ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к ряду азатрициклических соединений, которые выполняют функцию рецепторов фактора роста фибробластов (FGFR), а также к способу их получения и к фармацевтической композиции на их основе. Настоящее изобретение дополнительно относится к применению указанных выше азатрициклических соединений или фармацевтической композиции на их основе в лечении нарушений, опосредованных FGFR.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Протеинкиназы представляют собой ферменты, которые катализируют фосфорилирование белков; в большинстве случаев фосфорилирование происходит по остаткам серина (ser), треонина (thr) и тирозина (tyr) в белке. Многие аспекты процессов жизнедеятельности клетки (например, рост, дифференциация, пролиферация, клеточный цикл и выживаемость клетки) зависят от активности протеинкиназы. Кроме того, множество заболеваний (например, рак и воспаление) ассоциированы с аномальной активностью протеинкиназы.
На данный момент было обнаружено, что семейство протеин-тирозинкиназ (PTK) имеет более 100 представителей, которые играют важную роль в регуляции дифференциации, роста и пролиферации клеток. В соответствии со структурой PTK их можно разделить на два типа: PTK рецепторного типа и PTK нерецепторного типа. Первую также называют трансмембранной PTK, а последнюю также называют внутриклеточной PTK.
Рецепторы фактора роста фибробластов (FGFR), которые относятся к представителям надсемейства рецепторных тирозинкиназ (RTK), стали одной из мишеней международных фармацевтических компаний, разрабатывающих новые противоопухолевые лекарственные средства. FGFR вовлечен в модуляцию пролиферации, апоптоза, миграции клеток, в образование новых сосудов и в другие процессы. Вследствие широкого диапазона вариантов их применения FGFR и другие RTK жестко регулируются в обычных условиях. В опухоли, такой как рак молочной железы, рак мочевого пузыря, рак предстательной железы (в настоящее время исследуемые показания) и т. д., активирующая мутация FGFR или сверхэкспрессия лиганда/рецептора приводит к его непрерывной активации, которая не только тесно связана с частотой возникновения, развитием опухоли, неблагоприятным прогнозом в отношении опухоли, но также играет ключевую роль в ангиогенезе опухоли, инвазии опухоли и метастазировании. Следовательно, FGFR признан важным кандидатом для видов нацеленной терапии рака, и со временем все большее внимание получает разработка низкомолекулярных ингибиторов FGFR.
Семейство FGFR главным образом предусматривает FGFR1, FGFR2, FGFR3 и FGFR4, и, в частности, поделено на подтипы FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c и FGFR4. Они имеют общий домен, в том числе внеклеточный иммуноглобулиноподобный домен и внутриклеточный тирозинкиназный домен (Peijuan Du, Chemistry & Bioengineering, Vol. 31, No. 12, 2014, 5-8). Ген FGFR1 кодирует подтипы FGFR1b и FGFR1c на участке 8p12 хромосомы человека. Вследствие альтернативного сплайсинга они различаются по третьему иммуноглобулиноподобному домену. Ген FGFR2 расположен на участке 10q26 хромосомы человека, ген FGFR3 расположен на участке 4p16.3 хромосомы человека, и они также кодируют по два типа. В раковых клетках активация протоонкогенов человека путем амплификации генов, хромосомных транслокаций и точечных мутаций активирует ген FGFR. FGFR вовлечен в образование опухолей и ангиогенез в раковых клетках и эндотелиальных клетках соответственно, следовательно, нацеленные на FGFR лекарственные средства будут обеспечивать прямые или опосредованные противораковые эффекты.
Активация и трансдукция фактора роста фибробластов (FGF): FGF могут инициировать аутофосфорилирование FGFR по остаткам тирозина на ключевых активирующих петлях в тирозинкиназном домене, что обуславливает переход тирозинкиназного домена из неактивного состояния в активированное состояние (Bae J H, Schlessinger J. Molecules and Cells, 2010, 29(5): 443-448). Активированный тирозинкиназный домен в FGFR будет фосфорилировать другие остатки тирозина постепенно вдоль FGFR-связывающей адаптерной молекулы в участке связывания субстрата. Фосфорилирование остатков тирозина в C-концевой области FGFR обеспечивает возможность поглощения и активирования фосфатазы Cγ (PLCγ), в результате чего катализируется превращение фосфатидилинозитдифосфата (PIP2) в диглицерид (DAG) и инозиттрифосфат (IP3) (Dailey L, Ambrostti D, Mansukhani A, et al. Cytokine & Growth Factor Reviews, 2005, 16(2), 233-247). Активированный субстрат 2 для фосфорилирования FGFR (FRS2) способен поглощать адаптерную молекулу, которая представляет собой связывающий с рецептором фактора роста белок 2 (GRB2).
Сигнал FGF может передаваться в сигнальный путь Ras-митоген-активируемой протеинкиназы (Ras-MAPK) или PI3-киназы-протеинкиназы B (PI3K-AKT) посредством FRS2 и GRB2 и передаваться в сигнальный путь протеинкиназы C (PKC) или протеинкиназы D (PKD) посредством PLCγ и DAG-киназы, а также передаваться в каскадный путь высвобождения ионов кальция посредством PLCγ и IP3. FGF-индуцированная активация Ras-MAPK вовлечена в пролиферацию клетки, при этом FGF-индуцированная активация PI3K-AKT вовлечена в выживание клетки.
Сигнал FGF участвует в различных аспектах биологии опухоли, таких как антиапоптоз, ангиогенез, эпителиально-мезенхимальный переход (EMT), инвазия и т. д. Направленная на FGFR терапия стала актуальной проблемой в области клинической онкологии, и в терапии рака применялись разработанные и исследованные низкомолекулярные соединения для связывания с ATP-связывающим "карманом" в тирозинкиназном домене.
Разрабатываемые в настоящее время ингибиторы FGFR предусматривают AZD4547, BGJ398 (инфигратиниб), Debio-1347, JNJ42756493, FIIN-2, BLU-554, ARQ087 и PD173074 и т. д. Из них AZD4547, BGJ398 и Debio-1347 представляют собой ингибиторы FGFR1/2/3, и AZD4547 представляет собой многоцелевой ингибитор FGFR, колониестимулирующего фактора (CSF1R) и рецептора-2 фактора роста эндотелия сосудов (VEGFR-2); BLU-554 представляет собой селективный ингибитор FGFR4; JNJ42756493 и FIIN-2 представляют собой ингибиторы для всех FGFR; AZD4547, BGJ398 и Debio-1347 представляют собой обратимые ингибиторы FGFR; BLU-554 и FIIN-2 представляют собой необратимые ингибиторы FGFR (Masaru Katoh, Internatonal Journal of Molecular Medicine, 2016, 38:3-15).
В раковых клетках человека с аномальной активацией FGFR и антиапоптотическим потенциалом ингибирование передачи сигнала FGF может уменьшать нагрузку раковых клеток и при этом ингибировать ангиогенез, и ингибиторы FGFR могут усиливать чувствительность раковых клеток к традиционным противораковым лекарственным средствам (таким как 5-фторурацил, иринотекан, паклитаксел и т. д.). Учитывая глубокое понимание сигнальных сетей с участием FGF и интенсивные исследования механизмов действия FGF и FGFR, будут разработаны ингибиторы FGFR с высокой специфичностью и надлежащими терапевтическими эффектами, и, следовательно, применение направленно воздействующих на FGFR лекарственных средств при лечении опухолей обладает широкими перспективами.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к азатрициклическим соединениям, которые применяют в качестве ингибитора рецептора фактора роста фибробластов (FGFR), а также к их фармацевтически приемлемым солям, сольватам, хелатам, нековалентным комплексам или пролекарствам. Соединения по настоящему изобретению характеризуются общими структурами формулы (I),
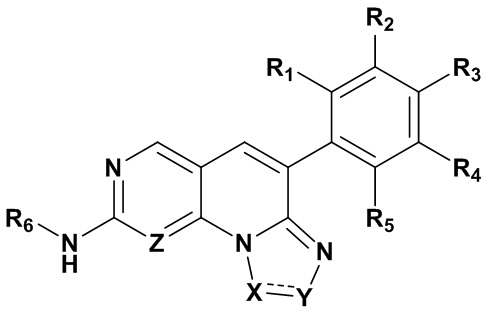
формула (I),
где
 представляет собой одинарную связь или двойную связь;
представляет собой одинарную связь или двойную связь;
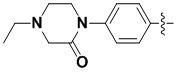
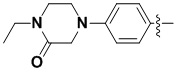
каждый из X и Y независимо выбран из N, NH, C=O и CR10;
Z представляет собой N или CR10;
каждый из R1, R2, R3, R4 и R5 независимо выбран из H, галогена, C1-8алкокси, замещенного C1-8алкокси, C1-8алкила, замещенного C1-8алкила, C2-8алкенила, замещенного C2-8алкенила, C2-8алкинила, замещенного C2-8алкинила, C3-8циклоалкила, замещенного C3-8циклоалкила, C6-10арила, замещенного C6-10арила, C5-10гетероарила, замещенного C5-10гетероарила, C3-10гетероциклила и замещенного C3-10гетероциклила; или
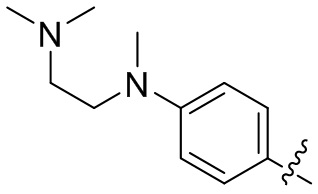
R2 и R3 или R3 и R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-8-членное замещенное или незамещенное гетероциклическое или гетероарильное кольцо, при этом гетероциклическое или гетероарильное кольцо содержит 1, 2 или 3 гетероатома, независимо выбранные из N, O или S;
R6 представляет собой C1-8алкил, замещенный C1-8алкил, C2-8алкенил, замещенный C2-8алкенил, C2-8алкинил, замещенный C2-8алкинил, C6-10арил, замещенный C6-10арил, C3-8циклоалкил, замещенный C3-8циклоалкил, C3-10гетероциклил, замещенный C3-10гетероциклил, C5-10гетероарил или замещенный C5-10гетероарил;
R6 может быть необязательно замещен R7;
R7 представляет собой гидроксил, галоген, C1-8алкил, замещенный C1-8алкил, C2-8алкенил, замещенный C2-8алкенил, C2-8алкинил, замещенный C2-8алкинил, C1-8алкокси, замещенный C1-8алкокси, C6-10арил, замещенный C6-10арил, C3-8циклоалкил, замещенный C3-8циклоалкил, C3-10гетероциклил, замещенный C3-10гетероциклил, C5-10гетероарил, замещенный C5-10гетероарил, гетероциклилалкил, замещенный гетероциклилалкил, гетероциклический карбонил, замещенный гетероциклический карбонил, -NR11R12, -NR11-C1-8алкилен-NR11R12, или R7 представляет собой замещенное или незамещенное C5-8гетероциклическое кольцо, конденсированное с R6;
R10 представляет собой H, галоген, амино, C1-6алкил, замещенный C1-6алкил, C2-8алкенил, замещенный C2-8алкенил, C2-8алкинил, замещенный C2-8алкинил, C3-8циклоалкил, замещенный C3-8циклоалкил, C6-10арил, замещенный C6-10арил, C5-10гетероарил, замещенный C5-10гетероарил, C3-10гетероциклил или замещенный C3-10гетероциклил;
R10 может быть необязательно замещен R8;
R8 представляет собой гидроксил, галоген, C1-8алкил, замещенный C1-8алкил, C2-8алкенил, замещенный C2-8алкенил, C2-8алкинил, замещенный C2-8алкинил, C3-10гетероциклоалкокси, замещенный C3-10гетероциклоалкокси, C6-10арил, замещенный C6-10арил, C3-8циклоалкил, замещенный C3-8циклоалкил, C3-10гетероциклил, замещенный C3-10гетероциклил, -S(O2)C3-10гетероциклил, замещенный -S(O2)C3-10гетероциклил, C5-10гетероарил, замещенный C5-10гетероарил или -NR11R12;
каждый из R11 и R12 независимо представляет собой H, C1-8алкил, замещенный C1-8алкил, C2-8алкенил, замещенный C2-8алкенил, C2-8алкинил, замещенный C2-8алкинил, C3-8циклоалкил, замещенный C3-8циклоалкил, C6-10арил, замещенный C6-10арил, C5-10гетероарил, замещенный C5-10гетероарил, C3-10гетероциклил или замещенный C3-10гетероциклил.
В настоящем изобретении дополнительно предусмотрены некоторые предпочтительные технические решения применительно к соединению формулы I.
В некоторых вариантах осуществления формулы I: X представляет собой N или CR10, R10 представляет собой H, амино, C1-6алкил, замещенный C1-6алкил, C3-6циклоалкил или замещенный C3-6циклоалкил.
В некоторых вариантах осуществления формулы I: X представляет собой CR10, R10 представляет собой H.
В некоторых вариантах осуществления формулы I: X представляет собой CR10, R10 представляет собой C1-6алкил, C1-6алкил, замещенный C5-10гетероциклилом, C1-6алкил, замещенный C6-10арилом, C1-6алкил, замещенный C3-6циклоалкилом, C1-6алкил или C3-6циклоалкил, замещенные амино, при этом C5-10гетероциклил, C6-10арил, C3-6циклоалкил или амино могут быть необязательно замещены.
В некоторых вариантах осуществления формулы I: X представляет собой CR10, R10 представляет собой C1-6алкил или C3-6циклоалкил, R10 может быть замещен R8, R8 представляет собой (R11)этилен-C(O)-N-фенил, этил-C(O)-N-фенил, морфолинил, -NR11R12, циклопропан, этилен-C(O)-пиперазинил, этилен-C(O)-азетидинокси, этилен-C(O)-пиперидинилокси, этилен-C(O)-аза-C6-10спироциклил, этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-N-пиперидинил, этилен-C(O)-пиперидинил, этилен-C(O)-C1-8алкилпиперазинил, -N(R11)этилен-C(O)-пиперидинил, -N(R11)этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-пиперидинил-S(O2)- или изопентенил-C(O)-пиперазинил, замещенный циано.
В некоторых вариантах осуществления формулы I: Y представляет собой N или CR10, R10 представляет собой H, амино, C1-6алкил, замещенный C1-6алкил, C3-6циклоалкил или замещенный C3-6циклоалкил.
В некоторых вариантах осуществления формулы I: Y представляет собой N.
В некоторых вариантах осуществления формулы I: Y представляет собой CR10, R10 представляет собой C1-6алкил, C1-6алкил, замещенный C5-10гетероциклилом, C1-6алкил, замещенный C6-10арилом, C1-6алкил, замещенный C3-6циклоалкилом, C1-6алкил, замещенный амино, или C3-6циклоалкил, при этом C5-10гетероциклил, C6-10арил, C3-6циклоалкил или амино могут быть необязательно замещены.
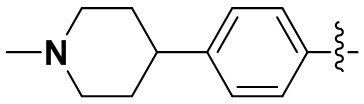
В некоторых вариантах осуществления формулы I: Y представляет собой CR10, R10 представляет собой C1-6алкил или C3-6циклоалкил, R10 может быть замещен R8, R8 представляет собой (R11)этилен-C(O)-N-фенил, этил-C(O)-N-фенил, морфолинил, -NR11R12, циклопропан, этилен-C(O)-пиперазинил, этилен-C(O)-азетидинокси, этилен-C(O)-пиперидинилокси, этилен-C(O)-аза-C6-10спироциклил, этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-N-пиперидинил, этилен-C(O)-пиперидинил, этилен-C(O)-C1-8алкилпиперазинил, -N(R11)этилен-C(O)-пиперидинил, -N(R11)этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-пиперидинил-S(O2)- или изопентенил-C(O)-пиперазинил, замещенный циано.
В некоторых вариантах осуществления формулы I: X представляет собой C=O, Y представляет собой NH.
В некоторых вариантах осуществления формулы I: Z представляет собой N.
В некоторых вариантах осуществления формулы I: каждый из R1, R3 и R5 независимо представляет собой H, F или Cl.
В некоторых вариантах осуществления формулы I: R1 и R5 выбраны из группы ниже:
(i) как R1, так и R5 представляют собой H;
(ii) как R1, так и R5 представляют собой Cl;
(iii) R1 представляет собой H, R5 представляет собой Cl;
(iv) R1 представляет собой Cl, R5 представляет собой H;
(v) как R1, так и R5 представляют собой F;
(vi) R1 представляет собой H, R5 представляет собой F; или
(vii) R1 представляет собой F, R5 представляет собой H.
В некоторых вариантах осуществления формулы I: R1 представляет собой Cl, как R3, так и R5 представляют собой H.
В некоторых вариантах осуществления формулы I: R3 представляет собой H.
В некоторых вариантах осуществления формулы I: каждый из R2 и R4 независимо выбран из H и C1-3алкокси.
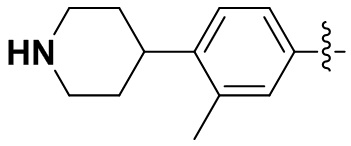
В некоторых вариантах осуществления формулы I: как R2, так и R4 представляют собой CH3O-.
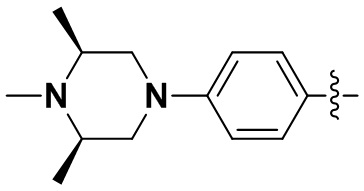
В некоторых вариантах осуществления формулы I: R2 и R3 или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют замещенное 5-членное гетероциклическое кольцо, содержащее 1-2 гетероатома, независимо выбранные из N, O или S, и 5-членное гетероциклическое кольцо замещено C1-3алкилом.
В некоторых вариантах осуществления формулы I: R2 и R3 или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют замещенное 5-членное гетероциклическое кольцо, содержащее один или два атома азота; или один атом азота и один атом серы; или один атом азота и один атом кислорода, и 5-членное гетероциклическое кольцо замещено метилом.
В некоторых вариантах осуществления формулы I: R2 и R3 или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют гетероциклическое кольцо, которое представляет собой  ,
,  ,
,  или
или 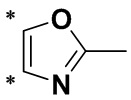 .
.
В некоторых вариантах осуществления формулы I: R6 представляет собой C1-6алкил, замещенный C1-6алкил, C6-10арил, замещенный C6-10арил, C3-6циклоалкил, замещенный C3-6циклоалкил, C5-10гетероциклил, замещенный C5-10гетероциклил, C6-10гетероарил или замещенный C6-10гетероарил.
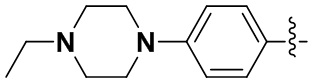
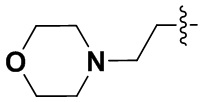
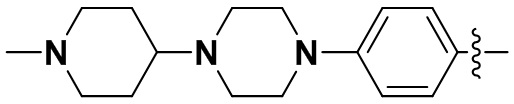
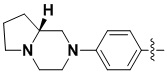
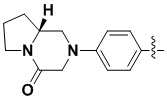
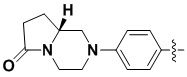
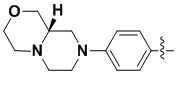
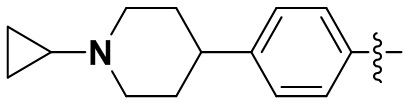
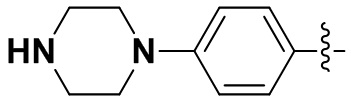
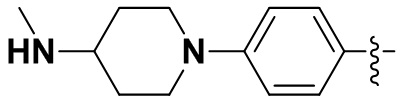
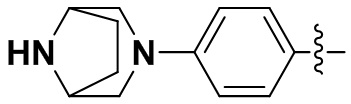
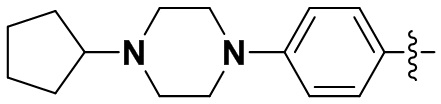
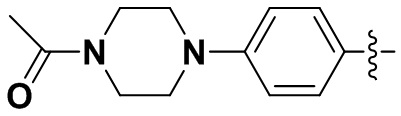
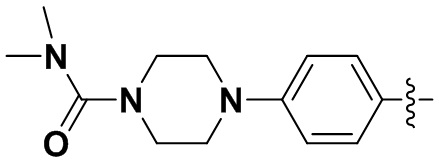
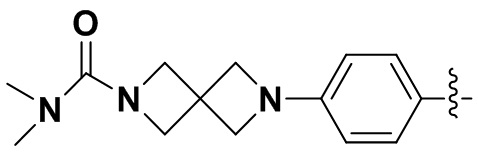
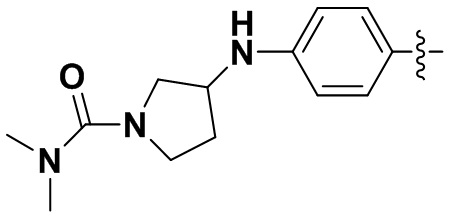
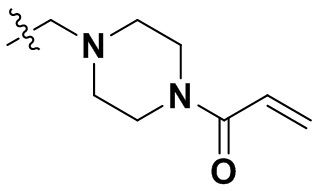
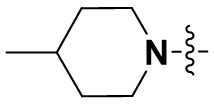
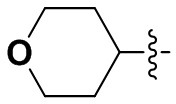
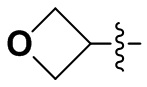
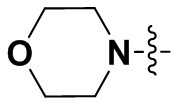
В некоторых вариантах осуществления формулы I: R6 представляет собой C1-4алкил, циклопентил, фенил, фенил, замещенный F, фенил, замещенный метокси, фенил, замещенный Cl, фенил, замещенный метилом, пиридил, тетрагидропиранил, R6 может быть замещен R7, при этом R7 представляет собой гидроксил, F, Cl, пиперазинил, замещенный этилом, морфолинил, пиперазинил, замещенный изопропилом, пиперазинил, замещенный оксетаном, пиперазинил, замещенный метилом, пиперазинил-CH2-, замещенный этилом, пиперазинил, замещенный этилом и оксилом, пиперазинил, замещенный триметилом, триметилэтилендиамин, пиперазинил, замещенный метилпиперидинилом, аза-C6-10бициклил, замещенный метилом, аза-C6-10бициклил, -N(метил)-C1-6метиленморфолинил, C4-10азациклоалкил, замещенный C2-6алкокси, пиперидинил, замещенный морфолинилом, пиперазинил, замещенный гидроксиэтилом, C2-6алкокси, замещенный морфолинилом, пиперидинил, замещенный этилом, пиперидинил, замещенный метилом, диметиламинопиперидинил, C6-10азабициклил, замещенный оксилом, нитроокса-C6-10бициклил, морфолинил-CH2-, метилпиперазинил-CH2-, пиперидинил, замещенный C3-10циклоалкилом, метиламинопиперидинил, пиперазинил, замещенный диметилом, пиперидинил, пиперазинил-CH2-, пиперазинил-C(O)-, замещенный диметилом, пиперазинил, замещенный гидроксициклобутаном, трифторметил-CH2-пиперазинил, пиперазинил, замещенный C3-10циклоалкилом, метил-C(O)-пиперазинил, (диметил)-N-C(O)-пиперазинил, (диметил)-N-C(O)-аза-C6-10спироциклил, (диметил)-N-C(O)-тетрагидропиррол-NH-, R7 представляет собой азотсодержащее 6-членное гетероциклическое кольцо, которое конденсировано с R6, при этом азотсодержащее 6-членное гетероциклическое кольцо является незамещенным или замещено этилом.
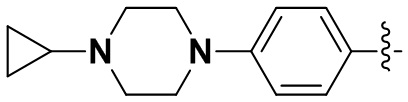
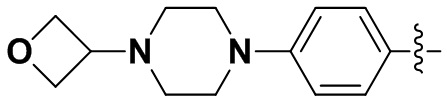
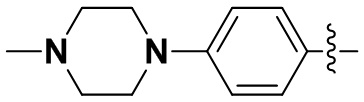
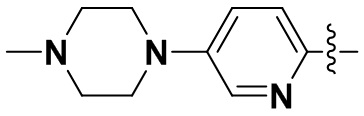
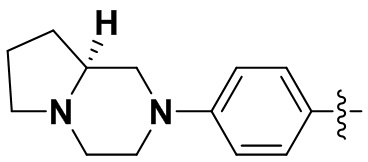
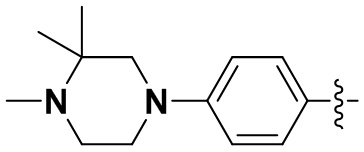
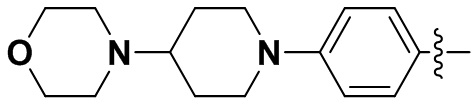
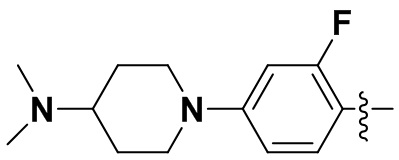
В некоторых вариантах осуществления формулы I: R6 представляет собой метил, 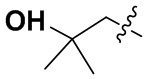 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
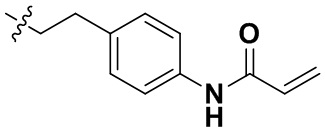
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 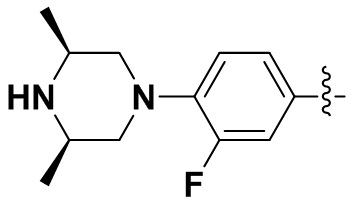 ,
, 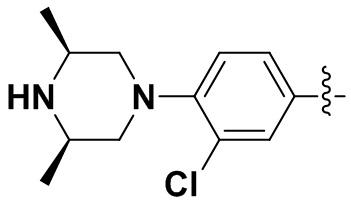 ,
, 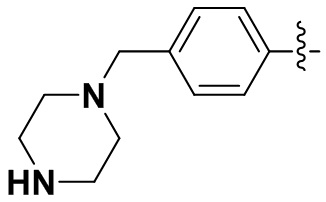 ,
, 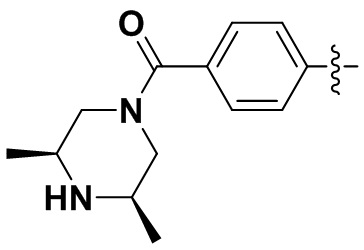 ,
, 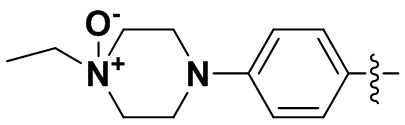 ,
, 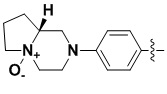 ,
, 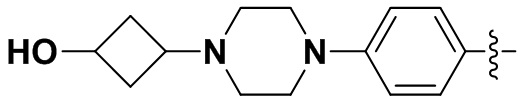 ,
, 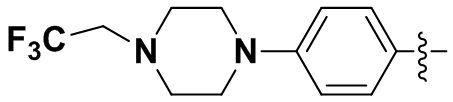 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  или
или  .
.
В некоторых вариантах осуществления формулы I: каждый из R11 и R12 независимо выбран из H, C1-6алкила, замещенного C1-6алкила, C3-6циклоалкила и замещенного C3-6циклоалкила.
В некоторых вариантах осуществления формулы I: каждый из R11 и R12 независимо выбран из H, метила и этила.
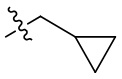
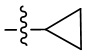
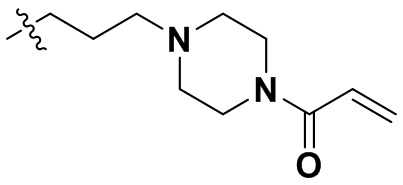
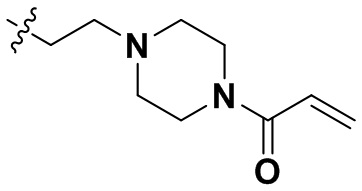
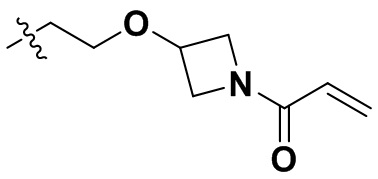
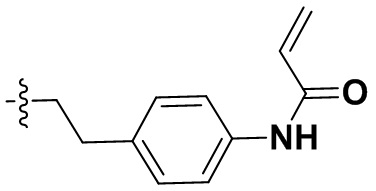
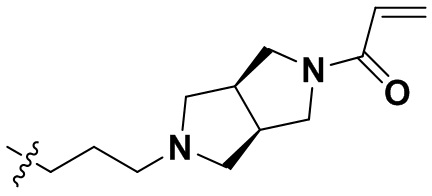
В некоторых вариантах осуществления формулы I: R10 представляет собой H, CH3, амино, 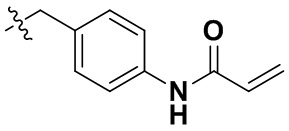 ,
, 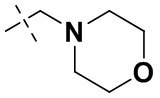 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 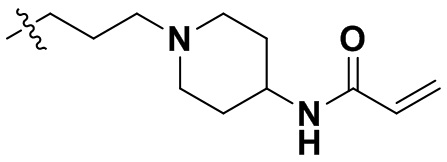 ,
, 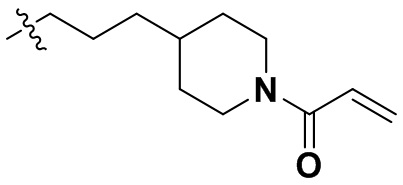 ,
, 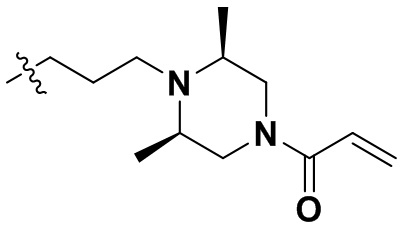 ,
, 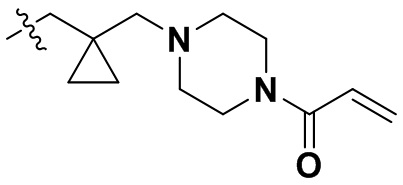 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 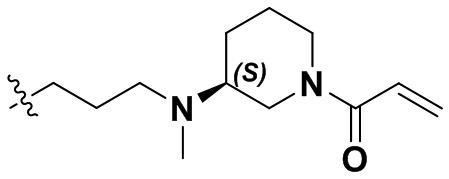 ,
, 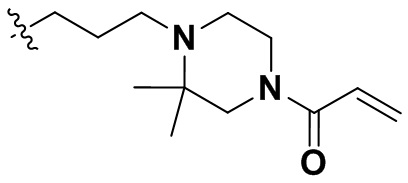 ,
, 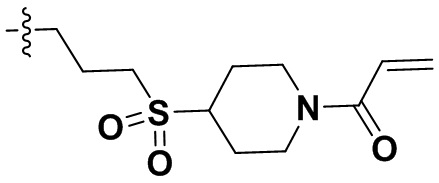 ,
, 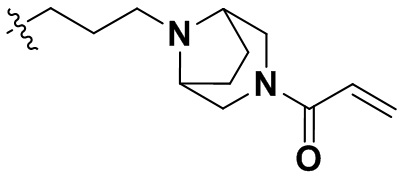 ,
,  или
или 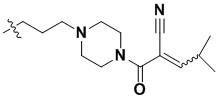 .
.
В некоторых вариантах осуществления формулы I: R10 представляет собой H или -CH3.
