Изобретение относится к органической химии, а именно к усовершенствованному способу синтеза 2'-дезоксиксилотимидина [1-(2'-дезокси- β -D-трео-пентофуранозил)тимина] . Известно, что это вещество или его 5'-защищенные производные можно синтезировать из дешевого и доступного сырья D-ксилозы [1,2] и использовать [1] в качестве исходных соединений в синтезе 3'-азидо 2', 3'-дидезокситимидина (азидотимидина) - эффективного лекарства против СПИДа.
Необходимо отметить, что в настоящее время сырьем для производства азидотимидина служит природный тимидин [1-(2'-дезокси- β -D-эритро-пентофуранозил)тимин] . Однако количество производимого тимидина ограничено (в частности из-за недостаточной доступности природной ДНК) и не может удовлетворить растущую потребность в нем медицинской промышленности.
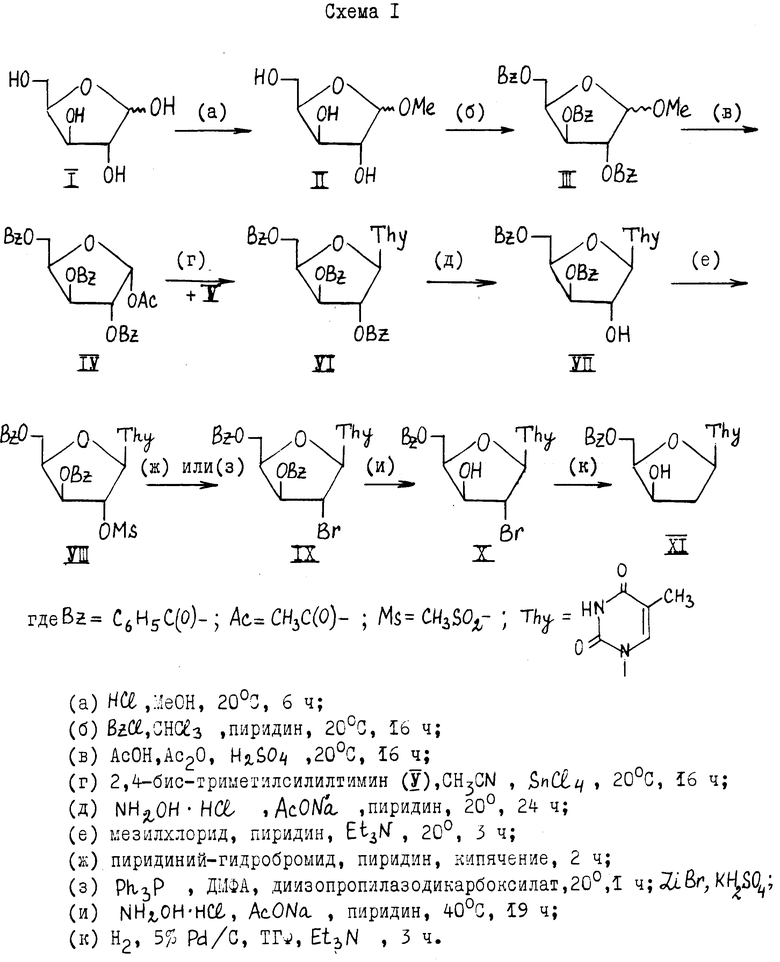
Изобретение расширяет сырьевую базу для синтеза азитодимидина, а также других соединений, которые используются в медицине в количестве лекарственных средств против ВИЧ-инфекции, например, 5'-фосфата 3' -азидо-2',3'-дидезокситимидина [3]. Известны два способа синтеза производных 2'-дезоксиксилотимидина как интермедиатов синтеза азидотимидина, описанные в патенте [1]. Оба способа основаны на конденсации D-ксилозы и тимина и включают следующие этапы синтеза (схемы 1,2), приведенные в конце описания: 1) синтез углеводной компоненты для нуклеозидной конденсации путем превращения D-ксилозы (I) через 1-0-метил-D-ксилофуранозид (II) и соединение III в 1-0-ацетил-2,3,5-три-0-бензоил α -ксилофуранозу (IV); 2) конденсация углеводной компоненты (IV) с силилированным тимином (V) с образованием нуклеозида 1-(2;3;5'-три-0-бензоил- β -D-ксилофуранозил)тимина (VI); 3) 2'-бромирование-получение 3', 5' -защищенного 2'-бром-2'-дезокси-D-ксилотимидина; 4) гидрогенолиз производного 2'-бром-2'-дезокси-D-ксилотимидина с образованием замещенного 2'-дезоксиксилотимидина; 5) 3'-азидирование, получение азидотимидина.
Этапы 1 и 2 идентичны в обоих способах [1]. В первом способе патента [1] на этапе 2'-бромирования сначала проводят избирательное 2'-дебензоилирование нуклеозида (VI), затем 3', 5'-ди-0-бензоил-D-ксилотимидин (VII) превращают в 3', 5'-ди -0-бензоил-2'-0-мезил-D-ксилотимидин (VIII), который бромируют (двумя способами) и получают 3', 5'-ди-0-бензоил-2'-бром-2'-дезоксиксилотимидин (IX). Избирательное деблокирование соединения IX приводит к 5'-0-бензоил-2'-бром-2'-дезокси-ксилотимидину (X), который гидрируют над палладием на угле Pd/C, получают 5'-0-бензоил-2'-дезокси- β -D-ксилотимидин (XI) с выходом 2,5-3% в расчете на D-ксилозу (схема 1).
Общий низкий выход замещенного 2'-дезокси-ксилотимидина (XI) определяется прежде всего низким выходом на первом этапе синтеза (39% соединения IV ( α -изомер) в расчете на D-ксилозу), что обусловлено выбором пути синтеза через 1-метилксилозид (II) и связано с выделением и использованием лишь одного кристаллического α -аномера соединения (IV) (а не смеси α , β -аномеров). Кроме того, недостаточно высокие выходы характерны и для других стадий рассматриваемого способа патента [1], например, для избирательных процессов (д, и) деблокирования положений 2' и 3' в нуклеозидах VI и IX, 2'-бромирования, гидрогенолиза. Удаление поочередно по одной из трех (2', 3', 5') или затем из двух (3', 5') одинаковых бензоильных защит, обладающих близкой устойчивостью, проводят, обрабатывая соединения VI и IX гидроксиламин-гидрохлоридом и ацетатом натрия в пиридине при 20 и 40oC, соответственно. Выходы при этом составляют 69 и 62%, соответственно.
