Область технического применения
Настоящее изобретение касается процесса получения гидрохлорида 2-амино-2-[2-[4(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропан-диола (который далее может называться просто «соединение I»), представляющего собой иммунодепрессант, обладающий незначительными побочными эффектами. Настоящее изобретение касается также новых промежуточных продуктов при получении соединения I.
Предпосылки создания изобретения
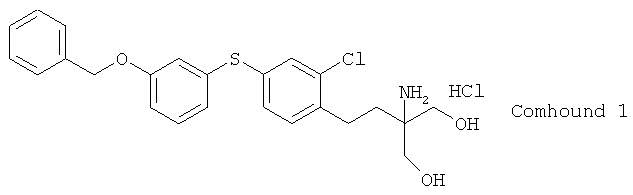
Соединение I представляет собой производное 2-амино-1,3-пропандиола, которое имеет структуру замещенного диарилсульфида и действует как иммунодепрессант. Оно является соединением, полезным с фармацевтической точки зрения при лечении различных заболеваний (включая аутоиммунные заболевания типа ревматоидного артрита, нефрита, артроза коленного сустава и системной красной волчанки; хронических воспалительных заболеваний типа воспалительного заболевания кишечника; а также аллергические заболевания типа астмы и дерматита). (Это соединение описано в примере 46 патента 1). Процесс получения соединения I отдельно описан в международном проспекте патента 1. Однако этот процесс трудно применим в промышленных масштабах, он нуждается в дальнейших усовершенствованиях по многим аспектам, включая эффективность получения и очистки, а также выхода готового продукта. Поэтому имеется существенная потребность в практическом процессе получения соединения I в промышленных масштабах.
Проспект патента 1 WO 03/029205.
Раскрытие настоящего изобретения
Задачи, которые должно решить настоящее изобретение.
Требования, касающиеся промышленного получения соединения I в качестве фармацевтического продукта высокого качества, включают: разработку новых полезных промежуточных продуктов, исключение стадии очистки колоночной хроматографией, усовершенствование процесса получения, увеличение выхода и степени чистоты продукта, а также снижение использования вредных растворителей.
Пути решения поставленных задач
В результате исследования способов, отвечающих указанным требованиям, заявителями было обнаружено, что возможно применение этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилата, 1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио]бензола и 2-[2-[4-(3-бензилокси-фенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола в качестве промежуточных продуктов при получении соединения I; а также то, что использование этих промежуточных упрощает и делает промышленно применимым получение соединения I. Именно этого открытие привело к настоящему изобретению.
В частности, настоящее изобретение касается:
1) Процесса получения гидрохлорида 2-амино-2-[2-[4(3-бензилокси-фенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрата, который включает стадии:
взаимодействия 4-(3-бензилоксифенилтио)-2-хлорбензальдегида с диэтилфосфоноацетатом этила, осуществляемого в растворителе в присутствии основания, с получением этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилата;
восстановления полученного этил 3-[4(3-бензилоксифенилтио)-2-хлорфенил] акрилата с последующими мезилированием, иодинированием и нитрованием, приводящими к 1-бензилокси-3-[3-хлор-4-(3-нитропропил-фенилтио]бензолу;
гидроксиметилирование полученного 1-бензилокси-3-[3-хлор-4-(3-нитропропилфенилтио]бензола формальдегидом, приводящее к 2-[2-[4(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиолу; а также
восстановление полученного 2-[2-[4(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола с получением целевого продукта.
2) промежуточного продукта при получении гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрохлорида, содержащего этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат или его гидрат в качестве нового соединения.
3) промежуточного продукта при получении гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрохлорида, содержащего 1-бензилокси-3-[3-хлор-4-(3-нитро-пропилфенилтио)]бензол или его гидрат в качестве нового соединения.
4) промежуточного продукта при получении гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрохлорида, содержащего гидрохлорид 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрат в качестве нового соединения.
Преимущества настоящего изобретения
Соединения по настоящему изобретению, служащие в качестве исходных, а именно этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат, 1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио]бензол и 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиол, а также их гидраты представляют собой новые соединения, которые никогда не были описаны и полезность которых ранее никогда не обсуждалась.
Каждое из веществ: этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат, 1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио]бензол и 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиол может быть промежуточным продуктом при получении соединения I; а также каждое из них допускает стабильное промышленное получение соединения I с высоким выходом и предусматривает простой способ очистки. Поэтому применение соединений по настоящему изобретению, прежде всего, делает возможным промышленное получение соединения I.
Наилучший способ реализации настоящего изобретения
Процесс по настоящему изобретению, делающий возможным промышленное получение соединения I, осуществляется следующим образом (смотри также схему I): этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат восстанавливают с помощью трихлорида висмута и борогидрида натрия. Восстановленный продукт далее восстанавливают с помощью борогидрида калия и хлорида лития. Полученное в результате спиртовое производное подвергают мезилированию путем взаимодействия метансульфонилхлоридом. Мезилированный продукт подвергают иодинированию с помощью иодида натрия при последующем взаимодействии с нитритом натрия с получением 1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио]бензола. Полученный продукт затем взаимодействует с формальдегидом, процесс проводят в присутствии основания с получением 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола. Восстановление этого соединения гидроксидом палладия приводит к соединению I.
