ТЕХНИЧЕСКАЯ ОБЛАСТЬ
Настоящее изобретение относится к новым соединениям и их фармацевтически приемлемым солям, которые ингибируют экзогенно или эндогенно стимулируемую секрецию желудочной кислоты и ввиду этого могут быть использованы для предупреждения и лечения желудочно-кишечных воспалительных заболеваний. Другие аспекты данного изобретения относятся к соединениям по изобретению для применения в терапии, к способам получения таких новых соединений, к фармацевтическим композициям, содержащим в качестве активного ингредиента по меньшей мере одно соединение по изобретению или его фармацевтически приемлемую соль, а также к использованию активных соединений в производстве лекарственных средств для указанного выше медицинского применения. Изобретение также относится к новым промежуточным соединениям для получения этих новых соединений.
ОПИСАНИЕ УРОВНЯ ТЕХНИКИ
Замещенные имидазо[1,2-a]пиридины, полезные при лечении язвы желудка, известны, например, из EP-B-0033094 и US 4450164 (Schering Corporation), из EP-B-0204285 и US 4725601 (Fujisawa Pharmaceutical Co.) и из публикаций J.J.Kaminski et al. в Journal of Medical Chemistry (vol.28, 876-892, 1985; vol.30, 2031-2046, 1987; vol.30, 2047-2051, 1987; vol.32, 1686-1700, 1989 и vol.34, 533-541, 1991).
Обзор вопросов, касающихся фармакологии желудочного кислотного насоса (Н+,К+-АТФазы), можно найти в Sachs et al. (1995) Annu. Rev. Pharmacol. Toxicol. 35: 277-305.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Неожиданно было установлено, что соединения формулы I, являющиеся производными имидазопиридина, в которых фенильная группировка является замещенной, а имидазопиридиновая группировка замещена в 6-ом положении карбоксамидной группой, являются особенно эффективными в качестве ингибиторов желудочно-кишечной Н+,К+-АТФазы и ввиду этого в качестве ингибиторов секреции желудочной кислоты. Карбоксамидная группа в 6-ом положении возможно выбрана так, чтобы давать соединения формулы I с молекулярной массой, не превышающей 600.
Таким образом, один из аспектов данного изобретения относится к производным имидазопиридина общей формулы I
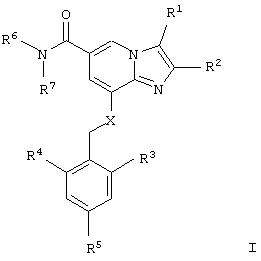
или их фармацевтически приемлемым солям,
где R1 представляет собой
Н,
CH3 или
CH2OH;
R2 представляет собой
CH3 или
CH2CH3;
R3 представляет собой
Н или
C1-C6алкил;
R4 представляет собой
Н или
C1-C6алкил;
R5 представляет собой
Н или
галоген;
R6 и R7 независимо представляют собой
Н,
C1-C6алкил,
гидроксилированный C1-C6алкил,
C1-C6алкоксизамещенный C1-C6алкил,
галогенированный C1-C6алкил,
арил, где арил представляет собой фенил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкила, C1-C6алкокси, (C1-C6алкил)2-N- или NH2SO2,
арилзамещенный C1-C6алкил, в котором арил представляет собой фенил, имидазолил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкокси или OH,
R8-(C1-C6)алкил-, в котором R8 представляет собой NH2С=O-, C1-C6 алкил-NHC=O-, (C1-C6алкил)2NC=O-, C1-C6алкил-OOC-, C1-C6алкил-CO-NH-, C1-C6алкил-OOCNH-, C1-C6алкил-O-, C7-C12алкил-O-, C1-C6алкил-SO-, C1-C6алкил-S-, C1-C6алкил-C=O-, ArCONH-, Ar(C1-C6алкил)CONH, ArC=O-, NH2CONH-, C1-C6алкил-NHCONH-, (C1-C6алкил)2-NCONH-, ArNHCONH-, гидроксилированный C1-C6алкил-O- или морфолинил; где Ar представляет собой фенил, пиридил или нафтил;
C7-C12алкил,
OH,

где R9 и R10 независимо представляют собой Н,
R11-(C1-C6)алкил-COO-(C1-C6)алкил-, где R11 представляет собой HOOC-, или
R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом, такое как морфолин, пиперазин, пирролидин, пиперидин, возможно замещенное одним или более чем одним заместителем, выбранным из галогена, C1-C6алкила, C1-C6алкокси, CF3, OH, фенила, NH2CO-, C1-C6алкил-CO-,
при условии, что по меньшей мере один из R6 и R7 не может быть Н, С1-С6алкилом, гидроксилированным C1-C6алкилом или С1-С6алкоксизамещенным С1-С6 алкилом; и
Х представляет собой NН или О.
Предпочтительными соединениями по изобретению являются такие соединения формулы I, в которых R1 представляет собой CH3 или CH2OH; R2 представляет собой CH3 или CH2CH3; R3 представляет собой CH3 или CH2CH3; R4 представляет собой CH3 или CH2CH3; R5 представляет собой H, Br, Cl или F; R6 и R7 независимо представляют собой
Н,
C1-C6алкил,
гидроксилированный C1-C6алкил,
C1-C6алкоксизамещенный C1-C6алкил,
галогенированный C1-C6алкил,
арил, где арил представляет собой фенил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкила, C1-C6алкокси, (C1-C6алкил)2-N- или NH2SO2,
арилзамещенный C1-C6алкил, в котором арил представляет собой фенил, имидазолил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкокси или OH,
R8-(C1-C6)алкил-, в котором R8 представляет собой NH2С=O-, C1-C6 алкил-NHC=O-, (C1-C6алкил)2NC=O-, C1-C6алкил-OOC-, C1-C6алкил-CO-NH-, C1-C6алкил-OOCNH-, C1-C6алкил-O-, C7-C12алкил-O-, C1-C6алкил-SO-, C1-C6алкил-S-, C1-C6алкил-C=O-, ArCONH-, Ar(C1-C6алкил)CONH, ArC=O-, NH2CONH-, C1-C6алкил-NHCONH-, (C1-C6алкил)2-NCONH-, ArNHCONH-, гидроксилированный C1-C6алкил-O- или морфолинил, где Ar представляет собой фенил, пиридил или нафтил;
C7-C12алкил,
OH,

