ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к способам получения кальципотриола {(5Z, 7E, 22E, 24S)-24-циклопропил-9,10-секохола-5,7,10(19),22-тетраен-1α-3β-24-триол} или моногидрата кальципотриола стереоселективным восстановлением. Настоящее изобретение дополнительно предлагает новые промежуточные соединения и способы синтеза промежуточных соединений, пригодных для получения кальципотриола или моногидрата кальципотриола.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Кальципотриол или кальципотриен (структура 1) [CAS 112965-21-6] показывает высокую активность в ингибировании нежелательного разрастания эпидермальных кератиноцитов [F.A.C.M. Castelijins, M.J. Gerritsen, I.M. J.J. van Erp, van de Kerkhof; Acta Derm. Venereol. 79, 11,1999]. Эффективность кальципотриола или моногидрата кальципотриола (II) при лечении псориаза была показана в ряде клинических испытаний [D.M.Ashcroft et al.; Brit. Med. J. 320, 963-7, 2000] и кальципотриол используют в нескольких коммерческих лекарственных рецептурах.
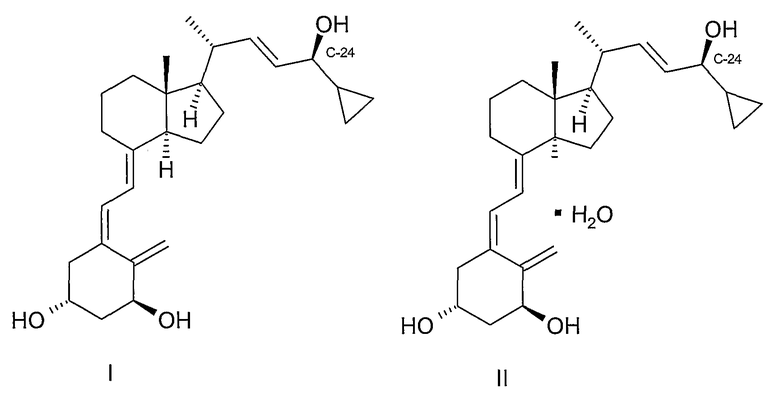
В получении кальципотриола необходима специфическая стереохимия гидроксильной группы при С-24 для полного проявления биологической активности. При действующей методологии требуемую стереохимию достигают одним из следующих способов:
(i) недиастереоселективным восстановлением С-24 кетотриенов с последующим разделением диастереомерных смесей С-24 гидроксилэпимеров с помощью хроматографии (WO 87/00834 & M.J. Calverley; Tetrahedron 43 (20), 4609-19, 1987);
(ii) присоединением энантиомерно-чистой С-24-гидроксилсодержащей боковой цепи к скелету витамина Д (M.J. Calverley, Synlett, 157-59, 1990);
(iii) селективной ферментативной этерификацией одного из С-24 гидроксилэпимеров с последующим хроматографическим разделением (WO 03/060094).
Недиастереоселективное восстановление С-24 кетотриенов с последующим разделением эпимерной смеси (i) представляет собой наиболее широко распространенную методику получения желаемого эпимера. Данный процесс восстановления дает, главным образом, нежелательный С-24 эпимерный спирт (обычно примерно 60% нежелательного 24-R эпимера) и в производственном масштабе трудно выделить желаемый S-эпимер из такой смеси с помощью хроматографии.
Стереоселективный синтез (ii) представляет собой неудобный способ для масштабного производства вследствие своей многостадийной природы и стоимости и вследствие факта использования токсичных промежуточных соединений. Способ ферментативной этерификации (iii) имеет неудобство, помимо высокой стоимости применяемых ферментов, способ включает, в зависимости от селективности применяемых ферментов, 1-2 дополнительных реакционных стадий, которые дополнительно увеличивают стоимость процесса.
Стереоселективное восстановление С-24 кетонов непосредственно до желательных С-24 гидроксилэпимеров было, например, описано для производных холестерина в WO 98/24800 и M. Ishiguro et al., J.C.S. Chem. Comm., 115-117,1981. Стереоселективное восстановление тройной связи в боковой цепи аналога кальципотриола c незащищенной триеновой системой, используя S-Alpine Borane, было описано M.J. Calvery et al. в Bioorg. Med. Chem. Lett., 1841-1844 3(9), 1993.
Главная техническая проблема использования способов стереоселективного восстановления для синтеза кальципотриола обусловлен тем, что ненасыщенная триеновая система известных до настоящего времени промежуточных соединений для синтеза кальципотриола химически лабильна в присутствии кислоты Льюиса, промежуточные соединения легко окисляются, и что они не совместимы с применением типичных условий реакции восстановления. Это приводит к уменьшению выхода, загрязнению продуктов и трудозатратным вспомогательным операциям, особенно в крупномасштабном производстве.
Целью данного изобретения является создание альтернативного способа синтеза кальципотриола, который может преодолеть одну или более различных проблем и неудобств, описанных выше.
Настоящее изобретение предлагает новый способ получения диастереомерно обогащенных С-24 гидроксилэпимеров производных кальципотриола с использованием нового пути синтеза, содержащего стадию стереоселективного восстановления. Настоящее изобретение дополнительно предлагает новые химически более устойчивые промежуточные соединения, в которых лабильная триеновая система защищена в виде аддукта с диоксидом серы. При получении диастереомерно обогащенных С-24 гидроксилэпимеров производных кальципотриола выход и эффективность последующего отделения желательного С-24 S-гидроксилэпимера могут быть значительно улучшены.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
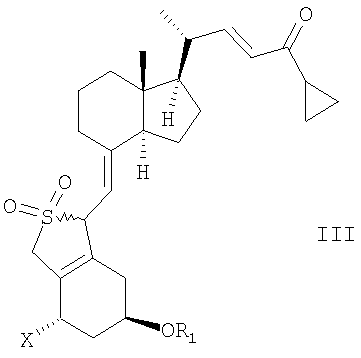
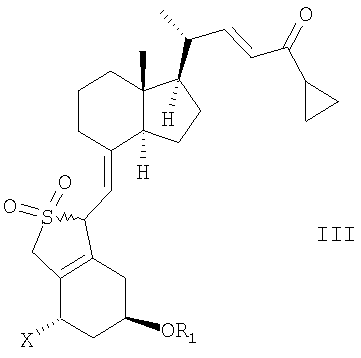
Неожиданно было найдено, что соединение общей структуры III,
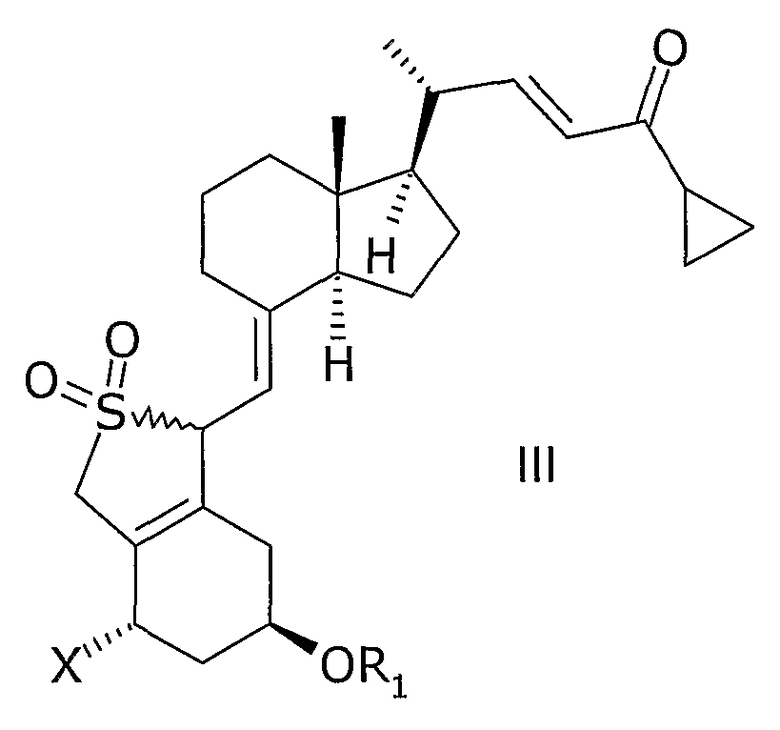
в которой Х представляет собой водород или ОR2,
и в которой R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу,
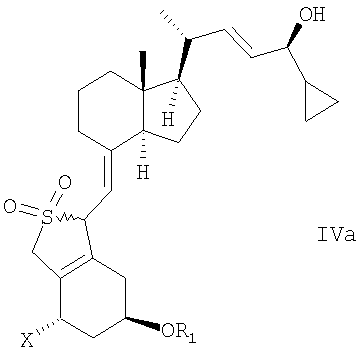
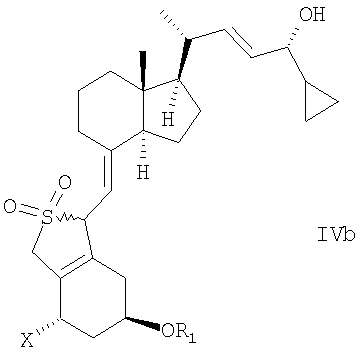
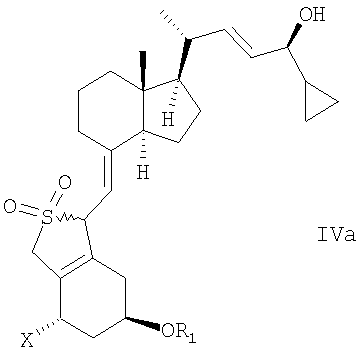
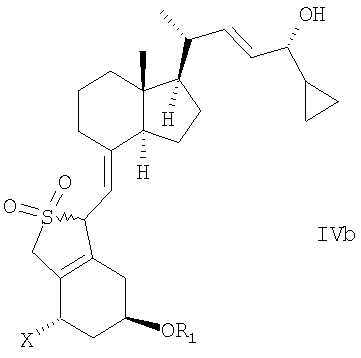
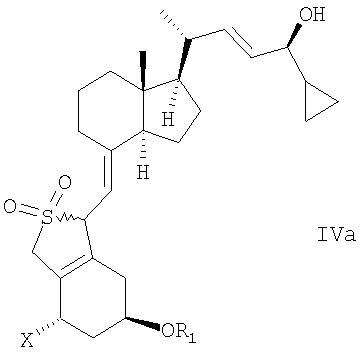
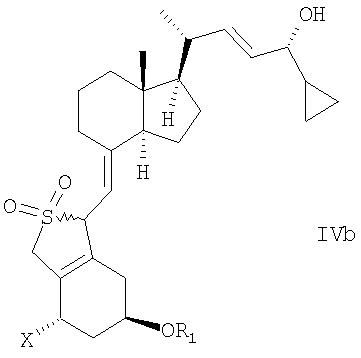
в инертном растворителе с восстановительным агентом или с восстановительным агентом в присутствии хирального вспомогательного вещества образует смесь соединений общей структуры IVa или IVb,
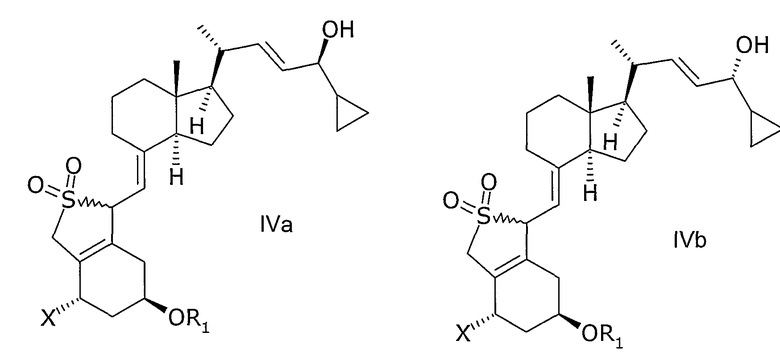
которая обогащена IVa, причем Х, R1 и R2 как определены выше.