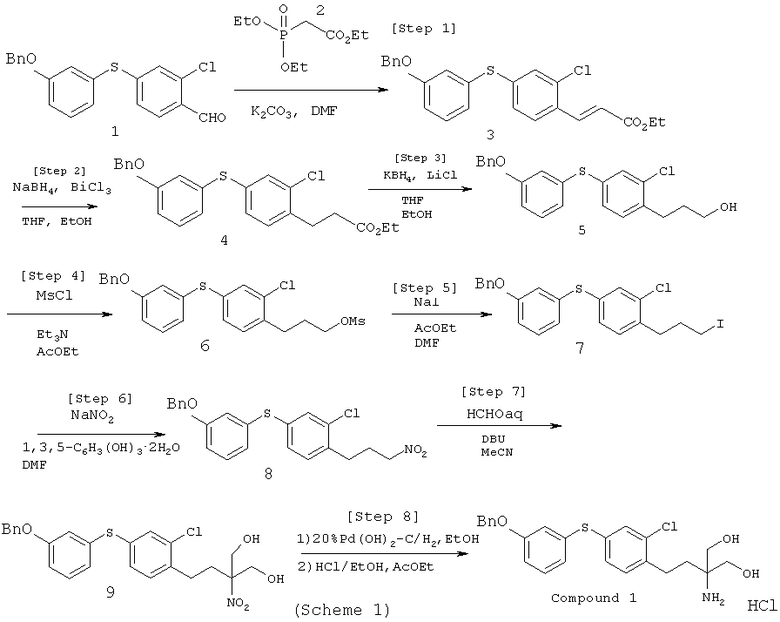
Опишем процесс получения новых соединений по настоящему изобретению, служащих в качестве исходных веществ, а именно этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилата (3), 1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио] бензола (8) и 2-[2-[4-(3-бензилокси-фенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола (9), а также их гидратов.
Смесь 4-(3-бензилоксифенилтио)-2-хлорбензальдегида (1) и диэтилфосфоноацетата этила (2) перемешивают в течение 3-8 ч при температуре в интервале от 50°С до 80°С. Реакцию проводят в органическом растворителе (типа диметилформамида или диметилсульфоксида) в присутствии основания (типа бикарбоната калия, бикарбоната натрия, карбоната калия или карбоната натрия). Затем реакционную смесь при перемешивании охлаждают на ледяной бане, а потом добавляют воду. Получившиеся в результате кристаллы собирают фильтрованием, промывают водным раствором 2-пропанола и получают этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат (3) в виде неочищенных кристаллов. Продукт растворяют в этилацетате, а смесь промывают водой. Органический слой упаривают при пониженном давлении, получая в результате масло бледно-желтого цвета. К этому продукту добавляют ацетон, смесь перемешивают при охлаждении в целях формирования кристаллов (при необходимости добавляют затравку). Затем добавляют воду и полученную смесь перемешивают при ее охлаждении. Образовавшиеся кристаллы собирают фильтрованием, промывают ацетоном-водой, а затем высушивают при температуре 40°С (или ниже) с получением этил 3-[4-(3-бензил-оксифенилтио)-2-хлорфенил]акрилата (3) или его гидрата.
После этого этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат (3) растворяют в тетрагидрофуране, или спирте (типа метанола или этанола), или их смеси. В процессе перемешивания, проводимого при температуре от 30°С до 40°С, добавляют трихлорид висмута или борогидрид натрия. Для того, чтобы завершить восстановление, вводят дополнительные количества трихлорида висмута и борогидрида натрия. Полученный продукт обрабатывают, при этом получают масло бледно-желтого цвета, которое растворяют в органическом растворителе (типа тетрагидрофурана). При выдержке полученной смеси при температуре, составляющей 25°С или ниже, добавляют борогидрид калия, хлорид лития и спирт (типа метанола и этанола). Затем смесь перемешивают в течение 1-3 ч при температуре, составляющей от 30°С до 50°С, а потом обрабатывают. Образовавшееся бледно-желтое масло растворяют в органическом растворителе (типа этилацетата). После этого добавляют метансульфонилхлорид, процесс проводят при температуре, составляющей 20°С или ниже, в присутствии основания (типа триэтиламина и пиридина). Затем смесь перемешивают в течение 1-3 ч и обрабатывают с получением бледно-желтого масла. Полученный продукт растворяют в смешанном растворителе, содержащем, например, этилацетат и диметилформамид. После этого в процессе перемешивания добавляют иодид натрия, а полученную смесь перемешивают в течение 3-5 ч при температуре, составляющей от 50°С до 80°С, обрабатывают и получают желтое масло. Это масло растворяют в органическом растворителе (типа диметилформамида), при перемешивании добавляют флороглюцин и нитрит натрия. Перемешивание продолжают в течение дополнительных 4-6 ч при температуре, составляющей от 20°С до 35°С. Затем добавляют пентагидрат тиосульфата натрия в соляном растворе, после обработки и очистки смеси получают 1-бензилокси-3-[3-хлор-4-(3-нитро-пропил)фенилтио] бензола (8) или его гидрат в виде бледно-желтого масла.
1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио]бензол (8) растворяют в органическом растворителе (типа ацетонитрила и тетрагидрофурана). В процессе охлаждения полученной смеси на водяной бане добавляют раствор формальдегида; реакция протекает в течение 1-3 ч при температуре, составляющей от 0 до 60°, в присутствии основания (типа 1,8-диазабицикло[5.4.0]ундека-7-ена, 1,5-диазабицикло[4.3.0]нон-5-ена, диазабицикло[2.2.2]октана, триалкиламина, гидроксида натрия и гидроксида калия). Затем полученный продукт обрабатывают с получением 2-[2-[4-(3-бензилокси-фенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола (9) или его гидрата.
