ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к серии производных аналога глюкагоноподобного пептида-1 (GLP-1) человека и к его фармацевтически приемлемым солям. Производные аналога GLP-1, предложенные в данном изобретении, обладают функцией GLP-1 человека и более длительным периодом полувыведения in vivo по сравнению с GLP-1 человека. Настоящее изобретение также относится к применению производных аналога GLP-1, их фармацевтически приемлемых солей или фармацевтических композиций, содержащих производные аналога GLP-1 или их фармацевтически приемлемые соли, при лечении инсулиннезависимого сахарного диабета, инсулинозависимого сахарного диабета или ожирения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Сахарный диабет является глобальным эпидемическим заболеванием и представляет собой синдром расстройства метаболизма глюкозы, белков и липидов вследствие абсолютного или относительного дефицита инсулина (Chen Ruijie. Status of research on diabetes drugs, Academic journal of Guangdong College of Pharmacy, 2001, 7(2): 131-133). Сахарный диабет можно разделить на сахарный диабет типа I и сахарный диабет типа II (сахарный диабет типа 2, далее - T2DM) в соответствии с его патогенезом. 90-95% всех пациентов, у которых диагностирован сахарный диабет, страдают T2DM, и у этих пациентов также наблюдается сопутствующее ожирение, недостаток физической активности (физическая неактивность), пресбиопия, семейная история сахарного диабета, нарушение метаболизма глюкозы и наличие семейной истории сахарного диабета и тому подобного. T2DM также является прогрессирующим заболеванием. Согласно статистическим данным 2000 г., Всемирной организацией здравоохранения установлено, что около 171 млн. человек во всем мире страдает сахарным диабетом; в 2005 г. Центрами по контролю и профилактике заболеваний США установлено, что 20,8 млн. американцев страдает сахарным диабетом, что доставляет около 7% населения Соединенных Штатов Америки; в 2006 г. согласно статистике Международной диабетической федерации число пациентов, страдающих сахарным диабетом, во всем мире составляет около 246 млн. (примерно 5,9% всего населения земного шара), и кроме того, 46% пациентов находятся в возрасте 40-59 лет. Исследования показали, что существует очень важное различие в том, как реагируют на глюкозу здоровые люди и пациенты с T2DM. Ответ на гипергликемию после приема пищи здоровых людей принадлежит к раннему ответу инсулина.
T2DM характеризуется ингибированием секреции инсулина и дисфункцией β-клеток поджелудочной железы, что приводит в результате к дефициту инсулина и гипергликемии (Ferrannini E. Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: problems and prospects. Endocr Rev. 1998, 19(4): 477-490). Пациенты с T2DM типично страдают гипергликемией после приема пищи и натощак (глюкоза натощак >125 мг/дл), и высокий уровень сахара в крови в основном является следствием того, что β-клетки поджелудочной железы неспособны секретировать достаточно инсулина, чтобы компенсировать дефицит инсулина, вызванный ингибированием инсулина в окружающих тканях (Weyer С., Bogardus С., Mott DM., et al. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Invest. 1999, 104(6): 787-794).
Одним из основных факторов риска T2DM является ожирение, которое очень вредно для здоровья человека. Возрастает риск развития сердечно-сосудистого заболевания и скоропостижной смерти пациентов, в то время как T2DM часто сосуществует с другими заболеваниями высокого риска, такими как гипертензия, дислипидемия и ожирение; у 60% пациентов с T2DM имеются сопутствующие микрососудистые осложнения, включая ретинопатию и невропатию, а также сопутствующая заболеваемость сердечно-сосудистыми заболеваниями, обусловленными T2DM, такими как коронарная болезнь сердца, инфаркт миокарда и удар и тому подобное. В США сердечно-сосудистые заболевания (ССЗ) являются основной причиной, приводящей в результате к заболеваемости и смертности, и T2DM является основным фактором риска, вызывающим макрососудистые осложнения, такие как атеросклероз, инфаркт миокарда, удар и периферические сосудистые заболевания. Риск смерти от сердечных заболеваний и удара у взрослых людей с диабетом в 2-4 раза выше, чем у человека, не страдающего диабетом. Кроме того, около 65% людей с диабетом умирает от сердечных заболеваний и удара.
Кроме физического и физиологического вреда для пациентов T2DM вызывает огромное экономическое бремя для общества. Согласно статистике в США стоимость лечения осложнений, обусловленных диабетом, составляет примерно $22,9 млрд., общая стоимость лечения T2DM и его осложнений составляет около $57,1 млрд., а общая стоимость, не включенная в бюджет, составляет более $8 млрд. ежегодно.
Лекарства для лечения T2DM были сфокусированы на таких лекарствах, как ранние пероральные гипогликемические лекарства сульфонильного класса и класса бигуанидов и недавние инсулиновые сенсибилизаторы и ингибиторы α-глюкозидазы, на разработке инсулинов животных и инсулинов человека, а также ряда новых методов изготовления препаратов, на исследовании новых механизмов лекарственной терапии от простого повышения инсулина до новых путей, действующих на продуцирование инсулина. Прибавление массы тела является распространенной побочной реакцией после введения пероральных или инъекционных гипогликемических агентов, что может снизить соблюдение пациентом режима и схемы лечения и может увеличить риск развития сердечно-сосудистого заболевания. Поэтому разработка новых типов лекарств для лечения T2DM, которые характеризуются высокой безопасностью, хорошим соблюдением пациентом режима и схемы лечения и низкой побочной реакцией, становится злободневной темой научно-исследовательских институтов и фармацевтических фирм против диабета.
Уже 100 лет назад Moore предположил, что двенадцатиперстная кишка может секретировать "химический стимулятор", который может стимулировать секрецию в поджелудочной железе, и попытался инъецировать кишечный экстракт для лечения диабета. Затем было сделано открытие, что гуморальные факторы, имеющие происхождение из секреции тонкого кишечника, могут усиливать эндокринную функцию поджелудочной железы, и примерно 50% секреции инсулина, индуцированной внутривенной или пероральной глюкозой, образуется в результате стимуляции пептидами, продуцируемыми кишечником. Таким образом, Zunz и Labarre создали концепцию "инкретина". К настоящему времени выделено два вида инкретинов, а именно глюкозозависимый инсулинотропный пептид (GIP) и глюкагоноподобный пептид-1 (GLP-1). И GIP, и GLP-1 секретируются специфичными нервными клетками тонкого кишечника, когда всасывается питательное вещество, причем GIP секретируется двенадцатиперстной кишкой и проксимальными тощекишечными К клетками, GLP-1 синтезируется в L клетках и в основном существует в дистальной тонкой кишке и в ободочной кишке (Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003, 26(10): 2929-2940).
GLP-1 существует в плазме в двух биологически активных формах, а именно амида GLP-1 (7-37) и GLP-1 (7-36), которые отличаются только одной аминокислотой, и их биологические эффекты и периоды полувыведения in vivo одинаковы (Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003, 26(10): 2929-2940).
Термином GLP-1 обычно называют амид GLP-1 (7-37) и GLP-1 (7-36). GIP и GLP-1 быстро расщепляются до неактивных форм дипептидилпептидазой-IV (DPP-IV) после высвобождения в желудочно-кишечном тракте, поэтому период полувыведения in vivo GIP и GLP-1 является очень коротким (период полувыведения in vivo GIP составляет примерно 5-7 мин, период полувыведения in vivo GLP-1 составляет примерно 2 мин) (Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003, 26(10): 2929-2940). Исследования показали, что большая часть процесса расщепления происходит, когда GIP и GLP-1 поступают в кровеносные сосуды, содержащие DPP-IV, и небольшое количество GLP-1 и GIP, которые не расщепились, поступит в поджелудочную железу и свяжется с их сайтами связывания для стимуляции β-клеток на высвобождение инсулина. В отличие от механизма действия сульфонилмочевины, которая непосредственно стимулирует высвобождение инсулина функциональными β-клетками, эффект инкретина в основном является глюкозозависимым. Кроме того, некоторые тесты на животных и на человека in vitro показали, что GLP-1 также обладает такими функциями, как подавление α-клеток и снижение гиперсекреции глюкагона и т.д.
Хотя уровни GIP в плазме у пациентов с T2DM являются нормальными, когда функция инкретина снижается или значительно утрачивается, уровни GLP-1 у пациентов T2DM снижаются, поэтому лекарства на основе GLP-1 вносят больший вклад в лечение T2DM. Хотя уровни как амида GLP-1 (7-37), так и амида GLP-1 (7-36) возрастут в течение нескольких минут после приема пищи, содержание амида GLP-1 (7-36) больше, поэтому секреция GLP-1 могла бы значительно возрасти за счет двойного эндокринного эффекта и эффекта передачи нервного сигнала перед тем, как переваренная пища поступит в тонкий кишечник и ободочную кишку со дна пищеварительного тракта. Уровень GLP-1 в плазме натощак является очень низким (примерно 5-10 пмоль/л) и быстро возрастает после еды (вплоть до 15-50 пмоль/л). При двойной функции DPP-IV и почечного клиренса уровень in vivo GLP-1 в кровообращении быстро снижается, хотя продолжается работа по исследованию того, играют ли также жизненно важную роль в инактивации активности GLP-1 другие ферменты, такие как нейтральная эндопептидаза 24 · 11 человека и тому подобное, поскольку вторым аминокислотным остатком GLP-1 является аланин, который является хорошим субстратом для DPP-IV, поэтому GLP-1 легко расщепляется до неактивных пептидных фрагментов. Действительно, DPP-IV in vivo является ключевой причиной утраты активности инкретина. Эксперименты показывают, что уровень GLP-1 у мышей, у которых ген DPP-IV инактивирован, значительно выше, чем у нормальных мышей, и секреция инсулина также повышена. Только благодаря присутствию DPP-IV содержание in vivo полноразмерного и биологически активного GLP-1 составляет только 10-20% от суммарного содержания GLP-1 в плазме (Deacon CF, Nauck MA, Toft-Nielsen M, et al. Both subcutaneously and intravenously administered glucagon-like peptide 1 are rapidly degraded from the NH2-terminus in type 2-diabetic patients and in healthy subjects. Diabetes. 1995, 44(9): 1126-1131).
GLP-1 и GIP выполняют соответствующие функции за счет связывания с рецепторами, связанными с G-белком (GPCR), которые имеют полностью различающиеся структуры. Большинство рецепторов GIP экспрессируется β-клетками поджелудочной железы, и меньшая часть рецепторов GIP экспрессируется в жировой ткани и в центральной нервной системе. Напротив, рецепторы GLP-1 в основном экспрессируются в α- и β-клетках поджелудочной железы и в периферических тканях, включая центральную и периферическую нервную систему, головной мозг, почки, легкие и желудочно-кишечный тракт и тому подобное. Активация двух инкретинов в β-клетках приведет в результате к быстрому возрастанию уровня цАМФ и внутриклеточного кальция в клетках, приводя посредством этого к их секреции во внеклеточную область глюкозозависимым путем. Пролонгированная передача сигнала от инкретиновых рецепторов связана с протеинкиназой А, приводящей в результате к транскрипции гена, повышающего биосинтез инсулина и стимулирующего пролиферацию β-клеток (Gallwitz В. Glucagon-like peptide-1-based therapies for the treatment of type 2 diabetes mellitus. Treat Endocrinol. 2005, 4(6): 361-370). Активация рецептора GLP-1 и рецептора GIP может также ингибировать апоптоз β-клеток поджелудочной железы грызунов и человека, в то же время, повышая их выживаемость (Li Y, Hansotia T, Yusta В, et al. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem. 2003, 278(1): 471-478). В соответствии с экспрессией рецептора GLP-1 GLP-1 может также ингибировать секрецию глюкагона, опорожнение желудка и прием пищи, а также усиливать расщепление глюкозы посредством неврального механизма. Следует отметить, что, как и другая реакция секреции инсулина, роль GLP-1 в регуляции уровня глюкозы является глюкагонозависимой, хотя роль глюкагона в антагонистичном высвобождении глюкагона, вызванном низким уровнем сахара в крови, полностью сохранена даже при фармакологическом уровне GLP-1.