где R9 и R10 независимо представляют собой Н,
R11-(C1-C6)алкил-COO-(C1-C6)алкил-, где R11 представляет собой HOOC-, или
R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом, такое как морфолин, пиперазин, пирролидин, пиперидин, возможно замещенное одним или более чем одним заместителем, выбранным из галогена, C1-C6алкила, C1-C6алкокси, CF3, OH, фенила, NH2CO-, C1-C6алкил-CO-,
при условии, что по меньшей мере один из R6 и R7 не может быть Н, С1-С6алкилом, гидроксилированным C1-C6алкилом или С1-С6алкоксизамещенным С1-С6 алкилом;
или их фармацевтически приемлемые соли.
Более предпочтительными соединениями по изобретению являются такие соединения формулы I, в которых R1 представляет собой CH3 или CH2OH; R2 представляет собой CH3; R3 представляет собой CH3 или CH2CH3; R4 представляет собой CH3 или CH2CH3; R5 представляет собой H, Br, Cl или F; R6 и R7 независимо представляют собой
Н,
C1-C6алкил,
гидроксилированный C1-C6алкил,
C1-C6алкоксизамещенный C1-C6алкил,
галогенированный C1-C6алкил,
арил, где арил представляет собой фенил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкила, C1-C6алкокси или (C1-C6алкил)2-N-, арилзамещенный C1-C6алкил, в котором арил представляет собой фенил, имидазолил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкокси или OH,
R8-(C1-C6)алкил-, в котором R8 представляет собой NH2С=O-, C1-C6 алкил-NHC=O-, (C1-C6алкил)2NC=O-, C1-C6алкил-OOC-, C1-C6алкил-CO-NH-, C1-C6алкил-OOCNH-, C1-C6алкил-O-, C7-C12алкил-O-, C1-C6алкил-SO-, C1-C6алкил-S-, C1-C6алкил-C=O-, ArCONH-, Ar(C1-C6алкил)CONH, ArC=O-, NH2CONH-, C1-C6алкил-NHCONH-, (C1-C6алкил)2-NCONH-, ArNHCONH-, гидроксилированный C1-C6алкил-O- или морфолинил, где Ar представляет собой фенил, пиридил или нафтил;
C7-C12алкил,
OH,
R11-(C1-C6)алкил-COO-(C1-C6)алкил-, где R11 представляет собой HOOC-, или
R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом, такое как морфолин, пиперазин, пирролидин, пиперидин, возможно замещенное одним или более чем одним заместителем, выбранным из галогена, C1-C6алкила, C1-C6алкокси, CF3, OH, фенила, NH2CO-, C1-C6алкил-CO-,
при условии, что по меньшей мере один из R6 и R7 не может быть Н, С1-С6алкилом, гидроксилированным C1-C6алкилом или С1-С6алкоксизамещенным С1-С6 алкилом.
Наиболее предпочтительные соединения по изобретению представляют собой:
- 2,3-диметил-8-(2-этил-6-метилбензиламино)-6-(морфолинокарбонил)-имидазо[1,2-a]пиридин;
- N-(4-этоксифенил)-8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-карбоксамид;
- N-[2-(диметиламин)-2-оксоэтил]-8-(2-этил-6-метилбензиламино)-N,2,3-триметил-имидазо[1,2-a]пиридин-6-карбоксамид;
- (8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридинил)(4-метилпиперазино)метанон;
- 1-((8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)-карбонил)-2-(s)-пирролидинкарбоксамид;
- 8-(2-этил-6-метилбензиламино)-N-гидрокси-2,3-диметилимидазо[1,2-a]пиридин-6-карбоксамид;
- (2-этил-6-метилбензиламино)-N-(2-(2-гидроксиэтокси)этил)-2,3-диметил-имидазо[1,2-a]пиридин-6-карбоксамид;
- (8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)(3-гидрокси-1-пирролидинил)метанон;
- N-(3,4-дигидроксифенетил)-8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-карбоксамид;
- 8-(2-этил-6-метилбензиламино-3-(гидроксиметил)-2-метил-6-(морфолино-карбонил)-имидазо[1,2-a]пиридин;
- N-((8-(2-этил-6-метилбензил)амино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)-карбонил)гуанидин;
- 4-(2-(((8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)-карбонил)амино)этокси)-4-оксобутановую кислоту.
Предпочтительными соединениями являются соединения в виде солей гидрохлорид или мезилат.
Настоящее изобретение также относится к комбинированному препарату для предупреждения или лечения желудочно-кишечных воспалительных заболеваний при одновременном, раздельном или последовательном применении, содержащему соединение по изобретению и по меньшей мере один противомикробный агент.
Настоящее изобретение также относится к способу получения соединений общей формулы I, где Х и R1-R7 такие, как определено для формулы I, заключающемуся в том, что соединение общей формулы II

где R1-R5 имеют указанные выше значения, гидролизуют в стандартных условиях с получением карбоновой кислоты общей формулы III
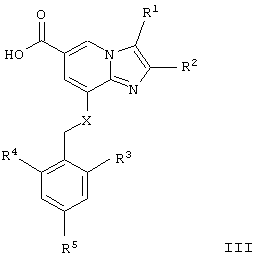
которую подвергают взаимодействию с аминосоединением формулы IV
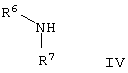
где R6 и R7 такие, как определено для формулы I, в присутствии агента сочетания в стандартных условиях в инертном растворителе.
Удобным является проведение этого взаимодействия в инертном растворителе, например ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле или диметилформамиде, в присутствии или в отсутствие основания. Основанием может быть, например, гидроксид щелочного металла, такой как гидроксид натрия и гидроксид калия, карбонат щелочного металла, такой как карбонат калия и карбонат натрия, или органический амин, такой как триэтиламин.
Согласно еще одному аспекту данное изобретение относится к фармацевтическим препаратам, обладающим ингибирующей секрецию желудочной кислоты активностью, содержащим в качестве активного ингредиента соединение по изобретению в комбинации с фармацевтически приемлемым разбавителем или носителем.
Кроме того, согласно данному изобретению предложено соединение формулы I для производства лекарства для ингибирования секреции желудочной кислоты или для лечения желудочно-кишечных воспалительных заболеваний.
Согласно еще одному аспекту, настоящее изобретение относится к способу ингибирования секреции желудочной кислоты, при котором млекопитающему, включая человека, при необходимости такого ингибирования вводят эффективное количество соединения по изобретению.
Настоящее изобретение также относится к способу лечения или профилактики состояний, в которые вовлечено инфицирование Helicobacter pylori слизистой оболочки желудка млекопитающего, включая человека, при котором указанному млекопитающему при необходимости такого лечения вводят эффективное количество соединения по изобретению в комбинации с по меньшей мере одним противомикробным агентом.
Кроме того, настоящее изобретение относится к фармацевтическому препарату для ингибирования секреции желудочной кислоты, в котором активный ингредиент представляет собой соединение по изобретению.
Данное изобретение также относится к фармацевтическому препарату для лечения или профилактики состояний, в которые вовлечено инфицирование Helicobacter pylori слизистой оболочки желудка человека, в котором активный ингредиент представляет собой соединение по изобретению, в комбинации для одновременного, раздельного или последовательного применения с по меньшей мере одним противомикробным агентом.
Еще одним аспектом настоящего изобретения являются новые промежуточные соединения, полезные для синтеза соединений по изобретению:
соединение формулы III

