Настоящее изобретение относится к группе производных тиазола, к способам получения этих соединений, к фармацевтическим композициям, содержащим, по меньшей мере, одно из этих соединений в качестве активного ингредиента, а также к применению этих композиций для лечения психиатрических и неврологических нарушений и других заболеваний, вызываемых нейротрансмиссией каннабиноида СВ. Производные тиазола согласно данному изобретению являются или антагонистами каннабиноидного (СВ) рецептора, агонистами СВ-рецептора, обратными агонистами СВ-рецептора или частичными агонистами СВ-рецептора. Производные тиазола согласно данному изобретению связывают либо СВ1-рецептор, либо СВ2-рецептор или оба СВ1- и СВ2-рецепторы.
Данное изобретение относится к применению раскрытого здесь соединения для получения лекарства, оказывающего полезное воздействие. Полезное воздействие раскрывается здесь или является очевидным для специалиста в данной области из описания изобретения и известного уровня техники. Данное изобретение также относится к применению соединения согласно данному изобретению для получения лекарства для лечения или предупреждения заболевания или состояния. Более конкретно, данное изобретение относится к новому применению для лечения заболевания или состояния, раскрытого здесь, или являющегося очевидным для специалиста в данной области из описания изобретения и известного уровня техники. Раскрытые здесь отдельные соединения в воплощениях данного изобретения применяют для получения лекарства.
Тиазолы заявлены в патенте WO0127094 как ингибиторы триглицерида. Патент WO0426863 раскрывает производные тиазола в качестве ингибиторов трансформирующего фактора роста (TgF). Производные 4,5-диарилтиазола раскрыты в патентах ЕР 388909 и ЕР 377457 как ингибиторы 5-липоксигеназы для лечения тромбоза, гипертензии, аллергии и воспаления. Все показанные там структуры содержат два фенильных кольца, которые замещены в пара-положении метоксигруппой, фтором, метилтиогруппой или метилсульфинильной группой. Патент WO 9603392 раскрывает сульфониларил-арилтиазолы для лечения воспаления и боли, артрита или лихорадки в качестве связанных с воспалением нарушений. Патент JP 05345772 относится к 4,5-диарилтиазолам в качестве ингибиторов ацетилхолинэстеразы и патент JP 04154773 раскрывает 4,5-диарилтиазолы, обладающие аналгезирующим, противовоспалительным и жаропонижающим действием.
В настоящий момент неожиданно найдено, что производные тиазола формулы (I), их пролекарства и их соли

где R и R1 являются одинаковыми или различными и представляют собой фенил или пиридинил, необязательно замещенный 1-3 заместителями Y, где Y представляет собой заместитель, выбираемый из группы, включающей метил, этил, пропил, метокси, этокси, гидрокси, гидроксиметил, гидроксиэтил, хлор, иод, бром, фтор, трифторметил, трифторметокси, метилсульфонил, метилсульфанил, трифторметилсульфонил, фенил или цианогруппу, при условии, что Х не представляет собой подгруппу (ii);
или один из радикалов R и R1 представляет собой фенильную или пиридинильную группу, необязательно замещенную 1-3 заместителями Y, где Y имеет вышеуказанное значение, а другой радикал представляет собой атом водорода или разветвленную или линейную С1-8-алкильную группу, разветвленную или линейную С3-8-гетероалкильную группу, содержащую один гетероатом из группы (N, O, S), С3-7-циклоалкильную группу, С3-7-циклоалкил-С1-3-алкильную группу, С3-7-гетероциклоалкил-С1-3-алкильную группу, причем эти группы могут быть замещены гидроксильной группой, метоксигруппой, метильной, трифторметилсульфонильной или трифторметильной группой или атомом фтора, а С3-7-гетероциклоалкил-С1-3-алкильная группа содержит один или два гетероатома из группы (O, N, S), или вышеуказанный другой радикал представляет собой бензильную группу, необязательно замещенную в фенильном кольце 1-3 заместителями Y, где Y имеет вышеуказанное значение;
Х представляет собой одну из подгрупп (i) или (ii):

