Область изобретения
Настоящее изобретение относится к новым соединениям, фармацевтическим композициям, содержащим их, применению указанных соединений в изготовлении лекарственного средства для лечения, в частности, шоковых состояний, а также к способу лечения указанных состояний, при котором вводят указанные соединения.
Предшествующий уровень техники
Пептидные агонисты вазопрессинового рецептора V1a, такие как терлипрессин, недавно (смотри, например, O'Brian et al., Lancet 359 (9313): 1209-10, June 4th, 2002) приобрели повышенное внимание для клинического применения в лечении критических заболеваний и состояний, включая шок гиповолемической (например, геморрагической) или вазодилататорной (например, септической) природы, варикозное расширение вен пищевода с кровотечением (ВРВПК), гепаторенальный синдром (ГРС), сердечно-легочную реанимацию и гипотензию, индуцированную анестезией. Также было показано, что они обладают клинической применимостью в лечении ортостатической гипотензии, дисфункции кровообращения, индуцированной парацентезом, кровопотери во время операции и кровопотери, связанной с обработкой ожогов и носовым кровотечением, и для лечения различных глазных заболеваний путем увеличения слезотечения/образования слез.
При лечении критических состояний контроль артериального кровяного давления является весьма желательным, а используемое лекарство обычно вводят внутривенно. Непрерывная внутривенная инфузия лекарства с увеличивающейся или уменьшающейся скоростью представляет собой практическое средство обеспечения желаемой степени контроля. Достижение так называемых стационарных концентраций лекарства в плазме зависит от периода полувыведения лекарства, вводимого инфузией. Обычно считают, что стационарная концентрация в плазме достигается после периода времени, эквивалентного трем периодам полувыведения лекарства. Для практического применения в клинических условиях следует достигать желаемого "стационарного" артериального кровяного давления в течение приблизительно двух часов, предпочтительно в течение одного часа или меньше. Поэтому агонисты V1a с периодом полувыведения более 1 часа обычно не рассматривают в качестве полезных для лечения критического состояния.
Недостаток терлипрессина при многих критических ситуациях заключается в большой продолжительности его действия, что затрудняет определение его воздействия при изменениях болезненного состояния. Эффективность терлипрессина в отношении рецептора человека V1a (hV1a) также нуждается в улучшении, например, в отношении обеспечения возможности использования меньших доз.
Соединение, известное как F180 (ср. пример 3 в патенте США №5459236), также обладает неудобным длительным периодом действия для того, чтобы его можно было рассматривать для лечения большинства критических состояний.
Основным недостатком других существующих соединений, например [Phe2, Orn8] ОТ (ср. пример 1f в патенте США №3352843) и аргининвазопрессина (AVP), является неспецифическая агонистическая активность в отношении рецептора. Активность в отношении родственных рецепторов, таких как V1b, V2 и окситоциновый (ОТ) рецепторы, потенциально может приводить к нежелательным побочным эффектам и беспокойству относительно безопасности. В качестве примера активация рецептора V2 может вызвать антидиурез (ср. десмопрессин), высвобождение факторов коагуляции/тромболиза и индуцировать вазодилатацию/гипотензию с ответной тахикардией. Последний побочный эффект также может быть вызван агонистической активностью в отношении ОТ рецептора.
Задача настоящего изобретения заключается в том, чтобы предложить соединения, которые были бы особенно полезными в лечении критических состояний.
Описание изобретения
Настоящее изобретение относится к соединениям, представленным общей формулой (I) (SEQ ID NO: 53);
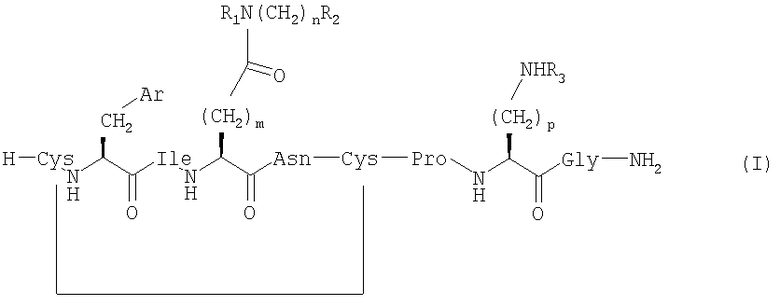 ,
,
где Ar представляет собой арильную группу, выбранную из ароматических карбоциклических кольцевых систем, пяти- или шестичленных гетероароматических кольцевых систем и бициклических гетероароматических кольцевых систем;
m выбран из 1, 2 и 3;
n выбран из 0, 1, 2, 3 и 4;
p выбран из 2, 3 и 4;
R1, R2 и R3 независимо выбраны из Н, ОН, алкила, групп O-алкил и ОС(O)-алкил;
алкил выбран из C1-6алкила с прямой цепью и С4-8алкила с разветвленной цепью и возможно имеет по меньшей мере один гидроксильный заместитель;
и если n=0, то R1 и R2 возможно вместе образуют азотсодержащую кольцевую структуру, включающую от 2 до 5 атомов углерода;
при условии, что когда Ar представляет собой фенил (аминокислотой №2 является Phe), m=2, n=0, R1=R2=H (аминокислотой №4 является Gln), тогда R3 не является Н, если p равен 3 или 4; и
их сольватам и фармацевтически приемлемым солям.
