Область техники
Данное изобретение относится к способу получения ингибитора ВИЧ-протеазы бисульфата атазанавира и его новых форм.
Уровень техники
Патент США № 5849911, Fässler et al. раскрывает ряд азапептидных ингибиторов ВИЧ-протеазы (который включает атазанавир), имеющих структуру:
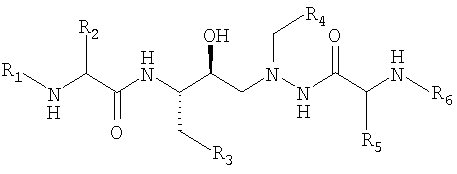
где
R1 обозначает низший алкоксикарбонил,
R2 обозначает вторичный или третичный алкил или низший алкилтио - низший алкил,
R3 обозначает фенил, который является незамещенным или замещенным одним или несколькими низшими алкоксильными радикалами, или С4-С8-циклоалкил,
R4 обозначает фенил или циклогексил, каждый из которых замещен в 4-положении ненасыщенным гетероциклилом, который присоединен через атом углерода в кольце, имеет 5-8 атомов в кольце, содержит от 1 до 4 гетероатомов, выбранных из азота, кислорода, серы, сульфинила (-SO-) и сульфонила (-SO2-), и является незамещенным или замещенным низшим алкилом или фенил - низшим алкилом,
R5, независимо от R2, имеет одно из значений, указанных для R2, и
R6, независимо от R1, обозначает низший алкоксикарбонил или его соль, при условии, что имеется, по меньшей мере, одна солеобразующая группа, которая включает различные группы фармацевтически приемлемых солей присоединения к кислоте.
Предусмотрены несколько способов получения азапептидов, включая получение соединения, где R1 и R6 и R2 и R5 обозначают в каждом случае два идентичных радикала, при этом диаминосоединение формулы:
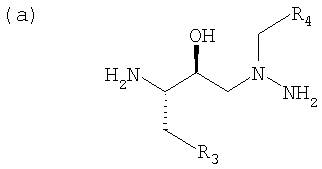
конденсируют с кислотой формулы:
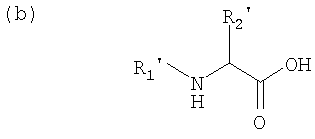
или ее реакционноспособным производным, где R′1 и R′2 и R2 и R5 имеют значение, указанное для R1 и R6 и для R2 и R5 соответственно.
При получении указанным выше способом атазанавира диаминосоединение (а) формулы:
получают путем сочетания эпоксида:
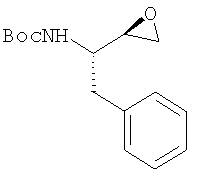
с гидразинкарбаматом:
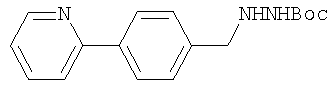
в присутствии изопропилового спирта с получением защищенного диамина:
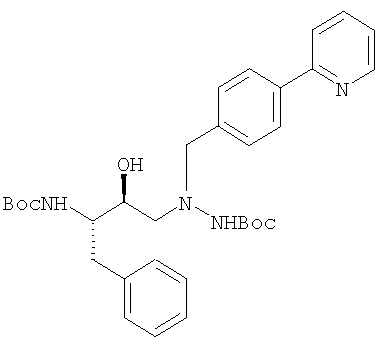
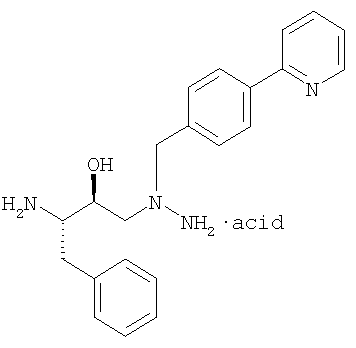
который обрабатывают соляной кислотой в присутствии растворителя, такого как тетрагидрафуран, с получением диамина (а):
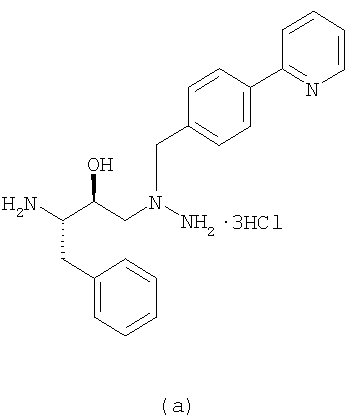
Диамин выделяют и используют на следующей стадии сочетания, когда он реагирует с кислотой (b):
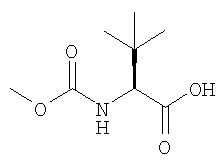
или ее реакционноспособным эфиром, с применением агента сочетания, такого как O-(1,2-дигидро-2-оксо-1-пиридил)-N,N,N′,N′-тетраметилуронийтертафторборат) (TPTU).
Было установлено, что свободное основание диамина нестабильно и поэтому его нежелательно применять при получении свободного основания атазанавира.
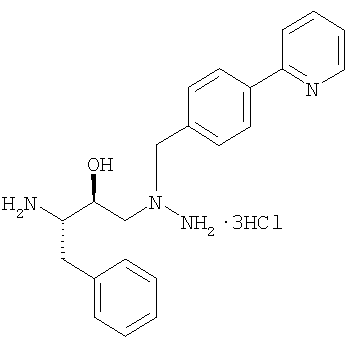
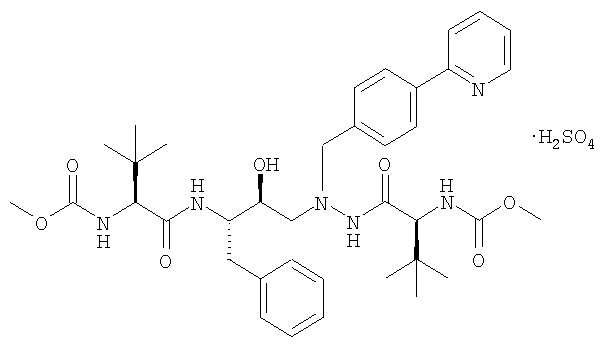
В патенте США № 6087383, Singh et al. описана бисульфатная соль азапептидного ингибитора ВИЧ-протеазы, известного как атазанавир, который имеет формулу:
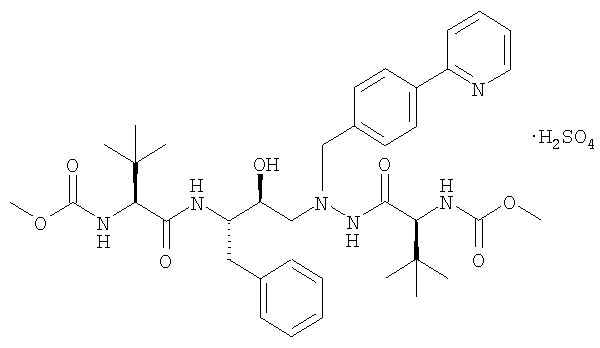
(называемый также бисульфатом атазанавира или сульфатом атазанавира).
В примере 3 Singh et al. описывают получение бисульфата атазанавира в виде кристаллов Типа-II, которые представляют собой гидратированную гигроскопичную и кристаллическую форму и в виде кристаллов Типа-I, которые представляют собой безводную/десольватированную кристаллическую форму.
Сущность изобретения
Согласно данному изобретению предусмотрены новые формы бисульфата атазанавира, которые включают Конфигурацию С и Форму Е3. Предпочтительной является Конфигурация С. Кроме того, в соответствии с данным изобретением предусмотрен способ получения бисульфата атазанавира в кристаллической Форме А (объемное лекарство) (которое называется кристаллами Типа-I в примере 3 патента США №6087383, Singh et al.). Кристаллическая Форма А, полученная по изобретению, имеет желательное однородное распределение частиц по размерам и практически постоянный средний размер частиц и применяется для превращения в Конфигурацию С, частично кристаллическое вещество, которое используют вместе с различными эксципиентами для приготовления лекарства.
Способ получения кристаллической Формы А бисульфата атазанавира по изобретению использует модифицированную методику кубической кристаллизации, когда серная кислота добавляется со все возрастающей скоростью в соответствии с кубическим уравнением (приведенным ниже), и включает стадии взаимодействия раствора свободного основания атазанавира в органическом растворителе (в котором бисульфатная соль атазанавира практически не растворяется) с первой порцией концентрированной серной кислоты в количестве, достаточном для реакции с менее, чем примерно 15%, предпочтительно, менее, чем примерно 12 вес.% свободного основания атазанавира, добавления кристаллических зародышей Формы А бисульфата атазанавира к реакционной смеси, добавления дополнительного количества концентрированной серной кислоты на многочисленных стадиях с возрастающей скоростью в соответствии с кубическим уравнением для образования кристаллов Формы А.