где R2 представляет собой разветвленную или линейную С1-8-алкильную группу, С3-7-циклоалкильную группу, С3-7-циклоалкил-С1-2-алкильную группу, С3-7-гетероциклоалкил-С1-2-алкильную группу, причем эти группы могут быть замещены гидроксильной, метильной или трифторметильной группой или атомом фтора, а С3-7-гетероциклоалкил-С1-2-алкильная группа содержит один или два гетероатома из группы (O, N, S),
или R2 представляет собой фенильную, бензильную, фенетильную или фенилпропильную группу, которая в своем фенильном кольце может быть замещена 1-3 заместителями Y, где Y имеет вышеуказанное значение,
или R2 представляет собой пиридильную, тиенильную или нафтильную группу, причем нафтильная группа может быть замещена атомом галогена, метильной группой или метоксигруппой или трифторметильной группой;
R3 представляет собой атом водорода или разветвленную или линейную С1-3-алкильную группу;
R4 представляет собой водород, разветвленную или линейную С1-10-алкильную группу или С3-8-циклоалкил-С1-2-алкильную группу, разветвленную или линейную С1-10-алкоксигруппу, С3-8-циклоалкильную группу, С5-10-бициклоалкильную группу, С5-10-бициклоалкил-С1-2-алкильную группу, С6-10-трициклоалкильную группу, С6-10-трициклоалкилметильную группу, разветвленную или линейную С3-10-алкенильную группу, С5-8-циклоалкенильную группу, причем эти группы могут содержать один или более гетероатомов из группы (O, N, S) и быть замещены гидроксильной, 1-3 метильными группами, этильной группой или 1-3 атомами фтора,
или R4 представляет собой фенильную группу, фениламиногруппу, феноксигруппу, бензильную, фенетильную или фенилпропильную группу, необязательно замещенную в фенильном кольце 1-3 заместителями Y, где Y имеет вышеуказанное значение,
или R4 представляет собой пиридильную или тиенильную группу,
или R4 представляет собой группу NR5R6,
где
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют насыщенную или ненасыщенную, моноциклическую или бициклическую, гетероциклическую группу, имеющую 4-10 атомов в кольце, причем гетероциклическая группа содержит один или более гетероатомов из группы (O, N, S) и указанная гетероциклическая группа может быть замещена разветвленной или линейной С1-3-алкильной, фенильной, гидроксильной или трифторметильной группой или атомом фтора, или
R3 и R4 вместе с атомом азота, к которому они присоединены, образуют насыщенную или ненасыщенную, моноциклическую или бициклическую, гетероциклическую группу, имеющую 4-10 атомов в кольце, причем гетероциклическая группа содержит один или более гетероатомов из группы (O, N, S) и указанная гетероциклическая группа может быть замещена разветвленной или линейной С1-3-алкильной группой, фенильной группой, аминогруппой, гидроксигруппой, метоксигруппой, цианогруппой или трифторметильной группой или атомом фтора или хлора;
являются модуляторами каннабиноидного (СВ) рецептора.
К данному изобретению относятся все соединения, имеющие формулу (I), рацематы, смеси диастереомеров и отдельные стереоизомеры. Таким образом, соединения, в которых заместители у потенциально асимметрических атомов углерода находятся или в R-конфигурации или в S-конфигурации, относятся к данному изобретению.
Также пролекарства, то есть соединения, которые при их введении людям любым известным путем метаболизируются в соединения формулы (I), относятся к данному изобретению. Пролекарства являются биообратимыми производными молекул лекарственного средства, используемыми для преодоления некоторых барьеров к достижению полезности исходной молекулы лекарственного средства. Эти барьеры включают, но не ограничиваясь этим, растворимость, проницаемость, устойчивость, предсистемный метаболизм и ограничения нацеливания (J. Stella, "Prodrugs as therapeutics", Expert Opin. Ther. Patents, 14(3), 277-280, 2004). В особенности, это относится к соединениям с первичными или вторичными аминогруппами или гидроксильными группами. Такие соединения могут вступать в реакцию с органическими кислотами с образованием соединений, имеющих формулу (I), где присутствует дополнительная группа, которая легко удаляется после введения (в организм) и представляющих собой, но не ограничиваясь этим, например, амидин, енамин, основание Манниха, гидроксилметиленовое производное, производное О-(ацилоксиметиленкарбамата), карбамат, сложный эфир, амид или енаминон. Пролекарство является неактивным соединением, которое переходит в активную форму при абсорбции (Medicinal Chemistry: Principles and Practice, 1994, ISBN 0-85186-494-5, изд.: F.D. King, с. 216).
Благодаря активности в отношении СВ-рецептора соединения согласно данному изобретению являются подходящими для применения в лечении психиатрических нарушений, таких как психоз, страх, депрессия, дефицит внимания, нарушения памяти, когнитивные нарушения, нарушения аппетита, ожирение, пристрастие, влечение, лекарственная зависимость, и неврологических нарушений, таких как нейродегенеративные нарушения, слабоумие, дистония, мышечная спастичность, тремор, эпилепсия, рассеянный склероз, травматическое повреждение головного мозга, инсульт, болезнь Паркинсона, болезнь Альцгеймера, болезнь Хантингтона, синдром Туретта, церебральная ишемия, церебральная апоплексия, черепно-мозговая травма, повреждение спинного мозга, нейровоспалительные заболевания, бляшковый склероз, вирусный энцефалит, заболевания, связанные с демиелинизацией, а также для лечения заболеваний, связанных с болью, включая невропатические расстройства, и другие заболевания, вызываемые каннабиноидной нейротрансмиссией, включая лечение септического шока, глаукомы, рака, диабета, рвоты, тошноты, астмы, респираторных заболеваний, желудочно-кишечных нарушений, сексуальных нарушений, язв желудка, поноса и сердечно-сосудистых нарушений.
ФАРМАКОЛОГИЧЕСКИЕ МЕТОДЫ
In vitro аффинность (сродство) к человеческим каннабиноидным рецепторам СВ1
Аффинность соединений согласно данному изобретению к каннабиноидным рецепторам СВ1 можно определять, используя мембранные препараты из клеток яичника китайского хомячка (СНО), в которые устойчиво трансфицирован человеческий каннабиноидный рецептор СВ1 конъюгированный с [3Н]СР-55,940 в качестве радиолиганда. После инкубации свежеприготовленного клеточного мембранного препарата с [3Н]-лигандом, с добавкой или без добавки соединений согласно данному изобретению разделение связанного и свободного лиганда осуществляют путем фильтрации через стекловолоконные фильтры. Радиоактивность на фильтре измеряют с помощью жидкостного сцинтилляционного счетчика.