Аминокислотой №8 является Orn, если R3=H и р=3, и Lys, если R3=Н и р=4.
Для задач настоящего изобретения использована следующая терминология.
Ароматические карбоциклические кольцевые системы включают фенил и нафтил.
Пятичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую пять кольцевых атомов, где 1, 2 или 3 кольцевых атома независимо выбраны из N, О и S. Предпочтительно такие кольцевые системы выбраны из группы, состоящей из тиенила, фурила, пирролила, имидазолила, тиазолила, оксазолила, пиразолила, изотиазолила, изоксазолила и тетразолила.
Шестичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую шесть кольцевых атомов, где 1, 2 или 3 кольцевых атома независимо выбраны из N, О и S. Она предпочтительно выбрана из пиридила, пиразинила, пиримидинила, триазинила и пиридазинила.
Бициклическая гетероароматическая кольцевая система представляет собой кольцевую систему, имеющую два пяти- или шестичленных гетероароматических кольца, или фенил и пяти- или шестичленное гетероароматическое кольцо, или фенил и гетероциклильное кольцо, или пяти- или шестичленное гетероароматическое кольцо и гетероциклильное кольцо; соединенные путем конденсации колец, причем указанная бициклическая гетероароматическая кольцевая система содержит 8-12 кольцевых атомов, где 1, 2 или 3 кольцевых атома независимо выбраны из N, О и S. Она предпочтительно выбрана из группы, состоящей из индола, хинолина, тетрагидрохинолина, изохинолина, тетрагидроизохинолина, 1,4-бензодиоксана, кумарина, бензофурана, 1,2-бензоизоксазола, бензотиофена, бензоксазола, бензотиазола, бензимидазола, бензотриазола, пирролизидина и хинолизидина.
Гетероциклильная или гетероциклическая группировка представляет собой насыщенную или частично насыщенную кольцевую систему, имеющую от 3 до 7 кольцевых атомов, где 1, 2 или 3 кольцевых атома независимо выбраны из N, О и S. Гетероциклильные группировки предпочтительно выбраны из группы, состоящей из азиридина, оксирана, тиирана, азетидина, оксетана, тиетана, пирролидина, пирролина, имидазолидина, пиразолидина, диоксолана, тетрагидрофуранила, пиперидина, пиперазина, морфолина, тетрагидропиранила, 1,4-диоксанила, гомопиперидинила, гомопиперазинила и гексаметиленоксида.
Следует упомянуть о том, что, например, изопропильная и 2-н-бутильная группы также охвачены выражением С1-6алкил с прямой цепью, поскольку указанное выражение не затрагивает место связывания рассматриваемой прямой цепи.
С1-6 означает наличие атомов углерода в количестве от одного до шести, включающем любое число между этими значениями, и эта номенклатура используется в этом описании изобретения аналогичным образом.
Примеры фармацевтически приемлемых солей включают соли присоединения кислоты, например соль, образующуюся путем взаимодействия с галогеноводородными кислотами, такими как соляная кислота, и минеральными кислотами, такими как серная кислота, фосфорная кислота и азотная кислота, а также алифатическими, алициклическими, ароматическими или гетероциклическими сульфоновыми или карбоновыми кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, пара-гидроксибензойная кислота, памовая (embonic) кислота, метансульфоновая кислота, этансульфоновая кислота, гидроксиэтансульфоновая кислота, галогенбензолсульфоновая кислота, толуолсульфоновая кислота и нафталинсульфоновая кислота.
Ar предпочтительно выбран из фенила, 2- или 3-тиенила, 2- или 3-фурила, 2-, 3- или 4-пиридила и 2-, 4- или 5-тиазолила. Особенно предпочтительно, чтобы R1 представлял собой Н.
В предпочтительных воплощениях p равен 2 или 3.
Предпочтительно R2 выбран из Н, ОН, СН3, СН2СН3, СН(СН3)2, CH(CH2OH)2, СН(ОН)СН3 (оба энантиомера), ОСН3 и ОСН2СН2OH.
Кроме того, R3 предпочтительно выбран из Н, метила, этила, н-пропила, изопропила и изоамила.