2-[2-[4-(3-бензилокси-фенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола (9) восстанавливают в органическом растворителе (типа этанола), процесс проводят в присутствии катализатора (типа гидроксид палладия-углерод) при температуре от комнатной до 60°С и под давлением водорода, значение которого составляло от атмосферной величины до 507 кПа. В результате был получен 2-[2-[4-(3-бензилокси-фенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиол. Это соединение растворили в органическом растворителе (типа этанола, 2-пропанола и этилацетата или их смеси) и осуществили его взаимодействие с соляной кислотой, получив при этом гидрохлорид 2-[2-[4-(3-бензилокси-фенилтио)-2-хлор-фенил]этил]-2-нитро-1,3-пропандиола (соединение I). Описанный выше процесс делает возможным простое получение соединения I с хорошим выходом и высокой чистотой.
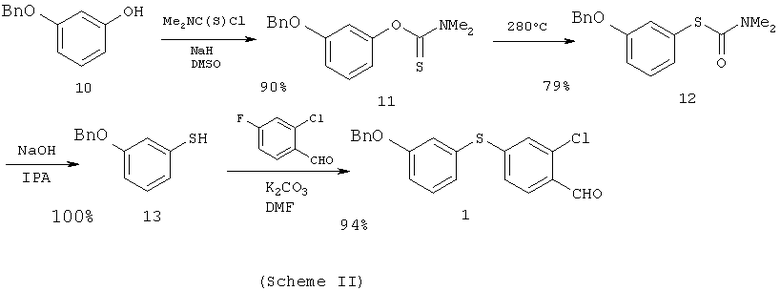
Исходное вещество - 4-(3-бензилоксифенилтио)-2-хлорбензальдегид (1) легко можно получить превращением коммерчески доступного 3-бензилоксифенола (10) в производное тиофенола. Для этого используют диметилтиокарбаматный способ (J.Org.Chem., 31, 3980 (1966)) при последующем восстановлении полученного тиофенольного производного 2-хлор-4-фторбензальдегидом (смотри схему II).
Как было описано выше, настоящее изобретение обеспечивает эффективный процесс промышленного получения соединения I.
Примеры
Ниже настоящее изобретение будет описано со ссылкой на специфичные примеры, которые не предназначены для какого-либо ограничения объема настоящего изобретения.
Контрольный пример 1
O-(3-бензилокси)фенилдиметилтиокарбамат (11)
60% гидрид натрия (19,9 г; 498 ммол) медленно добавили к раствору 3-бензилоксифенола (10) в обезвоженном диметилсульфоксиде (415 мл); процесс проводили при охлаждении водой и перемешивании. Перемешивание проводили в течение 30 мин, после этого при охлаждении водой и перемешивании медленно добавили хлорид диметилтио-карбамоила (61,5 г; 498 ммол). Затем смесь перемешивали при комнатной температуре в течение 1,5 ч, а после этого смесь медленно вылили в ледяную воду (2 л) и провели двукратное (1,5 л и 500 мл) экстрагирование этилацетатом. Органические слои объединили, последовательно промывали водой (2 л) и 28% солевым раствором (1 л), а потом высушили над безводным сульфатом натрия. Растворитель выпарили, а остаток очистили колоночной хроматографией на силикагеле (силикагель 60, 3 л, гексан:этилацетат = 10:15→1), получив в результате O-(3-бензилокси)фенилдиметилтиокарбамат (2) в виде масла бледно-желтого цвета (107 г; 90%).
400 MHz 1H-ЯМР (СDСl3) δ: 3.32 (3Н, s), 3.45 (3H, s), 5.04 (2H, s), 6.68-6.73 (2H, m), 6.86-6.89 (1H, m), 7.25-7.44 (6H, m).
EI-MS m/z: 287 (M+).
Контрольный пример 2
S-(3-бензилокси)фенилдиметилтиокарбамат (12)
O-(3-бензилокси)фенилдиметилтиокарбамат (11) (100 г; 348 ммол) перемешивали в течение 1 ч при внутриконтурной температуре, составляющей от 270°С до 285°С, и пониженном давлении (4,53 кПа). После того как смесь охладилась до комнатной температуры, добавили диизопропиловый эфир (200 мл), процесс проводили при внутриконтурной температуре, составляющей 50°С. В целях кристаллизации смесь перемешивали при комнатной температуре, затем добавили диизопропиловый эфир (100 мл) и смесь перемешивали в течение 1 ч при ее охлаждении в ледяной воде. Образовавшиеся кристаллы собрали фильтрованием и промыли диизопропиловым эфиром (50 мл), охлажденным с помощью льда. После этого промытый продукт высушивали в течение 3 ч при пониженном давлении, получив в результате S-(3-бензилокси)фенилдиметилтиокарбамат (3) в виде белого кристаллического порошка (79,3 г; 79%).
Температура плавления 68-69°С.
400 MHz 1Н-ЯМР (СDСl3) δ: 3.04 (3Н, br s), 3.09 (3H, br s), 5.05 (2H, s), 6.99-7.16 (3Н, m), 7.27-7.44 (6H, m).
EI-MS m/z: 287 (M+).
Контрольный пример 3
3-бензилоксибензолтиол (13)
К S-(3-бензилокси)фенилдиметилтиокарбамату (12) (40,0 г; 139 ммол) добавили 2-пропанол (160 мл), гидроксид натрия (22,2 г; 556 ммол) и очищенную воду (80 мл), полученную смесь перегоняли в колбе с обратным холодильником в атмосфере аргона. Затем смесь перемешали в ледяной воде и по каплям добавляли соляную кислоту (120 мл). После этого добавили очищенную воду (300 мл) и образовавшиеся кристаллы собрали фильтрованием. Полученный продукт промыли 20% водным пропанолом (300 мл), промытое соединение высушивали в течение 2 суток на воздухе при комнатной температуре. В результате был получен 3-бензилоксибензолтиол (4) в виде кристаллического порошка бледно-желтого цвета (30,1 г; 100%).