In vitro аффинность к человеческим каннабиноидным рецепторам СВ2
Аффинность соединений по данному изобретению к каннабиноидным рецепторам СВ2 можно определять, используя мембранные препараты из клеток яичника китайского хомячка (СНО), в которые устойчиво трансфицирован человеческий каннабиноидный рецептор СВ2, конъюгированный с [3Н]СР-55,940 в качестве радиолиганда. После инкубации свежеприготовленного клеточного мембранного препарата с [3Н]-лигандом, с добавкой или без добавки соединений согласно данному изобретению разделение связанного и свободного лиганда осуществляют путем фильтрации через стекловолоконные фильтры. Радиоактивность на фильтре измеряют с помощью жидкостного сцинтилляционного счетчика.
In vitro антагонизм по отношению к человеческим каннабиноидным рецепторам СВ1
In vitro антагонизм к СВ1-рецептору можно оценивать с помощью человеческого СВ1-рецептора, клонированного в клетках яичника китайского хомячка (СНО). СНО-клетки культивируют в питательной, модифицированной по Дульбекко среде Игла (DMEM), с добавлением 10% термоинактивированной фетальной телячьей сыворотки. Среду удаляют и заменяют DMEM без фетальной телячьей сыворотки, но содержащей [3Н]-арахидоновую кислоту, и инкубируют в течение ночи в шкафу для клеточной культуры (5% СО2/95% воздух; 37°С; водонасыщенная атмосфера). За этот период [3Н]-арахидоновая кислота внедряется в мембранные фосфолипиды. В день испытания среду удаляют и клетки промывают три раза, используя 0,5 мл DMEM, содержащей 0,2% бычьего сывороточного альбумина (BSA). Стимуляция СВ1-рецептора с помощью WIN 55,212-2 приводит к активации PLA2 с последующим высвобождением [3Н]-арахидоновой кислоты в среду. Этому WIN 55,212-2-индуцированному высвобождению в зависимости от концентрации противодействуют антагонисты СВ1-рецептора. Антагонистическое действие в отношении СВ1-тестируемых соединений выражают в виде рА2-величин.
In vivo антагонизм по отношению к человеческим каннабиноидным рецепторам СВ1
In vivo СВ1-антагонизм можно оценивать с помощью теста СР-55,940-индуцированной гипотензии у крысы. Самцов крыс с нормальным кровяным давлением (массой 225-300 г; Harlan, Horst, Нидерланды) анестезируют пентобарбиталом (80 мг/кг, внутрибрюшинно). Кровяное давление измеряют с помощью канюли, вставленной в левую сонную артерию, посредством датчика давления Spectramed DTX-plus (Spectramed B.V., Bilthoven, Нидерланды). После амплификации с помощью амплификатора Nihon Kohden Carrier Amplifier (тип АР-621G; Nihon Kohden B.V., Амстердам, Нидерланды), сигнал кровяного давления регистрируют на персональном компьютере (Compaq Deskpro 386s) с помощью программы сбора данных Po-Ne-Mah (Po-Ne-Mah Inc., Storrs, США). Сердечный ритм выводят из пульсового сигнала кровяного давления. Все соединения вводят перорально в виде микросуспензии с 1% метилцеллюлозы за 30 минут перед индукцией анестезии, которую осуществляют за 60 минут до введения агониста СВ1-рецептора СР-55,940. Объем инъекции составляет 10 мл·кг-1. После гемодинамической стабилизации вводят агонист СВ1-рецептора СР-55,940 (0,1 мг·кг-1, внутривенно) и устанавливают гипотензивный эффект (Wagner J.A. и др., Hemodynamic effects of cannabinoids: coronary and cerebral vasodilation mediated by cannabinoid CB1 receptors. Eur. J. Pharmacol., 2001, 423, 203-210).
Этот гипотензивный тест также можно использовать для оценки агонистических действий соединений в отношении СВ1-рецептора. Такому СВ1-агонистическому действию в отношении кровяного давления можно противодействовать с помощью селективного антагониста СВ1-рецептора, такого как римонабант.
Агонистическую или частично агонистическую активность соединений согласно данному изобретению по отношению к каннабиноидному рецептору можно определять с помощью опубликованных методов, таких как оценка in vivo каннабимиметических эффектов (Wiley J.L. и др., J. Pharmacol. Exp. Ther., 2001, 296, 1013).
Соединения согласно данному изобретению можно переводить в формы, пригодные для введения, с помощью обычных способов, используя вспомогательные вещества и/или жидкие или твердые носители.
Соединения согласно настоящему изобретению, как правило, вводят в виде фармацевтических композиций, которые являются важными и новыми воплощениями согласно данному изобретению вследствие наличия соединений, в частности специфических соединений, раскрытых в данном контексте. Типы фармацевтических композиций, которые можно использовать, включают, не ограничиваясь этим, таблетки, жевательные таблетки, капсулы, растворы, парентеральные растворы, суппозитории, суспензии и другие типы, раскрытые здесь или очевидные для специалиста в данной области из описания изобретения и известного уровня техники.
Воплощения согласно данному изобретению предусматривают фармацевтическую упаковку или набор, содержащая(ий) одну или более емкостей, заполненных одним или более ингредиентами фармацевтической композиции согласно данному изобретению. Связанными с такой емкостью (емкостями) могут быть различные письменные информационные материалы, такие как инструкции по применению или уведомление в форме, предписанной государственным учреждением по регулированию производства, применения или реализации фармацевтических продуктов, где отражено одобрение учреждением по производству, применению или реализации для введения людям или животным.
ОБЩИЕ АСПЕКТЫ СИНТЕЗА
Производные тиазола можно получать с помощью известных методов, например:
а) Organic Reactions, том VI, (1951), с. 367-409, изд. R. Adams, John Wiley and Sons Inc., Нью-Йорк;
b) J.S. Carter и др., Bioorg. Med. Chem. Lett. (1999), 9, 1167-1170;
с) T.T. Sakai и др., Bioorg. Med. Chem. (1999), 7, 1559-1566;
d) A. Tanaka и др., J. Med. Chem. (1994), 37, 1189-1199;
e) J.J. Talley и др., патент WO 9603392: Chem. Abstr., 125, 33628;
f) V. Cecchetti и др., Bioorg. Med. Chem. (1994), 2, 799-806;
g) T. Eicher и др., The Chemistry of Heterocycles, (1995), с. 149-155, Georg Thieme Verlag, Штутгарт, 1995, ISBN 313-100511-4, и приведенные здесь ссылки;
h) Gilchrist T.L., Heterocyclic Chemistry, 3-е изд., 1997, с. 319-327, Longman, Великобритания, ISBN 0-582-27843-0.
Альфа-галогенкетоны можно получать путем галогенирования соответствующего кетона. Реакция альфа-галогенкарбонильных соединений и тиоамида может приводить к большому ряду производных тиазола. Более конкретно, в результате конденсации альфа-бромкетонов с этилтиооксаматом получают (2-этоксикарбонил)тиазолы общей формулы (II).

