Область техники
Настоящее изобретение относится к новым соединениям и их терапевтически приемлемым солям, которые ингибируют экзогенную или эндогенную секрецию желудочной кислоты и, следовательно, могут быть использованы в предупреждении и лечении желудочно-кишечных воспалительных заболеваний. В следующих аспектах данное изобретение относится к соединениям по изобретению для использования в терапии, к способам получения таких новых соединений, к фармацевтическим композициям, содержащим в качестве активного ингредиента по меньшей мере одно соединение по изобретению или его терапевтически приемлемую соль, и к применению этих активных соединений в изготовлении лекарств для вышеуказанного медицинского применения.
Предпосылки изобретения
Замещенные имидазо[1,2-а]пиразины раскрыты в ЕР-А-0068378, US 4507294 и ЕР-А-0204285. Пирроло[2,3-d]пиридазины раскрыты в WO 91/17164, WO 92/06979, WO 93/08190 и WO 95/19980. Пирроло[1,2-а]пиразины раскрыты в US 5041442.
Производные бензимидазола и имидазопиридина, в которых фенильная группировка замещена низшим алкилом в положении 2 и 6 и которые эффективны как ингибиторы желудочно-кишечной Н+, К+-АТФазы, раскрыты в международной патентной заявке PCT/SE 97/00991 (дата подачи: 5 июня 1997) и в шведской патентной заявке №9700661-3 (дата подачи: 25 февраля 1997) соответственно.
Обзор по фармакологии насоса желудочной кислоты (H+, К+-АТФ-аза) опубликован в Sachs et al. (1995) Annu. Rev. Pharmacol. Toxicol. 35: 277-305.
Описание изобретения
Неожиданно было обнаружено, что соединения формулы I, представляющие собой замещенные гетероциклические соединения, в которых фенильная группировка замещена низшим (С1-С6)алкилом в положении 2 и 6, особенно эффективны в качестве ингибиторов желудочно-кишечной H+, К+-АТФ-азы и, следовательно, в качестве ингибиторов секреции желудочной кислоты.
Таким образом, данное изобретение относится к гетероциклическим соединениям с замещенной фенильной группировкой общей формулы I:
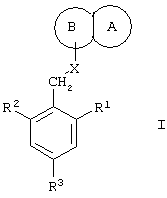
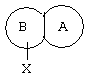
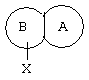
или их фармацевтически приемлемым солям, в которых
R1 представляет собой C1-С6алкил;
R2 представляет собой C1-С6алкил;
R3 представляет собой Н или галоген;
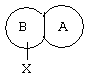
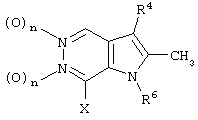
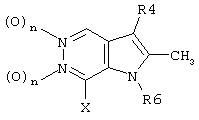
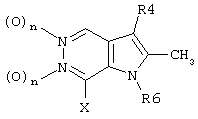
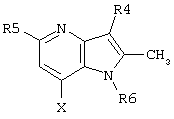
представляет собой замещенный гетероцикл, выбранный из
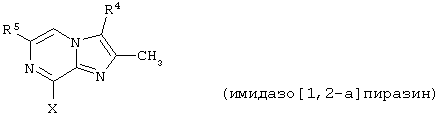
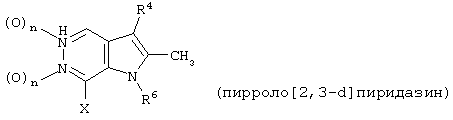
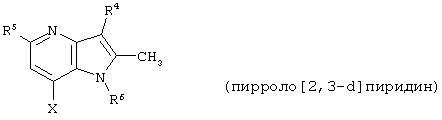

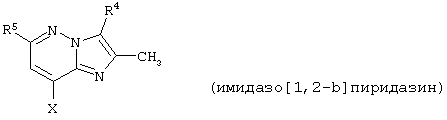
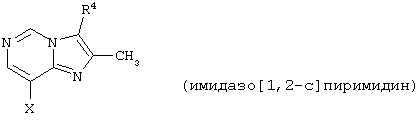
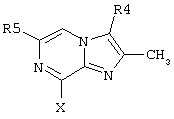
где R4 представляет собой Н, СН3, CH3OH или CH2CN;
R5 представляет собой Н или C1-С6алкил;
R6 представляет собой Н или C1-С6алкил;
R7 представляет собой Н или C1-С6алкил;
n равно 0 или 1;
Х представляет собой NH или О.