Кроме того, согласно данному изобретению предусмотрен способ получения формы атазанавира, которая получена из бисульфата атазанавира и включает бисульфат атазанавира и которая называется Конфигурацией С. Конфигурация С может быть получена путем суспендирования кристаллов Формы А в воде и сушки. Или же Конфигурация С может быть получена выдержкой кристаллов Формы А в условиях высокой относительной влажности, величина которой более примерно 95% RH (водяной пар), в течение, по меньшей мере, 24 ч. Конфигурация С может быть также получена путем мокрого гранулирования бисульфата атазанавира или комбинации бисульфата атазанавира и эксципиентов и сушки мокрого гранулята.
Согласно предпочтительному варианту кристаллы Формы А смешивают с эксципиентами, входящими в состав препарата, такими как один или несколько объемных агентов, например лактоза, одного или нескольких дезинтегрантов, таких как кросповидон, и подвергаются мокрому гранулированию с получением Конфигурации С в смеси с эксципиентами.
Далее, согласно данному изобретению предусмотрена новая форма бисульфата атазанавира, а именно, Форма Е3, которая является сильнокристаллической формой триэтанольного сольвата бисульфата атазанавира.
Форму Е3 получают суспендированием свободного основания атазанавира в этаноле, обработки суспензии концентрированной серной кислотой, нагревания, введения в полученный раствор зародышей мокрых (этанольных) кристаллов Е3, обработки смеси гептаном (или другим растворителем, таким как толуол или гексан), фильтрования и сушки.
Кроме того, в соответствии с данным изобретением предусмотрен способ получения кристаллов бисульфата атазанавира Формы А, который включает стадии получения триаминной соли формулы:
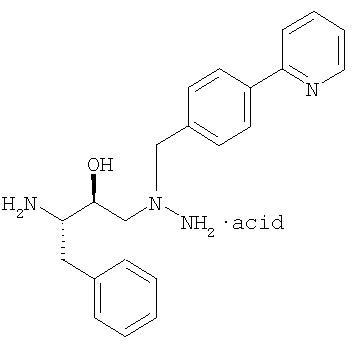
(предпочтительно, гидрохлоридная (3М) соль) и без выделения триаминной соли взаимодействие этой триаминной соли с активным сложным эфиром, предпочтительно, формулы:

в присутствии основания и органического растворителя с получением свободного основания атазанавира, которое, без выделения, превращается в бисульфат атазанавира путем модифицированной кубической кристаллизации, как описано в данной заявке.
Кроме того, в соответствии с данным изобретением предусмотрена новая композиция на основе бисульфата атазанавира, которая включает бисульфат атазанавира в виде кристаллической Формы А или Конфигурации С и фармацевтически приемлемый носитель для активного ингредиента. Фармацевтически приемлемый носитель может включать наполнители, связующее, дезинтегранты, смазывающие агенты и другие обычные эксципиенты.
Согласно изобретению различные формы бисульфата атазанавира могут быть охарактеризованы различными методами, которые хорошо известны специалистам. Эти формы могут быть охарактеризованы с использованием метода кристаллографии монокристалла, который основан на измерениях ячейки монокристалла Формы при фиксированной температуре определения. Подробное описание элементарных ячеек приведено в Stout and Jensen, X-Ray Structure Determination: A practical Guide, Macmillan Co., New York (1968), Chapter 3. Или же уникальное расположение атомов в пространстве внутри кристаллической решетки может быть охарактеризовано согласно наблюдаемым фракционным атомным координатам. Другим методом характеристики кристаллической структуры является порошковая кристаллография, когда экспериментальный или наблюдавшийся профиль дифракции сравнивается с имитированным профилем, характеризующим чистый порошковый материал, оба определяются при одной и той же температуре, но измерения нужной формы характеризуются как ряд величин 2Θ.
Могут быть использованы и другие средства, характеризующие форму, такие как ядерный магнитный резонанс в твердом состоянии (SSNMR), дифференциальная сканирующая калориметрия (DSC) и термогравиметрический анализ (TGA). Эти параметры также могут быть применены в комбинации для характеристики нужной формы.
Кристаллы Формы А могут быть охарактеризованы параметрами элементарной ячейки, равными:
Размеры ячейки:
а=9,86(5) Ǻ
b=29,245(6) Ǻ
с=8,327(2) Ǻ
α=93,56(2)°
β=114,77(3)°
γ=80,49(3)°
Пространственная группа 1
Молекулы/асимметричная единица 2,
при этом кристаллическая форма имеется при примерно 22°С.
Форму А можно охарактеризовать фракционными атомными координатами, указанными в Таблице 3, и кристаллической структурой, приведенной на Фиг.2.
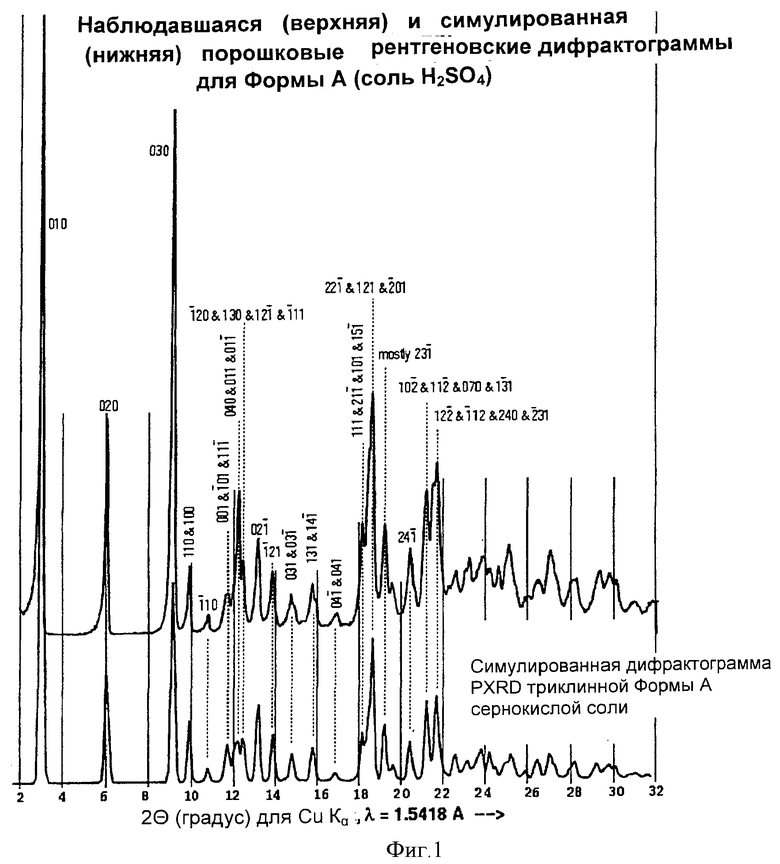
Форма А может быть охарактеризована имитированной и наблюдавшейся порошковой кристаллограммами, как показано на Фиг.1.
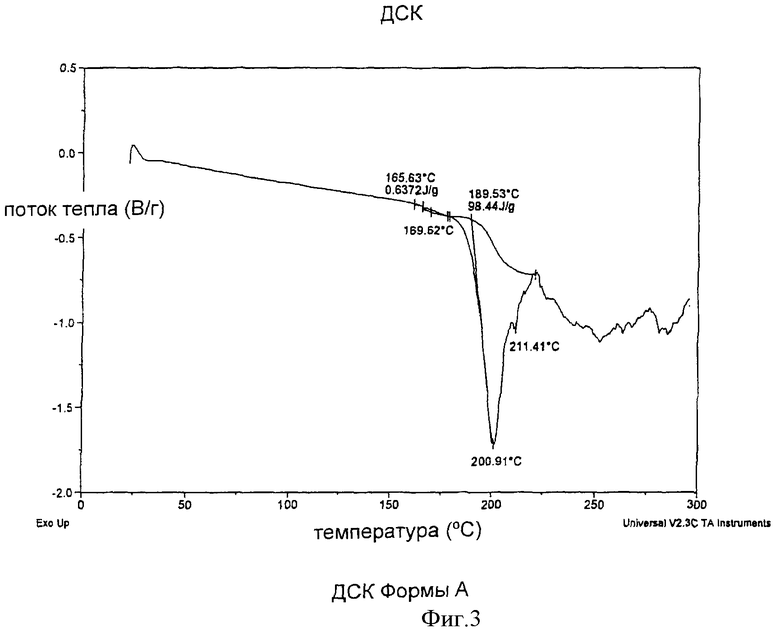
Форма А может быть также охарактеризована методом дифференциальной сканирующей калориметрии (DSC) с получением термограммы, имеющей эндотерму с началом пика при примерно 165,6°С, как показано на Фиг.3.