Соединения формулы (II) можно превращать в соответствующий N-метокси-N-метиламид (III) и затем вводить во взаимодействие с реагентом алкиллитием или ариллитием, получая соединение общей формулы (I), где Х представляет собой подгруппу (i).
Соединения общей формулы (II) можно амидировать с помощью амина общей формулы R3R4NH с получением соединения общей формулы (I), где Х представляет собой подгруппу (ii). Такие амидирования можно катализировать с помощью (СН3)3Al. (Для дополнительной информации об опосредованной алюминием конверсии сложных эфиров в амиды, см. J.I. Levin, E. Turos, S.M. Weinreb, Synth. Commun., (1982), 12, 989-993).
Альтернативно, соединение, имеющее формулу (II), превращают в соответствующую карбоновую кислоту и затем вводят во взаимодействие с так называемым галогенирующим агентом, таким как, например, тионилхлорид (SOCl2). При этой реакции получают соответствующий карбонилхлорид, который затем вводят во взаимодействие с соединением, имеющим формулу R3R4NH, где R3 иR4 имеют значения, как описано выше.
Альтернативно, сложно-эфирную группу в соединении (II) можно превращать в соответствующую карбоновую кислоту. Эту карбоновую кислоту можно вводить во взаимодействие с соединением, имеющим формулу R3R4NH, где R3 иR4 имеют значения, как описано здесь выше, посредством способов активации и связывания, таких как образование активного сложного эфира, или в присутствии так называемого связующего реагента, такого как, например, DCC, HBTU, HOAT (N-гидрокси-7-азабензотиазол), ВОР, CIP (2-хлор-1,3-диметилимидазолинийгексафторфосфат), PyAOP (7-азабензтриазол-1-илокситрис(пирролидино)фосфонийгексафторфосфат) и тому подобные. (Для дополнительной информации о способах активации и связывания см. а) M. Bodanszky, A. Bodanszky: The Practice of Peptide Synthesis, Springer-Verlag, Нью-Йорк, 1994; ISBN: 0-387-57505-7; b) K. Akaji и др., Tetrahedron Lett. (1994), 35, 3315-3318; c) F. Albericio и др., Tetrahedron Lett. (1997), 38, 4853-4856).
Следующие соединения можно получать в соответствии с этими методиками. Они предназначены для дальнейшей, более детальной иллюстрации данного изобретения, и поэтому не считаются ограничивающими объем охраны изобретения каким-либо образом.
СИНТЕЗЫ СОГЛАСНО ОТДЕЛЬНЫМ ПРИМЕРАМ
1Н-ЯМР-спектры регистрируют на приборе Varian UN400 (400 МГц) с тетраметилсиланом в качестве внутреннего стандарта. Химические сдвиги получают в м.д. (d-шкала) в сторону слабого поля от тетраметилсилана. Константы взаимодействия (J) выражают в Гц. Тонкослойную хроматографию осуществляют на предварительно покрытых F254 60 пластинах Merck и зоны визуализируют ультрафиолетовым излучением. Флэш-хроматографию осуществляют, используя силикагель 60 (0,040-0,063 мм, Merck). Колоночную хроматографию осуществляют, используя силикагель 60 (0,063-0,200 мм, Merck). Температуры плавления регистрируют на устройстве для определения температур плавления Büchi B-545 и не корректируют.
Пример 1
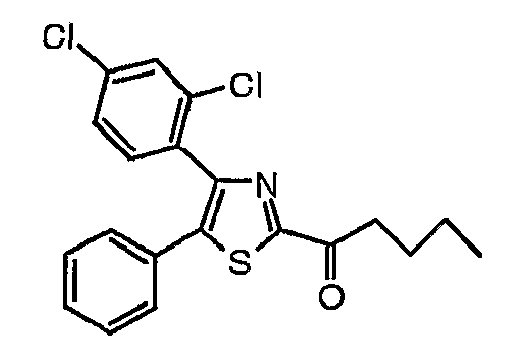
Пример 1
Часть А: К раствору 1-(2,4-дихлорфенил)-2-фенилэтанона (54,35 г, 0,205 моль) в бензоле (220 мл) медленно добавляют бром (10,6 мл, 0,205 моль) и полученный раствор перемешивают при комнатной температуре в течение 1 часа. Медленно добавляют водный 5%-ный раствор NaHCO3. Отделяют органический слой, сушат над MgSO4, отфильтровывают и выпаривают в вакууме с получением сырого 2-бром-1-(2,4-дихлорфенил)-2-фенилэтанона (69,4 г, выход 98%) в виде масла.
1H-ЯМР (400 МГц, CDCl3): δ 6,20 (с, 1H), 7,26 (дд, J=8 и 2 Гц, 1H), 7,31-7,50 (м, 7H).
Часть В: 2-Бром-1-(2,4-дихлорфенил)-2-фенилэтанон (25,83 г, 0,075 моль) и этилтиооксамат (15,0 г, 0,112 моль) растворяют в абсолютном этаноле (200 мл). Полученную смесь нагревают при температуре кипения с обратным холодильником в течение 16 часов. После выпаривания в вакууме сырое вещество растворяют в смеси воды и дихлорметана. Отделяют слой дихлорметана и водный слой экстрагируют три раза дихлорметаном. Собранные органические слои сушат (MgSO4), отфильтровывают и концентрируют. Полученное вещество очищают с помощью колоночной хроматографии (силикагель/дихлорметан) с получением 4-(2,4-дихлорфенил)-5-фенилтиазол-2-карбоксилата (10,5 г, выход 37%).
1H-ЯМР (400 МГц, CDCl3): δ Т,46 (т, J=7 Гц, 3H), 4,53 (кв, J=7 Гц, 2H), 7,24-7,38 (м, 7H), 7,43 (д, J=2 Гц, 1H).
Часть С: К раствору этил-4-(2,4-дихлорфенил)-5-фенилтиазол-2-карбоксилата (10,5 г, 0,028 моль) в метаноле (170 мл) медленно добавляют раствор КОН (8,9 г, 0,0896 моль) в воде (170 мл). Полученный раствор нагревают при 90°С в течение 2 часов и охлаждают до комнатной температуры. Добавляют смесь концентрированной HCl и льда. Образовавшийся осадок собирают, промывают водой и диэтиловым эфиром и сушат с получением 4-(2,4-дихлорфенил)-5-фенилтиазол-2-карбоновой кислоты (8,99 г, выход 92%). Температура плавления: 105оС.
Часть D: К перемешиваемой с помощью магнитной мешалки суспензии 4-(2,4-дихлорфенил)-5-фенилтиазол-2-карбоновой кислоты (4,2 г, 0,012 моль) в безводном дихлорметане (170 мл) последовательно добавляют 7-аза-1-гидроксибензтриазол (HOAT) (4,083 г, 0,030 моль), 7-азабензтриазол-1-илокситрис(пирролидино)фосфоний-гексафторфосфат (PyAOP) (15,64 г, 0,03 моль), диизопропилэтиламин (6,26 мл, 0,036 моль) и N-метокси-N-метиламин·HCl (2,925 г, 0,030 моль) и полученный раствор перемешивают в течение 16 часов при комнатной температуре. Медленно добавляют водный 5%-ный раствор NaHCO3 и полученную смесь экстрагируют (3 раза) дихлорметаном. Собранные органические слои сушат (MgSO4), отфильтровывают и концентрируют с получением сырого масла (18,9 г). Очищают с помощью флэш-хроматографии (силикагель; этилацетат/петролейный эфир = 1/1), получая N-метил-N-метокси-4-(2,4-дихлорфенил)-5-фенилтиазол-2-карбоксамид (4,0 г, выход 85%).
1H-ЯМР (400 МГц, CDCl3): δ 3,60 (шир.с, 3H), 3,90 (с, 3H), 7,21-7,33 (м, 7H), 7,45 (д, J=2 Гц, 1H).
Аналогично получают N-метил-N-метокси-4-(2-хлорфенил)-5-фенилтиазол-2-карбоксамид.
1H-ЯМР (400 МГц, CDCl3): δ 3,62 (шир.с, 3H), 3,90 (с, 3H), 7,22-7,45 (м, 9H).
Часть Е: К охлаждаемому (-70°С) и перемешиваемому раствору N-метил-N-метокси-4-(2,4-дихлорфенил)-5-фенилтиазол-2-карбоксамида (2,0 г, 0,005 моль) в ТГФ (20 мл) в атмосфере N2 добавляют н-BuLi (3,13 мл, 1,6 М раствор в гексане, 0,005 моль). После перемешивания в течение 30 минут раствор выдерживают до достижения комнатной температуры и перемешивают в течение 16 часов. Добавляют водный раствор HCl (20 мл, 1 н) и полученную смесь экстрагируют диэтиловым эфиром. Слои диэтилового эфира промывают водой (2 раза), сушат (MgSO4), отфильтровывают и концентрируют с получением сырого масла (2,03 г). Очищают с помощью флэш-хроматографии (силикагель/дихлорметан), получая 1-[4-(2,4-дихлорфенил)-5-фенилтиазол-2-ил]пентан-1-она (0,6 г, выход 31%).
1H-ЯМР (400 МГц, CDCl3): δ 0,95 (т, J=7 Гц, 3H), 1,38-1,48 (м, 2H), 1,72-1,80 (м, 2H), 3,16 (т, J=7 Гц, 2H), 7,20-7,35 (м, 7H), 7,46 (д, J=2 Гц, 1H).
Аналогично получают
Пример 2

