Настоящее изобретение относится к комплексам, содержащим бициклополиазамакроциклофосфоновую кислоту и ионы металлов, к их конъюгатам, и к их использованию в виде радиоактивных фармацевтических препаратов, в частности, для лечения и/или диагностики рака. В настоящей заявке описываются способы получения указанных комплексов и конъюгатов.
Многие исследования, ставившие перед собой диагностические и терапевтические цели, посвящены доставке радионуклидов к тканям и органам-мишеням. Для этого использовали различные молекулы, которые могли бы доставить активный компонент к нужному очагу и при этом обеспечить его стабильность, по крайней мере, до того, как эта система доставки сможет достигнуть нужный очаг. В этих целях были использованы галогенированные (например 131I и 88Br) органические молекулы. Так, например, иодированный гиппуран был использован для изучения функции почек (J. Nucl. Meo., 23, 377 - 380 (1982)). Для обнаружения и терапии рака предлагалось также использовать меченное 131I антитело, см., например, Cancer Res., 44, 5744 - 5751 (1984).
Металлические радионуклиды могут иметь различные ядерные свойства и структуру. Так, например, 201Tl, 67Cu, 99mTc, 90Y и различные изотопы In и Ga являются лишь немногими примерами радиоизотопов, которые использовались для диагностической визуализации и/или терапии. Из этих металлов, наибольшее распространение в качестве радиоактивных фармацевтических средств получила 99mTc-химия. Например, Tc-дифосфонаты используются для визуализации костной системы (см. , Subramanian и др., Radiology 149, 823 - 828, 1983). Loberg и др. [T.Nucl.Med., 16, 533, (1975) использовали для исследований функции печени липофильные 99mTc-комплексы, в которых Tc присутствует в состоянии окисления +3, а полный заряд комплекса иминодиуксусной кислоты составляет -1, Deutsch и др. [Science 213, 85 (1981)] смогли получить Tc-комплексы с As и P, содержащие лиганды, локализирующиеся в сердце. Эти соединения содержали Tc(lll)-сердцевину с полным зарядом комплекса + 1. Volkert и др. (Int'I. J. Appl. Rad. Isotopes 35, 467 - 470, 1974) успешно осуществили доставку Tc(lll) к тканям мозга.
Однако, поскольку 99mTc является плохим гамма-излучателем, то его использование ограничивается лишь диагностикой. Поэтому, необходимо получить такие комплексы и/или конъюгаты излучающих радиоизотопов, которые могли бы быть использованы в терапии. Deutsch и др. [Corina Int'l., Veronai and Raven Press, pp. 733 - 740, 1990) использовали комбинацию 186Re и дифосфоната для лечения опухолей кости. Кроме того, Simon и др. (патент США 4 898 724) описывают использование 153Sm и других редкоземельных радионуклидов в сочетании с аминофосфоновыми кислотами для лечения болей в костях и костных опухолей.
Метастазы в кости является распространенным и часто катастрофическим осложнением ракового заболевания. Боли, патологические переломы, частые неврологические расстройства и вынужденная неподвижность, вызываемые метастатическими поражениями, значительно ухудшают качество жизни ракового больного. Число пациентов, подвергающихся метастатическим заболеваниям, достаточно велико, поскольку приблизительно у 50% из всех пациентов, страдающих раком молочной железы, легких или предстательной железы, в конце концов, развиваются метастазы в костях. Метастазы костей также наблюдаются у пациентов с карциномой почек, желчного пузыря, щитовидной железы, шейки матки и другими опухолями, однако, в целом, метастазы костей развиваются у менее чем 20% пациентов, страдающих указанными видами опухолей. Метастазы костей, вызванные раковым заболеванием, редко угрожают жизни больного, и часто пациенты живут еще несколько лет после установления у них опухолевых поражений костей. На ранней стадии, непосредственная обработка очагов поражения для облегчения боли уменьшает потребность в наркотических средствах и повышает способность к передвижению. Поэтому, имеются все основания надеяться, что некоторые виды раковых заболеваний могут быть извлечены.
Использование радионуклидов для лечения метастазов в кости вследствие ракового заболевания восходит к началу 1950-х годов. Было предложено вводить нуклиды, излучающие радиоактивные частицы и изготовленные в соответствующей форме, для лечения кальцинозных поражений. Желательно, чтобы такие нуклиды концентрировались в области поражения кости с минимальным воздействием на мягкие ткани и нормальную кость. Было предложено использовать радиоактивные фосфорные (P-32 и P-33) соединения, однако, ядерные свойства и биолокализация этих соединений ограничивают их применение (E. Keplan и др., J. Nucl. Med. 1 (1) 1, 1960; патент США 3 965 254).
Другие попытки лечения рака кости были предприняты с использованием фосфорных соединений, содержащих группу бора. Эти соединения, которые вводили в организм внутривенно, аккумулировались в костной системе. После чего целевой участок облучали нейтронами в целях активации бора и обеспечения этого участка терапевтической дозой ионизирующего излучения (патент США 4399817).
Было также предложено использование Re-186-комплексов с дифосфонатом (L. Mathieu и др. , Int., J. Applied Pad. & Isotopes, 30, 725 -727 (1979); J. Weinenger A. R. Ketring и др. J. Nucl. Med., 245(5), P125, 1983). Однако, требования, предъявляемые к получению и очистке этих комплексов, снижают их ценность и диапазон применения.
Для лечения метастатических поражений кости было также предложено использовать стронций-89. Однако, продолжительный период полураспада (50,4 дня), высокие уровни в крови и низкая эффективность воздействия на пораженный участок по отношению к нормальной кости ограничивают возможности применения этого соединения (N. Firusian, P. Mellin, C. G. Schmidt, J. Urology, 116, 764 (1976); C.G. Schmidt, N. Firusian, Int. J. C1 Pharmacol., 93, 199 - 205, 1974).
Было описано паллиативное лечение метастатических опухолей кости, в котором использовался 1311-меченный α -амино-(3-иодо-4-гидркосибензилиден)дифосфонат M. Eisenhut, J. Nucl. Med., 25 (12), 1356 - 1361, 1984). Использование радиоактивного иода менее желательно, поскольку, как хорошо известно, он обнаруживает тенденцию локализоваться в щитовидной железе. Eisenhut упоминает иод как один из возможных метаболитов указанного соединения.
Использование радионуклидов для лечения опухолевого кальциноза или для ослабления болей обсуждается в опубликованной заявке на Европатент 176288, где описано применение комплексов Sm-153, Gd-159, Ho-166, Lu-177 или Yb-175 с лигандом, таким, как этилендиаминтетрауксусная кислота (ЭДТК) или гидроксиэтилендиаминтриуксусная кислота (ГЭДТК). Макроциклическая система, имеющая 1,4,7,10-тетраазациклододекановую часть, образующую комплекс с Sm-153, Gd-159, Ho-166, Lu-177 или Yb-175, и предназначенная для лечения опухолевого кальциноза и для ослабления боли, обсуждается в патенте США 5 059 412, причем, указанный комплекс является очень стабильным и имеет более низкий заряд, чем комплекс, раскрытый в опубликованной заявке на Европатент 176288.
Как известно, функционализированные хелаты, или бифункциональные координаторы способны ковалентно связываться с антителом, являющимся специфичным для эпитопов раковых или опухолевых клеток или антигенов. Радионуклидные комплексы таких антитело/хелатных конъюгатов могут быть использованы для диагностики и/или терапии в качестве средства для переноса радионуклина к раковой или опухолевой клетке (см., например, Meares и др., Anal Biochem. 142, 68 - 78, 1984; Krejcarek и др. Biochem and Biophys. Res. Comm. 77, 581 - 585, 1977).
Аминокарбоновые кислоты являются хорошо известными хелатообразующими агентами, и в течение многих лет представляют собой объекты научного исследования. Типичными представителями аминокарбоновых кислот являются нитрилотриуксусная кислота (NTA), этилендиаминтетрауксусная кислота EDTA), гидроксиэтилэтилендиаминтриуксусная кислота (HEEDTA), диэтилентриаминопентауксусная кислота (DTPA), транс-1,2-диаминоциклогексантетрауксусная кислота CDTA) и 1,4,7,10-тетраазациклододекантетрауксусная кислота (DOTA). На основе аминокарбоновых кислот было получено множество бифункциональных хелатообразующих агентов. Например, были получены циклический диангидрид DTPA (Hnatowich и др. Science 220, 613 - 615 (1983), патент США 4 479 930) и смешанные карбоксиугольные ангидриды DTPA (Gansow, патенты США 4 454 106 и 4 472 509; Krejcarek и др., Biochem. and Biophys. Res., Comm. 77, 581 - 585, 1977). При объединении ангидридов с белками связывание происходит посредством образования амидной связи с соответствующим удалением четырех из первоначальных пяти карбоксиметильных групп на диэтилентриаминовом (DETA) остове (Hnatowich и др., Int. J. Appl.Isot. 33, 327 - 332, 1982). Кроме того, в патентах США 4432907 и 4352751 раскрываются бифункциональные хелатообразующие агенты, используемые для связывания ионов металла с "органическими частицами, такими, как органические молекулы-мишени или антитела". Как и в вышеуказанных работах, в данном случае, связывание происходит с помощью амидной группы посредством утилизации диангидридов аминотетрауксусной кислоты. Примерами таких ангидридов являются диангидриды EDTA, DCTA, пропилендиаминотетрауксусной кислоты и фенилен 1,2-диаминотетрауксусной кислоты. Недавно, в патенте США 4 647 447 было раскрыто несколько комплексных солей, образованных из аниона комплексообразующей кислоты и предназначенных для использования в целях диагностики. В этой работе описывается конъюгирование посредством карбоксильной группы комплексообразующей кислоты, в результате чего связывание происходит с помощью амидной связи.
В работе Paik и др. J. Radioanal Chem. 57(12), 553 - 564, 1980, раскрывается использование п-нитробензилбромида в реакции с "блокированным" диэтилентриамином (т.е. бис-(2-фталимидоэтил)амином), с последующими процедурами разблокирования и карбоксиметилирования с использованием хлороуксусной кислоты, в результате чего получали N'-п-нитробензилдиэтилентриамин N,N,N'', N''-тетрауксусную кислоту. И также, поскольку связывание осуществляется с помощью атома азота, получали производное тетрауксусной кислоты. Конъюгирование бифункционального хелатообразующего агента и образование хелатного комплекса с индием также обсуждается в данной работе. Eckelman и др. в J. Pharm. Sci 64(4), 704 - 706 (1975) также описывают замещение на атоме азота, осуществляемое с помощью реакции аминов, таких, как этилендиамин или диэтилентриамин, с соответствующим алкилбромидом с последующим карбоксиметилированием. Эти соединения предлагаются в качестве потенциальных радиоактивных фармацевтических визуализирующих средств.
В литературе также хорошо описан и другой класс бифункциональных хелатообразующих средств на основе функциональных групп аминокарбоновых кислот. Так, например, Sundberg, Meares и др. в J. Med. Chem. 17 (12), 1304, 1974, раскрывают бифункциональные аналоги EDTA. Наиболее характерными из этих соединений являются 1-(п-аминофенил)-этилендиаминотетрауксусная кислота и 1-(п-бензолдиазоний)этилендиаминотетрауксусная кислота. Обсуждается конъюгирование с белками посредством пара-заместителя и связывание ионов радиоактивных металлов с хелатообразующей группой. Эти соединения также описаны в Biochem. and Biophys. Res. Comm. 75(1), 149 (1977) и в патентах США 3 994 966 и 4 043 998. Важно отметить, что присоединение ароматической группы к EDTA-структуре осуществляется посредством углерода этилендиаминового остова. В патенте США 4 622 420 описаны оптически активные бифункциональные хелатообразующие агенты на основе EDTA, HEDTA и DTPA. В этих соединениях, алкиленовая группа связывает ароматическую группу (которая содержит функциональную группу, необходимую для связывания с белком) с углеродом полиамина, который содержит хелатообразующую функциональную группу. Указанным соединениям посвящены и другие работы, например, Brechbiel и др., Inorg. Chem. 25, 2772 - 2781 (1986), патент США 4 647 447 и публикация Международного патента N WO 86/06384.
Позже, в патенте США 4 678 667 и Moi и др., Inorg. Chem. 26, 3458 - 3463 (1987) были раскрыты некоторые макроциклические бифункциональные хелатообразующие агенты и использование их хелатных конъюгатов с медью в диагностических или терапевтических целях. Связывание функциональной группы аминокарбоновой кислоты с остатком бифункциональной хелатообразующей молекулы происходит с помощью кольцевого углерода остова циклического полиамина. Так, например, линкер, связанный у одного конца с кольцевым углеродом циклического полиамина, у своего другого конца является также связанным с функциональной группой, способной реагировать с белком.
Другим классом бифункциональных хелатообразующих агентов, также заслуживающим внимания, являются соединения, в которых хелатообразующая часть (т.е. , аминокарбоновая кислота) молекулы связана посредством азота с функциональной группой молекулы, содержащей группу, способную реагировать с белком. Например, в патентной заявке (WO 84/03698, опубликованной 9.27.1984) Mikola и др. раскрывают бифункциональный хелатообразующий агент, полученный с помощью реакции п-нитробензилбромида с DETA с последующей реакцией с бромоуксусной кислотой и с получением аминокарбоновой кислоты. Нитрогруппу восстанавливают до соответствующей аминогруппы, которую затем превращают в изотиоцианатную группу путем реакции с тиофосгеном. Эти соединения являются бифункциональными хелатообразующими агентами, способными образовывать хелатные комплексы с лантанидами, которые могут быть затем конъюгированы с биоорганическими молекулами в целях использования их в качестве диагностических средств. Поскольку связывание линкерной части молекулы осуществляется посредством одного из атомов азота аминокарбоновой кислоты, то одна потенциальная аминокарбоксильная группа теряется для образования хелатного комплекса. Так, например, был получен бифункциональный хелатор на основе DETA, который содержит четыре (а не пять) кислотных групп. В этом отношении, этот класс бифункциональных хелаторов аналогичен классу соединений, где связывание с белком осуществляется посредством амидной группы с последующей потерей карбоксильной хелатирующей группы.
Недавно, Carney, Rogers и Johnson раскрывают (3-rd Int. Conf. on Monoclonal Antibodies For Cancer: Сан-Диего, Калифорния - 2/4 - 6/88), в статьях, озаглавленных "Absence of Intrisically Higher Tissue Uptake from Indium -111 Labeled Antibodies: Coаdministration of Indium-111 and Iodine-125 Labeled B72.3 in a Nude Mouse Model" и "Influence of Chelator Denticity on the Biodistribution of Indium-111 Labeled B72.3 Immunoconjugates in Nude Mice") биораспределение индия-111, образующего комплекс с EDTA- и DTPA-хелатообразующим бифункциональным агентом. Связывание ароматического кольца с EDTA/DTPA-частями осуществляется посредством ацетатметилена. Кроме того, на недавно имевшей место конференции (Florida Conf on Chem. in Biotechnology, April 26-29 (1988), Palm Coast, FL] D.K. Johnson и др., раскрыли бифункциональные производные EDTA и DTPA, где п-изотиоцианатобензильная часть связывается у метиленового атома углерода с одной из карбоксиметильных групп. Ранее, а патентах США 4 088 747 и 4 091 088 (1978) Hunt и др., раскрыли хелатообразующие агенты на основе этилендиаминдиуксусной кислоты (EDDA), где связывание ароматического кольца с частью EDDA осуществляется посредством алкилена или ацетатметилена. Было показано, что эти соединения могут быть использованы в качестве хелатов для исследования функций печени и желчного пузыря. Предпочтительным металлом является технеций-99m. Указывается, что в качестве радионуклидов для визуализации могут быть также использованы Индий-111 и индий-113m.
Известны и другие комплексы, где используются радиочастоты для индуцирования гипертермии (Japanese Kokai Tokkyo Kohc JP 61158931) и для флуоресцентно-иммунной терапии (FIGS) (K. Pettersson и др., Clinical Chem. 29(1) 60 - 64 (1983) и C. Meares и др., Acc. Chem. Res. 17, 202 -209, 1984).
Из вышеуказанного следует, что предпочтительным является комплекс, который не подвержен быстрой диссоциации; который быстро выводится из организма за исключением нужной ткани-мишени и который образует конъюгаты с антителом, предназначенные для использования в нужных целях.
Преимущественно, настоящее изобретение относится к новому типу стабильных комплексов металлов, особенно таких металлов, которые по своему строению относятся к редкоземельным или псевдоредкоземельным металлам. В настоящем изобретении раскрывается использование этих комплексов, и указывается, что изменения их заряда и липофильного характера могут благоприятным образом изменять биораспределение этих комплексов при их введении в организм животного. В объем настоящего изобретения также входят конъюгаты этих комплексов с биологически активным материалом, таким, как антитело. Указанные комплексы и конъюгаты могут входить в состав лекарственных форм вместе с фармацевтически приемлемыми носителями и могут быть введены млекопитающим в целях диагностики и/или терапии.