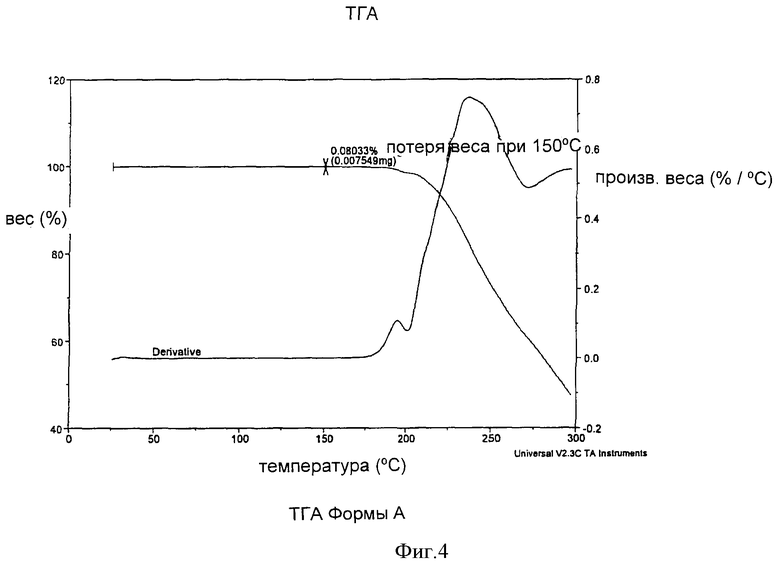
Форма А может быть охарактеризована кривой термогравиметрического анализа (TGA), которая показывает незначительную потерю веса при температуре около 100-150°С, как показано на Фиг.4.
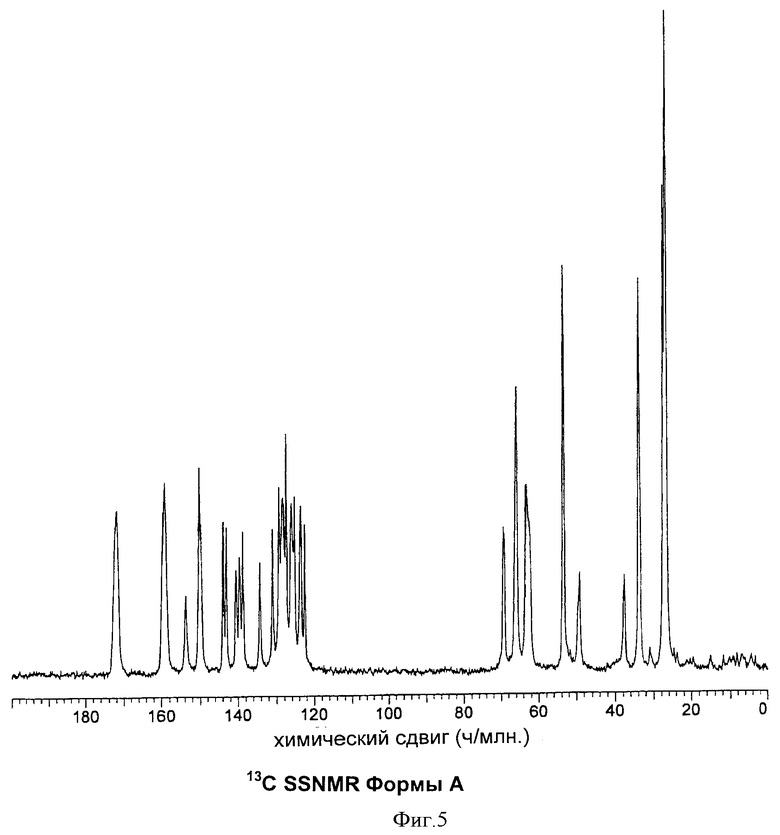
Форма А может быть охарактеризована химическими сдвигами при проведении ЯМР в твердом состоянии (SSNMR), как показано в Таблице 4, и спектром, приведенным на Фиг.5.
Форма А может быть охарактеризована фракционными атомными координатами, перечисленными в Таблице 5.
Форма А соли может быть охарактеризована изотермами влага - сорбция с увеличением веса, равным около 0,1% в интервале RH от 25 до 75% при 25°С.
Согласно одному аспекту изобретения Конфигурация С может быть охарактеризована методом порошковой кристаллографии, как показано на Фиг.5.
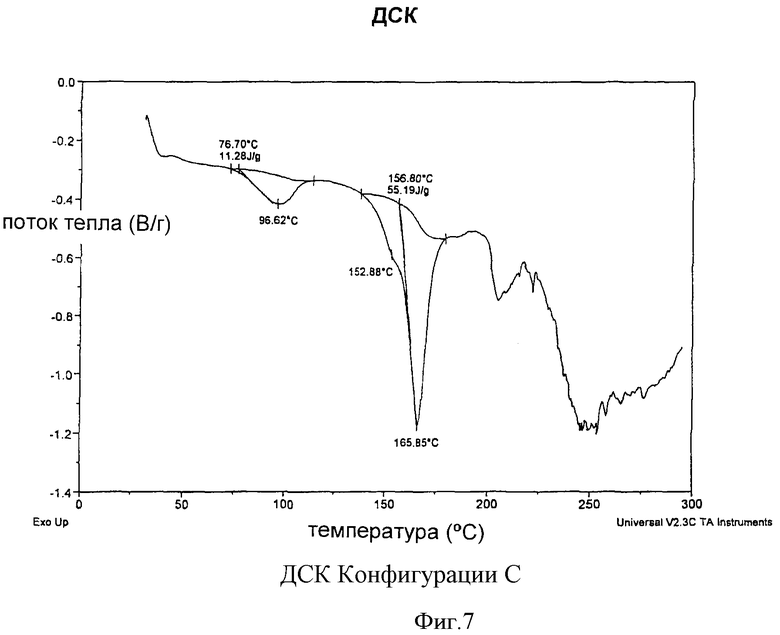
Согласно другому аспекту изобретения Конфигурация С может быть охарактеризована термограммой дифференциальной сканирующей калориметрии, как показано на Фиг.7, на термограмме видна эндотерма в интервале от примерно 76,7°С до примерно 96,6°С и от примерно 156,8°С до примерно 165,9°С.
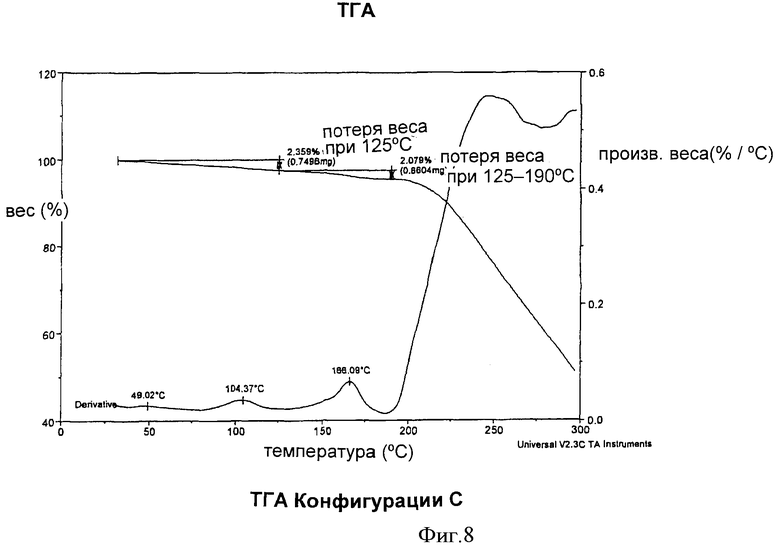
Согласно другому аспекту изобретения Конфигурация С может быть охарактеризована кривой термогравиметрического анализа; где видны потеря веса, равная примерно 2,4%, до примерно 125°С и потеря веса, равная примерно 4,4%, до примерно 190°С, как показано на Фиг.8.
Согласно данному изобретению Форма Е3 может быть охарактеризована кристаллографическими параметрами, как показано в Таблице 5, равными:
а=10,749(5) Ǻ
b=13,450(4) Ǻ
с=9,250(2) Ǻ
α=98,33(2)°
β=95,92(3)°
γ=102,83(3)°
Пространственная группа P1
Молекулы/асимметричная единица 1,
при этом кристаллическая форма наблюдается при примерно -23°С.
Согласно другому аспекту изобретения Форма Е3 может быть охарактеризована фракционными атомными координатами, перечисленными в Таблице 6.
