Настоящее изобретение относится к новым триарилимидазолам и их фармацевтически приемлемым солям. Указанные соединения являются ингибиторами протеинтирозинкиназы, прежде всего c-met киназы, и, следовательно, являются высоко эффективными лекарственными препаратами для лечения рака. Настоящее изобретение относится также к фармацевтическим композициям, которые содержат указанные новые соединения в качестве активных агентов, предназначенных для лечения рака и связанных с раком заболеваний.
Предпосылки создания изобретения
Протеинтирозинкиназы (РТК), ферменты, катализирующие перенос γ-фосфата из АТФ на остатки тирозина в составе белка-субстрата, являются основными компонентами путей передачи сигналов, которые контролируют пролиферацию и дифференциацию клеток. РТК подразделяются на два больших семейства, рецепторные тирозинкиназы (RTK) и нерецепторные тирозинкиназы (NRTK). RTK содержат домен, расположенный в плазматической мембране и включают внеклеточный домен, который связывается с лигандом, а также внутриклеточный фрагмент, который обладает каталитической активностью и включает регуляторные последовательности. Большинство RTK, подобных рецептору фактора роста гепатоцитов c-met, содержат единую полипептидную цепь и в отсутствие лиганда являются мономерами. Лиганд связывается с внеклеточным фрагментом RTK, при этом происходит димеризация мономерных рецепторов, что приводит к автофосфорилированию специфических остатков тирозина в цитоплазматическом фрагменте (см. обзоры Blume-Jensen P. и Hunter Т., Nature, 411, сс.355-365 (2001); Hubbard S.R. и др., J. Biol. Chem., 273, cc.11987-11990 (1998); Zwick E. и др., Trends Mol. Med. 8, cc.17-23 (2002)). В основном, при автофосфорилировании тирозина либо стимулируется внутренняя каталитическая киназная активность рецептора, либо становятся доступными участки связывания с сигнальными белками, включенными в последующие стадии передачи сигнала и содержащими домены, распознающие фосфотирозин, такие как гомологичный домен Src 2 (SH2) или домен, связывающий фосфотирозин (РТВ).
Протеинтирозинкиназы играют основную роль в механизме передачи внутриклеточного сигнала, в ответ на который происходят различные ответные реакции в клетке, такие как пролиферация, апоптоз и дифференциация. Следовательно, эти ферменты являются основной мишенью при разработке новых терапевтических препаратов, предназначенных для подавления пролиферации раковых клеток, метастазирования и ангиогенеза, а также для активации апоптоза. Наиболее исследованная стратегия лечения в клинике заключается в применении моноклональных антител для связывания с рецептором фактора роста тирозинкиназ. Однако использование низкомолекулярных ингибиторов тирозинкиназы теоретически имеет значительное преимущество по сравнению с моноклональными антителами. Низкомолекулярные ингибиторы могут легко проникать в ткани и обладать активностью в отношении внутриклеточных мишеней и мутантов таких мишеней, а также пероральной биодоступностью. Установлено, что некоторые производные свинца характеризуются высокой активностью в отношении таких мишеней, как EGFR, рецептор эндотелиального фактора роста сосудистых клеток и bcr-abl.
Рецептор фактора роста гепатоцитов c-met был впервые идентифицирован как активированный онкоген в обработанной N-метил-N'-нитрозогуанидином клеточной линии остеогенной саркомы человека (MUNG-HOS) по его способности вызывать трансформацию фибробластов NIH 3T3 мыши. Рецептор, кодируемый протоонкогеном c-met (расположенным на хромосоме 7), является двухцепочечным белком и содержит α-цепь с молекулярной массой 50 kDa, соединенную дисульфидной связью с β-цепью с молекулярной массой 145 kDa, которые образуют αβ-комплекс с молекулярной массой 190 kDa. Цепь α экспонирована на клеточной поверхности, в то время как цепь β расположена внутри клеточной мембраны и содержит внутриклеточный домен тирозинкиназы. Наличие указанного внутриклеточного домена тирозинкиназы позволяет включить рецептор c-met в семейство рецепторов тирозинкиназы (RTK), расположенных на клеточной поверхности.
Фактор роста гепатоцитов (HGF), известный также как фактор Скаттера (SF), является многофункциональным цитокином, который вызывает ответные реакции в различных клетках и тканях. Начиная с открытия и изучения его свойств, фактор HGF/SF становится объектом интенсивных исследований, прежде всего в связи с его ролью в развитии и прогрессии рака. В настоящее время существует множество доказательств его роли в качестве регулятора канцерогенеза, инвазии рака и метастазирования (см. обзоры Herynk M.H. и Radinsky R., In Vivo, 14, cc.587-596 (2000); Jiang W. и др., Crit. Rev. Oncol. Hematol., 29, cc.209-248 (1999); Longati Р. и др., Curr. Drug Targets, 2, cc.41-55 (2001); Maulik G. и др., Cytokine Growth Factor Rev., 13, cc.41-59 (2002); Parr С. и Jiang W.G., Histol. Histopathol, 16, cc.251-268 (2001)).
Фактор HGF/SF связывается с β-цепью зрелого рецептора c-met и индуцирует фосфорилирование остатков тирозина. Предполагается, что такие явления способствуют связыванию с активированным рецептором внутриклеточных сигнальных белков, содержащих гомологичные участки src (SH), такие как PLC-γ, Ras-GAP, PI-3 киназа pp60c-src и комплекс GRB-2 Socs. Каждый белок, содержащий SH2, активирует различную подгруппу сигнальных фосфопептидов и, таким образом, вызывает различные ответные реакции в клетке.
Мутации c-met подробно описаны для случаев наследственной и спорадической папиллярной карциномы почек человека и известны для случаев рака яичников, гепатоцеллюлярной карциномы у детей, метастазирующего плоскоклеточного рака головы и шеи, а также рака желудка. Сверхэкспрессия с-met наблюдается также при немелкоклеточном и мелкоклеточном раке легких, при опухолях молочной железы, ободочной кишки и предстательной железы. Так как установлено, что c-met играет важную роль в онкогенезе множества опухолей, для блокирования указанного рецептора тирозинкиназы применяются различные способы ингибирования.
Ингибирование протеинтирозинкиназы c-met можно использовать для подавления роста и инвазии опухоли, как описано в ряде документированных отчетов о доклинических испытаниях (см. Abounader R. и др., J. Natl. Cancer Inst., 91, cc.1548-1556 (1999); Laterra J. и др. Lab. Invest., 76, cc.565-577 (1997); Tomioka D., Cancer Res., 61, cc.7518-7524 (2001); Wang R. и др., J. Cell Biol, 153, cc.1023-1033 (2001)).
В заявке на выдачу патента WO 96/18626 описаны ингибиторы тирозинкиназ и c-met киназы, которые являются производными 2-(2,6-дихлорфенил)-4-фенил-5-(пиридин-4-ил)-1Н-имидазола (примеры 5, 6 и 55). Однако эти производные характеризуются отрицательным действием на цитохром Р450.