Один из недостатков первого способа [1] - невозможность достижения хорошего выхода при получении 3', 5'-дибензоил-2,2'-ангидросоединения формулы (A) из 2'-мезилата (VIII)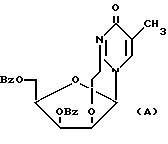
из-за неустойчивости бензоильной защиты в основных средах, применяемых для создания 2,2'-ангидросвязи. Поэтому использовались прямые методы бромирования соединения VIII: 1) обработка пиридинийбромидом в пиридине и 2) взаимодействие с трифенилфосфином в ДМФА в присутствии диизопропилазодикарбоксилата и литий бромида. Выходы, соответственно, только 45 и 54%.
Восстановление 2'-бромпроизводного в замещенный 2'-дезоксиксилотимидин каталическим гидрогенолизом требует обязательного введения дополнительной стадии (и) 3'-деблокирования во избежание образования побочного продукта с 2',3'-двойной связью. На стадии гидрогенолиза (к) выход составляет 58%.
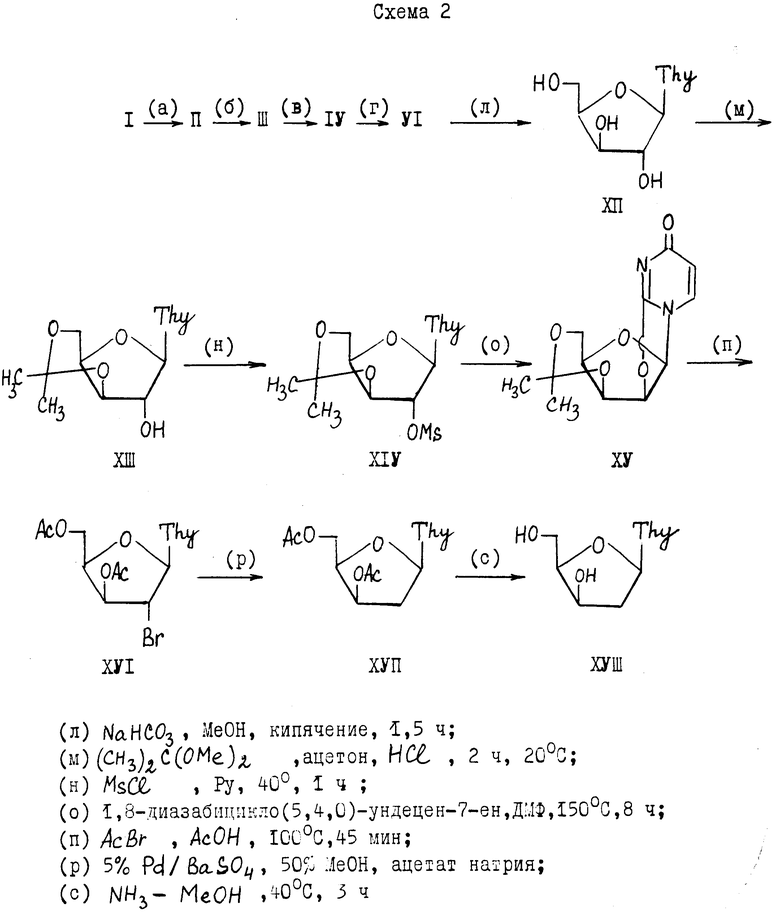
Во втором способе патента [1] проведена попытка преодоления недостатка первого способа [1] путем использования 3', 5'-изопропилиденовой защитной группы (устойчивой в основных средах) в процессе получения 2,2'-ангидросоединения (с последующей заменой этой защиты на 3',5'-диацетильные группы в процессе 2'-бромирования). Для этого проводят (см. схему 2) полное деблокирование нуклеозида (VI), полученный незащищенный ксилотимидин (XII) превращают в 3',5'-0-изопропилиден-D-ксилотимидин (XIII). Соединение (XIII) через 2'-мезилат (XIV) переводят в 2,2'-ангидридо- 3,5'-ди-О-изопропилиден-ксилотимидин (XV), который при обработке ацетилбромидом в уксусной кислоте дает 2'-бром-2'-дезокси-3', 5'-ди-О- ацетилксилотимидин (XVI). После каталитического гидрогенолиза (над Pd/C) получают 3',5'-ди-О-ацетил-2'-дезокси-ксилотимидин (XVII), причем чистота конечного продукта (XVII) составляет всего 70%. В результате второй способ изобретения [1] с двойной заменой защитных групп приводит к небольшому увеличению выхода (8,5% на исходную D-ксилозу) и одновременно к ухудшению качества ключевого интермедиата (XVII), из которого получают 2'-дезоксиксилотимидин (XVIII).
Таким образом, основным недостатком обоих способов [1] является низкий выход производных 2'-дезоксиксилотимидина - ключевых интермедиатов в синтезе азидотимидина.
