Настоящее изобретение относится к способу получения N-монофторалкилтропана, способу получения триалкилоловотропана, способу получения йодированного и/или йодированного радиоактивным йодом тропана и применению N-монофторалкилтропана в качестве предшественника в способе получения триалкилоловотропана и/или йодированного и/или йодированного радиоактивным йодом тропана.
Болезнь Паркинсона представляет собой хроническое дегенеративное заболевание центральной нервной системы, поражающее главным образом двигательную систему. Диагностические критерии для облегчения и стандартизации диагностического процесса, особенно на ранних стадиях заболевания, включают замедленность движений (брадикинезию) и ригидность, тремор покоя или постуральную нестабильность. К сожалению, при диагностике двигательных расстройств болезнь Паркинсона ошибочно диагностируется в от 6% до 25% случаев (Suwijn et al. 2015).
С другой стороны, было показано, что изменения в плотности и функции переносчика дофамина проявляются при нейродегенеративных и нейропсихиатрических заболеваниях, таких как болезнь Паркинсона (Niznik et al. 1991). Таким образом, диагноз болезни Паркинсона может быть подтвержден по метаболической активности переносчиков дофамина в базальных ганглиях, непосредственно измеренной с помощью позитронно-эмиссионной томографии (ПЭТ) и однофотонной эмиссионной компьютерной томографии (ОФЭКТ). Переносчик дофамина представляет собой мембраносвязанный белок с 12 предполагаемыми трансмембранными сегментами, расположенными с высокой плотностью в пресинаптических дофаминергических нервных окончаниях.
Обычно радиоизотопные индикаторы для ПЭТ, используемые в исследованиях относительного содержания переносчика дофамина и фармакологии, представляют собой производные кокаина, в частности замещенные тропаны, меченные углеродом-11 или фтором-18.
Мельтцер (Meltzer) и др. описывают синтез замещенных 3-фенилтропанов, в частности, метилового эфира 3β-(3,4-дихлорфенил)тропан-2β-карбоновой кислоты и его склонность к связыванию с сайтами узнавания кокаина или его использование в качестве лиганда для ПЭТ-визуализации, соответственно (Meltzer et al. 1993).
Кэрол (Carroll) и др. раскрывают ряд изомеров кокаина, в частности, метиловые эфиры 3β-(4-замещенного фенил)тропан-2β-карбоновой кислоты, и их связывание в месте связывания с переносчиком дофамина («кокаиновым рецептором») (Carroll et al. 1991).
Маевски (Majewski) и Лазны (Lazny) описывают синтез тропановых алкалоидов путем энантиоселективного депротонирования тропинона хиральными амидами лития в присутствии LiCl (Majewski and Lazny 1995).
Жоу (Zou) и др. раскрывают необратимые лиганды на основе тропана для переносчика дофамина, в частности, замещенный 3α-(дифенилметокси)тропан (бензтропин), такой как 3α-[бис(4'-фторфенил)метокси]тропан, и его синтез из кокаина (Zou et al. 2001).
Гу (Gu) и др. описывают синтез нескольких фторалкил-содержащих производных тропана с высоким сродством и селективностью в отношении переносчика дофамина и их применение для получения 18F-меченых ПЭТ-радиоизотопных индикаторов (Gu et al. 2001). Фторалкил-содержащие производные тропана получают из кокаина. Синтез включает реакцию дегидратации, введение 4-бромфенильного фрагмента, N-деметилирование и алкилирование с помощью алкилирующего агента формулы F-(CH2)n-Br в присутствии основания с получением N-монофторалкилтропана.
В ЕР 0703791 В1 описаны йодированные нейрозонды формулы
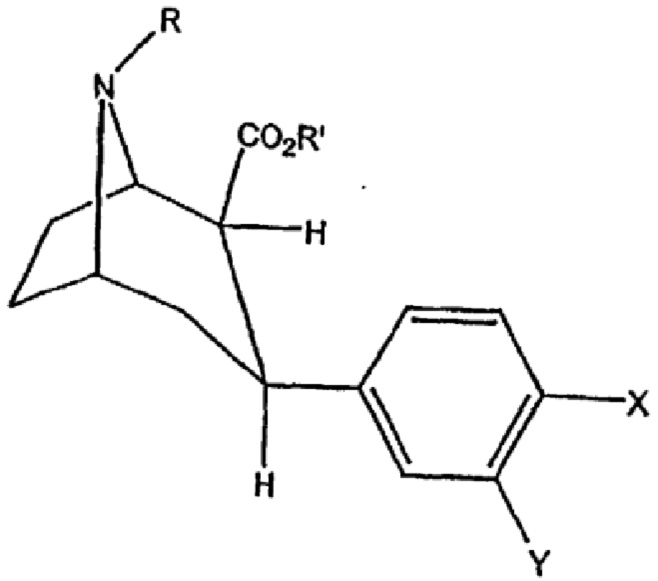
для картирования сайтов обратного захвата моноаминов, где R представляет собой монофторалкильную группу с 18F или 19F, R' представляет собой алкильную группу, Y представляет собой Н и X представляет собой изотоп йода, предпочтительно 2β-карбометокси-3β-(4-йодофенил)-8-(3-фторпропил)нортропан. Предпочтительно изотоп йода представляет собой 123I или 125I. Кроме того, в ЕР 0703791 В1 описаны соединения триалкилолова в качестве предшественников йодированных нейрозондов.
В ЕР 0988263 В1 описано получение йодированных радиоактивным йодом нейрорецепторных агентов, где предшественники нейрорецепторных агентов в виде триалкилолова превращают в йодированный радиоактивным йодом нейрорецепторный агент путем замены группы триалкилолова в предшественнике нейрорецепторного агента на группу радиоактивного йода, в частности 123I, в присутствии окислителя, предпочтительно хлорамина Т, при использовании диапазона рН, который регулируют с помощью буферной системы, с последующим отделением продукта радиоактивно меченного нейрорецепторного агента от побочных продуктов с помощью способов хроматографического разделения, предпочтительно ВЭЖХ, с использованием системы нетоксичных растворителей, содержащей этанол и воду.
Кроме того, в WO 2011/073256 А1 описано получение N-монофторалкилтропанов с использованием фторалкилйодидов или сложных эфиров фторалкилсульфонатов и их применение в качестве предшественников для получения нерадиоактивного тропана FP-CIT и 123I-меченого радиофармацевтического препарата DaTSCAN™ (123I-йофлупан или 123I-FP-CIT).
Рами-Марк (Rami-Mark) и др. описывают синтез ПЭТ-радиоизотопных индикаторов для переносчиков дофамина, начинающийся с кокаина, за шесть стадий реакции, включая введение 4-йодофенильного фрагмента и N-деметилирование (Rami-Mark et al. 2013).
Недостаток раскрытого синтеза состоит в применении кокаина в качестве реагента. Поскольку кокаин подпадает под действие закона об обращении с наркотическими средствами, требуются большие усилия в отношении применения и контроля за способами обращения наркотиков. Кроме того, путь синтеза, начинающийся с кокаина, требует более сложного синтеза.
Томпсон (Thompson) и др. раскрывают циклические аминокислоты, содержащие тропановый остов, в частности, для включения в синтетические пептиды. Синтез начинается с соединения (R)-2-карбометокси-3-тропинона или кокаина (Thompson et al. 1997). Начиная с (R)-2-карбометокси-3-тропинона, карбонильную группу восстанавливают до гидроксильной группы и затем проводят дегидратацию. (R)-2-карбометокси-3-тропинон получают путем разделения с помощью (+)- и (-)-винной кислоты.
Неймейер (Neumeyer) и др. описывают радио изотопные индикаторы для ПЭТ для визуализации переносчика дофамина, в частности, N-ω-фторалкильных аналогов (1R)-2β-карбометокси-3β-(4-йодофенил)тропана (β-CIT), и синтез триалкилоловотропана и йодированного радиоактивным йодом тропана, начиная с (1R)-2β-карбометокси-3β-(4-йодофенил)тропана (Neumeyer et al. 1994).
Задача настоящего изобретения состоит в разработке нового способа синтеза N-монофторалкилтропанов, в частности, исключающих применение кокаина в качестве исходного материала.
Кроме того, задача настоящего изобретения состоит в разработке способа с уменьшенным числом стадий синтеза.
