Область техники
Данное изобретение относится к новым агонистам β2 адренергического рецептора. Данное изобретение также относится к фармацевтическим композициям, содержащим такие соединения, способам применения таких соединений для лечения заболеваний, связанных с активностью β2 адренергического рецептора, и способам и промежуточным соединениям, применяемым для получения таких соединений.
Уровень техники
Агонисты β2 адренергического рецептора являются эффективными лекарственными средствами для лечения легочных заболеваний, таких как астма и хроническое обструктивное заболевание легких (включая хронические бронхиты и эмфизему). Агонисты β2 адренергического рецептора также полезны для лечения досрочных родов и потенциально применимы для лечения неврологических расстройств и сердечных заболеваний. Несмотря на успехи, которые были достигнуты с использованием для определенных агонистов β2 адренергического рецептора, современные агенты обладают меньшей, чем желательная, продолжительностью действия, мощностью, селективностью и/или началом действия. Таким образом, существует необходимость в дополнительных агонистах β2 адренергического рецептора, обладающих улучшенными свойствами, такими как мощность, селективность и/или начало действия.
Сущность изобретения
Данное изобретение относится к новым соединениям, которые обладают агонистическим действием в отношении β2 адренергического рецептора. Было обнаружено, что среди прочих свойств соединения в соответствии с данным изобретением являются мощными и селективными агонистами β2 адренергического рецептора. Кроме того, соединения в соответствии с данным изобретением обладают удивительной и неожиданной продолжительностью действия, что, как ожидается, позволит дозировать соединение один раз в сутки или даже реже.
Следовательно, данное изобретение относится к соединению формулы (I):
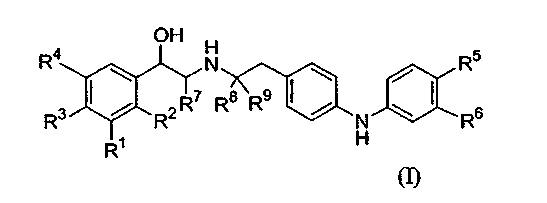
где
каждый из R1, R2, R3 и R4 независимо выбирают из водорода, гидрокси, амино, галогена, -CH2OH и -NHCHO или R1 и R2, взятые вместе, выбирают из -NHC(=O)CH=CH-, -CH=CHC(=O)NH-, -NHC(=O)S- и -SC(=O)NH-;
один из R5 и R6 является -[X-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13
и другой из R5 и R6 выбирают из водорода, гидрокси, C1-4алкокси и C1-4алкила, где C1-4алкил необязательно замещен галогеном,
где
каждый X независимо выбирают из -O-, -NH-, -S-, -NHSO2-, -SO2NH-, -NHC(=O)- и -C(=O)NH-;
каждый из R10, R11, R12 и R13 независимо является водородом или C1-4алкилом; или
R10 и R11, вместе с атомом азота, к которому они присоединены, или R10, вместе с атомом азота, к которому он присоединен, и атомом углерода соседнего C1-6алкиленила, или R12 и R13, вместе с атомом азота, к которому они присоединены, или R12, вместе с атомом азота, к которому он присоединен, и атомом углерода соседнего C1-6алкиленила, образуют гетероциклическое или гетероарильное кольцо, имеющее от 5 до 7 атомов в кольце, и необязательно содержащее дополнительный гетероатом, выбранный из кислорода, азота и серы, где атом азота необязательно замещен -S(O)2-C1-4алкилом; и
n равно 1, 2 или 3; и
каждый из R7, R8 и R9 независимо является водородом или C1-6алкилом;
или его фармацевтически приемлемая соль, или сольват, или стереоизомер.
В данном изобретении также представлены фармацевтические композиции, содержащие соединение в соответствии с данным изобретением и фармацевтически приемлемый носитель. В данном изобретении также представлены комбинации, включающие соединение в соответствии с данным изобретением и один или несколько других фармацевтических агентов, и фармацевтические композиции, содержащие такие комбинации.
В изобретении также представлен способ лечения млекопитающего, страдающего от заболевания или состояния, связанного с активностью β2 адренергического рецептора (например, легочного заболевания, такого как астма или хроническое обструктивное заболевание легких, преждевременных родов, неврологического расстройства, сердечного расстройства или воспаления), включающий введение млекопитающему терапевтически эффективного количества соединения в соответствии с данным изобретением. В изобретении также представлен способ лечения, включающий введение терапевтически эффективного количества комбинации соединения в соответствии с данным изобретением и одного или более других терапевтических агентов.
В данном изобретении также представлен способ лечения млекопитающего, страдающего заболеванием или состоянием, связанным с активностью β2 адренергического рецептора, включающий введение млекопитающему терапевтически эффективного количества фармацевтической композиции в соответствии с данным изобретением.
Соединения в соответствии с данным изобретением также могут применяться в качестве инструментов для исследований, например, при изучении биологических систем или образцов, или при изучении активности других химических соединений. Следовательно, в одном из аспектов данное изобретение относится к способу применения соединения формулы (I) или его фармацевтически приемлемой соли, или сольвата, или стереоизомера в качестве инструмента для исследований биологической системы или образца или для обнаружения новых агонистов β2 адренергического рецептора.
В отдельных и самостоятельных аспектах данное изобретение также относится к способам синтеза и промежуточным соединениям, описанным ниже, которые применяются для получения соединений в соответствии с данным изобретением.
В данном изобретении также представлено соединение в соответствии с данным изобретением, такое как описано здесь, для применения в медицинской терапии, а также применение соединения в соответствии с данным изобретением в производстве препаративной формы или лекарственного средства для лечения млекопитающего, страдающего от заболевания или состояния, связанного с активностью β2 адренергического рецептора (например, легочного заболевания, такого как астма или хроническое обструктивное заболевание легких, преждевременных родов, неврологического расстройства, сердечного расстройства или воспаления).
Краткое описание чертежей
На фиг.1 представлена порошковая рентгенограмма продукта из примера 17b.
На фиг.2 представлена кривая дифференциальной сканирующей калориметрии продукта примера 17b.
На фиг.3 представлена порошковая рентгенограмма продукта из примера 17c.
На фиг.4 представлена кривая дифференциальной сканирующей калориметрии продукта примера 17с.
На фиг.5 представлена порошковая рентгенограмма продукта из примера 17d.
На фиг.6 представлена кривая дифференциальной сканирующей калориметрии продукта примера 17d.
Подробное описание изобретения
В данном изобретении представлены новые ариланилиновые агонисты β2 адренергического рецептора формулы (I) или их фармацевтически приемлемые соли, или сольваты, или стереоизомеры. Представленные ниже заместители и значения дают характерные примеры различных аспектов данного изобретения. Эти характерные значения предназначены для более детального определения таких аспектов и не исключают другие значения и не ограничивают объем данного изобретения.
В одном аспекте данного изобретения R1 является галогеном, -CH2OH или -NHCHO.
В другом аспекте данного изобретения R1 является хлором, -CH2OH или -NHCHO или R1 является -CH2OH или -NHCHO.
В определенном аспекте R2 является водородом.
В определенном аспекте R3 является гидрокси или амино.
В определенных аспектах R4 является водородом или галогеном или R4 является водородом или хлором.
В определенном аспекте R1 является -NHCHO, R3 является гидрокси и R2 и R4, каждый, является водородом.
В другом определенном аспекте R1 и R2, взятые вместе, являются -NHC(=O)CH=CH- или -CH=CHC(=O)NH-, R3 является гидрокси и R4 является водородом.
В другом определенном аспекте R1 является -CH2OH, R3 является гидрокси и R2 и R4, каждый, является водородом.
В еще одном определенном аспекте R1 и R4 являются хлором, R3 является амино и R2 является водородом.
В еще одном определенном аспекте R1 и R2, взятые вместе, являются -NHC(=O)S- или -SC(=O)NH-, R3 является гидрокси и R4 является водородом.
В определенном аспекте R5 или R6 является -[X-C1-6алкиленил]n-NR10R11, где n, X, R10 и R11 такие, как определено в формуле (I).
В другом определенном аспекте R5 или R6 является -[O-C1-6алкиленил]n-NR10R11, где R10 и R11 такие, как определено в формуле (I), и n равно 1 или 2.
В другом определенном аспекте R5 или R6 является -O-C1-6алкиленил-NR10R11, где каждый из R10 и R11 независимо является водородом или C1-4алкилом. Характерные значения R5 или R6 включают, но не ограничены ими, -O(CH2)2NH2, -O(CH2)3N(CH3)2, -O(CH2)4NH2 и -OCH2C(CH3)2NH2.
В другом определенном аспекте R5 или R6 является -O-C1-6алкиленил-NR10R11, где R10 и R11, вместе с атомом азота, к которому они присоединены, образуют пиперазинильное кольцо. Например, R5 или R6 является -O(CH2)2-1-пиперазинилом.
В еще одном определенном аспекте R5 или R6 является C1-6алкиленил-NR12R13, где R12 и R13 такие, как определено в формуле (I).
В еще одном определенном аспекте R5 или R6 является C1-6алкиленил-NR12R13, где каждый из R12 и R13 независимо является водородом или C1-4алкилом. Характерные значения R5 или R6 в этом аспекте включают, но не ограничены ими, -(CH2)2NH2, -(CH2)2N(CH3)2 и -CH2C(CH3)2NH2.
В других определенных аспектах R5 или R6 является C1-4алкилом, необязательно замещенным галогеном, например, CF3; или R5 или R6 является C1-4алкокси, например, -OCH3; или R5 или R6 является водородом; или R5 или R6 является гидрокси.
В определенном аспекте R7 является водородом.
В определенном аспекте R8 является водородом.
В определенном аспекте R9 является водородом.
В одном аспекте данное изобретение относится к соединению формулы (II):
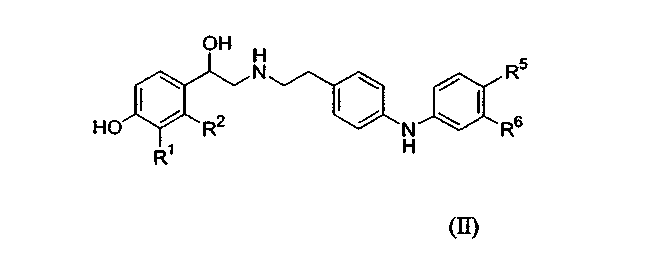
где
R1 является -CH2OH или -NHCHO и R2 является водородом; или R1 и R2, взятые вместе, являются -NHC(=O)CH=CH- или -CH=CHC(=O)NH-.
Один из R5 и R6 является -[O-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13,
и другой из R5 и R6 выбирают из водорода, гидрокси, C1-4алкокси и C1-4алкила, где C1-4алкил необязательно замещен галогеном,
где
каждый из R10, R11, R12 и R13 независимо является водородом или C1-4алкилом; или
R10 и R11, вместе с атомом азота, к которому они присоединены, или R10, вместе с атомом азота, к которому он присоединен, и атомом углерода соседнего C1-6алкиленила, или R12 и R13, вместе с атомом азота, к которому они присоединены, или R12, вместе с атомом азота, к которому он присоединен, и атомом углерода соседнего C1-6алкиленила образуют гетероциклическое или гетероарильное кольцо, имеющее от 5 до 7 атомов в кольце, и необязательно содержащее дополнительный гетероатом, выбранный из кислорода, азота и серы, где атом азота необязательно замещен -S(O)2-C1-4алкилом; и
n равно 1 или 2,
или его фармацевтически приемлемая соль, или сольват, или стереоизомер.
В другом аспекте данное изобретение относится к соединениям формулы (II), в которых R5 является -[O-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13 и R6 является водородом.
В другом аспекте данное изобретение относится к соединениями формулы (II), в которых R5 является C1-4алкокси и R6 является -[O-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13.
В другом аспекте данное изобретение относится к соединениям формулы (II), в которых
R5 выбирают из -O-C1-6алкиленил-NR10R11 и C1-6алкиленил-NR12R13 и R6 является водородом; или
R5 является C1-4алкокси и R6 является -C1-6алкиленил-NR12R13,
где каждый из R10, R11, R12 и R13 независимо является водородом или C1-4алкилом, или R10 и R11, вместе с атомом азота, к которому они присоединены, образуют пиперазинильное кольцо.
Определенная группа соединений в этом аспекте представляет собой группу, в которой R1 и R2, взятые вместе, являются -NHC(=O)CH=CH- или -CH=CHC(=O)NH-.
Другая определенная группа соединений в этом аспекте представляет собой группу, в которой R5 является -O-C1-6алкиленил-NR10R11 и R6 является водородом.
В других определенных аспектах данное изобретение относится к соединениям формулы (II), в которых переменные R1, R2, R5 и R6 имеют значения, указанные в таблице I ниже.
Особенно предпочтительными являются следующие соединения:
5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он;
8-гидрокси-5-[(R)-1-гидрокси-2-(2-{4-[4-(2-пиперазин-1-илэтокси)фениламино]фенил}этиламино)этил]-1H-хинолин-2-он;
5-[(R)-2-(2-{4-[4-(2-аминоэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он;
5-[(R)-2-(2-{4-[4-(2-диметиламиноэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он;
5-[(R)-2-(2-{4-[3-(2-аминоэтил)-4-метоксифениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он, где химическая номенклатура соответствует той, которая представлена в программе автоматического присваивания имен AutoNom, предоставляемой MDL Information Systems, GmbH (Frankfurt, Germany).
Как показано выше, соединения в соответствии с данным изобретением содержат один или несколько хиральных центров. Следовательно, данное изобретение включает рацемические смеси, чистые стереоизомеры (например, отдельные энантиомеры или диастереомеры) и насыщенные стереоизомерами смеси таких изомеров, если не указано иначе. Если показан конкретный стереоизомер, специалисту в данной области техники должно быть понятно, что незначительные количества других стереоизомеров могут присутствовать в композициях в соответствии с данным изобретением, если не указано иначе, при условии, что применение композиции в целом не ограничивается присутствием таких других изомеров.
В частности, соединения в соответствии с данным изобретением содержат хиральный центр на атоме углерода алкилена в формулах (I) и (II), к которому присоединена гидроксигруппа. Если применяется смесь стереоизомеров, предпочтительно, чтобы количество стереоизомера с (R)-ориентацией на хиральном центре, имеющем гидроксильную группу, было больше, чем количество соответствующего (S)-стереоизомера. При сравнении стереоизомеров одного и того же соединения (R)-стереоизомер считается предпочтительным перед (S) стереоизомером.
Определения
При описании соединений, композиций и способов в соответствии с данным изобретением представленные ниже термины имеют представленные ниже значения, если не указано иначе.
Термин «алкил» означает одновалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной, или их сочетание. Если не указано иначе, такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Характерные алкильные группы включают, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и подобные.
Термин «алкокси» означает одновалентную группу -O-алкил, в которой алкил такой, как определен выше. Характерные алкоксигруппы включают, например, метокси, этокси, пропокси, бутокси и подобные.
Термин «алкенил» означает одновалентную ненасыщенную углеводородную группу, содержащую, по крайней мере, одну двойную связь углерод-углерод, предпочтительно, 1 или 2 двойные связи углерод-углерод и которая может быть линейной или разветвленной, или их сочетание. Если не указано иначе, такие алкенильные группы обычно содержат от 2 до 10 атомов углерода. Характерные алкенильные группы включают, например, винил, аллил, изопропенил, бут-2-енил, н-пент-2-енил, н-гекс-2-енил, н-гепт-2-енил, н-окт-2-енил, н-нон-2-енил, н-дец-4-енил, н-дец-2,4-диенил и подобные.
Термин «алкинил» означает одновалентную ненасыщенную углеводородную группу, содержащую, по крайней мере, одну тройную связь углерод-углерод, предпочтительно, 1 тройную связь углерод-углерод, и которая может быть линейной или разветвленной, или их сочетание. Если не указано иначе, такие алкинильные группы обычно содержат от 2 до 10 атомов углерода. Характерные алкинильные группы включают, например, этинил, пропаргил, бут-2-инил и подобные.
Термин «алкиленил» означает двухвалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной, или их сочетание. Если не указано иначе, такие алкиленильные группы обычно содержат от 1 до 10 атомов углерода. Характерные алкиленильные группы включают, например, метилен, этилен, н-пропилен, н-бутилен, пропан-1,2-диил (1-метилэтилен), 2-метилпропан-1,2-диил(1,1-диметилэтилен) и подобные.
Термин «гетероарил» означает одновалентную ароматическую группу, имеющую одно кольцо или два сконденсированных кольца и содержащую в кольце, по крайней мере, один гетероатом (обычно от 1 до 3 гетероатомов), выбранных из азота, кислорода и серы. Если не указано иначе, такие гетероарильные группы обычно включают от 5 до 10 атомов в кольце. Характерные гетероарильные группы включают, например, пирролил, изоксазолил, изотиазолил, пиразолил, пиридил (или, эквивалентно, пиридинил), оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, пиридазинил, пиразинил, бензоксазолил, бензотиазолил, бензимидазолил, бензофуранил, бензотиофенил, хинолил, индолил, изохинолил и подобные, в которых место присоединения находится на любом свободном атоме углерода или азота в кольце.