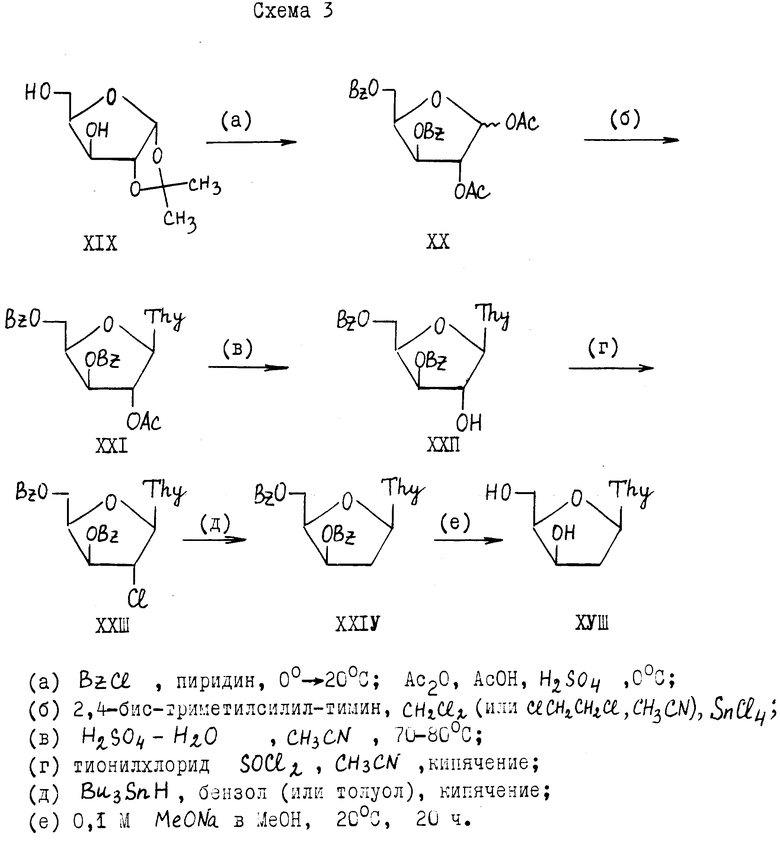
Наиболее близким к предлагаемому является способ получения 1-(2'-дезокси- -β- -D-трео-пентофуранозил) тимина (XVIII), описанный в патенте [2]. Основные этапы способа [2] : 1) избирательное блокирование двух пар 1,2- и 3,5-гидроксильных групп D-ксилозы с выходом на 1,2-ди-О-ацетил-3,5-О-бензоил- -α,β -D-ксилофуранозу; 2) нуклеозидная конденсация с силилированным тимином и последующее избирательное 2'-деблокирование; 3) получение 2'-галоид производного и последующего 2'-дезоксигенирование защищенного β- -D-ксилотимидина. По этому способу (см. схему 3, приведенную в конце текста описания). 1,2-О-изопропилиден- α -D-ксилофуранозу (XIX) превращают в 1,2-ди-О-ацетил-3,5-ди-О-бензоил- -α,β -D-ксилофуранозу (XX) обработкой последовательно бензоилхлоридом в пиридине и затем уксусным ангидридом в уксусной кислоте в присутствии серной кислоты при 0oC. Углеводную компоненту (XX) конденсируют с силилированным тимином, полученный нуклеозид (XXI) обрабатывают серной кислотой в смеси вода-ацетонитрил (избирательное 2'-дезацетилирование). Полученный 1-(3', 5'-ди-О-бензоил- -β- -D-трео- пентофуранозил)тимин (XXII) при действии на него хлористого тионила в ацетонитриле превращают в 2'-хлор-производное (XXIII), которое затем восстанавливают гидридом трибутилолова в толуоле в присутствии радиального катализатора и получают 1-(3',5'-ди-О-бензоил-2'-дезокси- β -D-трео-пентофуранозил) тимин (XXIV). Общий выход соединения XXIV в расчете на D-ксилозу составляет 31%. Соединение XXIV служило для синтеза 2'-д-ксилотимидина XVIII, который получали известным способом из XXIV. Выход 2'-д-ксилотимидина 29,5% (на ксилозу).
В способе [2] предусматривается, что исходную 1,2-О-изопропилиден- α -D-ксилофуранозу (XIX) можно синтезировать в две стадии из D-ксилозы по методу [4] через промежуточную 1,2:3,5-ди-О-изопропилиден- α -D-ксилофуранозу (XXV) [5] с выходом 64% на две стадии. При этом соединение XIX выделяют перегонкой в вакууме.
Выбор подходящей углеводной компоненты XX в способе [2] дает возможность получать нуклеозид (XXI), содержащий заметно различающиеся по устойчивости ациальные защитные группы в положениях 2' и 3' углеводного кольца (2'-ацетил, 3'-бензоил). Однако избирательное 2'-дезацетилирование нуклеозида (XXI) проводят в довольно жестких условиях: при нагревании его в смеси ацетонитрил-1 M H2SO4 (4: 0,6) при 70-80oC в течение 16 ч (причем продукт реакции (XXII) не кристаллизуют), что сказывается на выходе 2'-хлор-2'-дезоксинуклеозида (XXIII), составляющем всего 52,5% (на две стадии).
Недостатком способа [2] является то обстоятельство, что интермедиаты первых шести стадий синтеза, начиная с ксилозы, представляют собой некристаллизующиеся вещества. Эта масса (соединения XXV, XIX, XX) или технические аморфные вещества (XXI, XXII). Невозможность очистки указанных интермедиатов кристаллизацией наряду с необходимостью вакуумной перегонки ацетонированных углеводов может усложнить их получение в промышленном масштабе из-за снижения выходов и ухудшения качества промежуточных веществ.
Однако главным недостатком способа [2] является применение сильно ядовитого восстановителя гидрида трибутилолова Bu3SnH-маслообразного вещества с резким тяжелым запахом. Этот реагент очень неудобен для использования его в промышленности из-за трудности очистки от него нуклеозидного материала и по соображениям техники безопасности и экологии.
Мы усовершенствовали способ получения 2'-дезоксиксилотимидина (1) из D-ксилозы и тимина, включающий: 1) синтез производного D-ксилофуранозы, содержащего две пары различных ациальных защитных групп в 1,2- и 3,5-положениях (углеводная компонента); 2) нуклеозидную конденсацию углеводной компоненты с производным тимина с последующим избирательным деблокированием 2'-гидроксильной группы; 3) 2'-дезоксигенирование 3',5'-защищенного β -ксилотимидина с последующим деблокированием 3',5'-гидроксильных групп.
Согласно изобретению синтез 2'-дезоксиксилотимидина (1) из D-ксилозы и тимина состоит из следующих этапов и стадий.