Настоящее изобретение относится к новым комплексам, содержащим лиганд, который представляет собой соединение бициклополиазамакроциклофосфоновой кислоты формулы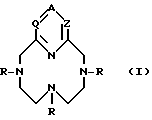
в котором: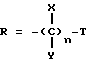
где
X и Y независимо представляют собой H, OH, C1-C3 алкил или COOH;
n является целым числом 1, 2 или 3, при условии, что если n = 2, то сумма X и Y должна быть равна двум или более H; а если n = 3, то сумма X и Y должна быть равна трем или более H;
T представляет собой H, C1-C18-алкил, COOH, OH, SO3H,
или
где
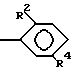
R1 представляет собой OH, C1-C5-алкил, или -O-(C1-C5-алкил);
R4 представляет собой H, NO2, NH2, изотиоцианато, семикарбазидо, тиосемикарбазидо, малеимидо, бромоацетамидо или карбоксил;
R2 представляет собой H или OH, при условии, что если R2 является OH, то R, содержащий R2, должен иметь все X и Y равные H; при условии, что, по крайней мере, один T должен быть P(O)R1OH, и при условии, что если один T является
то один X или Y в R может быть COOH, а все другие X и Y в R должны быть H;
A представляет собой CH, N, C-Br, C-Cl, C-OR3, C-OR8, N+-R5; 
R3 представляет собой H, C1-C5-алкил, бензил, или бензил, замещенный, по крайней мере, одним R4;
R4 определен выше;
R5 представляет собой C1-C16-алкил, бензил, или бензил, замещенный, по крайней мере, одним R4;
R8 представляет собой C1-C16-алкиламино;
X представляет собой Cl, Br, I или H3CCO2;
Q и Z независимо представляют собой CH, N, N+-R5X; C-CH2-OR3 или C-C(O)-R6;
R3 и R5 определены выше;
R6 представляет собой -O-(C1-C3-алкил), OH или NHR7;
R7 представляет собой C1-C5-алкил или биологически активный материал;
X определен выше;
или к их фармацевтически приемлемым солям;
при условии, что
a) если Q, A или Z являются N или N+-R5X, то две другие группы должны быть CH;
b) если A является C-B, C-Cl, C-OR3 или C-OR8, то оба Q и Z должны быть CH;
c) сумма R4, R7 и R8, если они присутствуют, не должна превышать 1; и
d) лишь один из Q или Z может быть C-C(O)-R6, и если один из Q или Z является C-C(O)-R6, то A должно быть CH;
и который образует комплексы с ионом металла 153Sm, 177Lu, 159Gd, 149Pm, 140La, 175Yb, 166Ho, 90Y, 47Sc, 186Re, 188Re, 142Pr, 99mTc, 67Ga, 68Ga, 105Ph, 97R, 111In, 113mIn или 115mIn.
Комплексы формулы (1) могут включать в себя различные ионы металлов, обычно в +3-состоянии, выбранные из самария (153Sm), лютеция (177Lu), гольмия (166Ho), иттрия (90Y), скандия (47Sc), рения (186Re) или (188Re), празеодима (142Pr), технеция (99mTc), галлия (67Ga) или (68Ga), или индия (111In) или (113mIn) или 115mIn; причем из них предпочтительными являются 153Sm, 177Lu, 159Go, 149Pm, 140La, 175Yb, 166Ho, 90Y, 47Sc, 142Pr, 99mTc, 67Ga, 68Ga, 111In или 115mIn; более предпочтительными являются 153Sm, 177Lu, 166Ho, 90Y, 99mTc, 67Ga, 68Ga, 111In, 113mIn или 115mIn; и наиболее предпочтительными являются 153Sm, 177Lu или 166Ho. Комплексы, испускающие гамма-излучение, такие, как 99mTc, 68Ga, 67Ga, 111In, 113mIn или 97Ru, могут быть использованы в качестве диагностических средств. Комплексы, обладающие корпускулярным излучением, такие, как 149Pm, 142Pr, 90Y или 175Yb, могут быть использованы в качестве терапевтических средств. Комплексы, обладающие как гамма-излучением, так и корпускулярным излучением, например такие, как 153Sm, 177Lu, 159Gd, 140La, 166Ho, 47Sc, 186Re, 188Re, 105Rh или 115mIn, могут быть использованы как диагностические, так и терапевтические средства. Полученные таким образом комплексы могут быть использованы как таковые, либо они могут быть ковалентно связаны с антителом или его фрагментом, и использованы в диагностических или терапевтических целях. Такие конъюгаты и комплексы могут быть использованы в качестве диагностические и/или терапевтических средств.
Особенно предпочтительными являются комплексы формулы 1 где: X и Y представляют собой H; n = 1; или A, Q и Z представляют собой CH.
Бифункциональные комплексы формулы (1) являются предпочтительными для получения конъюгатов настоящего изобретения. Такие комплексы содержат лиганд, который должен иметь: один R, в котором T-часть представляет собой: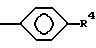
или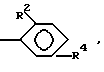
где
R2 и R4 определены выше, в частности, если в двух R не содержится R4, то оба T являются P(O)R1OH, где R1 определен выше, или если в двух R не содержится R4, то один из T является COOH, а другой является P(O)R1OH, где R1 определен выше; при этом, предпочтительной является та часть из вышеуказанных T, где один из X или Y является COOH; а также предпочтительными являются те лиганды, где n = 1 и/или остальные X и Y представляют собой H; или
A представляет собой C-OR3 или C-OR8, где R3 и R8 определены выше, или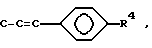
где
R4 определен выше, или A представляет собой CH, а один из Q или Z является CH, а другой является C-C(O)-R6, где R6 определен выше;
а особенно предпочтительными являются те лиганды, где R6 является NHR7, где R7 представляет собой биологически активный материал.
Комплексы или конъюгаты настоящего изобретения могут быть использованы для диагностики или лечения таких заболеваний, как рак.
Могут быть получены такие комплексы и конъюгаты настоящего изобретения, конкретный полный заряд которых будет благоприятным образом влиять на их биораспределение in vivo. Например, если ион металла имеет заряд +3, то могут быть получены следующие варианты:
(A) полный заряд составляет -2 или более, если
в трех R, T представляет собой P(O)R1OH, где R1 является OH, а n = 1; или
в двух R, T представляет собой P(O)R1OH, где R1 является OH, а в третьем R, T представляет собой COOH, и n = 1; или
в двух R, T представляет собой P(O)R1OH, где R1 является OH, в третьем R, T представляет собой P(O)R1OH, где R1 является C1-C5-алкилом, а n = 1; или
в двух R, T представляет собой P(O)R1OH, где R1 является OH, в третьем R, T представляет собой P(O)R1OH, где R1 является -O-(C1-C5-алкилом), а n = 1; или
(B) полный заряд составляет -1, если
в одном R, T представляет собой R(O)R1OH, где R1 является OH, а в двух других R, T представляет собой P(O)R1OH, где R1 является -O-(C1-C5-алкилом), а n = 1; или
в одном R, T представляет собой P(O)R1OH, где R1 является OH, а в двух других R, T представляет собой P(O)R1OH, где R1 представляет собой C1-C5-алкил, а n = 1; или
в одном R, T представляет собой R(O)R1OH, где R1 является OH, а двух других R, T представляет собой COOH, а n = 1; или
(C) полный нулевой заряд, если
в трех R, T представляет собой R(O)R1OH, где R1 является -O-(C1-C5-алкилом), а n = 1; или
в трех R, T представляет собой R(O)R1OH, где R1 является C1-C5-алкилом, а n = 1; или
(D) полный заряд составляет +1, если
один из A, Q или Z является N+-R5X, где R5 и X определены выше; в одном R, T-часть представляет собой R(O)R1OH, где R1 является C1-C5-алкилом или -O-(C1-C5-алкилом); а в двух других R, T-часть представляет собой COOH или P(O)R1OH, где R1 является C1-C5-алкилом или -O-(C1-C5-алкилом); а все X и Y являются H.
Комплексы и конъюгаты могут быть изготовлены в виде фармацевтически приемлемых форм для введения их животному.
Комплексы и конъюгаты настоящего изобретения с другими ионами металлов могут быть также использованы в качестве контрастных веществ, например, для ЯМР-визуализации. Использование таких комплексов и конъюгатов обсуждается в другой одновременно рассматриваемой заявке.
Комплексы настоящего изобретения содержат лиганд формулы (I), где по номенклатуре даны следующие числовые обозначения: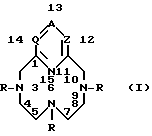
Настоящее изобретение относится к разработке радиоактивных фармацевтических средств, которые включают в себя модифицированный хелатный комплекс, обладающий способностью к специфической доставке радиоактивного фармацевтического агента к нужной ткани. Преимущество указанных радиоактивных фармацевтических средств заключается в повышенной доставке радиоактивного фармацевтического агента в нужные области, основанной на сродстве к данной ткани. Специфичность комплекса формулы (I) может регулироваться путем корректировки полного заряда и липофильных свойств этого комплекса. Полный заряд комплекса может варьироваться в пределах от -3 до +1. Например, для комплекса, имеющего 2 или более групп PO3H2, полный заряд будет высоко отрицательным, и он, по всей вероятности, будет поглощаться костью; если же полный заряд комплекса будет нулевым (т. е. комплекс будет нейтральным), то этот комплекс может обладать способностью переходить гематоэнцефалический барьер и поглощаться нормальным мозгом.
Тканеспецифичность может быть также реализована путем ионного и ковалентного связывания хелата с природной или синтетической молекулой, обладающей специфичностью к данной ткани-мишени. Одним из возможных применений этого способа является использование хелатов, конъюгированных с моноклональными антителами, которые транспортируют радиоактивный хелат к нужной ткани, что позволяет осуществлять соответствующий диагноз и терапию.
Кроме того, рассматриваемые радиоактивные фармацевтические агенты формулы (I), которые имеют нулевой заряд, являются особенно предпочтительными для получения конъюгатов настоящего изобретения, поскольку в данном случае минимизируются нежелательные ионные взаимодействия между хелатом и белком, что способствует сохранению иммунореактивности антитела.
Не претендуя на какую-либо конкретную теорию, можно ожидать, что при получении заряженного комплекса настоящего изобретения (например, возможно, -2 или -3 для кости, -1 для печени, или +1 для сердца), изменение ионного заряда хелата может оказывать влияние на биолокализацию данного комплекса. Так, например, если антитело или другая направляющая часть также является специфичной для той же самой мишени, то, в данном случае, конъюгат уже имеет две части, обеспечивающие специфическую доставку в нужный очаг.
Термины, используемые при определении формулы I и при описании настоящего изобретения, означают следующее. "C1-C3-алкил" "C1-C5-алкил", "C1-C18-алкил" означают прямые и разветвленные алкильные группы. Под термином "животное" следует понимать теплокровное млекопитающее, предпочтительно человека.
Термин "биологически активный материал" означает, например, декстран, пептид, или молекулы, которые обладают специфическим сродством к рецептору, или предпочтительно антитела или их фрагменты.
Термин "антитело" относится к любому поликлональному, моноклональному, химерному антителу или гетероантителу, а предпочтительно, к моноклональному антителу; термин "фрагмент антитела" включает в себя Fab-фрагменты и F(ab')2-фрагменты, и любую часть антитела, обладающую специфичностью по отношению к нужному эпитопу или эпитопам. При использовании термина "конъюгат хелата с радиоактивным металлом и антитела" или просто "конъюгат", понятие "антитело" означает целое антитело и/или его фрагменты, включая их полусинтетические или генетически сконструированные варианты. Предпочтительными антителами являются 1116-NS-19-9 (против карциномы прямой кишки), 1116-NS-3d (против карциноэмбрионального антигена, CEA), 703D4 (против рака легких человека), 704A1 (против рака легких человека), CC49 (анти-TAG-72), CC83 (анти-TAG-72) и B72.3. Клеточные линии гибридом 1116-NS-19-9, 1116-NS-3d, 703D4, 704A1, CC49, CC83 и B72 депонированы Американской коллекцией типовых культур, и имеют номера допуска ATCC HB 8059, ATCC CP1 8019, ATCC HB 8301, ATCC HB 8302, ATCC HB 9459, ATCC HB 9453 и ATCC HB 8108, соответственно.
Термин "комплекс", используемый в настоящем описании, относится к комплексу соединения настоящего изобретения, например, формулы (I), образованному с ионом металла, где, по крайней мере, один атом металла является хелатированным или секвестированным; термин "конъюгат" относится к конъюгату иона металла, ковалентно связанного с антителом или фрагментом антитела. Термины "бифункциональный координатор", "бифункциональный хелатообразующий агент" и "функционализированный хелатор" являются равноценными и относятся к соединениям, которые имеют хелирующую часть, обладающую способностью образовывать хелаты с ионом металла, и часть, ковалентно связанную с этой хелирующей частью и способную ковалентно связываться с антителом или фрагментом антитела.
Бифункциональные хелатообразующие агенты, описанные в настоящей заявке, представленные формулой (1), могут быть использованы для хелатированиия или секвестирования ионов металла с образованием хелатов с ионом металла называемых в настоящем описании "комплексами". Благодаря присутствию функционализирующей группы (представленной R4 в формуле 1), эти комплексы могут быть ковалентно связаны с биологически активными материалами, такими, как декстран, молекулами, обладающими специфическим сродством к рецептору, или предпочтительно антителами или их фрагментами. Таким образом, комплексы, описанные в настоящей заявке, могут быть ковалентно связаны с антителом или его фрагментом, или обладать специфическим сродством к рецептору, и в этом случае, они называются "конъюгатами".
Используемый в настоящем описании термин "фармацевтически приемлемая соль" означает любую соль (или смеси солей) комплекса или конъюгата формулы 1, являющуюся достаточно нетоксичной для использования ее при лечении или диагностики животных, предпочтительно млекопитающих. Таким образом, в соответствии с настоящим изобретением могут быть использованы соли. Типичными солями, образованными с помощью стандартных реакций из органических и неорганических источников, являются, например, соли серной, соляной, фосфорной, уксусной, янтарной, лимонной, молочной, малеиновой, фумаровой, пальмитиновой, холевой, пальмовой, слизевой, глутаминовой, глюконовой, о-камфорной, глутаровой, гликолевой, фталевой, винной, муравьиной, лауриновой, стеариновой, метансульфоновой, бензолсульфоновой, сорбиновой, пикриновой, бензойной, и коричной кислот и других подходящих кислот. Подходящими солями также являются соли, образованные с помощью стандартных реакций из органических и неорганических источников, таких, как ионы аммония, щелочных металлов, щелочноземельных металлов и других подходящих ионов. Особенно предпочтительными являются соли комплексов или конъюгатов формулы (I), которые являются солями калия, натрия или аммония. Могут быть также использованы и смеси вышеуказанных солей.
Лиганды для использования в комплексе или в конъюгате формулы (I) могут быть получены различными способами. Общие принципы таких способов синтеза представлены ниже в виде реакционных схем.
В схеме 1 получают лиганды формулы (I), где X и T = H, n = 1 (но при соответствующем изменении реагента может быть также n = 2 или 3, R имеет T = PO3H2, а Q, A и Z = CH.
В схеме 2 соединения формулы (I), где X и Y = H, n = 1 (но при соответствующем изменении реагента может быть также n = 2 или 3), а R имеет T, равное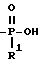
где
R1 = -O-(C1-C5-алкил);
Q, A и Z = CH.
В схеме 3 проиллюстрировано получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента может быть также n = 2 или 3), R имеет T, равное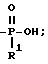
где
R1 = C1-C5-алкил;
Q, A, Z = CH.
Схема 4 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента может быть также n = 2 или 3), R имеет T, равное
где
R1 - -O-(C1-C5-алкил) или C1-C5-алкил;
A = C-Br; Q и Z = CH.
Схема 5 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R имеет T =
где
R1 = -O-(C1-C5-алкил) или C1-C3-алкил;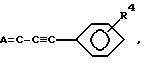
где
R4 = H; NO2, NH2 или SCL;
Q и Z = CH.
Схема 6 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R имеет T, равное
где
R1 = -O-(C1-C5-алкил) или C1-C5-алкил;
A = C-OR8, где R8 = C1-C5-алкиламино;
Q и Z = CH.
Схема 7 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R имеет T, равное
где
R1 = -OH, -O-(C1-C5-алкил) или C1-C5-алкил;
Z = C-C(O)-R6, где R6 = OH;
A и Q = CH.
Схема 8 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R имеет T, равное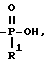
где
R1 = -OH, -O-(C1-C5-алкил) или C1-C5-алкил;
Z = C-CH2-OR3, где R3 = бензил;
Q и A = CH.
Схема 9 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R имеет T, равное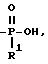
где
R1 = -OR, -O-(C1-C5-алкил) или C1-C5-алкил;
A = N или N+-R5X, где R5 = C1-C16-алкил;
X определен выше;
Q и Z = CH.
Схема 10 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R имеет T, равное
где
R1 = -OH, -O-(C1-C5-алкил) или C1-C5-алкил;
Q = N+-R5X, где R5 = C1-C16-алкил;
X определен выше;
A и Z = CH.
Схема 11 иллюстрирует получение соединений формулы I, в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 1 или 3), R имеет T, равное
где
R1 = -OH, -O-(C1-C5-алкил) или C1-C5-алкил;
Q = N или N+-R5X, где R5 = C1-C16-алкил;
X определен выше;
A и Z = CH.
Схема 12 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R имеет в 3-положении T, равное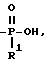
где
R1 = -OH или O-(C1-C5-алкил), а другой R имеет T = COOH, а A, Q и Z = CH.
Схема 13 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R в 3-положении имеет T, равное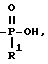
где
R1 = -OH или -O-(C1-C5-алкил),
R в 9-положении имеет T = COOH;
A, Q и Z = CH.
Схема 14 иллюстрирует получение соединений формулы (I), в которых X и Y = H, n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R в 3- и 9-положении имеют T, равное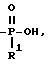
где
R1 = -OH или -O-(C1-C5-алкил);
R в 6-положении имеет T = COOH;
A, Q и Z = CH.
Схема 15 иллюстрирует получение соединений формулы (I), в которых n = 1 (но при соответствующем изменении реагента n может быть также равно 2 или 3), R в 3- и 9-положениях имеют T, равное
где
R1 = -OH или -O-(C1-C5-алкил;
X и Y = H;
R в 6-положении имеет 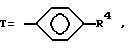
где
R4 = NO2 или NH2;
один из X или Y = H, а другой = COOH;
A, Q и Z = CH.
Схема 16 иллюстрирует получение соединений формулы (I), где n = 1 (но может быть также равно 2 или 3 при соответствующем изменении реагента), R в 3- и 6-положениях имеют T, равное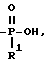
где
R1 = -OH или -O-(C1-C5-алкил);
X и Y = H;
R в 9-положении имеет 
где
R4 = NO2 или NH2;
один из X и Y = H, а другой = COOH;
A, Q и Z = CH.
В приведенных выше схемах, иллюстрирующих общий способ получения соединений, описаны конкретно стадии, которые могут быть использованы для осуществления нужной реакции. Описание этих стадий процесса в общих чертах приводится ниже.