В первом аспекте данное соединение относится к способу получения кальципотриола {(5Z, 7E, 22E, 24S)-24-циклопропил-9,10-секохола-5,7,10(19),22-тетраен-1α-3β-24-триол} или моногидрата кальципотриола, содержащему следующие стадии:
(а) восстановление соединения общей структуры III,
в которой Х представляет собой ОR2,
и в которой R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу,
в инертном растворителе с восстановительным агентом или с восстановительным агентом в присутствии хирального вспомогательного вещества для образования смеси соединений общей структуры IVa или IVb, которая обогащена IVa, причем Х, R1 и R2 определены выше;
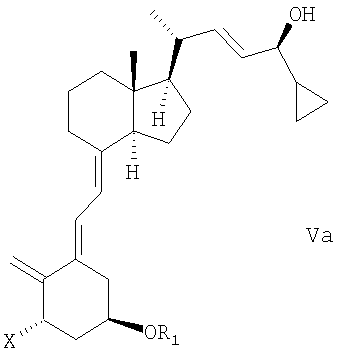
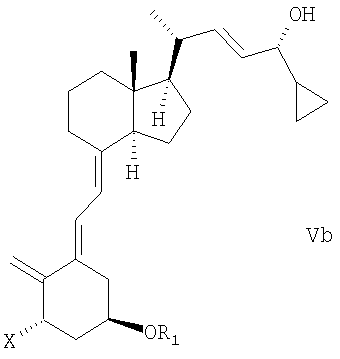
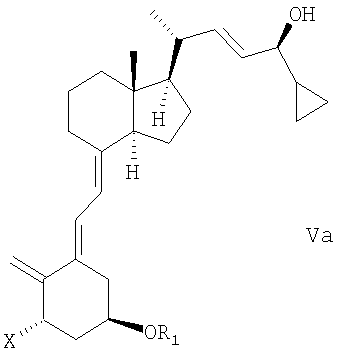
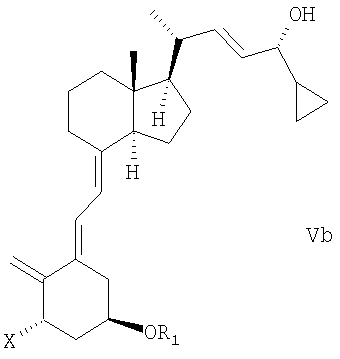
(b) взаимодействие смеси соединений общей структуры IVa или IVb, которая обогащена IVa, в присутствии основания с получением смеси соединений общей структуры Va и Vb, которая обогащена Va,
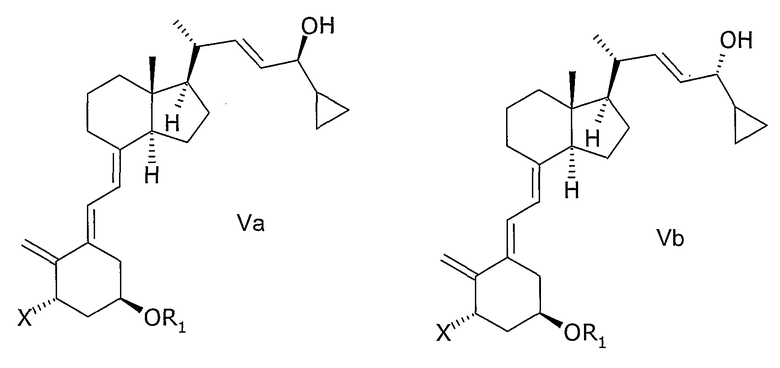
причем Х, R1 и R2 как определены выше;
(с) выделение соединения общей структуры Va из смеси соединений общей структуры Va и Vb, которая обогащена Va, причем Х, R1 и R2 определены выше;
(d) изомеризация соединения общей структуры Va в соединение общей структуры VIa,
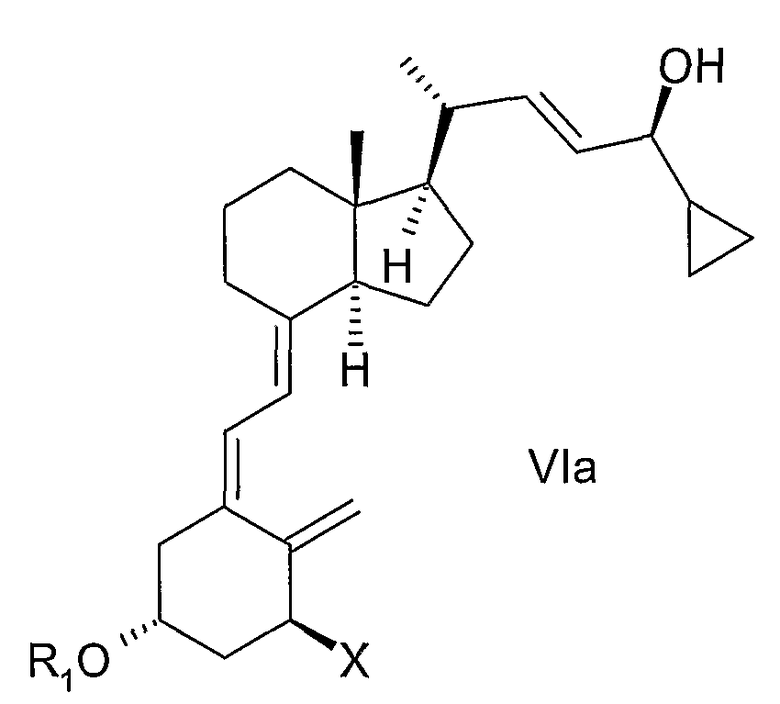
в которой Х, R1 и R2 определены выше; и
(е) когда R1 и/или R2 не является водородом, удаление гидроксизащитной группы R1 и/или R2 соединения общей структуры Va для получения кальципотриола или моногидрата кальципотриола.
В дополнительном аспекте данное изобретение относится к способу получения кальципотриола или моногидрата кальципотриола, содержащему стадии (а)-(b), упомянутые выше, и дополнительно содержащему следующие стадии:
(f) изомеризации смеси соединений общей структуры Va и Vb, причем Х, R1 и R2 как определены в пункте 2, которая обогащена Va до смеси соединений общей структуры VIa и VIb, которая обогащена VIa,
причем Х, R1 и R2 определены выше;
(g) выделение соединения общей структуры VIa из смеси соединений общей структуры VIa и VIb, которая обогащена VIa, причем Х, R1 и R2 определены выше;
(h) когда R1 и/или R2 не является водородом, удаление гидроксизащитной группы R1 и/или R2 соединения общей структуры VIa для получения кальципотриола или моногидрата кальципотриола.
В еще одном дополнительном аспекте данное изобретение относится к способу получения кальципотриола {(5Z, 7E, 22E, 24S)-24-циклопропил-9,10-секохола-5,7,10(19),22-тетраен-1α-3β-24-триол} или моногидрата кальципотриола, содержащему следующие стадии:
(j) восстановление соединения общей структуры III,
в котором Х представляет собой водород,
и в котором R1 представляет собой водород или гидроксизащитную группу, в инертном растворителе с восстановительным агентом или с восстановительным агентом в присутствии хирального вспомогательного вещества для образования смеси соединений общей структуры IVa или IVb,
которая обогащена IVa, причем Х, R1 и R2 определены выше;
(k) взаимодействие смеси соединений общей структуры IVa или IVb, которая обогащена IVa, в присутствии основания с получением смеси соединений общей структуры Va и Vb, которая обогащена Va, причем Х, R1 и R2 определены выше;
(l) выделение соединения общей структуры Va из смеси соединений общей структуры Va и Vb, которая обогащена Va, причем Х, R1 и R2 определены выше;
(m) гидроксилирование соединения общей структуры Va подходящим гидроксилирующим агентом, причем Х и R1 определены выше, с образованием соединения общей структуры Va, причем Х представляет собой ОR2 и R2 представляет собой водород, и при этом R1 определено выше;
(о) изомеризация соединения общей структуры Va в соединение общей структуры VIa,
причем Х, R1 и R2 определены выше; и
(p) когда R1 не является водородом, удаление гидроксизащитной группы R1 соединения общей структуры VIa для получения кальципотриола или моногидрата кальципотриола.
В еще одном дополнительном аспекте данное изобретение относится к способу получения кальципотриола или моногидрата кальципотриола, содержащему стадии (j)-(l) пункта 4 и дополнительно содержащему следующие стадии:
(q) защита С-24 гидроксигруппы соединения общей структуры Va, в которой Х представляет собой водород и в которой R1 представляет собой водород или гидроксизащитную группу, с гидроксизащитной группой;
(r) гидроксилирование С-24 гидроксизащищенного соединения общей структуры Va подходящим гидроксилирующим агентом, причем Х и R1 определены выше, для получения С-24 гидроксизащищенного соединения общей структуры Va, причем Х представляет собой ОR2 и R2 представляет собой водород, и при этом R1 определено выше;
(s) удаление С-24 гидроксизащитной группы соединения общей структуры Va;
(t) изомеризация соединения общей структуры Va в соединение общей структуры VIa,
причем Х, R1 и R2 определены выше; и
(u) когда R1 не является водородом, удаление гидроксизащитной группы R1 соединения общей структуры VIa для получения кальципотриола или моногидрата кальципотриола.
В еще одном дополнительном аспекте данное соединение относится к способу получения соединения общей структуры III,
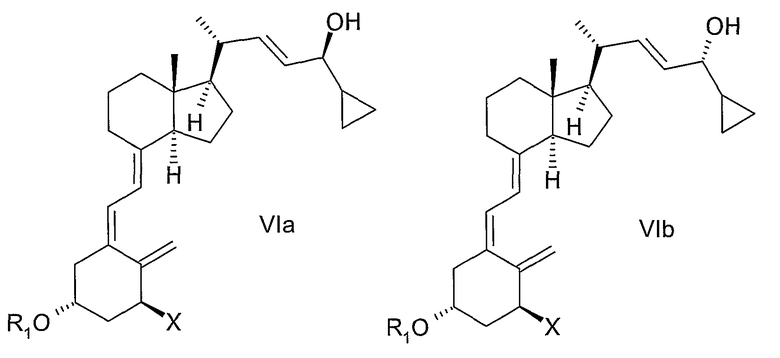
в которой Х представляет собой или водород, или ОR2, и причем R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу, взаимодействием соединения общей структуры VII или VIII,
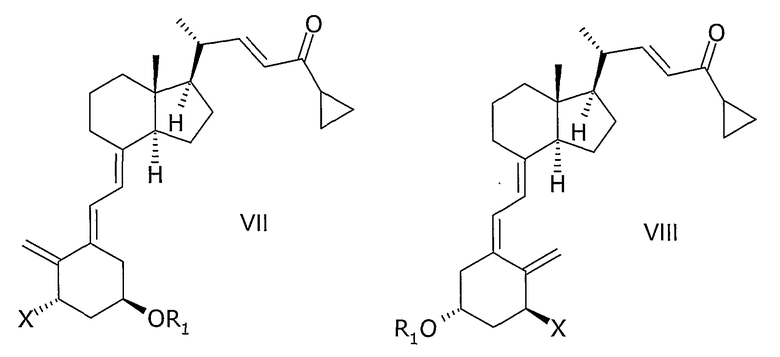
причем R1 и R2 как определены выше, с диоксидом серы.
В еще одном дополнительном аспекте данное изобретение относится к способу взаимодействия смеси соединений общей структуры IVa и IVb,
в которой Х представляет собой или водород, или ОR2,
и причем R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу, которая обогащена IVa, в присутствии основания с получением смеси соединений общей структуры Va и Vb, которая обогащена Va, при этом Х, R1 и R2 определены выше.
В еще одном дополнительном аспекте данное изобретение относится к способу получения кальципотриола {(5Z, 7E, 22E, 24S)-24-циклопропил-9,10-секохола-5,7,10(19),22-тетраен-1α-3β-24-триол} или моногидрата кальципотриола, содержащему любой из способов, описанных выше.
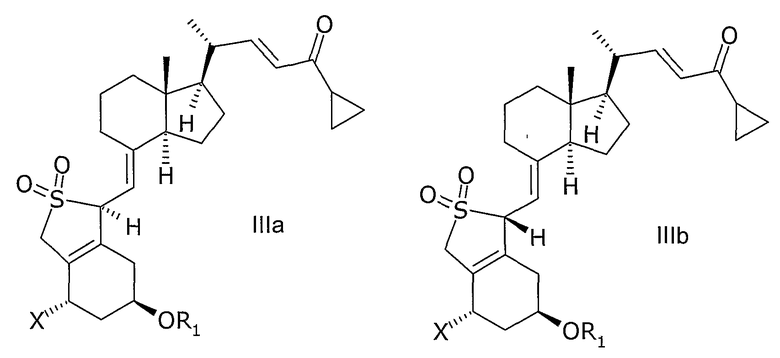
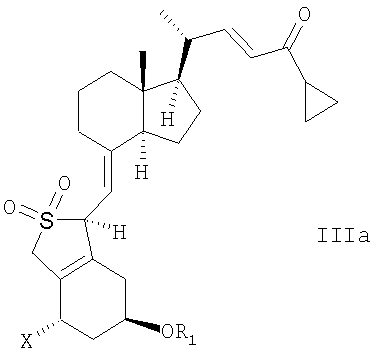
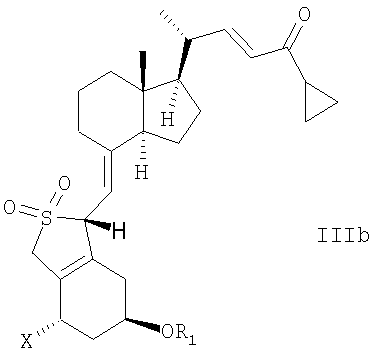
В еще одном дополнительном аспекте, данное изобретение относится к соединениям общей структуры IIIa или IIIb, или их смесям,
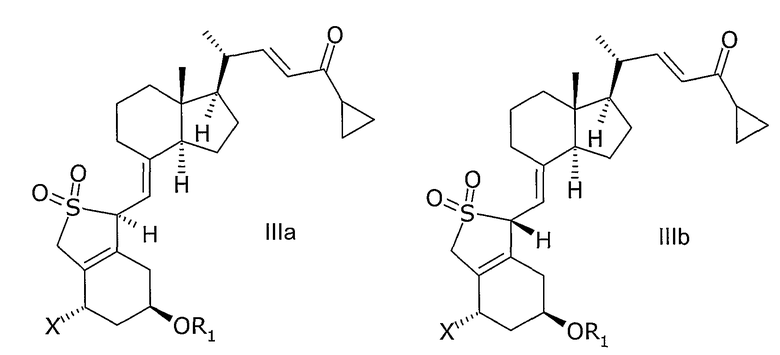
в которой Х представляет собой или водород, или ОR2,
и причем R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу.
