Настоящее изобретение относится к 1,2,4,5-тетразамещенным производным имидазола - селективным модуляторам рецепторов СВ1 каннабиноидов, конкретно антагонистам рецепторов СВ1 или обратным агонистам, обладающим высокой селективностью к подтипам рецепторов СВ1/СВ2, к способам получения указанных соединений и к новым полупродуктам, применяемым для синтеза указанных производных имидазола. Изобретение также относится к применению описанного здесь соединения для изготовления лекарственного средства, обладающего полезным действием. Полезное действие описано здесь в описании или является очевидным для специалистов в данной области из описания и общих знаний в данной области. Изобретение также относится к применению соединения изобретения для производства лекарственного средства для лечения или профилактики заболевания или патологического состояния. Более конкретно, изобретение относится к новому применению для лечения заболевания или патологического состояния, описываемого здесь или очевидного для специалиста в данной области из описания и общих знаний в данной области. В вариантах осуществления изобретения описываемые здесь конкретные соединения применяют для изготовления лекарственного средства, пригодного для лечения психических и неврологических нарушений.
Полизамещенные производные имидазола, обладающие сродством к рецепторам СВ1, известны из WO 03/027076 и WO 03/063781. Кроме того, в WO 03/040107 рассматриваются имидазолы для лечения ожирения. Кроме того, опубликована статья, в которой описаны имидазолы как антагонисты рецепторов СВ1 (Dyck et al., Bioorg. Med. Chem. Lett. 2004, 14, 1151-1154). В вышеуказанных патентных заявках и статье не рассматриваются данные о селективности к подтипам рецепторов СВ1/СВ2 описываемых здесь соединений.
Модуляторы рецепторов СВ1 имеют несколько возможных применений, таких как лекарственные средства для лечения психоза, чувства тревоги, депрессии, дефицита внимания, расстройства памяти, нарушений познавательной способности, нарушений аппетита, ожирения, наркомании, влечения (например, полового), лекарственной зависимости, нейродегенеративных расстройств, слабоумия, дистонии, мышечной спастичности, тремора, эпилепсии, рассеянного склероза, травматического повреждения головного мозга, удара, болезни Паркинсона, болезни Альцгеймера, эпилепсии, болезни Гентингтона, синдрома Туретта, церебральной ишемии, апоплексии мозга, черепно-мозговой травмы, инсульта, повреждения позвоночника, нейровоспалительных нарушений, склеротических бляшек, вирусного энцефалита, нарушений, связанных с демиелинизацией, а также для лечения нарушений, связанных с болью, включая расстройства, связанные с нейропатической болью, септического шока, глаукомы, диабета, рака, рвоты, тошноты, желудочно-кишечных расстройств, язвы желудка, диареи, половых нарушений, расстройства контроля над побуждениями и сердечно-сосудистых нарушений.
Рецепторы СВ2 находятся преимущественно в иммунной системе (селезенке, миндалинах, иммунных клетках), а также в микроглиальных клетках и астроцитах, и их связывают с ощущением нейропатической боли. Сильнодействующие модуляторы рецепторов СВ1, обладающие низким сродством к рецепторам СВ2 (т.е. соединения, обладающие высокой селективностью к подтипам рецепторов СВ1/СВ2), являются преимущественными соединениями по сравнению с неселективными или менее селективными модуляторами рецепторов каннабиноидов, поскольку они лишены возможных нежелательных побочных действий, опосредованных СВ2 рецепторами, таких как иммуннологических побочных действий или побочных действий, связанных с воспалением или действием, связанным с ощущением нейропатической боли.
Задачей настоящего изобретения является разработка производных имидазола с высокой селективностью к подтипам рецепторов СВ1/СВ2.
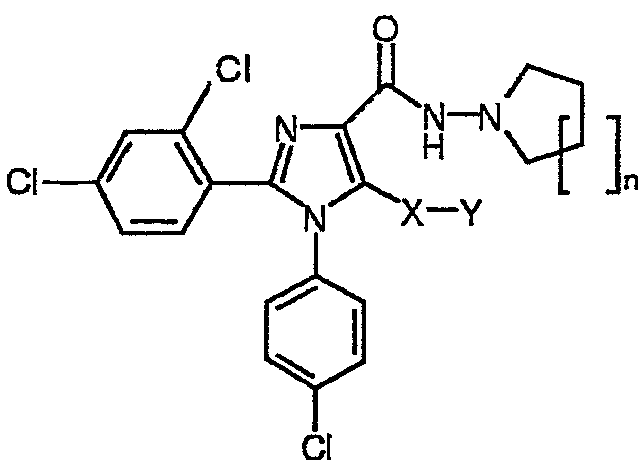
Неожиданно авторы обнаружили, что модификация исходной группы СН2Х в известных ранее имидазолах общей формулы (I) группой, содержащей атом серы, приводит в результате к новым соединениям, с коэффициентом усиления 10 селективности к подтипам рецепторов СВ1/СВ2, в результате чего отношение сродства СВ1/СВ2 значительно превысило 100. Соединения общей формулы (I):
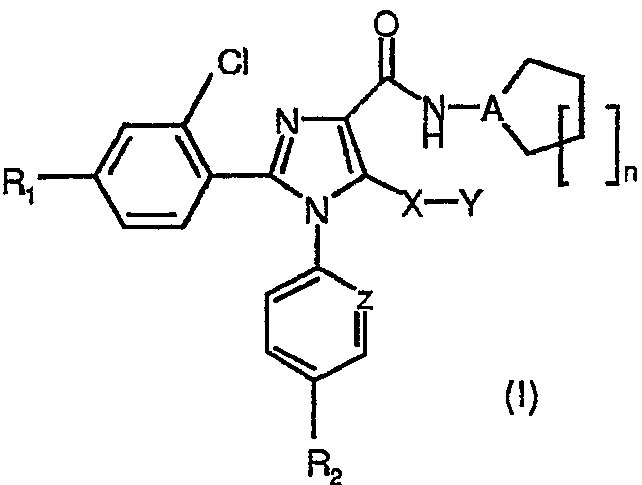
в которой
R1 представляет собой атом хлора, брома, фтора или водорода,
R2 представляет собой атом хлора или брома или группу CF3,
А представляет собой атом азота или группу СН,
Х представляет собой атом серы или сульфоксидную (S=O) группу, или сульфоновую (SO2) группу,
Y представляет собой атом водорода или группу: метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, трифторметил, фенил, бензил или пиридил,
Z представляет собой атом азота или группу СН,
n означает число 1, 2 или 3,
и таутомеры, стереоизомеры, пролекарства и фармакологически приемлемые соли соединения являются новыми и представляют собой сильнодействующие и СВ1/СВ2 селективные антагонисты рецепторов СВ1 или обратные агонисты.
Во всех сульфоксидах, описанных в данном изобретении, имеется хиральный центр. Изобретение относится к рацематам, смесям диастереомеров, а также к индивидуальным стереоизомерам соединений, имеющих формулу (I). Изобретение также относится к Е-изомерам, Z-изомерам и смесям Е/Z-изомеров соединений, имеющих формулу (I).