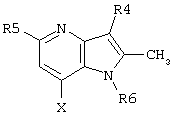
Предпочтительным является соединение по изобретению или его фармацевтически приемлемая соль, в котором
представляет собой
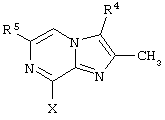
где R4, R5 и Х такие, как определено выше.
Также предпочтительным является соединение по изобретению или его фармацевтически приемлемая соль, в котором
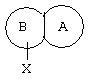
представляет собой
где R4, R6 и X такие, как определено выше.
Предпочтительным является соединение по изобретению или его фармацевтически приемлемая соль, в котором
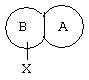
представляет собой
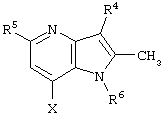
где R4, R5, R6 и Х такие, как определено выше.
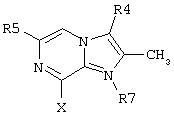
Предпочтительным является соединение по изобретению или его фармацевтически приемлемая соль, в котором
представляет собой
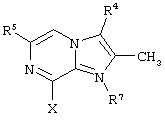
где R4, R5, R7 и Х такие, как определено выше.
Предпочтительным является соединение по п.1 или его фармацевтически приемлемая соль, в котором
представляет собой
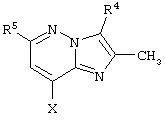
где R4, R5 и Х такие, как определено выше.
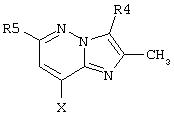
Предпочтительным является соединение по изобретению или его фармацевтически приемлемая соль, в котором
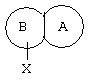
представляет собой
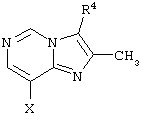
где R4 и Х такие, как определено выше.
Предпочтительным также является соединение по изобретению или его фармацевтически приемлемая соль, в котором R1 и R2 независимо представляют собой СН3 или СН2СН3.
Наиболее предпочтительным является соединение по изобретению, которое представляет собой 2,3-диметил-8-(2,6-диметилбензиламино)имидазо[1,2-а]пиразин
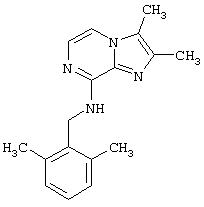
или его фармацевтически приемлемую соль.
Также наиболее предпочтительным является соединение по изобретению, которое представляет собой 2,3-диметил-8-(2,6-диметилбензилокси)имидазо[1,2-а]пиразин
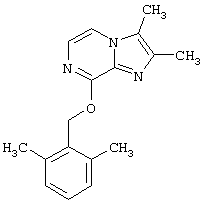
или его фармацевтически приемлемую соль.
Согласно изобретению предложен также способ получения соединения по изобретению, при котором соединения общей формулы II
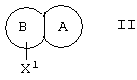
где X1 представляет собой ОН или NH2,
подвергают взаимодействию с соединением общей формулы III
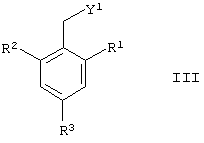
где R1, R3 и R4 такие, как определено для формулы I, a Y1 представляет собой отщепляемую группу, такую как галогенид, тозилокси или мезилокси.
Согласно изобретению предложен также способ получения соединения по изобретению, при котором соединения общей формулы IV
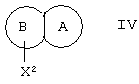
где X2 представляет собой отщепляемую группу, подвергают взаимодействию с соединением общей формулы V
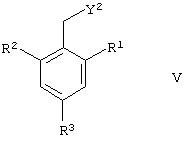
где R1, R3 и R4 такие, как определено для формулы I, a Y2 представляет собой NH2 или ОН.
Согласно изобретению предложено соединение по изобретению для ингибирования секреции желудочной кислоты.
В данном изобретении предложен также способ ингибирования секреции желудочной кислоты, при котором млекопитающему, включая человека, нуждающемуся в таком ингибировании, вводят эффективное количество соединения по изобретению.
Настоящее изобретение дополнительно относится к фармацевтическому препарату для использования в ингибировании секреции желудочной кислоты, в котором активный ингредиент представляет собой соединение по изобретению.
