ОПИСАНИЕ
Область изобретения
Настоящее изобретение связано с новыми агонистами β2-адренергических рецепторов. Изобретение связано также с фармацевтическими композициями, содержащими такие соединения, способами применения таких соединений для лечения заболеваний, ассоциированных с активностью β2-адренергических рецепторов, и способами получения таких соединений, а также с промежуточными соединениями, используемыми для получения таких соединений.
Основы создания изобретения
Агонисты β2-адренергических рецепторов, как было обнаружено, являются эффективными лекарственными средствами для лечения легочных заболеваний, таких как астма и хроническая обструктивная болезнь легких (включая хронический бронхит и эмфизему). Агонисты β2-адренергических рецепторов полезны также для лечения в случае преждевременных родов и потенциально применимы для лечения неврологических расстройств и кардиологических нарушений. В отличие от успехов, которые достигаются при использовании определенных агонистов β2-адренергических рецепторов, используемые в настоящее время лекарственные средства обладают менее чем удовлетворительными длительностью действия, силой, избирательностью и/или началом действия. Таким образом, существует потребность в новых агонистах β2-адренергических рецепторов, имеющих улучшенные свойства, такие как улучшенные продолжительность действия, сила, избирательность и/или начало действия.
Краткое содержание изобретения
В настоящем изобретении предложены новые соединения, которые обладают активностью агонистов β2-адренергических рецепторов. Помимо прочих свойств, соединения согласно изобретению, как было обнаружено, являются сильными и селективными агонистами β2-адренергических рецепторов. Кроме того, было обнаружено, что соединения согласно изобретению обладают неожиданным и необъяснимо продолжительным по длительности действием, что позволяет режим их дозирования свести до однократного - и даже еще менее частого - суточного введения.
Соответственно настоящее изобретение связано с соединением формулы (I):
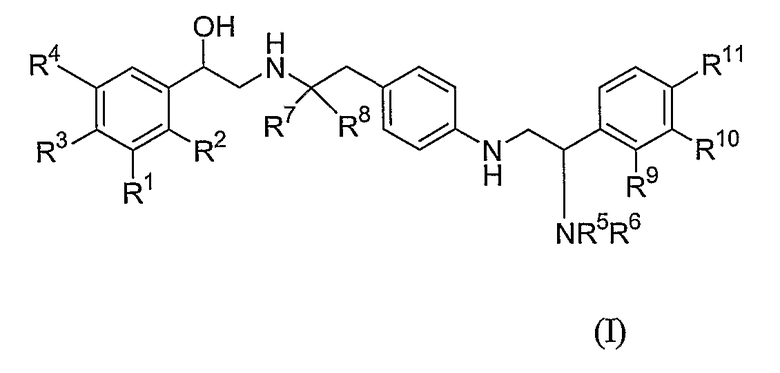
в которой
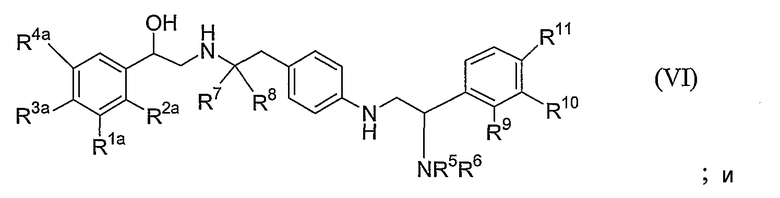
каждый из R1, R2, R3 и R4 независимо выбран из водорода, амино, галогена, гидрокси, -CH2OH и -NHCHO, или R1 и R2, взятые вместе, представляют собой -NHC(=O)CH=CH-, -CH=CHC(=O)NH-, -NHC(=O)S- или -SC(=O)NH-;
каждый из R5 и R6 независимо выбран из водорода, C1-6-алкила, -C(=O)Rd, C2-6-алкенила, C2-6-алкинила и C3-6-циклоалкила, где каждый C1-6-алкил, C2-6-алкенил, C2-6-алкинил и C3-6-циклоалкил необязательно замещены одним или более заместителями, независимо выбранными из арила, гетероарила, гетероциклила, -ORa и -NRbRc, где каждый арил, гетероарил и гетероциклил необязательно замещен одним или более заместителями, независимо выбранными из -ORa и -NRbRc,
или R5 и R6, совместно с атомом азота, к которому они прикреплены, образуют гетероциклическое кольцо, имеющее от 5 до 7 кольцевых атомов и содержащих 1 или 2 гетероатома, независимо выбранных из кислорода, азота и серы, где сера необязательно замещена одним или двумя атомами кислорода;
каждый из R7 и R8 независимо представляет собой водород или C1-6-алкил;
каждый из R9, R10 и R11 независимо выбран из водорода, C1-6-алкила, арила, галогена, -ORa и -NRbRc;
Rd представляет собой водород или C1-3-алкил, необязательно замещенные одним или более заместителями, независимо выбранными из -ORa, -NRbRc, пиперидинила и пирролидинила; и
каждый из Ra, Rb и Rc независимо представляет собой водород или C1-3-алкил;
или его фармакологически приемлемой солью или сольватом или стереоизомером.
Настоящее изобретение связано также с фармацевтическими композициями, включающими в себя соединение согласно изобретению и фармацевтически приемлемый носитель. Настоящее изобретение дополнительно связано с комбинациями, включающими в себя соединение согласно изобретению и один или более других терапевтических агентов, и фармацевтическими композициями, включающими в себя такие комбинации.
Настоящее изобретение связано также со способом лечения заболевания или состояния, ассоциированного с активностью β2-адренергических рецепторов (например, легочного заболевания, такого как астма или хроническая обструктивная болезнь легких, преждевременные роды, нервное расстройство, сердечное расстройство или воспаление) у млекопитающего, включающим в себя введение указанному млекопитающему терапевтически эффективного количества соединения согласно изобретению. Настоящее изобретение дополнительно связано со способом лечения, включающим в себя введение терапевтически эффективного количества комбинации соединения согласно изобретению с одним или более терапевтическими агентами.
Настоящее изобретение связано также со способом лечения заболевания или состояния, ассоциированного с активностью β2-адренергических рецепторов у млекопитающего, включающим в себя введение указанному млекопитающему терапевтически эффективного количества фармацевтической композиции согласно изобретению.
Соединения согласно изобретению могут быть использованы также в качестве средств для исследования, т.е. для изучения биологических систем или образцов, или для обнаружения новых агонистов β2-адренергических рецепторов. Соответственно в одном из аспектов, связанных со способом, данное изобретение направлено на способ агонистического воздействия на β2-адренергические рецепторы в биологической системе или образце, включающий в себя контактирование биологической системы или образца, содержащих β2-адренергический рецептор, с вызывающим агонистическое воздействие на β2-адренергические рецепторы количеством соединения формулы (I) или его фармацевтически приемлемой соли или сольвата или стереоизомера.
В отдельных и различных аспектах данное изобретение связано также со способами синтеза и описанными здесь промежуточными соединениями, которые используются для получения соединений согласно изобретению.
Настоящее изобретение связано также с описанным здесь соединением согласно изобретению для применения в медицине для лечения, а также для применения соединения согласно изобретению в производстве композиции или лекарственного средства для лечения заболевания или состояния, ассоциированного с активностью β2-адренергических рецепторов у млекопитающего, например, легочного заболевания, такого как астма или хроническая обструктивная болезнь легких, преждевременные роды, нервное расстройство, сердечное расстройство или воспаление.
Краткое описание чертежа
Настоящее изобретение иллюстрируется ссылкой на сопутствующий чертеж, на котором приведена картина порошковой дифракции рентгеновских лучей гидрохлорида N-{2-[4-((R)-2-гидрокси-2-фенилэтиламино)фенил]этил}-(R)-2-гидрокси-2-(3-формамидо-4-гидроксифенил)этиламина.
Подробное описание изобретения
Настоящее изобретение связано с новыми аминозамещенными этиламино-агонистами β2-адренергических рецепторов формулы (I) или их фармацевтически приемлемыми солями или сольватами или стереоизомерами. Последующие примеры и предпочтительные значения для радикалов и заместителей, а также диапазоны значений приведены исключительно с иллюстративными целями; они не исключают других определенных значений или других значений в рамках определенных диапазонов для радикалов и заместителей.
Примерами конкретных значений R1 являются галоген, -CH2OH и -NHCHO, включая хлор, -CH2OH и -NHCHO.
Другими конкретными значениями R1 являются -CH2OH или -NHCHO.
Конкретным значением для R2 является водород.
Конкретным значением для R1 и R2, взятых вместе, являются -NHC(=O)CH=CH- или -CH=CHC(=O)NH-.
Примерами конкретных значений для R3 являются гидрокси и амино.
Примерами конкретных значений для R4 являются водород и галоген, включая водород и хлор.
Одной из групп соединений формулы (I) являются соединения, где R1 представляет собой NHCHO, R3 представляет собой гидрокси, а каждый из R2 и R4 представляет собой водород.
Другой группой соединений формулы (I) являются соединения, в которых R1 и R2, совместно взятые, представляют собой -NHC(=O)CH=CH- или -CH=CHC(=O)NH-, R3 представляет собой гидрокси, а R4 представляет собой водород.
В другом аспекте изобретения R1 представляет собой -CH2OH, R3 представляет собой гидрокси, а каждый из R2 и R4 представляет собой водород.
В другом аспекте изобретения R1 и R4 представляют собой хлор, R3 представляет собой амино, а R2 представляет собой водород.
Примерами конкретных значений R5 и R6 являются R5 и R6, независимо выбранные из водорода, C1-6-алкила и C3-6-циклоалкила, где каждый C1-6-алкил необязательно замещен одним или более заместителями, независимо выбранными из гетероциклила, -ORa и -NRbRc. Другими примерами R5 и R6 являются R5 и R6, которые, совместно с атомом азота, к которому они прикреплены, образуют гетероциклическое кольцо, имеющее от 5 до 7 кольцевых атомов и содержащих от 1 до 2 гетероатомов, независимо выбранных из кислорода, азота и серы.
В одном из воплощений R5 и R6 независимо представляют собой водород или C1-3-алкил, где каждый C1-3-алкил необязательно замещен одним заместителем, независимо выбранным из гидроксила, амино, пиперидинила и пирролидинила. В другом воплощении R5 и R6, совместно с атомом азота, к которому они прикреплены, образуют морфолиниловое или пиперидиниловое кольцо.
В еще одном воплощении R5 и R6 независимо представляют собой водород или C1-3-алкил.
Конкретным значением R7 является водород.
Конкретным значением R8 является водород.
Примерами конкретных значений R9 являются водород, галоген и -ORa, где Ra представляет собой водород или C1-3-алкил.
Другим примером конкретных значений R9 являются гидрокси и метокси.
Другим конкретным значением R9 является водород.
Примерами конкретных значений R10 являются водород, галоген и -ORa, где Ra представляет собой водород или C1-3-алкил.
Другим примером конкретных значений R10 являются гидрокси и метокси.
Другим конкретным значением R10 является водород.
Примерами конкретных значений R11 являются водород, галоген и -ORa, где Ra представляет собой водород или C1-3-алкил.
Другим примером конкретных значений R11 являются гидрокси и метокси.
Другим конкретным значением R11 является водород.
В одном из воплощений настоящего изобретения соединение формулы (I) представляет собой соединение формулы (II):

в которой
R1 представляет собой -CH2OH или -NHCHO, и R2 является водородом; или R1 и R2, совместно взятые, представляют собой -NHC(=O)CH=CH- или -CH=CHC(=O)NH-;
каждый из R5 и R6 независимо выбран из водорода, C1-6-алкила, C2-6-алкенила, C2-6-алкинила и C3-6-циклоалкила, где каждый C1-6-алкил, C2-6-алкенил, C2-6-алкинил и C3-6-циклоалкил необязательно замещен одним или более заместителями, независимо выбранными из арила, гетероарила, гетероциклила, -ORa и -NRbRc, где каждый арил, гетероарил и гетероциклил необязательно замещен одним или более заместителями, независимо выбранными из -ORa и -NRbRc,
или R5 и R6, вместе с атомом азота, к которому они прикреплены, образуют гетероциклическое кольцо, имеющее от 5 до 7 кольцевых атомов и содержащее 1 или 2 гетероатома, независимо выбранных из кислорода, азота и серы, где сера необязательно замещена одним или двумя атомами кислорода; и
каждый Ra, Rb и Rc независимо выбран из водорода или C1-3-алкила;
или его фармацевтически приемлемую соль или сольват или стереоизомер.
Одной из групп соединений формулы (II) являются соединения, в которых R5 и R6 являются независимо выбранными из водорода, C1-6-алкила и C3-6-циклоалкила, где каждый C1-6-алкил необязательно замещен одним или более заместителями, независимо выбранными из гетероциклила, -ORa и -NRbRc, или R5 и R6, вместе с атомом азота, к которому они прикреплены, образуют гетероциклическое кольцо, имеющее от 5 до 7 кольцевых атомов и содержащее 1 или 2 гетероатома, независимо выбранных из кислорода, азота и серы.
Другой группой соединений формулы (II) являются соединения, где каждый R5 и R6 независимо представляют собой водород или C1-3-алкил, где каждый C1-3-алкил необязательно замещен одним заместителем, независимо выбранным из гидрокси, амино, пиперидинила и пирролидинила; или R5 и R6 вместе с атомом азота, к которому они прикреплены, образуют морфолиниловое или пиперидиниловое кольцо. В еще одной группе соединений формулы (II) каждый из R5 и R6 представляет собой водород или C1-3-алкил.
Особого внимания заслуживают следующие соединения:
5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-он:

N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамид:

5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-он;
N-[5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамид;
5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-он;
5-((R)-2-{2-[4-((R)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-он;
N-[5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамид;
N-[5-((R)-2-{2-[4-((R)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамид;
5-((R)-2-{2-[4-((S)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-он;
5-((R)-2-{2-[4-((S)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-он;
N-[5-((R)-2-{2-[4-((S)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамид и
N-[5-((R)-2-{2-[4-((S)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамид;
где химическая номенклатура соответствует номенклатуре автоматической программы наименований AutoNom, предоставленной информационными системами MDL, GmbH (Франкфурт, Германия).
Как было показано выше, соединения согласно изобретению содержат один или более хиральных центров. Соответственно изобретение, если особо не указано иное, включает в себя рацемические смеси, чистые стереоизомеры (т.е. индивидуальные энантиомеры или диастереомеры) и обогащенные стереоизомером смеси таких изомеров. Когда показан конкретный стереоизомер, специалистам в данной области следует понимать, что при этом подразумевается, что если особо не указано иное, минорные количества других стереоизомеров также могут присутствовать в композициях согласно изобретению при условии, что применимость композиции как целого не страдает от присутствия указанных других изомеров.
В частности, соединения согласно изобретению содержат хиральный центр на углероде алкилeновой группы в формулах (I) и (II), к которому присоединена гидроксигруппа. Когда используют смесь стереоизомеров, предпочтительно, чтобы количество стереоизомеров с (R)-ориентацией в хиральном центре, несущих гидроксигруппу, было больше, чем количество соответствующих (S)-стереоизомеров. При сравнении стереоизомеров одного и того же соединения (R)-стереоизомер является предпочтительнее, чем (S)-стереоизомер.
Определения
При описании соединений, композиций и способов согласно изобретению, если не указано иное, указанные ниже термины имеют указанные ниже значения.
Термин “алкил” означает моновалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной или же может быть их комбинацией. Репрезентативные алкильные группы включают в себя - в качестве примеров - метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и пр.
Когда подразумевается определенное количество атомов углерода в случае конкретного используемого здесь термина, число атомов углерода указывают перед самим термином. Например, термин "C1-6-алкил" означает алкильную группу, имеющую от 1 до 6 атомов углерода.
Термин “алкенил” означает моновалентную ненасыщенную углеводородную группу, которая содержит по меньшей мере одну двойную связь углерод-углерод, обычно 1 или 2 двойные связи углерод-углерод и которая может быть линейной или разветвленной или может быть их комбинацией. Репрезентативные алкенильные группы включают в себя - в качестве примеров - винил, аллил, изопропенил, бут-2-енил, н-пент-2-енил, н-гекс-2-енил, н-гепт-2-енил, н-окт-2-енил, н-нон-2-енил, н-дек-4-енил, н-дек-2,4-диенил и пр.
Термин “алкинил” означает моновалентную ненасыщенную углеводородную группу, которая содержит по меньшей мере одну тройную связь углерод-углерод, обычно 1 тройную связь углерод-углерод и которая может быть линейной или разветвленной или может быть их комбинацией. Репрезентативные алкинильные группы включают в себя - в качестве примеров - этинил, пропаргил, бут-2-инил и пр.
Термин “циклоалкил” означает моновалентную насыщенную карбоциклическую группу, которая может быть моноциклической или мультициклической. Репрезентативные циклоалкинильные группы включают в себя - в качестве примеров - циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и пр.
Термин “арил” означает моновалентный ароматический углеводород, имеющий единственное кольцо (т.е. фенил) или слитые кольца (т.е. нафталин). Если не указано иное, такие арильные группы обычно содержат от 6 до 10 кольцевых атомов углерода. Репрезентативные арильные группы включают в себя - в качестве примеров - фенил и нафталин-1-ил, нафталин-2-ил и пр.
Термин “гетероарил” означает моновалентную ароматическую группу, имеющую единственное кольцо или два слитых кольца и содержащую в этом кольце по меньшей мере один гетероатом (обычно от 1 до 3 гетероатомов), выбранный из азота, кислорода и серы. Если не указано иное, такие гетероарильные группы обычно содержат от 5 до 10 суммарных кольцевых атомов. Репрезентативные гетероарильные группы включают в себя - в качестве примеров - пирроил, изоксазолил, изотиазолил, пиразолил, пиридил (или, эквивалентно, пиридинил), оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, пиридазинил, пиразинил, бензоксазолил, бензотиазолил, бензимидазолил, бензофуранил, бензотиофенил, хинолил, индолил, изохинолил и пр., где местом присоединения является любой доступный кольцевой атом углерода или азота.