Пролекарства вышеуказанных соединений включены в объем настоящего изобретения. Пролекарства представляют собой терапевтические средства, которые неактивны per se, но превращаются в один или несколько активных метаболитов. Пролекарства являются биологически обратимыми производными лекарственных молекул, применяемыми для преодоления некоторых препятствий для применимости исходной лекарственной молекулы. Такие препятствия включают в себя, но не ограничиваются перечисленным, растворимость, проницаемость, стабильность, предсистемный метаболизм и ограничения направленной доставки (Medicinal Chemistry: Principles and Practice, 1994, ISBN 0-85186-494-5, Ed.: F.D.King, p.215; J.Stella, “Prodrugs as therapeutics”, Expert Opin. Ther. Patents, 14(3), 277-280, 2004; P. Ettmayer et.al., “Lessons learned from marketed and investigational prodrugs”, J.Med.Chem., 47, 2393-2404, 2004). Пролекарства, т.е. соединения, которые после введения в организм человека любым известным путем метаболизируют с образованием соединений, имеющих формулу (1), входят в объем изобретения. Конкретно, это имеет отношение к соединениям с первичными или вторичными амино- или гидроксигруппами. Указанные соединения могут взаимодействовать с органическими кислотами с образованием соединений, имеющих формулу (1), в которой присутствует дополнительная группа, легко удаляемая после введения в организм и представляющая собой, например, но не ограничивающаяся следующими примерами: амидин, енамин, основание Манниха, гидроксиметиленовое производное, производное О-(ацилоксиметиленкарбамата), карбамат, сложный эфир, амид или енаминон.
Конкретно, изобретение относится к соединениям общей формулы (I), в которой R1 представляет собой атом водорода или атом хлора и R2 является атомом хлора, Y представляет собой метильную или этильную группу, Z представляет собой СН, n означает 1, 2 или 3, А и Х имеют значения, как указано выше, и к таутомерам, стереоизомерам, пролекарствам или фармакологически приемлемым солям соединений.
Более конкретно, изобретение относится к соединениям общей формулы (I), в которой R1 и R2 представляют собой атомы хлора, Y представляет собой метил, Z представляет собой СН, n означает 1, 2 или 3, А представляет собой атом азота и Х имеет значения, указанные выше, к таутомерам, стереоизомерам, пролекарствам и фармакологически приемлемым солям соединений.
Общие аспекты синтезов
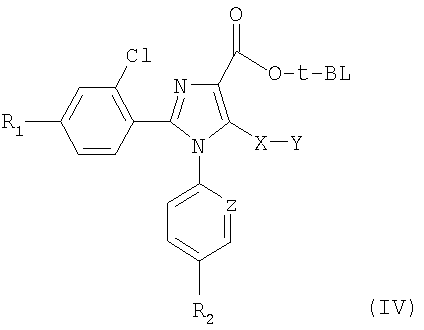
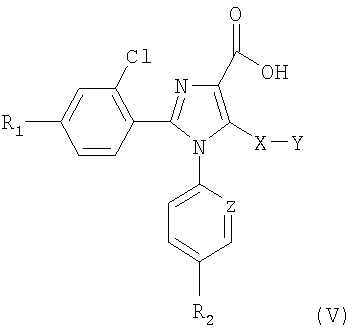
Синтез соединений, имеющих формулу (I), приведен на схеме 1. Карбоновую кислоту общей формулы (II) можно превратить в соответствующий трет-бутиловый эфир (III). Полученный сложный эфир (III) можно обработать сильным ненуклеофильным основанием в инертном безводном органическом растворителе и затем подвергнуть реакции с серосодержащим электрофилом YSSY, в котором Y представляет собой группу метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, трифторметил, фенил, бензил или пиридил, для получения соединения общей формулы (IV). Полученное соединение общей формулы (IV) можно окислить одним молярным эквивалентом м-хлорпербензойной кислоты для получения соответствующего сульфоксидного аналога. В альтернативном случае взаимодействие соединения общей формулы (IV) с двумя молярными эквивалентами м-хлорпербензойной кислоты может превратить серосодержащую группу в соответствующую сульфоновую группу. Сложный эфир общей формулы (IV) можно гидролизовать предпочтительно в кислотной среде для получения соответствующей карбоновой кислоты (V). К полученному в результате соединению общей формулы (V) можно присоединить амин в присутствии активирующего реагента или реагента сочетания для получения соединения общей формулы (I), в которой R1, R2, A, X, Y, Z и n имеют вышеуказанное значение.
Схема 1а
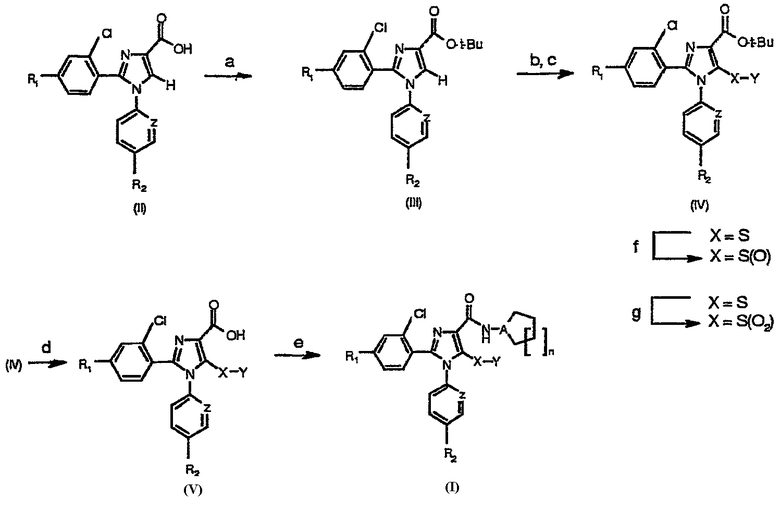
а Реагенты и условия: (а) Вос2О, DMAP, t-BuOH; b) LDA, ТГФ; (c) YSSY; (d) TFA, CH2Cl2; (e) производное амина, HBTU, DIPEA, CH3CN, комнатная температура; (f) 1 эквивалент м-СРВА, СН2Cl2, комнатная температура; (g) 2 эквивалента м-СВРА, СН2Cl2, комнатная температура.
В альтернативном случае соединения общей формулы (VI), в которой R1, R2, A, Z и n имеют вышеуказанное значение, могут быть подвержены взаимодействию с сильным ненуклеофильным основанием, например LDA, в инертном безводном органическом растворителе, с последующей обработкой серой (S8) или серосодержащим электрофилом YSSY, в котором Y представляет собой группу метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, трифторметил, фенил, бензил или пиридил для получения соединения общей формулы (I), в которой Х представляет собой атом серы, Y представляет собой атом водорода или группу метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, трифторметил, фенил, бензил или пиридил, и в которой R1, R2, A, Z и n имеют вышеуказанное значение (Схема 2).
Схема 2а
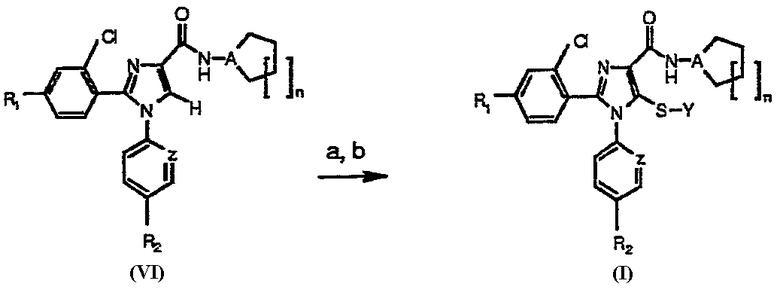
а Реагенты и условия: (а) LDA, Et2O; (b) S8 или YSSY
Дополнительную информацию по способам активации и присоединения аминов к карбоновым кислотам можно найти в:
а) M.Bondarzky and A.Bondarzky: The Practice of Peptide Synthesis, Springer-Verlag, New York, 1994; ISBN: 0-387-57505-7;
b) K.Akaji et al., Tetrahedron Lett. (1994), 35, 3315-3318);
c) F.Alberico et al., Tetrahedron Lett. (1997), 38, 4853-4856).