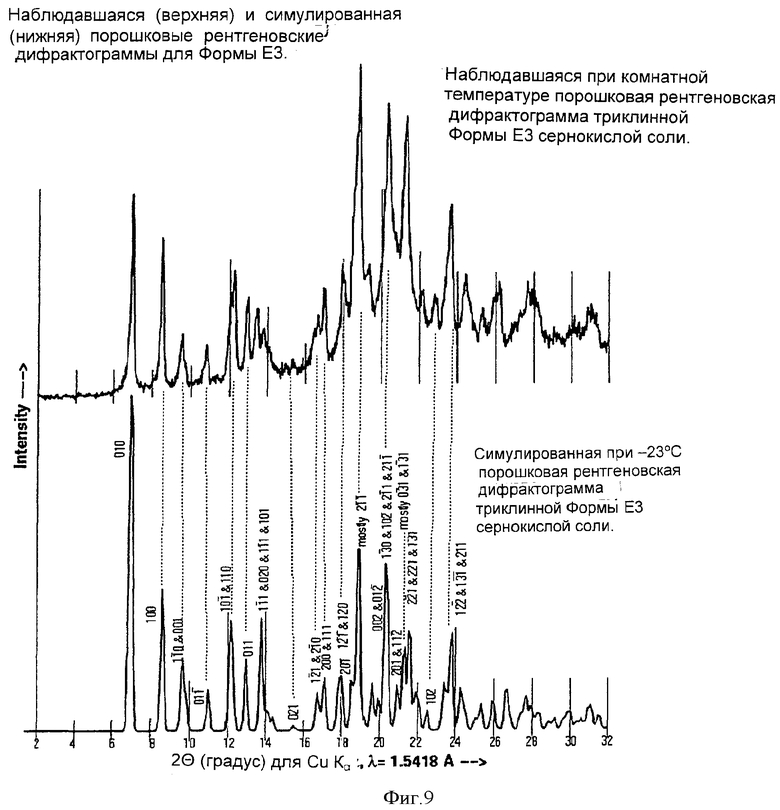
Согласно еще одному аспекту изобретения Форма Е3 может быть охарактеризована симулированной и наблюдавшейся дифрактограммами, показанными на Фиг.9.
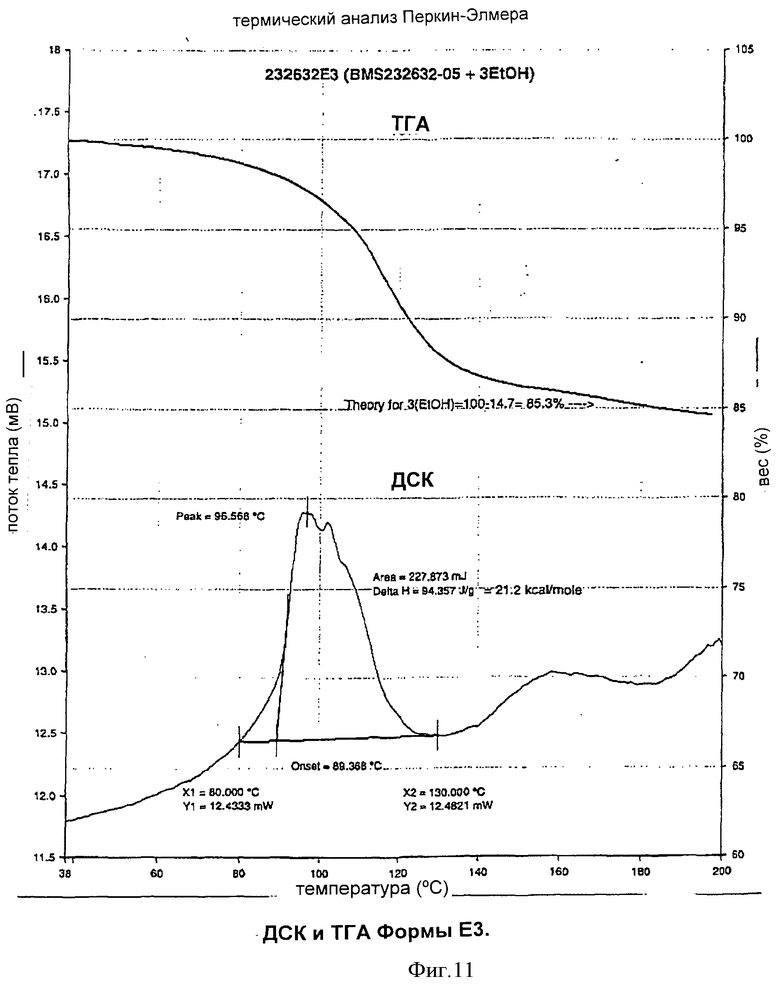
Согласно еще одному аспекту изобретения Форма Е3 может быть охарактеризована термограммой дифференциальной сканирующей калориметрии, на которой эндотерма обычно находится в пределах от примерно 89,4°С до примерно 96,6°С, как показано на Фиг.11.
Согласно еще одному аспекту изобретения Форма Е3 может быть охарактеризована кривой термогравиметрического анализа, на которой потеря веса, равная примерно 14,7%, наблюдается при примерно 150°С, как показано в Таблице 8.
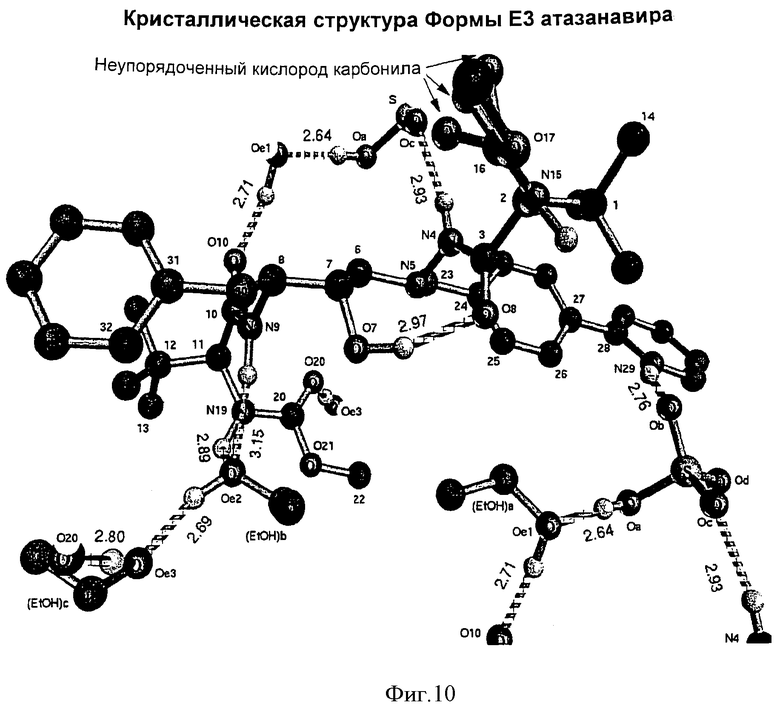
Согласно еще одному аспекту изобретения Форма Е3 может быть охарактеризована структурой кристаллов, как показано на Фиг.10.
Краткое описание фигур
На Фиг.1 показаны рассчитанная (симулированная) (22°С) и наблюдаемая (экспериментально при комнатной температуре) порошковые дифрактограммы (Cu Kα λ=1,5418 Ǻ) Формы А.
На Фиг.2 показана кристаллическая структура Формы А.
Фиг.3 представляет термограмму Формы А, полученную методом дифференциальной сканирующей калориметрии (DSC).
На Фиг.4 показана кривая, полученная методом дифференциальной сканирующей калориметрии (DSC), для образца Формы А.
На Фиг.5 представленные данные 13С ЯМР в твердом состоянии для Формы А.
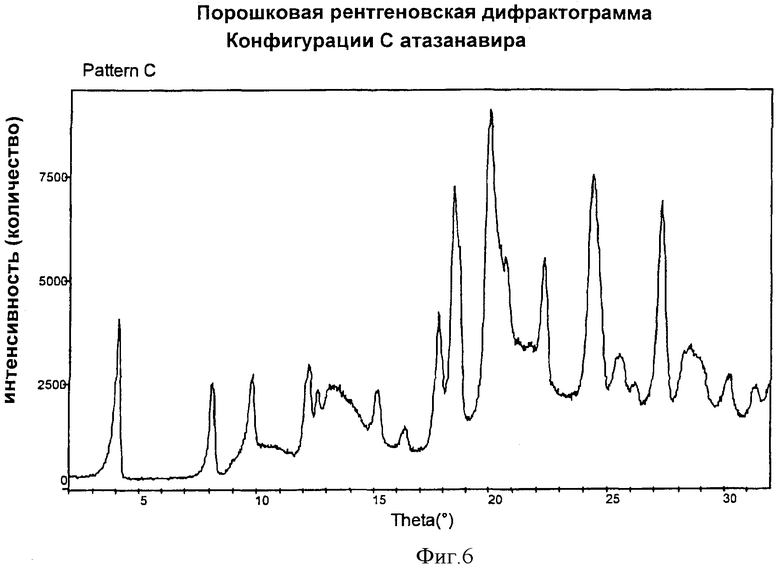
Фиг.6 представляет наблюдаемую (экспериментальную при комнатной температуре) дифрактограмму Конфигурации С, полученную методом порошковой кристаллографии (Cu Кα λ=1,5418 Ǻ).
На Фиг.7 показана термограмма Конфигурации С, полученная методом дифференциальной сканирующей калориметрии.
На Фиг.8 представлена кривая, полученная методом термогравиметрического анализа для Конфигурации С.
На Фиг.9 показана рассчитанная (симулированная) (22°С) и наблюдаемая (экспериментальная при комнатной температуре) дифрактограммы Формы Е3, полученные методом порошковой кристаллографии.
