Область техники
Настоящее изобретение относится к новым соединениям и их терапевтически приемлемым солям, которые ингибируют стимулируемую экзогенно или эндогенно секрецию желудочной кислоты и, таким образом, могут применяться для предотвращения и лечения желудочно-кишечных воспалительных заболеваний. В дополнительных аспектах данное изобретение относится к соединениям по настоящему изобретению для применения в терапии, к способам получения таких новых соединений, к фармацевтическим композициям, содержащим по меньшей мере одно соединение по данному изобретению или его терапевтически приемлемую соль в качестве активного ингредиента, и к применению активных соединений в изготовлении лекарств для указанного выше медицинского применения.
Предшествующий уровень техники
Замещенные имидазо[1,2-а]пиридины, полезные для лечения пептических язв, известны из уровня техники, например из ЕР-В-0033094 и US 4450164 (Schering Corporation); из ЕР-В-0204285 и US 4725601 (Fujisawa Pharmaceutical Co.); WO99/55706 и WO99/55705 (AstraZeneca) и из публикаций J. J. Kaminski et al. в Journal of Medicinal Chemistry (vol. 28, 876-892, 1985; vol. 30, 2031-2046, 1987; vol. 30, 2047-2051, 1987; vol. 32, 1686-1700, 1989; и vol. 34, 533-541, 1991).
Обзор фармакологии протонного насоса (Н+, K+-АТФазы) приведен в Sachs et al. (1995) Annu. Rev. Pharmacol. Toxicol. 35: 277-305.
В настоящее время авторы изобретения обнаружили дополнительные замещенные имидазо[1,2-а]пиридины, пригодные для лечения пептических язв, проявляющие благоприятные свойства и активность.
Описание изобретения
Неожиданно было обнаружено, что соединения формулы 1 особенно эффективны в качестве ингибиторов желудочно-кишечной Н+, К+-АТФазы и, следовательно, в качестве ингибиторов секреции желудочной кислоты.
В первом аспекте настоящее изобретение, таким образом, относится к соединениям общей формулы I
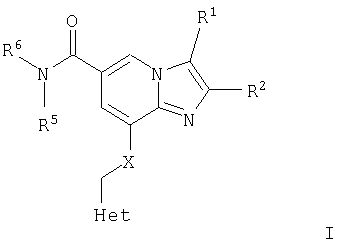
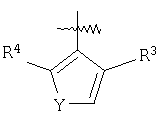
или их фармацевтически приемлемой соли, где Het представляет собой 4-, 5- или 6-членную ароматическую или алифатическую гетероциклическую группу, содержащую по меньшей мере один атом азота, кислорода или серы, замещенную группой R3 и группой R4 в орто-положениях;
R1 представляет собой
(а) Н,
(б) СН3 или
(в) СН2OH;
R2 представляет собой
(а) СН3 или
(б) СН2СН3;
R3 и R4 независимо выбраны из группы
(а) Н,
(б) C1-С6алкил,
(в) гидроксилированный С1-С6алкил или
(г) галоген;
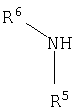
R5 и R6 представляют собой независимо выбранные заместители, содержащие атомы С, Н, N, О, S, Se, P и галогенов, которые дают соединения формулы I с молекулярной массой меньшей или равной 600;
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом; и
Х представляет собой
(а) NH или
(б) О.
Использованный здесь термин "С1-С6алкил" обозначает прямую или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода. Примеры указанного С1-С6алкила включают в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, а также пентил и гексил с прямой или разветвленной цепью, но не ограничиваются ими.
Термин "галогено" включает в себя фторо, хлоро, бромо и йодо.
Термин "4-, 5- или 6-членная ароматическая или алифатическая гетероциклическая группа, содержащая по меньшей мере один атом азота, кислорода или серы" включает в себя замещенные или незамещенные группы азетидин, фуран, тиофен, пиррол, пирролин, пирролидин, диоксолан, оксатиолан, оксазолан, оксазол, тиазол, имидазол, имидазолин, имидазолидин, пиразол, пиразолин, пиразолидин, изоксазол, изотиазол, оксадиазол, фуразан, триазол, тиадиазол, пиран, пиридин, пиперидин, диоксан, морфолин, дитиан, оксатиан, тиоморфолин, пиридазин, пиримидин, пиразин, пиперазин, триазин, тиадиазин, дитиазин, но не ограничивается ими, и следует понимать, что он включает в себя все изомеры идентифицированных выше групп. Термин "азетидинил" следует, например, понимать как включающий в себя 2- и 3-изомеры, а термины "пиридил" и "пиперидинил" следует, например, понимать как включающие в себя 2-, 3- и 4-изомеры.
Чистые энантиомеры, рацемические смеси и неэквимолярные смеси двух энантиомеров находятся в объеме данного изобретения. Следует понимать, что все возможные диастереомерные формы (чистые энантиомеры, рацемические смеси и неэквимолярныеные смеси двух энантиомеров) находятся в объеме данного изобретения. Также в данное изобретение включены производные соединений формулы I, которые обладают биологической функцией соединений формулы I.
В зависимости от условий способа конечные продукты формулы I получают либо в нейтральной форме, либо в форме соли. Как свободное основание, так и соли этих конечных продуктов находятся в объеме данного изобретения.