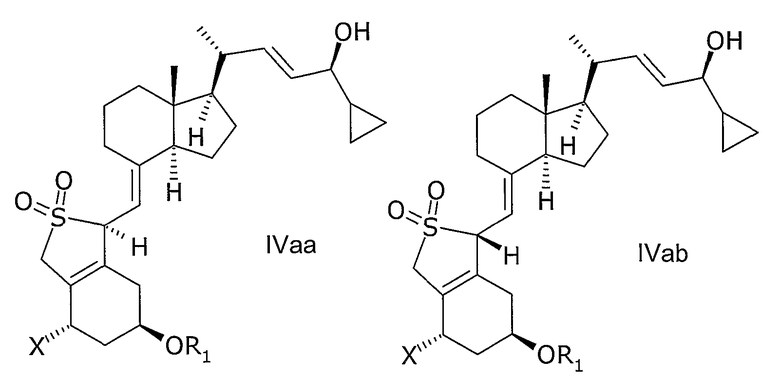
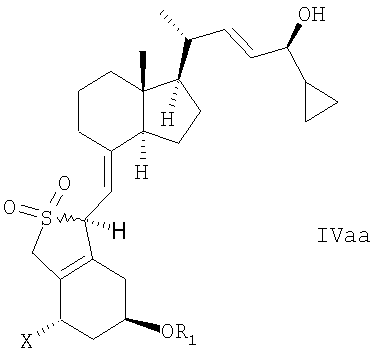
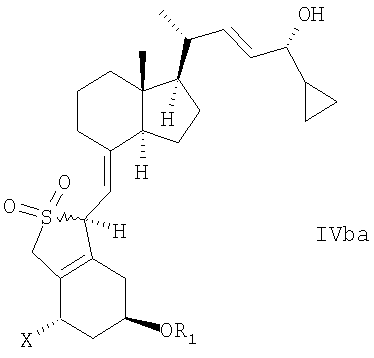
В еще одном дополнительном аспекте данное изобретение относится к соединениям общей структуры IVaa, IVab, IVba, IVbb, IVb или их смесям,
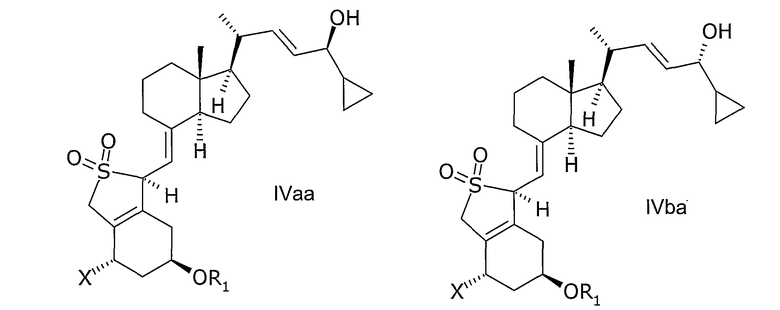
в которой Х представляет собой или водород или ОR2,
и причем R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу.
В еще одном дополнительном аспекте, данное изобретение относится к применению соединений общей структуры IIIa, IIIb, IVaa, IVab, IVba, IVbb в качестве промежуточных соединений в производстве кальципотриола или моногидрата кальципотриола.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Используемый здесь термин «гидроксизащитная группа» означает любую группу, которая образует производное, которое устойчиво к намеченной реакции, в которой упомянутая гидроксизащитная группа может быть селективно удалена реагентами, которые не атакуют регенерируемую гидроксигруппу. Упомянутое производное может быть получено селективной реакцией гидрокизащитного агента с гидроксигруппой. Силильные производные, такие как силильные эфиры, образующие трет.-бутилдиметилсилил, представляют собой примеры гидроксизащитных групп. Силилхлориды, такие как трет.-бутилдиметилсилилхлорид (ТБСХ), триметилсилилхлорид, триэтилсилилхлорид, дифенилметилсилилхлорид, триизопропилсилилхлорид, и трет.-бутилдифенилсилилхлорид представляют собой примеры гидроксизащитных агентов. Фтористый водород, такой как водный HF в ацетонитриле, или тетра-н-бутиламмонийфторид представляют собой примеры реагентов, которые могут удалять силильные группы. Другие гидроксизащитные группы включают в себя эфиры, такие как тетрагидропираниловый (ТГП) эфир, включая в себя алкоксиалкиловые эфиры (ацетали), такие как метоксиметиловый (МОМ) эфир, или сложные эфиры, такие как хлоруксусный эфир, триметилацетат, ацетат или бензоат. Примеры гидроксизащитных групп и способов защиты и удаления, включенные в рамки данной заявки (но не ограниченные ими) могут, например, быть найдены в “Protective Groups in Organic Synthesis” 3rd ed., T.W. Greene & P.G.M. Wuts eds., John Wiley 1999 и в “ Protecting Groups”, 1st ed., P.J. Kocienski, G. Thieme 2000.
Используемый здесь термин «алкил» означает линейную или разветвленную алкильную группу, которая может быть циклической или ациклической, имеющую от одного до двадцати атомов углерода, предпочтительно от одного до семи атомов углерода. Метильная группа, этильная группа, н-пропильная группа, изопропильная группа, пентильная группа, гексильная группа и трет.-бутилдиметильная группа представляют собой, не ограничиваясь ими, примеры алкильных групп.
Используемый здесь термин «восстановительный агент» означает любой агент, способный к восстановлению, включая в себя энантиоселектиное или диастереоселективное восстановление С-24 кетогруппы соединения общей структуры III, с получением соединения общей структуры IV. В одном варианте осуществления восстановительный агент может восстанавливать С-24 кетогруппу соединения общей структуры III без хирального вспомогательного соединения с образованием смеси соединений общей структуры IV, при этом упомянутая смесь обогащена желательным эпимером IVa (предпочтительно образующим 24-S изомер). В другом варианте осуществления восстановительный агент может восстанавливать С-24 кетогруппу соединения общей структуры III в присутствии хирального вспомогательного соединения с образованием смеси соединений общей структуры IV, при этом упомянутая смесь обогащена желательным эпимером IVa (предпочтительно образующим 24-S изомер). Восстановительный агент может быть хиральным или ахиральным. Примеры восстановительных агентов включают в себя (но не ограничиваются) борановые восстановительные агенты, гидриды металлов, такие как гидрид лития, гидрид алюминия, борогидрид натрия или AlH3, возможно в присутствии солей лантанидов (например, LaCl3, CeBr3, CeCl3) или NaBH3(OAc), Zn(BH4)2 и Et3SiH. Другие восстановительные агенты включают в себя (но не ограничиваются) водород в присутствии катализатора, такого как платина или рутений, натрий в этаноле, изопропиловый спирт и изопропоксид алюминия, и порошок цинка в воде или в спирте.
Используемый здесь термин «борановый восстановительный агент» включает в себя боран или любое производное борана, такое как борановые комплексы с аминами или эфирами. Не ограничивающие примеры борановых восстановительных агентов, например включают в себя N,N-диэтиланилинборан, боран-тетрагидрофуран, 9-боранбициклононан (9-ББН) или борандиметилсульфид.
Используемый здесь термин «хиральное вспомогательное соединение» означает хиральное соединение или оптически активный катализатор, например соединение, содержащее асимметрически замещенный атом углерода или вспомогательные хиральные соединения, или смеси хиральных соединений и/или оптически активных катализаторов, которые будут улучшать выход соединения общей структуры IVa по отношению к его эпимерам (увеличение молярного соотношения IVa:IVb) при восстановлении соединения общей формулы III с упомянутым восстановительным агентом. Упомянутые хиральные вспомогательные соединения могут, таким образом, быть любыми соединениями, которые способны увеличивать стереоселективность в реакции восстановления соединения общей структуры III по сравнению с выходом или стереоселективностью для IVa без присутствия или включения хирального вспомогательного соединения. Неограничивающие примеры хиральных вспомогательных соединений включают в себя хиральные 1,2-аминоспирты, такие как хиральные цис-1-амино-2-инданолпроизводные, такие как (1S,2R)-(-)-цис-1-амино-2-инданол или цис-1-амино-1,2,3,4-тетрагидронафталин-2-ол, такие как (1S,2R)-цис-1-амино-1,2,3,4-тетрагидронафталин-2-ол. Другие примеры представляют собой бинафтилпроизводные, такие как (R)-2,2'-бис(дифенилфосфино)-1,1'-бинафтилрутений ацетат 2,2'-дигидрокси-1,1'-бинафтилпроизводные. Дополнительные примеры включают в себя, но не ограничиваются, (R)-(+)-α,α-дифенил-2-пирролидинметанол,(R)-(+)-2-амино-4-метил-1,1-дифенил-1-пентанол,(R)-(-)-2-амино-3-метил-1,1-дифенил-1-бутанол, (R)-(+)-2-амино-1,1,3-трифенил-1-пропанол и (1S,2R)-(-)-2-амино-1,2-дифенилэтанол.
Используемый здесь термин «инертный растворитель» означает любой органический растворитель, совместимый с упомянутым восстановительным агентом в применяемых условиях реакции, или смеси таких растворителей. Выбор такого растворителя будет зависеть от специфики используемого восстановительного агента. Неограничивающие примеры инертных растворителей включают в себя углеводороды, такие как толуол, и эфиры, такие как трет.-бутилметиловый эфир или тетрагидрофуран.
Смесь соединений общей структуры IVa и IVb, которая обогащена IVa, означает смесь, возможно, содержащую другие соединения или растворители, с молярным соотношением (диастереомерное соотношение) IVa/IVb, равным единице (50:50) или большем, чем единица, таким образом, что смесь содержит, по меньшей мере, 50% соединения общей структуры IVa (с содержанием 50% или меньше соединения общей структуры IVb).
Смесь соединений общей структуры Va и Vb, которая обогащена Va, означает смесь, возможно, содержащую другие соединения или растворители, с молярным соотношением (диастереомерное соотношение) Va/Vb, равным единице (50:50) или большем, чем единица, таким образом, что смесь содержит, по меньшей мере, 50% соединения общей структуры Va (с содержанием 50% или меньше соединения общей структуры Vb).
Смесь соединений общей структуры VIa и VIb, которая обогащена VIa, означает смесь, возможно, содержащую другие соединения или растворители, с молярным соотношением (диастереомерное соотношение) VIa/VIb, равным единице (50:50) или большем, чем единица, таким образом, что смесь содержит, по меньшей мере, 50% соединения общей структуры VIa (с содержанием 50% или меньше соединения общей структуры VIb).
Используемый здесь термин «выделение соединения» включает в себя очистку и/или изоляцию соединения, например, по меньшей мере, 90% чистоты, такого как, по меньшей мере, 95% чистоты, такого как 97% чистоты, 98% чистоты, 99% чистоты. Термин «выделение соединения» также включает в себя значение увеличения концентрации соединения в смеси таких соединений, возможно содержащих растворители, таким образом, что смесь дополнительно обогащается желательным или предпочтительным соединением или изомером, таким как эпимер, после упомянутого выделения.
Варианты осуществления
В наиболее предпочтительном в настоящее время варианте осуществления настоящего изобретения Х представляет собой OR2.
В предпочтительном в настоящее время варианте осуществления настоящего изобретения R1 и/или R2 представляют собой алкилсилил, такой как трет.-бутилдиметилсилил, наиболее предпочтительно, когда R1 и R2 одинаковые.
В другом варианте осуществления настоящего изобретения R1 и R2 представляют собой водород.
В предпочтительном в настоящее время варианте осуществления настоящего изобретения восстановительный агент представляет собой борановый восстановительный агент, такой как N,N-диэтиланилинборан, борантетрагидрофуран, или борандиметилсульфид.
В предпочтительном в настоящее время варианте осуществления настоящего изобретения стадию восстановления проводят с хиральным восстановительным агентом или в присутствии хирального вспомогательного соединения.
В предпочтительном в настоящее время варианте осуществления настоящего изобретения хиральное вспомогательное соединение представляет собой хиральный 1,2-аминоспирт, такой как хиральное производное цис-1-амино-2-инданола, такое как (1S,2R)-(-)-цис-1-амино-2-инданол.
В предпочтительном в настоящее время варианте осуществления настоящего изобретения стадию восстановления проводят в интервале температур от 10 до 20°С, в частности 15-20°С.
В другом варианте осуществления настоящего изобретения молярное соотношение (диастереомерное соотношение IVa/IVb) смеси соединений общей структуры IVa и IVb, которая обогащена IVa, больше, чем 55:45, такое как 56:44, такое как 57:43, такое как 59:41, такое как 60:40, такое как 63:37, такое как 65:35, такое как 68:32, такое как 70:30, такое как 72:28, такое как 73:27, такое как 74:26, такое как 75:25, такое как 76:24, такое как 77:23, такое как 78:22, такое как 79:21, такое как 80:20.
В другом варианте осуществления настоящего изобретения молярное соотношение (диастереомерное соотношение Va/Vb) смеси соединений общей структуры Va и Vb, которая обогащена Va, больше, чем 55:45, такое как 56:44, такое как 57:43, такое как 59:41, такое как 60:40, такое как 63:37, такое как 65:35, такое как 68:32, такое как 70:30, такое как 72:28, такое как 73:27, такое как 74:26, такое как 75:25, такое как 76:24, такое как 77:23, такое как 78:22, такое как 79:21, такое как 80:20.
В другом варианте осуществления настоящего изобретения молярное соотношение (диастереомерное соотношение VIa/VIb) смеси соединений общей структуры VIa и VIb, которая обогащена VIa, больше, чем 55:45, такое как 56:44, такое как 57:43, такое как 59:41, такое как 60:40, такое как 63:37, такое как 65:35, такое как 68:32, такое как 70:30, такое как 72:28, такое как 73:27, такое как 74:26, такое как 75:25, такое как 76:24, такое как 77:23, такое как 78:22, такое как 79:21, такое как 80:20.
В одном варианте осуществления настоящего изобретения соединение общей структуры Va выделено, например, хроматографически из смеси соединений общей структуры Va и Vb, которая обогащена Va, при этом Х, R1 и R2 определены выше в (стадии (с)).
В другом варианте осуществления настоящего изобретения соединение общей структуры VIa выделено, например, хроматографически из смеси соединений общей структуры VIa и VIb, которая обогащена VIa, при этом Х, R1 и R2 определены выше в (стадии (g)).