Изобретение также относится к фармацевтическому препарату, обладающему противовоспалительной активностью в отношении желудочно-кишечных заболеваний, в котором активный ингредиент представляет собой соединение по изобретению.
Используемый здесь термин "С1-С6алкил" означает нормальную или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода. Примеры указанного низшего алкила включают в себя метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, а также пентил и гексил с нормальной и с разветвленной цепью.
Термин "галогено" включает в себя фтор, хлор, бром и йод.
Оба чистых энантиомера, рацемические смеси и неравные смеси двух энантиомеров входят в объем данного изобретения. Следует иметь в виду, что все возможные диастереомерные формы (чистые энантиомеры, рацемические смеси и неравные смеси двух энантиомеров) входят в объем данного изобретения. Данное изобретение охватывает также производные соединений формулы I, которые обладают биологической функцией соединений формулы I.
В зависимости от условий способа конечные продукты формулы I получают либо в нейтральной форме, либо в форме соли. Как свободные основания, так и соли этих конечных продуктов входят в объем данного изобретения.
Соли присоединения кислоты этих новых соединений способом, известным per se, могут быть превращены в свободное основание с использованием основных агентов, таких как щелочь, или посредством ионного обмена. Полученное свободное основание может образовывать соли с органическими или неорганическими кислотами.
При получении солей присоединения кислоты предпочтительно используют такие кислоты, которые соответственно образуют терапевтически приемлемые соли. Примерами таких кислот являются галогеноводородные кислоты, такие как хлороводородная кислота, серная кислота, фосфорная кислота, азотная кислота, алифатические, алициклические, ароматические или гетероциклические карбоновые или сульфоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, оксималеиновая кислота, пировиноградная кислота, п-оксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, оксиэтансульфоновая кислота, галогенбензолсульфоновая кислота, толуолсульфоновая кислота или нафталинсульфоновая кислота.
При осуществлении заявленных способов получения соединений по изобретению реакцию взаимодействия соединения общей формулы II с соединением общей формулы III удобно проводить в инертном растворителе, например в ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле, ксилоле или диметилформамиде, с основанием или без него.
Это основание представляет собой, например, гидроксид щелочного металла, такой как гидроксид натрия и гидроксид калия; алкоголят натрия, такой как метилат натрия и этилат натрия; гидрид щелочного металла, такой как гидрид натрия и гидрид калия; карбонат щелочного металла, такой как карбонат калия и карбонат натрия; или органический амин, такой как триэтиламин.
Реакцию взаимодействия соединения общей формулы IV с соединением общей формулы V удобно проводить в инертном растворителе, например в ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле, ксилоле или диметилформамиде, с основанием или без него. Основание представляет собой, например, гидроксид щелочного металла, такого как гидроксид натрия и гидроксид калия; алкоголят натрия, такой как метилат натрия и этилат натрия; гидрид щелочного металла, такой как гидрид натрия и гидрид калия; карбонат щелочного металла, такой как карбонат калия и карбонат натрия; или органический амин, такой как триэтиламин.
Фармацевтические препараты
В еще одном аспекте данное изобретение относится к фармацевтическим композициям, содержащим в качестве активного ингредиента по меньшей мере одно соединение по изобретению или его терапевтически приемлемую соль.
Соединения по изобретению также можно использовать в препаратах вместе с другими активными ингредиентами, например антибиотиками, такими как амоксициллин.
Для клинического применения соединения по изобретению готовят в виде фармацевтических препаратов для перорального, ректального, парентерального или другого способа введения. Фармацевтический препарат содержит соединение по изобретению в сочетании с одним или более чем одним фармацевтически приемлемым ингредиентом. Носитель может быть в форме твердого, полутвердого или жидкого разбавителя или капсулы. Эти фармацевтические препараты составляют следующий объект изобретения. Обычно количество активных соединений находится в пределах от 0,1 до 95% от массы препарата, предпочтительно от 0,1 до 20 мас.% в препаратах для парентерального применения и предпочтительно от 0,1 до 50 мас.% в препаратах для перорального введения.
При приготовлении фармацевтических препаратов, содержащих соединение по настоящему изобретению, в форме дозировочных единиц для перорального введения выбранное соединение может быть смешано с твердыми порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, производные целлюлозы, желатин, или с другими подходящими ингредиентами, а также с разрыхлителями и смазывающими агентами, такими как стеарат магния, стеарат кальция, стеарилфумарат натрия и полиэтиленгликолевые воски. Затем эту смесь перерабатывают в гранулы или прессуют в таблетки.