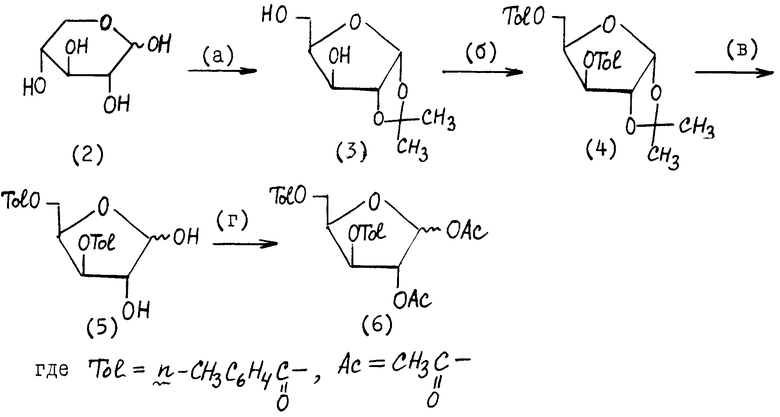
I. Превращение D-ксилозы (2) в 1,2-ди-О-ацетил-3,5-ди-О-п-толуил- α -D-ксилофуранозу (5), стадии (а) - (г) приведенной в конце текста описания.
На стадии (а) D-ксилозу (2) обрабатывают ацетоном в присутствии серной кислоты и воды на ходу при 2-8oC. В результате получают 1,2-О-изопропилиден- α -D-ксилофуранозу (3) с хорошим выходом.
На стадии (б) блокируют 3- и 5-гидроксильные группы путем обработки соединения формулы (3) n - толуилхлоридом в присутствии пиридина.
На стадии (в) проводят удаление 1,2-О-изопропилиденовой защиты при действии на соединение формулы (4) раствора сильной кислоты, такой как соляная кислота, в органическом растворителе, преимущественно диоксане. Соединение (4) получают в кристаллическом состоянии.
На стадии (г) соединение формулы (5) ацетилируют, обрабатывая его уксусным ангидридом или ацетилгалогенидом, например ацетилхлоридом в пиридине, и получают углеводную компоненту для нуклеозидной конденсации формулы (6).
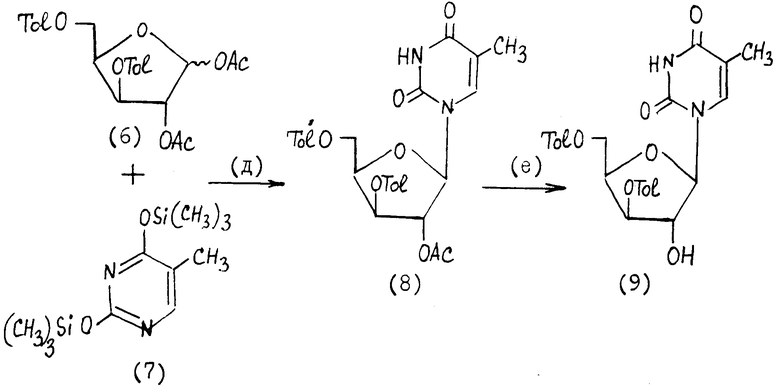
II. Получение 1-(3', 5'-ди-О-п-толуил- β -D-ксилофуранозил)тимина (9), стадии (д) - (е), приведенной в конце текста описания
На стадии (д) соединение формулы (6) вводят в реакцию с соединением формулы (7) в присутствии кислоты Льюиса, такой как тетрахлорид олова SnCl4, преимущественно в апротонном полярном растворителе, таком как ацетонитрил или галогенированный углеводород, например дихлорэтан.
На стадии (е) проводят избирательное удаление 2'-ацетильной защиты, обрабатывая соединение формулы (8) разбавленным спиртовым раствором аммиака в органическом растворителе, например толуоле, на холоду при 5 ± 3oC.
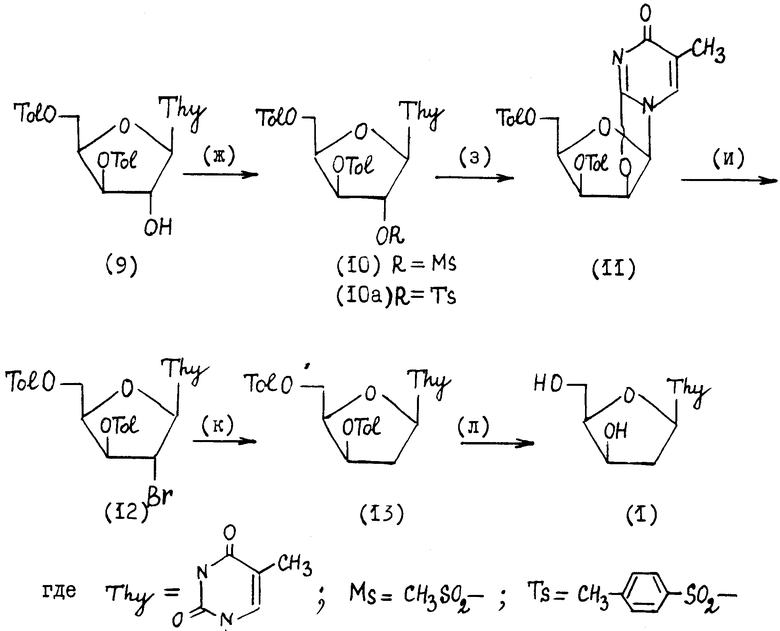
III. Образование 2'-бромпроизводного ксилофуранозилтимина, последующее его восстановление и деблокирование 3',5'-гидроксильных групп, стадии (ж) - (л), приведенной в конце текста описания.
На стадии (ж) соединение формулы (9) обрабатывают метансульфохлоридом (или п-толуолсульфохлоридом) в пиридине для введения уходящей группы по 2'-положению соединения формулы (9), получают 2'-мезилат (10) или 2'-тозилат (10а).
На стадии (з) нагревают смесь соединения формулы (10) или формулы (10а) с гидрокарбонатом натрия или калия в органическом растворителе, преимущественно в метаноле, этаноле. В результате получают хорошо кристаллизующееся 2,2'-ангидросоединение формулы (11).
На стадии (и) соединение формулы (11) вводят в реакцию с реагентом, который служит для замены гидроксильной группы в положении 2' на атом брома. Например, 2,2'-ангидросоединение формулы (11) вводят в реакцию с ацилбромидом (преимущественно ацетилбромидом) в органическом растворителе в присутствии спирта, или непосредственно обрабатывают раствором бромоводорода в органическом растворителе.
На стадии (к) проводят восстановление соединения формулы (12) под действием гипофосфористой (фосфиновой) кислоты в виде ее солей, преимущественно солей аминов, например триэтиламина, получают 5'-защищенный 2'-дезоксилотимидин (13) в кристаллическом состоянии.