В наиболее предпочтительном воплощении указанное соединение формулы (I) выбрано из группы, состоящей из (SEQ ID NO: 1-7, соответственно, в порядке появления):

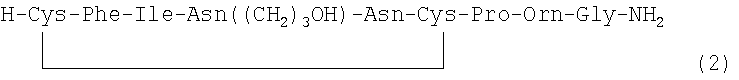
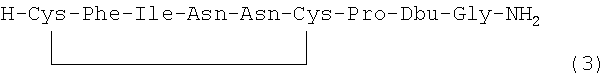
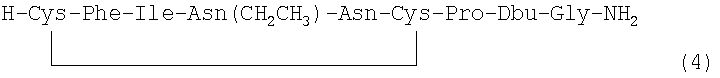
 и
и
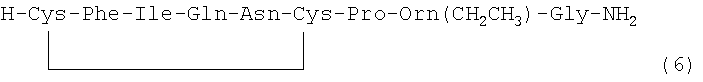 .
.
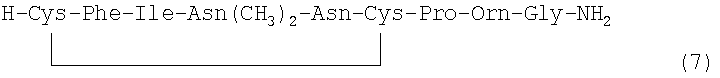
Номер в скобках указывает на то, как соединение обозначено далее.
Кроме того, настоящее изобретение относится к определенному выше соединению для применения в качестве фармацевтического препарата.
Соответственно настоящее изобретение также относится к фармацевтической композиции, содержащей определенное выше соединение в качестве активного ингредиента вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Фармацевтическая композиция может быть адаптирована для перорального, внутривенного, местного, внутриперитонеального, назального, трансбуккального, подъязычного или подкожного введения или для введения через дыхательные пути, например, в форме аэрозоля или мелкого порошка, суспендированного в воздухе. Таким образом, композиция может быть представлена, например, в форме таблеток, капсул, порошков, микрочастиц, гранул, сиропов, суспензий, растворов, трансдермальных пластырей или суппозиториев.
Следует отметить, что композиция по настоящему изобретению возможно может включать два или более чем два из вышеописанных соединений.
Фармацевтическая композиция по настоящему изобретению возможно может включать, например, по меньшей мере одну дополнительную добавку, выбранную из разрыхлителя, связующего вещества, смазывающего вещества, корригента, консерванта, красителя и любой их смеси. Примеры таких и других добавок приведены в "Handbook of Pharmaceutical Excipients"; Ed. A.H. Kibbe, 3rd Ed., American Pharmaceutical Association, USA and Pharmaceutical Press UK, 2000.
Наиболее предпочтительно фармацевтическая композиция по настоящему изобретению адаптирована для парентерального введения. Она может включать стерильный водный препарат соединений по изобретению, предпочтительно изотонический крови реципиента. Этот водный препарат может быть приготовлен в соответствии с известными способами с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Инъецируемый водный препарат Remestyp® (терлипрессин) представляет собой пример подходящего фармацевтического препарата. Препарат также может представлять собой стерильные инъецируемые раствор или суспензию в разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Примеры подходящих разбавителей представляют собой воду, раствор Рингера и изотонический раствор хлорида натрия. В качестве растворителя или суспендирующей среды могут быть использованы стерильные жирные масла. Также могут быть использованы мягкие жирные масла, в том числе синтетические моно- или диглицериды, и жирные кислоты, такие как олеиновая кислота.
Кроме того, настоящее изобретение относится к применению соединения, описанного выше, для приготовления лекарственного средства для лечения шока гиповолемической или вазодилататорной природы, ВРВПК, ГРС, сердечно-легочной реанимации, гипотензии, индуцированной анестезией, ортостатической гипотензии, дисфункции кровообращения, индуцированной парацентезом, кровопотери во время операции или кровопотери, связанной с обработкой ожогов и носовым кровотечением, и для лечения различных глазных заболеваний путем увеличения слезотечения/образования слез.
Еще одно воплощение изобретения относится к способу лечения шока гиповолемической или вазодилататорной природы, ВРВПК, ГРС, сердечно-легочной реанимации, гипотензии, индуцированной анестезией, ортостатической гипотензии, дисфункции кровообращения, индуцированной парацентезом, кровопотери во время операции или кровопотери, связанной с обработкой ожогов и носовым кровотечением, и различных глазных заболеваний путем увеличения слезотечения/образования слез, при котором пациенту-животному, включая человека, вводят терапевтически эффективное количество соединения, описанного выше.
Типичная доза соединений по настоящему изобретению находится в широком диапазоне и зависит от различных факторов, таких как индивидуальные потребности каждого пациента и путь введения. Доза, вводимая путем инфузии, обычно находится в диапазоне 0,01-200 мкг/кг массы тела в час. Лечащий врач-специалист способен оптимизировать дозу в зависимости от ситуации.
