Это изобретение относится к способам лечения вируса гепатита В (также упоминаемого как “HBV”), которые включают в себя введение хозяину в случае необходимости - либо отдельно, либо в сочетании - эффективного количества одного или более активных соединений, описываемых в заявке, или фармацевтически приемлемого пролекарства, или соли одного из этих соединений.
HBV находится на втором месте после табака как причина рака человека. Механизм, посредством которого HBV вызывает рак, не известен, хотя предполагают, что он может непосредственно запускать развитие опухоли или косвенно запускать развитие опухоли через хроническое воспаление, цирроз и клеточную регенерацию, связанную с инфекцией.
Вирус гепатита В достигает эпидемических уровней по всему миру. После инкубационного периода от двух до шести месяцев, в течение которого хозяин ничего не знает об инфекции, инфекция HBV может привести к острому гепатиту и поражению печени, что вызывает боль в брюшной полости, желтуху и повышенные уровни определенных ферментов в крови. HBV может вызывать молниеносный гепатит, быстро прогрессирующую и летальную форму заболевания, при котором разрушаются массивные участки печени.
Обычно после острого гепатита пациенты выздоравливают. Однако у некоторых пациентов высокие уровни вирусного антигена сохраняются в крови в течение длительного или неограниченного периода времени, вызывая хроническое инфекционное заболевание. Хронические инфекции могут привести к хроническому персистирующему гепатиту. Пациенты, инфицированные хроническим персистирующим HBV, встречаются больше всего в развивающихся странах. К середине 1991 в одной Азии имелось приблизительно 225 миллионов хронических носителей HBV, а во всем мире почти 300 миллионов носителей. Хронический персистирующий гепатит может быть причиной утомляемости, цирроза печени и гепатоцеллюлярной карциномы, первичного рака печени.
У жителей индустриальных стран группы высокого риска инфицирования HBV включают в себя группы людей, находившихся в контакте с носителями HBV или образцами их крови. Эпидемиология HBV очень похожа на эпидемиологию синдрома приобретенного иммунодефицита (AIDS; СПИД), что объясняет, почему инфекция HBV распространена среди пациентов со СПИДом или родственным СПИДу комплексом. Однако HBV является более заразным, чем HIV (ВИЧ).
Однако позднее с помощью генной инженерии были получены вакцины, которые широко используются в настоящее время. К сожалению, вакцины не могут помочь пациентам, которые уже инфицированы HBV. Также показано, что ежедневное лечение α-интерфероном, генно-инженерным белком обнадеживает, но эта терапия оказывается успешной приблизительно только у одной трети леченых пациентов. Кроме того, интерферон нельзя назначать перорально.
Идентифицирован ряд синтетических нуклеозидов, которые проявляют активность в отношении HBV. (-)-Энантиомер ВСН-189, известный как 3ТС, заявленный Liotta, et al. в Патенте США 5 539 116, одобрен U.S. Food and Drug Administration для лечения гепатита В. Также см. Европейский Патент ЕРА 0 494 119 А1, поданный BioChem Pharma, Inc.
Цис-2-гидроксиметил-5-(5-фторцитозин-1-ил)-1,3-окса-тиолан (“FTC”) проявляет активность в отношении HBV. См. Международную Заявку WO 92/15308; Furman, et al., “The Anti-Hepatitis B Virus Activities, Cytotoxicities, and Anabolic Profiles of the (-) and (+) Enantiomers of cis-5-Fluoro-1-[2-(Hydroxymethyl)-1,3-oxathiolane-5-yl]-Cytosine” Antimicrobial Agents and Chemotherapy, декабрь 1992, page 2686-2692; и Cheng, et al., Journal of Biological Chemistry, Volume 267(20), 13938-13942 (1992).
Von Janta-Lipinski et al., описывает применение L-энантиомеров 5'-трифосфатов 3'-фтор-модифицированных β-2'-дезоксирибонуклеозидов для ингибирования гепатит В-полимеразы (J. Med. Chem., 1998, 41, 2040-2046). В частности, 5'-трифосфаты 3'-дезокси-3'-фтор-β-L-тимидина (β-L-FTTP), 2',3'-дидезокси-3'-фтор-β-L-цитидина (β-L-FdCTP) и 2',3'-дидезокси-3'-фтор-β-L-5-метилцитидина (β-L-FMethCTP) описаны как эффективные ингибиторы ДНК-полимеразы HBV.
В Международной Заявке WO 96/13512 Genencor International, Inc. и Lipitek, Inc. заявляют, что некоторые L-рибофуранозил нуклеозиды можно использовать для лечения рака и вирусов. В частности, описывается применение этого класса соединений для лечения рака и ВИЧ.
В Патентах США №№ 5565438, 5567688 и 5587362 (Chu, et al.) описывается применение 2'-фтор-5-метил-β-L-арабинофуранолилуридина (L-FMAU) для лечения гепатита В и вируса Эпштейна-Барр (Epstein-Barr).
В Международной Заявке WO 92/18517 Yale University and University of Georgia Research Foundation, Inc., описано применение L-FddC (β-L-5-фтор-2',3'-дидезоксицитидин) для лечения вируса гепатита В.
В данной области известны синтетические нуклеозиды β-L-2'-дезоксицитидин (β-L-2'-dC), β-L-2'-дезокситимидин (β-L-dT) и β-L-2'-дезоксиаденозин (β-L-2'-dA). Antonin Holy впервые описал β-L-dC и β-L-dT в 1972, “Nucleic Acid Components and Their Analogs. CLIII. Preparation of 2'-deoxy-L-Ribonucleosides of the Pyrimidine Series,” Collect. Czech. Chem. Commun. (1972), 37(12), 4072-87. Morris S. Zedeck et al., впервые предложил β-L-dA для ингибирования синтеза индуцированных ферментов у Pseudomonas testosteroni, Mol. Phys. (1967), 3(4), 386-95.
Известно, что определенные 2'-дезокси-β-L-эритро-пентофуранонуклеозиды проявляют антинеопластическую и избирательную противовирусную активность. Verri et al. описывает применение 2'-дезокси-β-L-эритропентофуранонуклеозидов в качестве антинеопластических агентов и противогерпетических агентов (Mol. Pharmacol. (1997), 51(1), 132-138 и Biochem. J. (1997), 328(1), 317-20). Saneyoshi et al. описывают применение 2'-дезокси-L-рибонуклеозидов в качестве ингибиторов обратной транскриптазы (I) для контроля ретровирусов и для лечения СПИДа Jpn. Kokai Tokkyo Koho JPO6293645 (1994).
Giovanni et al. исследовал 2'-дезокси-β-L-эритропентофуранонуклеозиды отчасти против вируса псевдобешенства (PRV), Biochem. J. (1993), 294(2), 381-5.
Химиотерапевтическое применение 2'-дезокси-β-L-эритропентофуранонуклеозидов изучалось Tyrsred et al. (Biochim. Biophys. Acta (1968), 155(2), 619-22) и Bloch, et al. (J. Med. Chem. (1967), 10(5), 908-12).
В данной области известно, что β-L-2'-дезокситимидин (β-L-dT) ингибирует тимидинкиназу (ТК) вируса герпеса I типа (HSV-I). Iotti et al. в Международной Заявке WO 92/08727 описано, что β-L-dT избирательно ингибирует фосфорилирование D-тимидина посредством TK HSV-I, но не посредством ТК человека. Spaldari et al. сообщил, что L-тимидин фосфорилируется тимидинкиназой вируса герпеса I типа и ингибирует рост вирусов, J. Med. Chem. (1992), 35(22), 4214-20.
В свете того факта, что вирус гепатита В достигает эпидемических уровней во всем мире и оказывает тяжелое и часто трагическое воздействие на инфицированного пациента, существует серьезная потребность в обеспечении новыми эффективными фармацевтическими веществами для лечения человека, инфицированного вирусом, которые характеризуются низкой токсичностью для хозяина.
Поэтому целью данного изобретения является предоставление новых способов и композиций для лечения человека или других хозяев, инфицированных вирусом гепатита В.
Краткое описание изобретения
Описан способ лечения инфекции гепатита В у человека и других животных хозяев, который включает в себя введение эффективного количества биологически активного 2'-дезокси-β-L-эритропентофуранонуклеозида (альтернативно упоминаемого в описании как β-L-d-нуклеозид или β-L-2'-d-нуклеозид) или его фармацевтически приемлемой соли или пролекарства, вводимого либо отдельно, либо в сочетании, возможно, в фармацевтически приемлемом носителе. Термин 2'-дезокси, который использован в описании, относится к нуклеозиду, не содержащему никакой замещающей группы в 2'-положении.
Описываемые 2'-дезокси-β-L-эритропентофуранонуклеозиды или фармацевтически приемлемые пролекарства или соли, или фармацевтически приемлемые композиции, содержащие эти соединения, используются для предупреждения и лечения инфекций гепатита В и других родственных состояний, таких как анти-HBV-антитело-позитивное и HBV-позитивное состояния, хроническое воспаление печени, вызванное HBV, цирроз, острый гепатит, молниеносный гепатит, хронический персистирующий гепатит и утомляемость. Эти соединения или композиции также можно использовать профилактически для предупреждения или замедления прогрессирования клинического заболевания у индивидуумов, которые являются анти-HBV-антитело-позитивными или HBV-антиген-позитивными или которые подвергались воздействию HBV.
В одном из аспектов данного изобретения, производное 2'-дезокси-β-L-эритро-пентофуранонуклеозида является соединением формулы:

в которой R выбирается из группы, состоящей из Н, прямого, разветвленного или циклического алкила, СО-алкила, СО-арила, СО-алкоксиалкила, СО-арилоксиалкила, СО-замещенного арила, алкилсульфонила, арилсульфонила, аралкилсульфонил, остатка аминокислоты, моно-, ди- или трифосфата, или производного фосфата; и основание представляет собой пуриновое или пиримидиновое основание, которое необязательно замещено.
В другом аспекте производное 2'-дезокси-β-L-эритро-пентофуранонуклеозида представляет собой β-L-2'-дезоксиаденозин или его фармацевтически приемлемую соль или пролекарство формулы:
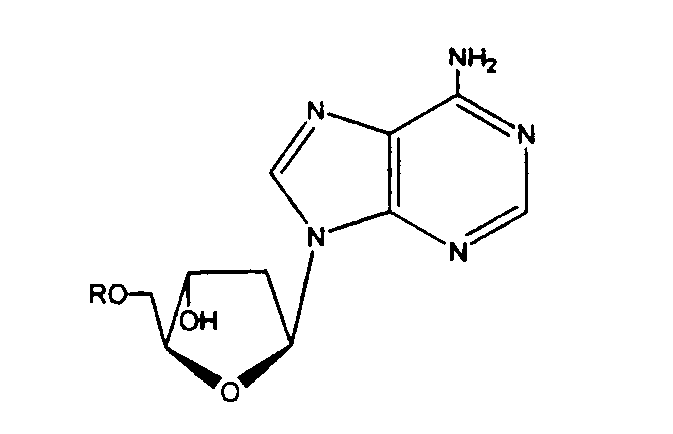
в которой R представляет собой Н, моно-, ди- или трифосфат, ацил или алкил, или стабилизированное фосфатное производное (для образования стабилизированного нуклеотидного пролекарства).
В еще одном аспекте, производное 2'-дезокси-β-L-эритро-пентофуранонуклеозида является β-L-2'-дезоксицитидином или его фармацевтически приемлемой солью или пролекарством формулы:
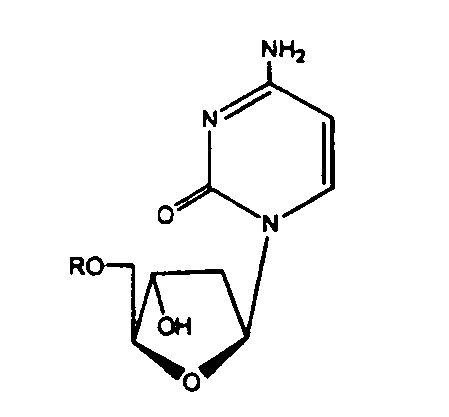
в которой R представляет собой Н, моно-, ди- или трифосфат, ацил или алкил, или стабилизированное фосфатное производное (для образования стабилизированного нуклеотидного пролекарства).
В следующем аспекте, производное 2'-дезокси-β-L-эритро-пентофуранонуклеозида является β-L-2'-дезоксиуридином или его фармацевтически приемлемой солью или пролекарством, описываемом формулой:
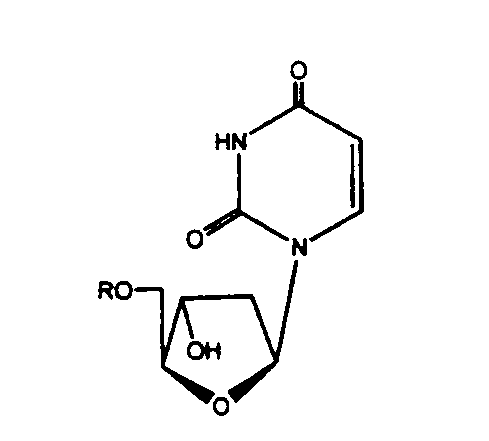
в которой R представляет собой Н, моно-, ди- или трифосфат, ацил или алкил, или стабилизированное фосфатное производное (для образования стабилизированного нуклеотидного пролекарства).
В другом аспекте, производное 2'-дезокси-β-L-эритро-пентофуранонуклеозида является β-L-2'-дезоксигуанозином или его фармацевтически приемлемой солью или пролекарством, имеющим формулу:
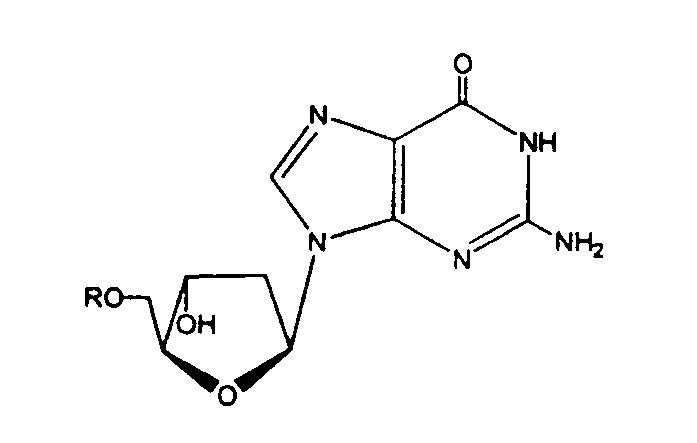
в которой R представляет собой Н, моно-, ди- или трифосфат, ацил или алкил, или стабилизированное фосфатное производное (для получения стабилизированного нуклеотидного пролекарства).
В следующем аспекте, производное 2'-дезокси-β-L-эритро-пентофуранонуклеозида является β-L-2'-дезоксиинозином или его фармацевтически приемлемой солью или пролекарством, имеющим формулу:

в которой R представляет собой Н, моно-, ди- или трифосфат, ацил или алкил, или стабилизированное фосфатное производное (для получения стабилизированного нуклеотидного пролекарства).
Еще в одном аспекте, производное 2'-дезокси-β-L-эритро-пентофуранонуклеозида является β-L-тимидином или его фармацевтически приемлемой солью или пролекарством формулы:
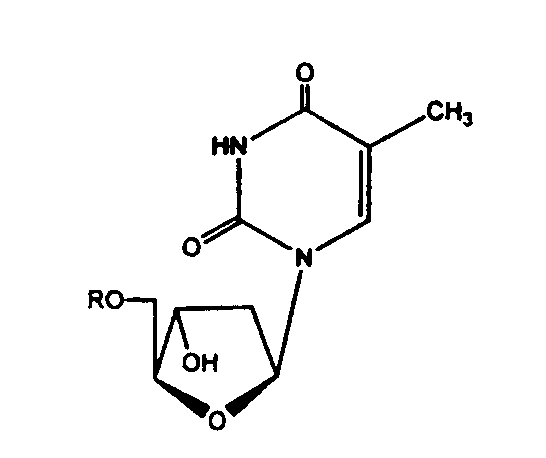
в которой R представляет собой Н, моно-, ди- трифосфат, ацил или алкил, или стабилизированное фосфатное производное (для получения стабилизированного нуклеотидного пролекарства).
В другом аспекте 2'-дезокси-β-L-эритро-пентофуранонуклеозид вводится в чередовании или в сочетании с одним или более 2'-дезокси-β-L-эритро-пентофуранонуклеозидами или с одним или более других соединений, которые проявляют активность в отношении вируса гепатита В. Вообще во время альтернирующей терапии эффективная доза каждого агента вводится по очереди, тогда как при сочетанной терапии эффективные дозы двух или более веществ вводятся вместе. Дозировки зависят от абсорбции, инактивации и скорости экскреции лекарственного вещества, а также других факторов, известных специалистам в данной области. Следует отметить, что величины доз также изменяют в зависимости от тяжести состояния для его облегчения. Кроме того, ясно, что для любого отдельного субъекта должны быть установлены специальные схемы и графики приема лекарственных средств, в зависимости от времени, согласно индивидуальной потребности и профессиональной оценке лицом, назначающим композиции и наблюдающим за их введением.
В следующем аспекте изобретение представляет способ лечения человека, инфицированного HBV, который включает в себя введение лечебной дозы пролекарства описанных производных 2'-дезокси-β-L-эритро-пентофуранонуклеозида. Пролекарство, которое используется в заявке, относится к соединению, которое превращается в нуклеозид при введении in vivo. Не ограничивающие примеры включают в себя фармацевтически приемлемую соль (альтернативно упоминаемую как «физиологически приемлемые соли»), 5' и N4 (цитидин)- или N6 (аденозин)-ацилированные или алкилированные производные активного соединения, или 5'-фосфолипид или 5'-эфиры липидов активного соединения.
Краткое описание чертежей
Фигура 1 иллюстрирует общий способ получения β-L-эритро-пентафуранонуклеозидов (β-L-dN), используя L-рибозу или L-ксилозу в качестве исходного материала.