Важная физиологическая роль эндогенных GLP-1 и GIP в гомеостазе глюкозы глубоко исследована посредством использования антагонистов рецепторов или нокаут-мышей по их генам. Острый антагонизм GLP-1 или GIP снижает секрецию инсулина in vivo грызунов и повышает содержание глюкозы в плазме. Подобным образом мутантные мыши, у которых рецептор GIP или GLP-1 инактивирован, также испытывали дефектную стимулируемую глюкозой секрецию инсулина и нарушенную толерантность к глюкозе. GLP-1 также обладал функцией регуляции глюкозы в крови натощак, поскольку острые антагонисты или повреждение гена GLP-1 вызывают повышение уровня глюкозы натощак у грызунов; в то же время, GLP-1 является основой контроля глюкозы в организме человека, и исследования на антагонисте эксендина (9-39) показали, что разрушение функции GLP-1 приведет в результате к дефектной стимулируемой глюкозой секреции инсулина, сниженной скорости клиренса глюкозы, повышенным уровням глюкагона и ускоренному опорожнению желудка. Физиологические роли GLP-1 (Deacon CF. Therapeutic strategies based on glucagon-like peptide 1. Diabetes. 2004, 53(9): 2181-2189) включают: (1) способствуя организации всасывания глюкозы, опосредует глюкозозависимую секрецию инсулина; (2) ингибирование секреции глюкагона после приема пищи, снижение высвобождения глюкозы в печени; (3) регуляция опорожнения желудка, предотвращение избыточной циркуляции глюкозы, когда пища всасывается в кишечнике; (4) ингибирование приема пищи (например, аппетита). Исследования на животных также показали физиологическую роль GLP-1 в стабилизации числа β-клеток поджелудочной железы in vivo.
GLP-1 и GIP привлекают интерес множества ученых благодаря их хорошим эффектам при регулировании сахара в крови и многим другим аспектам, в частности, их свойствам не вызывать гипогликемию и замедлять опорожнение желудка, чтобы контролировать массу тела. Люди пытаются исследовать лекарства, основанные на GLP-1 и GIP, для лечения T2DM. Хорошо известно, что у пациентов с T2DM отсутствует или утрачен инкретиновый эффект, где одна из причин состоит в том, что инкретиновый эффект GIP in vivo у пациента с T2DM значительно снижен; в то же время уровень GLP-1 in vivo у пациента с T2DM является очень низким, и уровень GLP-1, вызванный пищевыми стимулами, значительно снижен (Toft-Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab. 2001, 86(8): 3717-3723). Поскольку роль GLP-1 in vivo у пациентов с T2DM частично сохранена, синергист GLP-1 является одним из направлений исследований лекарств, предназначенных для усиления инкретинового эффекта у пациентов с T2DM.
Аналог GLP-1 также, как и эндогенный GLP или GIP, может глюкозозависимо ингибировать высвобождение глюкагона in vivo и стимулировать секрецию инсулина in vivo. Кроме того, аналоги GLP-1 играют роль при следующих симптомах:
(1) Низкий уровень сахара в крови. В отличие от других лекарств, усиливающих секрецию, аналоги GLP-1 стимулируют секрецию инсулина in vivo глюкозозависимо и, следовательно, их роль в снижении глюкозы в крови проявляет самоограничение, что, как правило, не вызывает гипогликемию в больших дозах. Несмотря на то, что в литературе описано, что GLP-1 может снизить уровень сахара в крови ниже нормального уровня, этот эффект является преходящим, и его считают естественным результатом стимуляции GLP-1 секреции инсулина. В связи с тем, что инактивация инсулина занимает некоторое время, когда стимулирующий эффект GLP-1 снижается вследствие снижения уровня сахара в крови, хотя новая секреция инсулина отсутствует, исходный инсулин все еще действует. Одним словом, GLP-1 может временно снизить сахар в крови до уровня ниже нормального уровня, но не вызывает серьезной и персистентной гипогликемии.
(2) Роль в отношении насыщения и снижения массы тела. Кроме непосредственного снижения сахара в крови GLP-1 может также снижать количество приема пищи, что подтверждено на грызунах и человеке. Следовательно, уровень глюкозы в крови может регулироваться косвенно за счет снижения массы тела. GLP-1 также обладает потенциальной ролью ингибирования секреции гастрина и желудочной кислоты, стимулируемой приемом пищи, и эти функции показывают, что GLP-1 может также играть роль в предупреждении пептической язвы желудка. Механизмы действия GLP-1 делают его не только идеальным лекарством для лечения пациентов с диабетом типа 2, но также лекарством для лечения пациентов с диабетом тучных. GLP-1 может усиливать чувство насыщения пациентов, снижать прием пищи и поддерживать массу тела или приводить к потере массы тела;
(3) Поддержание здоровья β-клеток. В нескольких исследованиях предположили, что GLP-1 может предупреждать переход от нарушенной толерантности к глюкозе к диабету, и в некоторых литературных источниках описано, что класс соединений GLP-1 обладает прямым эффектом на рост и пролиферацию β-клеток поджелудочной железы подопытных животных, и в некоторых экспериментах было обнаружено, что GLP-1 может стимулировать дифференциацию от стволовых клеток поджелудочной железы до функциональных β-клеток. Эти результаты позволяют предположить, что GLP-1 обладает функцией защиты островков Лангерганса поджелудочной железы и замедления прогрессирования диабета, и может поддерживать морфологию и функции β-клеток, при этом снижая апоптоз β-клеток;
(4) Эффект гипергликемии после приема пищи. Это явление представляет новое направление в лечении T2DM. Поскольку некоторые пероральные лекарства и экзогенные инсулины не могут ингибировать или снижать чрезмерную секрецию глюкагона у пациентов с T2DM, аналоги GLP-1 могут влиять на гиперсекрецию глюкагона посредством прямого ингибирования высвобождения глюкагона или паракринного ингибирования глюкагона в результате стимуляции секреции инсулина. Гипергликемия после приема пищи может быть эффективно снижена посредством этих двух механизмов; в то же время поддержание функции β-клеток может также играть роль в регулировании долговременной гипергликемии после приема пищи.
Кроме того, аналоги GLP-1 вводят посредством подкожной инъекции, что не требует вычисления количества углеводов для оценки оптимальной дозировки лекарства, и не требует самостоятельного мониторинга глюкозы, в результате чего эти виды лекарств более просты для применения, чем инсулин.
Ряд эффектов природного GLP-1 подтвержден, что приносит новую надежду на лечение T2DM, однако природный GLP-1 человека очень нестабилен и может расщепляться дипептидилпептидазой IV (DPP-IV), и его период полувыведения составляет только 1-2 минуты. При применении природного GLP-1 для снижения сахара в крови необходима непрерывная внутривенная инфузия или непрерывная подкожная инъекция, что приводит в результате к плохой клинической выполнимости. Столкнувшись с этой ситуацией, исследователи продолжают изучать способ продления времени действия GLP-1. Следовательно, разработка аналогов GLP-1 длительного действия или их производных становится важной областью интереса в области фармацевтики.
Эксенатид представляет собой синтетический эксендин-4, который разработан фирмой Eli Lilly и фирмой Amylin под торговым названием Баета®. Эксенатид одобрен для лечения T2DM FDA (Управление США по надзору за качеством пищевых продуктов и лекарственных средств) и ЕМЕА (Европейским агентством по оценке лекарственных средств). Он обладает 50% гомологией с GLP-1 млекопитающих по последовательности и имеет сходный сайт сродства к рецептору с GLP-1 (Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006, 368(9548): 1696-1705), и кодируется геном, специфичным для ящериц; по сравнению с GLP-1 аланин в положении 2 в GLP-1 заменен глицином в эксенатиде, что эффективно ингибирует энзимолиз ферментом DPP-IV, и его время полувыведения in vivo составляет примерно 60-90 мин (Kolterman OG, Kim DD, Shen L, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes melllitus. Am Health Syst Pharm. 2005, 62(2): 173-181), концентрация эксенатида in vivo после однократной подкожной инъекции постоянно возрастает и может достичь максимальной концентрации в плазме через 2 ч или около того, которая может сохраняться в течение 4-6 ч (Nielsen LL, Baron AD. Pharmacology of exenatide (synthetic exendin-4) for the treatment of type 2 diabetes. Curr Opin Investig Drugs. 2003, 4(4): 401-05). Следует отметить, что метаболизм эксенатида не происходит в печени, но он расщепляется в основном протеазой белка после фильтрации почечными гломерулами.
Эксенатид обладает специальными активностями, регулирующими глюкозу, включая глюкозозависимое усиление секреции инсулина, глюкозозависимое ингибирование неправильной избыточной секреции глюкагона, замедление опорожнения желудка и снижение приема пищи и тому подобное. В исследованиях in vitro и in vivo в моделях диабета обнаружено, что эксенатид также обладает эффектами запасания секреции инсулина первой стадии (первой фазы), стимуляции пролиферации β-клеток и стимуляции регенерации инсулина из их клеток-предшественников.
С целью достижения лучшего контроля глюкозы в крови необходима инъекция эксенатида дважды в сутки, что создает для пациентов большие неудобства. Кроме того, эксенатид вызывает тошноту от слабой до умеренной (эту реакцию имеет примерно 40% пациентов), диарею и рвоту (обе реакции имеет примерно 15% пациентов); примерно 50% пациентов, которых лечат эксенатидом, может образовывать антитела, хотя эти антитела не влияют на эффективность или не приводят к другим клиническим эффектам. Недавно обнаружено, что у шести пациентов встречалось кровотечение или симптомы некротизации поджелудочной железы после приема Биеты.
CJC-1131 представляет собой аналог GLP-1 с устойчивостью к пептидазе, разработанный фирмой ConjuChem Biotechnologies Inc., в котором Ala в положении 2 GLP-1 заменен D-Ala с целью усиления способности к устойчивости к энзимолизу DPP-IV, и структура которого содержит активный реакционный линкер, который может ковалентно (необратимо) связываться с сывороточным альбумином (Kim JG, Baggio LL, Bridon DP, et al. Development and characterization of a glucagon-like peptide-1 albumin conjugate: the ability to activate the glucagon-like peptide 1 receptor in vivo. Diabetes 2003, 52(3): 751-759), и комплекс GLP-1-сывороточный альбумин сохраняет активность GLP-1, хотя при этом повышает устойчивость к энзимолизу DPP-IV, продлевает действие in vivo, и его период полувыведения в плазме составляет примерно 20 суток.
В исследовании обнаружено, что Ki составляла примерно 12 нМ (Ki GLP-1 составляет 5,2 нМ), когда комплекс CJC-1131-сывороточный альбумин связывается с клетками яичника китайского хомячка, трансфицированными рекомбинантным рецептором GLP-1 поджелудочной железы человека; в то же время EC50 активации цАМФ этим комплексом составляет 11-13 нМ, где EC50 подобна GLP-1. Существующие литературные данные показывают, что эта связывающая молекула может снижать уровень глюкозы после приема пищи у мышей, у которых сахар крови является нормальным или высоким, и тесты показывают, что эта активность CJC-1131 действует на определенный функциональный рецептор GLP-1, в то же время у мышей CJC-1131 также обладает эффектом замедления опорожнения желудка и ингибирования приема пищи и тому подобным.
Завершена часть фазы II клинического испытания CJC-1131. В сентябре 2005 г. фирма ConjuChem пришла к заключению, что CJC-1131 может быть непригоден для режимов хронического дозирования, после анализа существующих результатов испытания, и приостановила клинические исследования CJC-1131. Клиническое испытание CJC-1131 еще не начато снова.