На стадии (л) 3', 5'-деблокирование проводят спиртовым раствором аммиака или амина, в результате получают 2'-дезоксиксилотимидин (1).
Преимуществом изобретения является выбор соответствующей углеводной компоненты для упомянутой нуклеозидной конденсации. Это 1,2-ди-0-ацетил-3,5-ди-O-п-толуил- -α,β- -D-ксилофураноза формулы (6), которую получают из 1,2-O-изопропилиден- -α- -D-ксилофуранозы (3). Использование п-толуильных защит вместо бензоильных, как в прототипе [2], значительно упрощает выделение и очистку большей части интермедиатов благодаря свойству соответствующих ди-п-толуильных производных хорошо кристаллизоваться. В результате эффективная очистка имеет место как в синтезе углеводной компоненты (в виде смеси α- - и β- -аномеров) на стадии получения кристаллической 3,5-ди-O-п-толуил- α,β -D-ксилофуранозы (5), так и при синтезе хорошо кристаллизующихся нуклеозидных интермедиатов (9) - (12). Это обстоятельство особенно важно при проведении технологического процесса.
Использование щелочелабильных и одновременно устойчивых в кислой среде ацильных (п-толуильной, ацетильной) защит гарантирует количественный выход на стадии нуклеозидной конденсации в технологическом процессе при возможном увлажнении растворителя и, как следствие, закислении среды в присутствии катализаторов Фриделя-Крафтса (SnCl4 и др.)
Большая устойчивость 3',5'-п-толуильных защит в основных средах (или при действии щелочных агентов) по сравнению с бензоильными как в способе [2], позволяет успешно проводить модификацию углеводной части нуклеозидных интермедиатов по атому C-2'. Так, в более мягких условиях с высокими выходами протекает избирательное 2'-дезацетилирование соединения (8) разбавленным раствором аммиака в органических растворителях при пониженных температурах, а также внутримолекулярное нуклеофильное замещение при непродолжительном кипячении с гидрокарбонатом натрия в спирте с образованием 2,2'-ангидропроизводного (11). Доступность такого 2,2'-ангидропроизводного (11) делает возможным получение соответствующего 2'-бромпроизводного формулы (12) - соединения, в котором бром-группа способна восстанавливаться. Выход ключевого 2'-бромпроизводного (12) из тетраацилксилофуранозы (6) составляет 60% (Cp. [2]: выход 1-(3',5'- -ди-O-бензоил-2'-хлор- β -D-ксилофуранозил)тимина равен 52% из тетраацилксилозы в расчете на 3 стадии).
Получение нового соединения формулы (13) согласно изобретению основано на оригинальном методе восстановления 2'-бромпроизводного нуклеозида (12) с помощью фосфинистой (гипофосфористой) кислоты. Фосфорная кислота (в виде своих солей) в отличие от высокотоксичного гидрида трибутиолова, применяемого в прототипе [2] на аналогичной стадии, является малотоксичным и дешевым реагентом, гораздо более безопасным при использовании его в промышленном производстве. Выход на стадии восстановления 95%.
Общий выход 2'-дезоксиксилотимидина (1) составляет 33% (в расчете на D-ксилозу). Чистота полученных продуктов доказана данными спектров ЯМР-1H и 13C.
Изобретение имеет следующие преимущества.
1) Предложен новый способ синтеза 2'-дезоксиксилотимидина, являющегося сырьем для синтеза лекарственных субстанций против ВИЧ-инфекции.
2) Способ включает синтез новых интермедиатов, получаемых из доступных D-ксилозы и тимина с использованием дешевых реагентов, обычных органических растворителей при замене части реагентов на более безопасные при промышленном производстве.
3) Все стадии данного многостадийного процесса характеризуются высокими выходами.
4) Технология выделения большинства интермедиатов значительно упрощается благодаря тому, что они представляют собой хорошо кристаллизующиеся вещества.
5) Первый интермедиат-1,2-O-изопропилиден- α -D-ксилофураноза синтезируют одностадийным способом без перегонки в вакууме, что значительно упрощает технологию его получения.
6) Получаемый согласно изобретению 2'-дезоксиксилотимидин (1) - чистое вещество, пригодное для использования его в синтезе 3'-азидо-2',3'-дидезокситимидина и его 5'-фосфоната.
Пример 1. 1,2-O-Изопропилиден- α -D-ксилофураноза (3).
Перемешивают 45 г (0,3 моль) D-ксилозы в растворе 22,5 мл (0,422 моль) конц. серной кислоты в 900 мл ацетона. Через 1 ч добавляют 200 мл воды и оставляют раствор при 2 - 8oC на 15 - 18 ч. Смесь нейтрализуют при перемешивании раствором гидрокарбоната натрия, фильтруют. Из фильтрата отгоняют ацетон, оставшийся водный раствор экстрагируют 3 раза по 70 мл хлороформа. Хлороформенный слой фильтруют, фильтрат упаривают в вакууме до густого масла получают 1,2-O-изопропилиден- α -D-ксилофуранозу (3). Выход ее составляет 54 г (94,6%).
Пример 2. 3,5-Ди-O-(п-толуил)- -α,β- -D-ксилофураноза (5).
Растворяют 46,9 г (0,247 моль) 1,2-O-изопропилиден-D-ксилофуранозы (3) в 200 мл абсолютированного пиридина, затем при охлаждении (4±2oC) и перемешивании приливают 72 мл (0,54 моль) п-толуилхлорида. После добавления реагента смесь перемешивают еще 2 ч при комнатной температуре, выливают в 800 мл воды, экстрагируют дихлорметаном (2•300 мл). Органический слой промывают 2 раза насыщенным раствором натрия гидрокарбоната, упаривают, переупаривают с толуолом (2•100 мл). Остаток растворяют в 500 мл диоксана, добавляют 8 мл 5%-ного раствора соляной кислоты и греют 4 ч при 60oC. Затем раствор упаривают в вакууме, остаток перемешивают с 1,5 л насыщенного раствора гидрокарбоната натрия, образующееся кристаллическое вещество отфильтровывают, получают 64,9 г соединения (5). Выход 68% в расчете на соединение (3).
Пример 3. 1-(3,5-Ди-O-п-толуил- -β- -D-ксилофуранозил)тимин (9).
