку. Смесь перемешивали с помощью вихревого встряхивателя с получением прозрачного раствора желтого цвета. Раствор FeCl3 добавляли по каплям к раствору Аро6619, приготовление которого описано выше. Смесь перемешивали при комнатной температуре в течение 6 дней. За это время образовывалось твердое вещество. Твердое вещество собирали фильтрованием с помощью вакуум-фильтра. Твердое вещество переносили обратно в круглодонную колбу. Затем добавляли 50 мл ацетона и 3 мл деионизированной воды. Смесь перемешивали в течение нескольких часов. Затем твердое вещество собирали фильтрацией с помощью вакуум-фильтра. Твердое вещество сушили на воздухе с получением 3,1 г (выход=66%). МС: 720,6 (М+1). Монокристаллы Fe(Apo6619)3 росли вследствие диффузии толуола во влажный ДМФ. Структура Fe(Apo6619)3, идентифицированная рентгеноструктурным анализом, показана на фиг.8.
ПРИМЕР 17: Определение Е1/2 системы Fe-Apo6619
А. Материалы и приборы
Железосинеродистый калий (III) приобретали у фирмы Aldrich. Мезилат дефероксамина (DFO) приобретали у фирмы Sigma. Атомно-абсорбционный стандартный раствор железа (содержит 1005 мкг/мл Fe в 1% масс. HCl) приобретали у фирмы Aldrich. Электрохимические измерения осуществляли с помощью циклического вольтамперометрического анализатора (BAS, CV-50W Potentiostat). Использовали программу BAS CV-50W Version 2.31. Следующие электроды были использованы для определения окислительно-восстановительных потенциалов комплексов железа: Ag/AgCl электрод сравнения (BAS, MF-2052); платиновый вспомогательный электрод (BAS, MW-1032) и стеклоуглеродный рабочий электрод (BAS, MF-2012). рН-метр (Accumet Research AR15, 13-636-AR15, Fisher Scientific) и рН-электрод (AccupHast комбинированный электрод, 13-620-297, Fisher Scientific) использовали для регулирования рН растворов образцов.
В. Приготовление растворов образцов
2,0 мМ раствор Fe(DFO) в 0,1 М NaCl (рН 7,4)
Мезилат дефероксамина (чистота = 95%) в количестве 148,1 мг точно отвешивали в 100-мл мерную колбу. Твердое вещество растворяли приблизительно в 30 мл 0,1 M NaCl с получением прозрачного бесцветного раствора. К раствору добавляли 11,114 мл стандартного раствора железа (содержит 1005 мкг/мл Fe в 1% масс. HCl). Раствор разбавляли посредством 0,1 М NaCl до метки 100 мл в мерной колбе. Образовавшийся раствор перемешивали с помощью вихревого встряхивателя до полного смешивания. Раствор переносили в 200-мл лабораторный стакан. Затем рН раствора доводили приблизительно до рН 7,1 путем добавления стандартных растворов гидроокиси натрия. Затем лабораторный стакан покрывали парафильмом и раствор оставляли перемешивать в течение ночи. рН раствора доводили до 7,40 на следующий день испытания. Рассчитанное молярное отношение между железомобщее и DFOобщий составляет 1:1,07.
2,0 мМ раствор Fe(Apo6619)3 в 0,1 M NaCl (pH 7,4)
Соединение Аро6619 в количестве 70,0 мг точно отвешивали в 50-мл мерную колбу. Твердое вещество растворяли приблизительно в 15 мл 0,1 М NaCl с получением прозрачного бесцветного раствора. К раствору добавляли 5557 мкл стандартного раствора железа (содержит 1005 мкг/мл Fe в 1% масс. HCl). Затем добавляли 0,1 M NaCl, чтобы довести общий объем до 50 мл. Образовавшийся раствор перемешивали с помощью вихревого встряхивателя до полного смешивания. Раствор переносили в 80-мл лабораторный стакан. Затем доводили рН раствора приблизительно до рН 7,1 путем добавления стандартных растворов гидроокиси натрия. Затем лабораторный стакан покрывали парафильмом, и раствор оставляли перемешивать в течение ночи. рН раствора доводили до 7,40 на следующий день испытания. Рассчитанное молярное отношение между железомобщее и соединением Аро6619общее составляет 1:3,15. Подобным образом, был приготовлен раствор 2,0 мМ Fe(деферипрон)3 в 0,1 M NaCl (рН 7,4).
