ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому соединению и его терапевтически приемлемым солям, которые ингибируют экзогенно или эндогенно стимулируемую секрецию кислоты желудочного сока и таким образом могут быть использованы для предупреждения и лечения желудочно-кишечных воспалительных заболеваний. В дополнительных аспектах настоящее изобретение относится к соединению по изобретению для применения в терапии, к способам получения такого нового соединения, к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение по изобретению или его фармацевтически приемлемую соль, и к применению соединения по изобретению для изготовления лекарств для указанного выше медицинского применения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Замещенные имидазо[1,2-а]пиридины, полезные для лечения пептических язвенных заболеваний, хорошо известны в данной области, например, из ЕР-В-0033094 и US 4450164 (Schering Corporation), из ЕР-В-0204285 и US 4725601 (Fujisawa Pharmaceutical Co.), из WO 99/55706 и WO 99/55705 (AstraZeneca), из WO 03/018582 (AstraZeneca) и из публикаций J.J.Kaminski и др. в Journal of Medical Chemistry (vol.28, 876-892, 1985; vol.30, 2031-2046, 1987; vol.30, 2047-2051, 1987; vol.32, 1686-1700, 1989 и vol.34, 533-541, 1991).
Обзор по фармакологии желудочного кислотного насоса (Н+,К+-АТФаза) смотри в Sachs et al. (1995) Annu. Rev. Pharmacol. Toxicol. 35: 277-305.
Авторы настоящего изобретения обнаружили, что замещенный имидазо[1,2-а]пиридин, пригодный для лечения пептических язвенных болезней, проявляет полезные свойства, например, быстрое начало действия, высокую эффективность и/или большую продолжительность, высокую растворимость и высокую скорость растворения.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Неожиданно было обнаружено, что соединение формулы I является очень эффективным ингибитором желудочно-кишечной Н+,К+-АТФазы и, следовательно, ингибитором секреции кислоты желудочного сока.
Таким образом, в одном аспекте настоящее изобретение относится к соединению формулы I

или его фармацевтически приемлемой соли.
В зависимости от условий процесса конечный продукт формулы I получают либо в нейтральной форме, либо в форме соли. Как свободное основание, так и соли конечного продукта находятся в рамках настоящего изобретения.
При получении солей присоединения кислот предпочтительно используют такие кислоты, которые образуют терапевтически приемлемые соли. Примерами таких кислот являются галогеноводородные кислоты, такие как соляная кислота, серная кислота, фосфорная кислота, азотная кислота, алифатические, алициклические, ароматические или гетероциклические карбоновые или сульфоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, п-гидроксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, гидроксиэтансульфоновая кислота, галогенбензолсульфоновая кислота, толуолсульфоновая кислота или нафталинсульфоновая кислота.
ПОЛУЧЕНИЕ
В настоящем изобретении также предложен следующий способ получения соединения формулы I.
Способ получения соединения формулы I включает в себя следующие стадии:
а) соединение формулы II

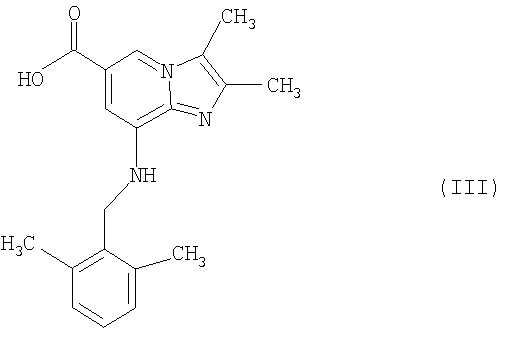
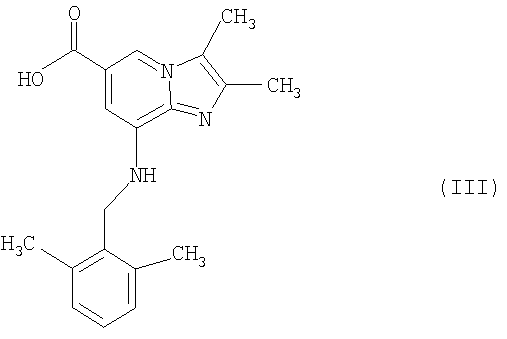
где R1 представляет собой C1-С6алкоксигруппу или -NH2, может быть гидролизовано в стандартных условиях в присутствии водной кислоты или основания до соответствующей карбоновой кислоты соединения формулы III

Кислота и основание могут быть выбраны из HCl, H2SO4 и NaOH.
