Область техники
Настоящее изобретение относится к способу получения производного дифенилметана и, в частности, к усовершенствованному способу получения производного дифенилметана, которое используется в качестве ингибитора натрий-зависимого котранспортера глюкозы (SGLT).
Уровень техники
Натрий-зависимый котранспортер глюкозы (SGLT) обеспечивает транспорт Na+, который осуществляется в соответствии с градиентом концентрации, и, в то же время, обеспечивает транспорт глюкозы, который не соответствует градиенту концентрации. В настоящее время были клонированы две важные изоформы SGLT, известные как SGLT1 и SGLT2. SGLT1 расположен в кишечнике, почках и сердце, и регулирует транспорт глюкозы в сердце посредством его экспрессии. Благодаря тому, что SGLT1 является высокоаффинным низкопроизводительным переносчиком, он отвечает только за реабсорбцию глюкозы в почках. С другой стороны, SGLT2 представляет собой низкоаффинный высокопроизводительный транспортер, который в основном расположен в апикальных доменах эпителиальных клеток в начальном отделе проксимального извитого канальца. У здоровых людей более 99% глюкозы в плазме, которая фильтруется в почечных клубочках, реабсорбируется, и менее 1% от общего количества отфильтрованной глюкозы выделяется с мочой. Предполагается, что SGLT2 обеспечивает 90% реабсорбции почечной глюкозы, а оставшиеся 10% опосредуются SGLT1 в конечном отделе проксимального прямого канальца. Генетическая мутация SGLT2 не оказывает каких-либо конкретных неблагоприятных воздействий на углеводный обмен. Однако повышенная секреция глюкозы в почках, составляющая около до 140 г/день, вызывается в зависимости от мутации. Согласно исследованиям мутации у людей, SGLT2 был предметом терапевтических исследований, потому что, по имеющимся оценкам, SGLT2 отвечает за большую часть реабсорбции глюкозы в почках.
В выложенной патентной публикации США №2015/0152075 описано соединение, имеющее дифенилметановый фрагмент, который обладает ингибирующей активностью в отношении SGLT2, а также способ его получения. В документе описано, что соединение производного дифенилметана является эффективным в лечении диабета, поскольку соединение производное дифенилметана демонстрирует превосходный ингибирующий эффект в отношении активности SGLT2 человека и значительно снижает экскрецию сахара с мочой у животных по сравнению с препаратом дапаглифлозином, который хорошо известен в качестве ингибитора SGLT2. Кроме того, в выложенной патентной публикации США №2014/0274918 описано производное дифенилметана, которое эффективно в качестве двойного ингибитора натрий-зависимого котранспортера глюкозы 1 (SGLT1) и натрий-зависимого котранспортера глюкозы 2 (SGLT2).
В примере 172 выложенной патентной публикации США №2015/0152075 или т.п. описан способ получения дифенилметанового соединения с28, аналогичный представленному на следующей реакционной схеме 1.
[Реакционная схема 1]
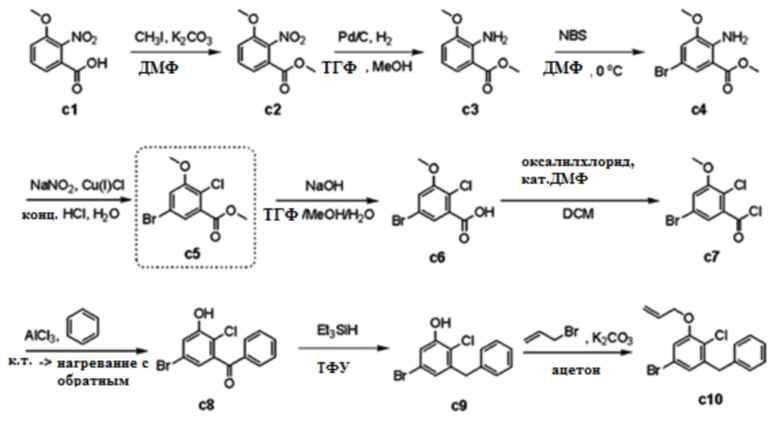

Однако, согласно общепринятому способу получения соединения с28, используется метод линейного синтеза, такой как образование пятиугольного кольца в агликоновой группе после связывания с глюкозной группой. В случае такого линейного синтеза, вследствие сложного пути синтеза, получается низкий конечный выход продукта. Кроме того, в случае, когда синтез заместителя глюкозной группы или циклопропилбензильной группы, которая связана с дигидробензофураном, протекает несоответствующим образом в середине синтеза, или, если подразумевается, что заместитель или циклопропилбензильная группа замещается на другую группу, имеет место недостаток, связанный с необходимостью проведения ее синтеза снова с самого начала. Кроме того, даже для процесса синтеза циклопропильной группы соединения с28, этот процесс осуществляют путем циклизации олефина посредством реакции Саймонса-Смита в конце пути синтеза, вследствие чего выход сильно изменяется в зависимости от состояния (чистота, безводный или тому подобное) реагента (диэтилцинк, растворитель) и тому подобное, и концентрации реакции.
На основании вышеизложенного, авторы настоящего изобретения обнаружили, что производное дифенилметана может быть эффективно получено способом конвергентного синтеза, в котором соответствующие основные группы синтезируются раздельно и затем соединяются друг с другом, в отличие от обычного способа линейного синтеза, и таким образом осуществляется настоящее изобретение.
Подробное описание изобретения
Техническая задача
Соответственно, целью настоящего изобретения является обеспечение усовершенствованного способа получения производного дифенилметана, который используется в качестве ингибитора SGLT.
Решение задачи
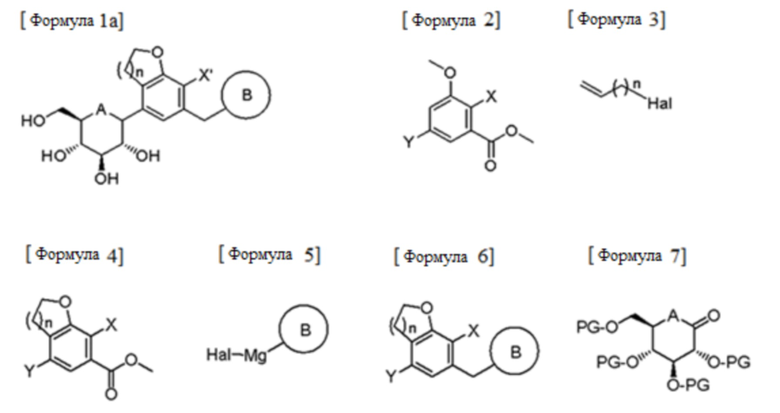
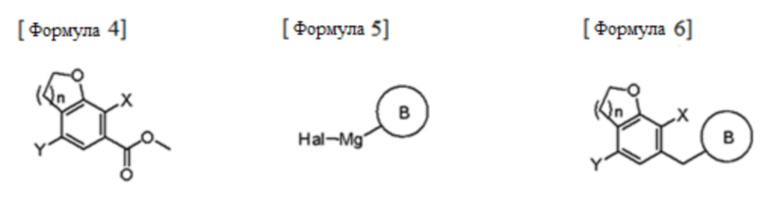
Согласно аспекту настоящего изобретения, предусмотрен способ получения соединения следующей формулы 1а, включающий стадии:
(1) взаимодействие соединения следующей формулы 2 с соединением следующей формулы 3 и циклизация полученного продукта взаимодействия с получением соединения следующей формулы 4;
(2) альдегидация или амидирование соединения формулы 4 с последующим взаимодействием полученного соединения с соединением следующей формулы 5 и осуществление восстановления с получением соединения следующей формулы 6; и
(3) взаимодействие соединения формулы 6 с соединением следующей формулы 7 и удаление защитных групп и восстановление,

в формулах,
А представляет собой кислород (О) или серу (S);
n равно 1 или 2;
PG представляет собой защитную группу;
X' представляет собой галоген или C1-7 алкил;
X, Y и Hal каждый независимо представляет собой галоген;
B представляет собой (B-1)  или (B-2)
или (B-2)  ,
,
где Ra, Rb, Rc и Rd каждый представляет собой независимо водород, галоген, гидрокси, меркапто, циано, нитро, амино, карбокси, оксо, C1-7 алкил, C1-7алкилтио, C2-7 алкенил, C2-7 алкинил, C1-7 алкокси, C1-7 алкокси-C1-7 алкил, C2-7 алкенил-C1-7алкилокси, C2-7 алкинил-C1-7алкилокси, C3-10 циклоалкил, C3-7циклоалкилтио, C5-10циклоалкенил, C3-10циклоалкилокси, C3-10циклоалкилокси-C1-7 алкокси, фенил-C1-7 алкил, C1-7алкилтио-фенил, фенил-C1-7 алкокси, моно- или ди-C1-7 алкиламино, моно- или ди-C1-7 алкиламино-C1-7 алкил, C1-7 алканоил, C1-7 алканоиламино, C1-7алкилкарбонил, C1-7алкоксикарбонил, карбамоил, моно- или ди-C1-7алкилкарбамоил, C1-7алкилсульфониламино, фенилсульфониламино, C1-7алкилсульфинил, C6-14арилсульфанил, C6-14арилсульфонил, C6-14 арил, 5-13-членный гетероарил, 5-10-членный гетероциклоалкил, 5-10-членный гетероциклоалкил-C1-7 алкил или 5-10-членный гетероциклоалкил-C1-7 алкокси;
кольцо С представляет собой C3-10 циклоалкил, C5-10циклоалкенил, C6-14 арил, 5-13-членный гетероарил или 5-10-членный гетероциклоалкил;
алкил, алкенил, алкинил и алкокси каждый независимо является незамещенным или имеет один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, С1-7 алкила и С2-7 алкинила;
циклоалкил, циклоалкенил, арил, гетероарил и гетероциклоалкил каждый независимо является незамещенным или имеет один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, C1-4 алкила и C1-4 алкокси; и
каждый гетероарил и гетероциклоалкил независимо содержит один или несколько гетероатомов, выбранных из группы, состоящей из N, S и O.
В соответствии с другим аспектом настоящего изобретения предложен способ получения соединения следующей формулы 1b, включающий стадии:
(1) взаимодействие соединения следующей формулы 2 с соединением следующей формулы 3 и циклизация полученного продукта взаимодействия с получением соединения следующей формулы 4;
(2) соединения формулы 4 подвергается альдегидации или амидированию с последующим взаимодействием полученного соединения с соединением формулы 5 и осуществлением восстановления с получением соединения следующей формулы 6;
(3) взаимодействие соединения формулы 6 с соединением следующей формулы 8 и затем осуществление восстановления с получением соединения следующей формулы 9;
(4) превращение фуранозного кольца соединения формулы 9 в пиранозное кольцо в кислотных условиях и затем введение в него защитной группы с получением соединения следующей формулы 10; и
(5) обработка соединения формулы 10 тиомочевиной, взаимодействие полученного продукта с С1-7 алкилгалогенидом и последующее проведение восстановления,



где в формулах,
R представляет собой C1-7алкилтио;
B, n, PG, X', X, Y и Hal являются такими, как определено выше в формуле 1.
Согласно еще одному аспекту настоящего изобретения предусмотрена кристаллическая форма соединения, полученного указанным выше способом, в частности кристаллическая форма соединения следующей формулы с28.
[Формула c28]
Преимущественные эффекты изобретения
Способ получения производного дифенилметана по настоящему изобретению осуществляется методом конвергентного синтеза, в котором основные группы синтезируются раздельно, а затем связываются друг с другом. Таким образом, по сравнению со способом линейного синтеза, описанным в документе из предшествующего уровня техники, могут быть достигнуты простой путь синтеза и высокий выход продукта, а воспроизводимость улучшается благодаря тому факту, что факторы риска (такие как возвращение в начало пути и повторение синтеза при сбое в середине синтеза), которые присущи пути линейного синтеза, могут быть уменьшены.
В частности, в соответствии со способом, описанным в документе из предшествующего уровня техники, остатки агликоновой группы должны быть синтезированы даже после связывания глюкозной группы с агликоновой группой. С другой стороны, согласно настоящему изобретению все остатки агликоновой группы могут быть образованы до того, как они будут связаны с глюкозной группой. Кроме того, арильная группа, связанная с концевой группой агликона, может быть легко синтезирована, вследствие чего возможны различные конструкции концевой группы.
Кроме того, кристаллическая форма соединения, полученного вышеуказанным способом, обладает превосходными физико-химическими свойствами и, таким образом, может быть эффективно использована в таких областях, как производство фармацевтических препаратов.
Краткое описание чертежей
На фиг.1 и 2 показаны спектры рентгенодифракции (XRD) и дифференциальной сканирующей калориметрии (DSC) кристаллической формы А, полученной в экспериментальном примере 4, соответственно.
На фиг.3 и 4 показаны спектры XRD и DSC кристаллической формы B, полученной в экспериментальном примере 4, соответственно.
На фиг.5 и 6 показаны спектры XRD и DSC кристаллической формы C, полученной в экспериментальном примере 4, соответственно.
На фиг.7 и 8 показаны спектры XRD и DSC кристаллической формы D, полученной в экспериментальном примере 4, соответственно.
Лучший способ осуществления изобретения
Настоящее изобретение относится к способу получения соединения формулы 1:
[Формула 1]

в формуле,
А представляет собой кислород (О) или серу (S);
R представляет собой гидроксиметил или C1-7алкилтио;
n равно 1 или 2;
X' представляет собой галоген (например, F, Cl, Br или I) или C1-7 алкил;
B представляет собой (B-1) или (B-2) ,
где Ra, Rb, Rc и Rd каждый представляет собой независимо водород, галоген, гидрокси, меркапто, циано, нитро, амино, карбокси, оксо, C1-7 алкил, C1-7алкилтио, C2-7 алкенил, C2-7 алкинил, C1-7 алкокси, C1-7 алкокси-C1-7 алкил, C2-7 алкенил-C1-7алкилокси, C2-7 алкинил-C1-7алкилокси, C3-10 циклоалкил, C3-7циклоалкилтио, C5-10циклоалкенил, C3-10циклоалкилокси, C3-10циклоалкилокси-C1-7 алкокси, фенил-C1-7 алкил, C1-7алкилтио-фенил, фенил-C1-7 алкокси, моно- или ди-C1-7 алкиламино, моно- или ди-C1-7 алкиламино-C1-7 алкил, C1-7 алканоил, C1-7 алканоиламино, C1-7алкилкарбонил, C1-7алкоксикарбонил, карбамоил, моно- или ди-C1-7алкилкарбамоил, C1-7алкилсульфониламино, фенилсульфониламино, C1-7алкилсульфинил, C6-14арилсульфанил, C6-14арилсульфонил, C6-14 арил, 5-13-членный гетероарил, 5-10-членный гетероциклоалкил, 5-10-членный гетероциклоалкил-C1-7 алкил или 5-10-членный гетероциклоалкил-C1-7 алкокси;
кольцо С представляет собой C3-10 циклоалкил, C5-10циклоалкенил, C6-14 арил, 5-13-членный гетероарил или 5-10-членный гетероциклоалкил;
алкил, алкенил, алкинил и алкокси каждый независимо является незамещенным или имеет один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, C1-7 алкила и C2-7 алкинил;
циклоалкил, циклоалкенил, арил, гетероарил и гетероциклоалкил каждый независимо является незамещенным или имеет один или несколько заместителей, выбранных из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, C1-4 алкила и C1-4 алкокси; и
каждый гетероарил и гетероциклоалкил независимо содержит один или несколько гетероатомов, выбранных из группы, состоящей из N, S и O.
В качестве конкретного примера, кольцо B-1 может быть выбрано из группы, состоящей из:

В формулах, R7 представляет собой водород или C1-7 алкил; R8a и R8b каждый представляет собой независимо C1-7 алкил или связаны друг с другом с образованием 5-10-членного гетероциклоалкила (который содержит, по меньшей мере, один гетероатом, выбранный из группы, состоящей из N, S и O).
В качестве другого конкретного примера, кольцо B-2 может быть выбрано из группы, состоящей из:

Предпочтительно соединение формулы 1 может представлять собой соединение, представленное следующей формулой 1a, или соединение, представленное следующей формулой 1b:
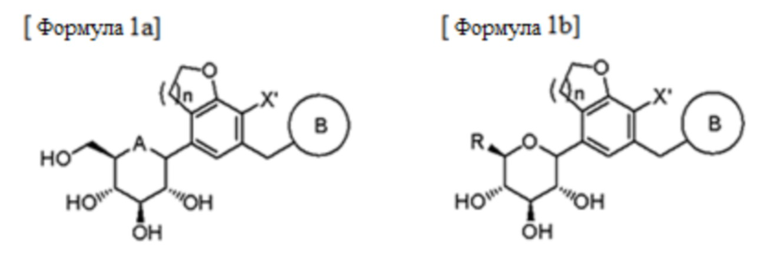
В формулах, A, B, R, X' и n являются такими, как определено выше в формуле 1.
Согласно предпочтительному примеру соединения формулы 1а, А может представлять собой кислород; n может быть равно 1; Х' может представлять собой галоген; и B может представлять собой фенил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, нитро, амино, меркапто, C1-7 алкила, C3-10 циклоалкила и C1-7 алкокси.
Кроме того, соединения формул 1a и 1b могут представлять собой соединения, в которых глюкоза находится в α-форме, β-форме или в рацемической форме.
Предпочтительно соединения формул 1a и 1b могут представлять собой соединения, в которых глюкоза находится в β-форме.
Способ получения соединения формулы 1a (формула 1, в которой R=гидроксиметил)
Согласно аспекту настоящего изобретения, предусмотрен способ получения соединения формулы 1а (формула 1, в которой R=гидроксиметил), включающий следующие стадии:
(1) взаимодействие соединения следующей формулы 2 с соединением следующей формулы 3 и циклизация полученного продукта с получением соединения следующей формулы 4;
(2) альдегидация или амидирование соединения формулы 4 с последующим взаимодействием полученного соединения с соединением формулы 5 и осуществлением восстановления с получением соединения следующей формулы 6; и
(3) взаимодействие соединения формулы 6 с соединением следующей формулы 7 и осуществление удаления защитных групп и восстановления.
в формулах,
А представляет собой кислород (О) или серу (S);
n равно 1 или 2;
PG представляет собой защитную группу;
X' представляет собой галоген или C1-7 алкил;
X, Y и Hal каждый независимо представляет собой галоген; и
B является таким, как определено выше в формуле 1.
Соединение формулы 2, используемое в качестве исходного вещества в вышеуказанном способе получения, может быть получено путем синтеза, опубликованным в документе из предшествующего уровня техники (публикация выложенного патента США №2015/0152075 А1). Например, соединение формулы 2 может быть получено путем включения следующих стадий:
(i) этерификация следующего соединения карбоновой кислоты формулы 2а с получением следующего соединения сложного метилового эфира формулы 2b;
(ii) гидрирование соединения формулы 2b с целью восстановления нитрогруппы с получением следующего соединения амина формулы 2с;
(iii) взаимодействие соединения формулы 2с с галогенирующим реагентом с получением следующего галогенированного соединения формулы 2d; и
(iv) соединение формулы 2d подвергается реакции Сандмейера. 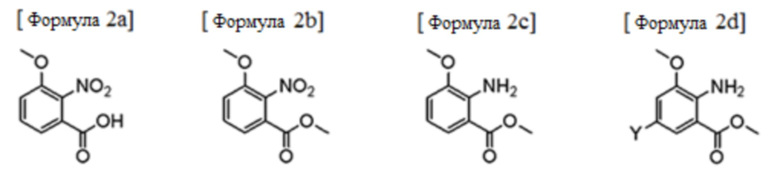
В формулах, Y представляет собой галоген.
Используемый в настоящем документе термин ʺгалогенʺ обозначает фтор (F), хлор (Cl), бром (Br) или йод (I).
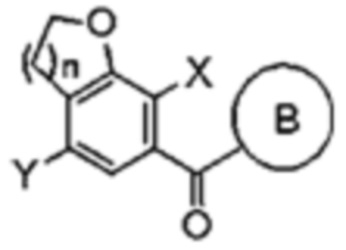
Стадия (1)
На стадии (1) соединение формулы 2 подвергают взаимодействию с соединением формулы 3, и полученный продукт подвергают реакции циклизации с получением соединения формулы 4.
Таким образом, перед связыванием с группой глюкозы, а также перед образованием концевого остатка (то есть кольца B) агликоновой группы, может быть предварительно образовано пентагональное или гексагональное кольцо, в котором содержится кислород в агликоновой группе.
В качестве конкретного примера, стадия (1) может включать этапы:
(i) взаимодействие соединения формулы 2 с соединением формулы 3 с получением соединения следующей формулы 3а;
(ii) аллильная группа соединения формулы 3а подвергается реакции перегруппировки, и полученный продукт подвергается реакции окисления или озонирования с последующим проведением восстановления с получением соединения следующей формулы 3d; и
(iii) соединение формулы 3d подвергается реакции циклизации с получением соединения формулы 4:
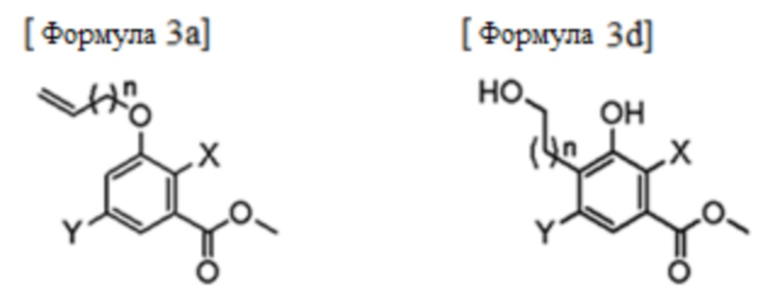
В формулах, n равно 1 или 2; и X и Y каждый независимо представляет собой галоген.
На стадии (i) соединение формулы 2 может быть подвергнуто стадии деметилирования перед реакцией с соединением формулы 3. Например, соединение формулы 2 может быть подвергнуто деметилированию с получением соединения следующей формулы 2e, и соединение формулы 2e может взаимодействовать с соединением формулы 3:
[Формула 2e]

В формуле, X и Y каждый независимо представляет собой галоген.
Реакция перегруппировки на стадии (ii) может проводиться, например, в реакции кляйзеновской перегруппировки.
Реакция перегруппировки может быть проведена путем добавления кислоты Льюиса. Кислота Льюиса может представлять собой, по меньшей мере, одну кислоту, выбранную из группы, состоящей из хлорида диизобутилалюминия, хлорида диэтилалюминия, хлорида алюминия и трихлорида бора.
Кроме того, реакцию перегруппировки можно проводить в реакции без растворителя или в условиях нагревания при высокой температуре (например, от 150°С до 170°С) в диэтиламине.
После проведения реакции перегруппировки на стадии (ii) соединение формулы 3a можно получить в виде соединения следующей формулы 3b:
[Формула 3b]

В формуле, n равно 1 или 2; и X и Y каждый независимо представляет собой галоген.
Реакция окисления или озонирования на стадии (ii) может быть осуществлена путем добавления тетроксида осмия (OsO4), калия осмата (VI) дигидрата или озона (O3).
После проведения реакции окисления или озонирования на стадии (ii), соединение формулы 3b можно получить в виде соединения следующей формулы 3с:
[Формула 3c]