Температура плавления 49-50°С.
400 MHz 1Н-ЯМР (d6-DMSO) δ: 5.06 (2Н, s), 5.41 (1H, s), 6.74-6.97 (3Н, m), 7.14 (1H, t, J=7.8 Hz), 7.30-7.44 (5H, m).
EI-MS m/z: 216 (M+).
Контрольный пример 4
4-(3-бензилоксифенилтио)-2хлорбензальдегид (1)
3-бензилоксибензолтиол (13) (75,0 г; 347 ммол) и 2-хлор-4-фторбензальдегид (55,0 г; 347 ммол) растворили в диметилформамиде (350 мл), процесс проводили при перемешивании. Затем полученный раствор поместили на водяную баню, имеющую температуру 30°С, и добавили карбонат калия (62,3 г; 451 ммол) и диметилформамид (25 мл). Образовавшуюся смесь перемешивали в течение 30 мин при температуре внутреннего контура, составляющей от 40°С до 42°С. После этого добавили водопроводную воду (125 мл) и смесь охладили в ледяной воде. В целях кристаллизации к смеси по каплям добавляли дополнительное количество водопроводной воды (625 мл). Смесь перемешивали в течение 30 мин, образовавшиеся кристаллы собрали фильтрованием и промыли 10% водным раствором 2-пропанола (188 мл). Влажные кристаллы растворили в этилацетате (750 мл) и раствор дважды (375 мл × 2) промывали водопроводной водой. Органический слой упарили при пониженном давлении, а к образовавшемуся остатку добавили ацетон (900 мл). Затем по каплям добавляли очищенную воду (386 мл), процесс проводили при температуре внутреннего контура, составляющей 23°С. Смесь перемешивали в течение 30 мин, образовавшиеся кристаллы собрали фильтрованием и промыли 50% водным раствором ацетона (400 мл). Промытые кристаллы высушивали на воздухе в течение ночи. После этого полученный продукт был высушен обдувкой воздушным потоком, процесс проводили при 45°С в течение 1 ч. В результате был получен 4-(3-бензилоксибензолтио)-2-хлорбензальдегид (1) в виде бесцветных призматических кристаллов (115 г; 94%).
Температура плавления 58-59°С.
400 MHz 1H-ЯМР (CDCl3) δ: 5.07 (2H, s), 7.04-7.14 (5H, m), 7.31-7.43 (6H, m), 7.75 (1H, d, J=8.3 Hz), 10.35 (1H, d, J=0.7 Hz).
EI-MS m/z: 354 (M+).
Пример 1
Этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат (3)
4-(3-бензилоксифенилтио)-2-хлорбензальдегид (1) (115 г; 325 ммол) и диэтилфосфоноацетат этила (2) (94,6 г; 422 ммол) добавили в диметилформамид (518 мл). В целях растворения всех соединений смесь перемешали. Затем добавили карбонат калия (58,3 г; 422 ммол) и полученную смесь перемешивали в течение 5,5 ч при температуре внешнего контура, составляющей от 70°С до 73°С.
При охлаждении этой смеси водой и ее перемешивании при температуре внешнего контура, составляющей 40°С, добавили водопроводную воду и в процессе охлаждения продолжали дальнейшее перемешивание. При температуре внешнего контура, составляющей 10°С, ввели затравку для усиления кристаллообразования. После этого по каплям медленно добавляли водопроводную воду и образовавшуюся смесь перемешивали в течение 30 мин при температуре внешнего контура, составляющей от 3°С до 10°С. Закристаллизовавшееся твердое соединение собрали при фильтровании, промыли 10% водным 2-пропанолом (518 мл), при этом в виде неочищенного продукта получили влажные кристаллы. Полученный продукт растворили в этилацетате (806 мл), а раствор дважды промывали водой (403 мл).
Органический слой упаривали при пониженном давлении, концентрат высушивали в течение 30 мин при температуре внешнего контура, составляющей 40°С, получив при этом масло бледно-желтого цвета. Полученный продукт растворили в ацетоне (691 мл). В процессе охлаждения смеси и ее перемешивании добавили очищенную воду (95 мл). При температуре внешнего контура, составляющей от 5°С до 10°С, ввели затравку в целях кристаллообразования. Смесь при ее охлаждении перемешивали в течение дополнительных 5 мин. Затем по каплям добавляли очищенную воду (366 мл) и смесь перемешивали в течение 30 мин при температуре внешнего контура, составляющей от 5°С до 10°С.
Закристаллизовавшееся твердое соединение собрали при фильтровании и промыли 40% водным ацетоном (461 мл); процесс проводили при температуре внешнего контура, составляющей 4°С. Промытый продукт высушивали в течение 50 мин в токе воздуха, а затем - на открытом воздухе при комнатной температуре. Дальнейшее высушивание проводили в токе воздуха в течение 8 ч при 38°С. В результате было получено соединение (3) в виде бледно-желтых сыпучих кристаллов (136 г; 320 ммол; выход 98%).
Температура плавления 45-47°С (метод горячей пластины).
EI-MS m/z: 424 (М+).
1Н-ЯМР (CDCl3, 400 MHz) δ: 1.34 (3H, t, J=7.1 Hz), 4.27 (2H, q, J=7.1 Hz), 5.05 (2H, s), 6.38 (1H, d, J=16.1 Hz), 6.92-7.11 (4H, m), 7.23-7.49 (8H, m), 8.02 (1H, d.J-15.9 Hz).
