Предпосылки создания изобретения
1. Область техники, к которой относится изобретение
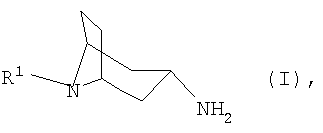
Настоящее изобретение относится к способу получения N-замещенных 3β-аминонортропанов формулы I
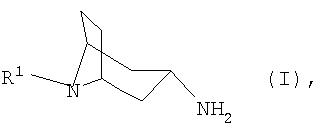
в которой R1 имеет указанные в формуле изобретения значения, из соответствующего 3-оксонортропана или соответствующего 3α-аминонортропана, заключающемуся в том, что либо 3-оксонортропан взаимодействием с арилметиламином, либо 3α-аминонортропан взаимодействием с арилальдегидом переводят в соответствующие имины, которые таутомеризуют, соответственно, изомеризуют и затем гидролизуют.
2. Уровень техники
Соединения формулы I являются ценными промежуточными продуктами в химическом синтезе различных фармацевтических действующих веществ (см., например, A.Nuhrich и др., Eur. Journ. Med. Chem. 31, 12, 1996, сс.957-964), или же сами они представляют собой фармацевтические действующие вещества, прежде всего в качестве модуляторов NMDA-рецептора (NMDA означает N-метил-D-аспартат) (см., например, А.Н.Lewin и др., Journ. Med. Chem, 41, 1998, сс.988-995).
Гидрированием оксимов тропанона в присутствии платинового катализатора и разбавителя при повышенном давлении (см., например, S. Archer, Journ. Am. Chem. Soc. 80, 1958, с.4677) получают N-замещенные 3-аминонортропаны преимущественно в α-форме.
Взаимодействие оксимов тропанона с металлическим натрием в спирте (см., например, A. Nuhrich и др., в указанном выше источнике) приводит преимущественно к образованию β-изомеров N-замещенных 3-аминонортропанов.
Следует, однако, отметить, что реализация этого способа в промышленном масштабе по соображениям техники безопасности возможна лишь в ограниченной степени.
С учетом вышеизложенного в основу настоящего изобретения была положена задача разработать способ получения N-замещенных 3β-аминонортропанов формулы I, который позволял бы получать эти соединения в промышленном масштабе с высоким общим выходом при одновременно высоком выходе с единицы объема в единицу времени. Еще одна задача изобретения заключалась в том, чтобы предложить 3β-аминонортропаны формулы I, которые в основном не содержат соответствующих 3α-изомеров.
Указанную задачу удается решить согласно изобретению с помощью предлагаемого в нем способа, обеспечивающего получение 3β-аминонортропана исходя из соответственно замещенного 3-оксонортропана или из соответственно замещенного 3α-аминонортропана.
Предлагаемый в изобретении способ позволяет получать 3β-аминонортропаны в промышленном масштабе с высоким выходом и с высокой степенью чистоты.