в формуле, n равно 1 или 2; и X и Y каждый независимо представляет собой галоген.
Затем соединение формулы 3с может быть восстановлено с получением соединения формулы 3d.
На стадии (iii) соединение формулы 3d может быть подвергнуто реакции циклизации с получением соединения формулы 4. В соответствии с таким способом, выход может быть улучшен по сравнению со способом циклизации, описанным в документе предшествующего уровня техники (публикация выложенного патента США №2015/0152075 A1).
Реакция циклизации может представлять собой реакцию циклизации с использованием реагента Вильсмейера, реакцию циклизации с использованием уходящей группы, реакцию циклизации с использованием галида или реакцию циклизации с использованием реакции Мицунобу.
В соответствии с примером, реакция циклизации может быть проведена путем добавления реагента Вильсмейера к соединению формулы 3d. Реакция в это время может проводиться при температуре от 0°C до нормальной температуры. С точки зрения выхода, предпочтительно, чтобы вышеуказанный реагент Вильсмейера получали непосредственно в процессе синтеза и использовали, и, например, может быть использован реагент Вильсмейера, полученный взаимодействием диметилформамида (ДМФА) с SOCl2 или PОCl3.
В соответствии с другим примером, реакция циклизации может быть проведена путем введения тозильной группы или мезильной группы в качестве уходящей группы. В соответствии с еще одним примером, реакцию циклизации можно проводить с использованием галида, такого как I2 и PBr3. В соответствии с еще одним примером, реакцию циклизации также можно проводить посредством реакции Мицунобу с использованием диизопропилазодикарбоксилата (DIAD) или тому подобного.
Эти реакции представляют собой реакции, в которых первичная спиртовая группа замещена группой, способной действовать в качестве уходящей группы, и замещенная группа действует как нуклеофил в отношении фенольной группы, в результате чего происходит реакция циклизации.
Стадия (2)
На стадии (2) соединение формулы 4 подвергают альдегидации или амидированию, а затем подвергают взаимодействию с соединением формулы 5. Полученное соединение восстанавливают, получая соединение формулы 6.
Например, стадия (2) может включать альдегидацию соединения формулы 4 с получением соединения следующей формулы 4а, а последующее взаимодействие соединений формулы 4а с соединением формулы 5:
[Формула 4a]

В формуле, n равно 1 или 2; и X и Y каждый независимо представляет собой галоген.
В частности, реакцию альдегидацию можно проводить путем восстановления соединения формулы 4 с получением соединения следующей формулы 4с с последующим взаимодействием соединений формулы 4 с хлорхроматом пиридиния (PCC), диоксидом магния, комплексом триоксида серы и пиридина или тому подобным. В результате, может быть получено соединение формулы 4а:
[Формула 4c]

В формуле, n равно 1 или 2; и X и Y каждый независимо представляет собой галоген.
При этом восстановитель, такой как NaBH4 и LiBH4, можно использовать во время восстановления соединения формулы 4. Кроме того, во время восстановления в качестве растворителя можно использовать спирт, тетрагидрофуран (ТГФ) или их смесь. В качестве предпочтительного примера во время восстановления можно использовать смешанный растворитель, состоящий из этанола и ТГФ, а его объемное соотношение при смешивании может составлять 1:1-1:3. Кроме того, во время восстановления можно дополнительно использовать кислоту Льюиса, и примеры кислоты Льюиса, которые можно использовать, включают LiCl и CaCl2.
Затем соединение формулы 4а может быть подвергнуто взаимодействию с соединением формулы 5 с получением соединения следующей формулы 6а:
[Формула 6a]

В формуле, n равно 1 или 2, X и Y каждый независимо представляет собой галоген, и B является таким, как определено выше в формуле 1.
Соединение формулы 6а может быть восстановлено с получением соединения формулы 6.
В качестве другого примера стадия (2) может включать амидирование соединения формулы 4 с получением соединения следующей формулы 4b, а затем взаимодействие соединения формулы 4b с соединением формулы 5:
[Формула 4b]

В формуле, n равно 1 или 2; и X и Y каждый независимо представляет собой галоген.
В частности, реакцию амидирования можно проводить, подвергая соединение формулы 4 гидролизации с последующей реакцией полученного продукта с гидрохлоридом N,O-диметилгидроксиамина (MeO(Me)NH⋅HCl) или тому подобным. В результате может быть получена форма амида Вайнреба, такая как формула 4b.
Затем соединение формулы 4b может быть подвергнуто взаимодействию с соединением формулы 5 с получением соединения следующей формулы 6b:
[Формула 6b]
В формуле, n равно 1 или 2; X и Y каждый независимо представляет собой галоген, и B является таким, как определено выше в формуле 1.
Затем соединение формулы 6b может быть восстановлено с получением соединения формулы 6.
Соединение формулы 5 может представлять собой реактив Гриньяра.
В соответствии с общим способом получения реактива Гриньяра, соединение следующей формулы 5а может быть подвергнуто взаимодействию с металлом магнием (Mg) с получением соединения формулы 5.
[Формула 5a]
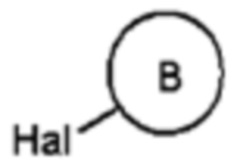
в формуле, B является таким, как определено выше в формуле 1, и Hal представляет собой галоген.
Как описано выше, согласно настоящему изобретению, группа В конечного соединения (соединение формулы 1а) может легко предварительно вводиться в соответствии со способом получения реактива Гриньяра перед сочетанием агликоновой группы с глюкозной группой, что не только обеспечивает различную дериватизацию, но также позволяет повысить конечный выход.
С другой стороны, согласно документу из предшествующего уровня техники (публикация выложенного патента США №2015/0152075 A1), чтобы завершить группу B конечного соединения, после соединения с группой глюкозы, требуется сложный процесс синтеза в конце пути синтеза, и, таким образом, существует проблема, заключающаяся в том, что выход реакции и воспроизводимость реакции значительно изменяются благодаря длительному процессу.
Стадия (3)
На стадии (3) соединение формулы 6 подвергают взаимодействию с соединением формулы 7, а затем проводят удаление защитных групп и восстановление.
Взаимодействие соединения формулы 6 с соединением формулы 7 можно проводить в присутствии н-бутиллития, втор-бутиллития, трет-бутиллития, изопропилмагнийхлорида (i-PrMgCl) или тому подобного.
Соединение формулы 6 можно подвергнуть взаимодействию с соединением формулы 7 с получением соединения следующей формулы 7а:
[Формула 7a]
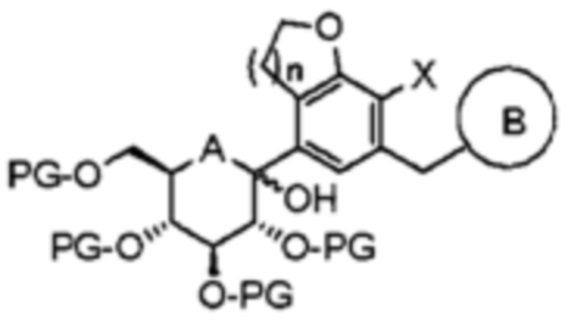
В формуле А представляет собой кислород или серу; n равно 1 или 2; Х является галогеном; PG представляет собой защитную группу; и B является таким, как определено выше в формуле 1.
Защитной группой может быть, например, триметилсилильная (TMS) группа, бензильная группа или ацетильная группа.
Затем из соединения формулы 7а можно удалить защитную группу для получения соединения формулы 1а. Например, в случае, когда защитная группа представляет собой триметилсилильную (TMS) группу, удаление защитной группы осуществляют путем добавления метансульфоновой кислоты (CH3SO3H) или триметилсилилтрифторметансульфоната (TMSOTf) к соединению формулы 7a, в результате чего может быть получено соединение формулы 1a.
Кроме того, после удаления защитной группы может быть дополнительно осуществлено восстановление с получением соединения формулы 1а. В это время дихлорметан (CH2Cl2) и ацетонитрил (CH3CN) могут использоваться в сочетании в качестве растворителя.
Соединение формулы 1а, полученное посредством вышеуказанных стадий, может представлять собой соединение, в котором смешаны α-форма и β-форма глюкозы.
Таким образом, дополнительное выделение может быть выполнено с получением только желаемой α-формы или β-формы. То есть после или во время процесса удаления защитной группы и восстановления можно дополнительно осуществлять выделение только соединения, в котором глюкоза находится в β-форме.
Например, защитная группа вводится в соединение, полученное путем снятия защиты и восстановления. Затем полученный продукт нагревают в спирте, этилацетате или дихлорметане, и полученный осадок выделяют и далее снимают защиту, после чего может быть получена только β-форма.
В частности, гидроксигруппа глюкозы в соединении, полученном путем снятия защиты и восстановления, защищена ацетильной группой или тому подобным. Затем полученный продукт нагревают и перемешивают в С1-6 спиртовом растворителе (этанол, изопропанол или тому подобное) и полученный осадок выделяют, в результате чего может быть получено только соединение следующей формулы 7b, в котором глюкоза находится в β-форме:
[Формула 7b]

В формуле, A представляет собой кислород или серу; n равно 1 или 2; X представляет собой галоген; PG представляет собой защитную группу; и B является таким, как определено выше в формуле 1.
Затем из соединения формулы 7b может снята защита с получением, в конечном итоге, только β-формы, которая может быть представлена следующей формулой 7с:
[Формула 7c]
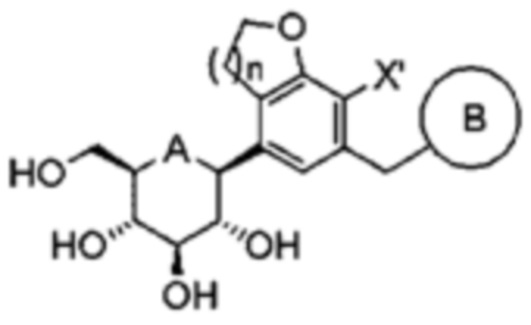
В формуле, A, B, n и X' являются такими, как определено выше в формуле 1.
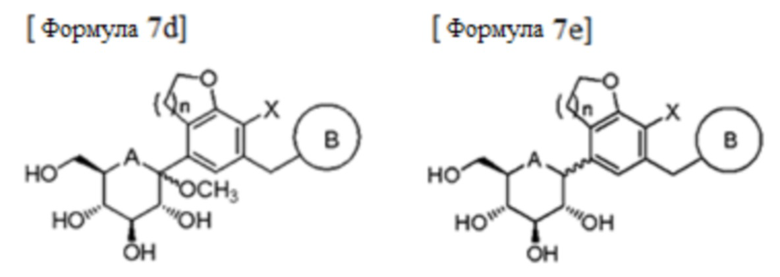
В соответствии с предпочтительным примером, стадия (3) может быть осуществлена способом, включающим стадии:
(3a-1) взаимодействие соединения формулы 6 с соединением формулы 7 в присутствии н-бутиллития, втор-бутиллития, трет-бутиллития или изопропилмагнийхлорида с получением соединения следующей формулы 7а;
(3a-2) удаление защитной группы и метилирование соединения формулы 7a в кислотных условиях в присутствии метанола с получением соединения следующей формулы 7d;
(3b) восстановление соединения формулы 7b с получением соединения следующей формулы 7e; и
(3c) введение защитной группы в соединение формулы 7e, нагревание полученного продукта в спирте, этилацетате или дихлорметане и выделение и удаление защитных групп полученного осадка с получением только β-формы:

В формулах, PG представляет собой защитную группу; и A, B, n и X являются такими, как определено выше в формуле 1a.
После взаимодействия на стадии (3a-1), предпочтительно, чтобы соединение формулы 7a было получено путем дальнейшего упаривания, экстракции, сушки, фильтрации или тому подобного, а затем использовалось на следующей стадии (3a-2),
Кислота, используемая на стадии (3a-2), может представлять собой хлористоводородную кислоту, серную кислоту, уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, п-толуолсульфоновую кислоту, газ хлороводород и т.п.
В соответствии с другим предпочтительным примером, стадия (3) может быть осуществлена способом, включающим стадии:
(3a') взаимодействие соединения формулы 6 с соединением формулы 7 в присутствии н-бутиллития, втор-бутиллития, трет-бутиллития или изопропилмагнийхлорида, и полученный продукт подвергается реакциям удаления защитной группы и метилирования в кислотных условиях в присутствии метанола без отдельной очистки с получением соединения следующей формулы 7d;
(3b') восстановление соединения формулы 7d с получением соединения следующей формулы 7e; и
(3c') введение защитной группы в соединение формулы 7e для выделения только β-формы и выполнение снятия защиты:
В формулах, A, B, n и X являются такими, как определено выше в формуле 1a.
На стадии (3a'), сначала проводят реакцию связывания и, на данном этапе, для реакции может быть использовано каждое из соединений формулы 7 и реакционный реагент (то есть н-бутиллитий, втор-бутиллитий, трет-бутиллитий или изопропилмагнийхлорид) в количестве от 1,5 до 2,5 эквивалентов, более предпочтительно, от 1,7 до 2,3 эквивалентов, и, в частности, около 2,0 эквивалентов, относительно 1 эквивалента соединения формулы 6. При этом реакция может проводиться при температуре в диапазоне от -80°С до -10°С и более предпочтительно от -70°С до -60°С, в течение от 1 до 12 часов или в течение от 1 до 3 часов. Кроме того, в качестве реакционного растворителя можно использовать отдельный растворитель - тетрагидрофуран или эфир, смешанный растворитель, состоящий из тетрагидрофурана и толуола (1:1) или тому подобное.
Кроме того, на стадии (3a'), реакции удаления защитных групп и метилирования осуществляют в кислотных условиях. Примеры используемой на данном этапе кислоты включают соляную кислоту, серную кислоту, уксусную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, п-толуолсульфокислоту и газ хлороводород. Кислота может быть использована в количестве от 2 до 5 эквивалентов и более предпочтительно в количестве 3 эквивалентов относительно 1 эквивалента соединения формулы 6. При этом реакция может проводиться при температуре в диапазоне от 0°С до 40°С и более предпочтительно от 20°С до 30°С, от 6 до 24 часов или от 6 до 12 часов. В качестве реакционного растворителя можно использовать метанол или тому подобное.
Затем на стадии (3b') осуществляется реакция восстановления, и на данном этапе можно использовать восстановитель и кислоту. Примеры восстановителя могут включать триэтилсилан, триизопропилсилан, трет-бутилдиметилсилан и борогидрид натрия. Примеры кислоты могут включать диэтиловый эфир трифторида бора, триметилсилилтрифторметансульфонат, хлорид алюминия, трифторуксусную кислоту и трифторметансульфоновую кислоту. Восстановитель можно использовать в количестве от 2 до 5 эквивалентов и более предпочтительно около 3 эквивалентов, и кислоту можно использовать в количестве от 1,5 до 3 эквивалентов и более предпочтительно около 2 эквивалентов. При этом реакцию можно проводить при температуре в диапазоне от -50 до 0°С и более предпочтительно в диапазоне от -20 до -10°С в течение от 2 до 12 часов или в течение от 2 часов до 5 часов. Кроме того, в качестве реакционного растворителя может быть использован отдельный растворитель, такой как дихлорметан, 1,2-дихлорэтан или ацетонитрил, или смешанный растворитель, такой как дихлорметан/ацетонитрил (1:1) и 1,2-дихлорметан/ацетонитрил (1:1).
Затем на стадии (3c') вводится защитная группа, и на этом этапе может быть проведена реакция с использованием ацетилирующего агента и основания. Примеры ацетилирующего агента включают ацетилхлорид, ацетилбромид и уксусный ангидрид, и примеры основания включают гидроксид натрия, карбонат натрия, триэтиламин, диизопропилэтиламин, пиридин, лутидин и 4-диметиламинопиридин. Ацетилирующий агент можно использовать в количестве от 4 до 12 эквивалентов и более предпочтительно около 8 эквивалентов, а основание можно использовать в количестве от 1 до 4 эквивалентов и более предпочтительно около 1,5 эквивалентов. При этом реакцию можно проводить при температуре в диапазоне от 0 до 50°С и более предпочтительно от 20 до 30°С в течение от 1 до 12 часов или от 1 до 3 часов. В качестве реакционного растворителя можно использовать ацетон, этилацетат, тетрагидрофуран, диметилформамид, диметилацетамид, дихлорметан, 1,2-дихлорэтан, хлороформ или тому подобное.
Наконец, реакция удаления защитных групп проводится на стадии (3c') и этом тапе можно использовать такие реагенты, как гидроксид лития, гидроксид натрия, гидроксид калия, метоксид натрия и этоксид натрия в количестве от 2 до 12 эквивалентов и более предпочтительно около 5 эквивалентов. При этом реакцию можно проводить при температуре в диапазоне от 0 до 50°С и более предпочтительно от 20 до 30°С в течение от 1 до 12 часов или от 1 до 3 часов. В качестве реакционного растворителя может быть использована смесь метанол/вода (1:1-3:1), дихлорметан/метанол (1:1-1:2), дихлорметан/этанол (1:1-1:2), тетрагидрофуран/метанол (1:1-1:2), тетрагидрофуран/этанол (1:1-1:2), тетрагидрофуран/метанол/вода (1:1:3-2:1:3), тетрагидрофуран/этанол/вода (1:1:3-2:1:3) или тому подобное.
В соответствии с еще одним предпочтительным примером, стадия (3) может быть осуществлена способом, включающим стадии:
(3a'') взаимодействие соединения формулы 6 с соединением формулы 7 в присутствии н-бутиллития, втор-бутиллития, трет-бутиллития или изопропилмагнийхлорида, и полученный продукт подвергается реакциям удаления защитной группы и метилирования в кислотных условиях в присутствии метанола без отдельной очистки с получением соединения следующей формулы 7d;
(3b'') введение защитной группы в соединение формулы 7d с получением соединения следующей формулы 7f; и
(3c") выделение только β-формы соединения формулы 7f и проведение реакции восстановления, а затем проведение реакции удаления защитной группы:
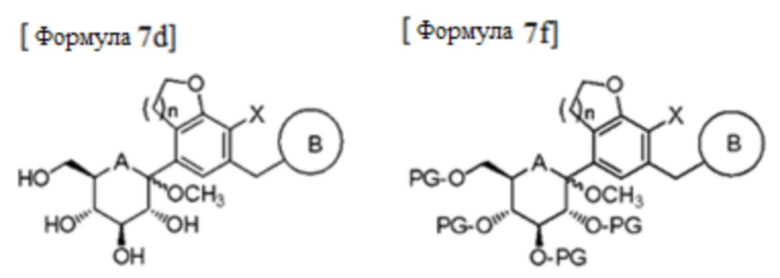
В формулах, PG представляет собой защитную группу; и A, B, n и X являются такими, как определено выше в формуле 1a.
На стадии (3a'') осуществляется реакция связывания, удаление защитных групп и метилирование. При этом предпочтительные условия, такие как эквивалентное соотношение, температура реакции и растворитель, являются такими, как проиллюстрировано на стадии (3a').
Затем на стадии (3b'') вводится защитная группа, и на этом этапе может быть проведена реакция с использованием ацетилирующего агента и основания. Предпочтительные условия, такие как тип ацетилирующего агента, тип основания, эквивалентного соотношения, температура реакции и растворитель, являются такими, как проиллюстрировано выше на стадии (3c').
Далее проводится реакция восстановления на стадии (3c''). На этом этапе могут использоваться восстановитель и кислота, и предпочтительные условия, такие как тип восстановителя, тип кислоты, эквивалентное соотношение, температура реакции и растворитель, являются такими, как проиллюстрировано выше на стадии (3b').
Кроме того, реакция удаления защитных групп проводится на стадии (3c''), и при этом предпочтительные условия, такие как тип реагента, эквивалентное соотношение, температура реакции и растворитель, являются такими, как проиллюстрировано выше на стадии (3c').
Как показано в вышеуказанных предпочтительных иллюстративных примерах, процесс получения соединения формулы 7d может быть осуществлен за две стадии или может быть осуществлен за одну стадию в виде реакции in-situ, в результате чего конечный выход дополнительно повышается. Кроме того, в случае осуществления получения за одну стадию в виде реакции in-situ, может быть получен неочищенный концентрированный остаток, содержащий соединение формулы 7b, или соединение формулы 7d может быть получено из него в виде твердого вещества путем кристаллизации и использовано на следующей стадии. В последнем случае можно легко добиться улучшения качества и контроля содержания влаги посредством удаления побочных продуктов реакции.
Кроме того, соединение формулы 7d может быть использовано на следующей стадии путем очистки его после синтеза. Например, (i) после синтеза, соединение формулы 7d может образовывать азеотропную смесь с органическим растворителем, таким как толуол, а остаток, полученный повторением процесса концентрирования для удаления остаточной влаги, может использоваться на следующей стадии, или (ii) после синтеза, соединение формулы 7d может быть подвергнуто кристаллизации, и объем твердого вещества, полученный путем удаления остаточной влаги вакуумной сушкой, может быть использован на следующей стадии.
Стадия алкилирования
Кроме того, согласно настоящему изобретению, можно дополнительно включать реакцию алкилирования после стадии (3), и в результате X' в формуле 1 может представлять собой С1-7 алкил.
Например, продукт после стадии (4) может взаимодействовать с метилбороновой кислотой с получением соединения формулы 1a, в которой X' замещен метилом.
Стадия кристаллизации
Соединение формулы 1а может быть получено в кристаллической форме, аморфной форме или в виде их смеси. Однако соединение формулы 1a в кристаллической форме является предпочтительным с точки зрения превосходной стабильности и негигроскопичности и, соответственно, наличия физико-химических свойств, которые облегчают получение препарата.
Следовательно, способ по настоящему изобретению может дополнительно включать кристаллизацию соединения формулы 1а после стадии (3). Кристаллизация может быть осуществлена с использованием различных растворителей, и, таким образом, могут быть получены различные кристаллические формы.
В качестве примера, растворитель, используемый для кристаллизации, может быть выбран из толуола; этилацетата; дихлорметана; ацетона; ацетонитрила; смеси 2-пропанола, тетрагидрофурана и дихлорметана; и смеси тетрагидрофурана и н-гексана, и в результате может быть получена кристаллическая форма А.
В качестве другого примера, растворитель, используемый для кристаллизации, может быть выбран из смеси метанола и дистиллированной воды; смеси метанола и н-гексана; и смеси метанола, дихлорметана и н-гексана, и в результате может быть получена кристаллическая форма В.
В качестве еще одного примера растворитель, используемый для кристаллизации, может быть выбран из смеси этанола, дистиллированной воды и н-гексана; и смеси тетрагидрофурана и толуола, и, в результате, может быть получена кристаллическая форма C.
В качестве еще одного примера, растворитель, используемый для кристаллизации, может представлять собой смесь этанола и н-гексана, и, в результате, может быть получена кристаллическая форма D.
В качестве предпочтительного примера, растворитель, используемый для кристаллизации, может быть выбран из группы, состоящей из толуола, этилацетата, дихлорметана, смеси тетрагидрофурана и дихлорметана, и смеси тетрагидрофурана и н-гексана.
Способ получения соединения формулы 1b (формула 1, в которой R=алкилтио и A=кислород)
В соответствии с еще одним аспектом настоящего изобретения, предложен способ получения соединения формулы 1b (формула 1, в которой R=C1-7алкилтио и A=кислород), причем способ включает следующие стадии:
(1) взаимодействие соединения следующей формулы 2 с соединением следующей формулы 3 и циклизация полученного продукта с получением соединения следующей формулы4;
(2) альдегидация или амидирование соединения формулы 4 с последующим взаимодействием полученного соединения с соединением формулы 5 и проведением восстановления с получением соединения следующей формулы 6;
(3) взаимодействие соединения формулы 6 с соединением следующей формулы 8 и затем осуществление восстановления с получением соединения следующей формулы 9;
(4) превращение фуранозного кольца соединения формулы 9 в пиранозное кольцо в кислых условиях и затем введение в него защитной группы с получением соединения следующей формулы 10; и
(5) обработка соединения формулы 10 тиомочевиной, взаимодействие полученного продукта с С1-7 галоидным алкилом и последующее восстановление.