Мягкие желатиновые капсулы могут быть приготовлены в виде капсул, содержащих смесь активного соединения или соединений по изобретению, растительного масла, жира или другой подходящей среды для мягких желатиновых капсул. Твердые желатиновые капсулы могут содержать гранулы активного соединения. Твердые желатиновые капсулы также могут содержать активное соединение в сочетании с твердыми порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, картофельный крахмал, кукурузный крахмал, амилопектин, производные целлюлозы или желатин.
Дозировочные единицы для ректального введения могут быть приготовлены (1) в форме суппозиториев, которые содержат активное вещество, смешанное с нейтральной жировой основой; (2) в форме желатиновых капсул для ректального введения, которые содержат активное вещество в смеси с растительным маслом, парафиновым маслом или другой подходящей средой для желатиновых капсул для ректального введения; (3) в форме готовых микроклизм или (4) в форме сухого препарата для приготовления микроклизм, который подлежит разведению подходящим растворителем непосредственно перед введением.
Жидкие препараты для перорального введения могут быть приготовлены в форме сиропов или суспензий, например растворов или суспензий, содержащих от 0,1 до 20 мас.% активного ингредиента, а остальное - сахар или сахарные спирты и смесь этанола, воды, глицерина, пропиленгликоля и полиэтиленгликоля. При желании такие жидкие препараты могут содержать красители, корригенты, сахарин и карбоксиметилцеллюлозу или другой загуститель. Жидкие препараты для перорального введения могут быть приготовлены также в форме сухого порошка, который подлежит разведению подходящим растворителем перед использованием.
Растворы для парентерального введения могут быть приготовлены в виде раствора соединения по изобретению в фармацевтически приемлемом растворителе, предпочтительно в концентрации от 0,1 до 10 мас.%. Эти растворы могут содержать также стабилизаторы и/или буферы, и их отпускают в стандартных дозах в форме ампул или флаконов.
Растворы для парентерального введения также могут быть приготовлены в виде сухого препарата, который подлежит разведению подходящим растворителем непосредственно перед использованием.
Типичная суточная доза активного вещества варьирует в широких пределах и зависит от различных факторов, таких как, например, индивидуальная потребность пациента, путь введения и заболевание. В общем случае пероральные и парентеральные дозировки должны быть в пределах от 5 до 1000 мг активного ингредиента в сутки.
Соединения по изобретению также могут быть использованы в препаратах вместе с другими активными ингредиентами, например для лечения или профилактики состояний, в которые вовлечена инфекция Helicobacter pylori слизистой оболочки желудка человека. Такими другими активными ингредиентами могут быть антимикробные средства, в частности:
- b-лактамовые антибиотики, такие как амоксициллин, ампициллин, цефалотин, цефаклор или цефиксим;
- макролиды, такие как эритромицин или кларитромицин;
- тетрациклины, такие как тетрациклин или доксициклин;
- аминогликозиды, такие как гентамицин, канамицин или амикацин;
- хинолоны, такие как норфлоксацин, ципрофлоксацин или эноксацин;
- другие средства, такие как метронидазол, нитрофурантоин или хлорамфеникол; или
- препараты, содержащие соли висмута, такие как основной цитрат висмута, основной салицилат висмута, основной карбонат висмута, основной нитрат висмута или основной галлат висмута.
ПРИМЕРЫ
Пример 1.1
Синтез 2,3-диметил-8-(2,6-диметилбензиламино)имидазо[1,2-а]пиразина
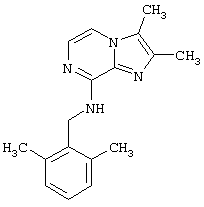
Перемешиваемую смесь 8-хлор-2,3-диметилимидазо[1,2-а]пиразина (0,5 г, 2,8 ммоль) и 2,6-диметилбензиламина (0,41 г, 3,0 ммоль) в ксилоле (10 мл) нагревают с обратным холодильником в течение 24 часов. Эту смесь выпаривают при пониженном давлении, растворяют в метиленхлориде (20 мл) и промывают 5% раствором карбоната натрия в воде (20 мл). Органический слой отделяют и выпаривают при пониженном давлении, а остаток очищают колоночной хроматографией на силикагеле. Перекристаллизацией из пентана получают 90 мг (23%) указанного в заголовке соединения.