Термин “гетероциклил” или “гетероциклическое кольцо” означает моновалентную насыщенную или частично ненасыщенную циклическую неароматическую группу, которая может быть моноциклической или мультициклической (например, слитой или соединенной мостиками) и которая содержит по меньшей мере один гетероатом (обычно от 1 до 3 гетероатомов), выбранный из азота, кислорода и серы. Если не указано иначе, такие гетероциклильные группы обычно содержат от 5 до 10 суммарных кольцевых атомов. Репрезентативные гетероциклические группы включают в себя - в качестве примеров - пирролидинил, пиперидинил, пиперазинил, имидазолидинил, морфолинил, индолил-3-ил, 2-имидазолинил, 1,2,3,4-тетрагидроизохинолин-2-ил, хинуклидинил и пр.
Термин “галоген” означает фтор, хлор, бром или иод.
Термин "амино" означает -NH2.
Термин “терапевтически эффективное количество” означает количество, достаточное для эффективного лечения при введении пациенту в случае необходимости лечения.
Термин “лечение” означает здесь лечение заболевания или медицинского показания у пациента, такого как млекопитающее (в частности, человека), которое включает в себя
(a) предотвращение возникновения заболевания или медицинского показания, т.е. профилактическое лечение пациента;
(b) улучшение состояния при заболевании или медицинском показании, т.е. удаление или регрессию заболевания или медицинского показания у пациента;
(c) супрессию заболевания или медицинского показания, т.е. замедление или остановку развития заболевания или медицинского показания у пациента; или
(d) облегчение симптомов заболевания или медицинского показания у пациента.
Фраза “заболевание или состояние, ассоциированное с активностью β2-адренергических рецепторов”, включает в себя все заболевания или состояния, которые подтверждены в настоящее время, или те, которые будут обнаружены в будущем, в том плане, что они ассоциированы с активностью β2-адренергических рецепторов. Такие болезненные состояния включают в себя, не ограничиваясь перечисленным, легочные заболевания, такие как астма или хроническая обструктивная болезнь легких (включая хронический бронхит и эмфизему), а также нервные расстройства и сердечные расстройства. Известно также, что активность β2-адренергических рецепторов ассоциирована с преждевременными родами (см. патент США номер 5872126) и некоторыми типами воспаления (см. публикацию международной патентной заявки номер WO 99/30703 и патент США номер 5290815).
Термин “фармацевтически приемлемая соль” означает соль, полученную из основания или кислоты, которая приемлема для введения пациенту, такому как млекопитающее. Такие соли могут быть получены из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот.
Соли, полученные из фармацевтически приемлемых кислот, включают в себя, не ограничиваясь перечисленным, соли уксусной, бензолсульфоновой, бензойной, камфосульфоновой, лимонной, этансульфоновой, фумаровой, глюконовой, глутаминовой, бромистоводородной, соляной, молочной, малеиновой, яблочной, миндальной, метансульфоновой, муциновой, азотной, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой, ксинафоевой (1-гидрокси-2-нафтойной кислоты) кислот и пр. Особенно предпочтительными являются соли, полученные из фумаровой, бромистоводородной, соляной, уксусной, серной, метансульфоновой, ксинафоевой и винной кислот.
Соли, полученные из фармацевтически приемлемых неорганических оснований, включают в себя соли алюминия, аммония, кальция, меди, железа(3), железа(2), лития, магния, марганца(3), марганца(2), калия, натрия, цинка и пр. Особо предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических оснований, включают в себя соли первичного, вторичного и третичного аминов, включая замещенные амины, циклические амины, природно образующиеся амины и пр., такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперадин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и пр.
Термин “сольват” означает комплекс или агрегат, образуемый одной или более молекул растворенного вещества, т.е. соединением согласно изобретению или его фармацевтически приемлемой солью и одной или более молекулами растворителя. Такие сольваты являются типичными твердыми кристаллами, имеющими по существу фиксированное молярное отношение растворенного вещества и растворителя. Репрезентативные растворители включают в себя, не ограничиваясь перечисленным, воду, метанол, этанол, изопропанол, уксусную кислоту и пр. Когда растворителем является вода, образуемый сольват представляет собой гидрат.
Предпочтительнее, если под термином «или фармцевтически приемлемая соль или сольват ее стереоизомера» подразумевается включение всех пермутаций солей, сольватов и стереоизомеров, таких как сольват фармацевтически приемлемой соли стереоизомера соединения формулы (I).
Термин “уходящая группа” означает функциональную группу атома, который может быть заменен другой функциональной группой или атомом в реакции замещения, такой как реакция нуклеофильного замещения. В качестве примера репрезентативные уходящие группы включают в себя группы хлоро, бромо и иодо; группы сложного сульфонового эфира, такие как мезилат, тозилат, брозилат, нозилат и пр.; и ацилоксигруппы, такие как ацетокси-, трифторацетокси- и пр.
Термин “аминозащитная группа” означает защитную группу, подходящую для предотвращения нежелательных реакций азота аминогруппы. Репрезентативные аминозащитные группы включают в себя, не ограничиваясь перечисленным, формильную группу; ацильные группы, например, алканоильные группы, такие как ацетильная; алкоксикарбонильные группы, такие как трет-бутоксикарбонильная (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонильная (Cbz) и 9-флуоренилметоксикарбонильная (Fmoc); арилметильные группы, такие как бензильная (Bn), тритильная (Tr) и 1,1-ди-(4'-метоксифенил)метильная; силильные группы, такие как триметилсилильная (TMS) и трет-бутилдиметилсилильная (TBS); и пр.
Термин “гидроксизащитная группа” означает защитную группу, подходящую для предотвращения нежелательных реакций гидроксигруппы. Репрезентативные гидроксизащитные группы включают в себя, не ограничиваясь перечисленным, алкильные группы, такие как метильная, этильная и трет-бутильная; ацильные группы, например, алканоильные группы, такие как ацетильная; арилметильные группы, такие как бензильная (Bn), п-метокисибензильная (PMB), 9-флуоренилметильная (Fm) и дифенилметильная (бензгидрильная, DPM); силильные группы, такие как триметилсилильная (TMS) и трет-бутилдиметилсилильная (TBS); и пр.
Общие приемы синтеза
Соединения согласно изобретению могут быть получены из легкодоступных исходных материалов с помощью следующих общих методов и приемов. Несмотря на то, что частный аспект настоящего изобретения иллюстрируется представленными ниже схемами, для специалистов в данной области должно быть очевидно, что все аспекты данного изобретения могут быть получены с помощью описанных здесь способов или с использованием других способов, реагентов и исходных материалов, известных специалистам в данной области. Следует понимать также и то, что если взяты типичные или предпочтительные условия процесса (т.е. температуры реакций, их длительность, молярное отношение реактантов, растворители, давления и т.д.), то, если не указано иначе, могут быть использованы и другие условия процесса. Оптимальные условия реакции могут варьировать в зависимости от конкретных используемых реактантов или растворителей, но такие условия могут определяться специалистами в данной области на основании общепринятых приемов оптимизации.
Кроме того, как будет, безусловно, понятно специалистам в данной области, могут быть необходимы обычно используемые защитные группы для предотвращения нежелательных реакций, связанных с определенными функциональными группами. Выбор подходящей защитной группы для конкретной функциональной группы, так же как и выбор подходящих условий для защиты и снятия защиты, хорошо известны в данной области. Например, целый ряд защитных групп, а также приемы их введения и удаления, описаны у T. W. Greene and G.M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, а также в ссылках, цитируемых в указанной публикации.
В одном из способов синтеза соединения формул (I) и (II) получены, как показано на схеме A. (Заместители и переменные, показанные в следующих схемах, соответствуют определениям, приведенным выше, если специально не указано иначе).
Схема А

где P1 представляет собой гидроксизащитную группу, P2 представляет собой гидроксизащитную группу, а L представляет собой уходящую группу, такую как бром.
Как показано на схеме A, соединение формулы 1 сначала реагирует с (R)-N2-[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамином (2), с получением промежуточного соединения формулы 3. Обычно такую реакцию проводят в апротонном растворителе в присутствии основания и при нагревании. Защитная группа P1 обычно представляет собой силильную защитную группу, которую обычно удаляют из промежуточного соединения формулы 3 с помощью фторида или кислого реагента с получением промежуточного соединения формулы 4. Защитная группа P2 обычно представляет собой бензильную защитную группу, которую обычно удаляют из промежуточного соединения формулы 4 путем гидрирования с помощью палладия на угле в качестве катализатора с получением продукта.
Соединения формулы 1, используемые в реакциях, описанных в настоящей заявке, легко получают с помощью методик, известных в данной области и описанных, например, в патентах США No 6653323 B2 и 6670376 B1, которые включены в настоящее описание в виде ссылки, вместе с цитируемыми в них ссылками. Промежуточное соединение 2 получено из легкодоступных исходных материалов, например, с помощью приемов, иллюстрируемых на схеме B.
Схема В
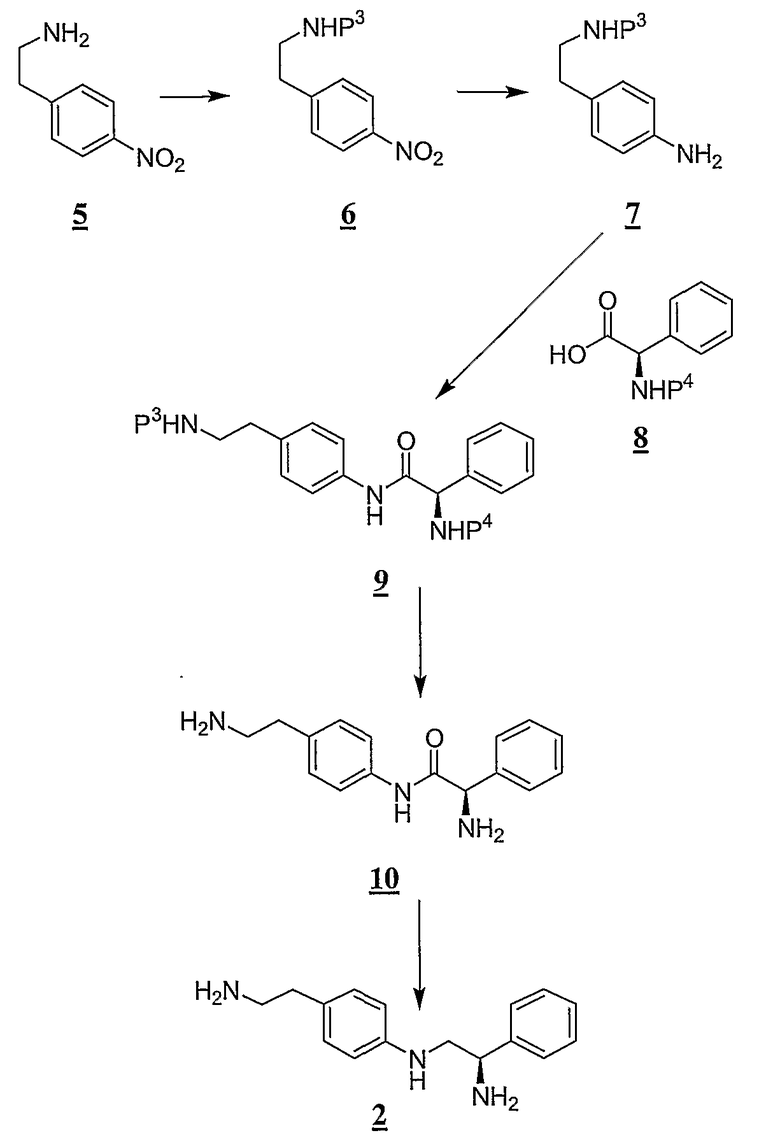
На схеме B P3 представляет собой аминозащитную группу и P4 представляет собой аминозащитную группу.
Как проиллюстрировано на схеме B, защитную группу, P3, добавляли к азоту в составе аминогруппы 2-(4-нитрофенил)этиламина, 5, с получением промежуточного соединения формулы 6. Защитная группа P3 обычно представляет собой трет-бутоксикарбонильную (Boc) группу, которая обычно добавляется в результате реакции ди-трет-бутилбикарбоната (Boc2O) в щелочных условиях. Промежуточное соединение 6 восстанавливают с получением промежуточного соединения формулы 7. Восстановление промежуточного соединения 6 обычно производят путем гидрирования с помощью палладия на угле в качестве катализатора. Амин промежуточного соединения 7 соединяется с защищенным фенилглицином 8 с получением промежуточного соединения формулы 9. Связывание промежуточного соединения 7 с соединением 8 может быть произведено с помощью спаривающего пептиды агента, например, 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDC), и можно использовать катализатор, например, 1-гидроксибензотриазолгидрат (HOBT) или 1-гидрокси-7-азабензотриазолгидрат (HOAT). С промежуточного соединения 9 снимают защиту обычно в кислых условиях с получением промежуточного соединения формулы 10, которое обычно восстанавливают с помощью боранового восстановителя с получением (R)-N2-[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамина (2).
Получение промежуточного соединения 2 дополнительно описано в примере 1, в частях a-e, ниже.
Альтернативно соединения согласно изобретению могут быть получены, как проиллюстрировано на схеме C.
Схема С
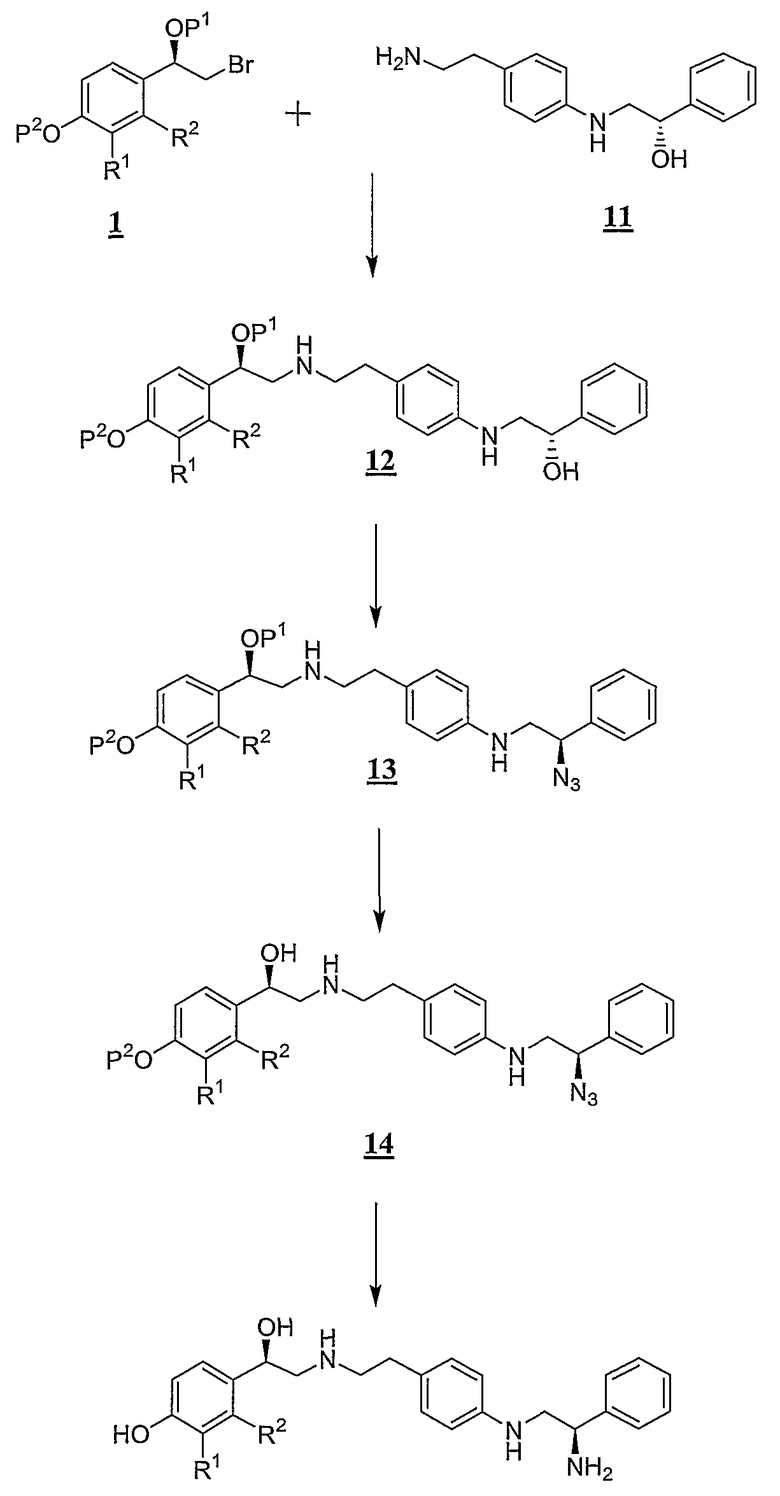
Согласно схеме C промежуточное соединение 1 соединяется с (S)-2-[4-(2-аминоэтил)фениламино]-1-фенилэтанолом (11) с получением промежуточного соединения формулы 12. Обычно такую реакцию проводят в полярном апротонном растворителе в присутствии основания и при нагревании. Промежуточное соединение 12 взаимодействует с реагентом, таким как дифенилфосфорилазид, который превращает спирт в уходящую группу, в результате образуется нуклеофильный азидный анион с получением промежуточного соединения формулы 13. Альтернативно могут быть использованы двухреагентные системы для превращения промежуточного соединения 12 в азид 13. Затем защитную группу P1, которая обычно представляет собой силильную защитную группу, удаляют, обычно с помощью фторида или кислого реагента, с получением промежуточного соединения формулы 14. Продукт может быть получен путем одновременного гидрирования азида и снятия защитной группы P2 промежуточного соединения формулы 14, когда P2 представляет собой группу, такую как бензил, которая удаляется путем гидрирования. Если защитная группа P2 является недостаточно подвижной, чтобы снять защиту, требуется дополнительная стадия снятия защиты.
Промежуточное соединение 11 легко получают в результате реакции 2-(4-аминофенил)этиламина с хиральным стиролоксидом, как описано в примере 3, часть a, ниже.
Дальнейшие подробности в отношении специфических условий реакции и других приемов для получения репрезентативного соединения согласно изобретению или его промежуточного соединения описаны ниже в примерах.