Способы синтеза
Соединения общей структуры III могут, например, быть синтезированы с помощью реакции Дильса-Альдера при обработке соединения общей структуры VII или VIII диоксидом серы. Используемый диоксид серы может быть жидким, газообразным или растворенным в подходящем растворителе. Подходящие растворители для реакции Дильса-Альдера представляют собой все растворители, которые совместимы с условиями реакции, такие как алканы, такие как гексан или гептан, углеводороды, такие как ксилолы, толуол, эфиры, такие как диэтиловый эфир или метил-трет.-бутиловый эфир (МТБЭ), ацетаты, такие как этилацетат или 2-пропилацетат, галогенированные растворители, такие как дихлорметан или смесь упомянутых растворителей. В предпочтительном варианте осуществления растворитель представляет собой толуол. В другом предпочтительном варианте осуществления растворитель представляет собой смесь не смешивающегося с водой растворителя и воды, такую как толуол и вода. Реакцию также можно проводить в чистом диоксиде серы без растворителя. Подходящая температура реакции способа находится в интервале от -50°С до 60°С, таком как от -30°С до 50°С, таком как от -15°С до 40°С, таком как от -5°С до 30°С, таком как от 0°С до 35°С, таком как от 5°С до 30°С, наиболее часто таком как от 10°С до 25°С, таком как от 15°С до 20°С. В одном варианте осуществления диоксид серы используют в избытке (моль/моль), таком как 5-100 молярный избыток, таком как 7-30 молярный избыток, таком как 10-15 молярный избыток. Любой избыток непрореагировавшего диоксида серы может быть удален из реакционной смеси, например, промывкой с водным основанием, таким как водный гидроксид натрия, или отгонкой диоксида серы, возможно, вместе с растворителем, возможно, при пониженном давлении. Соединения общей структуры III обычно получают как смесь их эпимеров IIIa и IIIb.
Молярное соотношение IIIa/IIIb смеси эпимеров, полученных по реакции Дильса-Альдера будет зависеть от групп Х, R1, R2 и от используемых условий реакции. Настоящее изобретение включает в себя смеси всех возможных композиций (молярное соотношение IIIa/IIIb), таких как 1:99, таких как 2:98, таких как 3:97, таких как 4:96, таких как 5:95, таких как 10:90, таких как 85:15, таких как 80:20, таких как 75:25, таких как 30:70, таких как 35:65, таких как 40:60, таких как 45:55, таких как 50:50, таких как 55:45, таких как 60:40, таких как 65:35, таких как 70:30, таких как 75:25, таких как 80:20, таких как 85:10, таких как 90:10, таких как 95:5, таких как 96:4, таких как 97:3, таких как 98:2, таких как 99:1.
Общая формула III включает в себя смеси всех возможных композиций, как перечислено выше.
В варианте осуществления настоящего изобретения соединения IIIa и IIIb используют как смесь, как показано в общей формуле III в последующей стадии восстановления. Смесь IIIa и IIIb может, но не обязательно, быть очищена или выделена хроматографией или кристаллизацией. В другом варианте осуществления соединение IIIa используют в последующей стадии восстановления. В еще одном варианте осуществления соединение IIIb используют в последующей стадии востановления.
Соединение общей структуры VII может, например, быть синтезировано согласно способам, раскрытым, например, M.J.Calverley, Tetragedron, Vol. 43, № 20, pp.4609-4619, 1987 или в WO 87/00834 и в ссылках, цитируемых там. Например, с соединения VII, в котором Х представляет собой OR2 и как R1, так и R2 представляют собой представляют собой трет.-бутилдиметилсилил, которое описано в данных ссылках, может быть снята защита водной гидрофтористой кислотой в ацетонитриле с получением смеси соединений, в которых Х представляет собой OR2, при этом или R1, или R2 представляют собой водород, или образуют соединение, в котором Х представляет собой OR2, и R1, и R2 - оба представляют собой водород. Данная смесь соединений может, например, быть разделена или хроматографией, или кристаллизацией, как, в общем, здесь описано. При взаимодействии упомянутых соединений общей структуры VII, в которой R1 и/или R2 представляют собой водород, с подходящим защитным агентом, новые группы R1 и/или R2 могут быть введены. В зависимости от стехиометрии используемого защитного агента и условий реакции может быть получена смесь незащищенных, монозащищенных и дизащищенных соединений. Любое промежуточное соединение в смеси, в котором Х представляет собой OR2 и или R1, или R2 представляет собой водород, может быть затем хроматографически выделено и может реагировать с подходящим защитным агентом, отличным от первого используемого агента, образуя соединения общей структуры VII, в которой Х представляет собой OR2 и R1 отличен от R2.
Соединения общей структуры VII, в которой Х представляет собой водород и R1 представляет собой водород или гидроксизащитную группу, могут, например, быть получены, начиная от соединений 7а и/или 7b, описанных M.J. Calverley, Tetrahedron, Vol. 43, № 20, p. 4610, 1987, последующими аналогичными процедурами и общими синтетическими способами, вышеописанным и цитированным выше в ссылках.
Способ восстановления по настоящему изобретению может, например, быть проведен при взаимодействии прохирального кетона общей структуры III с хиральным бораном как восстановительным агентом или с бораном как восстановительным агентом в присутствии хирального вспомогательного соединения. Способ приводит к энантиоселективному/диастереоселективному восстановлению прохирального кетона так, что из двух возможных эпимеров IVa или IVb образуется предпочтительно один соответствующий эпимер. Степень энантиоселективности/диастереоселективности будет зависеть от используемого восстановительного агента, хирального вспомогательного соединения и условий реакции.
Реакция восстановления соединения общей структуры III обычно протекает в температурном интервале от -80°С до 70°С, в таком как от -40°С до 60°С, в таком как от -15°С до 50°С, в таком как от -5°С до 40°С, например от 0°С до 5°С или от 5°С до 35°С. В одном варианте осуществления температурный интервал составляет от 10°С до 30°С, от 15°С до 25°С, от 15°С до 20°С. Оптимум температуры будет зависеть от специфических условий реакции и используемых реагентов. В одном варианте осуществления настоящего изобретения реакционную смесь немедленно охлаждали до 0-10°С после завершения, чтобы избежать образования побочных продуктов. Если N,N-диэтиланилин используют как восстановительный агент, то N,N-диэтиланилин может легко быть удален из реакционной смеси экстракцией с водной соляной кислотой. Одномолярный эквивалент по отношению к основанию, экстрагируемому 1 М соляной кислотой, является предпочтительным.
Восстановительный агент, возможно, растворенный или смешанный с инертным растворителем, может быть добавлен к соединению общей структуры III, возможно, растворенному или смешанному с инертным растворителем, например, в инертной атмосфере, такой как азот. Альтернативно, соединение общей структуры III, возможно, растворенное или смешанное с инертным растворителем, может быть добавлено к восстановительному агенту, возможно, растворенному или смешанному с инертным растворителем (обратный порядок).
В одном варианте осуществления настоящего изобретения восстановительный агент используют в эквимолярном количестве или в молярном избытке относительно соединения общей структуры III. В специфическом варианте осуществления настоящего изобретения молярное соотношение восстановительный агент/соединение общей структуры III составляет 1,0-5,0. В предпочтительном в настоящее время варианте осуществления настоящего изобретения молярное соотношение восстановительный агент/соединение общей структуры III составляет 1,8-3,0, такое как 2,3-3,9, такое как 2,5-2,7.
Хиральное вспомогательное соединение может реагировать с восстановительным агентом до восстановления in situ для образования хирального восстановительного агента или хиральное вспомогательное соединение может, например, служить как хиральный лиганд в комплексе с восстановительным агентом, то есть, например, образовывать хиральный восстановительный агент. Настоящее изобретение включает в себя использование таких хиральных восстановительных агентов или комплексов хиральный лиганд-восстановительный агент, которые получают и выделяют отдельно перед использованием для восстановления соединения общей структуры III.
Термин «восстановительный агент в присутствии хирального вспомогательного соединения», таким образом, включает в себя любой хиральный восстановительный агент. Например, хиральное вспомогательное соединение может реагировать с борановым восстановительным агентом до восстановления in situ с образованием хирального боранового восстановительного агента или хиральное вспомогательное соединение может служить как хиральный лиганд в борановом комплексе. Примеры таких хиральных борановых восстановительных агентов представляют собой хиральные оксаборолидины или оксазаборолидины, такие как хиральные оксазаборолидиновые реагенты, полученные из (1R,2S)-цис-1-aмино-2-инданола,(1S,2R)-цис-1-aмино-2-инданола, (S)-пролинола или B-(3-пинанил)-9-борабицикло[3.3.2]нонан(alpine-borane) или, например, 5,5-дифенил-2-метил-3,4-пропано-1,3,2-оксазаборолидина, (S)-2-метил-CBS-оксазаборолидина, (R)-2-метил-CBS-оксазаборолидина. Настоящее изобретение включает в себя поэтому использование таких хиральных восстановительных агентов как хиральные борановые восстановительные агенты, или как комплексы хиральный лиганд-восстановительный агент, такие как хиральный лиганд-борановые комплексы, которые получают и выделяют перед использованием для восстановления соединения общей структуры III.
Другой пример хирального лиганда в комплексе с восстановительным агентом представляет собой комплекс LiALH4 и 2,2'-дигидрокси-1,1'бинафтила.
Молярное соотношение восстановительный агент/хиральное вспомогательное соединение находится обычно в интервале 0,1-20,0, таком как 0,4-10, таком как 0,3-5,0, таком как 0,5-4,5, таком как 1,0-4,0, таком как 1,9-3,1, таком как 2,1-2,9, таком как 2,3-2,7, например 10,8, 5,4, 2,6, 2,5 или 1,6.
Хиральное вспомогательное соединение может присутствовать в каталитических количествах, таких как субстехиометрические, или в эквимолярных, или в молярном избытке по отношению к соединению общей структуры III или к восстановительному агенту. Например, соотношение хиральное вспомогательное соединение/соединение III может быть 0,25-2,5, таким как 0,5-2,0, таким как 0,8-1,3, таким как 0,9-1,2, таким как 1,0-1,1.
Выбор конкретного энантиомера хирального вспомогательного соединения будет определять стереоселективную ориентацию гидроксигруппы соединения общей структуры IV относительно C-24. Хиральное вспомогательное соединение, которое преимущественно приводит к S-конфигурации, при С-24 является предпочтительным.
Реакции, катализируемые боранами, были, например, рассмотрены Deloux and Srebnik [Chem. Rev.93, 763, 1993]. Примеры эффективных катализаторов, основанных на хирально модифицированных боранах, могут, например, быть найдены в [A.Hirao, J.Chem. Soc. Chem. Commun. 315,1981; E.J. Corey, J. Am. Chem. Soc. 109, 7925, 1987].
Примеры синтеза и/или использования, например, 1,2- и 1,3-аминоспиртов в стереоселективном восстановлении с бораном могут, например, быть найдены в [E.Didier et al.; Tetrahedron 47, 4941-4958, 1991; C.H. Senanayake et al., Tetrahedron Letters, 36(42), 7615-18, 1995, EP 0698028, EP 0640089, EP 0305180, WO 93/23408, WO 94/26751]. Синтез и/или использование хиральных цис-1-амино-2-инданолпроизводных в борановом восстановлении может, например, быть найден в [C.H. Senanayake, Aldrichimica Acta, 31(1), 1-15, 1998; A.K. Ghosh et al., Sinthesis, 937-961, 1998; Y. Hong et al., Tetrahedron Letters, 35(36), 6631-34, 1994; B.Di Simone, Tetrahedron Assimetry, 6(1) 301-06, 1995; Y. Hong et al., Tetrahedron Letters, 36(36), 6631-34, 1994; R. Hett et al., Org. Process. Res. Dev., 2, 96-99, 1998; или EP 0763005] и в ссылках, там цитируемых.
Способы получения кальципотриола, как здесь описаны, могут быть модифицированы в отношении последовательности реакционных стадий, пропуска одной или более реакционных стадий, или ввода дополнительных стадий очистки или взаимодействия на любой стадии последовательности реакций. Настоящее изобретение включает в себя все такие модификации.
Способ получения кальципотриола, как здесь описан, включает в себя дополнительно все варианты, в которых гидроксизащитные группы R1 и/или R2 для соединений или промежуточных соединений, где R1 и/или R2 не являются водородом, удаляются на любой стадии реакционной последовательности. Соединения или промежуточные соединения, в которых R1 и/или R2 представляют собой водород, могут быть защищены защитными агентами на любой стадии реакционной последовательности, включая в себя защитные агенты, которые образуют защитные группы, отличные от групп, удаленных ранее в реакционной последовательности.
Cоединения и промежуточные соединения по настоящему изобретению могут содержать асимметрично замещенные (хиральные) атомы углерода, и двойные связи углерод-углерод, которые могут вызывать существование изомерных форм, например, энантиомеров, диастереомеров и геометрических изомеров.
Эпимеры известны как диастереомеры, которые имеют противоположную конфигурацию (R или S) только у одного из тетраэдрических стереоспецифичесих центров в молекулах, имеющих множество стереоспецифических центров, такие как аналоги витамина Д, на которые ориентировано настоящее изобретение.
Обозначение, например, С-24 как эпимерного центра пары энантиомеров подразумевает, что конфигурация у других стереоспецифических центров пары является идентичной.
Настоящее изобретение относится ко всем изомерным формам, таким как эпимеры, или к чистым формам или к их смесям.
Указание специфических конформаций или конфигураций или в формулах, или в наименованиях соединений или промежуточных соединений настоящего изобретения будет показывать, что данная специфическая конформация или конфигурация является предпочтительным вариантом осуществления изобретения. Указание специфической конформации или конфигурации или в формулах, или в наименованиях соединений или промежуточных соединений настоящего изобретения будет указывать на то, что данная специфическая конформация или конфигурация представляет собой предпочтительный вариант осуществления изобретения. Указание специфической конформации или конфигурации или в формулах, или в наименованиях соединений или промежуточных соединений настоящего изобретения будет включать в себя любой другой изомер, отличный от специфически указанного, или в чистом виде, или в их смеси, как другой вариант осуществления настоящего изобретения.