Соли присоединения кислот новых соединений могут известным самим по себе способом быть превращены в свободное основание с использованием основных агентов, таких как щелочь, или путем ионного обмена. Полученное свободное основание может также образовывать соли с органическими или неорганическими кислотами.
При получении солей присоединения кислот предпочтительно используют такие кислоты, которые образуют пригодные терапевтически приемлемые соли. Примерами таких кислот являются галогеноводородные кислоты, такие как соляная кислота, серная кислота, ортофосфорная кислота, азотная кислота, алифатические, алициклические, ароматические или гетероциклические карбоновые или сульфоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, пара-гидроксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, гидроксиэтансульфоновая кислота, галогенобензолсульфоновая кислота, толуолсульфоновая кислота или нафталинсульфоновая кислота.
Предпочтительными соединениями по данному изобретению являются те соединения формулы I или их фармацевтически приемлемые соли, в которых Het представляет собой

R1 представляет собой
(а) Н,
(б) СН3 или
(в) СН2ОН;
R2 представляет собой
(а) СН3 или
(б) СН2СН3;
R3 и R4 независимо выбраны из группы
(а) Н,
(б) C1-С6алкил,
(в) гидроксилированный С1-С6алкил или
(г) галоген;
R5 и R6 представляют собой независимо выбранные заместители, содержащие атомы С, Н, N, О, S, Se, P и галогенов, которые дают соединения формулы I с молекулярной массой меньшей или равной 600;
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом;
Х представляет собой
(а) NH или
(б) О; и
Y представляет собой S, SO, SO2, О, NH, C=N и N=C.
Более предпочтительными соединениями по данному изобретению являются те соединения формулы I или их фармацевтически приемлемая соль, в которых Het представляет собой
R1 представляет собой СН3 или СН2ОН;
R2 представляет собой СН3 или СН2СН3;
R3 и R4 независимо выбраны из группы: Н, С1-С6алкил, гидроксилированный С1-С6алкил и галоген;
R5 и R6 независимо представляют собой
(а) Н,
(б) С1-С6алкил,
(в) моно- или дигидроксилированный С1-С6алкил,
(г) С1-С6алкокси-замещенный С1-С6алкил,
(д) C2-С6алкенил,
(е) C2-С6алкинил,
(ж) галогенированный C1-С6алкил,
(з) С3-С8циклоалкил,
(и) циклоалкил-замещенный C1-С6алкил,
(к) арил, где арил представляет собой фенил, пиридил, тиенил или фуранил, возможно замещенный одним или более чем одним заместителем, выбранным из галогена, C1-С6алкила, C1-С6алкокси, CF3, ОН, нитро, амино, C1-С6алкил-NH-, (C1-С6алкил)2-N- или CN,
(л) арил-замещенный C1-С6алкил, где арил представляет собой фенил, пиридил, тиенил или фуранил, возможно замещенный одним или более чем одним заместителем, выбранным из галогена, C1-С6алкила, C1-С6алкокси, CF3, ОН, нитро, амино, C1-С6алкил-NH-, (C1-С6алкил)2-N- или CN,
(м) R8-алкил, где R8 представляет собой NH2C=O-, C1-С6алкил-NHC=O-, (C1-С6алкил)2NC=O-, C1-С6алкил-ООС-, NH2SO2-, C1-С6алкил-SO2NH-, ArSO2NH-, циано, C1-С6алкил-CO-NH-, C1-С6алкил-OOCNH-, C1-С6алкил-О-, C1-С6алкил-SO-, C1-С6алкил-S-, C1-С6алкил-SO2-, C1-С6алкил-С=O-, NH2-, C1-С6алкил-NH-, (C1-С6алкил)2N-, ArCONH-, ArNHSO2-, (Ar)2-N-SO2-, C1-С6алкил-NHSO2-, ArS-, ArSO-, ArSO2-, ArC=O-, NH2CONH-C1-С6алкил-NHCONH-, (C1-С6алкил)2-NCONH-, ArNHCONH-, (C1-С6)2-N-SO2-, Ar-O-, Ar-NH-, Ar(C1-С6)N- или (C1-С6)2NSO2-; где Ar представляет собой фенил, пиридил, тиенил или фуранил, возможно замещенный одним или более чем одним заместителем, выбранным из галогена, C1-С6алкила, C1-С6алкокси, CF3, ОН, CN, нитро, амино, C1-С6алкил-NH- или (C1-С6алкил)2-N-;
R5 и R6 вместе с атомом азота, к которому они присоединены, могут образовывать насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом;
Х представляет собой
(а) NH или
(б) О; и
Y представляет собой S, SO, SO2, О, NH, C=N или N=C.
Особенно предпочтительными соединениями по данному изобретению являются те соединения формулы I или их фармацевтически приемлемая соль, в которых Het представляет собой
R1 представляет собой СН3 или CH2OH;
R2 представляет собой СН3 или СН2СН3;
R3 и R4 независимо представляют собой водород или C1-С6алкил;
R5 и R6 независимо представляют собой
(а) Н,
(б) C1-С6алкил,
(в) моно- или дигидроксилированный C1-С6алкил или
(г) C1-С6алкокси-замещенный C1-С6алкил;
Х представляет собой NH и Y представляет собой S, О, NH, C=N или N=C.