С. Определение окислительно-восстановительных потенциалов комплексов железа
Все потенциалы в тексте даны против Ag/AgCl электрода сравнения. Окислительно-восстановительные потенциалы 2,0 мМ K3Fe(CN)6 в 1,0 M нитрате калия измеряли в начале каждого рабочего дня, чтобы проверить точность работы циклического вольтамперметра. Определяли пики окислительно-восстановительных потенциалов 2,0 мМ растворов комплексов железа при рН 7,4, то есть Fe(DFO), Fe(L1)3 и Fe(Apo6619)3. Растворы образцов комплексов железа продували аргоном в течение 15 минут до сканирования посредством CV, и раствор находился под аргоном в течение измерений. Стеклоуглеродный рабочий электрод полировали окисью алюминия после каждого сканирования. Скорость сканирования составляла 300 мВ/с для раствора железосинеродистого калия (III) и 450 мВ/с для растворов Fe(DFO), Fe(L1)3 и Fe(Apo6619)3. На фиг.9 представлены данные вольтамперометрии комплексов железо(III)Ln при рН 7,4: a) K3Fe(CN)6; b) Fe(DFO); c) Fe(L1)3; {L1=деферипрон} и d) Fe(Apo6619)3. Измеряли пиковый восстановительный потенциал (Ер red), окислительный потенциал (Ep ox), абсолютную разницу между Ер red и Ep ox (р) и окислительно-восстановительный потенциал (Е1/2) четырех комплексов железа. Величину Е1/2 рассчитывали как (Ер red + Ep ox)/2, исходя из представленных на фиг.9 данных, которые приведены в таблице ниже.
Окислительно-восстановительные потенциалы 2,0 мМ K3Fe(CN)6 в 1,0 M нитрате калия измеряли в начале каждого рабочего дня, чтобы проверить точность работы циклического вольтамперметра. В обычном эксперименте, величины Ер red, Ep ox, р и Е1/2 для K3Fe(CN)6, определенные в данной лаборатории с использованием стеклоуглеродного рабочего электрода, составляли 197 мВ, 282 мВ, 85 мВ и 240 мВ, соответственно. Величины, полученные от Bioanalytical Systems Inc. (BAS) с использованием платинового рабочего электрода, составили 237 мВ, 306 мВ, 69 мВ и 272 мВ соответственно. Исходя из теоретического прогноза величина р должна составлять приблизительно 60 мВ для процесса переноса одного электрона. Экспериментальные величины хорошо согласуются с величинами, полученными из BAS.
В отличие от K3Fe(CN)6 окислительно-восстановительные свойства Fe(DFO), Fe(L1)3 и Fe(Apo6619)3 в значительной степени зависят от состояния поверхности рабочего электрода. Окислительно-восстановительные потенциалы воспроизводились только после тщательного полирования стеклоуглеродного рабочего электрода окисью алюминия после каждого сканирования.
Величины р для Fe(DFO), Fe(L1)3 и Fe(Apo6619)3 составили 112 мВ, 107 мВ и 85 мВ соответственно. Как можно увидеть (фиг.9), картины циклической вольтамперометрии Fe(DFO), Fe(L1)3 и Fe(Apo6619)3 являются, по существу, обратимыми. На основании приведенных двух наблюдений, разумно предположить, что картины циклической вольтамперометрии Fe(DFO), Fe(L1)3 и Fe(Apo6619)3 отражают обратимый процесс переноса одного электрона для каждого комплекса: Fe(III)Ln ∴ Fe(II)Ln. Величина Е1/2 для Fe(DFO), определенная в данной лаборатории, составляет -698 мВ против Ag/AgCl электрода сравнения, которая находится в прекрасном соответствии с литературной величиной (-688 мВ) (A.L. Crumbliss et al., Inorganic Chemistry, 2003, 42, 42-50). Величина Е1/2 для Fe(Apo6619)3 составляет -691 мВ, которая является подобной величине для Fe(DFO).