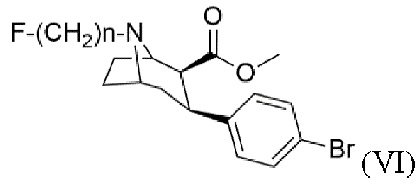
Задача была решена с помощью способа получения N-монофторалкилтропана формулы (VI)
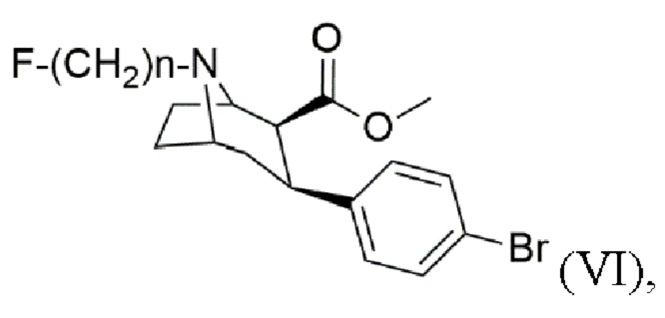
где n составляет 2, 3 или 4, включающего стадии:
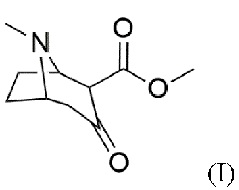
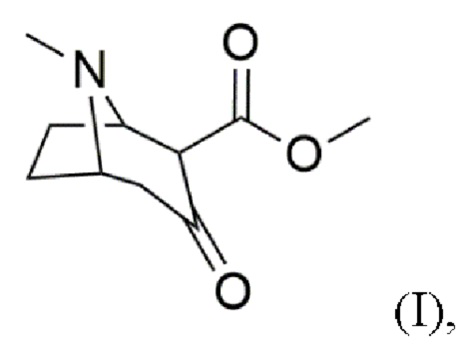
a) предоставление соединения формулы (I)
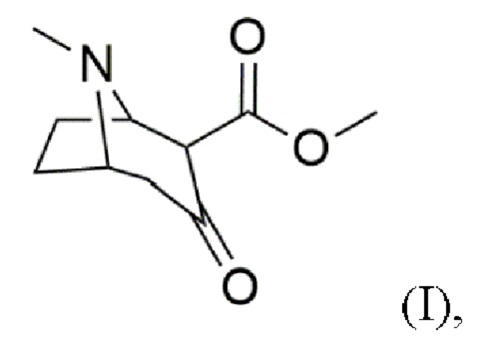
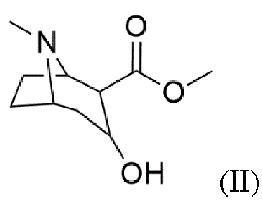
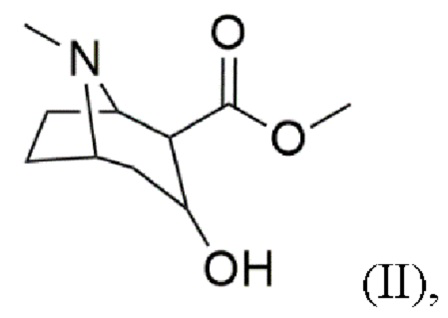
b) восстановление карбонильной группы в гидроксильную группу с получением соединения формулы (II)
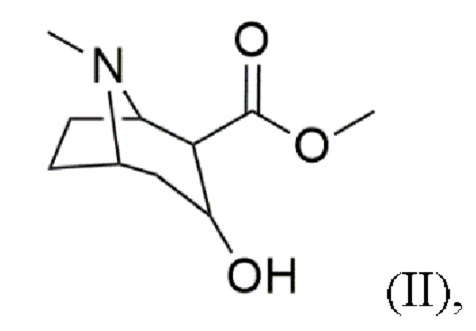
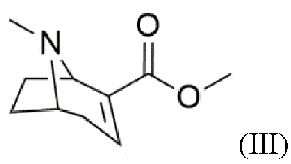
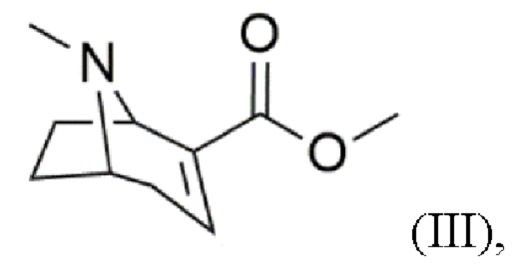
c) реакцию дегидратации с получением соединения формулы (III)
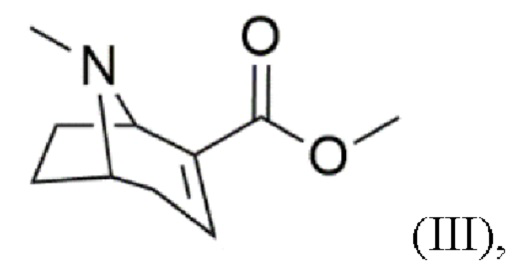
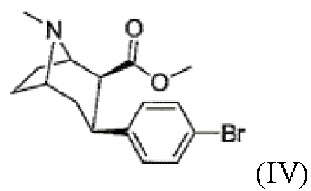
d) присоединение 4-бромфенильного фрагмента с получением соединения формулы
(IV)
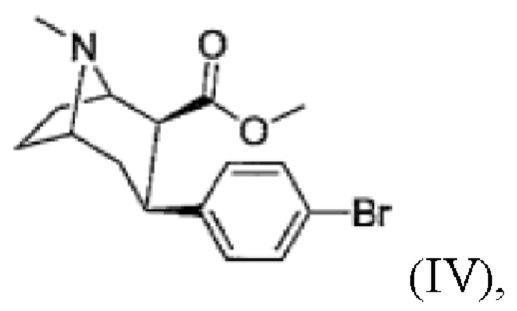
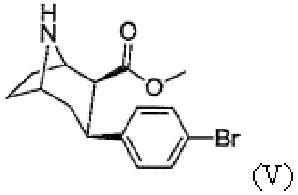
e) N-деметилирование с получением соединения формулы (V)

f) алкилирование с помощью алкилирующего агента формулы F-(CH2)n-Br в присутствии основания с получением N-монофторалкилтропана формулы (VI), где n составляет 2, 3 или 4.
Согласно изобретению способ проводят в порядке стадий (а), (b), (с), (d), (е) и (f).
Вместе с соединениями представленных формул, в частности, формул (I), (II), (III), (IV) и (V), включены все изомеры, в частности, диастереомеры, энантиомеры и таутомеры показанных химических структур; и показано целевое соединение, в частности, желательный энантиомер.
Разделение диастереомеров и/или энантиомеров происходит после каждой стадии или один раз после стадии (f). Предпочтительно диастереомеры разделяют после каждой стадии методом хроматографии. Предпочтительно энантиомеры разделяют один раз после стадии (f).
Преимущественно в изобретении предложен способ получения N-монофторалкилтропанов без использования кокаина. Кроме того, преимущественно получение N-монофторалкилтропанов проводят с сокращенным числом стадий синтеза, предпочтительно с 6 стадиями. Кроме того, способ согласно изобретению обеспечивает выход около 8% и, таким образом, соответствует синтезу, в котором исходным реагентом является кокаин.
Предпочтительно N-монофторалкилтропан формулы (VI), полученный способом согласно изобретению, имеет высокую энантиомерную чистоту, предпочтительно энантиомерную чистоту по меньшей мере 99:1, более предпочтительно по меньшей мере 99,5:0,5. Преимущественно энантиомерная чистота N-монофторалкилтропана формулы (VI) имеет решающее значение при использовании в качестве предшественника для получения йодированного радиоактивным йодом тропана формулы (VIII), поскольку этот энантиомер показывает наивысшую склонность к связыванию с переносчиком дофамина. Преимущественно, качество, в частности энантиомерная чистота, продукта, полученного способом согласно изобретению, соответствует продуктам, синтезированным, начиная с энантиомерно чистого кокаина.
N-монофторалкилтропан формулы (VI) более подходит в качестве предшественника для синтеза триалкилоловотропана формулы (VII) и/или йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII), чем соответствующее йодидное соединение, благодаря более высокой стабильности по сравнению с его йодсодержащим производным (VIII).
В воплощениях соединение формулы (I) предоставляют согласно стадии (а) путем добавления карбометоксигруппы в положение 2 в 3-тропинон согласно формуле (Ia)
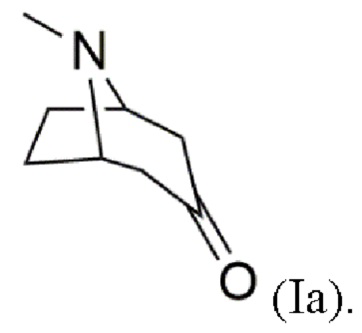
В других воплощениях карбометоксилирование проводят с использованием сильного основания, такого как гидрид натрия (NaH), диизопропиламид лития (LDA) или н-бутиллитий (н-BuLi), и карбометоксигруппы, такой как диметилкарбонат или метилцианоформиат (СН3ОС(O)CN).
Предпочтительно добавление карбометоксигруппы в положение 2 осуществляют с использованием гидрида натрия (NaH) и диметилкарбоната.
В некоторых воплощениях карбометоксилирование катализируется метанолом.
В других воплощениях соединение формулы (I) предоставляют в соответствии со стадией (а) с использованием одного из способов, описанных Финдлеем (Findlay 1957).
В других воплощениях соединение формулы (I) предоставляют в соответствии со стадией (а) путем реакции сукциндиальдегида с метиламином и монометиловым эфиром ацетондикарбоновой кислоты.
В воплощениях восстановление карбонильной группы в гидроксильную группу согласно стадии (b) проводят с использованием по меньшей мере одного восстановителя, выбранного из водорода (H2), амальгамы натрия/H2SO4 и гидридов, таких как боргидрид натрия (NaBH4). Предпочтительно восстановителем является водород (H2) или NaBH4, более предпочтительно NaBH4. Преимущественно эти восстановители восстанавливают карбонильную группу в гидроксильную группу без восстановления сложноэфирной группы.
В других воплощениях реакцию дегидратации согласно стадии (с) проводят с использованием реагента, выбранного из группы, включающей фосфорилхлорид (POCl3), соляную кислоту (HCl), бромоводородную кислоту (HBr) или 4-диметиламинопиридин (DMAP), триэтиламин и ангидрид трифторуксусной кислоты (TFAA). Примечательно, что в случае POCl3, HCl и HBr необходима последующая этерификация метанолом.
Предпочтительно реакцию дегидратации согласно стадии (с) проводят с использованием 4-диметиламинопиридина (DMAP), триэтиламина и ангидрида трифторуксусной кислоты (TFAA) по одностадийной методике.
В воплощениях присоединение 4-бромфенильного фрагмента согласно стадии (d) проводят с помощью реакции с металлоорганическим соединением, предпочтительно с помощью реакции Гриньяра.
В данной заявке термин «реакция с металлоорганическим соединением» относится к химической реакции, в которой металлоорганическое соединение используют для присоединения органической группы, предпочтительно алкильной, винильной или арильной группы, к другой органической молекуле путем образования углерод-углеродных связей. Используемый в данной заявке термин «металлоорганическое соединение» относится к соединению, имеющему по меньшей мере одну химическую связь между атомом углерода органической молекулы и металлом, предпочтительно щелочным, щелочноземельным или переходным металлом.
Используемый в данной заявке термин «реакция Гриньяра» относится к реакции с металлоорганическим соединением, в которой алкил-, винил- или арилгалогениды магния (реагенты Гриньяра) присоединяются в качестве нуклеофила к электрофильной группе, такой как карбонильная группа альдегида или кетона.
В альтернативных воплощениях присоединение 4-бромфенильного фрагмента согласно стадии (d) проводят путем арилирования производного тропана и затем реакции Зандмейера (Sandmeyer) или превращения в SiMe3-Br.