Еще одно воплощение настоящего изобретения представляет собой соединение формулы I, где Het представляет собой
R1 представляет собой Н, СН3 или СН2OH;
R2 представляет собой СН3 или СН2СН3;
R3 представляет собой C1-С6алкил;
R4 представляет собой C1-С6алкил;
R5 и R6 каждый независимо выбран из водорода, C1-С6алкила, моно- или дигидроксилированного C1-С6алкила, C1-С6алкокси-(C1-С6алкил), гидроксилированного C1-С6алкокси-(C1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин;
Х представляет собой NH; и
Y представляет собой S или О.
Еще одно воплощение настоящего изобретения представляет собой соединение формулы I, где Het представляет собой
R1 представляет собой СН3;
R2 представляет собой СН3;
R3 представляет собой C1-С6алкил;
R4 представляет собой C1-С6алкил;
R5 и R6 каждый независимо выбран из водорода, C1-С6алкила, моно- или дигидроксилированного C1-С6алкила, C1-С6алкокси-(C1-С6алкил), гидроксилированного C1-С6алкокси-(C1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин;
Х представляет собой NH; и
Y представляет собой S или О.
Наиболее предпочтительные соединения по настоящему изобретению представляют собой
2,3-диметил-8-[(2,4-диметил-тиофен-3-илметил)амино]имидазо[1,2-а]пиридин-6-карбоксамид или его фармацевтически приемлемую соль.
ПОЛУЧЕНИЕ
Настоящее изобретение также предлагает следующие способы получения соединений общей формулы I.
Способ А
Соединения формулы II
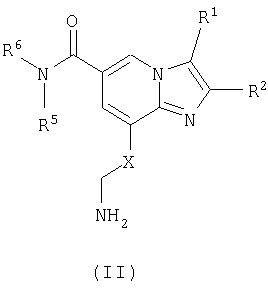
где R1, R2, R5 и R6 являются такими как определено для формулы I, могут быть подвергнуты взаимодействию с соединениями общей формулы III
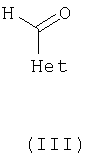
где Het определен для формулы I, в присутствии кислоты Льюиса, например хлорида цинка, с образованием соединений общей формулы IV
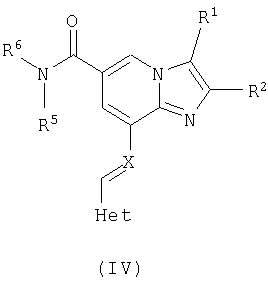
где R1, R2, R5, R6 и Het являются такими, как определено для формулы I, после чего эти соединения общей формулы IV восстанавливают, например, с помощью боргидрида натрия или цианоборгидрида натрия, с образованием соединений общей формулы I, где Х представляет собой NH. Эти реакции могут быть осуществлены в стандартных условиях в инертном растворителе, например метаноле или этаноле.
Способ Б
Соединения формулы V
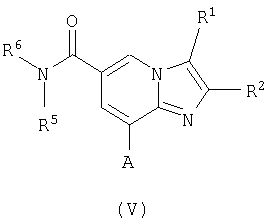
где R1, R2, R5, R6 являются такими, как определено для формулы I, a A представляет собой NH2 или ОН, могут быть подвернуты взаимодействию с соединениями формулы VI
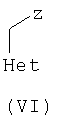
где Het является таким, как определено для формулы I, a Z представляет собой уходящую группу, такую как галогенид, тозил или мезил, с образованием соединений формулы I. Эту реакцию удобно проводить в инертном растворителе, например ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле или диметилформамиде, с основанием или без него. Основание представляет собой, например, гидроксид щелочного металла, такой как гидроксид натрия и гидроксид калия, карбонат щелочного металла, такой как карбонат калия и карбонат натрия; или органический амин, такой как триэтиламин.
Способ В
Способ получения соединений общей формулы I включает в себя следующие стадии:
а) Соединения формулы VII
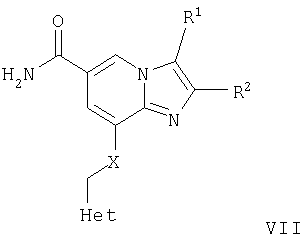
где R1, R2, Met и Х являются такими, как определено для формулы I, могут быть гидролизованы в стандартных условиях до соответствующей карбоновой кислоты формулы VIII
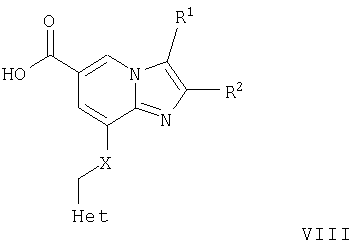
б) Соединения формулы VIII, где R1, R2, Het и Х являются такими, как определено для формулы I, могут быть подвергнуты взаимодействию с аминосоединениями формулы IX
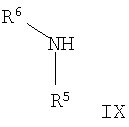
где R5 и R6 являются такими, как определено для формулы I, в присутствии агента сочетания, такого как тетрафторборат орто-бензотриазол-1-ил-N,N,N',N'-тетраметилурония (ТБТУ), с образованием соответствующих амидных соединений формулы I. Эта реакция может быть осуществлена в инертном растворители в стандартных условиях.
МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ
В еще одном аспекте данное изобретение относится к соединениям формулы I для применения в терапии, в частности, для применения против желудочно-кишечных воспалительных заболеваний. Данное изобретение также предлагает применение соединения формулы I в изготовлении лекарства для ингибирования секреции желудочной кислоты или для лечения желудочно-кишечных воспалительных заболеваний.