Использованные сокращения:
Abu 2-аминомасляная кислота
Boc трет-бутоксикарбонил
ВОР гексафторфосфат бензотриазол-1-илокси-трисдиметиламинофосфония
Dbu 2,4-диаминомасляная кислота
ДЦК N,N'-дициклогексилкарбодиимид
ДЦГА дициклогексиламин
ДХМ дихлорметан
DIAD диизопропилдиазодикарбоксилат
DIC N,N'-диизопропилкарбодиимид
DIEA N,N-диизопропил-N-этиламин
ДМФА N,N-диметилформамид
Fm 9-флуоренилметил
Fmoc 9-флуоренилметоксикарбонил
Hgn гомоглутамин
Hmp 2-гидрокси-3-меркаптопропионовая кислота
HOBt 1-гидроксибензотриазол
ВЭЖХ высокоэффективная жидкостная хроматография
I изо
Mmt 4-метокситритил
Mob пара-метоксибензил
МС масс-спектрометрия
Orn орнитин
Ph фенил
Pr пропил
PyBOP гексафторфосфат бензотриазол-1-илокси триспирролидинфосфония
o-NBS-CI 2-нитробензолсульфонилхлорид
ОТ окситоцин
Rt время удерживания
ТФУ трифторуксусная кислота
ТИПС триизопропилсилан
ТМОФ триметилортоформиат
ТФФ трифенилфосфин
Trt тритил
VT вазотоцин, [Ile3]вазопрессин
Если не указано иначе, использовали L-аминокислоты и соблюдена традиционная для аминокислот терминология.
Экспериментальная часть (синтез)
Производные аминокислот и смолы приобретены у коммерческих поставщиков (Novabiochem, Bachem Peptide International и PepTech Corporation). Fmoc-Hgn-OH синтезировали в соответствии с изложенным в литературе (Wisniewski, К., Kolodziejczyk, A.S. Org. Prep. Proced. Int. 1997, 29, 338-341). Другие химические вещества и растворители приобретены в Sigma-Aldrich, Fisher Scientific и VWR.
Представленные здесь соединения синтезировали стандартными способами твердофазной пептидной химии с использованием Fmoc- и Boc-методологии. Если не указано иначе, все реакции проводили при комнатной температуре. Кроме ссылок, цитированных выше, следующая стандартная справочная литература обеспечивает дополнительное руководство по общему описанию экспериментальных методик, а также по доступности требуемых исходных веществ и реагентов:
Kates, S.A., Albericio, F., Eds., Solid Phase Synthesis. A Practical Guide, Marcel Dekker, New York, Basel, 2000;
Stewart, J.M., Young, J.D. Solid Phase Synthesis, Pierce Chemical Company, 1984;
Bisello, et al., J. Biol. Chem. 1998, 273, 22498-22505; и
Merrifield, J. Am. Chem. Soc. 1963, 85, 2149-2154.
Чистота синтезированного пептида может быть определена при помощи аналитической ВЭЖХ с обращенной фазой. Структурная целостность пептидов может быть подтверждена с использованием аминокислотного анализа и масс-спектрометрии с электрораспылением.
Пептиды, синтезированные при помощи Fmoc-методологии, расщепляли с использованием раствора ТФУ/ТИПС/Н2О (96/2/2 об./об./об.), а расщепление в Boc-методологии осуществляли с использованием раствора 90% HF/10% анизол (об./об.). Образования дисульфидного мостика (кольца) достигали путем окисления линейных пептидов, растворенных в 10% ТФУ (водн.) с йодом. Пептиды очищали при помощи препаративной ВЭЖХ в триэтиламмонийфосфатных буферах (водн.). Окончательно соединения превращали в ацетатные соли с использованием обычной методологии ВЭЖХ. Фракции с чистотой более 97% объединяли и лиофилизировали.
Синтез пептидов с алкилированной боковой цепью в позиции №8:
Пептиды конструировали с использованием Fmoc-методологии. Остаток диаминокислоты в позиции №8 вводили при помощи кислотолабильной (т.е. удаляемой при помощи раствора, содержащего 1-2% ТФУ) защитной группы, такой как метокситритильная (Mmt; смотри Barlos, К. et al., Peptides 1992, Schneider, C.H., Eberle, A.M., Eds., ESCOM Science Publishers B.V., 1993, pp 283-284). Для удаления Mmt группы пептид, связанный со смолой, обрабатывали раствором ДХМ/ТИПС/ТФУ (93/5/2 об./об./об.). Восстановительное алкилирование смесью ацетон/NaBH(ОАс)3 позволило получить N-изопропиловый пептид.
Во избежание нежелательного N,N-диалкилирования при восстановительном алкилировании в описанном выше способе, которое может происходить при использовании прямоцепочечных алкилальдегидов, был разработан альтернативный способ, при котором аминогруппу после Mmt удаления сначала превращали в производное с использованием 2-нитробензолсульфонилхлорида (o-NBS-Cl; смотри Fukuyama, Т.; Jow, С.-К.; Cheung, M. Tetrahedron Lett. 1995, 36, 6373-6374). Получающийся в результате сульфонамид затем алкилировали с использованием подходящего спирта в традиционных условиях реакции Митсунобу, обычно с использованием ТФФ/DIAD в 1,2-диметоксиэтане (Mitsunobu, О. Synthesis 1981, 1-28). Затем с использованием 5%-ного тиофенолята калия в ДМФА удаляли группу o-NBS-Cl, после чего пептид отщепляли от смолы.