Пример 2
1H-ЯМР (400 МГц, CDCl3): δ 0,93 (т, J=7 Гц, 3H), 1,26-1,44 (м, 6H), 1,73-1,82 (м, 2H), 3,15 (т, J=7 Гц, 2H), 7,21-7,34 (м, 7H), 7,47 (д, J=2 Гц, 1H).
Пример 3

Пример 3
Температура плавления 78-80°С.
Пример 4

Пример 4
1H-ЯМР (400 МГц, CDCl3): δ 0,93 (т, J=7 Гц, 3H), 1,25-1,44 (м, 6H), 1,73-1,82 (м, 2H), 3,17 (т, J=7 Гц, 2H), 7,22-7,40 (м, 8H), 7,47 (дд, J=8 и 2 Гц, 1H).
Пример 5

Пример 5
Температура плавления 131-132°С.
Пример 6

Пример 6
Часть А: К перемешиваемому с помощью магнитной мешалки раствору 1-фенилгептан-1-он (23,7 г, 0,125 моль) в бензоле (160 мл) медленно добавляют бром (7,0 мл, 0,125 моль) и полученный раствор оставляют для взаимодействия при комнатной температуре в течение 1 часа. Медленно добавляют водный 5%-ный раствор NaHCO3 с последующим добавлением дихлорметана. Отделяют органический слой, сушат над MgSO4, отфильтровывают и выпаривают в вакууме с получением сырого 2-бром-1-фенилгептан-1-она (41,8 г, количественный выход) в виде масла.
1H-ЯМР (400 МГц, CDCl3): δ 0,90 (т, J=7 Гц, 3H), 1,28-1,78 (м, 6H), 2,04-2,25 (м, 2H), 5,11-5,16 (м, 1 H), 7,42-7,62 (м, 3H), 8,00-8,04 (м, 2H).
Часть В: 2-Бром-1-фенилгептан-1-он (20,17 г, 0,075 моль) и этилтиооксамат (15,0 г, 0,112 моль) растворяют в абсолютном этаноле (200 мл). Полученную смесь нагревают при температуре кипения с обратным холодильником в течение 16 часов. После выпаривания в вакууме сырое вещество растворяют в смеси воды и дихлорметана. Отделяют слой дихлорметана и водный слой экстрагируют три раза дихлорметаном. Собранные органические слои сушат (MgSO4), отфильтровывают и концентрируют. Полученное вещество очищают с помощью колоночной хроматографии (силикагель; дихлорметан/петролейный эфир = 1/1) с получением этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата (12,09 г, выход 53%) в виде медленно отверждающегося масла. Температура плавления 51-52°С.
Часть С: К перемешиваемому с помощью магнитной мешалки раствору этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата (12,09 г, 0,039 моль) в метаноле (240 мл) медленно добавляют раствор КОН (8,9 г) в воде (240 мл). Полученный раствор нагревают при температуре кипения с обратным холодильником в течение 2 часов и потом охлаждают до комнатной температуры. Добавляют смесь концентрированной HCl и льда. Образовавшийся осадок собирают, последовательно промывают водой и холодным диэтиловым эфиром и сушат с получением 5-(н-пентил)-4-фенилтиазол-2-карбоновой кислоты (3,54 г, выход 32%).
1H-ЯМР (400 МГц, CDCl3): δ 0,87 (т, J=7 Гц, 3H), 1,28-1,40 (м, 4H), 1,66-1,76 (м, 2H), 2,93-3,00 (м, 2H), 4,00 (шир.с, 1H), 7,35-7,46 (м, 3H), 7,58-7,64 (м, 2H).
Часть D: К перемешиваемой с помощью магнитной мешалки суспензии 5-(н-пентил)-4-фенилтиазол-2-карбоновой кислоты (1,18 г, 0,0043 моль) в безводном дихлорметане (35 мл) последовательно добавляют 7-аза-1-гидроксибензтриазол (HOAT) (1,46 г, 0,0107 моль), 7-азабензтриазол-1-илокситрис- и цис-миртаниламина (CAS (пирролидино)фосфонийгексафторфосфат (РуАОР) (5,59 г, 0,0107 моль), диизопропилэтиламин (2,24 мл, 0,0129 моль) и анилин (0,98 мл, 0,0107 моль) и полученный раствор перемешивают в течение 16 часов при комнатной температуре. Полученную смесь концентрируют и очищают с помощью флэш-хроматографии (силикагель/дихлорметан) с получением N-фенил-5-(н-пентил)-4-фенилтиазол-2-карбоксамида (0,89 г, выход 59%).
1H-ЯМР (400 МГц, CDCl3): δ 0,98 (т, J=7 Гц, 3Н), 1,26-1,41 (м, 4Н), 1,68-1,78 (м, 2Н), 2,97 (т, J=7 Гц, 2Н), 7,12-7,18 (м, 1Н), 7,34-7,52 (м, 5Н), 7,60-7,64 (м, 2Н), 7,69-7,74 (м, 2Н), 9,10 (шир.с 1Н).
Аналогично получают
Пример 7
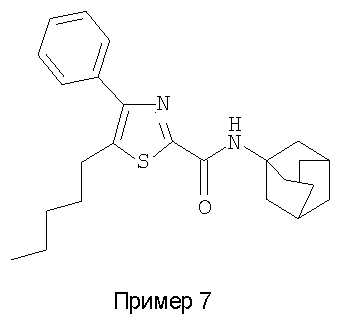
Получают из 5-(н-пентил)-4-фенилтиазол-2-карбоновой кислоты и 1-аминоадамантана.
Температура плавления 90-92°С.
Пример 8
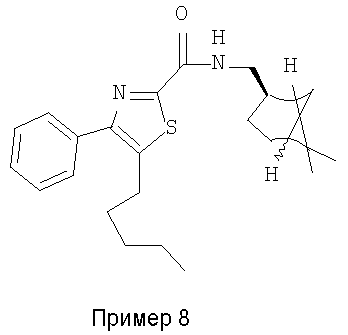
Получают из 5-(н-пентил)-4-фенилтиазол-2-карбоновой кислоты 38235-68-6).
1H-ЯМР (400 МГц, CDCl3): δ 0,89 (т, J=7 Гц, 3H), 1,08 (с, 3H), 1,20 (с, 3H), 1,26-1,38 (м, 4H), 1,50-1,62 (м, 1H), 1,66-1,74 (м, 2H), 1,82-2,04 (м, 5H), 2,28-2,40 (м, 2H), 2,94 (т, J=7 Гц, 2H), 3,39-3,50 (м, 2H), 7,29 (шир.т, J=7 Гц, 1H), 7,37-7,60 (м, 5H).
Пример 9
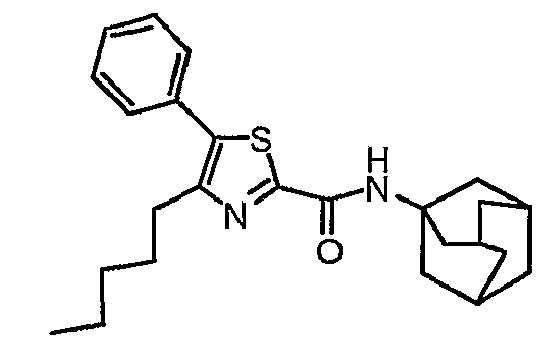
Пример 9
Часть А: 1-Бром-1-фенилгептан-2-он (19,98 г, 0,074 моль) и этилтиооксамат (15,0 г, 0,112 моль) растворяют в абсолютном этаноле (200 мл). Полученную смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов. После выпаривания в вакууме сырое вещество растворяют в смеси воды и дихлорметана. Отделяют слой дихлорметана и водный слой экстрагируют три раза дихлорметаном. Собранные органические слои сушат (MgSO4), отфильтровывают и концентрируют. Полученное вещество очищают с помощью колоночной хроматографии (силикагель; дихлорметан/петролейный эфир = 1/1) с получением этил-4-(н-пентил)-5-фенилтиазол-2-карбоксилата (5,24 г, выход 23%) в виде масла.
1H-ЯМР (400 МГц, CDCl3): δ 0,89 (т, J=7 Гц, 3H), 1,24-1,32 (м, 4H), 1,44 (т, J=7 Гц, 3H), 1,70-1,78 (м, 2H), 2,81-2,87 (м, 2H), 4,48 (кв, J=7 Гц, 2H), 7,40-7,48 (м, 5H).
Аналогично получают
Этил-4-бензил-5-фенилтиазол-2-карбоксилат в виде масла.
Этил-5-(н-пентил)-4-(2,4-дихлорфенил)тиазол-2-карбоксилат.
Температура плавления 92-93°С.
Часть В: К перемешиваемому с помощью магнитной мешалки раствору 1-аминоадамантана (1,607 г, 0,0086 моль) в безводном дихлорметане (10 мл) добавляют Al(СН3)3 (4,3 мл, 2 М раствор в гексане, 0,0086 моль) и полученный раствор оставляют для взаимодействия при комнатной температуре в течение 10 минут. Медленно добавляют водный 5%-ный раствор NaHCO3. Экстрагируют дихлорметаном, сушат над MgSO4, отфильтровывают и концентрируют в вакууме с последующей колоночной хроматографией (силикагель; дихлорметан), получая N-(адамант-1-ил)-4-(н-пентил)-5-фенилтиазол-2-карбоксамид (1,17 г, выход 72%).
1H-ЯМР (400 МГц, CDCl3): δ 0,88 (т, J=7 Гц, 3Н), 1,26-1,34 (м, 4Н), 1,68-1,78 (м, 8Н), 2,08-2,18 (м, 9Н), 2,71-2,76 (м, 2Н), 7,02 (шир.с, 1Н), 7,36-7,45 (м, 5Н).
Аналогично получают
Пример 10