1Н-ЯМР (300 МГц, CDCl3): δ 2,35 (s, 6Н), 2,45 (s, 6H), 4,70(d, 2H), 5,60 (bs, 1Н), 7,05-7,20 (m, 3Н), 7,25 (d, 1Н), 7,40 (d, 1H)
Пример 1.2
Синтез 2,3-диметил-8-(2,6-диметилбензилокси)имидазо[1,2-а]пиразина
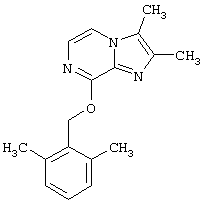
Гидрид натрия (0,15 г, 3 ммоль) (50% в масле) добавляют к перемешиваемому раствору 2,6-диметилбензилового спирта в ацетонитриле (10 мл). Затем добавляют 8-хлор-2,3-диметилимидазо[1,2-а]пиразина (0,4 г, 2,8 ммоль) и эту реакционную смесь нагревают с обратным холодильником в течение 20 часов. Растворитель выпаривают при пониженном давлении, остаток растворяют в метиленхлориде и промывают водой. Органический слой выпаривают при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетат:петролейный эфир (40-60) 1:1. Перекристаллизацией из петролейного эфира получают 0,42 г (50%) указанного в заголовке соединения.
1H-NMR (300 МГц, СDCl3): δ 2,35 (s, 3H), 2,40 (s, 3Н), 2,45 (s, 6H), 5,6 (s, 2Н) 6,95-7,15 (m, 3Н), 7,35-7,45 (m 2H).
БИОЛОГИЧЕСКИЕ ТЕСТЫ
1. Эксперименты in vitro
Ингибирование секреции кислоты в изолированных желудочных железах кролика
Ингибирующее действие на секрецию кислоты in vitro в изолированных желудочных железах кролика измеряли, как описано в Berglindh et al. (1976) Acta Physiol. Scand. 97, 401-414.
Определение H+, К+-АТФ-азной активности
Мембранные везикулы (от 2,5 до 5 мкг) инкубировали в течение 15 минут при +37°С в 18 мМ буфере Pipes/Tris, pH 7,4, содержащем 2 мМ MgCl2, 10 мМ KCl и 2 мМ АТФ. АТФ-азную активность оценивали по высвобождению неорганического фосфата из АТФ, как описано в LcBel et al. (1978) Anal. Biochem. 85, 86-89.
Соединение Примера 1 имело значение IC50, равное 0,16 мкМ, а соединение Примера 2 имело значение IC50, равное 2,78 мкМ.
2. Эксперименты in vivo
Ингибирующее действие на секрецию кислоты у самок крыс
Используют самок крыс линии Sprague-Dawly. Для сбора желудочных секретов и введения тестируемых веществ им устанавливают канюлированные фистулы в желудок (полость) и в верхнюю часть двенадцатиперстной кишки соответственно. До начала испытаний на восстановительный период после хирургической операции отводят 14 дней.
Перед тестами на секрецию животных на 20 часов лишают пищи, но не воды. Желудок периодически промывают (+37°С) через желудочную канюлю водопроводной водой и подкожно вводят 6 мл Ringer-Glucose. Секрецию кислоты стимулируют инфузией в течение 2,5-4 часов (1,2 мл/час, подкожно) пентагастрина и карбахола (20 и 110 нмоль/кг-час соответственно) и в течение этого времени собирают фракции желудочных секретов за отрезки времени 30 минут. Тестируемые вещества или носитель вводят либо через 60 минут после начала стимулирования (внутривенное и внутридуоденальное дозирование, 1 мл/кг), либо за 2 часа до начала стимулирования (пероральное дозирование, 5 мл/кг, желудочная канюля закрыта). Для исследования продолжительности действия временной интервал между дозированием и стимулированием может быть увеличен. Образцы желудочного сока титруют 0,1М NaOH до рН 7,0 и выход кислоты рассчитывают как произведение объема титранта на концентрацию.