Соединения по данному изобретению могут, таким образом, применяться для предотвращения и лечения желудочно-кишечных воспалительных заболеваний и связанных с желудочной кислотой заболеваний, таких как гастрит, язва желудка, язва двенадцатиперстной кишки, рефлюкс-эзофагит и синдром Золлингера-Эллисона, у млекопитающих, включая человека. Кроме того, соединения могут применяться для лечения других желудочно-кишечных расстройств, при которых желательно антисекреторное действие в желудке, например у пациентов, страдающих ульцерогенной аденомой поджелудочной железы, и у пациентов с острым кровотечением в верхнем отделе желудочно-кишечного тракта. Они также могут применяться у пациентов в случае интенсивной терапии, и до- и послеоперационно для предотвращения аспирации кислоты и стрессового изъязвления.
Типичная суточная доза активного вещества варьирует в широком диапазоне и зависит от различных факторов, таких как, например, индивидуальная потребность каждого пациента, путь введения и заболевание. В общем, дозировки для перорального и парентерального введения будут находиться в диапазоне от 5 до 1000 мг активного вещества в сутки.
ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ
В еще одном своем аспекте данное изобретение относится к фармацевтическим композициям, содержащим в качестве активного ингредиента по меньшей мере одно соединение по данному изобретению или его терапевтически приемлемую соль.
Соединения по данному изобретению могут также применяться в препаратах вместе с другими активными ингредиентами, например антибиотиками, такими как амоксициллин.
Для клинического применения соединения по данному изобретению готовят в виде фармацевтических препаратов для перорального, ректального, парентерального или другого способа введения. Фармацевтический препарат содержит соединение по данному изобретению в комбинации с одним или более чем одним фармацевтически приемлемым ингредиентом. Носитель может быть в форме твердого, полутвердого или жидкого разбавителя или капсулы. Эти фармацевтические препараты представляют собой еще один объект данного изобретения. Обычно количество активных соединений составляет от 0,1 до 95% по массе препарата, предпочтительно от 0,1 до 20% по массе в препаратах для парентерального применения и предпочтительно от 0,1 до 50% по массе в препаратах для перорального введения.
При приготовлении фармацевтических препаратов, содержащих соединение по настоящему изобретению в форме единиц дозирования для перорального введения, выбранное соединение может быть смешано с твердыми порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, производные целлюлозы, желатин или другой приемлемый ингредиент, а также с разрыхлителями и смазывающими агентами, такими как стеарат магния, стеарат кальция, стеарилфумарат натрия и полиэтиленгликолевые воски. Смесь затем гранулируют и прессуют в таблетки.
Могут быть приготовлены мягкие желатиновые капсулы, причем капсулы содержат смесь активного соединения или соединений по данному изобретению, растительного масла, жира или другого подходящего наполнителя для мягких желатиновых капсул. Твердые желатиновые капсулы могут содержать гранулы активного соединения. Твердые желатиновые капсулы могут также содержать активное соединение в комбинации с твердыми порошкообразными ингредиентами, таким как лактоза, сахароза, сорбит, маннит, картофельный крахмал, кукурузный крахмал, амилопектин, производные целлюлозы или желатин.
Лекарственные формы для ректального введения могут быть приготовлены (1) в форме суппозиториев, которые содержат активное вещество, смешанное с нейтральной жировой основой; (2) в форме желатиновой капсулы для ректального введения, которая содержит активное вещество в смеси с растительным маслом, парафиновым маслом или другим подходящим наполнителем для желатиновых капсул для ректального введения; (3) в форме готовой микроклизмы; или (4) в форме сухого препарата для микроклизмы для разведения в подходящем растворителе непосредственно перед введением.
Жидкие препараты для перорального введения могут быть приготовлены в форме сиропов или суспензий, например растворов или суспензий, содержащих от 0,1% до 20% по массе активного ингредиента, причем оставшаяся часть состоит из сахара или сахарных спиртов и смеси этанола, воды, глицерина, пропиленгликоля и полиэтиленгликоля. При желании такие жидкие препараты могут содержать красители, корригенты, сахарин и карбоксиметилцеллюлозу или другой загуститель. Жидкие препараты для перорального введения могут также быть приготовлены в форме сухого порошка для разведения подходящим растворителем перед использованием.
Растворы для парентерального введения могут быть приготовлены в виде раствора соединения по данному изобретению в фармацевтически приемлемом растворителе, предпочтительно в концентрации от 0,1 до 10% по массе. Эти растворы могут также содержать стабилизирующие ингредиенты и/или забуферивающие ингредиенты, и их распределяют на унифицированные дозы в форме ампул или флаконов. Растворы для парентерального введения могут также быть получены в виде сухого препарата для разведения подходящим растворителем непосредственно перед применением.
Соединения по данному могут также применяться в препаратах совместно с другими активными ингредиентами, например для лечения или профилактики состояний, в которые вовлечено инфицирование слизистой оболочки желудка человека Helicobacter pylori. Такие другие активные ингредиенты могут представлять собой антимикробные агенты, в частности:
- β-лактамовые антибиотики, такие как амоксициллин, ампициллин, цефалотин, цефаклор или цефиксим;
- макролиды, такие как эритромицин или кларитромицин;
- тетрациклины, такие как тетрациклин или доксициклин;
- аминогликозиды, такие как гентамицин, канамицин или амикацин;
- хинолоны, такие как норфлоксацин, ципрофлоксацин или эноксацин;
- другие, такие как метронидазол, нитрофурантоин или хлорамфеникол; или
- препараты, содержащие соли висмута, такие как субцитрат висмута, субсалицилат висмута, субкарбонат висмута, субнитрат висмута или субгаллат висмута.
