Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям, которые являются производными 3-фенилпропионовой кислоты, фармацевтическим композициям, включающим их, и их применению для лечения и/или предотвращения заболеваний и состояний, опосредованных гамма-рецептором пролифератора-активатора пероксисом (PPARγ). Соединения проявляют способность к связыванию с PPARγ рецептором и изменению его активности.
Уровень техники
Более 20 лет тому назад была открыта группа тиазолидиндионовых соединений, проявляющая активность на подопытных грызунах с диабетом типа 2 и резистентностью к инсулину. Хотя механизм их действия не был известен, соединения успешно использовались в терапии диабета типа 2. Статьи, показывающие, что они оказывают свое действие через ядерный PPARγ рецептор, были опубликованы лишь в середине девяностых годов. Сейчас хорошо известно, что внутриклеточные рецепторные белки семейства PPAR контролируют экспрессию генов, вовлеченных в регуляцию липидно-углеводного метаболизма.
Заболевания, такие как гиперлипидемия, атеросклероз, ожирение и диабет типа 2, становятся серьезной проблемой не только для промышленно развитых стран. Подсчитано, что более 150 миллионов человек во всем мире страдают от диабета типа 2, и ожидается, что это число удвоится к 2025 году. В Польше на текущий момент около двух миллионов людей страдают от этого заболевания и такое же число подвержено риску его развития. Цены на медицинское обслуживание пациентов-диабетиков достигают 6-8 процентов от общего бюджета медицинского обслуживания. На начальной стадии диабет может быть бессимптомным и может начаться в любом возрасте, однако чаще всего встречается в среднем возрасте и у пожилых людей. Прогрессирование диабета типа 2 является результатом наложения таких физиологических расстройств, как тканевая резистентность к инсулину, недостаточная выработка инсулина поджелудочной железой, повышенная выработка инсулина, следующая за усиленным глюконеогенезисом. Наиболее частыми осложнениями диабета являются микрососудистые изменения в сетчатке глаза, почках и нервной системе, что приводит к повышенному риску слепоты, почечной недостаточности и заболеваниям нервной системы. Диабет также является основной причиной инфаркта и инсульта.
PPARγ рецепторы, принадлежащие к семейству ядерных рецепторов, играют роль в регуляции метаболизма липидов и их депонирования. Они экспрессируются в жировой ткани и толстой кишке и вовлечены в процесс липогенеза. Лиганды, активирующие PPARγ рецепторы, могут усиливать действие инсулина и понижать уровень глюкозы в плазме. Также они могут быть полезны для регулирования и лечения расстройств метаболизма липидов и энергетического баланса.
Известны соединения, являющиеся производными или аналогами L-тирозина, которые оказывают свое действие через модуляцию PRAPγ рецепторного ответа, таким образом действуя на метаболизм глюкозы, липидный гемостаз и энергетический баланс.
В международных патентных заявках под номерами WO 03/011834 и WO 03/011814 описаны N-(2-бензоилфенил)-L-тирозиновые производные, которые имеют активность частичного PPARγ агониста и могут быть использованы в лечении и профилактике, в частности, нарушенной толерантности к инсулину, диабета типа 1 и 2, дислипидемии, расстройств, вызванных синдромом X, таких как гипертония, ожирение, резистентность к инсулину, гипергликемия, атеросклероз, ишемия миокарда, коронарная болезнь сердца, заболевания почек, а также для улучшения познавательных функций и для лечения осложнений, вызванных диабетом. Описанные соединения представляют собой производные L-тирозина, в которых гидроксильная группа тирозина замещена винильной группой и азот в аминогруппе тирозина замещен 2-бензоилфенильной группой.
В международной патентной заявке под номером WO 01/17994 описаны соединения оксазола - антагонисты PPARγ, которые могут быть полезны в лечении диабета, ожирения, метаболического синдрома, нарушенной инсулиновой толерантности, синдрома X и сердечно-сосудистых заболеваний, включая дислипидемию. Соединения представляют собой производные L-тирозина, в которых карбоксильная группа тирозина замещена пятичленным гетероциклом, гидроксильная группа тирозина замещена (5-метил-2-фенилоксазол-4-ил)этильной группой, а азот аминогруппы тирозина замещен 2-бензоилфенильной группой.
В международной патентной заявке под номером WO 97/31907 описаны производные 4-гидроксифенилалкановых кислот с агонистической активностью к PPARγ. Среди других описаны производные L-тирозина, в которых гидроксильная группа тирозина замещена пятичленным гетероциклом, который сам может быть замещен, и азот в аминогруппе тирозина замещен двузамещенной фенильной группой, включающей 2-бензоилфенильную группу.
В уровне техники все еще существует необходимость в новых соединениях - лигандах PPARγ, которые можно использовать в лечении и/или профилактике диабета и осложнений, являющихся результатом или связанных с диабетом, особенно расстройств липидного метаболизма и сердечно-сосудистых заболеваний.
Сущность изобретения
Настоящее изобретение относится к новым соединениям, являющимся производными 3-фенилпропионовой кислоты формулы (I):
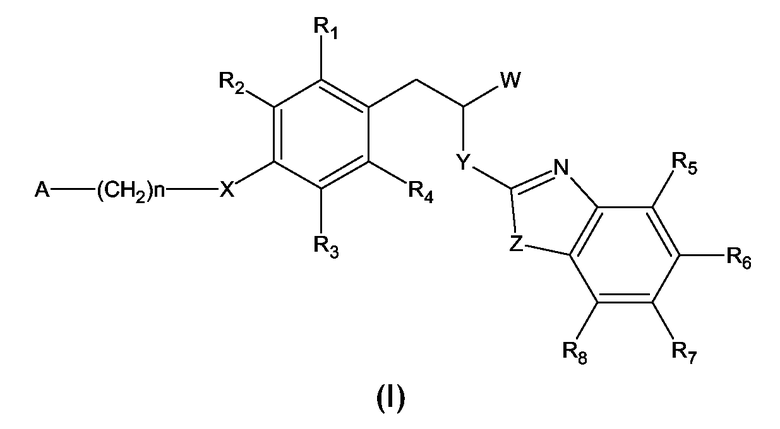
где
W представляет COOH группу или ее биоизостеры, или -COO-C1-C4-алкильную группу;
Y представляет NH, N-C1-C10-алкил, O, или S;
Z представляет NH, N-C1-C10-алкил, N-арил, N-гетероарил, S, или O;
X представляет O, S, NH, N-C1-C10-алкил, N-арил, NSO2-C1-C10-алкил, N-SO2-арил, или N-SO2-гетероарил;
R1, R2, R3, R4, R5, R6, R7 и R8 каждый независимо представляет атом водорода или заместитель, выбранный из группы, включающей
C1-C4-алкил, C1-C4-алкокси, C3-C7-циклоалкил, C3-C7-циклоалкокси, C1-C4-тиоалкокси, C3-C7-циклотиоалкокси, атом галогена, замещенный галогеном
C3-C7-циклоалкил, арил, гетероарил, -NO2, -CN, -SO2-NH2, -SO2-NH-C1-C4-алкил, -SO2-N(C1-C4-алкил)2, -CO-C1-C4-алкил, -O-CO-C1-C4-алкил, -CO-O-C1-C4-алкил, -CO-арил, -CO-NH2, -CO-NH-C1-C4-алкил, -CO-N(C1-C4-алкил)2;
А представляет C1-C4-алкил, C3-C7-циклоалкил, замещенный галогеном C3-C7-циклоалкил, арил, гетероарил, гетероциклил, -NH-CO-C1-C4-алкил, N(C1-C4-алкил)-CO-C1-C4-алкил, -NH-CO-арил, -N(C1-C4-алкил)-CO-арил, -N(C1-C4-алкил)-CO-C3-C7-циклоалкил, -NH-CO-NH2, -NH-CO-NH-C1-C4-алкил, -NH-CS-NH-C1-C4-алкил, -NH-CO-NH-арил, -NH-CS-NH-арил, -SO2-C1-C4-алкил, -SO2-арил, или -SO2-гетероарил; где арил, гетероарил и гетероциклил, необязательно, замещены одним или более заместителями, независимо выбранными из группы, включающей C1-C4-алкил, C1-C4-алкокси, C1-C4-тиоалкокси, этилендиокси, CN, галоген или фенил, где фенил необязательно замещен одним или более заместителями, независимо выбранными из C1-C4-алкила, C1-C4-алкокси и атома галогена; и
n представляет целое число от 0 до 4 включительно; и
их фармацевтически приемлемые соли.
Одна группа соединений по изобретению включает такие соединения, где W представляет COOH.
Другая группа соединений по изобретению включает такие соединения, где Y представляет NH.
Другая группа соединений по изобретению включает такие соединения, где Y представляет O.
Другая группа соединений по изобретению включает такие соединения, где Y представляет N-C1-C4-алкил, в особенности N-CH3.
Еще одна группа соединений по изобретению включает такие соединения, где Z представляет O.
Еще одна группа соединений по изобретению включает такие соединения, где Z представляет S.
Еще одна группа соединений по изобретению включает такие соединения, где Z представляет N-C1-C4-алкил, в особенности N-CH3.
Еще одна группа соединений по изобретению включает такие соединения, где Z представляет N-фенил.
Еще одна группа соединений по изобретению включает такие соединения, где X представляет O.
Еще одна группа соединений по изобретению включает такие соединения, где X представляет S.
Еще одна группа соединений по изобретению включает такие соединения, где X представляет NSO2-C1-C4-алкил, в особенности NSO2-CH3.
Еще одна группа соединений по изобретению включает такие соединения, где W представляет COOH, Y представляет NH, Z представляет O и X представляет O.
Еще одна группа соединений по изобретению включает такие соединения, где W представляет COOH, Y представляет O, Z представляет O и X представляет O.
Еще одна группа соединений по изобретению включает такие соединения, где W представляет COOH, Y представляет NH, Z представляет O и X представляет
NSO2-C1-C4-алкил, в особенности NSO2-CH3.
Еще одна группа соединений по изобретению включает такие соединения, где W представляет COOH, Y представляет NH, Z представляет S и X представляет NSO2-C1-C4-алкил, в особенности NSO2-CH3.
Частным вариантом воплощения определенных выше соединений формулы (I) являются такие соединения, где каждый из заместителей R1 - R8 представляет атом водорода.
Другим частным вариантом воплощения определенных выше соединений формулы (I) являются такие соединения, где n равняется 1 или 2.
Другая группа соединений по изобретению включает такие соединения, где A представляет гетероциклил, который необязательно замещен одним или более заместителями, независимо выбранными из группы, включающей C1-C4-алкил, C1-C4-алкокси, C1-C4-тиоалкокси, CN, атом водорода и фенил.