Установлено, что соединения по настоящему изобретению позволяют исключить указанные недостатки, являются активными ингибиторами c-met киназы и характеризуются достаточно высокой растворимостью.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединениям общей формулы (I)
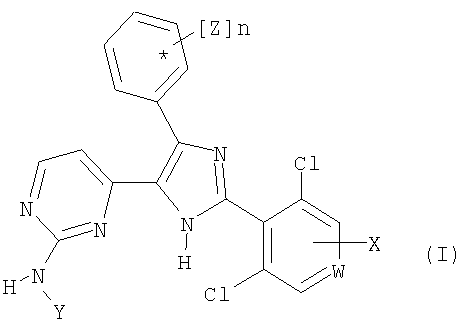
где W означает -N= и
X означает водород;
Y означает водород или группу A2-R;
А2 означает С1-С5алкилен, который необязательно замещен C1-С6алкилом, фенилом или гидроксилом;
R означает гидрокси, линейный или разветвленный C1-С6алкокси, амино, диметиламино, диэтиламино, трет-бутилоксикарбониламино, карбоксил, C1-С6алкоксикарбонил, триазолил, циано, пиперидино, 1-пирролидинил, морфолино, 4-метилпиперазин-1-ил, O-А1-NR3R4, S-A1-NR3R4, 4-карбоксифенил, фуран-3-ил, тиофен-2-ил или 3-метилтиофен-2-ил;
n равно 1 или 2 и
Z означает один или два заместителя, которые независимо выбирают из группы, включающей галоген, гидрокси, аллилокси, метил, C1-С3алкокси, которые необязательно замещены пиридинилом, метоксиметокси, (2-метоксиэтокси)метилокси, метилтио, этоксиметокси, метилендиокси, этинилом, триметилсилилэтинилом и бензилокси, которые необязательно содержат заместитель, выбранный из группы, включающей галоген, метокси, циано, нитро, метилендиокси, карбокси или этокси;
или альтернативно
W означает -СН=,
Х означает водород, OR1, SR2, (SO)R2, (SO2)R2, СН2-S-СН2-С(O)2-СН2-СН3, CH2-S-(CH2)2-OH или группу A1-Q;
А1 означает С1-С3алкиленовую группу;
Q означает OR1, SR2, SOR2, SO2R2, NR3R4, NHCH2CH2NR3R4 или галоген;
R1 выбирают из группы, включающей водород, C1-С3алкил, аллил, диметилфосфонилметил, 2,3-эпокси-1-пропил, (R)-2,3-дигидрокси-1-пропил, (S)-2,3-дигидрокси-1-пропил, 1,3-дигидрокси-2-пропил, 3-гидрокси-2-гидроксиметил-1-пропил, 2-метоксиэтоксиметил, 2,2-диметил-1,3-диоксолан-4-илметил или группу А1-Q1;
Q1 означает С1-С2алкокси, циано, карбоксил, C1-С6алкоксикарбонил, карбоксамид, -CO-NR3R4; C1-С6алкилсульфанил, C1-С6алкилсульфенил, С1-С6алкилсульфонил и
в случае, если А1 означает 1,2-этиленовую или 1,3-пропиленовыю группу,
Q1 означает гидрокси или NR3R4;
R2 означает C1-С6алкил, диметилфосфонилметил, 2,3-эпокси-1-пропил, 2,3-дигидрокси-1-пропил, 2,2-диметил-1,3-диоксолан-4-илметил или А1-Q1;
R3, R4 независимо выбирают из группы, включающей водород и C1-С6алкил, или
R3 и R4 вместе образуют 5-7-членное насыщенное или ненасыщенное кольцо, необязательно замещенное метильной группой и содержащее один или два гетероатома, которые независимо выбирают из N или О;
Y означает водород или группу А2-R;
А2 означает С1-С5алкилен, который необязательно замещен C1-С6алкилом, фенилом или гидроксилом;
R означает гидрокси, линейный или разветвленный C1-С6алкокси, амино, диметиламино, диэтиламино, трет-бутилоксикарбониламино, карбоксил, C1-С6алксоксикарбонил, триазолил, циано, пиперидино, 1-пирролидинил, морфолино, 4-метилпиперазин-1-ил, O-А1 -NR3R4, S-A1-NR3R4, 4-карбоксифенил, фуран-3-ил, тиофен-2-ил или 3-метилтиофен-2-ил;
n равно 1 или 2 и
Z означает C1-С3алкокси, замещенный пиридинилом, если n равно 1, причем
если n равно 2, один заместитель означает C1-С3алкокси, замещенный пиридинилом, а второй заместитель независимо выбирают из группы, включающей галоген, гидрокси, аллилокси, метил, C1-С3алкокси, метоксиметокси, (2-метоксиэтокси)метилокси, метилтио, этоксиметокси, метилендиокси, этинил, триметилсилилэтинил,
и к их фармацевтически приемлемым солям.
Неожиданно установлено, что фармацевтическая и противоопухолевая активность проявляется за счет ингибирования соединениями по настоящему изобретению киназы c-met, что, прежде всего, обусловлено присутствием 2,6-дихлорфенильного или пиридильного остатка в положении 2 имидазольного кольца.
Подробное описание вариантов осуществления изобретения
Предпочтительные C1-С6алкильные группы в составе R1, R2, R3, R4 и А2 означают метил, этил и пропил.
Предпочтительные группы C1-С6алкокси в составе Q1, R и Z означают метокси, этокси или изопропилокси.
Предпочтительные циклические системы, образованные группами R3 и R4, означают 1-пирролидинил-, пиперидино-, морфолино- или 4-метилпиперазин-1-ил.
Предпочтительно Х означает группу A1-Q, т.е. означает -CH2OH или -CH2-CH2-OH.
Предпочтительно Х означает группу -O-A1-Q1, т.е. означает -O-СН2-СН2-ОН,
-O-СН2-СООН или -O-CH2-CN.
Предпочтительно Y означает А2-R и включает 2-гидроксиэтил, 3-гидроксипропил, 2-метоксиэтил, 3-метоксипропил, (R)-2,3-дигидрокси-1 -пропил, (S)-2,3-дигидрокси-1-пропил, (R)-3-гидроксибутил, (R)-3-гидроксибутил, 2-морфолиноэтил, 3-морфолинопропил, (СН2)3СООН, 2-(4-метилпиперазин-1-ил)этил, 3-гидрокси-2,2-диметилпропил, 3-гидрокси-1-фенилпропил, 3-трет-бутилоксиэтил, 2-аминоэтил, 3-аминопропил, 4-аминобутил, 2-(N,N-диметиламино)этил, 3-(N,N-диметиламино)пропил, 3-(пирролидин-1-ил)пропил, CH2COOH, (CH2)2COOH, СН(С2Н5)COOH, (СН2)3COOC(CH3)3, (CH2)2-N-COOC(CH3)3, (CH2)3-N-COOC(CH3)3, (CH2)2-O-(CH2)2-H(CH3)2, (CH2)2-O-(CH2)2-NH2, (CH2)2-S-(CH2)2-N(CH3)2, (CH2)2-S-(CH2)3-N(CH3)2, (CH2)3-S-(CH2)2-N(CH3)2, (CH2)3-S-(CH2)3-Н(CH3)2, (1,2,4-триазол-1-ил)этил, 3-(1,2,4-триазол-3-ил)пропил.
Галоген означает фтор, хлор, бром или иод.
Предпочтительно n равно 1, а указанный заместитель Z расположен в положении 3 или 4. Если Z означает группу метокси, которая замещена пиридинилом, бензилокси или замещенным бензилокси, то Z предпочтительно расположен в положении 3.
Прежде всего предпочтительны соединения общей формулы (I) и их фармацевтически приемлемые соли, где W означает -N=, a Z выбирают группы, включающей 3-хлор, 4-хлор, 3-бром, 3-иод, 3-этинил, 3-метоксиметокси, 3-(2-метоксиэтокси)метилокси, 3-метилтио, 3-этоксиметокси, 3,4-мептилендиокси или 3-бензилокси, необязательно замещенной атомом галогена, группой метокси, циано, нитро, метилендиокси, карбокси или этокси.
Прежде всего, предпочтительны также соединения общей формулы (I), где
W означает -N=,
Х означает водород,
Y означает 2-гидроксиэтил, 3-гидроксипропил, 2-метоксиэтил, 3-метоксипропил, (R)-2,3-дигидрокси-1-пропил, (S)-2,3-дигидрокси-1-пропил, (R)-3-гидроксибутил, (S)-3-гидроксибутил, 3-гидрокси-2,2-диметилпропил, 2-морфолиноэтил, 3-морфолинопропил, 2-(4-метилпиперазин-1-ил)этил, 3-гидрокси-1-фенилпропил, 2-аминоэтил, 3-аминопропил, 4-аминобутил, 2-(N,N-диметиламино)этил, 3-(N,N-диметиламино)пропил, 3-(пирролидин-1-ил)пропил, СН2СООН, (СН2)2СООН, (CH2)3СООН, СН(С2Н5)СООН, (CH2)2-O-(CH2)2-N(CH3)2, (CH2)2-O-(CH2)2-NH2, (CH2)2-S-(CH2)2-N(CH3)2, (CH2)2-S-(CH2)3-N(СН3)2, (CH2)3-S-(CH2)2-N(CH3)2 или (CH2)3-S-(CH2)3-N(CH3)2;
n равно 1 и
Z выбирают из группы, включающей 3-хлор, 4-хлор, 3-бром, 3-иод, 3-этинил, 3-метоксиметокси или 3-бензилокси, необязательно замещенные атомом галогена, метокси, циано, нитро, метилендиокси, карбокси или этокси.
Прежде всего, предпочтительны также соединения общей формулы (I), где
W означает -N=,
X означает водород,
Y означает 2-гидроксиэтил, 3-гидроксипропил, (R)-2,3-дигидрокси-1-пропил, (S)-2,3-дигидрокси-1-пропил, 2-морфолиноэтил, 3-морфолинопропил, 2-(4-метилпиперазин-1-ил)этил, 2-аминоэтил, 3-аминопропил, 2-(N,N-диметиламино)этил, 3-(N,N-диметиламино)пропил или 3-(пирролидин-1-ил)пропил;
n равно 1 и
Z выбирают из группы, включающей 3-хлор, 4-хлор, 3-бром, 3-иод, 3-этинил, 3-метоксиметокси или 3-бензилокси, необязательно замещенной атомом галогена, метокси или циано.
Примеры такого соединения включают
2-(3,5-дихлорпиридин-4-ил)-4-(3-бензилоксифенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол.
Другой вариант осуществления настоящего изобретения относится к соединениям формулы (I), где
W означает -СН=,
Х означает водород, OR1, SR2, (SO)R2, (SO2)R2, CH2-S-CH2-С(O)2-CH2-CH3, CH2-S-(CH2)2-OH или группу A1-Q;
А1 означает C1-С3алкиленовую группу,
Q означает OR1, SR2, SOR2, SO2R2, NR3R4, NHCH2CH2NR3R4 или галоген;
R1 выбирают из группы, включающей водород, C1-C3алкил, аллил, диметилфосфонилметил, 2,3-эпокси-1-пропил, (R)-2,3-дигидрокси-1-пропил, (S)-2,3-дигидрокси-1-пропил, 1,3-дигидрокси-2-пропил, 3-гидрокси-2-гидроксиметил-1-пропил, 2-метоксиэтоксиметил, 2,2-диметил-1,3-диоксолан-4-илметил или группу А1-Q1;
Q1 означает C1-C2алкокси, циано, карбоксил, C1-С6алкоксикарбонил, карбоксамид, -CO-NR3R4, C1-C6алкилсульфанил, C1-C6алкилсульфенил, C1-C6алкилсульфонил и
в случае, если А1 означает 1,2-этиленовую или 1,3-пропиленовую группу, Q1 означает гидрокси или NR3R4;
R2 означает C1-C6алкил, диметилфосфонилметил, 2,3-эпокси-1-пропил, 2,3-дигидрокси-1-пропил, 2,2-диметил-1,3-диоксолан-4-илметил или A1-Q1;
R3, R4 независимо выбирают из группы, включающей водород и C1-C6алкил, или
R3 и R4 вместе образуют 5-7-членное насыщенное или ненасыщенное кольцо, необязательно замещенное метильной группой и содержащее один или два гетероатома, которые независимо выбирают из N или О;
Y означает 3-гидроксипропил, а
Z означает 3-бензилокси, который необязательно замещен атомом галогена, группой метокси или циано.