Указание неспецифической конформации или конфигурации или в формулах, или в наименованиях соединений или промежуточных соединений настоящего изобретения будет свидетельствовать, что смесь данных специфических конформаций или конфигураций представляет собой предпочтительный вариант осуществления изобретения. Например, соединение общей формулы III представляет собой смесь эпимеров общей формулы IIIa и IIIb.
Указание неспецифической конформации или конфигурации или в формулах, или в наименованиях, или в нумерациях соединений или промежуточных соединений настоящего изобретения будет включать в себя любой специфический изомер, хотя не указанный специфически в чистом виде, например, как другой вариант осуществления настоящего изобретения.
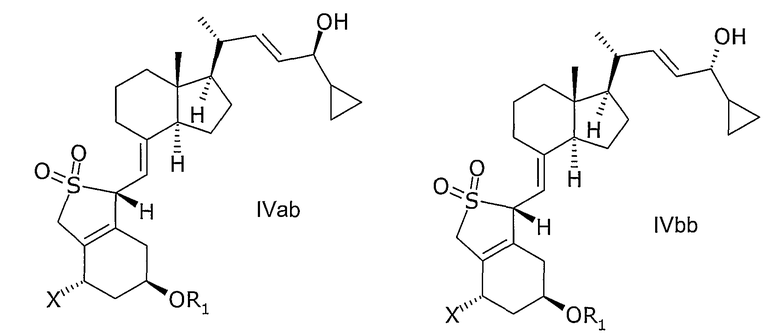
Например, соединение общей формулы IV включает в себя следующие два эпимера IVaa и IVab.
Обозначение соединения общей формулы III также включает в себя эпимеры IIIa и IIIb.
Чистые стереоизомерные формы соединений и промежуточных соединений по данному изобретению могут быть получены применением методик, известных в данной области, таких как хроматография или кристаллизация, или стереоселективным синтезом.
Способы разделения, выделения и очистки по настоящему изобретению включают в себя, но не ограничиваются, хроматографию, такую как адсорбционная хроматография (включая в себя колоночную хроматографию и хроматографию с псевдодвижущимся слоем (ПДС)), кристаллизацию или дистилляцию. Способы разделения, выделения и очистки могут быть использованы последовательно и в комбинациях.
Колоночная хроматография, пригодная для разделения аналогов витамина Д по настоящему изобретению, хорошо известна специалистам в данной области фармацевтической химии. Методика применяет колонки, заполненые стационарной фазой, например, кремнеземом, таким как предварительно обработанный кремнезем, на которые загружают образец для разделения. Образец затем элюируют подходящим элюентом. Элюирование может быть изократическим или с так называемым программированием растворителя (градиентным), когда состав элюента меняется регулярно, (например, линейно) или не регулярно (например, ступенчато через промежуток времени). Предварительно обработанный силикагель, хорошо известный специалистам в области хроматографии, представляет собой подходящую стационарную фазу. Элюирование с 5% (об.:об.) этилацетатом в гексане или в гептане с последующим элюированием чистым этилацетатом представляет собой не единственный пример программированного элюирования, которое приводит к желаемому разделению. Другие подходящие элюенты будут подобраны специалистами в данной области с помощью развития стандартных способов, например использованием смесей гептана и этилацетата подходящей полярности.
Для стадии хроматографирования может быть использована любая комбинация стационарной фазы (насадки) и элюента, которая способна разделить смесь эпимеров С-24. Такие комбинации могут быть легко определены специалистом в данной области с помощью типичного эксперимента. Например, предпочтительная стационарная фаза представляет собой кремнезем, такой как обработанный кремнезем.
Ретрореакция Дильса-Альдера смеси соединений общей структуры IVa и IVb, которая обогащена IVa, в присутствии основания для получения смеси соединений общей структуры Va и Vb, которая обогащена Va, при этом Х, R1 и R2 определены выше, может быть проведена во всех растворителях, которые совместимы с условиями реакции, таких как алканы, таких как гексан или гептан, в углеводородах, таких как ксилолы, толуол, в эфирах, таких как диэтиловый эфир или метил-трет.-бутиловый эфир (МТБЭ), в ацетатах, таких как этилацетат или 2-пропилацетат, в галогенированных растворителях, таких как дихлорметан, в воде или в смесях указанных растворителей.
Способы упомянутой ретрореакции Дильса-Альдера хорошо известны специалистам в области синтеза витамина Д (см., например, M.J. Calverley, Tetrahedron, Vol. 43, № 20, pp. 4609-4619, 1987 или в WO 87/00834).
Предпочтительные растворители представляют собой толуол, трет.-бутилметиловый эфир, воду или их смеси.
Основания, пригодные для использования в ретрореакции Дильса-Альдера включают в себя, но не ограничиваются ими, NaHCO3, KHCO3, Na2CO3 или K2CO3. В одном варианте осуществления настоящего изобретения основание представляет собой водный NaHCO3, и/или ретрореакцию Дильса-Альдера проводят при температуре выше 60°С, такой как 70°С, такой как от 70°С до 120°С, такой как от 74°С до 79°С, такой как от 72°С до 78°С.
В одном варианте осуществления настоящего изобретения температурный интервал экстракций и фазовых разделений после завершения ретрореакции Дильса-Альдера во время обработки реакционной массы составляет, примерно, 30-40°С.
Соединения общей структуры VIII могут быть получены изомеризацией соединений общей структуры VII.
Способы изомеризации соединений общей формулы Va и/или Vb до VIa и/или VIb, или VII до VIII хорошо известны специалистам в области синтеза витамина Д. Условия реакции могут, например, быть найдены в M.J.Calverley, Tetrahedron, Vol. 43, № 20, pp. 4609-4619, 1987 или в WO 87/00834 и в ссылках, там цитируемых. В предпочтительном варианте осуществления настоящего изобретения изомеризация представляет собой фотоизомеризацию, например УФ освещение в присутствии триплетного сенсибилизатора, например антрацена или 9-ацетилантрацена.
Соединения общей формулы III, IV, V, VI или VII, в которых Х=водород, могут быть гидроксилированы подходящим гидроксилирующим агентом, например селенитным промежуточным соединением при аллильном гидроксилировании, в условиях, найденных Гессе, например, с SeO2 и N-метилморфолин-N-оксидом при кипячении с обратным холодильником в метаноле и/или дихлорметане (D.R. Andrews et al., J. Org. Chem., 1986, 51, 1637) или как описано в M.J.Calverley, Tetrahedron, Vol. 43, № 20, pp. 4609-4619, 1987 или в WO 87/00834, для получения соединений общей формулы III, IV, V, VI или VII, в которой Х=гидрокси (Х=OR2 и R2=водород). Гидроксигруппы исходных материалов могут быть защищены подходящими защитными группами, такими, как определено выше, способами, такими как вышеописанные, например, чтобы избежать нежелательного окисления упомянутых гидроксигрупп.
Гидрат кальципотриола может быть получен кристаллизацией кальципотриола из водных растворителей так, как, например, в способах, описанных в WO 94/15912.
ПРИМЕРЫ
Общие замечания.
Все химические вещества, если не отмечено иначе, были получены из коммерческих источников. Все температуры плавления являются нескорректированными. Для спектра 1H ядерного магнитного резонанса (ЯМР) (300 МГц) и 13С ЯМР (75,6 МГц) значения химического сдвига (δ) приводятся в м.д., если не указано иное; для растворов в дейтерохлороформе относительно внутреннего тетраметилсилана (δ=0,00) или хлороформа (δ=7,26) или дейтерохлороформа (δ=76,81 для 13С ЯМР) как стандарта. Значение мультиплетности определено как дублет (д), триплет (т), квартет (кв) или не определено (м), и положение дается в приблизительной средней точке, если не приводится интервал. Все используемые органические растворители были технической марки. Хроматографию проводили на силикагеле, возможно, с использованием флеш-техники. Пластины для тонкослойной хроматографии, покрытые силикагелем, были выпущены Merck KGaA. Предпочтительно силикагель для хроматографии был выпущен Merck KGaA Германия: LiChroprep® Si60 (15-25 мкм). Этилацетат, дихлорметан, гексан, н-гексан, гептан или соответствующие смеси этилацетата, дихлорметана, метанола и петролейного эфира (40-60), гексана или гептана в качестве элюентов, если не отмечены другие. Все реакции можно легко проводить в инертной атмосфере, такой как атмосфера азота.
Соединения общей структуры III
Пример 1
III: Х=OR2, R1,R2=трет.-бутилдиметилсилил
1(S),3(R)-бис(трет.-бутилдиметилсилиокси)-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты
20(R),1(S),3(R)-бис(трет.-бутилдиметилсилиокси)-20-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен (приготовленный по способу, описанному M.J.Calverley, Tetrahedron, Vol. 43 № 20, pp. 4609-4619, 1987) (20,0 г) растворяли в толуоле (210 мл) при 20°С с последующим добавлением воды (40 мл) и SO2 (20 мл) при перемешивании. Когда на основании ВЭЖХ делался вывод, что реакция закончилась {Колонка LiChrosorb Si 60 5 мкм 250×4 мм Merck, поток 2 мл/мин, детектирование при 270 нм & детектирование массы, гексан/этилацетат 9:1 (об.:об.)}, обычно после 2-2,5 часов добавляли смесь гидроксида натрия (27,7%, 60 мл) и воды (80 мл) при 10-18°С до pH реакционной смеси, равного 6. Толуольную фазу отделяли и растворитель удаляли в вакууме без нагрева (предпочтительно ниже 30°С) с получением двух эпимерных SO2-аддуктов IIIa и IIIb в виде твердой смеси, содержащей в основном IIIа, как показано с помощью ТСХ. Два эпимерных SO2-аддукта IIIa и IIIb могут быть разделены с помощью хроматографии. Кристаллический IIIa может быть к тому же получен растиранием в порошок твердой смеси с метанолом.
1H ЯМР (CDCl3) IIIa/X=OR2, R1, R2 = трет.-бутилдиметилсилил = 6,73 (дд, 1Н), 6,14 (д, 1Н), 4,69 (д, 1Н), 4,62 (д, 1Н), 4,35 (с, 1Н), 4,17 (м, 1Н), 3,92 (д, 1Н), 3,58 (д, 1Н), 2,61 (м, 1Н), 2,29 (м, 1Н), 2,2-1,2 (м, 16H), 1,11 (д, 3H), 1,05 (м, 2H), 0,90 (м, 2H), 0,87 (с, 9H), 0,85 (с, 9H), 0,68 (с, 3H), 0,06 (с, 3H), 0,05 (с, 3H), 0,04 (с, 3H), 0,02 (с, 3H) м.д.
Получение 1
VII: Х=OR2, R2=водород
1(S),3(R)-бис(трет.-бутилдиметилсилиокси)-2(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен
20(R),1(S),3(R)-бис(трет.-бутилдиметилсилиокси)-20-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен, полученный по способу, описанному M.J.Calverley, Tetrahedron, Vol. 43 № 20, pp. 4609-4619, 1987) (20,0 г) растворяли в ацетонитриле. Добавляли водную фтористоводородную кислоту (40%) и смеcь перемешивали при комнатной температуре в течение 1 часа. Прохождение реакции можно легко контролировать с помощью ТСХ, используя этилацетат как элюент. Этилацетат добавляли к реакционной смеси и смесь промывали водным раствором гидрокарбоната натрия. Органическую фазу сушили с помощью MgSO4 и смесь концентрировали. Образовавшиеся кристаллы (белые иголки) отфильтровывали, промывали этилацетатом и сушили в вакууме, получая соединение VII, указанное в заголовке (Х=OR2, R1,R2=водород).
1H ЯМР (CDCl3) VII/X=OR2, R1, R2 = водород = 6,77 (дд, 1Н), 6,57 (д, 1Н), 6,15 (д, 1Н), 5,88 (дд, 1Н), 5,13 (дд, 1Н), 4,98 (с, 1Н), 4,50 (м, 1Н), 4,23 (м, 1Н), 2,86 (м, 2H), 2,29 (м, 2H), 2,14-1,20 (м, 16H), 1,14 (д, 3H), 1,08 (м, 2H), 0,89 (м, 2H), 0,61 (с, 3H) м.д.
Пример 2
III: Х=OR2, R1,R2=водород
1(S),3(R)-дигидрокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты.
Тот же способ, как и в Примере 1, за исключением того, что исходным материалом был 1(S),3(R)-дигидрокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен из приготовления 1.
1H ЯМР (CDCl3) III/X=OR2, R1, R2 = водород δ = 6,80 (дд, 1Н), 6,15 (д, 1Н), 4,75 (м, 2H), 4,5-3,9 (м, 4H), 3,70 (д, 1Н), 2,60 (м, 1Н), 2,5-0,8 (м, 25H), 0,68 (с, 3H) м.д.; 13C ЯМР (CDCl3) III/X=OR2, R1, R2 = водород δ = 201,0, 152,1, 151,0, 133,7, 129,2, 128,3, 108,8, 67,3, 65,1, 63,6, 56,1, 55,9, 55,5, 46,5, 40,1, 39,9, 33,9, 29,8, 27,4, 23,9, 22,1, 19,5, 18,9, 12,2, 11,2 м.д.
Получение 2
VII: X=OR2, R1=водород, R2=трет.-бутилдиметилсилил
1(S)-трет.-бутилдиметилсилил-3(R)-гидрокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен
20(R)1(S)3(R)-бис(трет.-бутилдиметилсилилокси)-20-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен был частично лишен защиты с использованием тех же условий снятия защиты, как и в Приготовлении 1, с получением смеси непрореагировавшего исходного материала, двух, частично лишенных защиты, промежуточных продуктов, и соединения Приготовления 1. Очистка хроматографией привела к получению чистого соединения, указанного в заголовке. 1H ЯМР, как было найдено, находится в соответствии со структурой.