В вышеуказанной группе A предпочтительно представляет изоксазолил, необязательно замещенный одним или более заместителями, независимо выбранными из C1-C4-алкила, в особенности -CH3.
Следующая группа соединений по изобретению включает такие соединения, где A представляет фенил, который необязательно замещен, в особенности этилендиоксигруппой.
Следующая группа соединений по изобретению включает такие соединения, где A представляет -N(C1-C4-алкил)-CO-C3-C7-циклоалкил, в особенности -N(CH3)-CO-циклогексил.
Следующая группа соединений по изобретению включает такие соединения, где A представляет -N(C1-C4-алкил)-CO-гетероарил, где гетероарил необязательно замещен одним или более заместителями, независимо выбранными из группы, включающей C1-C4-алкил, C1-C4-алкокси, C1-C4-тиоалкокси, CN, атом галогена, фенил, и фенил, необязательно замещеный одним или более заместителями, независимо выбранными из группы, включающей C1-C4-алкил, C1-C4-алкокси и атом галогена.
Предпочтительный гетероарил представляет пиримидинил, необязательно замещенный одним или более заместителями, независимо выбранными из группы, включающей C1-C4-алкил, C1-C4-алкокси, атом галогена, фенил, и фенил необязательно замещеный одним или более заместителями, независимо выбранными из группы, включающей C1-C4-алкил, C1-C4-алкокси и атом галогена.
Как примеры конкретных соединений изобретения могут быть упомянуты следующие:
1. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2,3-дигидро-1,4-бензодиоксин-6-илметокси)фенил]пропионовая кислота,
2. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-((3,5-диметилизоксазол-4-ил)метокси)фенил]пропионовая кислота,
3. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]этокси)фенил]пропионовая кислота,
4. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-[5-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-4-ил]этокси)фенил]пропионовая кислота,
5. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(4-{2-[[6-(2-хлорфенил)-5-циано-2-(метилтио)пиримидин-4-ил](метил)амино]этокси})фенил]пропионовая кислота,
6. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-(2-трет-бутил-5-метил-1,3-оксазол-4-ил)этокси)фенил]пропионовая кислота,
7. (2S)-2-(1,3-бензотиазол-2-иламино)-3-[4-(2-(2-трет-бутил-5-метил-1,3-оксазол-4-ил)этокси)фенил]пропионовая кислота,
8. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]тиоэтокси)фенил]пропионовая кислота,
9. (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]этилметансульфониламино)фенил]пропионовая кислота, и
10. (2S)-2-(1,3-бензоксазол-2-илокси)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]этокси)фенил]пропионовая кислота,
и его фармацевтически приемлемые соли.
Соединения по изобретению имеют высокое сродство к гамма-рецептору пролифератора-активатора пероксисом (PPARγ). Таким образом, соединения показывают способность к связыванию с PPARγ и изменению его активности.
Изобретение относится также к фармацевтической композиции, включающей по меньшей мере одно определенное выше соединение формулы (I) или его фармацевтически приемлемую соль, необязательно в сочетании с другими фармакологически активными компонентами, вместе с одним или более фармацевтически приемлемыми носителями и/или вспомогательными веществами.
Изобретение относится также к определенному выше соединению формулы (I) для применения в качестве лекарственного средства.
Изобретение далее относится к применению определенного выше соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения и/или профилактики заболеваний и состояний, опосредованных гамма-рецептором пролифератора-активатора пероксисом (PPARγ).
Изобретение далее относится к способу лечения и/или профилактики заболеваний и патологических состояний, опосредованных гамма-рецептором пролифератора-активатора пероксисом (PPARγ) у нуждающегося в этом млекопитающего, включающему введение указанному млекопитающему определенного выше соединения формулы (I) в терапевтически или профилактически эффективном количестве.
Так, в PPARγ-опосредованные заболевания и патологические состояния включают, в частности, нарушенную толерантность к инсулину, резистентность к инсулину, диабет типа 1 и 2 и осложнения, являющиеся результатом или связанные с диабетом, такие как периферическая невропатия, почечная недостаточность, ретинопатия, дислипидемия, расстройства, вызванные синдромом X, такие как гипертензия, ожирение, гипергликемия, атеросклероз, ишемия миокарда, коронарная болезнь сердца и другие сердечно-сосудистые заболевания и заболевания почек.
Соединения изобретения могут быть также полезны в улучшении познавательных функций.
Подробное описание изобретения
Определения
Термин “биоизостер”, который используется здесь, относится к химическому фрагменту, который замещает другую группу в молекуле активного соединения без значительного влияния на его биологическую активность. В этом случае могут быть затронуты другие свойства активного соединения, такие как, например, его стабильность, как лекарственного средства.
В качестве биоизостерных групп для карбоксильной (COOH) группы могут быть, в особенности, упомянуты 5-членные гетероциклические группы, имеющие от 1 до 4 гетероатомов, выбранных из азота, кислорода и серы, такие как, например, 1,3,4-оксадиазолил, 1,2,3-оксадиазолил, 1,2,5-оксадиазолил, 1,2,4-оксадиазолил, 1,3,4-тиодиазолил, 1,2,4-тиодиазолил, 1,2,3-тиодиазолил, 1,2,5-тиодиазолил, фурил, тиенил, пирролил, пиразолил, имидазолил, изоксазолил, изотиазолил и N-замещенный тетразолил. 5-членные гетероциклические группы могут быть необязательно замещены 1 или 2 заместителями, выбранными из группы, включающей фенил, пиридинил, линейную или разветвленную алкильную группу, аминогруппу, гидроксильную группу, фтор, хлор, бром, иод, трифторметил, трифторметокси, трифтортиометокси, алкокси и тиоалкокси.
В качестве биоизостерных групп для карбоксильной (COOH) группы могут быть также упомянуты фенил и 6-членные гетероциклические группы, имеющие от 1 до 4 гетероатомов, выбранных из азота, кислорода и серы, такие как, например, пиридинил, пиразинил, пиридазинил, пиримидинил, триазинил, тетразинил и другие. Фенил и 6-членные гетероциклические группы могут быть необязательно замещены 1 или 2 заместителями, выбранными из группы, включающей фенил, пиридинил, линейную или разветвленную алкильную группу, аминогруппу, гидроксильную группу, фтор, хлор, бром, иод, трифторметил, трифторметокси, трифтортиометокси, алкокси и тиоалкокси.
Термин “галоген” относится к атомам, выбранным из атомов фтора, хлора, брома и иода.
Термин “алкил” относится к насыщенным, линейным или разветвленным углеводородным группам, имеющим определенное число атомов углерода. Как конкретные алкильные заместители могут быть упомянуты следующие: метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил, пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, гексил, 1-метилпентил, 2-метилпентил, 1-этилбутил, 2-этилбутил, 3,3-диметилбутил, гептил, 1-этилпентил, октил, нонил и децил.
Термин “арил” относится к моно- и бициклическим ароматическим группам, имеющим от 6 до 14 атомов углерода. Примерами арильных групп являются фенил, толил, ксилил, нафтил, такие как нафтил-1-ил, нафтил-2-ил, 1,2,3,4-тетрагидранафт-5-ил и 1,2,3,4-тетрагидранафт-6-ил.
Термин “гетероарил” относится к моно- и бициклической гетероароматической группе, имеющей от 5 до 13 атомов углерода и от 1 до 4 гетероатомов, выбранных из N, O и S. Примерами гетероарильных групп являются пиррол-1-ил, пиррол-2-ил, пиррол-3-ил, фурил, тиенил, имидазолил, оксазолил, тиазолил, изоксазолил, 1,2,4-триазолил, оксадиазолил, тиодиазолил, тетразолил, пиридинил, пиримидинил, 1,3,5-триазинил, индолил, бензо[b]фурил, бензо[b]тиенил, индазолил, бензимидазолил, азаиндолил, циннолил, изохинолинил и карбазолил.
Термин “циклоалкил” относится к насыщенной или частично не насыщенной циклической углеводородной группе, имеющей от 3 до 7 атомов углерода. Примерами циклоалкильных групп являются циклопропил, циклобутил, циклопентил, циклопентенил, циклопентадиенил, циклогексил, циклогексенил и циклогептил.
Термин “гетероциклил” относится к насыщенной или частично ненасыщенной 5-, 6-членной циклической углеводородной группе, имеющей от 1 до 4 гетероатомов, выбранных из N, O и S. Предпочтительно насыщенный или частично не насыщенный циклический углеводород является моноциклическим и включает 4 или 5 атомов углерода и от 1 до 3 гетероатомов. Примерами гетероциклических групп являются пиперидинил, пиперазинил, морфолинил и пирролидинил.
Соединения изобретения содержат хиральный центр при атоме углерода, несущем W группу, и могут существовать в форме соответствующих энантиомеров, смесей энантиомеров, а также в виде рацемических смесей.
Следовательно, R и S энантиомеры, смеси энантиомеров, а также рацемические смеси соединений формулы (I) являются частью изобретения.
Таким образом, в одном варианте воплощения изобретение относится к соединениям формулы (I), имеющим стереохимическую конфигурацию, такую, как показано в формуле (IA):
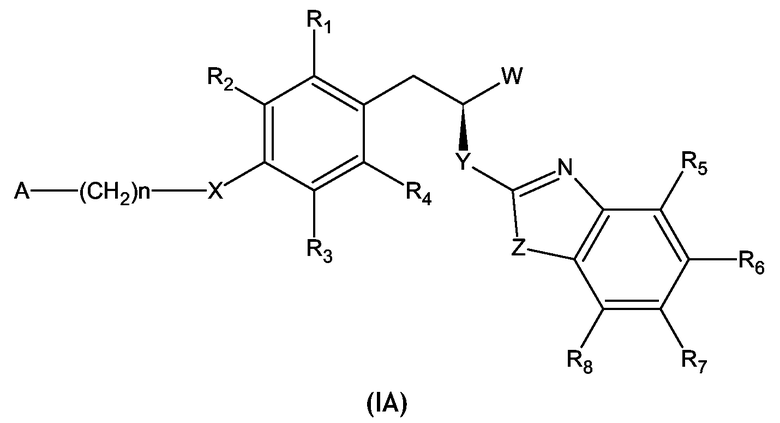
где W, X, Y, Z, A, n и R1 - R8 имеют значения, определенные выше для формулы (I),
и их фармацевтически приемлемым солям.