где R1, R2, R3, R4, R5 и Х такие, как определено для формулы I.
Используемый в данной заявке термин “С1-С6алкил” означает прямую или разветвленную алкильную группу, содержащую от 1 до 6 атомов углерода. Примеры указанного С1-С6алкила включают в себя метил, этил, н.-пропил, изопропил, н.-бутил, изобутил, втор-бутил, трет-бутил, а также пентил и гексил с прямой и разветвленной цепью.
Термин “галоген” включает в себя фтор, хлор, бром и иод.
Как чистые энантиомеры, рацемические смеси, так и неравные смеси двух энантиомеров находятся в пределах объема данного изобретения. Следует понимать, что все возможные диастереомерные формы (чистые энантиомеры, рацемические смеси и неравные смеси двух энантиомеров) находятся в пределах объема данного изобретения. Данным изобретением также охватываются производные соединений формулы I, которые обладают биологическим действием соединений формулы I, такие как пролекарства.
Кроме того, специалистам ясно, что хотя производные соединений формулы I могут не обладать фармакологической активностью сами по себе, они могут вводиться парентерально или перорально и после этого подвергаться метаболизму в организме с образованием фармакологически активных соединений по изобретению. Поэтому такие производные могут описываться как “пролекарства”. Все пролекарства соединений формулы I включены в пределы объема данного изобретения.
В зависимости от условий способа получения конечные продукты формулы I получают либо в нейтральной форме, либо в форме соли. Как свободное основание, так и соли этих конечных продуктов находятся в пределах объема данного изобретения.
Соли присоединения кислот новых соединений известным per se образом могут быть преобразованы в свободное основание либо с использованием основных реагентов, таких как щелочи, либо посредством ионного обмена. Кроме того, полученное свободное основание также может образовывать соли с органическими и неорганическими кислотами.
При получении солей присоединения кислот предпочтительно используются такие кислоты, которые образуют подходящие фармацевтически приемлемые соли. Примерами таких кислот являются галогеноводородные кислоты, такие как соляная кислота, серная кислота, фосфорная кислота, азотная кислота, алифатические, алициклические, ароматические или гетероциклические карбоновые либо сульфоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, п.-гидроксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, гидроксиэтансульфоновая кислота, галогенобензолсульфоновая кислота, толуолсульфоновая кислота или нафталинсульфоновая кислота.
Соединения по данному изобретению могут быть использованы для предупреждения и лечения у млекопитающих, включая человека, желудочно-кишечных воспалительных заболеваний и заболеваний, связанных с воздействием желудочной кислоты, таких как гастрит, язва желудка, язва двенадцатиперстной кишки, гастроэзофагеальный рефлюкс и синдром Золлинджера-Эллисона. Более того, данные соединения могут быть использованы для лечения других желудочно-кишечных расстройств, при которых желателен желудочный антисекреторный эффект, например, у пациентов с гастриномами и у пациентов с острым верхним желудочно-кишечным кровотечением. Они также могут быть использованы при интенсивной терапии пациентов и в пред- и пост-операционном периоде с целью предупреждения кислотной аспирации и образования язвы в результате стресса.
Типичная суточная доза активного вещества варьирует в широком интервале и будет зависеть от различных факторов, таких как, например, индивидуальные требования каждого пациента, путь введения и само заболевание. Обычно пероральные и парентеральные дозы активного вещества будут лежать в интервале от 5 до 1000 мг в сутки.
Кроме того, соединения по изобретению могут быть использованы в препаратах вместе с другими активными ингредиентами, например антибиотиками, такими как амоксициллин.
Для клинического применения соединения по изобретению изготавливают в виде фармацевтических препаратов для перорального, ректального, парентерального или других путей введения. Фармацевтический препарат содержит по меньшей мере одно соединение по изобретению в комбинации с одним или более чем одним фармацевтически приемлемым ингредиентом. Носитель может быть в виде твердого, полутвердого или жидкого разбавителя или в виде капсулы. Эти фармацевтические препараты являются еще одним объектом данного изобретения. Обычно количество активных соединений составляет 0,1-95% от массы препарата, предпочтительно 0,1-20% от массы препарата для парентерального применения и предпочтительно между 0,1 и 50 мас.% в препаратах для перорального введения.
При приготовлении фармацевтических препаратов, содержащих соединение по настоящему изобретению, в форме единиц дозирования для перорального введения выбранное соединение может быть смешано с твердыми, порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, производные целлюлозы, желатин или другой подходящий ингредиент, равно как и с разрыхлителями и смазывающими агентами, такими как магния стеарат, кальция стеарат, натрия стеарилфумарат и полиэтиленгликолевые воска. Затем смесь оформляют в виде гранул или прессуют в таблетки.
Мягкие желатиновые капсулы могут быть получены в виде капсул, содержащих смесь активного соединения или соединений по изобретению, растительного масла, жира или другого подходящего наполнителя для мягких желатиновых капсул. Твердые желатиновые капсулы могут содержать гранулы активного соединения. Кроме того, твердые желатиновые капсулы могут содержать активное соединение в комбинации с твердыми порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, картофельный крахмал, кукурузный крахмал, амилопектин, производные целлюлозы или желатин.
Единицы дозирования для ректального введения могут быть получены (1) в форме суппозиториев, которые содержат активное вещество, смешанное с нейтральной жировой основой; (2) в форме желатиновой ректальной капсулы, которая содержит активное вещество в смеси с растительным маслом, жидким парафином или другим подходящим наполнителем для желатиновых ректальных капсул; (3) в форме готовой к употреблению микроклизмы; или (4) в форме сухого препарата для микроклизмы, который следует перерастворить в подходящем растворителе прямо перед введением.
Жидкие препараты для перорального введения могут быть приготовлены в виде сиропов или суспензий, например растворов или суспензий, содержащих от 0,1 до 20 мас.% активного ингредиента и остаток, состоящий из сахара или сахарных спиртов и смеси этанола, воды, глицерина, пропиленгликоля и полиэтиленгликоля. Если желательно, такие жидкие препараты могут содержать красители, корригенты, сахарин и карбоксиметилцеллюлозу или другой загуститель. Кроме того, жидкие препараты для перорального введения могут быть приготовлены в виде сухого порошка, который следует перерастворить в подходящем растворителе перед использованием.
Растворы для парентерального введения могут быть получены в виде раствора соединения по изобретению в фармацевтически приемлемом растворителе, предпочтительно в концентрации от 0,1 до 10 мас.%. Кроме того, эти растворы могут содержать стабилизирующие ингредиенты и/или забуферивающие ингредиенты, и они могут быть представлены в стандартных дозах в виде ампул или флаконов. Растворы для парентерального введения также могут быть приготовлены в виде сухого препарата, который следует перерастворить в подходящем растворителе непосредственно перед использованием.
Кроме того, соединения по настоящему изобретению могут быть использованы для одновременного, раздельного или последовательного применения в препаратах в комбинации с другими активными ингредиентами, например, для лечения или профилактики состояний, включающих в себя инфицирование Helicobacter pylori слизистой оболочки желудка человека. Этими другими активными ингредиентами могут являться противомикробные агенты, в частности:
- β-лактамные антибиотики, такие как амоксициллин, ампициллин, цефалотин, цефаклор или цефиксим;
- макролиды, такие как эритромицин или кларитромицин;
- тетрациклины, такие как тетрациклин или доксициклин;
- аминогликозиды, такие как гентамицин, канамицин или амикацин;
- хинолоны, такие как норфлоксацин, ципрофлоксацин или эноксацин;
- другие агенты, такие как метронидазол, нитрофурантоин или хлорамфеникол; либо
- препараты, содержащие соли висмута, такие как висмута цитрат основной, висмута салицилат основной, висмута карбонат основной, висмута нитрат основной или висмута галлат основной.
Кроме того, соединения по настоящему изобретению могут быть использованы в комбинации для одновременного, раздельного или последовательного применения с антацидами, такими как алюминия гидроксид, магния карбонат и магния гидроксид или альгиновая кислота, либо совместно или в комбинации для одновременного, раздельного или последовательного применения с фармацевтическими средствами, которые ингибируют секрецию кислоты, такими как Н2-блокаторы (например, циметидин, ранитидин), ингибиторы Н+/К+-АТФазы (например, омепразол, пантопразол, лансопразол или рабепразол), либо совместно или в комбинации для одновременного, раздельного или последовательного применения с гастропрокинетиками (например цисаприд или мосаприд).
ПРИМЕРЫ
1. ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ
Пример 1.1
Синтез 2,3-диметил-8-(2-этил-6-метилбензиламино)-6-(морфолинокарбонил)-имидазо[1,2-а]пиридина