Еще один вариант осуществления настоящего изобретения относится к соединениям формулы (I), где
W означает -CH=,
X означает водород или OR1,
R1 выбирают из группы, включающей водород, C1-С3алкил, аллил, диметилфосфонилметил, 2,3-эпокси-1-пропил, (R)-2,3-дигидрокси-1-пропил, (S)-2,3-дигидрокси-1-пропил, 1,3-дигидрокси-2-пропил, 3-гидрокси-2-гидроксиметил-1-пропил, 2-метоксиэтоксиметил, 2,2-диметил-1,3-диоксолан-4-илметил или группу А1-Q1;
А1 означает C1-C3алкиленовую группу;
Q1 означает C1-C2алкокси, циано, карбоксил, C1-C6алкоксикарбонил, карбоксамид, C1-C6алкилсульфанил, C1-C6алкилсульфенил, C1-C6алкилсульфонил и
в случае, если А1 означает 1,2-этиленовую или 1,3-пропиленовую группу, Q1 означает гидрокси;
Y означает 3-гидроксипропил, а
Z означает 3-бензилокси.
Другой вариант осуществления настоящего изобретения относится к соединениям общей формулы (I), где
W означает -CH=,
Х означает водород, OR1, SR2, (SO)R2, (SO2)R2, CH2-S-CH2-C(O)2-CH2-CH3, CH2-S-(CH2)2-OH или группу A1-Q;
А1 означает C1-C3алкиленовую группу;
Q означает OR1, SR2, SOR2, SO2R2, NR3R4, NHCH2CH2NR3R4 или галоген;
R1 выбирают из группы, включающей водород, C1-C3алкил, аллил, диметилфосфонилметил, 2,3-эпокси-1-пропил, (R)-2,3-дигидрокси-1-пропил, (S)-2,3-дигидрокси-1-пропил, 1,3-дигидрокси-2-пропил, 3-гидрокси-2-гидроксиметил-1-пропил, 2-метоксиэтоксиметил, 2,2-диметил-1,3-диоксолан-4-илметил или группу А1-Q1;
Q1 означает С1-С2алкокси, циано, карбоксил, C1-С6алкоксикарбонил, карбоксамид, -CO-NR3R4, C1-C6алкилсульфанил, C1-С6алкилсульфенил, С1-С6алкилсульфонил и
в случае, если А1 означает 1,2-этиленовую или 1,3-пропиленовую группу, Q1 означает гидрокси или NR3R4;
R2 означает C1-С6алкил, диметилфосфонилметил, 2,3-эпокси-1-пропил, 2,3-дигидрокси-1-пропил, 2,2-диметил-1,3-диоксолан-4-илметил или А1-Q1;
R3, R4 независимо выбирают из группы, включающей водород и C1-С6алкил или
R3 и R4 вместе образуют 5-7-членное насыщенное или ненасыщенное кольцо, необязательно замещенное метильной группой и содержащее один или два гетероатома, которые независимо выбирают из N или О;
Y означает водород или группу А2-R;
А означает С1-С5алкилен, который необязательно замещен группой C1-С6алкил, фенил или гидрокси;
R означает гидрокси, линейный или разветвленный C1-С6алкокси, амино, диметиламино, диэтиламино, трет-бутилоксикарбониламино, карбоксил, C1-С6алкоксикарбонил, триазолил, циано, пиперидино, 1-пирролидинил, морфолино, 4-метилпиперазин-1-ил, O-A1-NR3R4, S-A1-NR3R4, 4-карбоксифенил, фуран-3-ил, тиофен-2-ил или 3-метилтиофен-2-ил;
n равно 1, а
Z означает C1-С3алкокси, замещенный группой пиридинил.
Еще один вариант осуществления настоящего изобретения относится к соединениям общей формулы (I), где
W означает -СН=,
Х означает водород, OR1, SR2, (SO)R2, (SO2)R2, СН2-S-СН2-С(O)2-СН2-СН3, CH2-S-(CH2)2-OH или группу A1-Q;
А1 означает C1-С3алкиленовую группу;
Q означает OR1, SR2, SOR2, SO2R2, NR3R4, NHCH2CH2NR3R4 или галоген;
R1 выбирают из группы, включающей водород, C1-С3алкил, аллил, диметилфосфонилметил, 2,3-эпокси-1-пропил, (R)-2,3-дигидрокси-1-пропил, (S)-2,3-дигидрокси-1-пропил, 1,3-дигидрокси-2-пропил, 3-гидрокси-2-гидроксиметил-1-пропил, 2-метоксиэтоксиметил, 2,2-диметил-1,3-диоксолан-4-илметил или группу А1-Q1;
Q1 означает С1-С2алкокси, циано, карбоксил, C1-С6алкоксикарбонил, карбоксамид, -CO-NR3R4, C1-С6алкилсульфанил, C1-С6алкилсульфенил, C1-С6алкилсульфонил, а
в случае, если А1 означает 1,2-этиленовую или 1,3-пропиленовую группу, Q1 означает гидрокси или NR3R4;
R2 означает C1-С6алкил, диметилфосфонилметил, 2,3-эпокси-1-пропил, 2,3-дигидрокси-1-пропил, 2,2-диметил-1,3-диоксолан-4-илметил или А1-Q1;
R3, R4 независимо выбирают из группы, включающей водород и C1-С6алкил или
R3 и R4 вместе образуют 5-7-членное насыщенное или ненасыщенное кольцо, необязательно замещенное метильной группой и содержащее один или два гетероатома, которые независимо выбирают из N или О;
Y означает 3-гидроксипропил;
n равно 1, а
Z означает пиридин-2-илметокси, пиридин-3-илметокси или пиридин-4-илметокси.
Примеры таких соединений включают
2-(2,6-дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(4-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол,
2-(2,6-дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(3-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол и
2-(2,6-дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(2-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол.
Формула (I) означает 2-(2,6-дихлорфенил)-4-фенил-5-(4-пиримидинил)-1Н-имидазолы, которые являются таутомерами 2-(2,6-дихлорфенил)-5-фенил-4-(4-пиримидинил)-1Н-имидазолов. Оба таутомера характеризуются одинаковой структурой, их номенклатура взаимозаменяема, и оба таутомера включены в объем настоящего изобретения. Соединения по настоящему изобретению содержат один или более асимметричных атомов углерода и существуют в виде рацематов, рацемических смесей, а также в виде индивидуальных диастереомеров, включая все возможные изомеры и оптические изомеры, которые включены в объем настоящего изобретения.
Соединения общей формулы (I) получают при взаимодействии соединения общей формулы (VI) или (VII) с амином Y-NH2, где W, X, Y, n и Z имеют значения, как определено выше в данном контексте, при температуре в диапазоне от 80 до 180°С с последующим выделением указанного соединения. Предпочтительно используют стехиометрические количества или избыток указанных аминов. Реакцию проводят в отсутствие растворителя или в растворителе, таком как диоксан, диметоксиэтан или толуол.
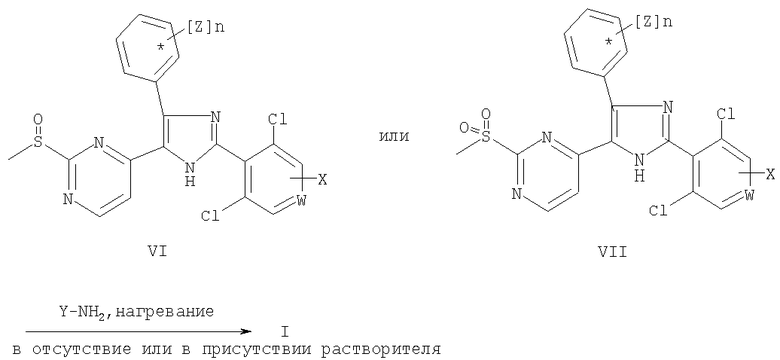
Соединения общей формулы (VI) и (VII) получают при окислении сульфидной группы в составе тиоэфиров общей формулы (V). Для получения сульфоксидов общей формулы (VI) окисление предпочтительно проводят с использованием 3-хлорпербензойной кислоты. Для синтеза сульфонов общей формулы (VII) предпочтительно используют оксон™.