Во втором варианте воплощения изобретение относится к соединениям формулы (I), имеющим стереохимическую конфигурацию, такую, как показано в формуле (IB):
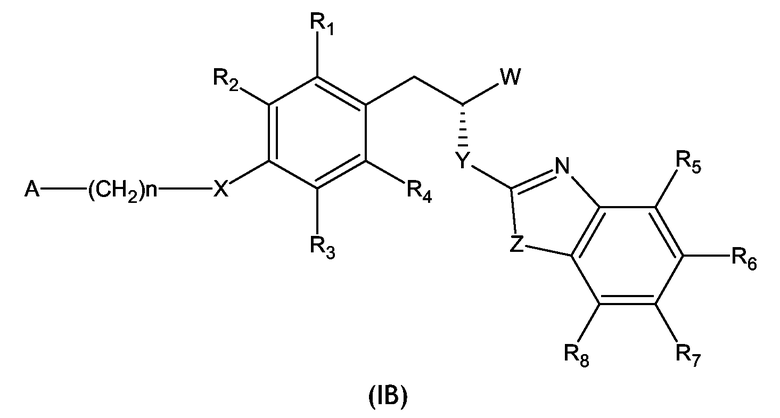
где W, X, Y, Z, A, n и R1 - R8 имеют значения, определенные выше для формулы (I),
и их фармацевтически приемлемым солям.
Соединения формулы (I), несущие остаток основания, могут быть превращены в соли с органическими и неорганическими кислотами общепринятым и известным способом посредством обработки подходящей кислотой в органическом растворителе, таком как спирт, кетон, эфир или хлорированный растворитель, и извлечения соли обычным способом. Примерами таких солей являются соли с фармацевтически приемлемыми неорганическими и органическими кислотами. В качестве примеров солей неорганических кислот могут быть упомянуты гидрохлорид, гидробромид, нитрат, сульфат, гидросульфат, пиросульфат, сульфит, пиросульфит, фосфат, моногидрофосфат, дигидрофосфат, метафосфат и пирофосфат. В качестве примеров солей органических кислот могут быть упомянуты ацетат, пропионат, акрилат, 4-гидроксибутират, каприлат, капронат, деканоат, оксалат, малонат, сукцинат, глутарат, адипат, пимелат, малеат, фумарат, цитрат, тартрат, лактат, фенилацетат, манделат, себацат, суберат, бензоат, фталат, алкил- и арилсульфонаты, такие как метансульфонат, пропансульфонат, п-толуолсульфонат, ксиленсульфонат, салицилат, циннамат, глутамат, аспартат, глюкуронат и галактуронат.
Соединения формулы (I), несущие кислотную группу, могут быть превращены в соли с органическими и неорганическими основаниями общепринятым и известным способом посредством реакции соединения формулы (I) с подходящим органическим или неорганическим основанием. Соли с фармацевтически приемлемыми основаниями включают соли щелочных и щелочно-земельных металлов, таких как Li, Na, K, Mg или Ca, соли аммония и соли с основными органическими соединениями, такими как, например, аргинин, гистидин, пиперидин, морфолин, пиперазин, этилендиамин или триэтиламин, а также четвертичные соли аммония.
Настоящее изобретение относится также к фармацевтическим композициям, включающим в себя соединения формулы (I) с фармацевтическими вспомогательными веществами, в зависимости от выбранного пути введения.
Одним из вариантов воплощения изобретения являются фармацевтические композиции, пригодные для перорального введения. Фармацевтические композиции, пригодные для перорального введения, могут быть в форме таблеток, капсул, пилюль, пастилок, порошков, или гранул, или растворов, или дисперсий в жидкости или им подобные. Каждая из вышеупомянутых форм будет заключать в себе предопределенное количество соединения изобретения в качестве активного компонента. Композиция в форме таблетки может быть приготовлена, используя любые фармацевтические вспомогательные вещества, известные в технике для этой цели и обычно используемые для приготовления твердых фармацевтических композиций. Примерами таких вспомогательных веществ являются крахмал, лактоза, микрокристаллическая целлюлоза, стеарат магния и связующие вещества, например поливинилпирролидон. Более того, активное соединение может быть включено в состав препарата с контролируемым высвобождением, такого как таблетки, включающие в себя гидрофильную или гидрофобную матрицы.
Фармацевтическая композиция в форме капсул может быть приготовлена с использованием общепринятых методов, например, посредством введения смеси активного соединения и вспомогательных веществ в твердые желатиновые капсулы. С другой стороны, может быть создана и заключена в твердые желатиновые капсулы полутвердая матрица активного соединения и высокомолекулярного полиэтиленгликоля или мягкие желатиновые капсулы могут быть наполнены раствором активного соединения в полиэтиленгликоле или его дисперсией в пищевом масле. Также рассматриваются порошковые формы для растворения перед использованием (например, лиофилизированные порошки). С другой стороны, для инъекционных лекарственных форм также могут быть использованы масляные наполнители.
Жидкие формы для парентерального введения могут быть составлены для введения инъекцией или продолжительной инфузией.
Приемлемыми путями введения посредствам инъекции являются внутривенное, внутрибрюшинное, внутримышечное и подкожное, причем внутривенные инъекции являются обычно предпочтительными. Обычная композиция для внутривенного введения включает стерильный изотонический водный раствор или дисперсию, включающую, например, активное вещество и декстрозу или хлорид натрия. Другими примерами подходящих вспомогательных веществ являются раствор лактата Рингера для инъекций, раствор лактата Рингера для инъекций с декстрозой, Нормосол-М с декстрозой, ацилированный раствор Рингера для инъекций. Лекарственная форма для инъекций может при желании включать вспомогательный растворитель, например полиэтиленгликоль; хелатирующий агент, например этилендиаминтетрауксусную кислоту; стабилизирующий агент, например циклодекстрин; и антиоксидант, например пиросульфат натрия.
Введенная доза будет зависеть от состояния пациента и выбранного пути введения и будет регулироваться врачом.
Соединения изобретения могут быть получены, используя процессы, описанные ниже и проиллюстрированные примерами.
Соединения формулы (I), где W имеет значение, отличное от -COOH и -COO-C1-C4-алкила, могут быть получены замещением атома водорода при X в молекуле соединения (II) A(CH2)n-группой.
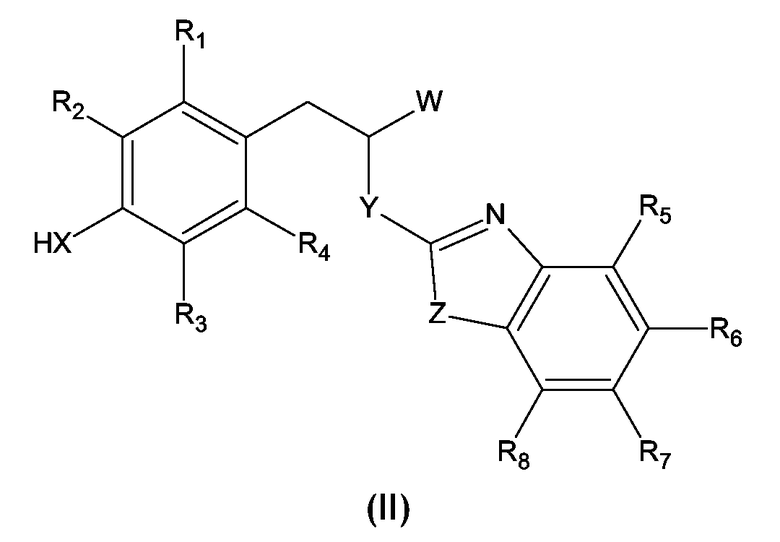
где X, Y, Z, A, n, и R1 - R8 имеют значения, выше определенные для формулы (I), и W имеет значение, отличное от -COOH и -COO-C1-C4-алкила.
Упомянутое замещение может быть выполнено посредством реакции Мицунобу описанного выше соединения формулы (II) с соединением формулы A(CH2)n-OH, где A и n имеют значения, описанные выше, согласно схеме 1
Схема 1
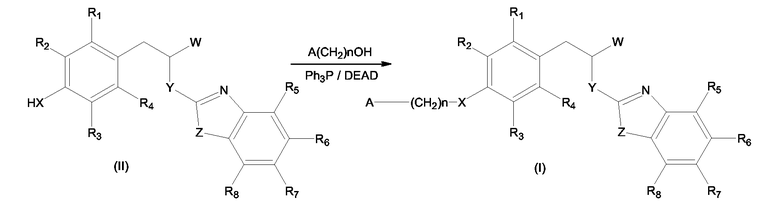
Реакция Мицунобу может быть проведена в безводных растворителях, таких как эфир или замещенный галогеном алкан, в присутствии диазосоединений, таких как DEAD, DIAD, ADDP, и трифенилфосфина, обычно в интервале температур от -20 до 20°С.
С другой стороны, упомянутое замещение атома водорода при X может быть выполнено алкилированием соединения формулы (II), где X, Y, Z и R1 - R8 имеют значения, определенные выше для формулы (I), и W имеет значение, отличное от -COOH и -COO-C1-C4-алкил, соединением формулы A(CH2)n-V, где A и n имеют значения, определенные выше для формулы (I), и V представляет собой уходящую группу, выбранную из галогенов и алкилсульфонильных или арилсульфонильных групп, в присутствии сильного основания, способного образовывать анион из соединения (II), такого как, например, гидрид натрия, с образованием соединения формулы (I) согласно схеме 2
Схема 2
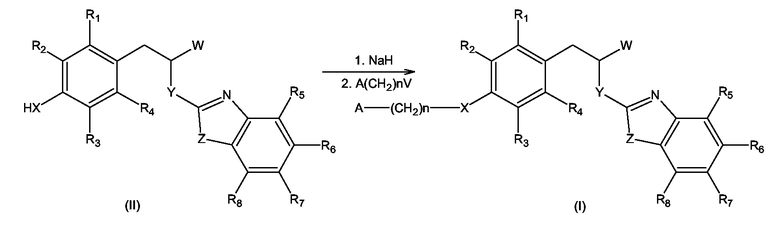
Реакция алкилирования может быть проведена в инертном органическом растворителе, таком как безводные ДМФА, ТГФ, ДМСО. Сильным основанием, способным образовывать анион, может быть гидрид натрия. Может быть использован сухой гидрид натрия или его суспензия в минеральном масле. Образование аниона проводится при комнатной температуре до полного прекращения выделения водорода. Далее, на второй стадии прибавляли алкилирующий реагент A(CH2)n-V, чистый или в виде раствора в инертном органическом растворителе, таком как ДМФА, ТГФ, ДМСО. Вторая стадия алкилирования проводится в интервале температур от 0 до 100°С.