В схеме 1 синтез начинается с галогенирования коммерчески доступного бипиридилового спирта (I) с использованием тионилхлорида. С помощью аналогичных процедур для превращения спирта в электрофильный субстрат, таких, как, например, обработка толуолсульфонилхлоридом, HBr или HCl, также может быть получен аналогичный реактивный продукт, который с успехом может быть использован в последующей реакции замыкания кольца. Процедуры макрокристаллизации много раз описывались в литературе, и нужный тетраазамакроцикл (3) был получен в соответствии с методом Stetter и др. Tetrahedron 37, 767 - 772 (1981). Затем было опубликовано описание более общих процедур, которые дают хороший выход аналогичных макроциклов с использованием более мягких условий [A. D.Sherry и др. J.Org. Chem. 54, 2990 - 2992 (1989). Детосилирование промежуточного макроцикла (из (3) с получением (4)) осуществляли в кислотных условиях и получали хороший выход. Процедуры восстановительного детосилирования также хорошо описаны в литературе, а поэтому могут быть адаптированы к рассматриваемой последовательности реакций. Фосфонометилирование с получением производного трис-аминофосфоновой кислоты (5; РСТМР) проводили в условиях типичной реакции Манниха с использованием фосфорной кислоты и формальдегида.
Помимо производных фосфоновой кислоты, могут быть также получены сложные эфиры фосфоновой кислоты [например, формулы (6)] в органических условиях с использованием спиртов или апротонных растворителей (например, ацетонитрила, бензола, толуола, тетрагидрофурана) и нужного диалкилфосфита в качестве нуклеофильных материалов (см. схему 2). В зависимости от реактивности амина эти реакции могут быть проведены при температуре от около 10 до около 100oC. Кроме того, в аналогичных условиях Манниха могут быть использованы триалкилфосфиты с получением сложного эфира фосфоновой кислоты посредством окисления фосфора (III) в фосфор (V) с одновременным удалением одного моля спирта (реакция Арбузова). Эти реакции могут быть осуществлены в присутствии или отсутствие растворителя. Если в реакциях с диалкил- или триалкилфосфитом в качестве растворителя используются спирты, то во избежание образования альтернативных продуктов в результате переэтерификации желательно использовать спирт, из которого получают соответствующий сложный эфир фосфоновой кислоты. Сложные эфиры такого типа могут быть также получены путем N-алкилирования α--галогенодиалкилфосфонатов в растворителях, таких, как ацетонитрил, хлороформ, диметилформамид, тетрагидрофуран или 1,4-диоксан без добавления или с добавлением ненуклеофильного основания, такого, как карбонат калия при комнатной температуре или выше. Полученное промежуточное соединение эфира перкислоты затем легко гидролизуется в основных условиях (водный гидроксид, pH 8 - 14, 30 - 110oC) с образованием производного полукислоты.
В схеме 3 макроциклические метилфосфиновые кислоты (10 и и 11) получают в условиях, аналогичных условиям, описанным в схеме 2. Используя диэтоксиметилфосфин в качестве нуклеофила, а также параформальдегид, можно проводить конденсацию в растворителях, таких, как тетрагидрофуран, диметилформамид, диоксан, ацетонитрил или спиртовая среда. Полученный сложный эфир фосфиновой кислоты может быть затем гидролизован в кислотных (6 н. HCl, 80 - 100oC) или основных (стехиометрические количества основания, 40 - 100oC) условиях с образованием соответствующей метилфосфоновой кислоты. Альтернативно, для получения производных фосфината, имеющих повышенные липофильные свойства, может быть применен метод, предложенный A.D. Sherry и др. (Inorg. Chem., 1991) с использованием этилфосфоновой кислоты, полученной in situ.
Схема 4 иллюстрирует способ введения дополнительной функциональной группы в пиридиновую единицу тетраазамакроцикла с 12 членами. Так, например, хелидамовая кислота (Sigma Chemical Company; 12) может быть превращена в бис-галогенометиловое производное (13), имеющее соответствующее замещение в 4-положении пиридила. Трансформации, приводящие к образованию этого промежуточного соединения, являются обычными в природе и получение этих соединений описано Takalo и др. [Acta Chemica Scadinavica B 42, 373 -377 (1988)]. Последующая макроциклизация с использованием этого промежуточного соединения (15) может быть осуществлена с помощью стандартной реакции (ДМФ, 100oC) с натрийтритосилированным триамином, или при комнатной температуре с тритосилированным свободным основанием и карбонатом калия, карбонатом натрия, или карбонатом цезия в качестве основания, с получением продуктов, аналогичных ранее описанным. Последующие реакции с получением фосфонат-полукислоты и фосфинатной функциональной группы являются идентичными трансформациям и условиям, описанным в предыдущих схемах.
В схеме 4 описаны 4-галогенопиридил-замещенные макроциклы (16), которые могут быть подвергнуты замещению в 4-положении пиридильной части, как описано в схеме 5. Так, например, металлоорганические Pd(11) комплексы могут быть использованы для катализа реакции сочетания между фенилацетиленом и фенилацетиленовыми производными и пиридильным макроциклом. Обычно эта реакция превращения протекает в безводных условиях, в растворителе, таком, как триэтиламин, и при температуре от около 10 до около 30oC для оптимального выхода. Идентичный продукт может быть также получен с использованием фенилацетилида Cu(1) в безводном пиридине при температуре от около 80 до около 110oC. Кроме того, для осуществления замещения на пиридиновом ядре, могут быть использованы процедуры стандартного анионного алкилирования с использованием, например, натрийалкоксидов в ДМФ или диоксане, при температуре от около 80 до около 100oC, и оснований, таких, как карбонат калия или гидроксид натрия. Макроциклические тетраазамакроциклы (24, 25, 26, 27, 28), дериватизированные таким образом, являются совместимыми с трансформациями, описанными в предыдущих схемах и приводящими к образованию аналогичных фосфонатных хелирующих агентов.
Вариант 4-пиридилового замещения описан в схеме 6, где 4-гидроксипиридильную группу (29) алкилируют с использованием бромоалкилнитрила, получая промежуточный нитрил с эфирной связью (31), который затем вводят в макроциклическую структуру. Процедуру алкилирования этого типа лучше всего осуществлять в безводных условиях в апротонном растворителе, таком, как тетрагидрофуран (ТГФ), и с использованием ненуклеофильного основания, такого, как гидрид натрия или бутиллития при температуре от около 30 до около 80oC. В общих чертах этот метод описан Chaubet и др. для ациклических аналогов (Tetrahedron Lette 31 (40), 5729 - 5732, 1990). Макроциклический нитрил, полученный таким образом, может быть восстановлен до первичного амина (36) с помощью стандартных процедур с последующим блокированием первичного амина с использованием 2-(т-бутоксикарбонилоксиимино)-2-фенилацетонитрила (BOC-ON; 37). Последующая функционализация макроциклических вторичных аминов (38, 39, 40, 41, 42, 43) может быть осуществлена с помощью обсуждавшихся процедур, но с тем лишь требованием, что защитная группа BOC будет затем удалена с использованием трифтороуксусной кислоты, как описано в схеме 6.
Функционализация может быть также осуществлена в 3-положении пиридилового кольца в макроциклической структуре, как показано на схеме 7. Newkome и др. (Tetrahedron 39 (12), 2001 - 2008, 1983) описали синтез этил 2,6-галогенометилникотината (45), который служит в качестве исходного материала в этом процессе. Так, например, трис-тозилированное макроциклическое промежуточное соединение (46) может быть детозилировано в кислотных условиях (HBr/AcOH, 25 - 115oC) с одновременным гидролизом и с получением производного никотиновой кислоты (48), или в результате восстановления сложного эфира в кипящем этаноле, осуществляемого перед детозилированием, может быть получено промежуточное 3-гидроксиметиловое соединение (47). Макроцикл никотиновой кислоты может быть затем замещен по общей схеме для функциализации вторичного амина с получением различных типов фосфонатных хелирующих агентов формулы (1) (49, 50, 51, 52, 53).
В противоположность этому, 3-гидроксиметиловый аналог предпочтительно блокировать до функционализации макроциклических аминов. На схеме 8 показана бензильная (Br) защитная группа, поскольку она должна быть устойчивой к жестким кислотным условиям, требуемым для стадии детосилирования. После соответствующей функционализации вторичных аминов, осуществляемой как описано в предыдущих схемах, бензильную группу удаляют в мягких условиях каталитической гидрогенизации (58).
Макроциклические производные могут быть также получены как описано в схемах 12 -14, где в одной и той же молекуле присутствуют как карбоксилатная, так и фосфонатная хелатирующие функциональные группы. Так, например, с помощью стандартных процедур водного алкилирования с использованием бромоуксусной кислоты могут быть введены различные количества карбоксилатных функциональных групп. После этой стадии, оставшиеся амины могут быть фосфонометилированы с помощью процедур, описанных в предыдущих схемах, с использованием формальдегида и фосфорной кислоты, диалкилфосфонатов или триалкилфосфитов.
Схемы 15 и 16 иллюстрируют метод синтеза, в котором в одно из положений макроциклического азота вводят ароматический нитробензильный заместитель. Обычно, макроциклический амин подвергают моно-N-функционализации в органическом растворителе, таком, как ацетонитрил или ДМФ, при комнатной температуре с использованием ненуклеофильного основания, такого, как карбонат калия. Затем осуществляют дополнительную функционализацию оставшихся положений азота с использованием методов и условий, описанных в предыдущих схемах. После введения нужных хелирующих групп, нитро-группу восстанавливают с использованием окиси платины и водорода в воде. В этой форме хелирующий агент является совместимым с техникой конъюгирования, с помощью которой может быть осуществлено связывание с более крупными синтетическими или натуральными молекулами.
Ионами металлов, используемых для получения комплексов настоящего изобретения, являются 153Sm, 177Lu, 159Gd, 149Pm, 140La, 175Yb, 166Ho, 90Y, 47Sc, 186Re, 188Re, 142Pr, 99mTc, 67Ga, 68Ga, 105Rh, 97Ru, 111In, 113mIn или 115mIn. Присутствующим анионом является галид, а предпочтительно хлорид, или свободная соль (окись металла).
Комплексы были получены способами, хорошо известными специалистам. Так, например, см. Chelating Agents and Metal Chelates, Dwyer & Mellor, Academic Press (1964), Глава 7. См., также способы получения аминокислот в Synthetic Production and Utilization of Amino Acids (изд. Kameko и др.) John Wiley and Sons (1974). Получение комплекса предусматривает реакцию бициклополиазамакроциклофосфоновой кислоты с ионом металла в водных условиях при pH от 5 до 7. Комплекс, образованный с помощью химической связи, является стабильной радионуклидной композицией, например, стабильной к диссоциации радионуклида, т.е., его отщепления от лиганда.
Комплексы настоящего изобретения обычно имеют молярное отношение лиганда к металлу, по крайней мере, около 1:1, предпочтительно от 1:1 до 3:1, а более предпочтительно от 1:1 до 5:1. Большой избыток лиганда является нежелательным, поскольку несвязанный с комплексом лиганд может быть токсичным для организма пациента, или он может привести к остановке сердца или к гипокальцинемическим судорогам.
Антитела или фрагменты антител, используемые в конъюгатах, описанных в настоящей заявке, могут быть получены с помощью традиционной техники. Высокоспецифичные моноклональные антитела могут быть продуцированы с помощью техники гибридизации, хорошо известной специалистам, см., например, Kohler и Milstein (Nature, 256, 495 - 497, 1975; Eur. J. Immunol., 6, 511 - 519, 1976). Такие антитела обычно обладают высокоспецифичной реактивностью. В антитело-меченных конъюгатах могут быть использованы антитела, направленные против любого нужного антигена или гаптена. В конъюгатах предпочтительно использовать моноклональные антитела или их фрагменты, обладающие высокой специфичностью к нужному эпитопу (эпитопам). Антитела, используемые в настоящем изобретении, могут быть направлены, например, против опухолей, бактерий, грибков, вирусов, паразитов, микоплазм, дифференцировочных антигенов или других антигенов клеточных мембран, поверхностных антигенов патогенов, токсинов, ферментов, аллергенов, лекарственных средств и любых биологически активных молекул. Примерами некоторых антител или их фрагментов являются 1116-NS-19-9, 1116-NS-3d, 703D4, 704A1 и B72.3. Все указанные антитела были депонированы в ATCC. Более полный перечень антигенов можно найти в патенте США 4 193 983, который вводится в настоящее описание посредством ссылки. Конъюгаты настоящего изобретения являются особенно предпочтительными для диагностики различных видов рака.
Настоящее изобретение предусматривает использование активного ингредиента в сочетании с физиологически приемлемым носителем, наполнителем или связующим. Способы получения таких композиций хорошо известны специалистам. Эти композиции могут быть изготовлены в виде суспензий, инъецируемых растворов или других подходящих препаратов. При этом могут быть использованы физиологически приемлемые суспендирующие среды с добавлением или без добавления адъювантов.
Для диагностики или терапии используется "эффективное количество" композиции. Это эффективное количество (или доза) может варьироваться в зависимости от заболевания и физических параметров животного, например, от его веса. Препараты настоящего изобретения могут быть использованы также в in vivo-диагностике.
Для более ясного понимания настоящего изобретения ниже приводятся конкретные примеры, которые, однако, носят лишь чисто иллюстративный характер.
В этих примерах используются термины, которые означают следующее:
ЖХ - жидкая хроматография; очистку проводили при низком давлении с использованием системы Dionex 2010i, снабженной анионообменной колонкой (23 х 2 см), заполненной вручную О-Сефарозой
ДМФ - диметилформамтд,
AcOH - уксусная кислота,
1CP - индуктивно связанная плазма,
г - грамм; мг - миллиграмм; кг - килограмм; мл - миллилитр; мкл - микролитр.
Общая процедура по стабилизации pH.
Маточный раствор 153SmCl3 получали путем добавления 2 мкл 3•10-4 М 153SmCl3 в 0,1 н. HCl к 2 миллитрам 3•10-4 М SmCl3- несущего раствора. Соответствующие лигандные растворы были затем получены в деионизованной воде. После этого получали комплекс "лиганд/металл, 1:1" путем объединения лигандов (растворенных в 100 - 500 мкл деионизованной воды) с 2 мл маточного 153SmCl3-раствора с последующим тщательным перемешиванием и получением кислотного раствора (pH 2). Затем pH раствора повышали до 7,0 с использованием 0,1 н. NaOH. Процент металла в виде комплекса определяли путем пропускания образца раствора комплекса через колонку G-50 с Сефадексомтм, элюируя солевым раствором (85% NaCl/NH4OH, 4:1), и собирая 2 х 3 мл-фракции. Затем сравнивали количество радиоактивности объединенных фракций, полученных в результате элюирования, и радиоактивности некомплексного металла, оставшегося на смоле. Профиль pH-стабильности строили путем корректировки pH аликвоты раствора комплекса с использованием 1М NaOH или 1М HCl и определения процента металла, присутствующего в виде комплекса, используя ионообменный метод, описанный выше.
Исходные материалы.
Пример A.
Получение 2,6-бис(хлорометил)пиридина.
К 100 мл тионилхлорида, который охлаждали в ледяной бане, добавляли 24 г (0,17 М) 2,6-бис(гидроксиметил)пиридина. Через 30 мин реакционную смесь нагревали до комнатной температуры, а затем нагревали с обратным холодильником в течение 1,5 ч. После охлаждения реакционной смеси до комнатной температуры образовавшийся твердый продукт фильтровали, промывали бензолом и осушали в вакууме. Затем твердый остаток нейтрализовали насыщенным NaHCO3, фильтровали и осушали, в результате чего получали 23,1 г (71,5%) целевого продукта в виде беловатого кристаллического твердого вещества, т.пл. 74,5 - 75,5oC, которое затем анализировали.
1H ЯМР (CDCl3) δ : 4,88 (с, 4H); 7,25 - 7,95 (м, 3H).
Пример B.
Получение 3,6,9-трис(п-толилсульфонил)-3,6,9,15-тетраазабицикло (9.3.1)пентадека-1(15),11,13-триена
ДМФ-раствор (92 мл) 6,9 г (11,4 мМ) 1,4,7-трис(п-толилсульфонил) диэтилентриаминдинатриевой соли размешивали и нагревали до 100oC в атмосфере азота. Затем к раствору по капле в течение 45 минут добавляли 2 г (11,4 мМ) 2,6-бис(хлорометил)пиридина (полученного способом, описанным в примере A) в 37 мл ДМФ. После завершения добавления реакционную смесь размешивали 12 ч при 40oC. Затем к реакционной смеси добавляли 50 - 75 мл воды, что сразу приводило к растворению NaCl с последующим осаждением целевого продукта. Полученную суспензию фильтровали, а твердый осадок промывали водой и осушали в вакууме. Целевой продукт был получен в виде желтовато-коричневого порошка [6,5 г, 86%, т. пл. 168 - 170oC (разлож.)], который затем анализировали и получали следующие данные:
1H-ЯМР CDCl3 : δ 2,40 (с, 3H); 2,44 (с, 6H); 2,75 (м. 4H); 3,3 (м, 4H); 4,28 (с, 4H); 7,27 (д, 2H); 7,34 (д, 4H); 7,43 (д, 2H); 7,65 (д, 4H); 7,75 (т, 1H); и
13C-ЯМР δ : 21,48; 47,29; 50,37; 54,86; 124,19; 127,00; 127,1; 129,73; 135,04; 135,74; 138,95; 143,42; 143,73; 155,15.
Пример C.
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15),11,13- триена
Раствор HBr и AcOH получали путем смешивания 48% HBr и ледяной AcOH в отношении 64: 35. К 112 мл HBr/AcOH-смеси добавляли 5,5 г (8,2 мМ) 3,6,9-трис(п-толилсульфонил)-3,6,9,15-тетраазабицикло(9.3.1) пентадека-1(15), 11,13-триена, полученного в соответствии с процедурой примера B, и реакционную смесь нагревали, постоянно размешивая, с обратным холодильником в мягких условиях в течение 72 часов. Затем реакционную смесь охлаждали до комнатной температуры и концентрировали до получения приблизительно 1/10 исходного объема. Остаточный раствор энергично размешивали, после чего добавляли 15 - 20 мл диэтилового эфира. Образовавшееся беловатое твердое вещество фильтровали, промывали диэтиловым эфиром и осушали в вакууме. Сухую тетрагидробромидную соль растворяли в 10 мл воды, доводили pH до 9,5 с использованием NaOH (50 мас.%) и непрерывно экстрагировали хлороформом в течение 4 ч. После осушки безводным сульфатом натрия хлороформ выпаривали и получали светло-коричневое маслообразное вещество, которое постепенно кристаллировалось при выдерживании при комнатной температуре, в результате чего получали 1,2 г (71%) целевого продукта (т.пл. 86 - 88oC), который затем анализировали.