Тиоэфиры общей формулы (V)
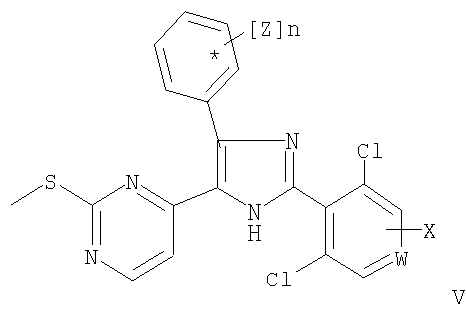
получают N-деоксигенированием соединений общей формулы (IV). Реакцию предпочтительно проводят с использованием этилбромацетата в присутствии триэтиламина (Somei М. и Tsuchiya M., Chem. Pharm. Bull., 29, сс.3145-3157 (1981)). В другом варианте указанное восстановление проводят при использовании триэтилфосфита в диметилформамиде.
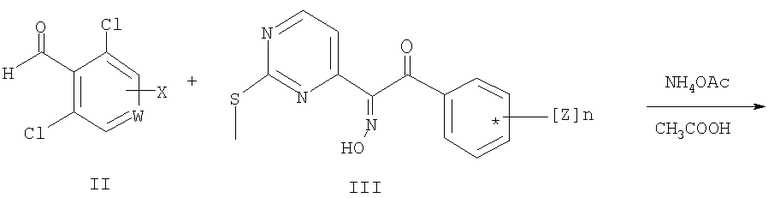
Соединение общей формулы (IV) получают при взаимодействии соединения общей формулы (III) с соединением общей формулы (II), где заместители W, Х и Z имеют значения, как определено выше в данном контексте. Указанная реакция является реакцией конденсации, и ее предпочтительно проводят в присутствии аммиака по известным методикам, описанным для других альдегидов.
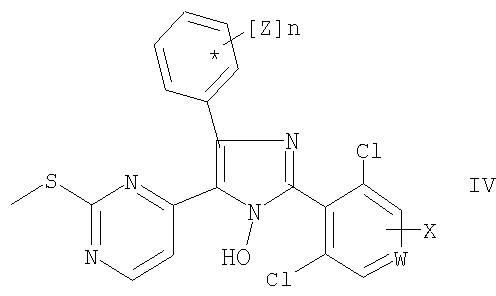
Другой вариант осуществления настоящего изобретения относится к применению соединения общей формулы (II), где заместитель Х имеет значение, как определено выше в данном контексте, для получения соединения общей формулы (I), как описано для упомянутого выше процесса.
2,6-Дихлорбензальдегиды являются ценными промежуточными соединениями при получении соединений общей формулы (I) по настоящему изобретению. 2,6-Дихлор-3-гидроксибензальдегид и 2,6-дихлор-4-гидроксибензальдегид являются известными соединениями. 2,6-Дихлор-3-гидроксибензальдегид получают из 3-гидроксибензальдегида (Gust R. и Schoenenberg H., Eur.J. Med.Chem., 28 сс.103-115 (1993)), но при этом требуется использование высокотоксичного газообразного хлора, что приводит к образованию побочных продуктов вследствие переокисления. Методика, описанная в настоящем изобретении (пример А2), позволяет исключить указанные недостатки. 2,6-Дихлор-4-гидроксибензальдегид получают из 3,5-дихлорфенола либо по реакции Реймера-Тимана (Baldwin J.J. и др., J. Med. Chem., 22, сс.687-693 (1979)), либо согласно последовательности реакций бромирования/реакции Гриньяра (заявка на выдачу патента WO 01/44154). Методика Реймера-Тимана не экономична из-за чрезвычайно низких выходов (<4%), кроме того, для этой реакции требуется использование хлороформа, что вызывает значительные проблемы с охраной окружающей среды. Другой известный способ синтеза с использованием бромирования/реакции Гриньяра включает 4 стадии, в том числе стехиометрическое бромирование бромом и использование токсичного хлорметилметилового эфира для защиты фенольной группы. Кроме того, общий выход составляет только 40%.
В настоящем изобретении предлагается усовершенствованный способ получения 2,6-дихлор-3-гидроксибензальдегида и 2,6-дихлор-4-гидроксибензальдегида. Указанный способ включает металлирование защищенного 2,4-дихлорфенола или 3,5-дихлорфенола литиевым основанием с последующим взаимодействием со сложным эфиром или амидом муравьиной кислоты, удалением защитной группы и выделением указанных соединений. Пригодные литиевые основания включают метиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий, литийдиизопропиламид или литийбистриметилсилиламид, предпочтительным является бутиллитий. Пригодные растворители включают диэтиловый эфир, тетрагидрофуран или 1,2-диметоксиэтан, предпочтительным является тетрагидрофуран. Стадию металлирования проводят при температуре от -100°С до -60°С, предпочтительно при температуре от -80°С до -70°С. Пригодные защитные группы включают триизопропилсиланил, трет-бутилдиметилсиланил или фенилдиметилсиланил, предпочтительной является триизопропилсиланильная группа. Пригодные производные муравьиной кислоты включают метилформиат, этилформиат, диметилформамид или N-формилпиперидин, предпочтительным является диметилформамид. Указанную методику применяют также для получения 2,6-дихлор-3-гидроксиметилбензальдегида и 2,6-дихлор-4-гидроксиметилбензальдегида по настоящему изобретению.
Термин «фармацевтически приемлемые соли», использованный ранее в данном контексте, означает стандартные кислотно-аддитивные соли или основно-аддитивные соли, которые сохраняют биологическую эффективность и свойства соединений формулы (I) и которые образуются при взаимодействии с пригодными нетоксичными органическими или неорганическими кислотами или органическими или неорганическими основаниями. Примеры кислотно-аддитивных солей включают соли неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота и азотная кислота, а также соли органических кислот, таких как пара-толуолсульфоновая кислота, салициловая кислота, метансульфоновая кислота, щавелевая кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота и т.п. Примеры основно-аддитивных солей включают соли, полученные с использованием гидроксидов аммония, калия, натрия и гидроксида четвертичного аммония, таких как, например, гидроксид тетраметиламмония. Химическое превращение фармацевтического соединения (т.е. лекарственного препарата) в соль используется в фармацевтике для повышения физической и химической стабильности, текучести и растворимостью, а также для снижения гигроскопичности соединений (см., например, сборник Н. Ansel и др., Pharmaceutical Dosage Forms and Drug Delivery Systems, 6th ed. (6oe изд.), с.196 и cc.1456-1457 (1995)).
Соединения формулы (I) и фармацевтически приемлемые соли соединений формулы (I) можно использовать в качестве лекарственных средств, например, в форме фармацевтических препаратов. Фармацевтические препараты вводят перорально, например, в форме таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако используют также и ректальное введение, например, в форме суппозиториев, парентеральное введение, например, в форме растворов для инъекций.
Соединения формулы (I) можно перерабатывать в смеси с фармацевтически инертными неорганическими или органическими носителями для получения фармацевтических препаратов. В качестве таких носителей для таблеток, таблеток с покрытием, драже и твердых желатиновых капсул используют, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновые кислоты или их соли и т.п. Пригодные носители для мягких желатиновых капсул включают, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т.п. В зависимости от природы активного соединения для получения мягких желатиновых капсул обычно не требуется никаких носителей. Пригодные носители для получения растворов и сиропов включают, например, воду, полиолы, глицерин, растительное масло и т.п. Пригодные носители для суппозиториев включают, например, природные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и т.п.
Кроме того, фармацевтические препараты содержат консерванты, солюбилизаторы, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферные вещества, маскирующие агенты или антиоксиданты. Фармацевтические препараты могут также содержать другие терапевтически ценные вещества.
Объект настоящего изобретения относится также к лекарственным средствам, содержащим соединение формулы (I) или его фармацевтически приемлемую соль в смеси с терапевтически инертным носителем, а также к способу их получения, который включает переработку одного или более соединений формулы (I) и/или их фармацевтически приемлемых солей, а также, при необходимости, одного или более других терапевтически ценных веществ и одного или более терапевтически инертных носителей в готовую галеновую форму.
Благодаря активности в качестве ингибиторов тирозинкиназ, предпочтительно c-met-киназ, соединения общей формулы (I) являются ценными ингредиентами лекарственных средств, предназначенных для лечения рака и других заболеваний, при которых наблюдается повышенная экспрессия рецептора c-met или других родственных рецепторов киназ.
Дозировка соединения по настоящему изобретению может изменяться в широких пределах в зависимости от индивидуальных требований в каждом конкретном случае. В случае перорального введения дозировка для взрослых пациентов изменяется от приблизительно 0,01 мг до приблизительно 1000 мг в сутки соединения общей формулы (I) или составляет соответствующее количество его фармацевтически приемлемой соли. Суточную дозу можно вводить в виде однократной дозы или в виде разделенных доз и, кроме того, верхний предел можно превысить в зависимости от показаний.
Следующие примеры и препараты представлены для иллюстрации изобретения и не ограничивают его объема.
Примеры
А. Синтез замещенных 2,6-дихлорбензальдегидов
Пример А1
2,6-Дихлор-4-гидроксибензальдегид (А1)
Получение 3,5-дихлортриизопропилсилилоксибензола (А1.1)
В раствор 3,5-дихлорфенола (200 г) и 2,6-лутидина (330 мл) в 3,0 л сухого CH2Cl2 при 0°С в течение 1 ч добавляли триизопропилсилилтрифлат (400 г) и полученную смесь перемешивали при указанной температуре в течение еще 3 ч. После гидролиза в 1,0 л воды органический слой промывали насыщенным раствором NaCl, сушили над MgSO4 и упаривали досуха (70°С/80 мбар). Остаток переносили в петролейный эфир и фильтровали через силикагель, при этом получали соединение А1.1 (360 г, 92%) в виде бесцветного масла.