Соединения по изобретению формулы (I), где W представляет -COOH или
-COO-C1-C4-алкил и X, Y, Z, A, n, и R1 - R8 имеют значения, определенные выше для формулы (I), могут быть получены посредством:
а) замещения атома водорода при X A(CH2)n-группой в соединении формулы (III)
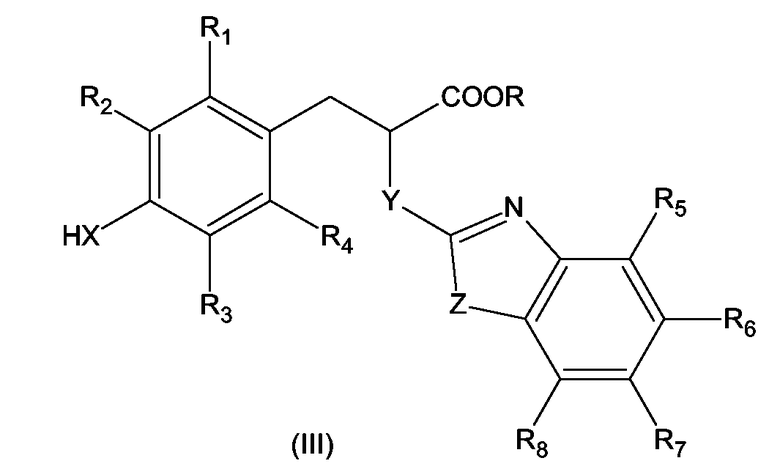
где R представляет C1-C4-алкильную группу и X, Y, Z и R1 - R8 имеют значения, определенные выше для формулы (I), чтобы образовать соединение формулы (I), где W представляет сложноэфирную группу -COOR, где R представляет C1-C4-алкильную группу и X, Y, Z, A, n и R1 - R8 имеют значения, определенные выше для формулы (I), за которым следует
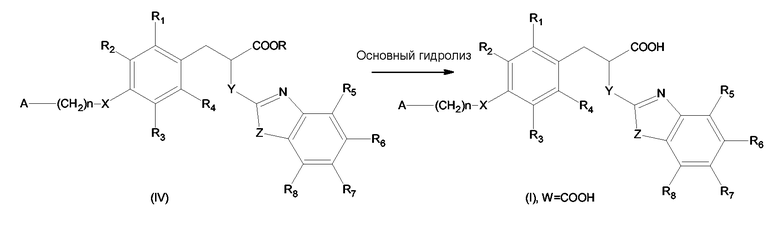
б) необязательно, основный гидролиз сложноэфирной группы -COOR до -COOH группы с образованием соединения формулы (I), где W представляет -COOH.
Вышеупомянутое замещение на стадии а) может быть выполнено по реакции Мицунобу соединения формулы (III) с соединением формулы A(CH2)n-OH, где A и n имеют значения, описанные выше для формулы (I), с образованием соединения формулы (IV) согласно схеме 3
Схема 3
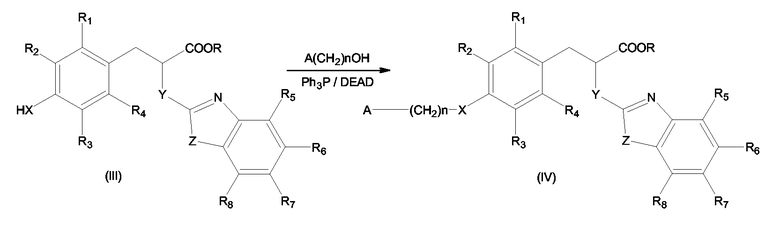
Описанная выше реакция Мицунобу может быть проведена в безводных растворителях, таких как эфир или замещенный галогеном алкан, в присутствии диазосоединений, таких как DEAD, DIAD, ADDP, и трифенилфосфина, обычно в интервале температур от -20 до 20°С.
С другой стороны, вышеупомянутое замещение атома водорода при X может быть проведено посредством реакции соединения формулы (III), где R представляет C1-C4-алкил и X, Y, Z и R1 - R8 имеют значения, определенные выше для формулы (I), с соединением формулы A(CH2)n-V, где A(CH2)n имеет значение, определенное выше для формулы (I), и V представляет уходящую группу, выбранную из галогенов и алкилсульфонильных или арилсульфонильных групп, в присутствии сильного основания, способного образовывать анион из соединения (III), такого как гидрид натрия, с образованием соединения формулы (IV) согласно схеме 4
Схема 4
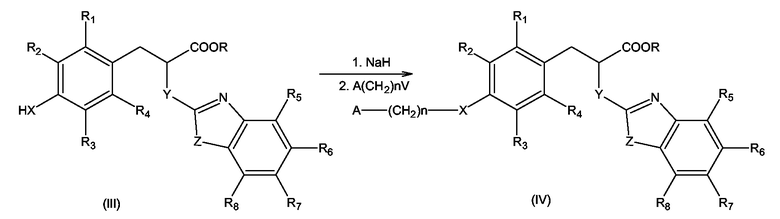
Реакция может быть проведена, как описано выше, для получения соединений формулы (I), где W имеет значение, отличное от -COOH и -COO-C1-C4-алкила.
Гидролиз сложноэфирной группы на стадии б) может быть проведен в основных условиях известным в области техники способом. В качестве примеров основания могут быть упомянуты гидроксиды щелочных металлов, такие как гидроксиды натрия, калия и лития. Для получения отдельных энантиомеров соединения формулы (I) предпочтительно проводить гидролиз с гидроксидом лития, который позволяет сохранить конфигурацию.
Основный гидролиз на стадии б) может быть, например, проведен в системе, состоящей из трех растворителей - ТГФ (тетрагидрофурана), метанола и воды, которая позволяет получить гомогенную реакционную смесь. По окончании гидролиза реакционная смесь может быть нейтрализована хлористоводородной кислотой и, если требуется, продукт в виде свободной кислоты может быть экстрагирован, например, этилацетатом согласно схеме 5, приведенной ниже:
Схема 5
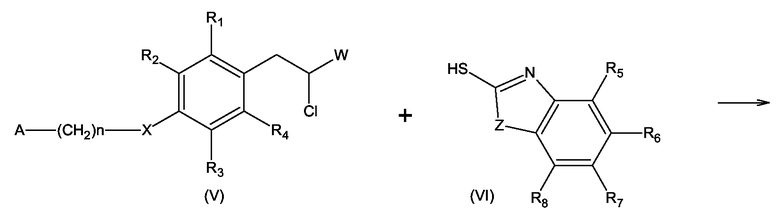
Соединения формулы (I), где Y = S и X, W, Z, A, n, и R1 - R8 имеют значения, определенные выше, могут быть получены по реакции соединения формулы (V), где W, X, A, n и R1 - R4 имеют значения, определенные выше для формулы (I), с соединением формулы (VI), где Z и R5 - R8 имеют значения, определенные выше для формулы (I), в присутствии основания в спиртовом растворе согласно схеме 6
Схема 6
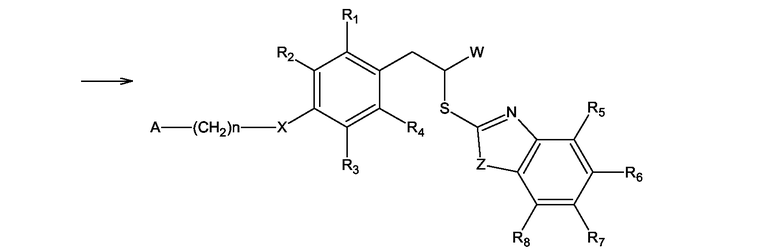
В случае получения соединений формулы (I), где W представляет COOH группу, исходным соединением в описанном выше процессе является соединение формулы (V), где W - защищенная COOH группа, представляющая собой сложный эфир, как показано на схеме 7. По окончании реакции COOH группа деблокируется посредством основного гидролиза.
Согласно схеме 7 первая стадия реакции, на которой образуются производные этил-2-хлор-3-фенилпропионата, осуществляется согласно методу, описанному Y.Kawamatsu, H.Asakawa, T.Saraie, E.Imamiya, K.Nishikawa, Y.Hamuro, Arzneim. Forsch./Drug Res./, 30(I), 4, 1980, 585-589. Полученный по реакции Меервейна хлорзамещенный сложный эфир реагирует с 1,3-бензоксазол-2-тиолом в присутствии основания в спиртовом растворе, давая соответствующий этиловый α-(1,3-бензоксазол-2-илтио)эфир. Сложный эфир гидролизуется в водно-спиртовом растворе NaOH или KOH. Свободные кислоты высвобождаются из солей при разбавлении хлористоводородной кислотой.
Схема 7
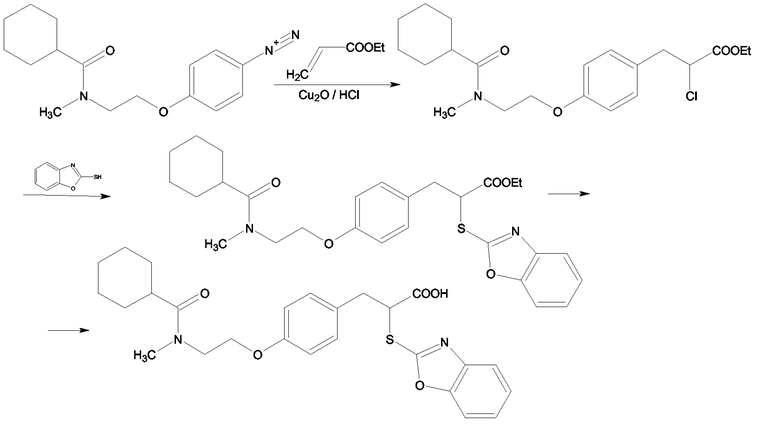
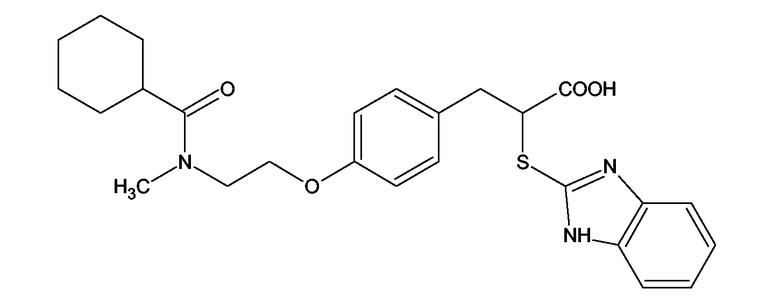
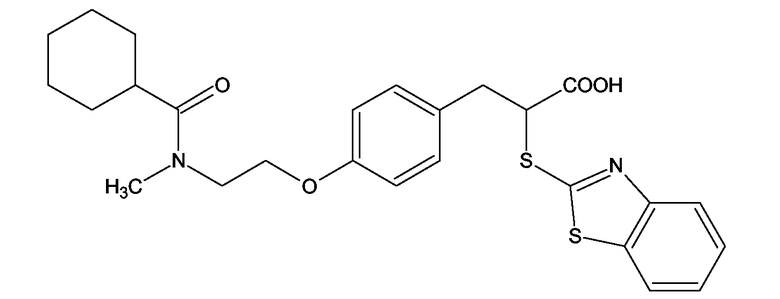
Аналогичным способом получали следующие примеры соединений:
Из оптически активных исходных веществ соединения формулы (I) могут быть получены как в рацемической форме, так и в форме отдельного энантиомера. С другой стороны, рацемические соединения формулы (I) могут быть разделены на энантиомеры, используя общепринятые методы, известные в области техники.