Используемый в данной заявке термин «реакция Зандмейера» относится к химической реакции, в частности, к радикально-нуклеофильному ароматическому замещению, в котором используют соли меди.
В других воплощениях N-деметилирование согласно стадии (е) проводят с использованием хлорэтилхлорформиатов или 2,2,2-трихлорэтоксикарбонилхлорида (TrocCl), цинка и уксусной кислоты. Предпочтительно N-деметилирование согласно стадии (е) проводят с использованием хлорэтилхлорформиатов.
Согласно изобретению алкилирование с помощью алкилирующего агента формулы F-(CH2)n-Br на стадии (f) проводят в присутствии основания с получением N-монофторалкилтропана формулы (VI), в которой п составляет 2, 3 или 4. Предпочтительно, алкилирование проводят с помощью алкилирующего агента 1-бром-3-фторпропана формулы F-(CH2)3-Br.
Наиболее предпочтительно, п составляет 3.
В данной заявке термин «основание» относится к соединению, которое принимает протоны от любого донора протонов. Подходящие основания известны специалистам в данной области. Предпочтительно основание на стадии (f) представляет собой карбонат калия.
В других воплощениях способ согласно изобретению дополнительно включает одно хиральное разделение, предпочтительно после алкилирования на стадии (f).
Преимущественно, хиральное разделение после алкилирования на стадии (f) требует меньших усилий и колонок меньшего размера или, соответственно, неподвижной фазы меньшего размера. Преимущественно, хиральное разделение после алкилирования на стадии (f) приводит к уменьшению количества анализов, в частности, анализов для определения энантиомерной чистоты проводят только после хирального разделения после стадии (f), а не после каждой стадии.
В других воплощениях хиральное разделение проводят методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хиральной неподвижной фазы. В данной заявке термин «хиральная неподвижная фаза» относится к неподвижной фазе для колоночной хроматографии, содержащей единственный энантиомер хирального соединения. Преимущественно, два энантиомера одного и того же соединения различаются по сродству к неподвижной фазе единственного энантиомера. Таким образом, использование хиральной неподвижной фазы приводит к высокой энантиомерной чистоте N-монофторалкилтропана формулы (VI), предпочтительно энантиомерной чистоте по меньшей мере 99:1, более предпочтительно по меньшей мере 99,5:0,5. Предпочтительно, хиральная неподвижная фаза состоит из материала на основе целлюлозы или амилозы.
В воплощениях подвижная фаза представляет собой органический растворитель, предпочтительно по меньшей мере один органический растворитель, выбранный из группы, включающей алканы, хлоралканы и алкиламины.
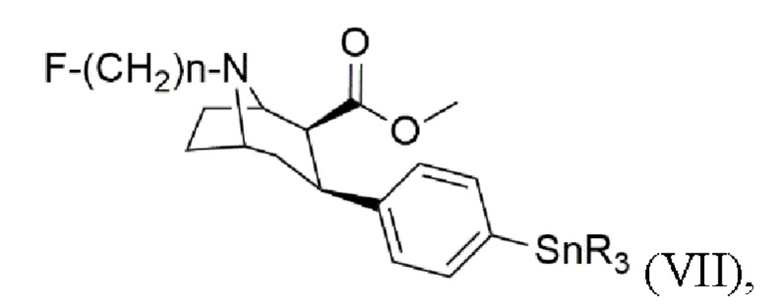
В других воплощениях способ согласно изобретению дополнительно включает стадии получения триалкилоловотропана формулы (VII)
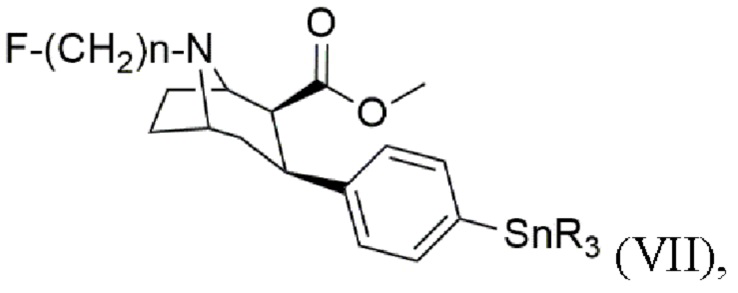
где n составляет 2, 3 или 4, при этом каждый R представляет собой С1-С4 алкильную группу, предпочтительно метальную или бутильную группу, группа, более предпочтительно метальную группу,
и/или получения йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII)

где n составляет 2, 3 или 4, при этом XI i представляет собой по меньшей мере один изотоп йода, предпочтительно 123I, 124I и/или 127I, более предпочтительно 123I или 124I; и/или получения фармацевтической композиции, предпочтительно способ согласно изобретению дополнительно включает стадии получения триалкилоловотропана формулы (VII) и получения йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII).
Согласно изобретению каждый R независимо выбирают из С1-С4 алкильной группы. Предпочтительно, все три заместителя R являются одинаковыми. В данной заявке термин «С1-С4 алкильная группа» относится к метальной, этильной, пропильной или бутильной группе.
Настоящее изобретение дополнительно включает способ получения триалкилоловотропана формулы (VII)
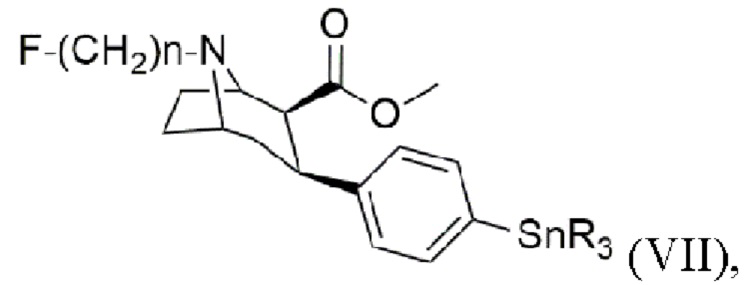
где n составляет 2, 3 или 4,
при этом каждый R представляет собой С1-С4 алкильную группу, предпочтительно метальную или бутильную группу, более предпочтительно метальную группу, включающий стадии:
(i) проведение способа по одному из пп. 1-9 с получением N-монофторалкилтропана формулы (VI),
(ii) взаимодействие соединения формулы (VI) со стадии (i) с Sn2R6 в присутствии по меньшей мере одного катализатора с получением триалкилоловотропана формулы (VII),
где каждый R представляет собой С1-С4 алкильную группу, предпочтительно метальную или бутильную группу, более предпочтительно метальную группу.
Настоящее изобретение также включает способ получения йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII)
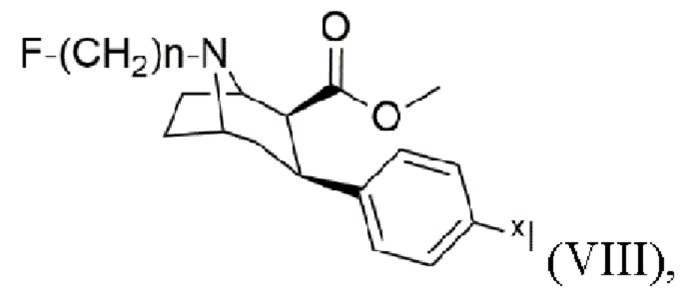
где n составляет 2, 3 или 4,
при этом xI представляет собой по меньшей мере один изотоп йода, предпочтительно 123I, 124I и/или 127I, более предпочтительно 123I или 124I, включающий стадии:
(1) проведение способа по одному из пп. 1-9 с получением N-монофторалкилтропана формулы (VI),
(2) проведение способа по п. 10 с получением триалкилоловотропана формулы (VII),
(3) взаимодействие триалкилоловотропана формулы (VII) со стадии (2) с источником xI в присутствии по меньшей мере одного окислителя с получением йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII),
причем xI представляет собой по меньшей мере один изотоп йода, предпочтительно 123I, 124I и/или 127I, более предпочтительно 123I или 124I.
В воплощениях xI представляет собой 123I или 124I или смесь 123I и 127I или смесь 124I и 127I.
Предпочтительно, xI представляет собой 123I.
В других воплощениях способ получения йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII) дополнительно включает приготовление фармацевтической композиции.
В воплощениях фармацевтическая композиция содержит фармацевтически приемлемый разбавитель или носитель. Предпочтительно фармацевтическая композиция представлена в форме, подходящей для внутривенного введения.
В качестве носителя предпочтительно используют воду, забуференную воду, 0,9%-ный солевой раствор, раствор глицина и подобные растворители. Растворы стерильны. Фармацевтические композиции стерилизуют обычными хорошо известными способами. Композиции содержат предпочтительно фармацевтически приемлемые эксципиенты, например, те, которые необходимы для обеспечения примерно физиологических условий и/или повышения стабильности, такие как агенты для регулирования значения рН и буферные агенты, предпочтительно выбранные из ацетата натрия, хлорида натрия, цитрата натрия, фосфата калия, хлорида калия, хлорида кальция, лактата натрия и гистидина.
Фармацевтические композиции должны быть стерильными и стабильными в условиях производства и хранения. Композиция может быть приготовлена в виде раствора, микроэмульсии, дисперсии, в липосомах или в других упорядоченных структурах, подходящих для этой цели и известных специалистам.
В предпочтительных воплощениях фармацевтическая композиция содержит смесь йодированного и йодированного радиоактивным йодом тропана формулы (VIII), где xI представляет собой 123I и 127I.
Доза йодированного и йодированного радиоактивным йодом тропана формулы (VIII) в фармацевтической композиции предпочтительно составляет от 0,07 мкг/мл до 0,13 мкг/мл.
Хотя изобретение описывает различные дозировки, специалисту в данной области будет понятно, что конкретный уровень дозы и частота дозирования для любого конкретного субъекта могут варьироваться и будут зависеть от множества факторов. Эти факторы включают возраст, массу тела, общее состояние здоровья, пол, диету, способ и время введения, скорость выведения, комбинацию лекарственных средств и организм, в отношении которого поставлен диагноз. Однако обычно дозировка будет приблизительно соответствовать той, которая типична для известных способов введения конкретного соединения. В воплощениях типичная доза йодированного радиоактивным йодом тропана формулы (VIII) будет находиться в диапазоне от 111 МБк до 185 МБк 123I.