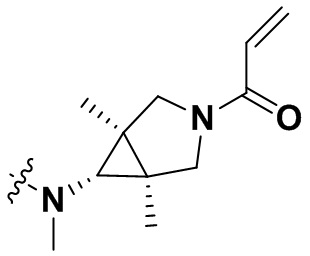
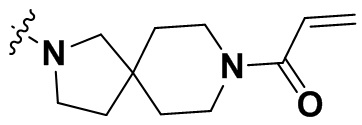
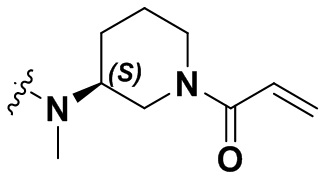
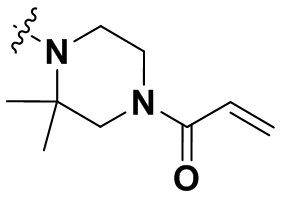
В настоящем изобретении дополнительно предусмотрены некоторые предпочтительные технические решения применительно к соединениям формулы I, где соединения представляют собой следующие:
(1) 1-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)-2-метилпропан-2-ол;
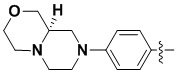
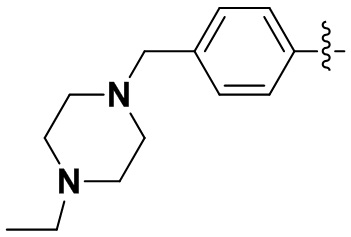
(2) 4-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-амин;
(3) 3-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)циклопентан-1-ол;
(4) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(5) 6-(2-хлор-3,5-диметоксифенил)-N-(2-морфолиноэтил)-[1,2,4]триазоло [4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(6) 1-((4-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-ил)амино)-2-метилпропан-2-ол;
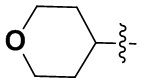
(7) 6-(2-хлор-3,5-диметоксифенил)-N-(тетрагидро-2H-пиран-4-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
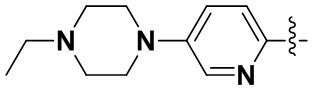
(8) 6-(2-хлор-3,5-диметоксифенил)-N-(5-(4-этилпиперазин-1-ил)пиридин-2-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(9) 6-(3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
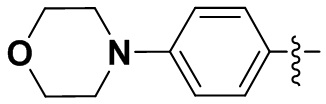
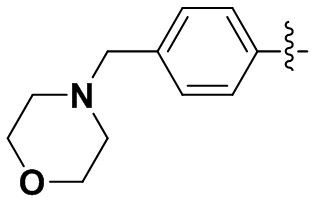
(10) 6-(2-хлор-3,5-диметоксифенил)-N-(4-морфолинофенил)-[1,2,4]триазоло [4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
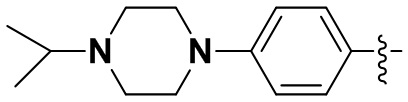
(11) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-изопропилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(12) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклопропилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(13) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(оксетан-3-ил)пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(14) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(диметиламино)пиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
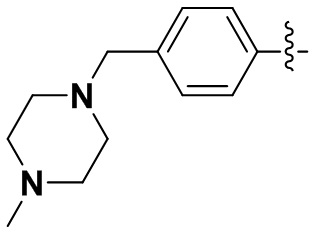
(15) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-метилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(16) 6-(2-хлор-3,5-диметоксифенил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
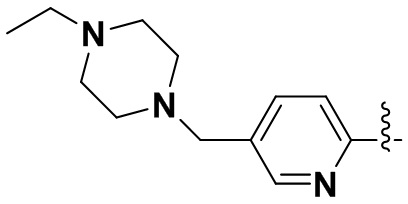
(17) 6-(2-хлор-3,5-диметоксифенил)-N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(18) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этил-2-оксопиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(19) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этил-3-оксопиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(20) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,4,5-триметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(21) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(N-(2-диметиламиноэтил-N-метиламино)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(22) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
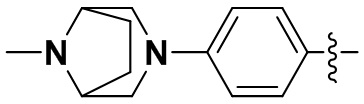
(23) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(8-метил-3,8-диазабицикло[3.2.1]октан-3-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
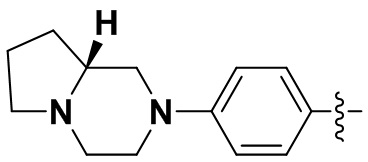
(24) (R)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(25) (S)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(26) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(3,3,4-триметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(27) N1-(6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)-N4-метил-N4-(2-морфолиноэтил)бензол-1,4-диамин;
(28) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(2-(пирролидин-1-ил)этокси)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(29) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-морфолинопиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(30) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(диметиламино)пиперидин-1-ил)-2-фторфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(31) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(32) 2-(4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)пиперазин-1-ил)этан-1-ол;
(33) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(2-морфолиноэтокси)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(34) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-этилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
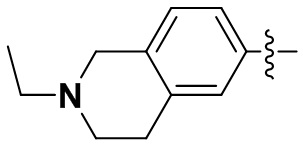
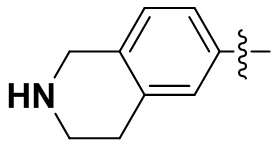
(35) 6-(2-хлор-3,5-диметоксифенил)-N-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
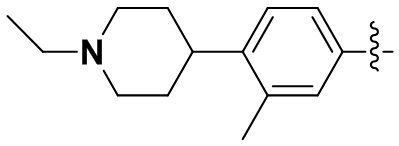
(36) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-этилпиперидин-4-ил)-3-метилфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(37) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-метилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
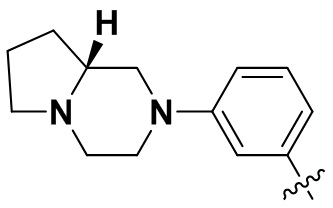
(38) (R)-6-(2-хлор-3,5-диметоксифенил)-N-(3-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(39) (R)-6-(3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(40) (R)-2-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)гексагидропирроло[1,2-a]пиразин-4(1H)-он;
(41) (R)-2-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)гексагидропирроло[1,2-a]пиразин-6(2H)-он;
(42) (S)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропиразино[2,1-c]-[1,4]оксазин-8(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(43) (R)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропиразино[2,1-c]-[1,4]оксазин-8(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(44) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((4-этилпиперазин-1-ил)метил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(45) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(морфолинометил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(46) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((4-метилпиперазин-1-ил)метил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(47) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-циклопропилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(48) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(49) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(метиламино)пиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(50) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(51) N-(4-(3,8-диазабицикло[3.2.1]октан-3-ил)фенил)-6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(52) 6-(2-хлор-3,5-диметоксифенил)-N-(3-метил-4-(пиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(53) 6-(2-хлор-3,5-диметоксифенил)-N-(1,2,3,4-тетрагидроизохинолин-6-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
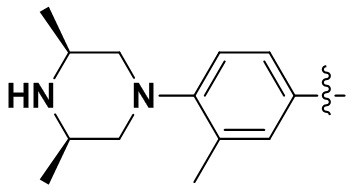
(54) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-метилфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
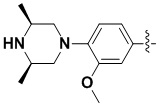
(55) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-метоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(56) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-фторфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(57) 6-(2-хлор-3,5-диметоксифенил)-N-(3-хлор-4-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(58) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(пиперазин-1-илметил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(59) (4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-((3S,5R)-3,5-диметилпиперазин-1-ил)метанон;
(60) (4-((6-(2,6-дихлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-((3S,5R)-3,5-диметилпиперазин-1-ил)метанон;
(61) 6-(2,6-дихлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(62) 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-морфолиноэтил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(63) 4-(2,6-дихлор-3,5-диметоксифенил)-N-(2-морфолиноэтил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-амин;
(64) (R)-6-(2,6-дихлор-3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(65) 6-(2,6-дихлор-3,5-диметоксифенил)-N-(4-(4-морфолинопиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(66) 6-(2-хлор-3,5-диметоксифенил)-N2-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2,9-диамин;
(67) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(68) N-(4-(4-этилпиперазин-1-ил)фенил)-6-(2-метил-1H-бензо[d]имидазол-6-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(69) 6-(4-хлор-2-метил-1H-бензо[d]имидазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(70) 6-(4,6-дихлор-2-метил-1H-бензо[d]имидазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(71) 6-(5-хлор-2-метил-1H-индол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(72) 6-(6-хлор-2-метилбензо[d]тиазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(73) 6-(4-хлор-2-метилбензо[d]тиазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(74) 6-(6-хлор-2-метилбензо[d]оксазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(75) 6-(4-хлор-2-метилбензо[d]оксазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(76) 6-(5-хлор-2-метилбензо[d]оксазол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(77) 6-(7-хлор-2-метилбензо[d]оксазол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(78) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-(4-(диметиламино)пиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(79) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-(1-этилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(80) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(81) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-морфолинофенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(82) 6-(5,7-дихлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-морфолинофенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(83) 6-(7-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-морфолинофенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(84) N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(85) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-9-метил-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
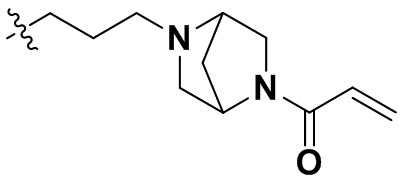
(86) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-((2-морфолиноэтил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)акриламид;
(87) 4-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-2-метил-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-амин;
(88) N-(4-((4-(2-хлор-3,5-диметоксифенил)-8-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)фенил)акриламид;
(89) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-9-(морфолинометил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(90) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)акриламид;
(91) N-(4-(2-(6-(2-хлор-3,5-диметоксифенил)-2-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)этил)фенил)акриламид;
(92) 6-(2-хлор-3,5-диметоксифенил)-9-((диметиламино)метил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(93) 6-(2-хлор-3,5-диметоксифенил)-9-(циклопропилметил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(94) 6-(2-хлор-3,5-диметоксифенил)-9-циклопропил-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(95) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)акриламид;
(96) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-((2-морфолиноэтил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)пропионамид;
(97) N-(4-(2-(6-(2-хлор-3,5-диметоксифенил)-2-((2-морфолиноэтил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)этил)фенил)акриламид;
(98) 6-(2-хлор-3,5-диметоксифенил)-2-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9(8H)-он;
(99) 4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-1-этилпиперазин-1-оксид;
(100) (8aR)-2-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)октагидро-5H-пирроло[1,2-a]пиразин-5-оксид;
(101) 3-(4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)пиперазин-1-ил)циклобутан-1-ол;
(102) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-этилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(103) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(2,2,2-трифторэтил)пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(104) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклопентилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(105) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклобутилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(106) (6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-ацетилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин);
(107) 4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-N,N-диметилпиперазин-1-карбоксамид;
(108) 6-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-N,N-диметил-2,6-диазаспиро[3.3]гептан-2-карбоксамид;
(109) (S)-3-((4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)амино)-N,N-диметилпирролидин-1-карбоксамид;
(110) 6-(2-хлор-3,5-диметоксифенил)-N-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(111) 6-(2-хлор-3,5-диметоксифенил)-N-(6-((4-этилпиперазин-1-ил)метил)пиридин-3-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(112) N-(4-((4-(2-хлор-3,5-диметоксифенил)-8-((2-морфолиноэтил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)фенил)акриламид;
(113) (1-(4-((6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он);
(114) 1-(4-(2-(6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)этил)пиперазин-1-ил)проп-2-ен-1-он;
(115) 1-(4-(3-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(116) 1-(4-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)пиперазин-1-ил)проп-2-ен-1-он;
(117) 1-(4-((4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(118) 1-(3-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этокси)азетидин-1-ил)проп-2-ен-1-он;
(119) 1-(3-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этокси)пиперидин-1-ил)проп-2-ен-1-он;
(120) N-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)фенил)акриламид;
(121) N-(3-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)фенил)акриламид;
(122) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(123) 1-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(124) 1-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)пиперазин-1-ил)проп-2-ен-1-он;
(125) 1-(6-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диазаспиро[3.3]гептан-2-ил)проп-2-ен-1-он;
(126) 1-(4-((2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)циклопропил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(127) 1-(4-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этокси)пиперидин-1-ил)проп-2-ен-1-он;
(128) 1-((3aR,6aS)-5-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)гексагидропирроло[3,4-c]пиррол-2(1H)-ил)проп-2-ен-1-он;
(129) N-(1-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперидин-4-ил)акриламид;
(130) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперидин-1-ил)проп-2-ен-1-он;
(131) 1-((3S,5R)-4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-3,5-диметилпиперазин-1-ил)проп-2-ен-1-он;
(132) 1-(4-((1-((4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)циклопропил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(133) 1-((2R,6S)-4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диметилпиперазин-1-ил)проп-2-ен-1-он;
(134) 1-(4-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)(метил)амино)пиперидин-1-ил)проп-2-ен-1-он;
(135) 1-((2R,6S)-4-(3-(8-((циклопропилметил)амино)-4-(2,6-дихлор-3,5-диметоксифенил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диметилпиперазин-1-ил)проп-2-ен-1-он;
(136) 1-((2R,6S)-4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-((2,2-дифторэтил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диметилпиперазин-1-ил)проп-2-ен-1-он;
(137) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-((2,2,2-трифторэтил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(138) 1-(2-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,7-диазаспиро[3.5]нонан-7-ил)проп-2-ен-1-он;
(139) 1-((1R,5S,6s)-6-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)(метил)амино)-3-азабицикло[3.1.0]гексан-3-ил)проп-2-ен-1-он;
(140) 1-(2-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,8-диазаспиро[4.5]декан-8-ил)проп-2-ен-1-он;
(141) (S)-1-(3-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)(метил)амино)пиперидин-1-ил)проп-2-ен-1-он;
(142) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-3,3-диметилпиперазин-1-ил)проп-2-ен-1-он;
(143) 1-(4-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)сульфонил)пиперидин-1-ил)проп-2-ен-1-он;
(144) 1-(8-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-3,8-диазабицикло[3.2.1]октан-3-ил)проп-2-ен-1-он;
(145) 1-(5-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,5-диазабицикло[2.2.1]гептан-2-ил)проп-2-ен-1-он;
(146) 1-(4-(3-(4-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(147) 2-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-карбонил)-4-метилпент-2-еннитрил;
(148) 2-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-карбонил)-4,4-диметилпент-2-еннитрил.
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая терапевтически эффективное количество по меньшей мере одного любого из соединений формулы (I) по настоящему изобретению и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
В настоящем изобретении дополнительно предусмотрена фармацевтическая композиция, в которой весовое отношение соединения формулы (I) к вспомогательному веществу может составлять 0,0001 к 10.
В настоящем изобретении предусмотрено применение соединения формулы (I) или фармацевтической композиции для получения лекарственного препарата.
В настоящем изобретении дополнительно предусмотрено следующее предпочтительное техническое решение в отношении применения.
Предпочтительно лекарственный препарат, полученный таким образом, можно применять для лечения, защиты, замедления или предупреждения проявления или прогрессирования рака или метастазов рака.
Предпочтительно применение предназначено для получения лекарственного препарата для лечения заболевания, опосредованного FGFR. Предпочтительно заболевание представляет собой рак.
Предпочтительно рак выбран из группы, состоящей из рака молочной железы, множественной миеломы, рака мочевого пузыря, рака эндометрия, рака желудка, рака шейки матки, рабдомиосаркомы, немелкоклеточного рака легкого, мелкоклеточного рака легкого, плеоморфного рака легкого, рака яичника, рака пищевода, меланомы, колоректального рака, гепатоцеллюлярной карциномы, рака головы и шеи, гепатохолангиокарциномы, миелодиспластического синдрома, злокачественной глиомы, рака предстательной железы, рака щитовидной железы, шванномы, плоскоклеточного рака легкого, лихеноидного кератоза, синовиальной саркомы, рака кожи, рака поджелудочной железы, рака яичка или липосаркомы.
Предпочтительно лекарственный препарат, полученный таким образом, можно применять в качестве ингибитора FGFR.
Предпочтительно в указанном выше применении FGFR предусматривает FGFR1, FGFR2, FGFR3 или FGFR4.
В настоящем изобретении также предусмотрен способ лечения и/или предупреждения заболевания, опосредованного FGFR, осуществляемый путем введения терапевтически эффективного количества по меньшей мере любого из соединений формулы (I) или фармацевтической композиции субъекту.
Предпочтительно в указанном выше применении FGFR предусматривает FGFR1, FGFR2, FGFR3 или FGFR4.
Предпочтительно в указанном выше применении заболевание, опосредованное FGFR, представляет собой рак.
Предпочтительно в указанном выше применении рак выбран из группы, состоящей из рака молочной железы, множественной миеломы, рака мочевого пузыря, рака эндометрия, рака желудка, рака шейки матки, рабдомиосаркомы, немелкоклеточного рака легкого, мелкоклеточного рака легкого, плеоморфного рака легкого, рака яичника, рака пищевода, меланомы, колоректального рака, гепатоцеллюлярной карциномы, рака головы и шеи, гепатохолангиокарциномы, миелодиспластического синдрома, злокачественной глиомы, рака предстательной железы, рака щитовидной железы, шванномы, плоскоклеточного рака легкого, лихеноидного кератоза, синовиальной саркомы, рака кожи, рака поджелудочной железы, рака яичка или липосаркомы.
В настоящем изобретении также предусмотрен способ лечения рака, предусматривающий введение субъекту терапевтически эффективного количества по меньшей мере любого из соединений формулы (I) или фармацевтической композиции, при этом рак выбран из группы, состоящей из рака молочной железы, множественной миеломы, рака мочевого пузыря, рака эндометрия, рака желудка, рака шейки матки, рабдомиосаркомы, немелкоклеточного рака легкого, мелкоклеточного рака легкого, плеоморфного рака легкого, рака яичника, рака пищевода, меланомы, колоректального рака, гепатоцеллюлярной карциномы, рака головы и шеи, гепатохолангиокарциномы, миелодиспластического синдрома, злокачественной глиомы, рака предстательной железы, рака щитовидной железы, шванномы, плоскоклеточного рака легкого, лихеноидного кератоза, синовиальной саркомы, рака кожи, рака поджелудочной железы, рака яичка или липосаркомы.
Предпочтительно в указанном выше применении субъектом, подлежащим лечению, является человек.
Настоящее изобретение относится к соединениям, применяемым в качестве ингибитора FGFR, и относится к применению таких соединений для получения лекарственного препарата, предназначенного для лечения или предупреждения заболевания, опосредованного FGFR, in vivo. Соединение характеризуется простой структурой, простым способом получения и надлежащим терапевтическим эффектом в качестве активного ингредиента. В качестве лекарственного средства, подлежащего выводу на рынок, соединение характеризуется низкой стоимостью и удобством приема, что является более благоприятным для широкого применения таких лекарственных средств, и может более эффективно помогать пациентам устранять боль и улучшать качество жизни.
Если не указано иное, термины, используемые в настоящем изобретении, имеют следующие значения.
Термин «алкил» включает насыщенные одновалентные углеводородные радикалы, имеющие прямые, разветвленные или циклические фрагменты. Например, алкильные радикалы включают без ограничения метил, этил, пропил, изопропил, циклопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклобутил, н-пентил, 3-(2-метил)бутил, 2-пентил, 2-метилбутил, неопентил, циклопентил, н-гексил, 2-гексил, 2-метилпентил и циклогексил. Подобным образом, C1-8, как в C1-8алкиле, определен как означающий группу, содержащую 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода в линейном, разветвленном или циклическом расположении.
Алкенильные и алкинильные группы включают алкены и алкины с прямой, разветвленной цепью или циклические алкены и алкины. Подобным образом «C2-8алкенил» и «C2-8алкинил» означают алкенильные или алкинильные радикалы, содержащие 2, 3, 4, 5, 6, 7 или 8 атомов углерода в линейном, разветвленном или циклическом расположении.
Алкоксирадикалы представляют собой простые эфиры на основе кислорода, образованные из ранее описанных алкильных групп с прямой, разветвленной цепью или циклических алкильных групп.
Термин «арил», используемый в данном документе, если не указано иное, означает незамещенную или замещенную моно- или полициклическую кольцевую систему, содержащую атомы кольца, представляющие собой углерод. Предпочтительные арилы представляют собой моноциклические или бициклические 6-10-членные ароматические кольцевые системы. Предпочтительные арилы представляют собой фенил и нафтил. Наиболее предпочтительный арил представляют собой фенил.
Термин «гетероарил» относится к одновалентной группе с гетероатомом, образованной путем удаления одного атома водорода от атома углерода в исходной гетероароматической кольцевой системе. Гетероарильная группа предусматривает 5-7-членное ароматическое моноциклическое кольцо, содержащее по меньшей мере один гетероатом, выбранный из N, O или S, например, 1-4 гетероатома или предпочтительно 1-3 гетероатома, а другие атомы в кольце представляют собой углерод; полигетероарильное кольцо предусматривает по меньшей мере один гетероатом, выбранный из N, O или S, например, 1-4 гетероатома или предпочтительно 1-3 гетероатома, а другие атомы в кольце представляют собой углерод, и по меньшей мере один из гетероатомов входит в состав ароматического кольца. В частности, предпочтительные гетероарильные группы представляют собой C3-10гетероарильные группы, включающие без ограничения пирролил, фурил, тиенил, пиридил, пиранил, пиразолил, пиримидинил, пиридазинил, пиразинил, имидазолил, тиазолил, оксазолил, изоксазолил, триазолил, индолил, бензофуранил, бензотиазолил, бензимидазолил, бензопиразолил, бензотриазолил, карбазолил, хинолил, изохинолинил, пуринил и подобные группы.
Однако, в любом случае, гетероарильная группа и арильная группа не пересекают и не содержат друг друга. Таким образом, в соответствии с указанным выше определением, если по меньшей мере одно ароматическое кольцо, остов которого состоит исключительно из атомов углерода, конденсировано с гетероциклической группой, вместо арильной группы получают гетероарильную группу.
«Циклоалкил» означает насыщенную или ненасыщенную циклическую группу без ароматичности. В зависимости от конкретного уровня насыщенности соответственно используют термины «циклоалкил», «циклоалкенил» или «циклоалкинил». Иллюстративные циклоалкильные группы включают без ограничения циклопропан, циклобутан, циклопентан, циклогексан или циклогексен и т. п. Конкретно циклоалкильная группа может представлять собой C3-10циклоалкильную группу, такую как C3-6циклоалкильная группа.
«Гетероциклоалкил» означает насыщенную или ненасыщенную циклическую группу без ароматичности, где один или более атомов углерода (и присоединенные атомы водорода) могут быть замещены одинаковыми или различными гетероатомами и соответствующими атомами водорода соответственно. Иллюстративные гетероатомы, которые замещают атомы углерода, включают без ограничения N, P, O, S и Si. Термины «гетероциклоалкил» или «гетероциклоалкенил» используют соответственно, если требуется описать определенную степень насыщенности. Иллюстративные гетероциклоалкильные группы включают без ограничения эпоксидные соединения, имидазолидины, морфолин, пиперазин, пиперидин, пиразолидин, пирролидин, хинуклидин, тетрагидрофуран или тетрагидропиран и т. п. Замещенная гетероциклоалкильная группа также включает кольцевую систему, замещенную по меньшей мере одним кислородсодержащим (=O) или оксидным (-O-) заместителем, таким как пиперидин-оксид-азота, морфолинил-оксид-азота, 1-оксо-1-тиоморфолинил и 1-диокси-1-тиоморфолинил.
Однако, в любом случае, гетероциклоалкильная группа и циклоалкильная группа не пересекают и не содержат друг друга. Таким образом, в соответствии с указанным выше определением, если по меньшей мере одно карбоциклическое кольцо конденсировано с гетероциклоалкильной группой с образованием ди-, поли- или спирокольца, оно все еще будет определяться как гетероциклоалкильная группа.
«Галоген» означает фтор (F), хлор (Cl), бром (Br) или йод (I). Предпочтительный галоген означает фтор, хлор и бром.
«Галоген» означает группу фтора, хлора, брома или йода.
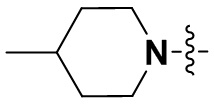
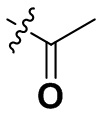
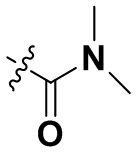
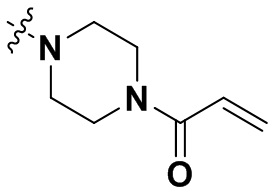
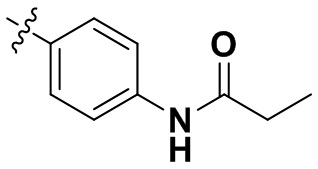
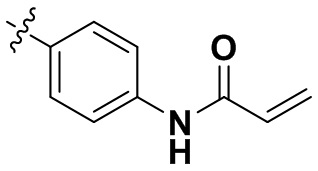
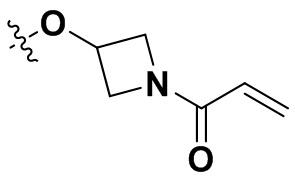
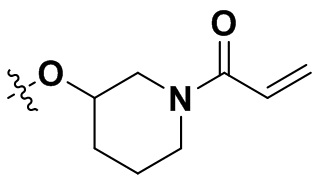
«Замещенный» означает, что каждый из одного или более атомов водорода в группе замещен одинаковыми или различными заместителями. Иллюстративные заместители включают без ограничения галоген, амино, гидрокси, оксо, карбонил, циано, алкил, алкокси, арил, циклоалкил, гетероциклил, гетероарил, алкилпиперазин, морфолинил. В некоторых вариантах осуществления заместители включают без ограничения галоген, амино, гидрокси, циано, циклопропил, фенил, диметиламино,  ,,
,,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  или
или  .
.
«*» в заместителе  , ,
, ,  или представляет собой участок, где кольцо присоединено к смежному кольцу.
или представляет собой участок, где кольцо присоединено к смежному кольцу.
В каждом случае, когда термин «алкил» или «арил» или соответствующие им приставки присутствуют в названии заместителя (такого как аралкильная группа или диалкиламиногруппа), заместители следует определять в соответствии с вышеуказанным «алкилом» и «арилом». Указанное количество атомов углерода (например, Cl-6) независимо будет представлять собой количество атомов углерода в алкильном фрагменте или в алкильном фрагменте в составе большего заместителя (где алкильная группа представляет собой приставку).
Термин «соединение», используемый в данном документе, включает соединение формулы (I) и все его фармацевтически приемлемые формы. Такие фармацевтически приемлемые формы включают его соли, сольваты, нековалентные комплексы, хелаты или пролекарства или любую смесь из всех из вышеуказанных.
Выражение «фармацевтически приемлемый» означает общеизвестный для применения у животных, в частности, для применения у людей.
Термин «композиция», используемый в настоящем изобретении, включает продукты, содержащие конкретное количество определенного компонента, а также любой продукт, полученный непосредственно или опосредованно из конкретного количества определенного компонента. Следовательно, сущность настоящего изобретения составляют фармацевтическая композиция, содержащая соединение по настоящему изобретению в качестве активного ингредиента, и способ ее получения.
«Терапевтически эффективное количество» означает дозу, достаточную для обеспечения определенного эффекта на лечение заболевания, состояния, симптома, показания или дискомфорта, когда соединение вводят субъекту для лечения и предупреждения и/или ингибирования по меньшей мере одного клинического состояния заболевания, состояния, симптома, показания и/или дискомфорта. Конкретное «эффективное терапевтическое количество» может варьироваться в зависимости от соединения, пути введения, возраста пациента, веса пациента, типа заболевания или дискомфорта, подлежащих лечению, симптомов и тяжести и т. п. Во всех случаях, если возможно, подходящая доза будет очевидной специалистам в данной области техники и может быть определена путем проведения обычного эксперимента.
Соединения по настоящему изобретению также могут быть представлены в форме фармацевтически приемлемых солей. Что касается применения в медицине, соли соединений по настоящему изобретению относятся к нетоксичным «фармацевтически приемлемым солям». Фармацевтически приемлемые солевые формы включают фармацевтически приемлемые кислотные/анионные или основные/катионные соли. Фармацевтически приемлемая кислотная/анионная соль обычно принимает форму, в которой основный азот протонирован неорганической или органической кислотой. Иллюстративные органические или неорганические кислоты включают хлористоводородную, бромистоводородную, йодистоводородную, перхлорную, серную, азотную, фосфорную, уксусную, пропионовую, гликолевую, молочную, янтарную, малеиновую, фумаровую, яблочную, винную, лимонную, бензойную, миндальную, метансульфоновую, гидроксиэтансульфоновую, бензолсульфоновую, щавелевую, памовую, 2-нафталинсульфоновую, п-толуолсульфоновую, циклогексиламинсульфоновую, салициловую, сахариновую или трифторуксусную кислоты. Фармацевтически приемлемые основные/катионные соли включают без ограничения соли алюминия, кальция, хлорпрокаина, холина, диэтаноламина, этилендиамина, лития, магния, калия, натрия и цинка.
В объем настоящего изобретения входят пролекарства соединений по настоящему изобретению. Как правило, такие пролекарства будут представлять собой функциональные производные соединений, которые in vivo легко превращаются в требуемое соединение. Таким образом, в способах лечения по настоящему изобретению термин «введение» будет охватывать лечение различных описанных нарушений с помощью соединения, которое явно предусматривается, или с помощью соединения, которое может конкретно не предусматриваться, но которое превращается в указанное соединение in vivo после введения субъекту. Традиционные процедуры выбора и получения подходящих производных пролекарств описаны, например, в “Design of Prodrugs”, ed. H. Bundgaard, Elsevier, 1985.
Предполагается, что определение любых заместителя или переменной в конкретном положении в молекуле не зависит от их определения где-либо еще в данной молекуле. Известно, что заместители и схемы замещения для соединений по настоящему изобретению может выбрать специалист в данной области техники для получения соединений, которые являются химически стойкими, и которые можно легко синтезировать с помощью методик, известных из уровня техники, а также способов, изложенных в данном документе.
Если соединение формулы (I) и его фармацевтически приемлемые соли находятся в форме сольватов или полиморфных форм, настоящее изобретение включает любые возможные сольваты и полиморфные формы. Тип растворителя, который образует сольват, конкретно не ограничен при условии, что растворитель является фармакологически приемлемым. Например, можно применять воду, этанол, пропанол, ацетон или т. п.
Термин «фармацевтически приемлемые соли» означает соли, полученные из фармацевтически приемлемых нетоксичных оснований или кислот. Если соединение по настоящему описанию является кислотным, его соответствующая соль может быть получена удобным способом из фармацевтически приемлемых нетоксичных оснований, в том числе неорганических оснований и органических оснований. Соли, полученные из таких неорганических оснований, включают соли алюминия, аммония, кальция, меди (двухвалентной и одновалентной), трехвалентного железа, двухвалентного железа, лития, магния, марганца (трехвалентного и двухвалентного), калия, натрия, цинка и т. п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как встречающиеся в природе и синтезированные замещенные амины. Другие фармацевтически приемлемые органические нетоксичные основания, из которых можно образовывать соли, включают ионообменные смолы, такие как, например, аргинин, бетаин, кофеин, холин, N’,N’-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т. п.
Если соединение по настоящему описанию является основным, его соответствующая соль может быть получена удобным способом из фармацевтически приемлемых нетоксичных кислот, в том числе неорганических кислот и органических кислот. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорасульфоновую, лимонную, этансульфоновую, муравьиную, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту и т. п. Предпочтительными кислотами являются лимонная, бромистоводородная, муравьиная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты. В частности, предпочтительными являются муравьиная и хлористоводородная кислоты. Поскольку соединения формулы (I) предназначены для фармацевтического применения, они предпочтительно представлены в практически чистой форме, например, характеризуются чистотой по меньшей мере 60%, более предпочтительно чистотой по меньшей мере 75%, особенно предпочтительно чистотой по меньшей мере 98% (% приведены в пересчете на вес).
Фармацевтические композиции по настоящему изобретению содержат соединение, представленное формулой I (или его фармацевтически приемлемую соль) в качестве активного ингредиента, фармацевтически приемлемый носитель и необязательно другие терапевтические ингредиенты или вспомогательные средства. Композиции включают композиции, подходящие для перорального, ректального, местного и парентерального (в том числе подкожного, внутримышечного и внутривенного) введения, хотя наиболее подходящий путь введения в любом конкретном случае будет зависеть от конкретного пациента, а также от природы и тяжести состояний, при которых вводят активный ингредиент. Фармацевтические композиции в целях удобства могут быть представлены в стандартной лекарственной форме и получены с помощью любого из способов, широко известных в области фармации.
На практике соединения, представленные формулой I, или их пролекарство, или метаболит, или фармацевтически приемлемые соли по настоящему изобретению можно в качестве активного ингредиента объединять в однородную смесь с фармацевтически приемлемым носителем в соответствии с традиционными методиками получения фармацевтических смесей. Носитель может быть представлен в широком спектре форм в зависимости от формы препарата, подлежащего введению, например, пероральному или парентеральному (в том числе внутривенному). Таким образом, фармацевтические композиции по настоящему изобретению могут быть представлены в виде дискретных единиц, подходящих для перорального введения, таких как капсулы, саше или таблетки, каждое из которых содержит предварительно заданное количество активного ингредиента. Дополнительно композиции могут быть представлены в виде порошка, в виде гранул, в виде раствора, в виде суспензии в водосодержащей жидкости, в виде не содержащей воды жидкости, в виде эмульсии типа «масло-в-воде» или в виде жидкой эмульсии типа «вода-в-масле». В дополнение к общепринятым лекарственным формам, изложенным выше, соединение, представленное формулой I, или его фармацевтически приемлемую соль также можно вводить с помощью средств контролируемого высвобождения и/или устройств доставки. Композиции могут быть получены с помощью любого из способов, известных в области фармации. Как правило, такие способы включают стадию смешивания активного ингредиента с носителем, который представляет собой один или более необходимых ингредиентов. Как правило, композиции получают путем однородного и тщательного смешивания активного ингредиента с жидкими носителями, или с тонкодисперсными твердыми носителями, или как с теми, так и с другими. Затем продукту в целях удобства можно придавать желаемый внешний вид.
Таким образом, фармацевтическая композиция по настоящему изобретению может содержать фармацевтически приемлемый носитель и соединение формулы I или его фармацевтически приемлемую соль. Соединения формулы I или их фармацевтически приемлемые соли также можно включать в фармацевтические композиции в комбинации с одним или более другими терапевтически активными соединениями.
Применяемый фармацевтически приемлемый носитель может представлять собой, например, твердое вещество, жидкость или газ. Примеры твердых носителей включают лактозу, порошкообразный гипс, сахарозу, тальк, желатин, агар, пектин, аравийскую камедь, стеарат магния, стеариновую кислоту, маннит, сорбит, микрокристаллическую целлюлозу, неорганические соли, крахмал, прежелатинизированный крахмал, порошкообразный сахар и т. п. Примерами жидких носителей являются сахарный сироп, арахисовое масло, оливковое масло и вода. Примеры газообразных носителей включают диоксид углерода и азот. При получении композиций, представляющих собой лекарственную форму для перорального применения, можно применять любые традиционные фармацевтические среды. Например, можно применять воду, гликоли, масла, спирты, ароматизирующие средства, консерванты, красящие средства и т. п. для получения жидких препаратов для перорального применения, таких как суспензии, настойки и растворы; при этом можно применять носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, средства, способствующие гранулированию, смазывающие средства, связующие, разрыхлители и т. п., для получения твердых препаратов для перорального применения, таких как порошки, капсулы и таблетки. Вследствие простоты их введения, таблетки и капсулы являются предпочтительными единичными лекарственными формами для перорального введения, в случае которых применяют твердые фармацевтические носители. Необязательно таблетки могут быть покрыты оболочкой с помощью стандартных методик с применением или без применения воды.
Таблетки, содержащие соединения или композицию по настоящему изобретению, можно получать путем прессования или формования, необязательно с одним или более вспомогательными ингредиентами или вспомогательными средствами. Прессованные таблетки можно получать путем прессования на подходящем устройстве, где активный ингредиент находится в сыпучей форме, такой как порошок или гранулы, необязательно в смеси со смазывающим средством, инертным разбавителем, поверхностно-активным или диспергирующим средством. Формованные таблетки можно получать путем формования в подходящем устройстве смеси порошкообразного соединения или композиции, увлажненных инертным жидким разбавителем. Каждая таблетка предпочтительно содержит от приблизительно 0,01 мг до приблизительно 5 г активного ингредиента, и каждые саше или капсула предпочтительно содержат от приблизительно 0,1 мг до приблизительно 0,5 г активного ингредиента. Например, состав, предназначенный для перорального введения людям, может содержать от приблизительно 0,1 мг до приблизительно 0,5 г активного средства, смешанного с соответствующим и подходящим количеством носителя, содержание которого может варьироваться от приблизительно 5 до приблизительно 99,99 процентов всей композиции. Стандартные лекарственные формы, как правило, будут содержать от приблизительно 0,1 мг до приблизительно 0,5 г активного ингредиента, как правило, 0,1 мг, 0,2 мг, 0,5 мг, 1 мг, 2 мг, 2,5 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг.
Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, можно получать в виде растворов или суспензий активных соединений в воде. Может быть включено подходящее поверхностно-активное вещество, такое как лаурилсульфат натрия, полисорбат-80 (Tween-80), полиоксиэтилен-гидрогенизированное касторовое масло, полоксамер. Также можно получать дисперсии в глицерине, в жидких полиэтиленгликолях и в их смесях в маслах. Кроме того, можно включать консервант для предотвращения нежелательного роста микроорганизмов.
Фармацевтические композиции по настоящему изобретению, подходящие для инъекционного применения, предусматривают стерильные водные растворы или дисперсии. Кроме того, композиции могут быть представлены в форме стерильных порошков для немедленного получения таких стерильных инъекционных растворов или дисперсий. Во всех случаях конечная инъекционная форма должна быть стерильной и должна обладать достаточной текучестью для легкого введения с помощью шприца. Фармацевтические композиции должны быть устойчивыми при условиях изготовления и хранения; таким образом, предпочтительно следует обеспечить их сохранность от контаминации микроорганизмами, такими как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащие, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и их подходящие смеси.
Фармацевтические композиции по настоящему изобретению могут быть представлены в форме, подходящей для местного применения, такой как, например, аэрозоль, крем, мазь, лосьон, опудривающий порошок или т. п. Кроме того, композиции могут быть представлены в форме, подходящей для применения в устройствах для трансдермальной доставки. Такие составы можно получать с использованием соединения, представленного формулой I, по настоящему изобретению или его фармацевтически приемлемой соли с помощью традиционных способов обработки. В качестве примера, крем или мазь получают путем смешивания гидрофильного материала и воды вместе с соединением в количестве от приблизительно 5 вес. % до приблизительно 50 вес. % с получением крема или мази с желаемой консистенцией.
Фармацевтические композиции по настоящему изобретению могут быть представлены в форме, подходящей для ректального введения, где носитель представляет собой твердое вещество. Предпочтительно, если смесь формируют в суппозитории со стандартной дозой. Подходящие носители включают масло какао и другие материалы, обычно применяемые в данной области техники. Суппозитории в целях удобства можно получать путем сначала смешивания композиции с размягченными или расплавленными носителями с последующим охлаждением и формованием в формах.
В дополнение к вышеупомянутым ингредиентам, представляющим собой носитель, описанные выше фармацевтические составы могут содержать при необходимости один или более дополнительных ингредиентов, представляющих собой носитель, таких как разбавители, буферы, ароматизирующие средства, связующие, поверхностно-активные средства, загустители, смазывающие средства, консерванты (в том числе антиоксиданты) и т. п. Кроме того, можно включать другие вспомогательные средства для придания составу изотоничности с кровью предполагаемого пациента. Композиции, содержащие соединение, описанное формулой I, или его фармацевтически приемлемые соли, также можно получать в форме порошка или жидкого концентрата.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фигура 1: кривая ингибирования для соединения 4 в клеточной линии NCI-H1581 человека при ксенотрансплантации бестимусным мышам; ось абсцисс представляет собой количество дней, прошедших после инокуляции клеток NCI-H1581, и ось ординат представляет собой объем опухоли.
Фигура 2: кривая ингибирования для соединения 50 в клеточной линии SNU-16 человека при ксенотрансплантации бестимусным мышам; ось абсцисс представляет собой количество дней, прошедших после инокуляции клеток NCI-H1581, и ось ординат представляет собой объем опухоли.
Фигура 3: кривая ингибирования для соединения 24, соединения 29 и соединения 64 в клеточной линии NCI-H1581 человека при ксенотрансплантации бестимусным мышам; ось абсцисс представляет собой количество дней, прошедших после инокуляции клеток NCI-H1581, и ось ординат представляет собой объем опухоли.
ПОДРОБНОЕ ОПИСАНИЕ
Настоящее изобретение дополнительно представлено без ограничения в виде следующих примеров, иллюстрирующих настоящее изобретение. В приведенных примерах настоящего изобретения методики или способы, если конкретно не указано иное, являются традиционными методиками или способами для данной области техники.
Если не указано иное, все доли и количества в процентах в настоящем изобретении приведены по весу, температура измерена в градусах Цельсия (°C).
В примерах использованы следующие сокращения:
BuN4F-THF: раствор фторида тетрабутиламмония в тетрагидрофуране;
2-BuOH: вторичный бутанол;
CH3CN: ацетонитрил;
DCM: дихлорметан;
DIEA: N,N-диизопропилэтиламин;
DMF: N,N-диметилформамид;
DMF-DMA: N,N-диметилформамид-диметилацеталь;
DMSO: диметилсульфоксид;
EA: этилакрилат;
EtOH: этанол;
ч.: час;
HATU: 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония гексафторфосфат;
hex или Hex: гексан;
HOAc: уксусная кислота;
i-PrOH: изопропанол;
KOAc: ацетат калия;
LiAlH4: алюмогидрид лития;
m-CPBA: M-хлорпероксибензойная кислота;
MeOH: метанол;
MeONa: метоксид натрия;
мин.: минуты;
NCS: N-хлорсукцинимид;
NMP: N-метилпирролидон;
Pd(dppf)Cl2: [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид;
к. т.: комнатная температура;
SEMCl: 2-(триметилсилил)этоксиметилхлорид;
TBAI: иодид тетрабутиламмония;
t-BuOK: трет-бутоксид калия;
TEA: триэтиламин;
TFA: трифторацетат;
TFAA: трифторуксусный ангидрид;
THF: тетрагидрофуран;
TMSCl: триметилхлорсилан;
TMSCN: триметилцианосилан;
xantPhos: 4,5-бис-дифенилфосфино-9,9-диметилоксазепин.