Краткое изложение сущности изобретения
Объектом изобретения является способ получения N-замещенных 3β-аминонортропанов формулы I или одной из их кислотно-аддитивных солей
где R1 представляет собой необязательно замещенный остаток, выбранный из группы, включающей С1-С8алкил, С2-С8алкенил, С3-С8циклоалкил и С6-С10арил-С1-С8алкил, при этом либо
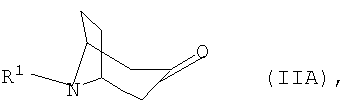
а) соответствующий 3-оксонортропан формулы IIА
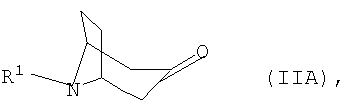
подвергают взаимодействию с арилметиламином формулы IIIA
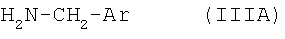 ,
,
в которой Ar обозначает необязательно замещенный фенильный остаток либо необязательно замещенный 5- или 6-членный гетероароматический остаток с по меньшей мере одним гетероатомом, выбранным из группы, включающей N, О и S, либо
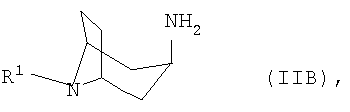
б) соответствующий 3β-аминонортропан формулы IIB
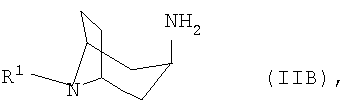
подвергают взаимодействию с арилальдегидом формулы IIIB

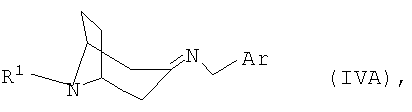
полученный в каждом случае имин формулы IVA или IVB
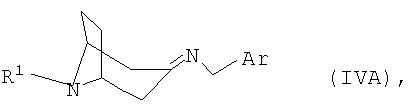
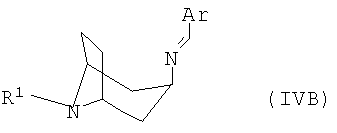
переводят в термодинамически стабильный таутомер, соответственно, изомер формулы V
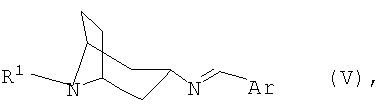
затем гидролизуют и при необходимости переводят в соответствующую кислотно-аддитивную соль.
Изобретение относится далее к новым соединениям формулы V
в которой R1 и Ar имеют указанные выше значения, а также к их таутомерам, соответственно, изомерам формул IVA и IVB.
Подробное описание изобретения
Под используемым выше и в последующем описании понятием "С1-С8алкил" индивидуально или в составе других групп либо остатков подразумевается прямоцепочечная или разветвленная алкильная группа с 1-8, предпочтительно 1-6, прежде всего 1-4, атомами углерода. В качестве примеров при этом можно назвать метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, неопентил и н-гексил. Наиболее предпочтительны метил и этил.
Под используемым выше и в последующем описании понятием "С2-С8алкенил" подразумевается прямоцепочечная или разветвленная группа с 2-8, предпочтительно 2-6, прежде всего 2-4, атомами углерода. В качестве примеров при этом можно назвать винил, проп-1-енил, проп-2-енил (аллил), бут-1-енил и пент-1-енил. Наиболее предпочтительны винил и аллил.
Под используемым выше и в последующем описании понятием "С3-С8циклоалкил" подразумевается циклоалкильная группа с 3-8, предпочтительно 3-6, прежде всего 5 или 6, атомами углерода. В качестве примеров при этом можно назвать циклопропил, циклобутил, цеклопентил и циклогексил.
Под используемым выше и в последующем описании понятием "С6-С10арил" индивидуально или в составе других групп либо остатков подразумевается необязательно замещенная C1-С6алкилом, C1-С6алкоксигруппой, аминогруппой, C1-С6алкиламиногруппой, ди(С1-С6алкил)аминогруппой или галогеном фенильная либо нафтильная группа, предпочтительно незамещенный фенил либо фенил, замещенный 1-3 заместителями, выбранными из группы, включающей С1-С4алкил, С1-С4алкоксигруппу, аминогруппу, С1-С4алкиламиногруппу и ди(С1-С4алкил)аминогруппу.
Под группой Ar подразумевается арильная группа с ее раскрытыми выше значениями или незамещенный либо замещенный C1-С6алкилом, C1-С6алкоксигруппой, аминогруппой, C1-С6алкиламиногруппой, ди(С1-С6алкил)аминогруппой или галогеном 5- или 6-членный гетероароматический остаток с по меньшей мере одним гетероатомом, выбранным из группы, включающей N, О и S.
Предпочтительно гетероароматический остаток представляет собой пиррол, фуран, тиофен, имидазол, пиразол, тиазол, тиадиазол, тетразол, пиридин, пиримидин или пиразин.
Предпочтительным является предлагаемый в изобретении способ, в котором используют соединение формулы I, где R1 представляет собой остаток, выбранный из группы, включающей C1-С6алкил, прежде всего метил или этил, и фенил-С1-С3алкил, прежде всего бензил.