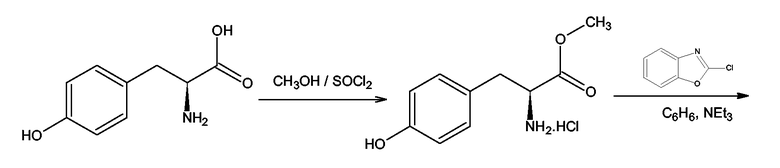
Производные тирозина формулы (III), где X = O, Y = NH и Z = O получали согласно Shyam B. Advani, Joseph Sam, Journal of Pharmaceutical Sciences, Vol.57, 10, 1968. Например, согласно схеме 8 гидрохлорид метилового эфира L-тирозина получали посредством этерификации L-тирозина с метанолом в присутствии тионил хлорида, за которой следовала реакция гидрохлорида метилового эфира L-тирозина с 2-хлор-1,3-бензоксазолом в бензоле в присутствии триэтиламина. Подобные методы использовались в случаях D-тирозина и D,L-тирозина.
Схема 8
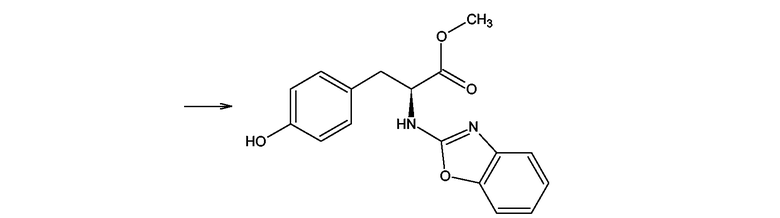
Тирозиновые соединения формулы (III), где X = O, Y = NH и Z = NH, N-алкил, N-арил, N-гетероарил или S могут быть получены посредством адаптации описанного выше метода Shyam B. Advani, Joseph Sam, Journal of Pharmaceutical Sciences, Vol.57, 10, 1968.
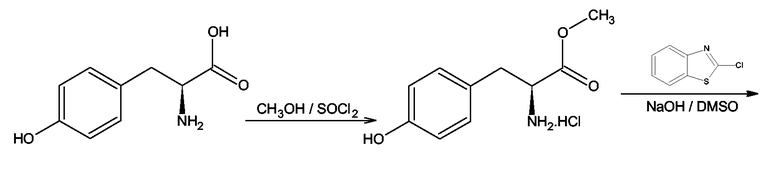
Тирозиновые производные формулы (III), где X = O, Y = NH и Z = S могут быть получены согласно методу, описанному Edward S. Lazer, Clara K.Miao, Hin-Chor Wong, Rondla Sorcek, Denice M. Spero, Alex Galman, Kollol Pal, Mark Behnke, Anne G. Graham, Jane M. Watrous, Carol A. Homon, Juergen Nagle, Arvind Shah, Yvan Guindon, Peter R.Farina, Julian Adams, J.Med.Chem., 1994, 37, 913-923, согласно схеме 9
Схема 9
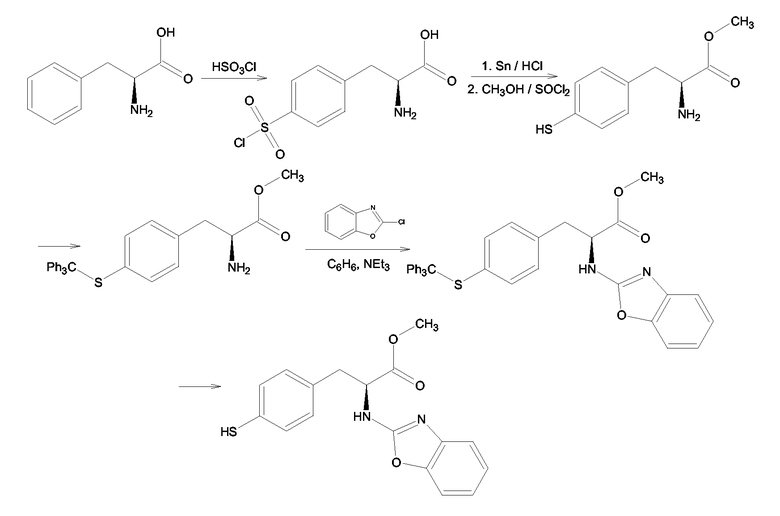
Производные 4-меркаптофенилаланина формулы (III), где Y = NH, Z = O и X = S получали согласно схеме 10, из 4-меркаптофенилаланина, который получали согласно Helen S.M. Lu, Martin Volk, Yuriy Kholodenko, Edward Gooding, Robin M. Hochstrasser, Willian F. DeGrado, Journal of the American Chemical Society, 119, 31, 1997, 7173-7180. Меркаптогруппу (SH) в 4-меркаптофенилаланине защищали тритильной группой с последующим замещением одного атома водорода при атоме азота α-аминогруппы 2-бензоксазолом. Последняя стадия синтеза - деблокирование SH группы.
Схема 10
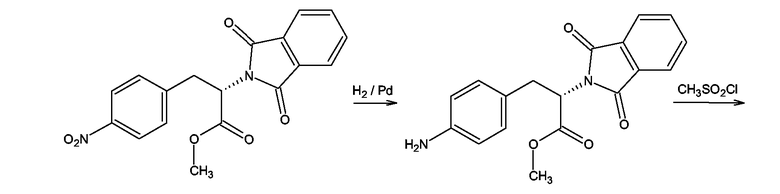
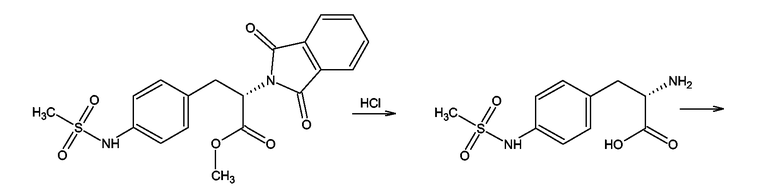
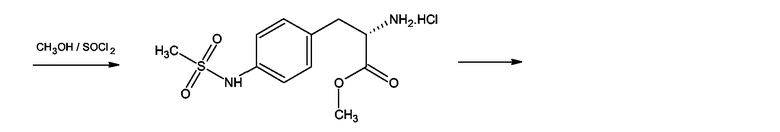
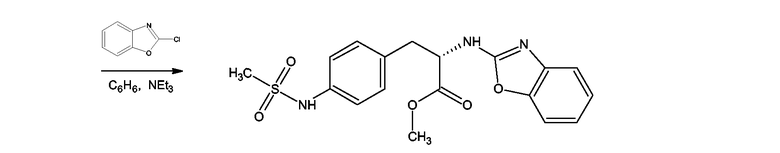
Производные 4-аминофенилаланина формулы (III), где Y = NH, Z = O и
X = NSO2-CH3 получали, как показано на схеме 11, для соединения, где X = NSO2-CH3, из метилового эфира 4-нитро-N-фталоилфенилаланина. Первую стадию синтеза проводили согласно F. Bergel, J.A.Stock, Journal of Organic Chemistry, 1956, 90-96. Полученный таким образом метиловый эфир 4-амино-N-фталоилфенилаланина мезилировали мезилхлоридом в пиридине в присутствии каталитических количеств ДМАП (DMAP). Следующей стадией было удаление фталоильной группы нагреванием с 6М водной соляной кислотой. Полученный таким образом 4-метансульфониламинофенилаланин превращали в гидрохлорид метилового эфира посредством этерификации в метаноле в присутствии тионилхлорида. Следующей стадией была реакция гидрохлорида метилового эфира 4-метансульфониламинофенилаланина с 2-хлорбензоксазолом в присутствии триэтиламина в бензоле
Схема 11
Исходные соединения формулы (VI), где Z = O, т.е. замещенные 2-меркаптобензоксазолы могут быть получены согласно Roger Lok, Rondla E. Leone, Antony J. Williams, J.Org.Chem., 61, 3289-3297, по реакции соединения формулы (VII), где R5 - R8 имеют значения, определенные выше для формулы (I), как показано на схеме 12
Схема 12
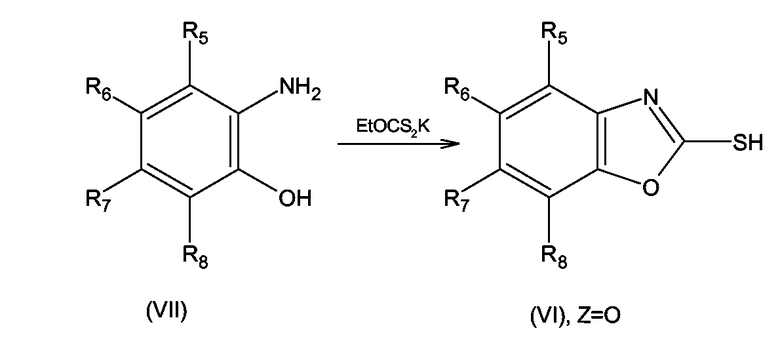
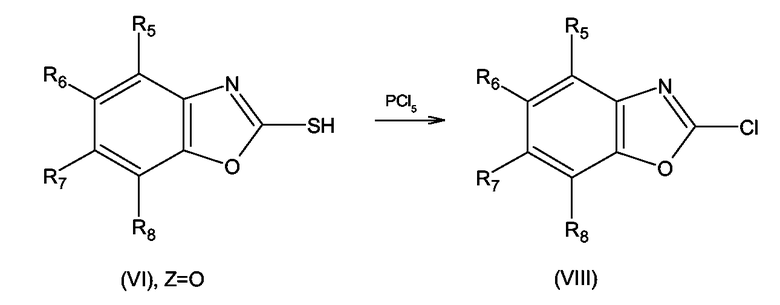
Исходные соединения формулы (VIII), т.е. замещенные 2-хлорбензоксазолы могут быть получены с использованием или адаптацией методов, описанных в Fortuna Haviv, James D. Ratajczyk, Robert W. DeNet, Francis A.Kerdesky, Rolad L.Walters, Steven P. Schmidt, James H. Holmes, Patrick R. Young, George W. Carter, J.Med. Chem., 1988, 31, 1719-1728, посредством реакции соединения формулы (VI), где R5 - R8 имеют значения, определенные выше для формулы (I), с пентахлоридом фосфора согласно схеме 13
Схема 13
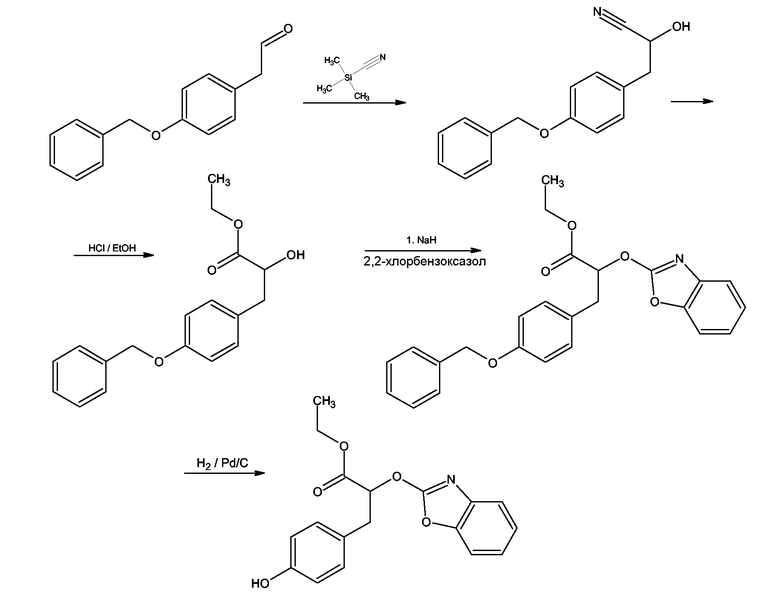
Этиловый эфир 3-[4-(бензилокси)фенил]-2-гидроксипропионовой кислоты получали согласно Takamura Makoto, Yanagisawa Hiroaki, Kanai Motoru, Shibasaki Masakatsu, Efficient Synthesis of Antihyperglycemic (S)-α-Aryloxy-β-phenylpropionic Amides Using a Bifunctional Asymmetric Catalyst, Chem. Pharm. Bull., 50, 8, 2002, 1118-1121. Впоследствии сложный эфир обрабатывали гидридом натрия и после этого 2-хлорбензоксазолом согласно схеме 14
Схема 14
Здесь используются следующие сокращения:
DIAD: диизопропил азодикарбоксилат
DEAD: диэтил азодикарбоксилат
ADDP: азодикарбонилдипиперидин
ПРИМЕРЫ
Пример 1
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(2,3-дигидро-1,4-бензодиоксин-6-илметокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = O, Y = NH, n = 1, A = 2,3-дигидро-1,4-бензодиоксин-6-ил формулы
Растворяли в 5 мл тетрагидрофурана (ТГФ) 2,3-дигидро-1,4-бензодиоксин-6-ил-метанол (0,25 г, 1,5 ммоль), метил (2S)-2-(1,3-бензоксазол-2-ил-амино)-3-(4-гидроксифенил)пропионат (0,31 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DIAD (0,61 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0.1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 50%. MC (ES) 446 (M+, 100%).