Соответственно в методическом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (I) или его соли или его стереоизомера или его защищенного производного, при этом способ включает в себя следующее:
введение в реакцию соединения формулы (III):

где P1 представляет собой гидроксизащитную группу, L представляет собой уходящую группу, каждый из R1a, R2a, R3a и R4a независимо определяются как такой же самый, как и R1, R2, R3 и R4 в формуле (I), или -OP2, где P2 представляет собой гидроксизащитную группу с соединением формулы (IV):
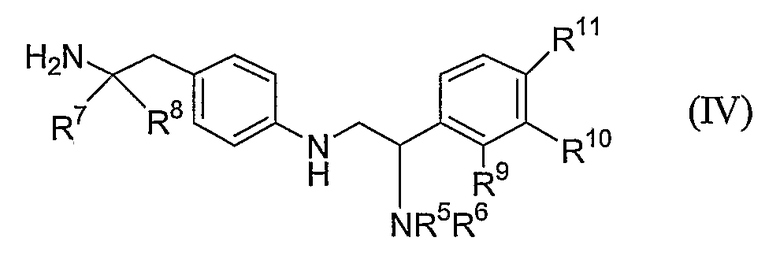
где R5, R6, R7, R8, R9, R10 и R11 определяют как в формуле (I) с получением соединения формулы (V):

удаление защитной группы P1 с получением соединения формулы (VI):
когда любой из R1a, R2a, R3a или R4a представляет собой -OP2, удаление защитной группы P2 с получением соединения формулы (I) или его соли или стереоизомера.
Фармацевтические композиции
Настоящее изобретение связано также с фармацевтическими композициями, включающими в себя соединение согласно изобретению. Соответственно такое соединение, предпочтительно в виде фармацевтически приемлемой соли, может быть составлено в композицию в любой подходящей для введения форме, такой как форма для перорального или парентерального введения или введения путем ингаляции.
В качестве иллюстрации такое соединение может быть смешано с подходящими фармацевтическими носителями и наполнителями и использовано в виде порошков, таблеток, капсул, эликсиров, суспензий, сиропов, облаток и пр. Такие фармацевтические композиции будут содержать приблизительно от 0,05 и примерно до 90% по весу активного соединения, а в более общем случае - от примерно 0,1 до примерно 30%. Фармацевтические композиции могут содержать обычно используемые носители и наполнители, такие как кукурузный крахмал или желатин, лактоза, сульфат магния, стеарат магния, сахароза, микрокристаллическая целлюлоза, каолин, маннит, дикальцийфосфат, хлорид натрия и альгиновая кислота. Дезинтеграторы, или разрыхлители, обычно используемые в композициях согласно изобретению, включают в себя кроскармелозу, микрокристаллическую целлюлозу, кукурузный крахмал, натрий-крахмал-гликолят и альгиновую кислоту.
Жидкая композиция будет, как правило, состоять из суспензии или раствора такого соединения или его фармацевтически приемлемой соли в подходящем жидком носителе(носителях), например, этаноле, глицерине, сорбите, неводном растворителе, таком как полиэтиленгликоль, маслах или воде, необязательно в присутствии суспендирующего агента, солюбилизирующего агента (такого как циклодекстрин), консерванта, поверхностно-активного агента, смачивающего агента, вкусовой добавки или окрашивающего агента. Альтернативно жидкая композиция может быть получена из восстанавливаемого порошка.
Например, порошок, содержащий активное соединение, суспендирующий агент, сахарозу и подсластитель, может быть восстановлен водой с образованием суспензии; сироп может быть получен из порошка, содержащего активный ингредиент, сахарозу и подсластитель.
Композиция в виде таблетки может быть получена с использованием любого подходящего фармацевтически приемлемого носителя (носителей), обычно используемого для приготовления твердых композиций. Примеры таких носителей включают в себя стеарат магния, крахмал, лактозу, сахарозу, микрокристаллическую целлюлозу и связующие вещества, например, поливинилпирролидон. Таблетка может быть получена также в виде таблетки, покрытой окрашенным слоем или же включенным в ее состав красителем в качестве носителя (носителей). Кроме того, активное соединение может быть составлено в композицию лекарственной формы с контролируемым высвобождением в виде таблетки, содержащей гидрофильный или гидрофобный матрикс.
Композиция в виде капсулы может быть получена с использованием обычных способов инкапсулирования, например, путем включения активного соединения и наполнителей в твердую желатиновую капсулу. Альтернативно, могут быть получены полутвердый матрикс активного соединения и высокомолекулярный полиэтиленгликоль с последующим заполнением ими твердых желатиновых капсул; или же могут быть получены раствор активного соединения или суспензия активного соединения в полиэтиленгликоле или суспензия в составе пищевого масла, например, жидкого парафина или фракционированного масла кокоса с последующим заполнением мягких желатиновых капсул.
Связующими веществами в таблетках могут служить гуммиарабик, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, поливинилпирролидон (повидон), гидроксипропилметилцеллюлоза, сахароза, крахмал и этилцеллюлоза. Скользящие вещества, которые могут быть использованы, могут включать в себя стеарат магния или стеараты других металлов, стеариновую кислоту, кремнийорганическую жидкость, тальк, воск, масла и коллоидную окись кремния.
Могут быть использованы ароматизирующие вещества, такие как мята, винтергреновое масло, вишневая отдушка и пр. Кроме того, для придания лекарственной форме более привлекательного вида или же для того, чтобы продукт легче было идентифицировать, может оказаться необходимым добавление красящего агента.
Соединения согласно изобретению и их фармацевтически приемлемые соли, которые активны при парентеральном введении, могут быть составлены в фармацевтические композиции для внутримышечного, интратекального (внутриоболочечного) или внутривенного введения.
Обычная композиция для внутримышечного или интратекального введения будет состоять из суспензии или раствора активного ингредиента в масле, например, арахисовом масле или кунжутном масле. Обычная композиция для внутривенного или интратекального введения будет состоять из стерильного изотонического водного раствора, содержащего, например, активный ингредиент и декстрозу или смесь декстрозы и хлорида натрия. Другими примерами являются лактат Рингера для инъекций, лактата Рингера плюс декстроза для инъекций, Нормозол-M и декстроза, Изолит E, ацилат Рингера для инъекций и т.п. В композиции может - хотя и не обязательно - быть использован вспомогательный растворитель, например, полиэтиленгликоль; хелатирующий агент, например, этилендиаминтетрауксусная кислота; солюбилизирующий агент, например, циклодекстрин; и антиоксидант, например, метабисульфит натрия. Альтернативно раствор может быть лиофильно высушен, а затем восстановлен соответствующим растворителем непосредственно перед введением.
Соединения согласно изобретению и их фармацевтически приемлемые соли, которые активны при местном введении, могут быть составлены в виде трансдермальных композиций или устройств для трансдермальной доставки ("накладок или пластырей"). Такие композиции включают в себя, например, вкладыш, резервуар для активного соединения, контрольную мембрану, прокладку и контактный клей. Такие трансдермальные пластыри могут быть использованы с целью обеспечения непрерывной или дискретной инфузии соединений согласно изобретению в контролируемых количествах. Конструирование и применение трансдермальных пластырей для доставки фармацевтических средств хорошо известно в данной области. См., например, патент США No. 5023252. Такие пластыри могут быть сконструированы для непрерывной, периодической доставки фармацевтических средств или же их доставки по мере необходимости.
Одним из предпочтительных способов введения соединений согласно изобретению является ингаляция. Ингаляция является эффективным приемом для доставки лекарственного средства непосредственно в дыхательные пути. Существует три основных типа устройств-ингаляторов для фармацевтических средств: ингаляторы-распылители, ингаляторы сухого порошка (DPI) и ингаляторы, снабженные дозаторами (MDI). Обычные устройства-распылители обеспечивают высокоскоростной поток воздуха, который заставляет терапевтическое средство распыляться в виде аэрозоля, который доставляется в дыхательные пути пациента. Композиция терапевтического средства составляется в жидкой форме, такой как раствор или суспензия микронизированных частиц, имеющих пригодный для их вдыхания размер, причем под микронизацией обычно подразумевается, что приблизительно 90% или более таких частиц имеют диаметр приблизительно менее чем 10 мкм.
Типичной композицией для применения в обычном устройстве-распылителе является изотонический водный раствор фармацевтической соли активного агента при концентрации активного агента, составляющей приблизительно от 0,05 мкг/мл до 1 мкг/мл. Соответствующие устройства-распылители являются коммерчески доступными, например, от производителя PARI GmbH (Старнберг, Германия). Другие устройства-распылители описаны, например, в патенте США 6123068.
Посредством ингаляторов сухого порошка, DPI, терапевтическое средство обычно вводят в виде свободного летучего порошка, который в процессе вдоха может быть диспергирован в дыхательных путях пациента. Разработаны также альтернативные устройства DPI, в которых для дисперсии порошка используются внешние источники энергии. Для получения свободного летучего порошка терапевтическое средство может быть составлено с соответствующим наполнителем (например, лактозой или крахмалом). Композицию сухого порошка можно получить, например, путем объединения частиц сухой лактозы с микронизированными частицами подходящей лекарственной формы, обычно фармацевтически приемлемой соли соединения согласно изобретению (т.е. активного агента) и сухого смешивания. В альтернативном варианте такой агент может быть составлен без наполнителей. Такую композицию загружают в распылитель сухого порошка, либо в картриджи ингалятора, либо в капсулы для применения в устройстве для доставки сухого порошка.
Примеры коммерчески доступных устройств DPI включают в себя Diskhaler (GlaxoSmithKline, Research Triangle Park, NC) (см., например, патент США No. 5035237); Diskus (GlaxoSmithKline) (см., например, патент США No. 6378519; Turbuhaler (AstraZeneca, Wilmington, DE) (см., например, патент США No. 4524769); и Rotahaler (GlaxoSmithKline) (см., например, патент США No. 4353365). Дополнительные примеры подходящих устройств DPI описаны в патентах США No. 5415162, 5239993 и 5715810, а также в приведенных в них ссылках.
Обычно из MDI высвобождается определенное количество терапевтического агента под действием сжатого вытеснительного газа. Композиции для введения с помощью MDI включают в себя раствор или суспензию активного ингредиента в ожиженном пропелленте. Несмотря на то что обычно в качестве пропеллентов используются хлорфторуглероды, такие как CCl3F, ввиду неблагоприятного воздействия, оказываемого такими агентами на озонный слой, получили развитие также композиции, в которых используются гидрофторалканы (HFA), такие как 1,1,1,2-тетрафторэтан (HFA 134a) и 1,1,1,2,3,3,3-гептафтор-н-пропан, (HFA 227). Дополнительные компоненты композиций HFA для введения с помощью MDI включают в себя сорастворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как сорбитантриолеат, олеиновая кислота, лецитин и глицерин. (См., например, патент США No. 5225183, европейский патент EP 0717987 A2 и международную заявку WO 92/22286.)
Таким образом, подходящая композиция для введения с помощью MDI может включать в себя от примерно 0,001% и до примерно 2% по весу кристаллической формы согласно изобретению, приблизительно от 0% и примерно до 20% по весу этанола и приблизительно от 0% примерно до 5% по весу поверхностно-активного вещества, при этом остальное количество составляет пропеллент HFA. При этом в одном из подходов для приготовления композиции к флакону, содержащему кристаллическую форму согласно изобретению, добавляют охлажденный или спрессованный гидрофторалкан, этанол (если он присутствует в композиции) и поверхностно-активное вещество (если оно присутствует в композиции). Для приготовления суспензии фармацевтическую соль используют в виде микронизированных частиц. Композицию загружают в аэрозольную канистру, которая представляет собой часть устройства MDI. Примерами устройств MDI, разработанных, в частности, для использования пропеллентов HFA, являются патенты США No. 6006745 и 6143227.
В альтернативном варианте композицию суспензии получают путем нанесения при распылении поверхностно-активного вещества на микронизированные частицы фармацевтической соли активного соединения. (См., например, WO 99/53901 и WO 00/61108). В качестве дополнительных примеров способа получения вдыхаемых частиц, а также композиций и устройств, подходящих для дозированной ингаляции, см. патенты США No. 6268533, 5983956, 5874063 и 6221398, а также WO 99/55319 и WO 00/30614.
Подразумевается, что любая форма соединений согласно изобретению (т.е. свободное основание, фармацевтическая соль или сольват), которая является подходящей для конкретного характера введения, может быть использована в описанных выше фармацевтических композициях.
Активные соединения используются в качестве агонистов β2-адренергических рецепторов, а следовательно, могут быть использованы для лечения заболеваний человека или состояний, опосредованных β2-адренергическими рецепторами или ассоциированных с активностью β2-адренергических рецепторов у млекопитающих, т.е. при медицинских показаниях, когда состояние может быть улучшено под действием лечения с помощью агонистов β2-адренергических рецепторов. Такие состояния включают в себя, не ограничиваясь перечисленным, легочные заболевания, такие как астма или хроническая обструктивная болезнь легких, преждевременные роды, неврологическое заболевание, сердечное расстройство или воспаление.
Активные соединения эффективны в широком интервале доз и обычно вводятся в терапевтически эффективном количестве. Однако должно быть понятно, что на самом деле количество вводимого соединения будет определяться лечащим врачом в зависимости от конкретных обстоятельств, включая состояние, которое должно быть подвержено лечению, выбранный путь введения, конкретно вводимое соединение и его относительная активность, возраст, вес и индивидуальная реакция пациента, тяжесть симптомов конкретного пациента и пр.
Подходящими дозами терапевтических агентов для введения путем ингаляции обычно являются дозы в интервале от примерно 0,05 мкг/день до примерно 1000 мкг/день, предпочтительно от примерно 0,1 мкг/день до примерно 500 мкг/день. Разумеется, что при определении доз, подходящих для введения путем ингаляции, следует учитывать характеристики конкретных устройств, связанные с тем, какая часть активного агента доставляется с их помощью в легкие пациента.
Соединение можно вводить с разной периодичностью: еженедельно, многократно в течение недели, ежедневно или по несколько раз в день. В курсе лечения может потребоваться введение в течение длительного периода, например, в течение нескольких недель или месяцев, или же курс лечения может потребовать хронического введения. Подходящими дозами для перорального введения обычно являются дозы от примерно 0,05 мкг/день до примерно 100 мг/день, предпочтительно от примерно 0,5 мкг/день до примерно 1000 мкг/день.
К другим свойствам соединений согласно изобретению относится то, что эти соединения, как было обнаружено, являются мощными и избирательными агонистами β2-адренергических рецепторов. В частности, соединения согласно изобретению проявляют исключительную избирательность в отношении β2-адренергических рецепторов по сравнению с β1- и β3-адренергическими рецепторами. Более того, как было обнаружено, соединения согласно изобретению оказывают поразительное и неожиданное по продолжительности действие. Как описано в приведенных ниже биологических исследованиях, длительность действия соединений согласно изобретению превышает 24 часа на животных моделях бронхопротекции.
Таким образом, настоящее изобретение обеспечивает способ лечения у млекопитающего заболевания или состояния, ассоциированного с активностью β2-адренергических рецепторов, включающий в себя введение млекопитающему терапевтически эффективного количества соединения согласно изобретению или фармацевтической композиции, включающей в себя соединение согласно изобретению.
Активные агенты согласно изобретению могут быть введены также совместно с одним или более другими терапевтическими агентами. Например, агенты согласно изобретению могут быть введены в сочетании с терапевтическими агентами, выбранными из противовоспалительных агентов (например, кортикостероидов и нестероидных противовоспалительных агентов (NSAID), антихолинергических агентов (в частности, антагонистов мускариновых рецепторов), других агонистов β2-адренергических рецепторов, противоинфекционных агентов (например, антибиотиков и противовирусных агентов) или антигистаминов. Настоящее изобретение, таким образом, связано, в следующем аспекте, с комбинацией, включающей в себя соединение согласно изобретению вместе с одним или более терапевтическими агентами, например, противовоспалительным агентом, антихолинергическим агентом, другим агонистом β2-адренергических рецепторов, противоинфекционным агентом или антигистамином.
Указанные другие терапевтические агенты могут быть использованы в виде фармацевтически приемлемых солей и сольватов. В случае необходимости эти другие терапевтические агенты могут быть использованы в виде оптически чистых стереоизомеров.
Подходящие противовоспалительные агенты включают в себя кортикостероиды и NSAID. Подходящими кортикостероидами, которые могут быть использованы в комбинации с соединениями согласно изобретению, являются такие перорально и путем ингаляции вводимые кортикостероиды и их пролекарственные формы, которые обладают противовоспалительной активностью. Примеры включают в себя метилпреднизолон, преднизолон, дексаметазон, флутиказонпропионат, S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-тиокарбоновой кислоты, S-(2-оксотетрагидрофуран-3S-ил)эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-пропионилокси-андроста-1,4-диен-17β-тиокарбоновой кислоты, сложные беклометазоновые эфиры (например, сложный эфир 17-пропионата или сложный эфир 17,21-дипропионата), будезонид, флунизолид, сложные эфиры мометазона (например, сложный эфир фуроата), триамцинолонацетонид, рофлепонид, циклезонид, бутиксокортпропионат, RPR-106541 и ST-126. Предпочтительные кортикостероиды включают в себя флутиказонпропионат, S-фторметиловый эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-17α-[(4-метил-1,3-тиазол-5-карбонил)окси]-3-оксо-андроста-1,4-диен-17β-тиокарбоновой кислоты и S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-тиокарбоновой кислоты, более предпочтительно S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-тиокарбоновой кислоты.
Подходящие NSAID включают в себя хромогликат натрия; недохромил натрия; ингибиторы фосфодиэстеразы (PDE) (например, теофиллин, ингибиторы PDE4 или смешанные ингибиторы PDE3/PDE4); антагонисты лейкотриена (например, монтелейкаст); ингибиторы синтеза лейкотриенов; ингибиторы iNOS; протеиназные ингибиторы, такие как ингибиторы триптазы и эластазы; антагонисты бета-2-интегрина и агонисты и антагонисты аденозинового рецептора (например, аденозиновые 2a-агонисты); антагонисты цитокинов (например, антагонисты хемокинов, такие как интерлейкиновое антитело (αIL-антитело), в частности, αIL-4-терапию, αIL-13-терапию или их комбинацию); или ингибиторы цитокинового синтеза. Подходящие другие агонисты β2-адренергического рецептора включают в себя сальметерол (такой, например, как ксинафоат), сальбутамол (такой, например, как сульфат или свободное основание), формотерол (такой, например, как фумарат), фенотерол или тербуталин или их соли.