В формулах,
R представляет собой C1-7 алкилтио;
n равно 1 или 2;
PG представляет собой защитную группу;
X' представляет собой галоген или C1-7 алкил;
X, Y и Hal каждый независимо представляет собой галоген; и
B является таким, как определено выше в формуле 1.
В вышеуказанных стадиях, стадии (1) и (2) могут быть выполнены таким же образом, как и стадии (1) и (2) способа получения соединения формулы 1a (формула 1, в которой R=гидроксиметил).
Стадии (3) -(5) будут подробно описаны ниже.
Стадия (3)
На стадии (3) соединение формулы 6 подвергают взаимодействию с соединением формулы 8 с получением соединения формулы 9.
Соединение формулы 8 может быть получено в соответствии с известным способом, например способом, описанным в WO 2009/014970. В частности, соединение формулы 8 может быть получено в соответствии со способом, описанным в WO 2009/014970, исходя из L-ксилозы.
В соответствии с примером, соединение формулы 6 можно подвергнуть взаимодействию с соединением формулы 8 с получением соединения следующей формулы 9а.
[Формула 9a]

В формуле, B, n и X являются такими, как определено выше в формуле 1.
Затем соединение формулы 9а может быть восстановлено с получением соединения формулы 9.
Стадия (4)
На стадии (4) фуранозное кольцо соединения формулы 9 преобразуется в пиранозное кольцо в кислотных условиях, и затем в него вводят защитную группу с получением соединения формулы 10. На этой стадии, пиранозное кольцо, составляющее группу глюкозы, может быть выполнено.
Защитной группой может быть, например, ацетильная группа.
Стадия (5)
В стадии (5) соединение формулы 10 обрабатывают тиомочевиной, подвергают взаимодействию с C1-7 галоидным алкилом, и затем восстанавливают. На этой стадии в конечное соединение может быть введена алкилтиогруппа (соединение формулы 1b).
С1-7 галоидным алкилом может быть, например, С1-7 алкилиодид.
Кроме того, после стадии (5) может быть дополнительно включена реакция алкилирования, и, в результате, может быть получено соединение формулы 1b, в которой X' представляет собой С1-7 алкил.
Кристаллическая форма
В соответствии с еще одним аспектом настоящего изобретения предложена кристаллическая форма соединения, полученного в соответствии с описанным выше способом получения.
В качестве примера, настоящее изобретение предусматривает кристаллическую форму соединения формулы 1а.
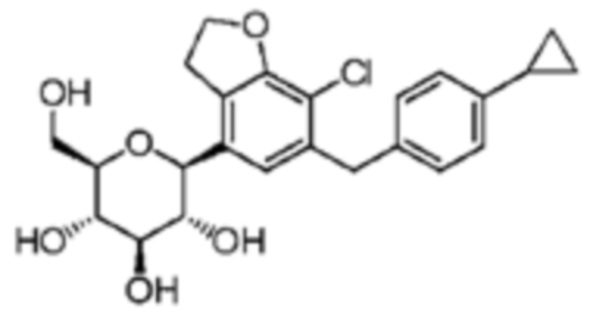
В качестве конкретного примера, настоящее изобретение предусматривает кристаллическую форму соединения формулы 1a, в которой A представляет собой O, B представляет собой циклопропилфенил, n равно 1, и X' представляет собой Cl, и которое находится в β-форме.
То есть, настоящее изобретение относится к кристаллической форме соединения следующей формулы с28.
[Формула c28]
Соединение формулы с28 может быть получено описанным выше способом получения соединения формулы 1а.
В соответствии с настоящим изобретением, возможно, что соединение формулы с28 находится в различных кристаллических формах, и соответствующие кристаллические формы будут подробно описаны ниже.
Далее термин ʺоколоʺ может означать отклонение, составляющее 5% и предпочтительно 2%, от предварительно определенного значения или диапазона. Например, ʺоколо 10%ʺ может означать от 9,5% до 10,5% и предпочтительно от 9,8% до 10,2%. В качестве другого примера, ʺоколо 100°Сʺ может означать от 95°С до 105°С и предпочтительно от 98°С до 102°С.
Во-первых, настоящее изобретение относится к кристаллической форме А соединения формулы с28. Кристаллическая форма A имеет рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ) 6,2° ± 0,2°, 7,2° ± 0,2°, 8,8° ± 0,2°, 17,6° ± 0,2°, 19,0° ± 0,2°, 22,5° ± 0,2° и 25,1° ± 0,2° в случае облучения с использованием Cu-Kα источника света. Эти пики могут быть пиками с относительной интенсивностью (I/Io), составляющей около 5% или выше и предпочтительно около 10% или выше.
Рентгеновский дифракционный спектр кристаллической формы А может дополнительно включать пики при углах дифракции (2θ) 15,4°±0,2°, 18,6°±0,2°, 21,6°±0,2° и 23,8°±0,2°.
Кроме того, кристаллическая форма может иметь эндотермический пик, в котором начальная точка соответствует около 157°С, а нижняя точка соответствует около 159°С при DSC (10°С/минуту).
Кроме того, настоящее изобретение относится к кристаллической форме B соединения формулы c28. Кристаллическая форма B имеет рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ), равные 7,0°±0,2°, 14,9°±0,2°, 17,7°±0,2°, 18,8°±0,2°, 20,6°±0,2°, 21,8°±0,2° и 23,5°±0,2° в случае облучения с использованием Cu-Kα источника излучения. Эти пики могут быть пиками с относительной интенсивностью (I/Io), составляющей около 5% или выше и предпочтительно около 10% или выше.
Рентгеновский дифракционный спектр кристаллической формы B может дополнительно включать пики при углах дифракции (2θ) 5,6°±0,2°, 9,4°±0,2° и 11,0°±0,2°.
Кроме того, кристаллическая форма может иметь эндотермический пик, в котором начальная точка соответствует около 79°С, и нижняя точка соответствует около 88°С, и эндотермический пик, в котором начальная точка соответствует около 103°С, и нижняя точка соответствует около 111°C, при DSC (10°C/мин).
Кроме того, настоящее изобретение относится к кристаллической форме C соединения формулы c28. Кристаллическая форма С имеет рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ) 5,6°±0,2°, 7,3°±0,2°, 15,7°±0,2°, 17,2°±0,2°, 18,9°±0,2°, 21,2°±0,2° и 21,9°±0,2° в случае облучения с использованием Cu-Kα источника излучения. Эти пики могут быть пиками с относительной интенсивностью (I/Io), составляющей около 5% или выше и предпочтительно около 10% или выше.
Рентгеновский дифракционный спектр кристаллической формы C может дополнительно включать пики при углах дифракции (2θ) 19,9°±0,2° и 23,1°±0,2°.
Кроме того, кристаллическая форма может иметь эндотермический пик, в котором начальная точка составляет около 157°С, а нижняя точка составляет около 159°С, при DSC (10°С/минуту).
Кроме того, настоящее изобретение относится к кристаллической форме D соединения формулы с28. Кристаллическая форма D имеет рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ) 5,5°±0,2°, 7,2°±0,2°, 15,3°±0,2°, 17,2°±0,2°, 17,6°±0,2°, 18,9°±0,2° и 21,1°±0,2° в случае облучения с использованием Cu-Kα источника излучения. Эти пики могут быть пиками с относительной интенсивностью (I/Io), составляющей около 5% или выше.
Рентгеновский дифракционный спектр кристаллической формы D может дополнительно включать пики при углах дифракции (2θ) 20,0°±0,2°, 22,5°±0,2° и 25,1°±0,2°.
Кроме того, кристаллическая форма может иметь эндотермический пик, в котором начальная точка соответствует около 157°С, а нижняя точка соответствует около 160°С, при DSC (10°С/минуту).
Такие кристаллические формы соединения формулы с28 являются превосходными с точки зрения физико-химических свойств (например, гигроскопичность и химическая стабильность) и, таким образом, могут легко обрабатываться в различных областях применения (например, при производстве фармацевтических препаратов).
Способ осуществления изобретения
Далее настоящее изобретение будет описано более подробно со ссылкой на примеры. Однако следующие примеры предназначены только для иллюстрации настоящего изобретения, и объем настоящего изобретения не ограничивается этими примерами.
Значения аббревиатур, указанных в следующих примерах, являются следующими.
- AcOH: Уксусная кислота
- ACN: Ацетонитрил
- Ac2O: Уксусный ангидрид
- BF3.OEt2: Эфират трифтористого бора
- DIPEA: N,N-диизопропилэтиламин
- DCM: Дихлорметан
- DMAP: 4-Диметиламинопиридин
- ДМФА: N,N-Диметилформамид
- EtOAc: Этилацетат
- EtOH: Этанол
- Et3SiH: Триэтилсилан
- HBTU: 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат
- Hex: Гексан
- i-PrOH: Изопропиловый спирт
- MeI: Иодметан
- MeOH: Метанол
- MsCl: Мезилхлорид
- NaOMe: Метоксид натрия
- NBS: N-бромсукцинимид
- PCC: Хлорхромат пиридиния
- Pd(PPh3)4: Тетракис(трифенилфосфин)палладий (0)
- TEA: Триэтиламин
- TEMPO: (2,2,6,6-Тетраметилпиперидин-1-ил)оксадинил
- ТГФ: Тетрагидрофуран
- TMSOTf: Триметилсилил трифторметансульфонат
- К.Т. или к.т.: Комнатная температура
Сравнительный пример 1: Синтез (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
Указанное в заголовке соединение получали способом, описанным в публикации выложенного патента США №2015/0152075. Для конкретных стадий синтеза сравнительного примера 1 делается ссылка на схему 1, как описано выше в разделе ʺУровень техникиʺ.
Стадия 1: Метил 3-метокси-2-нитробензоат (соединение c2 )
К смеси 3-метокси-2-нитробензойной кислоты (25,0 г, 126 ммоль) и K2CO3 (35,0 г, 253 ммоль) в ДМФА (126 мл) добавляли MeI (15,8 мл, 253 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2 часов. В смесь выливали воду (200 мл) и затем перемешивали при 5°С в течение 30 мин. Осажденное твердое вещество собирали фильтрацией, промывали водой и гексаном. Твердое вещество сушили при пониженном давлении с получением указанного в заголовке соединения в неочищенном виде (26,2 г, 98%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 7,60 (дд, J=8,2, 1,2 Гц, 1H), 7,50 (т, J=8,2 Гц, 1H), 7,26 (дд, J=8,2, 1,2 Гц, 1H), 3,39 (с, 3H), 3,99 (с, 3H); [M+Na]+ 235.
Стадия 2: Метил 2-амино-3-метоксибензоат (соединение c3 )
Суспензию метил 3-метокси-2-нитробензоата (26,2 г, 124 ммоль) и Pd/C (10 масс%, 6,0 г) в ТГФ (400 мл) и MeOH (200 мл) перемешивали в атмосфере Н2 при температуре 18 часов. К смеси добавляли EtOAc (300 мл) и фильтровали через слой целита. Фильтрат концентрировали в вакууме с получением указанного в заголовке соединения (22,4 г, 99%) в виде бесцветного масла.
1H ЯМР (400 МГц, CDCl3) δ 7,47 (дд, J=8,2, 1,2 Гц, 1H), 6,85 (дд, J=8,2, 1,2 Гц, 1H), 6,58 (т, J=8,2 Гц, 1H), 6,00 (ушир.с, 2H), 3,87 (с, 3H); [M+H]+ 182.
Стадия 3: Метил 2-амино-5-бром-3-метоксибензоат (соединение c4 )
К раствору метил 2-амино-3-метоксибензоата (22,4 г, 123 ммоль) в ДМФА (250 мл) частями добавляли N-бромсукцинимид (21,9 г, 123 ммоль) при 0°C. Смесь перемешивали при 0°С в течение 0,5 ч. К смеси добавляли воду и экстрагировали с помощью EtOAc (500 мл × 2). Объединенный органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (27,5 г, 86%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 7,60 (д, J=2,2 Гц, 1H), 6,90 (д, J=2,2 Гц, 1H), 6,03 (ушир.с, 1H), 3,87 (с, 3H); [M+H]+ 260.
Стадия 4: Метил 5-бром-2-хлор-3-метоксибензоат (соединение c5 )
К раствору метил 2-амино-5-бром-3-метоксибензоата (27,0 г, 103 ммоль) в H2O (70 мл) и конц. HCl (70 мл) по каплям добавляли раствор NaNO2 (21,5 г, 311 ммоль) в H2O (50 мл) при 0°C. После перемешивания в течение 1 часа, к реакционной смеси добавляли по каплям раствор Cu(I)Cl в конц. HCl (80 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 18 часов. К смеси добавляли воду (300 мл) и экстрагировали с помощью EtOAc (500 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенное указанное в заголовке соединение сушили в высоком вакууме и использовали на следующей стадии без дополнительной очистки (29,0 г, 100%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 7,49 (д, J=2,4 Гц, 1H), 7,16 (д, J=2,4 Гц, 1H), 3,93 (с, 36H), 3,92 (с, 3H); [M+H]+ 278
Стадия 5: 5-Бром-2-хлор-3-метоксибензойная кислота (соединение c6 )
К раствору метил 5-бром-2-хлор-3-метоксибензоата (25,0 г, 89,4 ммоль) в ТГФ (100 мл), H2O (100 мл) и MeOH (100 мл) добавляли водный 5н раствор NaOH по каплям при 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа. К смеси добавляли конц. HCl для подкисления, и смесь экстрагировали EtOAc (500 мл × 2). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (22,6 г, 96%) в виде твердого вещества оранжевого цвета.
1H ЯМР (400 МГц, CDCl3) δ 7,55 (с, 1H), 7,13 (с, 1H), 3,89 (с, 3H); [M+H]+ 265.
Стадия 6: 5-Бром-2-хлор-3-метоксибензоил хлорид (соединение c7 )
К суспензии 5-бром-2-хлор-3-метоксибензойной кислоты (6,0 г, 22,6 ммоль) в CH2Cl2 (100 мл) добавляли оксалилхлорид (2,4 мл, 27,1 ммоль) и каталитическое количество ДМФА при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2 часов. Смесь упаривали в вакууме и сушили в высоком вакууме с получением неочищенного указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3) δ 7,49 (д, J=2,4 Гц, 1H), 7,16 (д, J=2,4 Гц, 1H), 3,93 (с, 3H), 3,92 (с, 3H).
Стадия 7: (5-Бром-2-хлор-3-гидроксифенил)(фенил)метанон (соединение c8 )
Неочищенный 5-бром-2-хлор-3-метоксибензоил хлорид растворяли в бензоле (100 мл) и охлаждали до 0°C. К реакционной смеси добавляли AlCl3 (6,9 г, 52,0 ммоль) частями при 0°C. Смесь перемешивали при 90°С в течение 15 часов. Смесь охлаждали до комнатной температуры и упаривали в вакууме. Остаток охлаждали до 0°C и добавляли водный раствор 1н HCl. Смесь экстрагировали EtOAc (150 мл × 1). Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (7,33 г, количественный выход).
1H ЯМР (400 МГц, CDCl3) δ 7,85-7,82 (м, 2H), 7,70-7,64 (м, 1H), 7,55-7,49 (м, 2H), 7,37 (д, J=2,2 Гц, 1H), 7,13 (д, J=2,2 Гц, 1H), 5,94 (с, 1H).
Стадия 8: 3-Бензил-5-бром-2-хлорфенол (соединение c9 )
К смеси (5-бром-2-хлор-3-гидроксифенил)(фенил)метанон (362 мг, 1,16 ммоль) в трифторуксусной кислоте (3 мл) добавляли триэтилсилан (0,37 мл, 2,32 ммоль) и каталитическую трифлатную кислоту при 0°C. Смесь перемешивали при комнатной температуре в течение 12 часов. Полученную смесь гасили насыщенным раствором NaHCO3 при 0°C и экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (267 мг, 77%).
1H ЯМР (400 МГц, CDCl3) δ 7,37-7,32 (м, 2H), 7,30-7,27 (м, 1H), 7,22-7,19 (м, 2H), 7,13 (д, J=2,4 Гц, 1H), 6,92 (д, J=2,0 Гц, 1H), 4,07 (с, 2H). [M+H]+ 297.
Стадия 9: 1-(Аллилокси)-3-бензил-5-бром-2-хлорбензол (соединение c10 )
К смеси 3-бензил-5-бром-2-хлорфенола (1,72 г, 5,78 ммоль) и K2CO3 (1,6 г, 11,56 ммоль) в ацетоне (35 мл) добавляли аллилбромид (0,73 мл, 8,67 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 12 часов при 65°C. Полученную смесь фильтровали для удаления неорганических веществ. Фильтрат концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (1,96 г, 100%).
1H ЯМР (400 МГц, CDCl3) δ 7,32-7,27 (м, 2H), 7,25-7,22 (м, 1H), 7,21-7,17 (м, 2H), 6,92 (д, J=2,4 Гц, 1H), 6,92 (д, J=2,0 Гц, 1H), 6,10-6,00 (м, 1H), 5,48 (дкв, J=17,2 Гц, 1,6 Гц, 1H), 5,33 (дкв, J=12,4, 1,6 Гц, 1H), 4,59 (дт, J=4,4 Гц, 1,6 Гц, 2H), 4,08 (с, 2H). [M+H]+ 337.
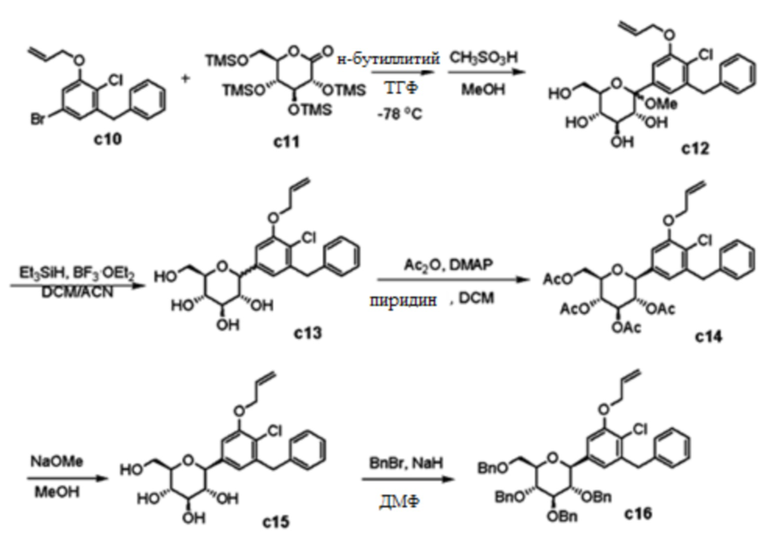
Стадия 10: (3R,4S,5S,6R)-2-(3-(Аллилокси)-5-бензил-4-хлорфенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триол (соединение c12 )
К раствору 1-(аллилокси)-3-бензил-5-бром-2-хлорбензола (1,96 г, 5,82 ммоль) в тетрагидрофуране (5,5 мл)/толуол (11 мл) добавляли по каплям н-бутиллитий (2,5 M в гексане, 2,6 мл, 6,41 ммоль) при -78°C в атмосфере азота. После перемешивания в течение 1 часа, к смеси по каплям добавляли раствор (3R,4S,5R,6R)-3,4,5-трис((триметилсилил)окси)-6-(((триметилсилил)окси)метил)тетрагидро-2H-пиран-2-она (c11; 3,54 г, 7,58 ммоль) в тетрагидрофуране (6,6 мл) с помощью канюли в течение 20 мин при -78°C. Реакционную смесь перемешивали в течение 3 часов при -78°C. К смеси по каплям добавляли CH3SO3H (0,6 мл, 9,25 ммоль) в MeOH (15 мл) при 0°C. Смесь нагревали до комнатной температуры в течение 18 часов, и затем гасили насыщенным раствором NaHCO3 при 0°C. Смесь упаривали при пониженном давлении с удалением летучих веществ. Водный остаток экстрагировали EtOAc (100 мл × 2). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке продукта в виде твердого вещества желтого цвета. [M+Na]+473.
Стадия 11: (3R,4R,5S,6R)-2-(3-(Аллилокси)-5-бензил-4-хлорфенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c13 )
К смеси (3R,4S,5S,6R)-2-(3-(аллилокси)-5-бензил-4-хлорфенил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола (2,55 г, 5,65 ммоль) в CH2Cl2 (30 мл) и CH3CN (30 мл) добавляли по каплям Et3SiH (1,82 мл, 11,3 ммоль) и BF3⋅Et2O (1,07 мл, 8,48 ммоль) при 0°C. Реакционную смесь перемешивали в течение 5 часов при комнатной температуре. Полученную смесь гасили насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
Стадия 12: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(3-(аллилокси)-5-бензил-4-хлорфенил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c14 )
К смеси (3R,4R,5S,6R)-2-(3-(аллилокси)-5-бензил-4-хлорфенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (c13) в CH2Cl2 (12 мл) добавляли Ac2O (4,7 мл, 49,72 ммоль), пиридин (4,0 мл, 49,45 ммоль) и DMAP (35 мг, 0,28 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь разбавляли EtOAc и промывали 1н раствором HCl. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (1,55 г, 47%).
1H ЯМР (400 МГц, DMSO-d6) δ 7,31-7,27 (м, 2H), 7,22-7,21 (м, 1H), 7,19-7,16 (м, 2H), 7,09 (д, J=1,6 Гц, 1H), 6,88 (д, J=1,6 Гц, 1H), 6,12-6,03 (м, 1H), 5,47 (дкв, J=17,6, 2,0 Гц, 1H), 5,35 (т, J=9,6 Гц, 1H), 5,30 (дкв, J=10,4, 1,6 Гц, 1H), 5,12 (т, J=9,6 Гц, 1H), 5,06 (т, J=9,6 Гц, 1H), 4,66-4,62 (m,3H), 4,13-4,03 (м, 5H), 2,04 (с, 3H), 2,03 (с, 3H), 1,95 (с, 3H), 1,70 (с, 3H); [M+Na]+ 611.
Стадия 13: (2S,3R,4R,5S,6R)-2-(3-(Аллилокси)-5-бензил-4-хлорфенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c15 )
К смеси (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(3-(аллилокси)-5-бензил-4-хлорфенил)тетрагидро-2H-пиран-3,4,5-триил триацетата (1,55 г, 2,63 ммоль) в MeOH (50 мл) добавляли NaOMe (25 масс% в MeOH, 2,34 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 12 часов. Полученную смесь нейтрализовали ледяной АсОН. Смесь разбавляли EtOAc и промывали насыщенным раствором NaHCO3. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт использовали на следующей стадии без дополнительной очистки. [M+Na]+ 443.
Стадия 14: (2S,3S,4R,5R,6R)-2-(3-(Аллилокси)-5-бензил-4-хлорфенил)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран (соединение c16 )
К смеси (2S,3R,4R,5S,6R)-2-(3-(аллилокси)-5-бензил-4-хлорфенил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола в ДМФА (26 мл) добавляли NaH (60% дисперсия в минеральном масле, 842 мг, 21,0 ммоль) при 0°C и перемешивали в течение 1 часа при комнатной температуре. К реакционной смеси добавляли по каплям бензилбромид (2,5 мл, 21,0 ммоль) при 0°C. Смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь гасили водой и экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (1,80 г, 88%).
1H ЯМР (400 МГц, CDCl3) δ 7,36-7,31 (м, 13H), 7,26-7,19 (м, 10H), 6,94 (д, J=1,6 Гц, 2H), 6,91 (дд, J=14,8, 2,0 Гц, 2H), 6,10-6,00 (м, 1H), 5,46 (дкв, J=17,2, 1,6 Гц, 1H), 5,31 (дкв, J=10,8, 1,6 Гц, 1H), 4,94 (ABкв, JAB=15,2 Гц, 2H), 4,90 (д, J=10,8 Гц, 1H), 4,70-4,64 (м, 2H), 4,57 (д, J=12,4 Гц, 1H), 4,52-4,49 (м, 2H), 4,46 (д, J=10,8 Гц, 1H), 4,23-4,16 (м, 2H), 4,08 (д, J=15,2 Гц, 1H), 3,89 (д, J=10,8 Гц, 1H), 3,84-3,75 (м, 4H), 3,61-3,57 (м, 1H), 3,48-3,44 (м, 1H); [M+Na]+ 803.
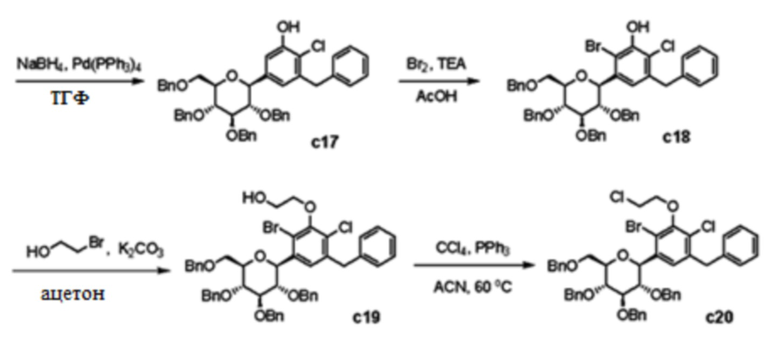
Стадия 15: 3-Бензил-2-хлор-5-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)фенол (соединение c17 )
К смеси ((2S,3S,4R,5R,6R)-2-(3-(Аллилокси)-5-бензил-4-хлорфенил)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пирана (1,80 г, 2,30 ммоль) в ТГФ (25 мл) добавляли NaBH4 (700 мг, 18,4 ммоль) и Pd(PPh3)4 (266 мг, 0,23 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь гасили насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (1,62 г, 95%).
1H ЯМР (400 МГц, CDCl3) δ 7,37-7,31 (м, 13H), 7,27-7,21 (м, 8H), 7,18-7,16 (м, 2H), 7,08 (д, J=2,0 Гц, 1H), 6,98 (дд, J=7,6, 2,0 Гц, 2H), 6,89 (д, J=2,0 Гц, 1H), 4,93 (ABкв, JAB=16,0 Гц, 2H), 4,89 (д, J=10,8 Гц, 1H), 4,67 (д, J=4,8 Гц, 1H), 4,64 (д, J=6,0 Гц, 1H), 4,57 (д, J=12,4 Гц, 1H), 4,46 (д, J=10,4 Гц, 1H), 4,19-4,12 (м, 2H), 4,03 (д, J=15,2 Гц, 1H), 3,95 (д, J=10,4 Гц, 1H), 3,82-3,75 (м, 4H), 3,61-3,57 (м, 1H), 3,49-3,45 (м, 1H); [M+Na]+ 763.
Стадия 16: 3-Бензил-6-бром-2-хлор-5-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)фенол (соединение c18 )
К смеси 3-бензил-2-хлор-5-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)фенола (1,62 г, 2,18 ммоль) в AcOH (11 мл) добавляли триэтиламин (0,46 мл, 3,27 ммоль) и бромин (0,11 мл, 2,18 ммоль) при 0°C. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь гасили насыщенным раствором NaHCO3 и экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (1,04 г, 58%). [M+Na]+ 841.
Стадия 17: 3-(3-Бензил-6-бром-2-хлор-5-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)фенокси)пропан-1-ол (соединение c19 )
К смеси 3-бензил-6-бром-2-хлор-5-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)фенола (1,04 г, 1,27 ммоль) и K2CO3 (0,35 г, 2,54 ммоль) в ацетоне (13 мл) добавляли 2-бромэтанол (0,14 мл, 1,90 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 12 часов при 50°C. Полученную смесь фильтровали для удаления неорганических веществ. Фильтрат концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением соединения c19 (1,10 г, 100%). [M+Na]+ 899.
Стадия 18: (2S,3S,4R,5R,6R)-2-(5-Бензил-2-бром-4-хлор-3-(2-хлорэтокси)фенил)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран (соединение c20 )
К смеси 3-(3-бензил-6-бром-2-хлор-5-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)фенокси)пропан-1-ола (1,09 г, 1,25 ммоль) и трифенилфосфина (1,64 г, 6,28 ммоль) в CH3CN (12 мл) добавляли четыреххлористый углерод (12 мл, 134 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 12 часов при 55°C. Полученную смесь упаривали с удалением растворителей. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (0,61 г, 55%). [M+Na]+ 903.
Стадия 19: 6-Бензил-7-хлор-4-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)-2,3-дигидробензофуран (соединение c21 )
К смеси (2S,3S,4R,5R,6R)-2-(5-бензил-2-бром-4-хлор-3-(2-хлорэтокси)фенил)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пирана (10,02 г, 11,4 ммоль) в ТГФ (114 мл) добавляли по каплям н-бутиллитий (2,5 M в гексане, 6,8 мл, 17,0 ммоль) при -78°C. Реакционную смесь перемешивали в течение 3 часов при -78°C. Полученную смесь гасили 1н раствором HCl (100 мл) и экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (6,0 г, 69%). [M+Na]+ 789.
Стадия 20: (2S,3R,4R,5S,6R)-2-(6-Бензил-7-хлор-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c22 )
Смесь 6-бензил-7-хлор-4-((2S,3S,4R,5R,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-пиран-2-ил)-2,3-дигидробензофурана (6,0 г, 7,82 ммоль) и Pd/C (0,35 г, 2,54 ммоль) в MeOH (220 мл)/ТГФ(220 мл) перемешивали в течение 5 часов при комнатной температуре в атмосфере H2. Полученную смесь фильтровали для удаления неорганических веществ через целит. Фильтрат концентрировали в вакууме с получением указанного в заголовке соединения (количественная величина). Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CD3OD) δ 7,28-7,14 (м, 5H), 6,89 (с, 1H), 4,65 (т, J=8,6 Гц, 2H), 4,17 (д, J=8,8 Гц, 1H), ), 4,07 (ABкв, ΔνAB=18,0 Гц, JAB=15,0 Гц, 2H), 3,90 (дд, J=11,8, 1,4 Гц, 1H), 3,72-3,67 (м, 1H), 3,53-3,42 (м, 3H), 3,40-3,37 (м, 2H); [M+Na]+ 507.
Стадия 21: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(6-бензил-7-хлор-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c23 )
К смеси (2S,3R,4R,5S,6R)-2-(6-бензил-7-хлор-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола в CH2Cl2 (78 мл) добавляли Ac2O (5,9 мл, 62,6 ммоль), пиридин (5,0 мл, 62,6 ммоль) и DMAP (48 мг, 0,39 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь разбавляли EtOAc, промывали 1н раствором HCl. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (4,54 г, 100%).
1H ЯМР (400 МГц, CDCl3) δ 7,32-7,28 (м, 2H), 7,25-7,18 (м, 3H), 6,59 (с, 1H), 5,30 (т, J=9,2 Гц, 2H), 5,19 (т, J=9,6 Гц, 1H), 4,77-4,68 (м, 2H), 4,35-4,32 (м, 1H), 4,31-4,26 (м, 1H), 4,21-4,14 (м, 1H), 4,11 (м, 1H), 4,02 (д, J=15,6 Гц, 1H), 3,83-3,79 (м, 1H), 3,42 (тд,, J=8,8, 1,6 Гц, 2H), 2,10 (с, 3H), 2,09 (с, 3H), 2,03 (с, 3H), 1,70 (с, 3H); [M+Na]+ 597.
Стадия 22: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(6-(4-ацетилбензил)-7-хлор-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c24 )
К смеси (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(6-бензил-7-хлор-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетата (3,75 г, 6,52 ммоль) в CH2Cl2 (78 мл) добавляли по каплям ацетилхлорид (3,71 мл, 52,16 ммоль) и хлорид алюминия (6,95 мг, 52,16 ммоль) при 0°C. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь гасили ледяной водой и экстрагировали с CH2Cl2. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (3,73 г, 93%).
1H ЯМР (400 МГц, CDCl3) δ 7,92-7,89 (м, 2H), 7,29-7,28 (м, 2H), 6,63 (с, 1H), 5,34-5,31 (м, 1H), 5,24-5,18 (м, 2H), 4,78-4,68 (м, 2H), 4,37-4,27 (м, 2H), 4,19-4,16 (м, 1H), 4,16-4,08 (м, 2H), 3,84-3,77 (м, 1H), 3,45-3,40 (м, 2H), 2,61 (с, 3H), 2,10 (с, 3H), 2,09 (с, 3H), 2,03 (с, 3H), 1,70 (с, 3H); [M+Na]+ 639.
Стадия 23: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(7-хлор-6-(4-(1-гидроксиэтил)бензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c25 )
К смеси (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(6-(4-ацетилбензил)-7-хлор-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетата (1,0 г, 1,62 ммоль) в ТГФ (7 мл) медленно добавляли борогидрид натрия (0,12 г, 3,24 ммоль) при -20°C, и затем к смеси добавляли по каплям MeOH (0,24 мл). Смесь перемешивали в течение 3 часов при комнатной температуре. Полученную смесь гасили насыщенным NaHCO3 и экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (0,52 г, 52%).
1H ЯМР (400 МГц, CDCl3) δ 7,29-7,27 (м, 2H), 7,15-7,12 (м, 2H), 6,54 (д, J=5,2 Гц, 1H), 5,29-5,26 (м, 1H), 5,18-5,13 (м, 2H), 4,89-4,84 (м, 2H), 4,71-4,66 (м, 2H), 4,32-4,29 (м, 1H), 4,27-4,22 (м, 1H), 4,15-4,11 (м, 1H), 4,04-3,96 (м, 2H), 3,80-3,75 (м, 1H), 3,40-3,35 (м, 2H), 2,06 (с, 3H), 2,05 (с, 3H), 1,99 (с, 3H), 1,68 (с, 3H), 1,47 (д, J=6,4 Гц, 3H); [M+Na]+ 641.
Стадия 24: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(7-хлор-6-(4-винилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c26 )
Смесь (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(7-хлор-6-(4-(1-гидроксиэтил)бензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетата (520 мг, 0,84 ммоль) и моногидрата п-толуолсульфоновой кислоты (16 мг, 0,084 ммоль) в толуоле (10 мл) перемешивали в течение 2 часов при 120°C. Полученную смесь разбавляли EtOAc и промывали водой. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (407 мг, 81%).
1H ЯМР (400 МГц, CDCl3) δ 7,32-7,30 (м, 2H), 7,12-7,10 (м, 2H), 6,71-6,63 (м, 1H), 6,55 (с, 1H), 5,71-5,66 (м, 1H), 5,29-5,25 (м, 1H), 5,25-5,13 (м, 3H), 4,71-4,66 (м, 2H), 4,33-4,29 (м, 1H), 4,28-4,22 (м, 1H), 4,15-4,11 (м, 1H), 4,08-3,96 (м, 2H), 3,79-3,75 (м, 1H), 3,41-3,35 (м, 2H), 2,06 (с, 3H), 2,05 (с, 3H), 1,99 (с, 3H), 1,68 (с, 3H); [M+Na]+ 623.
Стадия 25: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c27 )
К раствору диэтилцинка (1,1 M в толуоле, 1,74 мл, 1,91 ммоль) в CH2Cl2 (3 мл) добавляли по каплям трифторуксусную кислоту (0,15 мл, 1,91 ммоль) в CH2Cl2 (1,5 мл) при 0°C. Через 1 час к смеси добавляли по каплям диодметан (0,16 мл, 1,91 ммоль) в CH2Cl2 (1,5 мл) при 0°C. Через 1 час к смеси медленно добавляли (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(7-хлор-6-(4-винилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (460 мг, 0,77 ммоль) в CH2Cl2 (3 мл) при 0°C. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь гасили насыщенным раствором NH4Cl и экстрагировали с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage Isolera™ FLASH Purification System, EtOAc/Hex) с получением указанного в заголовке соединения (285 мг, 60%).
1H ЯМР (400 МГц, CDCl3) δ 7,04-7,02 (м, 2H), 6,98-6,95 (м, 2H), 6,53 (с, 1H), 5,29-5,24 (м, 1H), 5,18-5,12 (м, 2H), 4,71-4,65 (м, 2H), 4,31-4,26 (м, 1H), 4,25-4,22 (м, 1H), 4,15-4,11 (м, 1H), 4,05-3,91 (м, 2H), 3,79-3,74 (м, 1H), 3,40-3,35 (м, 2H), 2,06 (с, 3H), 2,05 (с, 3H), 1,99 (с, 3H), 1,88-1,81 (м, 1H), 1,66 (с, 3H), 0,94-0,89 (м, 2H), 0,66-0,61 (м, 2H); [M+Na]+ 637.
Стадия 26: (2S,3R,4R,5S,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c28)
Смесь (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетата (298 мг, 0,48 ммоль) и K2CO3 (536 мг, 3,88 ммоль) в MeOH (20 мл) перемешивали в течение 12 часов. Для удаления неорганических веществ полученную смесь фильтровали. Фильтрат концентрировали в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ (Gilson system, CH3CN/H2O) с получением указанного в заголовке соединения (101 мг, 47%).
Суммарный выход конечного соединения сравнительного примера 1 в соответствии с путем синтеза вышеуказанных стадий 1-26 определяли как составляющий 1% или меньше.
1H ЯМР (400 МГц, CD3OD) δ 7,02 (д, J=8,0 Гц, 2H), 6,92 (д, J=8,0 Гц, 2H), 6,81 (с, 1H), 4,59 (т, J=8,8 Гц, 2H), 4,11 (д, J=9,2 Гц, 1H), 3,96 (ABкв, ΔνAB=19,0 Гц, JAB=15,2 Гц, 2H), 3,87-3,84 (м, 1H), 3,67-3,63 (м, 1H), 3,47-3,37 (м, 3H), 3,35-3,33 (м, 3H), 1,85-1,79 (м, 1H), 0,91-0,86 (м, 2H), 0,61-0,57 (м, 2H); [M+Na]+ 469.
Пример 1: Синтез (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол


Стадия 1: Метил 5-бром-2-хлор-3-гидроксибензоат (соединение c29)
К раствору метил 5-бром-2-хлор-3-метоксибензоата (соединение c5; 30,0 г, 107,3 ммоль) в CH2Cl2 (300 мл) медленно добавляли BBr3 (25,9 мл, 268,3 ммоль) при 0°C в атмосфере азота. Смесь нагревали до комнатной температуры медленно и перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь гасили MeOH (100 мл) при 0°C. Смесь упаривали при пониженном давлении для удаления CH2Cl2, и затем туда добавляли MeOH (150 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали в вакууме с получением указанного в заголовке соединения (29,4 г, 110,8 ммоль, 103%).
1H ЯМР (400 МГц, CDCl3) δ 7,60 (д, J=2,4 Гц, 1H), 7,36 (д, J=2,4 Гц, 1H), 6,00 (с, 1H), 3,94 (с, 1H); [M+H]+ 265.
Стадия 2: Метил 3-(аллилокси)-5-бром-2-хлорбензоат (соединение c30)
К раствору метил 5-бром-2-хлор-3-гидроксибензоата (38,2 г, 143,9 ммоль) в ацетоне (700 мл) добавляли аллилбромид (14,9 мл, 172,7 ммоль) и K2CO3 (29,8 г, 215,9 ммоль) при комнатной температуре. Смесь перемешивали при 60 °С в течение 12 часов, и затем оставляли охлаждаться до комнатной температуры. После отфильтровывания нерастворимой соли через целит, фильтрат упаривали при пониженном давлении и остаток растворяли в EtOAc (500 мл). Органический раствор промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме (44,1 г, 144,3 ммоль, 100%). Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,49 (д, J=2,4 Гц, 1H), 7,16 (д, J=2,4 Гц, 1H), 6,09-6,00 (м, 1H), 5,48 (дд, J=17,2 Гц, 1,2 Гц, 1H), 5,35 (дд, J=10,6 Гц, 1,4 Гц, 1H), 4,63-4,61 (м, 2H), 3,93 (с, 3H); [M+H]+ 305.
Стадия 3: Метил 4-аллил-5-бром-2-хлор-3-гидроксибензоат (соединение c31)
К раствору метил 3-(аллилокси)-5-бром-2-хлорбензоата (10,0 г, 32,7 ммоль) в CH2Cl2 (150 мл) добавляли хлорид диизобутилалюминия (25% в гексане, 64,0 мл) по каплям (0,5-1 ч) при 0°C в атмосфере азота. Смесь нагревали до комнатной температуры медленно и дополнительно перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь охлаждали до 0°C и гасили 1 M HCl (50 мл), затем экстрагировали с EtOAc (150 мл × 2). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме (9,9 г, 32,3 ммоль, 99%). Неочищенный остаток использовали на следующей стадии без дополнительной очистки с получением указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3) δ 7,23 (с, 1H), 6,20 (с, 1H), 5,96-5,86 (м, 1H), 5,11-5,07 (м, 2H), 3,92 (с, 3H), 3,65 (дт, J=5,4 Гц, 1,4 Гц, 2H); [M+H]+ 305.
Стадия 4: Метил 5-бром-2-хлор-3-гидрокси-4-(2-гидроксиэтил)бензоат (соединение c33)
Метод A) Синтез путем восстановления альдегида
К смеси метил 4-аллил-5-бром-2-хлор-3-гидроксибензоата (9,9 г, 32,3 ммоль) в ТГФ/H2O (100 мл/100 мл) добавляли NaIO4 (20,8 г, 97,0 ммоль) и OsO4 (82 мг, 0,32 ммоль) при 0°C. После перемешивания при 0°С в течение 1 ч, реакционную смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 2 часов. Смесь фильтровали с удалением нерастворимых веществ. Фильтрат выливали в насыщенный раствор Na2S2O3 (100 мл) и смесь экстрагировали EtOAc (200 мл × 2). Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме (9,0 г, 29,4 ммоль, 91%). Неочищенный остаток использовали на следующей стадии без дополнительной очистки с получением метил 5-бром-2-хлор-3-гидрокси-4-(2-оксоэтил)бензоата (соединение c32). [M+H]+ 307.
К раствору метил 5-бром-2-хлор-3-гидрокси-4-(2-оксоэтил)бензоата (20,1 г, 65,3 ммоль) в ТГФ (200 мл) добавляли NaBH4 (2,72 г, 71,8 ммоль) при 0°C в атмосфере азота. Реакционную смесь перемешивали при 0°С в течение 2 часов. Смесь гасили насыщенным NH4Cl (100 мл) и экстрагировали с помощью EtOAc (100 мл × 2) [Одной экстракции может быть недостаточно]. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме (19,9 г). К суспензии остатка в EtOAc (20 мл) добавляли гексан (10-20 мл). Полученный осадок собирали фильтрацией и промывали гексаном (50 мл). Осадок сушили в высоком вакууме с получением указанного в заголовке соединения (14,3 г, 72%).
1H ЯМР (400 МГц, CDCl3) δ 7,68 (с, 1H), 7,35 (с, 1H), 3,97-3,93 (м, 2H), 3,92 (с, 3H), 3,21 (т, J=6,2 Гц, 2H); [M+H]+ 309.
Способ B) Синтез путем озонирования с последующим восстановлением
Газ озон барботировали через метил 4-аллил-5-бром-2-хлор-3-гидроксибензоат (10,2 г, 33,4 ммоль, чистота 80%) в CH2Cl2/MeOH (150 мл/35 мл) при -78°С в течение 4 ч (цвет раствора изменился с желтого на бледно-зеленый). После прекращения добавления озона реакционный раствор продували азотом до тех пор, пока зеленый цвет не изменился на другой (цвет раствора вернулся к желтому). Добавляли частями борогидрид натрия (2,5 г, 66,8 ммоль) при -78°C. Полученную смесь медленно нагревали до комнатной температуры в течение 2 ч, концентрировали, суспендировали в EtOAc и концентрировали. К остатку добавляли водный раствор 1н HCl (200 мл) и перемешивали в течение 30 мин. Осадок собирали фильтрацией (количественный, 80% чистота). Осадок суспендировали в EtOAc и перемешивали. К полученной смеси медленно добавляли гексан. Осадок получали путем фильтрации с получением указанного в заголовке соединения (7,9 г, 76,4%, 92% чистота).
1H ЯМР (400 МГц, MeOD) δ 7,57 (с, 1H), 3,93 (с, 3H), 3,76 (т, J=7,24 Гц, 2H), 3,20 (т, J=7,28 Гц, 2H); [M+H]+ 309.
Стадия 5: Метил 4-бром-7-хлор-2,3-дигидробензофуран-6-карбоксилат (соединение c34)
Получение реагента Вильсмейера; К раствору N,N-диметилформамида (7,9 мл, 102,2 ммоль) добавляли SOCl2 (7,5 мл, 102,2 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 2 часов при 40°C. Полученную смесь концентрировали в вакууме с получением гигроскопичного твердого вещества белого цвета.
К смеси реагента Вильсмейера (13,08 г, 102,2 ммоль) в ДМФА (100 мл) медленно добавляли метил 5-бром-2-хлор-3-гидрокси-4-(2-гидроксиэтил)бензоат (21,08 г, 68,10 ммоль) в ДМФА (130 мл) при 0°C. Смесь перемешивали в течение 1 ч при температуре 0°C - 15°C (нагревали равномерно). Реакционную смесь гасили триэтиламином (38 мл, 272,4 ммоль) в ДМФА (38 мл) при 0°C. После перемешивания в течение 10 мин, смесь выливали в воду (1400 мл) при 0°C и перемешивали в течение 2 часов при комнатной температуре. Полученный осадок собирали фильтрацией, промывали водой и сушили, концентрировали в вакууме с получением указанного в заголовке соединения (13,0 г, 44,6 ммоль, 65%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, CDCl3) δ 7,53 (с, 1H), 4,75 (т, J=8,8 Гц, 2H), 3,91 (с, 3H), 3,33 (т, J=8,8 Гц, 2H); [M+H]+ 291.
Стадия 6: (4-Бром-7-хлор-2,3-дигидробензофуран-6-ил)метанол (соединение c35)
К смеси метил 4-бром-7-хлор-2,3-дигидробензофуран-6-карбоксилата (13,0 г, 44,7 ммоль) в ТГФ/EtOH (150 мл/75 мл) медленно добавляли борогидрид натрия (5,07 г, 133,98 ммоль) при комнатной температуре. Смесь перемешивали в течение 12 часов при комнатной температуре. Полученную смесь гасили насыщенным раствором NH4Cl при 0°C и экстрагировали с помощью EtOAc (водный, pH ~7,0). Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (11,7 г, 44,4 ммоль, 99%) в виде твердого вещества белого цвета. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,15 (с, 1H), 4,73 (м, 4H), 3,29 (т, J=8,8 Гц, 2H), 1,91 (т, J=6,4 Гц, 1H); [M-H2O]+ 245.
Стадия 7: 4-Бром-7-хлор-2,3-дигидробензофуран-6-карбальдегид (соединение c36)
К раствору (4-бром-7-хлор-2,3-дигидробензофуран-6-ил)метанола (11,7 г, 44,4 ммоль) в CH2Cl2 (450 мл) медленно добавляли PCC (14,4 г, 66,6 ммоль, пиридиния хлорхромат) при комнатной температуре. После перемешивания в течение 8 часов, осадок фильтровали через слой из силикагеля и промывали CH2Cl2. Фильтрат концентрировали в вакууме с получением указанного в заголовке соединения (10,4 г, 39,8 ммоль, 90%) в виде твердого вещества белого цвета. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ 10,33 (с, 1H), 7,59 (с, 1H), 4,79 (т, J=8,8 Гц, 2H), 3,35 (т, J=8,8 Гц, 2H); [M+H]+ 261.
Стадия 8: (4-Бром-7-хлор-2,3-дигидробензофуран-6-ил)(4-циклопропилфенил)метанол (соединение c39)
Получение (4-циклопропилфенил)магния бромида (соединение c38); Трехгорлую колбу объемом 250 мл, содержащую магний (стружка, 1,1 г, 46,6 ммоль), сушили пламенем. Колба была снабжена конденсатором и дополнительной капельной воронкой в атмосфере азота. 4-Циклопропилфенилбромид (PepTech, USA) (6,0 мл, 42,4 ммоль) в безводном ТГФ (32,4 мл) переносили в капельную воронку. Реакцию Гриньяра инициировали приблизительно 5 мл раствора 4-циклопропилфенилбромида. Добавляли оставшийся раствор бромида при комнатной температуре в течение 4 часов. Полученный раствор непосредственно использовали на следующей стадии.
К раствору 4-бром-7-хлор-2,3-дигидробензофуран-6-карбальдегида (4,6 г, 17,6 ммоль) в безводном ТГФ (170 мл) добавляли свежеприготовленный раствор (4-циклопропилфенил)магния бромида (соединение c38) (30,0 мл 0,85 M в ТГФ, 26,4 ммоль) при 0°C в атмосфере азота. Реакционную смесь перемешивали при 0°С в течение 30 мин. Реакционную смесь гасили водой (100 мл) и экстрагировали с помощью EtOAc (100 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке продукта (7,6 г, 20,0 ммоль, 114%). Неочищенный остаток использовали на следующей стадии без дополнительной очистки с получением неочищенного указанного в заголовке соединения. [M-H2O]+ 361.
Стадия 9: 4-Бром-7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран (соединение c40)
К раствору (4-бром-7-хлор-2,3-дигидробензофуран-6-ил)(4-циклопропилфенил)метанола (7,6 г, 20,0 ммоль) в CH2Cl2/CH3CN (100 мл/ 100 мл) добавляли триэтилсилан (4,6 мл, 40 ммоль) и диэтилэфират трифторида бора (3,8 мл, 30 ммоль) при -20°C в атмосфере азота. Смесь постепенно прогревали до комнатной температуры и дополнительно перемешивали при комнатной температуре в течение 50 мин. Реакционную смесь гасили медленным добавлением насыщенного раствора NaHCO3 (200 мл) и экстрагировали с помощью EtOAc (100 мл). Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с получением указанного в заголовке продукта (4,4 г, 12,1 ммоль, 2 стадии 85%).
1H ЯМР (400 МГц, CDCl3) δ 7,07 (д, J=8,0 Гц, 2H), 6,99 (д, J=8,0 Гц, 2H), 6,80 (с, 1H), 4,70 (т, J=8,8 Гц, 2H), 3,97 (с, 2H), 3,26 (т, J=8,8 Гц, 2H), 1,88-1,84 (м, 1H), 0,95-0,90 (м, 2H), 0,68-0,64 (м, 2H).
Стадия 10: (3R,4S,5R,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2H-пиран-2-ол (соединение c41)
К раствору 4-бром-7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофурана (5,16 г, 14,2 ммоль) в тетрагидрофуране (80 мл) добавляли по каплям н-бутиллитий (2,5 M в гексане, 7,38 мл, 18,4 ммоль) при -78°C в атмосфере азота. После перемешивания в течение 40-60 мин при той же температуре (раствор желтоватого цвета), к смеси добавляли по каплям предварительно охлажденный (при -78°C) раствор (3R,4S,5R,6R)-3,4,5-трис((триметилсилил)окси)-6-(((триметилсилил)окси)метил)тетрагидро-2H-пиран-2-она (соединение c11; 8,6 г, 18,4 ммоль) в тетрагидрофуране (20 мл) с помощью канюли в течение 20 мин. Реакционную смесь перемешивали в течение 2-3 часов при той же температуре (раствор желтоватого цвета).
Реакционную смесь гасили 1% уксусной кислотой (20 мл) при -78°C, и затем упаривали при пониженном давлении с удалением летучих веществ. Водный остаток экстрагировали EtOAc (150 мл × 2). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (11,8 г, количественный) в виде масла бледно-желтого цвета. Неочищенный остаток использовали на следующей стадии без дополнительной очистки
Стадия 11: (3R,4S,5S,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триол (соединение c42)
К раствору (3R,4S,5R,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2H-пиран-2-ола (11,8 г) в MeOH (150 мл) добавляли CH3SO3H (1,5 мл, 23,5 ммоль) при 0°C по каплям. Смесь нагревали до комнатной температуры в течение 18 часов, и затем гасили насыщенным NaHCO3 при 0°C. Смесь упаривали при пониженном давлении с удалением летучих веществ. Водный остаток экстрагировали EtOAc (100 мл × 2). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (6,0 г, 88% 2 стадии) в виде твердого вещества желтого цвета. [M+Na]+499 и [M-OMe]+ 445.
Стадия 12: (3R,4R,5S,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c43 )
К перемешиваемому раствору (3R,4S,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола (6,0 г, 12,6 ммоль) в CH2Cl2/CH3CN (об/об=1:1, 120 мл) добавляли по каплям Et3SiH (6,0 мл, 37,8 ммоль), затем BF3.OEt2 (3,2 мл, 25,2 ммоль) при температуре от -50 до -45°C. Реакционную смесь нагревали до температуры -10-0°C в течение 3-3,5 часов перед гашением насыщенным NaHCO3 (130 мл). Смесь упаривали при пониженном давлении с удалением летучих веществ, и полученный остаток экстрагировали EtOAc (150 мл × 2). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (5,8 г, 12,9 ммоль, 102%) в виде твердого вещества желтого цвета. [M+Na]+ 469.
Стадия 13: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c27)
К раствору (3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (5,8 г, 12,9 ммоль) в CH2Cl2 (120 мл) добавляли DMAP (1,9 г, 15,5 ммоль) и Ac2O (9,7 мл, 103,76 ммоль) при комнатной температуре. После перемешивания в течение 18 часов при комнатной температуре, реакцию гасили водой (120 мл). Полученную смесь экстрагировали CH2Cl2 (100 мл × 2). После промывания 1 M HCl и насыщенным солевым раствором, объединенные органические слои сушили над MgSO4, фильтровали и упаривали при пониженном давлении (7,0 г, неочищенный). Суспендированный остаток в EtOH (45 мл) нагревали при 80°C до появления конденсации в течение 1 ч. Смесь охлаждали до комнатной температуры с перемешиванием в течение 18 часов. Полученный осадок фильтровали, промывали EtOH и сушили в вакууме с получением указанного в заголовке соединения (4,7 г, 7,6 ммоль, 59%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 7,04-7,02 (м, 2H), 6,98-6,95 (м, 2H), 6,53 (с, 1H), 5,29-5,24 (м, 1H), 5,18-5,12 (м, 2H), 4,71-4,65 (м, 2H), 4,31-4,26 (м, 1H), 4,25-4,22 (м, 1H), 4,15-4,11 (м, 1H), 4,05-3,91 (м, 2H), 3,79-3,74 (м, 1H), 3,40-3,35 (м, 2H), 2,06 (с, 3H), 2,05 (с, 3H), 1,99 (с, 3H), 1,88-1,81 (м, 1H), 1,66 (с, 3H), 0,94-0,89 (м, 2H), 0,66-0,61 (м, 2H); [M+Na]+ 637.
Стадия 14: (2S,3R,4R,5S,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c28)
К раствору (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетата (1,5 г, 2,44 ммоль) в ТГФ/MeOH (5,4 мл/10,8 мл; 0,15 M) добавляли 4 M водный раствор NaOH (2,8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Раствор охлаждали до 0°C перед нейтрализацией с помощью 1н HCl. Реакционный раствор разбавляли EtOAc и водой. Органический слой отделяли, и водный слой экстрагировали дважды с помощью EtOAc. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения.
Суспензию неочищенного указанного в заголовке соединения в толуоле (8 мл) нагревали при 40°С в течение 30 мин (липкий раствор -> прозрачный раствор -> образование твердого вещества белого цвета) и охлаждали до комнатной температуры. Взвесь фильтровали в фильтр-воронке, и отфильтрованный осадок промывали толуолом в объеме, равном двум объемам отфильтрованного осадка. Отфильтрованный осадок сушили в вакууме с получением 1,0 г (2,24 ммоль; количественный) указанного в заголовке соединения.
Определяли, что суммарный выход конечного соединения примера 1 в соответствии с путем синтеза вышеуказанных стадий 1-14 составляет около 12%.
1H ЯМР (400 МГц, CD3OD) δ 7,02 (д, J=8,0 Гц, 2H), 6,92 (д, J=8,0 Гц, 2H), 6,81 (с, 1H), 4,59 (т, J=8,8 Гц, 2H), 4,11 (д, J=9,2 Гц, 1H), 3,96 (ABкв, ΔνAB=19,0 Гц, JAB=15,2 Гц, 2H), 3,87-3,84 (м, 1H), 3,67-3,63 (м, 1H), 3,47-3,37 (м, 3H), 3,35-3,33 (м, 3H), 1,85-1,79 (м, 1H), 0,91-0,86 (м, 2H), 0,61-0,57 (м, 2H); [M+Na]+ 469.
Пример 2: Синтез (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола

Стадия 1: (3R,4S,5R,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2H-пиран-2-ол (соединение c41 )
К раствору 4-бром-7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофурана (соединение c40, 5,00 г, 13,8 ммоль) в тетрагидрофуране (140 мл) добавляли по каплям н-бутиллитий (2,5 M в гексане, 8,28 мл, 20,7 ммоль) при -78°C в атмосфере азота. После перемешивания при той же температуре в течение 5 минут, к смеси добавляли по каплям раствор TMS-защищенного лактона (соединение c11; 7,70 г, 16,6 ммоль) в тетрагидрофуране в течение 30 минут. Реакционную смесь перемешивали при той же температуре в течение 1 часа. Реакционную смесь гасили насыщенным водным раствором NH4Cl (300 мл) при 0°C и выполняли экстрагирование с помощью EtOAc. Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (10,3 г, количественный) в виде масла желтого цвета. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 2: (3R,4R,5S,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c43 )
К неочищенному (3R,4S,5R,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-3,4,5-трис(триметилсилилокси)-6-((триметилсилилокси)метил)тетрагидро-2H-пиран-2-олу (10,3 г) в CH2Cl2 (70 мл) и CH3CN (70 мл) добавляли триэтилсилан (8,8 мл, 55,2 ммоль) и TMSOTf (10 мл, 55,2 ммоль) при -78°C. После перемешивания при -78°C в течение 1 часа, реакционную смесь гасили водой (200 мл) при 0°C и выполняли экстрагирование с помощью CH2Cl2 (300 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением неочищенного указанного в заголовке соединения (6,3 г, количественный) в виде масла желтого цвета. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
Стадия 3: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c27 )
К раствору (3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (6,3 г, 13,8 ммоль) в CH2CL2 (140 мл) добавляли DMAP (0,84 г, 6,9 ммоль) и Ac2O (13,0 мл, 13,8 ммоль) при комнатной температуре. После осуществления перемешивания при комнатной температуре в течение 18 часов, реакционную смесь гасили водой (120 мл) и осуществляли экстрагирование с помощью DCM (200 мл). Органический слой промывали водным раствором NaHCO3 (100 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Суспендированный остаток в изопропиловом спирте (20 мл) нагревали при 80°C в течение 10 минут и охлаждали до комнатной температуры. Затем полученный осадок фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (4,52 г, 7,35 ммоль, 53%) в β-форме в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CDCl3) δ 7,04-7,02 (м, 2H), 6,98-6,95 (м, 2H), 6,53 (с, 1H), 5,29-5,24 (м, 1H), 5,18-5,12 (м, 2H), 4,71-4,65 (м, 2H), 4,31-4,26 (м, 1H), 4,25-4,22 (м, 1H), 4,15-4,11 (м, 1H), 4,05-3,91 (м, 2H), 3,79-3,74 (м, 1H), 3,40-3,35 (м, 2H), 2,06 (с, 3H), 2,05 (с, 3H), 1,99 (с, 3H), 1,88-1,81 (м, 1H), 1,66 (с, 3H), 0,94-0,89 (м, 2H), 0,66-0,61 (м, 2H); [M+Na]+ 637.
Стадия 4: (2S,3R,4R,5S,6R)-2-(7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c28 )
К раствору (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетата (4,52 г, 7,35 ммоль) в MeOH (70 мл) добавляли NaOMe (25 масс%, 0,35 мл). После перемешивания при комнатной температуре в течение 18 часов, реакционную смесь концентрировали в вакууме, разбавляли водой (200 мл) и экстрагировали с помощью EtOAc (300 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали перекристаллизацией в толуоле с получением указанного в заголовке соединения (3,16 г, 96%) в β-форме в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,02 (д, J=8,0 Гц, 2H), 6,92 (д, J=8,0 Гц, 2H), 6,81 (с, 1H), 4,59 (т, J=8,8 Гц, 2H), 4,11 (д, J=9,2 Гц, 1H), 3,96 (ABкв, ΔνAB=19,0 Гц, JAB=15,2 Гц, 2H), 3,87-3,84 (м, 1H), 3,67-3,63 (м, 1H), 3,47-3,37 (м, 3H), 3,35-3,33 (м, 3H), 1,85-1,79 (м, 1H), 0,91-0,86 (м, 2H), 0,61-0,57 (м, 2H); [M+Na]+ 469.
Пример 3: Синтез (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-метоксибензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-тиопиран-3,4,5-триола