Предпочтительным является далее предлагаемый в изобретении способ, в котором используют соединение формулы I, где Ar обозначает фенильный остаток, одно- или двузамещенный C1-С6алкоксигруппой, прежде всего метокси-, этокси- либо изопропоксигруппой, и/или ди(С1-С6алкил)аминогруппой, прежде всего диметиламино- либо диэтиламиногруппой. Наиболее предпочтительно эти заместители находятся в пара-положении относительно метиламиногруппы (формулы IIIA) или карбальдегидной группы (формула IIIB).
Каждую из отдельных стадий предлагаемого в изобретении способа предпочтительно проводить в инертном разбавителе, выбранном из группы, включающей необязательно галогенированные углеводороды, такие, например, как бензол, толуол, ксилол, метилциклогексан или дихлорметан, амиды, такие, например, как ацетамид или диметилацетамид, нитрилы, такие, например, как ацетонитрил или пропионитрил, сульфоксиды, такие, например, как диметилсульфоксид (ДМСО), простые эфиры, такие, например, как диэтиловый эфир, трет-бутилметиловый эфир (ТБМЭ) или тетрагидрофуран (ТГФ), и их смеси.
Согласно одному из предпочтительных вариантов осуществления предлагаемого в изобретении способа реакцию между соединением формулы IIА и соединением формулы IIIA, соответственно, между соединением формулы IIB и соединением формулы IIIB проводят в способствующих дегидратации условиях.
Как правило, в этой реакции используют 0,5-2,0 эквивалента, предпочтительно 0,8-1,2 эквивалента, соединения формулы IIА, соответственно, формулы IIB из расчета на один эквивалент соединения формулы IIIA, соответственно, формулы IIIB. При этом целесообразно использовать примерно эквимолярные количества соединений формулы IIА, соответственно, формулы IIB в пересчете на соответствующие количества соединений формулы IIIA, соответственно, формулы IIIB.
В качестве реакций, протекающих в способствующих дегидратации условиях, можно назвать среди прочих таковые, которые проводят в присутствии связывающих воду (так называемых дегидратирующих) агентов, таких, например, как пентаоксид фосфора, тетрахлорид титана и молекулярное сито, или реакцию, которую проводят в присутствии неорганической либо органической кислоты при повышенной температуре и удалении образовавшейся воды путем азеотропной перегонки. Особенно предпочтительно удалять воду с помощью n-толуолсульфоновой кислоты (n-TsOH) в толуоле.
Полученные имины формулы IVA или IVB предпочтительно переводить в таутомер формулы V в присутствии соответствующего основания.
Особенно предпочтительными являются новые имины формулы VI
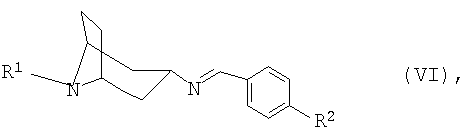
в которой R1 имеет указанные для формулы I значения, a R2 представляет собой Н, C1-С6алкоксигруппу, прежде всего метоксигруппу, или ди(С1-С6алкил)аминогруппу, прежде всего диметиламиногруппу.
Пригодными для использования в указанных целях основаниями являются сильные основания, прежде всего алкоголяты металлов, такие, например, как метанолят натрия, этанолят натрия, этанолят калия, трет-бутанолят натрия, трет-бутанолят калия, неопентанолят натрия, неопентанолят калия, прежде всего трет-бутанолят калия.
Как правило, изомеризацию осуществляют в присутствии инертного разбавителя. Предпочтительны для применения в этих целях полярные, апротонные растворители, такие, например, как диметилсульфоксид, ацетонитрил, диметилацетамид, N,N-диметилэтиленмочевина (ДЭМ), N,N- диметилпропиленмочевина (ДПМ) или их смеси. Согласно одному из особенно предпочтительных вариантов реакцию с трет-бутанолятом калия проводят в присутствии ТГФ и ДМСО.
Как правило, реакцию проводят при температуре в интервале от 0 до 120°С, предпочтительно от 25 до 100°С, прежде всего от 40 до 80°С. При соблюдении указанных условий изомеризация завершается через 1-15 ч.