На Фиг.10 показана структура кристаллов Формы Е3 и
На Фиг.11 представлены термограмма Формы Е3, полученная методом дифференциальной сканирующей калориметрии, и кривая термогравиметрического анализа для Формы Е3.
Подробное описание изобретения
Данное изобретение предусматривает, по меньшей мере частично, формы бисульфата атазанавира, а именно, форму Е3 и Конфигурацию С в качестве новых материалов, в частности, в фармацевтически приемлемой форме. Термин «фармацевтически приемлемая», используемый в данном описании, относится к таким соединениям, материалам, композициям и/или дозированным формам, которые, с точки зрения здравого медицинского суждения, пригодны для контактирования с тканями человека и животных без проявления сильной токсичности, раздражения, аллергической реакции или других проблемных осложнений при разумном отношении польза/риск. Согласно некоторым предпочтительным вариантам кристаллические формы свободного основания и его соли получают в практически чистом виде. Термин «практически чистый», используемый в данной заявке, означает соединение, имеющее степень чистоты более примерно 90%, включая, например, примерно 91%, примерно 92%, примерно 93%, примерно 94%, примерно 95%, примерно 96%, примерно 97%, примерно 98%, примерно 99% и примерно 100%.
Используемый термин «полиморф» относится к кристаллическим формам, имеющим одинаковый химический состав, но различное пространственное расположение молекул, атомов и/или ионов, образующих кристалл.
Используемый термин «сольват» относится к кристаллической форме молекулы, атома и/или ионов, которая содержит молекулы растворителя или растворителей, введенных в кристаллическую структуру. Молекулы растворителя в сольвате могут располагаться упорядочение и/или неупорядоченно. Сольват может содержать стехиометрическое или нестехиометрическое количество молекул растворителя. Например, сольват с нестехиометрическим количеством молекул растворителя может образоваться при частичной потере растворителя из сольвата.
Образцы кристаллических форм могут быть получены с практически чистой фазовой гомогенностью, что указывает на наличие доминирующего количества одной кристаллической формы и, возможно, незначительных количеств одной или нескольких кристаллических форм. Наличие более, чем одной кристаллической формы в образце может быть определено такими методами, как порошковая рентгеновская кристаллография (PXRD) или ядерный магнитный резонанс в твердом состоянии (SSNMR). Например, наличие экстрапиков на дифрактограмме, экспериментально измеренной, по сравнению с симулированной дифрактограммой, может свидетельствовать о более, чем одной кристаллической форме в образце. Симулированная дифрактограмма может быть рассчитана, исходя из данных для монокристалла (см. Smith D.K., "A FORTRAN Program for Calculating X-Ray Powder Diffraction Patterns" Lawrance Laboratory, Livermore, California, UCRL-7196 (April 1963). Кристаллическая форма предпочтительно имеет чисто гомогенную фазу, о чем свидетельствует менее 10%, предпочтительно, менее 5% и, более вероятно, менее 2% общей площади пиков в экспериментальной дифрактограмме, появляющейся от экстрапиков, которые отсутствуют на симулированной дифрактограмме (PXRD). Более предпочтительна кристаллическая форма, имеющая практически чистую гомогенную фазу с менее 1% общей площади пиков на экспериментальной дифрактограмме (PXRD), возникающей от экстрапиков, которые отсутствуют на симулированной дифрактограмме (PXRD).
Методы получения кристаллических форм известны из уровня техники. Кристаллические формы могут быть получены разными методами, включая, например, кристаллизацию перекристаллизацию из подходящего растворителя, сублимацию, рост из расплава, превращение в твердое состояние из другой фазы, кристаллизацию из суперкритической жидкости, а также струйное распыление. Методы кристаллизации или перекристаллизации кристаллических форм из смеси растворителей включают, например, выпаривание растворителя, уменьшение температуры смеси растворителей, введение зародышей кристаллов в сверхнасыщенную смесь соединения и/или соли в растворителе, сушку смеси при замораживании и добавление антирастворителей (противорастворителей) к смеси растворителей.
Кристаллы лекарств, включая полиморфы, методы их получения и характеристика кристаллов лекарств обсуждаются в Solid-State Chemistry of Drugs, S.R.Byrn, R.R.Pfeiffer, and J.G. Stowell, 2nd Edition, SSCI, West Lafayette, Indiana (1999).
В случае метода кристаллизации с применением растворителя выбор растворителя или растворителей обычно зависит от одного или нескольких факторов, таких как растворимость соединения, методика кристаллизации, давление паров растворителя. Можно применять комбинацию растворителей, например, соединение может быть солюбилизировано в первом растворителе с получением раствора с последующим добавлением антирастворителя для уменьшения растворимости соединения в растворе и образования кристаллов. Противорастворитель представляет собой растворитель, в котором соединение плохо растворяется. Подходящие растворители для получения кристаллов включают полярные и неполярные растворители.
По одному способу получения кристаллов бисульфат атазанавира суспендируют и/или перемешивают в подходящем растворителе с получением суспензии, которая может быть нагрета для ускорения растворения. Термин «суспензия» здесь обозначает насыщенный раствор бисульфата атазанавира или его соли, который может также содержать дополнительное количество бисульфата атазанавира или его соли с получением гетерогенной смеси бисульфата атазанавира или его соли и растворителя при данной температуре. Подходящие растворители в этом случае включают, например, полярные апротонные растворители и полярные протонные растворители и смеси двух или более этих растворителей, как описано в данной заявке.
Зародыши кристаллов могут быть добавлены к любой смеси для ускорения кристаллизации. Специалисту ясно, что введение зародышей используется как средство регулирования роста конкретной кристаллической формы или как средство регулирования распределения частиц по размерам в кристаллическом продукте. Соответственно, расчет количества необходимых зародышей зависит от размера доступных зародышей и желаемого размера средней частицы продукта, как описано, например, в «Programmed cooling of batch crystallizers», J.W.Mullin and J.Nyvlt, Chemical Engineering Science (1971) 26: 369-377. Вообще зародыши небольшого размера необходимы для эффективного регулирования роста кристаллов в партии. Зародыши небольшого размера могут быть получены пропусканием через сито, измельчением или микронизацией кристаллов большего размера или путем микрокристаллизации растворов. Следует обращать внимание, чтобы измельчение или микронизация кристаллов не приводили к любому изменению кристаллизации из желаемой кристаллической формы (а именно, изменению с получением аморфной формы или другого полиморфа).
Охлажденную смесь можно отфильтровать под вакуумом, выделенные твердые вещества можно промыть подходящим растворителем, таким как растворитель холодной перекристаллизации, и высушить в токе азота с получением желательной кристаллической формы. Выделенный твердый продукт можно подвергнуть анализу при помощи подходящего метода спектроскопии или анализа, такого как SSNMR, DSC, PXRD или т.п., для подтверждения образования предпочтительной кристаллической формы продукта.
Полученная кристаллическая форма обычно получается в количестве более примерно 70 вес.% выделенного продукта, предпочтительно более 90% в расчете на вес бисульфата атазанавира, первоначально применявшегося в способе кристаллизации. Продукт может быть измельчен или пропущен через сито с отверстиями для разделения кусков продукта, если это необходимо.
Кристаллические формы могут быть получены непосредственно из реакционной среды на последней стадии получения бисульфата атазанавира. Это может быть достигнуто, например, при применении на последней стадии способа растворителя или смеси растворителей, из которых может кристаллизоваться бисульфат атазанавира. Иначе кристаллические формы могут быть получены путем перегонки или добавления растворителя. Подходящие растворители для этой цели включают любой из растворителей, описанных в данной заявке, включая протонные полярные растворители, такие как спирты, и апротонные полярные растворители, такие как кетоны.
В общем реакционная смесь может быть отфильтрована для удаления любых нежелательных примесей, неорганических солей и т.п. с последующей промывкой растворителем, использованным для реакции или кристаллизации. Полученный раствор можно концентрировать для удаления избытка растворителя или газообразных компонентов. Если применяется отгонка, то конечное количество собранного дистиллята может меняться в зависимости от параметров процесса, например размера сосуда, степени перемешивания и т.п. В общем, реакционный раствор может быть отогнан до примерно {фракция (1/10)} от первоначального раствора перед заменой растворителя. Можно по ходу отбирать образцы и анализировать для определения степени реакции и вес.% продукта согласно стандартным методикам. Если желательно, дополнительно может быть добавлен или удален реакционный растворитель для оптимизации концентрации. Предпочтительно, чтобы конечная концентрация была примерно 50 вес.%, в этот момент обычно получается суспензия.
Может быть предпочтительно добавлять растворители в реакционный сосуд без отгонки реакционной смеси. Предпочтительными растворителями для этой цели являются такие растворители, которые могут в конечном счете оседать в кристаллической решетке, как обсуждалось выше в связи с обменом растворителями. Хотя конечно концентрация может меняться в зависимости от желательной степени чистоты, выделения и т.п., конечная концентрация свободного основания I в растворе, предпочтительно, равна от примерно 4% до примерно 7%. Реакционную смесь можно перемешивать после добавления растворителя и одновременно нагревать. Например, можно перемешивать реакционную смесь в течение примерно 1 ч, нагревая ее до примерно 70°С. Горячая смесь фильтруется и промывается реакционным растворителем, добавленным растворителем или их смесью. Для начала кристаллизации в любой раствор для кристаллизации можно добавить зародыши кристаллов.