Стадия 1: (3R,4S,5S,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)-2-(7-хлор-6-(4-метоксибензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-тиопиран-2-ол (соединение c52 )
Повторяли процедуру синтеза стадии 8 в примере 4, за исключением того, что в качестве исходного вещества использовали 4-бром-7-хлор-2,3-дигидробензофуран-6-карбальдегид, а в качестве реагента Гриньяра использовали (4-метоксифенил)магния бромид (соединение с48), с получением (4-бром-7-хлор-2,3-дигидробензофуран-6-ил)(4-метоксифенил)метанола (соединение c49). Затем повторяли процедуру синтеза стадии 9 в примере 4, за исключением того, что в качестве исходного вещества использовали (4-бром-7-хлор-2,3-дигидробензофуран-6-ил) (4-метоксифенил)метанол (соединение с49), с получением 4-бром-7-хлор-6-(4-метоксибензил)-2,3-дигидробензофурана (соединение c50).
К раствору 4-бром-7-хлор-6-(4-метоксибензил)-2,3-дигидробензофурана (соединение c50, 859 мг, 2,43 ммоль) в тетрагидрофуране (8 мл) при -78°C в атмосфере азота добавляли по каплям н-бутиллитий (2,5 M в гексане, 1,3 мл, 3,24 ммоль), и смесь перемешивали в течение 1,5 часов при той же температуре. Затем добавляли по каплям раствор (3R,4S,5S,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-тиопиран-2-она (соединение c51, 898 мг, 1,62 ммоль, это соединение синтезировали со ссылкой на Н. Driguez and B. Henrissat, Tetrahedron Lett. 1981, 22, 5061-5062, Kakinuma, H., et al., J. Med. Chem. 2010, 53, 3247-3261) в тетрагидрофуране (4 мл), и смесь перемешивали в течение 1,5 часов при той же температуре. Реакционную смесь гасили добавлением насыщенного раствора хлорида аммония. После завершения добавления, температуру раствора постепенно поднимали до значения комнатной температуры. Органический слой отделяли, и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением неочищенного соединения (3R,4S,5S,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)-2-(7-хлор-6-(4-метоксибензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-тиопиран-2-ола (количественный выход).
Стадия 2: 7-хлор-6-(4-метоксибензил)-4-((2S,3R,4R,5S,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-тиопиран-2-ил)-2,3-дигидробензофуран (соединение c53 )
К перемешиваемому при -20°C раствору лактола (c52) в дихлорметане (16 мл) добавляли триэтилсилан (1,6 мл, 9,72 ммоль) с последующим добавлением диэтилэфирата трифторида бора (0,8 мл, 6,48 ммоль) с такой скоростью, чтобы температура реакции поддерживалась между -20 и 0°C. Перед гашением насыщенным раствором бикарбоната натрия, раствор оставляли нагреваться до 0°C в течение 1,5 часов. После удаления органических летучих веществ при пониженном давлении, остаток распределяли между этилацетатом и водой. После экстрагирования водного слоя этилацетатом, объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфат магния, фильтровали и концентрировали в вакууме. Неочищенный остаток очищали хроматографией на силикагеле (силикагель, 3-25% этилацетат в гексане) с получением неочищенного соединения 7-хлор-6-(4-метоксибензил)-4-((2S,3R,4R,5S,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-тиопиран-2-ил)-2,3-дигидробензофурана (603 мг, 46%, 2 стадии) в виде твердого вещества белого цвета. [M+Na]+ 835.
Стадия 3: (2S,3R,4R,5S,6R)-2-(7-Хлор-6-(4-метоксибензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-тиопиран-3,4,5-триол (соединение c47 )
К раствору 7-хлор-6-(4-метоксибензил)-4-((2S,3R,4R,5S,6R)-3,4,5-трис(бензилокси)-6-((бензилокси)метил)тетрагидро-2H-тиопиран-2-ил)-2,3-дигидробензофурана (c53, 570 мг, 0,70 ммоль) в дихлорметане (8 мл) добавляли BCl3 (1,0 M в дихлорметане, 2,8 мл) при 0°C. Реакционную смесь перемешивали при 0°С в течение 1 часа. После гашения реакции метанолом, растворитель упаривали при пониженном давлении. Очистка препаративной ВЭЖХ с обращенной фазой (Gilson, SunFire™ Prep, градиент 5-50% ацетонитрил в воде) давала указанное в заголовке соединение (18 мг, 6%) в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, CD3OD) δ 7,07 (д, J=8,4 Гц, 2H), 6,79 (д, J=8,8 Гц, 2H), 6,76 (с, 1H), 4,63 (тд, J=8,0, 1,6 Гц, 2H), 3,95 (с, 2H), 3,92 (д, J=3,6 Гц, 1H), 3,79-3,75 (м, 3H), 3,74 (с, 3H), 3,71 (д, J=6,4 Гц, 1H), 3,56 (дд, J=10,0, 8,8 Гц, 1H), 3,42-3,35 (м, 2H), 3,24-3,20 (м, 1H), 3,01-2,96 (м, 1H), 0,90 (т, J=7,2 Гц, 3H); [M+Na]+ 475.
Пример 4: Получение (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)-6-(метилтио)тетрагидро-2H-пиран-3,4,5-триола
Стадия 1: ((3aS,5S,6R,6aS)-5-(Гидроксиметил)-2,2-диметилтетрагидрофуро[3,2-d][1,3]диоксол-6-ол
К суспензии L-(-)-ксилозы (19,15 г, 127,5 ммоль) и MgSO4 (30,72 г, 255,0 ммоль) в ацетоне (190 мл) добавляли концентрированную H2SO4 (1,9 мл) при комнатной температуре. Через 12 часов реакционную смесь (в которой все количество L-(-)-ксилозы было израсходовано) фильтровали и объединенное твердое вещество дважды промывали ацетоном (20 мл на промывку). Фильтрат желтого цвета нейтрализовали до значения pH, составляющего около 9, с помощью раствора NH4OH при перемешивании. Суспендированное твердое вещество удаляли фильтрованием. Фильтрат концентрировали с получением бис-ацетонидного промежуточного соединения в виде масла желтого цвета. Масло желтого цвета суспендировали в воде (5 мл), и затем значение pH корректировали с 9 до 2 с помощью 1н раствора HCl в воде. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Полученную смесь нейтрализовали добавлением 25% (масc/масс) K3PO4 в воде до тех пор, пока значение pH не стало составлять около 7. Смесь экстрагировали EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (12,63 г, 52%) в виде масла желтого цвета.
1H ЯМР (400 МГц, CD3OD) δ 5,88 (д, J=4,0 Гц, 1H), 4,47 (д, J=4,0 Гц, 1H), 4,18-4,14 (м, 1H), 4,11 (д, J=2,8 Гц, 1H), 3,83-3,71 (м, 2H), 1,45 (с, 3H), 1,29 (с, 3H).
Стадия 2: (3aS,5R,6S,6aS)-6-Гидрокси-2,2-диметилтетрагидрофуро[3,2-d][1,3]диоксолe-5-карбоновая кислота
К раствору ((3aS,5S,6R,6aS)-5-(гидроксиметил)-2,2-диметилтетрагидрофуро[3,2-d][1,3]диоксол-6-ола (14,6 г, 76,7 ммоль), NaHCO3 (19,3 г, 230,3 ммоль) и NaBr (1,6 г, 15,4 ммоль) в смеси ацетон/вода (120 мл/40 мл) добавляли TEMPO (0,24 г, 1,5 ммоль) при комнатной температуре. Смесь охлаждали до 0°C, и затем небольшими частями добавляли трихлоризоциануровую кислоту (17,8 г, 76,7 ммоль). Суспензию перемешивали при комнатной температуре в течение 12 часов. Добавляли метанол (2,0 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь фильтровали и промывали ацетоном (дважды, 20 мл на промывку). Органический растворитель удаляли в вакууме, водный слой экстрагировали EtOAc и органический слой концентрировали в вакууме. Добавляли ацетон и смесь фильтровали. Фильтрат концентрировали с получением желаемой кислоты (9,0 г, 58%) в виде твердого вещества светло-желтого цвета.
1H ЯМР (400 МГц, CD3OD) δ 5,98 (д, J=3,6 Гц, 1H), 4,71 (д, J=3,2 Гц, 1H), 4,51 (д, J=3,6 Гц, 1H), 4,36 (д, J=3,6 Гц, 1H), 1,45 (с, 3H), 1,31 (с, 3H).
Стадия 3: ((3aS,5R,6S,6aS)-6-Гидрокси-2,2-диметилтетрагидрофуро[3,2-d][1,3]диоксол-2-ил)(морфолино)метанон (соединение c56 )
К суспензии (3aS,5R,6S,6aS)-6-гидрокси-2,2-диметилтетрагидрофуро[3,2-d][1,3]диоксолe-5-карбоновой кислоты (9,0 г, 44,2 ммоль) и HBTU (25,1 г, 66,3 ммоль, N,N,N',N'-тетраметил-O-(1H-бензотриазол-1-ил)урония гексафторфосфат) в тетрагидрофуране добавляли 4-метилморфолин (7,3 мл, 66,3 ммоль) при комнатной температуре. Через 1 час к смеси добавляли морфолин (5,8 мл, 66,3 ммоль) при комнатной температуре. Через 12 часов полученную смесь фильтровали, и отфильтрованный осадок промывали тетрагидрофураном. Фильтрат концентрировали в вакууме, и неочищенное вещество очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (5,8 г, 48%) в виде твердого вещества желтого цвета.
1H ЯМР (400 МГц, CD3OD) δ 6,01 (д, J=3,6 Гц, 1H), 5,10 (с, 1H), 4,59 (д, J=2,4 Гц, 1H), 4,57 (д, J=3,6 Гц, 1H), 4,47 (д, J=2,4 Гц, 1H), 3,85-3,62 (м, 6H), 3,53-3,49 (м, 2H), 1,49 (с, 3H), 1,33 (с, 3H). [M+H]+ 274.
Стадия 4: (7-Хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)((3aS,5R,6S,6aS)-6-гидрокси-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-5-ил)метанон (соединение c58 )
К раствору 4-бром-7-хлор-6-(4-этоксибензил)-2,3-дигидробензофурана (соединение c57, 0,7 г, 1,90 ммоль) в ТГФ (17,5 мл) добавляли н-BuLi (2,5 M раствор в гексане, 0,9 мл, 2,28 ммоль) при -78°C. Через 1 час к смеси добавляли по каплям ((3aS,5R,6S,6aS)-6-гидрокси-2,2-диметилтетрагидрофуро[3,2-d][1,3]диоксол-5-ил)(морфолино)метанон (соединение c56, 0,17 г, 0,63 ммоль) в ТГФ (8,0 мл) при -78°C. Через 4 часа полученную смесь гасили насыщенным раствором NH4Cl, и выполняли экстрагирование с помощью EtOAc. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле (Biotage IsoleraTM FLASH Purification System) с получением указанного в заголовке соединения (0,13 г, 43%); [M+H]+ 475.
Стадия 5: (3aS,5S,6R,6aS)-5-((S)-(7-Хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)(гидроксил)метил)-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-6-ол (соединение c59 )
К раствору (7-хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)((3aS,5R,6S,6aS)-6-гидрокси-2,2-диметилтетрагидрофуран[2,3-d][1,3]диоксол-5-ил)метанона (0,13 г, 0,27 ммоль) в метаноле (18 мл) добавляли CeCl3⋅7H2O (0,12 г, 0,32 ммоль), и смесь перемешивали при комнатной температуре до тех пор, пока все твердые вещества не растворились. Затем смесь охлаждали до -78°C и добавляли небольшими порциями NaBH4 (0,012 г, 0,32 ммоль). Смесь перемешивали при -78°C в течение 2 часов, медленно нагревали до 0°C и гасили насыщенным раствором NH4Cl. Смесь концентрировали при пониженном давлении с удалением CH3OH, экстрагировали с помощью EtOAc и промывали насыщенным раствором NaCl. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенное указанное в заголовке соединение сушили в высоком вакууме и использовали на следующей стадии в виде твердого вещества белого цвета (0,13 г) без очистки.
[M+Na]+ 499.
Стадия 6: (3S,4R,5S,6S)-6-(7-Хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-2,3,4,5-тетраил тетраацетат (соединение c60 )
Раствор (3aS,5S,6R,6aS)-5-((S)-(7-хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)(гидроксил)метил]-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-6-ола (0,13 г, 0,27 ммоль) в смеси AcOH/вода (4,0/2,5 мл) перемешивали при 100°C в течение 12 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Неочищенный масло обрабатывали уксусным ангидридом (0,2 мл, 2,16 ммоль) в пиридине (0,7 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 8 часов. Полученную смесь гасили водой, экстрагировали с помощью EtOAc и промывали насыщенным солевым раствором. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (0,16 г, 96%) в виде твердого вещества белого цвета. [M+Na]+ 627.
Стадия 7: (2S,3S,4R,5S,6R)-2-(7-Хлор-6-(4-этоксибензил)-2,3- дигидробензофуран-4-ил)-6-(метилтио)тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c61 )
К раствору (3S,4R,5S,6S)-6-(7-хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-2,3,4,5-тетраил тетраацетата (160 мг, 0,26 ммоль) и тиомочевины (39 мг, 0,52 ммоль) в 1,4-диоксане (3,1 мл) добавляли TMSOTf (70 мкл, 0,39 ммоль) и реакционную смесь нагревали при 80°C в течение 4 часов. Смесь охлаждали до комнатной температуры, добавляли MeI (40 мкл, 0,65 ммоль) и DIPEA (452 мкл, 2,60 ммоль) и осуществляли перемешивали в течение 3 часов. Полученную смесь разбавляли EtOAc и промывали водой. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенное указанное в заголовке соединение сушили в высоком вакууме и использовали на следующей стадии в виде твердого вещества белого цвета (150 мг, 48%) без очистки.[M+Na]+ 615.
Стадия 8: (2S,3R,4R,5S,6R)-2-(7-Хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)-6-(метилтио)тетрагидро-2H-пиран-3,4,5-триол (соединение c54 )
К суспензии (2S,3S,4R,5S,6R)-2-(7-хлор-6-(4-этоксибензил)-2,3-дигидробензофуран-4-ил)-6-(метилтио)тетрагидро-2H-пиран-3,4,5-триил триацетата (150 мг, 0,25 ммоль) в CH3OH (0,7 мл) добавляли NaOMe (каталитическое количество, 25% раствор в CH3OH) при комнатной температуре. Через 20 часов полученную смесь концентрировали в вакууме. Неочищенный продукт разбавляли EtOAc и фильтровали через мембрану. Неочищенный продукт очищали препаративной ВЭЖХ (Gilson system, CH3CN/H2O) с получением указанного в заголовке соединения (41 мг, 35%).
1H ЯМР (400 МГц, CDCl3) δ 7,09 (д, J=8,4 Гц, 2H), 6,80 (д, J=8,8 Гц, 2H), 6,68 (с, 1H), 4,69-4,64 (м, 2H), 4,35 (д, J=10,0 Гц, 1H), 4,20 (д, J=9,2 Гц, 1H), 4,02-3,88 (м, 4H), 3,67-3,65 (м, 2H), 3,61-3,58 (м, 1H), 3,56-3,52 (м, 1H), 3,42-3,40 (м, 2H), 3,29-3,27 (м, 2H), 2,17 (с, 3H), 1,40 (т, J=7,0 Гц, 3H); [M+Na]+ 489.
Пример 5: Получение (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-этилбензил)-2,3-дигидробензофуран-4-ил)-6-(метилтио)тетрагидро-2H-пиран-3,4,5-триола
Указанное в заголовке соединение получали путем повторения процедуры синтеза примера 4, за исключением того, что на стадии 4 вместо 4-бром-7-хлор-6-(4-этоксибензил)-2,3-дигидробензофурана использовали 4-бром-7-хлор-6-(4-этилбензил)-2,3-дигидробензофуран.
1H ЯМР (400 МГц, CDCl3) δ 7,10-7,90(м, 4H), 6,71 (с, 1H), 4,68-4,62 (м, 2H), 4,33 (д, J=9,6 Гц, 1H), 4,18 (д, J=9,2 Гц, 1H), 4,07-4,00 (м, 2H), 3,63-3,57 (м, 3H), 3,52-3,49 (м, 1H), 3,39-3,37 (м, 2H), 3,27-3,25 (м, 2H), 2,60 (кв, J=7,4 Гц, 2H), 2,15 (с, 3H), 1,20 (т, J=7,6 Гц, 3H); [M+Na]+ 473.
Пример 6: Получение (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола
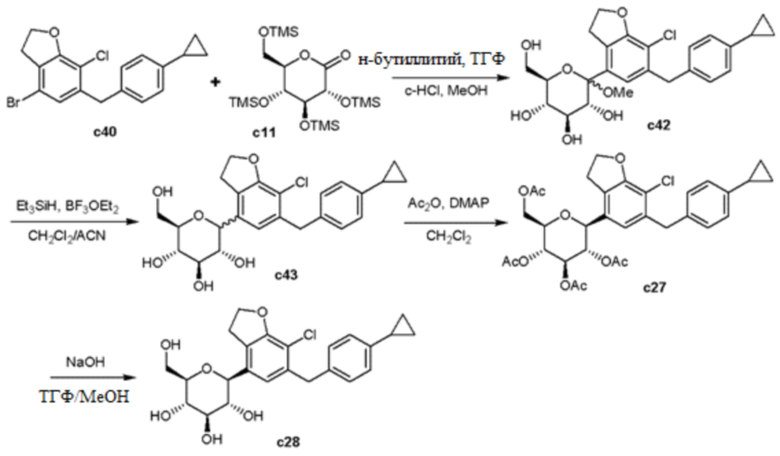
Стадия 1: (3R,4S,5S,6R)-2-[7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил])-6-(гидроксиeметил)-2-метокситетрагидро-2 H -пиран-3,4,5-триол (соединение c42 )
Указанное в заголовке соединение синтезировали следующим путем 1a или 1b.
(1a) В реакционный сосуд добавляли по каплям 4-бром-7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран (250 г, 0,687 моль), (3R,4S,5R,6R)-3,4,5-трис((триметилсилил)окси)-6-(((триметилсилил)окси)метил)тетрагидро-2H-пиран-2-он (642 г, 1,38 моль) и безводный тетрагидрофуран (2,00 л) при комнатной температуре в атмосфере азота, и смесь полностью растворяли. После охлаждения реакционного сосуда до -78°C, добавляли по каплям н-бутиллитий (550 мл, 2,0 M в гексане, 1,38 моль) в течение 1 часа, поддерживая внутреннюю температуру при -60°C или ниже. После завершения добавления н-бутиллития по каплям, смесь дополнительно перемешивали при -78°C в течение 40 минут. К реакционной смеси добавляли по каплям раствор концентрированной хлористоводородной кислоты в метаноле (152 мл/1,750 мл) в течение 20 минут, поддерживая внутреннюю температуру при -30°C или ниже. После завершения добавления по каплям, реакционный сосуд помещали в комнатную температуру и перемешивали в течение 18 часов. После подтверждения завершения реакции, реакционный сосуд охлаждали до 0°C, добавляли насыщенный водный раствор NaHCO3 (2,5 л), значение pH доводили до 9-10, используя прибор для измерения рН, и затем реакционный растворитель удаляли, используя вакуум-концентратор. Концентрат разбавляли EtOAc (2,5 л), дистиллированной водой (1,25 л) и насыщенным солевым раствором (1,25 л) и образовывались слои. Затем органические слои объединяли и водный слой экстрагировали EtOAc (2×1,25 л). Органические слои объединяли и промывали дистиллированной водой (2,5 л) и насыщенным солевым раствором (2,5 л). Органический слой сушили над MgSO4 (50 г) и фильтровали. Затем фильтрат концентрировали при пониженном давлении с удалением растворителя. Процедуру разбавления остатка толуолом (500 мл) и удаления отгонкой при пониженном давлении повторяли дважды с получением указанного в заголовке соединения (328 г) в виде жидкого вещества желтого цвета. Неочищенный остаток использовали на следующей стадии без дополнительной очистки.
(1b) В реакционный сосуд добавляли по каплям 4-бром-7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран (10,0 г, 27,5 моль), (3R,4S,5R,6R)-3,4,5-трис((триметилсилил)окси)-6-(((триметилсилил)окси)метил)тетрагидро-2H-пиран-2-он (25,7 г, 54,9 моль) и безводный тетрагидрофуран (80 мл) при комнатной температуре в атмосфере азота, и смесь полностью растворялась. После охлаждения реакционного сосуда до -78°C, добавляли по каплям н-бутиллитий (22,1 мл, 2,5 M в гексане, 54,9 ммоль) в течение 15 минут, поддерживая внутреннюю температуру при -60°C или ниже. После завершения добавления н-бутиллития по каплям, смесь дополнительно перемешивали при -78°C в течение 30 минут. К реакционной смеси добавляли по каплям раствор концентрированной хлористоводородной кислоты в метаноле (7,01 мл/70 мл) в течение 10 минут, поддерживая внутреннюю температуру при -30°C или ниже. После завершения добавления по каплям, реакционный сосуд помещали в комнатную температуру и перемешивали в течение 18 часов. После подтверждения завершения реакции, реакционный сосуд охлаждали до 0°C, добавляли насыщенный водный раствор NaHCO3 (60 мл), значение pH доводили до 9-10, используя прибор для измерения рН, и затем реакционный растворитель удаляли, используя вакуум-концентратор. Концентрат разбавляли EtOAc (60 мл), дистиллированной водой (60 мл) и насыщенным солевым раствором (60 мл), и образовывались слои. Затем органические слои объединяли и водный слой экстрагировали EtOAc (2 × 30 мл). Органические слои объединяли и промывали дистиллированной водой (60 мл) и насыщенным солевым раствором (60 мл). Органический слой сушили над MgSO4 (5 г), фильтровали и фильтрат концентрировали при пониженном давлении с удалением растворителя. Остаток разбавляли толуолом (50 мл), и затем медленно по каплям добавляли толуольный раствор к гексану (200 мл) при комнатной температуре при перемешивании гексана. Полученную суспензию перемешивали при той же температуре в течение 1 часа и далее фильтровали в вакууме. Полученный фильтрат промывали гексаном (10 мл) и затем сушили в вакуумной печи (40°C) до тех пор, пока содержание влаги в нем не составило, по результатам анализа Карла-Фишера, 1% или ниже, с получением указанного в заголовке соединения (12,6 г, 96%) в виде твердого вещества желтого цвета.
1H ЯМР (500 МГц, CDCl3): δ 7,02 (д, J=8,0 Гц, 2H), 6,92 (д, J=8,0 Гц, 2H), 6,81 (с, 1H), 4,64 (м, 1H), 4,57 (м, 1H), 4,05 (д, J=15,0 Гц, 1H), 3,96 (д, J=15,0 Гц, 1H), 3,93 (дд, J=11,8, 3,0 Гц, 1H), 3,87 (м, 2H), 3,65 (м, 2H), 3,51 (м, 1H), 3,30 (д, J=9,5 Гц, 1H), 3,14 (с, 3H), 1,83 (м, 1H), 0,91 (м, 2H), 0,63 (м, 2H); ЖХ-МС: [M-OMe]+ 445.
Стадия 2: (3R,4R,5S,6R)-2-[7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c43 )
В реакционном сосуде полностью растворяли неочищенный остаток (328 г, 0,687 моль), полученный в соответствии с путем реакции 1a на стадии 1, в CH2Cl2/CH3CN (= объем/объем, 1:1, 5,00 л) при перемешивании при комнатной температуре в атмосфере азота. Реакционный сосуд охлаждали до -50°C, и затем по каплям добавляли Et3SiH (329 мл, 2,08 моль) и BF3-OEt2 (170 мл, 1,37 моль) в течение 10 минут, поддерживая внутреннюю температуру при -45°C или ниже. Реакционную смесь медленно нагревали до -10°C в течение 1 часа и полученную смесь нагревали до 0°C. После осуществления перемешивания в течение 3 часов при 0°C, к реакционной смеси добавляли насыщенный водный раствор NaHCO3 (5,5 л), и значение pH доводили до 7,0-7,5, используя прибор для измерения рН. Органический растворитель удаляли из смеси, используя вакуум-концентратор, и концентрат разбавляли EtOAc (2,5 л). Затем органический слой выделяли. Водный слой разбавляли EtOAc (2 × 125 л) и выполняли экстрагирование. Все органические слои объединяли, сушили над безводным MgSO4 (50 г), фильтровали и затем фильтрат концентрировали при пониженном давлении с удалением растворителя. Остаток сушили в вакууме с получением указанного в заголовке соединения (307 г) в виде жидкого вещества желтого цвета. Полученный таким образом неочищенный остаток использовали на следующей стадии без дополнительной очистки.
1H ЯМР (500 МГц, CD3OD): δ 7,04 (д, J=8,0 Гц, 2H), 6,93 (д, J=8,0 Гц, 2H), 6,83 (с, 1H), 4,61 (т, J=9,0 Гц, 2H), 4,13 (д, J=9,0 Гц, 1H), 3,99 (д, J=15,0 Гц, 1H), 3,94 (д, J=15,0 Гц, 1H), 3,87 (д, J=12,0 Гц, 1H), 3,66 (м, 1H), 3,44 (м, 1H), 3,41 (т, J=9,0 Гц, 2H), 3,36 (м, 2H), 3,31 (м, 1H), 1,83 (м, 1H), 0,91 (м, 2H), 0,63 (м, 2H); ЖХ-МС: [M+Na]+ 469.
Стадия 3: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-[7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c27 )
В реакционный сосуд последовательно добавляли по каплям DMAP (101 г, 0,825 моль) и Ac2O (520 мл, 5,50 моль) при комнатной температуре при перемешивании раствора CH2Cl2 (5,00 л) неочищенного остатка (307 г, 0,687 моль) на стадии 2. Полученную реакционную смесь желтого цвета перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили дистиллированной водой (500 мл). Смесь разделялась на слои. Органический слой сохраняли, и водный слой экстрагировали дихлорметаном (2 × 1,25 л). Все органические слои объединяли и промывали водным раствором 1н HCl (2,5 л) и насыщенным солевым раствором (2,5 л). Органический слой сушили над MgSO4 (50 г), фильтровали и затем фильтрат концентрировали при пониженном давлении. Остаток разбавляли MeOH (2,5 л) и перемешивали при комнатной температуре в течение 30 мин. Полученное твердое вещество фильтровали при пониженном давлении, и фильтрат промывали MeOH (500 мл). Отфильтрованное твердое вещество сушили с получением указанного в заголовке соединения (357 г, выход: 84%, чистота: >97,6%) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ 7,04 (д, J=8,0 Гц, 2H), 6,95 (д, J=8,0 Гц, 2H), 6,53 (с, 1H), 5,24 (дд, J=9,5, 9,5 Гц, 1H), 5,12 (м, 2H), 4,67 (м, 2H), 4,29 (д, J=10,0 Гц, 1H), 4,24 (дд, J=12,5, 4,5 Гц, 1H), 4,13 (дд, J=12,5, 1,5 Гц, 1H), 4,02 (д, J=15,0 Гц, 1H), 3,92 (д, J=15,0 Гц, 1H), 3,77 (м, 1H), 3,38 (м, 2H), 2,07 (с, 3H), 2,06 (с, 3H), 1,99 (с, 3H), 1,84 (м, 1H), 1,66 (с, 3H), 0,92 (м, 2H), 0,63 (м, 2H); ЖХ-МС: [M+Na]+ 637.
Стадия 4: (2S,3R,4R,5S,6R)-2-[7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c28 )
К суспензии (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)тетрагидро-2H-пиран-3,4,5-триил триацетата (357 г, 0,580 моль) в ТГФ/MeOH (= объем/объем, 1:2, 4,28 л) добавляли по каплям 4 M водный раствор NaOH (668 мл, 2,67 моль) при комнатной температуре в течение 20 минут при перемешивании суспензии. Полученную суспензию дополнительно перемешивали при комнатной температуре в течение 2 часов. После охлаждения реакционного сосуда до 0°C к реакционной смеси медленно добавляли по каплям водный раствор 1н HCl (1,18 л) и pH доводили до значения 6,5-7,0, используя прибор для измерения рН. Реакционный растворитель удаляли, используя вакуум-концентратор, и концентрат разбавляли EtOAc (5,36 л) и дистиллированной водой (5,36 л). Смесь расслаивалась. Органический слой сохраняли, и водный слой экстрагировали EtOAc (2 × 1,79 л). Все органические слои объединяли и промывали дистиллированной водой (1,79 л). Органический слой сушили над MgSO4 (710 г), фильтровали и затем фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения.
Неочищенное указанное в заголовке соединение разбавляли EtOAc (3,89 л) и затем перемешивали при нагревании с обратным холодильником в течение 30 минут до полного растворения твердого вещества. Затем полученный продукт охлаждали до комнатной температуры. К полученной суспензии добавляли по каплям изопропиловый эфир (1,29 л) в течение 10 минут и перемешивали в течение 30 минут (включая время добавления по каплям). Процедуру добавления изопропилового эфира повторяли дважды. Потом реакционный сосуд охлаждали до 0°C и перемешивали в течение 30 минут. Полученное твердое вещество фильтровали при пониженном давлении, и фильтрат промывали смешанной жидкостью EtOAc/изопропиловый эфир (= объем/объем, 1:1, 357 мл). Отфильтрованное твердое вещество сушили в вакуумной печи (40°C, 18 часов) с получением указанного в заголовке соединения (236 г, выход: 92%, чистота: >99,7%) в виде твердого вещества белого цвета. Кроме того, определяли, что суммарный выход конечного соединения примера 6, в соответствии с путем синтеза вышеуказанных стадий 1-4, составляет около 77%.
1H ЯМР (500 МГц, CD3OD): δ 7,04 (д, J=8,0 Гц, 2H), 6,93 (д, J=8,0 Гц, 2H), 6,83 (с, 1H), 4,61 (т, J=9,0 Гц, 2H), 4,13 (д, J=9,0 Гц, 1H), 3,99 (д, J=15,0 Гц, 1H), 3,94 (д, J=15,0 Гц, 1H), 3,87 (д, J=12,0 Гц, 1H), 3,66 (м, 1H), 3,44 (м, 1H), 3,41 (т, J=9,0 Гц, 2H), 3,36 (м, 2H), 3,31 (м, 1H), 1,83 (м, 1H), 0,91 (м, 2H), 0,63 (м, 2H); ЖХ-МС: [M+Na]+ 469.
Пример 7: Получение (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола

Стадия 1: (3R,4S,5S,6R)-2-[7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триол (соединение c42 )
Согласно пути реакции 1b в стадии 1 примера 6 использовали 4-бром-7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран (21,6 г) с получением указанного в заголовке соединения (26,2 г, 93%) в виде твердого вещества светло-желтого цвета.
Стадия 2: (3R,4S,5R,6R)-6-(Ацетоксиметил)-2-[7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]-2-метокситетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c44 )
В реакционный сосуд последовательно добавляли по каплям DMAP (8,06 г, 66,0 ммоль) и Ac2O (51,8 мл, 550 моль) при комнатной температуре при перемешивании раствора (3R,4S,5S,6R)-2-[7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]-6-(гидроксиметил)-2-метокситетрагидро-2H-пиран-3,4,5-триола (26,2 г, 55,0 ммоль) в CH2Cl2 (65,5 л). Полученную реакционную смесь желтого цвета перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь гасили дистиллированной водой (50 мл). Смесь расслаивалась. Органический слой сохраняли, и водный слой экстрагировали CH2Cl2 (2 × 30 мл). Все органические слои объединяли и промывали 1н водным раствором HCl (50 мл) и насыщенным солевым раствором (30 мл). Органический слой сушили над MgSO4 (6 г), фильтровали, и затем фильтрат концентрировали при пониженном давлении. Остаток разбавляли MeOH (100 мл) и перемешивали при комнатной температуре в течение 12 часов. Полученное твердое вещество фильтровали при пониженном давлении, и фильтрат промывали MeOH (30 мл). Отфильтрованное твердое вещество сушили с получением указанного в заголовке соединения (29,2 г, в сумме 71% за две стадии) в виде твердого вещества белого цвета.
1H ЯМР (500 МГц, CDCl3): δ 7,01 (д, J=8,5 Гц, 2H), 6,95 (д, J=8,5 Гц, 2H), 6,65 (с, 1H), 5,53 (дд, J=10,0, 9,5 Гц, 1H), 5,19 (дд, J=10,0, 9,5 Гц, 1H), 4,99 (д, j=10,0 Гц, 1H), 4,63 (м, 2H), 4,34 (дд, J=12,0, 4,5 Гц, 1H), 4,14 (дд, J=12,0, 2,0 Гц, 1H), 4,05 (д, J=15,5 Гц, 1H), 4,01 (м, 1H), 3,99 (д, J=15,5 Гц, 1H), 3,49 (м, 1H), 3,33 (м, 1H), 3,17 (с, 3H), 2,08 (с, 3H), 2,05 (с, 3H), 1,94 (с, 3H), 1,83 (м, 1H), 1,61 (с, 3H), 0,90 (м, 2H), 0,62 (м, 2H).
Стадия 3: (2R,3R,4R,5S,6S)-2-(Ацетоксиметил)-6-[7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]тетрагидро-2H-пиран-3,4,5-триил триацетат (соединение c27 )
В реакционном сосуде полностью растворяли (3R,4S,5R,6R)-6-(ацетоксиметил)-2-[7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]-2-метокситетрагидро-2H-пиран-3,4,5-триил триацетат (5,00 г, 7,75 ммоль) в CH2Cl2/CH3CN (= объем/объем, 1:10, 550 мл), осуществляя перемешивание при комнатной температуре. После охлаждения реакционного сосуда до 0°C, последовательно добавляли по каплям Et3SiH (9,89 мл, 62,0 ммоль) и BF3-OEt2 (4,96 мл, 40,3 ммоль) в течение 10 минут, поддерживая внутреннюю температуру при 5°C или ниже. Реакционную смесь перемешивали при 5°C или ниже в течение 1 часа. Затем реакционную смесь нагревали до 10°C и дополнительно перемешивали в течение 2 часов. К реакционной смеси добавляли насыщенный водный раствор NaHCO3 и подтверждали, что значение pH составляло 7,0-7,5, используя прибор для измерения рН. Потом органический растворитель удаляли, используя вакуум-концентратор. Концентрат разбавляли EtOAc (50 мл) и органический слой выделяли. Водный слой экстрагировали EtOAc (2 × 30 мл). Все органические слои объединяли, сушили над MgSO4 (5 г), фильтровали и затем фильтрат концентрировали при пониженном давлении. Остаток растворяли в CH2Cl2 (5 мл), добавляли гексан (10 мл) и осуществляли перемешивали при комнатной температуре в течение 5 минут. К смеси добавляли изопропиловый эфир (20 мл) и осуществляли перемешивали в течение 30 минут. К полученной суспензии дополнительно добавили изопропиловый эфир (20 мл). Реакционный сосуд охлаждали до 0°C и перемешивали в течение 2 часов. Полученное твердое вещество фильтровали при пониженном давлении, и фильтрат промывали изопропиловым эфиром (10 мл). Отфильтрованное твердое вещество сушили в вакууме с получением указанного в заголовке соединения (4,38 г, 92%) в виде твердого вещества белого цвета.
Стадия 4: (2S,3R,4R,5S,6R)-2-[7-Хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триол (соединение c28 )
В соответствии со способом синтеза, описанным на стадии 4 примера 1, как указано выше, использовали (2R,3R,4R,5S,6S)-2-(ацетоксиметил)-6-[7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил]тетрагидро-2H-пиран-3,4,5-триил триацетат (4,38 г, 7,12 ммоль) с получением неочищенного указанного в заголовке соединения. Остаток разбавляли EtOAc (45,3 мл) и затем перемешивали при нагревании с обратным холодильником в течение 10 мин до полного растворения твердого вещества. Полученный продукт охлаждали до комнатной температуры. К полученной суспензии добавляли по каплям изопропиловый эфир (15,1 мл) в течение 10 минут и осуществляли перемешивали в течение 30 минут (включая время добавления по каплям). Процесс добавления изопропилового эфира повторяли дважды. Потом реакционный сосуд охлаждали до 0°C и перемешивали в течение 30 минут. Полученное твердое вещество фильтровали при пониженном давлении, и фильтрат промывали изопропиловым эфиром (10 мл). Отфильтрованное твердое вещество сушили в вакуумной печи (40°C, 18 часов) с получением указанного в заголовке соединения (2,93 г, выход: 92%, чистота: >99,5%) в виде твердого вещества белого цвета. Определяли, что суммарный выход конечного соединения примера 2, в соответствии с путем синтеза вышеуказанных стадий 1-4, составляет около 60%.
Экспериментальный пример 1: Оценка выхода в зависимости от условий реакции перегруппировки

(1) Реакция с использованием растворителя на основе амина или без использования растворителя
Метил 3-(аллилокси)-5-бром-2-хлорбензоат (соединение c30), в качестве исходного вещества, подвергали реакции перегруппировки при 160°С, в результате чего получали метил 4-аллил-5-бром-2-хлор-3-гидроксибензоат (соединение с31) и вычисляли его выход.
В это время количество исходного вещества и условия реакции корректировали, как показано в таблице 1 ниже. В качестве реакционного растворителя использовали 5 M диэтиламин (DEA) или реакцию проводили без какого-либо растворителя.
В результате выход составлял максимум 67% и больше не повышался.
(ч)
(2) Реакция с добавлением кислоты Льюиса
Метил 3-(аллилокси)-5-бром-2-хлорбензоат (соединение c30), в качестве исходного вещества, растворяли в растворителе и подвергали реакции перегруппировки. Затем остаток очищали колоночной хроматографией на силикагеле с получением метил 4-аллил-5-бром-2-хлор-3-гидроксибензоата (соединение c31) и вычисляли его выход.
При этом реакционный растворитель, температуру реакции и кислоту Льюиса корректировали, как показано в таблице 2 ниже, и реакцию проводили с добавлением или без добавления кислоты Льюиса.
В результате, в случае, когда добавляли диизобутилалюминий хлорид ((i-Bu)2AlCl) или хлорид диэтилалюминия (Et2AlCl), которым является кислота Льюиса, выход повышался до 80%.
Экспериментальный пример 2: Оценка выхода в зависимости от условий реакции циклизации
(1) Циклизация с использованием реагента Вильсмейера

Получение реагента Вильсмейера (производственный реагент A); К раствору N,N-диметилформамида (5,6 мл, 74,24 ммоль) при комнатной температуре добавляли SOCl2 (5,3 мл, 72,24 ммоль). Смесь перемешивали при 40°C в течение 2 часов и затем концентрировали в вакууме с получением реагента Вильсмейера, который представляет собой гигроскопичное твердое вещество белого цвета.
Получение реагента Вильсмейера (производственный реагент B); К раствору N,N-диметилформамида (2,9 мл, 37,07 ммоль) при комнатной температуре добавляли SOCl2 (2,7 мл, 37,07 ммоль). Смесь перемешивали при 40°C в течение 2 часов и затем концентрировали в вакууме с получением реагента Вильсмейера, который представляет собой гигроскопичное твердое вещество белого цвета.
Далее к смеси реагента Вильсмейера в ДМФА медленно добавляли метил 5-бром-2-хлор-3-гидрокси-4-(2-гидроксиэтил)бензоат (соединение c33) в ДМФА. Смесь подвергали реакции циклизации при перемешивании в течение 1 часа с получением метил 4-бром-7-хлор-2,3-дигидробензофуран-6-карбоксилата (соединение c34) и вычисляли его выход.
При этом исходное вещество, температуру реакции и тому подобное корректировали, как показано в приведенной ниже таблице 3, и осуществляли реакцию.
В результате получали циклизованный продукт с обычно высоким выходом. Однако в случае, когда исходное вещество было неочищенным, продукт извлекали с низким выходом.
(2) Циклизация с использованием уходящей группы
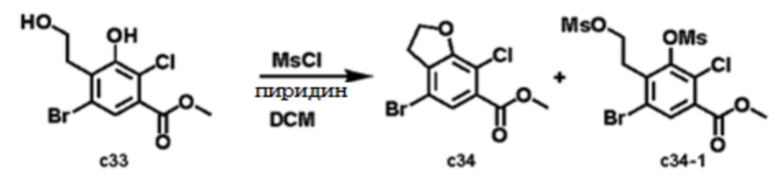
К метил 5-бром-2-хлор-3-гидрокси-4-(2-гидроксиэтил)бензоату (соединение c33), в качестве исходного вещества, добавляли мезилхлорид (MsCl) вместе с растворителем. Смесь подвергали реакции циклизации с получением метил 4-бром-7-хлор-2,3-дигидробензофуран-6-карбоксилата (соединение c34) и вычисляли его выход.
При этом корректировали количество используемого реагента, способ добавления и условия реакции, как показано в таблице 4 ниже, и осуществляли реакцию.
В результате иногда получали желаемое соединение с высоким выходом. Тем не менее, присутствовал недостаток, вызванный эквивалентным контролем MsCl и добавлением по каплям.
(3) Циклизация с использованием галида или реакции Мицунобу
Метил 5-бром-2-хлор-3-гидрокси-4-(2-гидроксиэтил)бензоат (соединение c33), в качестве исходного вещества, подвергали реакции циклизации с использованием галида или реакции Мицунобу с получением метил 4-бром-7-хлор-2,3-дигидробензофуран-6-карбоксилата (соединение c34) и вычисляли его выход.
При этом реагент, в котором объединены трифенилфосфин (PPh3), имидазол, иод (I2) и толуол, использовали для реакции циклизации с использованием галида, а реагент, в котором объединены трифенилфосфин (PPh3), диизопропилазодикарбоксилат (DIAD) и тетрагидрофуран (ТГФ), использовали для реакции Мицунобу.
В результате получали выход, составляющий примерно до 60%. Однако возникало затруднение, связанное с необходимостью удаления оксида трифенилфосфина, который является побочным продуктом, образующимся после реакции.
Экспериментальный пример 3: Оценка выхода в зависимости от условий реакции восстановления
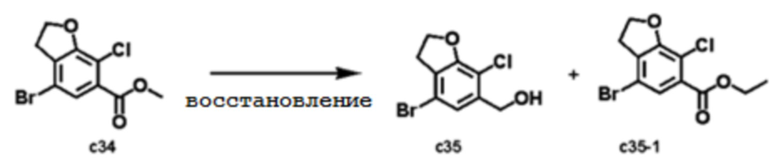
К метил 4-бром-7-хлор-2,3-дигидробензофуран-6-карбоксилату (соединение c34), в качестве исходного вещества, добавляли NaBH4 (3 экв.) и также добавляли или не добавляли кислоту Льюиса. Затем смесь подвергали реакции восстановления в растворителе при комнатной температуре с получением (4-бром-7-хлор-2,3-дигидробензофуран-6-ил)метанола (соединение c35) и вычисляли его выход. Кроме того, также измеряли количество побочного продукта 1 (соединение с35-1).
(1) Корректировали реакционный растворитель и добавляемое количество кислоты Льюиса, как показано в таблице 5 ниже, и осуществляли реакцию. В результате стало возможным получить продукт с самым высоким выходом в случае, когда в качестве реакционного растворителя использовали смесь ТГФ/EtOH (1:1), и получали хороший выход, составляющий 95%, даже в случае, когда использовали только NaBH4 без кислоты Льюиса.
(2) Без добавления кислоты Льюиса, корректировали реакционный растворитель и добавляемое количество NaBH4, как показано в таблице 6 ниже, и осуществляли реакцию. В результате, в случае, когда добавляли NaBH4 в количестве 3 экв. с использованием ТГФ/EtOH (1:1) или ТГФ/EtOH (2:1) в качестве реакционного растворителя, получали хороший выход, составляющий 95%, без побочного продукта.
(3) Корректировали реакционный растворитель, добавляемое количество NaBH4 и добавляемое количество кислоты Льюиса, как показано в таблице 7 ниже, и осуществляли реакцию. В результате реакция не протекала в случае, когда в качестве реакционного растворителя использовали только ТГФ или смесь ТГФ/i-PrOH. С другой стороны, в случае, когда в качестве реакционного растворителя использовали смесь ТГФ/этанол, получали хороший выход, составляющий 95% или выше, даже при использовании только NaBH4 без кислоты Льюиса.
Экспериментальный пример 4: Получение и анализ кристаллической формы
Из соединения, полученного в соответствии со способом по настоящему изобретению, в частности, неочищенного (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (соединение c28), который был получен в соответствии со стадиями 1-14 в примере 1, получали кристаллы посредством процесса кристаллизации в различных растворителях и анализировали.
Для XRD спектра, определяли порошковую рентгеновскую дифракцию с использованием рентгеновского дифрактометра путем облучения Cu-Kα лучами (длина волны 40=1,54056 Å) в соответствии с обычным методом.
Для дифференциальной сканирующей калориметрии (DSC) измерение проводили при скорости +1°С/мин с использованием дифференциального сканирующего калориметра.
(1) Получение кристалла с использованием растворителя толуола
Кристаллизация с использованием толуола аналогична описанной в более поздней части процедуры стадии 14 в примере 1. В частности, неочищенное соединение c28 растворяли в толуоле (8-кратное количество по массе c28) при нагревании при 40°C в течение 30 минут, и затем охлаждали до комнатной температуры. После образования суспензии при комнатной температуре, дополнительно выполняли перемешивание в течение 30 минут. Полученный осадок фильтровали, промывали толуолом (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 91,8%).
XRD спектр полученного кристалла показал кристаллическую форму (кристаллическая форма A), как показано на фиг.1, и углы дифракции (2θ), межплоскостные расстояния (d) и относительные интенсивности (I/Io× 100) характеристических пиков приведены в таблице 8.
Как показано на фиг.2, DSC спектр позволил подтвердить эндотермический пик плавления кристалла.
(2) Получение кристалла с использованием этилацетатного растворителя
Кристаллизация с использованием этилацетата аналогична описанной в более поздней части стадии 4 в примере 6. Конкретно, неочищенное соединение с28 растворяли в этилацетате (15-кратное количество по массе с28) путем перемешивания при нагревании с обратным холодильником и затем охлаждали до комнатной температуры. После образования суспензии при комнатной температуре, дополнительно выполняли перемешивание в течение 30 минут. К полученной смеси добавляли по каплям изопропиловый эфир (15-кратное количество по массе c28) в течение 30 минут и дополнительно выполняли перемешивание при комнатной температуре в течение 30 минут. Полученный осадок фильтровали, промывали 0°C этилацетатом (2-кратное количество по массе c28) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 88,3%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма A), аналогичную представленной в пункте (1) экспериментального примера 4.
(3) Получение кристалла с использованием дихлорметанового растворителя
Неочищенное соединение c28 растворяли в дихлорметане (170-кратное количество по массе c28) с нагреванием при 40°C и затем охлаждали до комнатной температуры. Смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавляли соединение c28 (10 мг, затравка), полученное на стадии 4 примера 6, и потом выполняли перемешивание в течение 12 часов. Полученный осадок фильтровали, промывали дихлорметаном (двукратно относительно массы c28) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 50,1%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма A), аналогичную представленной в пункте (1) экспериментального примера 4.
(4) Получение кристалла с использованием растворителя ацетона
Неочищенное соединение с28 растворяли в ацетоне (35-кратное количество по массе c28), выполняя нагревание с обратным холодильником, и затем охлаждали до комнатной температуры. После образования суспензии при комнатной температуре далее выполняли перемешивание в течение 3 часов. Полученный осадок фильтровали, промывали ацетоном (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 38,2%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма A), аналогичную представленной в пункте (1) экспериментального примера 4.
(5) Получение кристалла с использованием растворителя ацетонитрила
Неочищенное соединение c28 растворяли в ацетонитриле (10-кратное количество по массе c28) при нагревании при 60°C, и затем охлаждали до комнатной температуры. После образования суспензии при комнатной температуре, перемешивание далее выполняли в течение 1 часа. Полученный осадок фильтровали, промывали ацетонитрилом (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 39,4%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма A), аналогичную представленной в пункте (1) экспериментального примера 4.
(6) Получение кристалла с использованием растворителя 2-пропанола
Раствор неочищенного соединения c28 в 2-пропаноле (10-кратное количество по массе c28) растворяли при нагревании при 60°C и затем охлаждали до комнатной температуры. После образования суспензии при комнатной температуре, выполняли далее перемешивание в течение 30 минут. К реакционной смеси дополнительно добавляли по каплям 2-пропанол (5-кратное количество по массе c28) и затем выполняли перемешивание в течение 30 минут. Полученный осадок фильтровали, промывали 2-пропанолом (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 9,5%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма A), аналогичную представленной в пункте (1) экспериментального примера 4.
(7) Получение кристалла с использованием растворителя тетрагидрофуран/дихлорметан
Неочищенное соединение c28 растворяли в тетрагидрофуране (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении с удалением органического растворителя. Концентрированный остаток разбавляли дихлорметаном (30-кратное количество по массе c28) и перемешивали при комнатной температуре. После образования суспензии, дополнительно выполняли перемешивание в течение 30 минут. Полученный осадок фильтровали, промывали дихлорметаном (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 65,3%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма A), аналогичную представленной в пункте (1) экспериментального примера 4.
(8) Получение кристалла с использованием растворителя тетрагидрофуран/н-гексан
Неочищенное соединение c28 растворяли в тетрагидрофуране (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут. К реакционной смеси добавляли по каплям н-гексан (10-кратное количество по массе c28) и выполняли перемешивание в течение 1 часа. К полученной суспензии дополнительно добавили по каплям н-гексан (10-кратное количество по массе c28) и затем выполняли перемешивание в течение 30 минут. К реакционной суспензии многократно добавляли по каплям н-гексан (5-кратное количество по массе c28) и осуществляли перемешивали в течение 30 минут. Полученный осадок фильтровали, промывали н-гексаном (5-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 99,6%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма A), аналогичную представленной в пункте (1) экспериментального примера 4.
(9) Получение кристалла с использованием растворителя метанол/дистиллированная вода
Неочищенное соединение c28 растворяли в метаноле (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут. К реакционной смеси добавляли по каплям дистиллированную воду (10-кратное количество по массе c28) и затем выполняли перемешивание в течение 30 минут. К полученной суспензии дополнительно добавили по каплям дистиллированную воду (10-кратное количество по массе c28) и затем осуществляли перемешивание в течение 1 часа. Полученный осадок фильтровали, промывали дистиллированной водой (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 100%).
XRD спектр полученного кристалла показал кристаллическую форму (кристаллическая форма B), аналогичную показанной на фиг.3, и углы дифракции (2θ), межплоскостные расстояния (d) и относительные интенсивности (I/Io × 100) характеристических пиков приведены в таблице 9.
Как показано на фиг.4, DSC спектр позволил подтвердить эндотермический пик плавления кристалла.
(10) Получение кристалла с использованием растворителя метанол/н-гексан
Неочищенное соединение c28A растворяли в метаноле (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут. К реакционной смеси добавляли по каплям н-гексан (15-кратное количество по массе c28) и осуществляли перемешивали в течение 30 минут. К полученной суспензии дополнительно добавили по каплям н-гексан (10-кратное количество по массе c28) и осуществляли перемешивание в течение 1 часа. Полученный осадок фильтровали, промывали н-гексаном (5-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 97,1%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма B), аналогичную представленной в пункте (9) экспериментального примера 4.
(11) Получение кристалла с использованием растворителя метанол/дихлорметан/н-гексан
Неочищенное соединение c28 растворяли в смеси дихлорметан/метанол (20:1 по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут. К реакционной смеси добавляли по каплям н-гексан (10-кратное количество по массе c28) и осуществляли перемешивали в течение 1 часа. К полученной суспензии дополнительно добавили по каплям н-гексан (10-кратное количество по массе c28) и осуществляли перемешивание в течение 30 минут. К реакционной суспензии многократно добавляли по каплям н-гексан (5-кратное количество по массе c28) и осуществляли перемешивание в течение 30 минут. Полученный осадок фильтровали, промывали н-гексаном (5-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 99,2%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма B), аналогичную представленной в пункте (9) экспериментального примера 4.
(12) Получение кристалла с использованием растворителя тетрагидрофуран/толуол
Неочищенное соединение c28 растворяли в тетрагидрофуране (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении с удалением органического растворителя. Концентрированный остаток разбавляли толуол (30-кратное количество по массе c28) и затем перемешивали при комнатной температуре в течение 1 часа. Полученный осадок фильтровали, промывали толуолом (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 76,1%).
Рентгеновский дифракционный спектр полученного кристалла показал кристаллическую форму (кристаллическая форма C), аналогичную показанной на фиг.5, и в таблице 10 приведены углы дифракции (2θ), межплоскостные расстояния (d) и относительные интенсивности (I/Io×100) характеристических пиков.
Как показано на фиг.6, DSC спектр позволил подтвердить эндотермический пик плавления кристалла.
(13) Получение кристалла с использованием растворителя этанол/дистиллированная вода/н-гексан
Неочищенное соединение c28 растворяли этаноле (5-кратное количество по массе c28) при нагревании при 50°C и затем охлаждали до комнатной температуры. К реакционной смеси добавляли по каплям дистиллированную воду (10-кратное количество по массе c28) и перемешивание осуществляли в течение 1 часа. К полученной суспензии дополнительно добавили по каплям дистиллированную воду (10-кратное количество по массе c28) и осуществляли перемешивали в течение 30 минут. К реакционной суспензии добавляли по каплям н-гексан (1-кратное количество по массе c28) и осуществляли перемешивали в течение 1 часа. Полученный осадок фильтровали, промывали дистиллированной водой (2-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 95,8%). Спектральный анализ XRD полученного кристалла показал кристаллическую форму (кристаллическая форма C), аналогичную представленной в пункте (12) экспериментального примера 4.
(14) Получение кристалла с использованием растворителя этанол/н-гексан
Неочищенное соединение c28 растворяли в этаноле (5-кратное количество по массе c28) при нагревании при 50°C, и затем охлаждали до комнатной температуры. К реакционной смеси добавляли по каплям н-гексан (5-кратное количество по массе c28) и осуществляли перемешивали в течение 30 минут. К полученной суспензии дополнительно добавили по каплям н-гексан (10-кратное количество по массе c28) и осуществляли перемешивали в течение 30 минут. К реакционной суспензии многократно добавляли по каплям н-гексан (5-кратное количество по массе c28) и осуществляли перемешивали в течение 30 минут. Полученный осадок фильтровали, промывали н-гексаном (5-кратный объем фильтрата) и затем сушили в вакуумной печи (50°C, 12 часов) с получением белого кристалла (выход: 96,5%).
Рентгеновский дифракционный спектр полученных кристаллов показал аналогичную кристаллическую форму (кристаллическая форма D), как показано на рис.7, и углы дифракции (2θ), межплоскостные расстояния (d) и относительные интенсивности (I/Io×100) характеристических пиков приведены в таблице 11.
Как показано на фиг.8, DSC спектр позволил подтвердить эндотермический пик плавления кристалла.
Экспериментальный пример 5: Подтверждение стабильности кристаллической формы
Используя кристаллическую форму, полученную в пункте (2) экспериментального примера 4, проводили исследования подтверждения стабильности, связанные со свойствами, идентификационным анализом, содержанием влаги, удельным вращением, родственными веществами и содержанием в течение 3 месяцев в условиях для проведения ускоренного (температура: 40 ± 2°C, относительная влажность: 75 ± 5%) и долгосрочного (температура: 25 ± 2°С, относительная влажность: 60 ± 5%) исследований. Результаты исследований подтверждения стабильности приведены в таблице 12.
(1) Исследование свойств подтвердило отсутствие каких-либо изменений в свойствах кристаллической формы в условиях проведения ускоренного и долгосрочного исследований.
(2) Идентификационный анализ проводили с использованием инфракрасной спектрофотометрии и жидкостной хроматографии, которые находятся в числе общих методов исследований, предписанных Корейской фармакопеей, причем анализ подтвердил, что кристаллическая форма демонстрирует спектр, аналогичный спектру стандартного продукта, при обоих методах исследования.
<Аналитические условия>
- Колонка: Capcdell-pak C18 MG (USPL1), 250 × 4,6 мм, 5 мкм
- Температура: 35°C
- Детектор: детектор на светодиодах (PDA) (длина волны измерения: 225 нм)
- Скорость потока: 1,0 мл/мин
- Мобильная фаза: буфер/метанол (25:75)
- Буфер: буфер, полученный растворением 1,36 г дигидрофосфата калия в 1000 мл дистиллированной воды и затем доведением pH до 3,0 с помощью фосфорной кислоты.
(3) В случае спектра XRD, можно подтвердить, что не было изменений в кристаллической форме в условиях для проведения ускоренного и долгосрочного исследований.
(4) В случае измерения в соответствии с методом измерения влажности, который находится в числе общих методов исследований, предписанных Корейской фармакопеей, исследование на содержание влаги может подтвердить результаты, свидетельствующие о том, что кристаллическая форма лишь в незначительной степени обладает свойством содержать влагу в условиях для проведения ускоренного и долгосрочного исследований.
(5) Исследование, подтверждающее удельное вращение, может подтвердить структурную стабильность кристаллической формы в условиях для проведения ускоренного и долгосрочного исследований.
(6) Исследование, подтверждающее стабильность в отношении родственных веществ, проводили методом жидкостной хроматографии, который находится в числе общих методов исследований, предусмотренных Корейской фармакопеей. Время анализа раствора образца было таким, что измерение проводили до трех раз времени удержания основного пика. Площади пиков стандартного раствора (0,05 мг/мл) и раствора образца (1 мг/мл), за исключением всех пиков, присутствующих в холостом аналитическом растворе, определяли в соответствии с выражением для расчета. В результате стабильность кристаллической формы, применительно к родственным веществам в условиях для проведения ускоренного и долгосрочного исследований, может быть подтверждена.
Аналитические условия: аналитические условия жидкостной хроматографии для подтверждающего исследования в пункте (2) экспериментального примера 5
- Выражение для расчета: другие индивидуальные родственные вещества (%)=(площадь пика каждого родственного вещества в аналитическом образце × количество используемого стандартного продукта × чистота стандартного продукта)/(площадь пика основного пика стандартного раствора × количество используемого образца × коэффициент разбавления)
(7) Для исследования содержания, в качестве стандартного раствора использовали метанольный раствор стандартного продукта (0,2 мг/мл), а в качестве раствора образца (1 мг/мл) использовали метанольный раствор кристаллической формы. Раствор образца и стандартный раствор анализировали методом жидкостной хроматографии, который находится в числе общих методов исследований, предписанных Корейской фармакопеей, и площади пиков стандартного раствора и раствора образца использовали для выполнения вычисления с помощью приведенного ниже выражения. В результате может быть подтверждена стабильность кристаллической формы, которая практически не изменяется по содержанию в условиях для проведения ускоренного и долгосрочного исследований.
- Аналитические условия: аналитические условия жидкостной хроматографии для подтверждающего исследования в пункте (2) экспериментального примера 5
- Выражение для вычисления: другие индивидуальные родственные вещества (%) = (площадь пика основного пика исследуемого образца × количество используемого стандартного продукта × чистота стандартного продукта)/[площадь пика основного пика стандартного раствора × количество используемого образца × (100 - содержание влаги в образце)]
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИФЕНИЛМЕТАНА | 2017 |
|
RU2797392C2 |
| С-ГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ ФЕНИЛЬНОЕ КОЛЬЦО, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ПОЛУЧЕНИЯ ТАКОВЫХ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОВЫЕ | 2017 |
|
RU2739024C2 |
| ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ПРИМЕНЯЕМОЕ ДЛЯ СИНТЕЗА ИНГИБИТОРА SGLT, И СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА SGLT С ПРИМЕНЕНИЕМ УКАЗАННОГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2021 |
|
RU2802443C1 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| ТВЕРДЫЕ ФОРМЫ (2S,3R,4R,5S,6R)-2-(4-ХЛОР-3-(4-ЭТОКСИБЕНЗИЛ)ФЕНИЛ)-6-(МЕТИЛТИО)ТЕТРАГИДРО-2Н-ПИРАН-3,4,5-ТРИОЛА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2505543C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОРЫ КОТРАНСПОРТЕРОВ НАТРИЯ-ГЛЮКОЗЫ 1 И 2, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2669921C2 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗИЛБЕНЗОЛЬНЫХ ИНГИБИТОРОВ SGLT2 | 2013 |
|
RU2625795C2 |
| СОЕДИНЕНИЯ С-МАННОЗИДА, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МОЧЕВЫВОДЯЩИХ ПУТЕЙ | 2019 |
|
RU2790228C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
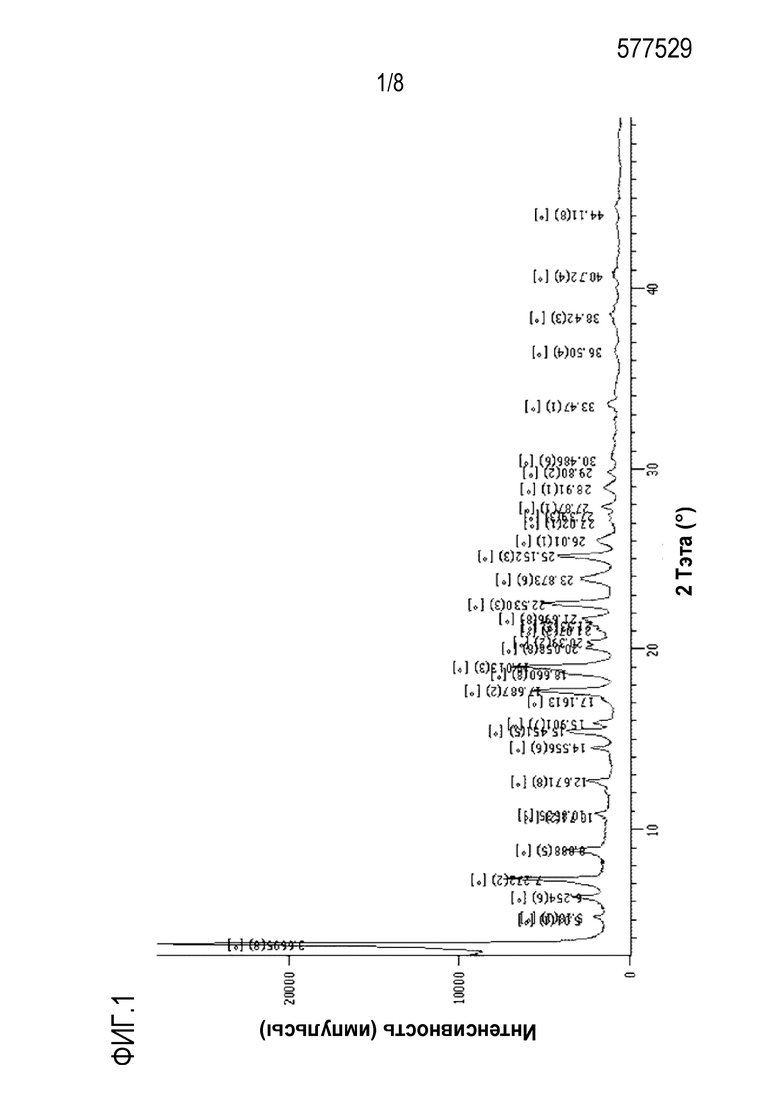
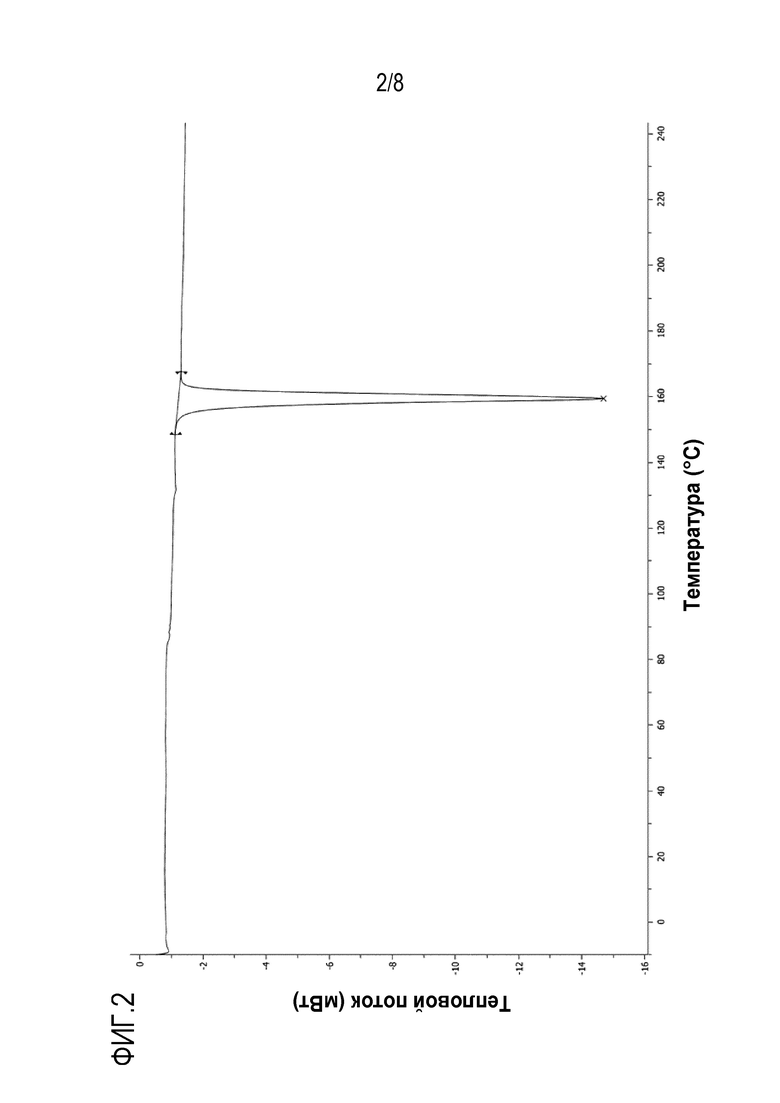