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,15 г; 0,44 ммоль) и о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,14 г; 0,44 ммоль) добавляют к метиленхлориду (10 мл). Добавляют морфолин (0,12 г; 1,4 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 1,5 ч. Реакционную смесь наносят на колонку с силикагелем и очищают хроматографией, используя в качестве элюента смесь этилацетат:метиленхлорид (1:1) и получая 0,12 г (66%) желаемого продукта.
1Н-ЯМР (300 МГц; CDCl3): δ 1.2 (t, 3H), 2.32 (s, 3H), 2.35 (s, 3H), 2.37 (s, 3H), 2.7 (q, 2H), 3.7 (s, 8H), 4.35 (d, 2H), 4.95 (bs, 1H), 6.15 (s, 1H), 7.0-7.2 (m, 3H), 7.4 (s, 1H).
Пример 1.2
Синтез N-(4-этоксифенил)-8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,15 г; 0,44 ммоль) и о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,14 г; 0,44 ммоль) добавляют к метиленхлориду (10 мл). Добавляют 4-этоксианилин (0,19 г; 1,4 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 72 ч. Растворитель упаривают при пониженном давлении, остаток наносят на колонку с силикагелем и очищают хроматографией, используя в качестве элюента смесь метиленхлорид:метанол (95:5). Остаток обрабатывают горячей смесью гексан:этилацетат (2:1), продукт отфильтровывают и высушивают, получая 0,14 г (74%) желаемого соединения в виде белых кристаллов.
1Н-ЯМР (300 МГц; CDCl3): δ 1.2 (t, 3H), 1.4 (t, 3H), 2.35 (s, 9H), 2.65 (q, 2H), 4.0 (q, 2H), 4.35 (d, 2H), 4.9 (t, 1H), 6.55 (s, 1H), 6.85 (d, 2H), 7.0-7.2 (m, 3H), 7.5 (d, 2H), 7.9 (s, 1H), 8.15 (s, 1H).
Пример 1.3
Синтез N-[2-(диметиламин)-2-оксоэтил]-8-(2-этил-6-метилбензиламино)-N,2,3-триметилимидазо[1,2-а]пиридин-6-карбоксамида
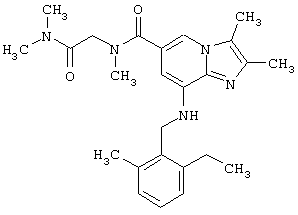
2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,13 г; 0,38 ммоль) и о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,12 г; 0,38 ммоль) добавляют к метиленхлориду (10 мл). Добавляют N,N-диметил-2-метиламино-ацетамид (0,088 г; 0,38 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. Растворитель упаривают при пониженном давлении, остаток очищают колоночной хроматографией, используя в качестве элюента смесь метиленхлорид:метанол (95:5), которая дает 80 мг (48%) указанного в заголовке продукта.
1Н-ЯМР (500 МГц; CDCl3): δ 1.2 (t, 3H), 2.3 (s, 6H), 2.35 (s, 3H), 2.65 (q, 2H), 2.75 (s, 6H), 2.95 (s, 3H), 3.15 (s, 2H), 4.35 (bs, 2H), 4.85 (bs, 1H), 6.25 (s, 1H), 7.0-7.2 (m, 3H), 7.45 (s, 1H).
Пример 1.4
Синтез (8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-ил)(4-метилпиперазин)метанона

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,5 г; 1,48 ммоль) и о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,48 г; 1,5 ммоль) добавляют к метиленхлориду (20 мл) и смесь перемешивают в течение 5 мин. Добавляют N-метилпиперазин (0,16 г; 1,6 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Растворитель упаривают при пониженном давлении, и очистка остатка колоночной хроматографией на силикагеле с использованием в качестве элюента смеси метиленхлорид:метанол (9:1) дает 0,46 г (74%) указанного в заголовке соединения.
1Н-ЯМР (500 МГц; CDCl3): δ 1.22 (t, 3H), 2.34 (s, 3H), 2.36 (s, 3H), 2.38 (s, 3H), 2.47 (bs, 4H), 2.71 (q, 2H), 2.80 (s, 3H), 3.65 (bs, 4H), 4.36 (d, 2H), 4.94 (t, 1H), 6.19 (s, 1H), 7.04-7.18 (m, 3H), 7.42 (s, 1H).
Пример 1.5
Синтез 1-((8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-ил)карбонил)-2-(s)-пирролидинкарбоксамида

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,15 г; 0,44 ммоль), о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,14 г; 0,45 ммоль) и триэтиламин (0,05 г; 0,5 ммоль) добавляют к метиленхлориду (10 мл) и смесь перемешивают в течение 10 мин. Добавляют (S)-пролинамид (0,016 г; 0,45 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. Растворитель упаривают при пониженном давлении, очистка остатка колоночной хроматографией на силикагеле с использованием в качестве элюента смеси метиленхлорид: метанол (9:1) и кристаллизация из диэтилового эфира дают 0,07 г (36%) указанного в заголовке соединения.
1Н-ЯМР (500 МГц; CDCl3): δ 1.21 (t, 3H), 2.1-2.2 (m, 4H), 2.33 (s, 3H), 2.35 (s, 3H), 2.37 (s, 3H), 2.70 (q, 2H), 3.65-3.75 (m, 2H), 4.36 (d, 2H), 4.80 (bs, 1H), 4.94 (bs, 1H), 5.88 (s, 1H), 6.33 (s, 1H), 6.98 (s, 1H), 7.04-7.19 (m, 3H), 7.54 (s, 1H).
Пример 1.6
Синтез 8-(2-этил-6-метилбензиламино)-N-гидрокси-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида
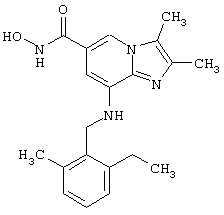
2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновая кислота (0,15 г; 0,45 ммоль), о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,14 г; 0,45 ммоль), триэтиламин (0,1 г; 0,99 ммоль) и гидроксиламина гидрохлорид (0,031 г; 0,46 ммоль) в диметилформамиде (5 мл).
Указанное в заголовке соединение получают в соответствии с примером 1.5 (выход: 0,016 г; 10%).
1Н-ЯМР (500 МГц; CDCl3): δ 1.15 (bs, 3H), 2.25 (bs, 9H), 2.6 (bs, 2H), 4.25 (bs, 2H), 4.95 (bs, 1H), 6.45 (bs, 1H), 6.9-7.1 (m, 3H), 7.75 (bs, 1H).
Пример 1.7
Синтез (2-этил-6-метилбензиламино)-N-(2-(2-гидроксиэтокси)этил)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновая кислота (0,3 г; 0,88 ммоль), о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,29 г; 0,90 ммоль) и 2-(2-аминоэтокси)этанол (0,2 г; 1,9 ммоль) в метиленхлориде (10 мл).
Указанное в заголовке соединение получают в соответствии с примером 1.5 (выход: 0,24 г; 80%).
1Н-ЯМР (500 МГц; CDCl3): δ 1.25 (t, 3H), 2.25 (s, 3H), 2.3 (s, 3H), 2.35 (s, 3H), 2.75 (q, 2H), 3.4-3.45 (m, 2H), 3.55-3.7 (m, 6H), 4.35 (d, 2H), 5.05 (t, 1H), 6.45 (s, 1H), 7.0-7.2 (m, 4H), 7.5 (s, 1H).
Пример 1.8
Синтез (8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-ил)(3-гидрокси-1-пирролидинил)метанона