Другим аспектом изобретения является применение N-монофторалкилтропана формулы (VI)
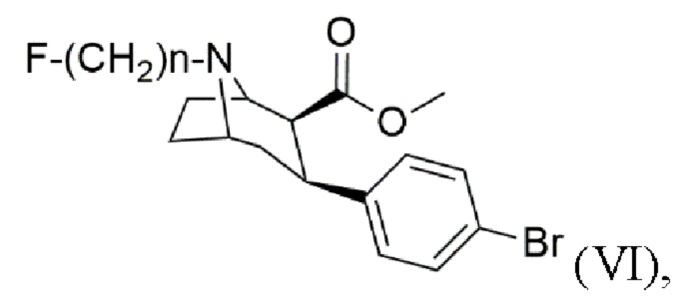
гдеп n составляет 2, 3 или 4;
в качестве предшественника в способе получения триалкилоловотропана формулы (VII)
где каждый R представляет собой С1-С4 алкильную группу, предпочтительно метальную или бутильную группу, более предпочтительно метальную группу; и/или йодированного и/или йодированного радиоактивным йодом тропана формулы (VII)

где xI представляет собой по меньшей мере один изотоп йода, предпочтительно 123I, 124I и/или 127I, более предпочтительно 123I или 124I.
N-монофторалкилтропан формулы (VI) является более подходящим в качестве предшественника для синтеза триалкилоловотропана формулы (VII) и/или йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII), чем соответствующее йодидное соединение.
Предпочтительно, N-монофторалкилтропан формулы (VI) используют в качестве предшественника для применения при диагностике болезни Паркинсона.
В воплощениях N-монофторалкилтропан формулы (VI), содержащий диастереомеры, энантиомеры и/или таутомеры, используют в качестве предшественника в способе получения триалкилоловотропана формулы (VII) и/или йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII). Предпочтительно, N-монофторалкилтропан формулы (VI) используют в качестве предшественника в способе получения триалкилоловотропана формулы (VII) и/или йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII) с чистотой по меньшей мере 98% ее, более предпочтительно по меньшей мере 99% ее.
Еще одним аспектом изобретения является способ диагностики болезни Паркинсона, включающий введение йодированного радиоактивным йодом и возможно йодированного тропана формулы (VIII), полученного способом согласно изобретению, предпочтительно путем введения йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII) или путем введения йодированного радиоактивным йодом тропана формулы (VIII).
В воплощениях способ диагностики болезни Паркинсона проводят с помощью следующих стадий:
(a) проведение способа по изобретению с получением N-монофторалкилтропана формулы (VI),
(b) взаимодействие соединения формулы (VI) со стадии (а) с Sn2R6 в присутствии по меньшей мере одного катализатора с получением триалкилоловотропана формулы (VII),
где каждый R представляет собой С1-С4 алкильную группу,
(c) взаимодействие триалкилоловотропана формулы (VII) со стадии (b) с источником xI в присутствии по меньшей мере одного окислителя с получением йодированного радиоактивным йодом и возможно йодированного тропана формулы (VIII),
где xI представляет собой по меньшей мере один изотоп йода, предпочтительно 123I или 124I и возможно 127I, более предпочтительно 123I или 124I,
(d) введение субъекту йодированного радиоактивным йодом и возможно йодированного тропана формулы (VIII).
В воплощениях способ диагностики болезни Паркинсона включает введение фармацевтической композиции йодированного радиоактивным йодом и возможно йодированного тропана формулы (VIII) на стадии (d).
В других воплощениях йодированный радиоактивным йодом тропан формулы (VIII) вводят в эффективном количестве.
В воплощениях объект представляет собой млекопитающее, предпочтительно человека.
В воплощениях фармацевтическую композицию вводят системно. Используемый в данной заявке термин «системное введение» или «вводимый системно» относится к фармацевтическим композициям, которые вводятся в кровоток субъекта и перемещаются по всему телу субъекта, достигая части тела субъекта, нуждающейся в диагностике, в эффективной дозе, прежде чем расщепляются путем метаболизма и выводятся из организма. Системное введение фармацевтических композиций можно осуществлять путем инъекции.
Фармацевтические композиции предпочтительно вводят парентерально, особенно предпочтительно внутривенно. В одном воплощении изобретения парентеральная фармацевтическая композиция существует в форме введения, подходящей для инъекции. Поэтому особенно предпочтительными композициями являются растворы, эмульсии или суспензии йодированного радиоактивным йодом тропана формулы (VIII), присутствующие в фармацевтически приемлемом разбавителе или носителе.
В другом воплощении можно объединять ранее описанные воплощения. Различные изменения и модификации в пределах сущности и объема раскрытого изобретения станут очевидными для специалистов в данной области техники после прочтения описания и других частей настоящей заявки.
Настоящее изобретение далее поясняется следующими неограничивающими чертежами и примерами.
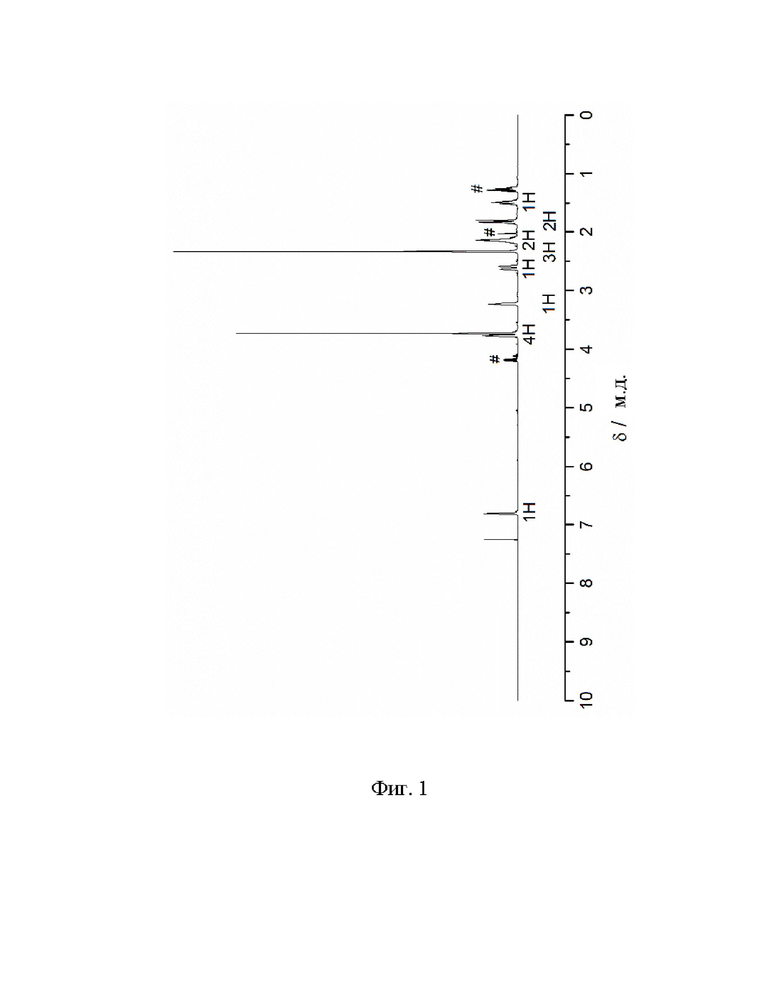
Фиг. 1: 1H ЯМР спектр метил-8-метил-8-азабицикло[3.2.1]окт-2-ен-2-карбоксилата (III) в CDCl3. Помеченные химические сдвиги относятся к остаточному растворителю.
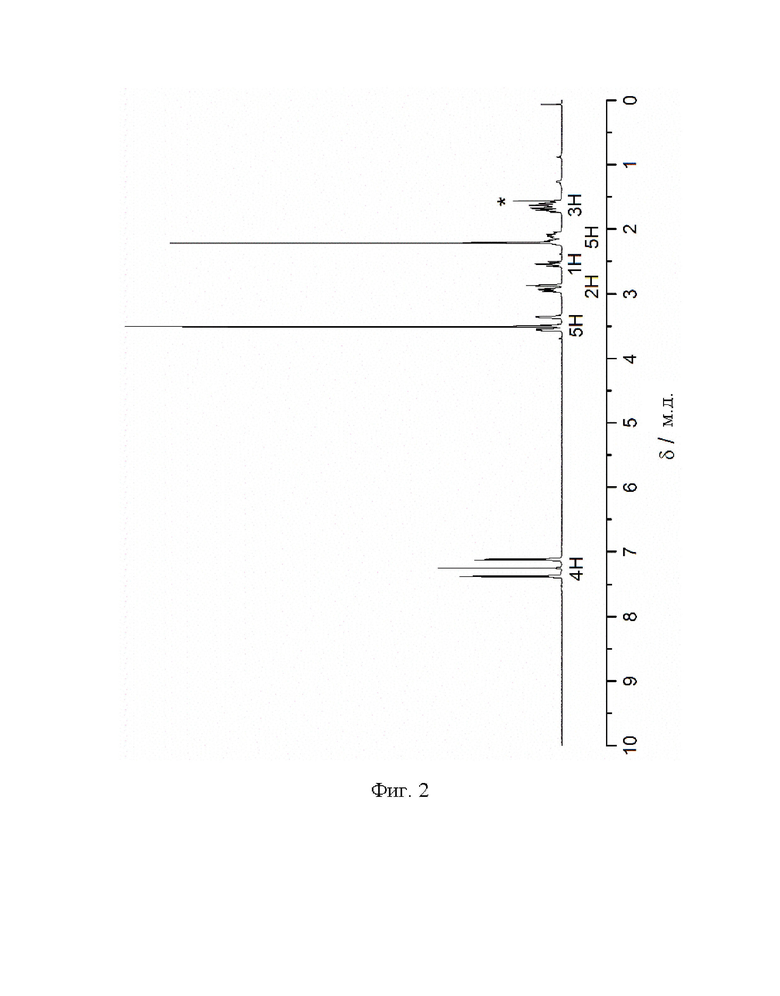
Фиг. 2: 1H ЯМР спектр метил-3-(4-бромфенил)-8-метил-8-азабицикло[3.2.1]октан-2-карбоксилата (IV) в CDCl3. Звездочкой отмечена вода.