Выбор конкретных синтетических методик зависит от факторов, известных для специалистов в данной области, таких как совместимость функциональных групп с применяемыми реагентами, возможность применения защитных групп, катализаторов, активирующих реагентов и реагентов сочетания, и структурных особенностей получаемого в конечном счете соединения.
Фармацевтически приемлемые соли можно получить с использованием стандартных методик, хорошо известных в данной области, например смешиванием соединения настоящего изобретения с подходящей кислотой, например, неорганической кислотой или органической кислотой.
Фармацевтические препараты
Соединения изобретения можно получать в формах, подходящих для введения обычными методами, с использованием вспомогательных веществ, таких как жидкий или твердый материал-носитель. Фармацевтические композиции изобретения можно вводить энтерально, перорально, парентерально (внутримышечно или внутривенно), ректально или локально (местно). Их можно вводить в форме растворов, порошков, таблеток, капсул (включая микрокапсулы), мазей (кремов или гелей) или суппозиториев. Подходящими наполнителями для указанных препаративных форм являются обычные для фармации жидкие или твердые наполнители и разбавители, растворители, эмульгаторы, смазывающие средства, корригенты, красители и/или буферные вещества. Часто применяемые вспомогательные вещества, которые можно упомянуть, представляют собой карбонат магния, диоксид титана, лактозу, маннит и другие сахара или сахара в форме спиртов, тальк, лактопротеин, желатин, крахмал, целлюлозу и ее производные, животные и растительные масла, как например масло из печени рыб, подсолнечное, арахисовое или кунжутное масло, полиэтиленгликоль, и растворители, такие, как например стерильная вода и одно- или многоатомные спирты, например глицерин.
Соединения настоящего изобретения обычно применяют как фармацевтические композиции, которые являются важными и усовершенствованными вариантами осуществления изобретения вследствие присутствия соединений, более конкретно описанных здесь. Типы фармацевтических композиций, которые можно применять, включают в себя, но не ограничиваются перечисленными, таблетки, жевательные таблетки, капсулы, растворы, парентеральные растворы, суппозитории, суспензии и другие типы, описанные здесь или очевидные для специалистов в данной области из описания и общих знаний в данной области. Изобретение также включает в себя получение или изготовление указанных фармацевтических композиций.
В вариантах осуществления изобретения предусмотрена фармацевтическая упаковка или набор, содержащий один или несколько контейнеров, наполненных одним или несколькими ингредиентами фармацевтической композиции данного изобретения. Вместе с такими контейнерами могут находиться письменные материалы, такие как инструкции для применения или уведомление в форме, рекомендованной правительственными организациями, регулирующими изготовление, применение или продажу фармацевтических продуктов, где указанное уведомление подтверждает одобрение учреждением изготовления, применения или продажи для человека или для применения в ветеринарии.
Вследствие высокой активности как антагонистов СВ1 или обратных агонистов соединения согласно изобретению являются подходящими для применения для лечения психических расстройств, таких как психоз, чувство тревоги, депрессия, нарушение внимания, расстройство памяти, нарушение познавательной способности, нарушение аппетита, ожирение, в особенности юношеское ожирение и ожирение, вызванное лекарственным средством, наркомания, влечение (например, половое), лекарственная зависимость, и неврологических расстройств, таких как нейродегенеративные расстройства, слабоумие, дистония, мышечная спастичность, тремор, эпилепсия, рассеянный склероз, травматическое повреждение головного мозга, внезапный приступ, болезнь Паркинсона, болезнь Альцгеймера, эпилепсия, болезнь Гентингтона, синдром Туретта, церебральная ишемия, апоплексия мозга, черепно-мозговая травма, инсульт, повреждение позвоночника, нейровоспалительные нарушения, склеротические бляшки, вирусный энцефалит, нарушений, связанных с демиелинизацией, а также для лечения нарушений, связанных с болью, включая нарушения, связанные с нейропатической болью, и другие заболевания, связанные с каннабиноидной трансмиссией, в том числе для лечения септического шока, глаукомы, рака, диабета, рвоты, тошноты, астмы, респираторных заболеваний, желудочно-кишечных расстройств, язвы желудка, диареи, половых нарушений, расстройства контроля над побуждениями и сердечно-сосудистых нарушений.
Активность соединений настоящего изобретения как модуляторов рецепторов каннабиноидов делает их особенно полезными для лечения ожирения, юношеского ожирения и ожирения, вызванного лекарственным средством, в особенности при применении в сочетании с ингибиторами липазы. Конкретными примерами соединений, которые можно применять в указанном сочетании препаратов, являются (но не ограничиваются перечисленными) синтетический ингибитор липазы орлистат, ингибиторы липазы, выделенные из микроорганизмов, таких как липстатин (из Streptomyces toxytricini), эбелактон В (из Streptomyces aburaviensis), синтетические производные указанных соединений, а также экстракты из растений, которые, как известно, обладают способностью ингибировать липазу, например, экстракты Alpinia officinarum, или соединения, выделенные из указанных экстрактов, подобные простому 3-метиловому эфиру галангина (из А, officinarum).
Фармакологические способы
Сродство к рецепторам СВ 1 каннабиноидов in vitro
Сродство соединений данного изобретения к рецепторам СВ1 каннабиноидов можно определять с использованием препаратов мембран клеток яичников китайских хомячков (СНО), в которых рецепторы СВ1 каннабиноидов человека стабильно трансфецируют с [3H]CP-55,940 в качестве радиоактивного лиганда. После инкубирования свежеприготовленного препарата клеточных мембран с [3H]-лигандом вместе с добавленным соединением, описанным в изобретении, или без него разделение связанного и свободного лиганда проводят фильтрованием через фильтры из стекловолокна. Радиоактивность на фильтре измеряют жидкостным сцинтилляционным счетчиком.
Сродство к рецепторам СВ 2 каннабиноидов in vitro
Сродство соединений данного изобретения к рецепторам СВ2 каннабиноидов можно определять с использованием препаратов мембран клеток яичников китайских хомячков (СНО), в которых стабильно трансфецируют рецепторы СВ2 каннабиноидов человека с [3H]CP-55,940 в качестве радиоактивного лиганда. После инкубирования свежеприготовленного препарата клеточных мембран с [3H]-лигандом вместе с добавленным соединением изобретения или без него, проводят разделение связанного и свободного лиганда фильтрованием через фильтры из стекловолокна. Радиоактивность на фильтре измеряют жидкостным сцинтилляционным счетчиком.