К 64,9 г (168, Оммоль) 3,5-ди-O-(п-толуил)- -α,β -D-ксилофуранозы (5) добавляют 120 мл уксусной кислоты, 60 мл уксусного ангидрида и 0,30 мл 1-метилимидазола. Раствор перемешивают при 50oC в течение 5 ч, затем упаривают в роторном испарителе и переупаривают 2 раза со 100 мл н-бутанола и 2 раза со 100 мл толуола, получают 79,9 г 3,5-ди-O-(п-толуил)-1,2-ди-O-ацетил- -α,β- -D-ксилофуранозы (6).
Смесь 23,5 г (0,186 моль) сухого тимина, 85,3 мл (0,41 моль) гексаметилдисилазана (ГМДС) и 18,8 мл (0,149 моль) триметилхлорсилана (ТМС) кипятят при перемешивании до полного растворения тимина, отгоняют избыток ГМДС и ТМС в вакууме, получают силилированный тимин (7) в виде масла. Растворяют 79,9 г 3,5-ди-O-(п-толуил)-1,2-ди-O-ацетил- α,β- -D-ксилофуранозы (6) в 500 мл абсолютированного 1,2-дихлорэтана. К раствору добавляют соединение (7), затем при перемешивании прибавляют 19,7 мл тетрахлорида олова. Смесь выдерживают при комнатной температуре в течение 16 ч, разбавляют хлороформом (500 мл) и выливают при перемешивании в 2 л 5%-ного водного раствора гидрокарбоната натрия. Смесь фильтруют через слой целита, промывают хлороформом (2•100 мл), объединенные фильтраты промывают 5%-ным раствором NaHCO3, сушат над сульфатом натрия, фильтруют, упаривают в вакууме при 40oC, получают соединение (8) в виде пены (89 г). Его растворяют в 400 мл толуола, добавляют 60 мл метанольного раствора аммиака, смесь выдерживают при 2 - 8oC в течение 16 - 18 ч, упаривают, остаток кристаллизуют из спирта, вещество сушат в вакууме (над P2O5), получают 67,0 г [80,7% на соединение (5)] 1-(3',5'-ди-O-(п-толуил)- β -D-ксилофуранозил)тимина (9).
Т. пл. 112 - 112, 5oC.
Спектр ЯМР-1H (в CDCl3), в м.д. δ :10,20 (ш.с 1H)3-H; 7,88 - 7,69 (м, 5H)6-H, аром. H; 7,19 - 7,17 (д, 4H) аром. H; 5,87 (c, 1H)H-1; 5,58(c, 1H)H-3', 4,98(ш.с., 1H) H-4'; 4,82-4,65 (м,H)H-5' и H-5''; 4,49(c, 1H)H-2'; 2,39(c,3H)Me(Tol); 1,87(c,3H)CH3тимина.
Пример 4.1-(2,2'-О-Цикло-3',5'-ди-О-n-толуил- β -D-ликсофуранозил)тимин (11).
Раствор 50,0 г (101,1 ммоль) нуклеозида (9) в 100 мл абсолютированного пиридина упаривают в вакууме роторного испарителя. Операцию сушки упариванием повторяют еще один раз. Полученное масло растворяют в 150 мл абс.пиридина, раствор охлаждают до 4-8oC и добавляют при перемешивании 11,8 мл (151,6 ммоль) мезилхлорида, смесь выдерживают 5 ч. при 22oC. Затем раствор медленно выливают при перемешивании в 700 мл воды. Смесь фильтруют, осадок промывают водой (3•20 мл).
Сырой осадок растворяют при нагревании в 800 мл этанола, добавляют 9,3 г (111 ммоль) гидроксикарбоната натрия NaHCO3. Смесь кипятят при интенсивном перемешивании в течение 1 ч., затем охлаждают, выдерживают при 4oC в течение 15 ч. , фильтруют. Осадок промывают этанолом и водой, сушат в вакууме над P2O5, получают 38,0 г 2,2' -ангидросоединения (11) (78,8%), т.пл. 260-261oC.
Спектр ЯМР-1H (в CDCl3): 7,87-7,78 (м,4H)аром; 7,26-7,16 (м, 5H)аром. +H-6; 6,18 (д, 1H, J1',2'= 5,80) H-1'; 5,77 (дд, 1H, J2',3'=6,05, J3',4'= 6,25)H-3'; 5,60 (дд, 1H)H-2'; 4,85 (м, 1H, J4'5'=5,34, J4'5''=7,63)H-4'; 4,52(дд, 1H, J5',5''= 12,2)H-5'; 4,34 (дд, 1H,)H-5''; 2,40 (3H,c)Me(Tol); 2,39(c, 3H)Me(Tol); 1,98 (д, J=1,08, 3H)5-Me.
Пример 5. 1-(3',5'-Ди-О-n-толуил-2'-бром-2'-дезокси- -β- -D-ксилофуранозил)тимин (12).
К суспензии 30 г (63 ммоль) (2,2'-О-цикло-3',5'-ди-О-n-толуил β - D-ликсофуранозил)тимина (11) в 300 мл этилацетата и 5 мл метанола добавляют при перемешивании 17,8 мл (240 ммоль) ацетилбромида и выдерживают еще 0,5 ч. Растворитель отгоняют, остаток кристаллизуют из этанола, получают 33,3 г (95%) соединения (12), т.пл. 129-130oC.
Спектр ЯМР-1H (в CDCl3); 8,58 (ш.с, 1H)NH; 7,91-7,76 и 7,26-7,21 (м, 8H) аромат; 7,58(д, J=1,34, 1H)H-6;6,31(д,J1',2'=1,83, 1H)H-1';5,76(дд, J2',3'= 1,34, J3',4'= 3,54,1H)H-3';5,09(м,J4',5'=6,96, J4',5'=4,27,1H)H-4'; 4,86(дд, J5',5''=12,09,1H)H-5'; 4,64(дд,1H)H-5'', 4,42(дд,1H)H-2'; 2,41(с,6H)Me(Tol); 1,87(д,J=1,10, 3H)5-Me.
Пример 6. 1-(3', 5'-Ди-0-п-толуил-2'-дезокси- β- -D-ксилофуранозил)тимин(13).