Фигура 2 представляет собой график, который иллюстрирует метаболизм L-dA, L-dC и L-dT в клетках Hep G2 человека в зависимости от аккумуляции и распада. Клетки инкубировали в присутствии с 10 мкМ соединения.
Фигура 3 представляет собой график, который иллюстрирует противовирусное действие β-L-dA, β-L-dT и β-L-dC на модели хронического гепатита сурка.
Используемый в описании термин “по существу в форме ординарного изомера” или “в изолированной форме” относится к 2'-дезокси-β-L-эритро-пентофуранонуклеозиду, который существует по крайней мере приблизительно на 95% в установленной стереоконфигурации. В предпочтительном варианте, активное соединение, по крайней мере этой степени чистоты, вводят хозяину при необходимости терапии.
Используемый в описании термин “гепатит В и родственные состояния” относится к гепатиту В и родственным состояниям таким как анти-HBV-антитело-позитивное и HBV-позитивное состояния, хроническое воспаление печени, вызванное HBV, цирроз, острый гепатит, молниеносный гепатит, хронический персистирующий гепатит и утомляемость. Способ согласно изобретению включает в себя применение производных 2'-дезокси-β-L-эритро-пентофуранонуклеозида профилактически для предупреждения или замедления прогрессирования клинического заболевания у индивидуумов, которые являются анти-HBV-антитело-позитивными или HBV-антиген-позитивными или которые подверглись воздействию HBV.
Используемый в описании термин алкил, если не указано особо, относится к насыщенному прямому, разветвленному или циклическому, первичному, вторичному или третичному углеводороду, обычно С1 до С18, предпочтительно С1 до С6 и, в частности, не ограничиваясь этим, включает в себя метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил, трет-бутил, изопентил, амил, трет-пентил, циклопентил и циклогексил.
Используемый в описании термин «ацил» относится к фрагменту формулы -С(О)R', где R' представляет собой алкил; арил, алкарил, аралкил, гетероароматику, алкоксиалкил, включая метоксиметил; арилалкил, включая бензил; арилоксиалкил, такой как феноксиметил; арил, включая фенил, необязательно замещенный галогеном, С1-С4 алкил или С1-С4 алкокси, или остаток аминокислоты. Термин ацил, не ограничиваясь этим, в частности, включает в себя ацетил, пропионил, бутирил, пентаноил, 3-метилбутирил, кислый сукцинат, 3-хлорбензоат, бензоил, ацетил, пивалоил, мезилат, пропионил, валерил, остаток капроновой, каприловой, каприновой, лауриновой, миристиновой, пальмитиновой, стеариновой и олеиновой кислот.
Используемый в описании термин “пуриновое или пиримидиновое основание”, не ограничиваясь этим включает в себя 6-алкилпурин и N6-алкилпурины, N6-ацилпурины, N6-бензилпурин, 6-галопурин, N6-винилпурин, N6-ацетиленовый пурин, N6-ацилпурин, N6-гидроксиалкилпурин, N6-тиоалкилпурин, N2-алкилпурины, N4-алкилпиримидины, N4-ацилпиримидины, 4-бензилпиримидины, N4-галопиримидины, N4-ацетиленовые пиримидины, 4-ацил- и N4-ацилпиримидины, 4-гидроксиалкилпиримидины, 4-тиоалкилпиримидины, тимин, цитозин, 6-азапиримидин, включая 6-азацитозин, 2- и/или 4-меркаптопиримидин, урацил, С5-алкилпиримидины, С5-бензилпиримидины, С5-галопиримидины, С5-винилпиримидин, С5-ацетиленовый пиримидин, С5-ацилпиримидин, С5-гидроксиалкилпурин, С5-амидопиримидин, С5-цианопиримидин, С5-нитропиримидин, С5-аминопиримидин, N2-алкилпурины, N2-алкил-6-тиопурины, 5-азацитидинил, 5-азаурацилил, триазолопиридинил, имидазолпиридинил, пирролопиримидинил и пиразолопиримидинил. Функциональные группы кислорода и азота на основании могут быть защищены по мере необходимости или по желанию. Подходящие защищающие группы хорошо известны специалистам в данной области и включают в себя триметилсилил, диметил-гексилсилил, трет-бутилдиметилсилил и трет-бутилдифенилсилил, тритил, алкильные группы, ацильные группы, такие как ацетил и пропионил, метансульфонил и п-толуолсульфонил.
Термин биологически активный нуклеозид, используемый в описании, относится к нуклеозиду, который характеризуется значением ЕС50, соответствующим 15 микромолям или менее при исследовании в клетках 2.2.15, трансфицированных вирионом гепатита.
Предпочтительные основания включают в себя цитозин, 5-фторцитозин, 5-бромцитозин, 5-иодцитозин, урацил, 5-фтор-урацил, 5-бромурацил, 5-иодурацил, 5-метилурацил, тимин, аденин, гуанин, инозин, ксантин, 2,6-диаминопурин, 6-аминопурин, 6-хлорпурин и 2,6-дихлорпурин, 6-бромпурин, 2,6-дибромпурин, 6-иодпурин, 2,6-дииодпурин, 5-бромвинилцитозин, 5-бромвинилурацил, 5-бромэтенилцитозин, 5-бромэтенилурацил, 5-трифторметилцитозин, 5-трифторметилурацил.
2'-дезокси-β-L-эритро-пентофуранонуклеозид может быть предоставлен в виде 5'-фосфолипида иои 5'-эфира липида, что описывается в следующих ссылках: Kucera, L.S., N. Lyer, E. Leake, A. Raben, Modest E.J., D.L.W., and C. Piantadosi. 1990. Novel membrane-interactive ether lipid analogs that inhibit infectious HIV-I production and induce defective virus formation. AIDS Res Hum Retroviruses. 6:491-501; Piantadosi, C., J.Marasco C.J., S.L. Morris-Natschke, K.L. Meyer, F. Gumus, J.R. Surles, K.S.Ishaq, L.S. Kusera, N. Iyer, C.A. Wallen, S.Piantadosi, and E.J. Modest. 1991-Synthesis and evaluation of novel ether lipid nucleoside conjugates for anti-HIV activity. J. Med. Chem. 34:1408-1414; Hostetler, K.Y., D.D. Richman, D.A. Carson, L.M. Stuhmiller, G.M.T. van Wijk, and H. van den Bosch. 1992. Greatly enhanced inhibition of human immunodeficiency virus type I replication in CEM and HT4-6C cells by 31-deoxythymidine dephosphate dimyristoylglycerol, a lipid prodrug of 31-deoxythymidine. Antimicrob Agents Chemother. 36:2025-2029; Hostetler, K.Y., L.M. Stuhmiller, H.B. Lenting, H. van den Bosch, and D.D.Richman. 1990. Synthesis and antiretroviral activity of phospholipid analogs of azidothymidine and other antiviral nucleosides. J. Biol. Chem. 265:6112-7.
2'-дезокси-β-L-эритро-пентофуранонуклеозид может быть превращен в фармацевтически приемлемый эфир посредством реакции с соответствующим эстерифицирующим агентом, например галогенангидридом или ангидридом. Нуклеозид или его фармацевтически приемлемое пролекарство может быть превращено в его фармацевтически приемлемую соль общепринятым способом, например с помощью обработки соответствующим основанием или кислотой. Эфир или соль может превращаться в исходный нуклеозид, например, в результате гидролиза.
Используемый в описании термин “фармацевтически приемлемые соли или комплексы” относится к солям или комплексам 2'-дезокси-β-L-эритро-пентофуранонуклозидов, которые сохраняют требуемую биологическую активность исходного соединения и проявляют минимальные, если вообще проявляют, нежелательные токсикологические воздействия. Не ограничивающими примерами таких солей являются (а) соли присоединения кислот, образованные неорганическими кислотами (например, хлористоводородная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, азотная кислота и тому подобные), и соли, образованные органическими кислотами такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, пальмовая кислоты, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновые кислоты, нафтaлиндисульфоновые кислоты и полигалактуроновая кислота; (b) соли присоединения основания, образованные катионами, такими как натрий, калий, цинк, кальций, висмут, барий, магний, алюминий, медь, кобальт, никель, кадмий, натрий, калий и тому подобное, или органическим катионом, полученным из N,N-дибензилэтилендиамина, аммония или этилендиамина; или (с) комбинациями (а) и (b); например, таннат цинка или тому подобное.
Используемый в описании термин «пролекарство» относится к соединению, которое превращается в нуклеозид при введении in vivo. Не ограничивающими примерами являются фармацевтически приемлемые соли (альтернативно упоминаемые как «физиологически приемлемые соли»), 5'- и N4- или N6- ацилированные или алкилированные производные активного соединения и 5'-фосфолипидные и 5'-эфирлипидные производные активного соединения.
Модификации активных соединений, особенно в N4, N6 и 5'-O положениях, могут воздействовать на биологическую доступность и скорость метаболизма активных веществ, обеспечивая таким образом контроль за доставкой активных соединений.
Предпочтительным аспектом данного изобретения является способ лечения инфекций HBV у человека или других животных хозяев, который включает в себя введение эффективного количества одного или более 2'-дезокси-β-L-эритро-пентофу-ранонуклеозидных производных, выбранных из группы, состоящей из β-L-2'-дезоксиаденозина, β-L-2'-дезоксицитидина, β-L-2'-дезоксиуридина, β-L-2'-гуанозина, β-L-2'-дезоксиинозина и β-L-2'дезокситимидина или их физиологически приемлемого пролекарства, включая фосфат, 5'- и или N6- алкилированное или ацилированное производное или его физиологически приемлемую соль, необязательно в фармацевтически подходящем носителе. Соединения этого изобретения или обладают анти-HBV-активностью или преобразуются в соединение или соединения, которые проявляют анти-HBV-активность. В предпочтительном аспекте, 2'-дезокси-β-L-эритро-пентофуранонуклеозид вводится в основном в форме ординарного изомера, то есть по крайней мере приблизительно на 95% в установленной стереоконфигурации.
Нуклеотидные пролекарства
Любой из описанных в описании нуклеозидов может быть введен в виде стабилизированного нуклеодитного пролекарства, чтобы увеличить активность, биологическую доступность, стабильность или, иначе говоря, чтобы изменить свойства нуклеозида. Известен ряд лигандов нуклеотидных пролекарств. Вообще алкилирование, ацилирование или другая липофильная модификация моно-, ди- или трифосфата нуклеозида будет увеличивать стабильность нуклеотида. Примерами заместительных групп, которые могут замещать один или более водородов на фосфатном фрагменте, являются алкил, арил, стероиды, углеводы, включая сахара, 1,2-диацилглицерин и спирты. Большинство описывается в публикации R. Jones and N. Bischofberger, Antiviral Research, 27 (1995) 1-17. Любые из них могут применяться в комбинации с описанными нуклеозидами для достижения желаемого действия.
В одном аспекте 2'-дезокси-β-L-эритро-пентофуранонуклеозид обеспечивается в виде 5'-гидроксильного липофильного пролекарства. Не ограничивающие примеры патентов США, описывающие подходящие липофильные заместители, которые можно ковалентно включить в нуклеозид, предпочтительно в 5'-OH положении нуклеозида, или липофильные препараты, включают в себя патенты США № 5149794 (22 сентября, 1992, Yatvin et al.); 5 194 654 (16 мартаб 1993, Hostetler et al.); 5223263 (29 июня, 1993, Hostetler et al.); 5256641 (26 октября, 1993, Yatvin et al.); 5411947 (2 мая, 1995, Hostetler et al.); 5463092 (31 октября, 1995, Hostetler et al.); 5543389 (6 августа, 1996, Yatvin et al.); 5543390 (6 августа 1996, Yatvin et al.), 5543391 (6 августа, 1996, Yatvin et al.); и 5554728 (10 сентября, 1996, Basava et al.).
Иностранные патентные заявки, которые описывают липофильные заместители, которые могут быть присоеденены к 2'-дезокси-β-L-эритро-пентофуранонуклеозидному производному данного изобретения или к липофильным препаратам, включают в себя WO 89/02733, WO 90/00555, WO 91/16920, WO 91/18914, WO 93/00910, WO 94/ 26273, WO 96/15132, EP 0 350 287, EP 93917054,4 и WO 91/19721.
Дополнительными не ограничивающими примерами 2'-дезокси-β-L-эритро-пентофуранонуклеозидов являются нуклеозиды, которые содержат заместители, описанные в следующих публикациях. Эти преобразованные 2'-дезокси-β-L-эритро-пентофуранонуклеозиды могут использоваться для целей, описанных в тексте, или, иначе говоря, как противовирусные агенты, включая анти-HBV агенты. D.H.W. (1973) Distribution of kinase and deaminase of 1 β-D-arabinofuranosylcytosine in tissues of man and mouse. Cancer Res. 33, 2816-2820; Holy, A. (1993) Isopolar phosphorous-modified nucleotide analogues. In: De Clercq (Ed.), Advances in Antiviiral Drug Design, Vol. I, JAI Press, pp. 179-231; Hong, C.I., Nechaev A., and West, C.R. (1979a) Synthesis and antitumor activity of 1β-D-arabinofuranosylcytosine conjugates of cortisol and cortisone. Biochem. Biophys. Rs. Commun. 88, 1223-1229; Hong, C.I., Nechaev, A., Kirisits, A.J. Buchheit, D.J. and West C.R. (1980) Nucleoside conjugates as potential antitumor agents. 3.Synthsis and antitumor activity of 1-(β-D-arabinofuranosyl)cytosine conjugates of corticosteroids and selected lipophilic alcohols. J. Med. Chem. 28, 171-177; Hostetler, K.Y., Stuhmiller, L.M., Lenting, H.B.M., van den Bosch, H. and Richmann, D.D. (1990) Synthesis and antiretroviral activity of phospholipid analogs of azidothymidine and other antiviral nucleosides. J. Biol. Chem. 265, 6112-6117; Hostetler, K.Y., Carson, D.A. and Richman, D.D. (1991); Phosphatidylazidothymidine: mechanism of antiretroviral action in CEM cells. J. Biol. Chem. 266, 11714-11717; Hostetler, K.Y., Korba, B., Sridhar, C., Gardener, M. (1994a) Antiviral activity of phosphatidyl-dideoxycytidine in hepatitis B-infected cells and enhanced hepatic uptake in mice. Antiviral Res. 24, 59-67; Hostetler, K.Y., Richman, D.D., Sridhar, C.N., Felgner, P.L., Felgner, J., Ricci, J., Gardener, M.F., Selleseth, D.W. and Ellis, M.N. (1994b) Phosphatidylazidothymidine and phosphatidyl-ddC: Assessment of uptake in mouse lymphoid tissues and antiviral activities in human immunodeficiency virus-infected cells and in rauscher leukemia virus-infected mice. Antimicrobial Agents Chemother. 38, 2792-2797; Hunston, R.N., Jones, A.A. McGuigan, C., Walker, R.T., Balzarini,J. and De Clercq, E. (1984) Synthesis and biological properties of some cyclic phosphotriesters derived from 2'deoxy-5-fluorouridine. J.Med. Chem. 27, 440-444; Ji, Y.H., Moog, C., Schmitt, G., Bischoff, P. and Luu, B. (1990); Monophosphoric acid diesters of 7β-hydroxycholesterol and of pyrimidine nucleosides as potential antitumor agents: synthesis and preliminary evaluation of antitumor activity. J. Med. Chem. 33, 2264-2270; Jones, A.S., McGuigan, C., Walker, R.T., Balzarini, J. and DeClercq, E. (1984) Synthesis, properties, and biological activity of some nucleoside cyclic phosphoramidates. J. Chem. Soc. Perkin Trans. I, 1471-1474; Juodka, B.A. and Smart, J. (1974) Synthesis of ditribonucleoside a(P→N) amino acid derivatives. Coll. Czech. Chem. Comm. 39, 363-968; Kataoka, S., Imai, J., Yamaji, N., Kato, M., Saito, M., Kawada, T. And Imai, S. (1989) Alkylated cAMP derivatives; selective synthesis and biological activities. Nucleic Acids Res. Sym. Ser., 21, 1-2; Kataoka, S., Uchida, R. and Yamaji, N. (1991) A convenient synthesis of adenosine 3',5'cyclic phosphate (cAMP) benzyl and methyl triesters. Heterocycles 32, 1351-1356; Kinchington, D., Harvey, J.J., O'Connor, T.J., Jones, B.C.N.M., Devine, K.G., Taylor-Robinson, D., Jeffries, D.J. and McGuigan, C. (1992) Comparison of antiviral effects of zidovudine phosphoramidate and phosphorodiamidate derivatives against HIV and MuLV in vitro. Antiviral Chem. Chemother. 3, 107-112; Kodama, K., Morozumi, M., Saitoh, K.I., Kuninaka, H., Yoshino, H. and Saneyoshi, M. (1989) Antitumor activity activity and pharmacology of 1-β-D-arabinofuranosylcytosine-5'-stearylphosphate; an orally active derivative of 1-β-D-arabinofuranosylcytosine. Jpn. J. Cancer Res. 80, 679-685; Korty, M. and Engels, J. (1979) The effects of adenosine- and guanosine 3',5'-phosphoric and acid benzyl esters on guinea-pig ventricular myocardium. Naunyn-Schmiedeberg's Arch. Pharmacol. 310, 103-111; Kumar, A., Goe, P.L., Jones, A.S. Walker, R.T. Balzarini, J. and De Clercq, E. (1990) Synthesis and biological evaluation of some cyclic phosphoramidate nucleoside derivatives. J. Med. Chem. 33, 2368-2375; LeBec, C., and Huynh-Dinh, T. (1991) Synthesis of lipophilic phosphate triester derivatives of 5-fluouridine and arabinocytidine as anticancer prodrugs. Tetrahedron Lett. 32, 6553-6556; Lichtenstein, J., Barner, H.D. and Cohen, S.S. (1960) The metabolism of exogenously supplied nucleotides by Escherichia coli., J. Biol. Chem. 235, 457-465; Lucthy, J., Von Daeniken, A., Friederich, J. Manthey, B., Zweifel, J., Schlatter, C. And Benn, M.H. (1981) Synthesis and toxicological properties of three naturally occurring cyanoepithioalkanes. Mitt. Geg. Lebensmittelunters. Hyg. 72, 131-133 (Chem. Abstr. 95, 127093); McGuigan, C. Tollerfield, S.M. and Riley, P.A. (1989) Synthesis and biological evaluation of some phosphate triester derivatives of the anti-viral drug Ara. Nucleic Acids Res. 17. 6065-6075; McGuigan, C., Devine, K.G., O'Connor, T.J., Galpin, S.A., Jeffries, D.J. and Kinchington, D. (1990a) Synthesis and evaluation of some novel phosphoramidate derivatives of 3'-azido-3'-deoxythymidine (AZT) as anti-HIV compounds. Antiviral Chem. Chemother. 1, 107-113; McGuigan, C., O'Connor, T.J., Nicholls, S.R., Nickson, C. and Kinchington, D. (1990b) Synthesis and anti-HIV activity of some novel substituted dialkyl phosphate derivatives of AZT and ddCyd. Antiviral. Chem. Chemother. 1, 355-360; McGuigan, C., Nicholls, S.R., O'Connor, T.J., and Kinchington, D. (1990c) Synthesis of some novel dialkyl phosphate derivative of 3'-modified nucleosides as potential anti-AIDS drug. Antiviral Chem. Chemother. 1, 25-33; McGuigan, C., Devine, K.G., O'Connor, T.J., and Kinchington, D. (1991) Synthesis and anti-HIV activity of some haloalkyl phosphoramidate derivatives of 3'-azido-3'-deoxythymidine (AZT); potent activity of the trichloroethyl methoxyalaninyl compound. Antiviral Res. 15, 255-263; McGuigan, C., Pathirana, R.N., Mahmood, N., Devine, K.G. and Hay, A.J. (1992) Aryl phoaphate derivatives of AZT retain activity against HIV-1 in cell lines which are resistant to the action of AZT. Antiviral Res. 17, 311-321; McGuigan, C., Pathirana, R.N., Choi, S.M., Kinchington, D. and O'Connor, T.J. (1993a) Phosphoramidate derivatives of AZT as inhibitors of HIV; studies on the carboxyl terminus, Antiviral Chem. Chemother. 4, 97-101; McGuigan, C., Pathirana, R.N., Balzarini, J. and De Clercq, E. (1993b) Intracellulal delivery of bioactive AZT nucleotides by aryl phosphate derivatives of AZT. J. Med. Chem.36, 1048-1052.