б) соединение формулы III может быть подвергнуто взаимодействию с аминосоединением формулы IV

в присутствии агента сочетания, такого как о-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU), с образованием соответствующего амида, т.е. соединения формулы I. Эта реакция может быть осуществлена в стандартных условиях в инертном растворителе, таком как диметилформамид (ДМФА), метиленхлорид и ацетонитрил или их смесь.
В одном из воплощений изобретения реакция согласно вышеупомянутой стадии (а) может быть осуществлена путем гидролиза соединения формулы II до соответствующей кислоты в присутствии NaOH в водном спирте, таком как водный метанол или этанол, при кипячении с обратным холодильником в течение 1-3 ч.
Другой способ получения соединения по изобретению включает в себя следующую стадию:
а) соединение формулы V

где R1 представляет собой С1-С6алкоксигруппу,
может быть подвергнуто взаимодействию с аминосоединением формулы IV

с получением соответствующего амида, т.е. соединения формулы I.
Эта реакция может быть осуществлена путем нагревания реагентов в чистом аминосоединении или растворенном в инертном растворителе в стандартных условиях, например в спирте, таком как метанол, при повышенной температуре, например при температуре от 40 до 60°С, или при кипячении с обратным холодильником. Эта реакция может быть осуществлена в присутствии основания или цианидной соли.
В одном из воплощений настоящего изобретения эту реакцию осуществляют в присутствии основания, выбранного из 1,8-диазабицикло(4.5.0)андец-7-ена (DBU) или 1,5-диазабицикло(4.3.0)нон-5-ена (DBN).
В другом воплощении настоящего изобретения эту реакцию осуществляют в присутствии алкоксида, такого как метоксид натрия, метоксид калия или этоксид калия.
В одном из воплощений настоящего изобретения соединение формулы V смешивают с соединением формулы IV в таком растворителе, как спирт, например метанол или этанол. К нагретой реакционной смеси добавляют основание и реакцию завершают при повышенной температуре, например при температуре от 40 до 60°С, или при кипячении с обратным холодильником. Основание может быть выбрано в том числе из метоксида калия или метоксида натрия.
ПРИМЕНЕНИЕ В МЕДИЦИНЕ
В дополнительном аспекте изобретение относится к соединению формулы I для применения в терапии. В частности, в изобретении предложено применение соединения формулы I для изготовления лекарства для ингибирования секреции кислоты желудочного сока или для лечения воспалительных заболеваний желудочно-кишечного тракта.
Таким образом, соединение согласно изобретению может быть использовано для предупреждения и лечения воспалительных заболеваний желудочно-кишечного тракта и заболеваний, связанных с кислотностью желудка, у млекопитающих, включая человека, таких как гастрит, рефлюксный эзофагит, синдром Золлингера-Эллисона и пептическая язвенная болезнь, включая язву желудка и язву двенадцатиперстной кишки. Кроме того, данное соединение может быть использовано для лечения других желудочно-кишечных расстройств, когда желателен эффект подавления желудочной секреции, например, у пациентов с гастриномой и у пациентов с острым кровотечением в верхнем отделе желудочно-кишечного тракта. Данное соединение также может быть использовано для эффективного контроля и лечения изжоги и других симптомов гастроэзофагеальной рефлюксной болезни (GERD) (острой и хронической симптоматической GERD, заживления и профилактики эрозивного эзофагита), регургитации, кратковременной и продолжительной терапии кислотного рефлюксного заболевания и тошноты. Они могут быть использованы также у пациентов в ситуациях интенсивной терапии и в период перед и после операции для предупреждения кислотной аспирации и стрессового изъязвления.