Синтез пептидов с N-алкилированной боковой цепью в позиции №4:
Пептиды конструировали с использованием Boc-методологии. Остаток в позиции №4 вводили в последовательность в виде Boc-Asp(OFm)-OH. После конструирования полного пептида удаляли защиту боковых цепей при помощи 30% пиперидина в ДМФА. Получающуюся в результате свободную карбоксильную группу превращали в желаемый амид путем сочетания с подходящим амином опосредованного PyBOP или BOP/DIEA. Затем удаляли N-концевую Boc группу с последующими расщеплением при помощи HF циклизацией и очисткой путем ВЭЖХ.
В Таблице 1 перечислены соединения, полученные по методике, описанной выше. R1 представляет собой Н для всех соединений, за исключением №7, где R1 представляет собой СН3. Звездочка * обозначает наиболее предпочтительные воплощения.
Следующие подробные примеры предложены для дополнительной иллюстрации синтеза:
Соединение 1; [Phe2, Hgn4, Orn (i-Pr)8]VT:
Использованные производные аминокислот представляли собой Вос-Cys(Trt)-OH, Fmoc-Phe-OH, Fmoc-Ile-OH, Fmoc-Hgn-OH, Fmoc-Asn(Trt)-OH, Fmoc-Cys(Trt)-OH, Fmoc-Pro-OH, Fmoc-Orn(Mmt)-OH и Fmoc-Gly-OH. Fmoc-Hgn-OH синтезировали как упомянуто выше. Аналитическую ВЭЖХ осуществляли на жидкостном хроматографе Waters 600 с использованием колонки Vydac C18, 5 мкм, 4.6×250 мм при скорости потока 2 мл/мин. Препаративную ВЭЖХ осуществляли на жидкостном хроматографе Waters 2000 с использованием картриджа Prepak, 47×300 мм при скорости потока 100 мл/мин. Анализ конечных соединений проводили на жидкостном хроматографе 1100 Agilent с использованием колонки Vydac C18, 5 мкм, 2.1×250 мм, при скорости потока 0,3 мл/мин. Масс-спектры регистрировали на масс-спектрометре Finnigan MAT.
На пептидном синтезаторе Applied Biosystems 9050 синтезировали полностью защищенную пептидную смолу, начиная с 2 г (0,5 ммоль) смолы Tentagel-S-RAM (Peptides International). Осуществляли опосредованные DIC/HOBt одиночные сочетания с 4-кратным избытком производных аминокислот. С использованием 20% пиперидина в ДМФА удаляли Fmoc группу. После завершения автоматического синтеза смолу переносили в пробирку для ручного синтеза и обрабатывали раствором ДХМ/ТИПС/ТФУ (93/5/2 об./об./об.) (30 мл) в течение 2×1,5 часа для удаления Mmt группы. Смолу тщательно промывали ДХМ и затем суспендировали в 15 мл смеси 1,2-дихлорэтан/TMOF (1:1 об./об.). Затем добавляли 0,2 мл ацетона с последующими 0,6 г NaBH(OAc)3. Суспензию встряхивали в течение ночи и смолу промывали метанолом, ДМФА и ДХМ и сушили в вакууме. Затем смолу обрабатывали 30 мл раствора ТФУ/ТИПС/Н2O (96/2/2 об./об./об.) в течение 1,5 часов и фильтровали. Фильтрат упаривали и неочищенный линейный пептид осаждали с использованием диэтилового эфира. Осадок сразу же растворяли в 500 мл 10%-ной ТФУ (водн.) и путем добавления 0,1 М I2 в метаноле к перемешиваемому на магнитной мешалке раствору до тех пор, пока сохранялось желтое окрашивание, окисляли пептид. Избыток йода восстанавливали аскорбиновой кислотой. Затем реакционную смесь охлаждали измельченным льдом и путем добавления концентрированного аммиака (водн.) доводили рН приблизительно до 5. Смесь наносили на колонку для ВЭЖХ и очищали с использованием триэтиламмонийфосфатного буфера с рН 5,2. Соединение элюировали с использованием градиента ацетонитрила. Фракции с чистотой, превышающей 97%, объединяли, и получающийся в результате раствор разбавляли 2 объемами воды. Раствор повторно наносили на колонку, которую затем промывали 2 л 0,1 М ацетата аммония (водн.) и уравновешивали с использованием 2%-ной уксусной кислоты (водн.). Соединение элюировали при помощи быстрого (3%/мин) градиента ацетонитрила. Фракции, содержащие желаемый продукт, объединяли и лиофилизировали. Получали 168 мг (приблизительно 30%-ный выход) белого аморфного порошка. ВЭЖХ: Rt=8,5 мин, градиент: 20→40% Б в течение 20 мин, t=40°C, растворитель А 0,01% ТФУ (водн.), растворитель Б 70% CH3CN, 0,01% ТФУ (водн.); чистота; 98,8%; МС (М+Н+): ожидалось 1048,5, обнаружено 1048,5.