Пример 2
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-((3,5-диметилизоксазол-4-ил)метокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = O, Y = NH, n = 1, A = 3,5-диметилизоксазол-4-ил формулы
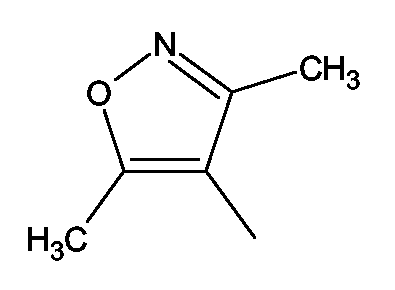
Растворяли в 5 мл тетрагидрофурана (ТГФ) (3,5-диметилизоксазол-4-ил)метанол (0,28 г, 1,5 ммоль), метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-(4-гидроксифенил)пропионат (0,31 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DEAD (0,52 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0.1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 60%. MC (ES) 407 (M+, 100%).
Пример 3
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]этокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = O, Y = NH, n = 2, A = (циклогексилкарбонил)метиоаминогруппа формулы
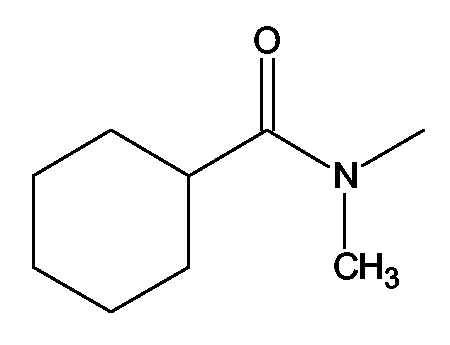
Растворяли в 5 мл тетрагидрофурана (ТГФ) N-(2-гидроксиэтил)-N-метилциклогексанкарбоксиамид (0,19 г, 1,5 ммоль), метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-(4-гидроксифенил)пропионат (0,31 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли ADDP (0,76 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0,1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 40%. MC (ES) 465 (M+, 100%).
Пример 4
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(2-[5-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-4-ил]этокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = O, Y = NH, n = 2, A = [5-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-4-ил] формулы
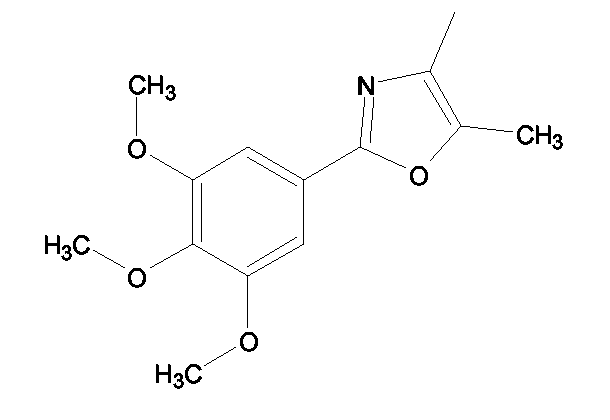
К раствору 2-[4-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-5-ил]этанола (2,93 г, 10 ммоль) в 30 мл пиридина прибавляли порциями при комнатной температуре 4-толуолсульфонилхлорид (1,9 г, 10 ммоль). Впоследствии реакционную смесь перемешивали в течение 5 ч при комнатной температуре, далее выливали ее в 200 мл воды и экстрагировали (3×) 50 мл дихлорметана. Объединенные вытяжки промывали 1М HCl, водным раствором бикарбоната натрия и солевым раствором. Чтобы получить продукт, 2-[4-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-5-ил]этил-4-толуолсульфонат, имеющий чистоту приблизительно 95%, органическую фазу сушили над сульфатом магния и упаривали растворитель.
К раствору 3,12 г метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-(4-гидроксифенил)пропионата в 50 мл диметилформамида прибавляли порциями при перемешивании и комнатной температуре под аргоном 60 % суспензию NaH в минеральном масле (0,4 г). После прекращения выделения газа прибавляли по каплям раствор 2-[4-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-5-ил]этил-4-толуолсульфоната (4,47 г, 10 ммоль) в диметилформамиде. Реакционную смесь нагревали при 80°С и перемешивании. После охлаждения реакционную смесь приливали в 1 л воды и экстрагировали несколько раз этилацетатом. Чтобы получить неочищенный метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-[5-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-4-ил]этокси)фенил]пропионат, объединенные вытяжки промывали солевым раствором, сушили над сульфатом магния и упаривали растворитель.
Неочищенный продукт (2,9 г), полученный выше, растворяли в смеси THF/MeOH/H2O (6:0,1:1, 20 мл). Прибавляли 1М LiOH (8 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 40%. MC (ES) 573 (M+, 100%).
Пример 5
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(4-{2-[[6-(2-хлорфенил)-5-циано-2-(метилтио)пиримидин-4-ил](метил)амино]этокси})фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = O, Y = NH, n = 2, A = [6-(2-хлорфенил)-5-циано-2-(метилтио)пиримидин-4-ил](метил)аминогруппа формулы
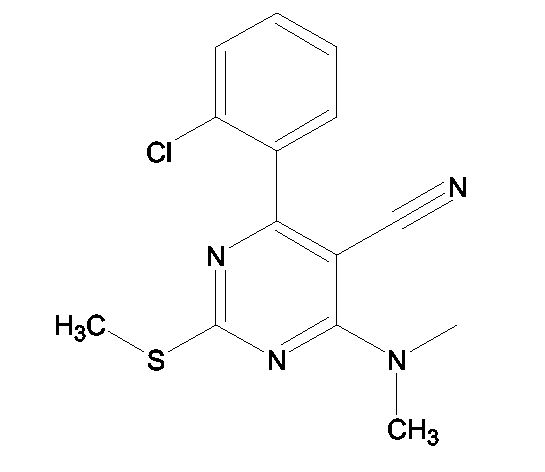
Растворяли в 5 мл тетрагидрофурана (ТГФ) 4-(2-хлорфенил)-6-[(гидроксиэтил)(метил)амино]-2-(метилтио)пиримидин-5-карбонитрил (0,50 г, 1,5 ммоль), метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-(4-гидроксифенил)пропионат (0,31 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DEAD (0,52 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0,1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 58%. MC (ES) 614 (M+, 100%).
Пример 6
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(2-(2-трет-бутил-5-метил-1,3-оксазол-4-ил)этокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = O, Y = NH, n = 2, A = 2-трет-бутил-5-метил-1,3-оксазол-4-ил формулы
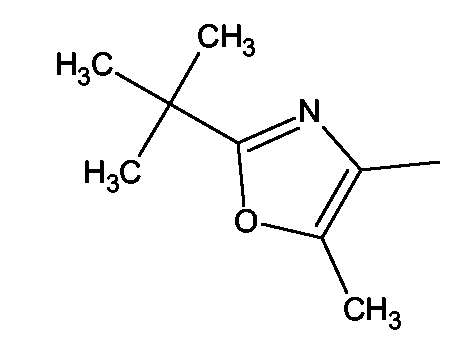
Растворяли в 5 мл тетрагидрофурана (ТГФ) 2-(2-трет-бутил-4-метил-1,3-оксазол-5-ил)этанол (0,27 г, 1,5 ммоль), метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-(4-гидроксифенил)пропионат (0,31 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DEAD (0,52 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0,1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 55%. MC (ES) 463 (M+, 100%).