Типичная суточная доза активного вещества варьирует в широком диапазоне и обычно зависит от различных факторов, таких как, например, индивидуальная потребность каждого пациента, путь введения и заболевание. Пероральные и парентеральные дозы обычно находятся в диапазоне от 5 до 1000 мг активного вещества в сутки, предпочтительно в диапазоне от 20 до 60 мг, например 50 мг. В зависимости от индивидуальной потребности и заболевания соединение по изобретению можно вводить пациенту при продолжительном лечении, а также при лечении по необходимости. С помощью соединения по изобретению предложены возможности для улучшения качества жизни индивидуумов, страдающих заболеваниями, связанными с желудочной кислотностью, и/или воспалительными заболеваниями желудочно-кишечного тракта.
ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ
В еще одном дополнительном аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение по изобретению или его терапевтически приемлемую соль.
Для клинического применения соединение по изобретению приготавливают в виде фармацевтических препаратов для перорального, ректального, парентерального или другого способа введения. Указанный фармацевтический препарат содержит соединение по изобретению в комбинации с одним или более фармацевтически приемлемыми ингредиентами. Носитель может находиться в форме твердого, полутвердого или жидкого разбавителя или капсулы. Эти фармацевтические препараты представляют собой дополнительный объект изобретения. Обычно количество соединения по изобретению составляет от 0,1 до 95 мас.% препарата, предпочтительно от 0,1 до 20 мас.% в препаратах для парентерального применения и предпочтительно от 0,1 до 50 мас.% в препаратах для перорального введения.
При приготовлении фармацевтических препаратов, содержащих соединение по изобретению, в форме стандартных доз для перорального введения выбранное соединение может быть смешано с твердыми, порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, производные целлюлозы, желатин или другой подходящий ингредиент, а также с разрыхлителями и скользящими агентами, такими как стеарат магния, стеарат кальция, стеарилфумарат натрия полиэтиленгликолевые воски. Затем смесь перерабатывают в гранулы или прессуют в таблетки.
Мягкие желатиновые капсулы могут быть приготовлены в виде капсул, содержащих активное соединение по изобретению, растительное масло, жир или другой подходящий наполнитель для мягких желатиновых капсул. Твердые желатиновые капсулы могут содержать гранулы соединения по изобретению. Твердые желатиновые капсулы также могут содержать соединение по изобретению в комбинации с твердыми порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, картофельный крахмал, кукурузный крахмал, амилопектин, производные целлюлозы или желатин.
Стандартные дозы для ректального введения могут быть приготовлены (1) в форме супозиториев, которые содержат соединение по изобретению, смешанное с нейтральной жировой основой; (2) в форме желатиновой ректальной капсулы, которая содержит соединение по изобретению в смеси с растительным маслом, парафиновым маслом или другим подходящим носителем для желатиновых ректальных капсул; (3) в форме готовой микроклизмы; или (4) в форме сухого препарата для микроклизмы, который подлежит растворению в подходящем растворителе непосредственно перед введением.
Жидкие препараты для перорального введения могут быть приготовлены в форме сиропов или суспензий, например растворов или суспензий, содержащих от 0,1 до 20 мас.% соединения по изобретению и остаток, состоящий из сахара или сахарных спиртов и смеси этанола, воды, глицерина, пропиленгликоля и полиэтиленгликоля. При желании, такие жидкие препараты могут содержать красители, корригенты, сахарин и карбоксиметилцеллюлозу или другой загуститель. Жидкие препараты для перорального введения могут быть приготовлены также в форме сухого порошка, подлежащего растворению в подходящем растворителе перед использованием.