Соединение 4 [Phe2, Asn(Et)4, Dbu8]VT:
Использованные производные аминокислот представляли собой Вос-Cys(Mob)-OH, Boc-Phe-OH, Boc-Ile-OH, Boc-Asp(OFm)-OH, Boc-Asn-OH, Boc-Pro-OH, Вос-Dbu(бензилоксикарбонил)-ОН ДЦГА соль и Boc-Gly-OH, все приобретены в Novabiochem и Bachem. Стадии ВЭЖХ и МС осуществляли как при синтезе соединения 1.
Вручную синтезировали полностью защищенную пептидную смолу, начиная с 0,6 г (0,4 ммоль) 4-метилбензгидриламиновой смолы (Novabiochem). Применяли опосредованные DCC, PyBOP или DIC/HOBt одиночные сочетания с 2,5-кратным избытком производных аминокислот. С использованием 50% ТФУ в ДХМ, содержащем 1% мета-крезола, удаляли Вос группу. После завершения синтеза 9-флуоренилметиловый эфир удаляли с β-карбоксильной группы аспарагиновой кислоты путем обработки 30% пиперидином в ДМФА в течение 2×30 мин. Смолу промывали 1М HOBt в ДМФА в течение 30 мин, а затем дважды только ДМФА. Свободную карбоксильную группу амидировали путем обработки в течение ночи 2 ммоль этиламин/PyBOP/DIEA в ДМФА. Конечную смолу промывали метанолом, ДМФА и ДХМ и сушили в вакууме. Пептид в течение 90 минут при 0°С отщепляли от смолы путем использования 30 мл безводного HF, содержащего 3 мл анизола. HF выпаривали, и неочищенный линейный пептид промывали диэтиловым эфиром. Пептид сразу же растворяли в 200 мл смеси 25% ацетонитрил/10% ТФУ (водн.) и окисляли, как описано выше. Получающуюся в результате смесь непосредственно наносили на колонку для ВЭЖХ и очищали с использованием триэтиламмонийфосфатного буфера при рН 2,3. Последующие стадии очистки аналогичны описанным в методике для соединения 1. Получали 41 мг (приблизительно 10%-ный выход) белого аморфного порошка. ВЭЖХ: Rt=10,0 мин, градиент: 20→40% Б в течение 20 мин, t=40°C, растворитель А 0,01% ТФУ (водн.), растворитель Б 70% СН3CN, 0,01% ТФУ (водн.); чистота: 100%; МС (M+H+): ожидалось 992,5, обнаружено 992,2.
Другие соединения получали аналогичным варьированием этих методик синтеза.
Экспериментальная часть (биологическое тестирование)
Анализы в отношении рецепторов in vitro:
Агонистическую активность соединений в отношении рецептора hV1a определяли в транскрипционном репортерном анализе путем временной трансфекции в клетки НЕК-293 ДНК, экспрессирующей рецептор hV1a, вместе с репортерной ДНК, содержащей промоторные элементы, чувствительные к внутриклеточному кальцию, регулирующие экспрессию люциферазы светлячка. Дополнительное руководство по проведению этого анализа смотри в Boss, V., Talpade, D.J., Murphy, T.J., J. Biol. Chem. 1996, May 3; 271 (18), 10429-10432. Клетки подвергали воздействию нескольких разведений соединений, разведенных 10-кратно на дозу, в течение 5 часов, с последующим лизисом клеток, определением активности люциферазы и определением эффективностей соединений и значений средней эффективной концентрации (EC50) путем нелинейной регрессии. В качестве внутреннего контроля в каждом эксперименте использовали аргинин-вазопрессин (AVP), и соединения тестировали по меньшей мере в трех независимых экспериментах. Для определения селективности соединения тестировали в транскрипционных репортерных анализах с использованием люциферазы, в которых экспрессировался рецептор окситоцина человека (hOT). Также проводили анализы в отношении других рецепторов (hV2, hV1b, V1a крысы и V2 крысы).
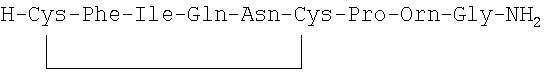
Дополнительно для задач сравнения другие использованные контрольные соединения представляли собой [Phe2, Orn8] ОТ, терлипрессин и F180.
Структура [Phe2, Orn8] ОТ представляет собой (SEQ ID NO: 51);
Структура F180 представляет собой (SEQ ID NO: 52):
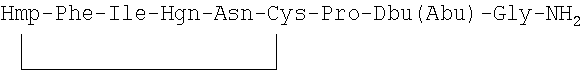
Результаты анализов in vitro представлены в таблице 2 ниже. Приведенное значение ЕС50 представляет собой среднее геометрическое, выраженное в нмоль/л (нМ). Значения селективности приведены в виде отношений ЕС50.