Получают из этил-4-(н-пентил)-5-фенилтиазол-2-карбоксилата и цис-миртаниламина (CAS 38235-68-6).
1H-ЯМР (400 МГц, CDCl3): δ 0,89 (т, J=7 Гц, 3Н), 1,10 (с, 3H), 1,22 (с, 3H), 1,25-1,34 (м, 4H), 1,54-1,78 (м, 3H), 1,84-2,06 (м, 5H), 2,31-2,42 (м, 2H), 2,72-2,78 (м, 2H), 3,44-3,50 (м, 2H), 7,24-7,28 (м, 1H), 7,37-7,46 (м, 5H).
Пример 11

Пример 11
Получают из этил-4-(н-пентил)-5-фенилтиазол-2-карбоксилата и циклогексиламина.
Температура плавления 84-85°С.
Пример 12

Пример 12 экзо
Получают из этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата и экзо-2-аминобицикло[2.2.1]гептана.
Температура плавления 64-65°С.
Пример 13

Пример 13 эндо
Получают из этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата и эндо-2-аминобицикло[2.2.1]гептана.
Температура плавления 80-82°С.
Пример 14

Пример 14
Получают из этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата и 4-изопропилпиперазина.
Температура плавления 84-85°С.
Пример 15

Пример 15
Получают из этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата и индан-2-иламина.
1H-ЯМР (400 МГц, CDCl3): δ 0,85 (т, J=7 Гц, 3H), 1,26-1,38 (м, 4H), 1,65-1,75 (м, 2H), 2,90-3,01 (м, 4H), 3,36-3,44 (м, 2H), 4,86-4,96 (м, 1H), 7,15-7,27 (м, 4H), 7,35-7,47 (м, 4H), 7,52-7,56 (м, 2H).
Пример 16

Пример 16
Получают из этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата и 3-амино-3-азабицикло[3.3.0]октана.
Температура плавления 86-87°С.
Пример 17

Пример 17
Получают из этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата и 1,2,3,4-тетрагидроизохинолина.
Температура плавления 50-51°С.
Пример 18

Пример 18
Получают из этил-5-(н-пентил)-4-фенилтиазол-2-карбоксилата и R-(+)-борниламина (CAS 32511-34-5).
1H-ЯМР (400 МГц, CDCl3): δ 0,90-1,02 (м, 13H), 1,22-1,47 (м, 6H), 1,60-1,84 (м, 5H), 2,36-2,45 (м, 1H), 2,94 (т, J=7 Гц, 2H), 4,36-4,44 (м, 1H), 7,32 (шир.д, J=7 Гц, 1H), 7,38-7,50 (м, 3H), 7,58-7,63 (м, 2H).
Пример 19