Описанные выше примеры даны только с целью иллюстрации и никаким образом не предназначены ограничивать объем изобретения. Специалист в данной области оценит, что изобретение может быть модифицировано различными путями без отклонения от сущности или основной идеи изобретения. Заявители утверждают все такие модификации.
Электрохимические свойства комплексов железо(III)Ln при рН 7,4 перечислены ниже:

Селективность связывания Аро6619 (рКа1=2,5, рКа2=8,6) с металлами
Эффективность Аро6619 и Аро6617, введенных в дозе 450 мкмоль/кг, в усилении экскреции железа с фекалиями у перегруженных железом крыс (n=6). Величины выражены в виде мкг/день/кг. Величины фекальной экскреции за три дня после введения предполагаемого комплексона представлены в таблице. Величины выражены в виде среднего значения ± 1SD.
(мкг/день/кг)
Эффективность Аро6619 и Аро6617 в усилении экскреции железа с мочой и фекалиями у перегруженных железом крыс (n=6/группу). Величины выражены в виде мкг/день/кг. Величины фекальной экскреции за 3 дня после введения комплексона представлены в таблице и сравнены с величинами базового уровня, определенными за 3 дня до введения комплексона. Величины выражены в виде среднего значения ± 1SD.
* р<0,05 против величины базового уровня в той же группе
τр<0,05 против Аро6617 при такой же дозе
Кристаллографические данные и уточнение структуры Fe(Apo6617)3
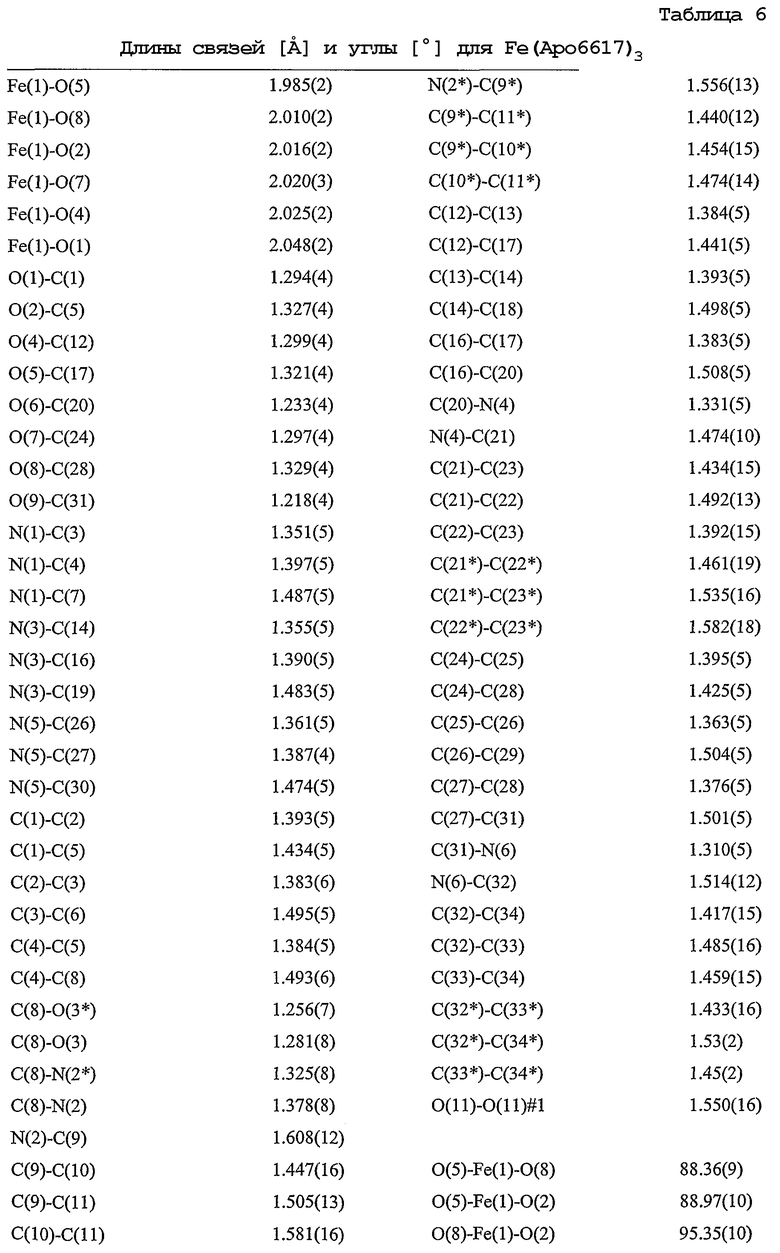


Кристаллографические данные и уточнение структуры Fe(Apo6619)3



| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА СОМАТОСТАТИНА ПОДТИПА 5 (SSTR5) | 2014 |