Albugon (альбумин-GLP-1) представляет собой лекарство длительного действия для лечения T2DM, разработанное фирмой GlaxoSmithKline, утвержденное фирмой Human Genome Sciences Inc., которое представляет собой слитый белок GLP-1 (с мутациями, повышающими устойчивость к DDP-IV) и альбумина. Его период полувыведения у обезьян составляет 3 суток. Основной идеей его разработки является сочетание рекомбинантного GLP-1 и сывороточного альбумина с образованием комплекса; посредством этого его период полувыведения in vivo значительно увеличен. Введение Albugon эффективно снижает уровень глюкозы в крови у мышей, повышает секрецию инсулина, замедляет опорожнение желудка и снижает прием пищи и т.д. (Baggio LL, Huang Q, Brown TJ, et al. A Recombinant Human Glucagon-Like Peptide (GLP)-1-Albumin Protein (Albugon) Mimics Peptidergic Activation of GLP-1 Receptor-Dependent Pathways Coupled With Satiety, Gastrointestinal Motility, and Glucose Homeostasis. Diabetes 2004, 53(9): 2492-2500). В настоящее время Albugon находится в фазе III клинического испытания.
В WO 9808871 раскрыто производное GLP-1, которое получено посредством модификации на GLP-1 (7-37) жирной кислотой, и период полувыведения in vivo GLP-1 значительно увеличен.
В WO 9943705 раскрыто производное GLP-1, которое химически модифицировано на N-конце, но в некоторых литературных источниках описано, что модификация аминокислот на N-конце значительно снизит активность всего производного GLP-1 (J. Med. Chem. 2000, 43, 1664 1669). Кроме того, в CN 200680006362, CN 200680006474, WO 2007113205, CN 200480004658, CN 200810152147 и WO 2006097538 и т.д. также раскрыта серия аналогов GLP-1 или их производных, полученных путем химической модификации или аминокислотной замены, из которых наиболее репрезентативным является лираглутид, разработанный фирмой Novo Nordisk, для которого завершено клиническое испытание фазы III. Лираглутид представляет собой производное GLP-1, структура которого содержит аналог GLP-1, последовательность которого на 97% гомологична последовательности GLP-1 человека, и этот аналог GLP-1 ковалентно сшит с пальмитиновой кислотой с образованием лираглутида, где пальмитиновая кислота структуры лираглутида нековалентно связана с сывороточным альбумином, и эти структурные характеристики определяют, что он может медленно высвобождаться из сайта инъекции без изменения активности GLP-1, и период его полувыведения in vivo пролонгирован; в то же время пальмитиновая кислота в его структуре образует определенное стерическое затруднение для предотвращения расщепления DPP-IV и для снижения почечного клиренса. В связи с этими вышеописанными характеристиками период полувыведения лираглутида в организме человека, введенного путем подкожной инъекции, составляет примерно 10-14 ч, теоретически его можно вводить один раз в сутки, и суточная доза составляет 0,6-1,8 мг. 23 апреля 2009 фирма Novo Nordisk объявила, что Комитет по лекарственным препаратам для применения у человека (СНМР), подчиненный ЕМЕА, дал положительную оценку лираглутиду и рекомендовал одобрение его включения в перечень. Фирма Novo Nordisk надеется, что Европейская комиссия одобрит его включение в перечень в течение двух месяцев.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является разработка серии производных аналога GLP-1, которые являются значительно более активными и имеют более длительный период полувыведения in vivo. Производное аналога GLP-1, предложенное данным изобретением, обладает такой же функцией, как GLP-1 человека, и имеет более длительный период полувыведения in vivo по сравнению с GLP-1 человека.
Целью настоящего изобретения также является разработка фармацевтической композиции, содержащей производное аналога GLP-1 или его фармацевтически приемлемую соль, для применения при лечении инсулиннезависимого сахарного диабета, инсулинозависимого сахарного диабета и ожирения.
Цели настоящего изобретения достигаются путем приведенных ниже технических решений.
В настоящем изобретении предложена серия производных аналога GLP-1, имеющих аминокислотную последовательность формулы (I), или их фармацевтически приемлемые соли:

где производные аналога GLP-1 содержат липофильный заместитель формулы R1(CH2)n-CO-, в котором R1 выбран из СН3- и НООС-, n представляет собой целое число от 8 до 25, Х1, Х2, Х10, Х12, Х13, Х14, Х16, Х17, Х19, Х20, Х21, Х24, Х27, Х28, Х29, Х30, Х31, Х32, Х33, Х34, Х35, Х36, Х37, Х38 и Х39 независимо выбраны из любой природной или неприродной аминокислоты или пептидных фрагментов, состоящих из любых природных или неприродных аминокислот.
Производные аналога GLP-1 относятся к новому пептиду GLP-1, полученного путем замены части аминокислот или удлинения цепи у С-конца пептида GLP-1 (7-37) человека, служащего в качестве предшественника, включающего амид GLP-1 (7-36) и GLP-1 (7-37), который обладает такой же функцией, как GLP-1 человека.
Производные относятся к созданию химической модификации аминокислотных остатков аналога GLP-1 путем использования липофильных заместителей, где типичной модификацией является образование амида или сложного эфира, предпочтительно образование амида.
В предпочтительной форме осуществления изобретения липофильный заместитель формулы R1(CH2)n-CO- и аминогруппа аминокислотных остатков аналога GLP-1 связаны амидной связью, в которой R1 выбран из СН3- и НООС-, и n представляет собой целое число от 8 до 25.
В другой предпочтительной форме осуществления изобретения липофильный заместитель формулы R1(CH2)n-CO- и ε аминогруппа Lys на С-конце аналога GLP-1 связаны амидной связью, в которой R1 выбран из СН3- и НООС-, и n представляет собой целое число от 8 до 25.
Еще в одной другой предпочтительной форме осуществления изобретения липофильный заместитель формулы R1(CH2)n-CO- и α аминогруппа Lys на С-конце аналога GLP-1 связаны амидной связью, в которой R1 выбран из СН3- и НООС-, и n представляет собой целое число от 8 до 25, и наиболее предпочтительно равно 14.
В другой предпочтительной форме осуществления изобретения Х1 в аминокислотной последовательности аналога GLP-1 выбран из L-His и D-His; X2 выбран из Ala, D-Ala, Gly, Val, Leu, Ile, Lys и Aib; X10 выбран из Val и Leu; X12 выбран из Ser, Lys и Arg; X13 выбран из Tyr и Gln; X14 выбран из Leu и Met; X16 выбран из Gly, Glu и Aib; X17 выбран из Gln, Glu, Lys и Arg; X19 выбран из Ala и Val; Х20 выбран из Lys, Glu и Arg; X21 выбран из Glu и Leu; X24 выбран из Val и Lys; Х27 выбран из Val и Lys; X28 выбран из Lys, Glu, Asn и Arg; X29 выбран из Gly и Aib; Х30 выбран из Arg, Gly и Lys; X31 выбран из Gly, Ala, Glu, Pro и Lys; X32 выбран из Lys и Ser; X33 выбран из Lys и Ser; X34 выбран из Gly, Ala и Sar; X35 выбран из Gly, Ala и Sar; X36 выбран из Pro и Gly; X37 выбран из Pro и Gly; X38 выбран из Pro и Gly; X39 выбран из Ser и Tyr.
В одной более предпочтительной форме осуществления настоящего изобретения аминокислотная последовательность аналога GLP-1 выбрана из группы, состоящей из SEQ ID NO: 1 - SEQ ID NO: 120.
В другой предпочтительной форме осуществления настоящего изобретения липофильный заместитель формулы R1(CH2)n-CO-, и аминогруппа аминокислотных остатков аналога GLP-1, последовательность которого выбрана из группы, состоящей из SEQ ID NO: 1 - SEQ ID NO: 120, связаны амидной связью, в которой R1 выбран из СН3- и НООС-, и n представляет собой целое число от 8 до 25.
В одной более предпочтительной форме осуществления настоящего изобретения липофильный заместитель формулы R1(CH2)n-CO- и ε аминогруппа С-концевого Lys аналога GLP-1, выбранного из группы, состоящей из SEQ ID NO: 1 - SEQ ID NO: 120, связаны амидной связью, в которой R1 выбран из СН3- и НООС-, и n представляет собой целое число от 8 до 25.
В одной более предпочтительной форме осуществления настоящего изобретения липофильный заместитель формулы R1(CH2)n-CO- и α аминогруппа С-концевого Lys аналога GLP-1, выбранного из группы, состоящей из SEQ ID NO: 1 - SEQ ID NO: 120, связаны амидной связью, в которой R1 выбран из СН3- и НООС-, и n представляет собой целое число от 8 до 25, предпочтительно n выбран из 8, 10, 12, 14, 16, 18, 20 и 22, наиболее предпочтительно n равно 14.
В одной более предпочтительной форме осуществления данного изобретения липофильный заместитель формулы R1(CH2)n-CO- и α аминогруппа С-концевого Lys аналога GLP-1, выбранного из группы, состоящей из SEQ ID NO: 1 - SEQ ID NO: 20, связаны амидной связью, в которой R1 выбран из СН3- и НООС-, и n представляет собой целое число от 8 до 25, предпочтительно n выбран из 8, 10, 12, 14, 16, 18, 20 и 22, наиболее предпочтительно n равно 14.
В другой более предпочтительной форме осуществления данного изобретения липофильный заместитель формулы R1(CH2)n-CO- и α аминогруппа С-концевого Lys аналога GLP-1, выбранного из группы, состоящей из SEQ ID NO: 1 - SEQ ID NO: 8, связаны амидной связью, в которой R1 представляет собой СН3, и n равно 14.
Производные аналогов GLP-1, предложенные в данном изобретении, принадлежат к амфотерным соединениям, и специалист в данной области техники может преобразовать их в соли путем использования кислотных или щелочных соединений известными методами, где кислотами, обычно используемыми для образования солей присоединения кислоты, являются: соляная кислота, бромисто-водородная кислота, йодисто-водородная кислота, серная кислота, фосфорная кислота, пара-толуолсульфоновая кислота, метансульфоновая кислота, щавелевая кислота, пара-бромфенилсульфоновая кислота, угольная кислота, янтарная кислота, лимонная кислота, бензойная кислота, уксусная кислота; эти соли включают сульфат, пиросульфат, трифторацетат, сульфит, бисульфит, фосфат, бифосфат, дигидрофосфат, метафосфат, пирофосфат, гидрохлорид, бромид, йодид, ацетат, пропионат, октаноаты, акрилат, формиат, изобутират, гексаноат, энантаты, пропиолат, оксалат, малонат, сукцинат, суберат, фумарат, малеат, 1,4-бутиндиоат, 1,6-гексиндиоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фенилацетат, фенпропионат, фенилбутират, цитрат, лактат, γ-гидроксибутират, гликолят, тартрат, метансульфонат, пропансульфонат, 1-нафтолсульфонат, 2-нафтолсульфонат, манделат и тому подобное, предпочтительно трифторацетат. Щелочные вещества можно также преобразовать в соли с производными аналогов GLP-1, где эти щелочные вещества включают аммоний, гидроксиды щелочных металлов или щелочноземельных металлов, а также карбонат, бикарбонат, типично гидроксид натрия, гидроксид калия, гидроксид аммония, карбонат натрия, карбонат калия и тому подобное.
Фармацевтические композиции, содержащие производные GLP-1 в соответствии с изобретением, можно применять для лечения пациентов, которые нуждаются в данном лечении, путем парентерального введения. Парентеральное введение может быть выбрано из подкожных, внутримышечных или внутривенных инъекций. Производные GLP-1 по изобретению можно также вводить посредством чрескожных путей, таких как введение посредством пластыря (предпочтительно пластыря для ионтофореза) и введение через слизистую оболочку.