Растворы для парентерального введения могут быть приготовлены в виде раствора соединения по изобретению в фармацевтически приемлемом растворителе, предпочтительно в концентрации от 0,1 до 10 мас.%. Эти растворы могут также содержать стабилизирующие ингредиенты и/или буферные ингредиенты и дозируются в стандартных дозах в форме ампул или флаконов. Растворы для парентерального введения могут быть приготовлены также в виде сухого препарата, подлежащего растворению в подходящем растворителе непосредственно перед использованием.
Соединение по изобретению также можно использовать в препаратах вместе с другими активными ингредиентами, например, для лечения или профилактики состояний, связанных с инфекцией слизистой оболочки желудка человека бактерией Helicobacter pylori. Такие другие активные ингредиенты могут представлять собой противомикробные агенты, в частности:
- β-лактамные антибиотики, такие как амоксициллин, ампициллин, цефалотин, цефаклор или цефиксим;
- макролиды, такие как эритромицин или кларитромицин;
- тетрациклины, такие как тетрациклин или доксициклин;
- аминогликозиды, такие как гентамицин, канамицин или амикацин;
- хинолоны, такие как норфлоксацин, ципрофлоксацин или эноксацин;
- другие, такие как метронидазол, нитрофурантоин или хлорамфеникол;
или
- препараты, содержащие соли висмута, такие как субцитрат висмута, субсалицилат висмута, субкарбонат висмута, субнитрат висмута или субгаллат висмута.
Соединение по изобретению может быть также использовано вместе или в комбинации с антацидами, такими как гидроксид алюминия, карбонат магния и гидроксид магния или альгиновая кислота, для одновременного, раздельного или последовательного применения, или вместе или в комбинации с фармацевтическими препаратами, которые ингибируют секрецию кислоты, такими как Н2-блокаторы (например, циметидин, ранитидин), ингибиторы Н+,К+-АТФазы (например, омепразол, пантопразол, лансопразол или рабепразол), для одновременного, раздельного или последовательного применения, или вместе или в комбинации с гастрокинетическими средствами (например, цисаприд или мосаприд) для одновременного, раздельного или последовательного применения.
Соединение по изобретению также может быть использовано вместе или в комбинации с другими активными ингредиентами для одновременного, раздельного или последовательного применения, например, для лечения или профилактики состояний, связанных с язвой желудка, индуцированной лекарствами. Такие другие активные ингредиенты могут представлять собой NSAID (НПВС, нестероидное противовоспалительное средство), NO-высвобождающии NSAID, ингибитор СОХ-2 (циклооксигеназы-2) или бисфосфонат.
Соединение по изобретению также может быть использовано вместе или в комбинации с антагонистом гастрина, таким как антагонист ССК2 (холецистокининового рецептора), для одновременного, раздельного или последовательного применения.
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
Другой аспект изобретения представляет собой новое промежуточное соединение, которое полезно в синтезе соединения по изобретению.
Таким образом, изобретение включает соединение формулы III
ПРИМЕРЫ
1. ПРИГОТОВЛЕНИЕ СОЕДИНЕНИЯ ПО ИЗОБРЕТЕНИЮ
Пример 1
Синтез 8-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоновой кислоты

8-Изопропил[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксилат (100 г, 0,274 моль) добавляли к метанолу (900 мл) и воде (100 мл). Добавляли гидроксид натрия (45 г, 1,13 моль) и смесь кипятили с обратным холодильником в течение 2 ч. Приблизительно половину растворителя испаряли и продукт осаждали путем добавления уксусной кислоты (100 мл) и воды (1000 мл). Смесь перемешивали в течение ночи при комнатной температуре. Продукт отфильтровывали и промывали водой и ацетоном. Получали 88 г (99%) соединения по изобретению.
1H-ЯМР (ДМСО, 400 МГц) δ 2,22 (s, 3H); 2,33 (s, 6H); 2,36 (s, 3H); 4,36 (d, 2Н); 5,03 (t, 1H); 6,64 (s, 1Н); 7,04-7,15 (m, 3H); 8,05 (s, 1H).