Фармакологические тесты in vivo:
Соединения тестировали in vivo в отношении продолжительности действия относительно стандартной дозы AVP. Тестирование кровяного давления осуществляли на анестезированных самцах крыс Sprague-Dawley (массой 270-300 г) с катетеризацией яремной вены и сонной артерии. Катетеризированную сонную артерию использовали для непрерывного контролирования кровяного давления, а яремную вену использовали для введения тестируемых соединений. Перед введением дозы крысам производили внутривенные инъекции дибенамина для усиления их восприимчивости к агонистам рецептора V1a (ср. Dekanski, J., Br. J. Pharmacol. 1952, 7, 567-572). Процедура введения дозы состояла из одной внутривенной инъекции физиологического раствора с последующими двумя последовательными инъекциями стандартной дозы AVP (0,1 нмоль/кг, приблизительно 70%-ная эффективная доза (ED70)) и трех-пяти увеличивающихся доз заданного соединения, выбранных для получения ответа, по меньшей мере сравнимого со стандартной дозой AVP. Интервалы дозирования устанавливали как время уменьшения кровяного давления до стабильного базового уровня.
Определение продолжительности действия основано на скорости убывания временно увеличенного диастолического артериального кровяного давления. Конкретно при экспоненциальном убывании плазменной концентрации можно показать, что если ответ измеряют после фазы распределения, то скорость убывания вблизи EC50 является линейной и обратно пропорциональной периоду полувыведения (Rowland, M. and Tozer, T. в "Clinical Pharmacokinetics, Concepts и Applications", 3rd ed., Lippincott Williams & Wilkins, Philadelphia, 1995).
Для измерения скорости убывания ответа для заданного соединения дозу выбирали таким образом, чтобы она обеспечивала амплитуду ответа, по возможности близкую к амплитуде ответа на вторую инъекцию стандартной дозы AVP. Для нормализации вариации между особями в отношении восприимчивости к V1a продолжительность действия для каждой тестируемой крысы выражали в виде отношения скорости убывания для контрольного ответа на AVP к скорости убывания для эффективной дозы соединения. Результаты, полученные для тестируемых соединений, представлены в таблице 2.
Все перечисленные ссылки следует рассматривать как неотъемлемую часть настоящей заявки на изобретение.






















| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2007 |
|
RU2415149C2 |
| Агонисты V1а-рецепторов | 2013 |
|
RU2634617C2 |
| АНАЛОГИ ОКСИТОЦИНА | 2009 |
|
RU2496788C2 |
| СИНТЕТИЧЕСКИЕ ПЕПТИДНЫЕ АМИДЫ И ИХ ДИМЕРЫ | 2007 |
|
RU2510399C2 |
| ПЕПТИДНЫЕ ВЕКТОРЫ | 2004 |
|
RU2361876C2 |
| СИНТЕТИЧЕСКИЕ ПЕПТИДНЫЕ АМИДЫ | 2007 |
|
RU2500685C2 |
| НОВЫЕ ОКТАПЕПТИДНЫЕ СОЕДИНЕНИЯ, ПРЕДСТАВЛЯЮЩИЕ СОБОЙ ПРОИЗВОДНЫЕ СОМАТОСТАТИНА | 2009 |
|
RU2547940C2 |
| ПРОИЗВОДНОЕ АНАЛОГА GLP-1 ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2565536C2 |
| ПРОИЗВОДНЫЕ МЕТАСТИНА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2454425C2 |
| ПЕПТИДНЫЕ КОМПОЗИЦИИ | 2014 |
|
RU2725150C2 |
Настоящее изобретение относится к новым соединениям, представляющим собой пептидные агонисты вазопрессинового рецептора VIa общей формулы (I). Также изобретение относится к фармацевтическим композициям, содержащим заявленные соединения в качестве активного ингредиента в терапевтически эффективном количестве, к применению указанных соединений в изготовлении лекарственного средства, обладающего агонистической активностью в отношении вазопрессинового рецептора V1a, и к способу лечения шока гиповолемической или вазодилататорной природы, варикозного расширения вен пищевода с кровотечением, гепаторенального синдрома, сердечно-легочной реанимации, гипотензии, индуцированной анастезией, ортостатической гипотензии, дисфункции кровообращения, индуцированной парацентезом, кровопотери во время операции или кровопотери, связанной с обработкой ожогов и носовым кровотечением. 4 н. и 8 з.п. ф-лы, 2 табл.