1Н-ЯМР (250 МГц, CDCl3): δ 1,03-1,15 (m, 18H, СН3), 1,16-1,35 (m, 3Н, СН), 6,73-6,80 (m, 2H, СНаром.), 6,92-6,98 (m, 1H, СНаром.).
13С-ЯМР (62,9 МГц, CDCl3): δ 12,7 (СН), 18,0 (СН3), 119,0, 121,6 (СНаром.), 135,2, 157,4 (Саром.).
Получение 2,6-дихлор-4-гидроксибензальдегида (А1) и 2,6-дихлор-4-триизопропилсилилоксибензальдегида (А1.2)
В раствор соединения А1.1 (360 г) в 2,6 л сухого тетрагидрофурана в атмосфере азота добавляли n-BuLi (440 мл, 2,7 M раствор в гексане), при этом температуру поддерживали ниже -65°С. После перемешивания в течение 2 ч при -70°С добавляли сухой диметилформамид (120 мл), при этом температуру поддерживали ниже -65°С. Смесь выдерживали при комнатной температуре в течение ночи, добавляли 4 M HCl (700 мл) и интенсивно перемешивали при комнатной температуре в течение 1 ч. Фазы разделяли (при необходимости добавляли твердый NaCl) и органический слой сушили над сульфатом натрия и упаривали в вакууме. Образовавшийся осадок перекристаллизовывали из толуола/тетрагидрофурана, при этом получали соединение Al (154 г, выход 70%), tпл 229-230°С.
1Н-ЯМР (250 МГц, ДМСО-D6): δ 6,94 (s, 2H, СНаром.), 10,25 (s, 1H, CH=O), 11,46 (ушир. s, 1H, ОН).
13С-ЯМР (62,9 МГц, ДМСО-D6): δ 117,0 (СНаром.), 120,7, 137,8, 162,1 (Саром.), 187,2 (СН=O).
Пример А2
Получение этилового эфира (3,5-дихлор-4-формилфенокси)уксусной кислоты (А2)
Смесь соединения А1 (2,87 г, 15 ммолей), этилбромацетата (2,76 г, 16,5 ммоля) и карбоната калия (2,90 г, 21 ммоль) в 50 мл сухого ацетона перемешивали при 60°С в течение 3 ч. После фильтрования и удаления растворителя остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/метанол, 100:2), при этом получали соединение А2 (3,55 г, выход 86%) в виде бесцветного твердого вещества.
1Н-ЯМР (250 МГц, CDCl3): δ 1,32 (t, 7,2 Гц, 3Н, СН3), 4,30 (q, 7,2 Гц, 2Н, СН2), 4,68 (s, 2Н, СН2), 6,92 (s, 2Н, СНаром.), 10,41 (s, 1Н, СН=O).
13С-ЯМР (62,9 МГц, CDCl3): δ 14,3 (СН3), 62,1, 65,5 (СН2), 116,4 (СНаром.), 123,8, 139,2, 160,9 (Саром.), 167,3 (С=O), 187,8 (СН=O).
В. Синтез амидов Вейнреба
Пример В1
3-Бензилокси-N-метокси-N-метилбензамид (В1)
В суспензию 3-бензилоксибензойной кислоты (136,8 г, 0,60 моля) в 1200 мл дихлорметана при 10°С добавляли триэтиламин (60,6 г, 0,6 моля). Через 15 мин добавляли раствор этилхлорформиата (64,8 г, 0,60 моля) в 100 мл дихлорметана, при этом температуру поддерживали в интервале от 10°С до 15°С. После перемешивания в течение 40 мин и добавления гидрохлорида N,O-диметилгидроксиламина (58,2 г, 0,60 моля) через 20 мин при температуре 10-15°С добавляли раствор триэтиламина (60,6 г, 0,60 моля). После перемешивания в течение еще 30 мин добавляли воду и органический слой сушили над сульфатом натрия. После фракционной перегонки в вакууме получали соединение В1 (131,9 г, выход 81%). МС (API+):273.
Пример В2
3-Гидрокси-N-метокси-N-метилбензамид (В2)
В раствор соединения В1 (100 г, 0,37 моля) в 750 мл тетрагидрофурана добавляли Pd/C (10 г, 10%) и смесь гидрировали при атмосферном давлении в течение 2 ч. Катализатор отфильтровывали и фильтрат упаривали, при этом получали соединение В2 (66,0 г, выход 98%).
МС (API+, API-):182, 180.
Пример В3
3-(2-Пиридинилметилокси)-N-метокси-N-метилбензамид (В3)
В раствор соединения В2 (1,21 г, 10,0 ммоля), трифенилфосфина (2,89 г, 11,0 ммоля) и пиридин-2-метанола (1,20 г, 11,0 ммоля) в 30 мл сухого тетрагидрофурана добавляли раствор диэтилазодикарбоксилата (1,92 г, 11,0 ммоля) в 5 мл сухого тетрагидрофурана и полученную смесь перемешивали при комнатной температуре в течение 2 ч. После удаления растворителя и хроматографии на колонке с силикагелем (элюент: гексан/этилацетат, 2:1) получали соединение В3 в виде масла светло-желтого цвета (2,81 г, выход 72% с учетом 30 мол.% примеси оксида трифенилфосфина (по данным ЯМР)). В связи с проблемами отделения примесей синтез повторяли с использованием PS-связанного трифенилфосфина, при этом получали чистое соединение В3 (выход 58%). МС (API+):М 273.
1Н-ЯМР (400 МГц, CDCl3): δ 3,25 (s, 3Н, СН3), 3,45 (s, 3Н, ОСН3), 5,15 (s, 2Н, ОСН2), 6,98-7,04 (m, 1H), 7,12-7,28 (m, 4H), 7,40-7,48 (m, 1H), 7,60-7,68 (m, 1H), 8,46-8,56 (m, 1H).
13С-ЯМР (100,6 МГц, CDCl3): δ 34,3 (СН3), 61,5 (ОСН3), 71,0 (ОСН2), 114,9, 117,6, 121,3, 121,8, 123,2, 129,7, 135,9*, 137,3, 149,6, 157,3*, 158,3* (Саром.Н), 169,9 (С=O) (*четвертичный углерод).
Пример В4
3-(3-Пиридинилметилокси)-N-метокси-N-метилбензамид (В4)
Указанное соединение (выход 81%) получали аналогично тому, как описано в примере В3, но при взаимодействии с 3-пиридинилметанолом. МС (API+): М 273.
1Н-ЯМР (400 МГц, ДМСО-D6): δ 3,24 (s, 3Н, СН3), 3,53 (s, 3Н, ОСН3), 5,20 (s, 2Н, ОСН2), 7,12-7,24 (m, 3Н), 7,34-7,48 (m, 2H), 7,84-7,92 (m, 1H), 8,52-8,60 (m, 1H), 8,68-8,72 (m, 1H).
13С-ЯМР (100,6 МГц, ДМСО-D6): δ 33,7 (СН3), 61,1 (ОСН3), 67,4 (ОСН2), 114,1, 117,3, 120,5, 124,0, 129,7, 132,8, 136,1, 136,2, 149,5, 149,6, 157,9 (Саром.Н), 169,0 (С=O).
Пример В5
3-(4-Пиридинилметилокси)-N-метокси-N-метилбензамид (В5)
Указанное соединение (выход 82%) получали аналогично тому, как описано в примере В3, но при взаимодействии с 4-пиридинилметанолом. МС (API+): М 273.
1Н-ЯМР (400 МГц, ДМСО-D6): δ 3,24 (s, 3Н, СН3), 3,51 (s, 3H, ОСН3), 5,24 (s, 2Н, ОСН2), 7,12-7,22 (m, 3Н), 7,34-7,41 (m, 1H), 7,42-7,48 (m, 2H), 8,52-8,64 (m. 2H).
13С-ЯМР (100,6 МГц, ДМСО-D6): δ 33,7 (СН3), 61,0 (ОСН3), 67,9 (OCH2), 114,1, 117,3. 120,7, 122,1, 129,8, 136,3, 146,4, 150,1, 157,7 (Саром.Н), 169,0 (С=O).
С. Синтез этанонов
Пример С1
1-(3-(2-Пиридинилметилокси)фенил)-2-(2-метилтиопиримидин-4-ил)этанон (С1)
Диизопропиламин (2,1 мл, 15 ммолей) растворяли в 70 мл сухого тетрагидрофурана, охлаждали до -75°С и через 10 мин добавляли раствор н-бутиллития (9,4 мл, 1,6 М, 15 ммолей). После перемешивания в течение 15 мин при -75°С в течение 10 мин при той же температуре добавляли раствор 2-метилтио-4-метилпиримидина (1,69 г, 12 ммолей) в 5 мл сухого тетрагидрофурана и полученную смесь перемешивали в течение еще 15 мин. Затем в течение 10 мин при -75°С добавляли раствор соединения В3 (2,73 г, 7 ммолей, чистота 70%) в 5 мл сухого тетрагидрофурана. Смесь перемешивали при -75°С в течение 1 ч, нагревали до комнатной температуры и затем выливали в этилацетат/воду (100 мл, 1:1). Водный слой экстрагировали 50 мл этилацетата и объединенные органические слои сушили над сульфатом натрия. После удаления растворителя в вакууме и хроматографии на колонке с силикагелем (элюент: н-гептан/этилацетат, 3:1) получали 1,52 г (выход 62%) соединения С1.