|
RU2671958C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ ТРАНСГЛУТАМИНАЗЫ ТКАНЕЙ | 2013 |
|
RU2652987C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| СОЕДИНЕНИЕ ЦЕФЕМА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1992 |
|
RU2024530C1 |
| ИНГИБИТОРЫ ПРОЛИЛГИДРОКСИЛАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2007 |
|
RU2429226C9 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ПРОИЗВОДНЫЕ 1-(4-ИЗОКСАЗОЛ-5-ИЛ)-1Н-ПИРАЗОЛ-1-ИЛ)-2-МЕТИЛПРОПАН-2-ОЛА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИРОРОВ ИЛ-17 И ИФН-ГАММА ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ И ХРОНИЧЕСКОГО ВОСПАЛЕНИЯ | 2018 |
|
RU2785342C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ПРОИЗВОДНЫЕ ТАКСОЛА С ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2419622C2 |


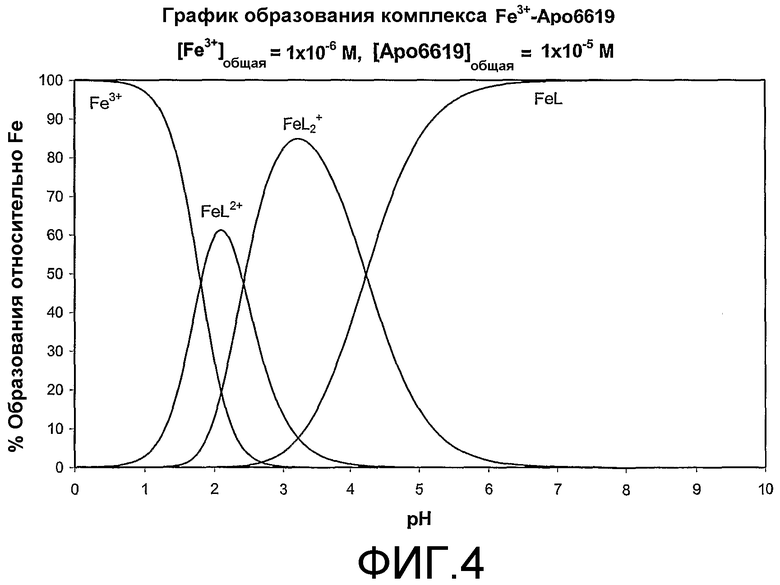

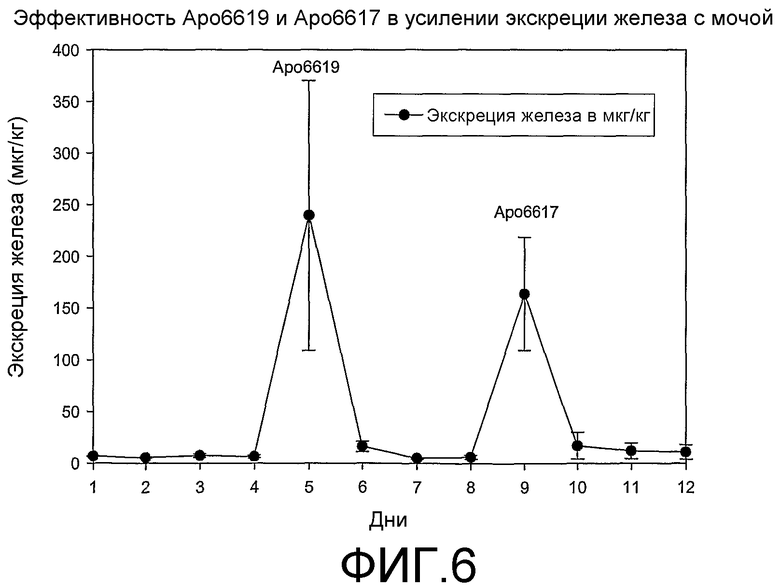
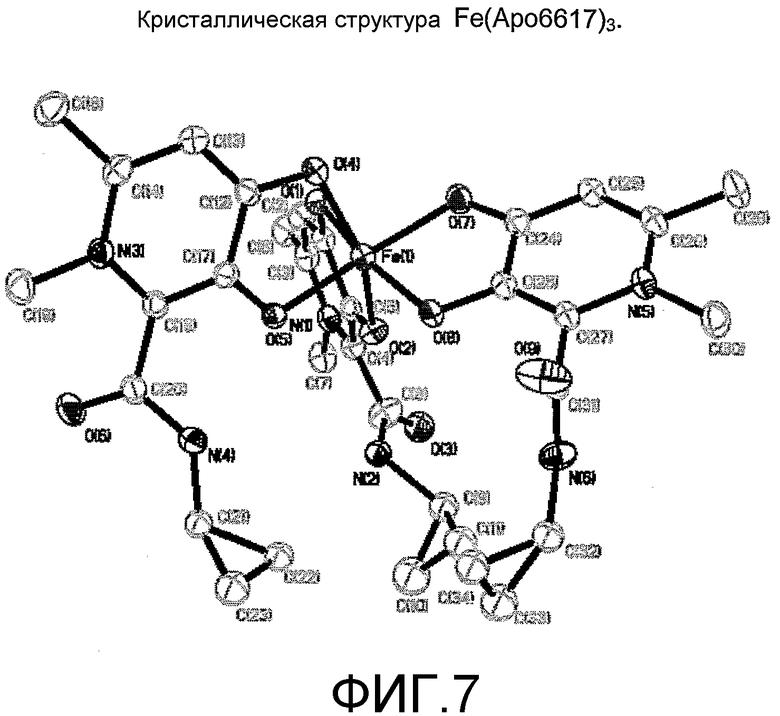
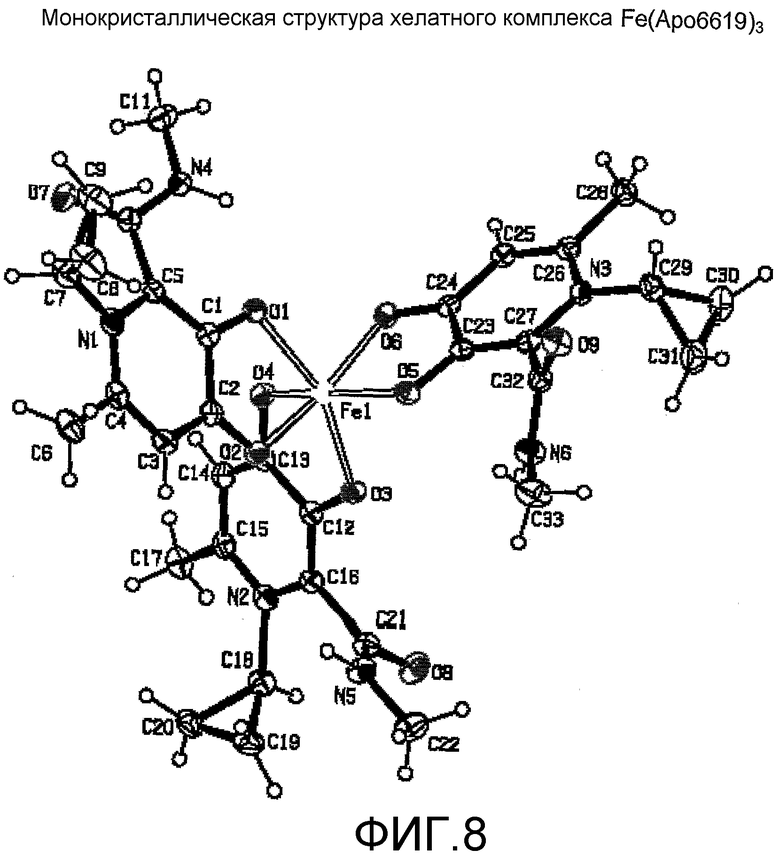

Настоящее изобретение относится к циклоалкильному производному 3-гидрокси-4-пиридинона общей формулы
 ,
,
где R1 является X при условии, что R2 является Y; или R1 является Т при условии, что R2 является W; или R1 является X при условии, что R2R5N, вместе взятые, образуют гетероциклическое кольцо, выбранное из морфолинила или пиперазинила, где морфолинильная или пиперазинильная группа является либо незамещенной, либо замещенной одной-тремя С1-С6алкильными группами; X является С3-С6циклоалкилом; Y выбирают из