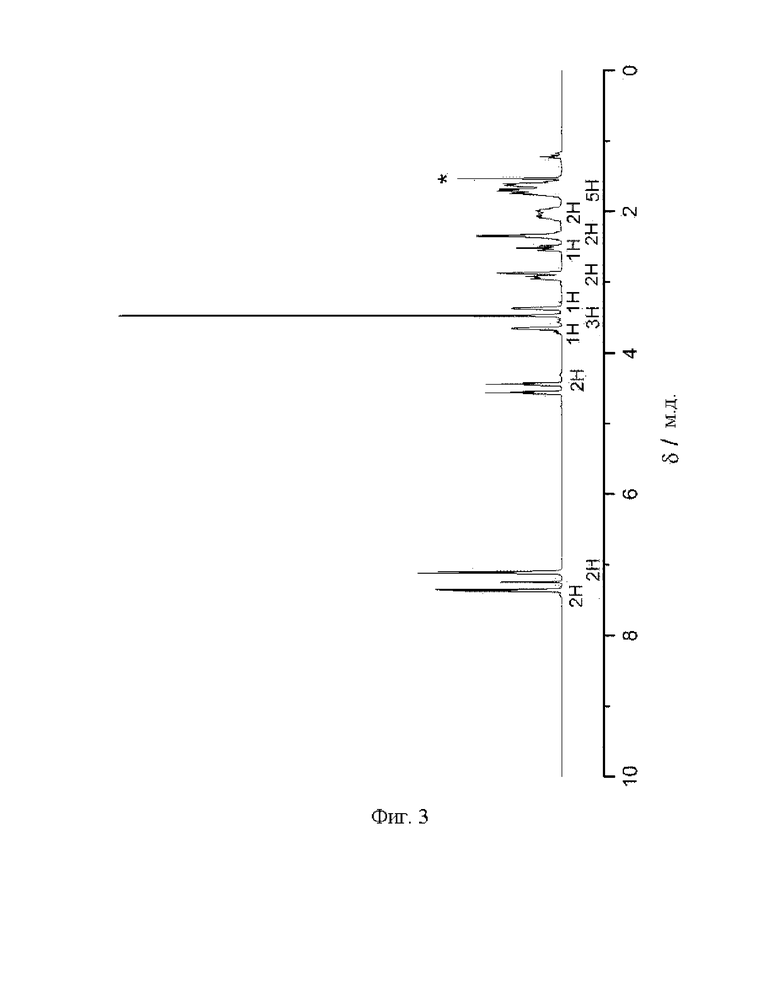
Фиг. 3: 1H ЯМР спектр (1R,2S,3S,5S)-метил-3-(4-бромфенил)-8-(3-фторпропил)-8-азабицикло[3.2.1]октан-2-карбоксилата (VI) в CDCl3. Звездочкой отмечена вода.
Фиг. 4: 19F ЯМР спектр (1R,2S,3S,5S)-метил-3-(4-бромфенил)-8-(3-фторпропил)-8-азабицикло[3.2.1]октан-2-карбоксилата (VI) в CDCl3.
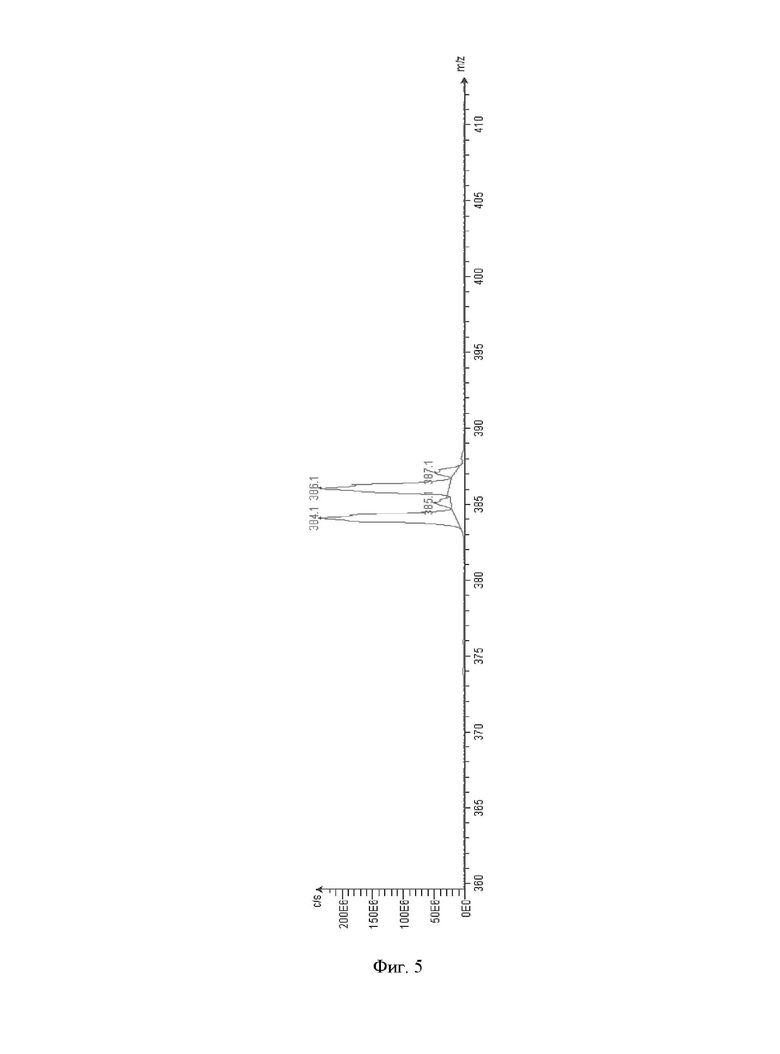
Фиг. 5: ESI-MS (1R,2S,3S,5S)-метил-3-(4-бромфенил)-8-(3-фторпропил)-8-азаби-цикло[3.2.1]октан-2-карбоксилата (VI).
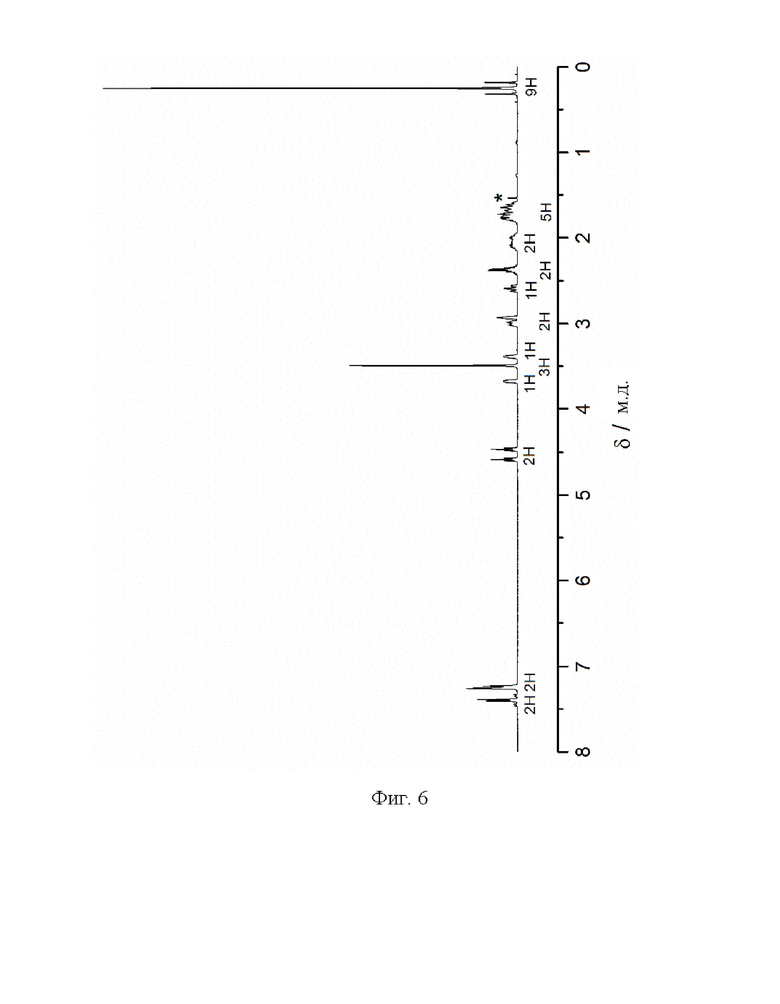
Фиг. 6: 1H ЯМР спектр (1R,2S,3S,5S)-метил-8-(3-фторпропил)-3-[4-(триметил-станнил)фенил]-8-азабицикло[3.2.1]октана-2-карбоксилата (VI) в CDCl3. Звездочкой отмечена вода.