Антагонизм к рецепторам СВ 1 каннабиноидов in vitro
Антагонизм in vitro к рецепторам СВ1 каннабиноидов можно определить с помощью рецептора СВ1 человека, клонированного в клетках яичников китайского хомячка (СНО). СНО клетки выращивают в культуральной среде, модифицированной по Дульбекко среде Игла (DMEM), с добавлением 10% инактивированной при нагревании фетальной телячьей сыворотки. Среду аспирируют и заменяют DMEM без фетальной телячьей сыворотки, но содержащей [3H]-арахидоновую кислоту, и инкубируют в течение ночи в камере для клеточной культуры (5% СО2/95% воздуха; 37°С; атмосфера, насыщенная парами воды). В течение указанного периода времени [3H]-арахидоновая кислота включается в фосфолипиды мембраны. В день испытания среду аспирируют и клетки промывают три раза 0,5 мл DMEM, содержащей 0,2% бычьего сывороточного альбумина (BSA). Стимуляция рецепторов СВ1 WIN 55,212-2 приводит к активации PLA2, за которой следует высвобождение [3H]-арахидоновой кислоты в среду. Это WIN 55,212-2-индуцируемое высвобождение антагонизируется антагонистами рецепторов СВ1 зависимым от концентрации образом.
Доза
Сродство соединений данного изобретения к рецепторам СВ1 определяли, как описано выше. Из сродства к связыванию, измеряемого для данного соединения формулы (1), можно оценивать самую низкую эффективную дозу. При концентрации соединения, равной двойной измеренной К-величине, почти 100% СВ1 рецепторов, вероятно, будут связаны с соединением. Пересчитывая полученную величину концентрации в мг соединения на 1 кг массы тела пациента, получают теоретическую самую низкую эффективную дозу, предполагая идеальную биодоступность. Фармакокинетические, фармакодинамические и другие показатели могут изменить фактически вводимую дозу в сторону более высокой или более низкой величины. Целесообразная вводимая доза составляет 0,001-1000 мг/кг, предпочтительно 0,1-100 мг/кг массы тела пациента.
Примеры
Пример 1. Вещества и способы
1Н и 13С ЯМР спектры регистрировали на приборе Bruker Avance DRX600 (600 МГц), на приборе Varian UN400 (400 МГц) или на приборе Varian VXR200 (200 МГц) с использованием ДМСО-d6 или CDCl3 в качестве растворителей, с тетраметилсиланом в качестве внутреннего стандарта. Химические сдвиги выражали в м.д. (шкала d) в сторону слабого поля от тетраметилсилана. Константы взаимодействия (J) выражали в Гц. Флэш-хроматографию осуществляли с использованием силикагеля 60 (0,040-0,063 мм, Merk). Хроматографию на колонке осуществляли с использованием силикагеля 60 (0,063-0,200 мм, Merk). Температуру плавления регистрировали на приборе для определения температуры плавления Buchi B-545. Масс-спектры регистрировали на приборе Micromass QTOF-2 с применением программного обеспечения MassLynx для обнаружения и обработки данных. Точное измерение массы производили исходя из квазимолекулярного иона [M+H]+.
Пример 2. Синтезы конкретных соединений
Соединения 1-3
1-(4-хлорфенил)-2-(2,4-дихлорфенил)-1Н-имидазол-4-карбоновая кислота
К перемешиваемому магнитной мешалкой раствору 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-1Н-имидазол-4-карбоксилата (18,44 г, 0,0466 мол) в ТГФ (240 мл) добавляли LiOH (2,24 г, 0,0932 мол) и Н2О (240 мл). Полученную в результате смесь перемешивали при 50°С в течение 16 ч для получения прозрачного раствора. После охлаждения до комнатной температуры добавляли HCl (1н. раствор, 95 мл) и Н2О (240 мл) для получения осадка, который отделяли фильтрованием, промывали водой и сушили в вакууме для получения 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-1Н-имидазол-4-карбоновой кислоты (16,83 г, выход 98%), температура плавления 138-142°С (с разложением); 1Н-ЯМР (600 МГц, ДМСО-d6) δ 7,08 (шир.д, J=8 Гц, 2Н), 7,31-7,37 (м, 4Н), 7,45 (д, J=8 Гц, 1Н), 7,96 (с, 1Н); 13С-ЯМР (150 МГц, ДМСО-d6) δ 126,87; 127,85; 127,91; 128,47; 129,36; 129,66; 133,56; 133,99; 134,44; 134,49; 135,54; 135,99; 143,77; 163,67.
Трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-1Н-имидазол-4-карбоксилат
К перемешиваемой магнитной мешалкой смеси 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-1Н-имидазол-4-карбоновой кислоты (20,77 г, 0,0565 мол) и Вос2О (24,63 г, 0,113 мол) в t-BuOH (275 мл) добавляли DMAP (2,07 г, 0,017 мол) и полученную в результате смесь перемешивали в течение 16 ч. После концентрирования в вакууме добавляли толуол и смесь снова концентрировали. Остаток очищали колоночной хроматографией (СН2Cl2/ацетон равно 95/5 (об./об.)) и перекристаллизовывали из простого диизопропилового эфира для получения трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-1Н-имидазол-4-карбоксилата (15,75 г, выход 66%), температура плавления 178-180°С; 1Н-ЯМР (200 МГц, CDCl3) δ 1,63 (с, 9Н), 7,05 (шир.д, J=8 Гц, 2Н), 7,25-7,37 (м, 4Н), 7,52 (д, J=8 Гц, 1H), 7,8 (с, 1Н).
Трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-1Н-имидазол-4-карбоксилат
К охлажденному (-20°С) и перемешиваемому магнитной мешалкой раствору трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-1Н-имидазол-4-карбоксилата (10,59 г, 25,0 ммол) в безводном ТГФ (100 мл) добавляли LDA (15,0 мл, 2 М раствор в смеси гептан/ТГФ, 30,0 ммол) и полученную в результате смесь перемешивали в течение 1 часа в атмосфере N2. Добавляли раствор (СН3S)2 (2,7 мл, 30,0 ммол) в ТГФ (20 мл) и полученный в результате раствор последовательно перемешивали при -40°С в течение 1 ч, позволяли достигать комнатной температуры и перемешивали в течение следующих 16 ч. Добавляли насыщенный водный раствор NH4Cl (250 мл) и полученный в результате раствор дважды экстрагировали этилацетатом (EtOAc). Объединенные органические слои промывали водой, сушили над MgSO4, фильтровали и концентрировали для получения трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-1Н-имидазол-4-карбоксилата с выходом 90% в виде масла, которое медленно отверждается; 1Н-ЯМР (200 МГц, CDCl3) δ 1,66 (с, 9Н), 2,28 (с, 3Н), 7,05 (шир.д, J=8 Гц, 2Н), 7,25 (дд, J=8 и 2 Гц, 1Н), 7,28 (д, J=2 Гц, 1H), 7,32-7,41 (м, 3Н).
Трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-1Н-имидазол-4-карбоксилат
К перемешиваемому магнитной мешалкой раствору трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-1Н-имидазол-4-карбоксилата (6,00 г, 12,8 ммол) в СН2Cl2 (25 мл) медленно добавляли раствор m-CPBA (6,90 г, с содержанием основного вещества 70%, 0,282 мол) в СН2Cl2 и полученную в результате смесь перемешивали в течение 16 час. Реакционную смесь дважды промывали 2н. раствором NaOH и сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (Et2O/петролейный эфир равно 2/1 (об./об.)) для получения трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульнонил-1Н-имидазол-4-карбоксилата (4,76 г, выход 74%) в виде белого твердого соединения, температура плавления 130°С; 1Н-ЯМР (400 МГц, CDCl3) δ 1,66 (с, 9Н), 3,34 (с, 3Н), 7,15 (шир.д, J=8 Гц, 2Н), 7,20-7,26 (м, 2Н), 7,32-7,41 (м, 3H).