Термин «гетероциклил» или «гетероциклическое кольцо» означает одновалентную насыщенную или частично ненасыщенную циклическую неароматическую группу, которая может быть моноциклической или полициклической (т.е. сконденсированной или мостиковой) и которая содержит, по крайней мере, один гетероатом (обычно от 1 до 3 гетероатомов), выбранных из азота, кислорода и серы. Если не указано иначе, такие гетероциклические группы обычно включают от 5 до 10 атомов в кольце. Характерные гетероциклические группы включают, например, пирролидинил, пиперидинил, пиперазинил, имидазолидинил, морфолинил, индолин-3-ил, 2-имидазолинил, 1,2,3,4-тетрагидроизохинолин-2-ил, хинуклидинил и подобные.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «лечение» в данном описании означает лечение заболевания или медицинского состояния у пациента, такого как млекопитающее (особенно человек), которое включает
(a) предотвращение возникновения заболевания или медицинского состояния, т.е. профилактическое лечение пациента;
(b) уменьшение заболевания или медицинского состояния, например, исключение или регрессию заболевания или медицинского состояния у пациента;
(c) подавление заболевания или медицинского состояния, например, замедление или остановку развития заболевания или медицинского состояния у пациента; или
(d) облегчение симптомов заболевания или медицинского состояния у пациента.
Термин «терапевтически эффективное количество» означает количество, достаточное для эффективного лечения при введении пациенту при необходимости такого лечения.
Фраза «заболевание или состояние, связанное с активностью β2 адренергического рецептора» включает все медицинские состояния, облегчаемые путем лечения агонистом β2 адренергического рецептора и включает все болезненные состояния и/или проявления, известные на данный момент, или которые будут обнаружены в будущем, связанные с активностью β2 адренергического рецептора. Такие болезненные состояния включают, но не ограничены ими, легочные заболевания, такие как астма и хроническое обструктивное заболевание легких (включая хронические бронхиты и эмфизему), а также неврологические заболевания и сердечные заболевания. Также известно, что активность β2 адренергического рецептора связана с преждевременными родами (см. патент США № 5872126) и некоторыми типами воспаления (см. публикацию международной заявки на патент № WO 99/30703 и патент США № 5290815).
Термин «фармацевтически приемлемая соль» относится к соли, полученной из основания или кислоты, которая приемлема для введения пациенту, такому как млекопитающее. Такие соли могут быть получены из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот.
Соли получают из фармацевтически приемлемых кислот, включающих, но не ограниченных ими, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую, ксинафоевую (1-гидрокси-2-нафтоевую кислоту), 1,5-нафталиндисульфоновую, коричную и подобные. Соли, полученные из фумаровой, бромистоводородной, хлористоводородной, уксусной, серной, метансульфоновой, 1,5-нафталиндисульфоновой, ксинафоевой, щавелевой, винной и 4-метилкоричной кислот, особенно предпочтительны.
Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца (III), марганца (II), калия, натрия, цинка и подобные. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклических аминов, природных аминов и подобных, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и подобные.
Термин «сольват» означает комплекс или агрегат, образованный одной или несколькими молекулами растворенного вещества, например, соединения в соответствии с данным изобретением или его фармацевтически приемлемой соли, и одной или несколькими молекулами растворителя. Такие сольваты обычно представляют собой кристаллические вещества, имеющие практически фиксированное молярное соотношение растворенного вещества и растворителя. Характерные растворители включают, например, воду, метанол, этанол, изопропанол, уксусную кислоту и подобные. Если растворителем является вода, сольват образуется в виде гидрата.
Должно быть понятно, что термин «или его фармацевтически приемлемая соль, или сольват, или стереоизомер» включает все вариации солей, сольватов и стереоизомеров, такие как сольват фармацевтически приемлемой соли или стереоизомера соединения формулы (I).
Термин «уходящая группа» означает функциональную группу или атом, которые могут быть замещены другой функциональной группой или атомом в реакции замещения, такой как реакция нуклеофильного замещения. Например, характерные уходящие группы включают группы хлора, брома и йода; группы сульфонового эфира, такие как мезилат, тозилат, брозилат, нозилат и подобные; и ацилоксигруппы, такие как ацетокси, трифторацетокси и подобные.
Термин «амино-защитная группа» означает защитную группу, подходящую для предотвращения нежелательных реакций по атому азота амино. Характерные амино-защитные группы включают, но не ограничены ими, формил; ацильные группы, например, алканоильные группы, такие как ацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы, такие как бензил (Bn), тритил (Tr) и 1,1-ди-(4'-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и подобные.
Термин «гидрокси-защитная группа» означает защитную группу, подходящую для предотвращения нежелательных реакций по гидроксигруппе. Характерные гидрокси-защитные группы включают, но не ограничены ими, алкильные группы, такие как метил, этил и трет-бутил; ацильные группы, например, алканоильные группы, такие как ацетил; арилметильные группы, такие как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и подобные.
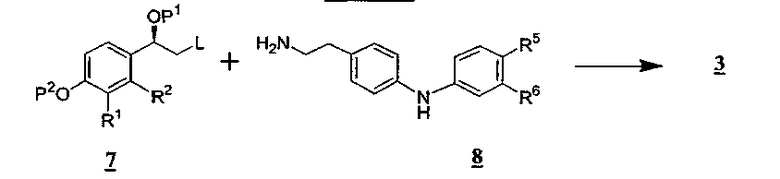
Общие методики синтеза
Соединения в соответствии с данным изобретением могут быть получены из легко доступных исходных материалов с применением представленных ниже общих способов и методик. Хотя на представленных ниже схемах проиллюстрирован определенный аспект данного изобретения, специалист в данной области техники поймет, что все аспекты данного изобретения могут быть получены с применением способов, описанных здесь, или с применением других способов, реагентов и исходных материалов, известных специалистам в данной области техники. Также должно быть понятно, что хотя даны типовые или предпочтительные условия процессов (т.е. температуры реакций, время, мольные соотношения реагентов, растворители, давление и т.д.), также могут применяться другие условия процессов, если не указано иначе. Оптимальные условия реакций могут варьироваться в зависимости от конкретных применяемых реагентов или растворителей, но такие условия могут быть определены специалистом в данной области техники обычной оптимизацией.
Кроме того, как очевидно специалисту в данной области техники, обычные защитные группы могут быть необходимы для предотвращения нежелательных реакций определенных функциональных групп. Выбор подходящей защитной группы для конкретной функциональной группы, а также подходящие условия для защиты и снятия защиты хорошо известны в данной области техники. Например, множество защитных групп и их введение и удаление описаны в T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, и представленных здесь ссылках.
В одном из способов синтеза соединения формул (I) и (II) получают, как показано на схеме А. (Заместители и переменные, показанные в представленных ниже схемах, имеют определения, данные выше, если не указано иначе.)
Схема А
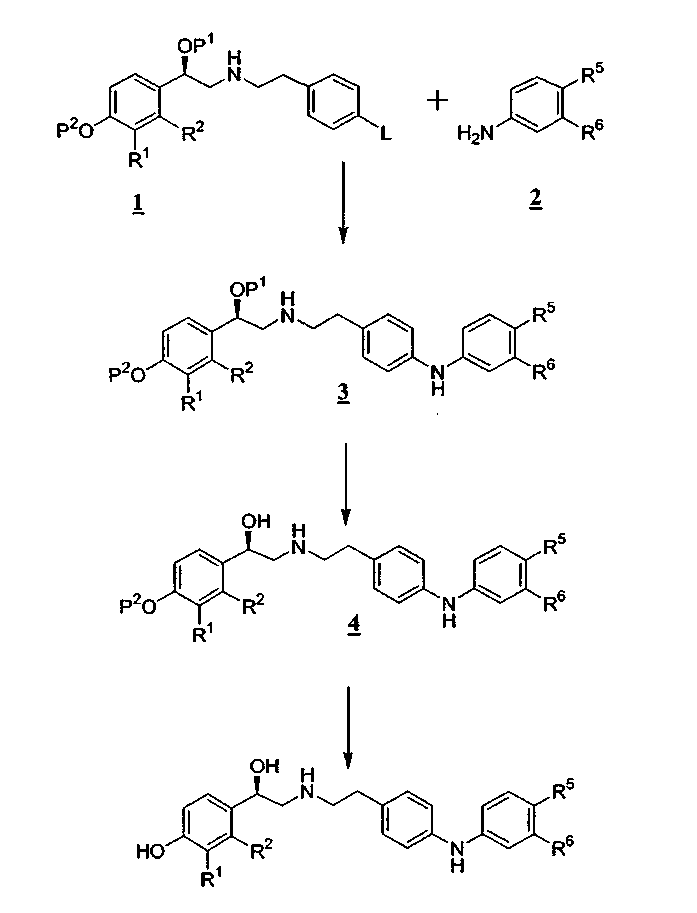
где P1 является гидрокси-защитной группой, P2 является гидрокси-защитной группой и L является уходящей группой, такой как бром.
Как показано на схеме А, соединение формулы 1 сначала подвергают взаимодействию с ариламином (2) с получением промежуточного соединения формулы 3. Обычно эту реакцию проводят в органическом растворителе в присутствии основания и катализатора на основе переходного металла и арилфосфинового лиганда при нагревании. Катализатором, применяемым для сочетания арильной группы с ариламином, является трис(дибензилиденацетон)дипалладий(0) вместе с рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтилом. Реакционную смесь обычно нагревают при температуре от около 50 до около 120°C в течение от около 0,25 до около 12 часов. Защитная группа P1 обычно является силильной защитной группой, которую обычно удаляют из промежуточного соединения формулы 3 с применением фторидного или кислотного реагента, с получением промежуточного соединения формулы 4. Защитная группа P2 обычно является бензильной защитной группой, которую обычно удаляют из промежуточного соединения формулы 4 гидрированием с применением катализатора палладий на угле, с получением продукта.
Альтернативный способ получения промежуточного соединения 3 иллюстрирован на схеме В.
Схема В
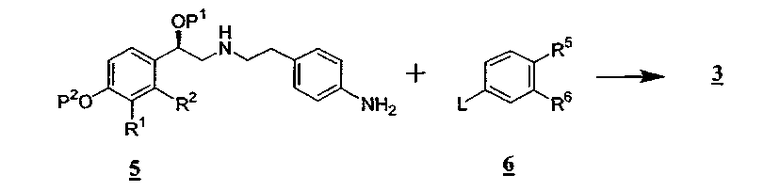
Условия сочетания промежуточных соединений 5 и 6 на схеме В для получения промежуточного соединения 3 обычно такие же, как те, которые применяются для сочетания промежуточных соединений 1 и 2 на схеме А.
Еще один альтернативный способ получения промежуточного соединения 3 иллюстрирован на схеме С.
Схема С
Реакцию на схеме С обычно проводят в полярном апротонном растворителе в присутствии основания. Типовые подходящие растворители включают диметилсульфоксид, диметилформамид, диметилацетамид и подобные. Реакционную смесь обычно нагревают при температуре от около 60 до около 140°C в течение от около 0,25 до около 4 часов.
Соединения формул 1 и 7, применяемые в реакциях, описанных в данной заявке, легко получают методами, известными в данной области техники, которые описаны, например, в патентах США № 6653323 B2 и 6670376 B1, которые включены сюда в качестве ссылок, и в представленных в них ссылках. Промежуточное соединение 5 может быть получено взаимодействием промежуточного соединения 7 с 2-(4-аминофенил)этиламином в апротонном растворителе при нагревании.
Промежуточные соединения 2 и 6 коммерчески доступны или могут быть получены из легко доступных исходных материалов. Например, если R5 является -[O-C1-6алкиленил]n-NR10R11 и R6 является водородом, промежуточное соединение 2', общей формулы 2, может быть получено методом, представленным на схеме D
Схема D
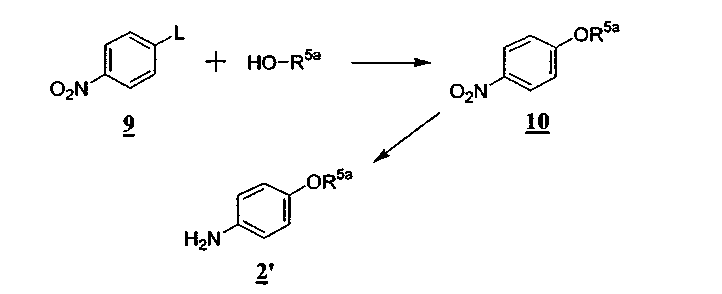
где R5a определен таким образом, что -OR5a является -[O-C1-6алкиленил]n-NR10R11. В качестве одного из примеров условий реакции, подходящих для схемы D, реакцию проводят в диметилсульфоксиде в присутствии гидрида натрия.
Если, например, R5 является C1-6алкиленил-NR12R13 и R6 является водородом, промежуточное соединение 2", общей формулы 2, может быть получено методом, представленным на схеме Е
Схема Е
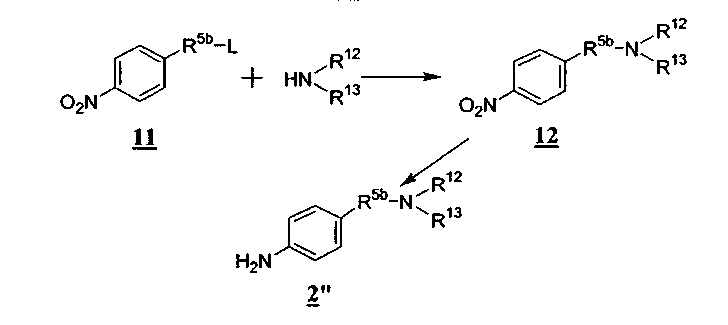
где R5b является C1-6алкиленилом.
Промежуточное соединение формулы 8 может быть получено взаимодействием промежуточного соединения формулы 2 с фенетиламином, замещенным уходящей группой в положении 4 фенильного кольца, например, 4-бромфенетиламином.
Другие подробности, касающиеся определенных условий реакции и других методик получения характерных соединений в соответствии с данным изобретением или их промежуточных соединений, описаны в представленных ниже примерах.
Следовательно, в относящемся к способу аспекте данное изобретение относится к способу получения соединения формулы (I) или его соли, или стереоизомера, или защитного производного, включающему
(a) взаимодействие
(i) соединения формулы (III):
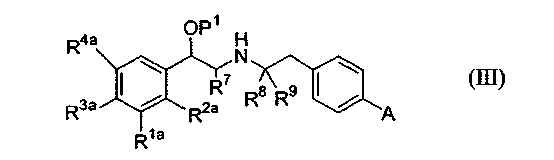
с соединением формулы (IV):
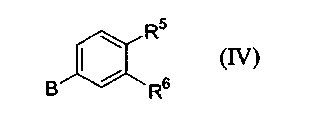
в присутствии катализатора на основе переходного металла; или
(ii) соединения формулы (V):
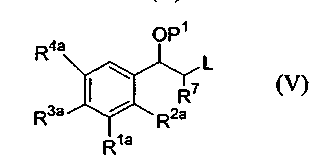
с соединением формулы (VI):
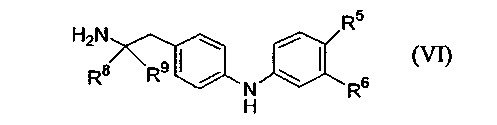
где P1 является гидрокси-защитной группой, каждый из R1a, R2a, R3a и R4a независимо либо определен так же, как R1, R2, R3 и R4 в формуле (I), либо является -OP2, где P2 является гидрокси-защитной группой; один из A и B является уходящей группой и другой из А и В является -NH2; L является уходящей группой и R5, R6, R7, R8 и R9 такие, как определены в формуле (I), с получением соединения формулы (VII):
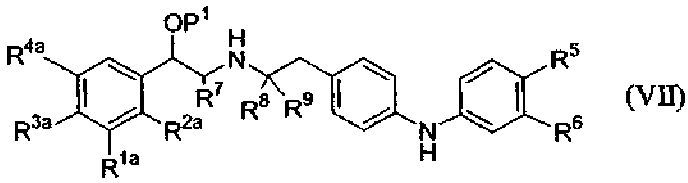
(b) удаление защитной группы P1 с получением соединения формулы (VIII):
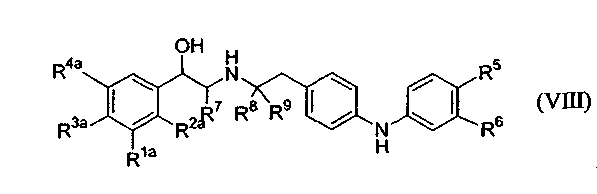
(c) если любой из R1a, R2a, R3a или R4a является -OP2, удаление защитной группы P2 с получением соединения формулы (I), или его соли, или стереоизомера.
Фармацевтические композиции
В данном изобретении также представлены фармацевтические композиции, содержащие соединение в соответствии с данным изобретением. Следовательно, соединение, предпочтительно в виде фармацевтически приемлемой соли, может быть изготовлено в любой подходящей для введения форме, такой как форма для перорального или парентерального введения или форма для введения ингаляцией.
В качестве иллюстрации, соединение может быть смешано с обычными фармацевтическими носителями и наполнителями и использовано в виде порошков, таблеток, капсул, эликсиров, суспензий, сиропов, вафлей и подобных. Такие фармацевтические композиции содержат от около 0,05 до 90 мас.% активного соединения, более предпочтительно от около 0,1 до около 30%. Фармацевтические композиции могут содержать обычные носители и наполнители, такие как кукурузный крахмал или желатин, лактозу, сульфат магния, стеарат магния, сахарозу, микрокристаллическую целлюлозу, каолин, маннит, фосфат дикальция, хлорид натрия и альгиновую кислоту. Разрыхлители, обычно применяемые в препаративных формах в соответствии с данным изобретением, включают кроскармеллозу, микрокристаллическую целлюлозу, кукурузный крахмал, гликолят натриевого крахмала и альгиновую кислоту.