Соединения по данному изобретению могут также применяться в препаратах совместно с другими активными ингредиентами, например для лечения или профилактики состояний, включающих в себя язву желудка, вызванную лекарствами. Такие другие активные ингредиенты могут представлять собой нестероидные противовоспалительные средства (НСПВС), не-НСПВС, ингибитор СОХ-2 (циклооксигеназа-2) или бифосфонат.
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
Еще один аспект данного изобретения представляет собой новые промежуточные соединения, которые пригодны для синтеза соединений по данному изобретению.
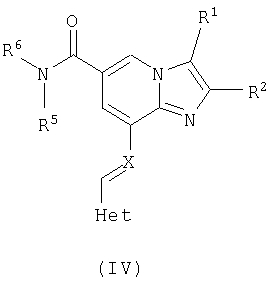
Таким образом, данное изобретение включает в себя соединение формулы (IV)
где R1, R2, R5, R6 и Het являются такими, как определено для формулы I выше.
ПРИМЕРЫ
1. Получение соединений по данному изобретению Пример 1.1
Синтез 8-{[(2,4-диметилтиен-3-ил)метил]амино}-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида
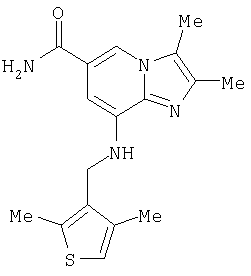
Мезилат 8-амино-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида (0,36 г, 1 ммоль), 2,4-диметилтиофен-3-карбальдегид (0,17 г, 1,2 ммоль), хлорид цинка (0,15 г, 1,1 ммоль) и цианоборгидрид натрия (0,14 г, 2,1 ммоль) добавляли к метанолу (20 мл) и смесь кипятили с обратным холодильником в течение 20 ч в атмосфере азота. После охлаждения до комнатной температуры к реакционной смеси добавляли триэтиламин (0,5 мл) и растворитель выпаривали при пониженном давлении. Очистка остатка посредством колоночной хроматографии на силикагеле с использованием в качестве элюэнта смеси метиленхлорид: метанол (10:1) дала 6 мг (2%) указанного в заголовке соединения.
1H-ЯМР (300 МГц, ДМСО-d6) 2,15 (s, 3Н), 2,25 (s, 3Н), 2,35 (s, 3H), 2,45(s, 3Н), 4,3 (d, 2H), 5,4 (t, 1H), 6,6 (s, 1H), 6.9 (s, 1H), 7,3 (bs, 1H), 7,95 (bs, 1H), 8,1 (s, 1Н).
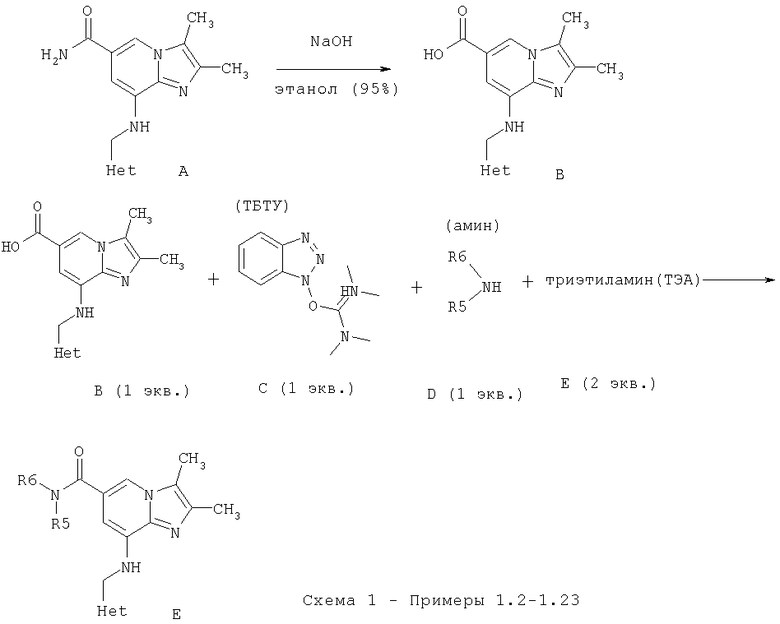
Примеры 1.2-1.23
Примеры 1.2-1.23 получали в соответствии со способом, представленным на Схеме 1 ниже
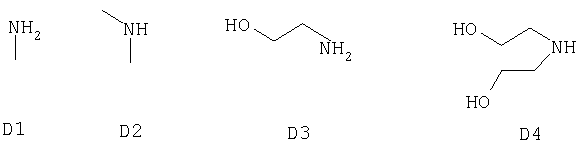
В качестве исходного соединения А в реакциях использовали следующие соединения.
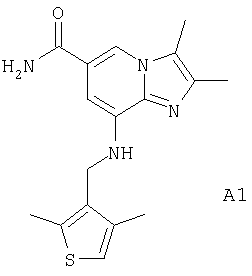 или
или 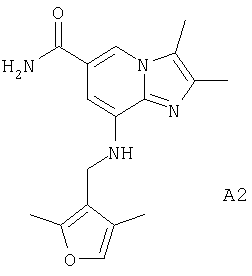
В качестве вступающего в реакцию соединения D использовали следующие соединения.