группы, состоящей из С3-С6циклоалкила, С1-С6алкила и С1-С6алкила, монозамещенного С3-С6циклоалкилом; Т означает С1-С6 алкил; W означает С3-С6циклоалкил; R3 означает водород; R4 выбирают из группы, состоящей из водорода и С1-С6алкила и; R5 выбирают из группы, состоящей из водорода и С1-С6алкила, и к его способу получения. Также изобретение относится к фармацевтической композиции на основе этих соединений и применению этих соединений в производстве лекарственного препарата. Технический результат: получены новые соединения, которые могут применяться для удаления избытка железа из организма больных с заболеваниями, связанными с перенасыщением железом. 5 н. и 12 з.п. ф-лы, 8 табл., 9 ил.

где R1 является X при условии, что R2 является Y; или
R1 является Т при условии, что R2 является W; или
R1 является X при условии, что R2R5N, вместе взятые, образуют гетероциклическое кольцо, выбранное из морфолинила или пиперазинила, где морфолинильная или пиперазинильная группа является либо незамещенной, либо замещенной одной-тремя С1-С6алкильными группами;
X является С3-С6циклоалкилом;
Y выбирают из группы, состоящей из С3-С6циклоалкила, C1-C6алкила и C1-С6алкила, монозамещенного С3-С6циклоалкилом;
Т означает С1-С6алкил;
W означает С3-С6циклоалкил;
R3 означает водород;
R4 выбирают из группы, состоящей из водорода и С1-С6алкила и;
R5 выбирают из группы, состоящей из водорода и C1-С6алкила;
и его фармацевтически приемлемая соль.

где R2 выбирают из группы, состоящей из С3-С6циклоалкила, С1-С6алкила и С1-С6алкила, монозамещенного С3-С6циклоалкилом;