Жидкая композиция обычно представляет собой суспензию, или раствор соединения, или фармацевтически приемлемой соли в подходящем жидком носителе (носителях), например, этаноле, глицерине, сорбите, неводном растворителе, таком как полиэтиленгликоль, маслах или воде, необязательно с применением суспендирующего агента, солюбилизатора (такого как циклодекстрин), консерванта, поверхностно-активного вещества, смачивающего агента, вкусовой добавки или красителя. Альтернативно, жидкая препаративная форма может быть получена из восстанавливаемого порошка.
Например, порошок, содержащий активное соединение, суспендирующий агент, сахарозу и подсластитель, может быть восстановлен водой с получением суспензии; сироп может быть получен из порошка, содержащего активный ингредиент, сахарозу и подсластитель.
Композиция в виде таблетки может быть получена с применением любого подходящего фармацевтического носителя (носителей) методами, применяемыми для получения твердых композиций. Примеры таких носителей включают стеарат магния, крахмал, лактозу, сахарозу, микрокристаллическую целлюлозу и связующие агенты, например, поливинилпирролидон. Также таблетка может иметь цветное пленочное покрытие, или краситель может быть включен как часть носителя. Кроме того, активное соединение может быть изготовлено в виде дозированной формы с контролируемым выделением, в виде таблетки, содержащей гидрофильную или гидрофобную матрицу.
Композиция в виде таблетки может быть получена с применением обычных методик инкапсулирования, например, введением активного соединения и наполнителей в твердую желатиновую капсулу. Альтернативно, полутвердая матрица из активного соединения и полиэтиленгликоля с высоким молекулярным весом может быть получена и засыпана в твердую желатиновую капсулу; или раствор активного соединения в полиэтиленгликоле или суспензия в съедобном масле, например, жидком парафине или фракционированном кокосовом масле, может быть получена и заполнена в мягкую желатиновую капсулу.
Связующие агенты для таблеток, которые могут применяться, включают аравийскую камедь, метилцеллюлозу, карбоксиметилцеллюлозу натрия, поливинилпирролидон (повидон), гидроксипропилметилцеллюлозу, сахарозу, крахмал и этилцеллюлозу. Применяемые смазывающие агенты включают стеарат магния или другие стеараты металлов, стеариновую кислоту, силиконовое масло, тальк, воски, масла и коллоидную двуокись кремния.
Также могут применяться вкусовые добавки, такие как перечная мята, масло зимолюбки, вишневый ароматизатор или подобные. Кроме того, может быть желательным добавление красителя для придания дозированной форме более привлекательного вида или для того, чтобы идентифицировать продукт.
Соединения в соответствии с данным изобретением и их фармацевтически приемлемые соли, которые являются активными при парентеральном введении, могут быть изготовлены для внутримышечного, интратекального или внутривенного введения.
Типовая композиция для внутримышечного или интратекального введения состоит из суспензии или раствора активного ингредиента в масле, например, арахисовом масле или конопляном масле. Типовая композиция для внутривенного или интратекального введения состоит из стерильного изотонического водного раствора, содержащего, например, активный ингредиент и декстрозу, или хлорид натрия, или смесь декстрозы и хлорида натрия. Другие примеры включают лактированную инъекцию Рингера, лактированную инъекцию Ригнера плюс декстроза, Normosol-M и декстрозу, Isolyte E, ацилированную инъекцию Рингера и подобные. Необязательно сорастворитель, например, полиэтиленгликоль; хелатирующий агент, например, этилендиаминтетрауксусная кислота; солюбилизирующий агент, например, циклодекстрин; и антиоксидант, например, метабисульфит натрия, могут быть включены в препаративную форму. Альтернативно, раствор может быть высушен с сублимацией и затем восстановлен подходящим растворителем непосредственно перед введением.
Соединения в соответствии с данным изобретением и их фармацевтически приемлемые соли, которые являются активными при местном введении, могут быть изготовлены в виде чрескожных композиций или устройств чрескожной доставки («пластырей»). Такие композиции включают, например, подложку, резервуар с активным соединением, контрольную мембрану, прокладку и контактный адгезив. Такие чрескожные пластыри могут применяться для обеспечения непрерывной или периодической подачи соединений в соответствии с данным изобретением в контролируемых количествах. Состав и применение чрескожных пластырей для доставки фармацевтических агентов хорошо известны в данной области техники. См., например, патент США № 5023252. Такие пластыри могут иметь конструкцию, позволяющую непрерывную, пульсирующую или «по требованию» доставку фармацевтических агентов.
Одним из предпочтительных способов введения соединения в соответствии с данным изобретением является ингаляция. Ингаляция является эффективным средством для доставки агента непосредственно в дыхательные пути. Существуют три основных типа фармацевтических ингаляционных устройств: ингаляторы с распылителем, ингаляторы сухих порошков (DPI) и дозирующие ингаляторы (MDI). Обычные ингаляторы с распылителем обеспечивают высокоскоростной поток воздуха, который позволяет распылять терапевтический агент в виде дымки, которая попадает в дыхательные пути пациента. Терапевтический агент изготовлен в виде жидкой формы, такой как раствор или суспензия тонкоизмельченных частиц вдыхаемого размера, где тонкоизмельченными обычно являются частицы, около 90% или более которых имеют диаметр менее около 10 мкм.
Типовая препаративная форма для применения в обычном распыляющем устройстве представляет собой изотонический водный раствор фармацевтической соли активного агента в концентрации активного агента от около 0,05 мкг/мл до около 1 мг/мл. Подходящие распыляющие устройства доступны коммерчески, например, от PARI GmbH (Starnberg, Germany). Другие распыляющие устройства описаны, например, в патенте США № 6123068.
DPI обычно подают терапевтический агент в виде свободнотекучего порошка, который может быть диспергирован в потоке вдыхаемого пациентом воздуха во время вдыхания. Альтернативные устройства DPI, в которых применяются внешние источники энергии для диспергирования порошка, также разработаны. Для получения свободнотекучего порошка терапевтический агент может быть изготовлен с применением подходящего наполнителя (например, лактозы или крахмала). Сухая порошковая препаративная форма может быть получена, например, объединением частиц сухой лактозы с тонкоизмельченными частицами подходящей формы, обычно фармацевтически приемлемой соли, соединения в соответствии с данным изобретением (т.е. активного агента) и сухим смешиванием. Альтернативно, агент может быть изготовлен без применения наполнителей. Препаративную форму загружают в диспенсер для сухого порошка, или в ингаляционные картриджи, или капсулы для применения в устройствах доставки сухого порошка.
Примеры устройств доставки DPI, которые являются коммерчески доступными, включают Diskhaler (GlaxoSmithKline, Research Triangle Park, NC) (см., например, патент США № 5035237); Diskus (GlaxoSmithKline) (см., например, патент США № 6378519; Turbuhaler (AstraZeneca, Wilmington, DE) (см., например, патент США № 4524769) и Rotahaler (GlaxoSmithKline) (см., например, патент США № 4353365). Другие примеры подходящих устройств DPI описаны в патентах США № 5415162, 5239993 и 5715810 и включенных в них ссылках.
MDI обычно высвобождают отмеренное количество терапевтического агента с применением сжатого газа-вытеснителя. Препаративные формы для введения посредством MDI включают раствор или суспензию активного ингредиента в сжиженном газе-вытеснителе. Хотя обычно в качестве газов-вытеснителей применяют хлорфторуглероды, такие как CCl3F, из-за негативного влияния таких агентов на озоновый слой были разработаны препаративные формы, в которых применяются гидрофторалканы (HFA), такие как 1,1,1,2-тетрафторэтан (HFA 134a) и 1,1,1,2,3,3,3,-гептафтор-н-пропан (HFA 227). Дополнительные компоненты препаративных форм с HFA для введения посредством MDI включают сорастворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как триолеат сорбитана, олеиновая кислота, лецитин и глицерин. (См., например, патент США № 5225183, EP 0717987 A2 и WO 92/22286.)
Таким образом, подходящие препаративные формы для введения посредством MDI могут включать от около 0,001% до около 2 мас.% кристаллической формы в соответствии с данным изобретением, от около 0% до около 20 мас.% этанола и от около 0% до около 5 мас.% поверхностно-активного вещества, а остаточное количество составляет газ-вытеснитель HFA. В одном варианте для получения препаративной формы охлажденный или сжатый гидрофторалкан добавляют во флакон, содержащий кристаллическую форму в соответствии с данным изобретением, этанол (если присутствует) и поверхностно-активное вещество (если присутствует). Для получения суспензии фармацевтическая соль должна быть в виде тонкоизмельченных частиц. Препаративную форму загружают в аэрозольный контейнер, который представляет собой часть устройства MDI. Примеры устройств MDI, разработанных специально для применения с газами-вытеснителями HFA, описаны в патентах США № 6006745 и 6143227.
В альтернативном способе получения суспензионную препаративную форму получают сушкой распылением оболочки из поверхностно-активного вещества на тонкоизмельченные частицы фармацевтической соли активного соединения. (См., например, WO 99/53901 и WO 00/61108.) Дополнительные примеры процессов получения вдыхаемых частиц, препаративных форм и устройств, подходящих для дозирования ингаляциями, описаны в патентах США № 6268533, 5983956, 5874063 и 6221398 и WO 99/55319 и WO 00/30614.
Должно быть понятно, что любая форма соединений в соответствии с данным изобретением (т.е. свободное основание, фармацевтическая соль или сольват), которая подходят для определенного способа введения, может применяться для фармацевтических композиций, описанных выше.
Ожидается, что активные соединения являются эффективными в большом интервале дозирования и вводятся в терапевтически эффективном количестве. Должно быть понятно, однако, что количество соединения, вводимое на практике, определяется лечащим терапевтом с учетом существующих условий, включая лечимое состояние, выбранный способ введения, конкретное вводимое соединение и его относительную активность, возраст, вес и реакцию конкретного пациента, тяжесть симптомов пациента и подобное.
Соединение может вводиться периодически: еженедельно, несколько раз в неделю, ежедневно или несколько раз в день. Режим лечения может потребовать введение в течение длительного периода времени, например, в течение нескольких недель или месяцев, или режим лечения может потребовать постоянное введение. Подходящие дозы для перорального введения в общем составляют от около 0,05 мкг/день до около 100 мг/день, предпочтительно от 0,5 до 1000 мкг/день.
Подходящие дозы терапевтических агентов для введения ингаляцией в общем составляют от около 0,05 мкг/день до около 1000 мкг/день, предпочтительно от около 0,1 мкг/день до около 500 мкг/день. Должно быть понятно, что при определении подходящей дозы для введения ингаляцией должны приниматься во внимание фракция активного агента, доставляемая в легкие, и характеристики конкретного устройства для доставки.
Среди других свойств было обнаружено, что соединения в соответствии с данным изобретением являются мощными и селективными агонистами β2 адренергического рецептора. В частности, соединения в соответствии с данным изобретением демонстрируют превосходную селективность в отношении β2 адренергического рецептора по сравнению с β1 и β3 адренергическими рецепторами. Более того, было обнаружено, что соединения в соответствии с данным изобретением обладают удивительной и неожиданной продолжительностью действия. Как описано в представленных ниже биологических исследованиях, соединения в соответствии с данным изобретением демонстрируют продолжительность действия более 24 часов на животной модели бронхопротекции.
Таким образом, в данном изобретении представлен способ лечения заболевания или состояния у млекопитающего, связанного с активностью β2 адренергического рецептора, включающий введение млекопитающему терапевтически эффективного количества соединения в соответствии с данным изобретением или фармацевтической композиции, включающей соединение в соответствии с данным изобретением.
Активные агенты в соответствии с данным изобретением также могут применяться как часть комбинации, включающей, кроме того, один или несколько других терапевтических агентов. Например, агенты в соответствии с данным изобретением могут вводиться вместе с одним или несколькими терапевтическими агентами, выбранными из противовоспалительных агентов (например, кортикостероидов и нестероидных противовоспалительных агентов (НСПВС), антихолинергических агентов (в частности, антагонистов мускаринового рецептора), других агонистов β2 адренергического рецептора, противоинфекционных агентов (например, антибиотиков или антивирусных средств) или антигистаминов. Следовательно, в другом аспекте данное изобретение относится к комбинации, включающей соединение в соответствии с данным изобретением и один или несколько терапевтических агентов, например, противовоспалительный агент, антихолинергический агент, другой агонист β2 адренергического рецептора, противоинфекционный агент или антигистамин.
Другие терапевтические агенты могут применяться в виде фармацевтически приемлемых солей или сольватов. Если применимо, другие терапевтические агенты могут применяться в виде оптически чистых стереоизомеров.
Подходящие противовоспалительные агенты включают кортикостероиды и НСПВС. Подходящие кортикостероиды, которые могут применяться в сочетании с соединениями в соответствии с данным изобретением, включают такие пероральные и ингалируемые кортикостероиды и их пролекарства, которые обладают противовоспалительным действием. Примеры включают метилпреднизолон, преднизолон, дексаметазон, флутиказон пропионат, 3-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил) окси]-11(3-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-тиокарбоновой кислоты, S-(2-оксотетрагидрофуран-3S-иловый) эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-пропионилоксиандроста-1,4-диен-17β-тиокарбоновой кислоты, сложные эфиры беклометазона (например, 17-пропионат или 17,21-дипропионат), будезонид, флунизолид, сложные эфиры мометазона (например, фуроат), ацетонид триамцинолона, рофлепонид, циклезонид, пропионат бутиксокорта, RPR-106541 и ST-126. Предпочтительные кортикостероиды включают пропионат флутиказона, S-фторметиловый эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-17α-[(4-метил-1,3-тиазол-5-карбонил)окси]-3-оксоандроста-1,4-диен-17β-тиокарбоновой кислоты и S-фторметиловый эфир 6α,9α-дифтор-17α- [(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-тиокарбоновой кислоты, более предпочтительно, 3-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-тиокарбоновой кислоты.
Подходящие НСПВС включают кромогликат натрия; недокромил натрия; ингибиторы фосфодиэстеразы (PDE) (например, теофиллин, ингибиторы PDE4 или смешанные ингибиторы PDE3/PDE4); антагонисты лейкотриена (например, монтелейкаст); ингибиторы синтеза лейкотриена; ингибиторы iNOS; ингибиторы протеазы, такие как ингибиторы триптазы и эластазы; антагонисты бета-2 интегрина и агонисты или антагонисты рецептора аденозина (например, агонисты аденозина 2а); антагонисты цитокина (например, антагонисты хемокина, такие как антитело интерлейкина (антитело αIL), особенно терапия αIL-4, терапия αIL-13 или их сочетание); или ингибиторы синтеза цитокина. Другие подходящие агонисты β2-адренорецептора включают салметерол (например, в виде ксинафоата), салбутамол (например, в виде сульфата или свободного основания), формотерол (например, в виде фумарата), фенотерол или тербуталин и их соли.
Также представляет интерес применение активного агента в соответствии с данным изобретением в комбинации с ингибитором фосфодиэстеразы 4 (PDE4) или смешанным ингибитором PDE3/PDE4. Характерные ингибиторы фосфодиэстеразы-4 (PDE4) или смешанные ингибиторы PDE3/PDE4 включают, но не ограничены ими, цис 4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту, 2-карбометокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он; цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол]; цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновую кислоту и подобные или их фармацевтически приемлемые соли. Другие характерные ингибиторы PDE4 или смешанные ингибиторы PDE4/PDE3 включают AWD-12-281 (elbion); NCS-613 (INSERM); D-4418 (Chiroscience и Schering-Plough); CI-1018 или PD-168787 (Pfizer); соединения бензодиоксола, описанные в WO 99/16766 (Kyowa Hakko); K-34 (Kyowa Hakko); V-11294A (Napp); рофлумиласт (Byk-Gulden); соединения фталазинона, описанные в WO 99/47505 (Byk-Gulden); Пумафентрин (Byk-Gulden, в настоящее время Altana); арофиллин (Almirall-Prodesfarma); VM554/UM565 (Vernalis); T-440 (Tanabe Seiyaku) и T2585 (Tanabe Seiyaku).
Подходящие антихолинергические агенты включают такие соединения, которые действуют как антагонисты мускаринового рецептора, в частности, такие соединения, которые являются антагонистами M1, M2 или M3 рецепторов, или их сочетания. Примеры соединений включают алкалоиды белладонны типа атропина, скополамина, гоматропина, гиосциамина; эти соединения обычно вводят в виде соли, являющейся солью третичных аминов. Эти лекарственные средства, предпочтительно в виде солей, легко доступны из множества коммерческих источников или могут быть получены с помощью данных, представленных в литературе, через:
Атропин - CAS-51-55-8 или CAS-51-48-1 (безводная форма), сульфат атропина - CAS-5908-99-6; оксид атропина - CAS-4438-22-6 или его HCl соль - CAS-4574-60-1 и нитрат метилатропина - CAS-52-88-0.
Гоматропин - CAS-87-00-3, гидрохлорид - CAS-51-56-9, метилбромид - CAS-80-49-9.
Гиосциамин (d, l) - CAS-101-31-5, гидробромид - CAS-306-03-6 и сульфат - CAS-6835-16-1.