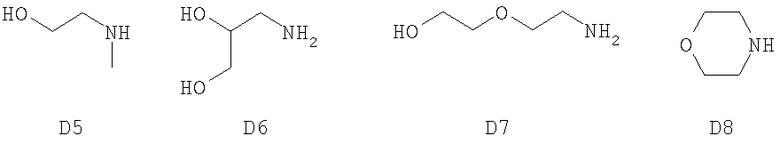
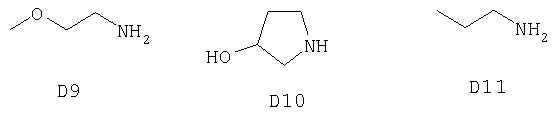
Таким образом получали следующие соединения F.
Общий способ: А (1 экв.) и гидроксид натрия (5 экв.) растворяли в этаноле (95%) и смесь кипятили с обратным холодильником в течение 24 ч. Растворитель выпаривали при пониженном давлении и к остатку добавляли воду. рН доводили путем добавления уксусной кислоты, а твердое вещество, которое выпадало в осадок, отделяли путем фильтрации, промывали водой и сушили с получением указанного в заголовке желаемого соединения (В).
В (1 экв.), С (1 экв.), D (1 экв.) и Е (1 экв.) добавляли к диметилформамиду и смесь перемешивали при к.т. в течение 24 ч. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографически с использованием силикагеля и смеси метиленхлорид/метанол в качестве элюента с получением желаемого соединения F.
Соединения согласно Примерам 1.2-1.23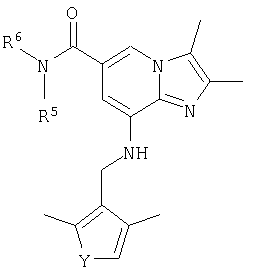
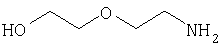
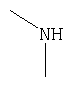

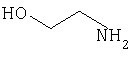

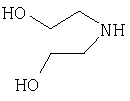
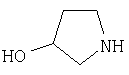
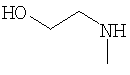

БИОЛОГИЧЕСКИЕ ТЕСТЫ
1. Эксперименты in vitro
Ингибирование секреции кислоты в изолированных желудочных железах кролика
Ингибирующее действие в отношении секреции кислоты in vitro в изолированных желудочных железах кролика измеряли в соответствии с тем, как описано в Berglindh et al. (1976) Acta Physiol. Scand. 97, 501-414.
Определение активности H+, K+-АТФазы
Мембранные везикулы (от 2,5 до 5 мкг) инкубировали в течение 15 мин при +37°С в 18 мМ буфере Pipes/Tris с рН 7,4, содержащем 2 мМ MgCl2, 10 мМ KCl и 2 мМ АТФ. Активность АТФазы оценивали по высвобождению неорганического фосфата из АТФ в соответствии с тем, как описано LeBel et al. (1978) Anal. Biochem. 85, 86-89.
2. Эксперименты in vivo
Ингибирующее действие в отношении секреции кислоты у самок крыс
Использовали самок крыс линии Sprague-Dawly. В их желудки (полость) и верхний отдел двенадцатиперстной кишки вставляли канюлированную фистулу для сбора желудочных секретов и введения тестируемых соединений, соответственно. После операции перед началом тестирования давали период восстановления, составлявший 14 дней.
Перед секреторными тестами животных лишали пищи, но давали воду в течение 20 ч. Желудок многократно промывали через желудочную канюлю водопроводной водой (+37°С) и подкожно вводили 6 мл раствора Рингер-глюкоза. Секрецию кислоты стимулировали путем инфузии в течение 2,5-4 ч пентагастрина (1,2 мл/ч, подкожно) и карбахола (20 и 110 нмоль/кг·ч, соответственно), при этом собирали желудочные секреты 30-минутными фракциями. Тестируемые вещества или носитель вводили либо через 60 мин после начала стимуляции (внутривенное и интрадуоденальное дозирование, 1 мл/кг), либо за 2 ч до начала стимуляции (пероральное дозирование, 5 мл/кг, желудочная канюля закрыта). Промежуток времени между введением дозы и стимуляцией может быть увеличен для того, чтобы исследовать продолжительность действия. Образцы желудочного сока титровали до рН 7,0 посредством NaOH, 0,1 М, и рассчитывали выход кислоты как произведение объема титранта и концентрации.
Дополнительные расчеты основаны на среднегрупповых ответах 4-6 крыс. В случае введения в процессе стимуляции выход кислоты в течение промежутков времени после введения тестируемого вещества или носителя выражают в виде дробных значений ответной реакции, принимая выход кислоты в течение 30-минутного периода, предшествующего введению, за 1,0. Процентное ингибирование рассчитывают, исходя из дробных значений ответной реакции, вызванной тестируемым соединением и носителем. В случае введения перед стимуляцией процентное ингибирование рассчитывают непосредственно по выходу кислоты, регистрируемому после тестируемого соединения и носителя.
Биологическая доступность у крыс
Используют взрослых крыс линии Sprague-Dawley. За один-три дня перед экспериментами всех крыс готовят путем канюлирования левой сонной артерии под анестезией. Крыс, используемых для экспериментов по внутривенному введению, также канюлируют в яремную вену (Popovic (1960) J. Appl. Physiol. 15, 727-728). Канюли выводят наружу в задней части шеи.