Пример 7
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]тиоэтокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = S, Z = O, Y = NH, n = 2, А = (циклогексилкарбонил)(метил)аминогруппа формулы
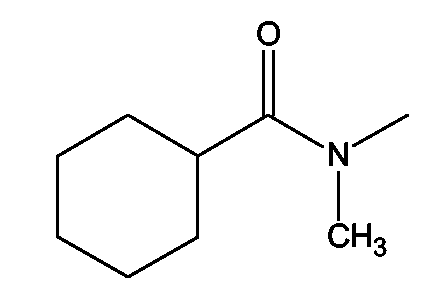
Растворяли в 5 мл тетрагидрофурана (ТГФ) N-(2-гидроксиэтил)-N-метилциклогексанкарбоксиамид (0,19 г, 1,5 ммоль), метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-(4-гидроксифенил)пропионат (0,33 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DEAD (0,52 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0,1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 46%. MC (ES) 481 (M+, 100%).
Пример 8
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]этилметансульфониламино) фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = CH3SO2N, Z = O, Y = NH, n = 2, А = (циклогексилкарбонил)(метил)аминогруппа формулы
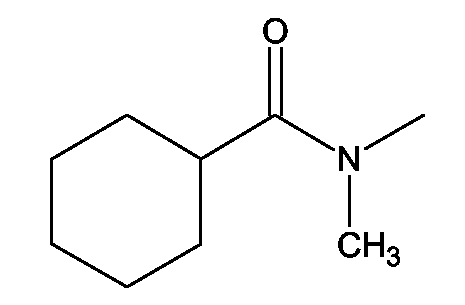
Растворяли в 5 мл тетрагидрофурана (ТГФ) N-(2-гидроксиэтил)-N-метилциклогексанкарбоксиамид (0,19 г, 1,5 ммоль), метил (2S)-2-(1,3-бензоксазол-2-иламино)-3-(4-метансульфониламинофенил)пропионат (0,39 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DEAD (0,52 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0,1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 35%. MC (ES) 542 (M+, 100%).
Пример 9
(2S)-2-(1,3-Бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]этокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = S, Y = NH, n = 2, А = (циклогексилкарбонил)(метил)аминогруппа формулы
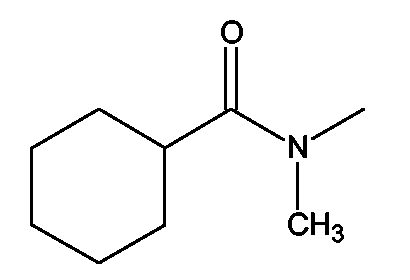
Растворяли в 5 мл тетрагидрофурана (ТГФ) N-(2-гидроксиэтил)-N-метилциклогексанкарбоксиамид (0,19 г, 1,5 ммоль), метил (2S)-2-(1,3-бензотиазол-2-иламино)-3-(4-гидроксифенил)пропионат (0,33 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DEAD (0,52 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0,1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 48%. MC (ES) 481 (M+, 100%).
Пример 10
(2S)-2-(1,3-Бензоксазол-2-илокси)-3-[4-(2-[(циклогексилкарбонил)(метил)амино]этокси)фенил]пропионовая кислота и ее метиловый эфир
R1 - R8 = H, W = COOH/COOCH3, X = O, Z = O, Y = O, n = 2, А = (циклогексилкарбонил)метиламиноэтил формулы
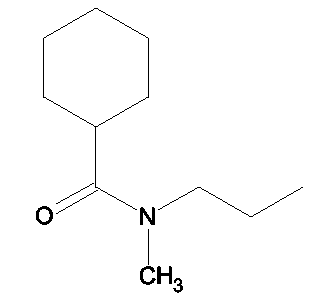
Растворяли в 5 мл тетрагидрофурана (ТГФ) N-(2-гидроксиэтил)-N-метилциклогексанкарбоксиамид (0,19 г, 1,5 ммоль), этил 2-(1,3-бензотиазол-2-илокси)-3-(4-гидроксифенил)пропионат (0,33 г, 1 ммоль) и трифенилфосфин (0,26 г, 1 ммоль). Реакционную смесь охлаждали до 5°С. Затем прибавляли DEAD (0,52 г, 3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18-24 ч. Впоследствии, чтобы получить продукт, метиловый эфир названной кислоты упаривали ТГФ.
Неочищенный продукт растворяли в смеси THF/MeOH/H2O (6:0,1:1, 2 мл). Прибавляли 1М LiOH (1,6 мл) и перемешивали реакционную смесь в течение 3 дней при комнатной температуре. Далее реакционную смесь нейтрализовали 1М соляной кислотой, прибавляли небольшое количество воды и смесь экстрагировали этилацетатом. Растворитель упаривали. Продукт очищали с помощью хроматографии. Выход 40%. MC (ES) 466 (M+, 100%).
Биологические тесты
Способность соединений изобретения связываться с PPAR гамма-рецептором и изменять его активность определяли, используя следующие методы.
Связывание in vitro
Способность соединений связываться с PPAR гамма-рецептором (in vitro) определяли согласно процедуре, описанной ниже, используя метод конкурентного вытеснения радиоактивно меченного лиганда из комплекса лиганд-рецептор. В качестве радиоактивно меченного лиганда использовали PPAR агонист - 3H-розиглитазон с конечной концентрацией 10нМ. К реакционной смеси также прибавляли избыток немеченых испытуемых веществ с конечной концентрацией 20 мкМ. Источником рецептора в испытаниях был человеческий рекомбинантный белок, содержащий LBD (лиганд-связывающий домен) PPAR гамма. Выделение несвязанного с рецептором радиоактивно меченного лиганда осуществляли с помощью декстран-угольного метода. Радиоактивность измеряли, используя сцинтилляционный счетчик LS 6500-Beckman Coulter. Полученные значения чисел сцинтилляций сравнивались со значениями, полученными для образцов, инкубированных с радиоактивно меченным лигандом (принимая за 0% вытеснения), и со значениями, полученными для образцов, содержащих как радиоактивно меченный лиганд, так и избыток не меченного радиоактивной меткой розиглитазона (принимая за 100% вытеснения). Полученные значения находились в диапазоне 0-130%.
Ссылки:
1. ADD1/SREBP1 activates PPAR gamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA. 1998 Apr 14; 95(8): 4333-7.
2. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995 Jun 2; 270(22): 12953-6.
3. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA. 1997 Apr. 29; 94(9): 4318-23.
Связывание в адипоцитах
Чтобы подтвердить способность испытуемых молекул связываться in vivo, выполняли аналогичные эксперименты с использованием клеточной линии мышиных фибробластов 3T3-L1, дифференцированных в адипоциты. Дифференцировку клеток фибробластов проводили в 12-луночных планшетах в течение 10-дневного периода. В день эксперимента клетки промывали дважды раствором PBS перед одночасовой инкубацией в среде DMEM, содержащей эталонное меченное тритием соединение (розиглитазон) с концентрацией 30 пкМ и различные концентрации испытуемых веществ (концентрация в пределах от 100 пкМ до 20 мкМ) при 37°С. Затем клетки промывали три раза раствором PBS и растворяли в 1М растворе NaOH. В приготовленном, как описано выше, лизате измеряли как радиактивность (используя сцинтилляционный счетчик LS 6500-Beckman Coulter), так и концентрацию белка (используя метод Бредфорда). Неспецифическое связывание оценивали в присутствии немеченого эталонного соединения (при концентрации 20 мкМ).
Полученные значения числа сцинтилляций сравнивались со значениями, полученными для образцов, инкубированных с радиоактивно меченным лигандом (принимая за 0% вытеснения), и со значениями, полученными для образцов, содержащих как радиоактивно меченный лиганд, так и избыток не меченного радиоактивной меткой розиглитазона (принимая за 100% вытеснения). Полученные значения находились в диапазоне 0-130%.
Ссылки:
1. Identification of high-affinity binding sites for the insulin sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a radioiodinated ligand for peroxisomal proliferator-activated receptor gamma. J. Pharmacol. Exp. Ther. 1998 Feb; 284(2): 751-9.
2. Differential regulation of the stearoyl-CoA desaturase genes by thiazolidinediones in 3T3-L1 adipocytes. J. Lipid Res. 2000 Aug; 41(8): 1310-6.
3. Distinct stages in adipogenesis revealed by retinoid inhibition of differentiation after induction of PPARgamma. Mol Cell Biol. 1996 Apr; 16(4): 1567-75.
4. Differentiation Kinetics of in vitro 3T3-L1 Preadipocyte Cultures. Tissue Eng. 2002 Dec; 8(6): 1071-1081.
5. Role of PPARgamma in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18(INK4c) and p21 (Waf1/Cip1), during adipogenesis. J. Biol. Chem. 1999 Jun 11; 274(24): 17088-97.
Адипогенез
Клетки клеточной линии 3T3-L1 (из ATCC) выдерживали в среде DMEM, дополненной 10% эмбриональной бычьей сывороткой и антибиотиками. За два дня до начала эксперимента клетки переносили в 12-луночные микропланшеты (30×104 клеток в лунке) и выдерживали в течение последующих 2 дней для слияния. После этого среду заменяли смесью DMEM, FBS и антибиотиков и прибавляли испытуемые вещества с конечной концентрацией 50 мкМ к клеткам. При этих условиях клетки выдерживались в течение 14 дней, при этом среду с испытуемыми соединениями меняли каждые 2 дня. После 10-14 дней дифференцированные клетки фотографировали с предварительным окрашиванием масляным красным О.
Ссылки:
1. Differential regulation of the stearoyl-CoA desaturase genes by thiazolidinediones in 3T3-L1 adipocytes. J. Lipid Res. 2000 Aug; 41(8): 1310-6.
Поглощение глюкозы
Дифференцированные 3T3-L1 фибробласты инкубировали в DMEM, дополненной 10% FBS и антибиотиками, с испытуемыми веществами (при концентрации 20 мкМ) в течение 48 ч. После этого клетки промывали PBS и далее прибавляли к клеткам свободный от сыворотки DMEM. Клетки хранились в инкубаторе в течение 3 ч (37°С/5% CO2), и далее среда заменялась KHR буферным раствором (25 мМ HEPES-NaOH; pH 7,4; 125 мМ NaCl; 5 мМ KCl; 1,2 мМ MgSO4; 1,3 мМ CaCl2; 1,3 мМ KH2PO4), и клетки инкубировали в течение 30 мин при 37°С. Поглощение глюкозы инициировали прибавлением в каждую испытуемую лунку 50 мкМ KRH буферного раствора, содержащего 0,5 мМ 2-дезокси-D-[1,2-3H]глюкозу (0,5 мкКи) и 100 нМ инсулин. После 10 мин инкубирования при 37°С среду удаляли и клетки промывали три раза ледяным KRH буферным раствором. Затем клетки растворяли в 1М NaOH. В приготовленном, как описано выше, лизате измеряли как радиактивность (используя сцинтилляционный счетчик LS 6500-Beckman Coulter), так и концентрацию белка (используя метод Бредфорда). Неспецифическое связывание оценивали в присутствии немеченого эталонного соединения (при концентрации 20 мкМ).