Cоотношение кето-енольных групп, определенное методом ЯМР в CDCl3 при 400 МГц составляло приблизительно 30:70. МС (API+, API-): M 352, 350.
Пример С2
1-(3-(3-Пиридинилметилокси)фенил)-2-(2-метилтиопиримидин-4-ил)этанон (С2)
Указанное соединение (выход 60%) получали аналогично тому, как описано в примере С1, но при использовании в качестве исходного материала соединения B4. МС (API+, API-): M 352, 350.
Пример С3
1-(3-(4-Пиридинилметилокси)фенил)-2-(2-метилтиопиримидин-4-ил)этанон (С3)
Указанное соединение (выход 55%) получали аналогично тому, как описано в примере С1, но при использовании в качестве исходного материала соединения B5. МС (API+, API-): M 352, 350.
Пример С4
1-(3-Бензилоксифенил)-2-(2-метилтиопиримидин-4-ил)этанон (С4)
Указанное соединение (выход 89%) получали аналогично тому, как описано в примере С1, но при использовании в качестве исходного материала соединения В1. МС (API+, API-): M 351, 349.
D. Синтез кетоксимов
Пример D1
1-(3-(2-Пиридинилметилокси)фенил)-2-(2-метилтиопиримидин-4-ил)-2-гидроксииминоэтанон (D1)
Соединение С1 (1,50 г, 4,30 ммоля) растворяли в ледяной уксусной кислоте (18,6 мл), тетрагидрофуране (15,0 мл) и воде (2,0 мл), после охлаждения до 5°С добавляли раствор нитрита натрия (353 мг, 5,1 ммоля) в 3,5 мл H2O, при этом температуру поддерживали в интервале от 5°С до 10°С. Охлаждающую баню удаляли и смесь перемешивали при комнатной температуре в течение 2 ч. После удаления растворителя в вакууме добавляли 35 мл воды и 240 мл этилацетата и смесь подщелачивали до рН 8 с использованием 3 н. NaOH. Фазы разделяли и водный слой экстрагировали 50 мл этилацетата. Объединенные органические слои сушили над сульфатом натрия и растворитель удаляли в вакууме, при этом получали 1,61 г соединения D1 (выход 99%). МС (API+), (API-): M 381, 379.
1Н-ЯМР (400 МГц, ДМСО-D6): δ 2,17 (s, 3Н, SCH3), 5,25 (s, 2H, OCH2), 7,30-7,35 (m, 1H), 7,36-7,42 (m, 3Н), 7,47-7,54 (m, 2H), 7,62-7,65 (m, 1H), 7,78-7,85 (m, 1H), 8,52-8,55 (m, 1H), 8,68-8,72 (m, 1H), 12,80 (ушир., 1H, ОН).
13С-ЯМР (100,6 МГц, ДМСО-D6): δ 13,6 (SCH3), 70,9 (ОСН2), 111,7, 113,8, 121,6, 122,0, 122,1, 123,4, 131,0, 136,4*, 137,3, 149,5, 154,0*, 156,5*, 158,7, 158,9*, 159,3*, 171,8*, 193,3* (С=O) (*четвертичный углерод).
Пример D2
1-(3-(3-Пиридинилметилокси)фенил)-2-(2-метилтиопиримидин-4-ил)-2-гидроксииминоэтанон (D2)
Указанное соединение (выход 92%) получали аналогично тому, как описано в примере С1, но при использовании в качестве исходного материала соединения С2. МС (API+, API-): M 381, 379.
1Н-ЯМР (400 МГц, ДМСО-D6): δ 2,19 (s, 3H, SCH3), 5,23 (s, 2H, OCH2), 7,32-7,46 (m, 4Н), 7,46-7,56 (m, 1H), 7,58-7,68 (m, 1H), 7,84-7,92 (m, 1H), 8,50-8,56 (m, 1H), 8,64-8,72 (m, 2H).
Пример D3
1-(3-(4-Пиридинилметилокси)фенил)-2-(2-метилтиопиримидин-4-ил)-2-гидроксииминоэтанон (D3)
Указанное соединение (выход 97%) получали аналогично тому, как описано в примере С1, но при использовании в качестве исходного материала соединения С3. МС (API+, API-): M 381, 379.
1Н-ЯМР (400 МГц, ДМСО-D6): δ 2,18 (s, 3H, SCH3), 5,27 (s, 2H, OCH2), 7,36-7,43 (m, 3H), 7,43-7,49 (m, 2H), 7,49-7,55 (m, 1H), 7,61-7,68 (m, 1H), 8,54-8,63 (m, 2H), 8,68-8,74 (m, 1H), 12,78 (s, 1H, ОН).
Пример D4
1-(3-Бензилоксифенил)-2-(2-метилтиопиримидин-4-ил)-2-гидроксииминоэтанон (D4)
Указанное соединение (выход 86%) получали аналогично тому, как описано в примере С1, но при использовании в качестве исходного материала соединения С4. МС (API+, API-): M 380, 378.
1Н-ЯМР (250 МГц, ДМСО-D6): δ 2,19 (s, 3H, SCH3), 5,17 (s, 2H, OCH2), 7,24-7,53 (m, 9H), 7,59-7,69 (m, 1H), 8,63-8,72 (m, 1H), 12,83 (ушир., 1H, ОН).
13С-ЯМР (62,9 МГц, ДМСО-D6): δ 13,6 (SCH3), 69,9 (OCH2), 111,7, 113,7, 121,6, 122,1, 128,1, 128,3, 128,8, 130,9, 136,5* 136,9*, 154,0*, 158,6, 159,1*, 159,4*, 171,8*, 193,5* (С=O) (*четвертичный углерод).
Е. Синтез N-гидроксиимидазолов
Пример Е1
2-(2,6-Дихлор-4-[этоксикарбонилметокси]фенил)-4-(3-(2-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-гидроксиимидазол (Е1)
Смесь соединения D1 (1,60 г, 4,2 ммоля), соединения А2 (1,40 г, 5,0 ммоля) и ацетата аммония (3,25 г, 42 ммоля) в 40 мл уксусной кислоты перемешивали при 105°С в течение 3,5 ч. Растворитель упаривали, остаток распределяли между 40 мл ледяной воды и 60 мл этилацетата и подщелачивали до рН 8 с использованием конц. водного аммиака. Водный слой экстрагировали этилацетатом и объединенные органические слои сушили над MgSO4, упаривали досуха и при этом получали 3,60 г медленно затвердевающего масла светло-коричневого цвета, которое использовали на следующей стадии (пример F1) без дополнительной очистки. МС (API+, API-): M 638, 636.
Пример Е2
2-(2,6-Дихлор-4-[этоксикарбонилметокси]фенил)-4-(3-(3-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-гидроксиимидазол (Е2)
Указанное соединение (Е2) в виде масла оранжевого цвета получали аналогично тому, как описано в примере Е1, но при использовании в качестве исходного материала соединения D2. МС (API+, API-): M 638, 636.
Пример Е3
2-(2,6-Дихлор-4-[этоксикарбонилметокси]фенил)-4-(3-(4-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-гидроксиимидазол (Е3)
Указанное соединение (Е3) в виде твердого вещества желтого цвета получали аналогично тому, как описано в примере Е1, но при использовании в качестве исходного материала соединения D3. МС (API+, API-): M 638, 636.
Пример Е4
2-(3,5-Дихлорпиридин-4-ил)-4-(3-бензилоксифенил)-5-(2-метилтиопиримидин-4-ил)-N-гидроксиимидазол (Е4)
Указанное соединение (Е4) в виде твердого вещества оранжевого цвета получали аналогично тому, как описано в примере Е1, но при использовании в качестве исходных материалов соединения D4 и 3,5-дихлорпиридин-4-карбоксальдегида (J. Med. Chem. 44, 997 (2001)). MC (API+, API-): M 536, 534.
F. Синтез N-H-имидазолов
Пример F1
2-(2,6-Дихлор-4-[метоксикарбонилметокси]фенил)-4-(3-(2-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-Н-имидазол (F1)
Смесь соединения Е1 (3,59 г, 4,2 ммоля), метилбромацетата (1,41 г, 8,4 ммоля) и триэтиламина (2,85 г, 28 ммолей) в 80 мл метанола перемешивали при 60°С в течение ночи. После удаления растворителя в вакууме и хроматографии на колонке с силикагелем (элюент: этилацетат/гексан, 3:1) получали очищенное соединение F1 (2,08 г, 81%, переэтерификация в метаноле). MC (API+, API-): M 608, 606.
Пример F2
2-(2,6-Дихлор-4-[метоксикарбонилметокси]фенил)-4-(3-(3-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-Н-имидазол (F2)
Указанное соединение (F2, выход 65%) получали аналогично тому, как описано в примере F1, но при использовании в качестве исходного материала соединения Е2. MC (API+, API-): M 608, 606.
Пример F3
2-(2,6-Дихлор-4-[метоксикарбонилметокси]фенил)-4-(3-(4-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-Н-имидазол (F3)
Указанное соединение (F3, выход 66%) получали аналогично тому, как описано в примере F1, но при использовании в качестве исходного материала соединения Е3. MC (API+, API-): M 608, 606.