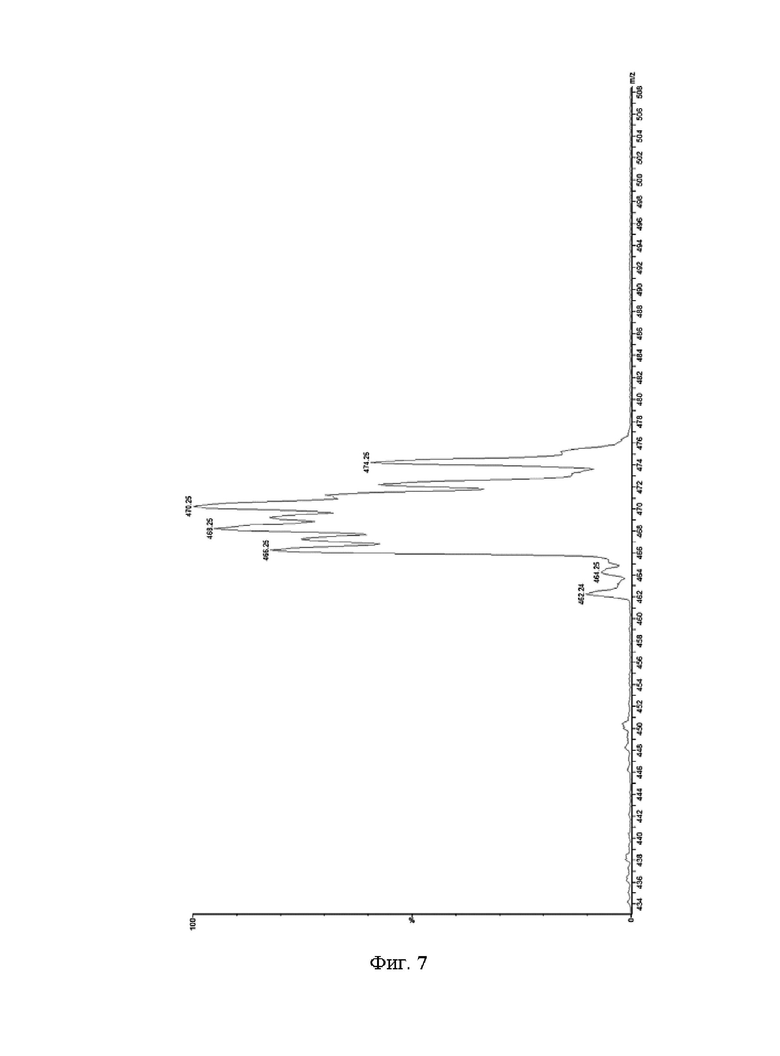
Фиг. 7: ESI-MS (1R,2S,3S,5S)-метил-8-(3-фторпропил)-3-[4-(триметилстаннил)-фенил]-8-азабицикло[3.2.1]октана-2-карбоксилата (VII).
Пример 1: Синтез 2-карбометокситропинона (I, метил-8-метил-3-оксо-8-азабицикло[3.2.1]октан-4-карбоксилата)
В атмосфере аргона раствор диметилкарбоната (2,2 экв.) в циклогексане добавляют к гидриду натрия (2 экв.) и нагревают до 70°С. После медленного добавления раствора тропинона (1 экв.) в циклогексане температуру повышают до температуры кипения с обратным холодильником. Суспензию кипятят с обратным холодильником в атмосфере аргона в течение 30 мин и затем осторожно добавляют каталитическое количество метанола. После этого реакционную смесь кипятят с обратным холодильником в течение ≥4 ч. После охлаждения до комнатной температуры суспензию гасят и экстрагируют водным раствором хлорида аммония. Циклогексановую фазу дополнительно экстрагируют водой. Затем водную фазу несколько раз экстрагируют хлороформом. Объединенные фазы хлороформа сушат над сульфатом магния и фильтруют. Использование роторного испарителя дает коричневый маслянистый продукт.
Выход: 89%
Пример 2: Синтез метил-3-гидрокси-8-метил-8-азабицикло[3.2.1]октан-2-карбоксилата (II)
Синтезированный 2-карбометокситропинон (I, 1 экв.) непосредственно используют в синтезе (II). Поэтому 2-карбометокситропинон растворяют в метаноле и охлаждают до ≤- 30°С, после чего добавляют борогидрид натрия (3 экв.). После реакции смесь медленно нагревают и добавляют к хлориду аммония. Использование роторного испарителя дает желтую суспензию, которая растворяется в воде. Затем для подщелачивания смеси используют концентрированный аммиак. После этого смесь экстрагируют хлороформом. Объединенные органические фазы сушат над сульфатом магния и фильтруют. Использование роторного испарителя дает маслянистый остаток.
Выход: 87%
Пример 3: Синтез метил-8-метил-8-азабицикло[3.2.1]окт-2-ен-2-карбоксилата (III) Диметиламинопиридин (0,02 экв.) и триэтиламин (2,9 экв.) добавляют в раствор метил-3-гидрокси-8-метил-8-азабицикло[3.2.1]октан-2-карбоксилата (II; 1 экв.) в дихлорметане. Реакционную смесь охлаждают (≤- 30°С) и по каплям добавляют ангидрид трифторуксусной кислоты (1,3 экв.). После перемешивания при комнатной температуре (>72 часов) добавляют водный раствор карбоната калия. Смесь несколько раз экстрагируют хлороформом. Объединенные органические фазы сушат над сульфатом магния, фильтруют и концентрируют на роторном испарителе. Остаток дважды экстрагируют смесью н-гексана, хлороформа и сульфата магния и затем фильтруют, соответственно. Объединенные экстракты концентрируют с помощью роторного испарителя. Очистку сырого продукта проводят методом колоночной хроматографии в основных условиях. Фракции продукта концентрируют с помощью роторного испарителя. Продукт охарактеризован методом 1Н ЯМР спектроскопии.
Выход: 33%
1Н ЯМР (400,0 МГц, CDCl3, 298 K): δ, м.д., 1,50 (м, 1H), 1,82 (м, 2Н), 2,15 (м, 2Н), 2,34 (с, 3Н), 2,62 (д, 1H), 3,23 (т, 1H), 3,73 (с, 3Н), 3,78 (д, 1H), 6,81 (т, 1H).
Пример 4: Синтез метил-3-(4-бромфенил)-8-метил-8-азабицикло[3.2.1]октан-2-карбоксилата (IV)
В атмосфере аргона 1,4-дибромбензол (2,1 экв.) растворяют в осушенном диэтиловом эфире и перемешивают, добавляя магний (2 экв.) в токе аргона. Смесь нагревают при кипении с обратным холодильником и перемешивают в течение ≥1 ч. Затем реакционную смесь охлаждают до 45°С и по каплям добавляют раствор метил-8-метил-8-азабицикло[3.2.1]окт-2-ен-2-карбоксилата (III, 1 экв.) в дихлорметане. После перемешивания в течение ≥3 ч при - 45°С реакционную смесь гасят раствором трифторуксусной кислоты (2,1 экв.) в дихлорметане при -78°С. После завершения добавления реакционную смесь медленно нагревают при перемешивании на охлаждающей бане. В реакционную смесь добавляют 1 н соляную кислоту и перемешивают в течение 15 мин, при этом рН составляет <2. Кислую водную фазу промывают диэтиловым эфиром. Затем для подщелачивания водной фазы используют концентрированный аммиак, который затем несколько раз экстрагируют дихлорметаном. Объединенную органическую фазу сушат с использованием сульфата магния, фильтруют и концентрируют с помощью роторного испарителя. Очистку сырого продукта проводят методом колоночной хроматографии в основных условиях. Фракции продукта концентрируют с помощью роторного испарителя. Окончательную очистку осуществляют путем кристаллизации. Бесцветное твердое вещество выделяют и сушат в вакууме. Продукт охарактеризован методом 1Н ЯМР спектроскопии. Выход: 40%
1Н ЯМР (400,0 МГц, CDCl3, 298 K): δ, м.д., 1,66 (м, 3Н), 2,15 (м, 5Н), 2,55 (т, 1H), 2,87 (т, 1H), 2,94 (м, 1H), 3,36 (м, 1H), 3,50 (с, 3Н), 3,56 (м, 1H), 7,13 (д, 2Н), 7,38 (д, 2Н).
Пример 5: Синтез метил-3-(4-бромфенил)-8-азабицикло[3.2.1]октан-2-карбоксилата (V)
В атмосфере аргона метил-3-(4-бромфенил)-8-метил-8-азабицикло[3.2.1]октан-2-карбоксилат (IV, 1 экв.) растворяют в осушенном 1,2-дихлорэтане и добавляют 1-хлорэтилхлорформиат (4 экв.). Раствор перемешивают в течение ≥16 ч при кипении с обратным холодильником. Затем добавляют вторую порцию 1-хлорэтилхлорформиата (1 экв.) и раствор кипятят с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры растворитель удаляют с помощью роторного испарителя. Остаток растворяют в метаноле и перемешивают в течение ≥2 ч при кипении с обратным холодильником. После упаривания растворителя добавляют насыщенный водный раствор гидрокарбоната натрия и водную фазу несколько раз экстрагируют дихлорметаном. Объединенную органическую фазу сушат с использованием сульфата магния, фильтруют и концентрируют с помощью роторного испарителя.
Выход: количественный.
Пример 6: Синтез (1R,2S,3S,5S)-метил-3-(4-бромфенил)-8-(3-фторпропил)-8-азабицикло[3.2.1]октан-2-карбоксилата (VI)
Карбонат калия (2,2 экв.) добавляют в раствор метил-3-(4-бромфенил)-8-азабицикло[3.2.1]октан-2-карбоксилата (V, 1 экв.) в ацетонитриле и перемешивают в течение ≥5 мин. Добавляют 1-бром-3-фторпропан (1,2 экв.) и перемешивают в течение ≥3 ч при кипении с обратным холодильником, после чего добавляют вторую порцию 1-бром-3-фторпропана (0,12 экв.) и перемешивают в течение дополнительного времени ≥1 ч при кипении с обратным холодильником. После охлаждения до комнатной температуры добавляют воду и водную фазу экстрагируют этилацетатом. Объединенную органическую фазу сушат с использованием сульфата магния, фильтруют и концентрируют с помощью роторного испарителя. Очистку сырого продукта проводят методом колоночной хроматографии в основных условиях. Фракции продукта концентрируют с помощью роторного испарителя. Окончательную очистку проводят путем кристаллизации. Бесцветное твердое вещество выделяют и сушат в вакууме.
Выход: 70%.
Рацемическое разделение проводят с помощью препаративной ВЭЖХ. Маслянистый энантиомерно чистый продукт затвердевает путем кристаллизации. Продукт охарактеризован методами 1H ЯМР и 19F ЯМР спектроскопии, по температуре плавления, ВЭЖХ для величины ее, масс-спектрометрии, а также qЯMP.
Выход: 72%.