Образцы крови (0,1-0,4 г) отбирают многократно из сонной артерии с интервалами вплоть до 5,5 часов после ввода дозы. Образцы замораживают до анализа тестируемого соединения.
Биологическую доступность оценивают путем расчета соотношения между площадью под кривой концентрация кровь/плазма (ППК) после (1) интрадуоденального (и.д.) или перорального (п.о.) введения и (2) внутривенного (в.в.) введения крысе или собаке, соответственно.
Площадь под кривой зависимости концентрации в крови от времени (ППК) определяют по логарифмическому/линейному правилу трапеций и экстраполируют в бесконечность путем деления последней определенной концентрации в крови на константу скорости элиминации в терминальной фазе. Системную биологическую доступность (F%) после интрадуоденального или перорального введения рассчитывают как
F(%)=(ППК (п.о. или и.д.)/ППК (в.в.))×100.
Ингибирование секреции желудочной кислоты и биологической доступности у собак, находящихся в сознании
Используют лабрадоров или гончих собак обоих полов. Их снабжают дуоденальной фистулой для введения тестируемых соединений или носителей и канюлированной желудочной фистулой или карманом Хейденхейма для сбора желудочного секрета.
Перед секреторными тестами животных не кормят в течение приблизительно 18 ч, но неограниченно дают воду. Секрецию желудочной кислоты стимулируют путем инфузии дигидрохлорида гистамина (12 мл/ч) в течение вплоть до 6,5 ч в дозе, приводящей к приблизительно 80% индивидуального максимального секреторного ответа, и желудочный сок собирают последовательными 30-минутными фракциями. Тестируемое вещество или носитель вводят перорально, и.д. или в.в. через 1 или 1,5 ч после начала инфузии гистамина, в объеме 0,5 мл/кг массы тела. В случае перорального введения следует отметить, что тестируемое соединение вводят в секретирующий кислоту желудок собак, снабженных карманом Хейденхейма.
Кислотность образцов желудочного сока определяют путем титрования до рН 7,0 и рассчитывают выход кислоты. Выход кислоты в периоды сбора после введения тестируемого вещества или носителя выражают как дробные значения ответной реакции, принимая выход кислоты во фракции перед введением за 1,0. Процентное ингибирование рассчитывают по дробным значениям ответной реакции, вызванной тестируемым соединением и носителем.
Образцы крови для анализа концентрации тестируемого соединения в плазме крови отбирают с интервалами вплоть до 4 ч после дозирования. Плазму крови отделяют и замораживают в течение 30 мин после сбора и позже анализируют. Системную биологическую доступность (F%) после перорального или и.д. введения рассчитывают в соответствии с тем, как описано выше для крысиной модели.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ ROR-ГАММА | 2017 |
|
RU2753490C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ЗАМЕЩЕННОЙ ФЕНИЛЬНОЙ ГРУППИРОВКОЙ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКИЕ ПРЕПАРАТЫ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ ЖЕЛУДОЧНОЙ КИСЛОТЫ | 1998 |
|
RU2241000C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ (S)-1-(3-ЭТОКСИ-4-МЕТОКСИФЕНИЛ)-2- МЕТАНСУЛЬФОНИЛЭТИЛАМИНА | 2013 |
|
RU2632875C2 |
| ПРОИЗВОДНЫЕ ИМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МИКРОБИОЦИДОВ | 2008 |
|
RU2532135C2 |
| ИНГИБИРУЮЩИЕ CDK-КИНАЗЫ ПИРИМИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2002 |
|
RU2330024C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ (ВАРИАНТЫ), СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ ЖЕЛУДОЧНОЙ КИСЛОТЫ И СПОСОБ ЛЕЧЕНИЯ СОСТОЯНИЙ, В КОТОРЫЕ ВОВЛЕЧЕНО ИНФИЦИРОВАНИЕ H.PYLORI | 1999 |
|
RU2235725C2 |
| ФУНГИЦИДНЫЕ КОМПОЗИЦИИ | 2012 |
|
RU2592554C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ВАНИЛОИДНЫХ АНТАГОНИСТОВ | 2005 |
|
RU2396261C2 |
| ИНГИБИТОРЫ HCV NS3 ПРОТЕАЗЫ | 2007 |
|
RU2468029C2 |
| СОЕДИНЕНИЯ, ВОЗДЕЙСТВУЮЩИЕ НА ГЛЮКОКИНАЗУ | 2002 |
|
RU2329043C9 |
Настоящее изобретение относится к имидазопиридиновым производным формулы (I), которые являются ингибиторами протонного насоса. Описывается соединение формулы I
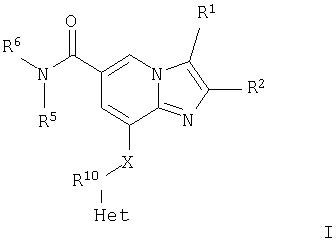
или его фармацевтически приемлемая соль, где Het представляет собой 5- или 6-членную ароматическую гетероциклическую группу, содержащую по меньшей мере один атом кислорода или серы, замещенную группой R3 и группой R4 в орто-положениях; R1 представляет собой Н, СН3 или СН2OH; R2 представляет собой СН3 или СН2СН3; R3 и R4 независимо выбраны из группы; Н, C1-С6алкил, гидроксилированный C1-С6алкил; R5 и R6 независимо выбраны из водорода, C1-С6алкила, гидроксилированного C1-С6алкила, С1-С6алкокси-(С1-С6алкил), гидроксилированного С1-С6алкокси-(С1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин; и Х представляет собой NH и R10 представляет собой СН2. Также описываются промежуточные соединения для их получения. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 2 н. и 7 з. п. ф-лы, 1 табл.