Фармацевтические композиции, содержащие производные GLP-1, по изобретению можно готовить посредством известных методов в области фармацевтической промышленности. Эти методы включают точное растворение и смешивание компонентов с получением желаемых конечных композиций. Например, производные GLP-1 растворяют в определенном количестве воды, где объем воды несколько меньше, чем конечный объем полученной композиции. По необходимости добавляют изотонические агенты, консерванты, сурфактанты и буферы, где изотоническими агентами являются хлорид натрия, маннит, глицерин, пропиленгликоль, сахар или альдит. Консервантами являются фенол, орто-крезол, пара-крезол, мета-крезол, сложный эфир метилпарагидроксибензоат, бензиловый спирт. Подходящими буферными агентами являются ацетат натрия, карбонат натрия, глицин, гистидин, лизин, дигидрофосфат натрия, динатрия гидрофосфат, фосфат натрия. Сурфактантами являются полоксамер, полоксамер-188, полоксамер-407, Твин 80 и Твин 20. При необходимости добавляют водные растворы кислот, таких как соляная кислота, или щелочей, таких как раствор гидроксида натрия, для доведения значений рН растворов, и, наконец, объем раствора доводят добавлением воды до получения необходимой концентрации. Кроме указанных компонентов, чтобы уменьшить агрегаты, образуемые композицией в процессе хранения, фармацевтические композиции по изобретению также содержат достаточное количество основных аминокислот или других щелочных реагентов, обладающих такой же функцией, таких как лизин, гистидин, аргинин, имидазол.
Производные аналогов GLP-1 по изобретению синтезируют вручную, где смола представляет собой смолу НМРА-АМ, α-аминогруппу производных аминокислот защищают Fmoc (флуоренилформилкарбонилом), тиол боковой цепи цистеина, амидо боковой цепи глутамина, имидазол боковой цепи гистидина защищают Trt (трифенилметилом), гуанидил боковой цепи аргинина защищают Pbf (2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонилом), индолил боковой цепи триптофана и аминогруппу боковой цепи лизина защищают Вос (трет-бутоксикарбонилом), гидроксил боковой цепи треонина, фенилол боковой цепи тирозина, гидроксил боковой цепи серина защищают tBu (трет-бутилом). Карбоксил С-концевых аминокислот единой пептидной цепи производных аналогов GLP-1, которые будут синтезированы, соединяют с нерастворимой высокомолекулярной смолой (амидной смолой Ринка) посредством ковалентных связей, а затем аминокислоты, связанные с твердофазным носителем, действуют в качестве амино-компонентов, защитную группу амино удаляют 20% раствором гексагидропиридин/ДМФ, а затем подвергают взаимодействию с избытком производных аминокислот для сшивания длинной пептидной цепи. Операцию (конденсация → отмывка → удаление защиты → отмывка → следующий раунд конденсации) повторяют до достижения желаемой длины пептидной цепи. Наконец, пептидную цепь отщепляют от смолы путем использования смеси ТФУ: вода: 1,2-дитиогликоль: триизопропилсилан (92,5:2,5:2,5:2,5) с получением сырых производных аналогов GLP-1 посредством осаждения эфиром. Сырые пептидные мономеры очищают посредством использования колонки с обращенной фазой С18, и в результате этого получают желаемые производные аналогов GLP-1. Способ нингидринового тестирования используют для мониторинга стадий конденсации и удаления защиты. То есть, когда на цепи смола-пептид присутствуют свободные аминокислоты, нингидриновый реагент покажет синее окрашивание, и не покажет никакого окрашивания, когда свободные аминокислоты отсутствуют (сам нингидриновый реагент имеет желтый цвет). Таким образом, когда реакция конденсации завершена, если во время нингидринового теста он показывает желтый цвет (цвет самого нингидринового реагента), то это позволяет предположить, что стадия сочетания завершена, и можно проводить операцию удаления защиты перед сочетанием следующей аминокислоты; если тестирование показывает синий цвет, это позволяет предположить, что на пептидных цепях еще имеется некоторое количество свободной аминокислоты, и необходимо еще раз повторить стадию сочетания или заменить существующий конденсирующий реагент, пока пептидные смолы не покажут желтый цвет в результате нингидринового теста.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Чтобы описать настоящее изобретение более подробно, приведены нижеследующие примеры. Однако настоящее изобретение не следует рассматривать как ограниченное формами осуществления, изложенными в данной заявке ниже.
Пример 1. Способ твердофазного синтеза HS-20001
1. Получение смолы Fmoc-Lys (Mtt)-HMP-AM
(1) Высушивание и набухание смолы НМР-АМ
50 г (30 ммоль) смолы НМР-АМ (0,6 ммоль/г), высушенной в течение 24 ч в вакууме, помещают в 2 л барботер, смолу подвергают набуханию с 500 мл N,N-диметилформамида (ДМФ) в течение 30 минут, ДМФ отсасывают, смолу промывают ДМФ в течение 1 мин, стадию отмывки повторяют дважды.
(2) Получение смолы Fmoc-Lys (Mtt)-HMP-AM1
1. Сочетание Fmoc-Lys(Mtt)-OH со смолой НМР-АМ. Смолу промывают 500 мл ДХМ, а затем стадию отмывки повторяют дважды. 56,2 г (90 ммоль) Fmoc-Lys(Mtt)-OH и 11,4 г (90 ммоль) DIC растворяют в 1 л ДХМ, затем добавляют в набухшую смолу НМР-АМ, затем добавляют 366 мг (3 ммоль) DMAP и подвергают взаимодействию в течение 24 ч.
2. Отмывка смолы
После взаимодействия смолу дважды промывают поочередно ДМФ и IPA, промывают ДМФ 3 раза.
3. Копирование гидроксила
15,3 г (150 ммоль) уксусного ангидрида и 19,4 г (150 ммоль) DIEA растворяют в 1 л ДМФ, добавляют в смолу и подвергают взаимодействию в течение 10 мин.
4. Отмывка смолы
Смолу промывают дважды 1 л 50% МеОН/ДМФ, 50% ДХМ/ДМФ, а затем промывают ДХМ три раза и обезвоженным этанолом три раза поочередно. После высушивания в вакууме получают смолу Fmoc-Lys (Mtt)-НМР-АМ.
(3) Анализы загрузки смолы Fmoc-Lys (Mtt)-HMP-AM
Точное количество 5-10 мг смолы помещают в 1 мл 20% раствора гексагидропиридин/ДМФ, перемешивают в течение 20 мин, затем 50 мкл супернатанта отбирают пипеткой и разбавляют в 2,5 мл ДМФ;
Чистые контрольные образцы: 50 мкл 20% раствора гексагидропиридин/ДМФ отбирают пипеткой и разбавляют в 2,5 мл ДМФ;
Степень замещения вычисляют, как описано ниже:
Sub=(A×51)/(7,8×m),
где А представляет собой значение поглощения в УФ при 301 нм; m представляет собой массу смолы, единицей является мг.
2. Высушивание и набухание смолы после твердофазного синтеза
50 г (20 ммоль) смолы Fmoc-Lys(Mtt)-HMPA-AM (0,4 ммоль/г), высушенной в течение 24 ч в вакууме, помещают в 2 л барботер, затем добавляют 500 мл N,N-диметилформамида (ДМФ) для набухания смолы в течение 30 мин, затем раствор ДМФ отсасывают.
3. Удаление защитной группы 4-метилтрифенилметил (Mtt) смолы Fmoc-Lys(Mtt)-HMPA-AM
Смолу дважды промывают 200 мл ДХМ, затем добавляют 1200 мл 1% ТФУ/ДХМ (ТФУ находится примерно в 8-кратном избытке) для удаления защитной группы Mtt в течение 1 ч, смолу поочередно промывают 200 мл 5% N,N-диизопропилэтиламина (DIEA)/ДМФ и ДМФ три раза, затем промывают ДМФ 3 раза.
4. Конденсация пальмитиновой кислоты
50 ммоль пальмитиновой кислоты и 50 ммоль 3-(диэтоксифосфорилокси)-1,2,3-фентриазин-4-кетона (DEPBT) растворяют в 400 мл ДМФ, затем добавляют 100 ммоль DIEA путем перемешивания для взаимодействия в течение 3 мин при комнатной температуре, этот раствор добавляют к смоле, подвергают взаимодействию в водяной бане при 37°С в течение 2 ч в атмосфере N2. После взаимодействия реакционный раствор отсасывают, смолу промывают ДМФ, изопропиловым спиртом (IPA) и ДМФ поочередно.
5. Удаление защитной группы 9-Fmoc (флуоренилметилоксикарбонил) смолы Fmoc-Lys (N-ε-пальмитиновая кислота)-НМРА-АМ
200 мл 20% раствора пиперидин/ДМФ помещают в барботер, наполненный смолой Fmoc-Lys(Mtt)-HMPA-AM, подвергают взаимодействию в течение 5 мин, а затем отсасывают, добавляют 200 мл 20% раствора пиперидин/ДМФ и подвергают взаимодействию в течение 20 мин при комнатной температуре. После взаимодействия смолу промывают 200 мл ДМФ четыре раза.
6. Твердофазный синтез части пептидной цепи HS-20001
1. Конденсация Fmoc-Ser(tBu)-OH
50 ммоль Fmoc-Ser(tBu)-OH растворяют в 125 мл 0,4 М 1-гидроксибензотриазола (HOBt)/ДМФ, затем добавляют 125 мл 0,4 М N,N'-диизопропилкарбодиимида (DIC)/ДХМ для активации и подвергают взаимодействию в течение 10 мин при комнатной температуре; этот раствор добавляют в смолу, подвергают взаимодействию под защитой азота при комнатной температуре, и нингидрин используют для обнаружения и контроля степени взаимодействия. После взаимодействия реакционный раствор удаляют; смолу промывают ДМФ, IPA и ДМФ поочередно.
2. Удлинение пептидной цепи
Пептид HS-20001 на смоле синтезируют в соответствии с последовательностью пептидной цепи HS-20001 от амино-конца (N-конца) к карбокси-концу (С-концу) (His-(D)-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Nle-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Gln-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser), где количества аминокислот и реагентов конденсации являются такими же, как количества для Fmoc-Ser(tBu)-OH, защищенные аминокислоты представляют собой Fmoc-Ser(tBu)-OH, Fmoc-Pro-OH, Fmoc-Ala-OH, Fmoc-Gly-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Ile-OH, Fmoc-Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Nle-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Thr(tBu)-OH, Fmoc-D-Ala-OH and Fmoc-His(Trt)-OH соответственно, и реакции конденсации и удаления защиты повторяют.
3. Последующая обработка смолы с пептидом HS-20001
Смолу с пептидом HS-20001, полученную на стадии 2, промывают ДМФ, IPA и ДМФ поочередно, дважды промывают абсолютным эфиром, затем высушивают в вакууме и получают смолу с пептидом HS-20001.
4. Получение сырого пептида HS-20001
Высушенную смолу с пептидом HS-20001 подвергают взаимодействию со свежим лизатом трифторуксусная кислота (ТФУ): триизопропилсилан (TIS): вода = 95:2,5:2,5 (об/об, и суммарно 10 мл лизата на грамм сухой смолы) в течение 4 ч при комнатной температуре. После взаимодействия реакционный раствор фильтруют, смолу дважды промывают ТФУ, фильтрат собирают и объединяют и концентрируют до 1/3 исходного объема посредством роторного выпаривания. HS-20001 осаждают и промывают холодным абсолютным эфиром, после центрифугирования и высушивания в вакууме получают белый сырой HS-20001.
5. Получение HS-20001 жидкостной хроматографией с обращенной фазой
10 г сырого HS-20001 растворяют в некотором количестве воды, фильтруют мембранным фильтром 0,45 мкм, затем очищают высокоэффективной жидкостной хроматографией с обращенной фазой (ОФ-ВЭЖХ), где подвижная фаза представляет собой А 0,1% ТФУ/H2O, В 0,1% ТФУ/ацетонитрил, где колонка представляет собой колонку Denali С-18 (диаметр частиц 8,3 мкм, 5×30 см), температура колонки составляет 45°С, длина волны обнаружения составляет 220 нм, скорость тока составляет 120 мл/мин. Пики продукта собирают, концентрируют в вакууме, чтобы удалить большую часть ацетонитрила, затем путем лиофилизации получают продукт HS-20001 2,25 г, чистота которого составляет 98,5%, выход 22,5%.