The question of chair-twist equilibria for the phosphate rings of nucleoside cyclic 3',5'-monophosphates. 1НNMR and x-ray crystallographic study of the diasteromers of thymidine phenyl cyclic 3',5'-monophosphate. J. Am. Chem. Soc. 109, 4058-4064; Nerbonne, J.M., Richard, S., Nargeot, J. and Lester, H.A. (1984) New photoactivatable cyclic nucleotides produce intracellular jumps in cyclic AMP and GMP concentrations. Nature 301, 74-76; Neumann, J.M., Herve, M., Debouzy, J.C., Guerra, F.I., Gouyette, C., Dupraz, B. and Huynh-Dinh, T. (1989) Synthesis and transmembrane transport studies by NMR of a glucosyl phospholipid of thymidine. J. Am. Chem. Soc. 111, 4270-4277; Ohno, R., Tatsumi, N., Hirano, M., Imai, K., Mizoguchi, H., Nakamura, T., Kosaka, M., Takatuski, K., Yamaya,T., Toyama, K., Yoshida, T., Masaoka, T., Hashimoto, S., Ohshima, T., Kimura, I., Yamada, K. and Kimura, J. (1991) Treatment of myelodysplastic syndromes with orally administered 1-β-D-rabinofuranosylcytosine-5'-srearylphosphate. Oncology 48, 451-455.
Palomino, E., Kessle, D. and Horwitz, J.P. (1989) A dihydropyridine carrier system for sustained delivery of 2',3'-dideoxynuclesides to the brain. J. Med. Chem. 32, 622-625; Perkins, R.M., Barney, S., Wittrock, R., Clark, P.H., Levin, R. Lambert, D.M., Petteway, S.R., Serafinowska, H.T., Bailey, S.M., Jackson, S., Harnden, M.R. Ashton, R., Sutton, D., Harvey, J.J. and Brown, A.G. (1993) Activity of BRL47923 and its oral prodrug, SB203657A against a rauscher murine leukemia virus infection in mice. Antiviral Res. 20 (Suppl. I), 84; Piantadosi, C., Marasco, C.J.,Jr. Morris-Natschke, S.L., Meyer, K.L., Gumus, F., Surles, J.R., Ishaq, K.S., Kucera, L.S., Iyer, N., Wallen, C.A., Piantadosi, S. and Modest, E.J. (1991) Synthesis and evaluation of novel ether lipid nucleoside conjugates for anti-HIV-1 activity. J. Med. Chem. 34, 1408-1414; Pompon, A., Lefebvre, I., Imbach, J.L., Kahn, S. and Farquhar, D. (1994) Decomposition pathways of the mono- and bis(pivaloyloxymethyl) esters of azidothymidine-5'-monophosphate in cell extract and in tissue culture medium; an application of the on-line ISRP-cleaning' HPLC technique. Antiviral Chem. Chemother. 5, 91-98; Postemark, T. (1974) Cyclic AMP and GMP. Annu. Rev. Pharmacol. 14, 23-33; Prisbe, E.J., Martin, J.C.M., McGee, D.P.C., Barker, M.F., Smee, D.F., Duke, A.E., Matthews, T.R. and Verheyden, J.P.J. (1986) Synthesis and antiherpes virus activity of phosphate and phosphonate derivatives of 9-[(1,3-dihydroxy-2-propoxy)methyl]guanine. J. Med. Chem. 29, 671-675; Puech, F., Gosselin, G., Lefebvre, I., Pompon, A., Aubertin, A.M., Dirn, A. And Imbach, J.L. (1993) Intracellular delivery of nucleoside monophosphate through a reductase-mediated activation process. Antivral Res. 22, 155-174; Pugaeva, V.P., Klochkeva, S.I., Mashbits, F.D. and Eizengart, R.S. (1969). Robins, R.K. (1984) The potential of nucleotide analogs as inhibitors of retroviruses and tumors. Pharm. Res. 11-18; Posowsky, A., Kim, S.H., Ross and J. Wick, M.M. (1982) Lipophilic 5'-(alkylphosphate) esters of 1-β-D-arabinofuranosylcytosine and its N4-acyl and 2.2'-anhydro-3'-O-acyl derivatives as potential prodrugs. J.Med. Chem. 25, 171-178; Ross, W. (1961) Increased sensitivity of the walker turnout towards aromatic nitrogen mustards carrying basic side chains following glucose pretreatment. Biochem. Pharm. 8, 235-240; Ryu, E.K., Ross, R.J., Matsushita, T., MacCoss, M., Hong, C.I. and West, C.R. (1982). Phospholipid-nucleoside conjugates. 3. Synthesis and preliminary biological evaluation of 1-β-D-arabinofuranosylcytosine 5'diphosphate{-},2-diacylglycerols. J. Med. Chem. 25, 1322-1329; Saffhill, R. And Hume, W.J. (1986) The degradation of 5-iododeoxyuridine and 5-bromodeoxyuridine by serum from different sources and its consequences for the use of these compounds for incorporation into DNA. Chem. Biol. Interact. 57, 347-355; Saneyoshi, M., Morozumi, M., Kodama, K., Machida, J., Kuninaka, A. and Yoshino, H. (1980) Synthetic nucleosides and nucleotides. XVI. Synthesis and biological evaluations of a series of 1-β-D-arabinofyranosylcytosine 5'-alkyl or arylphosphates. Chem. Pharm. Bull. 28, 2915-2923; Sastry, J.K., Nehete, P.N., Khan, S., Nowak, B.J., Plunkett, W., Arlinghaus, R.B. and Farquhar, D. (1992) Membrane-permeable dideoxyuridine 5'-monophosphate analogue inhibits human immunodeficiency virus infection. Mol. Pharmacol. 41, 441-445; Shaw, J.P., Jones, R.J., Arimilli, M.N., Louie, M.S., Lee, W.A. and Cundy, K.C. (1994) Oral bioavailaability of PMEA from PMEA prodrugs in male Sprague-Dawley rats. 9th Annbal AAPS Meeting. San Diego, CA (Abstract). Shuto, S., Ueda, S., Imamura, S., Fukukawa, K. Matsuda, A. and Ueda, T. (1987) A facile one-step synthesis of 5'-phosphatidyl-nucleosides by an enzymatic two-phase reaction. Tetrahedron Lett. 28, 199-202; Shuto, S., Itoh, H., Ueda, S., Imamura, S., Kukukawa, K., Tsujino, M., Matsuda, A. And Ueda, T. (1988) A facile enzymatic synthesis of 5'-(3-sn-phosphatidyl)nucleosides and antileukemic activities. Chem. Pharm. Bull. 36, 209-217. Одна предпочтительная группа фосфатного пролекарства представляет собой группу S-ацил-2-тиоэтила, также упоминаемую как “SATE”.
Сочетанная или альтернирующая терапия
Установлено, что резистентные к лекарству формы HBV могут появляться после пролонгированного лечения противовирусным агентом. Наиболее типично устойчивость к лекарственному средству возникает в результате мутации гена, который кодирует фермент, используемый в цикле жизни вируса, и чаще всего в случае ДНК полимеразы HBV. Недавно было продемонстрировано, что эффективность лекарственного средства против инфекции HBV может быть пролонгирована, увеличена или восстановлена введением соединения в сочетании или чередовании со вторым и, возможно, третьим противовирусным соединением, которое вызывает мутацию, иную, чем вызванная исходным лекарственным средством. Альтернативно, фармакокинетика, биологическое распределение или другие параметры лекарственного вещества могут быть изменены с помощью такой сочетанной или чередующейся терапии. Вообще, обычно сочетанная терапия является предпочтительной по сравнению с чередующейся терапией, так как она вызывает множественные одновременные стрессы у вируса.
Активность против вируса гепатита соединений В β-L-2'-dA, β-L-2'-dC, β-L-2'dU, β-L-2'dG, β-L-2'-dT, β-L-dI или других β-L-2'-нуклеозидов, представленных в описании, или пролекарств, фосфатов или солей этих соединений может быть увеличена введением двух или более этих нуклеозидов в cочетании или в чередовании. Альтернативно например, один или более из β-L-2'-dA, β-L-2'-dC, β-L-2'-dU, β-L-2'-dG, β-L-2'-dT, β-L-dI или других β-L-2'-нуклеозидов, представленных в описании, можно вводить в сочетании или в чередовании с 3ТС, FTC, L-FMAU, DAPD, фамцикловиром, пенцикловиром, BMS-200475, бис пом РМЕА (адефовир, дипивоксил); лобукавиром, ганцикловиром или рибаварином.
В любом из аспектов, описанных в заявке, если β-L-2'-нуклеозид данного изобретения вводится в сочетании или в чередовании со вторым нуклеозидным или ненуклеозидным ингибитором обратной транскриптазы, который фосфорилируется до активной формы, то предпочтительно, чтобы второе соединение фосфорилировалось бы ферментом, который отличается от фермента, фосфорилирующего in vivo выбранный β-L-2'-нуклеозид данного изобретения. Примерами киназных ферментов являются тимидинкиназа, цитозинкиназа, гуанозинкиназа, аденозинкиназа, дезоксицитидинкиназа, 5'-нуклеотидаза и дезоксигуанозин-киназа.
Получение активных соединений
Производные 2'-дезокси-β-L-эритро-пентофуранонуклеозида согласно изобретению известны в данной области и могут быть получены в соответствии со способом, описанным Holy, Collect. Czech. Chem. Commun. (1972), 37(12), 4072-87 и Mol. Phys. (1967), 3(4), 386-95.
Обычный способ получения β-L-эритро-пентофуранонуклеозидов (β-L-dN) с использованием L-рибозы или L-ксилозы в качестве исходного вещества представлен на Фигуре 1.
Моно-, ди- или трифосфатные производные активных нуклеозидов можно получать, как описано, в соответствии с опубликованными способами. Монофосфат может быть получен согласно способу Imai et al., J. Org. Chem., 34(6), 1547-1550 (июнь, 1969). Дифосфат может быть получен согласно способу Davisson et al., J.Org. Chem. 52(9), 1794-1801 (1987). Трифосфат может быть получен в соответствии со способом Hoard et al., J. Am. Chem.Soc., 87(8), 1785-1788 (1965).
Протоколы экспериментов
Точки плавления были определены в открытых капиллярных пробирках на аппарате Gallenkamp MFB-595-010 M и являются нескорректированными. Спектр поглощения в УФ регистировали на спектрофотометре Uvikon 931 (KONTRON) в этаноле. Спектры 1Н ЯМР сняты при комнатной температуре в DMSO-d6 на спектрометре Bruker AC 250 или 400. Химический сдвиг представлен в миллионных долях (м.д.), DMSO-d5 устанавливали при 2,49 м.д., как рекомендовано. Дейтерообмен, эксперименты по нарушению взаимодействия или 2D-COSY проводили для того, чтобы установить расположение протонов. Разнообразие сигналов представлено посредством s (синглет), d (дублет), dd (двойной дублет), t (триплет), q (квадруплет), br (широкий), m (мультиплет). Все J-величины представлены в Hz (Гц). Масс-спектры FAB регистрировали в положительном (FAB>0) или отрицательном (FAB<0) ионном режиме на масс спектрометре JEOL DX 300. Матриксом был 3-нитробензиловый спирт (NBA) или смесь (50:50, об/об) глицерина и тиоглицерина (GT). Удельное вращение измеряли на спектрополяриметре Perkin-Elmer 241 (длина хода 1 см) и выражали в единицах 10-1 градус.см2.г-1. Элементный анализ проводили с помощью микроанализатора “Microanalyses du CNRS, Division de Vernaison” (France). Анализы, представленные символами элементов или выходом, оказывались в пределах +/- 0,4% теоретических величин. Тонкослойную хроматографию проводили на предварительно покрытых алюминием пластинах силикагеля 60 F254 (Merck, Art 5554), визуализация продуктов осуществлялась по поглощению в УФ с последующим обугливанием с 10% спиртовой серной кислотой и нагреванием. Колоночную хроматографию проводили на силикагеле 60 (Merck, Art. 9385) под атмосферным давлением.
Пример 1
Стереоспецифический синтез 2'-дезокси-β-L-аденозина
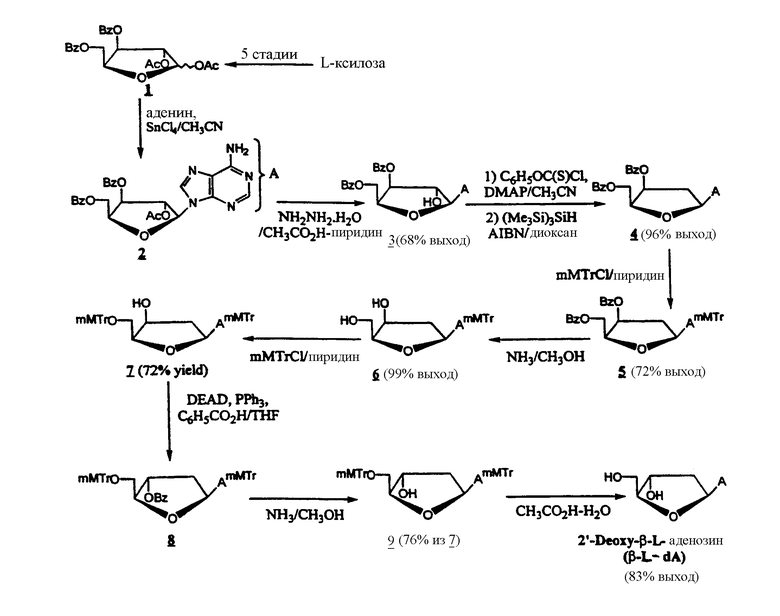
9-(3,5-Ди-О-бензоил-β-L-ксилофуранозил)аденин (3)
Раствор 9-(2-О-ацетил-3,5-ди-О-бензоил-β-L-ксилофуранозил) аденина 2 [ссылка: Gosselin, G., Bergogne, M.-C.; Imbach, J.-L., “Synthesis and Antiviral Evaluation of β-L-Xylofyranosyl Nucleosides of the Five Naturally Occuring Nucleic Acid Bases”, Journal of Heterocyclic Chemistry, 1993, 30 (октябрь-ноябрь), 1229-1233] (8,30 г, 16,05 ммоль) и 98% гидразин гидрат (234 мл, 48,5 ммоль) в смеси пиридина/ледяной уксусной кислоты (4/1, об/об, 170 мл) перемешивали при комнатной температуре в течение 22 часов. Реакцию гасили добавлением ацетона (40 мл) и перемешивали непрерывно в течение одного дополнительного часа. Реакционную смесь уменьшали на половину ее объема, разбавляли водой (250 мл) и экстрагировали хлороформом (2 х 150 мл). Органический слой промывали последовательно водным насыщенным раствором NaHCO3 (3 х 100 мл) и водой (3 х 100 мл), сушили, фильтровали, концентрировали и упаривали вместе с толуолом и метанолом. Остаток очищали хроматографией на колонке силикагеля (0-3% МеОН в дихлорметане), чтобы получить 3 (5,2 г, 68%), осажденного из диизопропилового эфира: 1Н ЯМР (DMSO-d6): δ 4,5-4,9 (m, 4H, H-2', H-4', H-5' и H-5''), 5,64 (t, 1H, H-3', J2',3' = J3',4' = 3,5 Hz), 6,3 (br s, 1H, OH-2'), 6,45 (d, 1H, H-1', J1',2' = 4,6 Hz), 7,3 (br s, 2H, NH2-6), 7,4-7,9 (m, 10H, 2 бензоилов), 8,07 и 8,34 (2s, 2H, H-2 и H-8); ms: матрикс G/T, (FAB+) m/z 476 [M+H]+, 136 [BH2]+, (FAB-) m/z 474 [M-H]-, 134 [B]-; УФ (95% этанол): λмаксим257 нм (ε 16400), 230 нм (ε 29300), λминим 246 нм (ε 14800); [α]D 20 = -64 (c 1,07, CHCl3 ). Аналитический расчет для С24Н21N5O4 (M=475,45): C,60,43; H, 4,45; N, 14,73. Обнаружено: С, 60,41; H, 4,68; N, 14,27.