Пример 19
Получают из этил-4-бензил-5-фенилтиазол-2-карбоксилата и циклогексиламина.
Температура плавления 104-106°С.
Пример 20
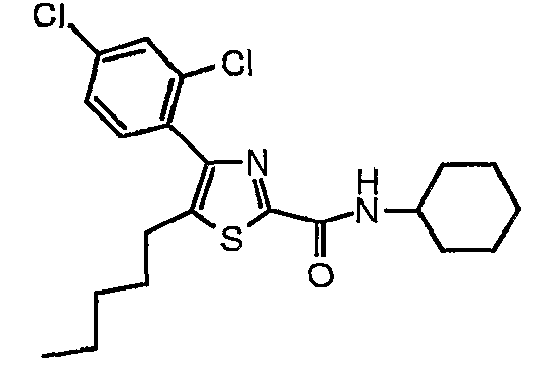
Пример 20
Получают из этил-5-(н-пентил)-4-(2,4-дихлорфенил)тиазол-2-карбоксилата и циклогексиламина.
1H-ЯМР (400 МГц, CDCl3): δ 0,85 (т, J=7 Гц, 3H), 1,13-1,46 (м, 8H), 1,55-1,68 (м, 4H), 1,71-1,80 (м, 2H), 1,96-2,06 (м, 2H), 2,67 (т, J=7 Гц, 2H), 3,86-3,98 (м, 1H), 7,05 (шир.д, J=7 Гц, 1H), 7,26-7,37 (м, 2H), 7,53 (д, J=2 Гц, 1H).
Пример 21
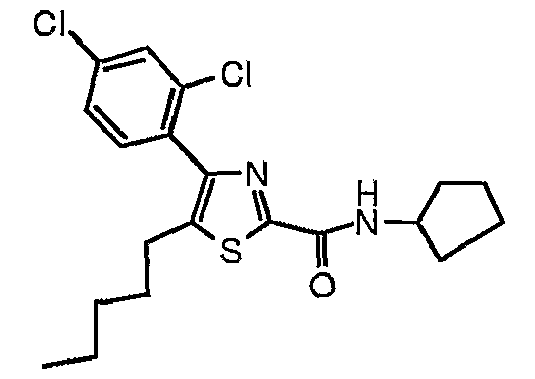
Пример 21
Получают из этил-5-(н-пентил)-4-(2,4-дихлорфенил)тиазол-2-карбоксилата и циклопентиламина.
1H-ЯМР (400 МГц, CDCl3): δ 0,85 (т, J=7 Гц, 3H), 1,20-1,30 (м, 4H), 1,48-1,77 (м, 8H), 2,01-2,10 (м, 2H), 2,67 (т, J=7 Гц, 2H), 4,31-4,41 (м, 1H), 7,09 (шир.д, J=7 Гц, 1H), 7,25-7,37 (м, 2H), 7,53 (д, J=2 Гц, 1H).
Пример 22
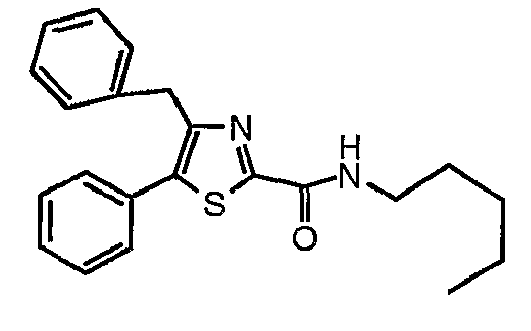
Пример 22
Получают из этил-4-бензил-5-фенилтиазол-2-карбоксилата и н-пентиламина.
1H-ЯМР (400 МГц, CDCl3): δ 0,89 (т, J=7 Гц, 3H), 1,33-1,40 (м, 4H), 1,59-1,67 (м, 2H), 3,40-3,47 (м, 2H), 4,16 (с, 2H), 7,15-7,32 (м, 6H), 7,39-7,42 (м, 5H).
Пример 23
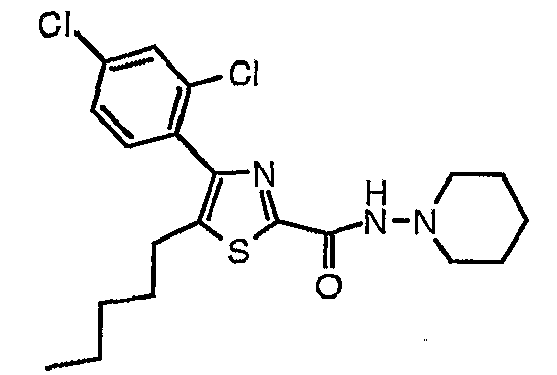
Пример 23
Получают из этил-5-(н-пентил)-4-(2,4-дихлорфенил)тиазол-2-карбоксилата и 1-аминопиперидина.
1H-ЯМР (400 МГц, CDCl3): δ 0,85 (т, J=7 Гц, 3H), 1,20-1,28 (м, 4H), 1,39-1,46 (м, 2H), 1,56-1,64 (м, 2H), 1,71-1,79 (м, 4H), 2,66 (т, J=7 Гц, 2H), 2,82-2,88 (м, 4H), 7,29 (д, J=8 Гц, 1H), 7,35 (дд, J=8 и 2 Гц, 1H), 7,53 (д, J=2 Гц, 1H), 7,88 (шир.с, 1H).
Пример 24

Пример 24
Получают из этил-5-(н-пентил)-4-(2,4-дихлорфенил)тиазол-2-карбоксилата и 4-аминоморфолина.
1H-ЯМР (400 МГц, CDCl3): δ 0,85 (т, J=7 Гц, 3H), 1,21-1,29 (м, 4H), 1,57-1,66 (м, 2H), 2,67 (т, J=7 Гц, 2H), 2,93-2,98 (м, 4H), 3,82-3,88 (м, 4H), 7,29 (д, J=8 Гц, 1H), 7,35 (дд, J=8 и 2 Гц, 1H), 7,54 (д, J=2 Гц, 1H), 7,95 (шир.с, 1H).
Пример 25