Скополамин - CAS-51-34-3, гидробромид - CAS-6533-68-2, метилбромид - CAS-155-41-9.
Предпочтительные антихолинергические агенты включают ипратропиум (например, в виде бромида), торговое наименование Atrovent, окситропиум (например, в виде бромида) и тиотропиум (например, в виде бромида) (CAS-139404-48-1). Также интересны метантелин (CAS-53-46-3), бромид пропантелина (CAS-50-34-9), метилбромид анизотропина или Valpin 50 (CAS-80-50-2), бромид клидиния (Quarzan, CAS-3485-62-9), копирролат (Robinul), йодид изопропамида (CAS-71-81-8), бромид мепензолата (патент США 2918408), хлорид тридигексетила (Pathilone, CAS-4310-35-4) и метилсульфат гексоциклия (Tral, CAS-115-63-9). См. также гидрохлорид циклопентолата (CAS-5870-29-1), тропикамид (CAS-1508-75-4), гидрохлорид тригексифенидила (CAS-144-11-6), пирензепин (CAS-29868-97-1), телензепин (CAS-80880-90-9), AF-DX 116 или метоктрамин и соединения, описанные в WO 01/04118, описание которых включено сюда в качестве ссылок.
Подходящие антигистамины (также обозначенные как антагонисты H1-рецептора) включают один или несколько из множества известных антагонистов, которые ингибируют H1-рецепторы и безопасны для применения у человека. Все они являются обратимыми конкурентными ингибиторами взаимодействия гистамина с H1-рецепторами. Большинство этих ингибиторов, в основном антагонисты первого поколения, характеризуются на основе их основных структур как этаноламины, этилендиамины и алкиламины. Кроме того, другие антигистамины первого поколения включают те, которые могут быть охарактеризованы на основе пиперазина и фенотиазинов. Антагонисты второго поколения, которые не являются седативными, имеют подобное взаимоотношение структура-активность в том, что они сохраняют основную этиленовую группу (алкиламины) или имитируют группу третичного амина с пиперизином или пиперидином. Примеры антагонистов включают
Этаноламины: малеат карбиноксамина, фумарат клемастина, гидрохлорид дифенилгидрамина и дименгидринат.
Этилендиамины: малеат пириламина, HCl трипеленнамина и цитрат трипеленнамина.
Алкиламины: хлорфенирамин и его соли, такие как малеат, и акривастин.
Пиперазины: HCl гидроксизина, памоат гидроксизина, HCl циклизина, лактат циклизина, HCl меклизина и HCl цетиризина.
Пиперидины: астемизол, HCl левокабастина, лоратадин и его дескарбоэтокси-аналог и гидрохлорид терфенадина и фексофенадина или другие фармацевтически приемлемые соли.
Гидрохлорид азеластина является еще одним антагонистом H1-рецептора, который может применяться в сочетании с соединением в соответствии с данным изобретением.
Примеры предпочтительных антигистаминов включают метапирилен и лоратадин.
Таким образом, в другом аспекте данное изобретение относится к комбинации, содержащей соединение формулы (I), или его фармацевтически приемлемую соль или сольват, или стереоизомер и кортикостероид. В частности, данное изобретение относится к комбинации, в которой кортикостероидом является пропионат флутиказона или в которой кортикостероидом является S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-тиокарбоновой кислоты или S-(2-оксотетрагидрофуран-3S-иловый) эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-пропионилоксиандроста-1,4-диен-17β-тиокарбоновой кислоты.
Таким образом, в другом аспекте данное изобретение относится к комбинации, содержащей соединение формулы (I), или его фармацевтически приемлемую соль, или сольват, или стереоизомер и ингибитор PDE4.
Таким образом, в другом аспекте данное изобретение относится к комбинации, содержащей соединение формулы (I), или его фармацевтически приемлемую соль, или сольват, или стереоизомер и антихолинергический агент.
Таким образом, в другом аспекте данное изобретение относится к комбинации, содержащей соединение формулы (I), или его фармацевтически приемлемую соль, или сольват, или стереоизомер и антигистамин.
Таким образом, в другом аспекте данное изобретение относится к комбинации, содержащей соединение формулы (I), или его фармацевтически приемлемую соль, или сольват, или стереоизомер, вместе с ингибитором PDE4 и кортикостероидом.
Таким образом, в другом аспекте данное изобретение относится к комбинации, содержащей соединение формулы (I), или его фармацевтически приемлемую соль, или сольват, или стереоизомер, вместе с антихолинергическим агентом и кортикостероидом.
В представленных выше сочетаниях термин «соединение формулы (I)» включает соединение формулы (II) и его предпочтительные группы и любое отдельно описанное соединение или соединения.
Следовательно, фармацевтические композиции в соответствии с данным изобретением могут необязательно содержать комбинации соединения формулы (I), или его фармацевтически приемлемой соли, или сольвата, или стереоизомера с одним или более другим терапевтическим агентом, как описано выше.
Отдельные соединения сочетаний в соответствии с данным изобретением могут быть изготовлены отдельно или изготовлены вместе в единой фармацевтической композиции. Отдельные соединения могут вводиться последовательно или одновременно, отдельными или объединенными фармацевтическими композициями. Подходящие дозы известных терапевтических агентов легко распознаются специалистами в данной области техники. Способы лечения в соответствии с данным изобретением поэтому включают введение отдельных соединений таких сочетаний либо последовательно, либо одновременно, отдельными или объединенными фармацевтическими композициями.
Таким образом, согласно другому аспекту данное изобретение относится к способу лечения заболевания или состояния, связанного с активностью β2 адренергического рецептора у млекопитающих, включающему введение млекопитающему терапевтически эффективного количества сочетания соединения формулы (I), или его фармацевтически приемлемой соли, или сольвата, или стереоизомера и одного или нескольких терапевтических агентов.
Так как соединения в соответствии с данным изобретением являются агонистами β2 адренергического рецептора, такие соединения также применяются в качестве инструментов для проведения исследований или изучения биологических систем или образцов, содержащих β2 адренергические рецепторы, или для открытия новых агонистов β2 адренергического рецептора. Более того, так как соединения в соответствии с данным изобретением обладают селективностью в отношении β2 адренергического рецептора по сравнению со связыванием и функциональной активностью с другими рецепторами β адренергических подтипов, такие соединения могут применяться для изучения эффектов селективного агонизма β2 адренергических рецепторов в биологической системе или образце. В таких исследованиях могут применяться любые подходящие биологические системы или образцы, содержащие β2 адренергические рецепторы, и такие исследования могут проводиться in vitro или in vivo. Характерные биологические системы или образцы, подходящие для таких исследований, включают, но не ограничены ими, экстракты клеток, мембраны плазмы, образцы тканей, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.) и подобные.
Биологическую систему или образец, содержащие β2 адренергический рецептор, подвергают контакту с оказывающим агонистическое воздействие на β2 адренергический рецептор количеством соединения в соответствии с данным изобретением. Эффективность агонистического воздействия на β2 адренергический рецептор определяют с помощью обычных методик и оборудования, таких как применяются для анализа радиолигандного связывания и функционального анализа, например, анализа опосредованных лигандом изменений во внутриклеточном циклическом монофосфате аденозина (cAMP), описанного ниже, или исследований сходного типа. Оказывающее агонистическое воздействие на β2 адренергический рецептор количество обычно составляет от около 1 наномоля до около 1000 наномолей. Если соединения в соответствии с данным изобретением применяют в качестве инструментов исследования для обнаружения новых агонистов β2 адренергического рецептора, данное изобретение также включает, как отдельные варианты, сбор сравнительных данных (с применением соответствующих анализов) и анализ данных тестирования для идентификации целевых тестируемых соединений.
Представленные ниже неограничивающие примеры иллюстрируют характерные фармацевтические композиции в соответствии с данным изобретением. Дополнительные подходящие носители для препаративных форм активных соединений в соответствии с данным изобретением представлены в Remington: The Science and Practice of Pharmacy, 20 th Edition, Lippincott Williams & Wilkins, Philadelphia, PA, 2000.
Пример препаративной формы А
Этот пример иллюстрирует получение характерной фармацевтической композиции для перорального введения соединения в соответствии с данным изобретением:
Указанные выше ингредиенты смешивают и засыпают в твердые желатиновые капсулы.
Пример препаративной формы В
Этот пример иллюстрирует получение другой характерной фармацевтической композиции для перорального введения соединения в соответствии с данным изобретением:
Указанные выше ингредиенты тщательно смешивают и прессуют в отдельные рифленые таблетки.
Пример препаративной формы С
Этот пример иллюстрирует получение характерной фармацевтической композиции для перорального введения соединения в соответствии с данным изобретением.
Пероральную суспензию готовят из следующей композиции.
Пример препаративной формы D
Этот пример иллюстрирует получение характерной фармацевтической композиции, содержащей соединение в соответствии с данным изобретением.
Препаративную форму для инъекций, буферизованную до pH 4, готовят из следующей композиции:
Пример препаративной формы Е
Этот пример иллюстрирует получение характерной фармацевтической композиции для инъекций соединения в соответствии с данным изобретением.
Восстанавливаемый раствор готовят добавлением 20 мл стерильной воды к 1 мг соединения в соответствии с данным изобретением. Перед применением раствор разбавляют 200 мл жидкости для внутривенного вливания, которая совместима с активным соединением. Такие жидкости выбирают из 5% раствора декстрозы, 0,9% хлорида натрия или смеси 5% декстрозы и 0,9% хлорида натрия. Другие примеры включают лактированный раствор для инъекций Рингера плюс 5% декстроза, Normosol-M и 5% декстроза, Isolyte E и ацилированный раствор для инъекций Рингера.
Пример препаративной формы F
Этот пример иллюстрирует получение характерной фармацевтической композиции для местного введения соединения в соответствии с данным изобретением.
Все указанные выше ингредиенты, за исключением воды, объединяют и нагревают до температуры 60°С при перемешивании. Затем добавляют достаточное для эмульгирования ингредиентов количество воды при температуре 60°С при энергичном перемешивании, затем добавляют воду до 100 г.
Пример препаративной формы G
Этот пример иллюстрирует получение характерной фармацевтической композиции, содержащей соединение в соответствии с данным изобретением.
Водную аэрозольную препаративную форму для применения в распылителе готовят растворением 0,1 мг фармацевтической соли активного соединения в 0,9% растворе хлорида натрия, подкисленном лимонной кислотой. Смесь перемешивают и обрабатывают ультразвуком до растворения активной соли. pH раствора доводят до значения в интервале от 3 до 8 медленным добавлением NaOH.
Пример препаративной формы Н
Этот пример иллюстрирует получение сухой порошковой препаративной формы, содержащей соединение в соответствии с данным изобретением.
Желатиновые картриджи для ингаляций заполняют фармацевтической композицией, содержащей следующие ингредиенты:
Фармацевтическую соль активного соединения тонко измельчают перед смешиванием с лактозой. Содержимое картриджей вводят с применением порошкового ингалятора.
Пример препаративной формы I
Этот пример иллюстрирует получение сухой порошковой препаративной формы, содержащей соединение в соответствии с данным изобретением, для применения в устройстве для ингаляции сухого порошка.
Готовят фармацевтическую композицию, имеющую композиционное объемное соотношение тонкоизмельченной фармацевтической соли к лактозе 1:200. Композицию пакуют в устройство для ингаляции сухого порошка, которое выдает от около 10 мкг до около 100 мкг активного лекарственного средства на дозу.
Пример препаративной формы J
Этот пример иллюстрирует получение препаративной формы, содержащей соединение в соответствии с данным изобретением, для применения в дозирующем ингаляторе.
Суспензию, содержащую 5% фармацевтической соли активного соединения, 0,5% лецитина и 0,5% трегалозы готовят диспегированием 5 г активного соединения в виде тонкоизмельченных частиц со средним размером менее 10 мкм в коллоидном растворе, полученном из 0,5 г трегалозы и 0,5 г лецитина, растворенного в 100 мл деминерализованной воды. Суспензию сушат распылением и полученный продукт тонко измельчают до частиц, имеющих средний диаметр менее 1,5 мкм. Частицы загружают в баллоны со сжатым 1,1,1,2-тетрафторэтаном.
Пример препаративной формы К
Этот пример иллюстрирует получение препаративной формы, содержащей соединение в соответствии с данным изобретением, для применения в дозирующем ингаляторе.
Суспензию, содержащую 5% фармацевтическую соль активного соединения и 0,1% лецитина, готовят диспегированием 10 г активного соединения в виде тонкоизмельченных частиц со средним размером менее 10 мкм в растворе, полученном из 0,1 г лецитина, растворенного в 200 мл деминерализованной воды. Суспензию сушат распылением и полученный продукт тонко измельчают до частиц, имеющих средний диаметр менее 1,5 мкм. Частицы загружают в баллоны со сжатым 1,1,1,2,3,3,3-гептафтор-н-пропаном.
Биологические анализы
Соединения в соответствии с данным изобретением и их фармацевтически приемлемые соли обладают биологической активностью и применяются для медикаментозного лечения. Способность соединения связываться с β2 адренергическим рецептором, а также его селективность, мощность в качестве агониста и свойственная ему активность могут быть продемонстрированы в представленных ниже тестах А-В или могут быть продемонстрированы с применением других тестов, известных в данной области техники.
Аббревиатуры
Получение мембран из клеток, экспрессирующих
человеческие β 1 или β 2 адренергические рецепторы
Колонии клеток, полученные из HEK-293, стабильно экспрессирующие клонированные человеческие β1 или β2 адренергические рецепторы, выращивают практически до конфлюэнтности в DMEM с 10% диализированной ЭБС в присутствии 500 мкг/мл Генетицина. Монослой клеток поднимают с применением Versene 1:5000 (0,2 г/л ЭДТК в ФРФБ) с применением скальпеля для клеток. Клетки осаждают центрифугированием при 1000 об/мин и осажденные клетки либо хранят замороженными при температуре -80°С, либо сразу же получают мембраны. Для получения осажденные клетки повторно суспендируют в лизирующем буфере (10 мM Tris/HCL pH 7,4 @ 4°C, одна таблетка “Complete Protease Inhibitor Cocktail Tablets with 2 мM EDTA” на 50 мл буфера (Roche кат.№ 1697498, Roche Molecular Biochemicals, Indianapolis, IN)) и гомогенизируют с применением плотно подогнанного стеклянного гомогенизатора Dounce (20 тактов) на льду. Гомогенат центрифугируют при 20000 Ч g, осадок промывают одиножды лизирующим буфером путем повторного суспендирования и центрифугируют, как указано выше. Конечный осадок повторно суспендируют в мембранном буфере (75 мM Tris/HCl pH 7,4, 12,5 мM MgCl2, 1 мM ЭДТК @ 25°C). Концентрацию белка в суспензии мембран определяют методом Брэдфорда (Bradford MM., Analytical Biochemistry, 1976, 72, 248-54). Мембраны хранят замороженными в аликвотах при температуре -80°C.
Тест A
Анализ связывания радиолиганда с человеческими β 1 и β 2 адренергическими рецепторами
Анализ связывания проводят в 96-ячеечных микротитровальных планшетах при общем тестируемом объеме 100 мкл с 5 мкг белка мембраны для мембран, содержащих человеческий β2 адренергический рецептор, или 2,5 мкг белка мембраны для мембран, содержащих человеческий β1 адренергический рецептор, в буфере для анализа (75 мM Tris/HCl pH 7,4 @ 25°C, 12,5 мM MgCl2, 1 мM ЭДТК, 0,2% АБС). Исследование связывания по насыщенности для определения значений Kd радиолиганда проводят с применением [3H]дигидроалпренолола (NET-720, 100 Ки/ммоль, PerkinElmer Life Sciences Inc., Boston, MA) в 10 различных концентрациях от 0,01 нM до 200 нM. Анализ замещения для определения значений pKi соединений проводят с [3H]дигидроалпренололом при 1 нM и 10 различных концентрациях соединения от 40 пM до 10 мкM. Соединения растворяют до концентрации 10 мM в буфере для растворения (25 мM Gly-HCl pH 3,0 с 50% ДМСО), затем разводят до 1 мM в 50 мM Gly-HCl pH 3,0 и затем периодически разводят в буфере для исследований. Неспецифическое связывание определяют в присутствии 10 мкм немеченого алпренолола. Тестируемые образцы инкубируют в течение 90 минут при комнатной температуре, реакции связывания останавливают быстрой фильтрацией через GF/B фильтровальные планшеты из стекловолокна (Packard BioScience Co., Meriden, CT), предварительно пропитанные 0,3% полиэтиленимином. Фильтровальные планшеты промывают трижды фильтрационным буфером (75 мM Tris/HCl pH 7,4 @ 4°C, 12,5 мM MgCl2, 1 мM ЭДТК) для удаления несвязанной радиоактивности. Планшеты сушат, добавляют 50 мкл Microscint-20 сцинтилляционной жидкости (Packard BioScience Co., Meriden, CT) и планшеты считывают в жидкостном сцинтилляционном счетчике Packard Topcount (Packard BioScience Co., Meriden, CT). Данные связывания анализируют с помощью анализа нелинейной регрессии с помощью комплекта GraphPad Prism Software (GraphPad Software, Inc., San Diego, CA) с применением 3-мерной модели для одноместного сравнения. Минимум кривой фиксируют как значение неспецифического связывания, определенного в присутствии 10 мкM алпренолола. Значения Ki для соединений рассчитывают из полученных значений IC50 и значения Kd радиолиганда с применением уравнения Cheng-Prusoff (Cheng Y, and Prusoff WH., Biochemical Pharmacology, 1973, 22, 23, 3099-108). Селективность подтипа рецептора рассчитывают как соотношение Ki(β1)/Ki(β2). Соединения в соответствии с данным изобретением демонстрируют большее связывание с β2 адренергическим рецептором по сравнению с β1 адренергическим рецептором, т.е. Ki(β1) > Ki(β2) с селективностью более около 30.