Гидролиз соединения формулы VI осуществляют, как правило, в присутствии соответствующей кислоты. Предпочтительны для применения в этих целях сильные неорганические или органические кислоты, такие, например, как серная кислота, соляная кислота, фосфорная кислота или трифторуксусная кислота. Особенно предпочтительно проводить гидролиз в двухфазной системе, состоящей из органического, плохо смешивающегося с водой растворителя, прежде всего толуола или дихлорметана, воды и соответствующей кислоты, прежде всего серной кислоты.
Гидролиз проводят, как правило, при температуре в интервале от 0 до 100°С, предпочтительно от 10 до 60°С, прежде всего от 15 до 30°С. При соблюдении указанных условий гидролиз завершается через 15-360 мин.
Согласно одному из особенно предпочтительных вариантов водную фазу полученного таким путем сырого продукта отделяют и с помощью соответствующего основания, предпочтительно гидроксида щелочного металла, прежде всего едкого натра, слегка подщелачивают, устанавливая значение рН на 7,5-9,5, прежде всего на примерно 8,0. Полученную таким путем водную фазу экстрагируют с помощью не смешивающегося с водой растворителя, предпочтительно алифатического либо ароматического углеводорода, прежде всего толуола. Полученная таким путем органическая фаза может использоваться для регенерации исходных материалов.
Водную фазу отделяют и с помощью соответствующего основания, предпочтительно гидроксида щелочного металла, прежде всего едкого натра, сильно подщелачивают, устанавливая значение рН предпочтительно на 10,0-14,0, прежде всего на примерно 12,7. Полученную таким путем водную фазу экстрагируют с помощью не смешивающегося с водой растворителя, предпочтительно необязательно галогенированного, алифатического либо ароматического углеводорода, прежде всего толуола или дихлорметана.
Полученные таким путем объединенные органические фазы концентрируют. В результате получают соединение формулы I в виде свободного основания.
Это основание по известным методам путем обработки неорганической либо органической кислотой можно переводить в соответствующую кислотно-аддитивную соль. С этой целью свободное основание целесообразно сначала растворять в полярном растворителе, предпочтительно в спирте, таком, например, как метанол, этанол или изопропанол, воде либо в смеси таких растворителей, прежде всего в смеси этанола и воды, а затем с помощью соответствующей кислоты, предпочтительно неорганической кислоты, такой как соляная кислота или серная кислота, устанавливать значение рН на слабоосновное.
Выпадающую при этом в осадок соль отделяют, промывают полярным растворителем и сушат. Таким путем получают продукт в чистой кристаллической форме, не требующей последующей очистки.
К другим преимуществам предлагаемого в изобретении способа следует отнести высокий выход продукта с единицы объема в единицу времени, достигаемый при осуществлении данного процесса, а также высокий выход и высокую степень чистоты получаемых в каждом случае промежуточных продуктов, которые можно подвергать дальнейшей переработке без хроматографической очистки.
Ниже способ получения соединений формулы I более подробно поясняется на примерах, которые лишь иллюстрируют некоторые возможные варианты осуществления изобретения, не ограничивая при этом его объем.
Пример 1
Гемисульфат N-метил-3β-аминонортропана
Смесь из 17,0 г N-метил-3α-аминонортропана, 18,7 г 4-диметиламино-бензальдегида, 125 мл толуола и 0,125 г n-TsOH кипятят в течение 5 ч на водоотделителе. Затем реакционную смесь концентрируют. В результате получают 36,2 г N-метил-3α-(4-диметиламинобензилиденамино)тропана, который без последующей очистки подвергают дальнейшей переработке.
Смесь из 36,2 г N-метил-3α-(4-диметиламинобензилиденамино)тропана, 100 мл ДМСО, 13,8 г 24%-ного раствора трет-бутанолята калия в ТГФ нагревают до 65-70°С с выдержкой при этой температуре в течение 10 ч. Затем реакционную смесь примешивают к 500 мл ацетонитрила, фильтруют и промывают ацетонитрилом. Остаток на фильтре растворяют в 300 мл толуола и медленно примешивают к смеси из 200 мл воды и 10 мл концентрированной серной кислоты.