По аналогии получали трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфинил-1Н-имидазол-4-карбоксилат взаимодействием с 1 молярным эквивалентом m-CPBA вместо двух молярных эквивалентов; 1Н-ЯМР (400 МГц, CDCl3) δ 1,64 (с, 9Н), 2,94 (с, 3Н), 7,20-7,36 (м, 7Н).
1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-1Н-имидазол-4-карбоновая кислота
К перемешиваемому магнитной мешалкой раствору трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-1Н-имидазол-4-карбоксилата (4,76 г, 9,49 ммол) в СН2Cl2 (60 мл) добавляли избыток TFA (9,40 мл, 0,2124 мол) и Et2SiH (3,8 мл, 0,0238 мол). Реакцию в растворе осуществляли при комнатной температуре в течение 16 ч и концентрировали в вакууме. Добавляли воду и выпавший осадок отделяли фильтрованием и затем сушили для получения 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-1Н-имидазол-4-карбоновой кислоты с количественным выходом, температура плавления ~130°С (с разложением); 1Н-ЯМР (400 МГц, CDCl3) δ 3,45 (с, 3Н), 3,50 (шир.с, 1Н), 7,40 (шир.д, J=8 Гц, 2Н), 7,42 (дд, J=8 и 2 Гц, 1Н), 7,50 (шир.д, J=8 Гц, 2H), 7,59 (д, J=2 Гц, 1Н), 7,61 (д, J=8 Гц, 1Н).
По аналогии получали 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфинил-1Н-имидазол-4-карбоновую кислоту.1Н-ЯМР (400 МГц, CDCl3) δ 2,99 (с, 3Н), 7,37-7,60 (м, 7Н), 13,20 (шир. с, 1H).
1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 1)
К перемешиваемой магнитной мешалкой суспензии 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-1Н-имидазол-4-карбоновой кислоты (2,23 г, 5,01 ммол) в безводном СН3CN (50 мл) последовательно добавляли N,N-диизопропилэтиламин (основание Хунига) (1,90 мл, 11,0 ммол), О-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийгексафторфосфат (HBTU) (2,27 г, 5,99 ммол) и 1-аминопиперидин (0,65 мл, 6,03 ммол). После перемешивания в течение 16 ч полученную смесь концентрировали в вакууме. Остаток растворяли в EtOAc, последовательно промывали водным раствором NaHCO3, водой и раствором соли, сушили над Na2SO4, фильтровали и концентрировали для получения неочищенного твердого соединения. Полученное твердое соединение дополнительно очищали флэш-хроматографией (силикагель EtOAc) и растирали с простым метил-трет-бутиловым эфиром для получения 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамида с выходом 84%, температура плавления 181-185°С (с разложением); 1Н-ЯМР (600 МГц, ДМСО-d6) δ 1,35-1,41 (м, 2Н), 1,61-1,66 (м, 4Н), 2,80-2,84 (м, 4Н), 3,52 (с,3Н) 7,38 (д, J=8 Гц, 2Н), 7,42 (дд, J=8 и 2 Гц 1Н), 7,46 (д, J=8 Гц, 2H), 7,57 (д, J=2 Гц, 1Н), 7,62 (д J=8 Гц, 1Н), 9,40 (с, 1Н); HRMS (C22H22Cl3N4O3S).[M+H]+ : найдено m/z 527,0469, вычислено 527,0478.
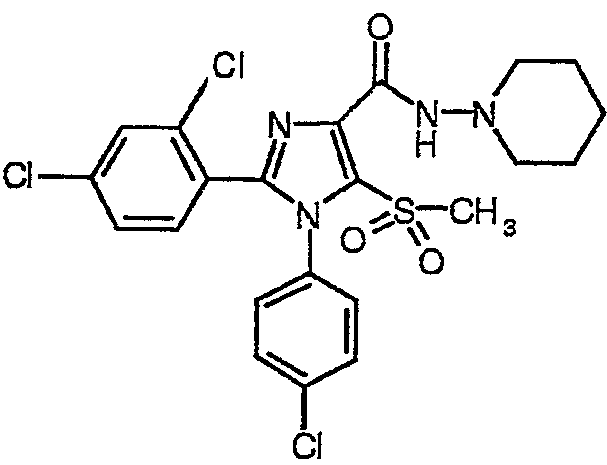
Соединение 1
По аналогии получали 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфонил-N-(циклогексил)-1Н-имидазол-4-карбоксамид (соединение 2). Температура плавления 191-192°С.
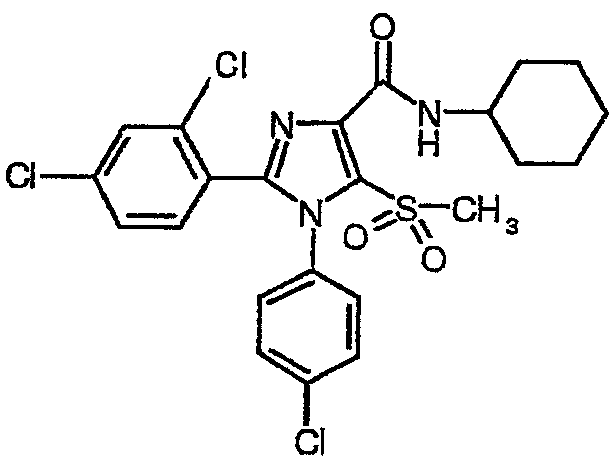
Соединение 2
По аналогии получали 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфинил-N-пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 3). Температура плавления 218-221°С.
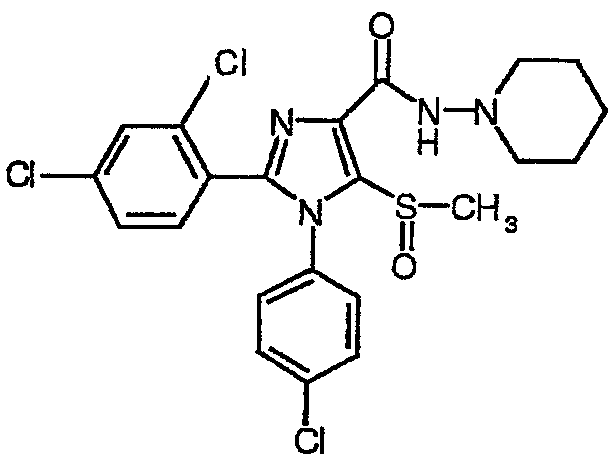
Соединение 3
Соединения 4-5
Рацемический 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфинил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 3, 3,86 грамм, 0,0075 мол) разделяли на энантиомеры, (-)-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфинил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 4, 1,3 грамм) ([aD 25]=-19°, c=0,94 (г/100 мл растворителя), метанол; энантиомерный избыток 97,2%; температура плавления: 242-244°С) и (+)-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфинил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 5, 1,4 грамма) ([aD 25]=+23°, с=0,94 (г/100 мл растворителя), метанол: энантиомерный избыток 99,5%; температура плавления: 243-245°С), соответственно с использованием препаративной ВЭЖХ и хиральной стационарной фазы Chiralpak AD 20 мкм. Подвижная фаза состоит из смеси 25% этанол/гептан (25/75 об./об.)).