9-(3,5-Ди-О-бензоил-2-дезокси-β-L-трео-пентофуранозил)-аденин (4)
К раствору соединения 3 (1,00 г, 2,11 ммоль) в сухом ацетонитриле (65 мл) добавляли 4-(диметиламино)пиридин (0,77 г, 6,32 ммоль) и фенокситиокарбонилхлорид (0,44 мл, 3,16 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. После концентрирования остаток растворяли в дихлорметане (50 мл) и промывали последовательно водой (2 х 30 мл), 0,5 N водным раствором хлористоводородной кислоты (30 мл) и водой (3 х 30 мл). Органический слой высушивали, фильтровали и концентрировали до сухого состояния. Неочищенное тиокарбонилированное промежуточное соединение непосредственно обрабатывали гидридом трис-(триметилсилил)силана (0,78 мл, 5,23 ммоль) и α,α'-азоизобутиронитрилом (AIBN, 0,112 г, 0,69 ммоль) в сухом диоксане (17 мл) при кипячении с обратным холодильником в течение 2 часов. Растворитель удаляли под вакуумом и остаток очищали хроматографией на колонке силикагеля (0-5% МеОН в дихлорметане), чтобы получить чистый 4 (0,93 г, 96%) в виде пенообразной массы: 1Н ЯМР (DMSO-d6): δ 2,9-3,1 (m, 2H, H-2' и Н-2''), 4,6-4,7 (m, 3H, H-4', H-5' и Н-5''), 5,8 (br s, 1H, H-3'), 6,43 (dd, 1H, H-1', J1',2' = 3,1 Hz, J1',2'' = 7,6 Hz), 7,3 (br s, 2H, NH2-6), 7,4-7,9 (m, 10H, 2 бензоила), 8,05 и 8,33 (2s, 2H, H-2 и H-8); ms: матрикс G/T, (FAB+) m/z 460 [M+H]+, 325 [S]+, 136 [BH2]+, (FAB-) m/z 458 [M-H]-, 134 [B]-; УФ (95% этанола): λмакс 261 нм (ε 14400), 231 нм (ε 26300), λминим 249 нм (ε 12000); [α]D 20 =-38 (c 1,04, DMSO).
6-N-(4-Монометокситритил)-9-(3,5-ди-О-бензоил-2-дезокси-β-L-трео-пентофуранозил)аденин (5)
К раствору соединения 4 (0,88 г, 1,92 ммоль) в сухом пиридине (40 мл) добавляли 4-монометокситритилхлорид (1,18 г, 3,84 ммоль). Смесь перемешивали при 60°С в течение 24 часов. После добавления метанола (5 мл) раствор концентрировали до сухого состояния, остаток растворяли в дихлорметане (50 мл) и промывали последовательно водой (30 мл), водным насыщенным NaHCO3 (30 мл) и водой (30 мл). Органический слой высушивали, фильтровали, концентрировали и упаривали с толуолом, чтобы получить 5 (1,01 г, 72%) в виде пенообразной массы: 1Н ЯМР (CDCl3): δ 2,9-3,0 (m, 2H, H-2' и Н-2''), 3,62 (s, 3H, OCH3), 4,6-4,8 (m, 3H, H-4', H-5' и Н-5''), 5,85 (pt, 1H, H-3'), 6,44 (dd, 1H, H-1', J1',2' =3,1 Hz, J 1',2''= 7,3 Hz), 6,9 (br s, 1H, NH-6), 6,7-6,8 и 7,2-7,4 (2m, 24H, 2 бензоила и ММТr), 7,97 и 8,13 (2s, 2H, H-2 и Н-8); ms: матрикс G/T, (FAB+) m/z 732 [M+H]+, (FAB-) m/z 730 [M-H]-; УФ (95% этанола): λмакс 274 нм (ε 12100), 225 нм (ε 24200), λминим 250 нм (ε 5900); [α]D 20 =-16 (c1,12, DMSO).
6-N-(4-Монометокситритил)-9-(2-дезокси-β-L-трео-пентофуранозил)-аденин (6)
Соединение 5 (0,95 г, 1,30 ммоль) обрабатывали метанольным раствором (насыщенным при -10°С) аммиака (40 мл) при комнатной температуре в течение ночи. После концентрирования остаток растворяли в дихлорметане (60 мл) и промывали водой (30 мл). Водный слой экстрагировали дважды дихлорметаном (10 мл). Объединенный органический слой высушивали, фильтровали и концентрировали. Остаток очищали хроматографией на колонке силикагеля (0-5% МеОН в дихлорметане), чтобы получить чистый 6 (0,67 г, 98%) в виде пенобразной массы: 1Н ЯМР (CDCl3): δ 2,6-2,9 (m, 2H, H-2' и H-2''), 3,5 (br s, 1H, OH-5'), 3,55 (s, 3H, OCH3), 3,9-4,0 (m, 3H, H-4', H-5' и H-5''), 4,5-4,6 (m, 1H, H-3'), 6,03 (dd, 1H, H-1', J1',2' = 4,0 Hz, J1',2'' = 8,8 Hz), 7,0 (br s, 1H, NH-6), 6,7-6,8 и 7,1-7,4 (2m, 14H, MMTr), 7,40 (d, 1H, OH-3', JH,OH = 10,6 Hz), 7,80 и 7,99 (2s, 2H, H-2 и Н-8); ms: матрикс G/T, (FAB+) m/z 524 [M+H]+, 408 [BH2]+, (FAB-) m/z 1045 [2M-H]-, 522 [M-H]-, 406 [B]-, УФ (95% этанол): λмакс 275 нм (ε 12300), λминим 247 нм (ε 3600); [α]D 20 =+ 28 (c 0,94, DMSO).
6-N-(4-монометокситритил)-9-(2-дезокси-5-О-(4-монометокситритил)-β-L-трео-пентофуранозил)аденин (7)
Соединение 6 (0,62 г, 1,24 ммоль) в сухом пиридине (25 мл) обрабатывали 4-монометокситритилхлоридом (0,46 г, 1,49 ммоль) при комнатной температуре в течение 16 часов. После добавления метанола (5 мл) смесь концентрировали до сухого состояния. Остаток растворяли в дихлорметане (60 мл) и промывали последовательно водой (40 мл), насыщенным водным раствором NaHCO3 (40 мл) и водой (3 х 40 мл). Органический слой высушивали, фильтровали, концентрировали и упаривали вместе с толуолом и метанолом. Остаток очищали хроматографией на колонке силикагеля (0-10% МеОН в дихлорметане), чтобы получить 7 (0,71 г, 72%) в виде пенообразной массы: 1Н ЯМР (DMSO-d6): δ 2,21 (d, 1H, H-2' J2',2''= 14,3 Hz), 2,6-2,7 (m, 1H, H-2'',), 3,1-3,3 (2m, 2H, H-5' и Н-5''), 3,64 и 3,65 (2s, 6H, 2 × OCH3), 4,1-4,2 (m, 1H, H-4'), 4,2-4,3 (m, 1H, H-3'), 5,68 (d, 1H, OH-3', JH,OH = 5,2 Hz), 6,24 (d, 1H, H-1', J1',2''=7,0 Hz), 6,7-6,8 и 7,1-7,3 (2m, 29H, 2 MMTr и NH-6), 7,83 и 8,21 (2s, 2H, H-2 и Н-8); ms: матрикс G/T, (FAB+) m/z 796 [M+H]+, 408 [BH2]+, (FAB-) m/z 794 [M-H]-, 406 [B]-; УФ (95% этанол): λмакс 275 нм (ε 30900), λминим 246 нм (ε 12800); [α]D 20=+14 (c 1,03, DMSO).
6-N-(4-Монометокситритил)-9-(3-О-бензоил-2-дезокси-5-О-(4-монометокситритил)-β-L-эритро-пентофуранозил)аденин (8)
Раствор диэтилазодикарбоксилата (0,38 мл, 2,49 ммоль) в сухом тетрагидрофуране (20 мл) добавляли по каплям к охлажденному раствору (0°С) нуклеозида 7 (0,66 г, 0.83 ммоль), трифенилфосфина (0,66 г, 2,49 ммоль) и бензойной кислоты (0,30 г, 2,49 ммоль) в сухом THF (20 мл). Смесь перемешивали при комнатной температуре в течение 18 часов и добавляли метанол (1 мл). Растворители удаляли при пониженном давлении и неочищенное вещество очищали хроматографией на колонке силикагеля (0-5% этилацетат в дихлорметане), чтобы получить соединение 8, немного загрязненное окисью трифенилфосфина.
6-N-(4-Монометокситритил)-9-(2-дезокси-5-О-(4-монометокситритил)-β-L-эритро-пентофуранозил)аденин (9)
Соединение 8 обрабатывали метанольным раствором (насыщенным при -10°С) аммиака (20 мл) при комнатной температуре в течение 24 часов, затем реакционную смесь концентрировали до сухого состояния. Остаток растворяли в дихлорметане (30 мл) и промывали водой (20 мл). Водный слой экстрагировали дихлорметаном (2 х 20 мл), а объединенную органическую фазу высушивали, фильтровали и концентрировали. После очистки хроматографией на колонке силикагеля (0-2% МеОН в дихлорметане) было получено чистое соединение 9 (0,50 г, 76% от 7) в виде пенки: 1Н ЯМР (DMSO-d6): δ 2,2-2,3 (m, 1H, H-2'), 2,8-2,9 (m, 1H, H-2''), 3,1-3,2 (m, 2H, H-5'и Н-5''), 3,64 и 3,65 (2s, 6H, 2 × OCH3), 3,97 (pq, 1H, H-4'), 4,4-4,5 (m, 1H, H-3'), 5,36 (d, 1H, OH-3', JH,OH=4,5 Hz), 6,34 (t, 1H, H-1', J1',2'=6,4 Hz), 6,8-6,9 и 7,1-7,4 (2m, 29H, 2 MMTr и NH-6), 7,81 и 8,32 (2s, 2H, H-2 и Н-8); ms: матрикс G/T, (FAB+) m/z 796 [M+H]+, 408 [BH2]+, (FAB-) m/z 794 [M-H]-, 406 [B]-; УФ (95% этанола): λмакс 276 нм (ε 42600)б λминим 248 нм (ε 23300); [α]D 20=+29 (c 1,05, DMSO).
2'-дезокси-β-L-аденозин(β-L-dA)
Соединение 9 (0,44 г, 0,56 ммоль) обрабатывали 80% водным раствором уксусной кислоты (17 мл) при комнатной температуре в течение 5 часов. Смесь концентрировали до сухого состояния, остаток растворяли в воде (20 мл) и промывали диэтиловым эфиром (2 × 15 мл). Водный слой концент-рировали и упаривали с толуолом и метанолом. Требуемый 2'-дезокси-β-L- аденозин (β-L-dA) (0,12 г, 83%) был получен после очистки хроматографией на колонке силикагеля (0-12% МеОН в дихлорметане) и фильтрации через ячейку Millex HV-4 (0,45 мк, Millipore): точка плавления 193-194°С (кристаллизован из воды) (Lit. 184-185°C для L-энантиомера [См. Robins, M.J., Khwaja, T.A., Robins, R.K. J. Org. Chem. 1970, 35, 636-639] и 187-189°С для D-энантиомера [См. Ness, R.K. in Synthetic Procedures in Nucleic Acid Chemistry; Zorbach, W.W., Tipson, R.S., Eds.; J. Wiley and sons: New York, 1968; Vol 1, pp 183-187]; 1H ЯМР (DMSO-d6): δ 2,2-2,3 и 2,6-2,7 (2m, 2H, H-2' и Н-2''), 3,4-3,6 (2m, 2H, H-5' и Н-5''), 3,86 (pq, 1H, H-4'), 4,3-4,4 (m, 1H, H-3'), 5,24 (t, 1H, OH-5', JH,OH=5,8 Hz), 5,30 (d, 1H, OH-3', JH,OH=4,0 Hz), 6,32 (dd, 1H, H-1', J1,2'=6,2 Hz, J1,2''=7,8 Hz), 7,3 (br s, 2H, NH2-6), 8,11 и 8,32 (2s, 2H, H-2 и Н-8); ms: матрикс G/T, (FAB+) m/z 252 [N+H]+, 136 [BH2]+, (FAB-) m/z 250 [M-H]-, 134 [B]-; УФ (95% этанол): λмакс 258 нм (ε 14300), λминим 226 нм (ε 2100); [α]D 20=+25 (c 1,03 H2O), (Lit. [α]D 20=+23 (c 1,0, H2O) для L-энантиомера [См. Robins, M.J., Khwaja, T.A., Robins, R.K. J. Org. Chem. 1970, 35, 636-639] и [α]D 20=-25 (c 0,47, H2O) для D-энантиомера [См. Ness, R.K. in Synthetic Procedures in Nucleic Acid Chemistry; Zorbach, W.W., Tipson, R.S., Eds.; J. Wiley and sons: New York, 1968; Vol 1, pp 183-187]). Аналитический расчет для С10Н13N5O3 + 1,5 H2O (M= 278,28): C, 43,16; H, 5,80; N, 25,17. Обнаружено: C, 43,63; H, 5,45; N, 25,33.
Пример 2
Стереоизбирательный синтез 2'-дезокси-β-L-аденозин (β-L-dA)
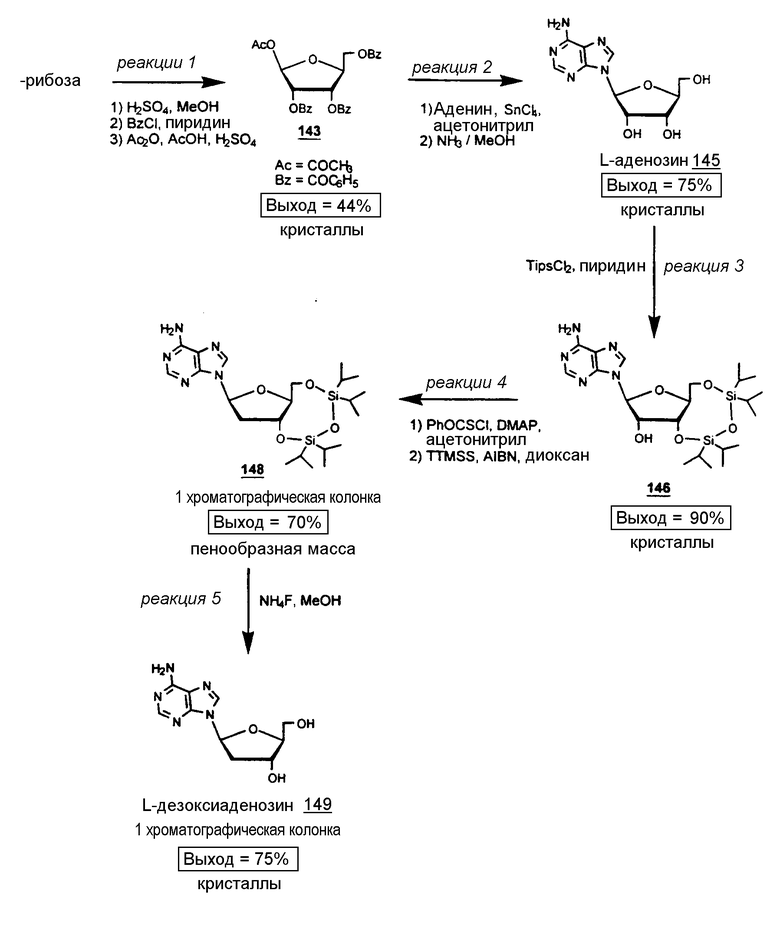
Реакция 1:
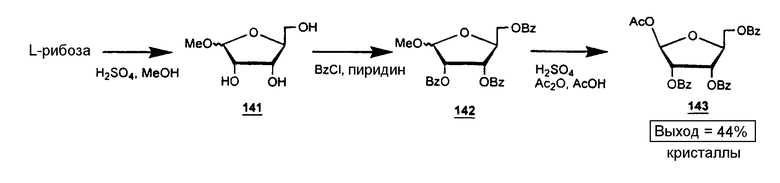
Исходное вещество: L-рибоза (Cultor Science Food, CAS [24259-59-4}, партия RIB9711013)
Реактивы: 95-97% серная кислота (Merck; ссылочный номер 1007311000); хлористый бензоил (Fluka; ссылочный номер 12930); сульфат натрия (Prolabo; ссылочный номер 28111365)
Растворители: Метанол, чистый для анализа (Prolabo; ссылочный номер 20847295); 99% пиридин (Acros; ссылочный номер 131780025); дихлорметан, чистый для анализа (Merck; ссылочный номер 1060506025); уксусная кислота, чистая для анализа (Carbo Erba; ссылочный номер 20104298); уксусный ангидрид (Fluka; ссылочный номер 45830); этанол 95 (Prolabo; ссылочный номер 20823293)
Ссылки: Recondo, E.F., and Rinderknecht, Eine neue, Einfache Synthese des 1-O-Acetyl-2,3,5-Tri-O-β-D-Ribofuranosides. Helv. Chim. Acta, 1171-1173 (1959).