1Н-ЯМР (CDCl3) δ : 2,21 (м, 4H); 2,59 (м, 4H); 3,06 (с, 3H); 3,85 (с, 4H); 6,89 (д, 2H); 7,44 (т, 1H); и
13C-ЯМР δ : 48,73; 49,01; 53,63; 119,67; 136,29; 159,54.
Целевые продукты
Пример 1.
Получение 3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15),11,13 триен-3,6,9-триметиленфосфоновой кислоты (РСТМР)
Смесь 2,06 г (10 мМ) 3,6,9,15-тетраазабицикдо[]9.3.1]пентадека- 1(15), 11,13-триена (полученного в соответствии с процедурой, описанной в примере C), 11,3 г (138 мМ) фосфорной кислоты и 15 г (152 мМ) концентрированной HCl нагревали в условиях мягкого орошения (103oC), постоянно размешивая при этом, после чего по капле добавляли (2 мл/мин) 12,2 г (150 мМ, 15 мл) водного формальдегида (37%). После завершения добавления реакционную смесь нагревали с обратным холодильником в течение 16 ч, охлаждали до комнатной температуры и концентрировали с получением густого вязкого маслообразного продукта. Этот продукт очищали путем анионообменной ЖХ (0 - 30% муравьиная кислота, 3 мл мин, время удерживания 32 мин). Объединенные фракции иофилизовали и получали 4,8 г (99%) целевого продукта в виде белого твердого вещества, т. пл. 275 - 280oC. Затем продукт анализировали и получали следующие данные:
1Н-ЯМР (D2O) δ : 2,83 (м, 6H); 3,46 (м, 10H), 7,28 (д, 2H), 7,78 (т, 1H); и
13C-ЯМР δ : 53,61; 53,81; 55,27; 57,93; 62,20; 125,48; 143,08; 152,31; и
31P-ЯМР δ : 8,12 (2P); 19,81 (1P).
Пример 2.
Получение комплекса 153Sm-3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15),11,13-триен-3,6,9-триметиленфосфоновой кислоты 153Sm-РСТМР
Раствор лиганда примера 1 получали путем растворения 3,8 мг лиганда в 0,517 мл деионизованной воды (pH 2). Затем получали комплекс лиганда/металла (1: 1) путем объединения 40 мкл лигандного раствора с 2 мл водного раствора SmCl2 • H2O (3• 10 М в 1 н. HCl), содержащего изотопный индикатор 153SmCl3. После тщательного смешивания процент комплексного металла определяли путем пропускания образца раствора комплекса через колонку с Сефадексомтм, элюируя солевым раствором (0,85% NaCl/MH4OH 4:1) и собирая 2 х 3 мл фракции. Количество радиоактивности в объединенных фракциях сравнивали с количеством радиоактивности металла, оставшегося на смоле. В этих условиях комплекс удалялся с элюентом, а металл, не связанный с комплексом, оставался на смоле. С помощью этого метода было определено, что степень комплексообразования составляет 98%. Образец раствора, который был пропущен через смолу, использовали для исследования pH. Затем определяли стабильность pH в соответствии с общей процедурой, описанной выше.
Пример 3.
Получение комплекса 166Ho-3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15),11,13-триен-3,6,9-триметиленфосфоновой кислоты 166Ho-РСТМР)
Повторяли процедуру примера 2, за исключением того, что вместо 153SmCl3 использовали 166HoCl3, в результате чего получали целевой продукт.
Пример 4.
Получение комплекса 90Y-3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триен-3,6,9-триметиленфосфоновой кислоты (90Y-РСТМР)
Повторяли процедуру примера 2, за исключением того, что вместо 153SmCl3 использовали 90YCl3, в результате чего получали целевой продукт.
Биораспределение.
Общая процедура.
Крысам Sprague Dawley давали 5 дней для акклиматизации, после чего им в хвостовую вену инъецировали 100 мкл раствора комплекса. На момент инъекции вес крыс составлял 150 - 200 г. Через 30 мин крыс забивали путем цервикальной дислокации и препарировали. Количество радиоактивности в каждой ткани определяли путем подсчета с помощью сцинтилляционного счетчика Na1, присоединенного к многоканальному анализатору. Для определения процентного содержания дозы в каждой ткани или органе полученное число отсчетов сравнивали с числом отсчета в 100 мкл-стандартах.
Процент дозы в крови определяли, исходя из того предположения, что кровь составляет 7% от массы тела. Процент дозы в кости определяли путем умножения процентной дозы, полученной для бедра, на 25. Процентную дозу в мышцах оценивали, исходя из предположения, что мышцы составляют 43% от массы тела.
Пример I.
Процент инъецированной дозы комплекса примера 2 (153Sm-РСТМР) в некоторых тканях приводятся в табл. I. Приведенные данные представляют собой средние величины, полученные по опорным точкам для 5 крыс.
Таблица I. Процент инъецированной дозы в некоторых тканях для 153Sm-РСТМР
Ткань - Средние величины
Кость - 60
Печень - 2,80
Почки - 0,38
Селезенка - 0,16
Мышцы - 0,40
Кровь - 0,29
Пример II.
Процент инъецированной дозы комплекса 166Ho с лигандом примера I (РСТМР) (полученного в примере 3) в некоторых тканях приводится в табл. II. Приведенные данные представляют собой средние величины, полученные по опорным точкам для 3 крыс.
Таблица II. % инъецированной дозы в некоторых тканях для 166Ho РСТМР
Ткань - Средняя величина
Кость - 43
Печень - 3,55
Почки - 0,33
Селезенка - Ниже фона
Мышцы - 0,34
Кровь - 0,12
Пример III.
Процент инъецированной дозы комплекса 90Y с лигандом примера I (РСТМР) (полученного в примере 4) в некоторых тканях приводится в табл. III. Приведенные данные представляют собой средние величины, полученные по опорным точкам для 3 крыс.
Таблица III. Процент инъецированной дозы в некоторых тканях для 90Y-РСТМР
Ткань - Средняя величина
Кость - 29
Печень - 0,21
Почки - 0,36
Селезенка - Ниже фона
Мышцы - 0,42
Кровь - 0,18
Отношение кости к крови (Q дозы/Г) составляет 140. Отношение кости к печени составляет 76. Отношение кости к мышцам составляет 400.
Пример IV.
Локализация комплекса 153Sm с лигандом примера I (РСТМР) (при отношении лиганда к металлу 5: 1) в кости составляла 60%. Этот результат позволяет предположить, что уменьшение заряда хелата способствует повышению специфичности доставки комплекса в кость.
Специалистам в данной области очевидны и другие варианты осуществления настоящего изобретения, которые вытекают из рассмотрения и практического применения изобретения, приведенных в настоящем описании. Причем подразумевается, что представленные примеры имеют лишь иллюстративный характер и возможны отклонения от них в пределах существа и объема изобретения, определенных в формуле изобретения, приведенной ниже.
Пример D.
Получение 2,6-бис(хлорметил)пиридина.
К 100 мл тионилхлорида, который охлаждался (на ледяной бане), добавлялось 24 г (0,17 моля) 2,6-бис(гидроксиметил)пиридина. Спустя 30 минут реакционная смесь подогревалась до комнатной температуры, затем нагревалась с обратным холодильником в течение 1,5 часов. После охлаждения реакционной смеси до комнатной температуры твердое вещество, которое образовалось, отфильтровывалось, промывалось бензолом и сушилось в вакууме. Твердое вещество затем нейтрализовалось насыщенным бикарбонатом натрия, отфильтровывалось и сушилось, давая 23,1 г (71,5%) целевого продукта в виде не совсем белого кристаллического твердого вещества, т.пл. 74,5 - 75,5oC, и далее характеризовалось следующими данными:
1Н ЯМР (CDCl3) δ 4,88 (с., 4H), 7,25 - 7,95 (м., 3H)
Пример E.
Получение 3,6,9-трис(п-толилсульфонил)-3,6,9,15-тетраазабицикло[9.3.1] пентадека 1-(15),11,13-триена
Раствор в диметилформамиде (92 мл) 6,9 г (11,4 ммоля) динатриевой соли 1,4,7-трис(п-толилсульфонил)диэтилентриамина перемешивался и нагревался до 100oC в атмосфере азота. К раствору добавлялось по каплям на протяжении 45 минут 2 г (11,4 ммоля) 2,6-бис(хлорметил)пиридина, полученного с помощью процедуры примера A, в 37 мл ДМФ. Когда добавление завершалось, реакционная смесь перемешивалась при 40oC в течение 12 часов. К реакционной смеси добавлялось затем 50 - 75 мл воды, что приводило в результате к немедленному растворению NaCl с последующим осаждением целевого продукта. Получающаяся в результате суспензия затем фильтровалась, и твердое вещество промывалось водой и сушилось в вакууме. Целевой продукт получался в виде светло-рыжевато-коричневого порошка, 6,5 г (86%), т.пл. 168 - 170oC разлож. и далее характеризовался следующим образом:
1H ЯМР (CDCl3) δ 2,40 (с., 3H), 2,44 (с.. 6H), 2,75 (м., 4H), 3,30 (м., 4H), 4,28 (с., 4H), 7,27 (д., 2H), 7,34 (д., 4H), 7,43 (д., 2H), 7,65 (д., 4H), 7,75 (т., 1H);
13С ЯМР δ 21,48, 47,29, 50,37, 54,86, 124,19, 127,00, 127,11, 129,73, 135,04, 135,74, 138,95, 143,42, 143,73, 155,15.
Пример F.
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадека- 1(15), 11, 13-триена.
Раствор HBr и AcOH приготавливался с помощью смешения 48% HBr и ледяной AcOH в соотношении 64: 35. К 112 мл HBr/AcOH смеси добавлялось 5,5 г (8,2 ммоля) 3,6,9-трис/п-толилсульфонил/- 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера B), и реакционная смесь нагревалась в условиях мягкой дефлегмации при постоянном перемешивании в течение 72 ч. Реакционная смесь затем охлаждалась до комнатной температуры и концентрировалась до приблизительно 1/10 первоначального объема. Оставшийся раствор перемешивался энергично и добавлялось 15 -20 мл диэтилового эфира. Образовывалось на совсем белое твердое вещество, которое отфильтровывалось, промывалось диэтиловым эфиром и сушилось в вакууме. Сухая тетрагидробромидная соль затем растворялась в 10 мл воды, доводилась до pH 9,5 с помощью NaOH (50% вес/вес) и непрерывно экстрагировалась хлороформом в течение 4 ч. После сушки над безводным сульфатом натрия хлороформ выпаривался, давая светло-рыжеватокоричневое масло, которое постепенно кристаллизовалось при стоянии при комнатной температуре, давая 1,2 г (71%) целевого продукта, т.пл. 86 - 88oC, которой характеризовался следующими данными:
1Н ЯМР (CDCl3) δ 2,21 (м., 4H), 2,59 (м., 4H), 3,06 (с., 3H), 3,85 (с.. 4H), 6,89 (д., 2H), 7,44 (т, 1H),
13C ЯМР δ 48,73, 49,01, 53,63, 119,67, 136,29, 159,54.
Пример G.
Получение 3,9-бис(натрий метиленсульфонат)-3,6,9,15- тетраазабицикло-(9.3.1)пентадека-1(15),11,13,-триена (PC2S)
Водный раствор (10,0 мл) 3,6,9,15-тетраазабицикло(9.3.1) пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C) 1.03 г (5,0 ммолей), добавлялось 0,5 мл концентрированной HCl, и смесь перемешивалась в течение 10 минут для обеспечения полного растворения. Получающийся в результате раствор имел величину pH 8,6. К раствору затем добавлялось 1,37 г (10,2 ммоля) HOCH2SO3Na с 5 мл деионизированной воды. Раствор нагревался при 60oC в течение 10 мин, и величина pH падала до 5,6. После охлаждения величина pH доводилась до 9,0 с помощью 1М водной гидроокиси натрия, затем осуществлялась лиофилизация, давая целевой продукт в виде белого твердого вещества с количественным выходом, которое характеризовалось следующими данными:
1Н ЯМР (D2O) δ 2,87 (т, 4H), 3,18 (т., 4H), 8,85 (с., 4H), 4,11 (с., 4H), 7,03 (д., 2H), 7,55 (т., 1H), и
13С ЯМР (D2O) δ 48,52, 54,04, 58,92, 79,09, 123,90, 141,37, 161,89.
Пример H.
Получение 3,9-бис(метиленнитрил)-3,6,9,15-тетраазабицикло (9.3.1)-пентадека-1(15),11,13-триена
К водному раствору, 10,0 мл, 3,9-бис(натрий метиленсульфонат)-3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15), 11,13- триена (полученного с помощью процедуры примера F), 2,26 г (5 ммолей) добавлялось 0,6 г (12,24 ммоля) цианида натрия. Смесь перемешивалась в течение 3 часов при комнатной температуре. Величина pH реакционной смеси составляла примерно 10. Величина pH доводилась до выше 13 с помощью концентрированной водной гидроокиси натрия. Продукт выпадал в осадок и экстрагировался хлороформом (3 • 20 мл), сушился над безводным сульфатом магния и фильтровался. После удаления растворителя и концентрирования в вакууме желаемый продукт выделялся в виде воскообразного белого порошка, 1,0 г (71%) и характеризовался следующими данными:
1Н ЯМР (CDCl3) δ 2,03 (шир., с., 4H), 2,64 (м., 4H), 3,82 (с., 4H), 3,90 (с., 4H), 7,14 (д., 2H), 7,62 (т., 1H); и
13С ЯМР (CDCl3) δ 46,08, 46,64, 52,89, 60,78, 115,31, 122,02, 137,57, 157,33.
Пример I
Получение 3,9-бис(метиленнитрил)-6-(метилендиметилфосфонат) 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15),11,13,-триена
3,9-бис(метиленнитрил)-3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триен (полученный с помощью процедуры примера G), 285 мг (1,0 ммоль), объединялся с 60 мл (2,0 ммоля, избыток) параформальдегида и 0,354 мл (372 мг, 3,0 ммоля, избыток) триметилфосфита. Смесь осторожно перемешивалась в течение 10 мин с получением суспензии. Затем нагревалась до 90oC в течение 1 ч. После того, как избыток реагентов и побочные продукты удалялись в вакууме (1 час при 125oC/0,01 мм рт.ст.) получающийся в результате темно-коричневый остаток растворялся в 20 мл хлороформа и промывался деионизированной водой (5•15 мл ). Органический слой сушился над безводным сульфатом магния, фильтровался, и избыток растворителей выпаривался в вакууме, давая требуемый продукт в виде желтого воскообразного твердого вещества, 168 мг (41%), которое характеризовалось следующими данными:
1Н ЯМР (CDCl3) δ 2,61 (шир., с., 8H), 2,73 (д., 2H), 3,62 и 3,68 (с., 6H), 3,73 (с., 4H), 3,84 (с., 4H), 7,06 (д., 2H), 7,57 (т., 1H); и
13С ЯМР (CDCl3) δ 44,44, 50,74, 51,03, 51,85, 52,51, 60,28, 115,61, 122,27, 137,24, 156,61.
Пример J.
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15), 11,13-триен-3,6,9- метилендиэтилфосфоната
Смесь 1 г (4,8 ммоля) 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C), 4,8 г (28,8 ммоля) триэтилфосфита и 864 мг (28,8 ммоля) параформальдегида нагревалась при 50oC при постоянном перемешивании в течение 45 мин. Реакционная смесь концентрировалась в вакууме, и вязкое масло хроматографировалось на колонке с основной окисью алюминия, при элюировании хлороформом. После концентрирования органического элюента желаемый продукт выделялся в виде бесцветного масла, 2,0 г (64%) и характеризовался следующими данными:
1Н ЯМР (CDCl3) δ 1,23 (м.. 18H), 2,77 (м., 12H), 3,04 (д., 6H),. 4,13 (м., 12H), 7,17 (д., 2H), 7,60 (т., 1H); и
13С ЯМР (CDCl3) δ 16,43, 50,03, 50,31, 50,43, 50,77, 51,23, 51,38, 52,63, 53,30, 60,86, 60,92, 61,63, 61,74, 61,83, 61,93, 62,32, 76,46, 76,97, 74,18, 77,48, 122,50, 137,10, 157,18; и
31Р ЯМР δ 24,92 (с., 2P), 24,97 (с., 1P).
Пример K
Получение 3,6,9,15-тетраазабицикло[9.3.1]пентадека-1(15),11,13- триен-3,6,9-метиленди/н-пропил/фосфоната
К 3 мл хлороформ (диоксанового раствора (1:1) добавлялось 100 мг (0,48 ммоля) 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15), 11,13- триена, полученного с помощью процедуры примера C), 318 мг (1,53 ммоля) трипропилфосфита и 46 мг (1,53 ммоля) параформальдегида. Реакционная смесь нагревалась при 90oC при перемешивании в течение 1 ч. Получающийся в результате гомогенный раствор концентрировался в вакууме, давая вязкое масло, которое хроматографировалось на колонке с нейтральной окисью алюминия, при элюировании хлороформом. После концентрирования органического элюента желаемый продукт выделялся в виде бесцветного масла, 320 мг (90%), и характеризовался следующими данными:
1Н ЯМР (CDCl3) δ 0,88 (м., 18H), 1,61 (м., 12H), 2,72 (м., 12H), 3,05 (д., 6H), 3,97 (м., 12H), 7,13 (д., 2H), 7,56 (т., 1H); и
13С ЯМР (CDCl3) δ 9,96, 23,73, 49,84, 50,14, 50,26, 50,57, 51,11, 51,23, 52,43, 53,01, 60,78, 60,84, 67,27, 67,40, 122,48, 137,04, 157,16; и
31Р ЯМР δ 24,98 (3P).