Получение промежуточного соединения M1
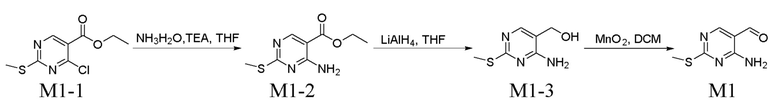
Стадия 1. Получение соединения M1-2
Растворяли 500 г соединения M1-1, 651 г TEA и 782 мл водного раствора аммиака (25%) в 2,4 л THF и перемешивали при к. т. в течение 12 ч. Реакционную смесь разбавляли водой, экстрагировали с помощью EA, промывали водой и затем высушивали с помощью безводного Na2SO4. Осадок собирали путем концентрирования в вакууме с получением соединения M1-2 (выход: 98,3%), которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 214.
Стадия 2. Получение соединения M1-3
В атмосфере азота при -20°C растворяли 250 г соединения M1-2 в 2 л THF и к полученному добавляли 570 мл LiAlH4 (2,5 н., суспендированный в THF) при перемешивании. Реакционную смесь перемешивали при -10°C в течение 3 ч., к реакционному раствору добавляли 50 мл воды для прекращения реакции, при температуре ниже 15°C добавляли 50 мл 15% раствора NaOH при перемешивании, затем добавляли 150 мл воды. В результате фильтрования получали твердое вещество, его промывали с помощью EA, фильтрат собирали и концентрировали при пониженном давлении с получением 180 г соединения M1-3 (выход: 89,7%). Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 172,1.
Стадия 3. Получение соединения M1
Перемешивали смесь 320 г M1-3, 1465 г MnO2 и 3 л DCM в течение 12 ч. при к. т., в результате фильтрования собирали твердое вещество, его промывали с помощью DCM, фильтрат собирали и концентрировали при пониженном давлении с получением 261 г соединения M1 (выход: 82,5%). Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 170,0.
Получение промежуточного соединения M2
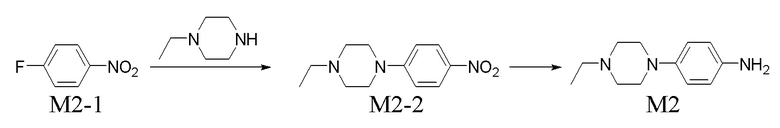
Стадия 1. Получение соединения M2-2
Перемешивали смесь 20 г соединения M2-1, 19 г 1-этилпиперазина, 39 г K2CO3 и 100 мл DMF в течение 12 ч. при к. т. Реакционную смесь добавляли в 250 мл воды, реакцию продолжали в течение 1 ч. и затем проводили фильтрование. Твердое вещество промывали водой и выпаривали до сухого состояния с получением 30 г M2-2 (выход: 90,0%). Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 236,1.
Стадия 2. Получение соединения M2
Подвергали реакции 30 г соединения M2-2, 500 мл метанола и 4,0 г палладия на угле (5% Pd) в течение 12 ч. в атмосфере водорода. В результате фильтрования собирали твердое вещество, его промывали метанолом и фильтрат собирали и концентрировали при пониженном давлении с получением 28,9 г неочищенного продукта M2, который представлял собой коричневое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 206,2.
Получение промежуточного соединения M3
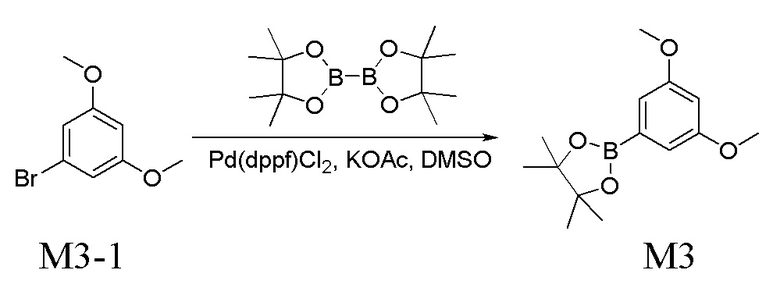
Подвергали реакции 1 г соединения M3-1, 1,4 г пинаколового сложного эфира бензойной кислоты, 337 мг Pd(dppf)Cl2, 903 мг CH3COOK и 20 мл DMSO в течение 2 ч. в атмосфере азота при 100°C. Реакционную смесь разбавляли водой, экстрагировали гексаном, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 1,4 г неочищенного продукта M3, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 265,2.
Получение промежуточного соединения M4
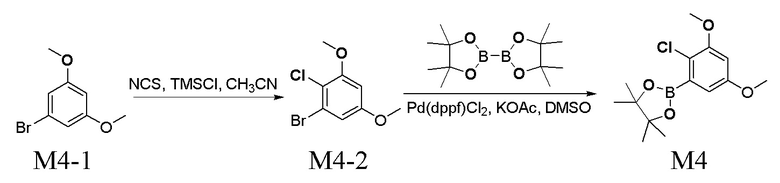
Стадия 1. Получение соединения M4-2
Перемешивали смесь 5,01 г соединения M4-1, 3,38 г N-хлорсукцинимида, 50 мл CH3CN, 0,25 г триметилхлорсилана в течение 1 ч. при к. т. Реакционную смесь гасили водой, экстрагировали с помощью EA, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 6,21 г неочищенного продукта M4-2, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
Стадия 2. Получение соединения M4
Перемешивали смесь 2,01 г соединения M4-2, 3,03 г пинаколового сложного эфира бензойной кислоты, 580 мг Pd(dppf)Cl2, 1,56 г CH3COOK и 20 мл 1,4-диоксана в течение 12 ч. в атмосфере азота при 80°C. Реакционную смесь разбавляли водой, экстрагировали гексаном, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 2,5 г неочищенного продукта M4, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 299,1.
Получение промежуточного соединения M5
Путь A
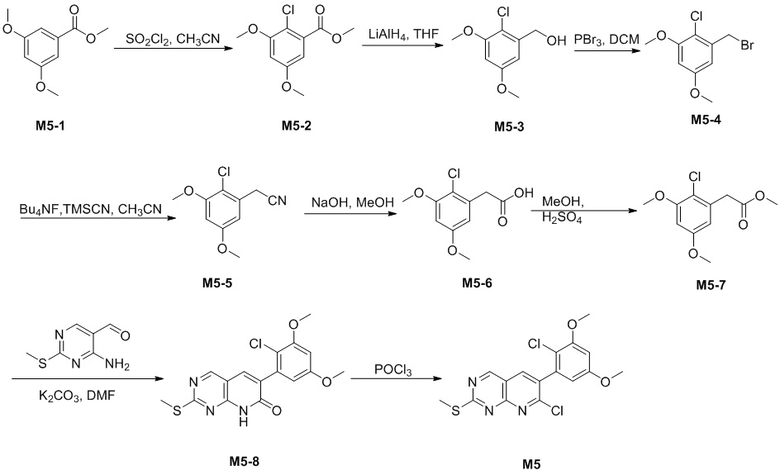
Стадия 1. Получение соединения M5-2
В атмосфере азота при -10°C медленно добавляли 86 г SO2Cl2 в раствор 100 г соединения M5-1, растворенного в 1000 мл CH3CN. Реакцию поддерживали при температуре ниже 0°C и перемешивали в течение 90 мин. Реакционную смесь гасили водой при 0°C и pH доводили до 7-8 с помощью 10% раствора NaOH. Реакционную смесь экстрагировали с помощью EA, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 129,5 г неочищенного продукта M5-2, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 231,0.
Стадия 2. Получение соединения M5-3
В атмосфере азота при -0°C медленно добавляли 24 мл LiAlH4 (2,5 н., суспендированный в THF) к раствору 129,5 г соединения M5-2, растворенного в 1,2 л THF, при перемешивании. Реакцию поддерживали в течение 90 мин. при 0°C, к реакционной смеси добавляли EtOAc и ее перемешивали в течение 30 мин. при 0°C, затем к полученному по каплям добавляли насыщенный раствор Na2SO4. Реакционную смесь фильтровали и фильтрат промывали с помощью EA, фильтрат объединяли, затем промывали насыщенным солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 168 г неочищенного продукта M5-3, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
Стадия 3. Получение соединения M5-4
В атмосфере азота при -10°C добавляли 94,7 мл трибромида фосфора к раствору 168 г соединения M5-3, растворенного в 1,1 л дихлорметана, при перемешивании. Реакцию поддерживали в течение 1,2 ч. при 0°C, затем реакционную смесь добавляли в ледяную воду и pH доводили до 7-8 с помощью 10% раствора NaOH. Реакционную смесь экстрагировали дихлорметаном, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/дихлорметан=10:1) с получением 91 г белого твердого вещества M5-4.
Стадия 4. Получение соединения M5-5
Растворяли 15,23 г соединения M5-4 и 11,38 г триметилцианосилана в 150 мл CH3CN, в который затем добавляли по каплям 114,7 мл 1,0 н. BuN4F-THF при 20°C. Реакцию поддерживали в течение 30 мин. при 70°C. Реакционную смесь нагревали до комнатной температуры и гасили водой. В результате фильтрования собирали твердое вещество, его промывали водой и выпаривали до сухого состояния с получением 11,22 г белого твердого вещества M5-25 (выход: 92,4%).
Стадия 5. Получение соединения M5-6
Смесь 11,22 г соединения M5-5, 10,60 г NaOH, 100 мл метанола и 150 мл воды нагревали с обратным холодильником в течение 3 ч. Реакционный раствор концентрировали при пониженном давлении и остаток растворяли в воде, затем экстрагировали с помощью EA, объединяли водную фазу, pH доводили до 2-3 с помощью 1 н. хлористоводородной кислоты. В результате фильтрования собирали твердое вещество, его промывали водой, высушивали при пониженном давлении с получением 8,06 г соединения M5-6, которое представляло собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 231,0.
Стадия 6. Получение соединения M5-7
Смесь 8,06 г соединения M5-6, 80 мл метанола и 8,0 мл концентрированной серной кислоты нагревали с обратным холодильником в течение 3 ч. Реакционную смесь нагревали до комнатной температуры и добавляли в ледяную воду. Реакционную смесь экстрагировали с помощью EA, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 4,02 г соединения M5-7, которое представляло собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 245,1.
Стадия 7. Получение соединения M5-8
Перемешивали смесь 9,42 г соединения M5-7, 5,20 г соединения M1, 5,85 г K2CO3 и 180 мл DMF в течение 8 ч. при 110°C. Реакционную смесь разбавляли водой, экстрагировали с помощью EA, промывали насыщенным раствором хлорида натрия, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат=3:1) с получением 2,30 г белого твердого вещества M5-8.
LC-MS [M+H+] 364,0.
Стадия 8. Получение соединения M5
Перемешивали смесь 2,71 г M5-8 и 50 мл оксихлорида фосфора в течение 3 ч. при 100°C. Реакционный раствор концентрировали и удаляли большую часть оксихлорида фосфора. Остаток гасили ледяной водой и pH доводили до 8 с помощью насыщенного раствора NaHCO3. Реакционную смесь экстрагировали с помощью EA, промывали насыщенным раствором хлорида натрия, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3,01 г соединения M5, которое представляло собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 382,0.
Путь B
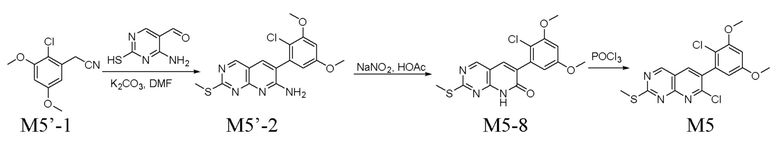
Стадия 1. Получение соединения M5’-2
Смесь 16,77 г соединения M5’-1, 11,17 г соединения M1, 27,33 г K2CO3 и 150 мл DMF перемешивали в течение 12 ч. при 110°C. Реакционную смесь нагревали до комнатной температуры и добавляли в ледяную воду. В результате фильтрования собирали твердое вещество, его промывали водой, высушивали при пониженном давлении с получением 17,21 г соединения M5’-2, которое представляло собой желтое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 363,1.
Стадия 2. Получение соединения M5-8
16,75 г соединения M5’-2 растворяли в 50 мл уксусной кислоты и смесь перемешивали при к. т., к полученному несколькими порциями добавляли 15,97 г NaNO2. Реакционную смесь перемешивали в течение 3 ч. при 70°C, охлаждали до к. т. и добавляли в ледяную воду. В результате фильтрования получали твердое вещество, его промывали водой, высушивали при пониженном давлении с получением 15,21 г соединения M5-8, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 364,0.
Стадия 3. Получение соединения M5
Получение является подобным стадии 8 пути A в M5.
Получение промежуточного соединения M6
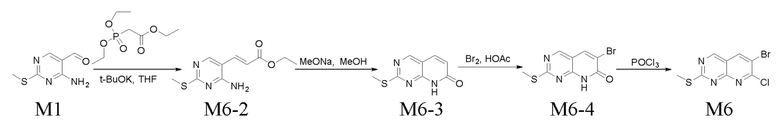
Стадия 1. Получение соединения M6-2
В атмосфере азота при 0°C добавляли 16 г триэтилфосфоноацетата в раствор 8,6 г t-BuOK, растворенного в 300 мл THF, при перемешивании. Реакцию поддерживали в течение 1 ч. при данной температуре. В реакционный раствор несколькими порциями добавляли 10 г соединения M1 при 0°C и его перемешивали в течение 12 ч. при к. т. Реакционную смесь гасили водой, экстрагировали с помощью EA, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 14 г неочищенного продукта M6-2, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 240,1.
Стадия 2. Получение соединения M6-3
14 г соединения M6-2, 200 мл метанола и 6,3 г метоксида натрия растворяли в 21 мл метанола, смесь нагревали с обратным холодильником в течение 4 ч. Растворитель удаляли при пониженном давлении, остаток суспендировали в 200 мл воды и pH доводили до 8 с помощью 2 н. раствора хлористоводородной кислоты. В результате фильтрования собирали твердое вещество, его промывали водой, высушивали при пониженном давлении с получением 9 г неочищенного продукта M6-3, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 194,0.
Стадия 3. Получение соединения M6-4
Смесь 9 г соединения M6-3, 300 мл уксусной кислоты и 4,8 мл жидкого брома перемешивали в течение 12 ч. при 50°C. Реакционный раствор разбавляли дихлорметаном, в результате фильтрования собирали твердое вещество и его промывали метанолом, высушивали при пониженном давлении с получением 12,9 г неочищенного продукта M6-4, который представлял собой сероватое белое твердое вещество.
LC-MS [M+H+] 271,9.
Стадия 4. Получение соединения M6
Смесь 12,9 г соединения M6-4 и 300 мл POCl3 перемешивали в течение 12 ч. при 110°C, удаляли большую часть POCl3 при пониженном давлении и остаток гасили ледяной водой. В результате фильтрования собирали твердое вещество, его промывали водой и высушивали при пониженном давлении с получением 13 г неочищенного продукта M6, который представлял собой белое твердое вещество.
LC-MS [M+H+] 289,9.
Получение промежуточного соединения M7
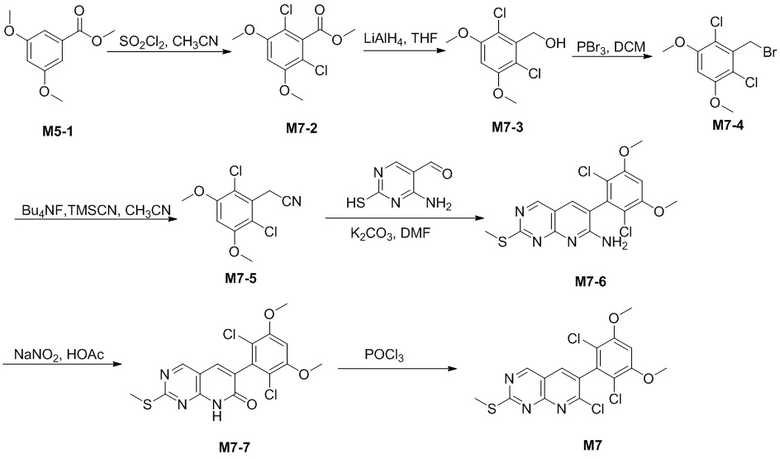
Стадия 1. Получение соединения M7-2
100 г соединения M5-1 растворяли в 1000 мл CH3CN в атмосфере азота при -10°C, к полученному медленно добавляли 172 г SO2Cl2 при перемешивании. Реакцию поддерживали в течение 90 мин. при температуре ниже 0°C. Реакционную смесь гасили водой при температуре ниже 0°C и pH доводили до 7-8 с помощью 10% NaOH. Реакционную смесь экстрагировали с помощью EA, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 148,8 г соединения M7-2, которое представляло собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 265,0.
Стадия 2. Получение соединения M7-3
149 г соединения M7-2 растворяли в 1,2 л THF в атмосфере азота при 0°C, к полученному добавляли 224 мл 2,5 н. раствора LiAlH4-THF при перемешивании. Реакцию поддерживали в течение 90 мин. при 0°C, в реакционный раствор добавляли EA и реакцию поддерживали в течение 30 мин. при 0°C, затем к полученному добавляли насыщенный раствор Na2SO4. В результате фильтрования собирали фильтрат, фильтрат промывали с помощью EA и фильтрат собирали, промывали насыщенным раствором хлорида натрия, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 196 г неочищенного продукта M7-3, который представлял собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
Стадия 3. Получение соединения M7-4
196 г соединения M7-3 растворяли в 1,1 л дихлорметана в атмосфере азота при -10°C, к полученному добавляли 94,7 мл трибромида фосфора при перемешивании. Смесь перемешивали в течение 1,2 ч. при 0°C, затем ее добавляли в ледяную воду и pH доводили до 7-8 с помощью 10% NaOH. Реакционную смесь экстрагировали дихлорметаном, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/дихлорметан=10:1) с получением 100 г белого твердого вещества M7-4.
Стадия 4. Получение соединения M7-5
Образовывали смесь из 17,19 г соединения M7-4, 11,38 г триметилцианосилана и 150 мл CH3CN, к полученному добавляли 114,7 мл 1,0 н. раствора BuN4F в THF при перемешивании при температуре ниже 20°C. Реакционный раствор перемешивали в течение 30 мин. при 70°C, охлаждали до к. т. и гасили водой. В результате фильтрования собирали твердое вещество, его промывали водой и высушивали при пониженном давлении с получением 14,34 г соединения M7-5 (выход: 90,0%), которое представляло собой белое твердое вещество.
Стадия 5. Получение соединения M7-6
Смесь 29,25 г соединения M7-5, 14,35 г 4-амино-2-метилпиримидин-5-формальдегида, 49,20 г K2CO3 и 400 мл DMF перемешивали в течение 12 ч. при 110°C. Реакционный раствор разбавляли водой, фильтровали, твердое вещество промывали водой и высушивали при пониженном давлении с получением 31,02 г соединения M7-6, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 397,0.
Стадия 6. Получение соединения M7-7
Несколькими порциями добавляли 22,06 г NaNO2 в раствор 25,41 г соединения M7-6, растворенного в 100 мл уксусной кислоты, при перемешивании при к. т. Реакционный раствор перемешивали в течение 3 ч. при 70°C, охлаждали до к. т., добавляли в ледяную воду и фильтровали. Твердое вещество промывали водой и высушивали при пониженном давлении с получением 23,42 г соединения M7-7, которое представляло собой желтое твердое вещество.
LC-MS [M+H+] 398,0.
Стадия 7. Получение соединения M7
Смесь 18,40 г соединения M7-7 и 100 мл оксихлорида фосфора перемешивали в течение 3 ч. при 100°C, реакционный раствор концентрировали и удаляли большую часть оксихлорида фосфора. Остаток гасили ледяной водой и pH доводили до 8 с помощью насыщенного раствора NaHCO3. Реакционную смесь экстрагировали с помощью EA, промывали насыщенным раствором хлорида натрия, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 15,01 г соединения M7, которое представляло собой белое твердое вещество, которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 416,0.
Пример 1. Получение 1-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)-2-метилпропан-2-ола
Путь A
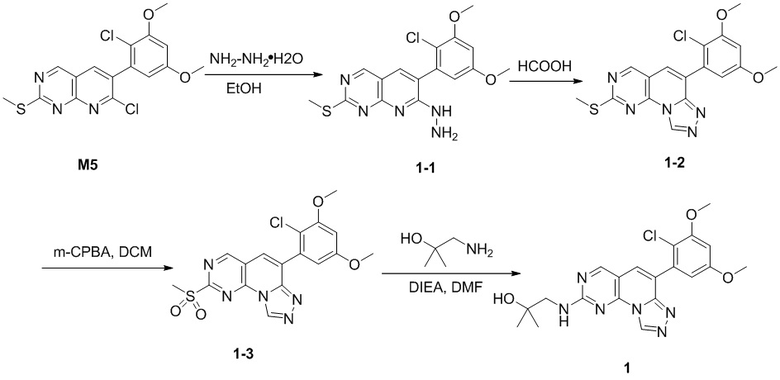
Стадия 1. Получение соединения 1-1
Смесь 500 мг соединения M5, 818 мг гидразина (80%) и 25 мл этанола нагревали с обратным холодильником в течение 2 ч. и концентрировали при пониженном давлении с получением 520 мг неочищенного продукта 1-1, который представлял собой желтое твердое вещество, которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 378,1.
Стадия 2. Получение соединения 1-2
Смесь 520 мг соединения 1-1 и 25 мл муравьиной кислоты нагревали с обратным холодильником в течение 1 ч. Реакционный раствор разбавляли водой и фильтровали. Твердое вещество промывали водой с получением 310 мг соединения 1-2 (выход: 58%), которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 388,1.
Стадия 3. Получение соединения 1-3
Несколькими порциями добавляли 378 мг м-хлорпероксибензойной кислоты в раствор 340 мг соединения 1-2, растворенного в 40 мл дихлорметана, при перемешивании при 0°C. Реакционный раствор перемешивали в течение 2 ч. при к. т. и гасили насыщенным раствором NaHCO3. Реакционную смесь экстрагировали дихлорметаном, промывали последовательно 5% раствором тиосульфата натрия и солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 340 мг неочищенного продукта 1-3, который представлял собой светло-желтое твердое вещество, которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 420,1.
Стадия 4. Получение соединения 1
Смесь 100 мг соединения 1-3, 60 мг 1-амино-2-метил-2-пропанола, 93 мг DIEA и 10 мл DMF перемешивали в течение 2 ч. при 80°C. Реакционный раствор охлаждали до к. т., разбавляли водой, затем экстрагировали с помощью EA, промывали насыщенным раствором хлорида натрия, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (дихлорметан/метанол=10:1) с получением 80 мг желтого твердого вещества, которое представляло собой соединение 1 (выход: 78,3%).
LC-MS [M+H+] 429,1.
1H ЯМР (DMSO-d6, 400 МГц) δ 9,74 (s, 1H), 9,02 (s, 1H), 7,61 (s, 1H), 6,83 (s, 1H), 6,73 (s, 1H), 3,91 (s, 3H), 3,80 (s, 3H), 3,52-3,50 (m, 2H), 3,37 (m, 2H), 1,14 (s, 6H).
Путь B
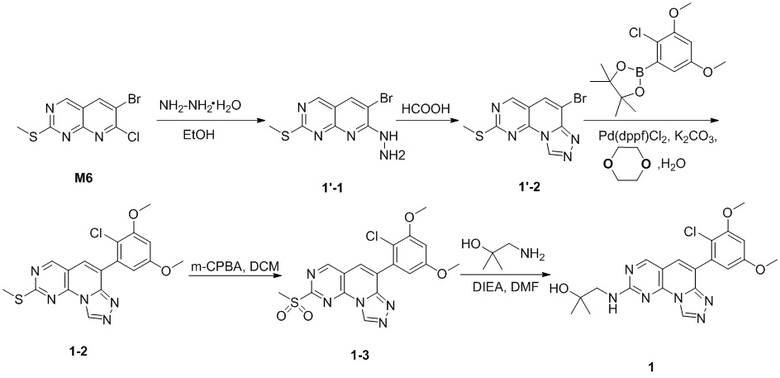
Стадия 1. Получение соединения 1’-1
Смесь 12,5 г соединения M6, 200 мл EtOH и 50 мл гидразина (80%) нагревали с обратным холодильником в течение 2 ч., реакционную смесь охлаждали до к. т. и фильтровали. Твердое вещество промывали с помощью EtOH и высушивали при пониженном давлении с получением 11 г соединения 1’-1, которое представляло собой сероватое белое твердое вещество, которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 286,0.
Стадия 2. Получение соединения 1’-2
Смесь 11 г соединения 1’-1 и 100 мл муравьиной кислоты нагревали с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали до к. т. и фильтровали. Твердое вещество промывали водой и высушивали при пониженном давлении с получением 5 г соединения 1’-2, которое представляло собой белое твердое вещество, которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 296,0.
Стадия 3. Получение соединения 1-2
В атмосфере азота смесь 16,2 г соединения 1’-2, 24,5 г соединения M4, 4,0 г Pd(dppf)Cl2, 22,6 г K2CO3, 320 мл 1,4-диоксана и 32 мл воды перемешивали в течение 12 ч. при 100°C. Реакционную смесь концентрировали при пониженном давлении и остаток очищали посредством колоночной хроматографии на силикагеле (DCM/EA=10:1) с получением 12 г соединения 1-2, которое представляло собой сероватое белое твердое вещество.
LC-MS [M+H+] 388,1.
Стадия 4. Получение соединения 1-3
Способ являлся подобным получению соединения 1-3 на стадии 3 пути A из примера 1.
Стадия 5. Получение соединения 1
Способ являлся подобным получению соединения 1 на стадии 4 пути A из примера 1.
Пример 2. Получение 4-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-амина
Путь A
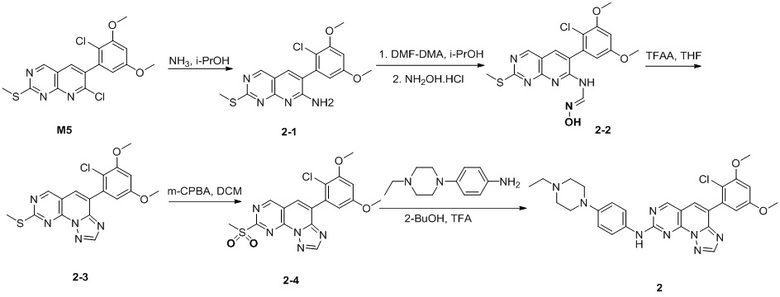
Стадия 1. Получение соединения 2-1
Смесь 100 мг соединения M5, 0,37 мл смеси аммиак-метанол и 5 мл изопропанола перемешивали в течение 24 ч. при 70°C. Реакционный раствор концентрировали при пониженном давлении с получением 90 мг неочищенного продукта 2-1, который представлял собой светло-желтое твердое вещество, которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 363,1.
Стадия 2. Получение соединения 2-2
Смесь 520 мг соединения 2-1, 25 мл изопропанола и 342 мг DMF-DMA нагревали с обратным холодильником в течение 12 ч. Реакционную смесь нагревали до 50°C, затем к полученному добавляли 199 мг гидрохлорида гидроксиламина. Реакционную смесь перемешивали в течение 12 ч. при 50°C и охлаждали до к. т. В результате фильтрования собирали твердое вещество, его промывали изопропанолом и высушивали при пониженном давлении с получением 450 мг соединения 2-2, которое представляло собой желтое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 406,1.
Стадия 3. Получение соединения 2-3
В атмосфере азота при 0°C добавляли 349 мг TFAA в раствор 450 мг соединения 2-2, растворенного в 120 мл THF. Реакционный раствор перемешивали в течение 12 ч. при к. т. и гасили насыщенным раствором NaHCO3, затем экстрагировали с помощью EA, промывали водой, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (гексан/этилацетат=10:1) с получением 450 мг желтого твердого вещества, которое представляло собой соединение 2-3.
LC-MS [M+H+] 388,1.
Стадия 4. Получение соединения 2-4
Смесь 420 мг соединения 2-3, 16 мл дихлорметана и 281 мг m-CPBA перемешивали в течение 2 ч. при к. т. Реакционный раствор гасили насыщенным раствором NaHCO3, затем экстрагировали дихлорметаном, промывали тиосульфатом натрия и солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 300 мг соединения 2-4, которое представляло собой желтое твердое вещество, которое применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 420,1.
Стадия 5. Получение соединения 2

Смесь 300 мг соединения 2-4, 176 мг соединения M2, 5 мл втор-бутанола и 0,5 мл TFA помещали в герметичную пробирку и перемешивали в течение 12 ч. при 120°C. Реакционный раствор концентрировали при пониженном давлении, остаток растворяли в дихлорметане и промывали последовательно насыщенным раствором Na2CO3 и солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/метанол=50:1) с получением 290 мг желтого твердого вещества, которое представляло собой соединение 2-3. Выход составлял 74,5%.
LC-MS [M+H+] 545,2.
1H ЯМР (DMSO-d6, 400 МГц) δ 9,28 (s, 1H), 8,61 (s, 1H), 7,97 (s, 1H), 7,92-7,67 (m, 2H), 6,99 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,76 (s, 1H), 6,68-6,47 (m, 1H), 3,92 (s, 3H), 3,81 (s, 3H), 3,13 (brs, 4H), 2,53 (brs, 4H), 2,41-2,39 (m, 2H), 1,04 (t, J = 6 Гц, 3H).
Путь B
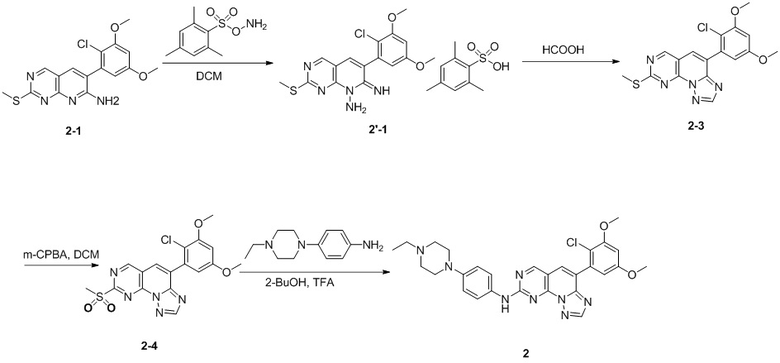
Стадия 1. Получение соединения 2’-1
3,56 г 2-[(аминоокси)сульфонил]-1,3,5-триметилбензола диспергировали в 60 мл дихлорметана и охлаждали до 0°C, к полученному добавляли 3,0 г соединения 2-1 при перемешивании и обеспечивали его естественное нагревание до комнатной температуры. Реакцию поддерживали в течение ночи, затем завершали, реакционную систему концентрировали до приблизительно 20 мл. В результате фильтрования собирали твердое вещество, его дважды промывали небольшим количеством DCM, затем высушивали с получением 3,5 г соединения 2’-1, которое представляло собой светло-желтое твердое вещество, при этом выход составил 73,2%.
LC-MS [M+H+] 378,1.
Стадия 2. Получение соединения 2-3
Смесь 600 мг соединения 2’-1 и 25 мл муравьиной кислоты нагревали с обратным холодильником в течение 1 ч. После завершения полученное охлаждали до комнатной температуры и добавляли в воду при перемешивании, затем осаждали большое количество светло-желтого твердого вещества. В результате фильтрования собирали твердое вещество, его три раза прополаскивали водой с получением нейтральной среды и высушивали в вакууме при 50°C в течение ночи с получением 300 мг соединения 2-3, которое представляло собой светло-желтое твердое вещество, при этом выход составлял 74,5%.
LC-MS [M+H+] 388,1.
Стадия 3. Получение соединения 2-4
Соединение 2-4 синтезировали аналогичным образом, как на стадии 4 пути A, с применением соединения 2-3 в качестве исходного материала.
LC-MS [M+H+] 420,1.
Стадия 4. Получение соединения 2
Соединение 2 синтезировали аналогичным образом, как на стадии 5 пути A, с применением соединения 2-4 в качестве материала.
LC-MS [M+H+] 545,2.
Соединения из таблицы 1 получали подобным образом, как в примерах 1 и 2, с применением других исходных материалов для реакции и подходящих реагентов.