Пример 25
Получают из этил-5-(н-пентил)-4-(2,4-дихлорфенил)тиазол-2-карбоксилата и N-метиланилина.
1H-ЯМР (400 МГц, CDCl3): δ 0,80 (шир.т, J=7 Гц, 3H), 1,14-1,28 (м, 4H), 1,50-1,62 (м, 2H), 2,56-2,66 (м, 2H), 3,56 (шир.с, 3H), 6,80-7,45 (м, 8H).
РЕЗУЛЬТАТЫ ФАРМАКОЛОГИЧЕСКИХ ИСПЫТАНИЙ
Данные по аффинности в отношении каннабиноидного рецептора, полученные в соответствии с приведенными выше протоколами, указаны в таблице ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТИАЗОЛА, ИМЕЮЩИЕ АНТАГОНИСТИЧЕСКУЮ, АГОНИСТИЧЕСКУЮ ИЛИ ЧАСТИЧНО АГОНИСТИЧЕСКУЮ СВ-АКТИВНОСТЬ | 2003 |
|
RU2301804C2 |
| ПРОИЗВОДНЫЕ 1Н-ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ КАННАБИНОИДНЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2337907C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИНА, ОБЛАДАЮЩИЕ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2357958C2 |
| ПРОИЗВОДНЫЕ 1H-1,2,4-ТРИАЗОЛ-3-КАРБОКСАМИДА В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРА КАННАБИНОИДОВ | 2003 |
|
RU2325382C2 |
| ПРОИЗВОДНЫЕ 1Н-ИМИДАЗОЛА КАК МОДУЛЯТОРЫ РЕЦЕПТОРА КАННАБИНОИДОВ СВ2 | 2006 |
|
RU2410377C2 |
| 1,3,5-ТРИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4,5-ДИГИДРО-1Н-ПИРАЗОЛА, ОБЛАДАЮЩИЕ СВ-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2360904C2 |
| ТЕТРАЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ CB КАННАБИНОИДОВ С ВЫСОКОЙ СЕЛЕКТИВНОСТЬЮ К ПОДТИПАМ РЕЦЕПТОРОВ CB/CB | 2005 |
|
RU2354650C2 |
| ПРОИЗВОДНЫЕ 1Н-ИМИДАЗОЛА, ОБЛАДАЮЩИЕ CB-АГОНИСТИЧЕСКОЙ, ЧАСТИЧНОЙ CB-АГОНИСТИЧЕСКОЙ ИЛИ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2299200C2 |
| ИНГИБИТОРЫ ТИРОЗИНФОСФАТАЗЫ БЕЛКА ЧЕЛОВЕКА И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2435763C2 |
| ПРОИЗВОДНЫЕ ТИАЗОЛА | 2005 |
|
RU2367661C2 |
Изобретение относится к новым соединениям формулы (I) и фармакологически приемлемым солям. Соединения настоящего изобретения обладают свойствами модулятора каннабиноидного СВ рецептора. В общей формуле (I)
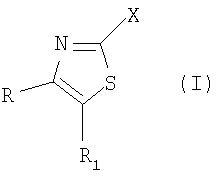


R и R1 являются одинаковыми или различными и представляют собой фенил, необязательно замещенный 1-3 заместителями Y, где Y представляет собой заместитель, выбираемый из группы, включающей хлор, иод, бром, фтор, при условии, что Х не представляет собой подгруппу (ii); или один из радикалов R и R1 представляет собой фенильную группу, а другой радикал представляет собой разветвленную или линейную С2-8-алкильную группу, или бензильную группу; Х представляет собой одну из подгрупп (i) или (ii). Изобретение также относится к применению соединений для получения фармацевтической композиции, к фармацевтической композиции, проявляющей активность модулятора каннабиноидного СВ рецептора, и к соединению общей формулы (IV), для которой значения радикалов представлены в формуле изобретения. 4 н. и 1 з.п. ф-лы, 1 табл.
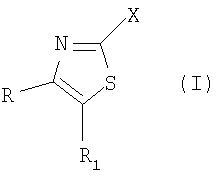
где R и R1 являются одинаковыми или различными и представляют собой фенил, необязательно замещенный 1-3 заместителями Y, где Y представляет собой заместитель, выбираемый из группы, включающей хлор, иод, бром, фтор, при условии, что Х не представляет собой подгруппу (ii);
или один из радикалов R и R1 представляет собой фенильную группу, а другой радикал представляет собой разветвленную или линейную С2-8-алкильную группу или бензильную группу;
Х представляет собой одну из подгрупп (i) или (ii)

где R2 представляет собой разветвленную или линейную С3-8-алкильную группу или фенильную группу при условии, что когда R2 представляет собой фенильную группу, R не является фенилом или 4-хлорфенилом,
R3 представляет собой водород, разветвленную или линейную C1-3-алкильную группу;
R4 представляет собой разветвленную или линейную С1-10-алкильную группу, С3-8-циклоалкильную группу, С5-10-бициклоалкильную группу, С5-10-бициклоалкил-С1-2-алкильную группу, С6-10-трициклоалкильную группу, С6-10-трициклоалкилметильную группу, причем группы могут быть замещены 1-3 метильными группами,
или R4 представляет собой фенильную группу, группу NR5R6,
где R5 и R6 вместе с атомом азота, к которому они присоединены, образуют насыщенную или ненасыщенную, моноциклическую или бициклическую, гетероциклическую группу, имеющую 4-10 атомов в кольце, причем гетероциклическая группа содержит один или два гетероатома из группы (О и N) и указанная гетероциклическая группа может быть замещена разветвленной или линейной C1-3-алкильной группой, или
R3 и R4 вместе с атомом азота, к которому они присоединены, образуют насыщенную или ненасыщенную, моноциклическую или бициклическую, гетероциклическую группу, имеющую 4-10 атомов в кольце, причем гетероциклическая группа содержит один или два N гетероатома;
и его фармакологически приемлемые соли.
и его фармакологически приемлемых солей, где
R, R1 и Х определены в п.1, для получения фармацевтической композиции, обладающей свойствами антагониста каннабиноидного СВ-рецептора.

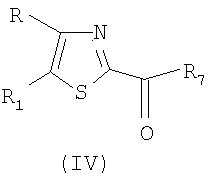
где один из радикалов R и R1 представляет собой фенильную группу, необязательно замещенную 1-3 заместителями Y, где Y имеет вышеуказанное значение, а другой радикал представляет собой разветвленную или линейную С2-8-алкильную группу, и R7 представляет собой гидроксильную группу, разветвленную или линейную С1-4-алкоксигруппу.
| RU 2059637 C1, 10.05.1996 | |||
| Всережимная туковая вибросеялка | 1959 |
|
SU127094A1 |
| СОСТАВ ДЛЯ УДАЛЕНИЯ БЕЛКОВЫХ ПЯТЕН | 0 |
|
SU377457A1 |
| WO 03007887 A2, 30.01.2003. | |||