Пример L
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15),11,13- триен-3,6,9-метиленди(н-бутил)фосфоната
Смесь 500 мг (2,4 ммоля) 3,6,9,15-тетраазабицикло(9.3.1) пентадека-1(15),11,13-триена, полученного с помощью процедуры примера C, 2,0 г (8 ммолей) трибутилфосфита и 240 мг (8 ммолей) параформальдегида нагревалась при 100oC при перемешивании в течение 1 ч. Получающийся в результате вязкий раствор концентрировался в вакууме, давая масло, которое хроматографировалось на колонке с основной окисью алюминия при элюировании хлороформом. После концентрирования органического элюента желаемый продукт выделялся в виде бесцветного масла, 1,25 г (65%), которое хараткеризовалось следующими данными:
1Н ЯМР (CDCl3) δ 0,84 (м., 18H), 1,27 (м.. 12H), 1,58 (м., 12H), 2,57 (м., 12H), 3,01 (д., 6H), 3,99 (м., 12H), 7,12 (д., 2H), 7,54 (т., 1H); и
13С ЯМР (CDCl3) δ 13,42, 13,46, 18,50, 18,59, 32,16, 32,43, 49,88, 50,03, 50,16, 50,63, 51,11, 51,27, 52,48, 53,16, 60,71, 60,78, 65,38, 65,48, 65,58, 122,46, 136,96, 157,14; и
31Р ЯМР δ 24,88 (2P), 24,93 (1P).
Пример M.
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15),11,13- триен-3[(4-нитрофенил)метилацетата]
К раствору 2,5 мл хлороформа, который быстро перемешивался, и 200 мг (0,97 ммоля) 3,66,9,15-тетраазабицикло(9.3.1)пентадека-1(19),11,13-триена, полученного с помощью процедуры примера C, добавлялось в виде одной порции 266 мг (0,97 ммоля) бром-(4-нитрофенил)метилацетата в 2,5 мл хлороформа. Реакционная смесь перемешивалась в течение 24 ч при комнатной температуре. Раствор концентрировался в вакууме, давая полутвердое вещество, которое хроматографировалось на силикагельной колонке при элюировании смесью хлороформ(метанол)гидроокись аммония (16: 4: 1). После концентрирования органического элюента выделялся желаемый продукт в виде светло-желтого твердого вещества, 250 мг (64%), которое хараткеризовалось следующими данными:
13С ЯМР (CDCl3) δ 45,67, 45,90, 45,97, 51,65, 52,08, 52,28, 53,78, 69,54, 119,00, 119,23, 122,85, 130,30, 137,06, 143,27, 147,05, 159,59, 160,41, 171,70.
Конечные продукты.
Пример 5.
Получение 3,9-диуксусная кислота -6-(метиленфосфоновая кислота)-3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15),11,13-триена (PC2AIP)
Концентрированный солянокислотный раствор (37%, 5 мл) 3,9-бис(метиленнитрил)-6-(метилендиметилфосфонат)-3,6,9,15- тетраазабицикло(9.3.1)пентадека-1(15), 11,13-триена, полученного в пример H, 168 мг (1,0 ммоль), нагревался в условиях дефлегмации в течение 16 ч. После охлаждения раствор выпаривался досуха, с последующим совместным выпариванием с деионизированной водой (2 х 10 мл) для удаления избытка соляной кислоты. Конечный продукт выделялся в виде темно-коричневого твердого вещества после лиофилизации концентрированного водного раствора, которое хараткеризовалось следующими данными:
1Н ЯМР (D2O) δ 2,68 (шир.с., 4H), 3,31 (шир.с., 4H), 4,08 (с., 4H), 4,55 (с., 4H), 7,16 (д., 2H), 7,68 (т., 1H); и
13С ЯМР (D2O) δ 52,35, 54,04, 57,02, 59,24, 62,26, 125,52, 143,64, 152,36, 171,54; и
31Р ЯМР (D2O) δ 20,03
Пример 6