Таблица 1
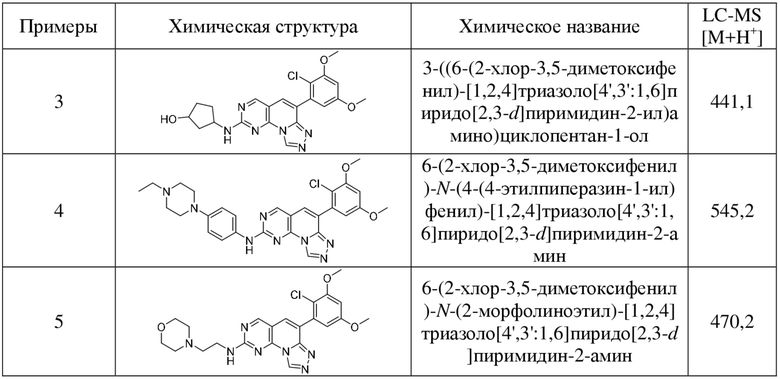
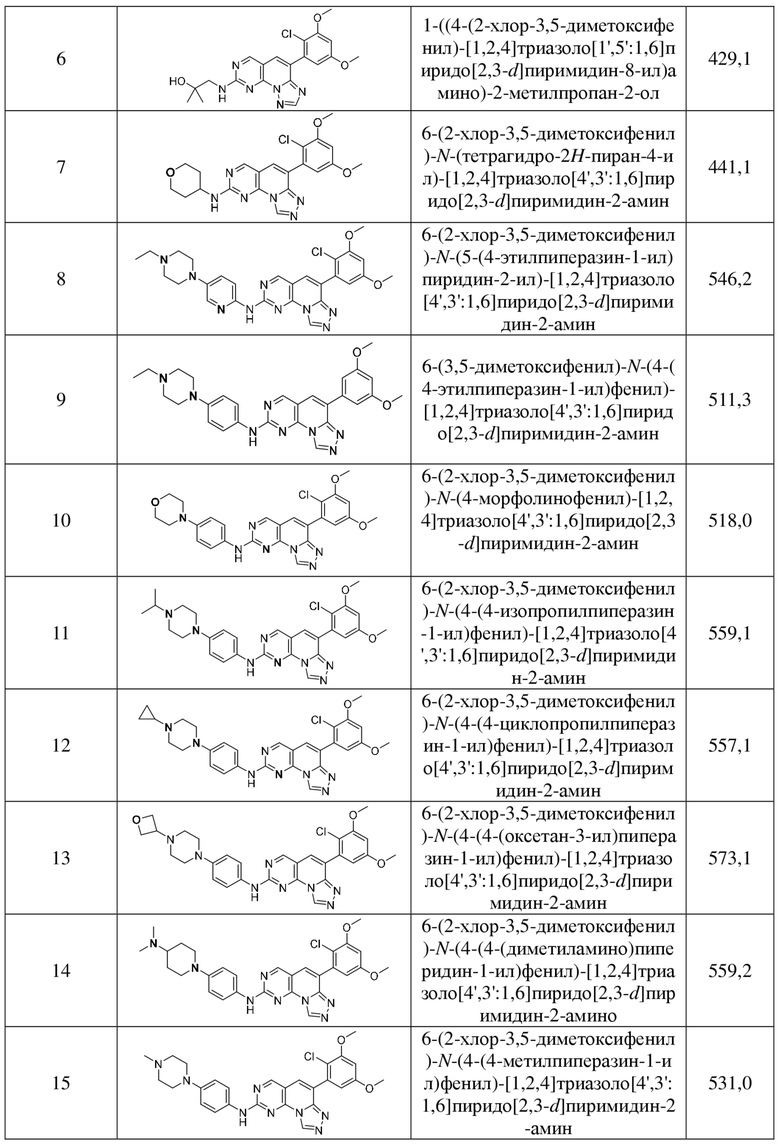
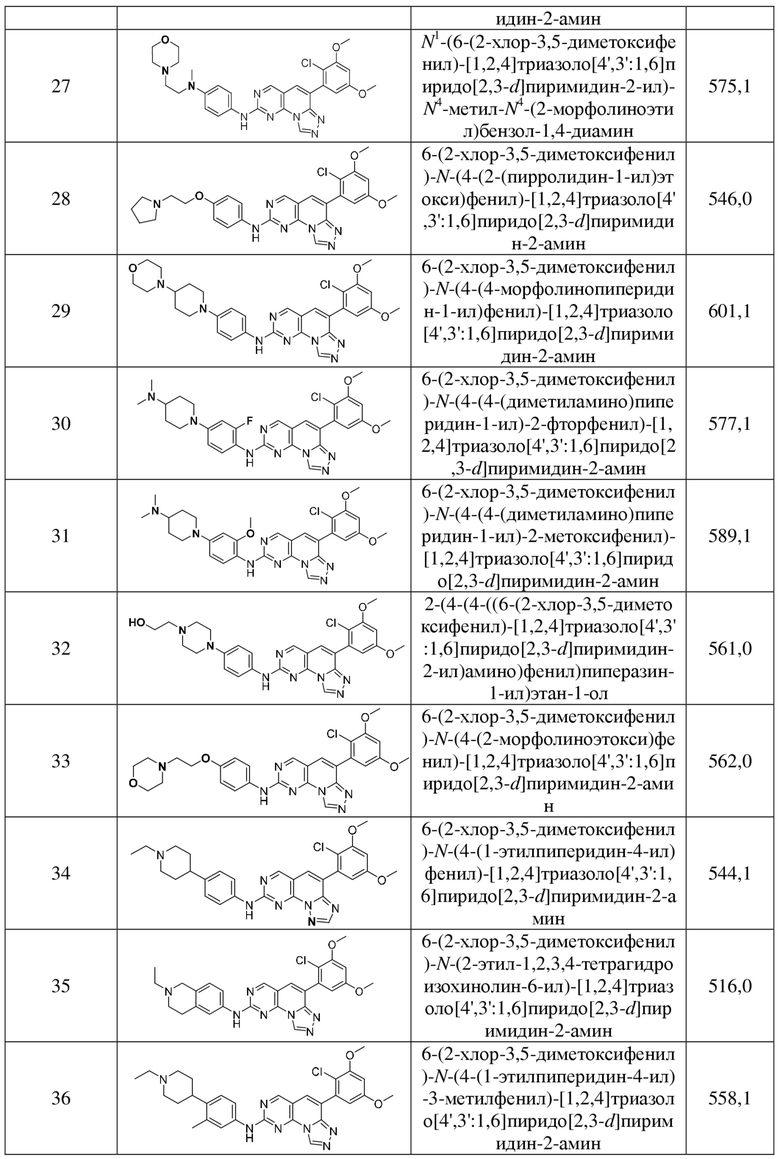
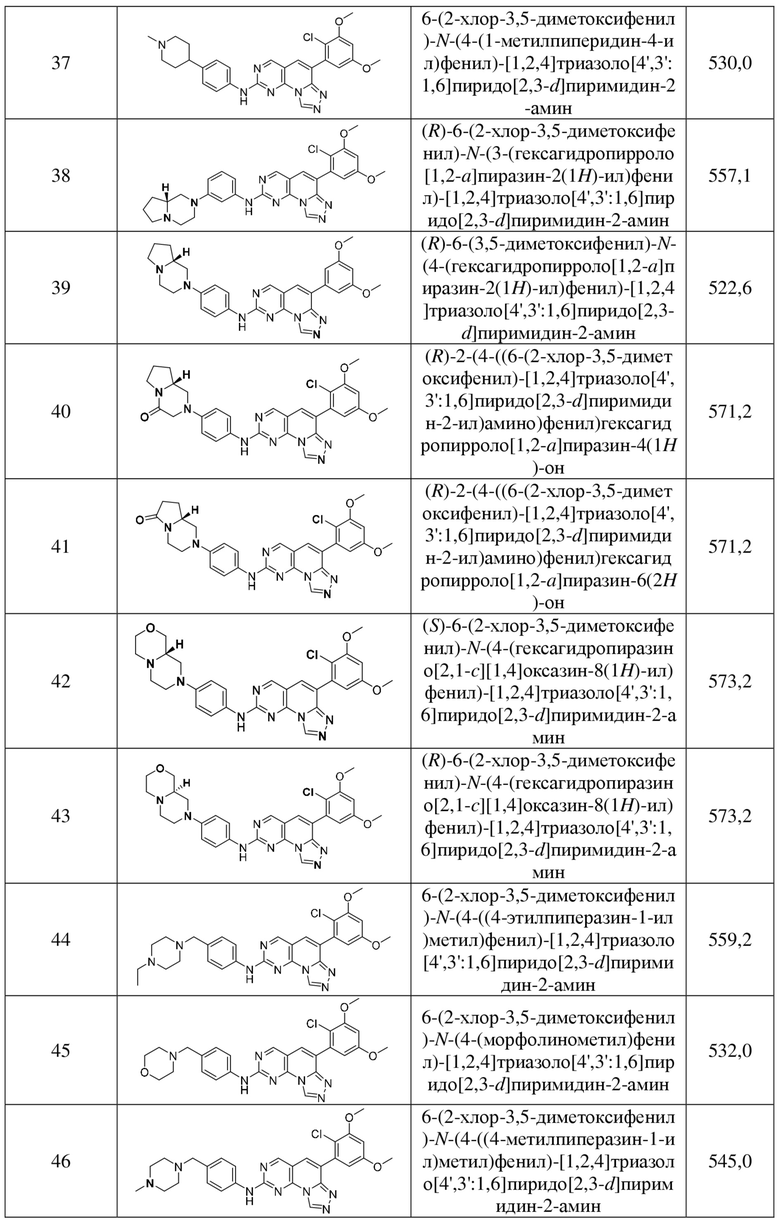
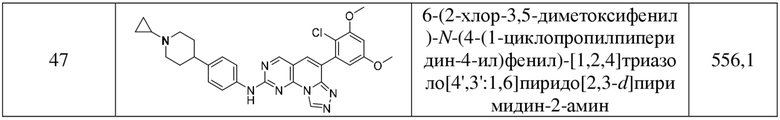
Данные ядерного магнитного резонанса для соединений 3, 4, 5, 22, 23, 24, 25, 26, 28, 29, 31, 32, 33, 34, 37, 42, 45, 46 и 52 показаны ниже:
1H ЯМР (DMSO-d6, 400 МГц) δ 9,70 (s, 1H), 9,01 (s, 1H), 7,63 (s, 1H), 6,83 (s, 1H), 6,73 (s, 1H), 4,47-4,29 (m, 1H), 4,13 (m, 1H), 3,91 (s, 3H), 3,80 (s, 3H), 3,38 (m, 2H), 2,36-2,11 (m, 1H), 2,11-1,84 (m, 1H), 1,77-1,53 (m, 4H) (соединение 3);
1H ЯМР (DMSO-d6, 400 МГц) δ 9,67 (s, 1H), 9,16 (s, 1H), 7,78 (brs, 2H), 7,68 (s, 1H), 7,02 (d, J = 8 Гц, 2H), 6,85 (s, 1H), 6,76 (s, 1H), 3,93 (s, 3H), 3,81 (s, 3H), 3,16 (brs, 4H), 2,75-2,59 (m, 4H), 2,44-2,33 (m, 2H), 1,09 (t, J = 6 Гц, 3H) (соединение 4);
1H ЯМР (DMSO-d6, 400 МГц) δ 9,68-9,47 (m, 1H), 9,01-9,01 (m, 1H), 7,61 (s, 1H), 6,83 (s, 1H), 6,74 (s, 1H), 3,92 (s, 3H), 3,90 (s, 3H), 3,63-3,56 (m, 8H), 2,60-2,56 (m, 2H), 2,43 (m, 2H) (соединение 5);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,35 (s, 1H), 9,66 (s, 1H), 9,17 (s, 1H), 7,77 (br, 2H), 7,69 (s, 1H), 7,01 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,75 (s, 1H), 3,95 (s, 3H), 3,81 (s, 3H), 3,18-2,92 (m, 13H), 2,44 (s, 3H), 2,01-1,65 (m, 4H) (соединение 22);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,31 (s, 1H), 9,64 (s, 1H), 9,15 (s, 1H), 7,74 (br, 2H), 7,67 (s, 1H), 6,90 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,75 (s, 1H), 3,91 (s, 3H), 3,80 (s, 3H), 3,44-3,39 (m, 2H), 3,09-3,09 (m, 2H), 2,50 (br, 2H), 2,45 (s, 3H), 2,07-2,05 (m, 2H), 1,80-1,78 (m, 2H) (соединение 23);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,31 (s, 1H), 9,65 (s, 1H), 9,14 (s, 1H), 7,75 (br, 2H), 7,67 (s, 1H), 7,00 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,75 (s, 1H), 3,92 (s, 3H), 3,81 (s, 3H), 3,75-3,58 (m, 2H), 3,04-2,72 (m, 3H), 2,44-2,08 (m, 4H), 1,87-1,34 (m, 4H) (соединение 24);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,34 (s, 1H), 9,66 (s, 1H), 9,16 (s, 1H), 7,77 (br, 2H), 7,68 (s, 1H), 7,03 (d, J = 8 Гц, 2H), 6,85 (s, 1H), 6,75 (s, 1H), 3,92 (s, 3H), 3,81 (s, 3H), 3,76-3,58 (m, 2H), 3,11-2,72 (m, 3H), 2,44-2,08 (m, 4H), 1,91-1,40 (m, 4H) (соединение 25);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,31 (s, 1H), 9,65 (s, 1H), 9,14 (s, 1H), 7,75 (br, 2H), 7,67 (s, 1H), 7,00 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,75 (s, 1H), 3,92 (s, 3H), 3,81 (s, 3H), 3,75-3,58 (m, 2H), 3,04-2,72 (m, 3H), 2,44-2,08 (m, 4H), 1,87-1,34 (m, 4H) (соединение 26);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,42 (s, 1H), 9,68 (s, 1H), 9,18 (s, 1H), 7,82 (br, 2H), 7,69 (s, 1H), 7,04 (d, J = 8 Гц, 2H), 6,85 (s, 1H), 6,76 (s, 1H), 4,22 (t, J = 6 Гц, 2H), 3,93 (s, 3H), 3,81 (s, 3H), 3,29 (t, J = 4 Гц, 2H), 3,05 (br, 4H), 1,88-1,85 (m, 4H) (соединение 28);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,31 (s, 1H), 9,65 (s, 1H), 9,15 (s, 1H), 7,76 (s, 2H), 7,68 (s, 1H), 7,00 (d, J = 8 Гц, 2H), 6,86 (s, 1H), 6,76 (s, 1H), 3,93 (s, 3H), 3,81 (s, 3H), 3,72-3,57 (m, 6H), 2,67-2,51 (m, 5H), 2,46-2,16 (m, 2H), 1,93-1,86 (m, 2H), 1,58-1,51 (m, 2H) (соединение 29);
1H ЯМР (DMSO-d6, 400 МГц) δ 9,53 (s, 1H), 9,10 (s, 1H), 9,15 (s, 1H), 7,76 (br, 1H), 7,66 (s, 1H), 6,85 (s, 1H), 6,75 (s, 1H), 6,71-6,70 (m, 1H), 6,62-6,60 (m, 1H), 3,92 (s, 3H), 3,83 (s, 3H), 3,80 (s, 3H), 3,53-3,23 (m, 5H), 2,71 (s, 6H), 2,15-2,07 (m, 2H), 1,83-1,71 (m, 2H) (соединение 31);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,35 (s, 1H), 9,66 (s, 1H), 9,16 (s, 1H), 7,76 (br, 2H), 7,68 (s, 1H), 6,99 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,75 (s, 1H), 4,62 (br, 1H), 3,92 (s, 3H), 3,80 (s, 3H), 3,57 (t, J = 6 Гц, 2H), 3,16-3,14 (m, 4H), 2,68-2,64 (m, 4H), 2,55-2,51 (m, 2H) (соединение 32);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,41 (s, 1H), 9,67 (s, 1H), 9,18 (s, 1H), 7,86-7,81 (m, 2H), 7,69 (s, 1H), 6,99 (d, J = 8 Гц, 2H), 6,85 (s, 1H), 6,75 (s, 1H), 4,08 (t, J = 6 Гц, 2H), 3,92 (s, 3H), 3,80 (s, 3H), 3,58 (t, J = 4 Гц, 4H), 2,69 (t, J = 6 Гц, 2H), 2,48 (t, J = 6 Гц, 4H) (соединение 33);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,60 (s, 1H), 9,35 (s, 1H), 8,64 (s, 1H), 8,02 (s, 2H), 7,74 (d, J = 8 Гц, 1H), 7,28 (d, J = 8 Гц, 2H), 6,85 (s, 1H), 6,76 (s, 1H), 3,92 (s, 3H), 3,80 (s, 3H), 3,29-3,24 (m, 1H), 3,12-3,05 (m, 2H), 3,01-2,96 (m, 4H), 2,17-1,93 (m, 4H), 1,27 (t, J = 6 Гц, 3H) (соединение 34);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,49 (s, 1H), 9,73 (s, 1H), 9,21 (s, 1H), 7,87 (d, J = 8 Гц, 2H), 7,71 (s, 1H), 7,29 (d, J = 8 Гц, 2H), 6,86 (d, J = 4 Гц, 1H), 6,77 (d J = 4 Гц, 1H), 3,93 (s, 3H), 3,82 (s, 3H), 3,34 (s, 3H), 3,20-3,17 (m, 2H), 2,64-2,55 (m, 3H), 1,87-1,74 (m, 4H) (соединение 37);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,35 (s, 1H), 9,64 (s, 1H), 9,13 (s, 1H), 7,78 (br, 2H), 7,65 (s, 1H), 7,02 (d, J = 8 Гц, 2H), 6,78 (s, 1H), 6,68 (s, 1H), 3,97-3,92 (m, 2H), 3,90 (s, 3H), 3,78 (s, 3H), 3,75-3,65 (m, 2H), 3,46-3,25 (m, 4H), 3,06-2,82 (m, 4H), 2,69-2,54 (m, 1H) (соединение 42);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,55 (s, 1H), 9,75 (s, 1H), 9,35 (s, 1H), 7,91 (d, J = 8 Гц, 2H), 7,72 (s, 1H), 7,38 (d, J = 8 Гц, 2H), 6,86 (s, 1H), 6,76 (s, 1H), 3,92 (s, 3H), 3,81 (s, 3H), 3,63 (br, 6H), 2,51-2,49 (m, 4H) (соединение 45);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,54 (s, 1H), 9,73 (s, 1H), 9,21 (s, 1H), 7,89 (d, J = 8 Гц, 2H), 7,71 (s, 1H), 7,34 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,75 (s, 1H), 3,91 (s, 3H), 3,81 (s, 3H), 3,54 (s, 2H), 2,84 (br, 4H), 2,59 (br, 4H), 2,50 (s, 3H) (соединение 46).
Пример 48. Получение соединения 48 (6-(2-хлор-3,5-диметоксифенил)-N-(4-(пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
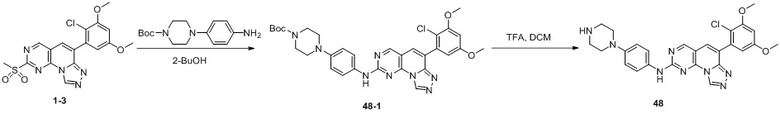
Стадия 1. Получение соединения 48-1
Смесь 1,0 г соединения 1-3, 1,03 г 1-Boc-4-(4-аминофенил)пиперазина и 20 мл втор-бутилового спирта нагревали до температуры возврата флегмы в течение 2,5 ч. Смесь охлаждали до комнатной температуры. Полученный в результате фильтрования осадок на фильтре три раза промывали втор-бутиловым спиртом, три раза промывали метил-бутиловым простым эфиром и высушивали над безводным сульфатом натрия с получением 1,16 г соединения 48-1, которое представляло собой желтое твердое вещество.
LC-MS [M+H+] 617,2.
Стадия 2. Получение соединения 48
Смесь 1,12 г соединения 48-1, 60 мл дихлорметана и 12 мл TFA перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении, полученное растворяли в 50 мл смеси DCM/MeOH=10/1. Смесь промывали насыщенным раствором бикарбоната натрия и насыщенным солевым раствором и высушивали над безводным сульфатом натрия. Полученное фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество растирали с 5 мл смеси EA/DCM=10/1, фильтровали и осадок на фильтре прополаскивали с помощью смеси EA/DCM=10/1 и высушивали с получением 1,02 г соединения 48, которое представляло собой желтое твердое вещество.
LC-MS [M+H+] 517,2.
1H ЯМР (DMSO-d6, 400 МГц) δ 10,33 (s, 1H), 9,65 (s, 1H), 9,15 (s, 1H), 7,77 (br, 2H), 7,67 (s, 1H), 7,00 (d, J = 8 Гц, 2H), 6,85 (d, J = 4 Гц, 1H), 6,76 (d J = 4 Гц, 1H), 3,92 (s, 3H), 3,81 (s, 3H), 3,72 (br, 1H), 3,12 (t, J = 6 Гц, 4H), 2,97 (t, J = 6 Гц, 4H).
Таблица 2

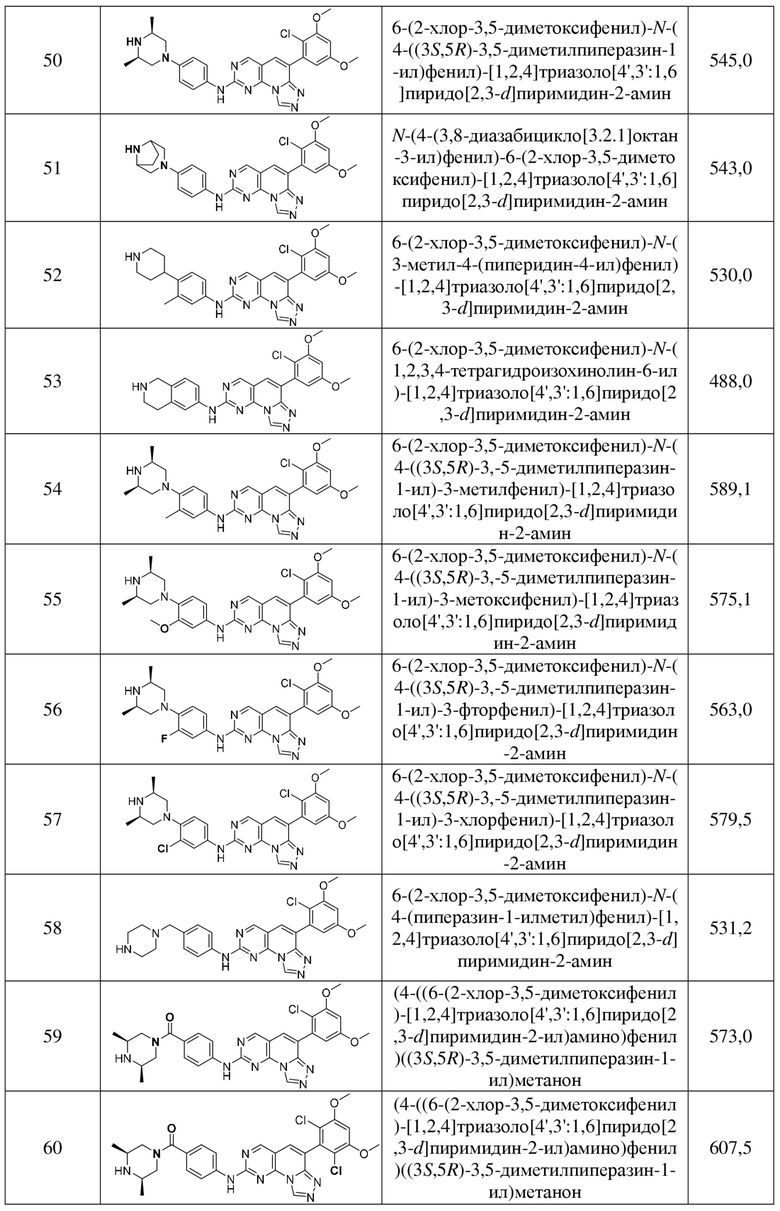
Данные ядерного магнитного резонанса для соединений 50, 52, 53, 56 и 59 показаны ниже:
1H ЯМР (DMSO-d6, 400 МГц) δ 10,30 (s, 1H), 9,65 (s, 1H), 9,13 (s, 1H), 7,75 (br, 2H), 7,66 (s, 1H), 6,98 (d, J = 8 Гц, 2H), 6,84 (s, 1H), 6,75 (s, 1H), 3,92 (s, 3H), 3,91 (s, 3H), 3,51 (d, J = 8 Гц, 2H), 2,93-2,88 (m, 2H), 2,50-2,49 (m, 1H), 2,15 (t, J = 12 Гц, 2H), 1,05 (d, J = 8 Гц, 6H) (соединение 50);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,42 (s, 1H), 9,66 (s, 1H), 9,20 (s, 1H), 7,86-7,84 (m, 1H), 7,71 (s, 1H), 7,59 (s, 1H), 7,22 (d, J = 8 Гц, 1H), 6,85 (s, 1H), 6,76 (s, 1H), 3,93 (s, 3H), 3,80 (s, 3H), 3,38 (br, 3H), 3,07-3,01 (m, 3H), 2,37 (s, 3H), 1,96-1,82 (m, 4H) (соединение 52);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,55 (s, 1H), 9,66 (s, 1H), 9,22 (s, 1H), 7,78-7,76 (m, 2H), 7,72 (s, 1H), 7,24 (d, J = 8 Гц, 1H), 6,85 (s, 1H), 6,76 (s, 1H), 4,28 (s, 2H), 3,93 (s, 3H), 3,80 (s, 3H), 3,49-3,42 (m, 3H), 3,09 (t, J = 6 Гц, 2H) (соединение 53);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,52 (s, 1H), 9,65 (s, 1H), 9,21 (s, 1H), 7,77 (br, 1H), 7,71 (s, 1H), 7,62 (d, J = 16 Гц, 1H), 7,10 (t, J = 8 Гц, 1H), 6,86 (s, 1H), 6,76 (s, 1H), 3,93 (s, 3H), 3,82 (s, 3H), 3,35-3,17 (m, 4H), 2,34-2,28 (m, 2H), 1,22 (s, 1H), 1,02 (d, J = 8 Гц, 6H) (соединение 56);
1H ЯМР (DMSO-d6, 400 МГц) δ 10,73 (s, 1H), 9,83 (s, 1H), 9,26 (s, 1H), 8,02 (d, J = 8 Гц, 2H), 7,74 (s, 1H), 7,46 (d, J = 8 Гц, 2H), 6,87 (d, J = 4 Гц, 1H), 6,78 (d J = 4 Гц, 1H), 3,93 (s, 3H), 3,82 (s, 3H), 3,57 (br, 1H), 3,39-3,32 (m, 4H), 2,73-2,64 (m, 2H), 1,22 (s, 1H), 0,92 (d, J = 8 Гц, 6H) (соединение 59).
Пример 61. Получение соединения 61 (6-(2,6-дихлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
Путь A
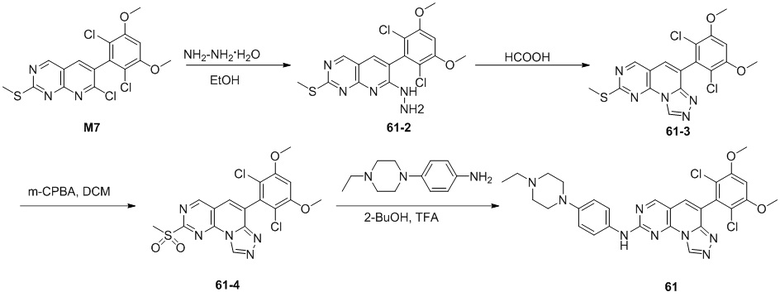
Стадия 1. Получение соединения 61-2
Соединение 61-2 получали с помощью процедуры, подобной описанной на стадии 1 пути A из примера 1.
LC-MS [M+H+] 412,0.
Стадия 2. Получение соединения 61-3
Соединение 61-3 получали с помощью процедуры, подобной описанной на стадии 2 пути A из примера 1.
LC-MS [M+H+] 422,0.
Стадия 3. Получение соединения 61-4
Соединение 61-4 получали с помощью процедуры, подобной описанной на стадии 3 пути A из примера 1.
LC-MS [M+H+] 454,0.
Стадия 4. Получение соединения 61
Соединение 61 получали с помощью процедуры, подобной описанной на стадии 5 из примера 2.
LC-MS [M+H+] 579,2.
1H ЯМР (DMSO-d6, 400 МГц) δ 9,67 (s, 1H), 9,17 (s, 1H), 7,80 (brs, 2H), 7,66 (s, 1H), 7,10 (s, 1H), 7,06 (d, J = 8 Гц, 2H), 4,00 (s, 6H), 3,23-2,76 (m, 8H), 2,76-2,59 (m, 2H), 1,18 (t, J = 6 Гц, 3H).
Путь B
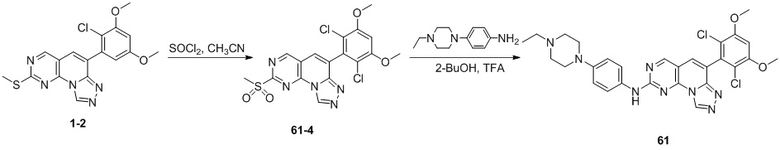
Стадия 1. Получение соединения 61-4
В атмосфере азота при 0°C добавляли 697 мг SO2Cl2 в смесь 500 мг соединения 1-2, 15 мл DCM и 5 мл NMP при перемешивании. Реакционную смесь перемешивали в течение 1 ч. при 0°C и гасили водой. Затем смесь экстрагировали с помощью DCM, промывали солевым раствором и высушивали над безводным Na2SO4. Смесь концентрировали при пониженном давлении с получением 700 мг неочищенного соединения 61-4. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 454,0.
Стадия 2. Получение соединения 61
Соединение 61 получали с помощью процедуры, подобной описанной на стадии 5 из примера 2.
LC-MS [M+H+] 579,2.
Путь C
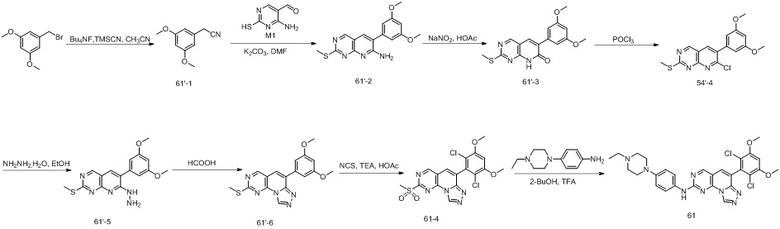
Стадия 1. Получение соединения 61’-1
40,0 г 3,5-диметоксибромбензила и 400 мл ацетонитрила охлаждали до 0°C при перемешивании и к полученному последовательно добавляли 34,3 г TMSCN и 346 мл 1 М Bu4NF-THF. После завершения добавления температуру смеси медленно повышали до 60°C и ее подвергали реакции в течение 1 ч. Смесь концентрировали при пониженном давлении после завершения реакции, остаток выливали в 500 мл ледяной воды и осаждали большое количество твердого вещества при перемешивании. Смесь фильтровали и твердое вещество промывали водой, высушивали с получением 30,9 г соединения 61’-1, которое представляло собой белое твердое вещество. Его применяли непосредственно на следующей стадии без очистки.
Стадия 2. Получение соединения 61’-2
Смесь 26,5 г соединения 61’-1 и 300 мл THF охлаждали до 0°C при перемешивании и к полученному порциями добавляли 6,4 г NaH (60% диспергированный в минеральном масле). Смесь подвергали реакции в течение 1,5 ч. при перемешивании при постоянной температуре. Порциями добавляли 18,1 г соединения M1, обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение ночи. Реакционную систему гасили путем добавления насыщенного водного раствора хлорида аммония и экстрагировали с помощью EA. Органическую фазу объединяли, промывали водой и высушивали. Смесь фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество высушивали в вакууме при 50°C в течение ночи с получением 24,7 г соединения 61'-2, которое представляло собой желтое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 329,1.
Стадия 3. Получение соединения 61’-3
К 31,2 г нитрита натрия порциями добавляли смесь 24,7 г соединения 61’-2 и 250 мл ледяной уксусной кислоты. После завершения добавления смесь медленно нагревали до 70°C и подвергали реакции в течение 5 ч. Смесь охлаждали до комнатной температуры и выливали в ледяную воду, затем осаждали большое количество твердого вещества. Смесь фильтровали и твердое вещество прополаскивали водой с получением нейтральной среды и высушивали в вакууме при 50°C в течение ночи с получением 24,6 г соединения 61’-3, которое представляло собой желтое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 330,1.
Стадия 4. Получение соединения 61’-4
Смесь 24,6 г соединения 61’-3, 250 мл ацетонитрила и 122,5 мл POCl3 нагревали до температуры возврата флегмы в течение 3 ч. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении с удалением большей части растворителя. Остаток гасили путем выливания в ледяную воду и экстрагировали с помощью EA. Органическую фазу объединяли, промывали водой и высушивали. Полученное фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество высушивали в течение ночи в вакууме при 50°C с получением 25,0 г соединения 61’-4, которое представляло собой светло-желтое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 348,1.
Стадия 5. Получение соединения 61’-5
Соединение 61’-5 получали с помощью процедуры, подобной описанной на стадии 1 пути A из примера 1.
LC-MS [M+H+] 344,1.
Стадия 6. Получение соединения 61’-6
Соединение 61’-6 получали с помощью процедуры, подобной описанной на стадии 1 пути A из примера 1.
LC-MS [M+H+] 354,1.
Стадия 7. Получение соединения 61-4
Порциями добавляли смесь 10,0 г соединения 61-4, 8,59 г TEA и 200 мл ледяной уксусной кислоты к 17,0 г NCS. Смесь подвергали реакции в течение 2 ч. при комнатной температуре. После завершения реакции смесь разбавляли с помощью EA и три раза промывали водой. Органическую фазу дважды промывали насыщенным водным раствором карбоната натрия, и дважды промывали водой, и высушивали. Фильтровали и концентрировали при пониженном давлении, полученное твердое вещество высушивали в течение ночи в вакууме при 50°C с получением 12,5 г соединения 61-4, которое представляло собой желтое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 454,0.
Стадия 8. Получение соединения 61
Соединение 61 получали с помощью процедуры, подобной стадии 5 из примера 2.
LC-MS [M+H+] 579,2.
Соединения из таблицы 3 получали с помощью процедуры, подобной описанной в примере 61, с применением других исходных материалов для реакции и подходящих реагентов.
Таблица 3
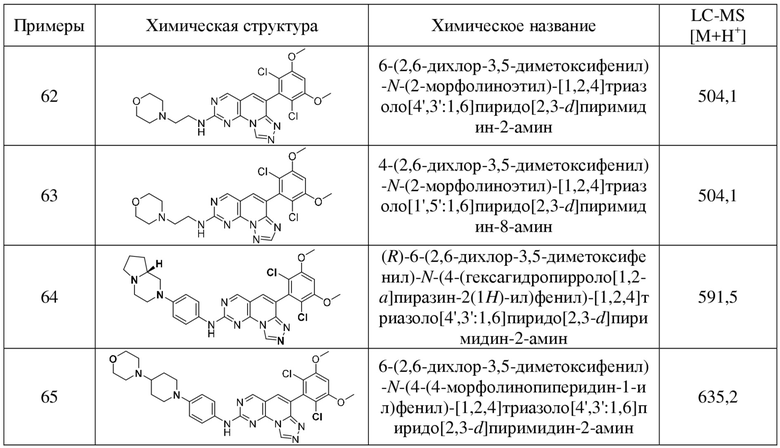
Данные ядерного магнитного резонанса для соединения 64 показаны ниже:
1H ЯМР (DMSO-d6, 400 МГц) δ 10,43 (s, 1H), 9,66 (s, 1H), 9,18 (s, 1H), 7,81 (br, 2H), 7,66 (s, 1H), 7,09 (s, 1H), 7,08-1,06 (m, 2H), 4,00 (s, 6H), 3,75-3,58 (m, 2H), 3,25-2,72 (m, 3H), 2,44-2,08 (m, 4H), 1,87-1,34 (m, 4H).
Пример 66. Получение соединения 66 (6-(2-хлор-3,5-диметоксифенил)-N2-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2,9-диамина)
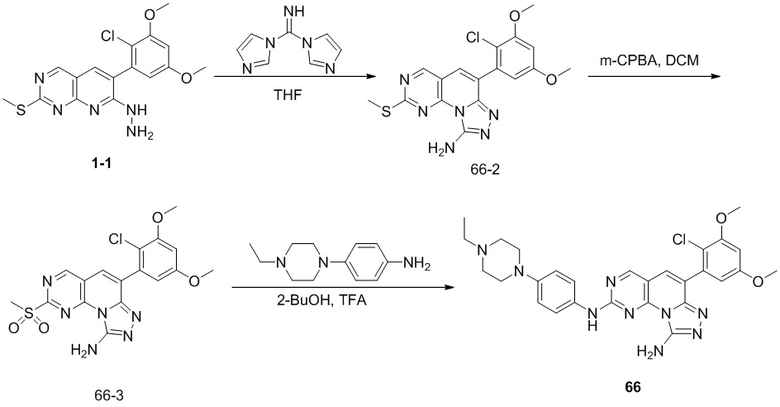
Стадия 1. Получение соединения 66-2
Смесь 100 мг соединения 1-1, 45 мг ди-(1H-имидазолил)имида и 10 мл THF нагревали до температуры возврата флегмы в течение 12 ч. Реакционную смесь разбавляли водой, экстрагировали с помощью EA, промывали солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/метанол=40/1) с получением 50 мг соединения 66-2, при этом выход составлял 47,0%, и соединение представляло собой бледно-желтое твердое вещество.
LC-MS [M+H+] 403,1.
Стадия 2. Получение соединения 66-3
Соединение 66-3 синтезировали с помощью процедуры, подобной описанной на стадии 4 из примера 2.
LC-MS [M+H+] 435,1.
Стадия 3. Получение соединения 66
Соединение 66 синтезировали с помощью процедуры, подобной описанной на стадии 5 из примера 2.
LC-MS [M+H+] 560,2.
Пример 67. Получение соединения 67 (6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
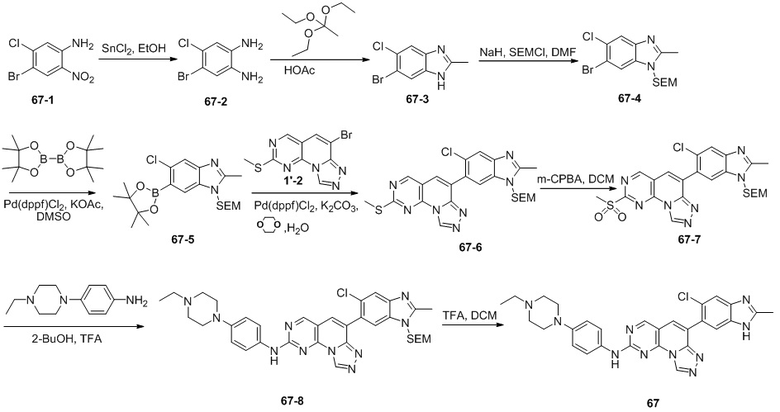
Стадия 1. Получение соединения 67-2
Смесь 5,23 г соединения 67-1, 18,77 г дихлорида олова и 125 мл EtOH подвергали реакции в течение 12 ч. при 70°C. Смесь концентрировали при пониженном давлении, остаток растворяли в воде и pH доводили до 8 с помощью насыщенного раствора NaHCO3. Смесь экстрагировали с помощью EA, промывали солевым раствором, высушивали над безводным Na2SO4 и концентрировали с получением 4,4 г соединения 67-2, которое представляло собой желтое твердое вещество.
LC-MS [M+H+] 220,9.
Стадия 2. Получение соединения 67-3
Смесь 4,4 г соединения 67-2, 50 мл уксусной кислоты и 4,83 г триэтилортоацетата перемешивали при 120°C в течение 1,5 ч. в герметичном контейнере, реакционный раствор концентрировали при пониженном давлении с получением 5,9 г соединения 67-3, которое представляло собой светло-желтое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 244,9.
Стадия 3. Получение соединения 67-4
В атмосфере азота при 0°C добавляли порциями 1,32 г NaH (60% в минеральном масле) в раствор 5,40 г соединения 67-3, растворенного в 100 мл THF, при перемешивании, реакционную смесь перемешивали в течение 1,5 ч. при 0°C. В реакционный раствор добавляли 7,33 г 2-(триметилсилил)этоксиметилхлорида при перемешивании при 0°C и реакционную смесь подвергали реакции в течение 12 ч. при к. т. Реакционную смесь гасили водой, экстрагировали с помощью EA, промывали солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 4,52 г неочищенного продукта, который представлял собой соединение 67-4, которое представляло собой серое твердое вещество.
LC-MS [M+H+] 375,0.
Стадия 4. Получение соединения 67-5
Смесь 1 г соединения 67-4, 811 мг пинаколового сложного эфира дибората, 195 мг Pd(dppf)Cl2, 1,0 г ацетата калия и 20 мл DMSO перемешивали в течение 3 ч. при 100°C в защитной атмосфере азота. Реакционную смесь охлаждали до к. т. и разбавляли водой. Смесь фильтровали и твердое вещество промывали с помощью смеси hex/EA (20/1). Органические фазы объединяли, высушивали над безводным Na2SO4 и концентрировали с получением 1 г неочищенного продукта, который представлял собой соединение 67-5, которое представляло собой светло-коричневое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 423,2.