Раствор L-рибозы 140 (150 г, 1 моль) в метаноле (2 литра) обрабатывали серной кислотой (12 мл) и оставляли при +4°С в течение 12 часов, а затем нейтрализовали пиридином (180 мл). Выпаривание дало α,β смесь метилрибофуранозидов 141 в виде сиропа. Раствор этой аномерной смеси в пиридине (1,3 литра) обрабатывали хлористым бензоилом (580 мл, 5 моль) с охлаждением и при механическом перемешивании. Раствор оставляли при комнатной температуре в течение 12 часов, а затем наливали на лед (приблизительно 10 литров) при непрерывном перемешивании. Смесь (масло в воде) фильтровали через слой целлита. Полученное масло на слое целлита промывали водой (3 × 3 литра), а затем растворяли этилацетатом (3 литра). Органическую фазу промывали 5% раствором NaHCO3 (2 литра) и водой (2 литра), высушивали над сульфатом натрия, фильтровали и выпаривали, чтобы получить 1-О-метил-2,3,5-три-О-бензоил-α/β-L-рибофуранозу 142 в виде густого сиропа. Масло растворяли в уксусном ангидриде (560 мл) и уксусной кислоте (240 мл). После добавления по каплям концентрированной кислоты (80 мл) раствор сохраняли на холоде (+4°С) при механическом перемешивании в течение 10 часов. Затем раствор наливали на лед (приблизительно 10 литров) при непрерывном перемешивании. Смесь (маслянистое соединение в воде) фильтровали через слой целлита. Полученное камедеобразное твердое вещество на слое целлита промывали водой (3 × 3 литра) и затем растворяли в дихлорметане (2,5 литра). Органическую фазу промывали 5% NaHCO3 (1 литр) и водой (2 × 2 литра), высушивали над сульфатом натрия, фильтровали и упаривали, чтобы получить камедеобразное твердое вещество 143, которое кристаллизовалось из этанола 95 (выход 225 г, 44%).
Анализ 1-О-ацетил-2,3,5-три-О-бензоил-β-L-рибофуранозы 143:
Точка плавления 129-130°С (EtOН 95) (lit.(1)точка плавления 130-131°С)
1Н ЯМР (200 MHz, CDCl3): δ 8,09-7,87 (m, 6H, Hаром), 7,62-7,31 (m, 9H, Hаром) 6,43 (s, 1H, H1), 5,91 (dd, 1H, H3, J3,4 = 6,7 Hz; J3,2 4,9 Hz), 5,79 (pd, 1H, H2, J2,3 = 4,9 Hz; J1,2<1), 4,78 (m, 2H, H4 и Н5), 4,51 (dd, 1H, H5, J5,5' = 13,1 Hz, J5',4 = 5,5 Hz), 2,00 (s, 3H, CH3CO); (идентичен коммерческой 1-О-ацетил-2,3,5-три-О-бензоил-β-D-рибофуранозе)
Масс-анализ (FAB+, GT) m/z 445 (M-OАc)+
Элементный анализ C28Н24О9 Рассчитано С 66,66; Н 4,79; Обнаружено С Н
Реакция 2:

Исходное вещество: Аденин (Pharma-Waldhof; ссылочный номер 400134001 партия 45276800)
Реагенты: Хлористое олово дымящее (Fluka; ссылочный номер 96558); NH3/метанол (метанол, насыщенный NH3; см. стр. 5); сульфат натрия (Prolabo; ссылочный номер 28111365)
Растворители: ацетонитрил (Riedel-de Hean; ссылочный номер 33019; перегнанный над CaH2); хлороформ, чистый (Acros; ссылочный номер 22706463); этилацетат, чистый (Carlo Еrba; ссылочный номер 528299)
Ссылки: Saneyoshi, M., and Satoh, E., Synthetic Nucleosides and Nucletides. XIII. Stannic Chloride Catalyzed Ribosylation of Several 6-Substituted Purines. Chem. Pharm. Bull., 27, 2518-2521 (1979); Nakayama, C., and Saneyoshi, M., Synthetic Nucleosides and Nucleotides. XX. Synthesis of Various 1-β-Xylofuranosyl-5-Alkyluracils and Related Nucleo-sides. Nucleosides, Nucleotides, 1, 139-146 (1982).
Аденин (19,6 г, 144 ммоль) суспендировали в ацетонитриле (400 мл) с 1-О-ацетил-2,3,5-три-О-бензоил-β-L-рибофуранозой 143 (60 г, 119 ммоль). К этой суспензии добавляли дымящееся хлористое олово (22 мл, 187 ммоль). Через 12 часов реакционную смесь концентрировали до небольшого объема (приблизительно 100 мл) и добавляли кислый карбонат натрия (110 г) и воду (120 мл). Полученное белое твердое вещество (соли олова) экстрагировали горячим хлороформом (5 х 200 мл). Объединенные экстракты фильтровали через слой целлита. Органическую фазу промывали 5% раствором NaHCO3 и водой, высушивали над сульфатом натрия, фильтровали и упаривали, чтобы получить соединение 144 (60 г, бесцветной пенки). Пенку обрабатывали метанолом, насыщенным аммиаком (220 мл) в закрытом сосуде при комнатной температуре при перемешивании в течение 4 дней. Растворитель выпаривали под пониженным давлением и полученный порошок суспендировали в этилацетате (400 мл) при кипячении с обратным холодильником в течение 1 часа. После фильтрования порошок перекристаллизовывали из воды (220 мл) для получения L-аденозина 145 (24 г, кристаллы, 75%).
Анализ β-L-аденозина:
Точка плавления 233-234°С (вода) (lit. (4) точка плавления 235-238°С)
1Н ЯМР (200 MHz, DMSO-D6): δ 8,34 и 8,12 (2s, 2H, H2 и Н8), 7,37 (1s, 2H, NH2), 5,86 (d, 1H, H1', J1',2' = 6,2 Hz), 5,43 (m, 2Н, OH2' и ОН5'), 5,19 (d, 1H, OH3', J = 3,7Hz), 4,60 (m, H2'), 4,13 (m, 1H, H3'), 3,94 (m, 1H, H4'), 3,69-3,49 (m, 2H, H5'a и Н5'b), (идентичен коммерческому D-аденозину)
Масс-анализ (FAB+, GT) m/z 268 (M+H)+, 136(BH2)+
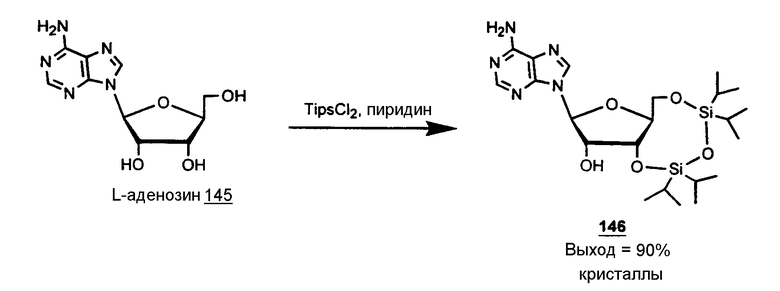
Реакция 3:
Реагенты: 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (Fluka; ссылочный номер 36520); сульфат натрия (Prolabo; ссылочный номер 28111,365)
Растворители: 99% пиридин (Acros; ссылочный номер 131780025); этилацетат, чистый (Carlo Еrba; ссылочный номер 528299); ацетонитрил (Riedel-de Haen; ссылочный номер 33019)
Ссылки: Robins, M.J., et al., Nucleic Acid Related Compounds. 42. A General Procedure for the Efficient Deoxygenation of Secondary Alcohols. Regiospecific and Stereoselective Conversion of Ribonucleosides to 2'-Deoxynucleosides. J. Am. Chem. Soc. 105, 4059-4065 (1983).
К L-аденозину 145 (47,2 г, 177 ммоль), суспендированному в пиридине (320 мл), добавляли 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (63 мл, 201 ммоль) и смесь перемешивали при комнатной температуре в течение 12 часов. Пиридин выпаривали, а остаток распределяли в этилацетате (1 литр) и 5% растворе NaHCO3 (600 мл). Органическую фазу промывали 0,5N раствором HCl (2 × 500 мл) и водой (500 мл), высушивали над сульфатом натрия, фильтровали и упаривали до сухого состояния, полученное сухое вещество кристаллизовали из ацетонитрила с получением соединения 146 (81 г, 90%).
Анализ 3',5'-O-(1,1,3,3-тетраизопропил-1,3-дисилоксанил)-β-L-аденозина 146
Точка плавления 97-98°C (ацетонитрил) (lit.(5) D энантиомер, точка плавления 98°С)1Н ЯМР (200 MHz, CDCl3): δ 8,28 и 7,95 (2s, 2H, H2 и Н8), 5,96 (d, 1H, J1',2' = 1,1 Hz), 5,63 (s, 2H, NH2), 5,10 (dd, 1H, H3', J3',4' = 7,6 Hz, J3',2' = 5,5 Hz), 4,57 (dd, 1H, H2', J2',1' = 1,2 Hz; J2',3' = 7,6 Hz), 4,15-3,99 (m, 3H, H4', H5'a и Н5'b), 3,31 (sl, 1H, OH2'), 1,06 (m, 28H, протоны изопропила)
Масс-анализ (FAB-, GT) m/z 508 (M-H)-, 134 (B)-, (FAB+, GT) m/z 510 (m+H)+, 136 (BH2)+
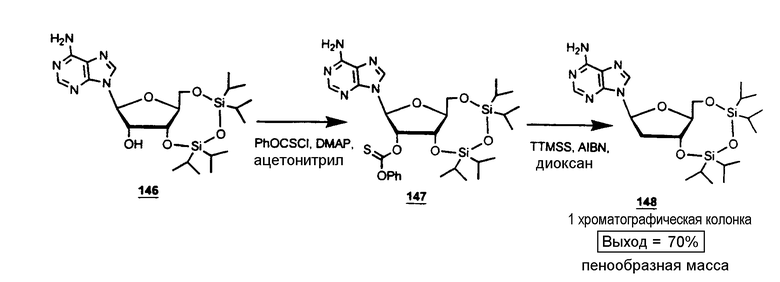
Реакция 4:
Реагенты: 99% Диметиламинопиридин (Acros; ссылочный номер 1482702050); 99% фенилхлортионокарбонат (Acros; ссылочный номер 215490050); трис(триметилсилил)силан “TTMSS” (Fluka; ссылочный номер 93411); α,α'-азоизобутиронитрил “AIBN” (Fluka, ссылочный номер 11630); сульфат натрия (Prolabo; ссылочный номер 28111365)
Растворители: ацетонитрил (Riedel-de Haen; ссылочный номер 33019); этилацетат, чистый (Carlo Erba; ссылочный номер 528299); диоксан, чистый для анализа (Merck; ссылочный номер 1096711000); дихлорметан (Merck; ссылочный номер 1060506025); метанол (Carbo Erba; ссылочный номер 309002);
Ссылки: Robins, M.J., Wilson, J.S., and Hansske, F., Nucleic Acid Related Compounds. 42. A General Procedure for the Efficient Deoxygenation of Secondary Alcohols. Regiospecific and Stereoselective Conversion of Ribonuc-leosides to 2'-Deoxynucleosides. J. Am. Chem. Soc. 105, 4059-4065 (1983).
К соединению 146 (34 г, 67 ммоль) добавили ацетонитрил (280 мл), DMAP (16,5 г, 135 ммоль) и фенилхлортионокарбонат (10,2 мл, 73 ммоль). Раствор перемешивали при комнатной температуре в течение 12 часов. Растворитель упаривали и остаток распределяли между этилацетатом (400 мл) и 0,5 N раствором HCl (400 мл). Органический слой промывали 0,5 N раствором HCl (400 мл) и водой (2 × 400 мл), высушивали над сульфатом натрия, фильтровали и упаривали до сухого состояния для получения промежуточного соединения в виде бледно-желтого твердого вещества. Неочищенный 147 растворяли в диоксане (мл) и добавляли AIBN (3,3 г, 20 ммоль) и TTMSS (33 мл, 107 ммоль). Раствор прогрессивно нагревали до кипячения с обратным холодильником и перемешивали в течение 2 часов. Реакционную смесь концентрировали до желтого масла, которое хроматографировали (элюент - дихлорметан/метанол 95/5)для получения соединения 148 (23 г, бесцветная пенообразная масса, 70%). Аликвоты кристаллизовали из этанола/петролейного эфира.
Анализ 3',5'-O-(1,1,3,3-тетраизопропил-1,3-дисилоксанил)-2'-дезокси-β-L-аденозина 148:
Точка плавления 110-111°С (EtOH/петролейный эфир) (Lit.(5) точка плавления 113-114°С (EtOH))
1H ЯМР (200 MHz, CDCl3): δ 8,33 и 8,03 (2s, 2H, Н2 и Н8), 6,30 (dd, 1H, H1', J = 2,85 Hz, J = 7,06 Hz), 5,63 (sl, 2H, NH2), 4,96 (m, 1H, H3'), 4,50 (m, 2Н, H5'aи Н5'b),2,68 (m, 2H, H2'a и Н2'b), 1,08(m, 28H, протоны изопропила)
Масс-анализ (FAB+, GT) m/z494 (M+H)+, 136 (BH2)+
Реакция 5:

Реагенты: фторид аммония (Fluka; ссылочный номер 09742); силикагель (Merck; ссылочный номер 1,07734,2500)
Растворители: Метанол, чистый для анализа (Prolabo; ссылочный номер 20847295); дихлорметан, чистый для анализа (Merck; ссылочный номер 1060506025); этанол 95 (Prolabo; ссылочный номер 20823,293)
Ссылки: Zhang, W., and Robins M.J., Removal of Silyl Protecting Groups from Hydroxyl Functions with Ammonium Fluoride in Methanol. Tetrahedron Lett., 33, 1177-1180 (1992).
Раствор 3',5'-O-(1,1,3,3-тетраизопропил-1,3-дисилоксанил)-2'-дезокси-L-аденозина 148 (32 г, 65 ммоль) и фторида аммония (32 г, ммоль) в метаноле перемешивали при кипячении с обратным холодильником в течение 2 часов. Добавляли силикагель и смесь осторожно упаривали для получения белого порошка. Этот порошок наносили на колонку силикагеля, которую элюировали дихлорметаном/метанолом 9/1. Соответствующие фракции объединяли и упаривали, чтобы получить белый порошок, который кристаллизовали из этанола 95(12,1 г, 75%).
Анализ 2'-дезокси-β-L-аденозина 149:
Точка плавления 189-190°С (EtOh 95) (идентичен коммерческому 2'-дезокси-D-аденозину)
1Н ЯМР (200 MHz, DMSO-D6): δ 8,35 и 8,14 (2s, 2H, H2 и Н8), 7,34 (sl, 2H, NH2), 6,35 (dd, 1H, H1', J = 6,1Hz, J = 7,85Hz), 5,33 (d, 1H, OH2', J = 4,0 Hz), 5,28 (dd, 1H, H3', J = 4,95 Hz, J 6,6 Hz), 4,42 (m, 1H, OH5'), 3,88 (m, 1H, H4'), 3,63-3,52 (m, 2H, H5'a и Н5'b), 2,71 (m, 1H, H2'a), 2,28(m, 1H, H2'b).(идентичен коммерческому 2'-дезокси-D-аденозину
αD+26° (c 0,5 вода) (коммерческий 2'-дезокси-D-аденозин -25° (с 0,5 вода)).
УФ λмакс 260 нм (ε 14100) (Н2О).
Масс-анализ (FAB+, GT) m/z 252 (M+H)+, 136 (BH2)+
Пример 3
Стереоспецифический синтез 2'-дезокси-β-L-цитидина
1-(3,5-Ди-О-бензоил-β-L-ксилофуранозил)урацил (11)
Гидрат гидразина (1,4 мл, 28,7 ммоль) добавляли к раствору 1-(2-О-ацетил-3,5-ди-О-бензоил-β-L-ксилофуранозил)-урацила 10 [См. Gosselin, G., Bergogne, M.-C., Imabach, J-L., “Synthesis and Antiviral Evaluation of β-L-Xylofuranosyl Nucleosides of the Five Naturally Occuring Nucleic Acid Bases”, Journal of Heterocyclic Chemistry, 1993, 30 (Oct.-Nov.), 1229-1233] (4,79 г, 9,68 ммоль) в пиридине (60 мл) и уксусной кислоте (15 мл). Раствор перемешивали в течение ночи при комнатной температуре. Добавляли ацетон (35 мл) и смесь перемешивали в течение 30 минут. Реакционную смесь упаривали при пониженном давлении. Полученный остаток очищали с помощью хроматографии на колонке силикагеля [элюент: ступенчатый градиент метанола (0-4%) в дихлорметане, чтобы получить 11 (3,0 г, 68%), которое кристаллизовали из циклогексана/дихлорметана: точка плавления = 111-114°С; 1Н ЯМР (DMSO-d6): δ 11,35 (br s, 1H, NH), 7,9-7,4 (m, 11H, 2C6H5Co, H-6), 6,38 (d, 1H, OH-2', JOH-2'=4,2 Hz), 5,77 (d, 1H, H-1', J1'-2'=1,9 Hz), 5,55 (d, 1H, H-5, J5-6=8 Hz), 5,54 (dd, 1H, H-3', J3'-2'= 3,9 Hz и J3'-4'=1,8 Hz), 4.8 (m, 1H, H-4'), 4,7 (m, 2H, H-5' и Н-5''), 4,3 (m, 1H, H-2'); MS: FAB>0 (матрикс GT) m/z 453 (M+H)+, 105 (C6H5CO)+; FAB<0 (матрикс GT) m/z 451 (М-Н)-, 121 (C6H5CO2)-, 111 (B)-; Аналитический расчет для С23Н20N2O8.H2O: C,58,09; H, 4,76; N,5,96. Обнаружено: С, 57,71; Н, 4,42; N, 5,70.