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15),11,13- триен-3,6,9-метиленэтилфосфонат трис (калиевой соли (PMEHE)
К водному 0,1 норм. раствору гидроокиси калия (2 мл) добавлялось 250 мг (0,38 ммоля) 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15), 11,13-триен-3,6,9- метилендиэтилфосфоната, полученного с помощью процедуры примера 1. Раствор нагревался при 90oC в течение 5 ч. Реакционная смесь охлаждалась до комнатной температуры, фильтровалась, сушилась вымораживанием, давая целевой продукт в виде не совсем белого твердого вещества, 252 мг (97%), который хараткеризовался следующими данными:
13С ЯМР (D2O) δ 18,98, 19,82, 51,78, 52,06, 53,08, 54,46, 54,68, 57,01, 58,22, 60,24, 63,19, 63,25, 63,36, 63,49, 63,59, 63,95, 64,18, 64,25, 66,80, 126,62, 141,63, 159,40; и
31Р ЯМР δ 20,58 (с., 2P), 20,78 (с., 1P)
Пример 7.
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15),11,13- триен-3,6,9-метилен/н-пропил(фосфонат трис) калиевой соли (PMPHE)
К водному раствору гидроокиси калия (0,5 мл 1 норм./диоксан 0,5 мл) добавлялось 81 мг (0,108 ммоля) 3,6,9,15-тетраазабицикло(9.3.1)-пентадека-1(15), 11,13-триен-3,6,9- метилен(н-пропил)фосфоната, полученного с помощью процедуры примера J. Раствор нагревался в условиях дефлегмации в течение 24 ч. Реакционная смесь охлаждалась до комнатной температуры и экстрагировалась диэтиловым эфиром. Эфирный экстракт затем концентрировался в вакууме, давая желаемый продукт в виде не совсем белого твердого вещества, 48,6 мг (60%), который хараткеризовался следующими данными:
31Р ЯМР δ 20,49 (с., 3P)
Пример 8
Получение 3,6,9,15-тетраазабицикло(9.3.1)пентадиен-1(15),11,13- триен-3,6,9-метилен/н-бутил/фосфоната трис/калиевой соли/ (PMRHE)
К водному раствору 35 мл 1 норм. гидроокиси калия добавлялось 3,21 г (3,88 ммоля) 3,6,9,15-тетраазабицикло/9.3.1/пентадека- 1(15),11,13-триен-3,6,9-метиленди/н-бутил/фосфоната /полученного с помощью процедуры примера K). Раствор нагревался в условиях дефлегмации в течение 5 дней. Реакционная смесь охлаждалась до комнатной температуры, фильтровалась, и фильтрат подвергался сушке вымораживанием, давая окрашенное в кремовый цвет твердое вещество. Данное вещество затем суспендировалось в 160 мл метанола и перемешивалось в течение 12 часов при комнатной температуре. Суспензия затем фильтровалась и фильтрат концентрировался, давая полутвердое вещество. Данное вещество бралось в 150 мл хлороформа и сушилось над безводным сульфатом натрия и фильтровалось. После концентрирования в вакууме продукт выделялся в виде не совсем белого твердого вещества, 1,86 г (62%), и характеризовался следующими данными:
1Н ЯМР (D2O) δ 0,68 (м., 9H), 1,14 (м., 6H), 1,37 (м., 6H), 2,76 (д., 6H), 3,41 (м., 12H), 3,73 (м., 6H), 7,24 (д., 2H), 7,76 (т., 1H); и
13С ЯМР (D2O) δ 15,76, 15,80, 21,12, 21,20, 34,96, 35,06, 35,14, 52,08, 52,53, 53,38, 53,48, 54,49, 54,75, 57,70, 57,76, 61,86, 67,65, 67,75, 67,98, 68,08, 125,15, 142,93, 152,25; и
31Р ЯМР δ 9,73 (с.. 2P), 21,00 (с., 1P).
Пример 9
Получение 3,6,9,15-тетраазабицикло[9.3.1]пентадека-1(15),11,13- триен-3[(4-нитрофенил)метилацетат]-6,9-метилендиэтилфосфоната