Пример 2. Способ твердофазного синтеза HS-20002
1. Получение смолы Fmoc-Lys (Mtt)-HMP-AM
См. Пример 1.
2. Высушивание и набухание смолы после твердофазного синтеза
50 г (20 ммоль) смолы Fmoc-Lys(Mtt)-HMPA-AM (0,4 ммоль/г), высушенной в течение 24 ч в вакууме, помещают в 2 л барботер, смолу подвергают набуханию с 500 мл ДМФ в течение 30 мин, затем раствор ДМФ отсасывают.
3. Удаление защитной группы Mtt смолы Fmoc-Lys(Mtt)-HMPA-AM
Смолу дважды промывают 200 мл ДХМ, защитную группу Mtt удаляют путем добавления 1200 мл 1% ТФУ/ДХМ (ТФУ находится примерно в 8-кратном избытке) в течение 1 ч, затем поочередно промывают 200 мл 5% DIEA/ДМФ и ДМФ три раза, затем промывают ДХМ 3 раза.
4. Конденсация пальмитиновой кислоты
50 ммоль пальмитиновой кислоты и 50 ммоль DEPBT растворяют в 400 мл ДМФ, а затем добавляют 100 ммоль DIEA путем перемешивания для взаимодействия в течение 3 мин при комнатной температуре, этот раствор добавляют к смоле, подвергают взаимодействию в водяной бане при 37°С в атмосфере N2 в течение 2 ч. После взаимодействия реакционный раствор удаляют и смолу промывают ДМФ, изопропиловым спиртом (IPA) и ДМФ поочередно.
5. Удаление защитной группы Fmoc смолы Fmoc-Lys (N-ε-пальмитиновая кислота)-НМРА-АМ
200 мл 20% раствора пиперидин/ДМФ помещают в барботер, наполненный смолой Fmoc-Lys(Mtt)-HMPA-AM, отсасывают после взаимодействия в течение 5 мин, добавляют 200 мл 20% раствора пиперидин/ДМФ для взаимодействия в течение 20 мин при комнатной температуре. После завершения реакции смолу промывают 200 мл ДМФ четыре раза.
6. Способ твердофазного синтеза части пептидной цепи HS-20002
1. Конденсация Fmoc-Ser(tBu)-OH
50 ммоль Fmoc-Ser(tBu)-OH растворяют в 125 мл 0,4 М HOBt/ДМФ, затем добавляют 125 мл 0,4 М DIC/ДХМ для активации и подвергают взаимодействию в течение 10 мин при комнатной температуре; раствор помещают в смолу и подвергают взаимодействию в атмосфере N2 при комнатной температуре, и нингидриновое тестирование используют для обнаружения и контроля степени взаимодействия. После взаимодействия реакционный раствор удаляют и смолу промывают ДМФ, IPA и ДМФ поочередно.
2. Удлинение пептидной цепи
Смолу с пептидом HS-20002 синтезируют в соответствии с последовательностью пептидной цепи HS-20002 с N-амино (N-конца) к карбокси-концу (С-концу) (His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp -Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser), где количества аминокислот и реагентов конденсации являются такими же, как количества для Fmoc-Ser(tBu)-OH, защищенные аминокислоты представляют собой Fmoc-Ser(tBu)-OH, Fmoc-Pro-OH, Fmoc-Ala-OH, Fmoc-Gly-OH, Fmoc-Asn(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Ile-OH, Fmoc-Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Met-OH, Fmoc-Gln(Trt)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Thr(tBu)-OH и Fmoc-His(Trt)-OH соответственно, и реакции конденсации и удаления защиты повторяют.
3. Последующая обработка смолы с пептидом HS-20002. Смолу с пептидом HS-20002, полученную на стадии 2, промывают ДМФ, IPA и ДМФ поочередно, дважды промывают абсолютным эфиром, затем высушивают в вакууме, наконец, получают смолу с пептидом HS-20002.
4. Получение сырого пептида HS-20002
Высушенную смолу с пептидом HS-20002 подвергают взаимодействию со свежим лизатом трифторуксусная кислота (ТФУ): триизопропилсилан (TIS): вода: 1,2-этандитиол (EDT) = 94:1:2,5:2,5 (об/об, и суммарно 10 мл лизата на грамм сухой смолы) в течение 4 ч при комнатной температуре. После взаимодействия реакционный раствор фильтруют, смолу дважды промывают ТФУ, фильтрат собирают и объединяют, и концентрируют до 1/3 исходного объема посредством роторного выпаривания. HS-20002 осаждают холодным абсолютным эфиром, после центрифугирования и высушивания в вакууме получают белый сырой HS-20002.
5. Получение HS-20002 жидкостной хроматографией с обращенной фазой
10 г сырого HS-20002 растворяют в некотором количестве воды, фильтруют мембранным фильтром 0,45 мкм, затем очищают высокоэффективной жидкостной хроматографией с обращенной фазой (ОФ-ВЭЖХ), где подвижная фаза представляет собой А 0,1% ТФУ/H2O, В 0,1% ТФУ/ацетонитрил, где колонка представляет собой колонку Denali С-18 (диаметр частиц 8,3 мкм, 5×30 см), температура колонки составляет 45°С, длина волны обнаружения составляет 220 нм, скорость тока составляет 120 мл/мин. Пики продукта собирают, концентрируют в вакууме, чтобы удалить большую часть ацетонитрила, затем путем лиофилизации получают продукт HS-20002 2,1 г, чистота которого составляет 98%, выход составляет 20,5%.
Пример 3. Способ твердофазного синтеза HS-20003
1. Получение смолы Fmoc-Lys (Mtt)-HMP-AM
См. Пример 1.
2. Высушивание и набухание смолы после твердофазного синтеза
50 г (20 ммоль) смолы Fmoc-Lys(Mtt)-HMPA-AM (0,4 ммоль/г), высушенной в течение 24 ч в вакууме, помещают в 2 л барботер, смолу подвергают набуханию с 500 мл ДМФ в течение 30 мин, затем раствор ДМФ отсасывают.
3. Удаление защитной группы Fmoc смолы Fmoc-Lys(Mtt)-HMPA-AM
200 мл 20% раствора пиперидин/ДМФ добавляют в барботер, наполненный смолой Fmoc-Lys(Mtt)-HMPA-AM, раствор отсасывают через 5 мин, а затем добавляют 200 мл 20% раствора пиперидин/ДМФ и дают возможность взаимодействовать еще в течение 20 мин при комнатной температуре. После взаимодействия смолу промывают 200 мл ДМФ четыре раза.
4. Конденсация пальмитиновой кислоты
50 ммоль пальмитиновой кислоты и 50 ммоль DEPBT растворяют в 400 мл ДМФ, затем добавляют 100 ммоль DIEA путем перемешивания для взаимодействия в течение 3 мин при комнатной температуре, этот раствор добавляют к смоле, подвергают взаимодействию в водяной бане при 37°С в атмосфере N2 в течение 2 ч. После взаимодействия реакционный раствор удаляют и смолу промывают ДМФ, изопропиловым спиртом (IPA) и ДМФ поочередно.
5. Удаление защитной группы Mtt смолы пальмитиновая кислота-Lys (Mtt)-НМРА-АМ
Смолу дважды промывают 200 мл ДХМ. Защитную группу Mtt удаляют путем добавления 1200 мл 1% ТФУ/ДХМ (ТФУ находится примерно в 8-кратном избытке) для взаимодействия в течение 1 ч. Смолу поочередно промывают 200 мл 5% DIEA/ДМФ и ДМФ три раза, затем промывают ДХМ 3 раза.
6. Способ твердофазного синтеза части пептидной цепи HS-20003
1. Конденсация Fmoc-Ser(tBu)-OH
50 ммоль Fmoc-Ser(tBu)-OH и 50 ммоль DEPBT растворяют в некотором количестве ДХМ, затем добавляют 100 ммоль DIEA для активации в течение 3 мин при комнатной температуре; раствор добавляют к смоле и подвергают взаимодействию в атмосфере N2 при комнатной температуре, и нингидрин используют для обнаружения и контроля степени взаимодействия. После взаимодействия реакционный раствор удаляют и смолу промывают ДМФ, IPA и ДМФ поочередно.
2. Удлинение пептидной цепи
Смолу с пептидом HS-20003 синтезируют в соответствии с последовательностью пептидной цепи HS-20003 с N-амино (N-конца) к карбокси-концу (С-концу) (His-(D)-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Glu-Glu-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser), где количества аминокислот и реагентов конденсации являются такими же, как для Fmoc-Ser(tBu)-OH, защищенные аминокислоты представляют собой Fmoc-Ser(tBu)-OH, Fmoc-Pro-OH, Fmoc-Ala-OH, Fmoc-Gly-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Ile-OH, Fmoc-Phe-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Thr(tBu)-OH соответственно, и реакции конденсации и удаления защиты повторяют.
3. Последующая обработка смолы с пептидом HS-20003
Смолу с пептидом HS-20003, полученную на стадии 2, промывают ДМФ, IPA и ДМФ поочередно, дважды промывают абсолютным эфиром, затем высушивают в вакууме, наконец, получают смолу с пептидом HS-20003.
4. Получение сырого пептида HS-20003
Высушенную смолу с пептидом HS-20003 подвергают взаимодействию со свежим лизатом трифторуксусная кислота (ТФУ): триизопропилсилан (TIS): вода = 95:2,5:2,5 (об/об, и суммарно 10 мл лизата на грамм сухой смолы) в течение 4 ч при комнатной температуре. После взаимодействия реакционный раствор фильтруют, смолу дважды промывают ТФУ, фильтрат собирают и объединяют и концентрируют до 1/3 исходного объема посредством роторного выпаривания. HS-20003 осаждают холодным эфиром при перемешивании, после центрифугирования и высушивания в вакууме в конечном счете получают белый сырой HS-20003.
5. Получение HS-20003 жидкостной хроматографией с обращенной фазой
10 г сырого HS-20003 растворяют в некотором количестве 20% уксусной кислоты в воде и перемешивают в течение по меньшей мере 4 ч, затем фильтруют мембранным фильтром 0,45 мкм, затем очищают высокоэффективной жидкостной хроматографией с обращенной фазой (ОФ-ВЭЖХ), где подвижная фаза представляет собой А 0,1% ТФУ/Н2О, В 0,1% ТФУ/ацетонитрил, где колонка представляет собой колонку Denali С-18 (диаметр частиц 8,3 мкм, 5×30 см), температура колонки составляет 45°С, длина волны обнаружения составляет 220 нм, скорость тока составляет 120 мл/мин. Пики продукта собирают, концентрируют при пониженном давлении, чтобы удалить большую часть ацетонитрила, затем путем лиофилизации получают продукт HS-20003 2,5 г, чистота которого составляет 98,5%, выход составляет 25%.
Пример 4. Способ твердофазного синтеза HS-20004
1. Получение смолы Fmoc-Lys (Mtt)-HMP-AM
См. Пример 1.
2. Высушивание и набухание смолы после твердофазного синтеза
50 г (20 ммоль) смолы Fmoc-Lys(Mtt)-HMPA-AM (0,4 ммоль/г), высушенной в течение 24 ч в вакууме, помещают в 2 л барботер, добавляют 500 мл ДМФ для набухания смолы в течение 30 мин, раствор ДМФ отсасывают.
3. Удаление защитной группы Fmoc смолы Fmoc-Lys(Mtt)-HMPA-AM
200 мл 20% раствора пиперидин/ДМФ добавляют в барботер, наполненный смолой Fmoc-Lys(Mtt)-HMPA-AM, а затем отсасывают через 5 мин, а затем добавляют 200 мл 20% раствора пиперидин/ДМФ для взаимодействия в течение 20 мин при комнатной температуре. После взаимодействия смолу промывают 200 мл ДМФ четыре раза.