Ссылки:
1. Role of peroxisome proliferator-activated receptor-gamma in maintenance of the characteristics of mature 3T3-L1 adipocytes. Diabetes. 2002 Jul; 51(7): 2045-55.
2. Identification of high-affinity binding sites for the insulin sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a radioiodinated ligand for peroxisomal proliferator-activated receptor gamma. J. Pharmacol. Exp. Ther. 1998 Feb; 284(2): 751-9.
3. Identification of bioactive molecules by adipogenesis profiling of organic compounds. J. Biol. Chem. 2003 Feb 28; 278(9): 7320-4. Epub 2002 Dec 19.
4. Evidence for the involvement of vicinal sulfhydryl groups in insulin-activated hexose transport by 3T3-L1 adipocytes. J. Biol. Chem. 1985 Mar 10; 260(5): 2646-52.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ 3-ФЕНИЛПРОПИОНОВОЙ КИСЛОТЫ | 2006 |
|
RU2369602C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ПУРИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1997 |
|
RU2228335C2 |
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
| ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНОВЫЕ И ИЗОХИНОЛИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2007 |
|
RU2455302C2 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛАЛКИЛПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2243970C1 |
| ЗАМЕЩЕННЫЕ АМИНОИНДАНЫ И ИХ АНАЛОГИ, И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2009 |
|
RU2522586C2 |
| АЦИЛИРОВАННЫЕ ГЕТЕРОАРИЛКОНДЕНСИРОВАННЫЕ ЦИКЛОАЛКЕНИЛАМИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2003 |
|
RU2338743C2 |
| БИ- И ПОЛИЦИКЛИЧЕСКИЕ ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА И ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2009 |
|
RU2532481C2 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ АМИДЫ, СОДЕРЖАЩИЕ НАСЫЩЕННУЮ СВЯЗЫВАЮЩУЮ ГРУППУ, И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2006 |
|
RU2412181C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ 1β-ДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2415855C2 |
Изобретение относится к производным 3-фенилпропионовой кислоты формулы (I) в качестве лиганда гамма-рецептора пролифератора-активатора пероксисом (PPARγ), к их фармацевтически приемлемым солям, а также к их применению, способу лечения и фармацевтической композиции на их основе. Соединения могут найти примененение для лечения и профилактики заболеваний, опосредованных гамма-рецептором пролифератора-активатора пероксисом (PPARγ), например диабета типа 2, резистентности к инсулину, метаболического синдрома, осложнений, являющихся результатом или связанных с диабетом, сердечно-сосудистых расстройств, атеросклероза, ожирения, когнитивных расстройств и расстройств метаболизма липидов. В общей формуле (I)
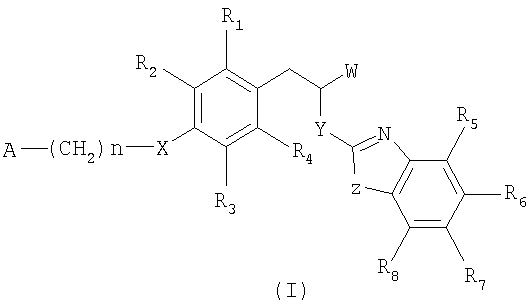
W представляет СООН или -СОО-С1-С4-алкильную группу; Y представляет NH; Z представляет S или О; Х представляет О; R1-R8 каждый независимо представляет атом водорода или атом галогена; А представляет моно-, би- или трициклический 5-13-членный гетероарил с 1 или 2 гетероатомами, выбранными из N, S и О, арил, выбранный из фенила и нафтила, или
-N(С1-С4-алкил)-СО-С3-С7-циклоалкил, где гетероарил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C4-алкила, CN, галогена фенила и фенила, необязательно замещеного 1-3 заместителями, независимо выбранными из С1-С4-алкокси, галогена и этилендиоксигруппы; и n представляет целое число от 0 до 3 включительно; и их фармацевтически приемлемые соли. 4 н. и 16 з.п. ф-лы.
1. Производные 3-фенилпропионовой кислоты формулы (I)
где W представляет СООН или -СОО-С1-С4-алкильную группу;
Y представляет NH;
Z представляет S или О;
Х представляет О;
R1-R8 каждый независимо представляет атом водорода или атом галогена;
А представляет моно-, би- или трициклический 5-13-членный гетероарил с 1 или 2 гетероатомами, выбранными из N, S и О, арил, выбранный из фенила и нафтила, или -N(C1-С4-алкил)-СО-С3-С7-циклоалкил, где гетероарил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из С1-С4-алкила, CN, галогена, фенила и фенила, необязательно замещенного галогеном или С1-С4-алкокси; и где арил необязательно замещен 1-3 заместителями, независимо выбранными из
С1-С4-алкокси, галогена и этилендиоксигруппы;
n представляет целое число от 1 до 3 включительно;
их фармацевтически приемлемые соли,
при условии, что исключаются следующие соединения:
N-(2-бензотиазолил)-O-[3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил]-L-тирозин,
этил N-(2-бензотиазолил)-O-[3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил]-L-тирозинат,
N-(бензоксазолил)-O-[3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил]-L-тирозин, и
этил-N-(бензоксазолил)-O-[3-(5,6,7,8-тетрагидро-1,8-нафтиридин-2-ил)пропил]-L-тирозинат.
2. Соединение по п.1, где W представляет СООН.
3. Соединение по п.1, где Z представляет О.
4. Соединение по п.1, где Z представляет S.
5. Соединение по п.1, где W представляет СООН, Y представляет NH, Z представляет О.
6. Соединение по любому из пп.1-5, где каждый из заместителей R1-R8 представляет атом водорода.
7. Соединение по любому из пп.1-5, где n равняется 1 или 2.
8. Соединение по п.1, где А представляет изоксазолил, необязательно замещенный
C1-С4-алкилом, в частности -СН3.
9. Соединение по любому из пп.1-5, где А представляет фенил, который необязательно замещен этилендиоксигруппой.
10. Соединение по любому из пп.1-5, где А представляет -N(С1-С4-алкил)-СО-С3-С7-циклоалкил.
11. Соединение по п.10, где А представляет -N(СН3)-СО-циклогексил.
12. Соединение по любому из пп.1-5, имеющее стереохимическую конфигурацию как показано в формуле (IA):
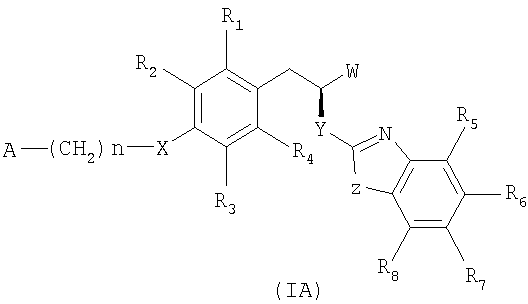
и его фармацевтически приемлемые соли.
13. Соединение по любому из пп.1-5, имеющее стереохимическую конфигурацию как показано в формуле (IB):
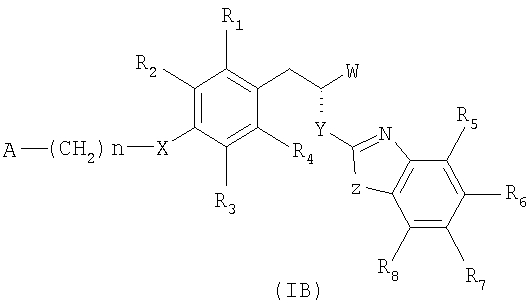
и его фармацевтически приемлемые соли.
14. Соединение по п.1, выбранное из следующих соединений:
-(2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2,3-дигидро-1,4-бензодиоксин-6-илметокси)фенил]пропионовая кислота,
-(2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-((3,5-диметилизоксазол-4-ил)метокси)фенил]пропионовая кислота,
-(2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-[(циклогексилкарбонил)-(метил)амино]этокси)фенил]пропионовая кислота,
-(2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-[5-метил-2-(3,4,5-триметоксифенил)-1,3-оксазол-4-ил]этокси)фенил]пропионовая кислота,
-(2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-(2-трет-бутил-5-метил-1,3-оксазол-4-ил)этокси)фенил]пропионовая кислота,
-(2S)-2-(1,3-бензоксазол-2-иламино)-3-[4-(2-(2-трет-бутил-5-метил-1,3-оксазол-4-ил)этокси)фенил]пропионовая кислота,
и их фармацевтически приемлемых солей.
15. Фармацевтическая композиция, обладающая сродством к гамма-рецептору пролифератора-активатора пероксисом (PPARγ), включающая соединение по любому из пп.1-14 или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемыми носителями и/или вспомогательными веществами.
16. Соединение по любому из пп.1-5 для применения в качестве лекарственного средства, обладающего сродством к гамма-рецептору пролифератора-активатора пероксисом (PPARγ).
17. Применение соединения формулы (I) по любому из пп.1-14 для изготовления лекарственного средства для лечения и/или предотвращения заболевания или состояния, опосредованного гамма-рецептором пролифератора-активатора пероксисом (PPARγ).
18. Применение по п.17, где указанное заболевание или состояние выбрано из группы, состоящей из диабета типа 2, резистентности к инсулину, метаболического синдрома, осложнений, являющихся результатом или связанных с диабетом сердечно-сосудистых расстройств, атеросклероза, ожирения, когнитивных расстройств и расстройств метаболизма липидов.
19. Способ лечения и/или профилактики заболеваний и состояний, опосредованных гамма-рецептором пролифератора-активатора пероксисом (PPARγ), у нуждающегося в этом млекопитающего, включающий введение указанному млекопитающему соединения формулы (I) по любому из пп.1-14 в терапевтически или профилактически эффективном количестве.
формулы (I) по любому из пп.1-14 в терапевтически или профилактически эффективном количестве.
20. Способ по п.19, где указанное заболевание или состояние выбрано из группы, состоящей из диабета типа 2, резистентности к инсулину, метаболического синдрома, осложнений, являющихся результатом или связанных с диабетом, сердечно-сосудистых расстройств, атеросклероза, ожирения, когнитивных расстройств и расстройств метаболизма липидов.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ИЛИ ЕГО ТАУТОМЕРНАЯ ФОРМА, И/ИЛИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, И/ИЛИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЙ СОЛЬВАТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СНИЖАЮЩАЯ СОДЕРЖАНИЕ ГЛЮКОЗЫ В КРОВИ, СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ГИПЕРГЛИКЕМИИ | 1993 |
|
RU2134686C1 |