Стадия 5. Получение соединения 67-6
Смесь 467 мг соединения 1’-2, 1 г соединения 67-5, 115 мг Pd(dppf)Cl2, 652 мг карбоната калия, 20 мл 1,4-диоксана и 2 мл воды перемешивали в течение 12 ч. при 100°C в защитной атмосфере азота. Реакционную смесь концентрировали при пониженном давлении, остаток очищали с помощью колонки с силикагелем (DCM/MeOH=20/1) с получением 650 мг соединения 67-6, которое представляло собой светло-коричневое твердое вещество, где выход составлял 80,5%.
LC-MS [M+H+] 512,1.
Стадия 6. Получение соединения 67-7
650 мг соединения 67-6 растворяли в 50 мл DCM, к полученному добавляли 329 мг м-хлорпероксибензойной кислоты. Реакционную смесь перемешивали в течение 1,5 ч. при к. т., затем концентрировали при пониженном давлении с получением 700 мг неочищенного продукта, который представлял собой соединение 67-7, которое представляло собой светло-желтое твердое вещество. Его применяли непосредственно при следующем получении без очистки.
LC-MS [M+H+] 544,1.
Стадия 7. Получение соединения 67-8
Смесь 700 мг соединения 67-7, 396 мг соединения M2, 50 мл 2-бутанола, 10 мл TFA перемешивали в течение 12 ч. при 120°C. Реакционный раствор концентрировали при пониженном давлении, остаток растворяли в 1 н. хлористоводородной кислоте. Смесь фильтровали, фильтрат экстрагировали с помощью EA и pH доводили до 8 с помощью насыщенного раствора NaHCO3. Реакционный раствор фильтровали и твердое вещество промывали водой, полученное твердое вещество высушивали в вакууме с получением 400 мг неочищенного продукта, который представлял собой соединение 67-8, которое представляло собой коричневое твердое вещество.
LC-MS [M+H+] 669,3.
Стадия 8. Получение соединения 67
Смесь 400 мг соединения 67-8, 40 мл DCM и 10 мл TFA перемешивали в течение 3 ч. при к. т., реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в воде и pH доводили до 8 с помощью насыщенного раствора NaHCO3. Смесь экстрагировали с помощью DCM, промывали солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=10/1) с получением 156 мг соединения 67, которое представляло собой желтое твердое вещество.
LC-MS [M+H+] 539,2.
Соединения из таблицы 4 получали с помощью процедуры, подобной описанной в примере 67, с применением других исходных материалов для реакции и подходящих реагентов.
Таблица 4
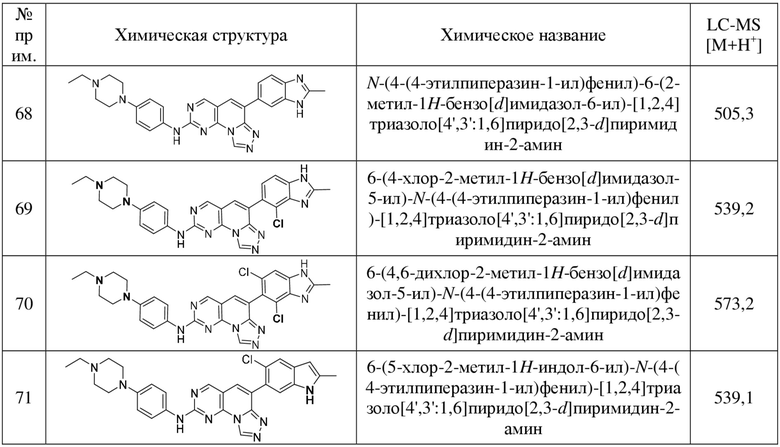
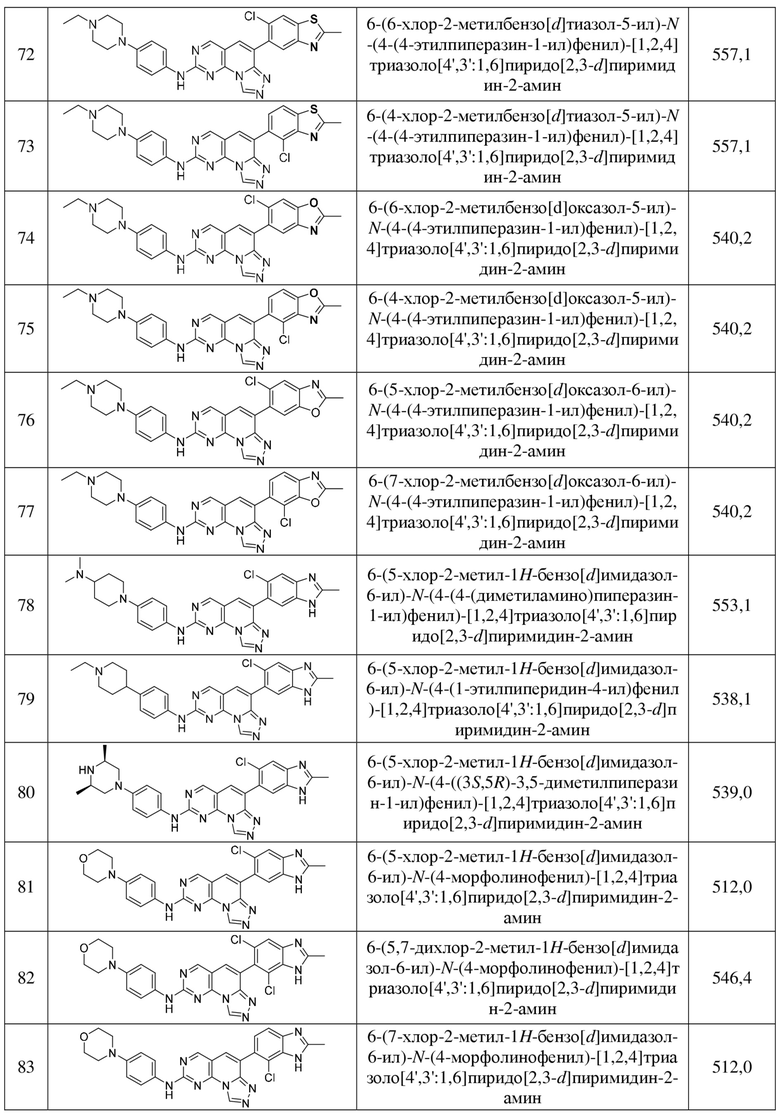

Данные ядерного магнитного резонанса для соединения 81 показаны ниже:
1H ЯМР (DMSO-d6, 400 МГц) δ 10,33 (s, 1H), 9,66 (s, 1H), 9,18 (s, 1H), 7,82-7,75 (m, 3H), 7,70-7,68 (m, 3H), 7,01 (d, J = 8 Гц, 2H), 3,76 (t, J = 4 Гц, 4H), 3,09 (t, J = 4 Гц, 4H), 2,55 (s, 3H).
Пример 85. Получение соединения 85 (6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-9-метил-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
Стадия 1. Получение соединения 85-2
Смесь 150 мг соединения 1-1, 5 мл н-бутилового спирта, 5 мл DMF и 65 мг триэтилортоацетата перемешивали в течение 3 ч. при 100°C. Реакционный раствор концентрировали при пониженном давлении и остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/MeOH=60/1) с получением 72 мг соединения 85-2 в виде желтого твердого вещества, при этом выход составлял 45,1%.
LC-MS [M+H+] 422,0.
Стадия 2. Получение соединения 85-3
Соединение 85-3 получали с помощью процедуры, подобной описанной на стадии 4 из примера 2.
LC-MS [M+H+] 454,0.
Стадия 3. Получение соединения 85
Соединение 85 получали с помощью процедуры, подобной описанной на стадии 5 из примера 2.
LC-MS [M+H+] 559,2.
1H ЯМР (DMSO-d6, 400 МГц) δ 9,07 (s, 1H), 7,56 (m, 2H), 7,56 (d, J = 11,8 Гц, 17H), 7,54 (s, 1H), 6,98 (d, 2H), 6,83 (s, 1H), 6,71 (s, 1H), 3,91 (s, 3H), 3,80 (s, 3H), 3,58 (s, 3H), 3,17 (m, 4H), 2,67 (m, 4H), 1,77 (q, 2H), 1,10 (t, 3H).
Пример 86. Получение соединения 86 (N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-((2-морфолиноэтил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)акриламида)
Стадия 1. Получение соединения 86-1
1,17 г 4-нитрофенилуксусной кислоты и 10 мл тионилхлорида нагревали до температуры возврата флегмы в течение 30 мин. Смесь концентрировали при пониженном давлении, добавляли 10 мл дихлорметана до растворения, снова концентрировали и операцию повторяли дважды с получением бесцветного масла, которое растворяли в 10 мл дихлорметана. Полученный раствор добавляли в раствор 2,03 г соединения 1-1, 1,63 г TEA и 40 мл дихлорметана при 0°C. После завершения добавления смесь подвергали реакции в течение ночи при перемешивании при комнатной температуре. Реакционную смесь гасили путем добавления воды и смесь экстрагировали дихлорметаном. Органическую фазу объединяли, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 3,42 г неочищенного продукта, который представлял собой соединение 86-1, которое представляло собой красновато-коричневое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 541,1.
Стадия 2. Получение соединения 86-2
Смесь 3,42 г соединения 86-1 и 20 мл POCl3 нагревали при 100°C в течение 3 ч. Смесь охлаждали до комнатной температуры и реакционную смесь гасили путем добавления в смесь льда и воды. Смесь экстрагировали дихлорметаном, органическую фазу объединяли, промывали последовательно насыщенным водным раствором NaHCO3 и водой и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH= от 100/1 до 50/1) с получением 1,31 г 86-2, которое представляло собой желтое твердое вещество.
LC-MS [M+H+] 523,1.
Стадия 3. Получение соединения 86-3
Систему суспензии 1,31 г соединения 86-2 и 39 мл дихлорметана охлаждали до 0°C и добавляли порциями в 0,76 г m-CPBA. Смесь подвергали реакции в течение 30 мин. при перемешивании при комнатной температуре и реакционную смесь гасили путем ее добавления в насыщенный водный раствор тиосульфата натрия. Смесь экстрагировали дихлорметаном, органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 1,25 г неочищенного продукта, который представлял собой соединение 86-3, которое представляло собой желтое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 555,1.
Стадия 4. Получение соединения 86-4
Смесь 600 мг соединения 86-3, 10 мл DMF, 432 мг DIEA и 217 мг N-(2-аминоэтил)морфолина нагревали в течение 2 ч. при 80°C. Смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном. Органическую фазу промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 450 мг соединения 86-4, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 605,2.
Стадия 5. Получение соединения 86-5
Смесь 450 мг соединения 86-4, 50 мл этанола, 10 мл воды и 1 мл концентрированной HCl добавляли в 463 мг SnCl2·2H2O. Полученную перемешанную систему нагревали в течение 2,5 ч. при 80°C. Прекращали нагревание, перемешанную систему охлаждали до комнатной температуры. Перемешанную систему разбавляли водой и pH доводили до 8~9 с помощью насыщенного водного раствора NaHCO3 и перемешанную систему экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 220 мг неочищенного продукта, который представлял собой соединение 86-5, которое представляло собой светло-желтое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 575,2.
Стадия 6. Получение соединения 86
Смесь 220 мг соединения 86-5, 30 мл дихлорметана и 116 мг TEA охлаждали до -5°C ~ 0°C и к полученному по каплям добавляли 52 мг акрилоилхлорида. После завершения добавления обеспечивали естественное повышение температуры и смесь подвергали реакции в течение 30 мин. при перемешивании. Смесь гасили водой, разбавляли дихлорметаном, промывали насыщенным водным раствором NaHCO3 и водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 155 мг соединения 86, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 629,1.
1H ЯМР (DMSO-d6, 400 МГц) δ 10,09-10,06 (m, 1H), 8,96 (s, 1H), 8,05-7,91 (m, 1H), 7,58-7,52 (m, 3H), 7,47-7,40 (m, 1H), 7,17-7,15 (m, 1H), 6,82 (s, 1H), 6,74-6,72 (m, 1H), 6,45-6,38 (m, 1H), 6,27-6,20 (m, 1H), 5,74-5,71 (m, 1H), 5,04 (s, 2H), 3,91 (s, 3H), 3,80 (s, 3H), 3,58-3,34 (m, 8H), 2,44-2,30 (m, 4H).
Соединения из таблицы 5 получали с помощью процедуры, подобной описанной в примерах 85 и 86, с применением других исходных материалов для реакции и подходящих реагентов.
Таблица 5
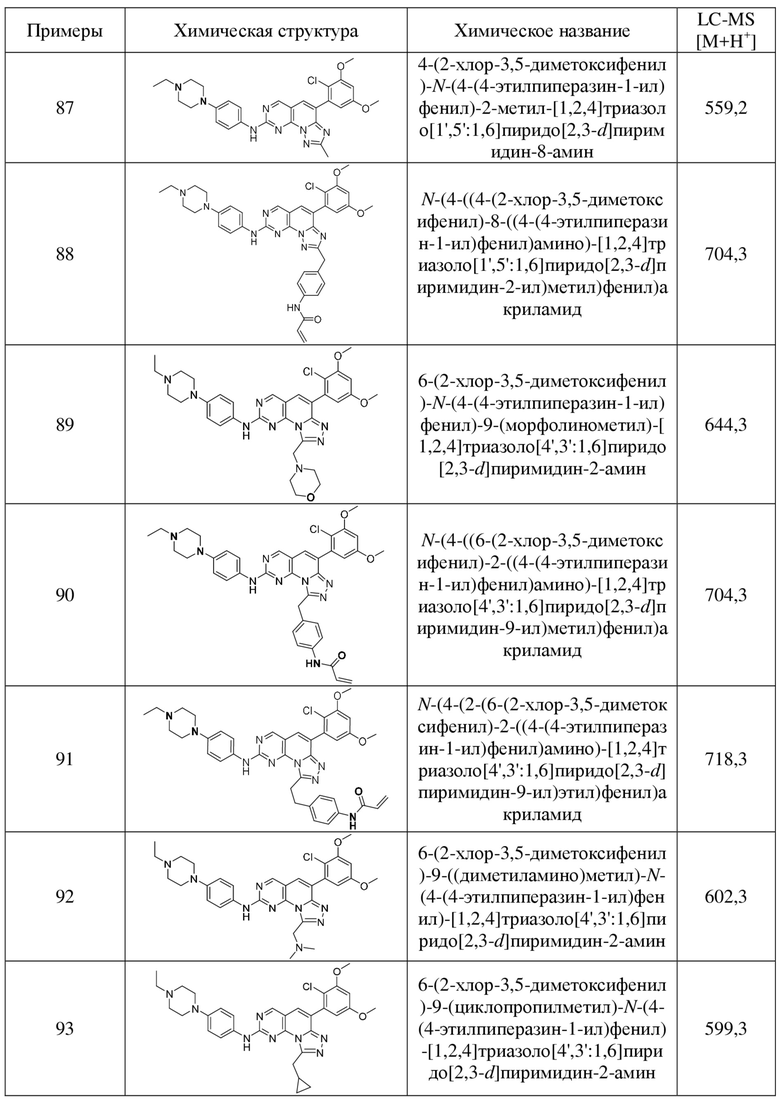
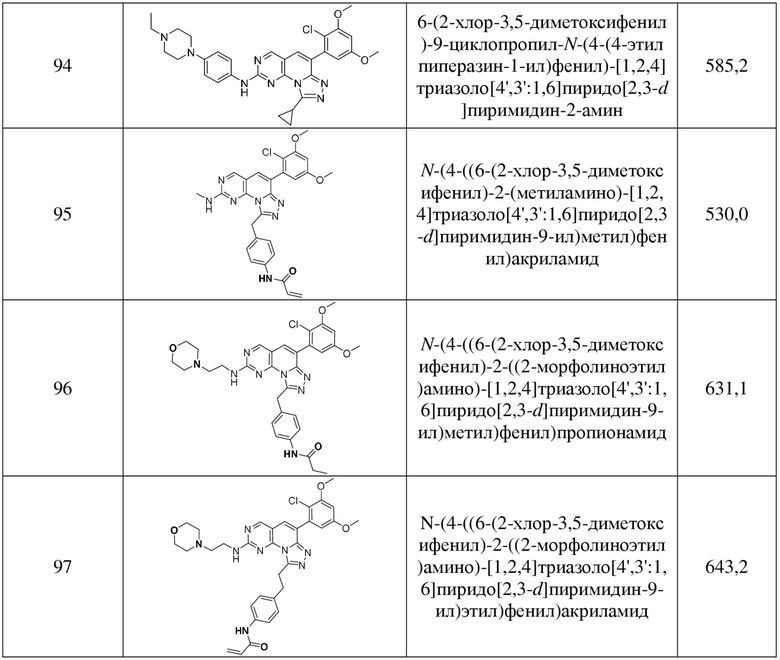
Данные ядерного магнитного резонанса для соединений 96 и 97 показаны ниже:
1H ЯМР (DMSO-d6, 400 МГц) δ 9,96 (s, 1H), 8,96 (s, 1H), 8,07-7,96 (m, 1H), 7,54-7,51 (m, 3H), 7,35-7,33 (m, 1H), 7,10 (d, J = 8 Гц, 1H), 6,82 (s, 1H), 6,73 (s, 1H), 5,01-4,91 (m, 2H), 3,90-3,79 (s, 3H), 3,79 (s, 3H), 3,57-3,49 (m, 4H), 3,34-3,32 (m, 2H), 2,51-2,49 (m, 2H), 2,43-2,39 (m, 2H), 2,31-2,30 (m, 4H), 1,04 (t, J = 6 Гц, 3H) (соединение 96).
1H ЯМР (DMSO-d6, 400 МГц) δ 10,08 (s, 1H), 8,96 (s, 1H), 8,16-8,07 (m, 1H), 7,60-7,58 (m, 2H), 7,50-7,48 (m, 1H), 7,23-7,21 (m, 2H), 7,07 (s, 1H), 6,49-6,40 (m, 1H), 6,27-6,22 (m, 1H), 5,74-5,72 (m, 1H), 3,99 (s, 3H), 3,90 (s, 3H), 3,56-3,43 (m, 6H), 3,33-3,18 (m, 4H), 2,47-2,27 (m, 4H) (соединение 97).
Пример 98. Получение соединения 98 (6-(2-хлор-3,5-диметоксифенил)-2-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9(8H)-она)
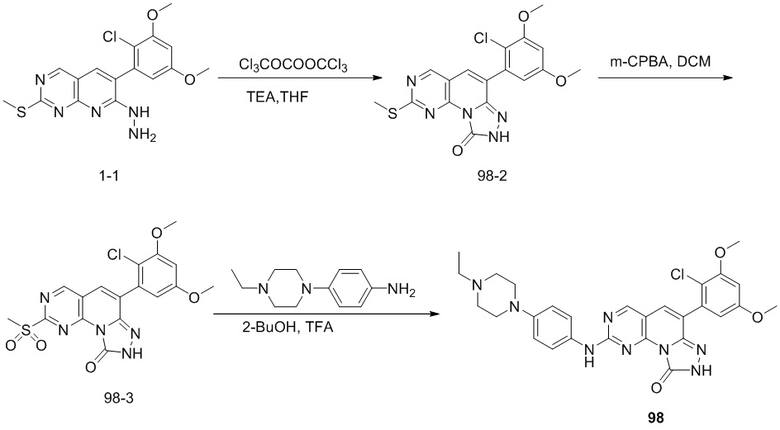
Стадия 1. Получение соединения 98-2
Смесь 100 мг соединения 1-1, 80 мг трифосгена, 5 мл THF и 0,11 мл TEA перемешивали в течение 1 ч. при 60°C. Реакционный раствор концентрировали в вакууме, остаток очищали посредством колоночной хроматографии на силикагеле (гексан/этилацетат=1/1) с получением 93 мг соединения 98-2 в виде светло-желтого твердого вещества. Выход составлял 87,0%.
LC-MS [M+H+] 404,1.
Стадия 2. Получение соединения 98-3
Способ является аналогичным стадии 4 из примера 2.
LC-MS [M+H+] 436,0.
Стадия 3. Получение соединения 98
Соединение 98 синтезировали с помощью процедуры, подобной описанной на стадии 5 из примера 2.
LC-MS [M+H+] 561,2.
Пример 99. Получение соединения 99 (4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-1-этилпиперазин-1-оксида)
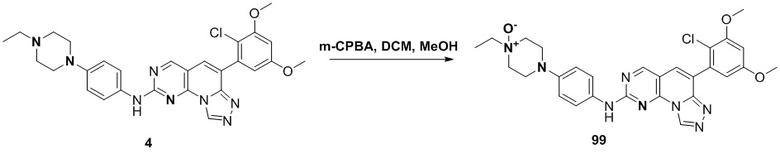
Растворяли 1,0 г соединения 4 в 30 мл дихлорметана и 3 мл метанола и охлаждали до 0°C. 0,47 г m-CPBA растворяли в 2 мл этилацетата и по каплям добавляли в указанный выше раствор. Обеспечивали естественное нагревание смеси при перемешивании в течение 30 мин, и реакция завершалась. Смесь гасили водой и экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором NaHCO3 и водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH= 20/1~5/1) с получением 410 мг соединения 99 в виде желтого твердого вещества.
LC-MS [M+H+] 561,2.
Пример 100. Получение соединения 100 ((8aR)-2-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)октагидро-5H-пирроло[1,2-a]пиразин-5-оксида)
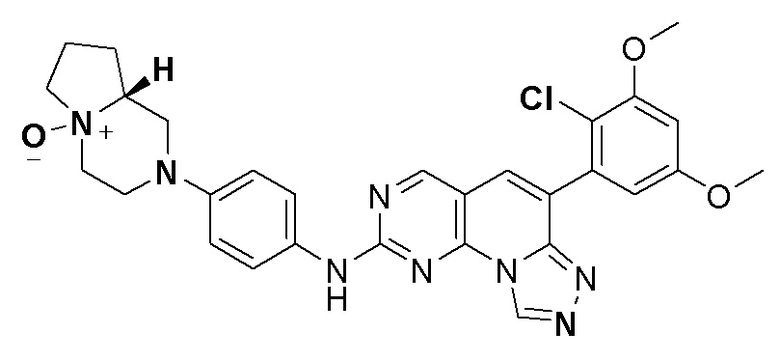
Соединение 100 синтезировали с помощью процедуры, подобной описанной в примере 99, с применением других исходных материалов и подходящего реагента.
LC-MS [M+H+] 573,2.
Пример 101. Получение соединения 101 (3-(4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)пиперазин-1-ил)циклобутан-1-ола)
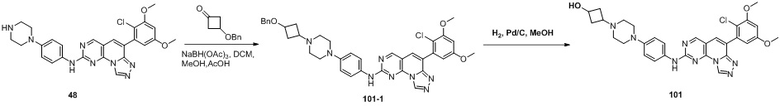
Стадия 1. Получение соединения 101-1
К смеси 500 мг соединения 48, 20 мл дихлорметана, 2 мл метанола, 0,5 мл уксусной кислоты добавляли 1,02 г триацетоксиборогидрида натрия. Смесь перемешивали в течение 3 ч. при комнатной температуре. После завершения реакции смесь разбавляли дихлорметаном, промывали насыщенным водным раствором NaHCO3 и водой, высушивали над безводным сульфатом натрия и высушивали в центробежной сушилке. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 550 мг соединения 101-1 в виде желтого твердого вещества.
LC-MS [M+H+] 677,3.
Стадия 2. Получение соединения 101-2
Смесь 550 мг соединения 101-1, 30 мл ледяной уксусной кислоты и Pd/C (100 мг) подвергали реакции в течение ночи в атмосфере водорода. Реакционную смесь фильтровали и концентрировали в вакууме. Твердое вещество разбавляли смесью DCM/MeOH=10/1, промывали насыщенным водным раствором NaHCO3 и водой, высушивали над безводным сульфатом натрия и высушивали в центробежной сушилке. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=10/1) с получением 120 мг соединения 101 в виде желтого твердого вещества.
LC-MS [M+H+] 587,2.
Пример 102. Получение соединения 102 (6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-этилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
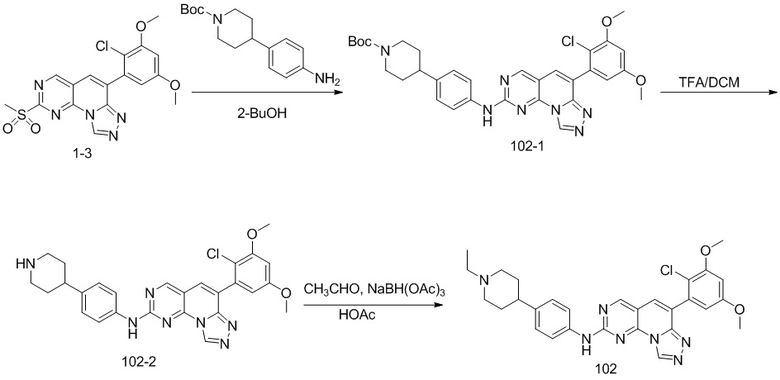
Стадия 1. Получение соединения 102-1
Смесь 2,57 г соединения 1-3, 1,82 г 4-(4-аминофенил)пиперидин-1-карбоксилата и 20 мл втор-бутилового спирта нагревали с обратным холодильником в течение 22 ч. Смесь охлаждали и фильтровали. Твердое вещество высушивали с получением 2,73 г соединения 102-1 в виде желтого твердого вещества.
LC-MS [M+H+] 616,2.
Стадия 2. Получение соединения 102-2
При температуре ниже 10°C добавляли 10 мл TFA в раствор 2,73 г соединения 102-1, растворенного в 100 мл дихлорметана. Смесь подвергали реакции в течение 2 ч. при комнатной температуре и концентрировали в вакууме. Твердое вещество растворяли в дихлорметане и pH доводили до 7-8 с помощью насыщенного водного раствора NaHCO3, затем жидкость отделяли. Органическую фазу промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Твердое вещество высушивали с получением 2,46 г соединения 102-2 в виде желтого твердого вещества. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 516,2.
Стадия 3. Получение соединения 102
Смесь 2,46 г соединения 102-2, 6,30 г паральдегида и 100 мл уксусной кислоты нагревали с обратным холодильником в течение 1 ч. и охлаждали до 25°C, к полученному добавляли 15,15 г триацетоксицианоборогидрида натрия. Смесь подвергали реакции в течение 3 ч. и гасили ледяной водой. Значение pH реакционной системы доводили до 7 с помощью 20% NaOH. Реакционную систему фильтровали, твердое вещество растворяли в 100 мл смеси DCM/MeOH=10/1, полученное промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=50/1-20/1) с получением 566 мг соединения 102 в виде желтого твердого вещества.
LC-MS [M+H+] 544,2.
1H ЯМР (DMSO-d6, 400 МГц) δ 10,47 (s, 1H), 9,71 (s, 1H), 9,18 (s, 1H), 7,85 (d, J = 8 Гц, 2H), 7,70 (s, 1H), 7,28 (d, J = 8 Гц, 2H), 6,86 (d, J = 4 Гц, 1H), 6,77 (d J = 4 Гц, 1H), 3,93 (s, 3H), 3,81 (s, 3H), 3,14-3,12 (m, 2H), 2,58-2,53 (m, 3H), 2,27-2,22 (m, 2H), 1,84-1,70 (m, 4H), 1,09 (t, J = 8 Гц, 3H).
Пример 103. Получение соединения 103 (6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(2,2,2-трифторфенил)пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
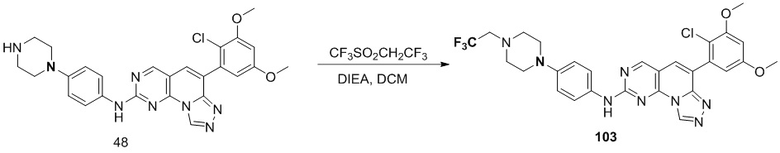
К смеси 50 мг соединения 48, 10 мл дихлорметана и 14 мг DIEA добавляли по каплям 25 мг трифторэтилтрифторметансульфоната. Реакционную систему нагревали с обратным холодильником в течение 3,5 ч. и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 18 мг соединения 103 в виде желтого твердого вещества.
LC-MS [M+H+] 599,2.
Пример 104. Получение соединения 104 (6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклопентилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
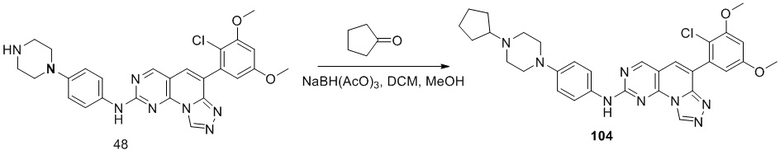
К смеси 100 мг соединения 48, 20 мл дихлорметана, 2 мл метанола и 1 мл циклопентанона порциями добавляли 122 мг триацетоксиборогидрида натрия. Смесь подвергали реакции в течение ночи при перемешивании при комнатной температуре. Реакционную смесь гасили водой и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=10/1) с получением 70 мг соединения 104 в виде желтого твердого вещества.
LC-MS [M+H+] 585,2.
Пример 105. Получение соединения 105 (6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклобутилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
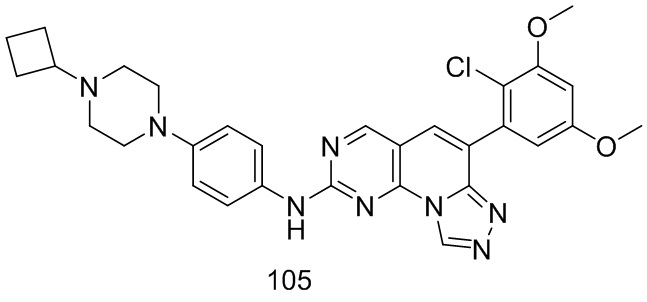
Соединение 105 синтезировали с помощью способа, подобного примеру 104, с применением других исходных материалов и подходящего реагента.
LC-MS [M+H+] 571,2.
Пример 106. Получение соединения 106 (6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-ацетилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
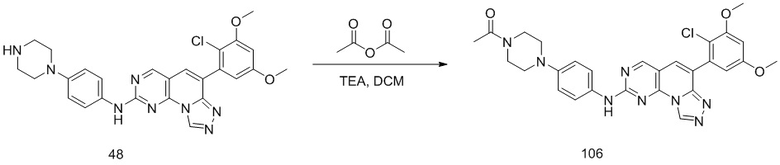
Смесь 100 мг соединения 48, 0,08 мл TEA и 20 мл дихлорметана охлаждали до 0°C. К смеси по каплям добавляли 24 мг уксусного ангидрида и обеспечивали ее естественное нагревание до комнатной температуры. Смесь подвергали реакции в течение 30 мин. при перемешивании, и реакция завершалась. Реакционную смесь гасили метанолом и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=12/1) с получением 45 мг соединения 106 в виде желтого твердого вещества.
LC-MS [M+H+] 559,2.
Пример 107. Получение соединения 107 (4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-N,N-диметилпиперазин-1-карбоксамида)
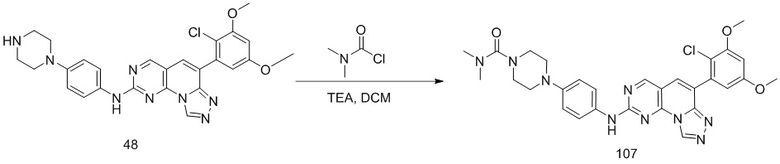
Смесь 500 мг соединения 48, 30 мл дихлорметана и 294 мг TEA охлаждали до 0°C и к смеси по каплям добавляли 156 мг диметилкарбамоилхлорида. Обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение 1 ч. при перемешивании. Реакционную смесь гасили водой и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 325 мг соединения 107 в виде желтого твердого вещества.
LC-MS [M+H+] 588,2.
Пример 108. Получение соединения 108 (6-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-N,N-диметил-2,6-диазаспиро[3.3]гептан-2-карбоксамид)
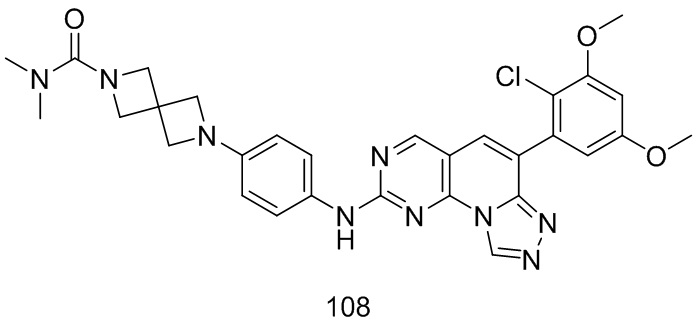
Соединение 108 синтезировали с помощью способа, подобного примеру 48 и примеру 107, с применением других исходных материалов и подходящего реагента.
LC-MS [M+H+] 600,2.
Пример 109. Получение соединения 109 ((S)-3-((4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)амино)-N,N-диметилпирролидин-1-карбоксамида)
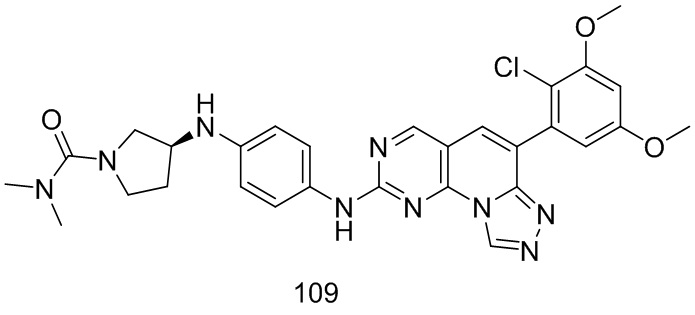
Соединение 109 синтезировали с помощью способа, подобного примеру 48 и примеру 107, с применением других исходных материалов и подходящего реагента.
LC-MS [M+H+] 588,1.
1H ЯМР (DMSO-d6, 400 МГц) δ 10,18 (s, 1H), 9,60 (s, 1H), 9,11 (s, 1H), 7,65-7,62 (m, 3H), 6,85 (s, 1H), 6,75 (s, 1H), 6,68-6,66 (m, 2H), 5,72 (d, J = 8 Гц, 1H), 3,92 (s, 3H), 3,79 (s, 3H), 3,65-3,61 (m, 2H), 3,15-3,11 (m, 2H), 2,73 (s, 6H), 2,12-2,07 (m, 1H), 1,80-1,77 (m, 1H).
Пример 110. Получение соединения 110 (6-(2-хлор-3,5-диметоксифенил)-N-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
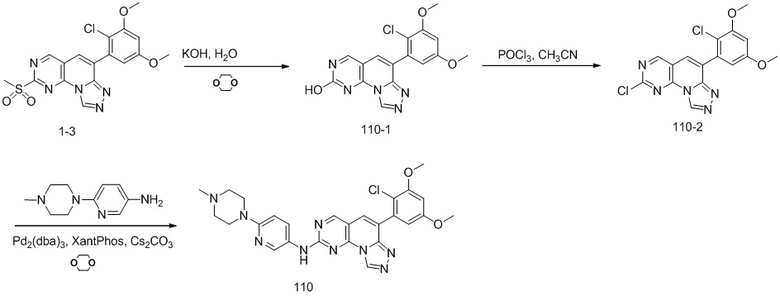
Стадия 1. Получение соединения 110-1
Смесь 10,01 г соединения 1-3, 100 мл воды, 25 мл диоксана и 6,68 г гидроксида калия перемешивали при 80°C и подвергали реакции в течение ночи. После завершения реакции смесь охлаждали до комнатной температуры, значение pH доводили до 3 с применением концентрированной хлористоводородной кислоты и большое количество твердого вещества отделяли. В результате фильтрования получали твердое вещество, его промывали водой и высушивали над безводным сульфатом натрия с получением 7,27 г соединения 110-1 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 358,1.
Стадия 2. Получение соединения 110-2
Смесь 7,27 г соединения 110-1, 120 мл ацетонитрила и 35 мл оксихлорида фосфора нагревали до температуры возврата флегмы и подвергали реакции в течение ночи. После завершения реакции смесь охлаждали до комнатной температуры. Реакционную смесь гасили путем добавления ледяной воды с последующим фильтрованием, твердое вещество три раза промывали водой и высушивали в течение ночи в вакууме при 50°C с получением 6,73 г соединения 110-2 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 376,0.
Стадия 3. Получение соединения 110
Смесь 240 мг соединения 110-2, 130 мг 5-амино-2-(4-метил-1-пиперазин)пиридина, 14 мг Pd(OAc)2, 7 мг XantPhos, 626 мг карбоната цезия и 15 мл диоксана подвергали реакции в течение 4 ч. при 100°C в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток растворяли с применением дихлорметана, полученное промывали водой и высушивали над безводным сульфатом натрия. Смесь фильтровали и фильтрат концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=5/1) с получением 120 мг соединения 110 в виде желтого твердого вещества.
LC-MS [M+H+] 532,2.