Раствор 250 мг (0,62 ммоля, 3,6,9,15-тетраазабицикло(9.3.1)пентадека-1(15), 11,13-триен-3-(4- нитрофенил)метилацетата, полученного с помощью процедуры примера L, 624 мг (3,7 ммоля) триэтилфосфита и 111 мг (3,7 ммоля) параформальдегида перемешивался при 100oC в течение 1 часа. Получающийся в результате гомогенный раствор концентрировался в вакууме, давая вязкое масло. Масло растворялось в 10 мл хлороформа и промывалось водой (3 х 5 мл). Органический слой сушился над безводным сульфатом магния, фильтровался, и фильтрат концентрировался в вакууме, давая продукт в виде вязкого масла, 326 мг (96%), который характеризовался следующими данными:
31Р ЯМР (CDCl3) δ 24,67, (с., 2P), 24,88 (с., 1P)
Биораспределение.
Общая процедура
Крысы Sprague Dawley оставались для акклиматизации в течение пяти дней, затем инъецировались 100 мкл раствора комплекса через хвостовую вену. Во время инъекции крысы весили между 150 и 200 г. Спустя 30 минут крыс убивали путем цервикального смещения и разрезали. Количество радиоактивности каждой ткани определялось путем подсчета в Nal сцинтилляционном счетчике, подсоединенном к многоканальному анализатору. Число подсчетов сравнивалось с подсчетами в 100 мкл стандартных пробах для того, чтобы определить процент дозы в каждой ткани
Пример V.
Процент инъецированной дозы комплекса примера 5 (153Sm-PMPHE) в нескольких тканях дан в табл. II. Числовые величины представляют среднее от минимум 3 крыс через 2 ч после инъекции.
Таблица II.
% инъецированной дозы 153Sm- PMPHE (2 ч)
Ткань - Среднее
Кости - 10,86
Печень - 4,14
Почки - 1,55
Селезенка - 0,05
Мышцы - 1,19
Кровь - 0,25
Сердце - 0,08
Легкие - 0,12
Головной мозг - 0,00
Желудок - 0,44
Тонкий кишечник - 10,71
Толстая кишка - 2,17
Пример VI.
Процент инъецированной дозы комплекса примера 6 (153Sm-PMPHE) в нескольких тканях дан в табл. III. Числовые величины представляют среднее от минимум 3 крыс через 2 ч после инъекции.
Таблица III.
% инъецированной дозы 153Sm-PMEHE (2ч).
Ткань - Среднее
Кости - 3,73
Печень - 2,70
Почки - 0,43
Селезенка - 0,05
Мышцы - 1,09
Кровь - 0,14
Сердце - 0,02
Легкие - 0,04
Головной мозг - 0,00
Желудок - 0,08
Тонкая кишка - 57,89
Толстая кишка - 0,77
Пример VII.
Процент инъецированной дозы комплекса примера 3 (153Sm-PC2A1) в некоторых тканях дан в табл. IV. Числовые значения представляют среднее от минимум 3 крыс через 2 ч после инъекции.
Таблица IV
% инъецированной дозы 153Sm-PC2A1P (2 ч).
Ткань - Среднее
Кости - 47,98
Печень - 1,46
Почки - 0,93
Селезенка - 0,02
Мышцы - 1,00
Кровь - 0,36
Сердце - 0,04
Легкие - 0,06
Головной мозг - 0,01
Желудок - 0,25
Тонкая кишка - 13,10
Толстая кишка - 0,12
Пример на фармацевтическую композицию
Ингредиент - Количество мг на капсулу
Соединение - 100
Лактоза - 146
Стеарат магния - 4
Капсула (N.1) - 250
Соединение с нейтральными компонентами измельчают в порошок, перемешивают и помещают в сухие желатиновые капсулы.
Типичную фармацевтическую композицию для инъекций готовят растворением 150 мг соединения Примера 2 в изотоническом растворе (1000 мл) с последующим запаиванием в 1 - 5 мл ампулы.
Соединение Примера 2 - 150 мг
Изотонический раствор для инъекций - 1000 мл