1H ЯМР (CDCl3) VII/X=OR2, R1 = водород, R2 = трет.-бутилдиметилсилил δ = 6,75 (дд, 1Н), 6,50 (д, 1Н), 6,14 (д, 1Н), 5,84 (д, 1Н), 5,00 (с, 1Н), 4,92 (с, 1Н), 4,47 (т, 1Н), 4,22 (м, 1Н), 2,85 (дд, 1Н), 2,62 (дд, 1Н), 2,43 (дд, 1Н), 2,29 (м, 1Н), 2,15-1,15 (м, 15H), 1,11 (д, 3H), 1,06 (м, 2H), 0,87 (с, 9H), 0,86 (м, 2H), 0,59 (с, 3H), 0,06 (с, 3H), 0,04 (с, 3H) м.д.
Пример 3
III: X=OR2, R1=водород, R2=трет.-бутилдиметилсилил
1(S)-трет.-бутилдиметилсилил-3(R)-гидрокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты.
Тот же способ как и в Примере 1, за исключением того, что исходным материалом был 1(S)-трет.-бутилдиметилсилил-3(R)-гидрокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен из Приготовления 2.
13С ЯМР (CDCl3) III/X=OR2, R1 = водород, R2 = трет.-бутилдиметилсилил δ = 200,3, 151,5, 150,4, 132,0, 129,5, 128,0, 108,5, 66,8, 65,5, 63,8, 56,1, 55,9, 55,2, 46,2, 39,8, 33,6, 29,5, 27,2, 25,4, 23,7, 21,8, 19,2, 18,5, 17,7, 11,8, 10,7, -4,7, -5,2 ppм; 1H ЯМР (CDCl3) IIIb/X=OR2, R1 = водород, R2 = трет.-бутилдиметилсилил δ = 6,75 (дд, 1Н), 6,14 (д, 1Н), 4,80 (д, 1Н), 4,65 (д, 1Н), 4,43 (м, 1Н), 4,25 (м, 1Н), 3,92 (д, 1Н), 3,63 (дд, 1Н), 2,60 (д, 1Н), 2,5-1,2 (м, 18H), 1,12 (д, 3H), 1,06 (м, 2H), 0,88 (с, 9H), 0,87 (м, 2H), 0,59 (с, 3H), 0,09 (с,3H), 0,07 (с, 3H) м.д.
Получение 3
VII: X=OR2, R1=COCMe3, R2=трет.-бутилдиметилсилил
1(S)-трет.-бутилдиметилсилил-3(R)-триметилацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен
1(S)-трет.-бутилдиметилсилил-3(R)-гидрокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен из Приготовления 2 может реагировать с хлоридом триметилуксусной кислоты в присутствии триэтиламина в дихлорметане. Полученный сырой продукт можно очистить хроматографически с получением чистого соединения, указанного в заголовке.
Получение 4
VII: X=OR2, R1=COCMe3, R2=водород
1(S)-гидрокси-3(R)-триметилацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен.
1(S)-трет.-бутилдиметилсилил-3(R)-триметилацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен может быть лишен защиты, используя тот же способ снятия защиты, как и в Приготовлении 1. Полученный сырой продукт можно очистить хроматографически с получением чистого соединения, указанного в заголовке.
Пример 4
III: X=OR2, R1=COCMe3, R2=водород
1(S)-гидрокси-3(R)-триметилацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты.
Тот же способ, как и в Примере 1, за исключением того, что исходным материалом был 1(S)-гидрокси-3(R)-триметилацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен из приготовления 4.
13C ЯМР (CDCl3) IIIa/X=OR2, R1 = COCMe3, R2 = водород δ = 200,4, 177,6, 151,6, 150,9, 132,8, 129,3, 128,1, 108,8, 66,9, 66,3, 64,6, 55,8, 55,5, 55,3, 46,3, 39,9, 38,5, 36,3, 30,2, 29,6, 27,2, 26,9, 23,7, 21,8, 19,3, 18,6, 11,9, 10,8 м.д.
Пример 5
III: X=OR2, R1=COCMe3, R2=трет.-бутилдиметилсилил
1(S)-трет.-бутилдиметилсилил-3(R)-триметилацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты.
Тот же способ как в Примере 1, за исключением того, что исходным материалом был 1(S)-трет.-бутилдиметилсилил-3(R)-триметилацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен, получаемый, например, из Приготовления 3.
Получение 5
VII: X=OR2, R1=COMe, R2=водород
1(S)-гидрокси-3(R)-ацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен.
1(S),3(R)-дигидрокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен (VII: X=OR2, R1, R2=водород) из Приготовления 1 может реагировать с эквивалентом ацетилхлорида в присутствии триэтиламина. Смесь продуктов можно очистить хроматографически на кремнеземе с получением чистого соединения, указанного в заголовке.
Пример 6
III: X=OR2, R1=COMe, R2=водород
1(S)-гидрокси-3(R)-ацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты.
Тот же способ, как в Примере 1, за исключением того, что исходным материалом был 1(S)-гидрокси-3(R)-ацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен (VII: X=OR2, R1=COMe, R2=водород), получаемый в Приготовлении 5.
13C ЯМР (CDCl3) IIIa/X=OR2, R1 = COMe, R2 = водород δ = 200,5, 170,3, 151,6, 150,9, 132,8, 129,2, 128,1, 108,3, 66,8, 66,4, 64,6, 55,9, 55,7, 55,3, 46,3, 39,9, 36,4, 30,4, 29,6, 27,2, 23,7, 21,8, 21,0, 19,3, 18,6, 11,9, 10,8.
Пример 7
III: X=OR2, R1=COMe, R2=трет.-бутилдиметилсилил
1(S)-трет.-бутилдиметилсилил-3(R)-ацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты.
Тот же способ, как в Примере 1, за исключением того, что исходным материалом был 1(S)-трет.-бутилдиметилсилил-3(R)-ацетокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен.
1H ЯМР (CDCl3) IIIa/X=OR2, R1 = COMe, R2 = трет.-бутилдиметилсилил δ = 6,75 (дд, 1Н), 6,16 (д, 1Н), 5,20 (м, 1Н), 4,71 (с, 2H), 4,33 (С, 1H), 3,95 (д, 1Н), 3,60 (д, 1Н), 2,61 (м, 1Н), 2,31 (м, 2H), 2,15-1,2 (м, 15H), 2,03 (с, 3H), 1,11 (д, 3H), 1,07 (м, 2H), 0,89 (м, 2H), 0,88 (с, 9H), 0,68 (с, 3H), 0,08 (с, 3H), 0,07 (с, 3H) м.д.
Пример 8
III: Х=водород, R1=трет.-бутилдиметилсилил
3(R)-трет.-бутилдиметилсилилокси-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддукты.
Исходный материал VII, Х=водород, R1=трет.-бутилдиметилсилил (полученный по способу, описанному M.J. Calverley, Tetragedron, Vol. 43, № 20, pp. 4609-4619, 1987) (38,5 г) растворяли в толуоле (550 мл) при 20°С с последующим добавлением воды (105 мл) и SO2 (50 мл) при перемешивании. Об окончании реакции судили по ВЭЖХ {Колонка LiChrosorb Si 60 5 мкм 250×4 мм Merck, поток 2 мл/мин, детектирование при 270 нм & детектирование массы, гексан/этилацетат 9:1 (об.:об.)}, обычно после 2-2,5 часов добавляли смесь гидроксида натрия (27,7%, 150 мл) и воды (480 мл) при 10-18°С до pH реакционной смеси 6. Толуольную фазу отделяли и растворитель удаляли в вакууме без нагрева (предпочтительно ниже 30°С) с получением двух эпимерных SO2-аддуктов IIIa и IIIb (Х=водород, R1=трет.-бутилдиметилсилил) в виде твердой смеси, содержащей в основном IIIа, как показано с помощью ТСХ. Два эпимерных SO2-аддукта IIIa и IIIb могут быть разделены хроматографией. Кристаллический IIIa может быть к тому же получен растиранием в порошок твердой смеси с метанолом.
13C ЯМР (CDCl3) (III: X = водород, R1 = трет.-бутилдиметилсилил, смесь изомеров IIIa и IIIb): 200,3, 151,6, 151,4, 149,8, 149,2, 130,5, 130,1, 128,3, 128,1, 126,6, 126,3, 110,5, 110,0, 67,4, 66,7, 66,6, 66,3, 58,0, 57,9, 55,8, 55,6, 55,3, 55,2, 46,3, 45,5, 39,9, 39,7, 34,4, 34,1, 33,9, 31,4, 30,8, 30,5, 29,6, 29,1, 27,3, 27,1, 26,7, 25,6, 25,1, 24,4, 24,1, 23,6, 23,2, 22,4, 21,9, 21,9, 19,4, 19,3, 18,6, 18,4, 17,9, 17,9, 13,9, 12,2, 11,9, 10,8, -5,0.
Соединения общей структуры IV
Пример 9
SO2-аддукт 1(S),3(R)-бис(трет.-бутилдиметилсилилокси)-20(R)-(3'-циклопропил-3'(S)-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена
(IVa: X=OR2, R1,R2=трет.-бутилдиметилсилил) и
SO2-аддукт 1(S),3(R)-бис(трет.-бутилдиметилсилилокси)-20(R)-(3'-циклопропил-3(R)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена
(IVb: X=OR2, R1,R2=трет.-бутилдиметилсилил)
(1S,2R)-(-)-цис-1-амино-2-инданол (5,0 г) смешивали с МТБЭ (160 мл) в атмосфере азота при 15-25°С с последующим добавлением N,N-диэтиланилинборана (16 мл) при той же температуре. Смесь перемешивали до прекращения выделения водорода. Смесь SO2-аддуктов 1(S),3(R)-бис(трет.-бутилдиметилсилиокси)-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена (III: X=OR2, R1,R2=трет.-бутилдиметилсилил), полученных в Примере 1, растворяли в толуоле (160 мл) и в МТБЭ (80 мл). Данный раствор по каплям добавляли к борансодержащей смеси при 15-25°С. Смесь перемешивали в течение 30-60 минут после завершения добавления, и затем гасили насыщенным водным NaHCO3 (110 мл) при 10-15°С. Органическую фазу отделяли и промывали 1 М соляной кислотой (100 мл) при 0-10°С с последующим промыванием насыщенным водным NaHCO3 (100 мл). Органическая фаза содержала SO2-аддукты 1(S),3(R)-бис(трет-бутилдиметилсилиокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена (IVa: X=OR2, R1, R2=трет.-бутилдиметилсилил) и SO2-аддукты 1(S),3(R)-бис(трет.-бутилдиметилсилиокси)-20(R)-(3'-циклопропил-3(R)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена (IVb: X=OR2, R1, R2=трет.-бутилдиметилсилил) в молярном соотношении 72-78:22-28 (IVa:IVb), как проверено ВЭЖХ анализом аликвоты после ретрореакции Дильса-Альдера и анализа в соответствии со способом, описанным в примере 10. Соединение IVaa выделяли хроматографией на кремнеземе.
13С ЯМР (CDCl3) IVa/X=OR2, R1 R2 = трет.-бутилдиметилсилил δ = 150,6, 137,6, 132,3, 129,3, 128,8, 109,0, 76,9, 67,3, 65,8, 64,5, 56,2, 56,1, 55,9, 46,0, 40,5, 40,0, 39,6, 34,1, 29,6, 27,4, 25,6, 25,5, 23,8, 21,8, 20,3, 17,8, 17,7, 17,4, 11,8, 2,8, 1,7, -4,7, -5,0/ -5,0, -5,2 м.д.
Пример 10
SO2-аддукты 3(R)-трет.-бутил-диметилсилиокси-20(R)-(3'-циклопропил-3'(S)-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена
(IVa: X=водород, R1=трет.-бутилдиметилсилил) и
SO2-аддукты 3(R)-трет.-бутил-диметилсилиокси-20(R)-(3'-циклопропил-3'(R)-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена
(IVb: X=водород, R1=трет.-бутилдиметилсилил),
(1S,2R)-(-)-цис-1-амино-2-инданол (1,22 г, 1,08 экв.) смешивали с МТБЭ (36 мл) в атмосфере азота при 15-25°С с последующим добавлением N,N-диэтиланилинборана (3,6 мл, 2,7 экв.) при той же температуре. Смесь перемешивали до прекращения выделения водорода. Смесь SO2-аддуктов 3(R)-бис(трет.-бутил-диметилсилиокси)-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена (III: X=водород, R1=трет.-бутилдиметилсилил), полученную в Примере 8 (4,32 г), растворяли в смеси МТБЭ (18 мл) и толуола (36 мл) при комнатной температуре и затем по каплям добавляли к борансодержащей смеси при 15-25°С в течение 15 мин. Смесь перемешивали в течение 60 минут после завершения добавления и затем гасили насыщенным водным NaHCO3 (25 мл). Органическую фазу отделяли и промывали 1 М соляной кислотой ( 25 мл) при 0-10°С с последующим промыванием насыщенным водным NaHCO3 (25 мл) при 10-20°С. Органическая фаза содержала SO2-аддукты 3(R)-бис(трет.-бутилдиметилсилиокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена (IVa: X=водород, R1=трет.-бутилдиметилсилил) и SO2-аддукты 3(R)-трет.-бутил-диметилсилиокси-20(R)-(3'-циклопропил-3(R)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триена (IVb: X=водород, R1=трет.-бутилдиметилсилил).