Различные формы, описанные здесь, можно отличить одну от другой при помощи различных методов анализа, известных специалисту в данной области. Такие методы включают, без ограничения, метод ядерного магнитного резонанса в твердом состоянии (SSNMR), метод порошковой рентгеновской кристаллографии (PXRD), метод дифференциальной сканирующей калориметрии (DSC) и/или метод термогравиметрического анализа (TGA).
Специалисту ясно, что рентгеновская дифрактограмма может быть получена с ошибкой измерения, которая зависит от применяемых условий измерения. В частности, общеизвестно, что интенсивности на дифрактограмме могут флуктуировать в зависимости от применяемых условий измерения и формы или морфологии кристалла. Далее, следует понимать, что относительные интенсивности могут также меняться в зависимости от условий эксперимента и, соответственно, точный порядок интенсивности не следует принимать во внимание. Дополнительно, ошибка в измерении угла дифракции в случае обычной рентгеновской дифрактограммы составляет обычно примерно 0,2% или менее, предпочтительно, около 0,1% (как указано ниже), и такая степень ошибки измерения должна приниматься во внимание как имеющая отношение к вышеупомянутым углам дифракции. Соответственно, следует иметь в виду, что кристаллические формы по изобретению не ограничены кристаллическими формами, которые обеспечивают получение рентгеновских дифрактограмм, полностью идентичных дифрактограммам, показанным на прилагаемых фигурах в данной заявке. Любые кристаллические формы, которые обеспечивают получение рентгеновских дифрактограмм, практически идентичных, показанным на прилагаемых фигурах, входят в объем данного изобретения. Способность оценить практическую идентичность рентгеновских дифрактограмм находится в компетенции специалиста в данной области.
Термин «Форма», используемый в отношении Формы А и Формы Е3, относится к гомогенной кристаллической структуре.
Термин «Конфигурация», используемый по отношению к Конфигурации С, относится к характерной рентгеновской дифрактограмме.
Термин «бисульфат атазанавира» относится к бисульфату атазанавира, а также к сульфату атазанавира.
При проведении способа получения кристаллов Формы А бисульфаты атазанавира по изобретению применяют модифицированный метод кубической кристаллизации, когда свободное основание атазанавира растворяют в органическом растворителе, в котором бисульфат атазанавира практически не растворяется, включающем ацетон, смесь ацетона и N-метилпирролидона, этанол, смесь этанола и ацетона и т.п., с получением раствора, в котором концентрация свободного основания атазанавира составляет от примерно 6,5 до примерно 9,7 вес.%, предпочтительно, от примерно 6,9 до примерно 8,1 вес.%.
Раствор свободного основания атазанавира нагревают при температуре в интервале от примерно 35°С до примерно 55°С, предпочтительно, от примерно 40°С до примерно 50°С и подвергают взаимодействию с таким количеством концентрированной серной кислоты (содержащей от примерно 95 до примерно 100% H2SO4), которое является достаточным для реакции с менее, чем 15%, предпочтительно, от примерно 5 до менее, чем примерно 12%, более предпочтительно, от примерно 8 до примерно 10 вес.% свободного основания атазанавира. Таким образом, исходный раствор свободного основания атазанавира вначале будет взаимодействовать с менее, чем 15%, предпочтительно, от примерно 5 до менее, чем примерно 12 вес.% применяемой серной кислоты. Во время реакции реакционную смесь поддерживают при температуре в пределах от примерно 35 до примерно 55°С, предпочтительно, от примерно 40°С до примерно 50°С.
Реакцию продолжают в течение промежутка времени, составляющего от примерно 12 до примерно 60 мин, предпочтительно, от примерно 15 до примерно 30 мин.
В реакционную смесь добавляют зародыши кристаллов Формы А бисульфата атазанавира в количестве, находящемся в пределах от примерно 0,1 до примерно 80%, предпочтительно, от примерно 3 до примерно 8% в расчете на вес свободного основания атазанавира, остающегося в реакционной смеси, поддерживая температуру реакционной смеси, равную от примерно 35 до примерно 55°С, предпочтительно, от примерно 40°С до примерно 50°С.
Реакцию продолжают до начала кристаллизации. Затем вводят серную кислоту в несколько стадий со все возрастающей скоростью согласно кубическому уравнению, описанному ниже, с получением бисульфата атазанавира, который в процессе сушки образует кристаллы Формы А.
Размер частиц кристаллов и морфология полученного бисульфата атазанавира зависят от скорости добавления серной кислоты, которая определяет скорость кристаллизации. Было установлено, что модифицированная методика «кубической» кристаллизации (кислота добавляется со все возрастающей скоростью согласно кубическому уравнению) обеспечивает образование сравнительно больших, более хорошо определенных кристаллов бисульфата атазанавира наряду с более узким интервалом размеров частиц и меньшим количеством мелких частиц, чем в случае кристаллизации при добавлении с постоянной скоростью. Было показано, что медленная начальная скорость подачи кислоты облегчает рост кристаллов без вторичного образования зародышей. Таким образом, поскольку площадь поверхности увеличивается с увеличением размера частиц, слой зародышей способен выдержать возрастающую скорость добавления кислоты без индуцирования вторичного образования зародышей. Медленная начальная скорость добавления дает кристаллам время стать больше, при этом возрастает их средний размер. Кубическая кристаллизация обеспечивает образование более плотного остатка на фильтре, что способствует удалению жидкости из осадка и промывке, а также образованию продукта, который легче поддается сушке с меньшим количеством твердых комков, чем при кристаллизации при постоянной скорости добавления.
Используемый метод кубической кристаллизации проводится при контролируемой температуре согласно публикации Mullin "Crystallization, 3rd Ed.", 1993, Butterworth - Heineman Pubs. и определяется следующим уравнением:
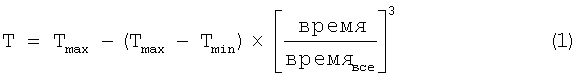
где Tmax = температура начала кристаллизации,
Tmin = температура окончания кристаллизации,
время = время прохождения кристаллизации,
времявсе = общее время кристаллизации.
Так как кристаллизация бисульфата атазанавира регулируется скоростью добавления серной кислоты, переменная величина температуры в уравнении (1) заменяется объемом кислоты. В этом уравнении переменная, обозначающая минимальный объем, отсутствует:

где Vвремя = объем серной кислоты, добавляемый во время прохождения кристаллизации,
Vвесь = общий объем кислоты, составляющий 90% от загрузки,
время = время прохождения кристаллизации,
времявсе = общее время кристаллизации.
Уравнение (2) называется «кубическим уравнением».
Регулируя скорость кристаллизации с помощью этого уравнения, можно регулировать в приемлемых пределах образование зародышей, так как в системе поддерживается постоянный низкий уровень перенасыщения.
Кристаллы Формы А идентифицируются методам рентгеновской порошковой кристаллографии и структурой кристаллов, как показано на Фиг.1 и 2, соответственно.
Кристаллическая Форма А бисульфата атазанавира или конфигурация С, а также Форма Е3, полученные, как описано выше, представляют собой конечный бисульфат атазанавира и могут применяться как лекарственные продукты для введения пациентам.
В соответствии со способом по изобретению Конфигурация С может быть получена путем действия воды на кристаллы Формы А с последующей сушкой.
Согласно другому способу по изобретению Конфигурация С может быть получена при выдержке кристаллов Формы А в условиях высокой относительной влажности, величина которой превышает примерно 95%, предпочтительно, она равна от примерно 95 до примерно 100% RH (водяной пар) в течение, по меньшей мере, 24 ч, предпочтительно, от примерно 24 до примерно 48 ч.
Согласно еще одному варианту изобретения Конфигурацию С получают мокрым гранулированием Формы А бисульфата атазанавира с получением гранул бисульфата атазанавира и затем гранулы высушивают.
При проведении процесса гранулирования бисульфат атазанавира гранулируют в воде и сушат при температуре в пределах от примерно 40°С до примерно 80°С, предпочтительно, в пределах от примерно 50°С до примерно 60°С. Стадию сушки предпочтительно проводить в течение примерно 2 ч, до примерно 20 ч, предпочтительно, в течение от примерно 8 ч до примерно 10 ч.
Конфигурация С может быть также получена мокрым гранулированием Формы А бисульфата атазанавира в присутствии обычных фармацевтических добавок, например, одного или нескольких объемных агентов, предпочтительно, лактозы, одного или нескольких дезинтегрантов, предпочтительно, кросповидона, и сушкой, как описано выше, с получением Конфигурации С в смеси с эксципиентами.
Именно Конфигурация С, Форма А или Форма Е3, предпочтительно Конфигурация С, используются в составе для введения при лечении болезней, вызванных вирусами, как описано ниже.