Также интересным является применение согласно изобретению активного агента в комбинации с ингибитором фосфодиэстеразы 4 (PDE4) или смешанными ингибиторами PDE3/PDE4. Репрезентативные ингибиторы фосфодиэстеразы-4 (PDE4) или смешанные ингибиторы PDE3/PDE4 включают в себя, не ограничиваясь перечисленным, цис- 4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту, 2-карбометокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он; цис- [4-циано-4-(3-циклопропилметокси-4-дифторометоксифенил)циклогексан-1-ол]; цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновую кислоту и т.п. или их фармацевтически приемлемые соли. Другие репрезентативные ингибиторы PDE4 или смешанные ингибиторы PDE4/PDE3 включают в себя AWD-12-281 (элбион); NCS-613 (INSERM); D-4418 (Chiroscience and Schering-Plough); CI-1018 или PD-168787 (Pfizer); бензодиоксоловые соединения, описанные в публикациях WO99/16766 (Kyowa Hakko); K-34 (Kyowa Hakko); V-11294A (Napp); рофлумиласт (Byk-Gulden); фталазиноновые соединения, описанные в публикациях WO 99/47505 (Byk-Gulden); Пумафентрин (Byk-Gulden, ныне Altana); арофиллин (Almirall-Prodesfarma); VM554/UM565 (Vernalis); T-440 (Tanabe Seiyaku); и T2585 (Tanabe Seiyaku).
Подходящими антихолинергическими агентами являются такие соединения, которые действуют в качестве антагонистов мускариновых рецепторов, в частности, те соединения, которые являются антагонистами M1-, M2- или M3-рецепторов, или их комбинации. Примеры соединений включают в себя алкалоиды из растений белладонны, как проиллюстрировано аналогами атропина, скополамина, гоматропина, хиосциамина; обычно эти соединения, которые являются третичными аминами, вводят в виде соли. Такие лекарственные средства, в частности, в виде солей, легко доступны из целого ряда коммерческих источников или же могут быть получены согласно литературным данным, а именно:
Атропин - CAS-51-55-8 или CAS-51-48-1 (безводная форма), атропинсульфат - CAS-5908-99-6; атропиноксид - CAS-4438-22-6 или его соль соляной кислоты - CAS-4574-60-1 и метилатропиннитрат - CAS-52-88-0.
Гоматропин - CAS-87-00-3, гидробромидная соль - CAS-51-56-9, метилбромидная соль - CAS-80-49-9.
Хиосциамин (d, l) - CAS-101-31-5, гидробромидная соль - CAS-306-03-6 и сульфатная соль - CAS-6835-16-1.
Скополамин - CAS-51-34-3, гидробромидная соль - CAS-6533-68-2, метилбромидная соль - CAS-155-41-9.
Предпочтительные антихолинергические средства включают в себя ипратропиум (такой, например, как бромид), имеющийся в продаже под названием Атровент, окситропиум (такой, например, как бромид) и тиотропиум (такой, например, как бромид) (CAS-139404-48-1). Представляют интерес также метантелин (CAS-53-46-3), пропантелина бромид (CAS-50-34-9), анизотропина метилбромид, или Валпин 50 (CAS-80-50-2), клидиниума бромид (Quarzan, CAS-3485-62-9), копирролат (Робинул), изопропамида иодид (CAS-71-81-8), мепензолата бромид (патент США No. 2918408), хлорид тридигексетила (Патилон, CAS-4310-35-4) и метилсульфат гексоциклиума (Tral, CAS-115-63-9). См. также циклопентолатгидрохлорид (CAS-5870-29-1), тропикамид (CAS-1508-75-4), гидрохлорид тригексифенидила (CAS-144-11-6), пирензепин (CAS-29868-97-1), телензепин (CAS-80880-90-9), AF-DX 116, или метоктрамин, и соединения, описанные в международной заявке WO 01/04118, описание которой включено в настоящее описание в виде ссылки.
Соответствующие антигистаминные средства (называемые также антагонистами H1-рецептора) включают в себя любой один или более из целого ряда известных антагонистов, которые ингибируют H1-рецепторы и применение которых является безопасным для человека. Все они являются обратимыми, конкурентными ингибиторами взаимодействия гистамина с H1-рецепторами. Большая часть этих ингибиторов, в основном антагонисты первого поколения, характеризуется, на основе структуры их корового ядра, как этаноламины, этилендиамины и алкиламины. Кроме того, другие антигистаминные средства первого поколения включают в себя такие, которые могут быть охарактеризованы как основанные на пиперизине и фенотиазинах. Антагонисты второго поколения, которые являются неседативными средствами, имеют сходное соотношение структура-активность, в рамках которого они сохраняют этиленовую группу корового ядра (алкиламины) или имитируют группу третичного амина пиперазином или пиперидином. Примерами антагонистов являются следующие вещества:
Этаноламины: карбиноксамина малеат, клемастина фумарат, дифенилгидрамина гидрохлорид и дименгидринат.
Этилендиамины: пириламина малеат, трипеленнамин-HCl и трипеленнамина цитрат.
Алкиламины: хлорфенирамин и его соли, такие как малеатная соль, и акривастин.
Пиперазины: гидроксизин-HCl, гидроксизина памоат, циклизин-HCl, циклизина лактат, меклизин-HCl и цетризин-HCl.
Пиперидины: Астемизол, левокабастин-HCl, лоратадин или его дескарбоэтокси-аналог и терфенадина и фексофенадина гидрохлорид или другая фармацевтически приемлемая соль.
Азеластина гидрохлорид является еще одним антагонистом H1-рецептора, который может быть использован в сочетании с соединением согласно изобретению.
Примеры предпочтительных антигистаминов включают в себя метапирилен и лоратадин.
Таким образом, в следующем аспекте настоящее изобретение связано с комбинацией, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль или сольват или стереоизомер, а также кортикостероид. В частности, настоящее изобретение связано с комбинацией, в которой кортикостероид представляет собой флутиказона пропионат или в которой кортикостероид представляет собой S-фторметильный эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-тиокарбоновой кислоты или S-(2-оксо-тетрагидрофуран-3S-ил)-эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-припионилокси-андроста-1,4-диен-17β-тиокарбоновой кислоты.
Таким образом, в следующем аспекте настоящее изобретение связано с комбинацией, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль или сольват или стереоизомер, а также ингибитор PDE4.
В следующем аспекте настоящее изобретение, таким образом, связано с комбинацией, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль или сольват или стереоизомер, а также антихолинергический агент.
В следующем аспекте настоящее изобретение связано, таким образом, с комбинацией, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль или сольват или стереоизомер, а также антигистамин.
В следующем аспекте настоящее изобретение связано, таким образом, с комбинацией, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль или сольват или стереоизомер, а также ингибитор PDE4 и кортикостероид.
В следующем аспекте настоящее изобретение связано, таким образом, с комбинацией, включающей в себя соединение формулы (I) или его фармацевтически приемлемую соль или сольват или стереоизомер, а также антихолинергический агент и кортикостероид.
В указанных выше комбинациях термин “соединение формулы (I)” включает в себя соединение формулы (II) и его предпочтительные группы, а также любое индивидуально описанное соединение или соединения.
Соответственно фармацевтические композиции согласно изобретению могут - необязательно - включать в себя соединение формулы (I) или его фармацевтически приемлемую соль или сольват или стереоизомер, а также один или более других терапевтических агентов, как описано выше.
Индивидуальные соединения таких комбинаций могут вводиться либо последовательно, либо одновременно в составе раздельных или комбинированных фармацевтических композиций. Соответствующие дозы известных терапевтических агентов хорошо известны специалистам в данной области. Следовательно, способы лечения согласно изобретению включают в себя введение индивидуальных соединений таких комбинаций либо последовательно, либо одновременно в составе раздельных или комбинированных фармацевтических композиций.
Таким образом, согласно следующему аспекту настоящее изобретение связано со способом лечения заболевания или состояния, ассоциированного у млекопитающего с активностью β2-адренергических рецепторов, включающим в себя введение млекопитающему фармацевтически эффективного количества комбинации соединения формулы (I) или его приемлемой соли или сольвата или стереоизомера с одним или более другими терапевтическими агентами.
Поскольку соединения согласно изобретению являются агонистами β2-адренергических рецепторов, такие соединения используются также в качестве инструмента для научных исследований или для изучения биологических систем или проб, содержащих β2-адренергические рецепторы, или для поисков с целью открытия новых агонистов β2-адренергических рецепторов. Более того, поскольку соединения согласно изобретению проявляют селективность в отношении β2-адренергических рецепторов по сравнению со связыванием и функциональной активностью в отношении других подтипов β-адренергических рецепторов, такие соединения используются также для изучения эффектов избирательного агонистического действия β2-адренергических рецепторов в биологической системе или биологической пробе. Любая подходящая биологическая система или биологическая проба, содержащая β2-адренергические рецепторы, может быть использована в таких исследованиях, которые могут проводиться либо in vitro, либо in vivo.
Репрезентативные биологические системы или пробы (образцы) включают в себя, не ограничиваясь перечисленным, клетки, клеточные экстракты, плазматические мембраны, образцы тканей, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.) и пр. Агонистические эффекты β2-адренергических рецепторов определяются с помощью обычных процедур и оборудования, таких как анализы связывания радиоактивных лигандов и функциональные анализы, например, описанный выше анализ опосредованных лигандом изменений уровня внутриклеточного циклического аденозинмонофосфата (цАМФ) или анализы аналогичного свойства. Количество соединения согласно изобретению, которое вызывает агонистическое действие в отношении β2-адренергических рецепторов, обычно колеблется приблизительно от 1 наномоля и примерно до 1000 наномолей. Когда соединения согласно изобретению используются в качестве инструмента для исследований в поисках новых агонистов β2-адренергических рецепторов, изобретение также включает в себя в качестве отдельных воплощений как наработку сравнительных данных (путем проведения соответствующих анализов), так и данные тест-анализа по идентификации представляющих интерес тест-соединений.
Следующие неограничивающие примеры иллюстрируют репрезентативные фармацевтические композиции согласно изобретению. Дополнительные подходящие носители для композиций активных соединений согласно изобретению можно найти в публикации Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & Wilkins, Philadelphia, PA, 2000.
Пример композиции A
В этом примере проиллюстрировано получение репрезентативной фармацевтической композиции для перорального введения соединения согласно изобретению:
Указанные выше ингредиенты смешивали и вносили в твердую желатиновую капсулу.
Пример композиции B
В этом примере проиллюстрировано получение другой репрезентативной фармацевтической композиции для перорального введения соединения согласно изобретению:
Указанные выше ингредиенты тщательно смешивали и спрессовывали в отдельные таблетки с насечкой.
Пример композиции C
В этом примере проиллюстрировано получение репрезентативной фармацевтической композиции для перорального введения соединения согласно изобретению.
Получена пероральная суспензия, имеющая следующий состав.
Пример композиции D
В этом примере проиллюстрировано получение репрезентативной фармацевтической композиции, содержащей соединение согласно изобретению.
Получена пероральная композиция, забуференная при рН 4, имеющая следующий состав:
Пример композиции E
В этом примере проиллюстрировано получение репрезентативной фармацевтической композиции для инъекции соединения согласно изобретению.
Восстановленный раствор получали путем добавления 20 мл стерильной воды к 1 мг соединения согласно изобретению. Перед употреблением раствор дополнительно разбавляли 200 мл жидкости для внутривенного введения, которая совместима с активным соединением. Такие жидкости выбирают из 5% раствора декстрозы, 0,9% хлорида натрия или смеси 5% декстрозы и 0,9% хлорида натрия. Другими примерами являются лактат Рингера для инъекций, лактат Рингера плюс 5% декстрозы для инъекций, Нормозол-M и 5% декстроза, Изолит E и ацилат Рингера для инъекций.
Пример композиции F
В этом примере проиллюстрировано получение репрезентативной фармацевтической композиции для местного применения соединения согласно изобретению.
Все из указанных выше ингредиентов, за исключением воды, объединяли и нагревали до 60°C при перемешивании. Затем добавляли необходимое количество воды при 60°C при тщательном перемешивании для получения эмульсии ингредиентов, а затем добавляли необходимое количество воды для доведения до 100 г.
Пример композиции G
В этом примере проиллюстрировано получение репрезентативной фармацевтической композиции, содержащей соединение согласно изобретению.
Водную аэрозольную композицию для использования в распылителе получали путем растворения 0,1 мг фармацевтически приемлемой соли активного соединения в 0,9% растворе хлорида натрия, подкисленного лимонной кислотой. Полученную смесь перемешивали и обрабатывали ультразвуком до тех пор, пока растворится активная соль. pH раствора доводили до значения в интервале от 3 до 8 путем медленного добавления NaOH.
Пример композиции H
В этом примере проиллюстрировано получение композиции сухого порошка, содержащего соединение согласно изобретению, для использования в ингаляционных картриджах.
Желатиновые картриджи для ингаляции наполняли фармацевтической композицией, содержащей следующие ингредиенты:
Фармацевтически приемлемую соль активного соединения микронизировали перед ее смешиванием с лактозой. Содержимое картриджей вводили с помощью порошкового ингалятора.
Пример композиции I
В этом примере проиллюстрировано получение композиции сухого порошка, содержащего соединение согласно изобретению, для использования в устройстве для ингаляции сухого порошка.
Фармацевтическую композицию получали из расчета, что отношение объема композиции микронизированной фармацевтически приемлемой соли к лактозе составляет 1:200. Композицию помещали в устройство для ингаляции сухого порошка, способное доставлять между примерно 10 мкг и примерно 100 мкг активного ингредиента лекарственного средства на дозу.
Пример композиции J
В этом примере проиллюстрировано получение композиции, содержащей соединение согласно изобретению, для использования в ингаляторе, снабженном дозатором.
Суспензию, содержащую 5% фармацевтически приемлемой соли активного соединения, 0,5% лецитина и 0,5% трегалозы, получали путем диспергирования 5 г активного соединения в виде микронизированных частиц со средним размером менее чем 10 мкм в коллоидном растворе, образованном из 0,5 г трегалозы и 0,5 г лецитина, растворенного в 100 мл деминерализованной воды. Суспензию сушили путем распыления, и полученный материал микронизировали до частиц, имеющих средний диаметр менее чем 1,5 мкм. Полученные частицы загружали в канистры со спрессованным 1,1,1,2-тетрафторэтаном.
Пример композиции K
В этом примере проиллюстрировано получение композиции, содержащей соединение согласно изобретению, для использования в ингаляторе, снабженном дозатором.
Суспензию, содержащую 5% фармацевтически приемлемой соли активного соединения и 0,1% лецитина, получали путем диспергирования 10 г активного соединения в виде микронизированных частиц со средним размером менее чем 10 мкм в растворе, образованном из 0,2 г лецитина, растворенного в 200 мл деминерализованной воды. Суспензию сушили путем распыления, и полученный материал микронизировали до частиц, имеющих средний диаметр менее чем 1,5 мкм. Полученные частицы загружали в канистры со спрессованным 1,1,1,2,3,3,3-гептафтор-н-пропаном.
Биологические анализы
Соединения согласно изобретению и их фармацевтически приемлемые соли обладают биологической активностью и используются в медицине для лечения заболеваний. Способность соединения связываться с β2-адренергическим рецептором, а также его избирательность, потенциал в качестве агониста и внутренняя активность могут быть продемонстрированы с помощью тестов A-B, представленных ниже, или же могут быть продемонстрированы с помощью других тестов, которые хорошо знакомы специалистам в данной области.
Используемые сокращения:
% эфф. -% эффективности
ATCC - Американская Коллекция Типовых Культур
БСА - бычий сывороточный альбумин
цАМФ - циклический аденозин 3':5'-монофосфат
DMEM - модифицированная Дульбекко среда Игла
ДМСО - диметилсульфоксид
EDTA - этилендиаминтетрауксусная кислота
Emax - максимальная эффективность
ЭБС - эмбриональная бычья сыворотка
Gly - глицин
HEK-293 - клетки - 293 эмбриональной почки человека
PBS - забуференный фосфатом физиологический раствор
об/мин - обороты в минуту
Tris - Трис(гидроксиметил)аминометан
Мембранный препарат из клеток, экспрессирующих человеческие β 1 -или β 2 -адренергические рецепторы
Полученную из клеток HEK-293 линию клеток, стабильно экспрессирующую клонированные человеческие β1- или β2-адренергические рецепторы соответственно, растили до достижения конфлуэнтности в среде DMEM, содержащей 10% диализированной ЭБС, в присутствии 500 мкг/мл генетицина. Клеточный монослой снимали механически с помощью скребка, используя версен, 1:5000 (0,2 г/л EDTA в PBS). Клетки осаждали путем центрифугирования, 1000 об/мин, и клеточные осадки либо замораживали при -80°C для хранения, либо из них сразу же выделяли клеточные мембраны. Для получения мембран клеточные осадки ресуспендировали в буфере для лизирования (10 мM Tris/HCl, pH 7,4, 4°C, одна таблетка из упаковки “Complete Protease Inhibitor Cocktail Tablets with 2 мM EDTA” на 50 мл буфера (Roche cat.# 1697498, Roche Molecular Biochemicals, Indianapolis, IN)) и гомогенизировали с помощью плотно пригнанного гомогенизатора Даунса (20 тактов) на льду. Гомогенат центрифугировали при 20000g, осадок единожды промывали буфером для лизирования путем ресуспендирования и центрифугирования, как указано выше. Конечный осадок ресуспендировали в мембранном буфере (75 мM Tris/HCl, pH 7,4, 12,5 мM MgCl2, 1 мM EDTA, 25°C). Концентрацию белка в мембранной суспензии определяли согласно методу Брэдфорда (Bradford MM., Analytical Biochemistry, 1976, 72, 248-54). Разделенные на аликвоты мембраны хранили при -80°C.