Пример 2
8-[(2,6-Диметилбензил)амино]-N-[(2S)-2-гидроксипропил]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамид
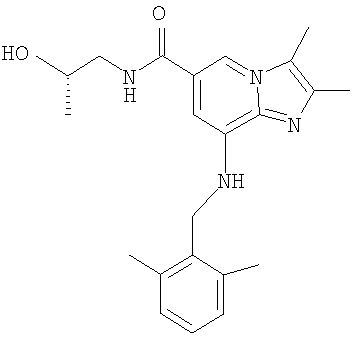
8-[(2,6-Диметилбензил)амино]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоновую кислоту (30 г, 0,0928 моль) и 30 г диизопропилэтиламина (30 г, 0,233 моль) добавляли к диметилформамиду (ДМФА) (250 мл). Добавляли о-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU) (36 г, 0,112 моль). Смесь перемешивали в течение 15 мин при комнатной температуре. Добавляли (S)-1-амино-2-пропанол (8,4 г, 0,112 моль) и перемешивание продолжали в течение 1 ч при комнатной температуре. Добавляли воду (1000 мл) и реакционную смесь нагревали до 70°С. Продукт осаждали, медленно добавляя еще воду (200 мл). Затем смесь перемешивали в течение 5 ч при 65-70°С. После охлаждения до комнатной температуры продукт отфильтровывали и промывали 40%-ным раствором метанола. Получали 33,7 г соединения по изобретению.
1H-ЯМР (CDCl3, 300 МГц) δ 1,20 (d, 3Н); 2,29 (s, 3Н); 2,32 (s, 3H); 2,35 (s, 6Н); 3,12-3,21 (m, 1Н); 3,58-3,66 (m, 2H); 3,96-4,06 (m, 1Н); 4,29 (d, 2H); 5,00 (t, 1H); 6,41 (s, 1Н); 6,89 (t, 1H); 7,02-7,15 (m, 3Н); 7,71 (s, 1H).
Пример 3
Мезилатная соль 8-[(2,6-диметилбензил)амино]-N-[(2S)-2-гидроксипропил]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида
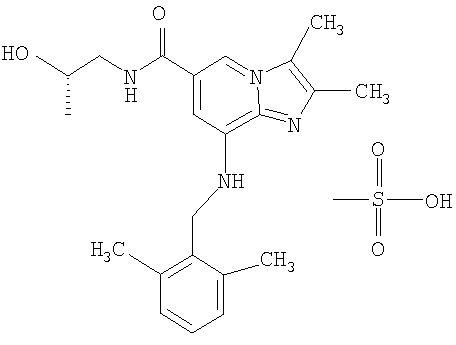
8-[(2,6-Диметилбензил)амино]-N-[(2S)-2-гидроксипропил]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамид (29,0 г, 0,0762 моль) растворяли в изопропаноле (280 мл), кипящем при температуре дефлегмации. К этому раствору добавляли метансульфоновую кислоту (7,4 г, 0,0770 моль), растворенную в изопропаноле (20 мл). После охлаждения в осадок выпадало кристаллическое вещество. Смесь оставляли на ночь при комнатной температуре. Продукт отфильтровывали и промывали изопропанолом. Получали 29,9 г соединения по изобретению в виде мезилатной соли.
1H-ЯМР (ДМСО, 500 МГц) δ 1,10 (d, 3Н); 2,25 (s, 3H); 2,36 (s, 6H); 2,42 (s, 3Н); 2,48 (s, 3Н); 3,21-3.33 (m, 2H); 3,82-3,85 (m, 1Н); 4,42 (d, 2Н); 6,16 (s, 1H); 7,13-7,21 (m, 3Н); 7,36 (s, 1H); 8,42 (s, 1H) 8,79 (t, 1H) 13,68(bs, 1H).