Тест B
Анализ суммарного цАМФ на Flashplate с применением колоний клеток, гетерологически экспрессирующих человеческий β 1 адренорецептор, β 2 адренорецептор и β 3 адренорецептор соответственно
Колонию клеток HEK-293, стабильно экспрессирующих клонированный человеческий β1 адренергический рецептор (клон H34.1), выращивают до около 70-90% конфлюэнтности в среде, состоящей из DMEM с добавлением 10% ЭБС и 500 мкг/мл Генецитина. Колонию клеток HEK-293, стабильно экспрессирующих клонированный человеческий β2-адренорецептор (клон H24.14), выращивают в той же среде до полной конфлюэнтности. Колонию клеток СНО-K1, стабильно экспрессирующих клонированный человеческий β3-адренорецептор, выращивают до около 70-90% конфлюэнтности в среде Ham F-12 с добавлением 10% ЭБС и 800 мкг/мл Генецитина, добавляемых каждый пятый пассаж. За день до исследования культуры переносят в ту же среду для выращивания без добавления антибиотиков.
Анализ цАМФ проводят в формате радиоиммунологического исследования с применением системы Flashplate Adenylyl Cyclase Activation Assay System с 125I-цАМФ (NEN SMP004, PerkinElmer Life Sciences Inc., Boston, MA) согласно инструкциям производителя.
В день исследования клетки одиножды промывают ФРФБ, поднимают добавлением Versene 1:5000 (0,2 г/л ЭДТК в ФРФБ) и подсчитывают. Клетки осаждают центрифугированием при 1000 об/мин и повторно суспендируют в стимулирующем буфере, предварительно нагретом до температуры 37°С. Для клеток, экспрессирующих β1-адренорецептор, 10 нМ ICI 118551 добавляют в стимулирующий буфер и клетки инкубируют в течение 10 мин при температуре 37°С. Клетки применяют в конечных концентрациях 30000, 40000 и 70000 клеток/ячейку для клеток, экспрессирующих β1-адренорецептор, β2-адренорецептор и β3-адренорецептор соответственно. Соединения растворяют до концентрации 10 мМ в ДМСО, затем разводят до 1 мМ в 50 мМ Gly-HCl pH 3,0 и затем периодически разводят в буфере для исследований (75 мМ Tris/HCl pH 7,4 @ 25°C, 12,5 мМ MgCl2, 1 мМ ЭДТК, 0,2% АБС). Соединения тестируют при 11 различных концентрациях, от 10 мкм до 9,5 пМ. Реакционные смеси инкубируют в течение 10 мин при температуре 37°С и реакции останавливают добавлением 100 мкл ледяного буфера для определения. Планшеты герметично закрывают, инкубируют в течение ночи при температуре 4°С и подсчитывают на следующее утро на сцинтилляционном счетчике с функцией «topcount» (Packard BioScience Co., Meriden, CT). Количество цАМФ, произведенной на миллилитр реакционной смеси, рассчитывают на основе подсчетов, выполненных для образцов и стандартов цАМФ, как описано производителем в инструкции для пользователя. Данные анализируют с помощью анализа нелинейной регрессии с применением программного обеспечения GraphPad Prism Software (GraphPad Software, Inc., San Diego, CA), используя 3-параметровую модель для сигмоидальной кривой доза-реакция (угол наклона Хилла = 1). Мощность агониста выражают в значениях pEC50.
Соединения в соответствии с данным изобретением в данном анализе демонстрируют мощную активность в отношении β2 адренергического рецептора, что подтверждается значениями pEC50 более около 9. Кроме того, тестируемые соединения демонстрируют селективность в функциональной активности по отношению к β2 рецептору по сравнению с функциональной активностью по отношению к β1 and β3 рецепторам. В частности, соединения в соответствии с данным изобретением демонстрируют EC50(β1)/EC50(β2) соотношения более чем около 10 и EC50(β3)/EC50(β2) соотношения более чем около 50.
Тест C
Анализ суммарного цАМФ на Flashplate с применением колоний клеток эпителия легких, гетерологически экспрессирующих человеческий β 2 адренорецептор
Для определения мощности и эффективности агониста (внутренней активности) в колонии клеток, экспрессирующих эндогенные уровни β2 адренергического рецептора, применяют колонию клеток эпителия легких (BEAS-2B) (ATCC CRL-9609, American Type Culture Collection, Manassas, VA) (January B, et al., British Journal of Pharmacology, 1998, 123, 4, 701-11). Клетки выращивают до 75-90% конфлюэнтности в полной, не содержащей сыворотки среде (LHC-9 MEDIUM, содержащей эпинефрин и ретиноевую кислоту, кат. № 181-500, Biosource International, Camarillo, CA). За день до исследования среду меняют на LHC-8 (без добавления эфедрина или ретиноевой кислоты, кат. № 141-500, Biosource International, Camarillo, CA).
Анализ цАМФ проводят в формате радиоиммунологического исследования с применением системы Flashplate Adenylyl Cyclase Activation Assay System с 125I-цАМФ (NEN SMP004, PerkinElmer Life Sciences Inc., Boston, MA) согласно инструкции производителя.
В день исследования клетки одиножды промывают ФРФБ, поднимают добавлением 5 мМ ЭДТК в ФРФБ и подсчитывают. Клетки осаждают центрифугированием при 1000 об/мин и повторно суспендируют в стимулирующем буфере, предварительно нагретом до температуры 37°С, в конечной концентрации 600000 клеток/мл. Клетки применяют в исследовании в конечной концентрации 30000. Соединения растворяют до концентрации 10 мМ в буфере для растворения (25 мM Gly-HCl, pH 3,0, с 50% ДМСО), затем разводят до 1 мМ в 50 мМ Gly-HCl, pH 3,0, и затем периодически разводят в буфере для исследований (75 мМ Tris/HCl, pH 7,4, @ 25°C, 12,5 мМ MgCl2, 1 мМ ЭДТК, 0,2% АБС).
Соединения тестируют при 10 различных концентрациях, от 10 мкМ до 40 пМ. Максимальную реакцию определяют в присутствии 10 мкм изопротеренола. Реакционные смесь инкубируют в течение 10 мин при температуре 37°С и реакции останавливают добавлением 100 мкл ледяного буфера. Планшеты герметично закрывают, инкубируют в течение ночи при температуре 4°С и подсчитывают на следующее утро на сцинтилляционном счетчике с функцией «topcount» (Packard BioScience Co., Meriden, CT). Количество цАМФ, произведенной на миллилитр реакционной смеси, рассчитывают на основе подсчетов, выполненных для образцов и стандартов цАМФ, как описано в инструкции пользователя производителя. Данные анализируют с помощью анализа нелинейной регрессии с применением программного обеспечения GraphPad Prism Software (GraphPad Software, Inc., San Diego, CA), используя 4-параметровую модель для сигмоидальной кривой доза-реакция с изменяющимся углом наклона. Соединения в соответствии с данным изобретением, тестированные в данном анализе, демонстрируют значения pEC50 более около 8.
Эффективность соединений (%эфф.) рассчитывают из соотношения полученного Emax (ВЕРХ выравнивающей кривой) и максимальной реакции, полученной для 10 мкM изопротеренола и выражают как %эфф. по отношению к изопротеренолу. Тестированные соединения демонстрируют %эфф. более около 50.
Тест D
Анализ бронхопротекции против вызванного ацетилхолином бронхоспазма на модели морских свинок
Группы из 6 самцов морских свинок (Duncan-Hartley (HsdPoc:DH) Harlan, Madison, WI), весящих от 250 до 350 г, индивидуально идентифицируют карточками на клетках. Во время исследования животным обеспечивают доступ к пище и воде без ограничения.
Тестируемые соединения вводят ингаляцией в течение 10 минут в дозирующей камере для обработки всего тела (R&S Molds, San Carlos, CA). Дозирующие камеры размещают таким образом, чтобы аэрозоль подавался одновременно в 6 отдельных камер из центрального коллектора. После 60-минутного периода аккумулирования и 10-минутного периода обработки распыленной водой для инъекций (ВДИ) морских свинок обрабатывают аэрозолем тестируемого соединения или носителя (ВДИ). Эти аэрозоли получают из водных растворов с применением LC Star Nebulizer Set (Model 22F51, PARI Respiratory Equipment, Inc. Midlothian, VA), подаваемых с помощью смеси газов (CO2 = 5%, O2 = 21% и N2 = 74%) под давлением 22 ф/д2. Поток газа через распылитель при указанном рабочем давлении составляет приблизительно 3 л/минуту. Полученные аэрозоли подают в камеры положительным давлением. Во время подачи аэрозольных растворов воздух для разбавления не применяют. Во время 10-минутного распыления распыляется приблизительно 1,8 мл раствора. Это количество получают гравиметрически, сравнением весов заполненного распылителя до и после распыления.
Бронхозащитное действие соединений, вводимых ингаляцией, оценивают с помощью плетизмографии всего тела через 1,5, 24, 48 и 72 часа после дозирования. За сорок пять минут до начала оценки легких каждую морскую свинку анестезируют внутримышечной инъекцией кетамина (43,75 мг/кг), ксилазина (3,50 мг/кг) и ацепромазина (1,05 мг/кг). После того, как место разреза выбрито и очищено 70% спиртом, делают 2-5 см срединный разрез вентральной части шеи. Затем выделяют яремную вену и катетеризуют наполненным физиологическим раствором полиэтиленовым катетером (PE-50, Becton Dickinson, Sparks, MD) для обеспечения внутривенного вливания 0,1 мг/мл раствора ацетилхолина (Ach) (Sigma-Aldrich, St. Louis, MO) в физиологическом растворе. Затем свободно вскрывают трахею и катетеризуют 14G тефлоновой трубкой (№ NE-014, Small Parts, Miami Lakes, FL). При необходимости анестезию поддерживают дополнительными внутримышечными инъекциями указанного выше анестезирующего коктейля. Глубину анестезии контролируют и корректируют, если животное начинает поджимать лапы или скорость дыхания составляет более 100 вдохов/минуту.
После завершения катетеризации животных помещают в плетизмограф (#PLY3114, Buxco Electronics, Inc., Sharon, CT) и вставляют пищеводный катетер под давлением для измерения легочного вытесняющего давления (давления). Трахеальную тефлоновую трубку присоединяют к отверстию плетизмографа для того, чтобы морская свинка могла дышать комнатным воздухом, находящимся вне камеры. Затем камеру герметично закрывают. Для поддержания температуры тела применяют нагревательную лампу и легкие морских свинок 3 раза наполняют 4 мл воздуха с применением 10 мл калиброванного шприца (#5520 Series, Hans Rudolph, Kansas City, MO) для того, чтобы не повредить нижние дыхательные пути, и для того, чтобы животное не пострадало от гипервентиляции.
Как только определено, что базовые значения находятся в интервале 0,3-0,9 мл/см H2O для коэффициента податливости, и в интервале 0,1-0,199 см H2O/мл в секунду для сопротивления, начинают легочную оценку. Компьютерная программа Buxco для легочных измерений обеспечивает сбор и дальнейшую обработку полученных в легких значений. Запуск этой программы инициирует протокол эксперимента и сбор данных. Изменение объема в течение времени, которое определяется плетизмографом при каждом вдохе, измеряют с помощью датчика давления Buxco. Интегрируя этот сигнал в течение времени, рассчитывают параметры потока для каждого вдоха. Этот сигнал вместе с изменением легочного вытесняющего давления, которое определяют с применением датчика давления Sensym (№ TRD4100), связывают через предусилитель Buxco (MAX 2270) с интерфейсом сбора данных (№ SFT3400 и SFT3813). Все другие легочные параметры получают из этих двух входных сигналов.
Базовые значения собирают в течение 5 минут, после чего морских свинок проверяют с применением Ach. Ach вливают внутривенно в течение 1 минуты из шприцевого насоса (sp210iw, World Precision Instruments, Inc., Sarasota, FL) в следующих дозах и предписанных интервалах времени с начала эксперимента: 1,9 мкг/минуту в 5 минут, 3,8 мкг/минуту в 10 минут, 7,5 мкг/минуту в 15 минут, 15,0 мкг/минуту в 20 минут, 30 мкг/минуту в 25 минут и 60 мкг/минуту в 30 минут. Если сопротивление или коэффициент податливости не возвращаются к базовым значениям через 3 минуты после каждого вливания Ach, легкие морской свинки 3 раза наполняют 4 мл воздуха из 10 мл калиброванного шприца. Записанные легочные параметры включают частоту дыхания (вдохи/минуту), коэффициент податливости (мл/см H2O) и легочное давление (см H2O/мл в секунду) (Giles et al., 1971). Как только измерения легочных функций завершаются на 35 минуте этого протокола, морских свинок вынимают из плетизмографа и умерщвляют удушением CO2.
Количество PD2, определяемое как количество Ach, необходимое для удваивания базового легочного сопротивления, рассчитывают с применением значений легочного сопротивления, полученных из потока, и давления сверх интервала при запросах Ach с применением следующего уравнения. Оно получено из уравнения, применяемого для расчета значений PC20 в клинике (Am. Thoracic Soc, 2000):
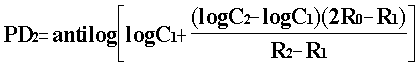
где
C1 = вторая от конца концентрация Ach (концентрация, предшествующая C2);
C2 = конечная концентрация Ach (концентрация, дающая двукратное увеличение легочного сопротивления (RL));
R0 = базовое значение RL;
R1 = значение RL после C1;
R2 = значение RL после C2.
Статистический анализ данных проводят с применением одностороннего анализа переменных с последующим вторичным анализом с применением теста Bonferroni/Dunn. Значение P<0,05 считается значительным.
Кривые доза-реакция сопоставляют 4-параметровым логистическим уравнением с применением GraphPad Prism, версия 3.00 для Windows (GraphPad Software, San Diego, California)
Y = Min + (Max-Min)/(1 + 10∧((log ED50-X)* Hillslope)),
где X является логарифмом дозы, Y является реакцией (PD2), и Y начинается при Min и асимптотически достигает Max с сигмоидальной формой.
Было обнаружено, что характерные соединения в соответствии с данным изобретением обладают значительным бронхозащитным действием в течение времени свыше 24 часов после дозирования.
Представленные ниже примеры синтеза иллюстрируют данное изобретение и не должны восприниматься как ограничивающие объем данного изобретения.
Примеры
Общее: Если не указано иначе, реагенты, исходные материалы и растворители получают из коммерческих источников, например, Sigma-Aldrich (St. Louis, MO), J. T. Baker (Phillipsburg, NJ) и Honeywell Burdick and Jackson (Muskegon, MI), и применяют без дальнейшей очистки; реакции проводят в атмосфере азота; реакционные смеси контролируют тонкослойной хроматографией (ТСХ на двуокиси кремния), аналитической высокоэффективной жидкостной хроматографией (анал. ВЭЖХ) или масс-спектрометрией; реакционные смеси обычно очищают флэш-хроматографией на колонке с силикагелем или препаративной ВЭЖХ с применением общего протокола, описанного ниже; образцы для ЯМР растворяют в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6) и спектр получают на инструменте Varian Gemini 2000 (300 МГц) при стандартных параметрах; масс-спектрометрическую идентификацию проводят методом ионизации электрораспылением (ЭРМС) с помощью инструмента Perkin Elmer (PE SCIEX API 150 EX).
Пример 1: Синтез 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
a. Получение гидрохлорида 4-(2-амино-2-метилпропокси)фениламина
Энергично перемешиваемую суспензию гидрида натрия (60% дисперсия в минеральном масле, 11,32 г, 0,28 моль) в диметилсульфоксиде (400 мл) нагревают до температуры 45°C в течение 1 ч. К этой суспензии затем добавляют чистый 2-амино-2-метил-1-пропанол (25,3 г, 1 экв.). Реакционную смесь нагревают до температуры 75°C в течение 1 ч, затем охлаждают до температуры 20°C на ледяной бане. Медленно добавляют 1-фтор-4-нитробензол (40 г, 1 экв.), сохраняя температуру ниже 30°C, и полученный темно-красный раствор перемешивают при комнатной температуре в течение еще 1 ч. Реакцию гасят водой (1000 мл), экстрагируют дихлорметаном (500 мл), и органический слой промывают (1:1 насыщенный водный хлорид натрия:вода, 1000 мл). Продукт осаждают добавлением 3M хлористоводородной кислоты (400 мл) к органическому слою. Затем полученное оранжевое твердое вещество фильтруют и промывают дихлорметаном до тех пор, пока фильтрат не станет бесцветным.