1-(3,5-Ди-О-бензоил-β-L-арабинофуранозил)урацил (12)
К раствору 1-(3,5-ди-О-бензоил-β-L-ксилофуранозил)-урацила 11 (8 г, 17,7 мл) в безводной смеси бензол-DMSO (265 мл, 6:4, об/об) добавляли безводный пиридин (1,4 мл), дициклогексилкарбодиимид (10,9 г, 53 ммоль) и дихлоруксусную кислоту (0,75 мл). Полученную смесь перемешивали при комнатной температуре в течение 4 часов, затем разбавляли этилацетатом (400 мл) и добавляли раствор щавелевой кислоты (4,8 г, 53 ммоль) в метаноле (14 мл). После перемешивания в течение 1 часа раствор фильтровали. Фильтрат промывали насыщенным раствором NaCl (2 × 500 мл), 3% раствором NaHCO3 (2 × 500 мл) и водой (2 × 500 мл). Органическую фазу высушивали над Na2SO4, затем упаривали при пониженном давлении. Полученный остаток солюбилизировали в смеси абсолютный EtOH-бензол (140 мл, 2:1, об/об). При 0°С к этому раствору добавляли NaBH4 (0,96 г, 26,5 ммоль). После перемешивания в течение 1 часа раствор разбавляли этилацетатом (400 мл), затем фильтровали. Фильтрат промывали насыщенным раствором NaCl (400 мл) и водой (400 мл). Органическую фазу высушивали над Na2SO4, затем упаривали при пониженном давлении. Полученное неочищенное вещество очищали хроматографией на колонке силикагеля [элюент: ступенчатый градиент метанола (0-3%) в дихлорметане для получения 12 (5,3 г, 66%), который кристаллизовали из ацетонитрила: точка плавления 182-183°С; 1Н-ЯМР (DMSO-d6): δ 11,35 (br s, 1H, NH), 8,0-7,5 (m, 11H, 2 C6H5CO, H-6), 6,23 (br s, 1H, OH-2'), 6,15 (d, 1H, H-1', J1'-2'=4 Hz), 5,54 (d, 1H, H-5,J5-6=8,1 Hz), 5,37 (t,1H, H-3', J3'-2'=J3'-4'=2,6 Hz), 4,7-4,6 (m, 2H, H-5' и Н-5''), 4,5 (m, 1H, H-4'), 4,4 (m,1H, H-2'); MS: FAB>0 (матрикс GT) m/z 453 (M+H)+, 341 (S)+, 113 (BH2)+, 105 (C6H5CO)+; FAB<0 (матрикс GT) m/z 451 (M-H)-,121 (C6H5CO2)-, 111 (B)-; Аналитический расчет для С23Н20N2O8; C, 61,06; H, 4,46; N, 6,19. Обнаружено С, 60,83; Н, 4,34; N,6,25.
1-(3,5-Ди-О-бензоил-2-дезокси-β-L-эритро-пентофуранозил)урацил (13)
К раствору 1-(3,5-ди-О-бензоил-β-L-арабинофуранозил)урацила 12 (5,2 г, 11,4 ммоль) в безводном 1,2-дихлорэтане (120 мл) добавляли фенокситиокарбонилхлорид (4,7 мл, 34,3 мл) и 4-(диметиламино)пиридин (DMAP, 12,5 г, 102,6 ммоль). Полученный раствор перемешивали при комнатной температуре в атмосфере аргона в течение 1 часа, а затем упаривали при пониженном давлении. Остаток растворяли в дихлорметане (300 мл) и органический раствор последовательно промывали охлажденным льдом 0,2 N раствором хлористо-водородной кислоты (3 × 200 мл) и водой (2 × 200 мл), высушивали над Na2SO4, затем упаривали при пониженном давлении. Неочищенное вещество упаривали несколько раз вместе с безводным диоксаном и растворяли в этом растворителе (110 мл). В атмосфере аргона к полученному раствору добавляли гидрид трис-(триметилсилил)силана (4,2 мл, 13,7 ммоль) и α,α'-азоизобутиронитрил ((AIBN, 0,6 г, 3,76 ммоль). Реакционную смесь нагревали и перемешивали при 100°С в течение 1 часа в атмосфере аргона, затем охлаждали до комнатной температуры и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке силикагеля [элюент:ступенчатый градиент метанола (0-5%)], чтобы получить 13 (2,78 г, 56%), который кристаллизовали из EtOH: точка плавления 223-225°С; Н-ЯМР (DMSO-d6): δ 11,4 (br s, 1H, NH), 8,0-7,5 (m, 11H,2 C6H5CO, H-6), 6,28 (t, 1H, H-1', J=7 Hz), 5,5 (m, 2H, H-1' и Н-5), 4,6-4,4 (m, 3H, H-4', H-5' и Н-5”), 2,6 (m, 2H, H-2' и Н-2''); MS: FAB>0 (матрикс GT) m/z437 (M+H)+, 3325 (S)+; FAB<0 (матрикс GT) m/z 435 (M-H)-, 111 (B)-; Аналитический расчет для С23Н20N2O7: C, 63,30; H, 4,62; N, 6,42. Обнаружено: С, 62,98; Н, 4,79; N,6,40.
2'-дезокси-β-L-цитидин (β-L-dC)
В атмосфере аргона к раствору 1-(3,5-ди-О-бензоил-2-дезокси-β-L-эритро-пентофуранозил)урацила 13 (2,66 г, 6,1 ммоль) в безводном 1,2-дихлорэтане (120 мл) добавляли реагент Лавессона (Lawesson) (1,72 г, 4,26 ммоль) и реакционную смесь перемешивали при кипячении с обратным холодильником в течение 2 часов. Растворитель затем выпаривали при пониженном давлении, а остаток очищали хроматографией на колонке силикагеля [элюент: ступенчатый градиент этилацетата (0-8%) в дихлорметане], чтобы получить 4-тиоинтермедиат в виде желтой пенообразной массы. Раствор этого тиоинтермедиата (1,5 г, 3,31 ммоль) в метанольном аммиаке (предварительно насыщенный при -10°С и герметично закрытый) (50 мл) нагревали при 1000С в баллоне из нержавеющей стали в течение 3 часов, а затем охлаждали до 0°С. Раствор упаривали при пониженном давлении. Полученное неочищенное вещество очищали хроматографией на колонке силикагеля [элюент: ступенчатый градиент метанола (0-20%) в дихлорметане]. Наконец, соответствующие фракции объединяли, фильтровали через ячейку Millex HV-4 (0,45 мкм, Millipore) и упаривали при пониженном давлении, чтобы получить требуемый 2'-дезокси-β-L-цитидин (β-L-dC) в виде пенообразной массы (0,6 г, 80%), который кристаллизовался из абсолютного EtOH: точка плавления = 198-199°С; 1Н-ЯМР (DMSO-d6): δ 7,77 (d, 1H, H-6, J6-5=7,4 Hz), 7,10 (br d, 2H, NH2), 6,13 (t, 1H, H-1', J=6,7 Hz), 5,69 (d, 1H, H-5, J5-6=7,4 Hz), 5,19 (d, 1H, OH-3', JOН-3' =4,1 Hz), 4,96 (t, 1H, OH-5', JOH-5'=JОH-5''=5,2 Hz), 4,1 (m, 1H, H-3', 3,75 (m, 1H, H-4'), 3,5 (m, 2H, H-5' и Н-5''), 2,0 (m, 1H, H-2'), 1,9 (m, 1Н< H-2''); MS: FAB>0 (матрикс GT) m/z 228 (M+H)+,112 (BH2)+; FAB<0 (матрикс GT) m/z 226 (M-H)-; [α}20 D= -69 (c 0,52, DMSO) [α]20 D=+76 (c 0,55, DMSO) для коммерчески доступного гидрохлорида D-энантиомера]. Аналитический расчет для С9Н13N3O4: C, 47,57; H, 5,77; N, 18,49. Обнаружено: С, 47,35; Н, 5,68; N,18,29.
Пример 4
Стереоизбирательный синтез 2'-дезокси-β-L-цитидина (β-L-dC)

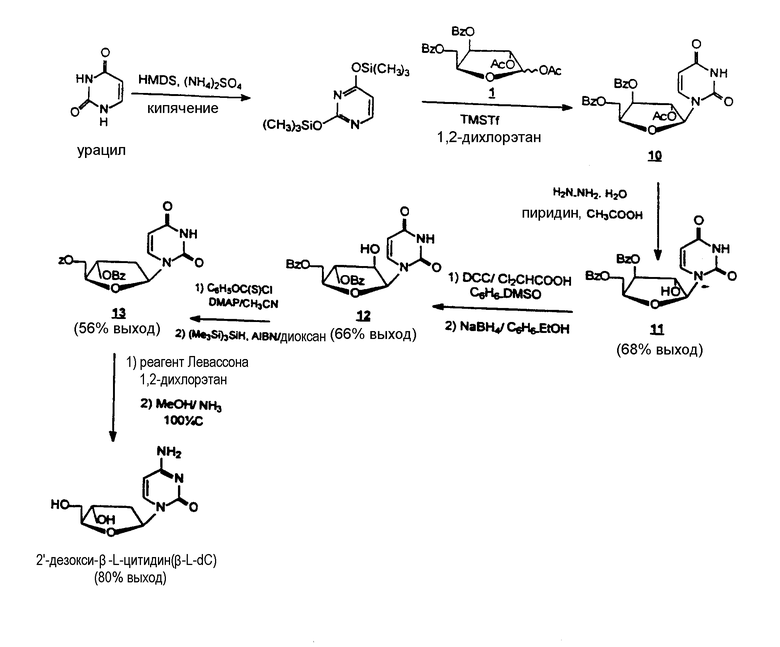
2-Амино-β-L-арабинофурано[1',2':4,5]оксазолин (1)
Смесь L-арабинозы (170 г, 1,1 моль), цианамида (100 г, 2,38 моль), метанола (300 мл) и 6 М NH4OH (50 мл)перемешивали при комнатной температуре в течение 3 дней, а затем содержали при -10°С в течение ночи. Продукт собирали отсасыванием, промывали последовательно метанолом и эфиром и высушивали под вакуумом. Выход, 130 г (66%) аналитически чистого соединения 1, точка плавления 170-172°С; 1Н ЯМР (DMSO-d6) δ м.д. 6,35 (br s, 2H, NH2), 5,15 (d, 1H, H-1, J=5,6 Hz), 5,45 (br s, 1H, OH-3), 4,70 (br s, 1H, OH-5), 4,55 (d, 1H, H-2, J=5,6 Hz), 4,00 (br s, 1H, H-3), 3,65 (m, 1H, H-4), 3,25 (m, 2H, H-5, H-5').
Реагенты:
L-арабиноза: Fluka, >99,5%, ссылочный номер 10839
Цианамид: Fluka, >98%, ссылочный номер 28330
O 2,2' -ангидро-β-L-уридин (2)
Раствор соединения 1 (98,8 г, 0,57 моль) и метилпропиолат (98 мл) в 50% водном этаноле (740 мл) кипятили с обратным холодильником в течение 5 часов, затем охлаждали и концентрировали при пониженном давлении до половины первоначального объема. После осаждения ацетоном (600 мл) продукт собирали отсасыванием, промывали этанолом и эфиром, высушивали. Маточный раствор частично концентрировали, концентрат осаждали ацетоном (1000 мл), твердое вещество собирали отсасыванием и промывали ацетоном и эфиром, чтобы получить еще одну массу продукта. Общий выход, 80 г (62%) соединения 2, точка плавления 236-240°С; 1Н ЯМР (DMSO-d6) δ м.д. 7,87 (d, 1H, H-6, J=7,4 Hz), 6,35 (d, 1H, H-1', J=5,7 Hz), 5,95 (d, 1H, H-5, J=7,4 Hz), 5,90(d, 1H, OH-3'), 5,20 (d, 1H, H-2', J=5,7 Hz), 5,00 (m, 1H, OH-3'), 4,44 (br s, 1H, H-3'), 4,05 (m, 1H, H-4'), 3,25 (m, 2H, H-5, H-5').
Реактив:
Метилпропиолат: Fluka, >97%, ссылочный номер 81863
3',5'-ди-О-бензоил-О 2,2' -ангидро-β-L-уридин (3)
К раствору соединения 2 (71,1 г, 0,31 моль) в безводном пиридине (1200 мл) добавляли хлорид бензоила (80,4 мл) при 0°С и в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов при исключении атмосферной влажности и останавливали добавлением этанола. Растворители упаривали при пониженном давлении и полученный остаток упаривали вместе с толуолом и абсолютным этанолом. Неочищенную смесь затем разбавляли этанолом, а осадок собирали отсасыванием, промывали последовательно этанолом и эфиром, высушивали. Выход 129 г (95,8%) соединения 3, точка плавления 254°С; 1Н ЯМР (DMSO-d6) δ частей на тысячу 8,1-7,4 (m, 11H, C6H5CO, H-6), 6,50 (d, 1H, H-1', J=5,7 Hz), 5,90 (d, 1H, H-5, J=7,5 Hz), 5,80 (d, 1H, H-2', J=5,8 Hz), 5,70 (d, 1H, H-3'), 4,90 (m,1H, H-4'), 4,35 (m, 2H, H-5, H-5').
Реактив:
Хлорид бензоила: Fluka, чистый для анализа, ссылочный номер 12930
3',5'-Ди-О-бензоил-2'-хлор-2'-дезокси-β,L-уридин (4)
При 0°С к раствору соединения 3 (60,3 г, 0,139 моль) в диметилформамиде (460) добавляли раствор 3,2 N-HCl/DMF (208 мл, приготовленный in situ добавлением 47,2 мл ацетилхлорида при 0°С к раствору 27,3 мл метанола и 133,5 мл диметилформамида). Реакционную смесь перемешивали при 100°С в течение 1 часа при устранении атмосферной влажности, охлаждали и вливали в воду (4000 мл). Осадок соединения 4 собирали отсасыванием, промывали водой и перекристаллизовывали из этанола. Кристаллы собирали, промывали охлажденным этанолом и эфиром, и высушивали при пониженном давлении. Выход, 60,6 г (92,6%) соединения 4, точка плавления 164-165°С; 1Н ЯМР (DMSO-d6) δ м.д. 8,7 (br s, 1H, NH), 8,1-7,3 (m, 11H, C6H5CO, H-6), 6,15 (d, 1H, H-1', J=4,8 Hz), 5,5 (m, 2H, H-5, H-2'), 4,65 (m, 4H, H-3', H-4', H-5', H-5'').
Реактивы:
Ацетилхолин: Fluka, чистый для анализа, ссылочный номер 00990
3',5'-Ди-О-бензоил-2'-дезокси-β,L-уридин (5)
Смесь соединения 4 (60,28 г, 0,128 моль)гидрида три-н-бутилолова (95 мл) и азабисизобутиронитрила (0,568 г) в сухом толуоле (720 мл) кипятили с обратным холодильником при перемешивании в течение 5 часов и охлаждали. Твердое вещество собирали отсасыванием и промывали холодным толуолом и петролейным эфиром. Фильтрат концентрировали при пониженном давлении и разбавляли петролейным эфиром, чтобы осадить дополнительную массу соединения 5. Выход 54,28 г (97,2%) соединения 5; точка плавления 220-221°С; 1Н ЯМР (CDCl3 δ м.д. 8,91 (br s, 1H, NH), 8,1-7,5 (m, 11H, C6H5CO и Н-6), 6,43 (q, 1H, H-1', J1',2'= 5,7 Hz и J1',2'' = 8,3 Hz), 5,7-5,6 (m, 2H, H-3' и Н-5), 4,8-4,6 (m, 3H,H-5', H-5'' и H-4'), 2,8-2,7 (m, 1H, H-2'), 2,4-2,3 (m, 1H, H-2'').
Реактивы:
Гидрид три-н-бутилолова: Fluka, >98%, cсылочный номер 90915
Азабисизобутиронитрил: Fluka, >98%, ссылочный номер 11630
3',5'-Ди-О-бензоил-2'-дезокси-β-L-4-тиоуридин (6)
Раствор соединения 5 (69 г, 0,158 моль) и реагент Лавессона (74 г) в безводном хлористом метилене (3900 мл) кипятили в атмосфере аргона в течение ночи. После упаривания растворителя неочищенный остаток очищали хроматографией на колонке силикагеля [элюент: градиент метанола (0-2%) в хлористом метилене], чтобы получить чистое соединение 6 (73 г) с количественным выходом; 1Н ЯМР (CDCl3) δ м.д. 9,5 (br s, 1H, NH), 8,1-7,4 (m, 10H, C6H5CO), 7,32 (d, 1H, H-6, J=7,7 Hz), 6,30 (dd, 1H, H-1', J=5,6 Hz и J= 8,2 Hz), 6,22 (d, 1H, H-5, J= 7,7 Hz), 5,6 (m, 1H, H-3'), 4,7 (m, 2H, H-5', H-5''), 4,5 (m, 1H, H-4'), 2,8 (m, 1H, H-2'), 2,3 (m, 1H, H-2'').
Реактивы:
Реагент Лавессона (Lawesson): Fluka, >98%, ссылочный номер 61750
2'-Дезокси-β-L-цитозин
Раствор соединения 6 (7,3 г, 0,016 моль) в метаноле, насыщенном аммиаком (73 мл), нагревали при 100°С в баллоне из нержавеющей стали в течение 3 часов. После осторожного охлаждения, растворитель выпаривали при пониженном давлении. Водный раствор остатка промывали этилацетатом и упаривали до сухого состояния. Такую процедуру осуществляли на 9 других образцах (каждый 7,3 г) соединения 6 (общее количество 6 = 73 г). 10 остатков объединяли, разбавляли абсолютным этанолом и охлаждали, чтобы получить 7 в виде кристаллов. Следы бензамида удаляли из кристаллов 6 экстракцией в системе твердое вещество-жидкость (при кипячении с обратным холодильником в этилацетате в течение 1 часа). Выход 28,75 г (78,6%) соединения 6; точка плавления 141-145°С; 1Н ЯМР (DMSO) δ м.д. 8,22 и 8,00 (2br s, 2H, NH2), 7,98 (d, 1H, H-6, J = 7,59 Hz), 6,12 (t, 1H, H-1', J =6,5 Hz и J = 7,6 Hz), 5,89 (d, 1H, H-5, J= 7,59 Hz), 5,3 (br s, 1H, OH-3'), 5,1 (br s, 1H, OH-5'), 4,2 (m, 1H, H-3'), 3,80 (q, 1H, H-4', J = 3,6 Hz и J = 6,9 Hz), 3,6-3,5 (m, 2H, H-5', H-5''), 2,2-2,0 (m, 2H, H-2', H-2''); FAB <0, (GT) m/e 226 (M-H)-, 110 (B)-; FAB>0 (GT) 228 (M+H)+, 112 (B+2H)+; [α]D 20 - 56,48 (c = 1,08 в DMSO); УФ (рН 7) λмакс = 270 нм (ε 10000).