Пример 2
1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио]бензол (8)
Тетрагидрофуран (ТГФ) (405 мл) и этанол (405 мл) добавили к этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилату (3) (135 г; 318 ммол). В процессе перемешивания смеси при температуре внешнего контура, составляющей 35°С, добавили трихлорид висмута (25,0 г; 79,4 ммол). Затем медленно добавили борогидрид натрия (15,0 г; 397 ммол). Перемешивание полученной смеси продолжали при ее охлаждении и медленно дополнительно добавили борогидрид натрия (15,0 г; 397 ммол). Продолжили перемешивание смеси при ее охлаждении. После этого при температуре внешнего контура, составляющей 40°С, добавили трихлорид висмута (5,01 г; 15,9 ммол) и борогидрид натрия (6,01 г; 159 ммол). Процесс проводили в течение 55 мин при температуре внешнего контура, составляющей от 40°С до 46°С.
В процессе перемешивания реакционной смеси и ее охлаждении добавили ацетон (41 мл), процесс проводили при температуре внешнего контура, составляющей 10°С, перемешивание осуществляли в течение 5 мин. Затем, для того чтобы довести значение рН до 1, добавили 1 мол/л соляной кислоты (810 мл) и этилацетат (675 мл) и в течение 30 мин проводили перемешивание.
Образовавшееся в результате реакции твердое вещество черного цвета отделили фильтрованием и промыли этилацетатом (135 мл). Органический слой отделили, собрали и последовательно промывали 0,1 мол/л соляной кислотой (405 мл), 5% водным раствором бикарбоната натрия (405 мл) и 28% солевым раствором (405 мл).
Органический слой упарили при пониженном давлении, высушивали в течение 30 мин при температуре внешнего контура, составляющей 50°С, получив в результате 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]пропионат этила (4) в виде масла бледно-желтого цвета (132 г; количество, эквивалентное 318 ммол). Без проведения очистки соединения (4) его растворили в тетрагидрофуране (949 мл). При перемешивании смеси добавили борогидрид калия (22,3 г; 413 ммол) и хлорид лития (17,5 г; 413 ммол); процесс проводили в течение 20 мин при температуре внутреннего контура, составляющей 20°С.
После этого добавили этанол (47,5 мл) и смесь перемешивали в течение 1 ч при температуре внутреннего контура, составляющей от 30°С до 33°С. Затем перемешивание проводили в течение дополнительных 40 мин, температура внутреннего контура составляла от 44°С до 45°С. Добавили этанол (23,7 мл) и для завершения реакции смесь перемешивали при температуре внутреннего контура, составляющей от 43°С до 45°С. В процессе охлаждения реакционной смеси и ее перемешивании добавили ацетон (71,2 мл), а затем воду (475 мл). Смесь перемешивали при охлаждении в течение 1 ч. После этого последовательно добавили соляную кислоту (217 мл; 434 ммол) и этилацетат (475 мл) при температуре внутреннего контура, составляющей 26°С, и продолжили перемешивание смеси.
Органический слой отделили, собрали и последовательно промывали 5% водным раствором бикарбоната натрия (475 мл) и 28% солевым раствором (475 мл). Затем органический слой упаривали при пониженном давлении и высушивали в течение 1 ч при температуре внешнего контура, составляющей 50°С. В результате был получен 3-[4-(3-бензилокси-фенилтио)-2-хлорфенил]пропанол (5) (124 г; количество, эквивалентное 318 ммол) в виде масла бледно-желтого цвета.
Без проведения очистки соединения (4) его растворили в этилацетате (1,22 л). Добавили триэтиламин (64,3 г; 635 ммол), и в процессе охлаждения смеси и при ее перемешивании медленно по каплям добавляли метансульфонилхлорид (54,6 г; 477 ммол); процесс проводили при температуре внутреннего контура, составляющей от 6°С до 15°С. После этого добавили водопроводную воду (1,22 л) и смесь перемешивали в течение 20 мин. Затем добавили 6 мол/л соляную кислоту (26,3 мл; 158 ммол), после этого органический слой отделили и собрали.
Органический слой последовательно промывали 2% водным раствором сульфата натрия (1,22 л) и 30% водным раствором сульфата натрия (612 мл). Затем органический слой упарили при пониженном давлении, а концентрат высушивали в течение 1 ч при пониженном давлении и при температуре внешнего контура, составляющей 50°С. В результате был получен 3-[4-(3-бензилоксифенилтио)-2-хлор-фенил]пропилметансульфонат (6) в виде масла бледно-желтого цвета (150 г; количество, эквивалентное 318 ммол).
Без проведения очистки соединение (6) растворили в этилацетате (441 мл) и диметилформамиде (441 мл). В процессе перемешивания полученной смеси добавили иодид натрия (61,9 г; 413 ммол), затем смесь перемешивали в течение 3 ч при температуре внутреннего контура, составляющей от 62°С до 65°С. При перемешивании и охлаждении смеси добавили 8% водный раствор сульфата натрия (1,32 л) и этилацетат (882 мл), процесс проводили при температуре внутреннего контура, составляющей 30°С. Органический слой отделили и собрали.
Образовавшийся органический слой промыли, добавив пентагидрат тиосульфата натрия (7,35 г) в 8% водном растворе сульфата натрия (1,32 л). Затем органический слой промыли 30% водным раствором сульфата натрия (662 мл), органический слой отделили, собрали и упарили при пониженном давлении. Получившийся концентрат высушивали в течение 1 ч при пониженном давлении и при температуре внешнего контура, составляющей 50°С. В результате получили 1-бензилокси-3-[3-хлор-4-(3-иодопропил)фенилтио]бензол (7) в виде желтого масла (160 г; количество, эквивалентное 318 ммол). Без проведения очистки соединения (7) его растворили в диметилформамиде (472 мл). При перемешивании смеси добавили дигидрат флороглюцина (12,9 г; 79,4 ммол), а затем - нитрит натрия (28,5 г; 413 ммол). Получившуюся смесь перемешивали при температуре внутреннего контура, составляющей от 29°С до 32°С.