Тест A
Анализ радиоактивного связывания с человеческими β 1 - и β 2 -адренергическими рецепторами
Анализы связывания были предприняты в 96-луночных микротитровальных планшетах, с общим объемом проб, составляющим 100 мкл, с 5 мкг мембранного белка в случае мембран, содержащих человеческий β2-адренергический рецептор, или 2,5 мкг мембранного белка в случае мембран, содержащих человеческий β1-адренергический рецептор, в аналитическом буфере (75 мM Tris/HCl pH 7,4, 25°C, 12,5 мM MgCl2, 1 мM EDTA, 0,2% БСА). Изучение насыщения связывания с целью определения значений Kd радиоактивного лиганда было проведено с использованием [3H]дигидроальпренолола (NET-720, 100 Ки/ммоль, Perkin Elmer Life Sciences Inc., Boston, MA) при 10 различных концентрациях соединения в интервале от 0,01 нM до 200 нM. Анализы распределения с целью определения значений pKi для соединений были произведены с использованием [3H]дигидроальпренолола в концентрации 1 нM и при 10 различных концентрациях соединения в интервале от 40 пM до 10 мкM. Соединения растворяли до концентрации 10 мM в буфере для растворения (25 мM Gly-HCl, pH 3,0, с 50% ДМСО), затем разбавляли до концентрации 1 мM в 50 мM Gly-HCl, pH 3,0, и из этого раствора делали серийные разведения буфером для анализа. Неспецифическое связывание определяли в присутствии 10 мкM немеченого альпренолола. Аналитические пробы инкубировали в течение 90 минут при комнатной температуре, реакции связывания останавливали путем быстрой фильтрации через пластинки стеклянного волокнистого фильтра GF/B (Packard BioScience Co., Meriden, CT), предварительно замоченные в 0,3% полиэтиленимине. Фильтровальные пластинки трижды промывали буфером для фильтрации (75 мM Tris/HCl, pH 7,4, 4˚C, 12,5 мM MgCl2, 1 мM EDTA) для удаления несвязанной радиоактивности. Пластинки сушили, добавляли 50 мкл сцинтилляционной жидкости Microscint-20 (Packard BioScience Co., Meriden, CT), и пластинки просчитывали в жидкостном сцинтилляционном счетчике Packard Topcount (Packard BioScience Co., Meriden, CT). Результаты связывания анализировали с помощью анализа нелинейной регрессии, используя пакет компьютерных программ GraphPad Prism (GraphPad Software, Inc., San Diego, CA) и используя 3-параметрическую модель односторонней конкуренции. Минимум кривой пришелся на значение неспецифического связывания, которое было получено в присутствии 10 мкM альпренолола. Значения Ki для соединений просчитывали для наблюдаемых значений IC50 и значений Kd радиолиганда с помощью уравнения Cheng-Prusoff (Cheng Y, and Prusoff WH., Biochemical Pharmacology, 1973, 22, 23, 3099-108). Селективность в отношении подтипа рецептора рассчитывали как отношение Ki(β1)/Ki(β2). Соединения согласно изобретению продемонстрировали большее сродство связывания с β2-адренергическим рецептором, чем с β1-адренергическим рецептором, т.е. Ki(β1)>Ki(β2), с селективностью, приблизительно составляющей больше чем 100.
Тест B
Цельноклеточный цАМФ-Flashplate-анализ с использованием линий клеток, гетерологично экспрессирующих, соответственно, человеческий β 1 -адренорецептор, β 2 -адренорецептор и β 3 -адренорецептор
Линию клеток HEK-293, стабильно экспрессирующих клонированный человеческий β1-адренорецептор (клон H34.1), выращивали приблизительно до состояния 70%-90% конфлуэнтности в среде, состоящей из DMEM, обогащенной 10% ЭБС и 500 мкг/мл генетицина. Линию клеток HEK-293, стабильно экспрессирующих клонированный человеческий β2-адренорецептор (клон H24.14), выращивали в той же самой среде до достижения полной конфлуэнтности. Линию клеток CHO-K1, стабильно экспрессирующих клонированный человеческий β3-адренорецептор, выращивали приблизительно до состояния 70%-90% конфлуэнтности в среде Ham F-12, обогащенной 10% ЭБС и 800 мкг/мл генетицина, добавляемого к каждому пятому пассажу. За день до анализа культуры переносили в такую же ростовую среду, но без содержания антибиотиков.
Анализы цАМФ были предприняты в формате радиоиммунного анализа с использованием Flashplate-системы анализа активации аденилилциклазы и с использованием 125I-цАМФ (NEN SMP004, PerkinElmer Life Sciences Inc., Boston, MA) в соответствии с инструкциями производителя.
В день анализа клетки единожды промывали в PBS, осадок поднимали версеном, 1:5000 (0,2 г/л EDTA в PBS), и считали. Клетки осаждали центрифугированием при 1000 об/мин и ресуспендировали в стимулирующем буфере, предварительно нагретом до 37°C. В случае клеток, экспрессирующих β1-адренорецептор, к стимулирующему буферу добавляли 10 нM ICI 118551, и клетки инкубировали в течение 10 мин при 37°C. Клетки использовали в конечных концентрациях 30000, 40000 и 70000 клеток/лунку соответственно для случаев клеток, экспрессирующих β1-адренорецептор, β2-адренорецептор и β3-адренорецептор. Соединения растворяли до концентрации 10 мM в ДМСО, затем разбавляли до 1 мM в 50 мM Gly-HCl, pH 3,0 и из этого раствора получали серийные разведения буфером для анализа (75 мM Tris/HCl, pH 7,4, 25°C, 12,5 мM MgCl2, 1 мM EDTA, 0,2% БСА). Соединения тестировали в анализе, насчитывающем 11 различных концентраций в интервале от 10 мкM до 9,5 пM. Реакционные смеси инкубировали в течение 10 мин при 37°C и реакцию останавливали путем добавления 100 мкл ледяного проявляющего буфера. Планшеты запечатывали, инкубировали в течение ночи при 4°C и на следующее утро просчитывали в сцинтилляционном счетчике (Packard BioScience Co., Meriden, CT). Количество цАМФ, продуцируемое на мл реакционной смеси, рассчитывали на основе счета, полученного для образцов и стандартов цАМФ, как описано в руководстве производителя, прилагаемом для пользователя. Данные анализировали с использованием анализов нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с использованием 3-параметрической модели для сигмоидальной кривой доза-ответ (с точкой перегиба, равной 1). Эффективность агонистической активности выражали в виде значений pEC50.
Соединения согласно изобретению продемонстрировали в этом анализе высокий уровень активности в отношении β2-адренергических рецепторов, как видно из величин pEC50, которые приблизительно выше 8,5. Кроме того, тестируемые соединения проявляли селективность (избирательность) функциональной активности в отношении рецептора β2 по сравнению с функциональной активностью в отношении рецепторов β1 и β3. В частности, было показано, что для соединений согласно изобретению отношения EC50(β1)/EC50(β2) приблизительно составляют более чем 50, а отношения EC50(β3)/EC50(β2) приблизительно составляют более чем 600.
Тест C
Цельноклеточный цАМФ-Flashplate-анализ линии эпителиальных клеток легкого, эндогенно экспрессирующих человеческий β 2 -адренергический рецептор
Для определения агонистических активностей и эффективности (внутренних, присущих активностей) в клеточной линии, экспрессирующей эндогенные уровни β2-адренергического рецептора, была использована человеческая линия клеток эпителия легких (BEAS-2B) (ATCC CRL-9609, Американская Коллекция Типовых Культур, Manassas, VA) (January B, et al., British Journal of Pharmacology, 1998, 123, 4, 701-11). Клетки выращивали до достижения 75-90% конфлуэнтности в полной, бессывороточной среде (среда LHC-9, содержащая эпинефрин и ретиноевую кислоту, cat # 181-500, Biosource International, Camarillo, CA). За день до анализа среду меняли на LHC-8 (без эпинефрина или ретиноевой кислоты, cat # 141-500, Biosource International, Camarillo, CA).
Анализы цАМФ предпринимали в формате радиоиммунного анализа, используя систему цАМФ-Flashplate-анализа активации аденилилциклазы, с использованием 125I-цАМФ (NEN SMP004, PerkinElmer Life Sciences Inc., Boston, MA), в соответствии с инструкциями производителя.
В день анализа клетки промывали в PBS, механически отделяли от подложки в присутствии 5 мM EDTA в PBS и считали. Клетки осаждали путем центрифугирования при 1000 об/мин и ресуспендировали в буфере для стимуляции, предварительно нагретом до 37°C, при конечной концентрации 600000 клеток/мл. В анализе использовали клетки при конечной концентрации 30000 клеток/лунку. Соединения растворяли в буфере для растворения (25 мM Gly-HCl, pH 3,0, с добавлением 50% ДМСО) до концентрации 10 мM, затем доводили до 1 мM в 50 мM Gly-HCl, pH 3,0, и из этого раствора делали серийные разведения буфером для анализа (75 мM Tris/HCl, pH 7,4, 25°C, 12,5 мM MgCl2, 1 мM EDTA, 0,2% БСА).
В этом анализе соединения тестировали при 10 различных концентрациях в интервале от 10 мкM до 40 пM. Максимальный ответ отмечали в присутствии 10 мкM изопротеренола. Реакционные смеси инкубировали в течение 10 мин при 37°C, и реакцию останавливали путем добавления 100 мкл ледяного проявляющего буфера. Планшеты запечатывали, инкубировали в течение ночи при 4°C и на следующее утро просчитывали в жидкостном сцинтилляционном счетчике (Packard BioScience Co., Meriden, CT). Количество цАМФ, продуцируемое на миллилитр реакционной смеси, рассчитывали на основе счета, полученного для проб и стандартов цАМФ, в соответствии с описанием производителя. Данные анализировали с использованием анализов нелинейной регрессии с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с использованием 4-параметрической модели при различных наклонах сигмоидальной кривой доза-ответ. Эффективность агонистической активности выражали в виде значений pEC50. Соединения согласно изобретению, тестируемые в этом анализе, демонстрировали значения pEC50, которые составляли более 8.
Эффективность соединений (% эфф.) рассчитывали из отношения наблюдаемого Emax (пик соответствующей кривой) и максимального ответа, полученного для случая 10 мкM изопротеренола, и были выражены в виде % эффективности относительно изопротеренола. Для тестируемых соединений было показано, что % эффективности приблизительно составляет более чем 50.
Тест D
Анализ защиты ткани бронхов от ацетилхолин-индуцированного бронхоспазма на модели морских свинок
Группы животных, по 6 самцов морских свинок в каждой (Duncan-Hartley (HsdPoc:DH) Harlan, Madison, WI), весом между 250 и 350 г, индивидуально идентифицировали по этикеткам на клетках. В процессе исследования животные имели свободный доступ к воде и пище, получая и то, и другое ad libitum.
Тестируемые соединения вводили путем ингаляции, производимой в течение 10 минут в камере с дозированным количеством средства, в которую животное помещали целиком (R&S Molds, San Carlos, CA). Камеры с дозированным количеством средства были сконструированы таким образом, чтобы аэрозоль из центрального коллектора одновременно доставлялся в шесть индивидуальных камер. После 60-минутного периода акклиматизации и 10-минутного периода нахождения в камере, в которой была распылена вода для инъекций (WFI), морских свинок подвергали действию аэрозоля, содержащего тестируемое соединение или носитель (WFI). Такие аэрозоли получали из водных растворов с помощью набора для распыления, LC Star Nebulizer Set (Model 22F51, PARI Respiratory Equipment, Inc. Midlothian, VA), приводимого в действие смесью газов CO2 = 5%, O2 = 21% и N2 = 74%) под давлением 22 фунт/кв.дюйм. Поток газа через распылитель при указанном оперативном давлении составлял приблизительно 3 л/минуту. Производимые аэрозоли подавались в камеры под действием положительного давления. В период доставки аэрозольных растворов никакого разбавления воздухом не происходит. В течение 10-минутного распыления распыляется приблизительно 1,8 мл раствора. Это определяли гравиметрически путем сравнения весов заполненного распылительного устройства перед распылением и после него.
Бронхозащитный эффект соединений, вводимых путем ингаляции, оценивали с помощью плетизмографии целого организма через 1,5, 24, 48 и 72 часа после введения дозировки. За сорок пять минут до начала оценки легочного статуса каждую морскую свинку анестезировали путем внутримышечной инъекции кетамина (43,75 мг/кг), ксилазина (3,50 мг/кг) и ацепромазина (1,05 мг/кг). После того как операционное поле очищали, сбривая шерстку и обрабатывая 70% спиртом, производили срединный разрез вентральной части шеи длиной 2-5 см. Затем высвобождали яремную вену и канюлировали ее полиэтиленовым катетером (PE-50, Becton Dickinson, Sparks, MD), заполненным физиологическим раствором, чтобы получить возможность для внутривенных инфузий раствора, содержащего 0,1 мг/мл ацетилхолина (Ach), (Sigma-Aldrich, St. Louis, MO) в физиологическом растворе. Затем свободно рассекали трахею, и в нее вводили канюлю из тефлоновой трубки с маркировкой 14G (#NE-014, Small Parts, Miami Lakes, FL). В случае необходимости анестезию поддерживали дополнительными внутримышечными инъекциями указанной выше анестезирующей смеси. Глубину анестезии контролировали, усиливая ее в случае, если животное реагировало на щипок или сжимание его лапки или если частота дыхания начинала превосходить 100 вдохов/минуту.
Сразу же по завершении канюлирования животное помещали в плетизмограф (#PLY3114, Buxco Electronics, Inc., Sharon, CT) и вводили канюлю пищеводного давления для измерения легочного давления вытеснения (давление). Тефлоновую трахеальную трубку присоединяли к отверстию плетизмографа, чтобы предоставить возможность морской свинке дышать комнатным воздухом снаружи камеры. Затем камеру запечатывали. Для поддержания температуры тела животного использовали нагревательную лампу, и в легкие морской свинки трижды нагнетали по 4 мл воздуха с помощью калиброванного 10-миллилитрового шприца (#5520 Series, Hans Rudolph, Kansas City, MO), чтобы обеспечить гарантию того, что нижние дыхательные пути не повреждены и что животное не страдает от гипервентиляции.
Сразу же как только убеждались, что базовые значения установились в интервале от 0,3 до 0,9 мл/см H2O (контроль соответствия) и в интервале от 0,1 до 0,199 см H2O/мл в секунду (контроль прочности), начинали производить легочную (дыхательную) оценку. Компьютерная программа Buxco для оценки легочного дыхания позволяла накапливать пульмональные данные и устанавливать их разброс и ошибку. Запуск этой программы инициировал протокол эксперимента и накопление данных (по снимаемым компьютером показателям). Изменения объема [вдыхаемого воздуха] во времени, которые имели место внутри плетизмографа при каждом вдохе, измеряли посредством передатчика давления Buxco. Путем интегрирования этого сигнала во времени рассчитывали для каждого вдоха размер воздушного потока. Этот сигнал наряду с изменениями показателей легочного давления вытеснения (давление), которые фиксировались и накапливались с помощью передатчика давления Sensym (#TRD4100), объединяли посредством преамплификатора Buxco (MAX 2270) с интерфейсом накопленных данных (# SFT3400 и SFT3813). Все остальные легочные параметры были получены (выведены) на основании этих двух показателей.
Базовые значения накапливали в течение 5 минут, и по прошествии указанного промежутка времени морских свинок сенсибилизировали ацетилхолином (Ach). Aцетилхолин инфузировали внутривенно в течение 1 минуты с помощью шприца (sp210iw, World Precision Instruments, Inc., Sarasota, FL) в следующих дозах и в течение следующих предварительно намеченных промежутков времени, считая от начала эксперимента: 1,9 мкг/минуту, 5 минут, 3,8 мкг/минуту, 10 минут, 7,5 мкг/минуту, 15 минут, 15,0 мкг/минуту, 20 минут, 30 мкг/минуту, 25 минут, и 60 мкг/минуту, 30 минут. Если резистентность (прочность) или контроль соответствия в течение 3 минут после каждой дозы ацетилхолина не возвращались на базовый уровень, легкие морских свинок трижды продували 4 миллилитрами воздуха из 10-миллилитрового калиброванного шприца. Регистрируемые легочные параметры включали в себя частоту дыхания (число вдохов/минуту), контроль соответствия (мл/см H2O) и легочную резистентность (см H2O/мл в секунду) (Giles et al., 1971). Сразу же по завершении измерений легочных функций, на 35-й минуте, в соответствии с протоколом морских свинок доставали из плетизмографа и умерщвляли под действием CO2.
Количество PD2, которое определяли как количество ацетилхолина (Ach), необходимое для достижения двойного - по сравнению с базовым - уровня легочной резистентности, рассчитывали на основании данных по легочной резистентности, полученных из величин воздушного потока и давления во всем интервале сенсибилизации ацетилхолином (Ach) с помощью следующего уравнения. Это уравнение было получено из уравнения, используемого для расчета значений PC20 в клинике (Am. Thoracic Soc, 2000).

где
C1 = концентрации Ach, предшествующей C2 (от второй до последней концентрации Ach)
C2 = конечной концентрации Ach (концентрации, приводящей к 2-кратному повышению легочной резистентности (RL))
R0 = базовое значение RL
R1 = значение RL после C1
R2 = значение RL после C2
Статистический анализ данных проводили с использованием однопараметрического дисперсионного анализа и последующего тест-анализа Bonferroni/Dunn. Значения P<0,05 считались значимыми.
Кривые доза-ответ, соответствующие четырехпараметрическому логистическому уравнению, были построены с помощью графической программы Prism, версия 3.00 для Windows (GraphPad Software, San Diego, California).
Y=Min+(Max-Min)/(1+10∧((logED50-X)*наклон)),
где X представляет собой логарифм дозы, Y представляет собой [величину] ответа (PD2), а Y, начинаясь у отметки Min и асимптотически приближаясь к отметке Max, имеет сигмоидальную форму.
Было показано, что репрезентативные соединения согласно изобретению обладают значительной бронхопротективной активностью во временном периоде после 24 часов после приема дозы.
Следующие примеры синтеза предлагаются исключительно с целью иллюстрации изобретения и ни в коем случае не должны быть восприняты как ограничение объема настоящего изобретения.