После 45-минутнуго перемешивания при комнатной температуре фазы разделяют, значение рН водной фазы с помощью едкого натра устанавливают на 8,0 и промывают ее 100 мл толуола. Фазы разделяют, водную фазу смешивают со 150 мл дихлорметана и с помощью едкого натра ее значение рН устанавливают на 12,9, после чего фазы разделяют и водную фазу трижды экстрагируют 150 мл дихлорметана. Объединенные органические фазы концентрируют. В результате получают 8,8 г коричневого масла. Это масло растворяют в 150 мл этанола, с помощью полуконцентрированной серной кислоты значение рН устанавливают на 7,5-6,5 и в течение 1 ч перемешивают при комнатной температуре. Таким путем получают 14,45 г соли с температурой разложения 320°С.
Пример 2
Гемисульфат N-метил-3β-аминонортропана
Смесь из 17,0 г N-метил-3α-аминонортропан, 17,0 г 4-метоксибензальдегида, 125 мл толуола и 0,125 г n-TsOH кипятят в течение 5 ч на водоотделителе. Затем реакционную смесь концентрируют. В результате получают 33,5 г N-метил-3α-(4-метоксибензилиденамино)тропана, который без последующей очистки подвергают дальнейшей переработке.
Смесь из 33,5 г N-метил-3α-(4-метоксибензилиденамино)тропана, 100 мл ДМСО, 8,5 г 24%-ного раствора трет-бутанолята калия в ТГФ нагревают до 65-70°С с выдержкой при этой температуре в течение 5 ч. Затем реакционную смесь примешивают к смеси из 500 мл ацетонитрила и 600 мл воды, трижды экстрагируют 300 мл толуола и концентрируют. Остаток растворяют в 200 мл воды и водную фазу трижды экстрагируют дихлорметаном порциями по 200 мл. Объединенные экстракты концентрируют, растворяют в 200 мл толуола и по каплям добавляют к раствору 10 мл концентрированной серной кислоты в 300 мл воды.
После 45-минутного перемешивания при комнатной температуре фазы разделяют, значение рН водной фазы с помощью едкого натра устанавливают на 8,0 и экстрагируют ее один раз 50 мл толуола и дважды дихлорметаном порциями по 50 мл. Затем водную фазу смешивают со 150 мл дихлорметана и с помощью едкого натра ее значение рН устанавливают на 12,9. Фазы разделяют и водную фазу 6 раз экстрагируют дихлорметаном порциями по 60 мл.
Объединенные органические фазы сушат над сульфатом натрия, фильтруют и концентрируют. В результате получают 17,1 г масла золотисто-коричневого цвета. Это масло растворяют в 200 мл этанола, с помощью полуконцентрированной серной кислоты значение рН устанавливают на 7,0-6,5 и в течение 30 мин перемешивают при комнатной температуре. Таким путем получают 17,5 г соли с температурой разложения 320°С.
Пример 3
Гемисульфат N-бензил-3β-аминонортропана
Смесь из 75,5 г гидрохлорида N-бензилнортропинона, 60 мл воды, 173 мл толуола и 22,5 мл едкого натра перемешивают при комнатной температуре. Затем фазы разделяют и водную фазу экстрагируют 87 мл толуола. Смесь толуольных фаз, 41,2 г 4-метоксибензиламина и 0,22 г n-TsOH кипятят в течение 5 ч на водоотделителе. Затем реакционную смесь концентрируют. В результате получают 95 г N-бензил-3-(4-метокси-бензилимино)тропана, который без последующей очистки подвергают дальнейшей переработке. Смесь из 95 г N-бензил-3-(4-метоксибензилимино)тропана, 240 мл ДМСО, 16,8 г 24%-ного раствора трет-бутанолята калия в ТГФ нагревают до 65-70°С с выдержкой при этой температуре в течение 5 ч. Затем реакционную смесь распределяют между 300 мл толуола и 900 мл воды и водную фазу дважды экстрагируют толуолом порциями по 300 мл. Объединенные толуольные фазы медленно примешивают к смеси из 960 мл воды и 21,7 мл концентрированной серной кислоты.