4. Конденсация пальмитиновой кислоты
50 ммоль пальмитиновой кислоты и 50 ммоль DEPBT растворяют в 400 мл ДМФ, затем добавляют 100 ммоль DIEA путем перемешивания в течение 3 мин при комнатной температуре, этот раствор добавляют к смоле, подвергают взаимодействию в водяной бане при 37°С в атмосфере N2 в течение 2 ч. После взаимодействия реакционный раствор удаляют и смолу промывают ДМФ, изопропиловым спиртом (IPA) и ДМФ поочередно.
5. Удаление защитной группы Mtt смолы пальмитиновая кислота-Lys(Mtt)-НМРА-АМ
Смолу дважды промывают 200 мл ДХМ. Защитную группу Mtt удаляют путем добавления 1200 мл 1% ТФУ/ДХМ (ТФУ находится примерно в 8-кратном избытке) для взаимодействия в течение 1 ч. Смолу поочередно промывают 200 мл 5% DIEA/ДМФ и ДМФ три раза, затем промывают ДХМ три раза.
6. Способ твердофазного синтеза части пептидной цепи HS-20004
1. Конденсация Fmoc-Ser(tBu)-OH
50 ммоль Fmoc-Ser(tBu)-OH и 50 ммоль DEPBT растворяют в некотором количестве ДХМ, затем добавляют 100 ммоль DIEA для активации в течение 3 мин при комнатной температуре; раствор добавляют к смоле, подвергают взаимодействию в атмосфере N2 при комнатной температуре и нингидрин используют для обнаружения и контроля степени взаимодействия. После взаимодействия реакционный раствор удаляют и смолу промывают ДМФ, IPA и ДМФ поочередно.
2. Удлинение пептидной цепи
Смолу с пептидом HS-20004 синтезируют в соответствии с последовательностью пептидной цепи HS-20004 с N-амино (N-конца) к карбокси-концу (С-концу) (His-Aib-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Glu-Glu-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser), где количества аминокислот и реагентов конденсации являются такими же, как для Fmoc-Ser(tBu)-OH, защищенные аминокислоты представляют собой Fmoc-Ser(tBu)-OH, Fmoc-Pro-OH, Fmoc-Ala-OH, Fmoc-Gly-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Ile-OH, Fmoc-Phe-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Aib-OH и Fmoc-His(Trt)-OH соответственно, и реакции конденсации и удаления защиты повторяют.
3. Последующая обработка смолы с пептидом HS-20004
Смолу с пептидом HS-20004, полученную на стадии 2, промывают ДМФ, IPA и ДМФ поочередно, дважды промывают абсолютным эфиром, затем высушивают в вакууме, наконец, получают смолу с пептидом HS-20004.
4. Получение сырого пептида HS-20004
Высушенную смолу с пептидом HS-20004 подвергают взаимодействию со свежим лизатом трифторуксусная кислота (ТФУ): триизопропилсилан (TIS): вода = 95:2,5:2,5 (об/об, и суммарно 10 мл лизата на грамм сухой смолы) в течение 4 ч при комнатной температуре. После взаимодействия реакционный раствор фильтруют, смолу дважды промывают ТФУ, фильтрат собирают и объединяют и концентрируют до 1/3 исходного объема посредством роторного выпаривания. HS-20004 осаждают холодным эфиром при перемешивании, после центрифугирования и высушивания в вакууме в конечном счете получают белый сырой HS-20004.
5. Получение HS-20004 жидкостной хроматографией с обращенной фазой
10 г сырого HS-20004 растворяют в некотором количестве 20% уксусной кислоты в воде и перемешивают в течение по меньшей мере 4 ч, затем фильтруют мембранным фильтром 0,45 мкм, затем очищают высокоэффективной жидкостной хроматографией с обращенной фазой (ОФ-ВЭЖХ), где подвижная фаза представляет собой А 0,1% ТФУ/H2O, В 0,1% ТФУ/ацетонитрил, где колонка представляет собой колонку Denali С-18 (диаметр частиц 8,3 мкм, 5×30 см), температура колонки составляет 45°С, длина волны обнаружения составляет 220 нм, скорость тока составляет 120 мл/мин. Пики продукта собирают, концентрируют в вакууме, чтобы удалить большую часть ацетонитрила, затем путем лиофилизации получают продукт HS-20004 2,25 г, чистота которого составляет 98,5%, выход составляет 22,5%.
Пример 5. Способ твердофазного синтеза HS-20005
Способ получения HS-20005 является таким же, как описано в Примере 4, где различие состоит в том, что аминокислотная последовательность заменена SEQ ID NO: 5, и получают 2,5 г продукта HS-20005, чистота которого составляет 98,5%, выход составляет 25%.
Пример 6. Способ твердофазного синтеза HS-20006
Способ получения HS-20006 является таким же, как описано в Примере 4, где различие состоит в том, что аминокислотная последовательность заменена SEQ ID NO: 6, и получают 2,25 г продукта HS-20006, чистота которого составляет 98,5%, выход составляет 22,5%.
Пример 7. Способ твердофазного синтеза HS-20007
Способ получения HS-20007 является таким же, как описано в Примере 4, где различие состоит в том, что аминокислотная последовательность заменена SEQ ID NO: 7, и получают 2,1 г продукта HS-20007, чистота которого составляет 98%, выход составляет 20,5%.
Пример 8. Способ твердофазного синтеза HS-20008
Способ получения HS-20008 является таким же, как описано в Примере 4, где различие состоит в том, что аминокислотная последовательность заменена SEQ ID NO: 8, и получают 2,5 г продукта HS-20008, чистота которого составляет 98,5%, выход составляет 25%.
Сравнительный пример твердофазного синтеза лираглутида
1. Получение смолы Fmoc-Lys(Mtt)-HMP-AM
(1) Высушивание и набухание смолы НМР-АМ
50 г (30 ммоль) смолы НМР-АМ (0,6 ммоль/г), высушенной в течение 24 ч в вакууме, помещают в 2 л барботер, добавляют 500 мл N,N-диметилформамида (ДМФ) для набухания смолы в течение 30 мин, раствор ДМФ отсасывают, добавляют ДМФ для отмывки смолы в течение 1 мин, стадию отмывки повторяют дважды.
(2) Получение смолы Fmoc-Lys(Mtt)-HMP-AM
1. Сочетание Fmoc-Lys(Mtt)-OH и смолы НМР-АМ. Смолу промывают 500 мл ДХМ три раза, 56,2 г (90 ммоль) Fmoc-Lys(Mtt)-OH и 11,4 г (90 ммоль) DIC растворяют в 1 л ДХМ, затем добавляют в набухшую смолу НМР-АМ, затем добавляют 366 мг (3 ммоль) DMAP и дают возможность взаимодействовать в течение 24 ч.
2. Отмывка смолы
После взаимодействия смолу дважды промывают поочередно ДМФ и IPA, промывают DMF 3 раза.
3. Кэпирование гидроксила
15,3 г (150 ммоль) уксусного ангидрида и 19,4 г (150 ммоль) DIEA растворяют в 1 л ДМФ и добавляют к смоле для взаимодействия в течение 10 мин.
4. Отмывка смолы
5 Смолу промывают 1 л 50% МеОН/ДМФ, дважды 50% ДХМ/ДМФ, промывают ДХМ три раза, промывают абсолютным этанолом три раза, затем высушивают в вакууме с получением смолы Fmoc-Lys(Mtt)-HMP-AM.
(3) Анализ загрузки смолы Fmoc-Lys(Mtt)-HMP-AM
Точное количество 5-10 мг смолы помещают в 1 мл 20% раствора гексагидропиридин/ДМФ, перемешивают в течение 20 мин, затем 50 мкл супернатанта отбирают пипеткой и разбавляют в 2,5 мл ДМФ;
Чистые контрольные образцы: 50 раствора гексагидропиридин/ДМФ отбирают пипеткой и разбавляют в 2,5 мл ДМФ. Степень замещения вычисляют, как описано ниже:
Sub=(A×51)/(7,8×m)
где А представляет собой значение поглощения УФ при 301 нм; m представляет собой количество смолы, единицей является мг.
2. Высушивание и набухание смолы твердофазного синтеза
50 г (20 ммоль) смолы Fmoc-Gly-HMP-AM (0,4 ммоль/г), высушенной в течение 24 ч в вакууме, помещают в 2 л барботер, затем добавляют 500 мл N,N-диметилформамида (ДМФ) для набухания смолы в течение 30 мин, затем раствор ДМФ отсасывают.
3. Способ твердофазного синтеза части пептидной цепи лираглутида
1. Конденсация Fmoc-Arg(Pbf)-OH
50 ммоль Fmoc-Arg(Pbf)-OH растворяют в 125 мл 0,4 М 1-5 гидроксибензотриазол (HOBt)/ДМФ, затем добавляют 125 мл 0,4 М N,N'-диизопропилкарбодиимида (DIC)/ДХМ для активации и подвергают взаимодействию в течение 10 мин при комнатной температуре; этот раствор добавляют к смоле, подвергают взаимодействию в атмосфере N2 при комнатной температуре, и нингидриновое тестирование используют для обнаружения и контроля степени взаимодействия. После взаимодействия реакционный раствор отсасывают; смолу промывают ДМФ, IPA и ДМФ поочередно.
2. Удлинение пептидной цепи
Пептид-предшественник лираглутида синтезируют в соответствии с 5 последовательностью пептидной цепи лираглутида с N-амидо (N-конца) к карбокси-концу (C-концу) (His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Arg-Gly), где количества аминокислот и реагентов конденсации являются такими же, как для Fmoc-Arg(Pbf)-OH, защищенные аминокислоты представляют собой Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Leu-OH, Fmoc-Trp(Boc)-OH, Fmoc-Ala-OH, Fmoc-Ile-OH, Fmoc-Phe-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Lys(Mtt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Ser(tBu)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Thr(tBu)-OH, Fmoc-His(Trt)-OH соответственно, и реакции конденсации и удаления защиты повторяют.
3. Удаление защитной группы Mtt пептида-предшественника лираглутида
Смолу дважды промывают 200 мл ДХМ, защитную группу Mtt удаляют дважды путем добавления 1200 мл 1% ТФУ/ДХМ (ТФУ находится примерно в 8-кратном избытке) для взаимодействия в течение 1 ч, смолу промывают поочередно 200 мл 5% N,N-диизопропилэтиламин (DIEA)/ДМФ и ДМФ три раза, промывают 3 раза ДМФ.
4. Модификация пептида-предшественника лираглутида пальмитиновой кислотой
50 ммоль Fmoc-Glu-OtBu растворяют в 125 мл 0,4 М 1-гидроксибензотриазола (HOBt)/ДМФ, затем добавляют 125 мл 0,4 М N,N'-диизопропилкарбодиимида (DIC)/ДХМ для активации и подвергают взаимодействию в течение 10 мин при комнатной температуре; этот раствор добавляют к смоле, подвергают взаимодействию в атмосфере N2 при комнатной температуре и нингидрин используют для обнаружения и контроля степени взаимодействия. После взаимодействия реакционный раствор отсасывают; смолу промывают ДМФ, IPA и ДМФ поочередно.
1 л 20% PIP/ДМФ добавляют для удаления защитной группы Fmoc в течение 5 мин, затем отсасывают, затем добавляют 1 л 20% PIP/ДМФ для удаления защитной группы Fmoc в течение 20 мин, затем отсасывают, смолу промывают ДМФ четыре раза.
50 ммоль пальмитиновой кислоты и 50 ммоль 3-(диэтоксифосфорилокси)-1,2,3-фентриазин-4-кетона (DEPBT) растворяют в 400 мл ДМФ, затем добавляют 100 ммоль DIEA для взаимодействия в течение 3 мин при перемешивании при комнатной температуре, этот раствор добавляют к смоле, подвергают взаимодействию в водяной бане на 37°С в атмосфере N2 в течение 2 ч. После взаимодействия реакционный раствор отсасывают, смолу промывают ДМФ, изопропиловым спиртом (IPA) и ДМФ поочередно.