Пример 111. Получение соединения 111 (6-(2-хлор-3,5-диметоксифенил)-N-(6-((4-этилпиперазин-1-ил)метил)пиридин-3-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амина)
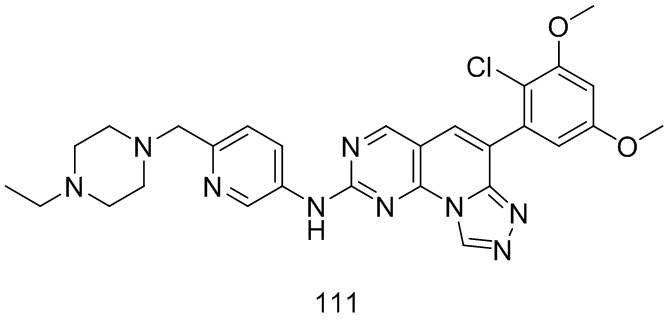
Соединение 111 синтезировали с помощью процедуры, подобной описанной в примере 110, с применением других исходных материалов и подходящего реагента.
LC-MS [M+H+] 560,2.
Пример 112. Получение соединения 112 (N-(4-((4-(2-хлор-3,5-диметоксифенил)-8-((2-морфолиноэтил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)фенил)акриламида)
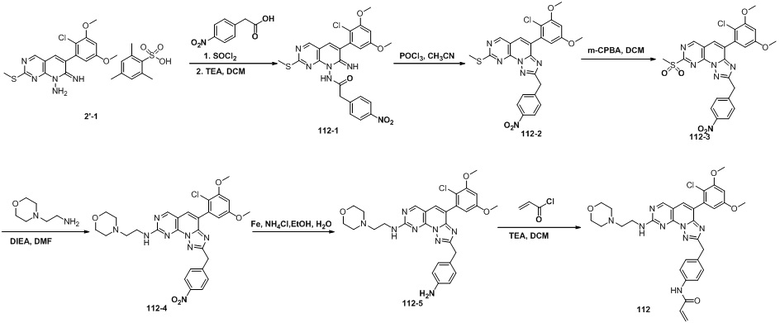
Стадия 1. Получение соединения 112-1
Смесь 575 мг 4-нитрофенилуксусной кислоты и 10 мл тионилхлорида нагревали до температуры возврата флегмы в течение 1 ч., удаляли тионилхлорид путем концентрирования при пониженном давлении, растворяли путем добавления 10 мл дихлорметана и концентрировали при пониженном давлении. Указанное действие повторяли дважды и концентрировали при пониженном давлении в течение 15 мин. Полученное маслянистое вещество растворяли в 3 мл дихлорметана, затем смесь по каплям добавляли в перемешанный раствор 800 мг соединения 2’-1, 20 мл дихлорметана и 643 мг TEA. После завершения добавления обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение ночи при перемешивании. После завершения реакции реакционную смесь гасили путем добавления воды, смесь экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 1,15 г неочищенного продукта 112-1 в виде красновато-коричневого твердого вещества. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 541,1.
Стадия 2. Получение соединения 112-2
Смесь 1,15 г соединения 112-1, 20 мл ацетонитрила и 2 мл оксихлорида фосфора нагревали до температуры возврата флегмы в течение 2 ч. и удаляли большую часть растворителей путем концентрирования при пониженном давлении. Остаток растворяли в дихлорметане, дважды промывали насыщенным водным раствором бикарбоната натрия и водой соответственно, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (Hex/EA=5/1) с получением 380 мг соединения 112-2 в виде светло-коричневого твердого вещества.
LC-MS [M+H+] 523,1.
Стадия 3. Получение соединения 112-3
380 мг соединения 112-2 в растворяли 10 мл дихлорметана, смесь охлаждали до 0°C и к полученному порциями добавляли 221 мг m-CPBA, обеспечивали естественное нагревание смеси и ее подвергали реакции в течение 1 ч. при перемешивании. Реакционную смесь гасили путем добавления насыщенного водного раствора бикарбоната натрия, экстрагировали дихлорметаном, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 400 мг неочищенного продукта 112-3 в виде желтого твердого вещества. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 555,1.
Стадия 4. Получение соединения 112-4
Смесь 400 мг соединения 112-3, 10 мл DMF, 273 мг DIEA и 138 мг N-(2-аминоэтил)морфолина подвергали реакции в течение 3,5 ч. при 80°C. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и высушивали в центробежной сушилке. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 150 мг соединения 112-4 в виде красновато-коричневого твердого вещества.
LC-MS [M+H+] 605,2.
Стадия 5. Получение соединения 112-5
Смесь 150 мг соединения 112-4, 20 мл этанола, 4 мл воды и 154 мг SnCl2·2H2O подвергали реакции в течение 2,5 ч. при 80°C. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали. Фильтрат разбавляли водой, экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 20 мг соединения 112-5 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 575,2.
Стадия 6. Получение соединения 112
Смесь 20 мг соединения 112-5, 10 мл дихлорметана и 11 мг TEA охлаждали до 0°C, к полученному по каплям добавляли 5 мг акрилоилхлорида. Обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение 1 ч. при перемешивании. Реакционную смесь гасили водой и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 15 мг соединения 112 в виде белого твердого вещества с выходом 68,6%.
LC-MS [M+H+] 629,2.
Пример 113. Получение соединения 113 (1-(4-((6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)пропил)пиперазин-1-ил)проп-2-ен-1-она)
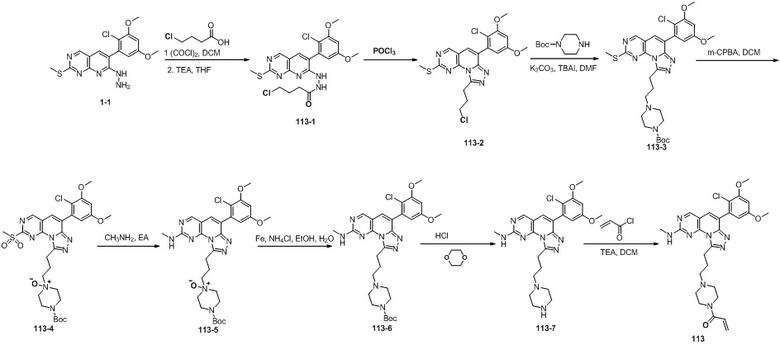
Стадия 1. Получение соединения 113-1
Добавляли каплю DMF в смесь 195 мг хлормасляной кислоты и 10 мл дихлорметана, смесь охлаждали до 0°C при перемешивании и к полученному добавляли 252 мг оксалилхлорида. После завершения добавления обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение 1 ч. при перемешивании. Реакционную систему концентрировали при пониженном давлении и полученное масло растворяли в 1 мл дихлорметана. Полученный выше раствор по каплям добавляли в смесь 500 мг соединения 1-1, 670 мг TEA и 30 мл THF при 0°C. После завершения добавления обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение 1 ч. при перемешивании. Реакционную смесь гасили путем добавления воды и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 700 мг соединения 113-1 в виде желтого твердого вещества. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 482,1.
Стадия 2. Получение соединения 113-2
Смесь 700 мг соединения 113-1 и 10 мл POCl3 подвергали реакции в течение 2 ч. при 100°C. После завершения реакции смесь охлаждали до комнатной температуры и гасили путем выливания в ледяную воду. Большое количество твердого вещества осаждали из смеси при перемешивании, его фильтровали, затем твердое вещество промывали водой и высушивали с получением 540 мг соединения 113-2 в виде белого твердого вещества. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 464,1.
Стадия 3. Получение соединения 113-3
Смесь 540 мг соединения 113-2, 482 мг карбоната калия, 433 мг 1-Boc-пиперазина, 43 мг иодида тетрабутиламмония и 30 мл DMF подвергали реакции в течение ночи при 80°C. Смесь разбавляли водой, экстрагировали дихлорметаном, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 470 мг соединения 113-3 в виде красновато-коричневого твердого вещества.
LC-MS [M+H+] 614,2.
Стадия 4. Получение соединения 113-4
Смесь 470 мг соединения 113-3 и 10 мл дихлорметана охлаждали до 0°C и к полученному порциями добавляли 461 мг m-CPBA. Обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение 1 ч. Реакционную смесь гасили путем добавления воды и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 650 мг соединения 113-4 в виде желтого твердого вещества. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 662,2.
Стадия 5. Получение соединения 113-5
Смесь 650 мг соединения 113-4, 2,5 мл раствора метиламина в тетрагидрофуране (2,0 М) и 10 мл этилацетата подвергали реакции в течение ночи при комнатной температуре. После завершения реакции смесь разбавляли дихлорметаном, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 340 мг соединения 113-5 в виде коричневого твердого вещества. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 613,3.
Стадия 6. Получение соединения 113-6
Смесь 340 мг соединения 113-5, 297 мг NH4Cl, 155 мг порошка железа, 10 мл безводного этанола и 3 мл воды подвергали реакции в течение 1 ч. при 80°C. Смесь охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в дихлорметане, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=25/1) с получением 50 мг соединения 113-6 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 597,3.
Стадия 7. Получение соединения 113-7
50 мг соединения 113-6 растворяли в 3 мл 1,4-диоксана и к полученному добавляли 3 мл 4 М раствора HCl в 1,4-диоксане при перемешивании. Смесь подвергали реакции в течение 30 мин. при комнатной температуре при перемешивании, концентрировали при пониженном давлении и высушивали в вакууме с получением 51 мг соединения 113-7 в виде светло-желтого твердого вещества. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 497,2.
Стадия 8. Получение соединения 113
Смесь 51 мг соединения 113-7, 52 мг TEA и 10 мл дихлорметана охлаждали до 0°C, к полученному добавляли раствор 9 мг акрилоилхлорида в 0,5 мл дихлорметана при перемешивании. После завершения добавления смесь подвергали реакции в течение 1 ч. при 0°C. Реакционную смесь гасили путем добавления воды, экстрагировали дихлорметаном, промывали насыщенным водным раствором бикарбоната натрия, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 38 мг соединения 113 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 551,2.
Пример 114. Получение соединения 114 (1-(4-((6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)этил)пиперазин-1-ил)проп-2-ен-1-она)
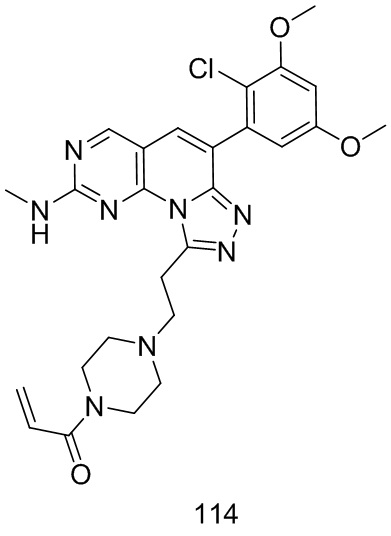
Соединение 114 получали с помощью процедуры, подобной описанной в примере 113, с применением других исходных материалов для реакции и подходящего реагента.
LC-MS [M+H+] 537,2.
Пример 115. Получение соединения 115 (1-(4-(3-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-она)
Путь A
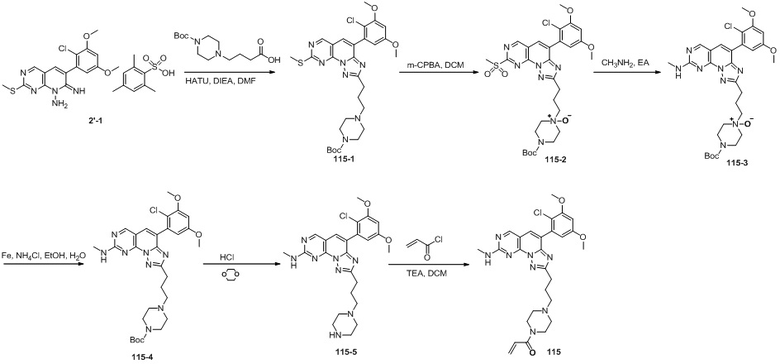
Стадия 1. Получение соединения 115-1
5,0 г соединения 2’-1, 2,24 г DIEA, 4,7 г 4-(4-Boc-пиперазин-1-ил)масляной кислоты, 6,58 г HATU и 60 мл DMF подвергали реакции в течение 30 мин. при комнатной температуре при перемешивании, затем смесь нагревали до 100°C и подвергали реакции в течение 2 ч. Смесь охлаждали до комнатной температуры, гасили путем добавления воды и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH= 0%~5%) с получением 3,4 г соединения 115-1 в виде красновато-коричневого масла.
LC-MS [M+H+] 614,2.
Стадия 2. Получение соединения 115-2
Применяя соединение 115-1 в качестве исходного материала, соединение 115-2 синтезировали с помощью процедуры, подобной описанной на стадии 4 из примера 112.
LC-MS [M+H+] 662,2.
Стадия 3. Получение соединения 115-3
Применяя соединение 115-2 в качестве исходного материала, соединение 115-3 синтезировали с помощью процедуры, подобной описанной на стадии 5 из примера 112.
LC-MS [M+H+] 613,3.
Стадия 4. Получение соединения 115-4
Применяя соединение 115-3 в качестве исходного материала, соединение 115-4 синтезировали с помощью процедуры, подобной описанной на стадии 6 из примера 112.
LC-MS [M+H+] 597,3.
Стадия 5. Получение соединения 115-5
Применяя соединение 115-4 в качестве исходного материала, соединение 115-5 синтезировали с помощью процедуры, подобной описанной на стадии 7 из примера 112.
LC-MS [M+H+] 497,2.
Стадия 6. Получение соединения 115
Применяя соединение 115-5 в качестве исходного материала, соединение 115 синтезировали с помощью процедуры, подобной описанной на стадии 7 из примера 115.
LC-MS [M+H+] 551,2.
Путь B
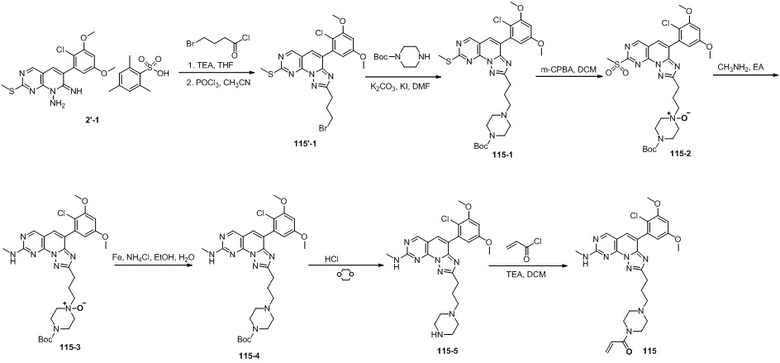
Стадия 1. Получение соединения 115’-1
Смесь 14,0 г соединения 2’-1, 8,6 г TEA и 150 мл THF охлаждали до 0°C при перемешивании, к полученному добавляли 6,7 г 4-бромбутирилхлорида, после завершения добавления обеспечивали естественное нагревание смеси до комнатной температуры. После завершения реакции реакционную смесь гасили путем добавления воды и смесь экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт растворяли в 240 мл ацетонитрила, добавляли 11 мл POCl3 и подвергали реакции в течение ночи при 80°C. После завершения реакции смесь охлаждали до комнатной температуры, реакционную систему гасили путем добавления ледяной воды и экстрагировали дихлорметаном. Органическую фазу объединяли, дважды промывали водой, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором соответственно, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/EA=10/1) с получением 5,3 г соединения 115’-1 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 508,0.
Стадия 2. Получение соединения 115-1
Смесь 100 мг соединения 115’-1, 41 мг 1-Boc-пиперазина, 83 мг карбоната калия, 4 мг иодида калия и 5 мл DMF подвергали реакции в течение ночи при 80°C. После завершения реакции смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 85 мг соединения 115-1 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 614,2.
Стадия 3. Получение соединения 115-2
Применяя соединение 115-1 в качестве исходного материала, соединение 115-2 синтезировали с помощью процедуры, подобной описанной на стадии 2 пути A из примера 115.
LC-MS [M+H+] 662,2.
Стадия 4. Получение соединения 115-3
Применяя соединение 115-2 в качестве исходного материала, соединение 115-3 синтезировали с помощью процедуры, подобной стадии 3 пути A из примера 115.
LC-MS [M+H+] 613,3.
Стадия 5. Получение соединения 115-4
Применяя соединение 115-3 в качестве исходного материала, соединение 115-4 синтезировали с помощью процедуры, подобной стадии 4 пути A из примера 115.
LC-MS [M+H+] 597,3.
Стадия 6. Получение соединения 115-5
Применяя соединение 115-4 в качестве исходного материала, соединение 115-5 синтезировали с помощью процедуры, подобной стадии 5 пути A из примера 115.
LC-MS [M+H+] 497,2.
Стадия 7. Получение соединения 115
Применяя соединение 115-5 в качестве исходного материала, соединение 115 синтезировали с помощью процедуры, подобной стадии 6 пути A из примера 115.
LC-MS [M+H+] 551,2.
Соединения из таблицы 6 получали с помощью процедуры, подобной описанной в примере 115, с применением других исходных материалов для реакции и соответствующего реагента.
Таблица 6
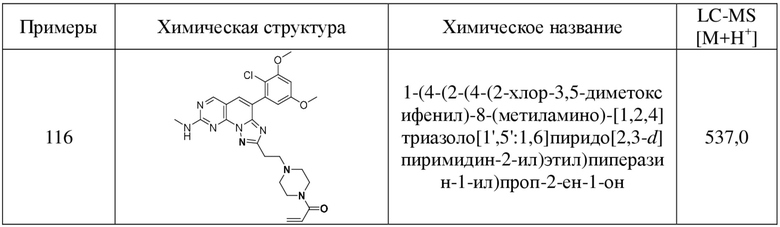

Пример 120. Получение соединения 120 (N-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)фенил)акриламида)
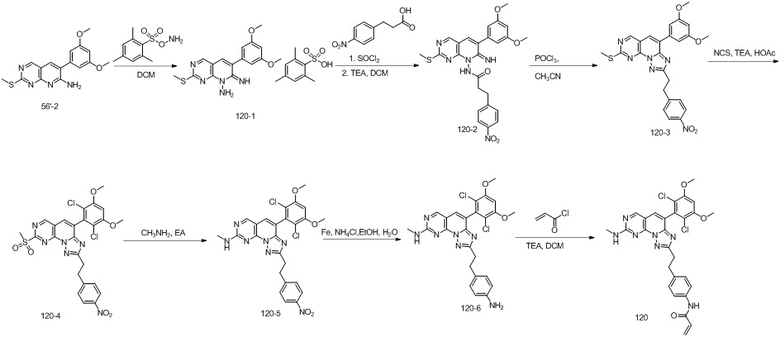
Стадия 1. Получение соединения 120-1
31,2 г 2-[(аминоокси)сульфонил]-1,3,5-триметилбензола диспергировали в 900 мл дихлорметана и охлаждали до 0°C. Добавляли порциями 23,8 г соединения 2-1 при перемешивании. Обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение ночи. После завершения реакции смесь фильтровали, твердое вещество дважды прополаскивали с помощью небольшого количества DCM и высушивали, затем получали 34,9 г соединения 120-1, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 344,1.
Стадия 2. Получение соединения 120-2
Смесь 162 мг 4-нитрофенилпропионовой кислоты и 5 мл тионилхлорида нагревали с обратным холодильником в течение 1 ч. Смесь концентрировали при пониженном давлении с удалением тионилхлорида, затем добавляли 10 мл дихлорметана, смесь концентрировали при пониженном давлении и операцию повторяли дважды. Смесь концентрировали при пониженном давлении в вакууме в течение 15 мин. Полученное масло растворяли в 2 мл дихлорметана и по каплям добавляли к смеси 300 мг соединения 120-1, 15 мл дихлорметана и 0,38 мл TEA при 0°C. После завершения добавления обеспечивали естественное нагревание смеси до комнатной температуры и ее подвергали реакции в течение ночи при перемешивании. После завершения реакции реакционную смесь гасили путем добавления воды и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 300 мг соединения 120-2 в виде красновато-коричневого твердого вещества. Его применяли непосредственно на следующей стадии без очистки.
LC-MS [M+H+] 521,2.
Стадия 3. Получение соединения 120-3
Смесь 300 мг соединения 120-2, 1 мл оксихлорида фосфора и 10 мл ацетонитрила нагревали с обратным холодильником в течение 1 ч. при 80°C, реакционную смесь суспендировали и смесь охлаждали до комнатной температуры. Реакционную смесь гасили ледяной водой, экстрагировали дихлорметаном, промывали насыщенным солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/метанол=25/1) с получением 277 мг соединения 120-3 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 503,1.
Стадия 4. Получение соединения 120-4
368 мг NCS порциями добавляли в смесь 277 мг соединения 120-3, 0,23 мл TEA и 4 мл уксусной кислоты, смесь перемешивали в течение 1 ч. при комнатной температуре. После завершения смесь разбавляли этилацетатом, дважды промывали водой, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором соответственно, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 332 мг соединения 120-4 в виде желтого твердого вещества. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 603,0.
Стадия 5. Получение соединения 120-5
Смесь 332 мг соединения 120-4, 2,75 мл раствора метиламина в тетрагидрофуране и 10 мл этилацетата перемешивали в течение 1 ч. при комнатной температуре. Реакционную смесь гасили водой, экстрагировали этилацетатом, промывали водой, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/метанол =25/1) с получением 305 мг соединения 120-4 в виде желтого твердого вещества.
LC-MS [M+H+] 554,1.
Стадия 6. Получение соединения 120-6
Смесь 305 мг соединения 120-4, 50 мг Pd/C и 40 мл метанола подвергали реакции в течение 1 ч. при перемешивании при комнатной температуре в атмосфере водорода. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением 99 мг соединения 120-5 в виде светло-желтого твердого вещества. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 524,1.
Стадия 7. Получение соединения 120
26 мг акрилоилхлорида по каплям добавляли к смеси 99 мг соединения 120-5, 0,08 мл триэтиламина и 10 мл дихлорметана при перемешивании при 0°C, после завершения добавления смесь подвергали реакции в течение 1 ч. при 0°C. Реакционную смесь гасили насыщенным водным раствором бикарбоната натрия и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали водой, высушивали с помощью безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (дихлорметан/метанол =25/1) с получением 25 мг соединения 120 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 578,1.
Пример 121. Получение соединения 121 (N-(3-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)фенил)акриламида)
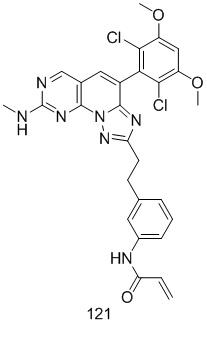
Соединение 121 получали с помощью процедуры, подобной примеру 120, с применением другого исходного материала и подходящего реагента.
LC-MS [M+H+] 578,1.
Пример 122. Получение соединения 122 (1-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-она)
Путь A
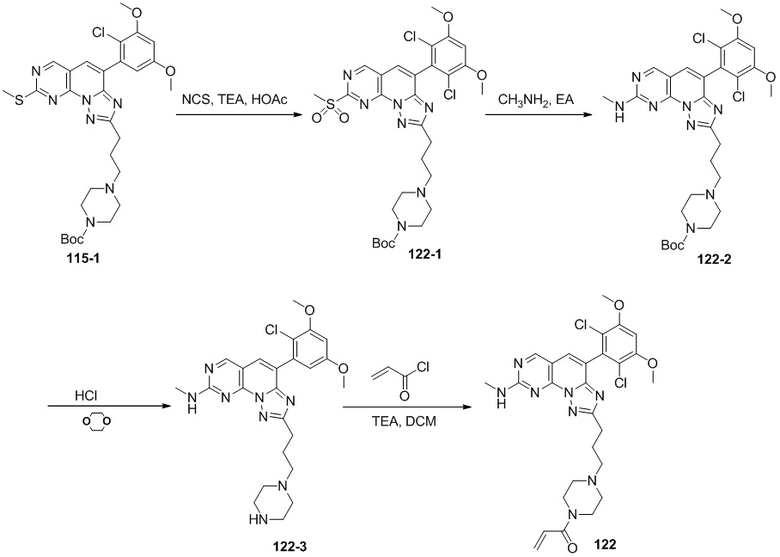
Стадия 1. Получение соединения 122-1
Смесь 176 мг соединения 122-1, 67 мг TEA, 134 мг NCS и 10 мл ледяной уксусной кислоты подвергали реакции в течение 1 ч. при перемешивании при комнатной температуре. Смесь разбавляли этилацетатом, дважды промывали водой, насыщенным водным раствором карбоната натрия и насыщенным солевым раствором соответственно, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 180 мг соединения 122-1 в виде светло-желтого твердого вещества. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 680,2.
Стадия 2. Получение соединения 122-2
Смесь 180 мг соединения 122-1, 1,3 мл раствора метиламина в тетрагидрофуране и 10 мл этилацетата подвергали реакции в течение 1 ч. при перемешивании при комнатной температуре. Смесь разбавляли этилацетатом, промывали водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH= 0%~5%) с получением 140 мг соединения 122-2 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 631,2.
Стадия 3. Получение соединения 122-3
Применяя соединение 122-2 в качестве исходного материала, соединение 122-3 синтезировали с помощью процедуры, подобной стадии 5 пути A из примера 122.
LC-MS [M+H+] 497,2.
Стадия 4. Получение соединения 122
Применяя соединение 122-3 в качестве исходного материала, соединение 122 синтезировали с помощью процедуры, подобной стадии 6 пути A из примера 122.
LC-MS [M+H+] 585,2.
Путь B
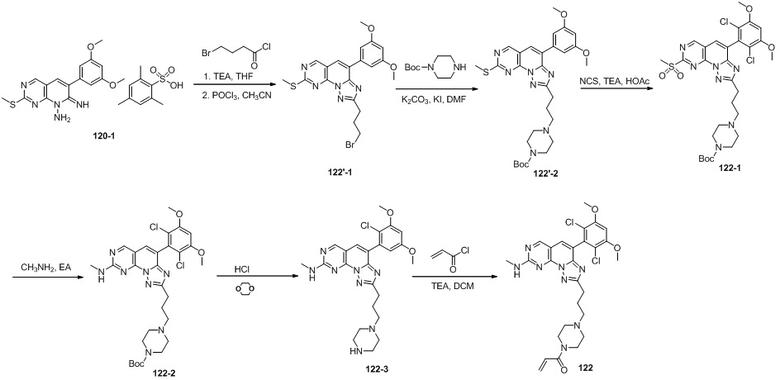
Стадия 1. Получение соединения 122’-1
Смесь 59,8 г соединения 120-1, 35,4 г TEA и 500 мл THF охлаждали до 0°C при перемешивании. К смеси по каплям добавляли 51,0 г 4-бромбутирилхлорида; после завершения добавления смесь нагревали до комнатной температуры и смесь подвергали реакции в течение ночи при перемешивании. Реакционную смесь гасили водой и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали насыщенным раствором бикарбоната натрия и водой, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в смеси 500 мл ацетонитрила и 50 мл POCl3 и подвергали реакции в течение 3 ч. при 80°C. Смесь охлаждали до комнатной температуры, гасили ледяной водой и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали дважды водой, насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором соответственно, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (Hex/EA=1/1) с получением 15,7 г соединения 121’-1 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 474,1.
Стадия 2. Получение соединения 122’-2
Смесь 8,2 г соединения 122’-1, 4,8 г 1-Boc-пиперазина, 7,2 г карбоната калия, 0,29 г иодида калия и 50 мл DMF перемешивали при 80°C и подвергали реакции в течение 3 ч. Смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом. Органическую фазу объединяли, три раза промывали насыщенным солевым раствором, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (DCM/MeOH=20/1) с получением 7,8 г соединения 122’-2 в виде светло-желтого твердого вещества.
LC-MS [M+H+] 580,3.
Стадия 3. Получение соединения 122-1
Контролировали температуру смеси 7,8 г соединения 122’-2, 4,1 г TEA и 80 мл ледяной уксусной кислоты на уровне ниже 5°C и к смеси порциями добавляли 8,1 г NCS. Смесь перемешивали при комнатной температуре и подвергали реакции в течение 2 ч. Смесь гасили водой и экстрагировали этилацетатом. Органическую фазу объединяли, дважды промывали водой, насыщенным водным раствором карбоната натрия и насыщенным солевым раствором соответственно, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 9,92 г соединения 122-1 в виде желтого твердого вещества.
LC-MS [M+H+] 680,2.
Стадии 4, 5 и 6 являются аналогичными стадиям 2, 3 и 4 пути A из примера 122 соответственно.
Соединения из таблицы 7 получали с помощью процедуры, подобной примеру 122, с применением других исходных материалов для реакции и соответствующего реагента.
Таблица 7
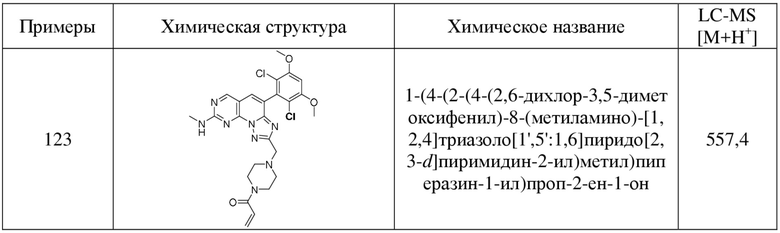
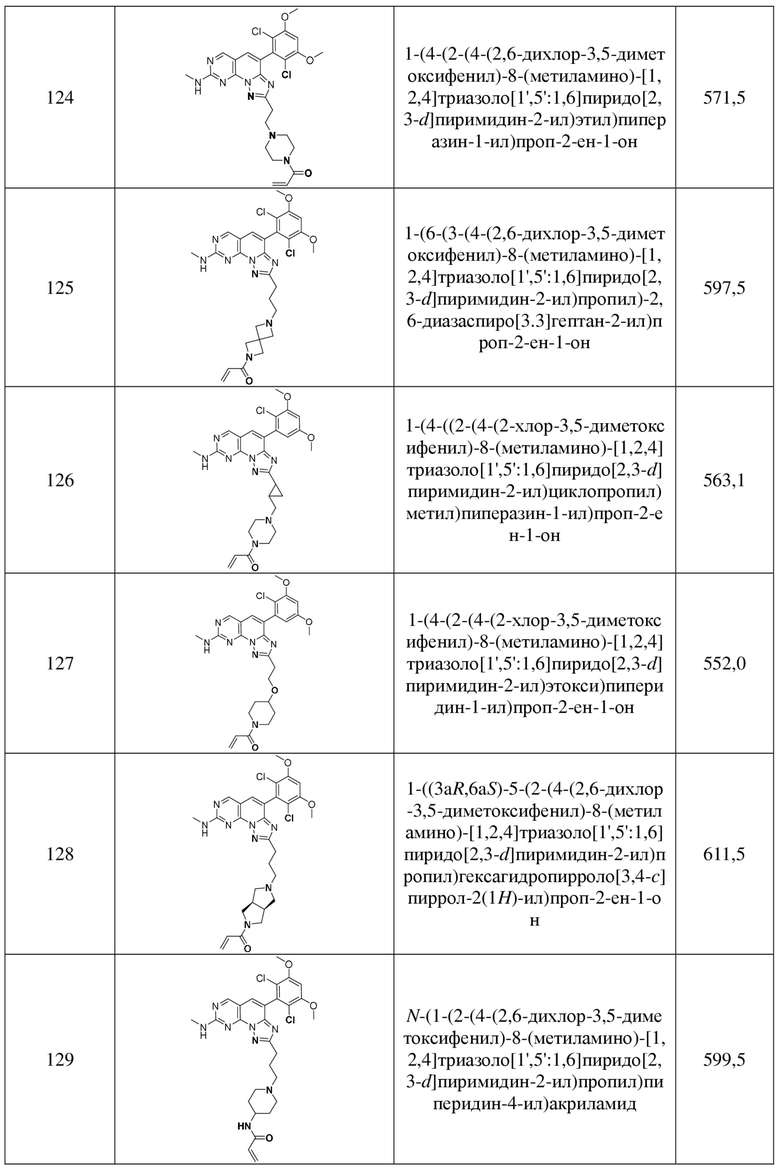
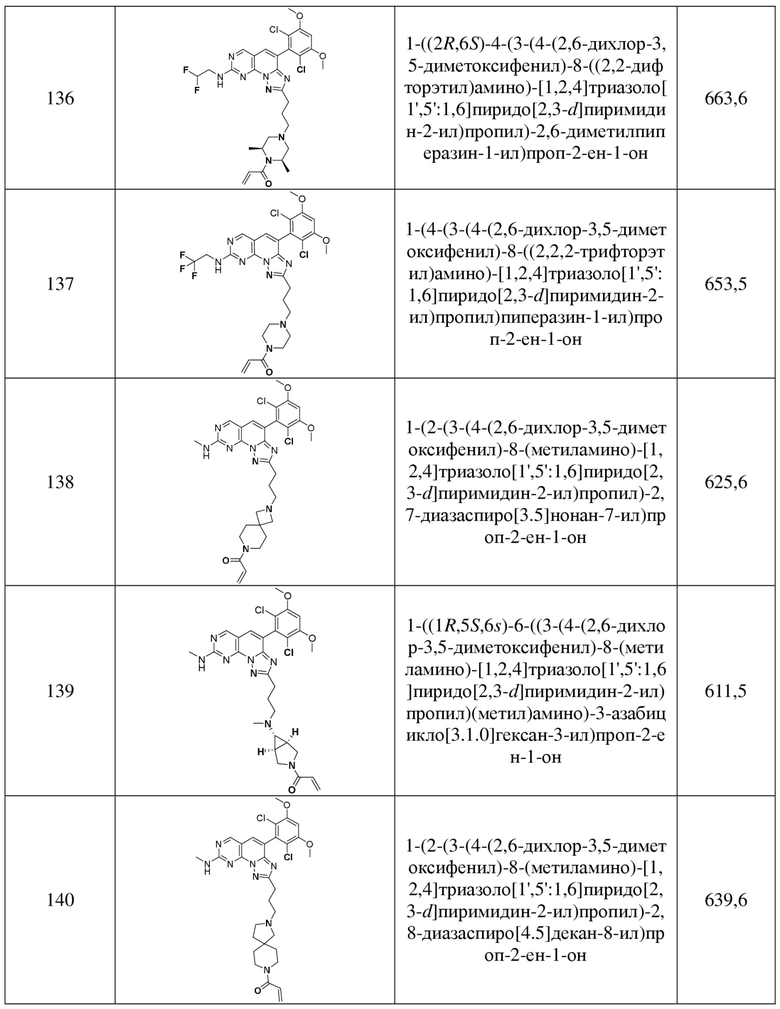
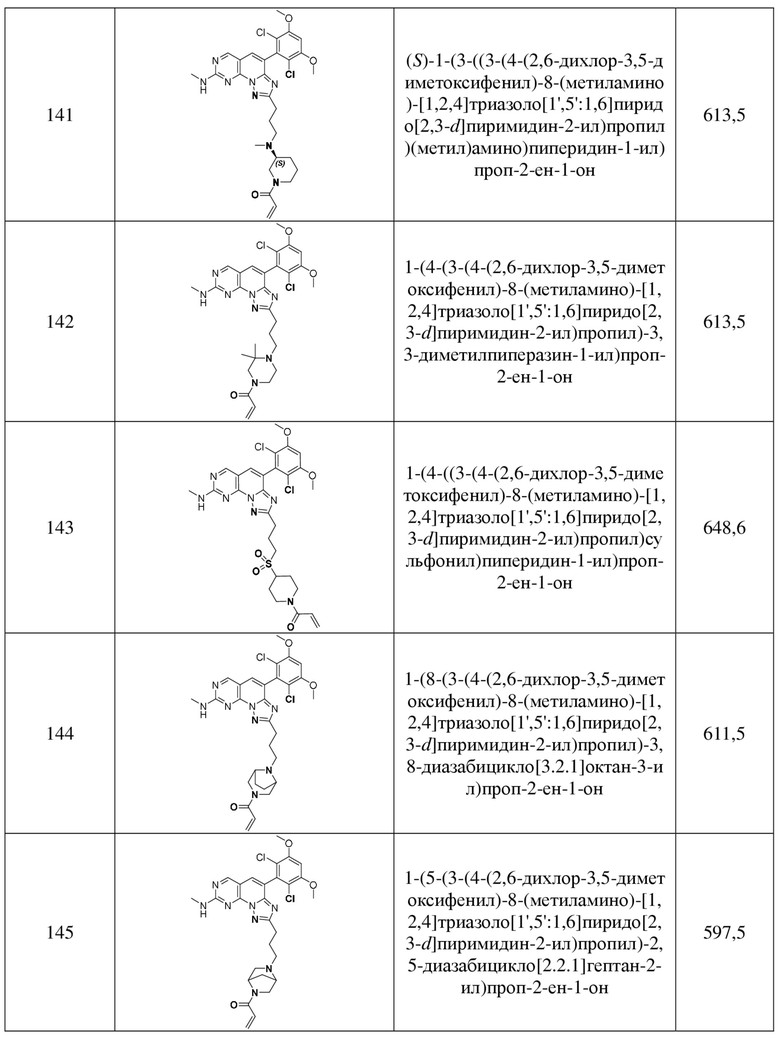
Пример 146. Получение соединения 146 (1-(4-(3-(4-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-она)
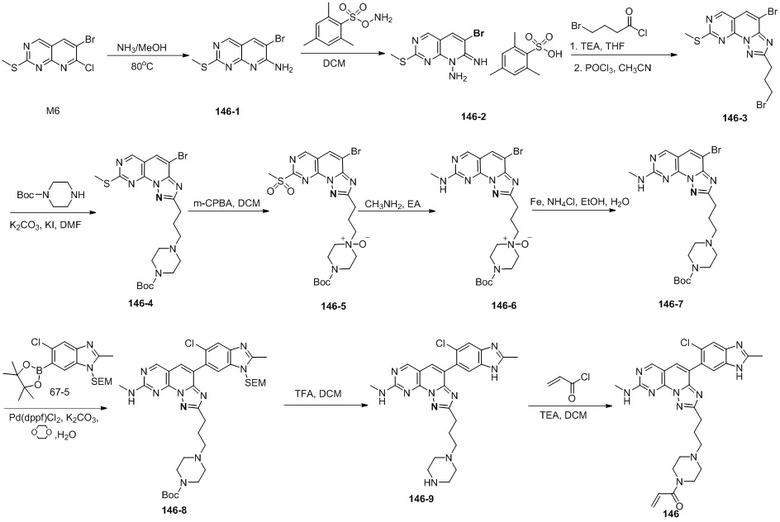
Стадия 1. Получение соединения 146-1
Смесь 500 мг соединения M6 и 5 мл раствора аммиака в метаноле (7 М) подвергали реакции в течение 24 ч. при 80°C. Реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли с помощью 50 мл дихлорметана, промывали три раза водой и высушивали над безводным сульфатом натрия. Раствор фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии (Hex/EA=1/1) с получением 440 мг соединения 146-1, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 270,1.