Соединения 6-11
1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-1Н-имидазол-4-карбоновая кислота
К перемешиваемому магнитной мешалкой раствору трет-бутил-1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-1Н-имидазол-4-карбоксилата (4,00 г, 8,53 ммол) в СН2Cl2 (60 мл) добавляли избыток TFA (8,40 мл, 0,111 мол). Реакцию в растворе проводили при комнатной температуре в течение 16 час и затем концентрировали в вакууме. Добавляли воду и образовавшийся осадок отделяли фильтрованием и потом сушили для получения 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-1Н-имидазол-4-карбоновой кислоты с выходом 98%, температура плавления ~100°С (с разложением); 1Н-ЯМР (400 МГц, CDCl3) δ 2,41 (с, 3Н), 3,60 (шир. с, 1Н), 7,08 (шир.д, J=8 Гц, 2Н), 7,26 (дд, J=8 и 2 Гц, 1Н), 7,30 (д, J=8 Гц 1H), 7,35 (д, J=2 Гц, 1Н), 7,37 (шир.д, J=8 Гц, 2Н).
1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 6)
К перемешиваемой магнитной мешалкой суспензии 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-1Н-имидазол-4-карбоновой кислоты (1,72 г, 4,16 ммол) в безводном СН3CN (45 мл) последовательно добавляли N,N-диизопропилэтиламин (основание Хунига) (1,60 мл, 9,20 ммол), О-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийгексафторфосфат (HBTU) (1,89 г, 4,99 ммол) и 1-аминопиперидин (0,54 мл, 5,01 ммол). После перемешивания в течение 40 ч добавляли воду и полученную в результате смесь экстрагировали дихлорметаном. Дихлорметановый слой последовательно дважды промывали 1н. раствором HCl и водой, сушили над MgSO4, фильтровали и концентрировали для получения неочищенного масла. Полученное масло дополнительно очищали флэш-хроматографией (силикагель EtOAc) и растирали с простым диэтиловым эфиром для получения 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамида с выходом 72%, температура плавления 170°С (с разложением); 1Н-ЯМР (600 МГц, ДМСО-d6) δ 1,35-1,42 (м, 2Н), 1,62-1,67 (м, 4Н), 2,35 (с, 3Н) 2,80-2,84 (м, 4Н), 7,29 (д, J=8 Гц, 2Н), 7,42 (дд, J=8 и 2 Гц 1Н), 7,45 (д, J=8 Гц, 2H), 7,52 (д, J=2 Гц, 1Н), 7,62 (д J=8 Гц, 1Н), 8,90 (с, 1Н); 13С-ЯМР (150 МГц, ДМСО-d6) δ 19,26; 23,32; 25,63; 55,98; 127,52; 128,61; 129,14; 129,23; 129,86; 130,12; 130,18; 134,45; 134,66; 136,01; 137,12; 144,04; 158,98; HRMS (C22H22Cl3N4OS) [M+H]+: найдено m/z 495,0592, вычислено 495,0580.
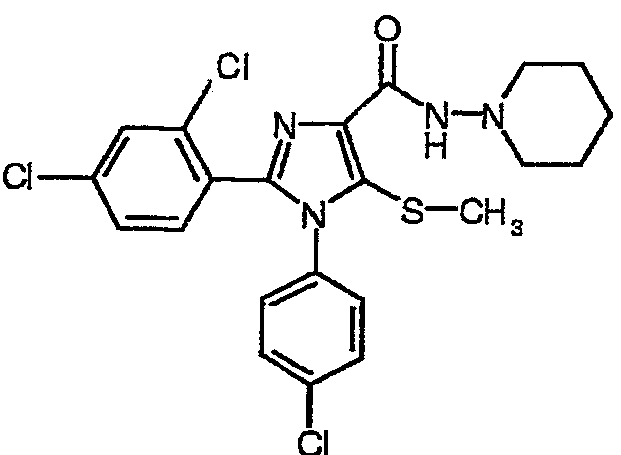
Соединение 6
По аналогии получали 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-N-(циклогексил)-1Н-имидазол-4-карбоксамид (соединение 7). Температура плавления 152-154°С.
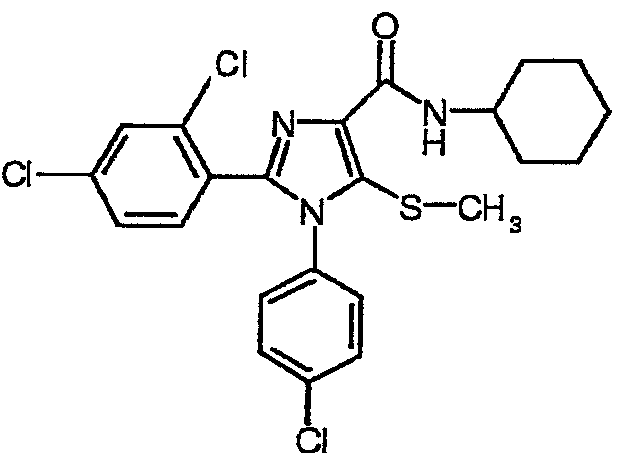
Соединение 7
По аналогии получали 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-этилсульфанил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 8). 1Н-ЯМР (400 МГц, CDCl3) δ 1,09 (т, J=7 Гц, 3Н), 1,40-1,48 (м, 2Н), 1,72-1,80 (м, 4Н), 2,84-2,92 (м, 4Н), 3,00 (к, J=7 Гц, 2Н), 7,03 (дт, J=8 и 2 Гц 2H), 7,23-7,35 (м, 5Н), 8,02 (шир.с, 1Н).
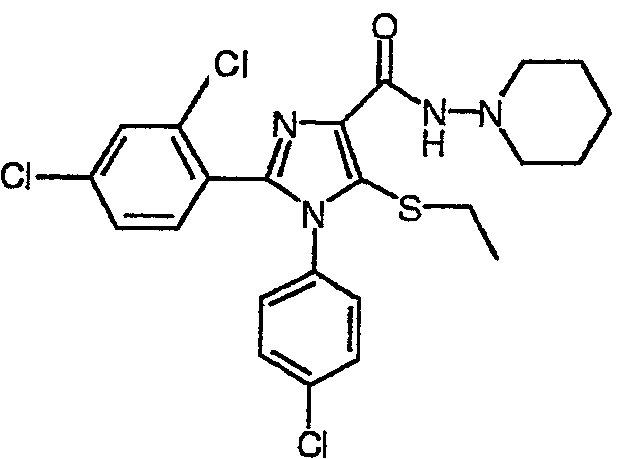
Соединение 8
По аналогии получали 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-N-(пирролидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 9). Температура плавления 158°С.