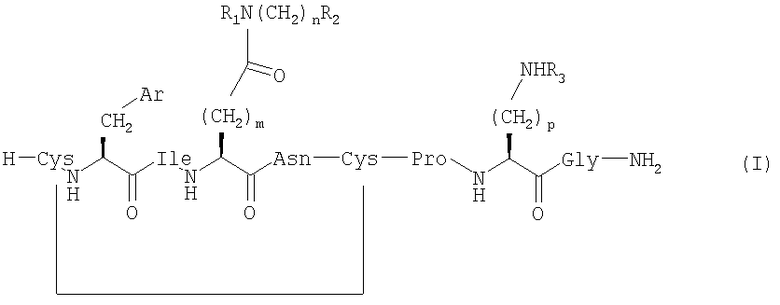
1. Соединение формулы (I) (SEQ ID NO: 53)
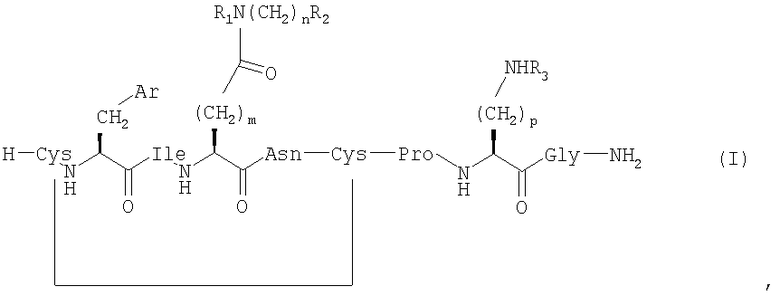
где Ar представляет собой арильную группу, выбранную из фенила, пятичленных гетероароматических кольцевых систем, имеющих в качестве гетероатома 1 или 2 атома N, О или S, или шестичленных гетероароматических кольцевых систем, имеющих в качестве гетероатома 1 атом N;
m выбран из 1, 2 и 3;
n выбран из 0, 1, 2, 3 и 4;
р выбран из 2, 3 и 4;
R1, R2 и R3 независимо выбраны из Н, ОН, алкила и групп O-алкил;
алкил выбран из C1-6алкила с прямой цепью и С4-8алкила с разветвленной цепью и возможно имеет по меньшей мере один гидроксильный заместитель;
при условии, что если Ar представляет собой фенил, m=2, n=0, R1=R2=H и р равен 3 или 4, то R3 не является Н,
и его фармацевтически приемлемые соли.
2. Соединение по п.1, где Ar выбран из фенила, 2- или 3-тиенила, 2- или 3-фурила, 2-, 3- или 4-пиридила и 2-, 4- или 5-тиазолила.
3. Соединение по п.1, где R1 представляет собой Н.
4. Соединение по п.1, где р равен 2 или 3.
5. Соединение по п.1, где R2 выбран из Н, ОН, СН3, СН2СН3, СН(СН3)2,
СН(СН2OH)2, СН(ОН)СН3, ОСН3 и ОСН2СН2OH.
6. Соединение по п.1, где R3 выбран из Н, метила, этила, н-пропила, изопропила и изоамила.
7. Соединение по п.1, где соединение выбрано из группы, состоящей из (SEQ ID NO: 1-7, соответственно, в порядке появления)
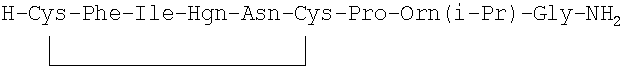
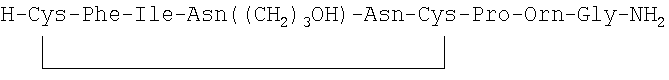
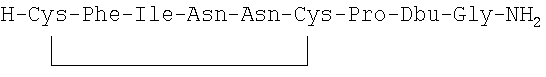
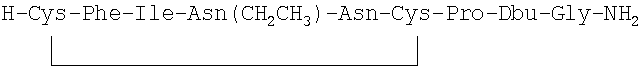
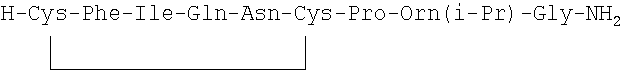
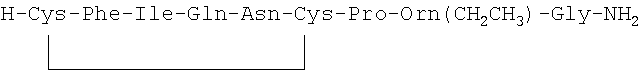
8. Соединение по п.1, имеющее структурную формулу

9. Соединение по любому из пп.1-8, обладающее агонистической активностью в отношении вазопрессивного рецептора V1a.
10. Фармацевтическая композиция, обладающая агонистической активностью в отношении вазопрессивного рецептора V1a, содержащая соединение по любому из пп.1-8 в качестве активного ингредиента в терапевтически эффективном количестве вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
11. Применение соединения по любому из пп.1-8 для изготовления лекарственного средства, обладающего агонистической активностью в отношении вазопрессивного рецептора V1a.
12. Способ лечения шока гиповолемической или вазодилататорной природы, варикозного расширения вен пищевода с кровотечением, гепаторенального синдрома, сердечно-легочной реанимации, гипотензии, индуцированной анестезией, ортостатической гипотензии, дисфункции кровообращения, индуцированной парацентезом, кровопотери во время операции или кровопотери, связанной с обработкой ожогов и носовым кровотечением, при котором пациенту-животному, включая человека, вводят терапевтически эффективное количество соединения по любому из пп.1-8.
| ПРОИЗВОДНЫЕ ВАЗОТОЦИНА | 1991 |
|
RU2067586C1 |
| RU 2073010 C1, 10.02.1997 | |||
| US 5459236 A, 17.10.1995 | |||
| Yves de Keyzer et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