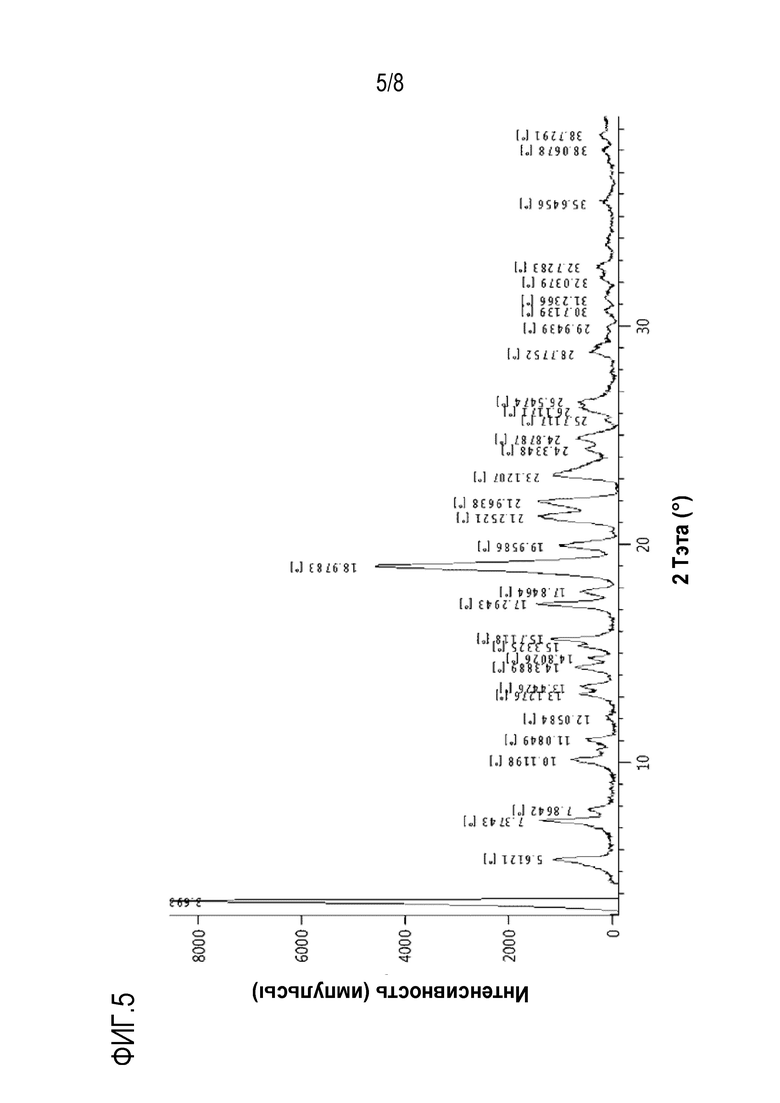

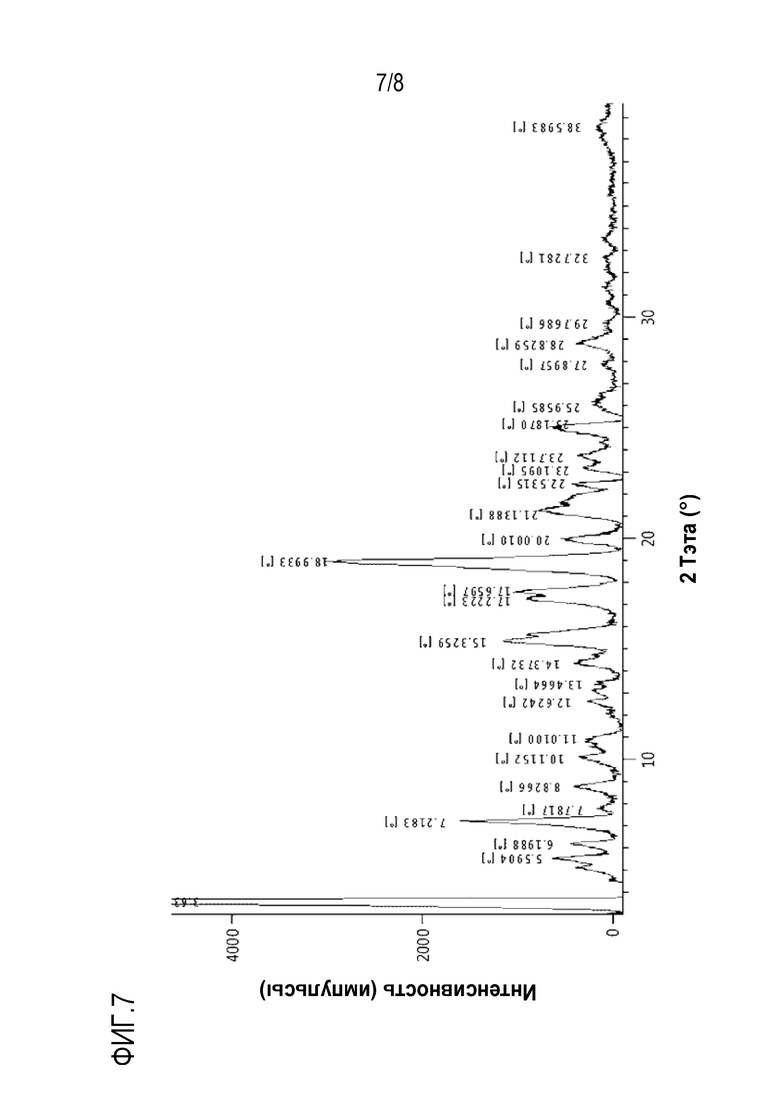

Настоящее изобретение относится к вариантам способа получения кристаллической формы В соединения (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (соединение c28), имеющей рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ), равные 7,0°±0,2°, 14,9°±0,2°, 17,7°±0,2°, 18,8°±0,2°, 20,6°±0,2° 21,8°±0,2° и 23,5°±0,2° в случае облучения с использованием Cu-Kα источника излучения. Один из вариантов способа включает следующие стадии: (1) растворение неочищенного соединения c28 в метаноле (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут; (2) добавление к реакционной смеси по каплям дистиллированной воды (10-кратное количество по массе c28) и затем осуществление перемешивания в течение 30 минут; (3) дополнительное добавление к полученной суспензии по каплям дистиллированной воды (10-кратное количество по массе c28); (4) перемешивание в течение 1 часа; (5) фильтрацию и промывание полученного осадка дистиллированной водой и (6) сушку полученного фильтрата в вакуумной печи (50°C, 12 часов) с получением белого кристалла формы В. Технический результат – получение кристаллической формы соединения c28. 3 н.п. ф-лы, 8 ил., 12 табл., 13 пр.
1. Способ получения кристаллической формы В соединения (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (соединение c28), имеющей рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ), равные 7,0°±0,2°, 14,9°±0,2°, 17,7°±0,2°, 18,8°±0,2°, 20,6°±0,2°, 21,8°±0,2° и 23,5°±0,2° в случае облучения с использованием Cu-Kα источника излучения, где способ включает стадии:
(1) растворение неочищенного соединения c28 в метаноле (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут;
(2) добавление к реакционной смеси по каплям дистиллированной воды (10-кратное количество по массе c28) и затем осуществление перемешивания в течение 30 минут;
(3) дополнительное добавление к полученной суспензии по каплям дистиллированной воды (10-кратное количество по массе c28);
(4) перемешивание в течение 1 часа;
(5) фильтрацию и промывание полученного осадка дистиллированной водой; и
(6) сушку полученного фильтрата в вакуумной печи (50°C, 12 часов) с получением белого кристалла формы В.
2. Способ получения кристаллической формы В соединения (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (соединение c28), имеющей рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ), равные 7,0°±0,2°, 14,9°±0,2°, 17,7°±0,2°, 18,8°±0,2°, 20,6°±0,2°, 21,8°±0,2° и 23,5°±0,2° в случае облучения с использованием Cu-Kα источника излучения, где способ включает стадии:
(1) растворение неочищенного соединения c28 в метаноле (5-кратное количество по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут;
(2) добавление к реакционной смеси по каплям н-гексана (15-кратное количество по массе c28) и осуществление перемешивания в течение 30 минут;
(3) дополнительное добавление к полученной суспензии по каплям н-гексана (10-кратное количество по массе c28);
(4) перемешивание в течение 1 часа:
(5) фильтрацию и промывание полученного осадка н-гексаном; и
(6) сушку полученного фильтрата в вакуумной печи (50°C, 12 часов) с получением белого кристалла формы В.
3. Способ получения кристаллической формы В соединения (2S,3R,4R,5S,6R)-2-(7-хлор-6-(4-циклопропилбензил)-2,3-дигидробензофуран-4-ил)-6-(гидроксиметил)тетрагидро-2H-пиран-3,4,5-триола (соединение c28), имеющей рентгеновский дифракционный спектр, который включает пики при углах дифракции (2θ), равные 7,0°±0,2°, 14,9°±0,2°, 17,7°±0,2°, 18,8°±0,2°, 20,6°±0,2°, 21,8°±0,2° и 23,5°±0,2° в случае облучения с использованием Cu-Kα источника излучения, где способ включает стадии:
(1) растворение неочищенного соединения c28 в смеси дихлорметан/метанол (20:1 по массе c28), осуществляя перемешивание при комнатной температуре в течение 30 минут;
(2) добавление к реакционной смеси по каплям н-гексана (10-кратное количество по массе c28);
(3) осуществление перемешивания в течение 1 часа;
(4) дополнительное добавление к полученной суспензии по каплям н-гексана (10-кратное количество по массе c28);
(5) перемешивание результирующей суспензии в течение 30 минут;
(6) многократное добавление к реакционной суспензии по каплям н-гексана (5-кратное количество по массе c28);
(7) перемешивание;
(8) фильтрация и промывание полученного фильтрата н-гексаном; и
(9) сушка в вакуумной печи (50°C, 12 часов) с получением белого кристалла формы B.
| US 20150152075 A1, 04.06.2015 | |||
| WO 2012041898 A1, 05.04.2012 | |||
| EA 201200600 A1, 28.12.2012 | |||
| N | |||
| Variankaval et al | |||
| From Form to Function: Crystallization of Active Pharmaceutical Ingredients | |||
| AIChE Journal, 2008, 54(7), 1682-1688. |