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновая кислота (0,15 г; 0,44 ммоль), о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,14 г; 0,44 ммоль) и 3-пирролидинол (0,12 г; 1,4 ммоль) в метиленхлориде (10 мл).
Указанное в заголовке соединение получают в соответствии с примером 1.4. Кристаллизация из смеси этилацетат:гексан (2:1) (выход: 0,24 г; 80%).
1Н-ЯМР (300 МГц; CDCl3): δ 1.23 (t, 3H),1.93 (bs, 2H), 2.33 (s, 3H), 2.34 (s, 3H), 2.41 (s, 3H), 2.70 (q, 2H), 3.51-3.89 (m, 4H), 4,35 (d, 2H), 4.38-4.55 (m, 1H), 5 .04 (bs, 1H), 6.35 (s, 1H), 7.01-7.16 (m, 3H), 7.51 (s, 1H).
Пример 1.9
Синтез N-(3,4-дигидроксифенетил)-8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,15 г; 0,44 ммоль) и о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,14 г; 0,45 ммоль) добавляют к диметилформамиду (10 мл) и смесь перемешивают в течение 5 мин. Добавляют 3,4-дигидроксифенетиламин (0,27 г; 1,4 ммоль) и триэтиламин (0,28 г; 1,4 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 72 ч. Растворитель упаривают при пониженном давлении, очистка остатка колоночной хроматографией на силикагеле с использованием в качестве элюента смеси метиленхлорид:метанол (9:1) и кристаллизация из ацетонитрила дают 0,059 г (28%) указанного в заголовке соединения.
1Н-ЯМР (400 МГц; ДМСО-d6): δ 1.15 (t, 1H), 2.22 (s, 3H), 2.33 (s, 3H), 2.37 (s, 3H), 2.65-2.74 (m, 4H), 3.41 (q, 2H), 4.37 (d, 2H), 4.85 (t, 1H), 6.48 (dd, 1H), 6.63-6.66 (m, 2H), 6.70 (d, 1H), 7.07-7.21 (m, 3H), 8.04 (d, 1H), 8.49 (t, 1H), 8.63 (s, 1H), 8.75 (s, 1H).
Пример 1.10
Синтез (8-(2-этил-6-метилбензиламино)-3-(гидроксиметил)-2-метил-6-(морфолинокарбонил)-имидазо[1,2-а]пиридина

8-(2-Этил-6-метилбензиламино)-3-гидроксиметил-2-метилимидазо[1,2-а]пиридин-6-карбоновая кислота (0,012 г; 0,034 ммоль), о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,011 г; 0,034 ммоль) и морфолин (0,009 г; 0,1 ммоль) в метиленхлориде (1 мл).
Указанное в заголовке соединение получают в соответствии с примером 1.1 (выход: 0,008 г; 56%).
1Н-ЯМР (300 МГц; ДМСО-d6): δ 1.23 (t, 3H), 2.33 (s, 3H), 2.39 (s, 3H), 2.72 (q, 2H), 3.74 (bs, 8H), 4.37 (d, 2H), 4.85 (s, 2H), 5.02 (t, 1H), 6.27 (d, 1H), 7.06-7.22 (m, 3H), 7.75 (d, 1H).
Пример 1.86
Синтез N-((8-(2-этил-6-метилбензил)амино)-2,3-диметилимидазо[1,2-а]пиридин-6-ил)карбонил)гуанидина

2,3-Диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,2-а]пиридин-6-карбоновую кислоту (0,5 г; 1,5 ммоль), диизопропилэтиламин (0,57 г; 1,5 ммоль) и гуанидина карбонат (0,53 г; 2,9 ммоль) добавляют к диметилформамиду (10 мл). Добавляют о-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийтетрафторборат (TBTU) (0,48 г; 1,5 ммоль) и реакционную смесь перемешивают при 50оС в течение 3 ч. Растворитель упаривают при пониженном давлении, очистка остатка колоночной хроматографией на силикагеле с использованием в качестве элюента смеси метиленхлорид: метанол (100:15) и кристаллизация из диэтилового эфира дает 0,12 г (21%) указанного в заголовке соединения.
1Н-ЯМР (500 МГц; CDCl3): δ 1.1 (t, 3H), 2.25 (s, 3H), 2.3 (s, 3H), 2.35 (s, 3H), 2.7 (q, 2H), 4.35 (d, 2H), 4.8 (bs, 1H), 6.9 (s, 1H), 7.05-7.2 (m, 3H), 8.25 (s, 1H).
Пример 1.87
Синтез 4-(2-(((8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-ил)карбонил)амино)этокси)-4-оксобутановой кислоты

2,3-Диметил-8-(2-этил-6-метилбензиламино)-N-гидроксиэтилимидазо[1,2-а]пиридин-6-карбоксамид (250 мг; 0,263 ммоль) и янтарный ангидрид (100 мг; 1,00 ммоль) добавляют к 7 мл ацетона. Смесь кипятят с обратным холодильником в течение 48 ч. Выпавший в осадок продукт отфильтровывают и промывают ацетоном и эфиром, получая 288 мг (91%) указанного в заголовке соединения.
1Н-ЯМР (500 МГц; ДМСО): δ 1.16 (t, 3H), 2.24 (s, 3H), 2.35 (s, 3H), 2.39 (s, 3H), 2.48-2.58 (m, 4H), 2.70 (q, 2H), 3.54 (q, 2H), 4.19 (t, 2H), 4,39 (d, 2H), 4.90 (t, 1H), 6.72 (s, 1H), 7.09-7.22 (m, 3H), 8.08 (s, 1H), 8.59 (t, 1H), 12,25 (s, 1H).
Соединения из примеров 11-85 получают в параллельных синтезах, используя приведенный в схеме 1 способ.