Пример 4
8-[(2,6-Диметилбензил)амино]-N-[(2S)-2-гидроксипропил]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамид
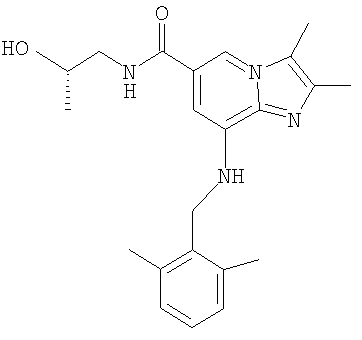
Смесь 8-изопропил-[(2,6-диметилбензил)амино]-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксилата (5 г, 13 ммоль) и (S)-1-амино-2-пропанола (3,8 г, 50 ммоль) в метаноле (10 мл) и толуоле (10 мл) нагревали до 40-45°С. Добавляли метоксид калия (0,57 г, 2,6 ммоль) в виде раствора в метаноле (32%, мас./мас.). Реакционную смесь нагревали до температуры дефлегмации (67-68°С) и поддерживали при этой температуре до желательного превращения (2-4 ч), контролируемого с помощью ВЭЖХ.
После охлаждения и добавления воды (15 мл) продукт выпадал в осадок. Продукт отфильтровывали, промывали водой и высушивали под вакуумом. Получали 4,1 г (80%) соединения по изобретению в виде белого твердого вещества.
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
Биологические анализы соединения по изобретению были осуществлены, как описано ниже. Эти анализы также были осуществлены с 8-[(2,6-диметилбензил)амино]-N-(2-гидроксиэтил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамидом, в качестве, контрольного соединения. 8-[(2,6-Диметилбензил)амино]-N-(2-гидроксиэтил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамид, в частности, раскрыт в WO 99/55705.
1. Эксперименты in vitro
Ингибирование секреции кислоты в изолированных желудочных железах кролика
Ингибирующее действие на секрецию кислоты in vitro в изолированных желудочных железах кролика определяли, как описано в Berglindh et al. (1976) Acta Physiol. Scand. 97, 401-414.
Определили, что коэффициент ингибирования (IC50) для соединения по изобретению составляет 0,26 мкмоль/л. Измеренный IC50 для 8-[(2,6-диметилбензил)амино]-N-(2-гидроксиэтил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида составлял 0,28 мкмоль/л.
Определение Н+,К+-АТФазной активности
Мембранные везикулы (2,5-5 мкг) инкубировали в течение 15 мин при +37°С в 18 мМ Pipes/Трис-буфере, рН 7,4, содержащем 2 мМ MgCl2, 10 мМ KCl и 2 мМ АТФ. АТФазную активность оценивали по высвобождению неорганического фосфата из АТФ, как описано в LeBel et al. (1978) Anal. Biochem. 85, 86-89.
Измеренный коэффициент ингибирования (IC50) для соединения по изобретению составлял 0,69 мкмоль/л. Измеренное значение IC50 для 8-[(2,6-диметилбензил)амино]-N-(2-гидроксиэтил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида составляло 0,75 мкмоль/л.
2. Эксперименты in vivo
Ингибирующее действие на секрецию кислоты у самок крыс
Используют самок крыс линии Sprague-Dawley. Им вводят канюлированную фистулу в желудок (просвет) и в верхнюю часть двенадцатиперстной кишки соответственно для сбора желудочных секретов и введения тестируемых веществ. Перед началом тестирования дается восстановительный период продолжительностью 14 суток после операции.
Перед секреторными анализами животных на 20 ч лишают корма, но не воды. Желудок многократно промывают через желудочную канюлю водопроводной водой (+37°С) и подкожно вводят 6 мл раствора Рингера с глюкозой. Секрецию кислоты стимулируют инфузией пентагастрина и карбохолина (20 и 110 нмоль/кг·час соответственно) в течение 2,5-4 ч (1,2 мл/час, подкожно), в течение которых желудочные секреты собирают в виде 30-мин фракций. Тестируемое вещество или носитель вводят либо через 60 мин после начала стимуляции (внутривенное и интрадуоденальное дозирование, 1 мл/кг), либо за 2 ч до начала стимуляции (пероральное дозирование, 5 мл/кг, желудочная канюля закрыта). Временной интервал между дозированием и стимуляцией может быть увеличен, для того чтобы проанализировать длительность действия. Образцы желудочного сока титруют до рН 7,0 с помощью 0,1 М NaOH и выход кислоты рассчитывают как произведение объема и концентрации титранта.