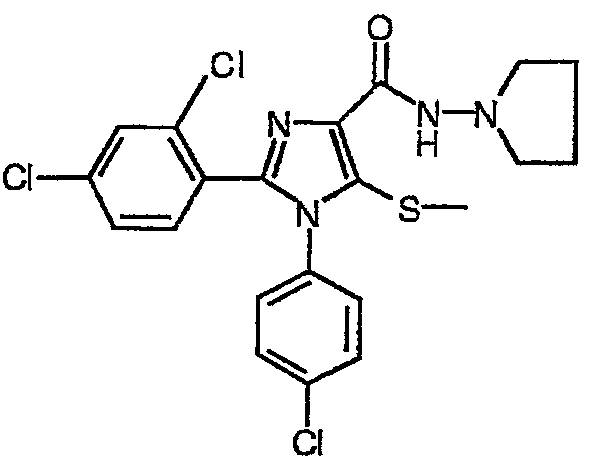
Соединение 9
По аналогии получали 1-(4-хлорфенил)-2-(2,4-дихлорфенил)-5-метилсульфанил-N-(азепан-1-ил)-1Н-имидазол-4-карбоксамид (соединение 10). 1Н-ЯМР (400 МГц, CDCl3) δ 1,63-1,68 (м, 4Н), 1,74-1,81 (м, 4Н), 2,42 (с, 3Н), 3,17-3,22 (м, 4Н), 7,04 (дт, J=8 и 2 Гц, 2Н), 7,23-7,36 (м, 5H), 8,50 (шир.с, 1Н).
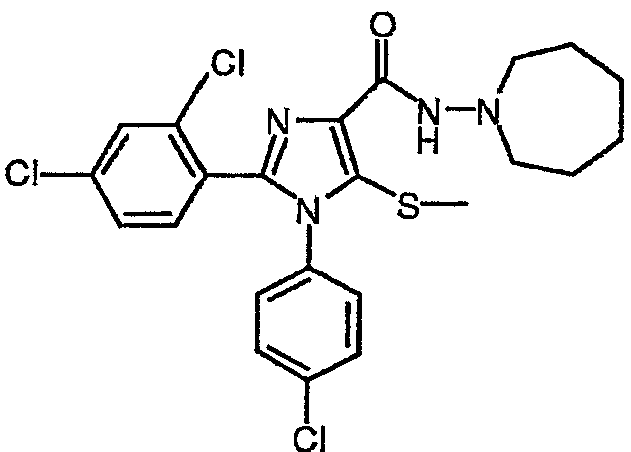
Соединение 10
По аналогии получали 1-(4-хлорфенил)-2-(2-хлорфенил)-5-метилсульфанил-N-(пиперидин-1-ил)-1Н-имидазол-4-карбоксамид (соединение 11). Температура плавления 192-193°С.
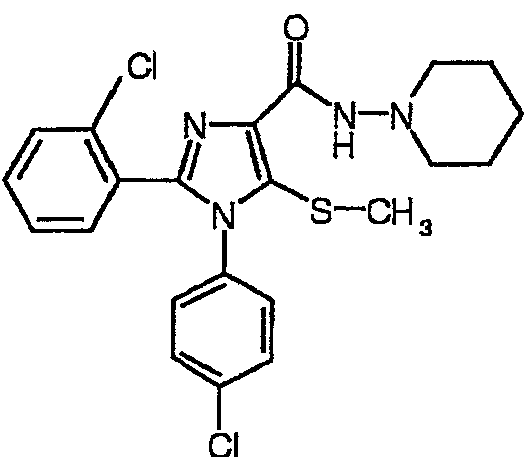
Соединение 11
Конкретные соединения, синтез которых описан выше, предназначены для дополнительной более детальной иллюстрации изобретения и поэтому не могут ограничивать объем изобретения никоим образом. Другие варианты осуществления изобретения будут очевидны для тех специалистов в данной области из рассмотрения описания и практического применения представленного здесь изобретения. Таким образом, подразумевается, что описание и примеры предназначены только в качестве иллюстрации.
Пример 4: Препаративные формы, применяемые в исследованиях на животных
Для перорального введения (р.о.): к требуемому количеству (0,05-5 мг) твердого соединения 1 в стеклянной пробирке добавляли несколько стеклянных шариков и твердое соединение измельчали энергичным встряхиванием в течение 2 минут. После добавления 1 мл раствора 1% метилцеллюлозы в воде и 2% (об./об.) полоксамера 188 (Lutrol F68) соединение суспендировали энергичным встряхиванием в течение 10 минут. Величину рН регулировали добавлением нескольких капель водного раствора NaOH (0,1н.). Остальные частицы в суспензии дополнительно суспендировали с использованием ультразвуковой бани.
Для внутрибрюшинного (i.p.) введения: к требуемому количеству (0,5-15 мг) твердого соединения 1 в стеклянной пробирке добавляли несколько стеклянных шариков и твердое соединение измельчали энергичным встряхиванием в течение 2 минут. После добавления 1 мл раствора 1% метилцеллюлозы и 5% маннита в воде соединение суспендировали энергичным встряхиванием в течение 10 мин. После этого устанавливали рН 7.
Пример 5: результаты фармакологических испытаний
Некоторые данные сродства к рецепторам СВ1/СВ2 (средние результаты по меньшей мере трех независимых экспериментов, произведенных согласно вышеописанным протоколам) для ранее известных соединений и репрезентативных соединений данного изобретения представлены в нижеприведенной таблице. Приведенные данные иллюстрируют воздействие на отношения селективности рецепторов СВ1/2, достигаемые структурной модификацией соединений, которая составляет основу настоящего изобретения: новые соединения сохраняют свое высокое сродство к рецепторам СВ1 каннабиноидов, в то же время сродство к рецепторам СВ2 весьма существенно уменьшается.
Соединения в таблице 1 имеют следующую общую структурную формулу:
Сродство к рецепторам СВ1 и СВ2 известных ранее соединений (строчки 1 и 2) и соединений, представленных в изобретении (строчки 3-6)
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1Н-ИМИДАЗОЛА, ОБЛАДАЮЩИЕ CB-АГОНИСТИЧЕСКОЙ, ЧАСТИЧНОЙ CB-АГОНИСТИЧЕСКОЙ ИЛИ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2299200C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИНА, ОБЛАДАЮЩИЕ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2357958C2 |
| ПРОИЗВОДНЫЕ 1Н-ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ КАННАБИНОИДНЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2337907C2 |
| ПРОИЗВОДНЫЕ ТИАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ КАННАБИНОИДНОГО РЕЦЕПТОРА | 2004 |
|
RU2348620C2 |
| ПРОИЗВОДНЫЕ ТИАЗОЛА, ИМЕЮЩИЕ АНТАГОНИСТИЧЕСКУЮ, АГОНИСТИЧЕСКУЮ ИЛИ ЧАСТИЧНО АГОНИСТИЧЕСКУЮ СВ-АКТИВНОСТЬ | 2003 |
|
RU2301804C2 |
| ПРОИЗВОДНЫЕ 1H-1,2,4-ТРИАЗОЛ-3-КАРБОКСАМИДА В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРА КАННАБИНОИДОВ | 2003 |
|
RU2325382C2 |
| 2-АМИНОБЕНЗОТИАЗОЛЫ В КАЧЕСТВЕ ОБРАТНЫХ АГОНИСТОВ РЕЦЕПТОРОВ CB | 2004 |
|
RU2344132C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ1 | 2005 |
|
RU2377238C2 |
| ПРОИЗВОДНЫЕ 4,5-ДИГИДРО-1H-ПИРАЗОЛА, ОБЛАДАЮЩИЕ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2281941C2 |
| НОВЫЕ ЛИГАНДЫ КАННАБИОИДНЫХ РЕЦЕПТОРОВ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2006 |
|
RU2420518C2 |
Настоящее изобретение относится к 1,2,4,5-тетразамещенным производным имидазола общей формулы (I), в которой R1 представляет собой атом хлора, брома, фтора или водорода, R2 представляет собой атом хлора или брома, А представляет собой атом азота или группу СН, Х представляет собой атом серы или сульфоксидную (S=O) группу, или сульфоновую (SO2) группу, Y представляет собой атом водорода или метильную, этильную, н-пропильную, изопропильную, н-бутильную, трет-бутильную, Z представляет собой группу СН, n представляет собой число 1, 2 или 3, и фармакологически приемлемые соли соединений. Также изобретение относится к фармацевтической композиции на основе соединения общей формулы (I), к соединениям, имеющим общие формулы (IV) и (V), и к применению соединения общей формулы (I). Технический результат: получены новые производные имидазола, которые являются селективными модуляторами рецепторов СВ1 каннабиноидов, конкретно агонистами рецепторов СВ1 или обратными агонистами, обладающими высокой селективностью к подтипам рецепторов СВ1/СВ2. 6 н. и 6 з.п. ф-лы, 1 табл.