Пример F4
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(2-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-Н-имидазол (F4)
В раствор соединения F1 (2,07 г, 3,3 ммоля) в 50 мл сухого ТГФ в атмосфере азота при 0°С добавляли 1 М раствор LiAlH4 в ТГФ до отсутствия в смеси F1 по данным ЖХВР. После гидролиза в присутствии 0,5 мл воды и удаления растворителя остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/метанол, 9:1), при этом получали соединение F4 (выход 93%), MC (API+, API-): M 580, 578.
Пример F5
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(3-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-Н-имидазол (F5)
Указанное соединение (F5, выход 64%) получали аналогично тому, как описано в примере F4, но при использовании в качестве исходного материала соединения F2. МС (API+, API-): M 580, 578.
Пример F6
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(4-пиридинилметилокси)фенил)-5-(2-метилтиопиримидин-4-ил)-N-Н-имидазол (F6)
Указанное соединение (F6, выход 69%) получали аналогично тому, как описано в примере F4, но при использовании в качестве исходного материала соединения F3. МС (API+, API-): M 580, 578.
Пример F7
2-(3,5-Дихлорпиридин-4-ил)-4-(3-бензилоксифенил)-5-(2-метилтиопиримидин-4-ил)-N-Н-имидазол (F7)
Указанное соединение (F7, выход 66%) получали аналогично тому, как описано в примере F1, но при использовании в качестве исходного материала соединения Е4. МС (API+, API-): M 520, 518.
G. Синтез сульфинил- и сульфанил-N-Н-имидазолов
Пример G1
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(2-пиридинилметилокси)фенил)-5-(2-метансульфинилпиримидин-4-ил)-N-Н-имидазол (G1)
В раствор соединения F4 (1,20 г, 2,1 ммоля) в 300 мл этилацетата и 50 мл раствора дихлорметана при 40°С в течение 10 мин добавляли раствор мета-хлорпербензойной кислоты (0,70 г, 3,1 ммоля) в 20 мл этилацетата и выдерживали при указанной температуре в течение 1 ч. Затем смесь нагревали до комнатной температуры и перемешивали в течение ночи, промывали (насыщенный водный NaHCO3/насыщенный водный Na2CO3, 1:1), сушили над MgSO4 и упаривали досуха, при этом получали 1,21 г неочищенного соединения G1, которое использовали без дальнейшей очистки. МС (API+, API-): M 596, 594.
Пример G2
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(3-пиридинилметилокси)фенил)-5-(2-метансульфинилпиримидин-4-ил)-N-Н-имидазол (G2)
Указанное соединение G2 получали аналогично тому, как описано в примере G1, но при использовании в качестве исходного материала соединения F5. МС (API+, API-): M 596, 594.
Пример G3
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(4-пиридинилметилокси)фенил)-5-(2-метансульфинилпиримидин-4-ил)-N-Н-имидазол (G3)
Указанное соединение G3 получали аналогично тому, как описано в примере G1, но при использовании в качестве исходного материала соединения F6. МС (API+, API-): M 596, 594.
Пример G4
2-(3,5-Дихлорпиридин-4-ил)-4-(3-бензилоксифенил)-5-(2-метансульфанилпиримидин-4-ил)-N-Н-имидазол (G4)
Соединение F7 (2,38 г, 4,6 ммоля) суспендировали в 200 мл метанола и при комнатной температуре в течение 20 мин добавляли раствор оксона (5,62 г, 9,1 ммоля), перемешивали при комнатной температуре в течение ночи, метанол удаляли в вакууме, а остаток переносили в этилацетат. Органический слой промывали водным NaHCO3, сушили над Na2SO4 и упаривали досуха. Неочищенное соединение G4 (47%) использовали без дальнейшей очистки. МС (API+, API-): M 552, 550.
Н. Синтез аминопиримидинил-N-Н-имидазолов
Пример H1
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(2-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1H-имидазол (H1)
Соединение G1 (1,21 г, 2,0 ммоля) и 3-амино-1-пропанол (3,1 г, 40,6 ммоля) нагревали при 110°С в течение 60 мин. После очистки препаративной ЖХВР/МС на колонке RP 18 (элюент: градиент метанол/вода) получали соединение H1 (510 мг, выход 42%). МС (API+, API-): M 607, 605.
Пример Н2
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(3-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1H-имидазол (Н2)
Указанное соединение (Н2, выход 50%) получали аналогично тому, как описано в примере H1, но при использовании в качестве исходного материала соединения G2. МС (API+, API-): M 607, 605.
Пример Н3
2-(2,6-Дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(4-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1H-имидазол (Н3)
Указанное соединение (Н3, выход 32%) получали аналогично тому, как описано в примере H1, но при использовании в качестве исходного материала соединения G3. МС (API+, API-): M 607, 605.
Пример Н4
2-(3,5-Дихлорпиридин-4-ил)-4-(3-бензилоксифенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол (Н4)
Указанное соединение (Н4, выход 68%) получали аналогично тому, как описано в примере H1, но при использовании в качестве исходного материала соединения G4. МС (API+, API-): M 547, 545.
Пример J
Анализ автоактивации киназы c-met (AAK)
Принцип анализа
C-met является типичной тирозинкиназой, которая включена в процессы метастазирования, пролиферации/апоптоза и ангиогенеза опухолей. Анализ относится к типу ИФА, при котором измеряют фосфорилирование c-met с использованием антител, специфичных к фосфотирозину.
Лизатом клеток аденокарциномы НТ29 ободочной кишки человека, характеризующихся высоким содержанием c-met, покрывали лунки микротитрационного планшета с использованием антител против рецептора hHGF (anti-hHGFR). АТФ-фосфорилирование c-met определяли в присутствии или в отсутствие исследуемых соединений с использованием системы для детектирования, включающей мышиные IgG к фосфотирозину и меченый POD антимышиный IgG козы. Ферментативную активность определяли с использованием стандартного субстрата POD - ТМБ, при 450/620 нм.
Материалы
Планшеты: 96-луночные полистирольные планшеты (NUNC), покрытые стрептавидином микротитрационные планшеты.
Клеточная линия/лизат: клетки НТ29 (АТСС НТВ-38) аденокарциномы ободочной кишки человека (конфлуентность 2,5×105 клеток/см2) промывали фосфатно-солевым буферным раствором (ФСБ) и инкубировали в буферном растворе для лизиса в течение 10 мин при охлаждении льдом. Супернатант собирали и разбавляли буферным раствором ТСБ. Лизат быстро замораживали в жидком азоте и хранили при -80°С.
Реагенты (если не указано иное, все рабочие растворы хранили при 4°С):
исходный раствор для детектирования anti-hHGFR: 50 мкг/мл (фирмы R&D Systems, номер по каталогу BAF 358), конечная концентрация антител 1 мкг/мл,
исходный раствор мышиных антител p-Tyr (PY99): 200 мкг/мл (фирмы Santa Cruz Biotechnology, моноклональные IgG2b, номер по каталогу SC-7020), конечная концентрация 0,2 мкг/мл,
антимышиный IgG козы: 2 мл (фирмы Bio-Rad, номер по каталогу 170-6516), коньюгат (H+L)-HRP, конечная концентрация 1:2000,
блокирующий реагент (фирмы Roche Diagnostics GmbH, номер по каталогу 1112589) для ИФА, разбавление в ТСБ 1:10,
АТФ, аденозин-5'-трифосфат, исходный раствор 10 мМ (фирмы Roche Diagnostics GmbH, номер по каталогу 127531), конечная концентрация 40 мкМ,
ТСБ, трис-солевой буферный раствор: 50 мМ трис, рН 7,5 (фирмы Roche Diagnostics GmbH, номер по каталогу 708976), 150 мМ NaCl (фирмы Sigma, номер по каталогу S-3014),
буферный раствор ТСБ-Т для промывки: трис-солевой буферный раствор, 50 мМ трис, рН 7,5, 150 мМ NaCl, содержащий 0,5% твин-20,
буферный раствор для киназы: трис-солевой буферный раствор, 50 мМ трис, рН 7,5, 100 мМ NaCl, 60 мМ MgCl2 (фирмы Sigma Chemical Company, номер по каталогу М-1028),
буферный раствор для лизиса: 50 мМ трис, рН 7,5, содержащий 1% нонидет Р40 (фирмы Roche Diagnostics GmbH, номер по каталогу 1754599), 0,5% дезоксихолевой кислоты (фирмы Sigma Chemical Company, номер по каталогу D-6750), конечная концентрация 1 мМ PMSF, исходный раствор 70 мМ (фирмы Roche Diagnostics GmbH, номер по каталогу 837091), 40 мкл/мл Complete (фирмы Roche Diagnostics GmbH, номер по каталогу 1836145), конечная концентрация 40 мкл/мл,
ТМБ: тетраметилбензидин (фирмы Intergen Company, номер по каталогу 910000).
Образцы: 10 мМ в ДМСО (хранят при -20°С), размораживают при комнатной температуре.
Методика
1. В лунки аналитического планшета добавляли по 50 мкл раствора антител для детектирования anti-hHGFR в растворе блокирующего реагента (конечная концентрация 1 мкг/мл), планшет инкубировали в течение 60 мин при комнатной температуре при встряхивании на качалке для микропланшетов.
2. Раствор антител для детектирования anti-hHGFR удаляли из лунок аналитического планшета.