После того как смесь перемешали и охладили, в целях экстракции в нее добавили пентагидрат тиосульфата натрия (78,9 г; 318 ммол) в 5% солевом растворе (2,36 л) и этилацетат (1,57 л). Органический слой отделили, собрали и промывали вначале 5% солевым раствором (1,57 л), затем дважды 5% водным раствором бикарбоната натрия (1,57 л), а в завершение - 28% солевым раствором (786 мл). После этого органический слой упарили при пониженном давлении, а концентрат высушивали в течение 15 мин при температуре внешнего контура, составляющей 50°С. Образовавшийся маслянистый остаток растворили в толуоле (157 мл), а полученный раствор упарили при пониженном давлении.
К полученному концентрату добавили дополнительное количество толуола (157 мл) и раствор упарили при пониженном давлении. Концентрат высушивали в течение 30 мин при температуре внешнего контура, составляющей 50°С. В результате получили масло коричневого цвета (134 г), которое растворили в толуоле (236 мл). Образовавшийся раствор очищали с помощью колоночной хроматографии на силикагеле (силикагель = 393 г); используя в качестве элюанта толуол. Целевые фракции объединили и упарили при пониженном давлении. Концентрат высушивали в течение 15 мин при температуре внешнего контура, составляющей 50°С. Полученный в результате маслянистый остаток растворили в этилацетате (157 мл) и этот раствор упаривали при пониженном давлении. Затем ввели дополнительное количество этилацетата (157 мл) и раствор вновь упаривали при пониженном давлении. Потом концентрат высушивали при пониженном давлении в течение 30 мин при температуре внешнего контура, составляющей 50°С. В результате был получен 1-бензилокси-3-[3-хлор-4-(3-нитро-пропил)фенилтио]бензол (8) в виде масла бледно-желтого цвета (72,4 г; 175 ммол; выход 55%).
EI-MS m/z: 413 (M+).
1H-ЯМР (CDCl3, 400 MHz) δ: 2.32 (2H, quint., J=7.1 Hz), 2.81 (2H, t, J=7.3 Hz), 4.40 (2H, t, J=7.1 Hz), 5.03 (2H, s), 6.89-6.98 (3H, m), 7.11-7.41 (9H, m).
Пример 3
2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил1-2-нитро-1,3-пропандиол (9)
1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио] бензол (8) (72,3 г; 175 ммол) растворили в ацетонитриле (217 мл). В процессе охлаждения полученного раствора и его перемешивании добавили 37% раствор формальдегида (14,5 мл; 179 ммол). Затем по каплям медленно добавляли 1,8-диазабицикло[5.4.0]ундека-7-ен (DBU) (2,66 г; 17,5 ммол), процесс проводили при температуре внешнего контура, составляющей от 0°С до 8°С. Получившуюся смесь перемешивали в течение 10 мин. После этого добавили 37% раствор формальдегида (14,6 мл; 180 ммол), процесс проводили при температуре внешнего контура, составляющей 10°С или ниже. Смесь перемешивали в течение 1,5 ч при температуре внешнего контура, составляющей от 2°С до 3°С.
К реакционной смеси добавили 1 мол/л соляной кислоты (17,5 мл) для того, чтобы довести рН до значения от 3 до 4; смесь перемешивали в течение 15 мин. Затем реакционную смесь упаривали при пониженном давлении в течение 15 мин. Образовавшийся маслянистый остаток растворили в этаноле (217 мл) и 2 мол/л соляной кислоте (108 мл). Этот раствор перемешивали, процесс проводили в течение 30 мин при температуре внешнего контура, составляющей 45°С. Затем реакционную смесь упаривали при пониженном давлении. К образовавшемуся маслянистому остатку в целях экстрагирования добавили этилацетат (723 мл) и 2% солевой раствор, органический слой отделили и собрали.
Органический слой последовательно промывали 5% водным раствором бикарбоната натрия (361 мл) и 2% солевым раствором (361 мл), а затем упарили при пониженном давлении. Полученный концентрат высушивали в течение 15 мин при пониженном давлении и при температуре внешнего контура, составляющей 50°С. К образовавшемуся маслянистому остатку добавили метанол (72 мл) и полученный раствор упаривали при пониженном давлении. Затем ввели дополнительное количество метанола (72 мл) и этот раствор вновь упаривали при пониженном давлении. После этого концентрат высушивали при пониженном давлении в течение 30 мин при температуре внешнего контура, составляющей 50°С. В результате было получено масло бледно-желтого цвета (83,8 г).
Полученный продукт растворили в метаноле (723 мл). В процессе охлаждения и перемешивания образовавшегося раствора добавили водопроводную воду, процесс проводили при температуре внутреннего контура, составляющей от 5°С до 10°С. К полученному мутному раствору добавили кристаллическую затравку. Когда произошло усиление образования кристаллов, смесь перемешивали в течение приблизительно 15 мин. При перемешивании по каплям добавляли водопроводную воду (593 мл). В процессе перемешивания произошло охлаждение смеси, ее перемешивание дополнительно продолжали в течение 1 ч и при температуре внутреннего контура, составляющей от 5°С до 10°С. Затем смесь выдерживали в течение ночи при комнатной температуре. Закристаллизовавшееся твердое соединение собрали при фильтровании и промыли 50% водным метанолом (145 мл). Образовавшиеся влажные кристаллы высушивали вначале путем обдува током воздуха при комнатной температуре, а затем - обдувом током воздуха при 30°С в течение 16 ч. В результате было получено соединение (4) в виде кристаллического порошка бледно-желтого цвета (80,7 г; 170 ммол; выход 98%).