Нейтрализуют 27, 9 мл 50%-ного раствора гипофосфористой кислоты (0,267 моль), добавляя к нему 37,2 мл (0,267 моль) триэтиламина, раствор упаривают досуха, переупаривают с диоксаном (2•50 мл) и остаток растворяют в 250 мл диоксана. К полученному раствору добавляют 30 г 2'-бромпроизводного (12) и кипятят 20 мин, добавляя вначале и через 10 мин по 10 мг азобисизобутиронитрила. Затем отгоняют растворитель, к остатку добавляют по 150 мл воды и экстрагируют вещество хлористым метиленом (2•100 мл). Объединенный органический слой промывают насыщенным раствором гидрокарбоната натрия, упаривают, кристаллизуют из спирта, получают 23,7 г (92,1%) соединения (13), т.пл. 130,5-131,0oC.
Спектр ЯМР-1H(CDCl3): 8,60(ш. с 1H), NH; 7,89-7,60 и 7,25-7,19(м, 9H) аромат. +H-6; 6,32(дд, J1',2'= 7,84, J1',2''=2,68, 1H)H1'; 5,77 (м,1H)H-3'; 4,77 (дд, J4',5'=6,89, J5',5=11,96, 1H)H-5'; 4,68(дд, J4',5''=4,34, 1H)H-5''; 4,52(м, 1H)H-4'; 2,92(ддд, J2',2''=15,72, J2',3'=5,74, 1H)H-2'; 2,42(c, 3H)Me; 2,40(c,3H),Me; 2,32(ддд, J3',2''=0,85, 1H)H-2''; 1,85 (д, J=1,1)5-Me.
Пример 7. 1-(2'-дезокси- β -D-ксилофуранозил)тимин(1).
Растворяют при перемешивании 23,7 г(49,5 ммоля) 1-(3',5'-д-0-п-толуил-2'-дезокси- -β- - D-ксилофуранозил)тимина(13) в 170 мл насыщенного раствора аммиака в метаноле, раствор выдерживают 2 сут при комнатной температуре. Затем остаток кристаллизуют из метанола, получают 11,39 г соединения (1), выход 95,0%, т.пл. 170-172oC.
Спектр ЯМР1H (CDCl3-CD3OD) : 7,92(д,1H)H-6;6,17(дд. J1',2'=8,2, J1',2''= 2,6,1H)H-1'; 4,43(м,1H)H-3';3,95(ш.с.3H)H-4',H-5', H-5''; 2,64 (ддд, J2',3'= 5,5, J2',2''= 14,9,1H)H-2'; 2,06(дд, J3',2''<0,5,1H) H-2'',1,96(д, J=0,7, 3H)5-Me.
Источники информации:
1. EP-A2-0292101, 23,11.88, кл. C 07 H 19/06.
2. EP-A2-0301908, 01.02.89, кл. C 07 H 19/073.
3. а) Патент СССР N 1548182; б) Патент США N 5043437.
4. A.Holy, Coll. Cz. Chem. Comms., (1977), v.42, p.902.
5. P.A.Levene, A.L. Raymond, J.Biol. Chem., (1933), v.102, p.317.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 3'-АЗИДО-2',3'-ДИДЕЗОКСИТИМИДИНА | 1994 |
|
RU2102399C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2′, 3′ -ДИДЕГИДРО- 3′ -ДЕЗОКСИТИМИДИНА | 1993 |
|
RU2047619C1 |
| СИНТЕЗ β-L-2'-ДЕЗОКСИНУКЛЕОЗИДОВ | 2004 |
|
RU2361875C2 |
| L-НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АНТИ-HBV ИЛИ АНТИ-EBV АКТИВНОСТЬЮ, СПОСОБ ИНГИБИРОВАНИЯ HBV ИЛИ EBV ИНФЕКЦИИ | 1995 |
|
RU2171809C2 |
| МОДИФИЦИРОВАННЫЕ НУКЛЕОЗИД-5'-ТРИФОСФАТЫ КАК АНТИВИРУСНЫЕ АГЕНТЫ | 1996 |
|
RU2183213C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЗАМЕЩЕННЫХ 2-ДЕЗОКСИ-2-ФТОР-D-РИБОФУРАНОЗИЛ-ПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2407747C2 |
| ПРОИЗВОДНЫЕ 5'-H-ФОСФОНАТА 3'-АЗИДО-3'-ДЕЗОКСИТИМИДИНА И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2187509C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ФТОР-2'-АЛКИЛЗАМЕЩЕННЫХ ИЛИ ДРУГИХ ЗАМЕЩЕННЫХ РИБОФУРАНОЗИЛПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2433124C2 |
| БЕТА-L-2' ДЕЗОКСИНУКЛЕОЗИДЫ ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА В | 1999 |
|
RU2424016C2 |
| β-L-2'-ДЕЗОКСИНУКЛЕОЗИДЫ ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА В | 1999 |
|
RU2300381C2 |
Использование относится к органической химии, а именно к способу синтеза 2-дезоксилотимидина, производным Д-ксилофуранозы и производным 2-дезоксиксилотимидина, которые могут быть использованы в качестве исходных и промежуточных соединений в синтезе 3-азидо-2,3-дидезокситимидина (азидотимидина)-эффективного лекарства против СПИДа, а также 5-фосфоната 3-азидо-2,3-дидезокситимидина. Сущность: способ получения 2-дезоксиксилотимидина, включающий обработку Д-ксилозы ацетоном в присутствии концентрированной серной кислоты, обработку полученного соединения (3) п-толуилхлоридом в присутствии пиридина с образованием соединения (4), удаление 1,2-0-изопропилиденовой защитной группы при действии на соединение формулы (4) раствора минеральной кислоты с образованием соединения формулы (5), ацетилирование соединения формулы (5) путем обработки его уксусным ангидридом в среде уксусной кислоты в присутствии нуклеофильного катализатора или путем обработки его уксусным ангидридом или ацетилгалогенидом в пиридине с образованием соединения формулы (6), реакцию соединения формулы (6) с производным тимина формулы (7) с образованием соединения формулы (8), избирательный гидролиз 2-ацетата в соединении формулы (8) путем обработки последнего разбавленным спиртовым раствором аммиака с образованием соединения формулы (9), обработку соединения формулы (9) метансульфохлоридом или п-толуолсульфохлоридом с образованием 2-мезилата формулы (10) или 2-тозилата формулы (10а), обработку соединения формулы (10) или (10а) гидрокарбонатом натрия или калия в органическом растворителе с образованием 2,2-ангидросоединения формулы (11), взаимодействие соединения формулы (11) с реагентом, который служит для замены гидроксильной группы на атом брома с образованием соединения формулы (12), восстановление соединения формулы (12) с образованием соединения формулы (13), удаление 3- и 5-п-толуильных защитных групп в соединении формулы (13). Соединения формулы (А)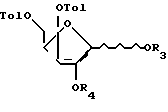 в которой Tol - n - толуильная защитная группа n-СН3-С6Н4-С(О) -, R3 и R4 вместе составляют одну изопропилиденовую защитную группу, или R3 = R4-водород. Соединения общей формулы (Б):
в которой Tol - n - толуильная защитная группа n-СН3-С6Н4-С(О) -, R3 и R4 вместе составляют одну изопропилиденовую защитную группу, или R3 = R4-водород. Соединения общей формулы (Б):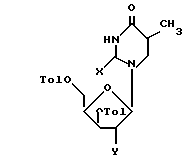
в которой Х представляет собой оксогруппу, а У-ацетогруппу, или У - гидроксильная группа, или У-мезильная (или п-тозильная) группа. Предложенные соединения могут быть использованы в качестве промежуточных соединений для получения 3'-азидо-2', 3'-дидезокситимидина или 5'-фосфоната 3'-азидо-2', 3'- дидезокситимидина. 3 с.п.ф-лы.