Примеры
Общие замечания: Если не указано иначе, реагенты, стартовый материал и растворители были получены из коммерческих источников, например, Sigma-Aldrich (St. Louis, MO), J. T. Baker (Phillipsburg, NJ), а также Honeywell Burdick и Jackson (Muskegon, MI), и использованы без дальнейшей очистки; реакции проводили в атмосфере азота; реакционные смеси контролировали с помощью тонкослойной хроматографии (TLC на основе кремнезема), аналитической жидкостной хроматографии высокого разрешения (анал. HPLC) или масс-спектрометрии; реакционные смеси обычно очищали с использованием колоночной флэш-хроматографии на силикагеле или с использованием HPLC, пользуясь описанной ниже общей методикой; образцы ЯМР растворяли в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6), и спектры были получены на приборе Varian Gemini 2000 (300 MГц) в условиях стандартных параметров; и масс-спектрометрическую идентификацию осуществляли, используя метод электрораспылительной ионизации (ESMS) на приборе Perkin Elmer (PE SCIEX API 150 EX).
Пример 1: Синтез 5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она
a. Получение трет-бутилового эфира [2-(4-нитрофенил)этил]карбаминовой кислоты
Ди-трет-бутилбикарбонат (20 г, 92 ммоль) суспендировали в насыщенном гидрокарбонате (200 мл) и охлаждали до 0°C. Добавляли диоксан (10 мл). Раствор гидрохлорида 2-(4-нитрофенил)этиламина (20 г, 99 ммоль) получали путем покапельного добавления в воду (150 мл). В процессе добавления имела место кристаллизация. После окончания процесса добавления смесь перемешивали при 0°C в течение 15 минут, далее - при комнатной температуре в течение 16 часов. Продукт, полученный в результате фильтрации, промывали в воде (500 мл), затем сушили на воздухе.
b. Получение трет-бутилового эфира [2-(4-нитрофенил)этил]карбаминовой кислоты
К частично высушенному осадку, полученному на предыдущей стадии, добавляли палладий на угле (2 г, 10% Pd), с последующим добавлением метанола (250 мл, в атмосфере азота). Атмосферу заменяли атмосферой водорода и смесь перемешивали при атмосферном давлении в течение 24 часов. Осадок палладия удаляли посредством фильтрации и растворитель удаляли при пониженном давлении. Указанное в заголовке промежуточное соединение получали в виде грязно-белого твердого вещества (20 г, 85 ммоль, 93% после двух стадий).
c. Получение трет-бутилового эфира (R)-{[4-(2-трет-бутоксикарбониламиноэтил)фенилкарбамоил]фенилметилкарбаминовой кислоты
Трет-бутиловый эфир [2-(4-аминофенил)этил]карбаминовой кислоты (1 г, 4,2 ммоль), ((R)-трет-бутоксикарбониламино)фенилацетиловую кислоту (920 мг, 3,7 ммоль) и 1-гидроксибензотриазол (600 мг, 4,4 ммоль) растворяли в N,N-диметилформамиде (10 мл) в атмосфере азота и охлаждали до 0°C. Добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (850 мг, 4,4 ммоль) и полученную смесь перемешивали при температуре 0°C в течение 10 минут, далее при комнатной температуре в течение 21 часа. Смесь распределяли между водой и этилацетатом и органическую фазу промывали 0,5M лимонной кислотой, насыщенной гидрокарбонатом натрия и насыщенной хлоридом натрия. Затем органическую фазу сушили над сульфатом натрия и выпаривали досуха. Указанный в заголовке промежуточный продукт использовали без дальнейшей очистки.
d. Получение (R)-2-амино-N-[4-(2-аминоэтил)фенил]-2-фенилaцетамида
Сырой продукт, полученный на предыдущей стадии, растворяли в дихлорметане (10 мл) и добавляли трифторуксусную кислоту (10 мл). Смесь перемешивали в течение одного часа, затем летучие составляющие удаляли при пониженном давлении. Масло извлекали в дихлорметане и промывали 1н гидроксидом натрия. Дихлорметановую фазу сушили над сульфатом натрия и выпаривали досуха, получая в результате указанный в заголовке промежуточный продукт (690 мг, 2,6 ммоль), который использовали без дальнейшей очистки.
e. Получение (R)-N 2 -[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамина
Сырой продукт (690 мг, 2,6 ммоль), полученный на предыдущей стадии, растворяли в тетрагидрофуране (7 мл) и обрабатывали комплексом боран-диметилсульфида (1 мл). Смесь кипятили с обратным холодильником в течение 5 часов и затем охлаждали до комнатной температуры. Добавляли метанол (30 мл) и смесь выпаривали досуха. Снова добавляли метанол (30 мл) и смесь выпаривали досуха. Указанный в заголовке промежуточный продукт использовали без дальнейшей очистки.
f. Получение 5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-8-бензилокси-1H-хинолин-2-она
Сырой продукт (330 мг, 1,3 ммоль), полученный на предыдущей стадии, 8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1H-хинолин-2-он (490 мг, 1,0 ммоль), иодид натрия (150 мг, 1,0 ммоль) и гидрокарбонат натрия (250 мг, 3,0 ммоль) обрабатывали диметилсульфоксидом (2,5 мл) в атмосфере азота и нагревали до 140°C в течение 15 минут. Полученную смесь охлаждали до комнатной температуры и распределяли между водой и этилацетатом. Органическую фазу промывали насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Продукт очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая его в виде трифторацетатной соли этого соединения.
g. Получение 5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-бензилокси-1H-хинолин-2-она
Полученный на предыдущей стадии продукт растворяли в тетрагидрофуране (1,5 мл) и обрабатывали триэтиламин-тригидрофторидом (160 мкл) в течение 24 часов. Смесь распределяли между 1н гидроксидом натрия и этилацетатом. Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха, получая указанный в заголовке промежуточный продукт, который использовали без дальнейшей очистки.
h. Синтез 5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она
Сырой продукт (42 мг), полученный на предыдущей стадии, растворяли в дихлорметане (1 мл), и добавляли трихлорид бора (1,0M в дихлорметане, 400 мкл). Через 15 минут добавляли воду (5 мл) и ацетонитрил (500 мкл) и удаляли дихлорметан при пониженном давлении. Соединение, указанное в заголовке, очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая его в виде трифторацетатной соли указанного соединения. 1H ЯМР (300 MГц, ДМСО-d6): 10,4 (ушир.с, 2H), 9,2 (ушир.с, 1H), 8,6 (ушир.с, 3H), 8,1 (д, 1H, J=10,2 Гц), 7,2-7,4 (м, 5H), 7,0 (д, 1H, 8,2 Гц), 6,8-6,9 (м, 3H), 6,5 (д, 2H, J=8,0 Гц), 6,4 (с, 1H, J=10,2 Гц), 5,3 (ушир.д, 1H, J=7,7 Гц), 4,3 (м, 1H), 3,4 (дд, 1H, J=7,1, 13,9 Гц), 3,3 (дд, 1H, J=6,5, 13,9 Гц), 2,8-3,0 (м, 4H), 2,6-2,8 (м, 2H). m/z: [M+H+] вычислено для C27H30N4O3: 459,2; найдено 459,4.
Пример 2: Синтез N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
a. Получение N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-2-бензилоксифенил]формамида
В атмосфере азота сырой (R)-N2-[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамин (пример 1, стадия e) (320 мг, 1,3 ммоль), N-[2-бензилокси-5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)фенил]формамид (450 мг, 1,0 ммоль), иодид натрия (150 мг, 1,0 ммоль) и карбонат калия (550 мг, 4,0 ммоль) обрабатывали диметилсульфоксидом (2,5 мл) и нагревали до температуры 140°C в течение 15 минут. Полученную смесь охлаждали до комнатной температуры и распределяли между водой и этилацетатом. Органическую фазу промывали насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Продукт очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая указанный в заголовке промежуточный продукт в виде его трифторацетатной соли.
b. Получение N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-бензилоксифенил]формамида
Продукт, полученный на предыдущей стадии, растворяли в тетрагидрофуране (2,0 мл) и обрабатывали триэтиламинтригидрофторидом (200 мкл) в течение 23 часов. Полученную смесь распределяли между 1н хлоридом натрия и этилацетатом. Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха, получая указанный в заголовке промежуточный продукт (55 мг), который использовали без дальнейшей очистки.
c. Синтез N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
К смеси продукта (55 мг), полученного на предыдущей стадии, и палладия на угле (10% Pd, 11 мг) добавляли метанол (2 мл) в атмосфере азота. Полученную эмульсию интенсивно перемешивали в атмосфере водорода (атмосферное давление) в течение 23 часов. Катализатор удаляли посредством фильтрации и раствор подкисляли уксусной кислотой и разбавляли водой. Соединение, указанное в заголовке, очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая его в виде трифторацетатной соли этого соединения. 1H ЯМР (300 MГц, ДМСО-d6): 10,0 (с, 1H), 9,5 (с, 1H), 8,5 (ушир.с, 2H), 8,3 (ушир.с, 3H), 8,2 (с, 1H), 8,0 (с, 1H), 7,2-7,4 (м, 5H), 6,9 (д, 2H, J=8,0 Гц), 6,8 (дд, 1H), 6,7 (д, 1H, J=8,2 Гц), 6,5 (д, 2H, J=8,2 Гц), 6,0 (м, 1H), 5,6 (м, 1H), 4,6 (м, 1H), 4,2 (м, 1H), 2,8-3 (м, 4H), 2,6-2,7 (м, 2H). m/z: [M+H+] вычислено для C25H30N4O3: 435,2; найдено 435,3.
Пример 3: Альтернативный синтез 5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она
a. Получение (S)-2-[4-(2-аминоэтил)фениламино]-1-фенилэтанола
В 3-горлую колбу объемом 1000 мл помещали 20 г (147 ммоль) 2-(4-аминофенил)этиламина и 30 мл 1,3-диметил-3,4,5,6-тетрагидро-2(1H)пиримидинона (DMPU). Реакционная колба была снабжена расположенной сверху мешалкой и термометром. Реакционную колбу продували азотом и помещали в баню с холодной водой. Реакционную смесь обогащали 165 мл (165 ммоль) 1,0M раствора натрий-бис(триметилсилил)амида в тетрагидрофуране (температура поддерживалась ниже 30°C). Раствор натрий-бис(триметилсилил)амида добавляли одной порцией при интенсивном перемешивании. Реакционную смесь охлаждали до -10°C и добавляли (S)-оксид стирола (17 мл, 150 ммоль). Скорость добавления контролировали путем поддержания температуры ниже -10°C. Давали возможность реакционной смеси прогреться до 20°C в течение 15 минут после добавления (S)-стиролоксида, затем доводили температуру до 28°C в течение 30 минут. Реакционную смесь охлаждали до 25°C, а затем гасили реакцию путем покапельного добавления 90 мл воды. Затем реакционную смесь переносили в отдельную воронку, разбавляли 100 мл изопропилацетата и промывали насыщенным водным хлоридом натрия (90 мл). Органический слой трижды промывали смесью 90 мл воды и 90 мл насыщенного водного раствора хлорида натрия и в конце - насыщенным водным хлоридом натрия (180 мл). Органический слой концентрировали под вакуумом. Остаток дважды концентрировали из изопропанола (порцией в 100 мл)) и затем вновь растворяли в изопропаноле (500 мл) и нагревали при перемешивании до 70°C. Через две минуты добавляли концентрированную соляную кислоту (27 мл, 327 ммоль). Смеси давали остыть до комнатной температуры и перемешивали в течение 14 часов. Выпавший в осадок продукт отделяли путем фильтрации и промывали изопропанолом и изопропилацетатом. Полученный продукт сушили под вакуумом в водяной бане при температуре 50°C в течение 1 часа, а затем растворяли в 80 мл воды и перемещали в разделительную воронку. Добавляли изопропилацетат (80 мл) и 10н водный раствор хлорида натрия (40 мл, 400 ммоль). Разделительную воронку трясли и фазы разделяли. Органический слой промывали единожды 40 мл насыщенного NaCl и сушили над сульфатом магния. Твердые вещества собирали и фильтрат концентрировали. Осадок дважды концентрировали из толуола с получением указанного в заголовке промежуточного продукта в виде масла (14,7 г, 59 ммоль, 40%).
b. Получение 5-((R)-2-{2-[4-((S)-2-гидрокси-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-8-бензилокси-1H-хинолин-2-она
В атмосфере азота (S)-2-[4-(2-аминоэтил)фениламино]-1-фенилэтанол (1,7 г, 6,6 ммоль) и 8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1H-хинолин-2-он (1,5 г, 3,1 ммоль) обрабатывали диметилсульфоксидом (4,0 мл) и нагревали до 120°C в течение 40 минут. Смесь медленно охлаждали до комнатной температуры и распределяли между водой и этилацетатом (после удаления нерастворимых остатков смолы посредством декантации). Органическую фазу промывали 0,9M раствором ацетата натрия/уксусной кислоты, насыщенным гидрокарбонатом натрия и насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Указанный в заголовке промежуточный продукт использовали без дальнейшей очистки.
c. Получение 5-((R)-2-{2-[4-((R)-2-азидо-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-8-бензилокси-1H-хинолин-2-она
Продукт (500 г, 0,75 ммоль), полученный на предыдущей стадии, в атмосфере азота растворяли в тетрагидрофуране (9 мл) и обрабатывали дифенилфосфорилазидом (325 мкл, 1,5 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-еном (226 мкл, 1,5 ммоль). Смесь кипятили с обратным холодильником в течение 3,5 часов, затем охлаждали до комнатной температуры в течение 16 часов. Дополнительно к смеси добавляли дифенилфосфорилазид (160 мкл, 0,75 ммоль) и 1,8-диазабицикло [5.4.0]ундец-7-ен (113 мкл, 0,75 ммоль) и полученную смесь кипятили с обратным холодильником в течение последующих 3 часов, затем охлаждали до комнатной температуры. Смесь распределяли между этилацетатом и водой. Органическую фазу промывали ацетатом натрия/уксусной кислотой, насыщенным гидрокарбонатом натрия и насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Полученный продукт очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая указанный в заголовке промежуточный продукт в виде его трифторацетатной соли (110 мг).
d. Получение 5-((R)-2-{2-[4-((R)-2-азидо-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-бензилокси-1H-хинолин-2-она
Продукт, полученный на предыдущей стадии (110 мг), растворяли в тетрагидрофуране (2,0 мл) и обрабатывали триэтиламинтригидрофторидом (200 мкл) в течение 23 часов. Полученную смесь распределяли между 1н гидроксидом натрия и этилацетатом. Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха, получая указанный в заголовке промежуточный продукт (50 мг), который использовали без дальнейшей очистки.
e. Синтез 5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она
К смеси продукта (50 мг), полученного на предыдущей стадии, и палладия на угле (10% Pd, 10 мг) добавляли в атмосфере азота дихлорметан (500 мкл) и этанол (500 мкл). Полученную эмульсию интенсивно перемешивали в атмосфере водорода (при атмосферном давлении) в течение 23 часов. Дополнительно добавляли катализатор (10 мг) и N,N-диметилформамид (1 мл) и эмульсию интенсивно перемешивали в течение последующих 24 часов. Катализатор удаляли посредством фильтрации и смесь концентрировали при пониженном давлении. Соединение, указанное в заголовке, очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации в виде трифторацетатной соли этого соединения.
Пример 4: Альтернативный синтез N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
a. Получение N-[5-((R)-2-{2-[4-((S)-2-гидрокси-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-2-бензилоксифенил]формамида
В атмосфере азота (S)-2-[4-(2-аминоэтил)фениламино]-1-фенилэтанол (пример 3, часть a) (1,7 г, 6,6 ммоль), N-[2-бензилокси-5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)фенил]формамид (2,4 г, 5,2 ммоль), карбонат калия (2,8 г, 20 ммоль) и иодид натрия (860 мг, 5,7 ммоль) обрабатывали диметилсульфоксидом (4,7 мл) и нагревали до 140°C в течение 12 минут. Смесь охлаждали до комнатной температуры и распределяли между водой и 50%-ным этилацетатом/изопропилацетатом (после удаления нерастворимых остатков смолы посредством декантации). Органическую фазу промывали водой, 0,9M раствором ацетат натрия/уксусная кислота, насыщенным гидрокарбонатом натрия и насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Указанное в заголовке промежуточное соединение использовали без дальнейшей очистки.
b. Получение N-[5-((R)-2-{2-[4-((R)-2-азидо-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилметилсиланилокси)этил)-2-бензилоксифенил]формамида
В атмосфере азота продукт, полученный на предыдущей стадии (900 мг, 1,4 ммоль), растворяли в тетрагидрофуране (9 мл) и обрабатывали дифенилфосфорилазидом (610 мкл, 2,8 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-еном (420 мкл, 2,8 ммоль). Смесь кипятили с обратным холодильником в течение 3,5 часов, затем охлаждали до комнатной температуры в течение 16 часов. Дополнительно добавляли дифенилфосфорилазид (305 мкл, 1,4 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (210 мкл, 1,4 ммоль) и смесь кипятили с обратным холодильником в течение последующих 3 часов, затем охлаждали до комнатной температуры. Полученную смесь распределяли между ацетатом и водой. Органическую фазу промывали 0,9M раствором ацетата натрия/уксусной кислоты, насыщенным гидрокарбонатом натрия и насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Полученный продукт очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая указанное в заголовке промежуточное соединение в виде его трифторацетатной соли (180 мг).
c. Получение N-[5-((R)-2-{2-[4-((R)-2-азидо-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-бензилоксифенил]формамида
Продукт, полученный на предыдущей стадии (130 мг), растворяли в тетрагидрофуране (2,0 мл) и обрабатывали триэтиламинтригидрофторидом (200 мкл) в течение 23 часов. Полученную смесь распределяли между 1н гидроксидом натрия и этилацетатом. Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха, получая указанный в заголовке промежуточный продукт (90 мг), который использовали без дальнейшей очистки.
d. Синтез N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
К смеси продукта (90 мг), полученного на предыдущей стадии, и палладия на угле (10% Pd, 18 мг) в атмосфере азота добавляли дихлорметан (1 мл) и этанол (1 мл). Полученную суспензию интенсивно перемешивали в атмосфере азота (при атмосферном давлении) в течение 23 часов. Дополнительно добавляли катализатор (18 мг) и N,N-диметилформамид (1 мл) и суспензию интенсивно перемешивали в течение последующих 24 часов. Катализатор удаляли посредством фильтрации и смесь концентрировали при пониженном давлении. Соединение, указанное в заголовке, очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации в виде его трифторацетатной соли.