Соединения общей структуры V
Пример 11
1(S),3(R)-бис(трет.-бутилдиметилсилилокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)триен
(Va: X=OR2, R1, R2=трет.-бутилдиметилсилил) и
1(S),3(R)-бис(трет.-бутил-диметилсилилокси)-20(R)-(3'-циклопропил-3(R)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)триен
(Vb: X=OR2, R1,R2=трет.-бутилдиметилсилил)
Органическую фазу из Примера 9, содержащую SO2-аддукты IVa ( X=OR2, R1, R2=трет.-бутилдиметилсилил) и Vb (X=OR2, R1, R2=трет.-бутилдиметилсилил) энергично перемешивали с насыщенным водным раствором NaHCO3 (110 мл) (температура бани примерно 90°С), где отгонялся МТБЭ. Ретрореакцию Дильса-Альдера можно было легко контролировать с помощью ВЭЖХ {Колонка LiChrosorb Si 60 250×4 мм Merck, расход 1 мл/мин, детектирование при 270 нм, гексан/этилацетат 9:1,5 (об.:об.)}. После завершения (обычно 2-2,5 часа) реакционную смесь охлаждали до 30-40°С и органическую фазу отделяли, промывали насыщенным водным раствором NaHCO3 (110 мл) и водой (100 мл). Растворитель удаляли в вакууме и полученное масло (29 г) растворяли в гексане (200 мл). Органическую смесь охлаждали до примерно -15°С, фильтровали через короткий слой кремнезема, и остаток промывали гексаном (примерно 100 мл). Гексановую фазу промывали смесью метанол и вода (1:2) и органический растворитель удаляли в вакууме. Оставшееся масло, содержащее смесь 1(S),3(R)-бис(трет.-бутилдиметилсилилокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)триена (Va: X=OR2, R1, R2=трет.-бутилдиметилсилил) и 1(S),3(R)-бис(трет.-бутил-диметилсилилокси)-20(R)-(3'-циклопропил-3(R)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)триена (Vb: X=OR2, R1, R2=трет.-бутилдиметилсилил) в молярном соотношении в интервале 72-78:22-28 (Va:Vb), как проверено с помощью ВЭЖХ {Колонка LiChrosorb Si 60 5 мкм 250×4 мм Merck, расход 1 мл/мин, детектирование при 270 нм, н-гептан/2-пропанол 100:0,25 (об.:об.): время удерживания Va примерно 14,3 мин, Vb: 11,9 мин; или гексан/этилацетат 90:15 (об.:об.): время удерживания Va примерно 7,6 мин, Vb: 6,4 мин}, очищали хроматографией, как описано ранее M.J. Calverley, Tetrahedron, Vol. 43, № 20, pp. 4609-4619, 1987 или WO 87/00834, получая 10,9 г (98,9% чистоты ВЭЖХ) Va/X=ОR2, R1, R2=трет.-бутилдиметилсилил после перекристаллизации из смеси гексана и метанола и небольшого количества триэтиламина (при медленном испарении гексана с последующим охлаждением до -15°С) в полном соответствии с данными, описанными M.J. Calverley, Tetrahedron, Vol. 43, № 20, pp. 4617, 1987 для соединения 22.
13C ЯМР (CDCl3) Va/X=OR2, R1, R2 = трет.-бутилдиметилсилил δ = 153,4, 142,9, 137,9, 135,2, 128,7, 121,5, 116,3, 106,4, 77,1, 70,0, 67,0, 56,2, 55,8, 45,7, 43,7, 40,2, 39,8, 36,3, 28,7, 27,5, 25,6, 25,6, 23,3, 22,0, 20,3, 18,0, 17,9, 17,4, 12,1, 2,9, 1,6, -5,0, -5,0, -5,1; Vb/X=OR2, R1, R2 = трет.-бутилдиметилсилил δ = 153,5, 142,9, 137,6, 135,3, 128,7, 121,5, 116,3, 106,4, 76,8, 70,0, 67,0, 56,2, 56,0, 45,7, 43,8, 40,2, 39,7, 36,4, 28,7, 27,6, 25,7, 25,6, 23,3, 22,0, 20,3, 18,0, 17,9, 17,3, 12,1, 2,8, 1,6, -5,0, -5,1, -5,1 м.д.
Пример 12
3(R)-трет.-бутилдиметилсилилокси-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)триен
(Va: X=водород, R1=трет.-бутилдиметилсилил) и
3(R)-трет.-бутилдиметилсилилокси-20(R)-(3'-циклопропил-3(R)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)триен
(Vb: X=водород, R1=трет.-бутилдиметилсилил)
Органический раствор из Примера 10, содержащий SO2-аддукты IVa (X=водород, R1=трет.-бутилдиметилсилил) и IVb (X=водород, R1=трет.-бутилдиметилсилил), энергично перемешивали с насыщенным водным раствором NaHCO3 (25 мл) (температура бани примерно 90°С), при этом отгонялся МТБЭ. Ретрореакцию Дильса-Альдера можно было легко контролировать с помощью ВЭЖХ {Колонка LiChrosorb Si 60 250×4 мм Merck, расход 1 мл/мин, детектирование при 270 нм, гексан/этилацетат 9:1,5 (об.:об.)}. После завершения (примерно 2 часа) реакционную смесь охлаждали до 15-25°С и органическую фазу отделяли, промывали водой (25 мл), содержание в смеси Va:Vb (Х=водород, R1=трет.-бутилдиметилсилил) в молярном соотношении (75:25), как проверено ВЭЖХ { Колонка LiChrosorb Si 60 5 мкм 250×4 мм Merck, расход 1 мл/мин, детектирование при 270 нм, гексан/этилацетат 90:15 (об.:об.): время удерживания Vb примерно 6,1 мин, время удерживания Va примерно 7,4 мин.
1H ЯМР (CDCl3) Va/X = водород, R1 = трет.-бутилдиметилсилил δ = 6,45 (д, 1Н), 5,84 (д, 1Н), 5,46 (м, 2H), 4,92 (с, 1Н), 4,63 (с, 1Н), 3,84 (м, 1Н), 3,42 (м, 1Н), 2,85 (д, 1Н), 2,64 (д, 1Н), 2,45 (м, 1Н), 2,32-1,18 (м, 17H), 1,04 (д, 3H), 0,98 (м, 1Н), 0,87 (с, 9H), 0,56 (с, 3H), 0,51 (м, 2H), 0,32 (м, 1Н), 0,22 (м, 1Н), 0,05 (с, 3H), 0,04 (с, 3H); Vb/X = водород, R1 = трет.-бутилдиметилсилил δ = 6,45 (д, 1Н), 5,83 (д, 1Н), 5,47 (м, 2H), 4,90 (с, 1Н), 4,62 (с, 1Н), 3,83 (м, 1Н), 3,45 (м, 1Н), 2,83 (д, 1Н), 2,62 (д, 1Н), 2,44 (м, 1Н), 2,24 (м, 1Н), 2,18-1,17 (м, 16H), 1,03 (д, 3H), 0,96 (м, 1Н), 0,86 (с, 9H), 0,55 (с, 3H), 0,50 (м, 2H), 0,30 (м, 1Н), 0,20 (м, 1Н), 0,05 (с, 3H), 0,04 (с, 3H).
Соединения общей структуры VI
Пример 13
1(S),3(R)-бис(трет.-бутил-диметилсилилокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Z),7(Е),10(19)триен
(X=OR2, VIa: R1,R2=трет.-бутилдиметилсилил)
1(S),3(R)-бис(трет.-бутил-диметилсилилокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Z),7(Е),10(19)триен (X=OR2, VIa: R1, R2=трет.-бутилдиметилсилил), полученный в Примере 11, фотоизомеризовали в толуоле с использованием ультрафиолетовой лампы высокого давления, как описано ранее M.J. Calverley, Tetrahedron, Vol. 43, № 20, pp. 4609-4619, 1987 или WO 87/00834, за исключением того, что 9-ацетилантрацен использовали вместо антранцена, получая 1(S),3(R)-бис(трет.-бутилдиметилсилилокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Z),7(Е),10(19)триен (VIa X=OR2, R1, R2=трет.-бутилдиметилсилил) после хроматографии в полном соответствии с данными, описанными M.J. Calverley в Tetrahedron, Vol. 43, № 20, pp. 4618, 1987 для соединения 28.
Кальципотриол
Пример 14
1(S),3(R)-дигидрокси-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Z),7(Е),10(19)-триен
1(S),3(R)-бис(трет.-бутил-диметилсилилокси)-20(R)-(3'-циклопропил-3(S)'-гидроксипроп-1'(Е)-енил)-9,10-секопрегна-5(Z),7(Е),10(19)триен (VIa X=OR2, R1, R2=трет.-бутилдиметилсилил), полученный в Примере 13, был лишен защиты, используя тетрабутилфторид аммония в тетрагидрофуране, с последующим хроматографированием, как описано раньше M.J. Calverley, Tetrahedron, Vol. 43, № 20, pp. 4609-4619, 1987 или WO 87/00834. Кристаллизация из смеси этилацетат/гексан, содержащей несколько капель диэтиламина, привела к получению кальципотриола в полном соответствии с данными, описанными M.J. Calverley в Tetrahedron, Vol. 43, № 20, pp. 4618, 1987 для соединения 4.
Моногидрат кальципотриола
Пример 15
Кальципотриол, полученный в примере 14, кристаллизовали из смеси этилацетат/вода, как описано в WO 94/15912, получая моногидрат кальципотриола в полном соответствии с характеристическими данными, описанными в данном патенте.
Диастереоселективное восстановление в различных условиях восстановления
Пример 16
соединение
(экв.)
(°С)
Va:Vb (%)
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(1,1 экв.)
(2,7 экв)
толуол
(0,5 экв.)
(2,7 экв)
толуол
(0,5 экв.)
(2,7 экв)
толуол
трифенил-1-пропанол
(0,5 экв.)
(2,7 экв)
толуол
дифенилэтанол
(0,5 экв.)
(2,7 экв)
толуол
Таблица 1
Диастереоселективное восстановление соединений общей структуры III, где Х=OR2 и R1 и R2=трет.-бутилдиметилсилил (смесь 1(S),3(R)-бис(трет.-бутилдиметилсилиокси)-20(R)-(3'-циклопропил-3'-оксопроп-1'(Е)-енил)-9,10-секопрегна-5(Е),7(Е),10(19)-триен SO2-аддуктов из Примера 1 с последующей процедурой, аналогичной Примеру 9, с последующей хелеотропной экструзией диоксида серы, с последующей процедурой, аналогичной Примеру 11, приводит к образованию соединений общей структуры Va: X=OR2, R1, R2=трет.-бутилдиметилсилил при различных условиях (экв.=молярные эквиваленты относительно III; МТБЭ=трет.-бутилметиловый эфир; ДЭАБ=N,N-диэтиланилинборан).
Настоящее изобретение относится к промежуточным соединениям, используемым в синтезе кальципотриола или моногидрата кальципотриола, к способам получения упомянутых промежуточных соединений и к способам стереоселективного восстановления упомянутых промежуточных соединений. Способ позволяет повысить выход С-24 гидроксилэпимеров производных кальципотриола. 11 н. и 16 з.п. ф-лы, 1 табл.
1. Способ восстановления соединения общей структуры III,
в которой Х представляет собой или водород или OR2,
и в которой R1 и R2 могут быть одинаковыми или различными и представляют собой или водород или гидроксизащитную группу, в инертном растворителе с хиральным восстановительным агентом или с восстановительным агентом в присутствии хирального вспомогательного вещества,
где восстановительный агент представляет собой борановое производное и где хиральное вспомогательное вещество представляет собой хиральный 1,2-аминоспирт,
с получением смеси соединений общей структуры IVa и IVb,
которая обогащена IVa, при этом X, R1 и R2 определены выше.
2. Способ получения кальципотриола {(5Z, 7Е, 22Е, 24S)-24-циклопропил-9,10-секохола-5, 7, 10(19), 22-тетраен-1α-3β-24-триол} или моногидрата кальципотриола, включающий следующие стадии:
(а) восстановление соединения общей структуры III,
в которой Х представляет собой OR2,
и в которой R1 и R2 могут быть одинаковыми или различными и представляют собой или водород или гидроксизащитную группу, в инертном растворителе с хиральным восстановительным агентом или с восстановительным агентом в присутствии хирального вспомогательного вещества,
где восстановительный агент представляет собой борановое производное и где хиральное вспомогательное вещество представляет собой хиральный 1,2-аминоспирт,
с получением смеси соединений общей структуры IVa и IVb, которая обогащена IVa,
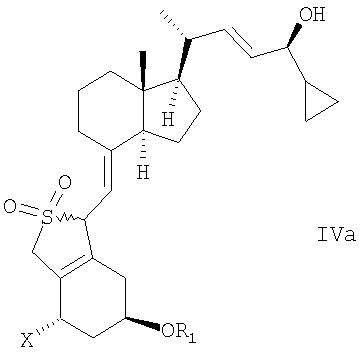
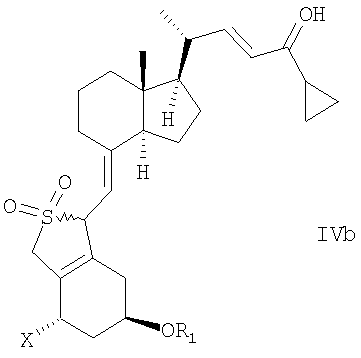
при этом X, R1 и R2 определены выше;
(b) взаимодействие смеси соединений общей, структуры IVa и IVb, которая обогащена IVa, в присутствии основания с получением смеси соединений общей структуры Va и Vb, которая обогащена Va,
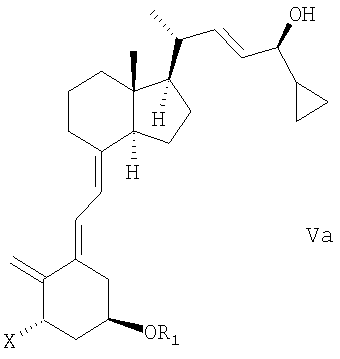
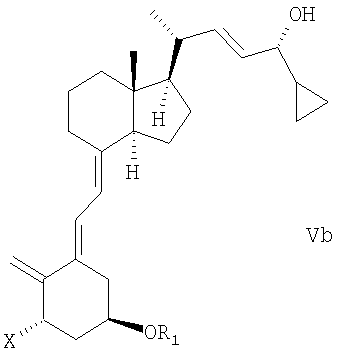
при этом X, R1 и R2 определены выше;
(c) выделение соединения общей структуры Va из смеси соединений общей структуры Va и Vb, которая обогащена Va, при этом X, R1 и R2 определены выше;
(d) изомеризация соединения общей структуры Va в соединение общей структуры VIa,
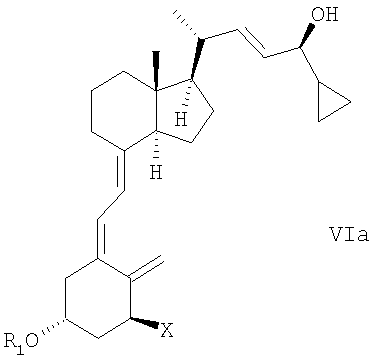
при этом X, R1 и R2 определены выше; и
(e) когда R1 и/или R2 не является водородом, удаление гидроксизащитной группы (групп) R1 и/или R2 соединения общей структуры VIa для получения кальципотриола или моногидрата кальципотриола.