Дальнейшие вычисления основаны на средних значениях ответной реакции для группы из 4-6 крыс. В случае введения во время стимулирования выход кислоты в течение промежутков времени после введения тестируемого вещества или носителя выражают в виде дробных значений ответной реакции, принимая за 1,0 выход кислоты за период 30 минут, предшествующий введению. Процент ингибирования вычисляют из дробных значений ответной реакции, вызванной тестируемым соединением и носителем. В случае введения до стимулирования процент ингибирования вычисляют непосредственно из выхода кислоты, зафиксированного после введения тестируемого соединения и носителя.
Биодоступность у крысы
Используют взрослых крыс линии Sprague-Dawly. За один-три дня до экспериментов всем крысам под наркозом устанавливают канюли в левую каротидную артерию. Крысам, используемым для внутривенных экспериментов, вводят канюли также в шейную вену (Popovic (1960) J. Appl. Physiol. 15, 727-728). Канюли располагают в затылочной части шеи.
Образцы крови (0,1-0,4 г) многократно отбирают из каротидной артерии с интервалами до 5,5 часа после введения дозы. До анализа тестируемого соединения образцы замораживают.
Биодоступность определяют посредством вычисления отношения между площадью под кривой концентрации в крови/плазме (AUC) после (1) внутридуоденального (в.д.) или перорального (п.о.) введения и (2) внутривенного (в.в.) введения крысе или собаке соответственно.
Площадь под кривой зависимости концентрации в крови от времени, AUC, определяют по логарифмическому/линейному правилу трапеций и экстраполируют в бесконечность путем деления последней установленной концентрации в крови на константу скорости элиминации в конечной фазе. Системную биодоступность (F%) после внутридуоденального или перорального введения вычисляют как
F(%)=(AUC (п.о. или в.д.)/АUС (в.в.))×100.
Ингибирование секреции желудочной кислоты и биодоступность у собак, находящихся в сознании
Используют собак породы Лабрадор или Гончая обоих полов. Им устанавливают дуоденальную фистулу для введения тестируемых соединений или носителя и канюлированную желудочную фистулу или карман Хейденхейма для сбора желудочного секрета.
Перед тестами на секрецию животных держат без пищи в течение около 18 часов, но воду дают без ограничений.
Секрецию желудочной кислоты стимулируют инфузией продолжительностью до 6,5 часа гистамина дигидрохлорида (12 мл/час) в дозе, которая вызывает ответную реакцию, составляющую примерно 80% от индивидуального максимального секреторного ответа, и желудочный сок собирают в последующие 30-минутные отрезки времени. Тестируемое вещество или наполнитель вводят перорально, в.д. или в.в. через 1 час или 1,5 часа после начала инфузии гистамина в объеме 0,5 мл/кг массы тела. В случае перорального введения следует отметить, что тестируемое соединение вводят в секретирующий кислоту желудок собак, снабженных карманом Хейденхейма.
Кислотность образцов желудочного сока определяют титрованием до рН 7,0 и вычисляют выход кислоты. Выход кислоты в промежутки времени сбора после введения тестируемого вещества или носителя выражают в виде дробных значений ответной реакции, принимая за 1,0 выход кислоты за отрезок времени, предшествующий введению. Процент ингибирования вычисляют из дробных значений ответной реакции, вызванной тестируемым соединением и носителем.
Образцы крови для анализа на концентрацию тестируемого соединения в плазме отбирают с интервалами времени до 4 часов после дозирования. После отбора плазму отделяют и замораживают в течение 30 минут и позднее анализируют. Системную биодоступность (F%) после перорального или в.д. введения рассчитывают, как описано выше в модели на крысах.
Изобретение относится к гетероциклическим соединениям с замещенной фенильной группировкой формулы I 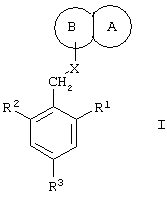 или его фармацевтически приемлемая соль, в которой R1 представляет собой C1-С6алкил; R2 представляет собой C1-С6алкил; R3 представляет собой Н или галоген и
или его фармацевтически приемлемая соль, в которой R1 представляет собой C1-С6алкил; R2 представляет собой C1-С6алкил; R3 представляет собой Н или галоген и 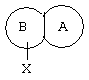 представляет собой замещенный гетероцикл, как определено в п.1 формулы изобретения; и Х представляет собой NH или O. Изобретение также относится к способу получения соединений формулы I и к фармацевтическим препаратам для использования в ингибировании секреции желудочной кислоты на основе этих соединений. Технический результат - получение новых соединений и фармацевтических препаратов на их основе, обладающих противовоспалительной активностью в отношении желудочно-кишечных заболеваний. 6 с. и 10 з.п. ф-лы.