Стадия 2. Получение соединения 146-2
Применяя соединение 146-1 в качестве исходного материала, соединение 146-2 синтезировали с помощью процедуры, аналогичной описанной на стадии 1 пути B из примера 2.
LC-MS [M+H+] 286,0.
Стадия 3. Получение соединения 146-3
Применяя соединение 146-2 в качестве исходного материала, соединение 146-3 синтезировали с помощью процедуры, аналогичной описанной на стадии 1 пути B из примера 115.
LC-MS [M+H+] 417,9.
Стадия 4. Получение соединения 146-4
Применяя соединение 146-3 в качестве исходного материала, соединение 146-4 синтезировали с помощью процедуры, аналогичной описанной на стадии 2 пути B из примера 115.
LC-MS [M+H+] 522,1.
Стадия 5. Получение соединения 146-5
Применяя соединение 146-4 в качестве исходного материала, соединение 146-5 синтезировали с помощью процедуры, аналогичной описанной на стадии 3 пути B из примера 115.
LC-MS [M+H+] 570,1.
Стадия 6. Получение соединения 146-6
Применяя соединение 146-5 в качестве исходного материала, соединение 146-6 синтезировали с помощью процедуры, аналогичной описанной на стадии 4 пути B из примера 115.
LC-MS [M+H+] 521,2.
Стадия 7. Получение соединения 146-7
Применяя соединение 146-6 в качестве исходного материала, соединение 146-7 синтезировали с помощью процедуры, аналогичной описанной на стадии 5 пути B из примера 115.
LC-MS [M+H+] 505,2.
Стадия 8. Получение соединения 146-8
Применяя соединение 146-7 и соединение 67-5 в качестве исходных материалов, соединение 146-8 синтезировали с помощью процедуры, аналогичной описанной на стадии 5 из примера 67.
LC-MS [M+H+] 721,3.
Стадия 9. Получение соединения 146-9
Смесь 50 мг соединения 146-8, 5 мл трифторацетата и 5 мл дихлорметана подвергали реакции в течение 2 ч. при комнатной температуре. Раствор концентрировали при пониженном давлении с удалением растворителя. Полученный остаток разбавляли с помощью 50 мл дихлорметана, промывали насыщенным водным раствором гидрокарбоната натрия и водой и высушивали над безводным сульфатом натрия. Раствор фильтровали и высушивали в центробежной сушилке с получением 35 мг соединения 146-9, которое представляло собой коричневое твердое вещество. Его применяли непосредственно в следующей реакции без очистки.
LC-MS [M+H+] 491,2.
Стадия 10. Получение соединения 146
Смесь 35 мг соединения 146-9, 22 мг TEA и 10 мл дихлорметана охлаждали до 0°C, к полученному добавляли 13 мг акрилоилхлорида и его подвергали реакции в течение 30 мин. при 0°C. Реакционную смесь гасили водой и экстрагировали дихлорметаном. Органическую фазу объединяли, промывали насыщенным раствором бикарбоната натрия и водой и высушивали над безводным сульфатом натрия. Раствор фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством колоночной хроматографии (DCM/MeOH=15/1) с получением 18 мг соединения 146, которое представляло собой белое твердое вещество.
LC-MS [M+H+] 545,2.
Пример 147. Получение соединения 147 (2-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-карбонил)-4-метилпент-2-еннитрила)
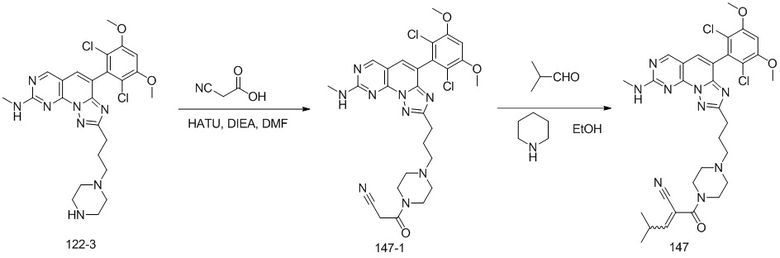
Стадия 1. Получение соединения 147-1
Смесь 388 мг соединения 147-3, 10 мл DMF, 472 мг DIEA, 94 мг цианоуксусной кислоты и 833 мг HATU подвергали реакции в течение ночи при перемешивании при комнатной температуре. Реакционную смесь гасили водой и экстрагировали этилацетатом. Органическую фазу объединяли, промывали водой и высушивали над безводным сульфатом натрия. Раствор фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали посредством колоночной хроматографии (DCM/MeOH=15/1) с получением 64 мг соединения 147-1, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 598,2.
Стадия 2. Получение соединения 147-2
Смесь 64 мг соединения 147-1, 20 мг пиперидина и 6 мл абсолютного этанола, в который добавляли 42 мг изобутиральдегида, подвергали реакции в течение ночи при перемешивании при комнатной температуре. Раствор концентрировали при пониженном давлении, остаток очищали посредством колоночной хроматографии (DCM/MeOH=15/1) с получением 14 мг соединения 147, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 652,2.
Пример 148. Получение соединения 148 (2-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-карбонил)-4,4-диметилпент-2-еннитрила)

К смеси 29 мг соединения 147-1, 8 мг пиперидина и 5 мл абсолютного этанола добавляли 21 мг пентанальдегида. Смесь нагревали с обратным холодильником, подвергали реакции в течение ночи при перемешивании и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии (DCM/MeOH=15/1) с получением 10 мг соединения 148, которое представляло собой светло-желтое твердое вещество.
LC-MS [M+H+] 666,2.
Фармакологический анализ
Пример A. Анализ киназы
Способ 1. Ингибирующую активность некоторых соединений по настоящему изобретению в отношении FGFR1, FGFR2, FGFR3, FGFR4, KDR тестировали путем анализа изменения подвижности (концентрация ATP представляет собой значение Km).
Аналитический метод:
Реагент: основный киназный буфер: 50 мМ HEPES (pH 7,5); 0,0015% Brij-35
Стоп-раствор: 100 мМ HEPES (pH 7,5); 0,015% Brij-35; 0,2% реагента для нанесения покрытия №3; 50 мМ EDTA
Получение соединений: разбавляют анализируемые соединения до конкретной концентрации с применением 100% DMSO
Способ проведения реакции:
1) получают 2,5X раствор фермента:
добавляют киназу в 1X основный киназный буфер;
2) получают 2,5X раствор пептида:
добавляют FAM-меченный пептид и ATP в 1X основный киназный буфер;
3) получают анализируемый планшет:
переносят 10 мкл исследуемого соединения в 384-луночный планшет, добавляют 90 мкл 1X основного киназного буфера;
4) добавляют 10 мкл 2,5X раствора фермента в каждую лунку анализируемого планшета и инкубируют в течение 10 мин. при комнатной температуре;
5) добавляют 10 мкл 2,5X раствора фермента в каждую лунку анализируемого планшета и инкубируют в течение заданного времени при 28°C; 6) останавливают реакцию путем добавления 25 мкл стоп-раствора в каждую лунку;
7) считывают данные с помощью Caliper и рассчитывают значение IC50.
Значения IC50 для вариантов осуществления показаны в таблице 5, где A означает IC50 < 1 нМ; B означает IC50 в диапазоне 1-10 нМ; C означает IC50 > 10 нМ; D означает IC50 > 100 нМ.
Таблица 5
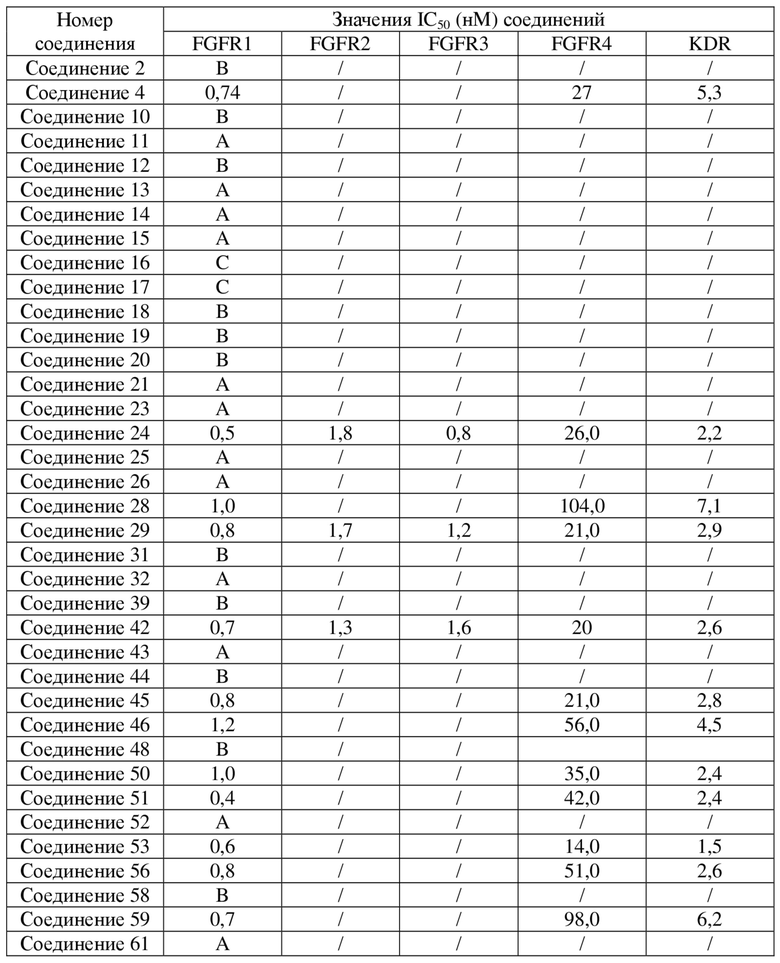
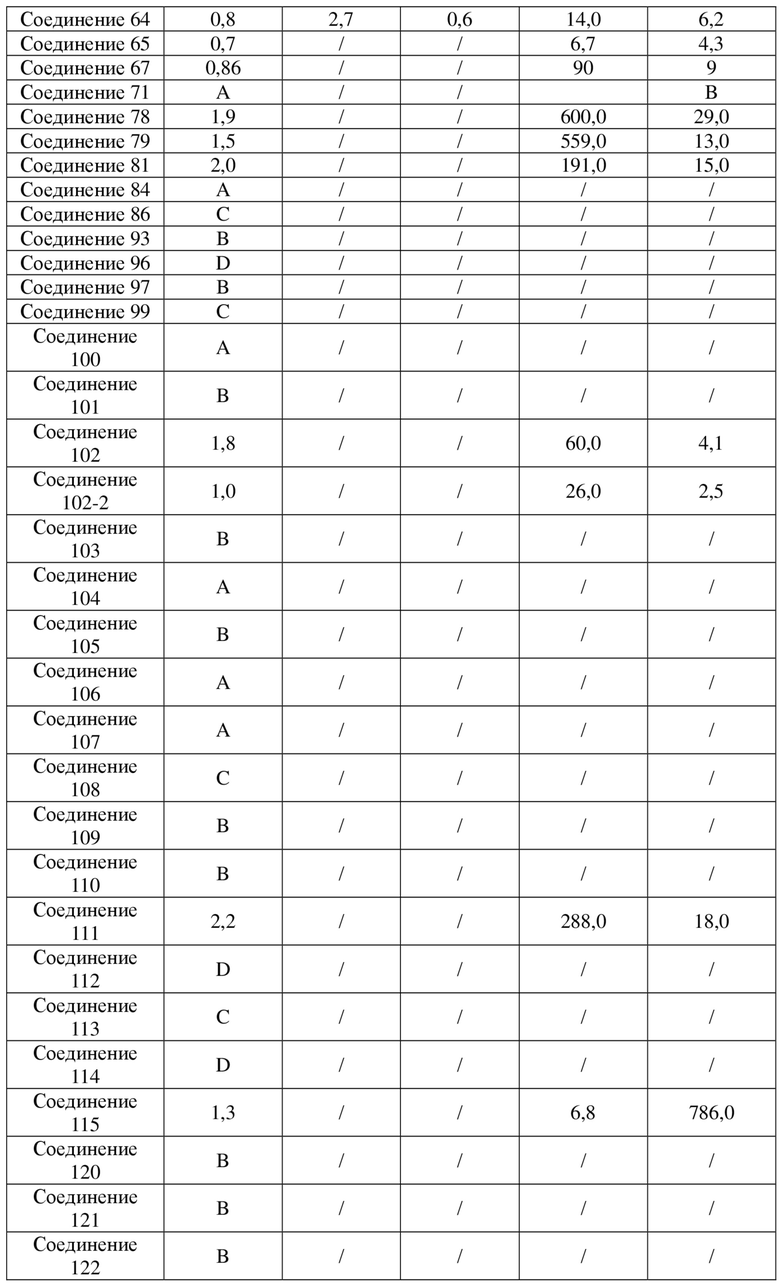
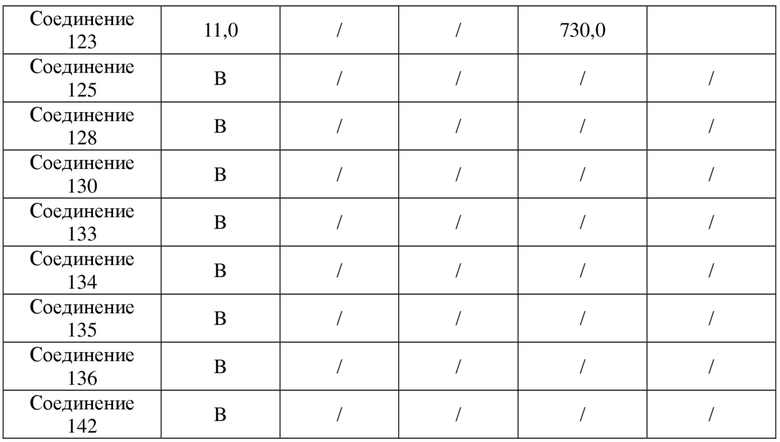
Способ 2. Ингибирующую активность соединения 4 по настоящему изобретению в отношении FGFR1, FGFR2, FGFR3, FGFR4 тестировали с помощью способа изменения меченного изотопом 33P-ATP (концентрация ATP представляет собой значение Km).
Аналитический метод:
Реагент: основный киназный буфер: 20 мМ Hepes (pH 7,5), 10 мМ MgCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT, 1% DMSO
Добавление соответствующего коферментного фактора для каждой киназной реакции
Получение соединений: разбавляют анализируемые соединения до конкретной концентрации с применением 100% DMSO и процесс разбавления заканчивают с помощью epMotion 5070
Способ проведения реакции:
1) составляют субстрат в свежеполученном реакционном буфере;
2) добавляют необходимое количество коферментного фактора в раствор указанного выше субстрата;
3) добавляют киназу в раствор субстрата и осторожно смешивают;
4) добавляют соединения, растворенные в 100% DMSO, в указанную выше киназную реакционную смесь, тщательно перемешивают и инкубируют в течение 20 мин. при комнатной температуре;
5) добавляют 33P-ATP (удельная активность 10 мкКи/мкл) к реакционной смеси, чтобы инициировать реакцию, и инкубируют в течение 2 ч. при комнатной температуре;
6) регистрируют активность киназы по связыванию на фильтре.
Экспериментальные данные показаны в таблице 6.
Таблица 6
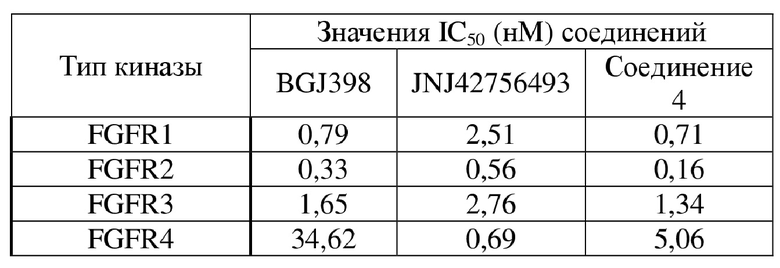
Соединение 4 было способно ингибировать активность киназы FGFR1, 2 и 3 дикого типа, при этом активность примерно эквивалентна подобным лекарственным средствам BGJ398 и JNJ42756493, и соединение 4 характеризуется немного меньшей ингибирующей активностью в отношении FGFR4.
Пример B. Анализ пролиферации клеток
Способ 1. Эффект, заключающийся в ингибировании роста, некоторых соединений по настоящему изобретению в отношении опухолевых клеток человека NCI-H1581, культивированных in vitro, наблюдали с помощью способа, в котором применяется набор для анализа пролиферации клеток CellTiter 96® AQueous One Solution.
Аналитический метод. 180 мкл клеточной суспензии добавляли в 96-луночный планшет и помещали в инкубатор с CO2 в течение ночи. Анализируемые соединения растворяли в DMSO и подвергали 3-кратному градиентному разбавлению с получением всего 10 концентраций. 20 мкл среды, содержащей анализируемое соединение или DMSO, переносили в соответствующие лунки с клетками соответственно и инкубировали в течение 144 ч. в 5% CO2 при 37°C. 40 мкл реагента для анализа пролиферации клеток CellTiter 96® AQueous One Solution добавляли в анализируемый планшет и инкубировали в течение 2 ч. в 5% CO2 при 37°C. Значение IC50 рассчитывали путем регистрирования значения поглощения света (OD490) при 490 нм с применением устройства VICTOR TM X5.
Способ 2. Эффект, заключающийся в ингибировании роста, некоторых соединений по настоящему изобретению в отношении опухолевых клеток человека NCI-H1581 и SNU-16, культивированных in vitro, наблюдали с применением способа анализа CellTiter Glo.
Аналитический метод. Добавляли подходящий объем цельной среды и клетки суспендировали. 180 мкл клеточной суспензии добавляли в 96-луночный планшет и помещали в инкубатор с CO2 в течение ночи. Анализируемые соединения растворяли в DMSO и подвергали 3-кратному градиентному разбавлению с получением всего 10 концентраций. Анализируемые соединения или DMSO в качестве контрольного вещества переносили в соответствующие лунки, содержащие 100 мкл среды соответственно и инкубировали в течение 96 ч. в 5% CO2 при 37°C. 100 мкл реагента CellTiter-Glo добавляли в анализируемый планшет и инкубировали в течение 10 мин. при комнатной температуре для стабилизации люминесцентного сигнала. Значение IC50 рассчитывали путем регистрации RLU (относительных единиц люминесценции) с применением устройства VICTOR TM X5.
Экспериментальные данные показаны в таблице 7 и таблице 8, где A означает IC50 < 1 нМ; B означает IC50 в диапазоне 1-10 нМ; C означает IC50 > 10-100 нМ; D означает IC50 > 100 нМ.
Таблица 7
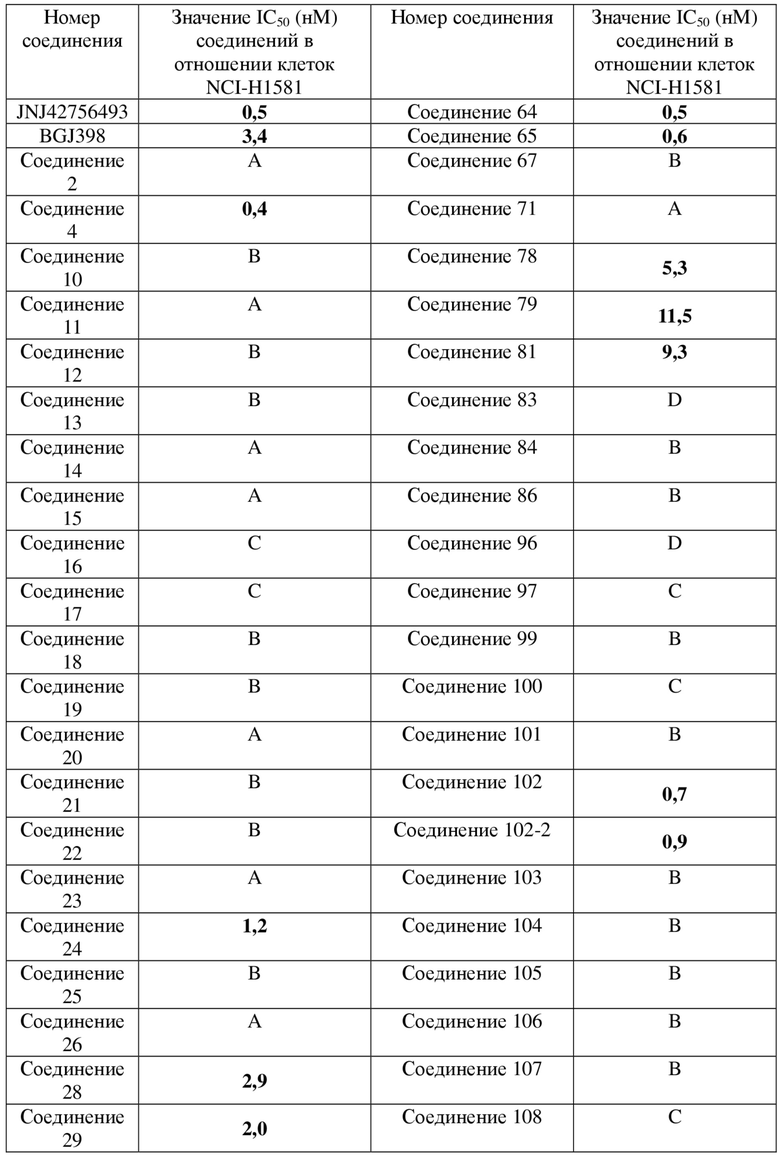
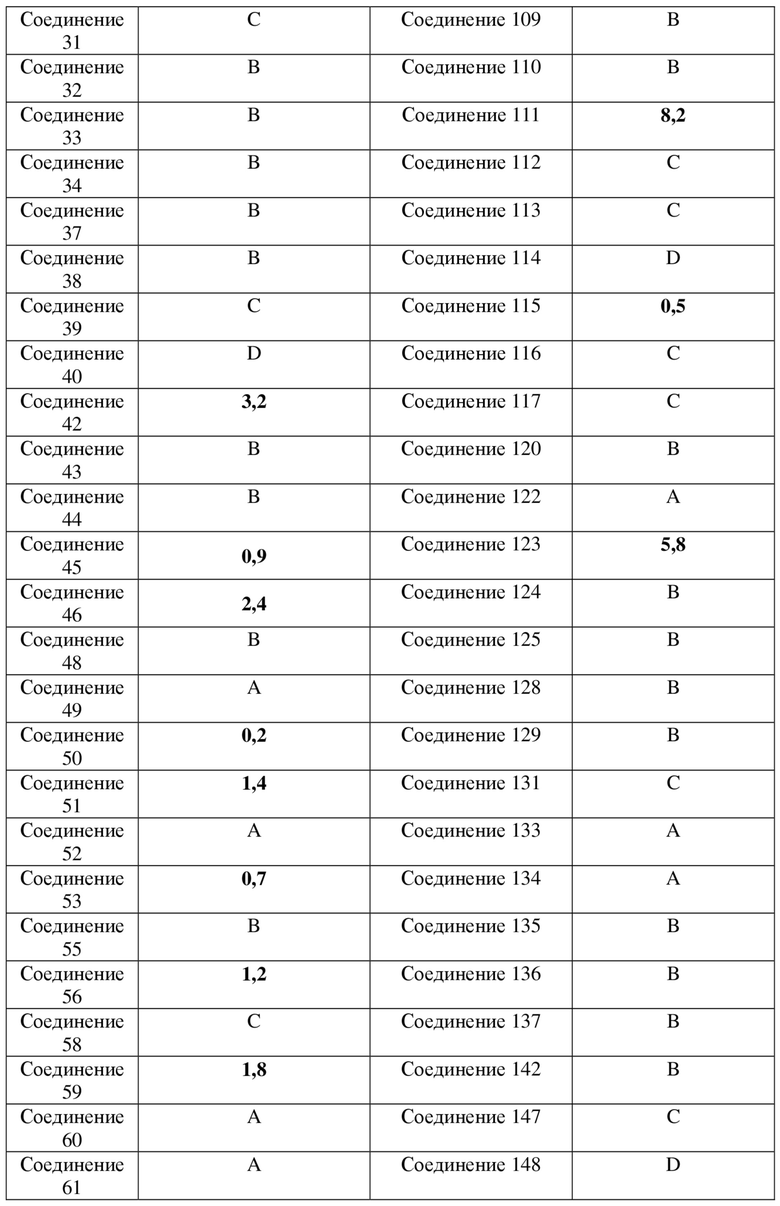
Таблица 8

Эффект, заключающийся в ингибировании роста, соединения 4 в отношении опухолевых клеток человека NCI-H1581 (клетки немелкоклеточного рака легкого человека), RT-112 (клетки рака мочевого пузыря человека), OPM-2 (клетки миеломы человека), NCI-H716 (клетки колоректального рака человека), MFE-296 (клетки рака эндометрия человека), JHH-7 (клетки гепатомы человека), DMS 114 (клетки рака легкого человека), SNU-16 (клетки рака желудка человека), MDA-MB-453 (клетки рака молочной железы человека) и RT4 (клетки рака мочевого пузыря человека), культивированных in vitro, наблюдали с применением способа анализа CellTiter Glo.
Аналитический метод. 100 мкл клеточной суспензии добавляли в 96-луночный планшет и помещали в инкубатор с CO2 в течение ночи. Анализируемые соединения растворяли в DMSO и подвергали 3-кратному градиентному разбавлению с получением всего 10 концентраций. Анализируемое соединение или DMSO переносили в соответствующие лунки с клетками соответственно и инкубировали в течение 96 ч. в 5% CO2 при 37°C. 100 мкл реагента CellTiter-Glo добавляли в анализируемый планшет и инкубировали в течение 10 мин. при комнатной температуре для стабилизации люминесцентного сигнала. Значение IC50 рассчитывали путем регистрации RLU (относительных единиц люминесценции) с применением Enspire. Экспериментальные данные показаны в таблице 9.
Таблица 9
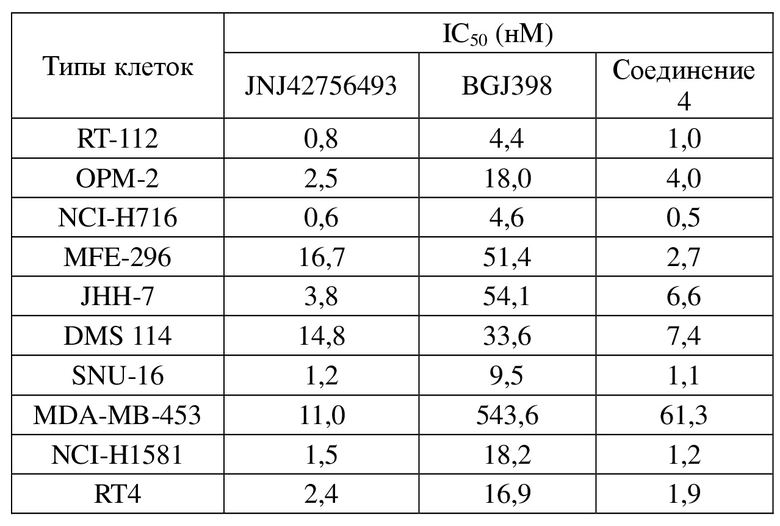
Соединение 4 ингибировало рост опухолевых клеток человека in vitro дозозависимым образом, и особенно чувствительными были клеточные линии (NCI-H1581, RT-112, OPM-2, NCI-H716, MFE-296, JHH-7, DMS 114 и SNU-16) с аномальными изменениями в сигнальном пути FGFR, при этом значение IC50 составляло 0,5-2,7 нМ.
Пример C. Модели ксенотрансплантатов опухолей
Реагенты: DMSO, полиэтиленгликоль-15-гидроксистеарат (солютол), солевой раствор.
Животные: для инокуляции клеточной линии NCI-H1581: линия бестимусных мышей BALB/C-nude: животные, свободные от специфической патогенной микрофлоры, вес 18~22 г, самки, получены от Vitrallihua Experimental Animal Technology Co., Ltd., кормили пищей, свободной от специфической патогенной микрофлоры, и обеспечивали возможность пить дистиллированную воду в неограниченном количестве.
Для инокуляции клеточной линии SNU-16: линия бестимусных мышей BALB/C-nude: животные, свободные от специфической патогенной микрофлоры, вес 18~22 г, самки, получены от Shanghai Xipuer-Beikai Experimental Animal Technology Co., Ltd., кормили пищей, свободной от специфической патогенной микрофлоры, и обеспечивали возможность пить дистиллированную воду в неограниченном количестве.
Линия раковых клеток человека: линия клеток немелкоклеточного рака легкого человека NCI-H1581, получена от Shanghai Ruizhi Chemical Research Co., Ltd.; линия клеток рака желудка человека SNU-16, получена от ATCC.
Способ анализа трансплантата опухоли. Клеточную линию NCI-H1581 инокулировали подкожно в правую подмышечную впадину бестимусных мышей в ламинарном боксе в асептических условиях в количестве 1 x 107 клеток/100 мкл/особь; клеточную линию SNU-16 инокулировали подкожно в правую подмышечную впадину бестимусных мышей в количестве 0,5 x 107 клеток/100 мкл/особь. Через 10 дней опухоли вырастали и были прощупываемыми (приблизительно 100-200 мм3). Животных произвольным образом делили на группы по 6 особей и каждое животное в группе взвешивали. Группе положительного контроля проводили введение внутрижелудочно один раз в день. Экспериментальной группе проводили введение внутрижелудочно один или два раза в день, и длительность введения была такой же, как и в группе положительного контроля лекарственного средства. Бестимусных мышей содержали при комнатной температуре, составляющей 20-22°C, относительной влажности, составляющей 40-60%, и защитную систему дополняли средой стерильного шкафа с ламинарным потоком. Объем подкожной опухоли измеряли штангенциркулем через каждые 3-4 дня после начала эксперимента и строили кривую роста опухоли для расчета степени ингибирования роста опухоли. Проводили статистический анализ результатов испытаний с применением программного обеспечения GraphPad Prism 5, и экспериментальные данные показаны на фигурах 1-3.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРИДИН АЗОЛОПИРИМИДИН-5-(6Н)-ОНА | 2013 |
|
RU2653054C2 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ HDAC6 | 2018 |
|
RU2764718C2 |
| ДИАМИНОТРИАЗОЛЫ, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗ | 2003 |
|
RU2350606C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
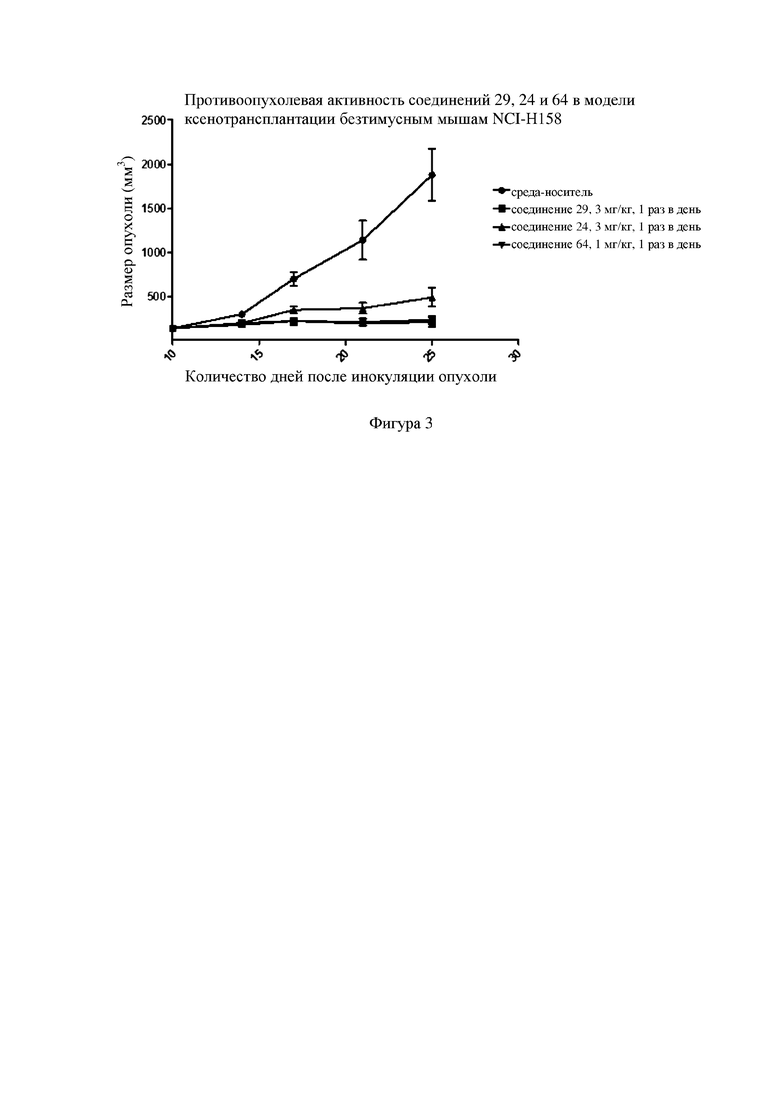
Изобретение относится к азатрициклическому соединению формулы I
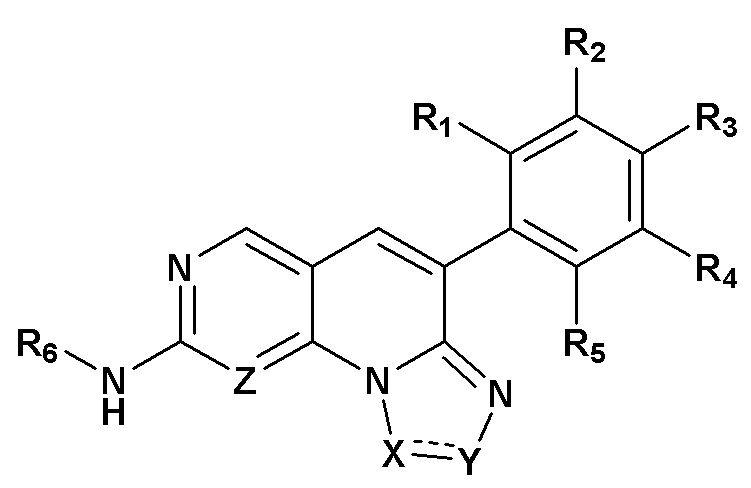
Формула (I),
где  представляет собой двойную связь; один из X и Y выбран из N и другой из CR10; Z представляет собой N; каждый из R1, R2, R3, R4 и R5 независимо выбран из H, галогена, C1-8алкокси; или R3 и R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-членное замещенное C1-4алкилом или галогеном или незамещенное гетероарильное кольцо, при этом 5-членное гетероарильное кольцо содержит 1, 2 или 3 гетероатома, независимо выбранных из N, O или S; R6 представляет собой C1-8алкил, фенил, пиридил, C3-8циклоалкил, C5-6 гетероциклил, содержащий 1-2 гетероатома в цикле, выбранных из N и O; R6 необязательно замещен R7; R7 представляет собой гидроксил, галоген, C1-8алкил, C1-8алкокси, C3-8циклоалкил, C3-8циклоалкил, замещенный С1-4алкилом, C5-10гетероциклил, содержащий 1-2 гетероатома, независимо выбранных из азота и кислорода, замещенный С1-4алкилом, C1-4алкилен-OH, -NR11R12, -оксо, -(CO)-C1-4алкилом, -(CO)-NR11R12, C1-4галогеналкилом, C3-6циклоалкилом, C3-6циклоалкил-OH, С4-6гетероциклилом, содержащим 1-2 гетероатома в цикле, выбранных из азота и кислорода, или С4-6гетероциклил-С1-4алкилом, содержащим 1-2 гетероатома в цикле, выбранных из N и O, C5-6гетероциклилом, содержащим 1-2 гетероатома, выбранных из азота и кислорода, C6гетероциклический карбонил, содержащий 1-2 гетероатома азота в цикле, замещенный С1-4алкилом, -NR11R12, -NR11-C1-8алкилен-NR11R12, -C1-4алкилен-C5-6гетероциклил, содержащий 1-2 гетероатома, выбранных из азота и кислорода, C1-4алкилен-C5-6гетероциклил-С1-4алкил, содержащий 1-2 гетероатома азота в цикле, -C1-8алкокси-C5-6гетероциклил, содержащий 1-2 гетероатома в цикле, выбранных из азота и кислорода, или в случае, где R6 представляет собой фенил, R7 представляет собой незамещенное C5-6гетероциклическое кольцо, содержащее атом азота в кольце, конденсированное с R6; R10 представляет собой H, амино, C1-6алкил, C3-8циклоалкил; R10 необязательно замещен R8; R8 представляет собой C1-8алкил, C4-6гетероцикло-C1-6алкокси, замещенный -(CO)-C2-8алкенилом, содержащий 1-2 гетероатома, выбранных из атомов азота и кислорода в цикле, фенил, замещенный -NH-(CO)-C2-4алкенилом или -NH-(CO)-C1-4алкилом, C3-8циклоалкил, C5-6гетероциклил, содержащий 1-2 гетероатома, выбранных из азота и кислорода; моно-, би- или спиро-C5-10гетероциклил, содержащий 1-2 атома азота; замещенный C1-4алкилом, C1-4алкил(CO)-, C2-4алкенил-C(O), C1-4алкил (CO)-NH-, C2-4алкенил-C(O)-NH-группы или -(CO)-C2-8алкенил-CN, моно-, би- или спирогетероциклил, содержащий 1-2 атома азота, -S(O2)C4-6гетероциклил-(CO)-C2-4алкенил, -NR11R12, замещенный C4-6 гетероциклил-(CO)-C2-4 алкенилом, содержащий 1-2 гетероатома азота в цикле, или -NR11R12; каждый из R11 и R12 независимо представляет собой H, C1-8алкил, которое выполняет функцию ингибитора рецепторов фактора роста фибробластов (FGFR), а также фармацевтической композиции на его основе, и применению при лечении заболеваний, опосредованных FGFR. 5 н. и 29 з.п. ф-лы, 3 ил., 12 табл., 148 пр.