4. Последующая обработка смолы с пептидом лираглутидом
Смолу с пептидом лираглутидом, полученную на стадии 2, промывают ДМФ, IPA и ДМФ поочередно, промывают три раза ДХМ, дважды промывают абсолютным эфиром, затем высушивают в вакууме, наконец, получают смолу с пептидом лираглутидом.
5. Получение сырого пептида лираглутида
Высушенную смолу с пептидом лираглутидом подвергают взаимодействию со свежим лизатом трифторуксусная кислота (ТФУ): триизопропилсилан (TIS): вода = 95:2,5:2,5 (об/об, и суммарно 10 мл лизата на грамм сухой смолы) в течение 4 ч при комнатной температуре. После взаимодействия реакционный раствор фильтруют, смолу дважды промывают ТФУ, фильтрат собирают и объединяют, концентрируют до 1/3 исходного объема посредством роторного выпаривания, лираглутид осаждают холодным абсолютным эфиром, после центрифугирования и высушивания в вакууме получают белый сырой лираглутид.
6. Получение лираглутида жидкостной хроматографией с обращенной фазой
10 г сырого лираглутида растворяют в определенном количестве раствора NH4HCO3, фильтрованного 0,45 мкм мембранным фильтром, затем очищают высокоэффективной жидкостной хроматографией с обращенной фазой (ОФ-ВЭЖХ), где подвижная фаза представляет собой А 0,1% ТФУ/H2O, В 0,1% ТФУ/ацетонитрил, где колонка представляет собой колонку Denali С-18 (диаметр частиц 8,3 мкм, 5×30 см), температура колонки составляет 45°С, длина волны обнаружения составляет 220 нм, скорость тока составляет 120 мл/мин. Пики продукта собирают, концентрируют при пониженном давлении, чтобы удалить большую часть ацетонитрила, затем путем лиофилизации получают 2,25 г продукта лираглутида, чистота которого составляет 98%, выход составляет 12,5%.
Экспериментальный пример 1: Тестирование агонистической активности соединений на рецепторе глюкагоноподобного пептида-1 (GLP1R)
GLP1R представляет собой рецептор, сопряженный с G белком, связывание которого с агонистами приведет в результате к повышению внутриклеточной концентрации цАМФ. В настоящем эксперименте GLP1R и плазмиду с геном-репортером люциферазы, регулируемым элементами цАМФ ответа, котрансфицируют в клетки НЕК293. Когда соединение связывается с рецептором и активирует рецепторы, экспрессия люциферазы повысится. Состояние активации соединения на GLP1R можно исследовать путем тестирования активности люциферазы.
Экспериментальные методики:
1. Клетки НЕК293, стабильно трансфицированные GLP1R и плазмидой pCRE-Luc, высевают в 96-луночный планшет в количестве 40000 клеток/лунка/100 мкл и инкубируют при 37°С в течение 24 ч.
2. Соединения или положительные контрольные лекарства, имеющие определенный градиент концентрации (3 лунки на концентрацию), добавляют и инкубируют при 37°С в течение 5 ч. Отрицательным контролем является растворитель ДМСО.
3. 50 мкл культуральной среды отбирают из каждой лунки и добавляют 50 мкл субстрата люциферазы, а затем перемешивают на вортексе в течение 10 мин.
4. 80 мкл реакционного раствора отбирают и переносят в белый 96-луночный планшет, затем определяют на считывающем устройстве для микропланшетов Invision (инструмент измерения ферментативного мечения).
Результаты эксперимента: по сравнению с положительным контрольным соединением лираглутидом активность соединения HS-20001 по изобретению приблизительно равна активности положительного соединения, но HS-20002-20008 проявляют значительно лучшую агонистическую активность.
Экспериментальный пример 2. Тест активности in vivo
Мышей db/db с диабетом типа 2 делят на шесть групп на основе случайного уровня глюкозы в крови и массы тела (8 на группу). Физиологический раствор, 3 или 10 мкг/кг новых соединений серии HS (лираглутид, 20001, 20002, 20003, 20004, 2005, 2006, 2007, 2008) вводят путем однократной подкожной инъекции. Случайный уровень глюкозы в крови определяют в различные моменты времени после введения.
Субъектами животных, используемыми в данном эксперименте, являются мыши db/db, которые были получены корпорацией США, называемой Jackson, и содержатся и размножаются Шанхайским институтом фармакологии Китайской Академии Наук, сертификатом соответствия которых является: SCXK (HU) 2008-0017, масса тела: 35-50 г; пол: самец 85, самка 86, содержатся в виварии степени SPF (свободном от специфической патогенной микрофлоры); Температура: 22-24°С; Влажность: 45-80%; Освещенность: 150-300LX, чередование день-ночь 12 ч.
Тестируемыми лекарствами в эксперименте являются HS-20001, HS-20002, HS-20003, HS-20004, HS-20005, HS-20006, HS-20007, HS-20008, лираглутид (разработанный фирмой Novo Nordisk, в качестве положительного контроля).
Способ приготовления: 1 флакон соединения (2 мг/флакон) растворяют дважды дистиллированной водой с получением бесцветного и прозрачного раствора, концентрация которого составляет 2 мг/мл, затем этот раствор разводят до 0,6 мкг/мл и 2 мкг/мл физиологическим раствором (хлорид натрия для инъекций, Double-Crane Pharmaceutical Co., Ltd. Anhui, номер партии: 0807286С). Глюкометр "ACCU-CHEK® Advantage" от фирмы Roche используют для определения глюкозы в крови.
Установка дозы и группа
Подопытная группа 1:
Контрольная группа: физиологический раствор
Группа лираглутида: 3 мкг/кг
Группа HS-20001: 3 мкг/кг
Группа HS-20002: 3 мкг/кг
Группа HS-20003: 3 мкг/кг
Группа HS-20004: 3 мкг/кг
Группа HS-20005: 3 мкг/кг
Группа HS-20006: 3 мкг/кг
Группа HS-20007: 3 мкг/кг
Группа HS-20008: 3 мкг/кг
Подопытная группа 2:
Контрольная группа: физиологический раствор
Группа лираглутида: 10 мкг/кг
Группа HS-20001: 10 мкг/кг
Группа HS-20002: 10 мкг/кг
Группа HS-20003: 10 мкг/кг
Группа HS-20004: 10 мкг/кг
Группа HS-20005: 10 мкг/кг
Группа HS-20006: 10 мкг/кг
Группа HS-20007: 10 мкг/кг
Группа HS-20008: 10 мкг/кг
Путь и объем введения: Однократная подкожная инъекционная доза, объем дозы составляет 5 мл/кг.
Способ теста
Скрининг, группирование и введение мышей db/db с диабетом типа 2
Подопытная группа 1:
171 мышь db/db (самцы 85, самки 86) выращивают в отдельных клетках после отлучения от матери, кормят рационом с высоким содержанием жиров. Случайный уровень глюкозы в крови и уровень глюкозы в крови натощак измеряют после того, как мыши db/db достигают возраста семь недель. 80 мышей db/db, которые заболели, собирают и делят на 10 групп в соответствии со случайным уровнем глюкозы в крови, уровнем глюкозы в крови натощак и массой тела, как описано ниже: группа контроля модели, группа лираглутида - 3 мкг/кг, группа HS-20001 - 3 мкг/кг, группа HS-20002 - 3 мкг/кг, группа HS-20003 - 3 мкг/кг, группа HS-20004 - 3 мкг/кг, группа HS-20005 - 3 мкг/кг, группа HS-20006 - 3 мкг/кг, группа HS-20007 - 3 мкг/кг и группа HS-20008 - 3 мкг/кг.
Подопытная группа 2:
Измеряют случайные уровни глюкозы в крови мышей db/db. 80 мышей db/db, которые заболели, собирают и делят на 10 групп в соответствии со случайными уровнями глюкозы в крови, уровнями глюкозы в крови натощак и массой тела, как описано ниже: группа контроля модели, группа лираглутида - 10 мкг/кг, группа HS-20001 - 10 мкг/кг, группа HS-20002 - 10 мкг/кг, группа HS-20003 - 10 мкг/кг, группа HS-20004 - 10 мкг/кг, группа HS-20005 - 10 мкг/кг, группа HS-20006 - 10 мкг/кг, группа HS-20007 - 10 мкг/кг и группа HS-20008 - 10 мкг/кг.
Каждая группа содержит 8 мышей, половину самцов и половину самок. Животным каждой группы вводят тестируемые соединения или контрольный растворитель соответственно посредством однократной подкожной инъекции. Случайный уровень глюкозы в крови определяют через 1 ч, 2 ч, 4 ч, 8 ч и 24 ч после введения и вычисляют процент снижения глюкозы в крови:
Скорость снижения глюкозы в крови = (глюкоза в крови контрольной группы - глюкоза в крови группы обработки)/глюкоза в крови контрольной группы * 100%.
Экспериментальные результаты
Тест 1: Эффект новых соединений в низкой дозе, вводимых путем однократной дозы, на случайный уровень глюкозы в крови мышей db/db.
Результаты можно видеть в таблице 2 и в таблице 3. У мышей db/db, которым вводят 3 мкг/кг HS-20002, 20004, 20005, 20006, 20007 или 20008 посредством однократной подкожной инъекции, через час значения случайного уровня глюкозы в крови значимо снижаются по сравнению с мышами контрольной группы (Р<0,05), проценты снижения составляют 24,51%, 15,00%, 14,00%, 14,25%, 13,98% и 13,90% соответственно; через 2 ч и через 4 ч после введения значения случайного уровня глюкозы в крови поддерживаются на более низком уровне и имеют значимое отличие от таковых контрольной группы (Р<0,05); через 8 ч после введения значения случайного уровня глюкозы в крови не имеют значимого отличия от таковых контрольной группы. У мышей, которым вводят 3 мкг/кг HS-20003 посредством однократной подкожной инъекции, через час значения случайного уровня глюкозы в крови значимо снижаются по сравнению с мышами контрольной группы (Р<0,05) вплоть до 17,33%, через 2 ч, 4 ч и 8 ч после введения значения случайного уровня глюкозы в крови не проявляют значимого отличия от таковых контрольной группы. После введения 3 мкг/кг HS-20001 мышам db/db посредством однократной подкожной инъекции значения случайного уровня глюкозы в крови несколько снижаются по сравнению с мышами контрольной группы, но без значимого различия. Значения случайного уровня глюкозы в крови группы мышей, которым вводили лираглутид, не имеют значимого снижения.
Тест 2: Эффект новых соединений в высокой дозе, вводимых посредством однократной дозы, на случайный уровень глюкозы в крови мышей db/db
Результаты можно видеть в таблице 4 и в таблице 5. У мышей db/db, которым вводили 10 мкг/кг HS-20002 посредством однократной подкожной инъекции, через час значения случайного уровня глюкозы в крови значимо снижаются по сравнению с таковыми контрольной группы (Р<0,01); через 2 ч, 4 ч и 8 ч после введения значения случайного уровня глюкозы в крови поддерживаются на более низком уровне, где значения через 4 ч после введения наиболее очевидны, где процент их снижения составляет вплоть до 40,67%, и значимо отличаются от таковых контрольной группы (Р<0,001) до 24 ч после введения значения случайного уровня глюкозы в крови все еще значимо ниже, чем значения контрольной группы. У мышей db/db, которым вводили 10 мкг/кг HS-20003 посредством однократной подкожной инъекции, через час значения случайного уровня глюкозы в крови значимо снижаются по сравнению с таковыми контрольной группы (Р<0,01), и снижение составляет вплоть до 23,62%; через 2 ч, 4 ч и 8 ч после введения значения случайного уровня глюкозы в крови все еще поддерживаются на более низком уровне, через 24 ч после введения нет значимого отличия по сравнению с контрольной группой. У мышей db/db, которым вводили 10 мкг/кг HS-20001 посредством однократной подкожной инъекции, через 2 ч значения случайного уровня глюкозы в крови значимо снижаются по сравнению с таковыми контрольной группы, через 4 ч и 8 ч после введения значения случайного уровня глюкозы в крови все еще поддерживаются на более низком уровне, через 24 ч после введения значения случайного уровня глюкозы в крови не проявляют значимого отличия по сравнению с контрольной группой. HS-20002, HS-20004, HS-20005, HS-20006, HS-20007 или HS-20008 вводят мышам посредством однократной подкожной инъекции, и значения случайного уровня глюкозы в крови снижаются сразу и значимо, процент снижения составляет вплоть до 36,20%, через 2 ч, через 4 ч и 8 ч после введения значения случайного уровня глюкозы в крови все еще поддерживаются на более низком уровне, через 24 ч после введения нет значимого отличия по сравнению с контрольной группой. Значения случайного уровня глюкозы в крови мышей группы, которой вводили лираглутид, не имеют значимого снижения.