После 60-минутного перемешивания при комнатной температуре фазы разделяют, значение рН водной фазы с помощью едкого натра устанавливают на 8,0, промывают ее 300 мл толуола и насыщают поваренной солью. Затем водную фазу смешивают со 150 мл дихлорметана и ее значение рН с помощью едкого натра устанавливают на 12,9. Фазы разделяют и водную фазу трижды экстрагируют 150 мл толуола. Объединенные органические фазы концентрируют. В результате получают 54,5 г продукта в виде коричневого масла. Это масло растворяют в смеси из 330 мл этанола и 6,67 мл воды и значение рН с помощью 35 мл 32%-ной серной кислоты устанавливают на 8,6. Образовавшийся осадок фильтруют и промывают 265 мл этанола. Полученное твердое вещество сушат при 50°С. Таким путем получают 53,8 г соли в виде белых кристаллов с температурой плавления 283,4-289,4°С.
Пример 4
Гемисульфат N-бензил-3β-аминонортропана
Смесь из 89,5 г полугидрата дигидрохлорида N-бензил-3α-аминонортропана, 100 мл воды и 172 мл толуола смешивают с 45 мл 45%-ного едкого натра и полученную смесь перемешивают. Затем фазы разделяют и водную фазу экстрагируют 86 мл толуола. Объединенные толуольные фазы смешивают с 44,8 г 4-диметиламинобензальдегида и 0,22 г n-TsOH и в течение 4 ч кипятят на водоотделителе. После этого реакционную смесь концентрируют. В результате получают 104,2 г N-бензил-3α-(4-диметиламинобензилиден-амино)тропана, который без последующей очистки подвергают дальнейшей переработке.
Смесь из 104,2 г N-бензил-3α-(4-диметиламинобензилиденамино)тропана, 239 мл ДМСО, 24 г 24%-ного раствора трет-бутанолята калия в ТГФ нагревают до 65-70°С с выдержкой при этой температуре в течение 4 ч 15 мин. Затем реакционную смесь распределяют между 300 мл толуола и 900 мл воды, в которой растворен 21 г поваренной соли, после чего водную фазу дважды экстрагируют толуолом порциями по 300 мл. Объединенные толуольные фазы промывают 300 мл воды, в которой растворены 7 г поваренной соли. Полученную таким путем органическую фазу медленно примешивают к смеси из 875 мл воды и 21,8 мл концентрированной серной кислоты.
После 60-минутного перемешивания при комнатной температуре фазы разделяют, значение рН водной фазы с помощью едкого натра устанавливают на 8,0 и промывают ее 300 мл толуола. Далее фазы разделяют, водную фазу смешивают с 300 мл толуола и ее значение рН с помощью едкого натра устанавливают на 12,7. Органическую фазу отделяют, а водную фазу дважды экстрагируют 300 мл толуола. Объединенные органические фазы после экстракции при рН 12,7 концентрируют. В результате получают 58,77 г (90,6% от теории) продукта в виде масла желтоватого цвета. Это масло растворяют в смеси из 330 мл этанола и 6,67 мл воды и значение рН с помощью 35 мл 32%- ной серной кислоты устанавливают на 8,6. Образовавшийся остаток фильтруют и промывают 265 мл этанола. Полученное твердое вещество сушат при 50°С. Таким путем получают 66,65 г (83,7% от теории) соли в виде белых кристаллов с температурой плавления 283,4-284,9°С.