Раствор А: 0,149 ммоль в 1 мл диметилформамида.
Раствор B (TBTU): 0,297 ммоль в 1 мл диметилформамида.
Раствор C+D: Амин (C) (0,297 ммоль в 1 мл диметилформамида) + TEA (D) (0,594 ммоль в 1 мл диметиламина).
К раствору А (300 мкл) добавляют раствор B (150 мкл) и раствор C + D (150 мкл). Реакционную смесь перемешивают, встряхивая при комнатной температуре в течение ночи. Растворитель упаривают при пониженном давлении. Остаток растворяют в смеси дихлорметан/метанол (9/1) (600 мкл) и фильтруют через набивку с силикагелем (100 г); гель промывают смесью дихлорметан/метанол (9/1) (0,5-1,0 мл). Фильтрат упаривают при пониженном давлении, получая желаемые соединения. (При необходимости соединения очищают с помощью препаративной ВЭЖХ).
Анализ соединений из примеров осуществляют с помощью ВЭЖХ и соединения идентифицируют с помощью ЖХ-масс-спектрометрии. Все соединения, полученные в примерах 11-85, демонстрируют масс-спектры, подтверждающие предложенную структуру.
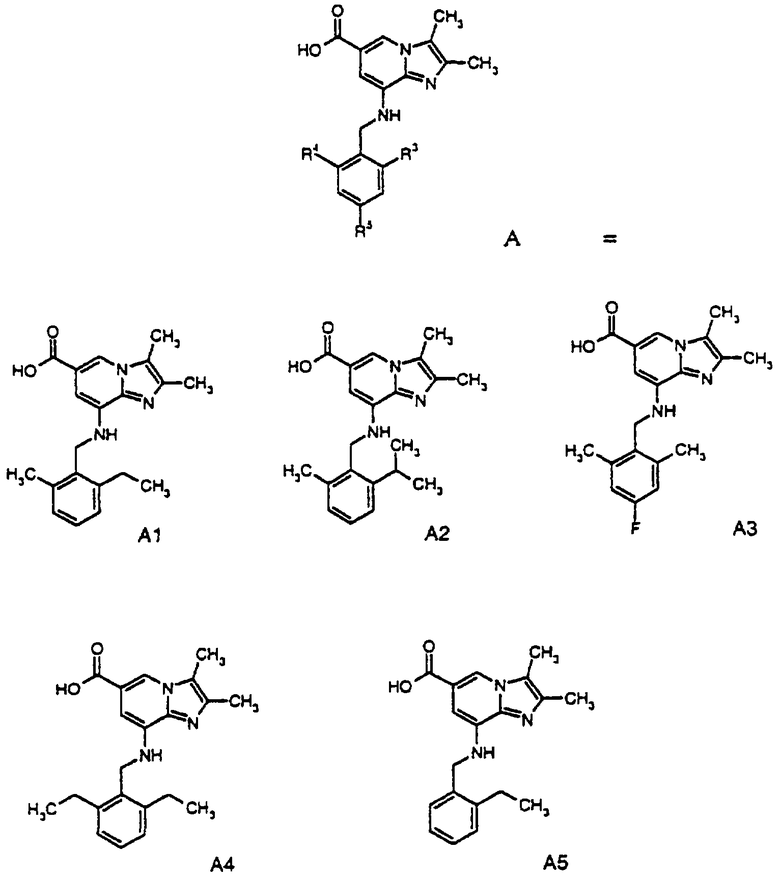
В качестве исходного соединения А в реакциях используют следующие соединения:
В качестве исходного соединения C в реакции используют следующие амины:

Соединения из примеров 11-85 получают в соответствии со схемой 1.
Первичный или вторичный аминный азот представляет собой азот, вовлеченный во взаимодействие.
Например, А1+C5→пример 27


2. ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ
Пример 2.1
Синтез 8-(2-этилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоновой кислоты
8-(2-Этилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамид (1,0 г; 0,0031 моль) и гидроксид натрия (1,2 г; 0,031 моль) растворяют в этаноле (95%-нsq; 30 мл) и кипятят с обратным холодильником в течение ночи. Растворитель упаривают при пониженном давлении и к остатку добавляют воду. рН подводят до 7, добавляя концентрированную HCl (2,6 мл), и выпавшие в осадок твердые частицы отделяют фильтрованием, промывают водой и высушивают, получая 1,0 г (99%) указанного в заголовке соединения.
1Н-ЯМР (300 МГц; ДМСО-d6): δ 1.2 (t, 3H), 2.25 (s, 3H), 2.35 (s, 3H), 2.7 (q, 2H), 4.45 (d, 2H), 6.3 (s, 1H), 6.45 (t, 1H), 7.05-7.25 (m, 4H), 7.95 (s, 1H).
Пример 2.2
Синтез 8-(2,6-диэтилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоновой кислоты
8-(2,6-Диэтилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамид (1,5 г; 0,0043 моль) и гидроксид натрия (1,7 г; 0,043 моль) растворяют в этаноле (95%-ный; 30 мл).
Указанное в заголовке соединение получают в соответствии с примером 1.4 (выход: 1,5 г; 99%).
1Н-ЯМР (400 МГц; ДМСО-d6): δ 1.14 (t, 6H), 2.22 (s, 3H), 2.37 (s, 3H), 2.67 (q, 4H), 4.37 (d, 2H), 4.89 (t, 1H), 6.68 (s, 1H), 7.11 (d, 2H), 7.23 (t, 1H), 8.09 (s, 1H).
Пример 2.3
Синтез 8-(2,6-диметил-4-фторбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоновой кислоты
8-(2,6-Диметил-4-фторбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида мезилат (1,47 г; 0,0034 моль) и гидроксид натрия (1,7 г; 0,034 моль) растворяют в этаноле (95%-ный; 30 мл).
Указанное в заголовке соединение получают в соответствии с примером 2.1 (выход: 1,1 г; 95%).
1Н-ЯМР (400 МГц; ДМСО-d6): δ 2.23 (s, 3H), 2.34 (s, 6H), 2.36 (s, 3H), 4.31 (d, 2H), 5.04 (bs, 1H), 6.70 (s, 1H), 6.90 (d, 2H), 8.02 (s, 1H).
Пример 2.4
Синтез 8-(2-изопропил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоновой кислоты
8-(2-Изопропил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида мезилат (1,2 г; 0,0027 моль) и гидроксид натрия (1,1 г; 0,027 моль) растворяют в этаноле (95%-ный; 25 мл).
Указанное в заголовке соединение получают в соответствии с примером 2.1 (выход: 1,1 г; 95%).
1Н-ЯМР (300 МГц; ДМСО-d6): δ 1.69 (d, 6H), 2.74 (s, 3H), 2.85 (s, 3H), 2.89 (s, 3H), 3.73 (m, 1H), 4.90 (d, 2H), 5.48 (t, 1H), 7.19 (s, 1H), 7.55-7.61 (m, 1H), 7.70-7.76 (m, 2H), 8.60 (s, 1H).
Пример 2.5
Синтез 8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоновой кислоты
8-(2-Этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-а]пиридин-6-карбоксамида мезилат (11,0 г; 0,025 моль) и гидроксид натрия (7,0 г; 0,17 моль) растворяют в этаноле (95%-ный; 120 мл) и кипятят с обратным холодильником в течение 20 ч. Растворитель упаривают при пониженном давлении и к остатку добавляют воду (150 мл). рН подводят до 5, добавляя концентрированную HCl и уксусную кислоту, и выпавшие в осадок твердые частицы отделяют фильтрованием, промывают водой и ацетоном и высушивают, получая 7,6 г (88%) указанного в заголовке соединения.
1Н-ЯМР (500 МГц; ДМСО-d6): δ 1.15 (t, 3H), 2.26 (s, 3H), 2.34 (s, 3H), 2.39 (s, 3H), 2.69 (q, 2H), 4.38 (d, 2H), 5.2 (bs, 1H), 6.73 (s, 1H), 7.07-7.2 (m, 3H), 8.12 (s, 1H).
Пример 2.6
Синтез 8-(2-этил-6-метилбензиламино)-3-гидроксиметил-2-метилимидазо[1,2-а]пиридин-6-карбоновой кислоты
8-(2-Этил-6-метилбензиламино)-3-гидроксиметил-2-метилимидазо[1,2-а]пиридин-6-карбоксамид (0,02 г; 0,057 ммоль) и гидроксид натрия (0,02 г; 0,29 ммоль) растворяют в этаноле (95%-ный; 1 мл) и кипятят с обратным холодильником в течение 20 ч. Растворитель упаривают при пониженном давлении и к остатку добавляют воду (1 мл). рН подводят до 5, добавляя уксусную кислоту, и выпавшие в осадок твердые частицы отделяют фильтрованием, промывают водой и высушивают, получая 0,012 г (60%) указанного в заголовке соединения.
1Н-ЯМР (300 МГц; ДМСО-d6): δ 1.14 (t, 3H), 2.22 (s, 3H), 2.33 (s, 3H), 2.67 (q, 2H), 4.33 (d, 2H), 4.55 (bs, 1H), 4.67 (s, 2H), 6.83 (s, 1H), 7.06-7.24 (m, 3H), 8.15 (s, 1H).
БИОЛОГИЧЕСКИЕ ТЕСТЫ
1. Эксперименты in vitro
Ингибирование секреции кислоты в изолированных желудочных железах кролика
Ингибирующее действие на секрецию кислоты in vitro в изолированных желудочных железах кролика измеряют, как описано Berglindh et. al. (1976) Acta Physiol. Scand. 97, 401-414.
Определение активности Н+,К+-АТФазы
Мембранные везикулы (от 2,5 до 5 мкг) инкубируют в течение 15 мин при +37оС в 18 мМ Pipes/Трис-буфере рН 7,4, содержащем 2 мМ MgCl2, 10 мМ КСl и 2 мМ АТФ. Активность АТФазы оценивают по высвобождению неорганического фосфата из АТФ, как описано в работе LeBel et al. (1978) Anal. Biochem. 85, 86-89.
2. Эксперименты in vivo
Ингибирующее действие на секрецию кислоты у самок крыс
Используют самок крыс линии Sprague-Dawly. Для сбора желудочных секретов и введения тестируемых веществ им вводят канюлированную фистулу в желудок (просвет) и верхнюю часть двенадцатиперстной кишки соответственно. Восстановительный период после операции перед началом тестирования составляет 14 дней.
Перед проведением тестов на секрецию животных на 20 ч лишают пищи, но не воды. Желудок неоднократно промывают через желудочную канюлю водопроводной водой (+37 оС) и подкожно вводят 6 мл глюкозы Рингера (Ringer-Glucose). Кислотную секрецию стимулируют инфузией в течение 2,5-4 ч (1,2 мл/ч, подкожно) пентагастрина и карбахола (20 и 110 нмоль/кг·ч, соответственно) и в продолжение этого периода времени отбирают желудочные секреты в виде 30-минутных фракций. Тестируемые вещества или наполнитель дают или через 60 мин после начала стимулирования (внутривенное и интрадуоденальное дозирование, 1 мл/кг), или за 2 ч до начала стимулирования (пероральное дозирование, 5 мл/кг, желудочная канюля закрыта). Временной интервал между дозированием и стимулированием может быть увеличен с целью изучения продолжительности действия. Образцы желудочного сока титруют до рН 7.0 с помощью 0,1 М NaOH и выход кислоты рассчитывают как произведение объема титранта и его концентрации.
Дальнейшие расчеты основываются на средних групповых ответах 4-6 крыс. В том случае, когда введение осуществляют во время стимулирования, выход кислоты во время периодов после введения тестируемого вещества или наполнителя выражают в виде фракционных ответов, принимая выход кислоты в 30-минутный период, предшествующий введению, равным 1,0. Процент ингибирования рассчитывают из фракционных ответов, вызываемых тестируемым соединением и наполнителем. В том случае, когда введение осуществляют до стимулирования, процент ингибирования рассчитывают непосредственно из выхода кислоты, зарегистрированного после введения тестируемого соединения и наполнителя.
Биодоступность у крыс
Используют взрослых крыс линии Sprague-Dawley. За один-три дня до экспериментов всем крысам под анестезией вводят канюли в левую сонную артерию. Крысам, используемым для внутривенных экспериментов, также вводят канюли в шейную вену (Popovic (1960) J. Appl. Physiol. 15, 727-728). Канюли выводят наружу на затылке.
Образцы крови (0,1-0,4 г) неоднократно через интервалы времени отбирают из сонной артерии в течение 5,5 часов после введения дозы. Образцы замораживают до анализа тестируемого соединения.
Биодоступность оценивают путем подсчета соотношения между площадями под кривыми концентрации в крови/плазме (AUC) после (1) интрадуоденального (и.д.) или перорального (п.о.) введения и (2) внутривенного (в.в.) введения для крысы или собаки соответственно.
Площадь под кривой зависимости концентрации от времени, AUC, определяют по логарифмической/линейной формуле трапеций и экстраполируют к бесконечности в результате деления последней концентрации, определенной в крови, на константу скорости элиминирования в заключительной фазе. Системную биодоступность (F%) после интрадуоденального и перорального введения рассчитывают как F(%)=(AUC (п.о. или и.д.)/AUC (в.в.))×100.
Ингибирование секреции желудочной кислоты и биодоступность у находящейся в сознании собаки
Используют охотничьих собак лабрадор и гончих собак обоих полов. Им вводят дуоденальные фистулы, необходимые для введения тестируемых соединений или наполнителя, и канюлированные желудочные фистулы, либо мешки Хейденхейма (Heidenhaim-pouch) для сбора желудочного секрета.
Перед проведением тестов на секрецию животные голодают в течение приблизительно 18 ч при свободном доступе к воде. Желудочную кислотную секрецию стимулируют инфузией гистамина гидрохлорида (12 мл/ч) (вплоть до 6,5 ч) в дозе, вызывающей приблизительно 80% индивидуального максимального секреторного ответа, и желудочный сок собирают в виде последовательных 30-минутных фракций. Вещество или наполнитель дают перорально, и.д. или в.в. через 1 или 1,5 ч после начала гистаминовой инфузии в объеме 0,5 мл/кг массы тела. Следует отметить, что в случае перорального введения тестируемое соединение вводят в секретирующий кислоту главный желудок собаки с мешком Хейденхейма (Heidenhaim-pouch).
Кислотность образцов желудочного сока определяют титрованием до рН 7,0 и производят расчет выхода кислоты. Выход кислоты в периоды сбора после введения тестируемого соединения или наполнителя выражают в виде фракционных ответов, принимая выход кислоты во фракции, предшествующей введению, за 1,0. Процент ингибирования рассчитывают исходя из фракционных ответов, вызванных тестируемым соединением и наполнителем.
Образцы крови для анализа концентрации тестируемого соединения в плазме отбирают с интервалами во времени вплоть до 4 ч после дозирования. Плазму отделяют и замораживают в течение 30 мин после сбора и позже анализируют. Системную биодоступность (F%) после перорального или и.д. введения рассчитывают как описано выше для модели на крысах.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО ИМИДАЗОПИРИДИНА | 2001 |
|
RU2275372C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-A]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2725147C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-А]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ, СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ ЖЕЛУДОЧНОЙ КИСЛОТЫ, СПОСОБ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И СПОСОБ ЛЕЧЕНИЯ СОСТОЯНИЙ, В КОТОРЫЕ ВОВЛЕЧЕНО ИНФИЦИРОВАНИЕ H.PYLORI | 1998 |
|
RU2193036C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ, АССОЦИИРОВАННЫХ С АКТИВНОСТЬЮ NLRP | 2019 |
|
RU2795108C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗО[1,2-b]ПИРИДАЗИНЫ, ЗАМЕЩЕННЫЕ ИМИДАЗО[1,5-b]ПИРИДАЗИНЫ, РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2017 |
|
RU2773333C2 |
| ПРОИЗВОДНОЕ ИМИДАЗОПИРИДИНА, ИСПОЛЬЗУЕМОЕ ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА | 2013 |
|
RU2648242C2 |
| ИНГИБИТОРЫ ГИСТОНДЕАЦЕТИЛАЗЫ | 2014 |
|
RU2673819C2 |
| СОЕДИНЕННЫЕ МОСТИКОВОЙ СВЯЗЬЮ N-ЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДО-ИНГИБИТОРЫ ГАММА-СЕКРЕТАЗЫ | 2006 |
|
RU2422443C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2550352C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА КОЛОНИЕСТИМУЛИРУЮЩЕГО ФАКТОРА-1 (CSF-1R) | 2016 |
|
RU2748884C2 |
Описываются производные имидазопиридина общей формулы I