Конфигурация С характеризуется методом рентгеновской порошковой кристаллографии, как показано на Фиг.3.
Форму Е3 получают путем суспендирования свободного основания атазанавира в этаноле, обработки суспензии концентрированной серной кислотой при мольном отношении кислота: свободное основание в пределах от примерно 1:1 до примерно 1,1:1, нагревания полученного раствора при температуре от примерно 30°С до примерно 40°С, введения зародышей мокрых (этанол) кристаллов Е3 сульфата атазанавира, обработки смеси гептаном (или другим растворителем, таким как гексан или толуол), фильтрования и сушки формы Е3 бисульфата атазанавира (триэтанольный сольват).
Стадия зародышеобразования включает применение определенного количества зародышей для осуществления образования кристаллов Е3, например, мольное отношение зародышей Формы Е3 бисульфата атазанавира к свободному основанию составляет от примерно 0,02:1 до примерно 0,04:1.
Форму Е3 идентифицируют методом рентгеновской порошковой кристаллографии, что показано на Фиг.7, и структурой кристаллов, как показано на Фиг.6.
Согласно данному изобретению атазанавир в форме его свободного основания получают путем обработки раствора защищенной триаминной соли формулы
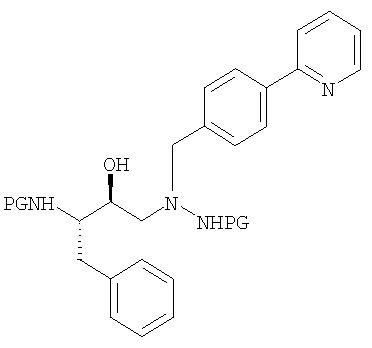
(где PG обозначает защитную группу, такую как трет.бутилоксикарбонил (Boc) или трифторацетил, предпочтительно Boc), кислотой, предпочтительно, соляной (когда применяют Boc) или основанием (когда применяют трифторацетил) в присутствии органического растворителя, такого как метиленхлорид, тетрагидрофуран или метанол, причем этот растворитель, предпочтительно, является метиленхлоридом, при температуре в пределах от примерно 25°С до примерно 50°С, предпочтительно, от примерно 30°С до примерно 40°С с образованием триаминной соли кислоты, предпочтительно, соляной, формулы:
и без выделения триаминной соли, взаимодействия этой соли с активным эфиром кислоты формулы:
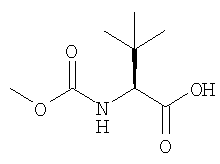
предпочтительно, активным эфиром формулы:
в присутствии основания, такого как K2HPO4, диизопропилэтиламин, N-метилморфолин, карбонат натрия или карбонат калия, предпочтительно K2HPO4, в присутствии органического растворителя, такого как метиленхлорид, смесь этилацетата и бутилацетата, ацетонитрил или этилацетат, предпочтительно, метиленхлорид, при температуре в пределах от примерно 25°С до примерно 50°С, предпочтительно, от примерно 30°С до примерно 40°С с получением свободного основания атазанавира.
Защищенный исходный триамин получают по реакции эпоксида:

где PG обозначает, предпочтительно. Boc, такого как N-(трет.бутилоксикарбонил)-2-(S)-амино-1-фенил-3(R)-3,4-эпоксибутан, с гидразинкарбаматом:

где PG, предпочтительно, обозначает Boc, в присутствии изопропилового спирта или другого спирта, такого как этанол или бутанол.
Бисульфат атазанавира пригоден для введения теплокровному животному, особенно человеку, для лечения или профилактики болезни, которая отвечает на ингибирование ретровирусной протеазы, особенно ретровирусной аспартат- протеазы, такой как ВИЧ-1- или ВИЧ-2-gag-протеазы, например ретровирусного заболевания, такого как СПИД или его предварительные стадии.
Бисульфат атазанавира, особенно Конфигурация С, Форма А или Форма Е3, предпочтительно, Конфигурация С или Форма А, можно применять в способе лечения болезней, вызванных вирусами, особенно ретровирусами, особенно СПИД′а или его предварительных стадий, когда терапевтически эффективное количество Конфигурации С бисульфата атазанавира. Формы А или Формы Е3 вводится в дозе, являющейся эффективной при лечении указанной болезни, особенно у теплокровного животного, например человека, который вследствие наличия одной из упомянутых болезней, особенно СПИД′а или его предварительных стадий, требует такого лечения. Предпочтительная доза, которую нужно ввести теплокровным животным, например людям, вес которых равен примерно 70 кг, составляет от примерно 3 мг до примерно 1,5 г, предпочтительно, от примерно 10 мг до примерно 1,25 г, например, от примерно 50 мг до примерно 600 мг в день, делится предпочтительно на 1-4 единичные дозы, которые могут быть, например, одинаковыми. Обычно дети получают половину дозы для взрослых. Лекарство, предпочтительно, вводить перорально.
Конфигурация С, Форма А или Форма Е3 бисульфата атазанавира применяются для описанных выше фармацевтических целей. Подходящие композиции, содержащие Конфигурацию С или Форму А или Форму Е3 для орального применения включают таблетки, порошки, капсулы и эликсиры. Около 10-600 мг активного ингредиента смешивают с физиологически приемлемым носителем, средством для доставки, эксципиентом, связующим, консервантом, стабилизатором, вкусовой добавкой и т.д. и получают единичную дозированную форму, как она называется в фармацевтической практике.
Фармацевтические композиции для орального введения могут быть получены путем соединения активного ингредиента с твердыми носителями, гранулирования полученной смеси, если это желательно, и после добавления соответствующих эксципиентов, если это желательно или необходимо, переработки смеси в таблетки, ядра, драже, капсулы или порошки для орального применения. Можно также вводить активные ингредиенты в полимерные носители, которые дают возможность активным ингредиентам диффундировать или высвобождаться в измеримых количествах.
Объемные агенты или наполнители содержаться в фармацевтической композиции по изобретению в количестве, находящемся в пределах от примерно 0 до примерно 95 вес.% и, предпочтительно, от примерно 10 до примерно 85 вес.% в расчете на композицию. Примеры объемных агентов или наполнителей, подходящих для применения, включают, но не ограничиваются этим, производные целлюлозы, такие как микрокристаллическая целлюлоза или древесная целлюлоза, лактоза, сахароза, крахмал, желатинизированный крахмал, декстроза, маннит, фруктоза, ксилит, сорбит, кукурузный крахмал, неорганические соли, такие как карбонат кальция, фосфат кальция, дикальцийфосфат, сульфат кальция, декстрин/декстраты, мальтодекстрин, прессуемые сахара и другие известные объемные агенты или наполнители и/или смеси двух или более этих компонентов, предпочтительно, лактоза.
Связующее может содержаться в фармацевтических композициях по изобретению в количестве, которое находится в пределах от примерно 0 до примерно 20 вес.%, предпочтительно, от примерно 1 до примерно 10 вес.% в расчете на композицию. Примеры связующих, подходящих для применения, включают, но не ограничиваются этим, гидроксипропилцеллюлозу, кукурузный крахмал, желатинизированный крахмал, модифицированный кукурузный крахмал, поливинилпирролидон (PVP) (молекулярный вес которого колеблется от примерно 5000 до примерно 80000, предпочтительно, он равен примерно 40000), гидроксипропилметилцеллюлозу (НРМС), лактозу, смолу акации, этилцеллюлозу, ацетат целлюлозы, а также восковые связующие, такие как карнаубский воск, парафин, спермацет, полиэтилены или микрокристаллический воск, а также другие обычные связующие и/или смеси двух или более связующих, предпочтительно, гидроксипропилцеллюлозу.
Дезинтегрант может содержаться в фармацевтической композиции по изобретению в количестве, которое находится в пределах от примерно 0 до примерно 20% по весу, предпочтительно, от примерно 0,25 до примерно 15% по весу в расчете на композицию. Примеры дезинтегрантов, подходящих для применения, включают, но не ограничиваются этим, натриевую соль кроскармелозы, кросповидон, картофельный крахмал, желатинизированный крахмал, кукурузный крахмал, натриевую соль гликолята крахмала, микрокристаллическую целлюлозу или другой известный дезинтегрант, предпочтительно, натриевую соль кроскармелозы.
Смазывающий агент может содержаться в фармацевтической композиции по изобретению в количестве, находящемся в пределах от примерно 0,1 до примерно 4% по весу, предпочтительно, от примерно 0,2 до примерно 2% по весу в расчете на композицию. Примеры смазывающих агентов для получения таблеток, пригодных для применения, включают, но не ограничиваются этим, стеарат магния, стеарат цинка, стеарат кальция, тальк, карнаубский воск, стеариновую кислоту, пальмитиновую кислоту, стеарилфумарат натрия или гидрированные растительные жиры и масла или другие известные смазывающие агенты и/или смеси двух или более смазывающих агентов, предпочтительно, стеарат магния.