Твердое вещество немедленно переносят в колбу для гидрирования. Добавляют палладий (10% об./об. на угле, 50% об./об. вода) с последующим добавлением метанола (500 мл). Суспензию энергично встряхивают под давлением 3 атмосфер водорода в течение 16 ч. Затем катализатор фильтруют, растворитель удаляют при пониженном давлении и полученное твердое вещество сушат азеотропной перегонкой с толуолом (3Ч150 мл) с получение указанного в заголовке соединения в виде белого твердого вещества (4,0 г, 0,18 моль, 65%).
b. Получение 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-(трет-бутилдиметилсиланилокси)этил]-8-бензилокси-1H-хинолин-2-он
Смесь продукта со стадии а (23,2 г, 1,1 экв.), гидрохлорида 8-бензилокси-5-{(R)-2-[2-(4-бромфенил)этиламино-1-(трет-бутилдиметилсиланилокси)этил}-1H-хинолин-2-она (66,0 г, 0,1 моль), и трет-бутоксида натрия (54,0 г, 5,5 экв.) в толуоле (600 мл) перемешивают при температуре 90°C до получения гомогенного раствора. Добавляют трис(дибензилиденацетон) палладия (1,4 г, 0,015 экв.) с последующим добавлением рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (2,87 г, 0,045 экв.). Реакционную смесь перемешивают при температуре 90єC в течение 3 ч, затем охлаждают. Раствор промывают водой (100 мл), 1:1 насыщенным водным хлоридом натрия: водой (100 мл), затем сушат над сульфатом натрия. Растворитель удаляют при пониженном давлении с получением указанного в заголовке промежуточного соединения в виде темно-коричневого твердого вещества (40 г, неочищенное), которое применяют без дальнейшей очистки.
c. Получение 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-бензилокси-1H-хинолин-2-она
Продукт с предыдущей стадии обрабатывают тригидрофторидом триэтиламина (36 г) в 2-пропаноле (500 мл)/этаноле (100 мл) при комнатной температуре в течение 16 ч. Смесь концентрируют при пониженном давлении до одной трети от исходного объема. Добавляют 1M водный гидроксид натрия (500 мл) с последующим добавлением ацетонитрила (500 мл) и изопропилацетата (500 мл). Водный слой удаляют и органическую фазу промывают 1:1 насыщенным водным хлоридом натрия:водой (400 мл), затем насыщенным водным хлоридом натрия (400 мл). Органические слои сушат над сульфатом натрия, и растворитель удаляют в вакууме с получением указанного в заголовке промежуточного соединения (50 г, неочищенное) в виде коричневого твердого вещества, которое применяют без дальнейшей очистки.
d. Синтез 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
Гидроксид палладия (10 г, 20% об./об. на угле, 50% об./об. вода) добавляют к продукту с предыдущей стадии, с последующим добавлением этанола (500 мл). Суспензию энергично перемешивают в атмосфере водорода в течение 8 ч. Катализатор отфильтровывают и фильтрат концентрируют при пониженном давлении с получением указанного в заголовке соединения (40 г), которое очищают ВЭЖХ с обращенной фазой и выделяют в виде трифторацетата лиофилизацией. 1H ЯМР (300 МГц, ДМСО-d6): 10,4 (с, 1H), 9,3 (шс, 1H), 8,7 (шс, 1H), 8,15 (м, 2H), 7,8 (шс, 1H), 7,03 (д, 1H, J= 8,2), 6,76-7,01 (м, 10H), 6,42 (д, 1H, J= 9,6), 6,1 (шс, 1H), 5,33 (д, 1H, J= 9,1), 3,8 (с, 2H), 2,7-3,1 (м, 6H), 1,21 (с, 6H); m/z: [M+H+] рассчитано для C29H34N4O4, 503,3; найдено 503,5.
Примеры 2-8: Синтез соединений 2-8
По методике примера 1, за исключением замены 2-амино-2-метил-1-пропанола соответствующим спиртом на стадии а, получают трифторацетаты соединений 2-8.
Соединение 2: 5-[(R)-2-(2-{4-[4-(2-аминоэтокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он: m/z: [M+H+] рассчитано для C27H31N4O4, 475,2; найдено 475,3.
Соединение 3: 5-[(R)-2-(2-{4-[4-(3-аминопропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он: m/z: [M+H+] рассчитано для C28H33N4O4, 489,2; найдено 489,5.
Соединение 4: 5-[(R)-2-(2-{4-[4-(4-аминобутокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он: m/z: [M+H+] рассчитано для C29H35N4O4, 503,3; найдено 503,5.
Соединение 5: 5-{(R)-2-[2-(4-{4-[2-(2-аминоэтокси)этокси]фениламино]фенил)этиламино]-1-гидроксиэтил}-8-гидрокси-1H-хинолин-2-он: m/z: [M+H+] рассчитано для C29H35N4O5, 519,3; найдено 519,5.
Соединение 6: 8-гидрокси-5-[(R)-1-гидрокси-2-(2-{4-[4-(2-морфолин-4-илэтокси)фениламино]фенил}этиламино)этил]-1H-хинолин-2-он: m/z: [M+H+] рассчитано для C31H37N4O5, 545,3; найдено 545,6.
Соединение 7: 8-гидрокси-5-[(R)-1-гидрокси-2-(2-{4-[4-(2-пиперазин-1-илэтокси)фениламино]фенил}этиламино)этил]-1H-хинолин-2-он: m/z: [M+H+] рассчитано для C31H38N5O4, 544,7; найдено 544,7.
Соединение 8: 8-гидрокси-5-[(R)-1-гидрокси-2-(2-{4-[4-пиридин-2-илметокси)фениламино]фенил}этиламино)этил]-1H-хинолин-2-он: m/z: [M+H+] рассчитано для C31H31N4O4, 523,1; найдено 523,2.
Пример 9: Синтез 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)-3-трифторметилфениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
По методике, описанной в примере 1, за исключением замены 1-фтор-4-нитробензола 1-фтор-4-нитро-2-трифторметилбензолом на стадии а, получают указанное в заголовке соединение. m/z: [M+H+] рассчитано для C30H34F3N4O4, 571,3; найдено 571,3.
Пример 10: Синтез 8-гидрокси-5-{(R)-1-гидрокси-2-[2-(4-{4-[2-(4-метансульфонилпиперазин-1-ил)-этокси]фениламино}фенил)этиламино]-этил}-1H-хинолин-2-она
a. Получение 8-бензилокси-5-[(R)-1-(трет-бутилдиметилсиланилокси)-2-(2-{4-[4-(2-пиперазин-1-илэтокси)фениламино]фенил}этиламино)этил]-1H-хинолин-2-она
По методике, описанной в примере 1, стадии а и b, за исключением замены 2-амино-2-метил-1-пропанола на N-(2-гидроксиэтил)пиперазин на стадии а, получают указанное в заголовке соединение.
b. Получение 8-бензилокси-5-{(R)-1-(трет-бутилдиметилсиланилокси)-2-[2-(4-{4-[2-(4-метансульфонилпиперазин-1-ил)этокси]фениламино}фенил)этиламино]этил}-1H-хинолин-2-она
Продукт со стадии а (100 мг) обрабатывают метансульфонилхлоридом (15,3 мг, 1 экв.) в смеси диизопропилэтиламина (0,14 мл) и тетрагидрофурана (2 мл) при комнатной температуре в течение 1 ч. Затем смесь выпаривают с получением указанного в заголовке промежуточного соединения.
c. Синтез 8-гидрокси-5-{(R)-1-гидрокси-2-[2-(4-{4-[2-(4-метансульфонилпиперазин-1-ил)этокси]фениламино}фенил)этиламино]этил}-1H-хинолин-2-она
По методике примера 1, стадии с и d, промежуточное соединение со стадии b превращают в указанное в заголовке соединение. m/z: [M+H+] рассчитано для C32H40N5O6S, 622,3; найдено 622,5.
Пример 11: Синтез 5-[(R)-2-(2-{4-[4-(2-аминоэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
a. Получение 5-[(R)-2-(2-{4-[4-(2-аминоэтил)фениламино]фенил}этиламино)-1-(трет-бутилдиметилсиланилокси)этил]-8-бензилокси-1H-хинолин-2-она
Трис(дибензилиденацетон)дипалладий (0) (0,028 г, 0,031 ммоль), а затем рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (0,057 г, 0,092 ммоль) добавляют к раствору 2-(4-аминофенил)этиламина (0,100 г, 0,740 ммоль), гидрохлорида 8-бензилокси-5-{(R)-2-[2-(4-бромфенил)этиламино-1-(трет-бутилдиметилсиланилокси)этил}-1H-хинолин-2-она (0,393 г, 0,612 ммоль) и трет-бутоксида натрия (0,265 г, 2,75 ммоль) в толуоле при комнатной температуре. Полученную смесь нагревают при температуре 90°C в течение 2 ч, затем охлаждают. Раствор промывают водой (100 мл), 1:1 насыщенным водным хлоридом натрия:водой (100 мл), затем сушат над сульфатом магния. Растворитель удаляют при пониженном давлении с получением указанного в заголовке промежуточного соединения в виде темно-коричневого твердого вещества (0,473 г), которое применяют без дальнейшей очистки.
b. Получение 5-[(R)-2-(2-{4-[4-(2-аминоэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-бензилокси-1H-хинолин-2-она
Продукт с предыдущей стадии (0,473 г, 0,714 ммоль) обрабатывают тригидрофторидом триэтиламина (0,173 г, 1,07 ммоль) в тетрагидрофуране (20 мл) при комнатной температуре в течение 16 ч. Смесь разбавляют дихлорметаном (100 мл) и водой (100 мл). Полученную смесь энергично перемешивают и подщелачивают (pH >10) добавлением 1н. водного гидроксида натрия. Органическую фазу промывают водой (200 мл) и затем промывают насыщенным водным хлоридом натрия (200 мл). Органические слои сушат над сульфатом магния и растворитель удаляют при пониженном давлении с получением указанного в заголовке промежуточного соединения (0,500 г) в виде коричневого твердого вещества, которое применяют без дальнейшей очистки.
c. Синтез 5-[(R)-2-(2-{4-[4-(2-аминоэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
Палладий (0,100 г, 10 мас.% на активированном угле) добавляют к раствору продукта предыдущей стадии в 1:1 метаноле:дихлорметане (20 мл). Суспензию энергично перемешивают под давлением 1 атмосфера водорода в течение 16 ч. Реакционную смесь фильтруют через целит и концентрируют при пониженном давлении с получением указанного в заголовке соединения, которое очищают ВЭЖХ с обращенной фазой, и выделяют в виде трифторацетата лиофилизацией. 1H ЯМР (300 МГц, ДМСО-d6): δ 10,4 (д, 2H), 8,6 (шс, 2H), 8,0 (д, 2H), 7,7 (шс, 3H), 7,0 (д, 1H), 6,96 (д, 4H), 6,84-6,88 (м, 4H), 6,45 (д, 1H), 6,08 (с, 1 H), 5,19 (д, 1H), 3,0-3,2 (м, 2H), 2,8 (шс, 2H), 2,72-2,75 (м, 2H), 2,62 (м, 2H); m/z: [M+H+] рассчитано для C27H30N4O3, 458,56; найдено 459,4.
Пример 12: Синтез 5-[(R)-2-(2-{4-[4-(2-диметиламиноэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
a. Получение диметил-[2-(4-нитрофенил)этил]амина
Гидрохлорид диметиламина (0,390 г, 4,78 ммоль) добавляют к раствору, содержащему бромид 4-нитрофенэтила (1,0 г, 4,35 ммоль) и N,N-диизопропилэтиламин (1,69 г, 13,05 ммоль) в безводном диметилформамиде (20 мл). Реакционную смесь нагревают до температуры 60°C в течение 5 часов, затем охлаждают. Раствор разбавляют 1:1 водой:дихлорметаном (200 мл), затем выливают в делительную воронку. Органические слои собирают и продукт экстрагируют 1н. водным гидрохлоридом. Органические слои отделяют и водный слой подщелачивают добавлением 1н. водного гидроксида натрия. Продукт экстрагируют дихлорметаном (100 мл) и промывают насыщенным водным хлоридом натрия (200 мл). Органические слои сушат над сульфатом магния и растворитель удаляют при пониженном давлении с получением указанного в заголовке промежуточного соединения (0,426 г, неочищенный) в виде прозрачного масла, которое применяют без дальнейшей очистки.
b. Получение 4-(2-диметиламиноэтил)фениламина
Палладий (0,043 г, 10 мас.% на активированном угле) добавляют к раствору продукта предыдущей стадии (0,430 г, 2,2 ммоль) в метаноле (20 мл). Суспензию энергично перемешивают в атмосфере водорода в течение 6 часов. Реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением указанного в заголовке промежуточного соединения (0,307 г, неочищенное), которое применяют без дальнейшей очистки.
c. Синтез 5-[(R)-2-(2-{4-[4-(2-диметиламиноэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
По методике, описанной в примере 11, за исключением замены 2-(4-аминофенил)этиламина в примере 11, стадия а, на продукт предыдущей стадии, получают трифторацетат указанного в заголовке соединения. 1H ЯМР (300 МГц, ДМСО-d6): δ 10,4 (с, 2H), 9,8 (шс, 1H), 8,9 (шс, 1H), 8,7 (шс, 1H), 8,0 (д, 2H), 6,8-7,2 (м, 10H), 6,4 (д, 1H), 6,2 (шс, 1H), 5,2 (д, 1H), 2,8-3,1 (м, 6H), 2,3-2,7(м, 8H); m/z: [M+H+] рассчитано для C29H34N4O3, 486,61; найдено 487,5.
Пример 13: Синтез 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
a. Получение 1,1-диметил-2-(4-нитрофенил)этиламина
Гидрохлорид α,α-диметилфенетиламина (20 г, 108 ммоль) растворяют в концентрированной серной кислоте (40 мл). Раствор охлаждают до температуры -10°C. По каплям добавляют азотную кислоту (31 мл, 90%) в течение 30-минутного периода, сохраняя температуру реакции ниже -5°C. Раствор перемешивают дополнительные 45 минут, затем выливают в лед и нагревают до комнатной температуры в течение ночи. Продукт собирают фильтрацией (13,7 г). К фильтрату добавляют водный гидроксид натрия (1н.) и указанное в заголовке соединение выделяют в виде прозрачного масла.
b. Получение 4-(2-амино-2-метилпропил)фениламина
Палладий (0,136 г, 10 мас.% на активированном угле) добавляют к раствору продукта стадии а (1,36 г, 7,0 ммоль) в метаноле (20 мл). Суспензию энергично перемешивают под давлением 1 атмосферы водорода в течение 6 ч. Реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением указанного в заголовке промежуточного соединения (1,04 г, неочищенное), которое применяют без дальнейшей очистки.
c. Синтез 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
По методике, описанной в примере 11, за исключением замены 2-(4-аминофенил)этиламина в примере 11, стадия а, на продукт предыдущей стадии, получают трифторацетат указанного в заголовке соединения. 1H ЯМР (300 МГц, ДМСО-d6): δ 10,4 (д, 2H), 8,8 (шс, 1H), 8,65 (шс, 1H), 8,0 (д, 2H), 7,7 (с, 3H), 6,8-7,0 (м, 8H), 6,4 (д, 1H), 6,1 (шс, 1H), 5,2 (д, 1H), 3 (шс, 4H), 2,7 (м, 2H), 2,6 (с, 2H), 1,0 (с, 6H); m/z: [M+H+] рассчитано для C29H34N4O3, 486,61; найдено 487,5.
Пример 14: Синтез N-{5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-2-гидроксифенил}формамида
a. Получение [4-(2-амино-2-метилпропоксифенил]-[4-(2-аминоэтил)фенил]амина
Смесь 4-(2-амино-2-метилпропокси)фениламина (1,0 г, 5,5 ммоль) (свободное основание промежуточного соединения примера 1, стадия а), 4-бромфенетиламина (1,1 г, 1 экв.) и трет-бутоксида натрия (1,9 г, 3,5 экв.) в толуоле (30 мл) перемешивают при температуре 90°C до получения гомогенного раствора. Добавляют трис(дибензилиденацетон)дипалладий(0) (252 мг, 0,05 экв.) с последующим добавлением рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (516 мг, 0,015 экв.). Реакционную смесь перемешивают при температуре 90°C в течение 16 ч, затем охлаждают. Раствор разбавляют этилацетатом (100 мл), промывают (1:1 насыщенный водный хлорид натрия:вода (100 мл), затем экстрагируют в 6M хлористоводородной кислоте (100 мл). Водный слой промывают этилацетатом (2Ч100 мл), затем разбавляют изопропилацетатом (100 мл). Смесь охлаждают до температуры 0°C и нейтрализуют гидроксидом натрия (13 г в 20 мл воды). Водный слой удаляют и органический слой промывают (1:1 насыщенный водный хлорид натрия:вода 100 мл), сушат над сульфатом магния и выпаривают с получением указанного в заголовке промежуточного соединения (1,3 г, неочищенное).
b. Получение N-{5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-(трет-бутилдиметилсиланилокси)этил]-2-бензилоксифенил}формамида
Смесь N-[5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)-2-бензилоксифенил]формамида (2,0 г, 4,35 ммоль), продукта предыдущей стадии (1,3 г, 1 экв.), карбоната калия (2,4 г, 4 экв.) и йодида натрия (718 мг, 1,1 экв.) в диметилсульфоксиде (8 мл) нагревают при температуре 140єC в течение 20 мин. Смесь охлаждают, разбавляют водой (20 мл) и экстрагируют этилацетатом (2×20 мл). Органические слои промывают насыщенным водным хлоридом натрия (20 мл), сушат над сульфатом натрия и выпаривают. Остаток очищают ВЭЖХ с обращенной фазой с получением указанного в заголовке промежуточного соединения (500 мг, 0,73 ммоль, 17% выход).
c. Синтез N-{5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-2-гидроксифенил}формамида
По методике примера 1, стадии с и d, промежуточное соединение стадии b превращают в указанное в заголовке соединение. m/z: [M+H+] рассчитано для C27H35N4O4, 479,3; найдено 479,3.