или их фармацевтически приемлемые соли, где R1 представляет собой Н, CH3 или CH2OH; R2 представляет собой CH3 или CH2CH3; R3 представляет собой Н или C1-C6алкил; R4 представляет собой Н или C1-C6алкил; R5 представляет собой Н или галоген; R6 и R7 независимо представляют собой Н, C1-C6алкил, гидроксилированный C1-C6алкил, C1-C6алкоксизамещенный C1-C6алкил, галогенированный C1-C6алкил, фенил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкила, C1-C6алкокси, (C1-C6алкил)2-N- или NH2SO2, арил-замещенный C1-C6алкил, в котором арил представляет собой фенил, имидазолил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкокси или OH, R8-(C1-C6)алкил-, в котором R8 представляет собой NH2С=O-, C1-C6 алкил-NHC=O-, (C1-C6алкил)2NC=O-, C1-C6алкил-OOC- и другие при условии, что по меньшей мере один из R6 и R7 не может быть Н, С1-С6алкилом, гидроксилированным C1-C6алкилом или С1-С6алкоксизамещенным С1-С6алкилом; и Х представляет собой NH или О; а также промежуточные соединения, фармацевтические препараты на их основе, способ ингибирования секреции желудочной кислоты и способ лечения состояний, в которые вовлечено инфицирование H.YLORI. Новые соединения ингибируют экзогенно или эндогенно стимулируемую секрецию желудочной кислоты и ввиду этого могут быть использованы для предупреждения и лечения желудочно-кишечных воспалительных заболеваний. 9 н. и 6 з.п. ф-лы.

или их фармацевтически приемлемые соли,
где R1 представляет собой Н, CH3 или CH2OH;
R2 представляет собой CH3 или CH2CH3;
R3 представляет собой Н или C1-C6алкил;
R4 представляет собой Н или C1-C6алкил;
R5 представляет собой Н или галоген;
R6 и R7 независимо представляют собой Н, C1-C6алкил, гидроксилированный C1-C6алкил, C1-C6алкоксизамещенный C1-C6алкил, галогенированный C1-C6алкил, арил, где арил представляет собой фенил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкила, C1-C6алкокси, (C1-C6алкил)2-N- или NH2SO2, арилзамещенный C1-C6алкил, в котором арил представляет собой фенил, имидазолил, возможно замещенный одним или более чем одним заместителем, выбранным из C1-C6алкокси или OH, R8-(C1-C6)алкил-, в котором R8 представляет собой NH2С=O-, C1-C6 алкил-NHC=O-, (C1-C6алкил)2NC=O-, C1-C6алкил-OOC-, C1-C6алкил-CO-NH-, C1-C6алкил-OOCNH-, C1-C6алкил-O-, C7-C12алкил-O-, C1-C6алкил-SO-, C1-C6алкил-S-, C1-C6алкил-C=O-, ArCONH-, Ar(C1-C6алкил)CONH, ArC=O-, NH2CONH-, C1-C6алкил-NHCONH-, (C1-C6алкил)2-NCONH-, ArNHCONH-, гидроксилированный C1-C6алкил-O- или морфолинил; где Ar представляет собой фенил, пиридил или нафтил; C7-C12алкил, OH,

где R9 и R10 независимо представляют собой Н, R11-(C1-C6)алкил-COO-(C1-C6)алкил-, где R11 представляет собой HOOC-, или
R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом, такое как морфолин, пиперазин, пирролидин, пиперидин, возможно замещенное одним или более чем одним заместителем, выбранным из галогена, C1-C6алкила, C1-C6алкокси, CF3, OH, фенила, NH2CO-, C1-C6алкил-CO-, при условии, что по меньшей мере один из R6 и R7 не может быть Н, С1-С6алкилом, гидроксилированным C1-C6алкилом или С1-С6алкокси-замещенным С1-С6 алкилом; и
Х представляет собой NН или О.

где R9 и R10 независимо представляют собой Н, R11-(C1-C6)алкил-COO-(C1-C6)алкил-, где R11 представляет собой HOOC-, или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное или ненасыщенное кольцо, возможно содержащее один или более чем один дополнительный гетероатом, такое как морфолин, пиперазин, пирролидин, пиперидин, возможно замещенное одним или более чем одним заместителем, выбранным из галогена, C1-C6алкила, C1-C6алкокси, CF3, OH, фенила, NH2CO-, C1-C6алкил-CO-, при условии, что по меньшей мере один из R6 и R7 не может быть Н, С1-С6алкилом, гидроксилированным C1-C6алкилом или С1-С6алкоксизамещенным С1-С6алкилом, или его фармацевтически приемлемая соль.
2,3-диметил-8-(2-этил-6-метилбензиламино)-6-(морфолинокарбонил)-имидазо[1,2-a]пиридин;
N-(4-этоксифенил)-8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-карбоксамид;
N-[2-(диметиламин)-2-оксоэтил]-8-(2-этил-6-метилбензиламино)-N,2,3-триметилимидазо[1,2-a]пиридин-6-карбоксамид;
(8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-ил)(4-метилпиперазино)метанон;
1-((8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)карбонил)-2-(s)-пирролидинкарбоксамид;
8-(2-этил-6-метилбензиламино)-N-гидрокси-2,3-диметилимидазо[1,2-a]пиридин-6-карбоксамид;
(2-этил-6-метилбензиламино)-N-(2-(2-гидроксиэтокси)этил)-2,3-диметил-имидазо[1,2-a]пиридин-6-карбоксамид;
(8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)(3-гидрокси-1-пирролидинил)метанон;
N-(3,4-дигидроксифенетил)-8-(2-этил-6-метилбензиламино)-2,3-диметил-имидазо[1,2-a]пиридин-6-карбоксамид;
8-(2-этил-6-метилбензиламино-3-(гидроксиметил)-2-метил-6-(морфолинокарбонил)-имидазо[1,2-a]пиридин;
N-((8-(2-этил-6-метилбензил)амино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)карбонил)гуанидин;
4-(2-(((8-(2-этил-6-метилбензиламино)-2,3-диметилимидазо[1,2-a]пиридин-6-ил)карбонил)амино)этокси)-4-оксобутановую кислоту
или их фармацевтически приемлемую соль.

где R1 - R5 имеют указанные выше значения, гидролизуют в стандартных условиях с получением карбоновой кислоты общей формулы III

которую подвергают взаимодействию с аминосоединением формулы IV

где R6 и R7 имеют указанные в п.1 значения, в присутствии агента сочетания в стандартных условиях в инертном растворителе.

где R1, R2, R3, R4, R5 и Хтакие, как определено в п.1.
| ЗАПОЛНИТЕЛЬ | 0 |
|
SU308917A1 |
| Прибор для отмучивания формовских песков при определении степени их коллоидальности | 1931 |
|
SU33094A1 |
| EP 0228006 A1, 08.07.1987 | |||
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНТАГОНИСТИЧЕСКУЮ АКТИВНОСТЬ В ОТНОШЕНИИ ГИСТАМИНА (H) | 1992 |
|
RU2114845C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО [1,5-А] ПИРИДИНА, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2069662C1 |