Пример 5: Альтернативное получение (R)-N 2 -[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамина
a. Получение трет-бутилового сложного эфира [2-(4- аминофенил)этил]карбаминовой кислоты
К суспензии 4-аминофенэтиламина (65,1 г, 1,0 экв.) в дихлорметане (1,5 л) при температуре 0°C покапельным путем добавляли ди-трет-бутилбикарбонат (99,2 г, 0,95 экв.) в дихлорметане (300 мл). Полученный раствор медленно нагревали до комнатной температуры и перемешивали в течение 18 часов. Добавляли воду (200 мл), растворитель выпаривали при пониженном давлении до получения объема приблизительно в 1 л, водный и органический слои разделяли и органический слой промывали водой (200 мл), далее промывали насыщенным водным хлоридом натрия (100 мл). Органический слой сушили над безводным сульфатом натрия (40 г). Твердые вещества фильтровали и фильтрат концентрировали, получая сырой продукт (100,7 г), указанный в заголовке, который суспендировали в смеси гексана (745 мл) и этилацетата (150 мл). Суспензию нагревали до получения чистого раствора, затем раствор медленно охлаждали до комнатной температуры. Полученные кристаллы фильтровали, промывали 10% раствором этилaцетат/гексан (100 мл) и сушили под вакуумом, получая указанный в заголовке промежуточный продукт (55,1 г, выход 48%).
b. Получение трет-бутилового эфира (R)-{[4-(2-трет-бутоксикарбониламиноэтил)фенилкарбонил]фенилметилкарбаминовой кислоты
Колбу объемом 1 л загружали продуктом, полученным на предыдущей стадии (30,0 г, 1,07 экв.), ((R)-трет-бутоксикарбониламино)фенилуксусной кислотой (30,0 г, 1,0 экв.) и раствором 1-гидрокси-7-азабензотриазола (16,3 г, 1,01 экв.) в N,N-диметилформамиде (240 мл). Полученный раствор перемешивали до полного растворения твердых веществ. Раствор охлаждали на ледяной бане в течение 15 минут и добавляли 1-[3-диметиламинопропил]-3-этилкарбодиимида гидрохлорид (26,9 г, 1,18 экв.). Реакционную смесь перемешивали при температуре 0°C в течение 80 минут. Реакционную смесь распределяли между водой и этилацетатом и органическую фазу промывали последовательно водой, 1н соляной кислотой, насыщенным водным бикарбонатом натрия и насыщенным водным хлоридом натрия. Органическую фазу сушили над безводным сульфатом натрия. Твердые вещества фильтровали и фильтрат концентрировали, получая указанный в заголовке промежуточный продукт в виде твердого вещества (количественный выход 57 г), которое использовали без дальнейшей очистки.
c. Получение (R)-2-амино-N-[4-(2-аминоэтил)фенил]-2-фенилацетамида
Сырой продукт (57 г), полученный на предыдущей стадии, объединяли с дихлорметаном (100 мл). Полученную смесь охлаждали до 0°C и через 15 минут добавляли трифторуксусную кислоту (150 мл). Смесь перемешивали в течение 1,5 часов при комнатной температуре, затем летучие составляющие удаляли при пониженном давлении. Полученное масло разбавляли дихлорметаном (300 мл) и добавляли 1н гидроксид натрия (200 мл), далее добавляли 10н гидроксид натрия (50 мл). Слои разделяли и основный водный слой экстрагировали при помощи дихлорметана (3×200 мл). Органические слои объединяли и сушили над безводным сульфатом натрия (20 г). Твердые вещества удаляли посредством фильтрации и фильтрат концентрировали, получая указанный в заголовке промежуточный продукт (количественный выход 33,3 г), который использовали без дальнейшей очистки.
d. Получение (R)-N 2 -[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамина
Сырой продукт (33,3 г, 1,0 экв.), полученный на предыдущей стадии, растворяли в тетрагидрофуране (250 мл) и охлаждали на ледяной бане. Добавляли комплекс борана с диметилсульфидом (45,5 мл, 4,0 экв.). Полученную смесь нагревали до 65˚C, перемешивали в течение 2 часов, затем охлаждали до 0°C. Добавляли метанол (650 мл), далее добавляли трифторуксусную кислоту (5 мл) и смесь выпаривали досуха. Осадок растворяли в метаноле (200 мл), далее растворяли в трифторуксусной кислоте (5 мл) и снова выпаривали досуха. Полученный осадок растворяли в метаноле (150 мл), и добавляли 1н гидроксид натрия (150 мл), далее добавляли 10н гидроксид натрия (40 мл). Раствор концентрировали для удаления органического растворителя и оставшийся водный слой экстрагировали дихлорметаном (400 мл, а затем 3×100 мл). Органические слои объединяли и сушили над безводным сульфатом натрия. Твердые вещества удаляли путем фильтрации и фильтрат концентрировали, получая промежуточный сырой продукт (29,8 г), указанный в заголовке.
Сырой продукт растворяли в этаноле (600 мл) и перемешивали при 80°C в течение 30 минут. Добавляли раствор L-яблочной кислоты (16,6 г, 1,06 экв.) в воде (90 мл), далее покапельно добавляли этанол (350 мл). Полученную смесь перемешивали при 80°C в течение 30 минут и далее - при комнатной температуре в течение 12 часов. Твердые вещества собирали посредством вакуумной фильтрации, промывали 10% раствором вода/этанол (130 мл), затем - этанолом (130 мл). Кристаллы сушили над вакуумом, получая соль L-яблочной кислоты промежуточного соединения, указанного в заголовке (38,6 г). Соль L-яблочной кислоты растворяли в воде (150 мл) и добавляли дихлорметан (175 мл). Полученную смесь перемешивали и охлаждали до 0°C, и добавляли 10,0н гидроксид натрия (50 мл). Слои разделяли и водный слой экстрагировали дихлорметаном (2Ч175 мл). Органические слои объединяли и сушили над безводным сульфатом натрия (20 г). Твердые вещества удаляли путем фильтрации и фильтрат концентрировали, получая на выходе указанный в заголовке промежуточный продукт (24,9 г, е.e. >99%) в виде бесцветного масла.
Пример 6: Альтернативный синтез N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
a. Получение N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-2-бензилоксифенил]формамида
В атмосфере азота (R)-N2-[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамин (пример 5) (22,4 г, 1,4 экв.), N-[2-бензилокси -5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)фенил]формамид (29,2 г, 1,0 экв.), карбонат калия (34,7 г, 4,0 экв.) объединяли с диметилсульфоксидом (35 мл). Полученную суспензию перемешивали при температуре 100°C в течение 85 минут. Реакционную смесь охлаждали до комнатной температуры и добавляли воду (200 мл) и изопропилацетат (200 мл). Слои разделяли, и органический слой промывали водой (200 мл), далее - насыщенным водным хлоридом натрия (150 мл), и сушили над безводным сульфатом магния (20 г). Твердые вещества удаляли путем фильтрации и фильтрат концентрировали, получая на выходе указанный в заголовке сырой промежуточный продукт в виде масла янтарного цвета.
b. Получение N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-бензилоксифенил]формамида
Продукт, полученный на предыдущей стадии (50,5 г, 1,0 экв.), растворяли в тетрагидрофуране (300 мл) и обрабатывали триэтиламинтригидрофторидом (19,1 г, 1,5 экв.) в течение 12 часов при комнатной температуре. Органический супернатант сливали, а к образовавшемуся остаточному продукту, содержащему смолянистое твердое вещество, добавляли изопропилацетат (200 мл), а затем 1,0н водный раствор гидроксида натрия (200 мл). Полученную смесь перемешивали до растворения основной части твердого вещества. Верхний слой двухфазной смеси декантировали и сохраняли. Изопропилацетат (150 мл) добавляли к водному слою и перемешивали до полного растворения всех твердых веществ, затем двухфазную смесь объединяли с отделенным ранее органическим слоем. Слои разделяли и основный водный слой снова экстрагировали изопропилацетатом (150 мл). Органические слои объединяли и сушили над безводным сульфатом натрия. Твердые вещества удаляли путем фильтрации и растворитель выпаривали, получая указанный в заголовке сырой промежуточный продукт (38,3 г) в виде осадка янтарного цвета.
Сырой продукт разделяли на три части. Сырой продукт репрезентативной части (18,1 г) растворяли в ацетонитриле (125 мл) и раствор выпаривали досуха. Осадок разбавляли водой (40 мл), ацетонитрилом (20 мл) и уксусной кислотой (4 мл). Раствор фильтровали, очищали с помощью препаративной ВЭЖХ, и чистые фракции объединяли и концентрировали путем лиофилизации, обеспечивая получение указанного в заголовке промежуточного продукта в виде его трифторацетатной соли в виде аморфного твердого вещества. Общий выход для трех частей составляет 20 г, выход 34%.
c. Синтез N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
Продукт (16,0 г, 1,0 экв.), полученный на предыдущей стадии, растворяли в тетрагидрофуране (640 мл) и добавляли гидроксид палладия на угле (3,2 г, 0,2 экв.) при продувке азотом. Раствор перемешивали в атмосфере водорода в течение 3-5 часов. Реакционную колбу продували азотом и реакционную смесь фильтровали через целит (30,0 г) и промывали тетрагидрофураном (100 мл). Растворитель удаляли под вакуумом, получая на выходе указанный в заголовке сырой промежуточный продукт (16,0 г) в виде масла.
Сырой продукт разделяли на три части. В репрезентативной части сырой продукт (4,0 г) растворяли в воде (10 мл) и перемешивали в течение 10 минут до растворения. Раствор фильтровали, очищали с помощью препаративной ВЭЖХ и чистые фракции объединяли и концентрировали путем лиофилизации, обеспечивая получение указанного в заголовке промежуточного соединения в виде его трифторацетатной соли в виде аморфного твердого вещества. Общий выход для трех частей составляет 8,4 г, выход 60%.
Пример 7: Синтез кристаллического гидрохлорида N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
a. Получение N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-бензилоксифенил]формамида в виде свободного основания
Трифторацетатную соль N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-бензилоксифенил]формамида (пример 6, часть b) распределяли между дихлохметаном (100 мл) и 1,0н водным гидроксидом натрия (100 мл). Органический слой дополнительно промывали 1,0н водным гидроксидом натрия (100 мл), а затем водой (100 мл). Органический слой сушили над безводным сульфатом натрия в течение 15 минут. Твердые вещества собирали посредством вакуумной фильтрации, и растворитель выпаривали, получая указанное в заголовке промежуточное соединение в виде масла.
b. Получение N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида в виде свободного основания
К продукту, полученному на предыдущей стадии (1,5 г), добавляли гидроксид палладия 20 мас.% на угле (300 мг), далее добавляли смесь тетрагидрофуран:этанол (1:1) (60 мл). Полученную взвесь интенсивно перемешивали в атмосфере азота в течение ночи. Катализатор удаляли путем фильтрации и фильтрат концентрировали под вакуумом, получая указанный в заголовке сырой продукт (1,2 г).
c. Получение затравочных кристаллов гидрохлорида N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
В круглодонной колбе продукт в виде свободного основания, полученный на предыдущей стадии (120 мг), перемешивали с изопропиловым спиртом (3,6 мл) при температуре 50°C до получения гомогенного раствора, далее добавляли 0,5н HCl (0,58 мл), и полученную смесь перемешивали в течение дополнительных 5 минут при температуре 50°C. Раствор постепенно охлаждали до комнатной температуры в течение 1,5 часов и затем перемешивали в течение ночи. Полученные кристаллы фильтровали и сушили в вакууме, получая на выходе указанный в заголовке кристаллический продукт (78,0 мг).
d. Синтез кристаллического гидрохлорида N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
В круглодонной колбе продукт (1,0 г) в виде свободного основания, полученный на стадии b, растворяли в изопропиловом спирте (30 мл) при температуре 45°C до получения гомогенного раствора, затем добавляли 0,5н HCl (4,8 мл). Полученный раствор нагревали в течение нескольких минут и добавляли затравочные кристаллы, полученные на предыдущей стадии (приблизительно 5 мг). Раствор охлаждали до 35°C и перемешивали в течение 2 часов. Раствор медленно охлаждали до комнатной температуры в течение двухчасового периода. Полученные кристаллы отделяли и сушили посредством воздушной фильтрации, получая указанную в заголовке соль гидрохлорида (690 мг). Добавляли изопропиловый спирт (7 мл) и воду (3,36 мл), и кристаллы снова нагревали до 45°C. Добавляли изопропиловый спирт (14 мл), и взвесь перемешивали в течение 1 часа. Раствор медленно охлаждали до комнатной температуры и далее снова нагревали до температуры 40°C в течение 5 часов. Раствор медленно охлаждали до комнатной температуры и перемешивали в течение ночи. Кристаллы отделяли путем фильтрации и сушили на воздухе, получая указанную в заголовке кристаллическую гидрохлоридную соль (550 мг).
Пример 8: Исследование кристаллического гидрохлорида N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
Образец кристаллической соли гидрохлорида, полученный в примере 7, стадия d, охарактеризован следующим образом: 1H ЯМР (300 MГц): 9,5 (с, 1H), 8,2 (с, 1H), 8,0 (ушир.с, 1H), 7,1-7,4 (м, 6H), 6,7-6,9 (м, 5H), 6,4 (д, 2H), 5,5 (ушир.с, 2H), 4,5 (д, 1H), 4,0 (т, 1H), 3,1 (ушир.с, 2H), 2,6-2,8 (м, 4H), 2,3 (м, 5H); m/z: [M+H+] вычислено для C25H30N4O3: 435,4; найдено 435,5. Анализ элементов (масс.%) вычислен для C25H30N4O3·HCl: C, 63,8, H, 6,6, N, 11,9, O 10,2, Cl 7,5; найдено: C, 63,7, H, 6,8, N, 11,8, O, 9,7, Cl 8,1. Содержание воды, вычисленное по методу Карла Фишера, составляет 0,9%.
На кривой, полученной при сканирующей дифференциальной калориметрии (прибор TA, модель DSC2010, уравновешивали при 30°C и нагревали на 5°C в минуту, вплоть до 300°C), при эндотермическом тепловом потоке наблюдался острый пик приблизительно между 185°C и примерно 200°C.
На чертеже показана картина порошковой дифракции рентгеновских лучей, полученная на дифрактометре Rigaku X-Ray Miniflex, с использованием эмиссии Cu Kα (30 кв, 15 мA) при скорости сканирования порядка 3° в минуту и величиной шага порядка 0,03° на точку.
Пример 9: Синтез N-[5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
a. Получение трет-бутилового эфира (S)-{[4-(2-трет-бутоксикарбониламиноэтил)фенилкарбамоил]фенилметилкарбаминовой кислоты
Трет-бутиловый эфир [2-(4-аминофенил)этил]карбаминовой кислоты (3,95 г, 16,7 ммоль) и ((S)-трет-бутоксикарбониламино)фенилацетиловую кислоту (3,97 г, 15,7 ммоль) растворяли с помощью 0,5M раствора 1-гидрокси-7-азабензотриазола в N,N-диметилформамиде (31,76 мл) в атмосфере азота и охлаждали до 0°C. Добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (3,57 г, 18,6 ммоль) и смесь перемешивали при температуре 0°C в течение 10 минут, затем - при комнатной температуре в течение 1,5 часа. Полученную смесь распределяли между водой и этилацетатом и органическую фазу промывали 1,0н HCl, насыщенным гидрокарбонатом натрия и насыщенным хлоридом натрия. Затем органическую фазу сушили над сульфатом натрия и выпаривали досуха. Указанный в заголовке промежуточный продукт использовали без дальнейшей очистки.
b. Получение(S)-2-амино-N-[4-(2-аминоэтил)фенил]-2-фенилацетамида
Сырой продукт, полученный на предыдущей стадии, растворяли в дихлорметане (15 мл) и охлаждали до 0°C. Добавляли трифторуксусную кислоту (15 мл), и смесь перемешивали в течение 30 минут при температуре 0°C. Раствор нагревали до комнатной температуры и перемешивали в течение 1 часа, затем летучие составляющие удаляли при пониженном давлении. Масло извлекали в дихлорметане и промывали 1н гидроксидом натрия. Органическую фазу сушили над сульфатом натрия и выпаривали досуха, получая указанный в заголовке промежуточный продукт (4,3 г, 16,2 ммоль), который использовали без дальнейшей очистки.
c. Получение(S)-N 2 -[4-(2-аминоэтил)фенил]-1-фенилэтан-1,2-диамина
Сырой продукт (4,3 г, 16,2 ммоль), полученный на предыдущей стадии, растворяли в тетрагидрофуране (50 мл) и обрабатывали комплексом боран-диметилсульфид (5,7 мл). Смесь кипятили с обратным холодильником при температуре 65°C в течение 2 часов, и затем охлаждали до комнатной температуры. Через 30 минут добавляли метанол (50 мл), затем добавляли трифторуксусную кислоту (3 мл) и полученную смесь выпаривали досуха. Полученное вещество растворяли в метаноле (50 мл) и трифторуксусной кислоте (1 мл) и снова выпаривали досуха. Полученное масло далее растворяли в метаноле (30 мл), 1,0н гидроксиде натрия (30 мл), затем добавляли 10,0н гидроксид натрия (5 мл). Раствор перемешивали в течение 10 минут, разбавляли водой и экстрагировали дихлорметаном. Органический слой сушили над сульфатом натрия и выпаривали досуха. Сырой промежуточный продукт (3,7 г, 14,4 ммоль), указанный в заголовке, растворяли в этиловом спирте (105 мл) и нагревали до 80°C. К полученному нагретому раствору добавляли раствор D-яблочной кислоты (2,16 г, 16,1 ммоль) в H2O (5,3 мл), далее добавляли этиловый спирт (45 мл). Полученный раствор охлаждали до комнатной температуры и перемешивали в течение 15 часов. Осадок фильтровали и распределяли между водой и дихлорметаном, и органическую фазу промывали 1,0н NaOH и насыщенным хлоридом натрия. Органическую фазу сушили над сульфатом натрия и выпаривали досуха, получая указанный в заголовке промежуточный продукт (2,0 г, 7,8 ммоль).
d. Получение N-[5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-2-бензилоксифенил]формамида
В атмосфере азота сырой продукт (431 мг, 1,7 ммоль), полученный на предыдущей стадии, N-[2-бензилокси-5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)фенил]формамид (471 мг, 1,0 ммоль) и гидрокарбонат натрия (300 мг, 3,5 ммоль) обрабатывали диметилсульфоксидом (1,2 мл) и нагревали до 100°C в течение 1 часа. Полученный раствор охлаждали до комнатной температуры и распределяли между водой и этилацетатом. Органическую фазу промывали насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Указанный в заголовке промежуточный продукт использовали без дальнейшей очистки.
e. Получение N-[5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-бензилоксифенил]формамида
Продукт, полученный на предыдущей стадии, растворяли в тетрагидрофуране (5 мл) и обрабатывали триэтиламинтригидрофторидом (823 мкл) в течение 8 часов. Полученную смесь распределяли между 1н гидроксидом натрия и дихлорметаном. Органическую фазу сушили над сульфатом натрия и выпаривали досуха. Продукт очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая указанное в заголовке промежуточное соединение в виде его трифторацетатной соли (150 мг, 0,2 ммоль).
f. Синтез N-[5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
Продукт (150 мг, 0,2 ммоль), полученный на предыдущей стадии, растворяли в этиловом спирте и добавляли в атмосфере азота уксусную кислоту (5 мл) и 20% Pd(OH)2 (24 мг). Реакционную колбу продували водородом при атмосферном давлении и реакционную смесь перемешивали в атмосфере водорода в течение 20 часов. Катализатор фильтровали и летучие составляющие выпаривали. Соединение, указанное в заголовке, очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации в виде его трифторацетатной соли (79,0 мг, 0,12 ммоль). m/z: [M+H+] вычислено для C25H30N4O3: 435,2; найдено: 435,8.