R5 выбирают из группы, состоящей из водорода и C1-С6алкила или R5R2N, которые, взятые вместе, образуют гетероциклическое кольцо, выбранное из морфолинила или пиперазинила, где морфолинильная или пиперазинильная группа является либо незамещенной, либо замещенной одной-тремя С1-С6алкильными группами;
R3 означает водород; и
R4 выбирают из группы, состоящей из водорода и C1-С6алкила.
где R2 выбирают из группы, состоящей из С3-С6циклоалкила, С1-С6алкила и C1-С6алкила, монозамещенного С3-С6циклоалкилом;
R5 выбирают из группы, состоящей из водорода и C1-C6алкила или R5R2N, которые, взятые вместе, образуют гетероциклическое кольцо, выбранное из морфолинила или пиперазинила, где морфолинильная или пиперазинильная группа является либо незамещенной, либо замещенной одной-тремя С1-С6алкильными группами;
R3 означает водород; и
R4 выбирают из группы, состоящей из водорода и С1-С6алкила;
который включает стадию удаления защитной бензильной группы посредством реакции гидрирования производного 3-бензилоксипиридин-4-она общей формулы, или его хлористоводородной соли,

где R2, R5, R5R2N, R3, R4 являются такими, как определено в п.1.
| US 6472532, 29.10.2002 | |||
| US 6476229, 05.11.2002 | |||
| СПОСОБ СОХРАНЕНИЯ СЖАТЫХ ЦИФРОВЫХ АУДИО- И ВИДЕОСИГНАЛОВ | 1999 |
|
RU2287907C2 |
| US 5688815, 18.11.1997 | |||
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1,4-ДИГИДРОПИРИДИН-3,5-ДИКАРБОНОВОЙ КИСЛОТЫ В ВИДЕ R-ИЗОМЕРОВ И ИХ СОЛЕЙ, ИСХОДНЫЕ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2155752C2 |