Соотношение лиганд/металл = 300/1 при молярной концентрации 153Sm 3 • 10-4 М. Вводимое количество составляет 1 мл.
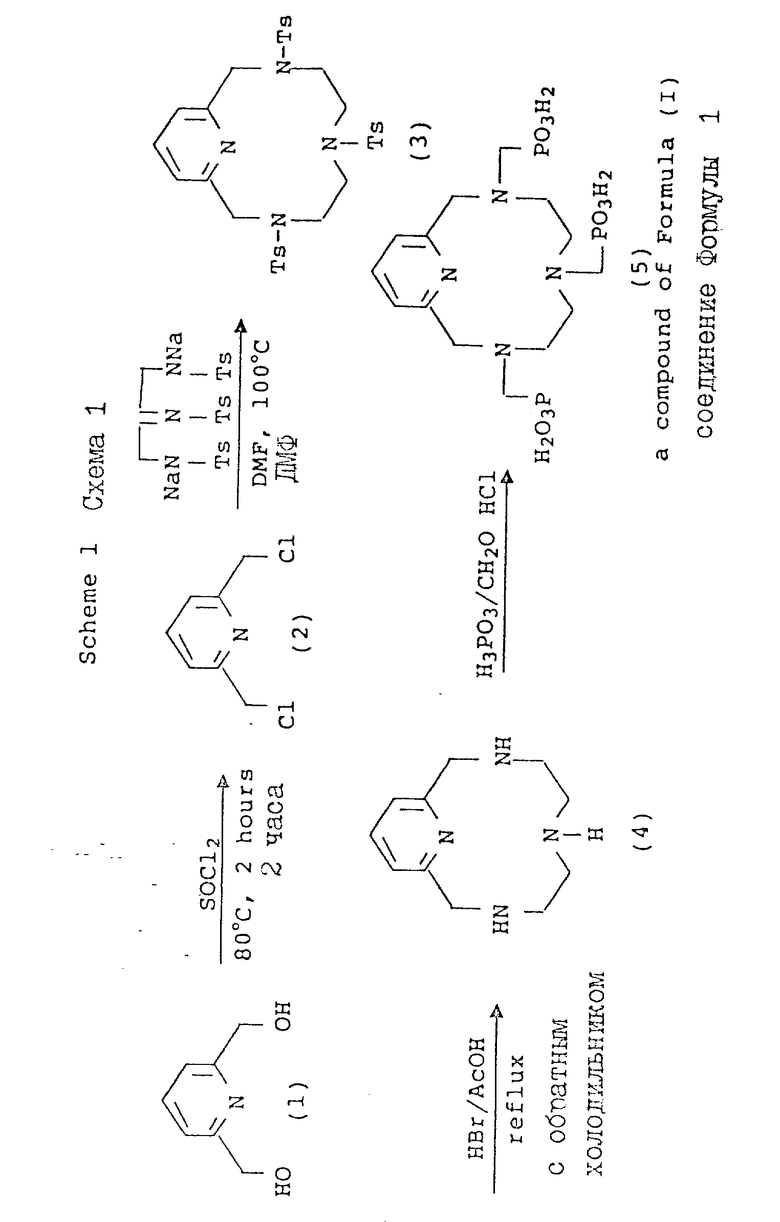
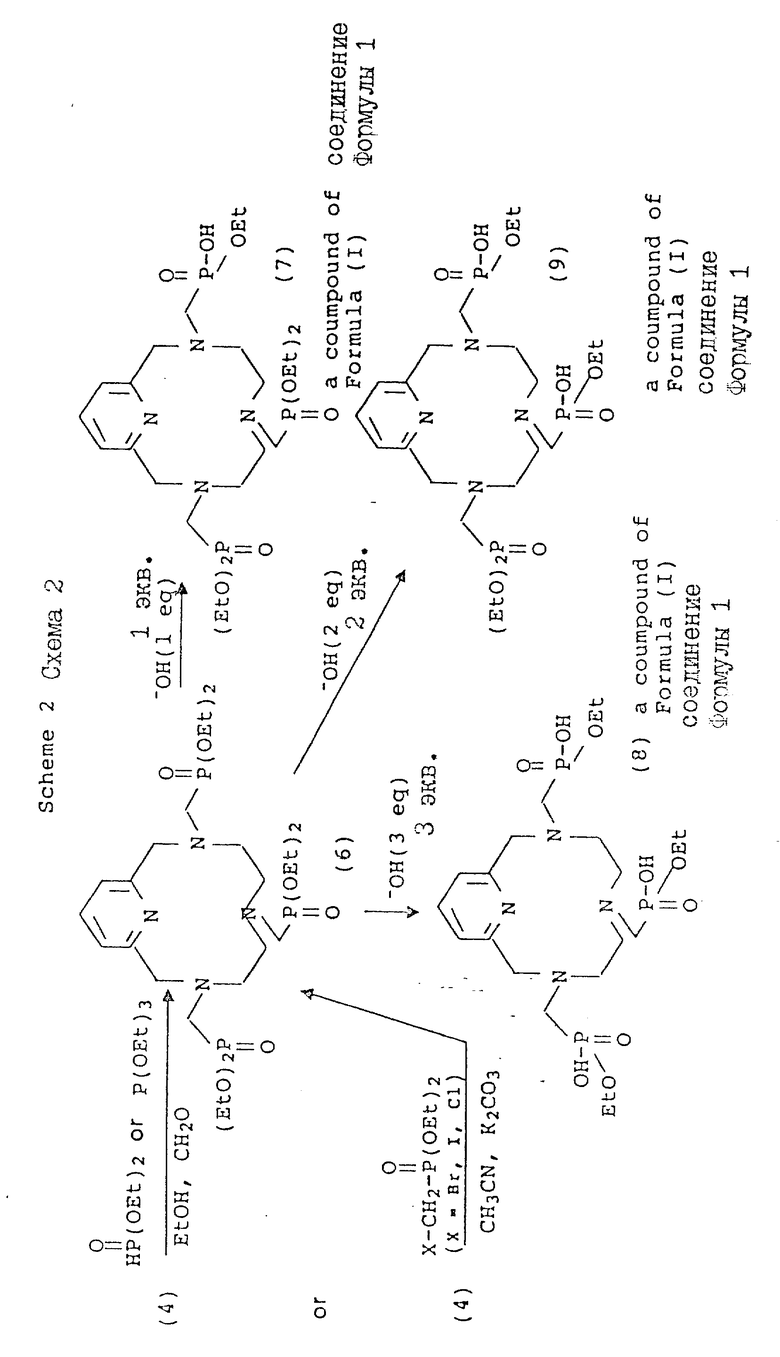
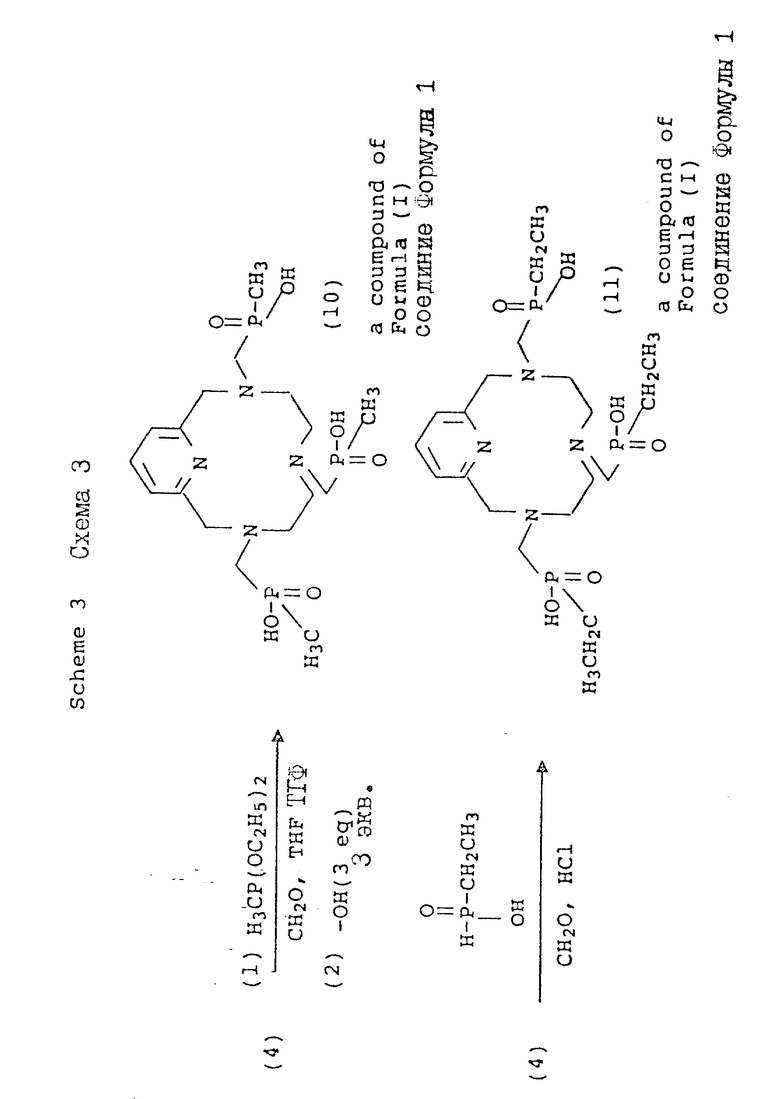
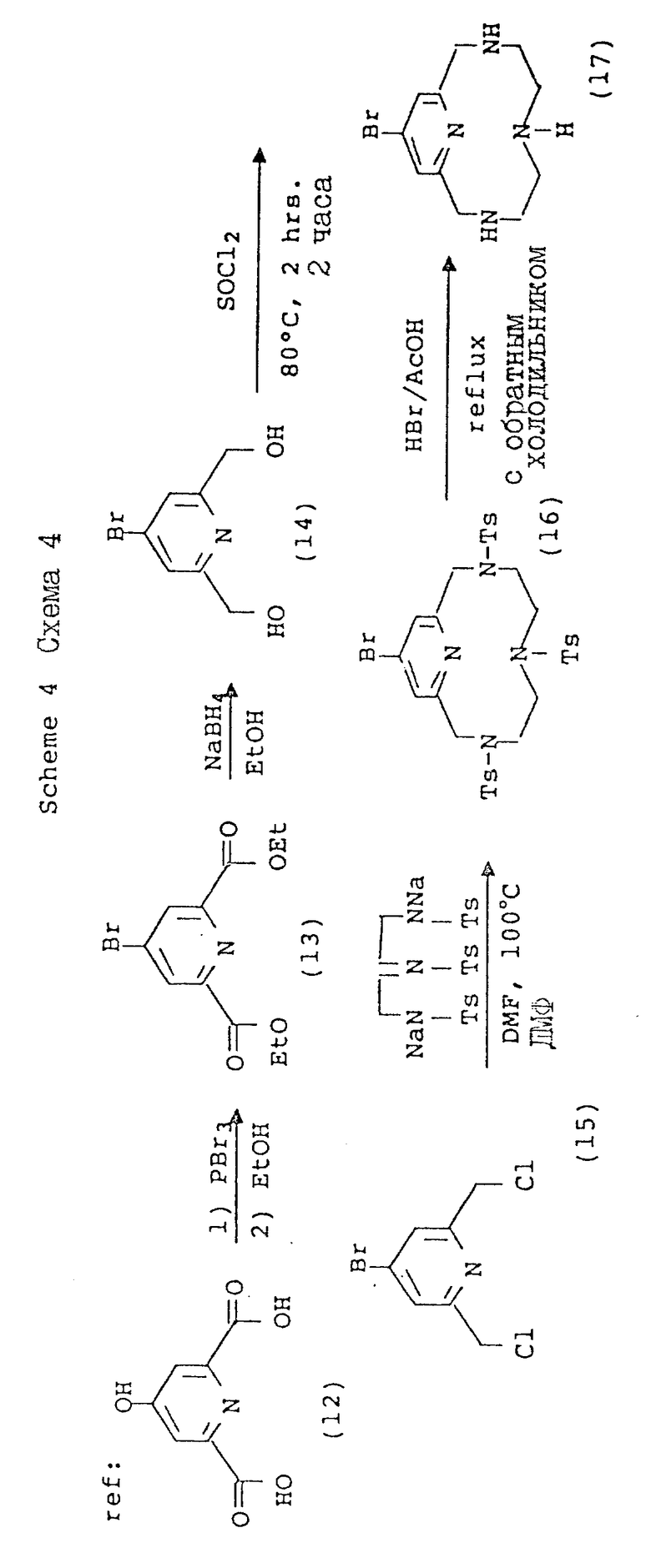


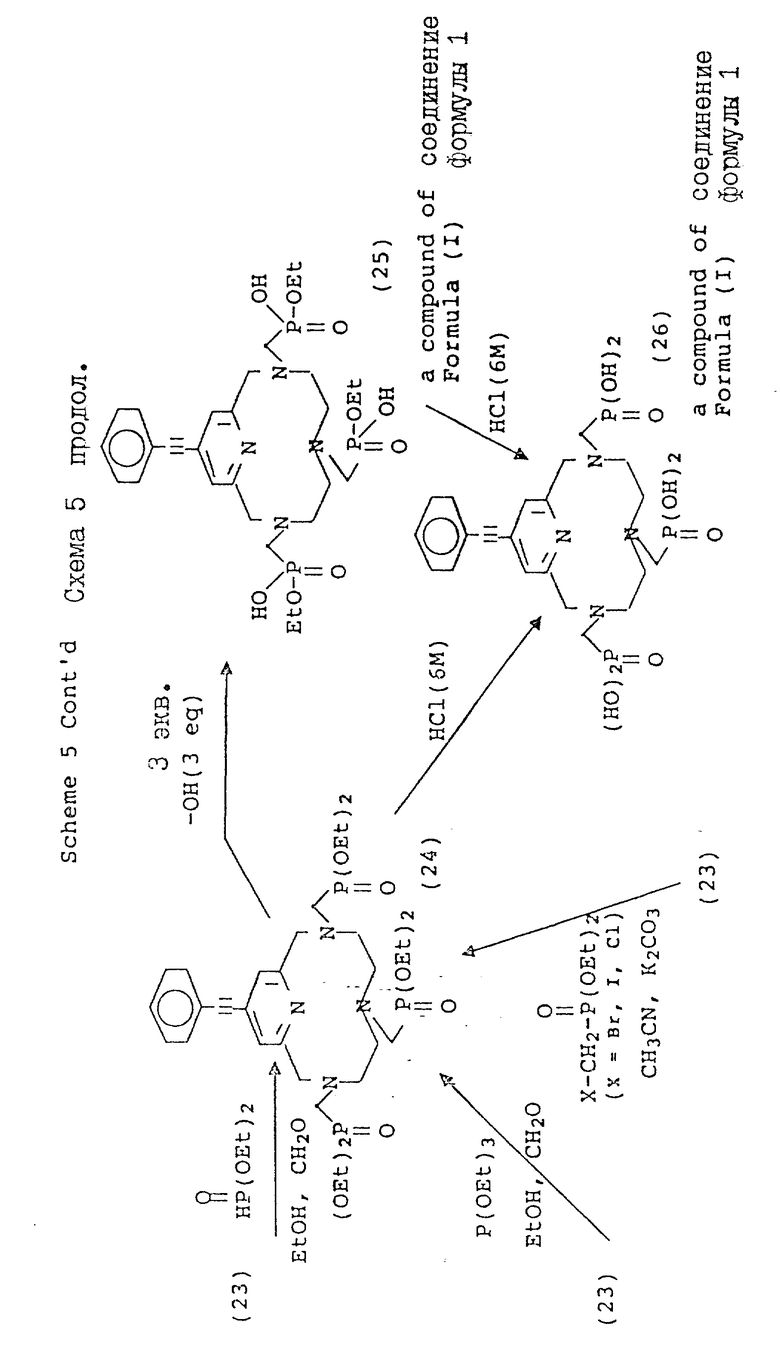
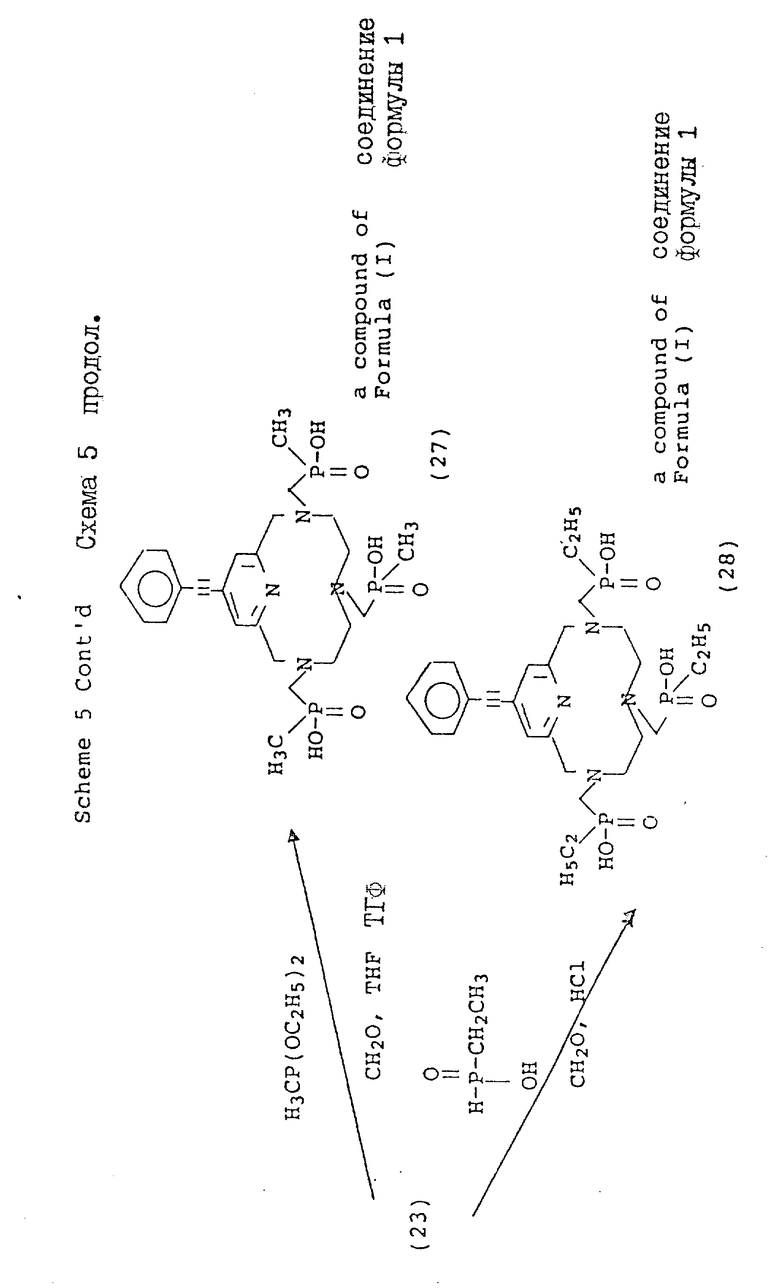

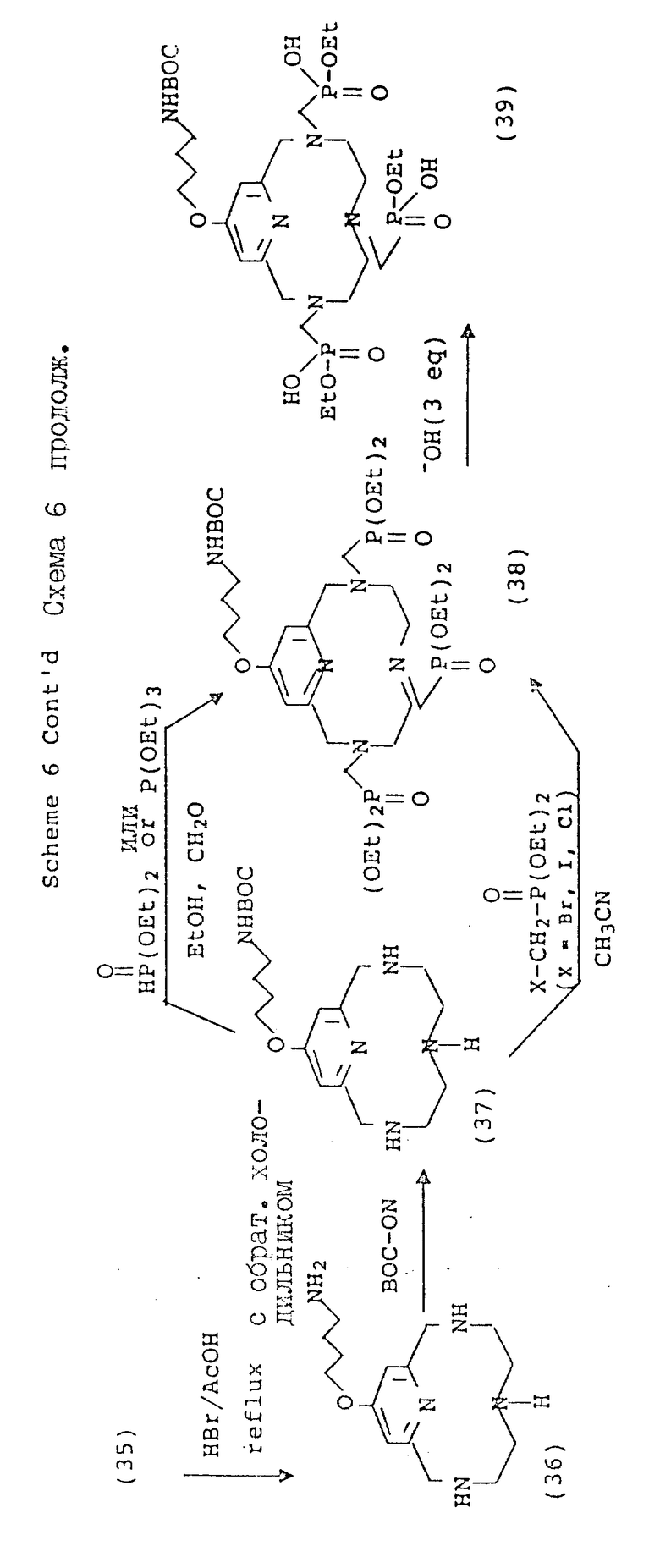
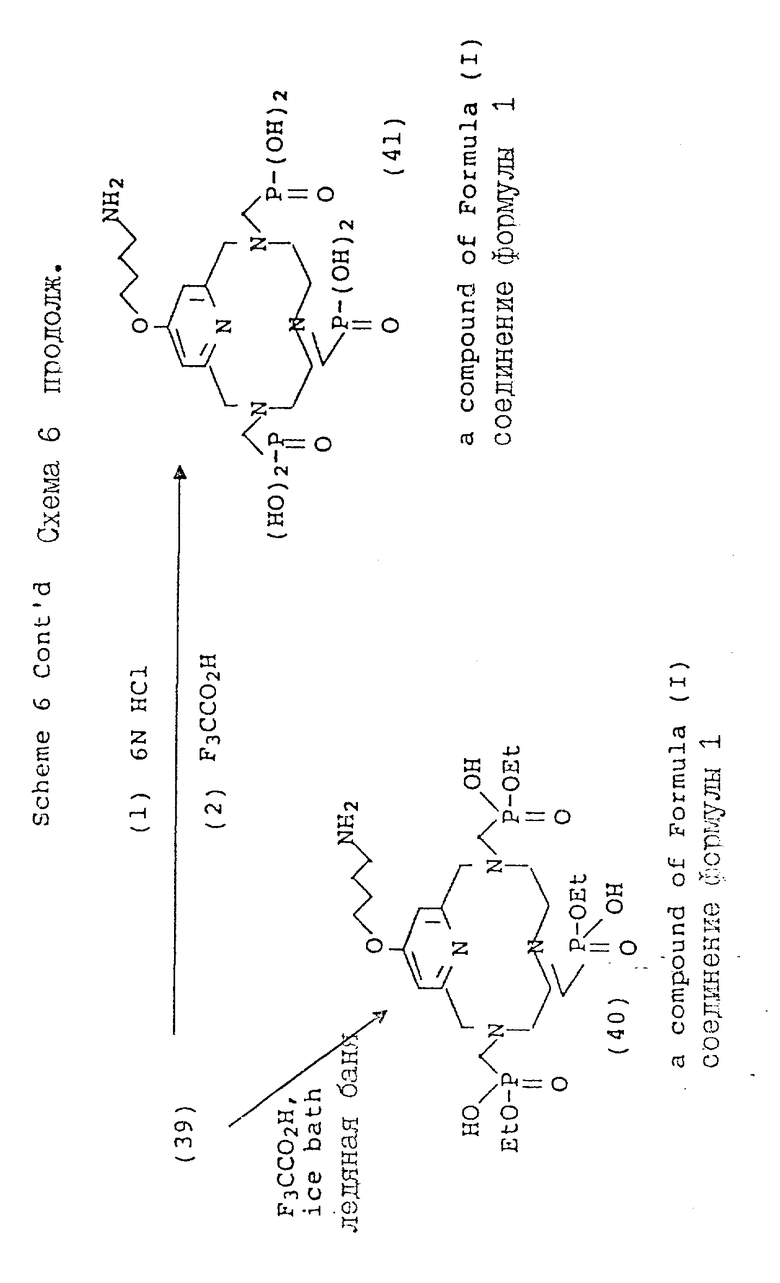
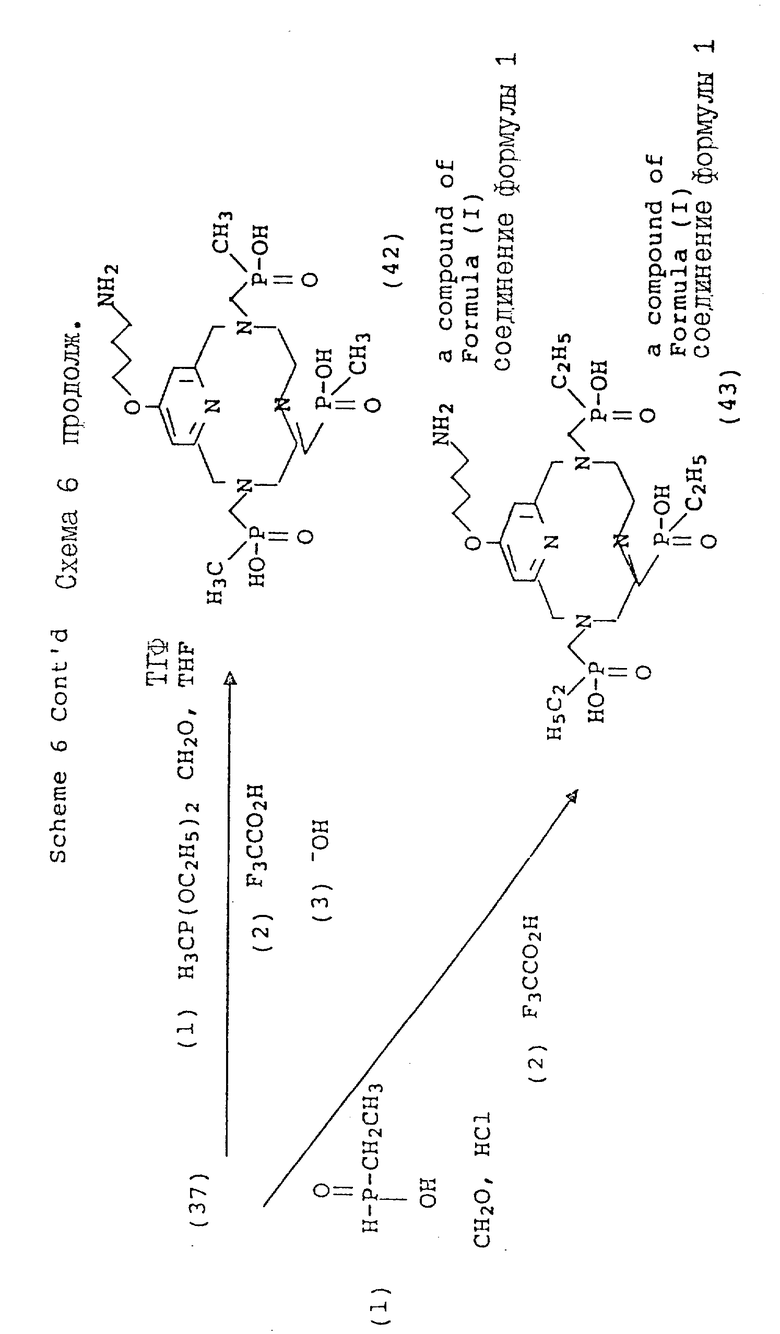



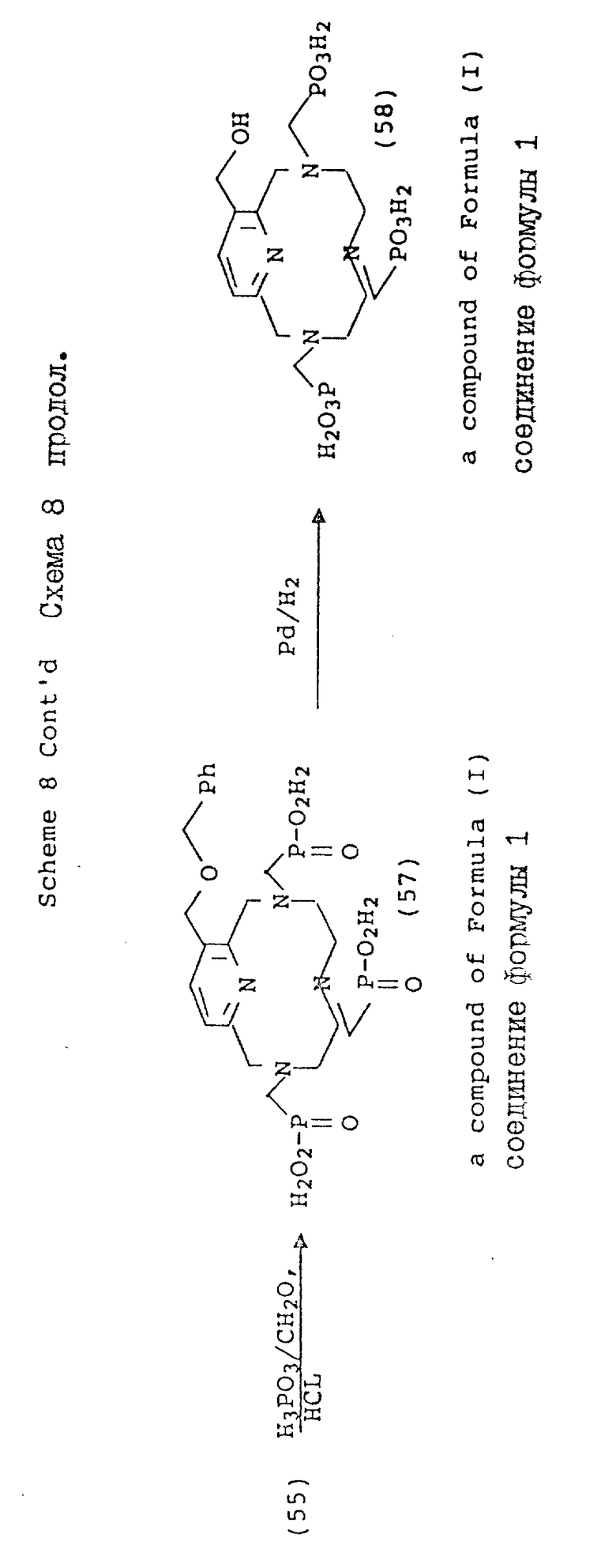


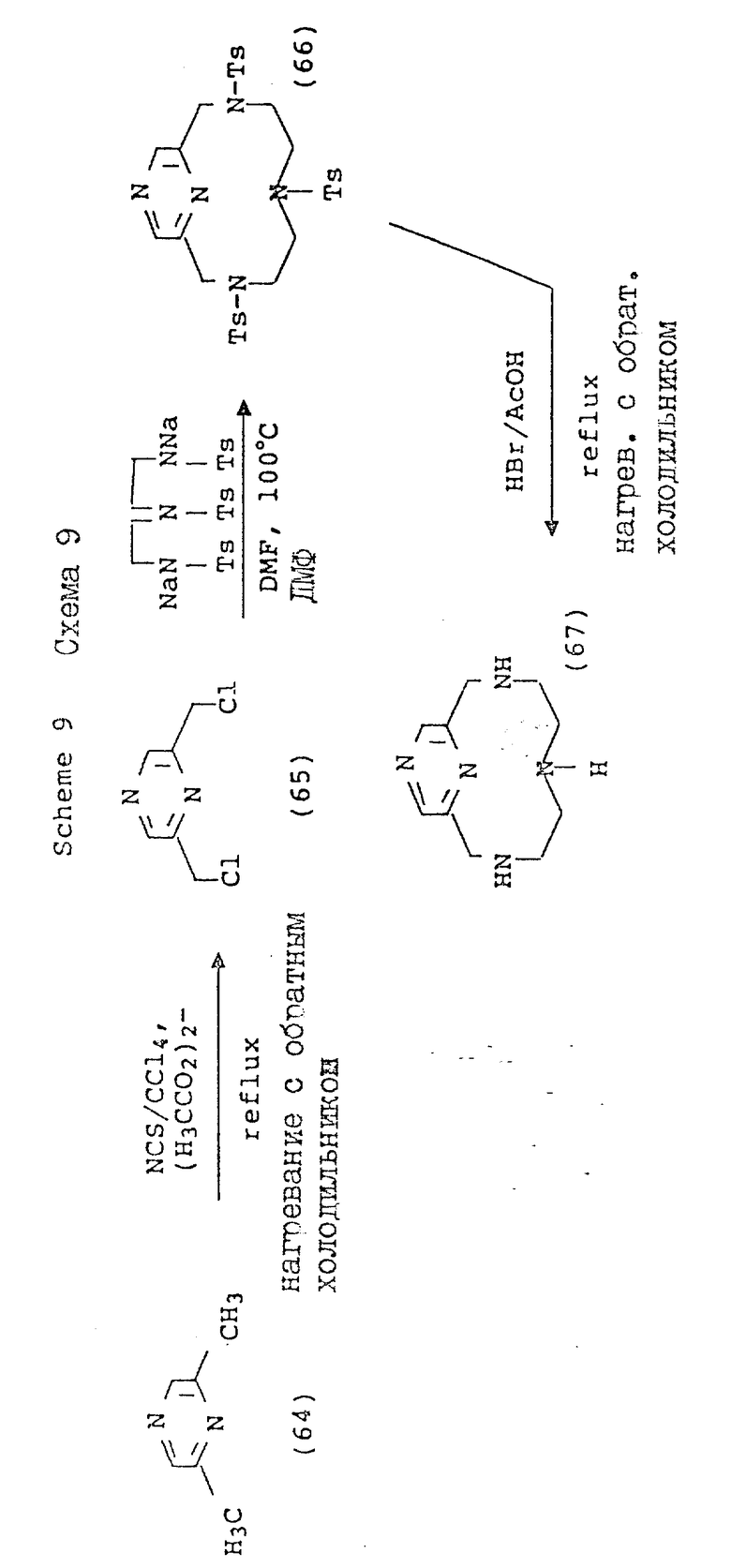

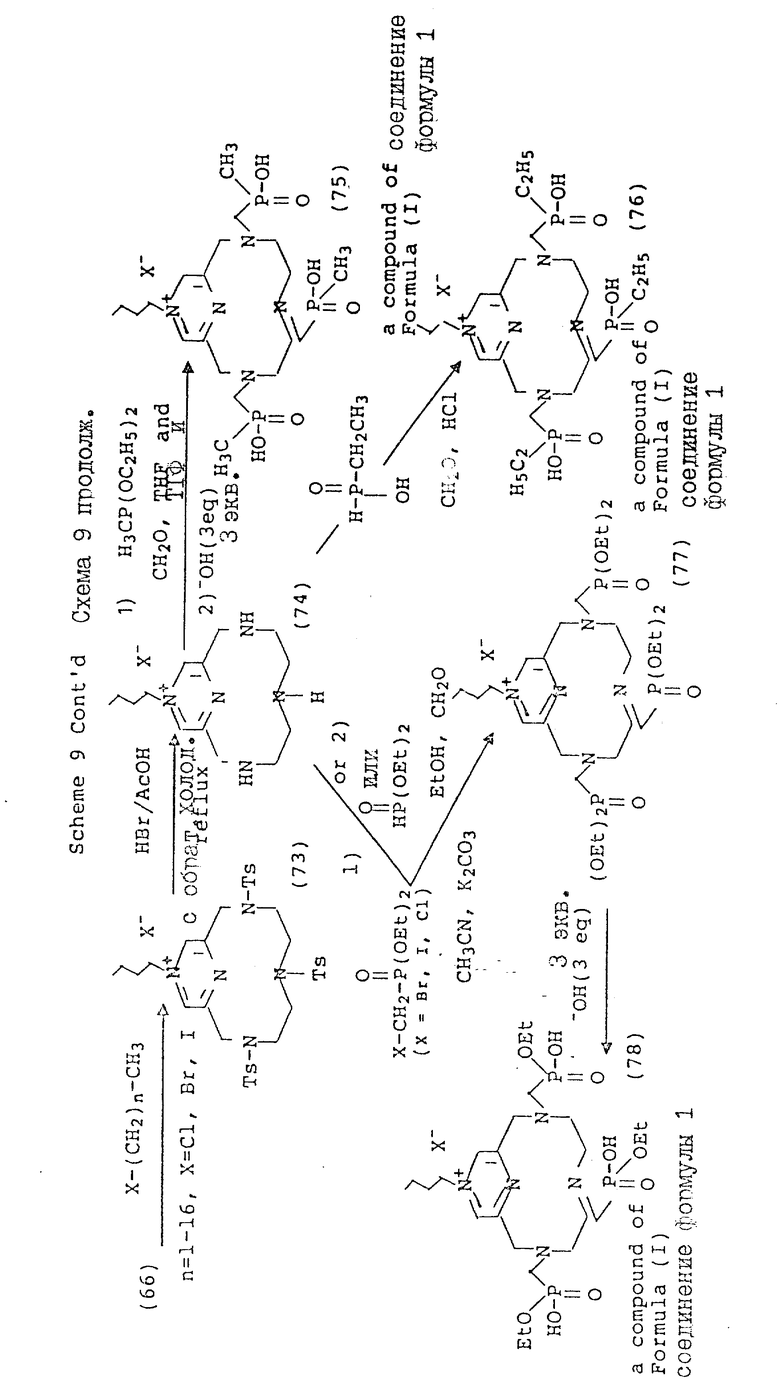
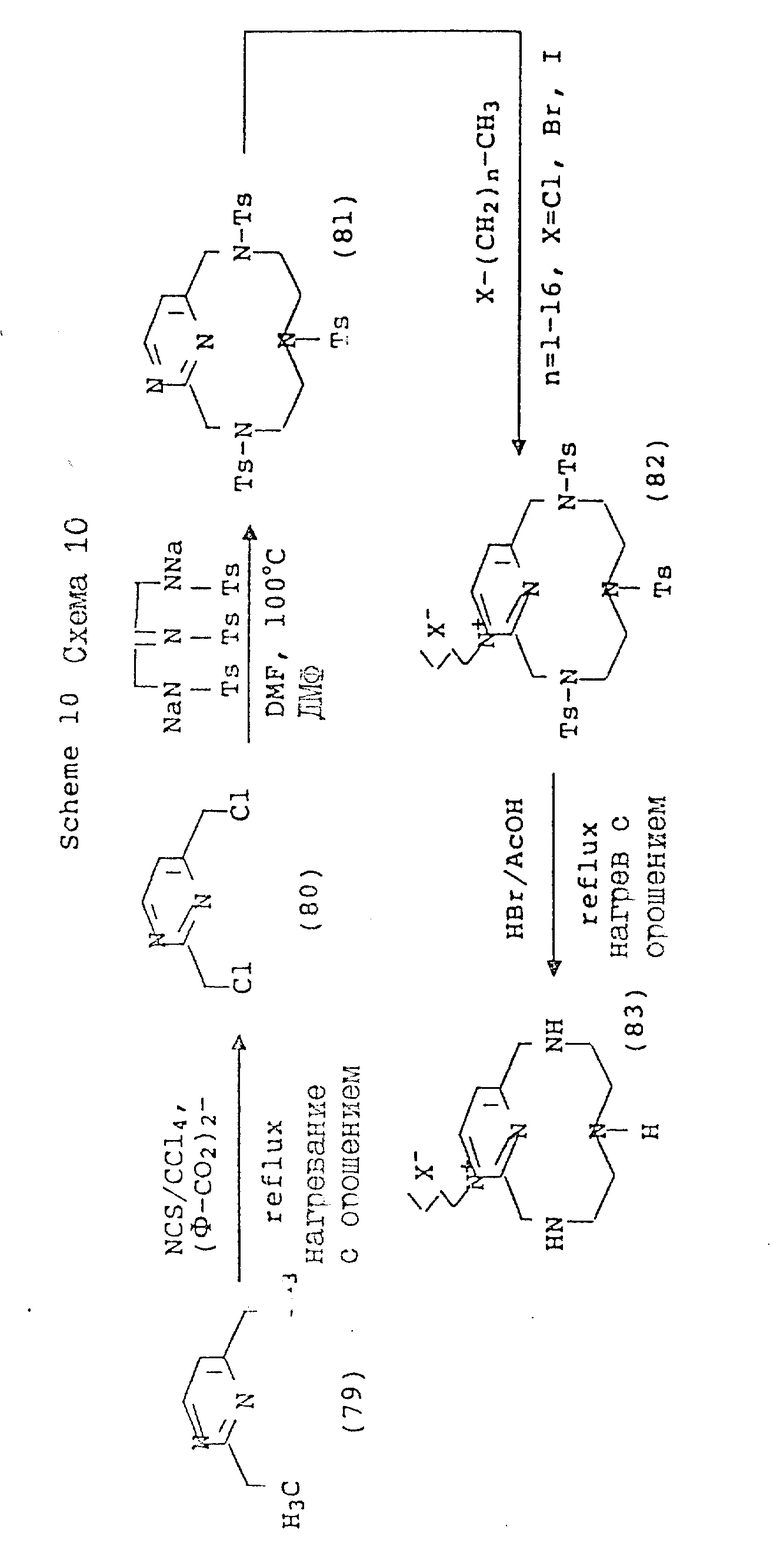
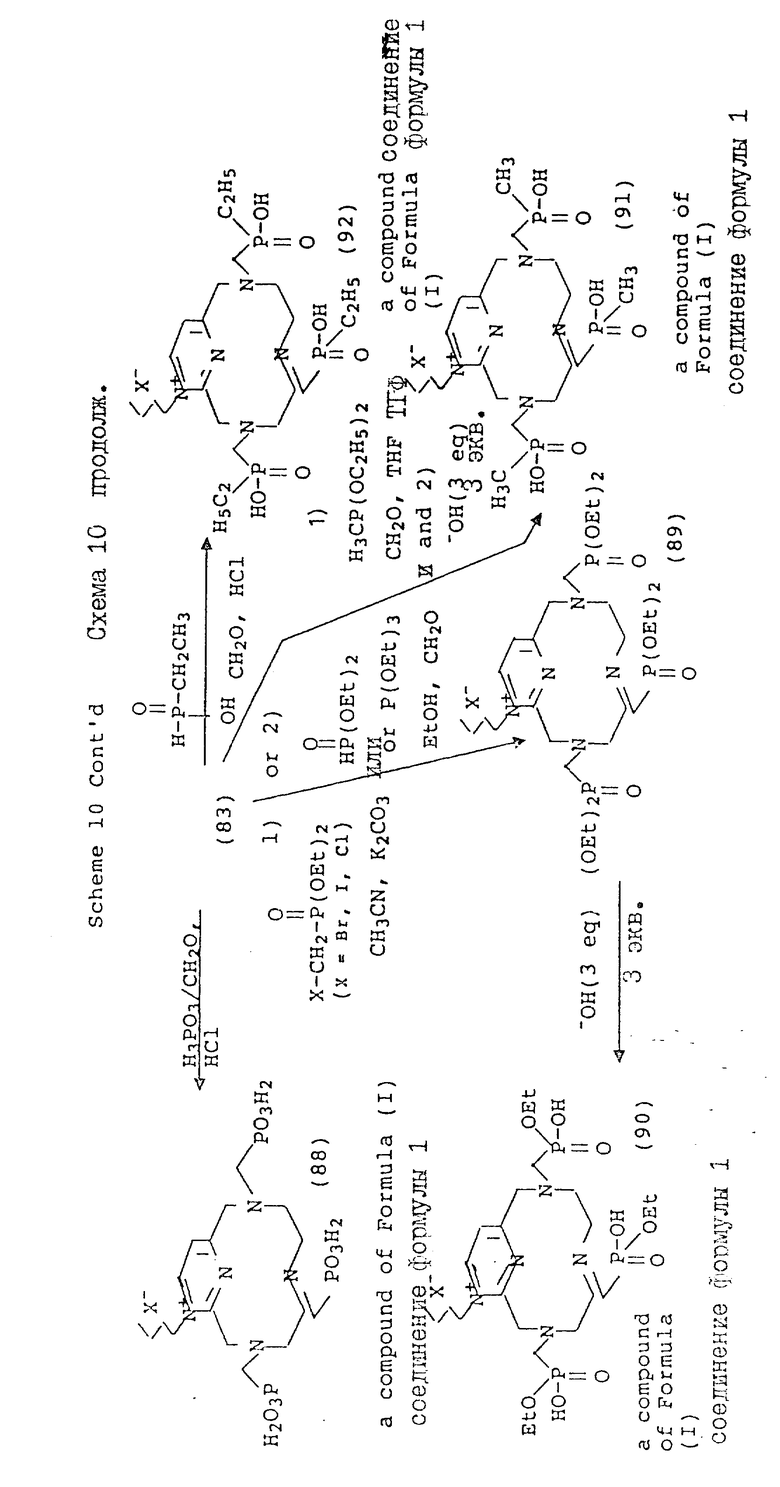




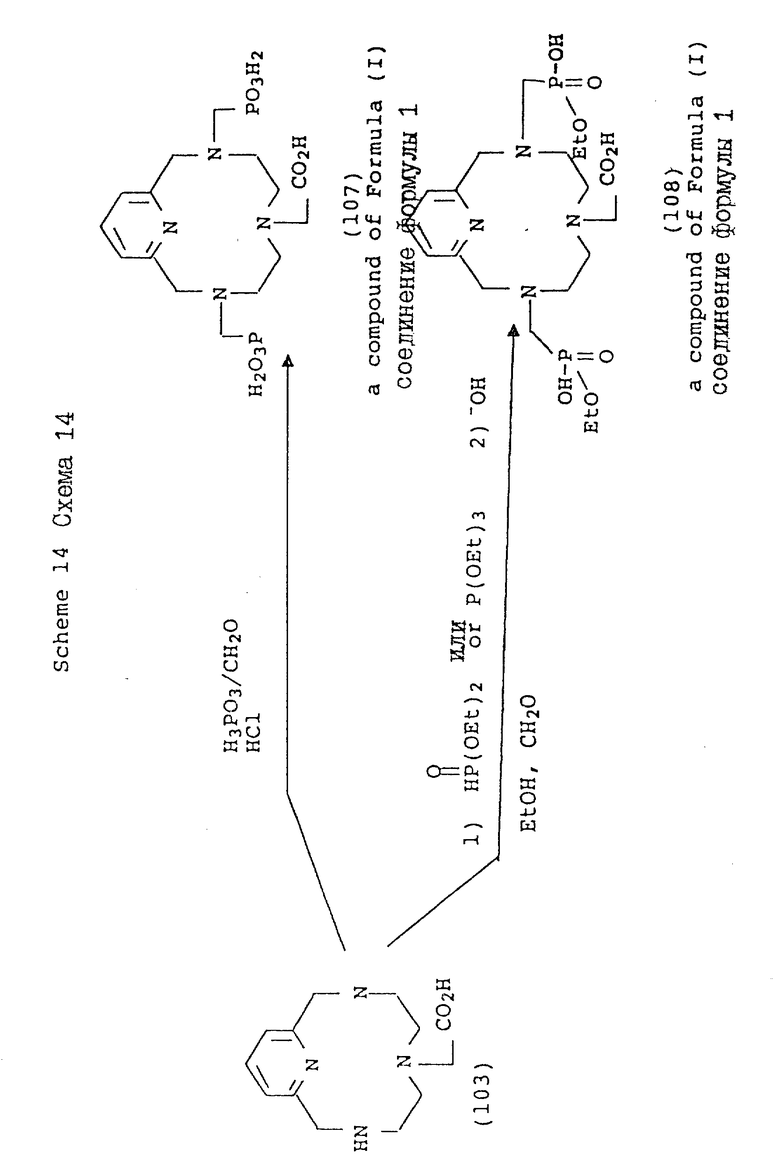

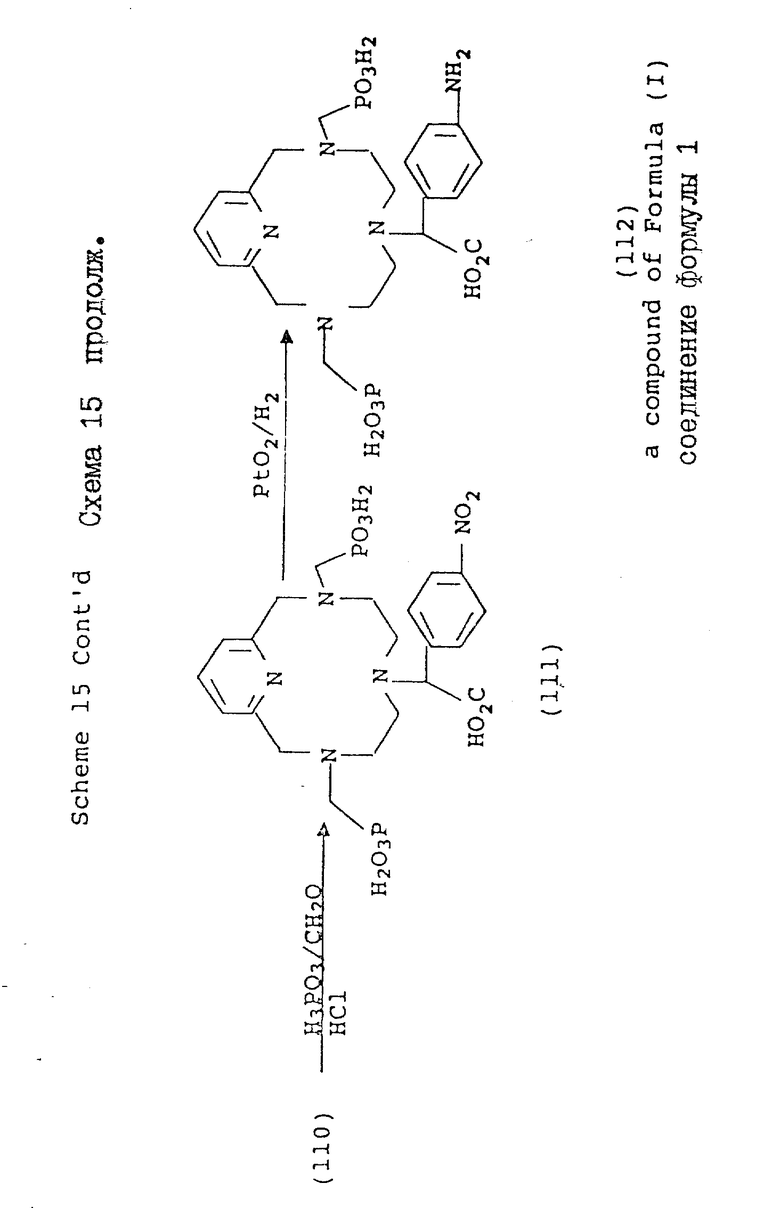
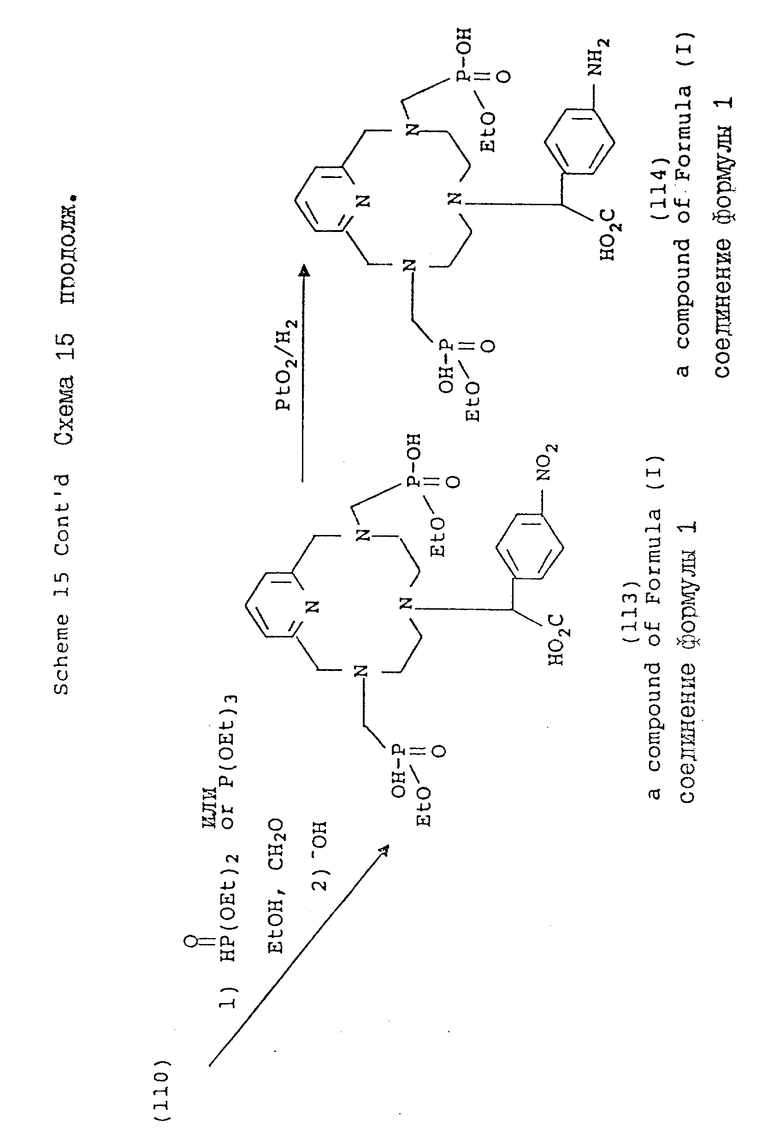
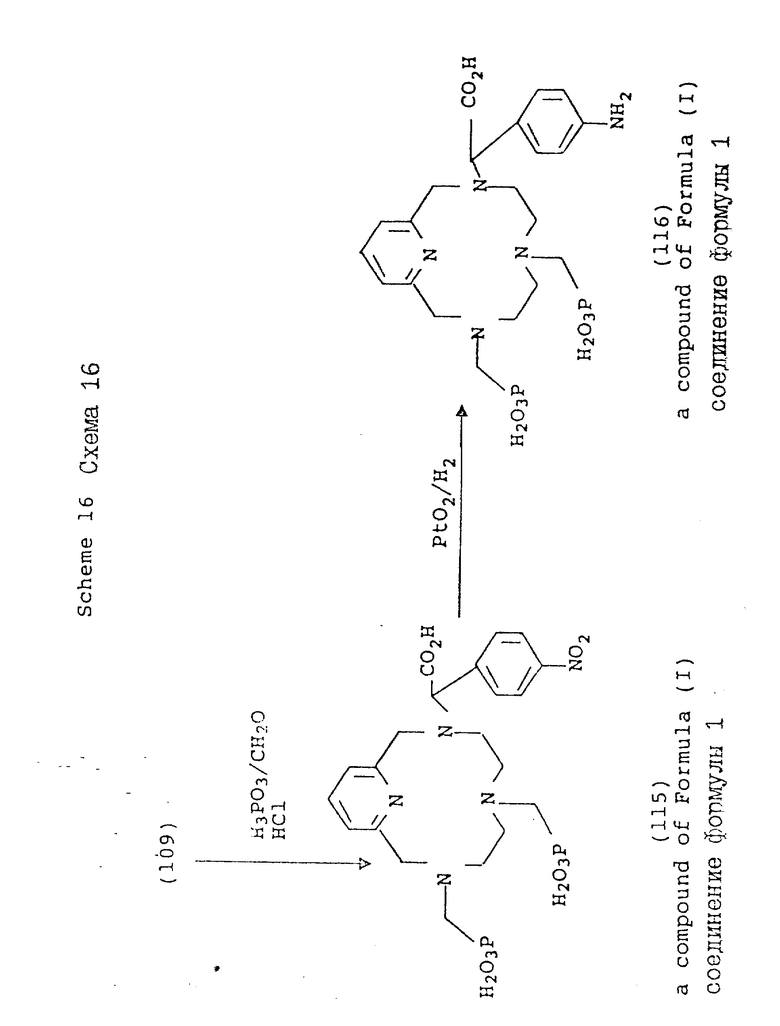

Предложены комплексы бициклополиазамакроциклофосфоновой кислоты с ионами металлов, в частности с 153Sm, 177Lu, 159Gd, 149Pm, 140La, 175Yb, 166Ho, 90Y, 47Sc, 186Re, 188Re, 142Pr, 99mTc, 67Ga, 68Ga, 105Ru, 111In, 113mIn или 115mIn. Комплексы ковалентно связаны с биологической активной молекулой, например с антителом или его фрагментом, с образованием конъюгатов. Полученные комплексы и конъюгаты полезны для использования в качестве радиофармацевтических препаратов для лечения и/или диагностики. Предложены также способы получения данных соединений. 3 с. и 5 з.п. ф-лы.
\ \ \ 1 1. Комплексы металлов, выбранных из группы <M^>153<D>Sm, <M^ >166<D>Ho, <M^ >90<D>Y, с бициклополиазамакроциклом формулы I \\\6 $$$ \\\1 где $$$ \ \ \4 X и Y - независимо H, C<Mv>1<D> - C<Mv>3<D>-алкил; \\\4 T - группа COOH, \\\6 $$$ \\\1 где R<M^>1<D> - OH, C<Mv>1<D> - C<Mv>5<D>-алкил или -O-(C<Mv>1<D> - C<Mv>5<D>-алкил), \\\1 или их фармацевтически приемлемые соли. \\\2 2. Комплексы по п.1, где X и Y - водород. \\\2 3. Комплексы по п. 1, где присутствует по крайней мере один радикал R<M^>1<D>, представляющий -O-(C<Mv>1<D> - C<Mv>5<D>-алкил). \\\2 4. Комплексы по п.1, у которых бициклополиазамакроцикл представляет собой 3,6,9,15-тетраазабицикло (9,3,1) пентадека-1(15), 11,13-триен-3,6,9-триметилфосфоновую кислоту. \ \ \ 2 5. Комплексы по пп.1 - 4, проявляющие противораковую активность. \\\2 6. Способ получения комплексов металлов, выбранных из группы <M^ >153<D>Sm, <M^ >166<D>Ho, <M^ >90<D>Y, с бициклополиазамакроциклом формулы I, где R имеет указанные значения, отличающийся тем, что бициклополиазамакроцикл формулы I подвергают взаимодействию с солью соответствующего металла в водной среде при pH 5 - 7. \\\2 7. Способ по п.6, отличающийся тем, что в качестве бициклополиазамакроцикла используют 3,6,9,15-тетраазабицикло (9,3,1) пентадека-1(15), 11,13-триен-3,6,9-триметиленфосфоновую кислоту. \\\2 8. Фармацевтическая композиция для лечения рака, включающая активный ингредиент и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного ингредиента содержит комплекс по пп.1 - 4 в эффективном количестве.
| EP, 0391766, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Jnorg | |||
| Chem, N 26, p.3458 - 3463, 1987. | |||