3. Способ получения кальципотриола или моногидрата кальципотриола, включающий стадии (а)-(b) по п.2 и дополнительно включающий следующие стадии:
(f) изомеризация смеси соединений общей структуры Va и Vb, при этом X, R1 и R2, как определены в п.2, которая обогащена Va, в смесь соединений общей структуры VIa и VIb, которая обогащена VIa,
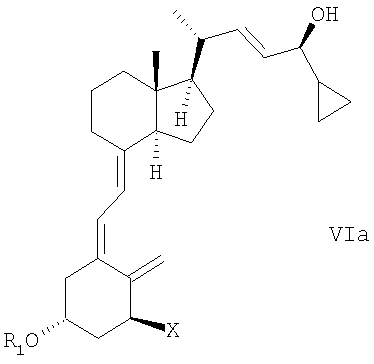
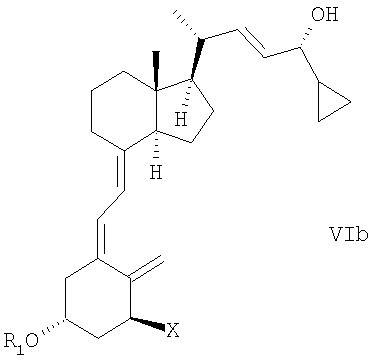
при этом X, R1 и R2 определены выше;
(g) выделение соединения общей структуры VIa из смеси соединений общей структуры VIa и VIb, которая обогащена VIa, при этом X, R1 и R2, определены выше;
(h) когда R1 и/или R2 не является водородом, удаление гидроксизащитной группы (групп) R1 и/или R2 соединения общей структуры VIa для получения кальципотриола или моногидрата кальципотриола.
4. Способ получения кальципотриола {(5Z, 7Е, 22Е, 24S)-24-циклопропил-9,10-секохола-5, 7, 10(19), 22-тетраен-1α-3β-24-триол} или моногидрата кальципотриола, включающий следующие стадии:
(j) восстановление соединения общей структуры III,
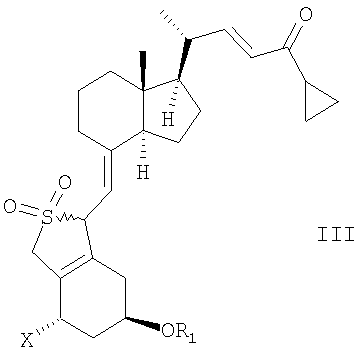
в которой Х представляет собой водород,
и в которой R1 представляет собой водород или гидроксизащитную группу, в инертном растворителе с хиральным восстановительным агентом или с восстановительным агентом в присутствии хирального вспомогательного вещества,
где восстановительный агент представляет собой борановое производное и где хиральное вспомогательное вещество представляет собой хиральный 1,2-аминоспирт,
получая смесь соединений общей структуры IVa или IVb, которая обогащена IVa,
при этом X и R1 определены выше;
(k) взаимодействие смеси соединений общей структуры IVa и IVb, которая обогащена IVa, в присутствии основания с получением смеси соединений общей структуры Va и Vb, которая обогащена Va,
при этом Х и R1 определены выше;
(l) выделение соединения общей структуры Va из смеси соединений общей структуры Va и Vb, которая обогащена Va, при этом Х и R1 определены выше;
(m) гидроксилирование соединения общей структуры Va подходящим гидрооксилирующим агентом, при этом Х и R1 определены выше, с получением соединения общей структуры Va, в которой Х представляет собой OR2 и R2 представляет собой водород, и при этом R1 определено выше;
(о) изомеризация соединения общей структуры Va в соединение общей структуры VIa,
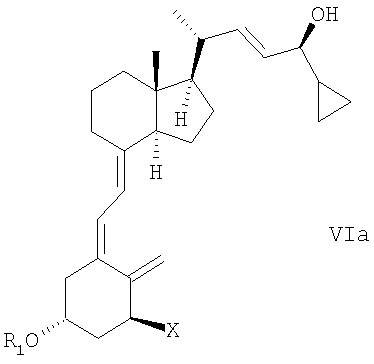
при этом X, R1 и R2 определены выше; и
(р) когда R1 не является водородом, удаление гидроксизащитной группы R1 соединения общей структуры VIa для получения кальципотриола или моногидрата кальципотриола.
5. Способ получения кальципотриола или моногидрата кальципотриола, включающий стадии (j)-(l) п.4 и дополнительно включающий следующие стадии:
(q) защита С-24 гидроксигруппы соединения общей структуры Va,
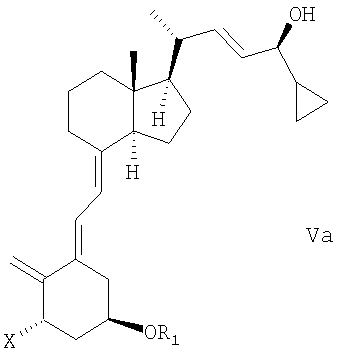
в которой Х представляет собой водород, и в которой R1 представляет собой водород или гидроксизащитную группу, с гидроксизащитной группой;
(r) гидроксилирование С-24 гидроксизащищенного соединения общей структуры Va подходящим гидроксилирующим агентом, при этом Х и R1 определены выше, с получением С-24 гидроксизащищенного соединения общей структуры Va, в которой Х представляет собой OR2 и R2 представляет собой водород, и при этом R1 определен выше;
(s) удаление С-24 гидроксизащитной группы соединения общей структуры Va;
(t) изомеризация соединения общей структуры Va в соединение общей структуры VIa,
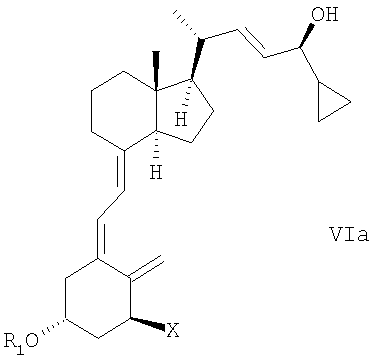
при этом X, R1 и R2 определены выше;
(u) когда R1 не является водородом, удаление гидроксизащитной группы R1 соединения общей структуры VIa для получения кальципотриола или моногидрата кальципотриола.
6. Способ по любому из пп.1-5, в котором стадию восстановления восстановительным агентом осуществляются в присутствии хирального вспомогательного вещества.
7. Способ по п.6, в котором восстановительный агент представляет собой N,N-диэтиланилинборан, борантетрагидрофуран или борандиметилсульфид.
8. Способ по п.6, в котором хиральное вспомогательное соединение представляет собой хиральное цис-1-амино-2-инданолпроизводное.
9. Способ по п.6, в котором хиральное вспомогательное соединение представляет собой (1S, 2R)-(-)-цис-1-амино-2-инданол.
10. Способ по любому из пп.1-5, в котором инертный растворитель представляет собой толуол, трет.-бутилметиловый эфир, тетрагидрофуран или их смеси.
11. Способ по любому из пп.1-5, в котором смесь соединений общей структуры IVa и IVb, полученная восстановлением соединения общей структуры III, имеет молярное соотношение IVa:IVb, которое составляет, по меньшей мере, 56:44.
12. Способ по п.11, в котором стадию восстановления проводят при температуре от 10 до 20°С.
13. Способ получения соединения общей структуры III,
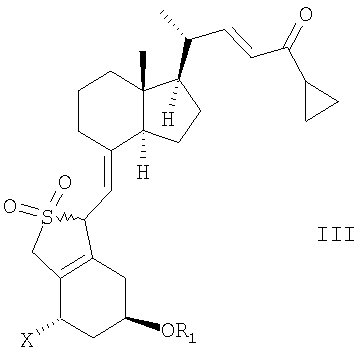
в которой Х представляет собой или водород или OR2,
и в которой R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу, реакцией соединения общей структуры VII или VIII,
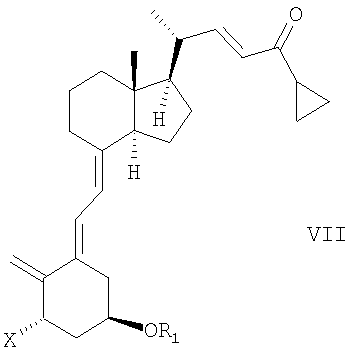
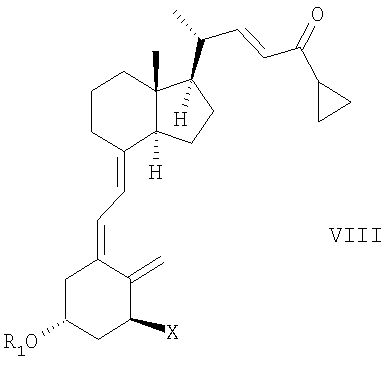
при этом R1 и R2 определены выше, с диоксидом серы.
14. Способ по любому из пп.1-5 или 13, в котором соединение общей структуры III представляет собой эпимер общей структуры IIIa
15. Способ по любому из пп.1-5 или 13, в котором соединение общей структуры III представляет собой эпимер общей структуры IIIb
16. Способ взаимодействия смеси соединений общей структуры IVa и IVb,
в которой Х представляет собой или водород или OR2
при этом R1 и R2 могут быть одинаковыми или различными и представляют собой водород или гидроксизащитную группу,
которая обогащена IVa, в присутствии основания с получением смеси соединений общей структуры Va и Vb, которая обогащена Va,
при этом X, R1 и R2 определены выше.
17. Способ по пп.1-3, 13 или 16, в котором Х представляет собой OR2.
18. Способ по п.17, в котором R1 и/или R2 представляют собой алкилсилил.
19. Способ по п.18, в котором R1 и/или R2 представляют собой трет.-бутилдиметилсилил.
20. Способ получения кальципотриола {(5Z, 7Е, 22Е, 24S)-24-циклопропил-9, 10-секохола-5, 7, 10(19), 22-тетраен-1α-3β-24-триол} или моногидрата кальципотриола, который включает способ по любому из пп.1-19.
21. Соединение общей структуры IIIа или IIIb или их смеси,
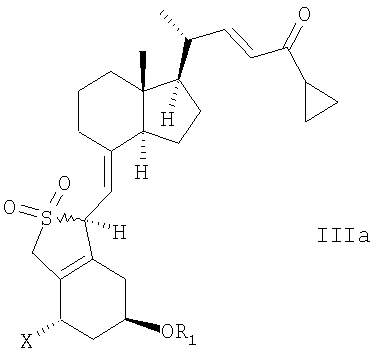
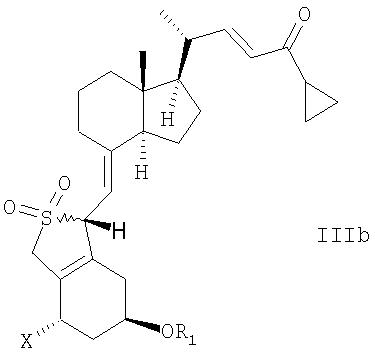
в которой Х представляет собой водород или OR2, и в которой R1 и R2 могут быть одинаковыми или различными, и представлять собой водород или гидроксизащитную группу.
22. Соединение общей структуры IVaa, IVab, IVba, IVbb, IVb или их смеси,
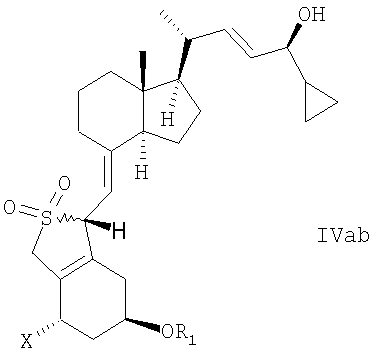
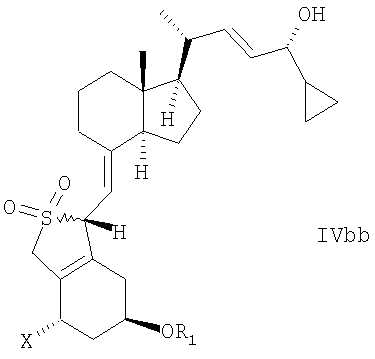
в которой Х представляет собой или водород или OR2, и в которой R1 и R2 могут быть одинаковыми или различными, и представлять собой водород или гидроксизащитную группу.
23. Соединение по п.21 или 22, в котором Х представляет собой OR2.
24. Соединение по п.23, в котором R1 и R2 представляют собой алкилсилил.
25. Соединение по п.24, в котором R1 и R2 представляют собой трет.-бутилдиметилсилил.
26. Соединение по любому из пп.21 и 22, в котором R1 и R2 представляют собой водород.
27. Применение соединения по любому из пп.21-26 в качестве промежуточного соединения для получения кальципотриола или моногидрата кальципотриола.
| US 6531460 B1, 11.03.2003 | |||
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| ГИДРАТ КАЛЬЦИПОТРИОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2128646C1 |