Реактивы:
Метанольный раствор аммиака: заранее насыщали при -5°С, герметично закрывали и хранили в холодильнике.
Пример 5
Стереоизбирательный синтез 2'-дезокси-β-L-тимидина (β-L-dT)
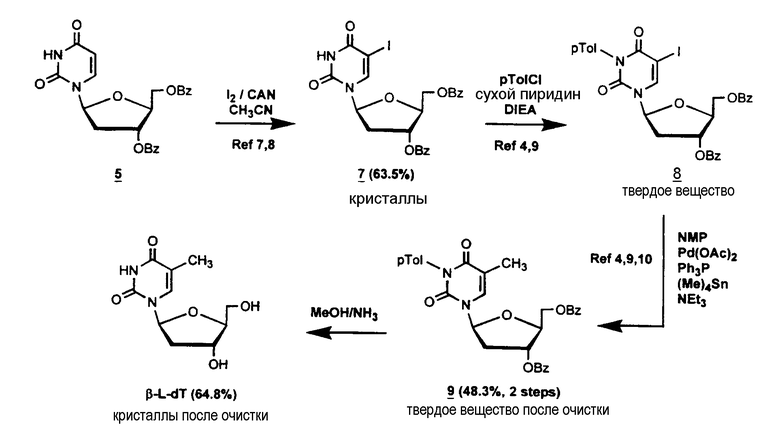
3',5'-Ди-О-бензоил-2'-дезокси-5-иод-β-L-уридин (7)
Смесь соединения 5 (105,8 г, 0,242 моль), иода (76,8 г), CAN (66,4 г) и ацетонитрила (2550 мл) перемешивали при 80°С в течение 3 часов, затем реакционную смесь охлаждали при комнатной температуре, приводя к кристаллизации соединения 7 (86,6 г, 63,5%); точка плавления 192-194°С; 1Н ЯМР (DMSO) δ м.д. 8,34 (s, 1H, NH), 8,2-7,2 (m, 11H, 2 C6H5CO, H-6), 6,31 (q, 1H, H-1', J = 5,5 Hz и J = 8,7 Hz), 5,5 (m, 1H, H-3'), 4,7 (m, 2H, H-5', H-5''), 4,5 (m, 1H, H-4'), 2,7 (m, 1H, H-2'), 2,3 (m, 1H, H-2''); FAB<0, (GT) m/e 561 (M-H)-, 237 (B)-; FAB>0 (GT) 563(M+H)+; [α]D 20 + 39,05 (c = 1,05 в DMSO); УФ (EtOH 95) νмакс =281 нм (ε 9000), νминим= 254 нм (ε 4000), νмакс= 229 нм (ε 31000); Аналитический расчет для С23Н19IN2O7: C, 49,13; H, 3,41; N, 4,98; I, 22,57; Обнаружено: С, 49,31; Н, 3,53; N,5,05; I, 22,36.
Реактивы:
Иод: Fluka, 99,8%, ссылочный номер 57650
Нитрат церия аммония (CAN): Aldrich, >98,5%, ссылочный номер 21547-3
3',5'-Ди-О-бензоил-2'-дезокси-3-N-толуоил-β-L-тимидин (9)
К раствору соединения 7 (86,6 г, 0,154 моль) в безводном пиридине (1530 мл), содержащем N-этилдиизопропиламин (53,6 мл), добавляли порциями при 0°С п-толуоилхлорид (40,6 мл). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, затем добавляли воду, чтобы остановить реакцию, а реакционную смесь экстрагировали хлористым метиленом. Органическую фазу промывали водой, высушивали над сульфатом аммония и упаривали до сухого состояния, чтобы получить неочищенный 3',5'-ди-О-бензоил-2'-дезокси-3-N-толуоил-5-иод-β-L-уридин (8), который можно использовать для следующей стадии без дальнейшей очистки.
Раствор неочищенной смеси 8, ацетата палладия (3,44 г), трифенилфосфина (8,0 г) в N-метилпиролидиноне (1375 мл) с триэтиламином (4,3 мл) перемешивали при комнатной температуре в течение 45 минут. Затем добавляли тетраметилолово (42,4 мл) по каплям при 0°С в атмосфере аргона. После перемешивания при 100-110°С в течение ночи реакционную смесь вливали в воду и экстрагировали диэтиловым эфиром. Органический раствор высушивали над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке силикагеля [элюент: ступенчатый градиент этилацетата (0-10%) в толуоле], чтобы получить соединение 9 в виде пенообразного вещества (42,3 г, 48,3% для 2 стадий). 1Н ЯМР (DMSO) δ м.д. 8,3-7,2 (m, 15H, 2 C6H5CO, 1 CH3C6H4CO, H-6), 6,29 (t, 1H, H-1', J = 7,0 Hz), 5,7 (m, 1H, H-3'), 4,7-4,5 (m, 3H, H-5', H-5'', H-4'), 2,7-2,6 (m, 2H, H-2', H-2''); FAB<0, (GT) m/e 567 (M-H)-, 449 (M-CH3C6H4CO)-, 243 (B)-, 121 (C6H5COO)-; FAB>0 (GT) 1137 (2M+H)+, 569 (M+H)+, 325 (M-B)_, 245 (B+2H)+, 119 (CH3C6H5CO)+.
Реактивы:
п-Толуоилхлорид, Aldrich, 98%, ссылочный номер 10663-1
Диизопропилэтиламин: Aldrich, >99,5%, ссылочный номер 38764-9
N-метилпиролидинон: Aldrich, >99%, ссылочный номер 44377-8
Ацетат палладия: Aldrich, >99,98%, ссылочный номер 37987-5
Трифенилфосфин: Fluka, >97%, ссылочный номер 93092
Тетраметилолово: Aldrich, >99%, ссылочный номер 14647-1
2'-Дезокси-β-L-тимидин
Раствор соединения 9 (42,3 г, 0,074 моль) в метаноле, насыщенном аммиаком (1850 мл), перемешивали при комнатной температуре в течение двух дней. После упаривания растворителя остаток разбавляли водой и промывали несколько раз этилацетатом. Водный слой отделяли, упаривали при пониженном давлении и остаток очищали хроматографией на колонке силикагеля [элюент: ступенчатый градиент метанола (0-10%) в хлористом метилене], чтобы получить чистый 2'-дезокси-β-L-тимидин (11,62 г, 64,8%), который кристаллизовался из этанола; точка плавления 185-188°С; 1Н ЯМР (DMSO) δ м.д. 11,3 (s, 1H, NH), 7,70 (s, 1H, H-6), 6,2 (pt, 1H, H-1'), 5,24 (d, 1H, OH-3', J = 4,2 Hz), 5,08 (t, 1H, OH-5', J = 5,1 Hz), 4,2 (m, 1H, H-3”), 3,7 (m, 1H, H-4'), 3,5-3,6 (m, 2H, H-5', H-5''), 2,1-2,0 (m, 2H, H-2', H-2''); FAB<0, (GT) m/e 483 (2M-H)-, 349 (M+T-H)-, 241 (M-H)-, 125 (B)-; FAB>0 (GT) 243 (M+H)+, 127 (B+2H)+; )+; [α]D 20 - 13,0 (c = 1,0 в DMSO); УФ (рН 1) νмакс = 267 нм (ε 9700)б νминим = 234 нм (ε 2000).
Реактив:
Метанольный раствор аммиака: заранее насыщали при -5°С, герметично закрывали и хранили в холодильнике.
Пример 6
Стереоизбирательный синтез 2'-дезокси-β-L-инозина (β-L-dI)
β-L-dI синтезировали посредством деаминирования 2'-дезокси-β-L-аденозина (β-L-dA) по способу, ранее описанному в разделе 9-D-глюкопиранозила (См. I. Iwai, T. Nishimura and B. Shimizu, Synthetic Procedures in Nucleic Acid Chemistry, W.W. Aorbach and R.S. Tipson, eds., Jоhn Wiley & Sons, Inc. New York, vol. 1, pp 135-138 (1968)).
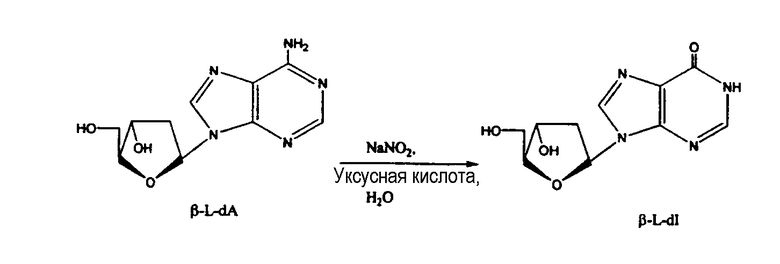
Таким образом, раствор β-L-dA (200 мг) в смеси уксусной кислоты (0,61 мл) и воды (19 мл) нагревали с нитритом натрия (495 мг) и смесь перемешивали при комнатной температуре в течение ночи. Раствор затем упаривали до сухого состояния при пониженном давлении. Водный раствор остатка наносили на колонку с ионообменной смолой IR-120 (H+) и колонку элюировали водой. Соответствующие фракции собирали и упаривали до сухого состояния, чтобы получить чистый β-L-dI, который кристаллизовался из метанола (106 мг, 53%, выход не оптимальный): точка плавления = 209-211°С; УФ (Н2О), λмакс = 247 нм; 1Н-ЯМР (DMSO-d6)= 8,32 и 8,07 (2s, 1H каждый, Н-2 и Н-8), 6,32 (pt, 1H, H-1; J= 6,7 Hz), 4,4 (m, 1H, H-3'), 3,9 (m, 1H, H-4'), 3,7-3,4 (m, 2H частично закрыт HOD, H-5',5''), 2,6 и 2,3 (2m, 1H каждый, H-2' и H-2''); спектр масс (матрикс, глицерин-тиоглицерин, 1:1, об/об), FAB>0: 253 (m+H)+, 137 (основание +2Н)+; FAB<0: 251 (m-H)-, 135 (основание)-; [α]D 20=+19,3 (-c 0,88, H2O).
Анти-HBV активность активных соединений
Способность активных соединений ингибировать рост вируса в культурах клеток 2.2.15 (клетки HepG2, трансформированные вирионом гепатита) можно оценивать по способу, детально описанному ниже.
Выводы и описание исследования противовирусного эффекта в этих культуральных системах и анализа ДНК HBV описаны Korba and Milman (1991, Antiviral Res.,15:217). Оценку противовирусного действия проводили на двух отдельных пассажах клеток. Все лунки во всех планшетах засевались с одинаковой плотностью и в одно и то же время.
Вследствие наследственной изменчивости в уровнях как внутриклеточной, так и внеклеточной ДНК HBV, только снижения больше чем в 3,5 раз (для ДНК вириона HBV) или 3,0 раза (для интермедиатов репликации ДНК HBV) ниже средних уровней этих форм ДНК HBV в необработанных клетках рассматривались как статистически значимые (Р<0,05). Уровни интегрированной ДНК HBV в каждый клеточный препарат ДНК (который оставался постоянным на клетку в этих экспериментах) использовались для расчета уровней внутриклеточных форм ДНК HBV, таким образом, обеспечивая то, что одинаковые количества клеточной ДНК сравниваются между отдельными образцами.
Типичные величины для внеклеточной ДНК вириона HBV в необработанных клетках колебались от 50 до 150 пг/мл культуральной среды (в среднем приблизительно 76 пг/мл). Внутриклеточные интермедиаты репликации ДНК HBV в необработанных клетках колебались от 50 до 100 мкг/пг клеточной ДНК (в среднем приблизительно 74 пг/мкг клеточной ДНК). Вообще снижение уровней внутриклеточной ДНК HBV вследствие обработки противовирусными соединениями оказалось менее выраженным и происходило медленее, чем снижение уровней ДНК вириона HBV (Korba and Milman, 1991, Antiviral Res., 15: 217).
Способ, посредством которого в этих экспериментах проведен анализ гибридизации, дает в результате эквивалентность, приблизительно равную 1,0 пг внутриклеточной ДНК HBV на 2-3 геномные копии на клетку и 1,0 пг/мл внеклеточной ДНК HBV на 3 × 105 вирусных частиц/мл.
Пример 7
Исследовали способность трифосфатных производных β-L-dA, β-L-dC, β-L-dU, β-L-2'-dG, β-L-dI, и β-L-dT ингибировать вирус гепатита В. В Таблице 1 описываются сравнительные ингибиторные активности трифосфатов β-L-dT (β-L-dT-TP), β-L-dC (β-L-dC-TP), β-L-dU (β-L-dU-TP) и β-L-dA (β-L-dA-TP) в отношении ДНК-полимеразы вируса гепатита сурка (WHV), ДНК- α-,β- и γ-полимераз человека.
aIC50: концентрация, вызывающая 50% ингибирование
bKi определяли, используя активированную ДНК тимуса теленка в качестве матрицы для затравки и dATP (dАТФ) - в качестве субстрата. Ингибиторы анализировали с помощью графического метода Диксона (Dixon). При этих условиях средняя Km ДНК-α-полимеразы человека для dATP рассчитана приблизительно как 2,6 мкМ. ДНК-β-полимеразы человека характеризовалась Km cтационарного состояния 3,33 мкМ для dATP. ДНК-γ-полимеразы человека характеризуется Km устойчивого состояния 5,2 мкМ.
Пример 8
Исследовали активность против вируса гепатита В - β-L-dA, β-L-dC, β-L-dU, β-L-2'-dG и β-L-dT - на трансфицированных клетках Hep G-2 (2.2.15). Таблица 2 иллюстрирует действие β-L-dA, β-L-dC, β-L-dU и β-L-dT на репликацию вируса гепатита В в трансфицированных клетках Hep G-2 (2.2.15).
aВнеклеточная ДНК
b Интермедиат репликации (внутриклеточная ДНК)
Пример 9
Определяли влияние сочетаний β-L-dA, β-L-dC и β-L-dT на рост вируса гепатита В в клетках 2.2.15. Результаты представлены в Таблице 3.
Каждое сочетание проявляло анти-HBV активность, которая была синергической. Кроме того, сочетания L-dA + L-dC + L-dT также были синергическими в этой модели.
Пример 10
Определяли ингибирование репликации вируса гепатита В в клетках 2.2.15 посредством β-L-dA и β-L-dC, в отдельности и в сочетаниях. Результаты представлены в Таблице 4.
аβ-L-2'-дезоксиаденозин: IC50 = 0,09 мкМ
bβ-L-2'-дезоксицитидин: IC50 = 0,06 мкМ
Величины показателей сочетаний свидетельствуют о синергическом действии (<1), аддитивном действии (=1) и антагонистическом действии (>1)
Пример 11
Определяли эффективность L-dA, L-dT и L-dC в отношении гепаднавирусной (hepadnavirus) инфекции у сурков (Marmota monax), хронически инфицированных вирусом гепатита сурков (WHV). Эта животная модель инфекции HBV является общепринятой и используется для оценки противовирусных агентов, направленных против HBV.
Протокол:
Экспериментальные группы (n = 3 животным/группу, получавшую лекарство, n = 4 животным/контроль)
Группа 1 контроль наполнителем
Группа 2 ламивудин (3ТС) (10 мг/кг/день)
Группы 3-6 L-dA (0,01; 0,1; 1,0; 10 мг/кг/день)
Группы 7-10 L-dt (0,01; 0,1; 1,0; 10 мг/кг/день)
Группы 11-14 L-dC (0,01; 0,1; 1,0; 10 мг/кг/день)
Лекарственные средства вводили перорально через желудочный зонд один раз ежедневно и брали образцы крови на 0, 1, 3, 7, 14, 21, 28 день и после лечения +1, +3, +7, +14, +28, и +56 дней. Оценка активности и токсичности базировалась на снижении ДНК WHV в сыворотке: дот-блоттинг, количественная PCR (полимеразно-цепная реакция, ПЦР). Результаты представлены на Фигуре 3 и в Таблице 5.
Противовирусная активность LdA, Ldt, LdC на модели хронической инфекции HBV сурка
1LdA, LdT, LdC вводили 10 мг/кг перорально один раз в день
2Предел определения составляет 1 нг/мл ДНК-WHV на мл сыворотки
Данные показывают, что L-dA, L-dT и L-dC высоко активны на этой модели in vivo. Первое, вирусная нагрузка снижается до неопределяемых (L-dT) или почти неопределяемых (L-dA, L-dC) уровней. Второе, показано, что L-dA, L-dT и L-dC более активны, чем 3ТС (ламивудин) на этой модели. Третье, повторное появление вируса не определялось по меньшей мере в течение двух недель после отмены L-dT. Четвертое, кривая «доза-ответ» позволяет предположить, что способы увеличения доз L-dA и L-dC будут проявляться в антивирусной активности подобной активности L-dT. Пятое, у всех животных, получавших лекарственные средства, увеличивался вес и не наблюдалось токсичности, связанной с лекарственным средством.