Другие расчеты основаны на совокупных средних ответах от 4-6 крыс. В случае введения во время стимуляции выход кислоты в течение периодов после введения тестируемого вещества или носителя выражают в виде относительных ответов, принимая за 1,0 выход кислоты за 30-мин период, предшествующий введению. Процент ингибирования рассчитывают из относительных ответов, вызываемых тестируемым соединением и носителем. В случае введения до стимуляции процент ингибирования рассчитывают непосредственно из выхода кислоты, зарегистрированного после введения тестируемого соединения и носителя.
Биодоступность в организме крысы
Используют взрослых крыс линии Sprague-Dawley. За один-три дня до экспериментов всех крыс подготавливают путем канюлирования левой сонной артерии под наркозом. Крысам, используемым для внутривенных экспериментов, канюлю вводят также в яремную вену (Popovic (1960) J.Appl. Physiol. 15, 727-728). Канюли выводят на заднюю часть шеи.
После введения дозы из сонной артерии многократно, с интервалами до 5,5 ч, отбирают образцы крови (0,1-0,4 г). Эти образцы замораживают до анализа тестируемого соединения.
Биодоступность определяют путем вычисления отношения между площадью под кривой (AUC) концентрации в крови/плазме после (1) интрадуоденального (и/д) или перорального (п/о) введения и (2) внутривенного (в/в) введения соответственно у крысы или собаки.
Площадь под кривой зависимости концентрации в крови от времени (AUC) определяют, используя линейно-логарифмическую формулу трапеций и экстраполируя на бесконечность путем деления последней установленной концентрации в крови на константу скорости элиминации на конечной стадии. Системную биодоступность (F%) после интрадуоденального или перорального введения рассчитывают как
F(%)=(AUC (п/о или и/д) /AUC (в/в))×100.
Ингибирование секреции кислоты желудочного сока и биодоступность у находящейся в сознании собаки
Использовали собак породы Лабрадор или гончая любого пола. Им вводили дуоденальную фистулу для введения тестируемых соединений или носителя и канюлированную желудочную фистулу или мешок Гейденгайна (Heidenhain-pouch) для сбора желудочного секрета.
Перед секреторными тестами животных лишали пищи на примерно 18 ч, но не ограничивали доступ к воде. Секрецию кислоты желудочного сока стимулировали в течение периода времени вплоть до 6,5 ч путем инфузии дигидрохлорида гистамина (12 мл/ч) в дозе, вызывающей примерно 80% индивидуального максимального секреторного ответа, и желудочный сок собирали последовательно в 30-мин фракции. Тестируемое вещество или носитель вводили перорольно, и/д или в/в через 1 или 1,5 ч после начала инфузии гистамина в объеме 0,5 мл/кг массы тела. Следует отметить, что в случае перорольного введения тестируемое соединение вводят в большой желудок, секретирующий кислоту, собаки с мешком Гейденгайна.
Кислотность в образцах желудочного сока определяют титрованием до рН 7,0 и рассчитывают выход кислоты. Выход кислоты в периоды сбора после введения тестируемого соединения или носителя выражают в виде относительных ответов, принимая выход кислоты во фракции, предшествующей введению, за 1,0. Процент ингибирования рассчитывают из относительных ответов, вызываемых тестируемым соединением и носителем.
Для анализа концентрации тестируемого соединения в плазме после дозирования берут образцы крови с интервалами до 4 ч. Плазму отделяют и замораживают в течение 30 мин после сбора и позже анализируют. Системную биодоступность (F%) после перорального или и/д введения рассчитывают, как описано выше в крысиной модели.