Пример 15: Синтез 5-[(R)-2-(2-{4-[3-(2-аминоэтил)-4-метоксифениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
a. Получение 5-{(R)-2-[2-(4-аминофенил)этиламино]-1-(трет-бутилдиметилcиланилокси)этил]-8-бензилокси-1H-хинолин-2-она
Смесь 5-(2-бром-1-(трет-бутилдиметилсиланилокси)этил)-8-бензилокси-1H-хинолин-2-она (5,0 г, 1,0 экв.) и 2-(4-аминофенил)этиламина (2,8 г, 2,0 экв.) в диметилсульфоксиде (10 мл) нагревают при температуре 100°C в течение 12 ч. Реакционную смесь охлаждают до температуры 20°C и полученное красное масло разбавляют этилацетатом (200 мл) и экстрагируют двумя порциями воды (200 мл). Органический слой промывают двумя порциями (1:1 10% водной уксусной кислоты и насыщенного водного хлорида натрия (200 мл). Органический слой подщелачивают осторожным экстрагированием двумя порциями насыщенного бикарбоната натрия (200 мл), затем насыщенного хлорида натрия (200 мл). Полученный органический раствор обрабатывают безводным сульфатом натрия и растворитель удаляют при пониженном давлении с получением указанного в заголовке промежуточного соединения (5,1 г), которое применяют без дальнейшей очистки.
b. Получение 5-[(R)-2-(2-{4-[3-(2-аминоэтил)-4-метоксифениламино]фенил}этиламино)-1-(трет-бутилдиметилсиланилокси)этил]-8-бензилокси-1H-хинолин-2-она
Смесь продукта предыдущей стадии (0,500 г, 1,0 экв.), гидробромида 5-бром-2-метоксифенетиламина (0,343 г, 1,2 экв.) и трет-бутоксида натрия (0,397 г, 4,5 экв.) в толуоле (10 мл) перемешивают при температуре 90°C до получения гомогенного раствора. Добавляют трис(дибензилиденацетон) палладия (0,0042 г, 0,05 экв.) с последующим добавлением рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтила (0,0086 г, 0,15 экв.). Реакционную смесь перемешивают при температуре 90°C в течение 5 ч, затем охлаждают до температуры 20°C. Раствор разбавляют дихлорметаном (100 мл) и промывают водой (2×100 мл). Органический раствор сушат над сульфатом натрия. Реакционную смесь фильтруют и растворитель удаляют при пониженном давлении с получением указанного в заголовке промежуточного соединения (1,0 г) в виде темно-коричневого твердого вещества, которое применяют без дальнейшей очистки.
c. Получение 5-[(R)-2-(2-{4-[3-(2-аминоэтил)-4-метоксифениламино]фенил}этиламино)-1-гидрокси)этил-8-бензилокси-1H-хинолин-2-она
Неочищенное твердое вещество (1,0 г) с предыдущей стадии обрабатывают тригидрофторидом триэтиламина (0,70 мл) в тетрагидрофуране (10 мл) при комнатной температуре в течение 5 ч. Смесь разбавляют дихлорметаном (100 мл) и экстрагируют 0,1 M NaOH (2×100 мл), затем насыщенным водным хлоридом натрия (100 мл). Органический слой обрабатывают безводным сульфатом натрия, и растворитель сушат при пониженном давлении. Полученное твердое вещество очищают ВЭЖХ с обращенной фазой и выделяют в виде трифторацетата лиофилизацией с получением указанного в заголовке промежуточного соединения (200 мг).
d. Синтез 5-[(R)-2-(2-{4-[3-(2-аминоэтил)-4-метоксифениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
Палладий (10 мг, 10% об./об. на угле), а затем метанол (5 мл) добавляют к промежуточному соединению предыдущей стадии (50 мг) и суспензию энергично перемешивают в атмосфере водорода в течение 5 ч. Реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением указанного в заголовке соединения (42,0 мг), которое очищают ВЭЖХ с обращенной фазой и выделяют в виде трифторацетата лиофилизацией. m/z: [M+H+] рассчитано для C28H32N4O4, 489,3; найдено 489,5.
Пример 16: Синтез N-{5-[(R)-2-(2-{4-[3-(2-аминоэтил)-4-метоксифениламино]фенил}этиламино)-1-гидроксиэтил]-2-гидроксифенил}формамида
По методике примера 15, за исключением замены 5-(2-бром-1-(трет-бутилдиметилсиланилокси)этил)-8-бензилокси-1H-хинолин-2-она на N-[5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)-2-бензилоксифенил]формамид, получают указанное в заголовке соединение. m/z: [M+H+] рассчитано для C26H32N4O4, 465,3; найдено 465,3.
Пример 17: Соли 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
a. Получение свободного основания 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
Водный (10%) раствор бикарбоната аммония (50 мл) добавляют одной порцией к раствору продукта примера 1, трифторацетата 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она (1,80 г, 2,1 ммоль) в этаноле (6 мл). Раствор перемешивают при комнатной температуре в течение 1 ч. Полученное твердое вещество фильтруют и сушат при пониженном давлении с получением указанного в заголовке соединения (0,84 г, 1,67 ммоль, 80%) в виде желтого твердого вещества.
b. Синтез сульфата 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
Свободное основание, полученное на предыдущей стадии (5,85 г, 11,6 ммоль), растворяют в ацетонитриле:воде (200 мл:60 мл) при температуре 90°C и фильтруют для удаления оставшихся твердых частиц. Фильтрат повторно нагревают до температуры 90°C и добавляют раствор концентрированной серной кислоты (2 мл) в ацетонитриле:воде (18 мл:2 мл). Раствор охлаждают до комнатной температуры в течение 2 ч, затем охлаждают на бане лед/вода до температуры 10°C. Твердое вещество фильтруют и сушат при пониженном давлении с получением неочищенной указанной в заголовке соли (5,70 г, 82%). Этот продукт (5,70 г) повторно растворяют в ацетонитриле:воде (120 мл:410 мл) при температуре 90°C и снова охлаждают до комнатной температуры в течение 2 ч. Суспензию охлаждают на бане лед/вода до температуры 10°C, твердое вещество фильтруют и сушат при пониженном давлении с получением гидрата указанной в заголовке соли (4,40 г, 63% общий выход) в виде не совсем белого порошка. Порошковая рентгенограмма (XRPD) продукта показана на фиг.1. Кривая дифференциальной сканирующей калориметрии продукта показана на фиг.2.
c. Синтез нападизилата 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
К раствору свободного основания, полученного на стадии а, (23 мг, 0,046 ммоль) в изопропаноле:воде (1,8 мл:0,2 мл) при температуре 60°C добавляют тетрагидрат 1,5-нафталиндисульфоновой кислоты (33 мг, 2 экв.) с получением белого осадка. Температуру увеличивают до 70°C и добавляют еще 1 мл воды с получением прозрачного раствора. После охлаждения до комнатной температуры суспензию фильтруют и сушат с получением указанной в заголовке соли (25 мг, 63%) в виде белого порошка. XRPD продукта показана на фиг.3. Кривая дифференциальной сканирующей калориметрии продукта показана на фиг.4.
d. Синтез оксалата 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
К раствору свободного основания, полученного на стадии а, (100 мг, 0,2 ммоль) в изопропаноле:воде (3,6 мл:0,4 мл) при температуре 60°C добавляют щавелевую кислоту (50 мг, 2 экв.) с получением белого осадка. Температуру увеличивают до 70°C и добавляют еще 4 мл воды с получением прозрачного раствора. После охлаждения до комнатной температуры суспензию фильтруют и сушат с получением указанной в заголовке соли (82 мг, 65%) в виде не совсем белого порошка. XRPD продукта показана на фиг.5. Кривая дифференциальной сканирующей калориметрии продукта показана на фиг.6.
e. Синтез 4-метилциннамата 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он (0,21 г) растворяют в смеси тетрагидрофурана (1,2 мл) и воды (1,2 мл). 4-Метилкоричную кислоту (0,07 г, преимущественно транс) добавляют к перемешиваемому раствору при комнатной температуре, и она растворяется. Через приблизительно 10 мин начинается кристаллизация. Суспензию перемешивают в течение ночи и фильтруют. Фильтровальную лепешку промывают водным тетрагидрофураном (1:1 ТГФ:вода, 2×0,4 мл, 1×0,2 мл) с получением указанного в заголовке соединения, которое сушат в течение ночи в вакууме при температуре 45°C (выход: 0,207 г). 1H ЯМР (400 МГц, ДМСО-d6; ДМСО-d5 ссылочно при δ (ч./млн.) 2,5). δ (ч./млн.): 1,22 (6H) с; 2,30 (3H) с; 2,64 (2H) т J=6,6 Гц; 2,72-2,86 (4H) м; 3,74 (2H) с; 5,09 (1H) м; 6,43 (1H) д J=15,9 Гц; 6,49 (1H) д J=10,0 Гц; 6,86 (4H) м; 6,93-7,03 (5H) м; 7,06 (1H) д J=7,8 Гц; 7,17 (2H) д J=7,8 Гц; 7,34 (1H) д J=15,9 Гц; 7,45 (2H) д J=7,8 Гц; 7,75 (1H) с; 8,19 (1H) д J=10,0 Гц. Кристаллический продукт характеризуют XRPD и ДСК.
f. Синтез 4-метилциннамата 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-она
5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1H-хинолин-2-он (17,77 г) растворяют в смеси тетрагидрофурана (89 мл) и воды (89 мл). 4-Метилкоричную кислоту (6,02 г, преимущественно транс) взвешивают и около одной четверти добавляют к раствору свободного основания, затем засевают кристаллы, полученные на стадии е. Смесь перемешивают и оставшуюся кислоту добавляют порциями в течение последующих 1,5 ч. Суспензию перемешивают в течение еще 4 ч и затем суспензию фильтруют. Фильтровальную лепешку промывают водным тетрагидрофураном (1:1 ТГФ:вода, 36 мл) и затем тетрагидрофураном (2Ч18 мл) с получением указанного в заголовке соединения, которое сушат в течение ночи в вакууме при температуре 40-50°C (выход: 16,916 г). 1H ЯМР (400 МГц, CD3OD; TMS ссылочно при δ ( ч./млн.) 0) δ (ч./млн.): 1,39 (6H) с; 2,32 (3H) с; 2,79 (2H) т J=7,2 Гц; 2,92-3,03 (4H) м; 3,87 (2H) с; 5,25 (1H) дд J=3,9 и 8,8 Гц; 6,44 (1H) д J=15,9 Гц; 6,63 (1H) д J=9,8 Гц; 6,86-6,93 (4H) м; 6,96 (1H) д J=8,3 Гц; 6,99-7,05 (4H) м; 7,15 (2H) д J=7,8 Гц; 7,19 (1H) д J=8,3 Гц; 7,36 (1H) д J=15,9 Гц; 7,38 (2H) д J=7,6 Гц; 8,34 (1H) д J=9,8 Гц. Кристаллический продукт характеризуют XRPD и ДСК.
g. Аналитические методы
Порошковые рентгенограммы на фиг.1, 3 и 5 получают на диффрактометре Rigaku с применением Cu Kα (30,0 кВ, 15,0 мА) излучения. Анализ проводят с применением гониометра, работающего в режиме непрерывного сканирования 3° в минуту с шагом 0,03° в интервале от 2 до 45°. Образцы получают на кварцевых держателях испытательной машины в виде тонкого слоя порошкообразного продукта. Инструмент откалиброван по стандарту металла кремния.
Кривые дифференциальной сканирующей калориметрии на фиг.2, 4 и 6 получают на инструментах TA модели DSCQ10. Образцы помещают в герметично закрытые алюминиевые поддоны для анализа, ссылочным образцом служит пустой поддон. Образцы уравновешивают при температуре 30°C и нагревают со скоростью 5°C в минуту до температуры 300°C. Инструмент откалиброван по стандарту индия.
Хотя данное изобретение описано в определенных вариантах, специалисту в данной области техники должно быть понятно, что могут быть проведены различные изменения и заменены различные эквиваленты, не выходя за рамки объема данного изобретения. Кроме того, многие модификации могут быть проведены для адаптации к определенной ситуации, материалу, композиции, процессу, стадиям процесса, цели, идее и объему данного изобретения. Кроме того, все публикации, патенты и патентные документы, цитированные в описании, включены сюда в качестве ссылок в полном объеме, так как если бы они были отдельно включены сюда в качестве ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОЗАМЕЩЕННЫЕ ЭТИЛАМИНО-АГОНИСТЫ β-АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2004 |