3. В каждую лунку аналитического планшета добавляли по 250 мкл раствора блокирующего реагента, инкубировали планшет в течение 20 ч при 4°С.
4. Раствор блокирующего реагента удаляли из лунок аналитического планшета.
5. В лунки добавляли по 50 мкл лизата НТ29, аналитический планшет инкубировали в течение 180 мин при 4°С на качалке для микропланшетов.
6. Лунки аналитического планшета промывали ТСБ 2 раза по 200 мкл.
7. В лунки добавляли по 40 мкл 0,2% ДМСО в буферном растворе для киназы.
8. Добавляли по 40 мкл раствора образца (разбавленного в буферном растворе для киназы, конечная концентрация 22,5 мкМ).
9. Образцы растворяли (соотношение 1:3) при встряхивании на качалке для микропланшетов.
10. К образцам добавляли по 10 мкл АТФ, растворенного в буферном растворе для киназы (200 мкМ), до конечной концентрации 40 мкМ АТФ. В лунки с положительным контролем добавляли по 40 мкл буферного раствора для киназы и 10 мкл 200 мкМ раствора АТФ. В лунки с отрицательным контролем добавляли по 40 мкл буферного раствора для киназы и 10 мкл буферного раствора для киназы, не содержащего АТФ. Планшет инкубировали в течение 60 мин при комнатной температуре при встряхивании на качалке для микропланшетов.
11. Аналитический планшет промывали буферным раствором ТСБ 2 раза по 200 мкл и блокирующим реагентом 2 раза по 200 мкл в лунку.
12. В лунки планшета добавляли по 50 мкл мышиных моноклональных антител P-Tyr (PY99), IgG2b (конечная концентрация 200 нг/мл) в растворе блокирующего реагента, инкубировали планшет в течение ночи при 4°С при встряхивании на качалке для микропланшетов.
13. Планшет промывали буферным раствором ТСБ 2 раза по 200 мкл и блокирующим реагентом 2 раза по 200 мкл.
14. В каждую лунку добавляли по 50 мкл коньюгата антимышиных IgG (H+L)-HRP козы в растворе блокирующего реагента (соотношение 1:2000), планшет инкубировали в течение 60 мин при комнатной температуре при встряхивании на качалке для микропланшетов.
15. Лунки промывали буферным раствором ТСБ-Т 6 раз по 200 мкл.
16. В лунки добавляли по 50 мкл раствора ТМБ, инкубировали в течение 30 мин при комнатной температуре при встряхивании на качалке микропланшетов, затем добавляли по 25 мкл 1 М H2SO4.
17. Измеряли оптическую плотность (Е) при 450/620 нм.
18. Ингибирование в % рассчитывали по уравнению:
1-[(Eобразец-Eотриц.контроль)/(Eположит.контроль-Eотриц.контроль)×1000]
Данные описанного выше анализа свидетельствуют о том, что агенты по настоящему изобретению в типичном случае характеризуются величиной IC50 (ингибирование киназы) в диапазоне от приблизительно 1 нМ до приблизительно 100 нМ.
Пример К
Состав таблетки (влажное гранулирование)
Методика получения
1. Ингредиенты 1, 2, 3 и 4 смешивали и гранулировали в смеси с очищенной водой.
2. Гранулы сушили при 50°С.
3. Гранулы измельчали на пригодном оборудовании для измельчения.
4. Добавляли ингредиент 5 и перемешивали в течение 3 мин, затем прессовали на пригодном прессе.
Пример L
Состав капсулы
Методика получения
1. Ингредиенты 1, 2 и 3 смешивали в пригодном смесителе в течение 30 мин.
2. К смеси добавляли ингредиенты 4 и 5 и перемешивали в течение 3 мин.
3. Полученной смесью заполняли капсулы пригодного типа и размера.

| название | год | авторы | номер документа |
|---|---|---|---|
| 2-(2,6-ДИХЛОРФЕНИЛ)ДИАРИЛИМИДАЗОЛЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2003 |
|
RU2320645C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1993 |
|
RU2130453C1 |
| ПРОИЗВОДНЫЕ НАФТИЛЕНА КАК ИНГИБИТОРЫ ЦИТОХРОМА Р450 | 2004 |
|
RU2363696C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ОПИОИДНЫХ РЕЦЕПТОРОВ | 2003 |
|
RU2332411C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСИМЕТИЛФУРАЗАНКАРБОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1994 |
|
RU2134687C1 |
| ЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ КОНДЕНСИРОВАННОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2815814C1 |
| ПРИМЕНЕНИЕ РЕЦЕПТОРА АНТАГОНИСТА ЕР4 ДЛЯ ЛЕЧЕНИЯ ХРЯЩЕВОЙ БОЛЕЗНИ | 2014 |
|
RU2663620C2 |
| ПРОИЗВОДНЫЕ БИСФЕНОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРА АНДРОГЕНА | 2017 |
|
RU2767257C2 |
| ИНГИБИТОР FGFR И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2745035C1 |
| НЕСИММЕТРИЧНЫЕ АРИЛФОСФИТЫ, ПОЛИМЕРНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1992 |
|
RU2071478C1 |
Изобретение относится к новым соединениям общей формулы (I) и фармацевтически приемлемым солям указанного соединения, обладающим свойствами ингибитора c-met киназы, которые могут найти применение для лечения рака и связанных с раком заболеваний. В общей формуле (I)
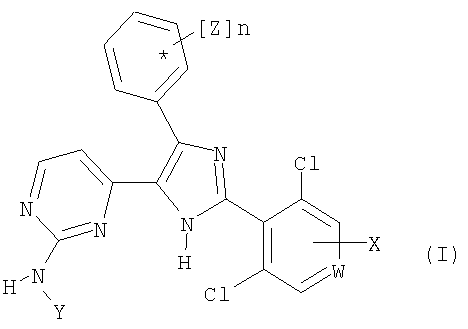
W означает -N= и Х означает водород, Y означает водород или группу A2-R; А2 означает С1-С5алкилен; R означает гидрокси; n равно 1 и Z означает бензилокси;
или альтернативно W означает -СН= и Х означает водород, OR1; R1 выбирают из группы A-Q1; А1 означает 1,2-этиленовую или 1,3-пропиленовую группу; Q1 означает гидрокси;
Y означает группу A2-R; А2 означает C1-С5алкилен; R означает гидрокси; n равно 1 и Z означает C1-С3алкокси, замещенный пиридинилом, и фармацевтически приемлемые соли указанного соединения. 2 н. и 6 з. п. ф-лы.
1. Соединение общей формулы (I)
где W означает -N= и
Х означает водород,
Y означает водород или группу A2-R;
A2 означает С1-С5алкилен;
R означает гидрокси;
n равно 1 и
Z означает бензилокси;
или альтернативно
W означает -СН= и
Х означает водород, OR1;
R1 выбирают из группы A1-Q1;
А1 означает 1,2-этиленовую или 1,3-пропиленовую группу;
Q1 означает гидрокси;
Y означает группу A2-R;
A2 означает С1-С5алкилен;
R означает гидрокси;
n равно 1 и
Z означает C1-С3алкокси, замещенный пиридинилом, и фармацевтически приемлемые соли указанного соединения.
2. Соединение по п.1, где W означает -N=,
Х означает водород,
Y означает 2-гидроксиэтил, 3-гидроксипропил, (R)-3-гидроксибутил, (S)-3-гидроксибутил;
n равно 1 и Z означает 3-бензилокси.
3. Соединение по п.1, где W означает -N=,
Х означает водород,
Y означает 2-гидроксиэтил, 3-гидроксипропил;
n равно 1 и
Z означает 3-бензилокси.
4. Соединение по п.3, которое представляет собой 2-(3,5-дихлорпиридин-4-ил)-4-(3-бензилоксифенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол.
5. Соединения по п.1,
где W означает -СН=,
Х означает водород, OR1;
R1 выбирают из группы A1-Q1;
А1 означает 1,2-этиленовую или 1,3-пропиленовую группу, Q1 означает гидрокси;
Y означает группу А2-R;
А2 означает С1-С5алкилен;
R означает гидрокси;
n равно 1 и
Z означает C1-С3алкокси, замещенный пиридинилом.
6. Соединения по п.1, где W означает -СН=,
Х означает водород, OR1;
R1 выбирают из группы A1-Q1;
А1 означает 1,2-этиленовую или 1,3-пропиленовую группу, Q1 бозначает гидрокси;
Y означает 3-гидроксипропил;
n равно 1 и
Z означает пиридин-2-илметокси, пиридин-3-илметокси или пиридин-4-илметокси.
7. Соединения по п.6, которые представляют собой
2-(2,6-дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(4-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол,
2-(2,6-дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(3-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1H-имидазол или
2-(2,6-дихлор-4-[2-гидроксиэтокси]фенил)-4-(3-(2-пиридинилметилокси)фенил)-5-(2-[3-гидроксипропиламино]пиримидин-4-ил)-N-1Н-имидазол.
8. Фармацевтическая композиция, обладающая инигибирующим действием в отношении c-met киназы, содержащая одно или более соединений по любому из пп.1-7 в качестве активного ингредиента(ов) в смеси с фармацевтически приемлемыми адъювантами.
| RU 2001101820 A1, 20.11.2001 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| US 6610695 В1, 26.08.2003 | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| US 5656644 A, 12.08.1997. | |||