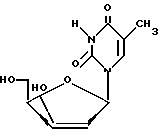
включающий нуклеозидную конденсацию производного D-ксилофуранозы, содержащего две пары различных ацильных групп в 1,2 и 3,5-положениях и полученного из D-ксилозы через 1,2-O-изопропилиден -α- D-ксилофуранозу формулы II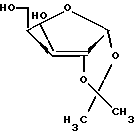
с 2,4-бис(триметилсилил)тимином формулы III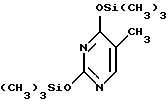
с последующим избирательным 2'-деблокирование, и 2'-дезоксигенирование 3', 5'-защищенного β- D-ксилотимидина с последующим 3',5'-деблокированием, отличающийся тем, что проводят обработку 1,2-O-изопропилиден -α- D-ксилофуранозы формулы II п-толуилхлоридом в присутствии пиридина с образованием соединения формулы IV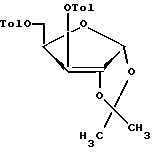
в которой Tol - п-толуильная группа п-CH3-C6H4C(O),
удаление 1,2-O-изопропилиденовой защитной группы при действии на соединение формулы IV раствора минеральной кислоты в органическом растворителе или органической кислоты с образованием соединения формулы V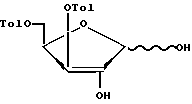
ацетилирование соединения формулы V путем обработки его уксусным ангидридом в среде уксусной кислоты в присутствии нуклеофильного катализатора или путем обработки его уксусным ангидридом или ацетилгалогенидом в пиридине с образованием соединения формулы VI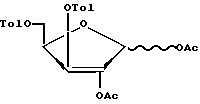
реакцию соединения формулы VI с 2,4-бис(триметилсилил)тимином формулы III с образованием соединения формулы VII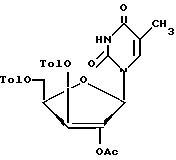
изюирательный гидролиз соединения формулы VIII разбавленным спиртовым раствором аммиака в органическом растворителе с образованием соединения формулы VIII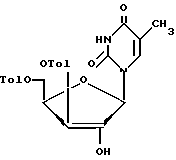
обработку соединения формулы VIII метансульфохлоридом или п-толуолсульфохлоридом в пиридине для введения уходящей группы в положении 2' с образованием 2'-мезилата формулы IX или 2'-тозилата формулы IXа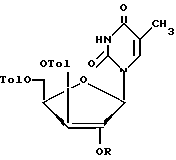
в которых R = CH3SO2 - соединение формулы IX или R - п-CH3C6H4SO2 - соединение формулы IXа,
обработку соединения формулы IX или IXа гидрокарбонатом натрия или калия в полярном органическом растворителе с образованием 2,2-ангидросоединения формулы X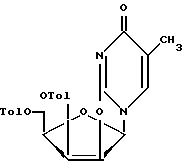
взаимодействие соединения формулы X с реагентом, который служит для замены гидроксильной группы в положении 2 на атом брома, что приводит к образованию соединения формулы II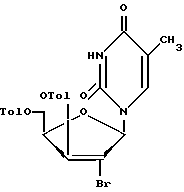
восстановление соединения формулы II при взаимодействии с гипофосфористой кислотой или ее солями с образованием соединения формулы XII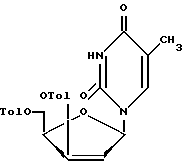
удаление 3'- и 5'-п-толуильных защитных групп в соединении формулы XII при обработке его основными реагентами в органическом растворителе с образованием 2'-дезоксиксилотимидина формулы I.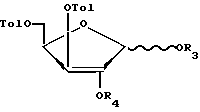
в которой Tol - п-толуильная защитная группа п-CH3C6H4C(O);
R3 и R4 вместе составляют одну изопропилиденовую защитную группу, или R3 = R4 - водород, или R3 = R4 - ацетильная защитная группа,
в качестве промежуточных соединений для получения 3'-азидо-2',3'-дидезоксимитидина или 5'-фосфоната 3'-азидо-2',3'-дидезокситимидина.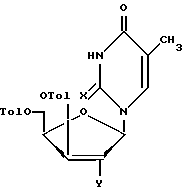
в которой Х - оксогруппа;
Y - ацетоксигруппа, гидроксильная группа, или мезильная (или п-тозильная) группа, или бром, или водород, или Х и Y вместе представляют собой -О- связь между положениями 2 и 2' соединения формулы Б,
в качестве промежуточных соединений для получения 3'-азидо-2',3'-дидезокситимидина или 5'-фосфоната 3'-азидо-2',3'-дидезокситимидина.