Вывод из тестов:
Случайный уровень глюкозы в крови мышей db/db, которым вводили серию новых соединений по изобретению посредством однократной подкожной инъекции, может быть значимо снижен; случайный уровень глюкозы в крови может очевидно снижаться соединениями HS-20002, HS-20003, HS-20004, HS-20005, HS-20006, HS-20007 и HS-20008 в дозе 3 мкг/кг. Причем HS-20002 и HS-20004 проявляют значительно лучший эффект на снижение случайного уровня глюкозы в крови, продолжительность гипогликемического эффекта после однократной подкожной инъекции дозозависима, продолжительность эффекта HS-20002 и HS-20004 на снижение случайного уровня глюкозы в крови в дозе 3 мкг/кг составляет более 4 ч, и продолжительность эффекта HS-20001, HS-20002, HS-20003, HS-20004, HS-20005, HS-20006, HS-20007 и HS-20008 на снижение случайного уровня глюкозы в крови в дозе 10 мкг/кг составляет более 8 ч.
| название | год | авторы | номер документа |
|---|---|---|---|
| Сайт-специфически монопегилированные аналоги эксендина и способ их получения | 2012 |
|
RU2625015C2 |
| ДВОЙНЫЕ АГОНИСТЫ GLP1/GIP ИЛИ ТРОЙНЫЕ АГОНИСТЫ GLP1/GIP/ГЛЮКАГОНА | 2013 |
|
RU2652783C2 |
| МУЛЬТИРЕЦЕПТОРНЫЙ АГОНИСТ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2816492C2 |
| НОВЫЕ КОМПОЗИЦИИ АГОНИСТОВ ЭКСЕНДИНА И СПОСОБЫ ИХ ВВЕДЕНИЯ | 2000 |
|
RU2242244C2 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
| МУЛЬТИАГОНИСТЫ И ИХ ПРИМЕНЕНИЕ | 2022 |
|
RU2833609C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЬЮГАТ ГИАЛУРОНОВОЙ КИСЛОТЫ, ЛИНКЕРА И ДВОЙНОГО АГОНИСТА GLP-1/ГЛЮКАГОНА | 2016 |
|
RU2719482C2 |
| ПРИМЕНЕНИЕ ДОЛГОДЕЙСТВУЮЩИХ ПЕПТИДОВ GLP-1 | 2013 |
|
RU2657573C2 |
| НОВЫЙ БЕЛКОВЫЙ КОНЪЮГАТ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ НЕАЛКОГОЛЬНОГО СТЕАТОГЕПАТИТА, ОЖИРЕНИЯ И ДИАБЕТА | 2021 |
|
RU2822001C1 |
| ЛИПОФИЛЬНЫЕ ПРОИЗВОДНЫЕ ПЕПТИДНЫХ ГОРМОНОВ | 1996 |
|
RU2171261C2 |








































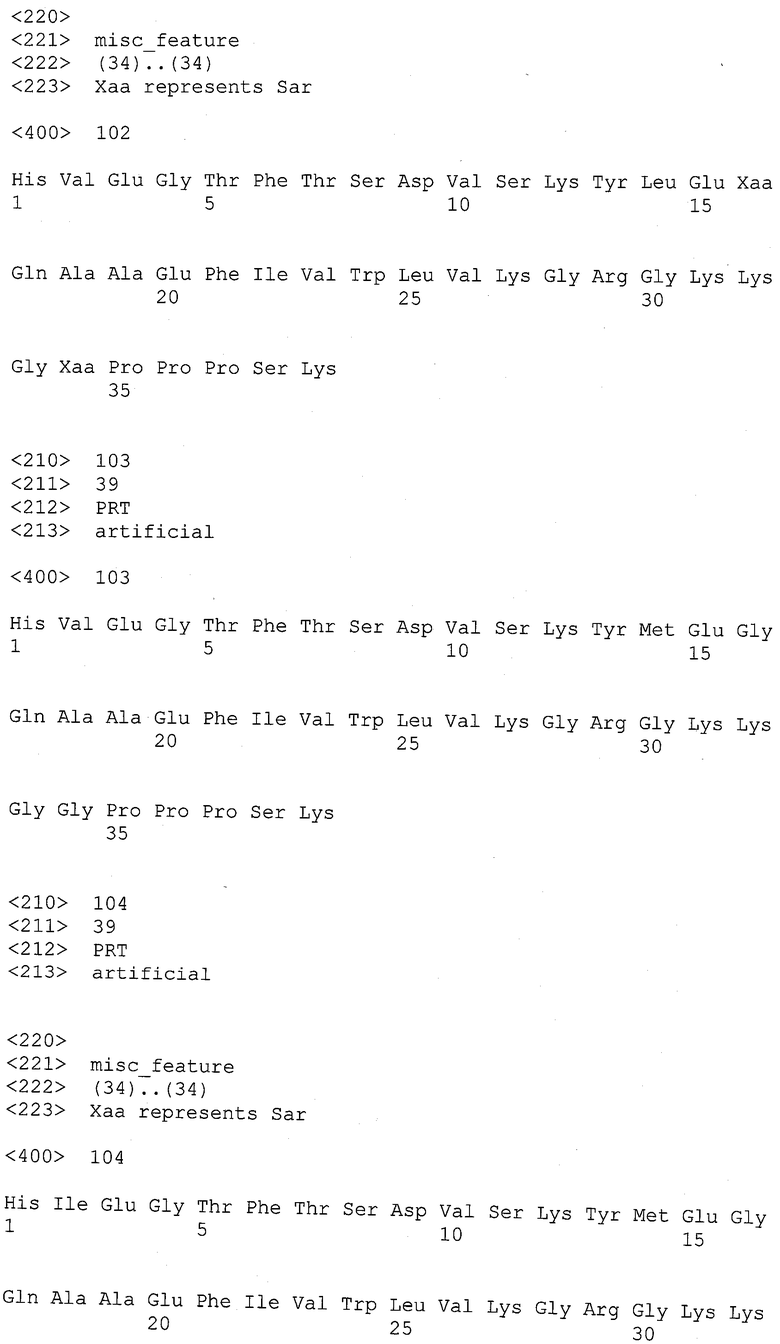







Изобретение относится к области биотехнологии, конкретно к получению производных аналогов GLP-1, и может быть использовано в медицине для лечения инсулиннезависимого сахарного диабета, инсулинозависимого сахарного диабета или ожирения. Получают аминокислотные последовательности аналогов GLP-1, содержащие липофильный заместитель формулы R1(CH2)n-CO-, в котором R1 представляет собой СН3- или НООС-, n представляет собой целое число от 8 до 25. При этом липофильный заместитель связан амидной связью с ε аминогруппой или α аминогруппой Lys на С-конце аналога GLP-1. Изобретение позволяет сохранить активность GLP-1 человека и увеличить период полувыведения in vivo по сравнению с GLP-1 человека. 3 н. и 9 з.п. ф-лы, 6 табл., 2 пр.
1. Производное аналога GLP-1 или его фармацевтически приемлемая соль, обладающие функцией GLP-1 человека, где аминокислотная последовательность аналога GLP-1 выбрана из:
His-(D)-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Nle-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-lle-Glu-Trp-Leu-Lys-Gln-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-Lys (SEQ ID No. 1);
His-(D)-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Glu-Glu-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Arg-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-Lys (SEQ ID No. 3);
His-Aib-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Glu-Glu-Ala-Ala-Lys-Glu-Phe-lle-Ala-Trp-Leu-Val-Arg-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-Lys (SEQ ID No. 4);
His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Glu-Phe-lle-Val-Trp-Leu-Val-Lys-Gly-Arg-Gly-Lys-Lys-Gly-Gly-Pro-Pro-Pro-Ser-Lys (SEQ ID No. 5);
His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Glu-Phe-lle-Val-Trp-Leu-Val-Lys-Gly-Arg-Gly-Lys-Lys-Gly-Sar-Pro-Pro-Pro-Ser-Lys (SEQ ID No. 6);
His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Glu-Gln-Ala-Ala-Glu-Phe-lle-Val-Trp-Leu-Val-Lys-Gly-Arg-Gly-Lys-Lys-Gly-Gly-Pro-Pro-Pro-Ser-Lys (SEQ ID No. 7); и
His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Glu-Gln-Ala-Ala-Glu-Phe-lle-Val-Trp-Leu-Val-Lys-Gly-Arg-Gly-Lys-Lys-Gly-Sar-Pro-Pro-Pro-Ser-Lys (SEQ ID No. 8),
где производное аналога GLP-1 содержит липофильный заместитель формулы R1(CH2)n-CO-, в котором R1 представляет собой СН3- или НООС-, n представляет собой целое число от 8 до 25, и где липофильный заместитель связан амидной связью с ε аминогруппой или α аминогруппой Lys на С-конце аналога GLP-1.
2. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 1, где липофильный заместитель формулы R1(СН2)n-СО- и аминогруппа аминокислотных остатков аналога GLP-1 связаны амидной связью, R1 представляет собой СН3- или НООС-, и n представляет собой целое число от 8 до 25.
3. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 2, где липофильный заместитель формулы R1(CH2)n-CO- и ε аминогруппа Lys на С-конце аналога GLP-1 связаны амидной связью, R1 представляет собой СН3- или НООС-, и n представляет собой целое число от 8 до 25.
4. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 2, где липофильный заместитель формулы R1(CH2)n-CO- и α аминогруппа Lys на С-конце аналога GLP-1 связаны амидной связью, R1 представляет собой СН3- или НООС-, и n представляет собой целое число от 8 до 25.
5. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 4, где R1 представляет собой СН3-, и n представляет собой целое число, выбранное из 8, 10, 12, 14, 16, 18, 20 и 22.
6. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 5, где R1 представляет собой СН3-, и n равно 14.
7. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 4, где R1 представляет собой НООС-, и n представляет собой целое число, выбранное из 14, 16, 18, 20 и 22.
8. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 7, где R1 представляет собой НООС-, и n равно 14.
9. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 1, где R1 представляет собой СН3-, и n равно 14.
10. Производное аналога GLP-1 или его фармацевтически приемлемая соль по п. 1, где аминокислотная последовательность аналога GLP-1 представляет собой SEQ ID No. 4.
11. Фармацевтическая композиция для лечения инсулиннезависимого сахарного диабета, инсулинозависимого сахарного диабета или ожирения, содержащая:
(1) терапевтически эффективное количество производных аналога GLP-1 или их фармацевтически приемлемых солей по любому из пп. 1-10, и
(2) фармацевтически приемлемый эксципиент или носитель.
12. Применение производного аналога GLP-1 или его фармацевтически приемлемой соли по любому из пп. 1-10 или фармацевтической композиции по п. 11 для получения лекарственного средства для лечения инсулиннезависимого сахарного диабета, инсулинозависимого сахарного диабета или ожирения.
| US 2003199672 A1, 23.10.2003 | |||
| Рамный фильтрпресс | 1955 |
|
SU104156A1 |
| Щит для прокладки тоннелей | 1934 |
|
SU41548A1 |
| US 5424286, 13.06.1995 | |||
| WO 2008058461 A1, 22.05.2008 | |||
| RU 2007112115 A1, 10.10.2008 | |||