представляет собой замещенный гетероцикл, как определено в п.1 формулы изобретения; и Х представляет собой NH или O. Изобретение также относится к способу получения соединений формулы I и к фармацевтическим препаратам для использования в ингибировании секреции желудочной кислоты на основе этих соединений. Технический результат - получение новых соединений и фармацевтических препаратов на их основе, обладающих противовоспалительной активностью в отношении желудочно-кишечных заболеваний. 6 с. и 10 з.п. ф-лы.
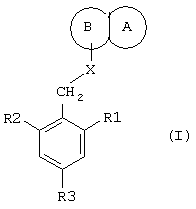
или его фармацевтически приемлемая соль,
где R1 представляет собой C1-С6алкил;
R2 представляет собой С1-С6алкил;
R3 представляет собой Н или галоген;
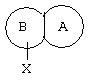
представляет собой замещенный гетероцикл, выбранный из
или
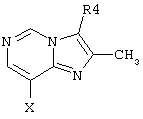
где R4 представляет собой Н, СН3, СН2ОН или CH2CN;
R5 представляет собой Н или C1-С6алкил;
R6 представляет собой Н или С1-С6алкил;
R7 представляет собой Н или С1-С6алкил;
n = 0 или 1;
Х представляет собой NH или О.
 представляет собой
представляет собой
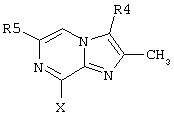
где R4, R5 и Х такие, как определено в п.1.
 представляет собой
представляет собой
где R4, R6 и Х такие, как определено в п.1.
 представляет собой
представляет собой
где R4, R5, R6 и Х такие, как определено в п.1.
 представляет собой
представляет собой
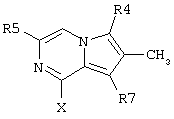
где R4, R5, R7 и Х такие, как определено в п.1.
представляет собой
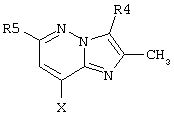
где R4, R5 и X такие, как определено в п.1.
 представляет собой
представляет собой
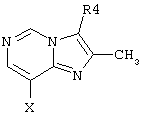
где R4 и Х такие, как определено в п.1.
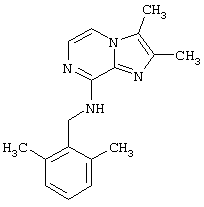
или его фармацевтически приемлемую соль.
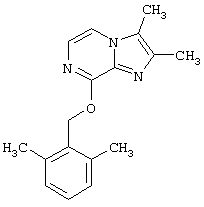
или его фармацевтически приемлемую соль.
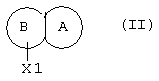
где X1 представляет собой ОН или NH2,
подвергают взаимодействию с соединением общей формулы III
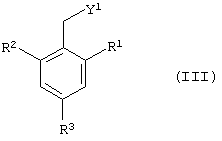
где R1, R3 и R4 такие, как определено для формулы I;
Y1 представляет собой отщепляемую группу.
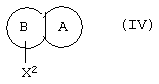
где X2 представляет собой отщепляемую группу,
подвергают взаимодействию с соединением общей формулы V
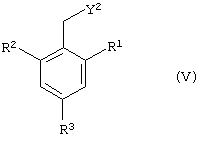
где R1, R3 и R4 такие, как определено для формулы I;
a Y2 представляет собой NH2 или ОН.
| ВОЗДУХОРАСПРЕДЕЛИТЕЛЬ АВТОМАТИЧЕСКОГО ЛОКОМОТИВНОГО ТОРМОЗА | 1945 |
|
SU68378A1 |
| US 4507294 А, 26.03.85 | |||
| Устройство для автоматической расцепки железнодорожного подвижного состава | 1978 |
|
SU742218A1 |
| US 5041442 А, 20.08.91 | |||
| WO 9308190 A1, 29.04.93 | |||
| WO 9206979 A1, 30.04.92 | |||
| WО 9117164 A1, 14.11.91 | |||
| US 5534515 А, 09.07.96 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРРОЛО- | 0 |
|
SU237153A1 |