Температура плавления 69-70°С (метод горячей пластины).
FAB-MS (положительный) m/z: 473[M-H]+.
1H-ЯМР (CDCl3, 400 MHz) δ: 2.12-2.17 (2H, m), 2.54 (2H, t, J=6.8 Hz), 2.65-2.69 (2H, m), 4.08 (2H, dd, J=6.3, 12.2 Hz), 4.28 (2H, dd, J=7.1, 12.5 Hz), 5.02 (2H, s), 6.88-6.96 (3H, m), 7.08-7.41 (9H, m).
Пример 4
Гидрохлорид 2-амино-2-[2[4-(3-бензилоксифенилтио)-2-хлор-
фенил]этил]-1,3-пропандиола (соединение II)
2-амино-2-[2[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиол (9) (80,2 г; 169 ммол) растворили в этаноле (802 мл). После того как атмосферу внутреннего контура заменили на атмосферу инертного газа (аргона), добавили 20% гидроксид палладия на углероде (61,5 г; 32,1 г сухого продукта). Затем атмосферу заменили на водородную. При присоединенном баллоне водорода полученную реакционную смесь энергично перемешивали в течение 24 ч и при температуре внешнего контура, составляющей 50°С.
Атмосферу заменили на атмосферу инертного газа (аргона), а катализатор отделили фильтрованием, процесс проводили при температуре внешнего контура, составляющей 30°С. Оставшуюся смесь промыли 90% водным этанолом (401 мл). Фильтрат упаривали при пониженном давлении, а потом концентрат высушивали в течение 15 мин при пониженном давлении и при температуре внешнего контура, составляющей 50°С. К полученному продукту добавили ацетонитрил (80 мл) и смесь упаривали при пониженном давлении. Ввели дополнительное количество ацетонитрила (80 мл) и при пониженном давлении смесь упарили еще раз. Полученный концентрат высушивали при пониженном давлении в течение 30 мин и при температуре внешнего контура, составляющей 50°С.
К образовавшемуся остатку добавили ацетонитрил (401 мл) и смесь перемешивали в течение 30 мин при температуре внутреннего контура, составляющей от 65°С до 70°С. После этого смесь отставили для охлаждения, при этом перемешивание продолжалось, а затем охладили при перемешивании. После этого смесь перемешивали в течение 30 мин при температуре внутреннего контура, составляющей от 5°С до 10°С. Закристаллизовавшееся твердое вещество собрали при фильтровании, промыли ацетонитрилом (160 мл, температура внутреннего контура 1°С) и высушивали при комнатной температуре, получив в результате неочищенные кристаллы (44,9 г). К полученному продукту добавили ацетонитрил (201 мл) и смесь перемешивали в течение 30 мин при температуре внутреннего контура, составляющей от 65°С до 70°С. После этого смесь отставили для охлаждения, при этом перемешивание продолжалось, а затем смесь перемешивали в течение 1 ч при температуре внутреннего контура, составляющей от 3°С до 10°С. Закристаллизовавшийся продукт собрали при фильтровании и промыли ацетонитрилом (120 мл, температура внутреннего контура 2°С).
Промытый продукт высушивали под током воздуха, а затем в течение 3 ч - путем обдува воздушным потоком при 50°С. В результате был получен порошок белого цвета (42,4 г). К этому продукту добавили ацетонитрил (170 мл) и очищенную воду (170 мл), смесь перемешивали и нагревали для того, чтобы растворить полученное твердое вещество. Раствор охладили при перемешивании, в результате этого при температуре внутреннего контура, составляющей 40°С, произошло кристаллообразование. Смесь вновь охладили при перемешивании, а затем перемешивание продолжали в течение 1 ч при температуре внешнего контура, составляющей от 3°С до 10°С. Закристаллизовавшееся твердое вещество отделили фильтрованием и промыли 50% водным ацетонитрилом (127 мл; внутренняя температура 2°С). Промытый продукт высушивали в токе воздуха в течение 1 ч. Затем провели дальнейшее высушивание в течение 14 ч путем обдува током воздуха, процесс проводили при 50°С. В результате был получен белый кристаллический порошок (38,4 г). К этому продукту (38,2 г) добавили этанол (229 мл), и полученную смесь перемешивали и нагревали для растворения твердого вещества. Раствор отфильтровали и промыли этанолом (38 мл).
Фильтрат перемешали при его одновременном нагревании и добавили 6 мол/л соляную кислоту (15,8 мл; 94,8 ммол), процесс проводили при температуре внутреннего контура, составляющей 60°С (кристаллизация выросла). Затем провели добавку этилацетата (535 мл) и в течение 10 мин проводили перемешивание при температуре внутреннего контура, составляющей от 50°С до 58°С. После этого смесь охлаждали, продолжая ее перемешивание, а затем дальнейшее перемешивание проводили при температуре внутреннего контура, составляющей от 1°С до 10°С. Закристаллизовавшийся продукт собрали при фильтровании, и промывали смесью этанола (57 мл) и этилацетата (57 мл). Промытый продукт высушивали в течение 1 ч под током воздуха; в результате были получены влажные кристаллы, которые затем высушивали в течение 15 ч путем обдува током воздуха при 60°С. Высушенный продукт был измельчен, в результате было получено соединение I в виде белого кристаллического порошка (38,3 г; 79,7 ммол; 47% выход).
Температура плавления 158-160°С (метод горячей пластины).
FAB-MS (положительный) m/z: 444 [С24Н26СlNО3S+Н]+.
Элементный анализ: С24Н26СlNО3S·НСl, вычислено: С 60.00, Н 5.66, N 2.92; найдено: С 59.83, Н 5.51, N 2.85.