Токсичность соединений
Для выяснения того, не являются ли любые наблюдаемые противовирусные эффекты следствием общего воздействия на клеточную жизнеспособность, проводили исследование токсичности. Использованный способ представляет собой определение влияния β-L-dA, β-L-dC и β-L-dT на рост клеток в клорогенном анализе костного мозга человека, при сравнении с ламувидином. Результаты представлены в Таблице 6.
Получение фармацевтических композиций
Человека, страдающего любым описанным в заявке нарушением, включая гепатит В, можно лечить введением пациенту эффективного количества β-2'-дезокси-β-L-эритро-пентофуранонуклеозида, например, β-L-2'-дезоксиаденозина, β-L-2'-дезоксицитидина, β-L-2'-дезоксиуридина, β-L-2'-дезоксигуанозина или β-L-2'-дезокситимидина или их фармацевтически приемлемого пролекарства или соли в присутствии фармацевтически приемлемого носителя или разбавителя. Активные вещества можно вводить с помощью любого соответствующего способа, например, перорального, парентерального, внутривенного, внутрикожного, подкожно или местно в жидкой или твердой форме.
Активное соединение включается в фармацевтически приемлемый носитель или разбавитель в количестве, достаточном, чтобы обеспечить пациента терапевтически эффективным количеством соединения для ингибирования вирусной репликации in vivo, не оказывая серьезного токсического воздействия на пациента, которого лечат. Термин «ингибиторное количество» подразумевает количество активного ингредиента, достаточное, чтобы оказать ингибирующее действие, которое определяется посредством, например, такого способа как способы, описанные в заявке.
Предпочтительная доза соединения для всех вышеупомянутых состояний располагается в области приблизительно от 1 до 50 мг/кг, предпочтительно от 1 до 20 мг/кг веса тела в день, большей частью приблизительно от 0,1 до 100 мг на килограмм веса тела реципиента в день. Диапазон эффективных дозировок фрамацевтически приемлемого пролекарства можно рассчитать на основании веса родственного нуклеозида, который должен быть доставлен. Если пролекарство проявляет активность само по себе, эффективная доза может быть установлена, как упомянуто выше, с помощью веса пролекарства, или другими способами, известными специалистам в данной области.
Соединение удобно вводится в любой приемлемой стандартной дозированной форме, включая форму, содержащую от 7 до 3000 мг, предпочтительно от 70 до 1400 мг активного ингредиента на стандартную дозированную форму, но не ограничивается этим. Обычно общепринята пероральная доза 50-1000 мг.
В идеальном случае активный ингредиент следует вводить, чтобы достичь пиковых концентраций активного соединения в плазме приблизительно от 0,2 до 70 мкМ, предпочтительно примерно от 1,0 до 10 мкМ. Это может быть достигнуто, например, при внутривенной инъекции 0,1 до 5% раствора активного ингредиента, необязательно в физиологическом растворе, или введено в виде болюса активного ингредиента.
Концентрация активного соединения в лекарственной композиции зависит от абсорпции, инактивации и скорости экскреции лекарственного вещества, а также других факторов, известных специалистам в данной области. Следует отметить, что величины доз также изменяются в зависимости от тяжести состояния, которое следует облегчить. Кроме того, понятно, что для любого отдельного субъекта следует установить специальную схему приема лекарственного средства в зависимости от времени в соответствии с индивидуальной потребностью и профессиональной оценкой лица, назначающего композиции или наблюдающего за их введением, и что приведенные здесь диапазоны концентраций являются только примерами и не предназначены для ограничения сферы действия или применения заявленной композиции. Активный ингредиент можно вводить сразу или можно разделить на ряд небольших доз для введения с различными интервалами времени.
Предпочтительным способом введения активного соединения является пероральный. Пероральные композиции обычно включают в себя инертный растроритель или съедобный носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для целей перорального терапевтического введения активное соединение может вводиться с наполнителями и применяться в форме таблеток, пастилок или капсул. Фармацевтически совместимые связующие агенты и/или адъювантные вещества могут быть включены как часть композиции.
Таблетки, пилюли, капсулы, пастилки и тому подобное могут содержать любой из следующих ингредиентов или соединений подобной природы: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; наполнитель, такой как крахмал или лактоза, дезинтегратор, такой как альгиновая кислота, примогель или кукурузный крахмал; смазки, такие как стеарат магния или стероты; вещество, обеспечивающее скольжение, такое как коллоидная двуокись кремния; подсластители, такие как сахароза или сахарин; или вкусовое вещество, такое как перечная мята, метилсалицилат или апельсиновый аромат. Если стандартная лекарственная форма является капсулой, она может содержать, кроме веществ вышеназванного типа, жидкий носитель, такой как жирное масло. Кроме того, стандартные формы могут содержать различные другие вещества, которые изменяют физическую форму препарата, например покрытия сахаром, шеллак или другие энтеросолюбильные вещества.
Соединение можно вводить как компонент эликсира, суспензии, сиропа, воды, жевательной резинки и тому подобного. В дополнение к активным соединениям, сироп может содержать сахарозу как подсластитель и некоторые консерванты, красители или окрашивающие вещества или ароматизаторы.
Соединение или его фармацевтически приемлемое производное или соли можно также смешивать с другими активными веществами, которые не ухудшают требуемое действие, или с веществами, которые дополняют требуемое действие, такие как антибиотики, противогрибковые вещества, противовоспалительные вещества, ингибиторы протеаз или другие нуклеозидные или ненуклеозидные противовирусные агенты. Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного применения, могут включать в себя следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислоты или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и вещества для поддержания тонуса, такие как хлорид натрия или декстроза. Парентеральные препараты могут быть заключены в ампулы, одноразовые шприцы или виалы многократной дозы, сделанные из стекла или пластика.
При внутривенном введении предпочтительными носителями являются физиологический раствор или забуференный фосфатом физиологический раствор (PBS).
В предпочтительном аспекте активные соединения готовят с носителями, которые защищают соединение от быстрой элиминации из организма, как, например, композиции с контролируемым высвобождением, включающие имплантаты и микроинкапсулированные системы доставки. Можно использовать биодеградируемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полиуксусная кислота. Способы получения таких композиций, очевидно, известны специалистам в данной области. Также вещества могут быть получены коммерчески от Alza Corporation.
Липосомные суспензии (включающие в себя липосомы, таргетирующие инфицированные клетки моноклональными антителами к вирусным антигенам) также предпочтительны в качестве фармацевтически приемлемых носителей. Их можно получать в соответствии со способами, известными специалистам в данной области, например, как описано в Патенте США № 4522811. Например, липосомные композиции можно проготовить растворением соответствующего липида(ов) (такого как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, арахадоил-фосфатидилхолин и холестерин) в неорганическом растворителе, который затем упаривается, оставляя тонкий слой высушенного липида на поверхности резервуара. Затем водный раствор активного соединения или его монофосфатное, дифосфатное и/или трифосфатное производное вносят в резервуар. Резервуар затем вращают рукой, чтобы отделить липидное вещество от стенок резервуара и диспергировать липидные агрегаты, создавая, таким образом, липосомную суспензию.
Это изобретение описано с упоминанием его предпочтительных осуществлений. Изменения и модификации изобретения очевидны специалистам в данной области из предшествующего подробного описания изобретения. Полагают, что все эти изменения и модификации находятся в пределах сферы действия этого изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| β-L-2'-ДЕЗОКСИНУКЛЕОЗИДЫ ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА В | 1999 |
|
RU2300381C2 |
| НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ПРОТИВ ВИРУСА ГЕПАТИТА В | 1999 |
|
RU2237479C2 |
| МОНОЦИКЛИЧЕСКИЕ L-НУКЛЕОЗИДЫ, ИХ АНАЛОГИ И ПРИМЕНЕНИЯ | 1997 |
|
RU2188828C2 |
| Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение | 2017 |
|
RU2659388C1 |
| Циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноаты, способ их получения и применения | 2017 |
|
RU2647576C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ФТОР-2'-АЛКИЛЗАМЕЩЕННЫХ ИЛИ ДРУГИХ ЗАМЕЩЕННЫХ РИБОФУРАНОЗИЛПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2433124C2 |
| Противовирусная композиция и способ ее применения | 2017 |
|
RU2650610C1 |
| 2`-АМИНО-2`-ДЕЗОКСИНУКЛЕОЗИДЫ - ИНГИБИТОРЫ РЕПРОДУКЦИИ ВИРУСОВ КОРИ И МАРБУРГ | 2003 |
|
RU2264409C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |
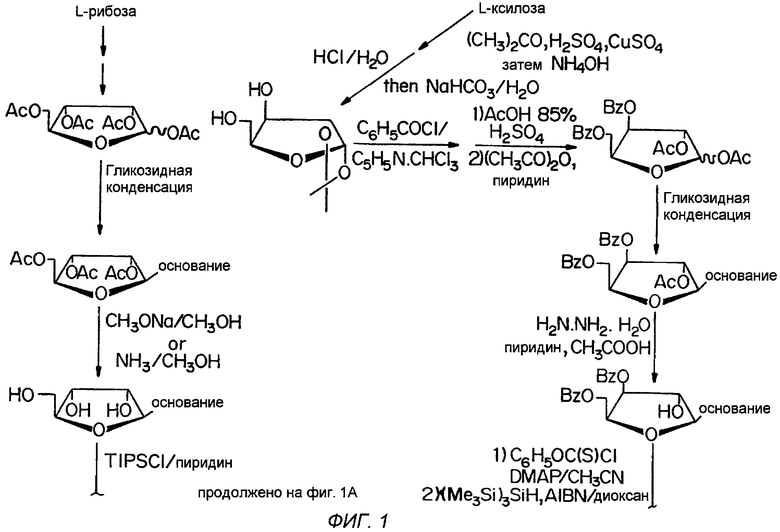
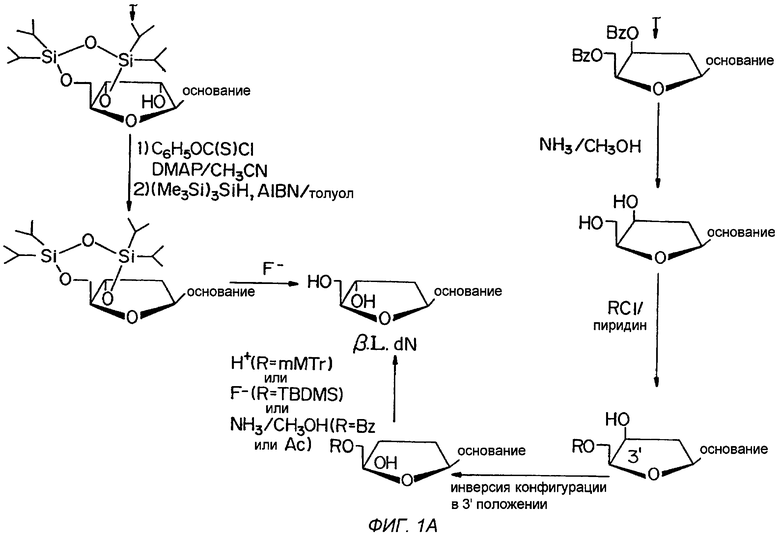

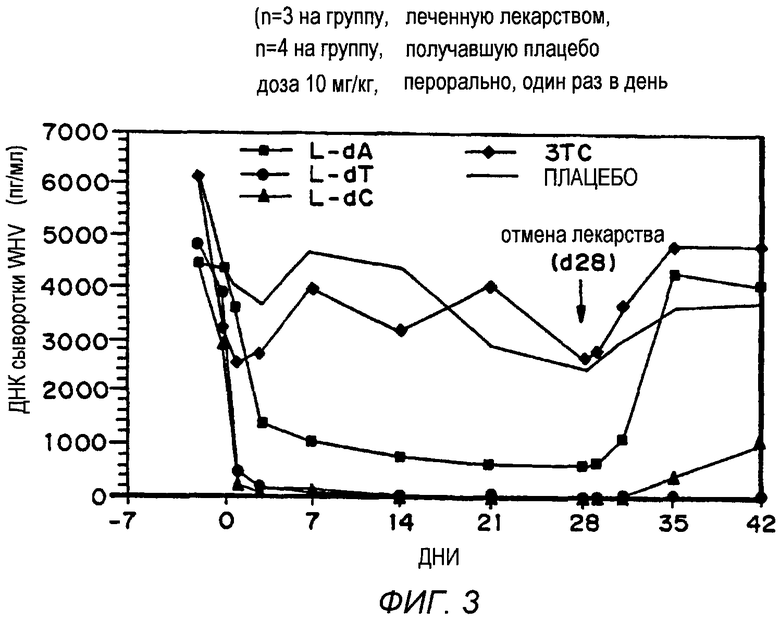
Данное изобретение относится к лекарственным средствам для лечения хозяина, инфицированного вирусом гепатита В, содержащим соединения указанных структурных формулы или к их фармацевтически приемлемым солям. Кроме того, изобретение относится к фармацевтическим композициям для лечения хозяина, инфицированного вирусом гепатита В, на основе каждого из данных соединений в отдельности (или их фармацевтически приемлемой соли), а также в их комбинации с эффективным количеством соединения, выбранного из группы, состоящей из β-L-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолана (3ТС), цис-2-гидроксиметил-5-(5-фторцитозина-1-ил)-1,3-оксатиолана (FTC), β-L-2'-фтор-5-метил-арабинофуранозил-уридин (L-FMAU), β-D-2,6-диаминопуриндиоксолана (DAPD), фамцикловира, пенцикловира, 2-амино-1,9-дигидро-9-[4-гидрокси-3-(гидроксиметил)2-метиленциклопентил]-6Н-пурин-6-она (энтекавир, ВMS-200475), 9-[2-(фосфоно-метокси)этил]аденина (РМЕА, адефовир, дипивоксил), лобукавира, ганцикловира и рибавирина. 6 н. и 6 з.п. ф-лы, 6 табл., 4 ил.
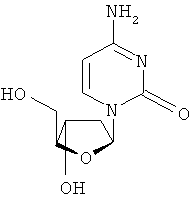
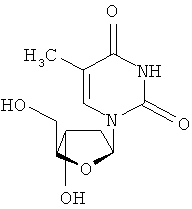
1. Лекарственное средство для лечения хозяина, инфицированного вирусом гепатита В, содержащее соединение формулы
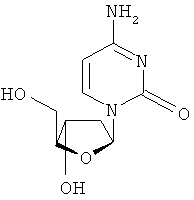
или его фармацевтически приемлемую соль.
2. Лекарственное средство для лечения хозяина, инфицированного вирусом гепатита В, содержащее соединение формулы
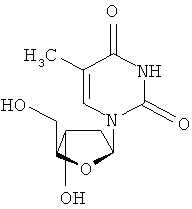
или его фармацевтически приемлемую соль.
3. Фармацевтическая композиция для лечения хозяина, инфицированного вирусом гепатита В, содержащая терапевтически эффективное количество соединения формулы
или его фармацевтически приемлемую соль;
и фармацевтически приемлемый носитель или разбавитель.
4. Фармацевтическая композиция для лечения хозяина, инфицированного вирусом гепатита В, содержащая терапевтически эффективное количество соединения формулы
или его фармацевтически приемлемую соль;
и фармацевтически приемлемый носитель или разбавитель.
5. Фармацевтическая композиция для лечения хозяина, инфицированного вирусом гепатита В, содержащая терапевтически эффективное количество соединения формулы
или его фармацевтически приемлемую соль в комбинации с эффективным количеством соединения, выбранного из группы, состоящей из β-L-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолана (3ТС), цис-2-гидроксиметил-5-(5-фторцитозина-1-ил)-1,3-оксатиолана (FTC), β-L-2'-фтор-5-метиларабинофуранозилуридин (L-FMAU), (β-D-2,6-диаминопуриндиоксолана (DAPD), фамцикловира, пенцикловира, 2-амино-1,9-дигидро-9-[4-гидрокси-3-(гидроксиметил)2-метиленциклопентил]-6Н-пурин-6-она (энтекавир, ВMS-200475), 9-[2-(фосфонометокси)этил]аденина (РМЕА, адефовир, дипивоксил), лобукавира, ганцикловира и рибавирина;
и фармацевтически приемлемый носитель или разбавитель.
6. Фармацевтическая композиция для лечения хозяина, инфицированного вирусом гепатита В, содержащая терапевтически эффективное количество соединения формулы
или его фармацевтически приемлемую соль в комбинации с эффективным количеством соединения, выбранного из группы, состоящей из β-L-2-гидроксиметил-5-(цитозин-1-ил)-1,3-оксатиолана (3ТС), цис-2-гидроксиметил-5-(5-фторцитозина-1-ил)-1,3-оксатиолана (FTC), β-L-2'-фтор-5-метиларабинофуранозил-уридин (L-FMAU), β-D-2,6-диаминопуриндиоксолана (DAPD), фамцикловира, пенцикловира, 2-амино-1,9-дигидро-9-[4-гидрокси-3-(гидроксиметил)2-метиленциклопентил]-6Н-пурин-6-она (энтекавир, BMS-200475), 9-[2-(фосфонометокси)этил]аденина (РМЕА, адефовир, дипивоксил), лобукавира, ганцикловира и рибавирина;
и фармацевтически приемлемый носитель или разбавитель.
7. Фармацевтическая композиция по пп.3, 4, 5 и 6, где соединение по меньшей мере на 95% представлено в указанной энантиомерной форме.
8. Фармацевтическая композиция по пп.3, 4, 5 и 6, содержащая фармацевтически приемлемый носитель.
9. Фармацевтическая композиция по п.8, где фармацевтически приемлемый носитель является подходящим для перорального введения.
10. Фармацевтическая композиция по п.9, где соединение представлено в виде единичной дозы.
11. Фармацевтическая композиция по п.10, где единичная доза содержит от 7 до 3000 мг соединения.
12. Фармацевтическая композиция по п.10, где единичная доза представляет собой таблетку или капсулу.
| Yong-Lian Zhu et al | |||
| Antimicrobial Agents and Chemotherapy | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Annalisa VERRI et al | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |
| RU 95119386 A, 20.09.1997. | |||