Определяли эффект на стимулированную гистамином секрецию кислоты у собаки с мешком Гейденгайна после введения 0,25 мкмоль/л соединения по изобретению, средний процент ингибирования через 1-3 ч (среднее, n=2) составляет 91%.
Бидоступность 8-[(2,6-диметилбензил)амино]-N-(2-гидроксиэтил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида (0,25 мкмоль/л) составляла 61%.
Растворимость в искусственной кишечной жидкости в состоянии голодания (FaSSIF)
В данной методике описана процедура измерения растворимости твердого вещества в FaSSIF (искусственной кишечной жидкости в состоянии голодания). Раствор FaSSIF представляет собой изотонический фосфатный буфер, рН которого подведено до 6,5 для имитации рН в тощей кишке. Таурохолевую кислоту и лецитин добавляют до концентрации 3 ммоль/л и 0,75 ммоль/л соответственно.
1 мг твердого вещества, т.е. соединения по изобретению, добавляют к 1 мл раствора FaSSIF и уравновешивают при 37°С. Образцы отбирают через 1 и 24 ч. Образцы переносят в пробирки "эппендорф" и центрифугируют при 10000 g и 37°С в течение 10 мин. Отбирают подходящий объем супернатанта и разбавляют его до соответствующей концентрации. Концентрации соединений в образцах анализируют с помощью ЖХ/УФ/МС.
Растворимость соединения по изобретению, измеренная через 24 ч, составляла 48 мкмоль/л, а растворимость соединения по изобретению в форме мезилатной соли, измеренная через 24 ч, составляла 218 мкмоль/л.
ПОЛУЧЕНИЕ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ
ПРИМЕР: Получение таблеток
Активный ингредиент 1 смешивают с ингредиентами 2, 3, 4 и 5 в течение 10 минут. Затем добавляют стеарат магния, полученную смесь затем перемешивают в течение примерно 5 минут и прессуют в таблет-форме с пленочным покрытием или без него.
Описывается новое производное имидазопиридина формулы I, способ его получения, промежуточные соединения, фармацевтический препарат, которые ингибируют экзогенно или эндогенно стимулируемую секрецию желудочной кислоты и таким образом могут быть использованы для предупреждения и лечения заболеваний, связанных с желудочной кислотностью, или воспалительных заболеваний желудочно-кишечного тракта. 2 н. и 6 з.п. ф-лы.
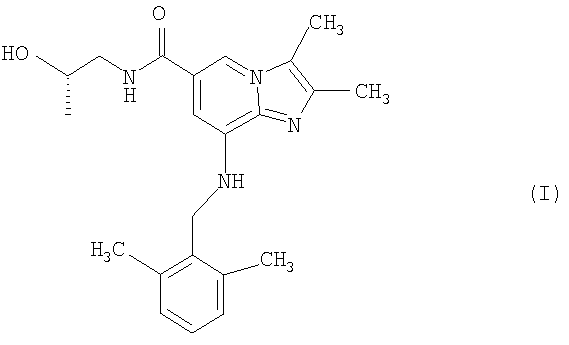

или его фармацевтически приемлемая соль.
а) обработки соединения формулы II

где R1 представляет собой C1-С6-алкоксигруппу или -NH2, в присутствии водной кислоты или основания в стандартных условиях соединением формулы III
б) взаимодействия соединения формулы III с соединением формулы IV
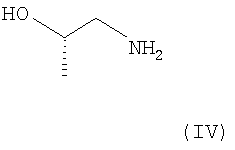
в присутствии агента сочетания в инертном растворителе в стандартных условиях с образованием соединения формулы I.
| WO 9955705 А1, 04.11.1999 | |||
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-А]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ, СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ ЖЕЛУДОЧНОЙ КИСЛОТЫ, СПОСОБ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И СПОСОБ ЛЕЧЕНИЯ СОСТОЯНИЙ, В КОТОРЫЕ ВОВЛЕЧЕНО ИНФИЦИРОВАНИЕ H.PYLORI | 1998 |
|
RU2193036C2 |