Изобретение относится к способу получения N-замещенных 3β-аминонортропанов формулы I или к одной из их кислотно-аддитивной солей
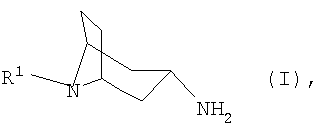
где R1 представляет собой необязательно замещенный остаток, выбранный из группы, включающей С1-С8алкил, С2-С8алкенил, С3-С8циклоалкил и С6-С10арил-С1-С8алкил, отличающийся тем, что либо
а) соответствующий 3-оксонортропан формулы IIА
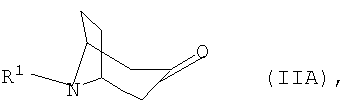 ,
,
подвергают взаимодействию с арилметиламином формулы IIIA
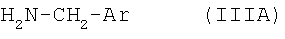 ,
,
в которой Ar обозначает необязательно замещенный фенильный остаток либо необязательно замещенный 5- или 6-членный гетероароматический остаток с по меньшей мере одним гетероатомом, выбранным из группы, включающей N, О и S, либо
б) соответствующий 3α-аминонортропан формулы IIB
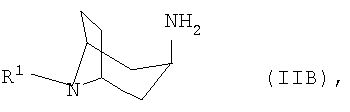
подвергают взаимодействию с арилальдегидом формулы IIIB
 ,
,
полученный в каждом случае имин формулы IVA или IVB
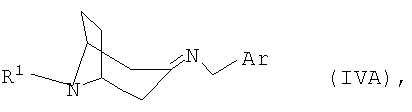
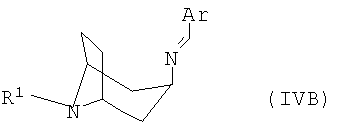
переводят в термодинамически стабильный таутомер, соответственно изомер формулы V
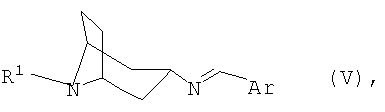
затем гидролизуют и при необходимости переводят в соответствующую кислотно-аддитивную соль.
Полученные соединения формулы I являются ценными промежуточными продуктами в химическом синтезе различных фармацевтических действующих веществ или же сами они представляют собой фармацевтические действующие вещества, прежде всего в качестве модуляторов NMDA-рецептора. Технический результат - способ позволяет получать 3β-аминонортропаны в промышленном масштабе с высоким выходом и с высокой степенью чистоты. 2 н. и 7 з.п. ф-лы.
где R1 представляет собой необязательно замещенный остаток, выбранный из группы, включающей С1-С8алкил, С2-С8алкенил, С3-С8циклоалкил и С6-С10арил-С1-С8алкил, отличающийся тем, что либо
а) соответствующий 3-оксонортропан формулы IIA
 ,
,
подвергают взаимодействию с арилметиламином формулы IIIA
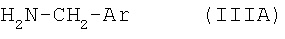 ,
,
в которой Ar обозначает необязательно замещенный фенильный остаток либо необязательно замещенный 5- или 6-членный гетероароматический остаток с по меньшей мере одним гетероатомом, выбранным из группы, включающей N, О и S, либо
б) соответствующий 3α-аминонортропан формулы IIB
подвергают взаимодействию с арилальдегидом формулы IIIB

полученный в каждом случае имин формулы IVA или IVB
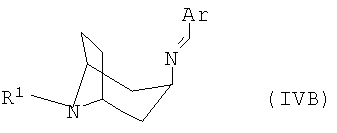
переводят в термодинамически стабильный таутомер, соответственно изомер, формулы V
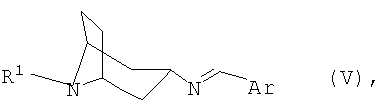
затем гидролизуют и при необходимости переводят в соответствующую кислотно-аддитивную соль.
в которой R1 представляет собой остаток, выбранный из группы, включающей С1 -С8алкил и С6-С10арил-С1-С8алкил, а Ar обозначает незамещенный фенил, либо фенильный остаток одно- или двузамещенный C1-С6алкоксигруппой, или его таутомер, соответственно изомер формулы IVA или IVB.
| Burks E | |||
| John et al | |||
| Organic process research & development, 1(3), 198-210, 1997 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Устройство допускового контроля | 1983 |
|
SU1129571A1 |
| RU 99123933 A, 10.08.2001. | |||