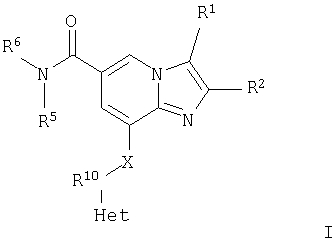
или его фармацевтически приемлемая соль,
где Het представляет собой 5- или 6-членную ароматическую гетероциклическую группу, содержащую по меньшей мере один атом кислорода или серы, замещенную группой R3 и группой R4 в ортоположениях;
R1 представляет собой Н, СН3 или СН2ОН;
R2 представляет собой СН3 или СН2СН3;
R3 и R4 независимо выбраны из группы: Н, С1-С6алкил, гидроксилированный C1-С6алкил;
R5 и R6 независимо выбраны из водорода, C1-С6алкила, гидроксилированного C1-С6алкила, C1-С6алкокси-(C1-С6алкил), гидроксилированного C1-С6алкокси-(C1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин;
Х представляет собой NH и R10 представляет собой CH2.
R1 представляет собой Н, СН3 или СН2ОН;
R2 представляет собой СН3 или СН2СН3;
R3 и R4 независимо представляют собой водород, C1-С6алкил или гидроксилированный C1-С6алкил;
R5 и R6 независимо выбраны из водорода, C1-С6алкила, гидроксилированного C1-С6алкила, C1-С6алкокси-(C1-С6алкил), гидроксилированного C1-С6алкокси-(C1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин;
Х представляет собой NH и R10 представляет собой СН2;
Y представляет собой S, SO, SO2, О.
R1 представляет собой СН3 или СН2OH;
R2 представляет собой СН3 или СН2СН3;
R3 и R4 независимо представляют собой Н, C1-С6алкил или гидроксилированный C1-С6алкил;
R5 и R6 независимо представляют собой
(а) Н,
(б) С1-С6алкил,
(в) гидроксилированный C1-С6алкил,
(г) C1-С6алкоксизамещенный C1-С6алкил,
Х представляет собой NH и R10 представляет собой СН2;
Y представляет собой S, SO, SO2, О.
R1 представляет собой СН3 или СН2OH;
R2 представляет собой СН3 или СН2СН3;
R3 и R4 независимо представляют собой водород и C1-С6алкил;
R5 и R6 независимо представляют собой Н, C1-С6алкил, гидроксилированный C1-С6алкил или C1-С6алкокси-замещенный C1-С6алкил;
Х представляет собой NH и R10 представляет собой СН2;
Y представляет собой S или О.
R1 представляет собой Н, СН3 или СН2ОН;
R2 представляет собой СН3 или СН2СН3;
R3 представляет собой C1-С6алкил;
R4 представляет собой C1-С6алкил;
R5 и R6 каждый независимо выбран из водорода, C1-С6алкила, гидроксилированного C1-С6алкила, C1-С6алкокси-(C1-С6алкил), гидроксилированного C1-С6алкокси-(C1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин;
Х представляет собой NH и R10 представляет собой СН2;
Y представляет собой S или О.
R1 представляет собой СН3;
R2 представляет собой СН3;
R3 представляет собой C1-С6алкил;
R4 представляет собой C1-С6алкил;
R5 и R6 каждый независимо выбран из водорода, С1-С6алкила, гидроксилированного C1-С6алкила, C1-С6алкокси-(C1-С6алкил), гидроксилированного С1-С6алкокси-(С1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин;
Х представляет собой NH и R10 представляет собой СН2;
Y представляет собой S или О.
где Het представляет собой 5- или 6-членную ароматическую гетероциклическую группу, содержащую по меньшей мере один атом кислорода или серы, замещенную группой R3 и группой R4 в ортоположениях;
R1 представляет собой Н, СН3 или СН3ОН;
R2 представляет собой СН3 или СН2СН3;
R3 и R4 независимо выбраны из группы: Н, С1-С6алкил, гидроксилированный C1-С6алкил;
R5 и R6 независимо выбраны из водорода, C1-С6алкила, гидроксилированного C1-С6алкила, C1-С6алкокси-(C1-С6алкил), гидроксилированного C1-С6алкокси-(С1-С6алкил), либо R5 и R6 могут вместе с атомом азота, к которому они присоединены, образовывать морфолин или гидроксилированный пирролидин.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| WO 00/11000 A2, 02.03.2000 | |||
| Прибор для отмучивания формовских песков при определении степени их коллоидальности | 1931 |
|
SU33094A1 |
| ИМИДАЗОПИРИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ | 1994 |
|
RU2136682C1 |
| АЛКОКСИАЛКИЛКАРБАМАТЫ ИМИДАЗО[1,2-A]ПИРИДИНОВ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2139288C1 |
| ИМИДАЗОПИРИДИНАЗОЛИДИНОНЫ, ИХ СОЛИ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2138501C1 |