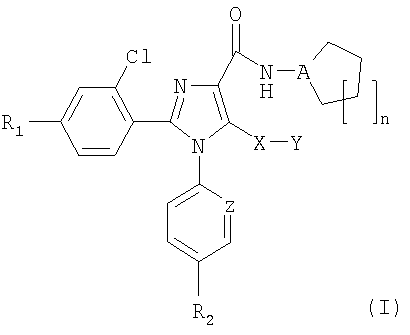
1. Соединение общей формулы (I)
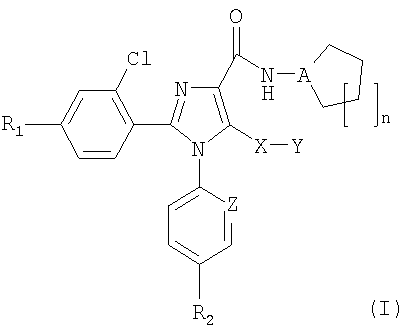
в которой R1 представляет собой атом хлора, брома, фтора или водорода,
R2 представляет собой атом хлора или брома,
А представляет собой атом азота или группу СН,
Х представляет собой атом серы или сульфоксидную (S=O) группу, или сульфоновую (SO2) группу,
Y представляет собой атом водорода или метильную, этильную, н-пропильную, изопропильную, н-бутильную, трет-бутильную группу,
Z представляет собой группу СН,
n представляет собой число 1, 2 или 3,
и фармакологически приемлемые соли соединений.
2. Соединения по п.1 общей формулы (I), в которой R1 представляет собой атом водорода или атом хлора, R2 представляет собой атом хлора, Y представляет собой метильную или этильную группу, Z представляет собой СН, n обозначает 1, 2 или 3, А и Х имеют значения как в п.1, и фармакологически приемлемые соли соединений.
3. Соединение по п.2 общей формулы (I), в которой R1 представляет собой атом хлора, Y представляет собой метильную группу, А представляет собой атом азота, и фармакологически приемлемые соли соединений.
4. Фармацевтические композиции, проявляющие свойства антагониста рецепторов СВ1, обладающие высокой селективностью к подтипам рецепторов СВ1/СВ2, включающие фармакологически активное количество по меньшей мере одного соединения по пп.1-3 или соль такого соединения в качестве активного ингредиента, а также фармацевтически приемлемый носитель и/или по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
5. Способ получения фармацевтических композиций по п.4, отличающийся тем, что по меньшей мере одно соединение по пп.1-3 объединяют с фармацевтически приемлемым носителем и/или вспомогательным веществом и таким образом переводят в форму, подходящую для введения.
6. Соединение по любому из пп.1-3 или его соль в качестве активного ингредиента лекарственного средства для лечения заболеваний, связанных с каннабиноидной нейротрансмиссией.
7. Соединение общей формулы (IV)
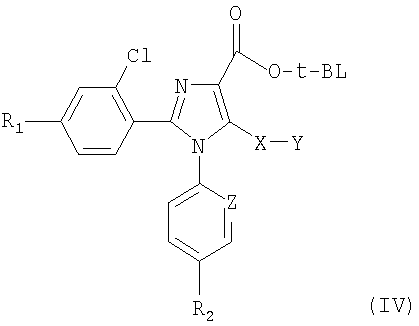
в которой Y представляет собой метильную, этильную, н-пропильную, изопропильную, н-бутильную, трет-бутильную группу, и R1, R2, Х и Z имеют значения, указанные в п.1.
8. Соединения общей формулы (V)
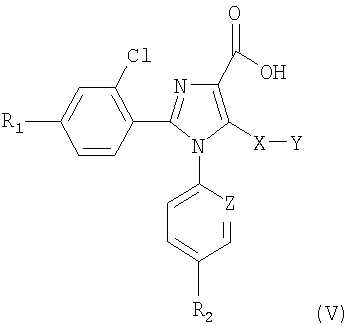
в которой Y представляет собой метильную, этильную, н-пропильную, изопропильную, н-бутильную, трет-бутильную группу, и R1, R2, Х и Z имеют значения, указанные в п.1.
9. Применение соединения по любому из пп.1-3 для получения фармацевтической композиции для лечения психоза, чувства тревоги, депрессии, дефицита внимания, расстройства памяти, нарушений познавательной способности, нарушений аппетита, ожирения, в особенности юношеского ожирения и ожирения, вызванного лекарственными средствами, наркомании, влечения (например, полового), лекарственной зависимости и неврологических расстройств, таких как нейродегенеративные нарушения, слабоумие, дистония, мышечная спастичность, тремор, эпилепсия, рассеянный склероз, травматическое повреждение головного мозга, удар, болезнь Паркинсона, болезнь Альцгеймера, эпилепсия, болезнь Гентингтона, синдром Туретта, церебральная ишемия, апоплексия мозга, черепно-мозговая травма, инсульт, повреждение позвоночника, нейровоспалительные нарушения, склеротические бляшки, вирусный энцефалит, нарушения, связанные с демиелинизацией, а также для лечения нарушений, связанных с болью, включая нарушения, связанные с нейропатической болью, и других заболеваний, связанных с каннабиноидной нейротрансмиссией, в том числе для лечения септического шока, глаукомы, рака, диабета, рвоты, тошноты, астмы, респираторных заболеваний, желудочно-кишечных расстройств, язвы желудка, диареи, половых нарушений и сердечно-сосудистых нарушений.
10. Применение по п.9 соединения по любому из пп.1-3 для получения фармацевтической композиции для лечения нарушений аппетита, в особенности ожирения, юношеского ожирения и ожирения, индуцированного лекарственным средством.
11. Применение по п.10 соединения по любому из пп.1-3 для получения фармацевтической композиции для лечения нарушений, связанных с питанием, в особенности ожирения, юношеского ожирения и ожирения, индуцированного лекарственным средством, отличающееся тем, что указанная фармацевтическая композиция содержит также по меньшей мере один ингибитор липазы.
12. Применение по п.11, отличающееся тем, что указанный ингибитор липазы представляет собой орлистат или липстатин.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 5616601 А, 01.04.1997. | |||