представляет собой двойную связь; один из X и Y выбран из N и другой из CR10; Z представляет собой N; каждый из R1, R2, R3, R4 и R5 независимо выбран из H, галогена, C1-8алкокси; или R3 и R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-членное замещенное C1-4алкилом или галогеном или незамещенное гетероарильное кольцо, при этом 5-членное гетероарильное кольцо содержит 1, 2 или 3 гетероатома, независимо выбранных из N, O или S; R6 представляет собой C1-8алкил, фенил, пиридил, C3-8циклоалкил, C5-6 гетероциклил, содержащий 1-2 гетероатома в цикле, выбранных из N и O; R6 необязательно замещен R7; R7 представляет собой гидроксил, галоген, C1-8алкил, C1-8алкокси, C3-8циклоалкил, C3-8циклоалкил, замещенный С1-4алкилом, C5-10гетероциклил, содержащий 1-2 гетероатома, независимо выбранных из азота и кислорода, замещенный С1-4алкилом, C1-4алкилен-OH, -NR11R12, -оксо, -(CO)-C1-4алкилом, -(CO)-NR11R12, C1-4галогеналкилом, C3-6циклоалкилом, C3-6циклоалкил-OH, С4-6гетероциклилом, содержащим 1-2 гетероатома в цикле, выбранных из азота и кислорода, или С4-6гетероциклил-С1-4алкилом, содержащим 1-2 гетероатома в цикле, выбранных из N и O, C5-6гетероциклилом, содержащим 1-2 гетероатома, выбранных из азота и кислорода, C6гетероциклический карбонил, содержащий 1-2 гетероатома азота в цикле, замещенный С1-4алкилом, -NR11R12, -NR11-C1-8алкилен-NR11R12, -C1-4алкилен-C5-6гетероциклил, содержащий 1-2 гетероатома, выбранных из азота и кислорода, C1-4алкилен-C5-6гетероциклил-С1-4алкил, содержащий 1-2 гетероатома азота в цикле, -C1-8алкокси-C5-6гетероциклил, содержащий 1-2 гетероатома в цикле, выбранных из азота и кислорода, или в случае, где R6 представляет собой фенил, R7 представляет собой незамещенное C5-6гетероциклическое кольцо, содержащее атом азота в кольце, конденсированное с R6; R10 представляет собой H, амино, C1-6алкил, C3-8циклоалкил; R10 необязательно замещен R8; R8 представляет собой C1-8алкил, C4-6гетероцикло-C1-6алкокси, замещенный -(CO)-C2-8алкенилом, содержащий 1-2 гетероатома, выбранных из атомов азота и кислорода в цикле, фенил, замещенный -NH-(CO)-C2-4алкенилом или -NH-(CO)-C1-4алкилом, C3-8циклоалкил, C5-6гетероциклил, содержащий 1-2 гетероатома, выбранных из азота и кислорода; моно-, би- или спиро-C5-10гетероциклил, содержащий 1-2 атома азота; замещенный C1-4алкилом, C1-4алкил(CO)-, C2-4алкенил-C(O), C1-4алкил (CO)-NH-, C2-4алкенил-C(O)-NH-группы или -(CO)-C2-8алкенил-CN, моно-, би- или спирогетероциклил, содержащий 1-2 атома азота, -S(O2)C4-6гетероциклил-(CO)-C2-4алкенил, -NR11R12, замещенный C4-6 гетероциклил-(CO)-C2-4 алкенилом, содержащий 1-2 гетероатома азота в цикле, или -NR11R12; каждый из R11 и R12 независимо представляет собой H, C1-8алкил, которое выполняет функцию ингибитора рецепторов фактора роста фибробластов (FGFR), а также фармацевтической композиции на его основе, и применению при лечении заболеваний, опосредованных FGFR. 5 н. и 29 з.п. ф-лы, 3 ил., 12 табл., 148 пр.
1. Соединение формулы I
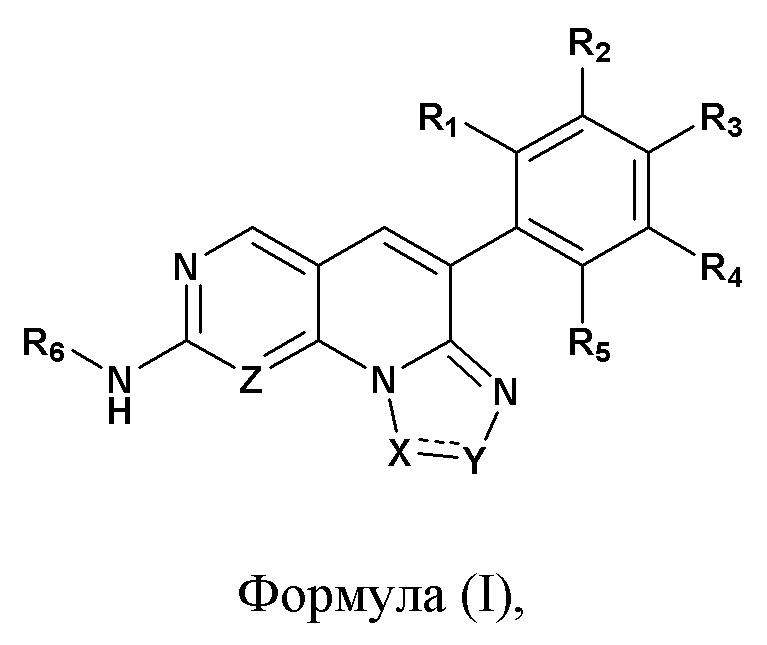
где  представляет собой двойную связь;
представляет собой двойную связь;
один из X и Y выбран из N и другой из CR10;
Z представляет собой N;
каждый из R1, R2, R3, R4 и R5 независимо выбран из H, галогена, C1-8алкокси; или
R3 и R4 вместе с атомом углерода, к которому они присоединены, могут образовывать 5-членное замещенное C1-4алкилом или галогеном или незамещенное гетероарильное кольцо, при этом 5-членное гетероарильное кольцо содержит 1, 2 или 3 гетероатома, независимо выбранных из N, O или S;
R6 представляет собой C1-8алкил, фенил, пиридил, C3-8циклоалкил, C5-6гетероциклил, содержащий 1-2 гетероатома в цикле, выбранных из N и O;
R6 необязательно замещен R7;
R7 представляет собой гидроксил, галоген, C1-8алкил, C1-8алкокси, C3-8циклоалкил, C3-8циклоалкил, замещенный С1-4алкилом, C5-10гетероциклил, содержащий 1-2 гетероатома, независимо выбранных из азота и кислорода, замещенный С1-4алкилом, C1-4алкилен-OH, -NR11R12, -оксо, -(CO)-C1-4алкилом, -(CO)-NR11R12, C1-4галогеналкилом, C3-6циклоалкилом, C3-6циклоалкил-OH, С4-6гетероциклилом, содержащим 1-2 гетероатома в цикле, выбранных из азота и кислорода, или С4-6гетероциклил-С1-4алкилом, содержащим 1-2 гетероатома в цикле, выбранных из N и O, C5-6гетероциклилом, содержащим 1-2 гетероатома, выбранных из азота и кислорода, C6гетероциклический карбонил, содержащий 1-2 гетероатома азота в цикле, замещенный С1-4алкилом, -NR11R12, -NR11-C1-8алкилен-NR11R12, -C1-4алкилен-C5-6гетероциклил, содержащий 1-2 гетероатома, выбранных из азота и кислорода, C1-4алкилен-C5-6гетероциклил-С1-4алкил, содержащий 1-2 гетероатома азота в цикле, -C1-8алкокси-C5-6гетероциклил, содержащий 1-2 гетероатома в цикле, выбранных из азота и кислорода, или в случае, где R6 представляет собой фенил, R7 представляет собой незамещенное C5-6гетероциклическое кольцо, содержащее атом азота в кольце, конденсированное с R6;
R10 представляет собой H, амино, C1-6алкил, C3-8циклоалкил;
R10 необязательно замещен R8;
R8 представляет собой C1-8алкил, C4-6гетероцикло-C1-6алкокси, замещенный -(CO)-C2-8 алкенилом, содержащий 1-2 гетероатома, выбранных из атомов азота и кислорода в цикле, фенил, замещенный -NH-(CO)-C2-4алкенилом или -NH-(CO)-C1-4алкилом, C3-8циклоалкил, C5-6гетероциклил, содержащий 1-2 гетероатома, выбранных из азота и кислорода; моно-, би- или спиро-C5-10гетероциклил, содержащий 1-2 атома азота; замещенный C1-4алкилом, C1-4алкил(CO)-, C2-4алкенил-C(O), C1-4алкил (CO)-NH-, C2-4алкенил-C(O)-NH-группы или -(CO)-C2-8алкенил-CN, моно-, би- или спирогетероциклил, содержащий 1-2 атома азота, -S(O2)C4-6гетероциклил-(CO)-C2-4алкенил, -NR11R12, замещенный C4-6 гетероциклил-(CO)-C2-4 алкенилом, содержащий 1-2 гетероатома азота в цикле, или -NR11R12;
каждый из R11 и R12 независимо представляет собой H, C1-8алкил.
2. Соединение по п. 1, где X представляет собой N или CR10, R10 представляет собой H, амино, C1-6алкил или C3-6циклоалкил.
3. Соединение по п. 1 или 2, где X представляет собой CR10, R10 представляет собой H.
4. Соединение по п. 1 или 2, где X представляет собой CR10, R10 представляет собой C1-6алкил.
5. Соединение по п. 1 или 2, где X представляет собой CR10, R10 представляет собой C1-6алкил или C3-6циклоалкил, R10 может быть замещен R8, R8 представляет собой этил-C(O)-N-фенил, морфолинил, -NR11R12, циклопропан, этилен-C(O)-пиперазинил, этилен-C(O)-азетидинокси, этилен-C(O)-пиперидинилокси, этилен-C(O)-аза-C6-10спироциклил, этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-N-пиперидинил, этилен-C(O)-пиперидинил, этилен-C(O)-C1-8алкилпиперазинил, -N(R11)этилен-C(O)-пиперидинил, -N(R11)этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-пиперидинил-S(O2)- или изопентенил-C(O)-пиперазинил, замещенный циано.
6. Соединение по любому из пп. 1-5, где Y представляет собой N или CR10, R10 представляет собой H, амино, C1-6алкил или C3-6циклоалкил.
7. Соединение по любому из пп. 1-6, где Y представляет собой N.
8. Соединение по любому из пп. 1-6, где Y представляет собой CR10, R10 представляет собой C1-6алкил.
9. Соединение по любому из пп. 1-6, где Y представляет собой CR10, R10 представляет собой C1-6алкил или C3-6циклоалкил, R10 может быть замещен R8, R8 представляет собой этил-C(O)-N-фенил, морфолинил, -NR11R12, циклопропан, этилен-C(O)-пиперазинил, этилен-C(O)-азетидинокси, этилен-C(O)-пиперидинилокси, этилен-C(O)-аза-C6-10спироциклил, этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-N-пиперидинил, этилен-C(O)-пиперидинил, этилен-C(O)-C1-8алкилпиперазинил, -N(R11)этилен-C(O)-пиперидинил, -N(R11)этилен-C(O)-аза-C6-10бициклил, этилен-C(O)-пиперидинил-S(O2)- или изопентенил-C(O)-пиперазинил, замещенный циано.
10. Соединение по любому из пп. 1-9, где каждый из R1, R3 и R5 независимо представляет собой H, F или Cl.
11. Соединение по любому из пп. 1-10, где R1 и R5 выбраны из группы ниже:
(i) как R1, так и R5 представляют собой H;
(ii) как R1, так и R5 представляют собой Cl;
(iii) R1 представляет собой H, R5 представляет собой Cl;
(iv) R1 представляет собой Cl, R5 представляет собой H;
(v) как R1, так и R5 представляют собой F;
(vi) R1 представляет собой H, R5 представляет собой F; или
(vii) R1 представляет собой F, R5 представляет собой H.
12. Соединение по любому из пп. 1-10, где R1 представляет собой Cl, как R3, так и R5 представляют собой H.
13. Соединение по любому из пп. 1-12, где R3 представляет собой H.
14. Соединение по любому из пп. 1-13, где каждый из R2 и R4 независимо выбран из H и C1-3алкокси.
15. Соединение по любому из пп. 1-14, где как R2, так и R4 представляют собой CH3O-.
16. Соединение по любому из пп. 1-9, где R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 5-членное гетероарильное кольцо, содержащее 1-2 гетероатома, независимо выбранных из N, O или S, и 5-членное гетероарильное кольцо замещено C1-3алкилом.
17. Соединение по п. 16, где R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют 5-членное гетероарильное кольцо, содержащее один или два атома азота, или один атом азота и один атом серы, или один атом азота и один атом кислорода, и при этом 5-членное гетероарильное кольцо замещено метилом.
18. Соединение по п. 16 или 17, где R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют гетероарильное кольцо, которое представляет собой  ,
,  ,
, 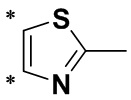 или
или 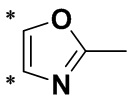 .
.
19. Соединение по любому из пп. 1-18, где R6 представляет собой C1-4алкил, циклопентил, фенил, фенил, замещенный F, фенил, замещенный метокси, фенил, замещенный Cl, фенил, замещенный метилом, пиридил, тетрагидропиранил, R6 замещен R7, при этом R7 представляет собой гидроксил, F, Cl, пиперазинил, замещенный этилом, морфолинил, пиперазинил, замещенный изопропилом, пиперазинил, замещенный оксетаном, пиперазинил, замещенный метилом, пиперазинил-CH2-, замещенный этилом, пиперазинил, замещенный этилом и оксилом, пиперазинил, замещенный триметилом, триметилэтилендиамин, пиперазинил, замещенный метилпиперидинилом, аза-C6-10бициклил, замещенный метилом, аза-C6-10бициклил, -N(метил)-C1-6метиленморфолинил, C4-10азациклоалкил, замещенный C2-6алкокси, пиперидинил, замещенный морфолинилом, пиперазинил, замещенный гидроксиэтилом, C2-6алкокси, замещенный морфолинилом, пиперидинил, замещенный этилом, пиперидинил, замещенный метилом, диметиламинопиперидинил, C6-10азабициклил, замещенный оксилом, нитроокса-C6-10бициклил, морфолинил-CH2-, метилпиперазинил-CH2-, пиперидинил, замещенный C3-10циклоалкилом, метиламинопиперидинил, пиперазинил, замещенный диметилом, пиперидинил, пиперазинил-CH2-, пиперазинил-C(O)-, замещенный диметилом, пиперазинил, замещенный гидроксициклобутаном, трифторметил-CH2-пиперазинил, пиперазинил, замещенный C3-10циклоалкилом, метил-C(O)-пиперазинил, (диметил)-N-C(O)-пиперазинил, (диметил)-N-C(O)-аза-C6-10спироциклил, (диметил)-N-C(O)-тетрагидропиррол-NH-, R7 представляет собой незамещенное азотсодержащее 6-членное гетероциклическое кольцо, которое конденсировано с R6.
20. Соединение по любому из пп. 1-19, где R6 представляет собой метил, 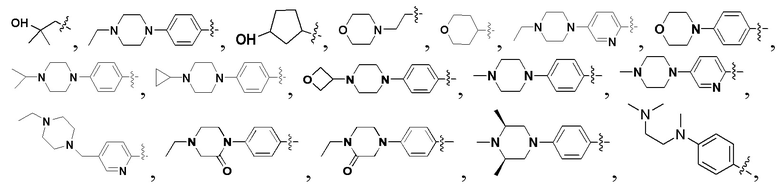
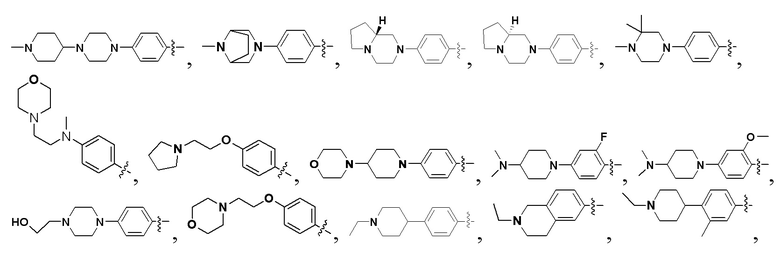
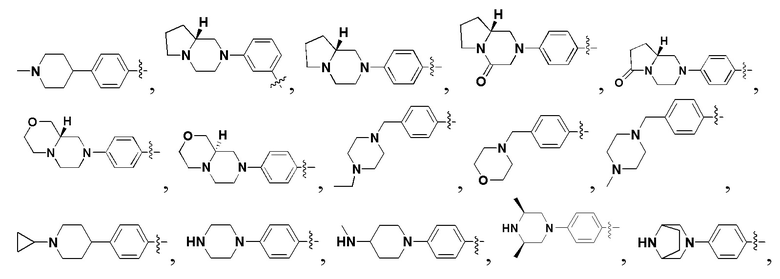
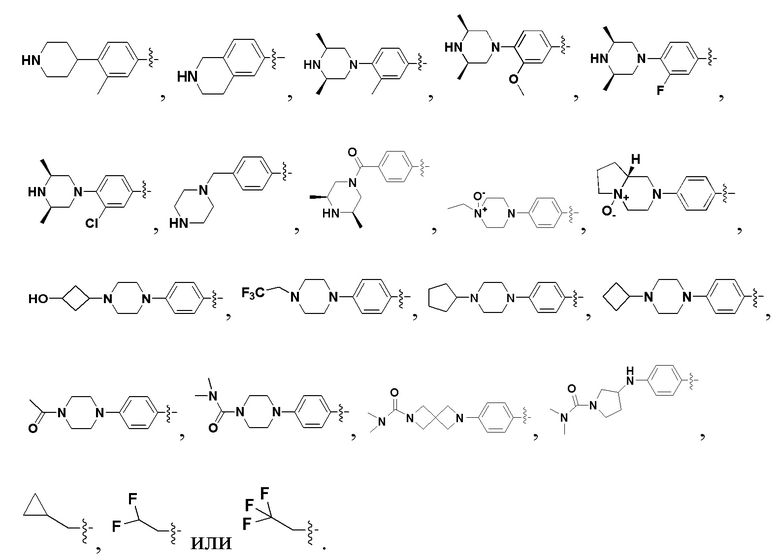
21. Соединение по любому из пп. 1-20, где каждый из R11 и R12 независимо выбран из H, C1-6алкила.
22. Соединение по любому из пп. 1-20, где каждый из R11 и R12 независимо выбран из H, метила и этила.
23. Соединение по любому из пп. 1-21, где R10 представляет собой H, -CH3, амино,
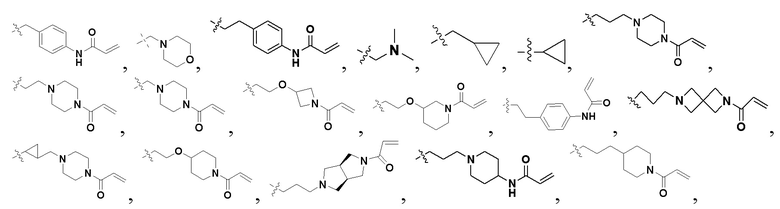
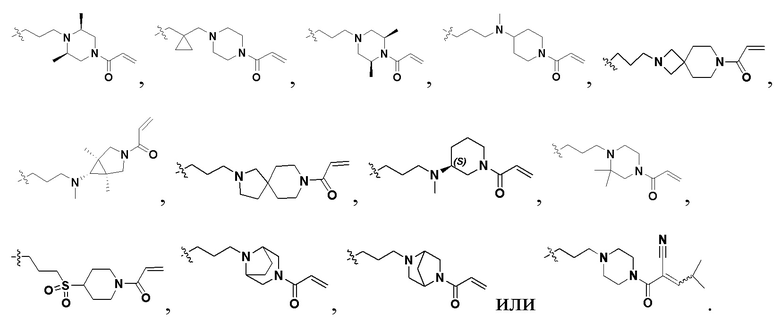
24. Соединение по любому из пп. 1-22, где R10 представляет собой H или -CH3.
25. Соединение по п. 1, где соединение представляет собой:
(1) 1-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)-2-метилпропан-2-ол;
(2) 4-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-амин;
(3) 3-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)циклопентан-1-ол;
(4) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(5) 6-(2-хлор-3,5-диметоксифенил)-N-(2-морфолиноэтил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(6) 1-((4-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-ил)амино)-2-метилпропан-2-ол;
(7) 6-(2-хлор-3,5-диметоксифенил)-N-(тетрагидро-2H-пиран-4-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(8) 6-(2-хлор-3,5-диметоксифенил)-N-(5-(4-этилпиперазин-1-ил)пиридин-2-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(9) 6-(3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(10) 6-(2-хлор-3,5-диметоксифенил)-N-(4-морфолинофенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(11) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-изопропилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(12) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклопропилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(13) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(оксетан-3-ил)пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(14) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(диметиламино)пиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(15) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-метилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(16) 6-(2-хлор-3,5-диметоксифенил)-N-(5-(4-метилпиперазин-1-ил)пиридин-2-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(17) 6-(2-хлор-3,5-диметоксифенил)-N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(18) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этил-2-оксопиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(19) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этил-3-оксопиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(20) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,4,5-триметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(21) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(N-(2-диметиламиноэтил-N-метиламино)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(22) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(23) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(8-метил-3,8-диазабицикло[3.2.1]октан-3-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(24) (R)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(25) (S)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(26) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(3,3,4-триметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(27) N1-(6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)-N4-метил-N4-(2-морфолиноэтил)бензол-1,4-диамин;
(28) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(2-(пирролидин-1-ил)этокси)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(29) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-морфолинопиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(30) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(диметиламино)пиперидин-1-ил)-2-фторфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(31) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(32) 2-(4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)пиперазин-1-ил)этан-1-ол;
(33) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(2-морфолиноэтокси)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(34) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-этилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(35) 6-(2-хлор-3,5-диметоксифенил)-N-(2-этил-1,2,3,4-тетрагидроизохинолин-6-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(36) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-этилпиперидин-4-ил)-3-метилфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(37) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-метилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(38) (R)-6-(2-хлор-3,5-диметоксифенил)-N-(3-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(39) (R)-6-(3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(40) (R)-2-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)гексагидропирроло[1,2-a]пиразин-4(1H)-он;
(41) (R)-2-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)гексагидропирроло[1,2-a]пиразин-6(2H)-он;
(42) (S)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропиразино[2,1-c][1,4]оксазин-8(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(43) (R)-6-(2-хлор-3,5-диметоксифенил)-N-(4-(гексагидропиразино[2,1-c][1,4]оксазин-8(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(44) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((4-этилпиперазин-1-ил)метил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(45) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(морфолинометил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(46) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((4-метилпиперазин-1-ил)метил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(47) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-циклопропилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(48) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(49) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(метиламино)пиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(50) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(51) N-(4-(3,8-диазабицикло[3.2.1]октан-3-ил)фенил)-6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(52) 6-(2-хлор-3,5-диметоксифенил)-N-(3-метил-4-(пиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(53) 6-(2-хлор-3,5-диметоксифенил)-N-(1,2,3,4-тетрагидроизохинолин-6-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(54) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-метилфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(55) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-метоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(56) 6-(2-хлор-3,5-диметоксифенил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)-3-фторфенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(57) 6-(2-хлор-3,5-диметоксифенил)-N-(3-хлор-4-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(58) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(пиперазин-1-илметил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(59) (4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-((3S,5R)-3,5-диметилпиперазин-1-ил)метанон;
(60) (4-((6-(2,6-дихлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)((3S,5R)-3,5-диметилпиперазин-1-ил)метанон;
(61) 6-(2,6-дихлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(62) 6-(2,6-дихлор-3,5-диметоксифенил)-N-(2-морфолиноэтил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(63) 4-(2,6-дихлор-3,5-диметоксифенил)-N-(2-морфолиноэтил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-амин;
(64) (R)-6-(2,6-дихлор-3,5-диметоксифенил)-N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(65) 6-(2,6-дихлор-3,5-диметоксифенил)-N-(4-(4-морфолинопиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(66) 6-(2-хлор-3,5-диметоксифенил)-N2-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2,9-диамин;
(67) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(68) N-(4-(4-этилпиперазин-1-ил)фенил)-6-(2-метил-1H-бензо[d]имидазол-6-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(69) 6-(4-хлор-2-метил-1H-бензо[d]имидазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(70) 6-(4,6-дихлор-2-метил-1H-бензо[d]имидазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(71) 6-(5-хлор-2-метил-1H-индол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(72) 6-(6-хлор-2-метилбензо[d]тиазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(73) 6-(4-хлор-2-метилбензо[d]тиазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(74) 6-(6-хлор-2-метилбензо[d]оксазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(75) 6-(4-хлор-2-метилбензо[d]оксазол-5-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(76) 6-(5-хлор-2-метилбензо[d]оксазол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(77) 6-(7-хлор-2-метилбензо[d]оксазол-6-ил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(78) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-(4-(диметиламино)пиперидин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(79) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-(1-этилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(80) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-((3S,5R)-3,5-диметилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(81) 6-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-морфолинофенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(82) 6-(5,7-дихлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-морфолинофенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(83) 6-(7-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-N-(4-морфолинофенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(84) N-(4-(гексагидропирроло[1,2-a]пиразин-2(1H)-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(85) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-9-метил-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(86) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-((2-морфолиноэтил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)акриламид;
(87) 4-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-2-метил-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-8-амин;
(88) N-(4-((4-(2-хлор-3,5-диметоксифенил)-8-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)фенил)акриламид;
(89) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-этилпиперазин-1-ил)фенил)-9-(морфолинометил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(90) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)акриламид;
(91) N-(4-(2-(6-(2-хлор-3,5-диметоксифенил)-2-((4-(4-этилпиперазин-1-ил)фенил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)этил)фенил)акриламид;
(92) 6-(2-хлор-3,5-диметоксифенил)-9-((диметиламино)метил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(93) 6-(2-хлор-3,5-диметоксифенил)-9-(циклопропилметил)-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(94) 6-(2-хлор-3,5-диметоксифенил)-9-циклопропил-N-(4-(4-этилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(95) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)акриламид;
(96) N-(4-((6-(2-хлор-3,5-диметоксифенил)-2-((2-морфолиноэтил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)метил)фенил)пропионамид;
(97) N-(4-(2-(6-(2-хлор-3,5-диметоксифенил)-2-((2-морфолиноэтил)амино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)этил)фенил)акриламид;
(98) 4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-1-этилпиперазин-1-оксид;
(99) (8aR)-2-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)октагидро-5H-пирроло[1,2-a]пиразин-5-оксид;
(100) 3-(4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)пиперазин-1-ил)циклобутан-1-ол;
(101) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(1-этилпиперидин-4-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(102) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-(2,2,2-трифторэтил)пиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(103) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклопентилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(104) 6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-циклобутилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(105) (6-(2-хлор-3,5-диметоксифенил)-N-(4-(4-ацетилпиперазин-1-ил)фенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин);
(106) 4-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-N,N-диметилпиперазин-1-карбоксамид;
(107) 6-(4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)-N,N-диметил-2,6-диазаспиро[3.3]гептан-2-карбоксамид;
(108) (S)-3-((4-((6-(2-хлор-3,5-диметоксифенил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-ил)амино)фенил)амино)-N,N-диметилпирролидин-1-карбоксамид;
(109) 6-(2-хлор-3,5-диметоксифенил)-N-(6-(4-метилпиперазин-1-ил)пиридин-3-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(110) 6-(2-хлор-3,5-диметоксифенил)-N-(6-((4-этилпиперазин-1-ил)метил)пиридин-3-ил)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-2-амин;
(111) N-(4-((4-(2-хлор-3,5-диметоксифенил)-8-((2-морфолиноэтил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)фенил)акриламид;
(112) (1-(4-((6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он);
(113) 1-(4-(2-(6-(2-хлор-3,5-диметоксифенил)-2-(метиламино)-[1,2,4]триазоло[4',3':1,6]пиридо[2,3-d]пиримидин-9-ил)этил)пиперазин-1-ил)проп-2-ен-1-он;
(114) 1-(4-(3-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(115) 1-(4-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)пиперазин-1-ил)проп-2-ен-1-он;
(116) 1-(4-((4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(117) 1-(3-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этокси)азетидин-1-ил)проп-2-ен-1-он;
(118) 1-(3-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этокси)пиперидин-1-ил)проп-2-ен-1-он;
(119) N-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)фенил)акриламид;
(120) N-(3-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)фенил)акриламид;
(121) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(122) 1-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(123) 1-(4-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этил)пиперазин-1-ил)проп-2-ен-1-он;
(124) 1-(6-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диазаспиро[3.3]гептан-2-ил)проп-2-ен-1-он;
(125) 1-(4-((2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)циклопропил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(126) 1-(4-(2-(4-(2-хлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)этокси)пиперидин-1-ил)проп-2-ен-1-он;
(127) 1-((3aR,6aS)-5-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)гексагидропирроло[3,4-c]пиррол-2(1H)-ил)проп-2-ен-1-он;
(128) N-(1-(2-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперидин-4-ил)акриламид;
(129) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперидин-1-ил)проп-2-ен-1-он;
(130) 1-((3S,5R)-4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-3,5-диметилпиперазин-1-ил)проп-2-ен-1-он;
(131) 1-(4-((1-((4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)метил)циклопропил)метил)пиперазин-1-ил)проп-2-ен-1-он;
(132) 1-((2R,6S)-4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диметилпиперазин-1-ил)проп-2-ен-1-он;
(133) 1-(4-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)(метил)амино)пиперидин-1-ил)проп-2-ен-1-он;
(134) 1-((2R,6S)-4-(3-(8-((циклопропилметил)амино)-4-(2,6-дихлор-3,5-диметоксифенил)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диметилпиперазин-1-ил)проп-2-ен-1-он;
(135) 1-((2R,6S)-4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-((2,2-дифторэтил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,6-диметилпиперазин-1-ил)проп-2-ен-1-он;
(136) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-((2,2,2-трифторэтил)амино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(137) 1-(2-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,7-диазаспиро[3.5]нонан-7-ил)проп-2-ен-1-он;
(138) 1-((1R,5S,6S)-6-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)(метил)амино)-3-азабицикло[3.1.0]гексан-3-ил)проп-2-ен-1-он;
(139) 1-(2-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,8-диазаспиро[4.5]декан-8-ил)проп-2-ен-1-он;
(140) (S)-1-(3-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)(метил)амино)пиперидин-1-ил)проп-2-ен-1-он;
(141) 1-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-3,3-диметилпиперазин-1-ил)проп-2-ен-1-он;
(142) 1-(4-((3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)сульфонил)пиперидин-1-ил)проп-2-ен-1-он;
(143) 1-(8-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-3,8-диазабицикло[3.2.1]октан-3-ил)проп-2-ен-1-он;
(144) 1-(5-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)-2,5-диазабицикло[2.2.1]гептан-2-ил)проп-2-ен-1-он;
(145) 1-(4-(3-(4-(5-хлор-2-метил-1H-бензо[d]имидазол-6-ил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-ил)проп-2-ен-1-он;
(146) 2-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-карбонил)-4-метилпент-2-еннитрил или
(147) 2-(4-(3-(4-(2,6-дихлор-3,5-диметоксифенил)-8-(метиламино)-[1,2,4]триазоло[1',5':1,6]пиридо[2,3-d]пиримидин-2-ил)пропил)пиперазин-1-карбонил)-4,4-диметилпент-2-еннитрил.
26. Лекарственное средство для лечения заболевания, опосредованного FGFR, содержащее соединение по любому из пп. 1-25 и фармацевтически приемлемое вспомогательное вещество.
27. Лекарственное средство по п. 26, где указанное соединение присутствует в весовом отношении к вспомогательному веществу в диапазоне приблизительно 0,0001:1-10.
28. Применение соединения по любому из пп. 1-25 для получения лекарственного средства для лечения заболевания, опосредованного FGFR.
29. Применение по п. 28, где заболевание представляет собой рак, при этом рак представляет собой множественную миелому, рак мочевого пузыря, рак эндометрия, рак желудка, немелкоклеточный рак легкого, мелкоклеточный рак легкого, плеоморфный рак легкого, колоректальный рак, гепатоцеллюлярную карциному, плоскоклеточный рак легкого.
30. Применение по п. 28, где указанный FGFR включает FGFR1, FGFR2, FGFR3 или FGFR4.
31. Применение соединения по любому из пп. 1-25 для получения лекарственного средства, где лекарственное средство применяется для лечения или предупреждения либо для замедления или предупреждения проявления или прогрессирования рака или метастазов рака, при этом рак представляет собой множественную миелому, рак мочевого пузыря, рак эндометрия, рак желудка, немелкоклеточный рак легкого, мелкоклеточный рак легкого, плеоморфный рак легкого, колоректальный рак, гепатоцеллюлярную карциному, плоскоклеточный рак легкого.
32. Способ лечения состояния, опосредованного FGFR, предусматривающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1-25 или лекарственного средства по п. 26 или 27.
33. Способ по п. 32, где указанный FGFR включает FGFR1, FGFR2, FGFR3 или FGFR4.
34. Способ по п. 32 или 33, где состояние, опосредованное FGFR, представляет собой рак, при этом рак представляет собой множественную миелому, рак мочевого пузыря, рак эндометрия, рак желудка, немелкоклеточный рак легкого, мелкоклеточный рак легкого, плеоморфный рак легкого, колоректальный рак, гепатоцеллюлярную карциному, плоскоклеточный рак легкого.
| WO 2003082871 A1, 20.11.2007 | |||
| CN 105481858 A, 13.04.2016 | |||
| DE 2929414 A1, 05.02.1981 | |||
| US 4666812 A, 19.05.1987 | |||
| ДВУХДИАФРАГМЕННЫЙ НАСОС С ПНЕВМОПРИВОДОМ | 2011 |
|
RU2543372C2 |
| WO 2013007708 A1, 01.17.2013 | |||
| RU 2015141138 A, 03.02.2017 | |||
| RU 2014115478 A, 10.11.2015. | |||