Пример 10: Синтез 5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она
Используя процедуры, аналогичные процедурам, описанным на стадиях d, e, и f примера 9, и заменяя на стадии d 8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1H-хинолин-2-он на N-[2-бензилокси-5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)фенил]формамид, получали указанное в заголовке соединение в виде его трифторацетатной соли m/z: [M+H+] вычислено для C27H30N4O3: 459,2; найдено: 459,4.
Пример 11: Синтез 5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она
a. Получение(R)-(бензилоксикарбонилметиламино)фенилуксусной кислоты
(R)-(бензилоксикарбониламино)фенилуксусную кислоту (2,0 г, 7,0 ммоль) в атмосфере азота растворяли в тетрагидрофуране и добавляли гидрид натрия (60% дисперсия в минеральном масле, 840 мг, 21 ммоль). К раствору добавляли метилиодид (737 мг, 50 ммоль) и реакционную смесь перемешивали в течение 30 минут. Добавляли к реакционной смеси воду (1 мл), и летучие компоненты выпаривали. Продукт очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая указанное в заголовке промежуточное соединение (1,66 г, 5,5 ммоль).
b. Получение бензилового эфира {(R)-[4-(2-трет-бутоксикарбониламиноэтил)фенилкарбамоил]фенилметил}метил-карбаминовой кислоты
Трет-бутиловый эфир [2-(4-аминофенил)этил]карбаминовой кислоты (1,31 г, 5,6 ммоль), продукт, полученный на предыдущей стадии (1,66 г, 5,6 ммоль), 1-гидрокси-7-азабензотриазол (1,46 г, 10,5 ммоль), растворяли в N,N-диметилформамиде (10 мл) в атмосфере азота и охлаждали до 0°C. Добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (2,02 г, 10,5 ммоль), и раствор перемешивали при температуре 0°C в течение 10 минут, затем - при комнатной температуре в течение 1,5 часов. Смесь распределяли между водой и этилацетатом и органическую фазу промывали 1,0н HCl, насыщенным гидрокарбонатом натрия и насыщенным хлоридом натрия. Органическую фазу сушили над сульфатом натрия и выпаривали досуха. Указанный в заголовке промежуточный продукт использовали без дальнейшей очистки.
c. Получение бензилового эфира {(R)-[4-(2-аминоэтил)фенилкарбамоил]фенилметил}метилкарбаминовой кислоты
Сырой продукт, полученный на предыдущей стадии, растворяли в дихлорметане (5 мл) и добавляли трифторуксусную кислоту (5 мл). Смесь перемешивали в течение 30 минут, затем летучие составляющие удаляли при пониженном давлении. Масло извлекали в дихлорметане и промывали 1н гидроксидом натрия. Дихлорметановую фазу сушили над сульфатом натрия и выпаривали досуха, получая указанный в заголовке промежуточный продукт, который использовали без дальнейшей очистки.
d. Получение (R)-N-[4-(2-аминоэтил)фенил]-2-метиламино-2-фенилацетамида
Сырой продукт (800 мг, 1,9 ммоль), полученный на предыдущей стадии, растворяли в метаноле (5 мл) и добавляли в атмосфере азота дихлорметан (5 мл) и 10% палладий на угле (200 мг). Реакционную колбу продували водородом при атмосферном давлении и реакционную смесь перемешивали в атмосфере водорода в течение 2 часов. Палладиевый катализатор удаляли путем фильтрации, и летучие составляющие выпаривали, получая указанный в заголовке промежуточный продукт.
e. Получение(R)-N 2 -[4-(2-аминоэтил)фенил]-N 1 -метил-1-фенилэтан-1,2-диамина
Сырой продукт (523 мг, 1,84 ммоль), полученный на предыдущей стадии, растворяли в тетрагидрофуране (50 мл), обрабатывали комплексом боран-диметилсульфид (0,7 мл). Смесь кипятили с обратным холодильником при температуре 65°C в течение 2 часов и затем охлаждали до комнатной температуры. Добавляли метанол (10 мл), далее добавляли 4,0н HCl в диоксане (1,4 мл), смесь перемешивали в течение 10 минут и затем выпаривали досуха. Масло вновь извлекали в метаноле (50 мл) и трифторуксусной кислоте (1 мл) и выпаривали досуха. Полученное масло далее растворяли в метаноле (10 мл) и KOH (10 мл 20% раствора в H2O) и перемешивали в течение 10 минут. Раствор разбавляли водой и экстрагировали дихлорметаном. Органический слой сушили над сульфатом натрия и выпаривали досуха. Указанный в заголовке промежуточный продукт получали в виде масла, которое использовали без дальнейшей очистки.
f. Получение 5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-(трет-бутилдиметилсиланилокси)этил)-8-бензилокси-1H-хинолин-2-она
В атмосфере азота сырой продукт (250 мг, 0,93 ммоль), полученный на предыдущей стадии, 8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1H-хинолин-2-он (454 мг, 0,93 ммоль) и гидрокарбонат натрия (234 мг, 2,8 ммоль) обрабатывали диметилсульфоксидом (10 мл) и нагревали до 100°C в течение 3 часов. Полученную смесь охлаждали до комнатной температуры и распределяли между водой и этилацетатом. Органическую фазу промывали насыщенным хлоридом натрия, сушили над сульфатом натрия и выпаривали досуха. Указанный в заголовке промежуточный продукт использовали без дальнейшей очистки.
g. Получение 5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-бензилокси-1H-хинолин-2-она
Продукт (80 мг, 0,12 ммоль), полученный на предыдущей стадии, растворяли в тетрагидрофуране (5 мл) и обрабатывали триэтиламинтригидрофторидом (21 мкл) в течение 5 часов. Раствор выпаривали досуха и полученный продукт очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации, получая указанный в заголовке промежуточный продукт в виде его трифторацетатной соли.
h. Синтез 5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она
Продукт, полученный на предыдущей стадии (70 мг), растворяли в этиловом спирте (2 мл), и в атмосфере азота добавляли 10% палладий на угле (14 мг). Реакционную колбу продували водородом при атмосферном давлении, и реакционную смесь перемешивали в атмосфере водорода в течение 2 часов. Катализатор удаляли путем фильтрации, и летучие составляющие выпаривали. Соединение, указанное в заголовке, очищали с помощью ВЭЖХ с обращенной фазой и выделяли путем лиофилизации в виде его трифторацетатной соли (40 мг, 0,057 ммоль). 1H ЯМР (300 MГц): 10,6 (ушир.с, 2H), 9,2 (ушир.д, 2H), 8,8 (ушир.д, 2H), 8,2 (д, 1H, J=10,2 Гц), 7,4-7,6 (м, 5H), 7,2 (д, 1H, 8,2 Гц), 6,9-7,0 (м, 3H), 6,5-6,6 (м, 3H), 6,2 (ушир.с, 1H,), 5,3 (ушир.д, 1H, J=7,1 Гц), 4,3 (м, 1H), 3,6 (дд, 1H, J=7,0, 14,0 Гц), 3,3 (дд, 1H, J=6,3, 14,0 Гц), 2,8-3,0 (м, 4H), 2,6-2,8 (м, 2H), 2,4 (с, 3H). m/z: [M+H+] вычислено для C28H32N4O3: 473,3; найдено: 473,3.
Пример 12: Синтез N-[5-((R)-2-{2-[4-((S)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида
Используя процедуры, аналогичные процедурам, описанным на стадиях f, g, и h примера 11, и заменяя на стадии f N-[2-бензилокси-5-((R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил)фенил]формамид на 8-бензилокси-5-[(R)-2-бром-1-(трет-бутилдиметилсиланилокси)этил]-1H-хинолин-2-он, получали указанное в заголовке соединение в виде его трифторацетатной соли. 1H ЯМР (300 MГц): 10,0 (с, 1H), 9,5 (с, 1H), 8,5 (ушир.с, 2H), 8,5 (ушир.с, 2H), 8,2 (с, 1H), 8,0 (с, 1H), 7,2-7,4 (м, 5H), 6,7-6,9 (м, 4H), 6,4 (д, 2H), 6,0 (м, 1H), 5,6 (м, 1H), 4,6 (м, 1H), 4,2 (м, 1H), 3,6 (дд, 1H, J=7,1, 14,0 Гц), 3,3 (дд, 1H, J=6,0, 14,0 Гц), 2,8-3 (м, 4H), 2,6-2,7 (м, 2H), 2,3 (с, 3H). m/z: [M+H+] вычислено для C26H32N4O3: 449,3; найдено: 449,5.
Несмотря на то что настоящее изобретение было описано со ссылками на его специфические воплощения, для специалистов в данной области должно быть очевидно, что в объеме, охватываемом настоящим изобретением, допустимы различные изменения и эквивалентные замены, не меняющие суть настоящего изобретения. Кроме того, может быть произведено множество модификаций, с тем чтобы адаптировать настоящее изобретение к конкретной ситуации, материалу, представляющей интерес композиции, способу, стадии или стадиям способа. Подразумевается, что все указанные модификации входят в объем заявленного изобретения и прилагаемой здесь формулы изобретения. Кроме того, все публикации, патенты и патентные документы, цитируемые в настоящем описании, считаются включенными в него путем ссылок во всей их полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АРИЛАНИЛИНА В КАЧЕСТВЕ АГОНИСТОВ β АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2005 |
|
RU2370490C2 |
| ДИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АКТИВНОСТЬЮ АГОНИСТА В2 АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2010 |
|
RU2543716C2 |
| ДИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АКТИВНОСТЬЮ АГОНИСТА β АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2010 |
|
RU2676686C2 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА | 2004 |
|
RU2330841C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИФЕНИЛЬНОГО СОЕДИНЕНИЯ | 2005 |
|
RU2381223C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ СОЕДИНЕНИЙ СО СТРУКТУРОЙ СОЛИ ЧЕТВЕРТИЧНОГО АММОНИЯ | 2017 |
|
RU2722720C1 |
| БИФЕНИЛЬНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2005 |
|
RU2366656C2 |
| СОЛИ МОНТЕЛУКАСТА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2020 |
|
RU2840778C2 |
| КОМБИНАЦИЯ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АГОНИСТА БЕТА-2-АДРЕНОРЕЦЕПТОРА | 2008 |
|
RU2460527C2 |
| ГУАНИДИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2008 |
|
RU2480458C2 |

Изобретение относится к соединениям общей формулы (II) и их фармацевтически приемлемым солям и стереоизомерам в качестве агонистов β2-адренергического рецептора, к фармацевтической композиции на их основе, способу лечения, к способу их получения, их применению, а также кристаллическому гидрохлориду N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2гидроксифенил]формамида. Соединения могут найти применение для лечения и профилактики заболеваний, опосредованных β2-адренергическим рецептором, таких как астма и хроническая обструктивная болезнь легких. В общей формуле (II)
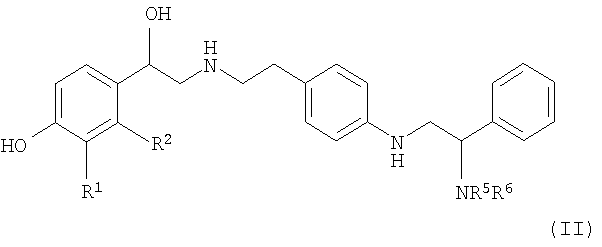
R1 представляет собой -NHCHO, и R2 является водородом; или R1 и R2, взятые совместно, представляют собой -NHC(=O)CH=CH- или -CH=CHC(=O)NH-; каждый из R5 и R6 независимо выбирают из водорода или C1-6-алкила. 6 н. и 8 з.п. ф-лы, 1 ил., 6 табл.
1. Соединение формулы (II):

в которой R1 представляет собой -NHCHO, и R2 является водородом; или
R1 и R2, взятые совместно, представляют собой -NHC(=O)CH=CH- или -CH=CHC(=O)NH-;
каждый из R5 и R6 независимо выбирают из водорода или C1-6-алкила;
или его фармацевтически приемлемая соль или стереоизомер.
2. Соединение по п.1, где пространственная конфигурация при атоме углерода алкиленовой группы, несущей гидроксильную группу, представляет собой (R).
3. Соединение по п.1, выбранное из
5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1H-хинолин-2-она;
N-[5-((R)-2-{2-[4-((Е)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил] формамида;
5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она;
N-[5-((R)-2-{2-[4-((8)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида;
5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она;
5-((R)-2-{2-[4-((R)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она;
N-[5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида;
N-[5-((R)-2-{2-[4-((R)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида;
5-((R)-2-{2-[4-((S)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она;
5-((R)-2-{2-[4-((S)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она;
N-[5-((R)-2-{2-[4-((8)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида; и
N-[5-((R)-2-{2-[4-((S)-2-диметиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида; и
их фармацевтически приемлемых солей.
4. Соединение по п.3, выбранное из
5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она;
N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида;
5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она;
N-[5-((R)-2-{2-[4-((S)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида;
5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-8-гидрокси-1Н-хинолин-2-она; и
N-[5-((R)-2-{2-[4-((R)-2-метиламино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида; и
их фармацевтически приемлемых солей.
5. Кристаллический гидрохлорид N-[5-((R)-2-{2-[4-((R)-2-амино-2-фенилэтиламино)фенил]этиламино}-1-гидроксиэтил)-2-гидроксифенил]формамида, где кристаллическая соль охарактеризована порошковой рентгенограммой, в которой положения пиков соответствуют по существу положениям пиков картины, показанной на рисунке.
6. Фармацевтическая композиция, обладающая свойством агонизировать β2-адренергический рецептор, содержащая терапевтически эффективное количество соединений по любому из пп.1-5 и фармацевтически приемлемый носитель.
7. Фармацевтическая композиция по п.6, где композиция дополнительно содержит терапевтически эффективное количество кортикостероида.
8. Фармацевтическая композиция по п.6 или 7, где композиция составлена для введения путем ингаляции.
9. Способ лечения астмы или хронической обструктивной болезни легких у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества соединения по любому из пп.1-5.
10. Способ по п.9, дополнительно включающий введение терапевтически эффективного количества кортикостероида.
11. Способ получения соединения формулы (II), включающий реакцию соединения формулы (III'):

где P1 представляет собой гидроксизащитную группу, L представляет собой уходящую группу, R1 и R2 определены в п.1, и R313 представляет собой -ОН или -ОР2, где P2 представляет собой гидроксизащитную группу с соединением формулы (IV'):

в которой R5 и R6 определены в п.1, с получением соединения формулы (V'):
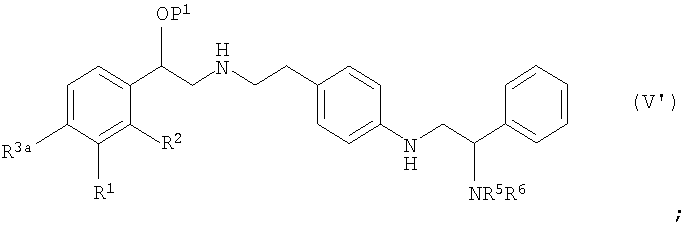
удаление защитной группы P1 с получением соединения формулы (VI'):
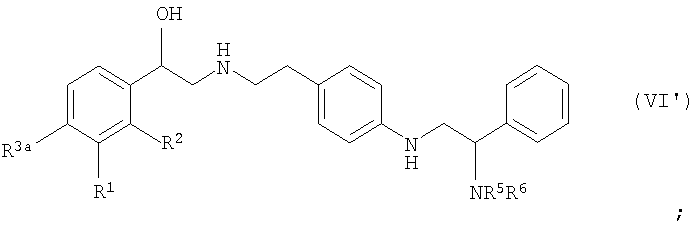
и когда R3a представляет собой -ОР2, удаление защитной группы Р2 с получением соединения формулы (II) или его соли или стереоизомера.
12. Соединение по любому из пп.1-5, обладающее свойством агонизировать β2-адренергический рецептор.
13. Применение соединения по любому из пп.1-5 для производства лекарственного средства для лечения заболевания или состояния у млекопитающего, ассоциированного с активностью β2-адренергического рецептора.
14. Применение по п.13, где заболеванием или состоянием является астма или хроническая обструктивная болезнь легких.
| RU 97100652 А, 10.02.1999 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