1Н ЯМР (400,0 МГц, CDCl3, 298 K): δ, м.д., 1,70 (м, 5Н), 2,05 (м, 2Н), 2,37 (м, 2Н), 2,54 (т, 1H), 2,89 (с, 1H), 2,96 (м, 1H), 3,38 (с, 1H), 3,49 (с, 3Н), 3,68 (с, 1H), 4,52 (дт, 2Н), 7,13 (д, 2Н),7,38(д, 2Н);
19F ЯМР (376,3 МГц, CDCl3, 298 K): δ, м.д., -221,2;
ESI-MS: m/z=384, 386;
m.p. 86°С;
величина ее методом ВЭЖХ: 100%; количественный анализ методом qЯМР: ≥99,7%
Пример 7: Синтез (1R,2S,3S,5S)-метил-8-(3-фторпропил)-3-[4-(триметилстаннил)-фенил]-8-азабицикло[3.2.1]октан-2-карбоксилата (VII)
В атмосфере аргона в раствор метил-3-(4-бромфенил)-8-(3-фторпропил)-8-азабицикло[3.2.1]октан-2-карбоксилата (VI, 1 экв.) в толуоле (25±5 мл⋅г-1) добавляют гексаметилдиолово (1,9 экв.), растворенный в толуоле (45±5 мл⋅г-1). В полученную смесь добавляют тетракис(трифенилфосфин)палладий (0) (0,18 экв.) и раствор кипятят с обратным холодильником в течение по меньшей мере 1 часа. Реакционную смесь охлаждают до комнатной температуры и затем фильтруют, в результате чего полученный раствор концентрируют с помощью роторного испарителя. Неочищенный продукт очищают методом препаративной ВЭЖХ в основных условиях. Затем фракции, содержащие целевое соединение, сушат в вакууме на роторном испарителе, а затем лиофилизуют, при этом получают бесцветное масло.
Выход: от 15% до 40%.
1Н ЯМР (400,0 МГц, CDCl3, 298 K): δ, м.д., 0,25 (с, 9Н), 1,70 (м, 5Н), 2,05 (м, 2Н), 2,37 (м, 2Н), 2,59 (дт, 1H), 2,93 (м, 1H), 3,00 (м, 1H), 3,39 (м, 1H), 3,49 (с, 3Н), 3,67 (м, 1H), 4,52 (дт, 2Н), 7,24 (д, 2Н), 7,40 (д, 2Н);
19F ЯМР (376,3 МГц, CDCl3, 298 K): δ, м.д., -221,1;
ESI-MS: m/z=466, 468, 470;
величина ее методом ВЭЖХ: 100%
Пример 8: Синтез (1R,2S,3S,5S)-метил-8-(3-фторпропил)-3-(4-[123I]йодофенил)-8-азабицикло[3.2.1]октан-2-карбоксилата (VIII)
Щелочной раствор [123I]йодида переносят из блока концентрирования в реакционный сосуд. Значение рН регулируют и добавляют носитель путем добавления [127I] йодида, растворенного в фосфатном буфере (общее количество йодида меняется). Затем в реакционный сосуд последовательно добавляют (1R,2S,3S,5S)-метил-8-(3-фторпропил)-3-[4-(триметилстаннил)фенил]-8-азабицикло[3.2.1]-октан-2-карбоксилат (VII, 1 экв.), растворенный в этаноле, и хлорамин-Т (55 экв.), растворенный в воде для инъекций (WFI). Реакционную смесь перемешивают при 30°С в течениег ≥1 минуты. Затем в реакционный сосуд добавляют метабисульфит натрия (279 экв.), растворенный в воде, и перемешивают. Очистку проводят методом препаративной ВЭЖХ. Фракцию продукта выделяют, составляют композицию и затем распределяют по флаконам.
Цитируемая не патентная литература
Carrol FI, Lewin АН, Abraham Р, Parham K, Boja JW, Kuhar MJ (1991) Synthesis and Ligand Binding of Cocaine Isomers at the Cocaine Receptor [Синтез и связывание лигандов изомеров кокаина на кокаиновом рецепторе]. J. Med. Chem. 34, 2719-2725.
Findlay SP (1957) Concerning 2-Carbomethoxytropinone. Contribution from the National Institute of Arthritis and Metabolic Diseases, National Institutes of Health, Public Health Service, U. S. Department of Health, Education and Welfare [О 2-карбометокситропиноне. Вклад Национального института артрита и метаболических заболеваний, Национальных институтов здравоохранения, Службы общественного здравоохранения, Министерства здравоохранения, образования и социального обеспечения США]. 22, 1385-1394.
Gu Х-Н, Zong R, Kula NS, Baldessarini RJ, Neumeyer JL (2001) Synthesis and biological evaluation of a series of novel N- or O-fluoroalkyl derivatives of tropane: potential positron emission tomography (PET) imaging agents for the dopamine transporter [Синтез и биологическая оценка ряда новых N- или О-фторалкильных производных тропана: потенциальные агенты для визуализации для позитронно-эмиссионной томографии (ПЭТ) для переносчика дофамина], BIOORGANIC & MEDICINAL CHEMISTRY LETTERS 11 (23), 3049-3053.
Majewski M, Lazny R (1995) Synthesis of Tropane Alkaloids via Enantioselective Deprotonation of Tropinone [Синтез тропановых алкалоидов посредством энантиоселективного депротонирования тропинона]. J. Org. Chem. 60, 5825-5830.
Meltzer PC, Liang AY, Brownell A-L, Elmaleh DR, Madras BK (1993) Substituted 3-Phenyltropane Analogs of Cocaine: Synthesis, Inhibition of Binding at Cocaine Recognition Sites, and Positron Emission Tomography Imaging [Замещенные 3-фенилтропановые аналоги кокаина: синтез, ингибирование связывания в местах распознавания кокаина и позитронно-эмиссионная томография]. J. Med. Chem. 36, 855-862.
Neumeyer J L, Wang S, Gao Y, Milius RA, Kula NS, Campbell A, Baldessarini RJ, Zea-Ponce Y, Baldwin RM, Innis RB (1994) N-ω-Fluoroalkyl Analogs of (l1R)-2β-Carbomethoxy-3β-(4-iodophenyl)-tropane (β-CIT): Radiotracers for Positron Emission Tomography and Single Photon Emission Computed Tomography Imaging of Dopamine Transporters [N-ω-фторалкильные аналоги (1R)-2β-карбометокси-3β-(4)-йодофенил)-тропан (β-CIT): радиофармацевтические индикаторы для визуализации методами позитронно-эмиссионной томографии и однофотонной эмиссионной компьютерной томографии переносчиков дофамина], JOURNAL OF MEDICINAL CHEMISTRY 37 (11), 1558-1561.
Niznik HB, Fogel EF, Fassos FF, Seeman P (1991) The Dopamine Transporter Is Absent in Parkinsonian Putamen and Reduced in the Caudate Nucleus [Переносчик дофамина отсутствует в скорлупе при паркинсонизме и снижен в хвостатом ядре]. J. Neurochem. 56, 192.
Rami-Mark С, Bornat В, Fink С, Otter Р, Ungersboeck J, Vraka С, Haeusler D, Nics L, Spreitzer H, Hacker M, Mitterhauser M, Wadsak W (2013) Synthesis, radiosynthesis and first in vitro evaluation of novel PET-tracers for the dopamine transporter: [11C]IPCIT and [18F]FE@IPCIT [Синтез, радиосинтез и первая оценка in vitro новых ПЭТ-радиоффармацевтических индикаторов переносчика дофамина: [11С]IPCIT и [18F]FE@IPCIT]. BIOORGANIC & MEDICINAL CHEMISTRY 21 (24), 7562-7569.
Suwijn SR, van Boheemen CJM, de Haan RJ, Tissingh G, Booij J, de Bie RMA (2015) The diagnostic accuracy of dopamine transporter SPECT imaging to detect nigrostriatal cell loss in patients with Parkinson's disease or clinically uncertain parkinsonism: a systematic review [Диагностическая точность SPECT визуализации переносчика дофамина для выявления потери нигростриарных клеток у пациентов с болезнью Паркинсона или клинически неопределенным паркинсонизмом: систематический обзор]. EJNMMI Research. 5, 12.
Thompson РЕ, Hearn MTW (1997) Fmoc-protected Tropane-based Amino Acids for Peptide Structure-Function Studies [Fmoc-защищенные аминокислоты на основе тропана для изучения структуры и функции пептидов], TETRAHEDRON LETTERS 38 (16), 2907-2910.
Zou M-F, Kopajtic T, Katz JL, Wirtz S, Justice JB, Newman AH (2001) Novel Tropane-Based Irreversible Ligands for the Dopamine Transporter [Новые необратимые лиганды на основе тропана для переносчика дофамина]. J. Med. Chem. 44, 4453-4461.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОСТОЙ СПОСОБ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2711358C1 |
| ХИНОЛИНОНКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ 5-HT РЕЦЕПТОРОВ | 2005 |
|
RU2394033C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| ЗАМЕЩЕННЫЕ ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2840772C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| АЗАБИЦИКЛИЧЕСКИЕ И ДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2018 |
|
RU2795521C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| ОКСАДИАЗОЛЫ В КАЧЕСТВЕ АГОНИСТОВ МУСКАРИНОВОГО РЕЦЕПТОРА M1 И/ИЛИ M4 | 2019 |
|
RU2817018C2 |

Изобретение относится к способам получения N-монофторалкилтропана, триалкилоловотропана, йодированного тропана, а также к применению N-монофторалкилтропана в качестве предшественника для получения триалкилоловотропана и/или йодированного тропана. Способ получения N-монофторалкилтропана формулы (VI) включает стадии (a)-(f). На этапе (a) предоставляют соединение формулы (I), после чего на этапе (b) восстанавливают его карбонильную группу в гидроксильную группу с получением соединения формулы (II). На стадии (c) осуществляют реакцию дегидратации соединения (II) с использованием 4-диметиламинопиридина, триэтиламина и ангидрида трифторуксусной кислоты с получением соединения формулы (III). На этапе (d) к соединению (III) присоединяют 4-бромфенильный фрагмент с получением соединения (IV). На стадии (e) производят N-деметилирование соединения (IV) с получением смеси, включающей соединение формулы (V) и его энантиомер. На этапе (f) в присутствии основания осуществляют алкилирование производного (V) с помощью F-(CH2)n-Br, где n составляет 2, 3 или 4, с получением смеси, включающей N-монофторалкилтропан формулы (VI) и его энантиомер, при этом энантиомеры разделяют один раз после стадии (f). Технический результат заключается в предоставлении способа получения N-монофторалкилтропанов с уменьшенным числом стадий синтеза, исключающего применение кокаина в качестве исходного материала. 4 н. и 8 з.п. ф-лы, 7 ил., 8 пр.