1Н-ЯМР (DMSO-d6, 400 MHz) δ: 1.75-1.79 (2H, m), 2.71-2.75 (2H, m), 3.54 (4H, d, J=5.1 Hz), 5.08 (2H, s), 5.42 (2H, t, J=4.9 Hz), 6.88-7.00 (3H, m), 7.23-7.41 (9H, m), 7.93 (3H, brs).
Промышленная применимость
Применение одного из новых соединений по настоящему изобретению (а именно этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат, 1-бензилокси-3-[3-хлор-4-(3-нитропропил)фенилтио]бензол и 2-[2-[4-(3-бензилокси-фенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиол) в качестве промежуточных продуктов при получении соединения I; эффективного иммунодепрессанта делает возможным простое и промышленно применимое получение соединения I с высокой степенью чистоты и стабильным выходом. Было показано, что настоящее изобретение предлагает путь обеспечения соединения I с высоким качеством и при высоком выходе.
Настоящим изобретением установлено промышленное преимущество процесса получения соединения I в качестве эффективного иммунодепрессанта. Соединение I, полученное согласно процессу по настоящему изобретению, можно использовать как фармацевтический продукт, обладающий высокой степенью чистоты и высоким качеством.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КРИСТАЛЛИЗАЦИИ ГИДРОХЛОРИДА 2-АМИНО-2-[2-[4-(3-БЕНЗИЛОКСИФЕНИЛТИО)-2-ХЛОРФЕНИЛ]ЭТИЛ]-1,3-ПРОПАНДИОЛА | 2009 |
|
RU2482110C2 |
| 5-[3-(4-БЕНЗИЛОКСИФЕНИЛТИО)-ФУР-2-ИЛ]-ИМИДАЗОЛИДИН-2,4-ДИОН И АНАЛОГИ В КАЧЕСТВЕ ИНГИБИТОРОВ ЭЛАСТАЗЫ МАКРОФАГОВ | 2005 |
|
RU2391339C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ КИШЕЧНИКА, СОДЕРЖАЩЕЕ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА ПРОИЗВОДНОЕ 2-АМИНО-1,3-ПРОПАНДИОЛА, ИЛИ СПОСОБ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ КИШЕЧНИКА | 2007 |
|
RU2428976C2 |
| КОМБИНАЦИЯ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АГОНИСТА БЕТА-2-АДРЕНОРЕЦЕПТОРА | 2008 |
|
RU2460527C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| КОМПОЗИЦИИ НА ОСНОВЕ СОЕДИНЕНИЙ 2-АМИНО-1,3-ПРОПАНДИОЛА | 2010 |
|
RU2468793C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2023 |
|
RU2814846C1 |
| ПРОИЗВОДНЫЕ 3-ФЕНИЛПИРАЗОЛА, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ФЕНИЛПИРАЗОЛА И ФУНГИЦИДНАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2072991C1 |
| ЗАМЕЩЕННЫЕ ПИРРОЛИДИН-2-КАРБОКСАМИДЫ | 2011 |
|
RU2564022C2 |
| ПРОИЗВОДНЫЕ 2-ПИПЕРИДИНО-1-АЛКАНОЛА | 1991 |
|
RU2029769C1 |
Изобретение относится к способу получения гидрохлорида 2-амино-2-[2-[4-(3-бензилокси-фенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрата, включающий стадии: взаимодействия 4-(3-бензилоксифенилтио)-2-хлорбензальдегида и диэтилфосфоноацетата этила в растворителе в присутствии основания с получением этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил] акрилата; восстановления образовавшегося этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилата при последующих мезилировании, иодинировании и нитровании с получением 1-бензилокси-3-[3-хлор-4-(3-нитропропил-фенилтио]бензола; гидроксиметилирования образовавшегося 1-бензилокси-3-[3-хлор-4-(3-нитропропилфенилтио]бензола формальдегидом с получением 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола; а также восстановления образовавшегося 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола с получением целевого продукта. Изобретение также относится к промежуточным продуктам: этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилату, 1-бензилокси-3-[3-хлор-4-(3-нитропропилфенилтио)]бензолу, 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиолу или к их гидратам. Техническим результатом является промышленное получение гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола с хорошим выходом и высокой чистотой. 4 н.п. ф-лы.
1. Процесс получения гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрата, включающий стадии:
взаимодействия 4-(3-бензилоксифенилтио)-2-хлорбензальдегида и диэтилфосфоноацетата этила в растворителе в присутствии основания с получением этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилата;
восстановления образовавшегося этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилата при последующих мезилировании, иодинировании и нитровании с получением 1-бензилокси-3-[3-хлор-4-(3-нитропропил-фенилтио] бензола;
гидроксиметилирования образовавшегося 1-бензилокси-3-[3-хлор-4-(3-нитропропилфенилтио] бензола формальдегидом с получением 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола, а также восстановления образовавшегося 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиола с получением целевого продукта.
2. Промежуточный продукт получения гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрата, содержащий новое соединение - этил 3-[4-(3-бензилоксифенилтио)-2-хлорфенил]акрилат или его гидрат.
3. Промежуточный продукт получения гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрата, содержащий новое соединение - 1-бензилокси-3-[3-хлор-4-(3-нитропропилфенилтио)] бензол или его гидрат.
4. Промежуточный продукт получения гидрохлорида 2-амино-2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-1,3-пропандиола или его гидрата, содержащий новое соединение - 2-[2-[4-(3-бензилоксифенилтио)-2-хлорфенил]этил]-2-нитро-1,3-пропандиол или его гидрат.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ получения замещенных дифенилсульфидов или их солей | 1976 |
|
SU697050A3 |