|
RU2385317C2 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА | 2004 |
|
RU2330841C2 |
| ДИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АКТИВНОСТЬЮ АГОНИСТА В2 АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2010 |
|
RU2543716C2 |
| ДИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АКТИВНОСТЬЮ АГОНИСТА β АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2010 |
|
RU2676686C2 |
| ПРОИЗВОДНЫЕ 4-(2-АМИНО-1-ГИДРОКСИЭТИЛ)ФЕНОЛА В КАЧЕСТВЕ АГОНИСТОВ β2-АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2007 |
|
RU2451675C2 |
| ГУАНИДИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2008 |
|
RU2480458C2 |
| ПРОИЗВОДНЫЕ 4-(2-АМИНО-1-ГИДРОКСИЭТИЛ)ФЕНОЛА, КАК АГОНИСТЫ β2 АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2440330C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2605557C2 |
| ИНГИБИТОРЫ НЕПРИЛИЗИНА | 2011 |
|
RU2622288C2 |
| СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2244709C2 |
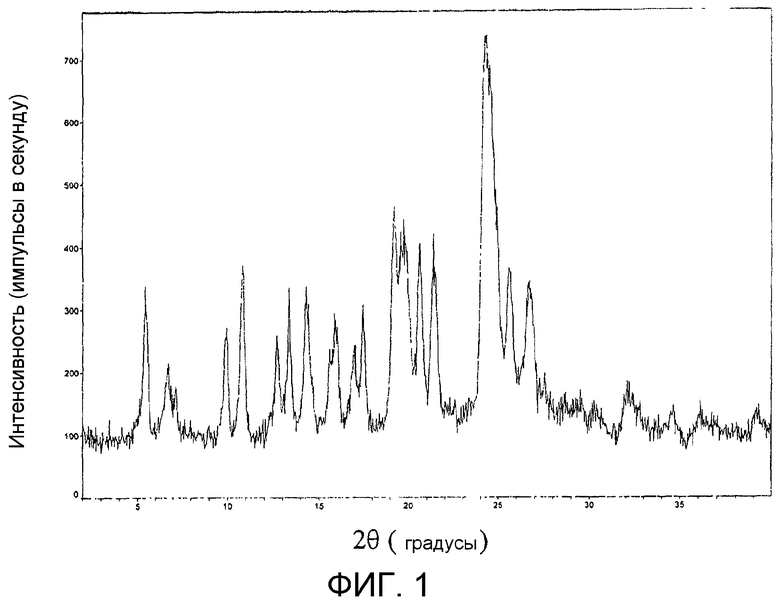
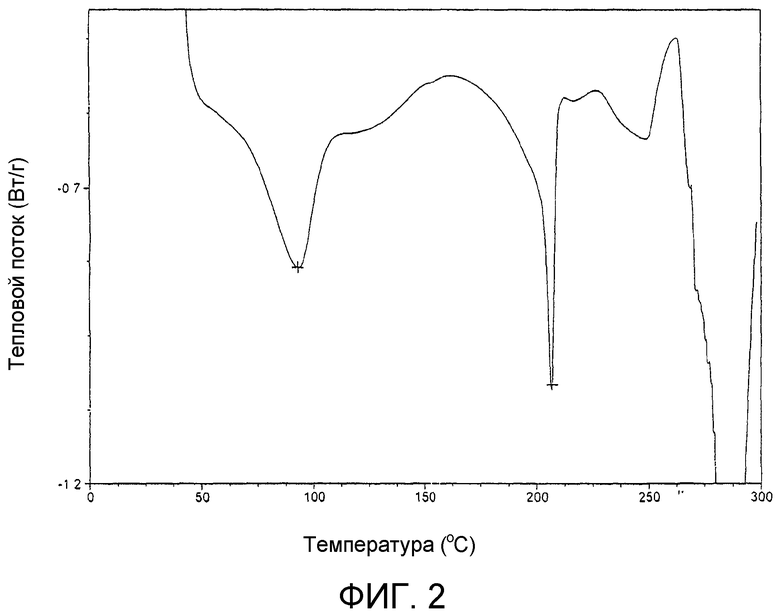
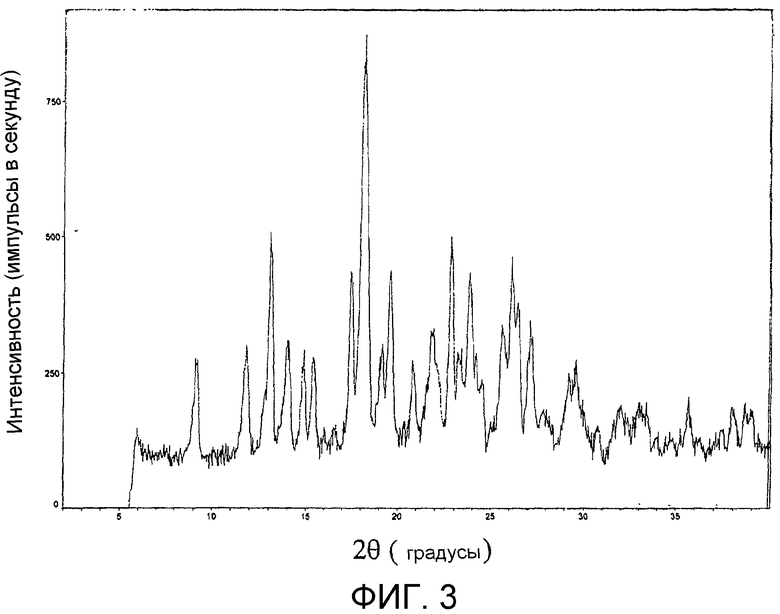
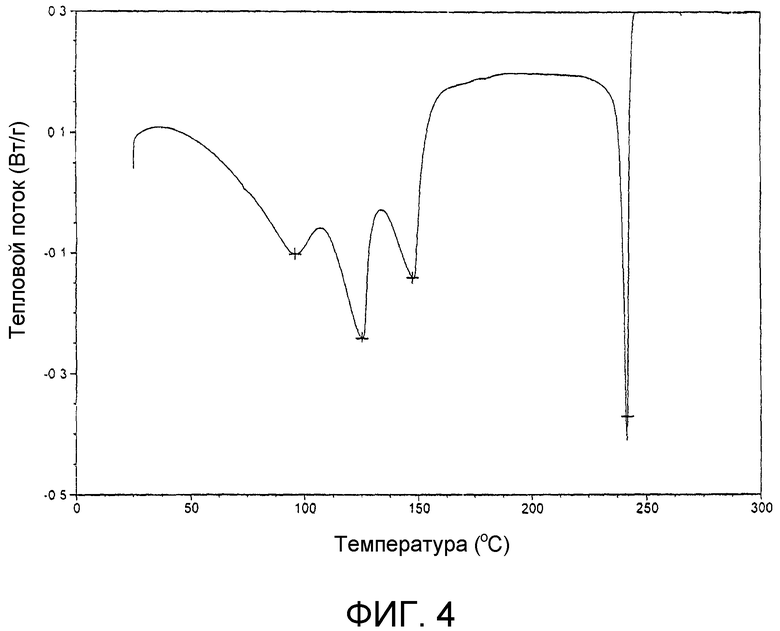
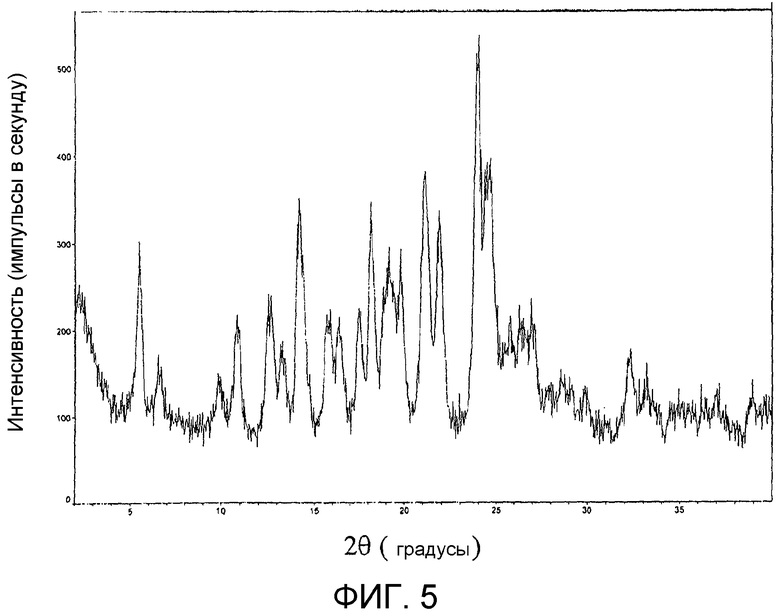
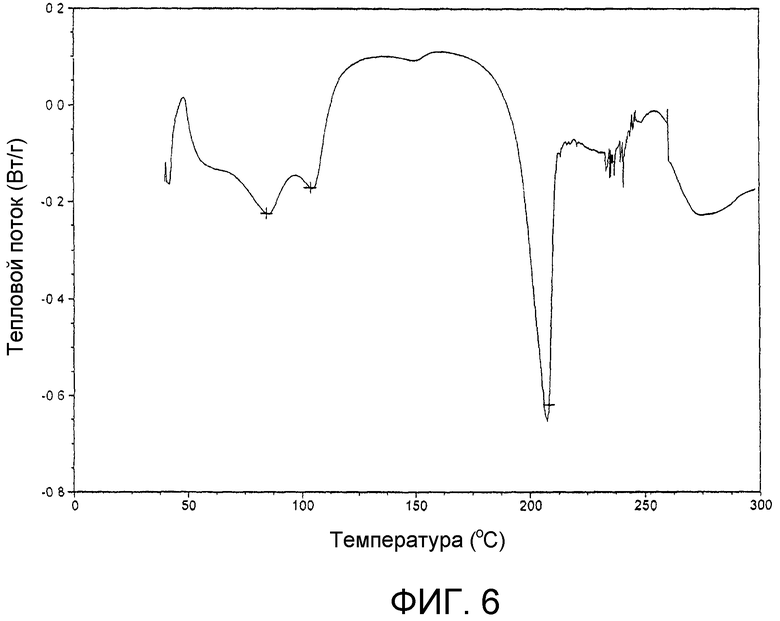
(57)Изобретение относится к новым соединениям формулы (I)
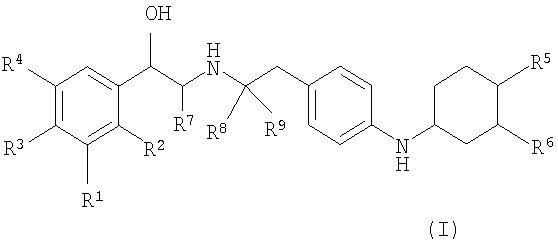
где каждый из R1, R2, R3 и R4 независимо выбирают из водорода, гидрокси и -NHCHO или R1 и R2, взятые вместе, выбирают из -NHC(=O)CH=CH- и -CH=CHC(=O)NH-; R1 также может представлять собой -СН2ОН; один из R5 и R6 представляет собой -[X-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13 и другой из R5 и R6 выбирают из водорода, С1-6алкокси и С1-4алкила, где С1-4алкил необязательно замещен галогеном, где каждый Х представляет собой -О-; каждый из R10, R11, R12 и R13 независимо является водородом или С1-4алкилом; или R10 и R11, вместе с атомом азота, к которому они присоединены, или R10 вместе с атомом азота, к которому он присоединен, и атомом углерода соседнего C1-6алкиленила образуют гетероциклическое или гетероарильное кольцо, имеющее от 5 до 7 атомов в кольце и необязательно содержащее дополнительный гетероатом, выбранный из кислорода и азота, где атом азота необязательно замещен -S(O)2-С1-4алкилом; и n равно 1 или 2; и каждый из R7, R8 и R9 независимо является водородом; или к его фармацевтически приемлемой соли, или сольвату, или стереоизомеру. Изобретение также относится к применению и фармацевтической композиции на их основе, обладающей агонистическим действием в отношении β2 адренергического рецептора. Технический результат - получение новых соединений для применения в терапии для лечения астмы или хронического обструктивного заболевания легких. 6 н. и 19 з.п. ф-лы, 1 табл., 6 ил.
1. Соединение формулы (I):
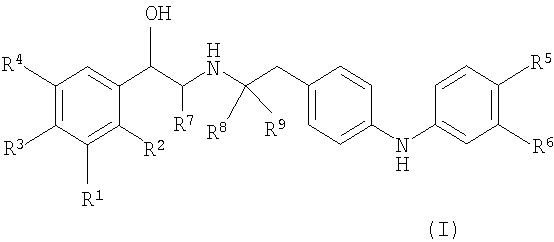
где каждый из R1, R2, R3 и R4 независимо выбирают из водорода, гидрокси, и -NHCHO, или R1 и R2, взятые вместе, выбирают из -NHC(=O)СH=СН- и -CH=CHC(=O)NH-; R1 также может представлять собой -СН2ОН;
один из R5 и R6 представляет собой -[X-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13,
и другой из R5 и R6 выбирают из водорода, С1-4алкокси и С1-4алкила, где C1-4алкил необязательно замещен галогеном,
где каждый Х представляет собой -О-;
каждый из R10, R11, R12 и R13 независимо является водородом или С1-4алкилом; или
R10 и R11, вместе с атомом азота, к которому они присоединены, или R10, вместе с атомом азота, к которому он присоединен, и атомом углерода соседнего С1-6алкиленила, образуют гетероциклическое или гетероарильное кольцо, имеющее от 5 до 7 атомов в кольце, и необязательно содержащее дополнительный гетероатом, выбранный из кислорода и азота, где атом азота необязательно замещен -S(O)2-С1-4алкилом; и
n равно 1 или 2; и
каждый из R7, R8 и R9 независимо является водородом;
или его фармацевтически приемлемая соль или сольват или стереоизомер.
2. Соединение по п.1, которое является соединением формулы (II):
где R1 является -NHCHO, и R2 является водородом; или R1 и R2, взятые вместе, являются -NHC(=O)CH=CH- или -CH=CHC(=O)NH-;
один из R5 и R6 является -[О-С1-6алкиленил]n-NR10R11 или С1-6алкиленил-NR12R13,
и другой из R5 и R6 выбирают из водорода, С1-4алкокси и С1-4алкила, где С1-4алкил необязательно замещен галогеном,
где каждый из R10, R11, R12 и R13 независимо является водородом или С1-4алкилом; или
R10 и R11, вместе с атомом азота, к которому они присоединены, или R10, вместе с атомом азота, к которому он присоединен, и атомом углерода соседнего С1-6алкиленила, образуют гетероциклическое или гетероарильное кольцо, имеющее от 5 до 7 атомов в кольце, и необязательно содержащее дополнительный гетероатом, выбранный из кислорода и азота, где атом азота необязательно замещен -S(O)2-С1-4алкилом; и
n равно 1 или 2;
или его фармацевтически приемлемая соль или сольват или стереоизомер.
3. Соединение по п.2, в котором R5 является -[O-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13 и R6 является водородом.
4. Соединение по п.2, в котором R5 является С1-4алкокси и R6 является -[O-C1-6алкиленил]n-NR10R11 или C1-6алкиленил-NR12R13.
5. Соединение по п.2, в котором
R5 выбирают из -О-C1-6алкиленил-NR10R11 и С1-6алкиленил-NR12R13, и R6 является водородом; или
R5 является С1-4алкокси, и R6 является -С1-6алкиленил-NR12R13,
где каждый из R10, R11, R12 и R13 независимо является водородом или С1-4алкилом, или R10 и R11, вместе с атомом азота, к которому они присоединены, образуют пиперазинильное кольцо.
6. Соединение по п.5, в котором R1 и R2, взятые вместе, являются -NHC(=O)CH=CH- или -CH=CHC(=O)NH-.
7. Соединение по п.6, в котором R5 является -О-С1-6алкиленил-NR10R11 и R6 является водородом.
8. Соединение по п.2, в котором атом углерода алкилена, содержащий гидроксильную группу, имеет стереохимическую конфигурацию (R).
9. Соединение по п.2, которое выбирают из следующих соединений:
5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-он;
8-гидрокси-5-[(R)-1-гидрокси-2-(2-{4-[4-(2-пиперазин-1-илэтокси)фениламино]фенил}этиламино)этил]-1Н-хинолин-2-он;
5-[(R)-2-(2-{4-[4-(2-аминоэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-он;
5-[(R)-2-(2-{4-[4-(2-диметиламиноэтил)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-он;
5-[(R)-2-(2-{4-[3-(2-аминоэтил)-4-метоксифениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-он; и
их фармацевтически приемлемых солей или сольватов или стереоизомеров.
10. Соединение по п.9, которое является 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино] фенил} этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-оном или его фармацевтически приемлемой солью или сольватом или стереоизомером.
11. Соединение по п.10, которое является сульфатом 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-она.
12. Соединение по п.10, которое является нападизилатом 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-она.
13. Соединение по п.10, которое является оксалатом 5-[(R)-2-(2-{4-[4-(2-амино-2-метилпропокси)фениламино]фенил}этиламино)-1-гидроксиэтил]-8-гидрокси-1Н-хинолин-2-она.
14. Фармацевтическая композиция, обладающая агонистическим действием в отношении β2 адренергического рецептора, содержащая терапевтически эффективное количество соединения по любому из пп.1-13 и фармацевтически приемлемый носитель.
15. Фармацевтическая композиция по п.14, которая дополнительно содержит терапевтически эффективное количество кортикостероида.
16. Фармацевтическая композиция по п.14, которая дополнительно содержит терапевтически эффективное количество S-фторметилового эфира 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-тиокарбоновой кислоты.
17. Фармацевтическая композиция по любому из пп.14-16, которая предназначена для введения ингаляцией.
18. Соединение по любому из пп.1-13, обладающее агонистическим действием в отношении β2 адренергических рецепторов.
19. Применение соединения по любому из пп.1-13 для получения лекарственного средства для лечения заболевания или состояния, связанного с активностью β2 адренергического рецептора, у млекопитающего.
20. Применение по п.19, в котором заболеванием или состоянием является астма или хроническое обструктивное заболевание легких.
21. Применение по п.19, в котором лекарственное средство предназначено для введения ингаляцией.
22. Способ получения соединения формулы (I) по любому из пп.1-13, включающий
(а) взаимодействие
(i) соединения формулы (III):
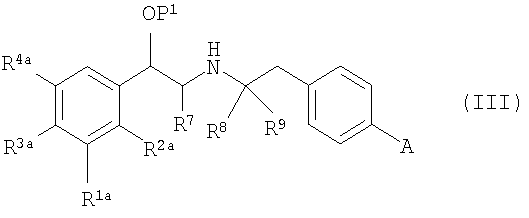
с соединением формулы (IV):
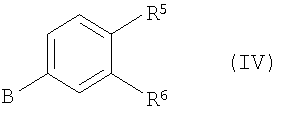
в присутствии катализатора на основе переходного металла; или
(ii) соединения формулы (V):
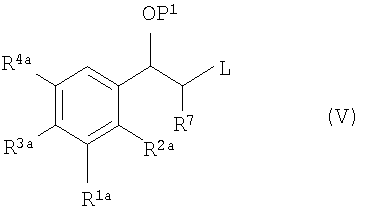
с соединением формулы (VI):
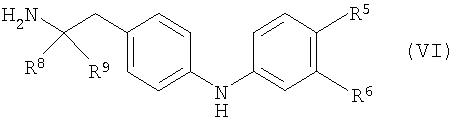
где P1 является гидроксизащитной группой, каждый из R1a, R2a, R3а и R4а, независимо, либо имеет такое же значение, как R1, R2, R3 и R4 в формуле (I), либо является -ОР2, где Р2 является гидроксизащитной группой; один из А и В является уходящей группой и другой из А и В является -NH2; L является уходящей группой; и R5, R6, R7, R8 и R9 такие, как определено в формуле (I), с получением соединения формулы (VII):
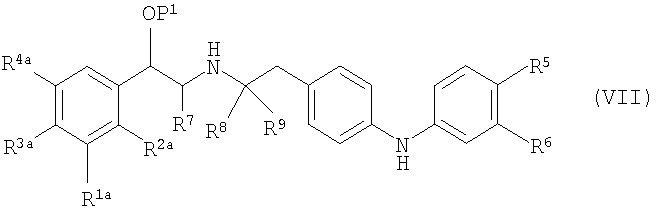
(b) удаление защитной группы Р1 с получением соединения формулы (VIII):
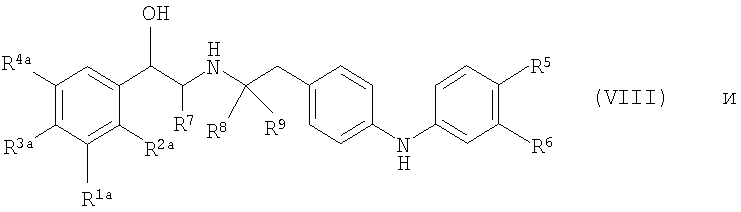
(с) если любой из R1a, R2a, R3а или R4a является -ОР2, удаление защитной группы Р2 с получением соединения формулы (I), или его соли или стереоизомера.
23. Соединение по пп.1-13, полученное способом по п.22.
24. Способ лечения астмы или хронического обструктивного заболевания легких у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пп.1-13.
25. Способ исследования воздействия соединений по пп.1-13 на β2 адренергический рецептор биологической системы или образца, включающий
(a) контактирование биологической системы или образца с соединением по любому из пп.1-13; и
(b) определения воздействия, оказываемого соединением на биологическую систему или образец.
| US 6670376 B1, 30.12.2003 | |||
| WO 03042164 A1, 22.05.2003 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WO 03042160 A1, 22.05.2003 | |||
| ПЛЕНОЧНАЯ МАГНИТНАЯ МАТРИЦА | 0 |
|
SU200622A1 |
| ПРОИЗВОДНЫЕ 1-АМИНО-2-ЗАМЕЩЕННОГО ФЕНИЛЭТАНОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1992 |
|
RU2095344C1 |