1. Способ получения N-монофторалкилтропана формулы (VI)
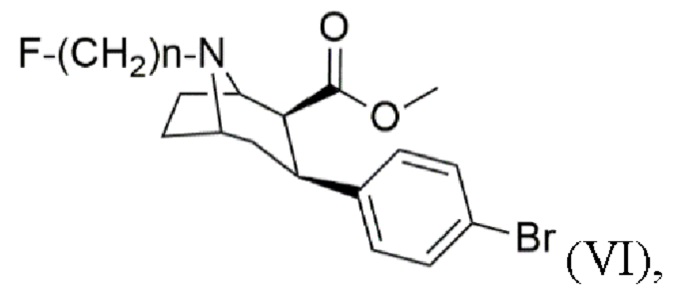
где n составляет 2, 3 или 4, включающий стадии:
(a) предоставление соединения формулы (I)
(b) восстановление карбонильной группы в гидроксильную группу с получением соединения формулы (II) с использованием по меньшей мере одного восстановителя, выбранного из водорода, амальгамы натрия/H2SO4 и гидридов,
(c) реакция дегидратации с получением соединения формулы (III) с использованием 4-диметиламинопиридина, триэтиламина и ангидрида трифторуксусной кислоты,
(d) присоединение 4-бромфенильного фрагмента с получением смеси, включающей соединение формулы (IV) и его энантиомер
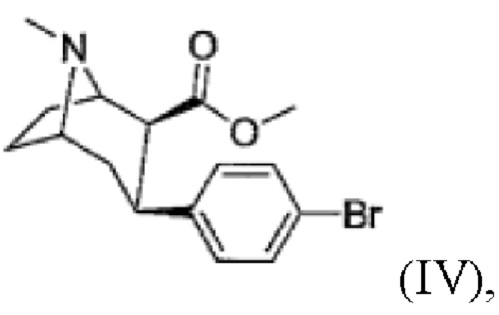
(e) N-деметилирование с получением смеси, включающей соединение формулы (V) и его энантиомер,
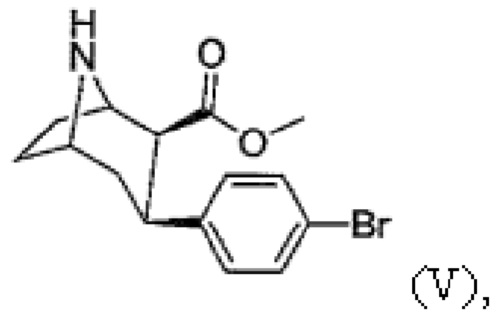
(f) алкилирование с помощью алкилирующего агента формулы F-(CH2)n-Br в присутствии основания с получением смеси, включающей N-монофторалкилтропан формулы (VI) и его энантиомер, где n составляет 2, 3 или 4, в котором энантиомеры разделяют один раз после стадии (f).
2. Способ по п. 1, в котором соединение формулы (I) предоставляют согласно стадии (a) путем присоединения карбометоксигруппы в положение 2 3-тропинона формулы (Ia)
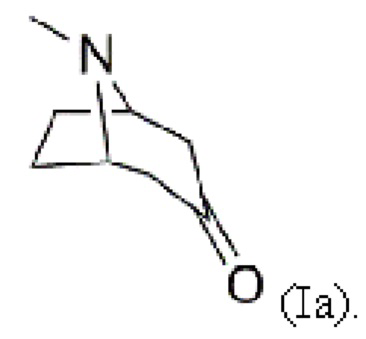
3. Способ по п. 1 или 2, в котором присоединение 4-бромфенильного фрагмента согласно стадии (d) проводят с помощью реакции с металлорганическим соединением, предпочтительно реакции Гриньяра.
4. Способ по одному из пп. 1-3, в котором N-деметилирование согласно стадии (e) проводят с использованием хлорэтилхлорформиатов или 2,2,2-трихлорэтоксикарбонилхлорида, цинка и уксусной кислоты.
5. Способ по одному из пп. 1-4, в котором основание на стадии (f) представляет собой карбонат калия.
6. Способ по одному из пп. 1-5, в котором энантиомеры разделяют посредством хирального разделения.
7. Способ по п. 6, в котором хиральное разделение проводят методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хиральной неподвижной фазы.
8. Способ получения триалкилоловотропана формулы (VII)
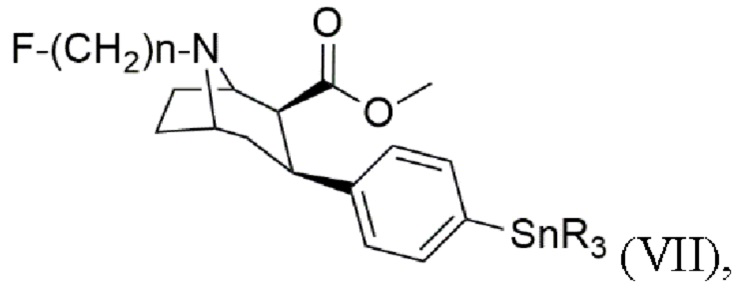
где n составляет 2, 3 или 4,
при этом каждый R представляет собой C1-C4 алкильную группу, включающий стадии:
(i) проведение способа по одному из пп. 1-7 с получением N-монофторалкилтропана формулы (VI),
(ii) реакция соединения формулы (VI) со стадии (i) с Sn2R6 в присутствии по меньшей мере одного катализатора с получением триалкилоловотропана формулы (VII), где каждый R представляет собой C1-C4 алкильную группу.
9. Способ получения йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII)
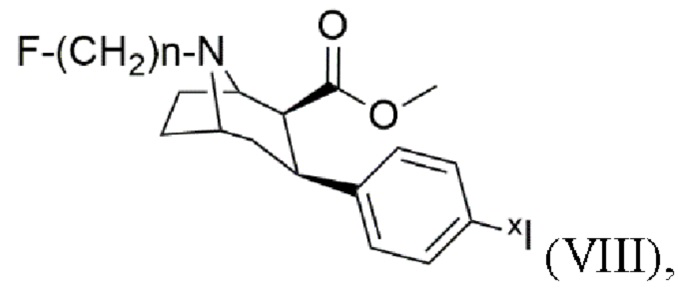
где n составляет 2, 3 или 4,
при этом xI представляет собой по меньшей мере один изотоп йода, включающий стадии:
(1) проведение способа по одному из пп. 1-7 с получением N-монофторалкилтропана формулы (VI),
(2) проведение способа по п. 8 с получением триалкилоловотропана формулы (VII),
(3) взаимодействие триалкилоловотропана формулы (VII) со стадии (2) с источником xI в присутствии по меньшей мере одного окислителя с получением йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII), где xI представляет собой по меньшей мере один изотоп йода.
10. Способ по п. 9, в котором xI представляет собой 123I.
11. Способ по п. 9 или 10, дополнительно включающий приготовление фармацевтической композиции.
12. Применение N-монофторалкилтропана формулы (VI)
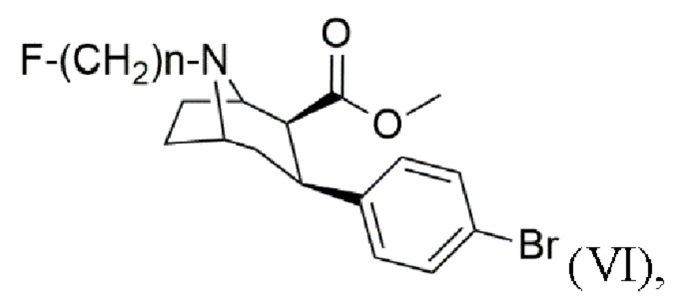
где n составляет 2, 3 или 4, в качестве предшественника в способе получения триалкилоловотропана формулы (VII)
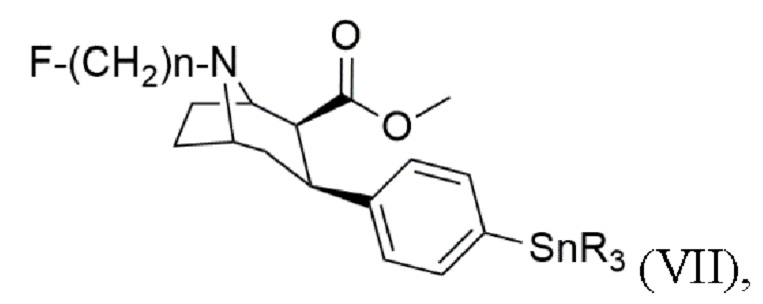
где каждый R представляет собой С1-С4 алкильную группу, предпочтительно метильную или бутильную группу, более предпочтительно метильную группу;
и/или йодированного и/или йодированного радиоактивным йодом тропана формулы (VIII)
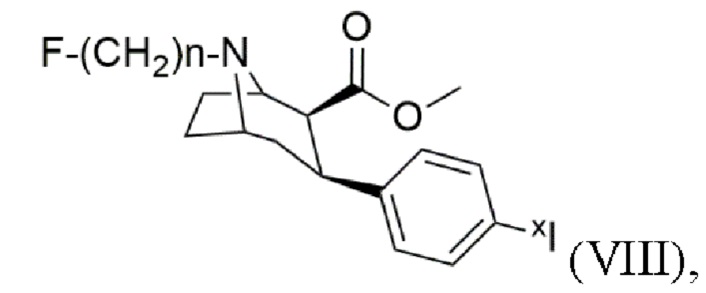
где xI представляет собой по меньшей мере один изотоп йода.
| Thompson P | |||
| E | |||
| et al | |||
| Fmoc-protected Tropane-based Amino Acids for Peptide Structure-Function Studies, Tetrahedron Letters, 1997, 38 (16), pp | |||
| Прибор для механического отбирания средней пробы зерна | 1923 |
|
SU2907A1 |
| Rami-Mark C | |||
| et al | |||
| Synthesis, radiosynthesis and first in vitro evaluation of novelvPET-tracers for the dopamine transporter: [11C]IPCIT and [18F]FE@IPCIT, Bioorganic & Medicinal | |||