Капсулы представляют собой твердые желатиновые капсулы и также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Твердые желатиновые капсулы могут включать активный ингредиент в виде гранул, например, с наполнителями, такими как лактоза, связующими, такими как крахмалы, кросповидон, и/или добавками, способствующими скольжению, такими как тальк или стеарат магния, и, если желательно, стабилизаторами. В мягких желатиновых капсулах, активный ингредиент, предпочтительно, растворен или суспендирован в подходящих масляных эксципиентах, таких как жирные масла, парафиновое масло или жидкие полиэтиленгликоли, могут быть также добавлены стабилизаторы и/или антибактериальные агенты.
Следующие ниже примеры отражают предпочтительные варианты изобретения.
Пример 1.
1-[4-(пирвдин-2-ил)фенил]-5(S)-2,5-бис{[N-(метоксикарбонил)-L-трет.лейцинил]амино}-4(S)-гидрокси-6-фенил-2-азагексана бисульфат (Форма А) (Форма А бисульфата атазанавира).
А.
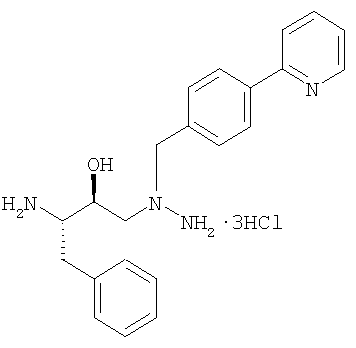
1-[4-(пиридин-2-ил)фенил]-5(S)-2,5-бис[трет.бутилоксикарбонил)амино]-4(S)-гидрокси-6-фенил-2-азагексан · 3HCl (триаминная·3HCl соль).
В трехгорлую круглодонную колбу объемом 1000 мл с механической мешалкой, трубкой для ввода азота и датчиком температуры добавляли защищенный триамин 1-[4-(пиридин-2-ил)фенил]-5(S)-2,5-бис[трет.бутилоксикарбонил)-амино]-4(S)-гидрокси-6-фенил-2-азагексана:
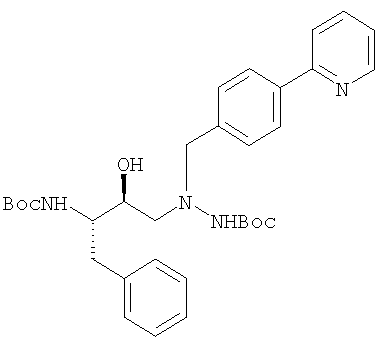
(100 г, 0,178 мол) и CH2Cl2 (500 мл, 5 мл/г защищенного триамина) (получено, как описано Z.Xu et al., Process Research and Development for an Efficient Synthesis of the НIV Protease Inhibitor BMS-232,632, Organic Process Research and Development, 6, 323-328 (2002)) и полученную суспензию перемешивали, поддерживая температуру, равной от примерно 5°С до примерно 22°С.
К реакционной смеси добавляли концентрированную соляную кислоту (68 мл, 0,82 мол, 4,6 экв.) с такой скоростью, чтобы температура реакционной смеси оставалась равной от 5°С до 30°С. Реакционную смесь нагревали до 30-40°С и перемешивали до завершения реакции по данным ЖХВР.
Добавляли воду (70-210 мл, 0,7-2,1 мл/г защищенного триамина) и перемешивали реакционную смесь в течение 15 мин и давали фазам разделиться. Верхний слой (обогащенное триамином ·3HCl солью) водное масло помещали в воронку.
В.
(Активный эфир N-метоксикарбонил-L-трет.лейцина )
В трехгорлую круглодонную колбу объемом 3000 мл, снабженную механической мешалкой, воронкой, трубкой для ввода азота и датчиком температуры добавляли N-метоксикарбонил-L-трет.лейцин (77,2 г, 0,408 мол, 2,30 экв.), 1-гидрокси- бензотиазол (НОВТ) (60,8 г, 0,450 мол, 2,53 экв.) и N-этил-N′-диметиламинопропилкарбодиимид (EDAC) (82,0 г, 0,430 мол, 2,42 экв.) с последующим введением CH2Cl2 (880 мл, 8,8 мл/г защищенного триамина) и полученную смесь перемешивали при комнатной температуре (18-25°С) до завершения образования активного эфира по данным ЖХВР.
С. 1-[4-(пиридин-2-ил)фенил]-5(S)-2,5-бис{[N-(метоксикарбонил)-L-трет.лейцинил]амино}-4(S)-гидрокси-6-фенил-2-азагексан (свободное основание атазанавира).
Безводный двухосновный фосфат калия (K2HPO4, 226 г, 1,30 мол, 7,30 экв. на защищенный триамин) растворяли в 1130 мл воды (11,3 мл/г защищенного амина, 5 мл/г K2HPO4).
К раствору активного эфира, полученного в разделе В, добавляли раствор K2HPO4. К перемешиваемой смеси активный эфир/водный раствор K2HPO4 медленно добавляли водный раствор гидрохлоридной соли из раздела А в течение 1,5-2,0 ч, продолжая перемешивание и поддерживая температуру в колбе от 5 до 20°С. После завершения добавления раствора гидрохлоридной соли из раздела А реакционную смесь (реакция сочетания) нагревали до 30-40°С и перемешивали до окончания реакции сочетания по данным ЖХВР.
Продукт сочетания охлаждали до 15-20°С и нижний, обогащенный продуктом органический слой, отделяли от верхнего водного слоя.
Органическую фазу, обогащенную продуктом, промывали 1 М NaH2PO4 (880 мл, рН 1,5, 8,8 мл/г защищенного триамина, 5 мол. экв. на защищенный триамин), фазам давали разделиться и удаляли отработанный водный слой.
Промытый обогащенный продуктом органический слой перемешивали с 0,5N NaOH (800 мл, 8 мл/г защищенного триамина) до тех пор, пока ЖХВР обогащенной органической фазы не покажет, что содержание активных эфиров составляет 0,3 I.I. каждого. Фазам давали разделиться и отработанный водный слой удаляли.
Обогащенную органическую фазу промывали 5% NaH2PO4 (450 мл, 4,5 мл/г защищенного триамина, рН 4,3), фазам давали разделиться и отработанный водный слой удаляли.
Обогащенную органическую фазу промывали 10 вес./об.% NaCl (475 мл, 4,75 мл/г защищенного триамина) и отработанный водный слой удаляли.
Концентрация свободного основания в растворе составляла 120-150 мг/мл и выход (расчетный) составлял 95-100 мол.%.
D. Обмен растворителей: CH2Cl2 на смесь ацетон/N-метилпирролидон.
К обогащенному раствору свободного основания из раздела С в трехгорлой круглодонной колбе объемом 3000 мл, снабженной механической мешалкой, датчиком температуры и конденсатором, добавляли N-метилпирролидон (148 мл, 1,25 мл/г свободного основания на основе количественного анализа в ходе процесса). Раствор концентрировали до примерно 360 мл (2,5-3,5 мл/г свободного основания из раздела С) при температуре в рубашке, равной 70°С или менее; к концентрированному раствору добавляли 500 мл ацетона (4-5 мл/г свободного основания из раздела С) и отгоняли смесь до получения объема, равного 400 мл или менее.
Добавление ацетона и отгонку повторяли до тех пор, пока анализ по ходу процесса не показал, что содержание CH2Cl2 достигло целевого значения. В момент кристаллизации объем CH2Cl2 в обогащенном органическом растворителе составлял 0,77 об/об %. К концентрированному раствору свободного основания добавляли ацетон до получения раствора 16 мл/г свободного основания. Температуру бани поддерживали равной 40-50°С для предотвращения кристаллизации свободного основания. Раствор отфильтровывали через фильтр 10 мкм или с меньшей пористостью, поддерживая температуру, равной 40-50°С. Фильтр промывали ацетоном (125 мл, 1,0 мл/г свободного основания) и промывную воду добавляли к раствору обогащенного свободного основания в ацетоне/N-метилпирролидоне, который использовали на следующей стадии.
Е. 1-[4-(пиридин-2-ил)фенил]-5(S)-2,5-бис{[N-(метоксикарбонил)-L-трет.лейцинил]амино}-4(S)-гидрокси-6-фенил-2-азагексана бисульфат.
Около 10% (2 г) общей загрузки концентрированной серной кислоты (19 г, 1,10 экв.) добавляли под поверхностью к раствору свободного основания в ацетоне/N-метилпирролидоне из раздела D, поддерживая температуру при 40-50°С.
В реакционную смесь добавляли зародыши с 5,0 вес.% (на свободное основание в растворе) бисульфата. Полученную смесь перемешивали при 40-50°С в течение, по меньшей мере, 30 мин, в это время бисульфат начал кристаллизоваться, о чем свидетельствует увеличение мутности смеси. Остающаяся серная кислота (17,8 г) добавлялась через примерно 5 ч в пять стадий согласно протоколу, определенному кубическим уравнением, при этом поддерживалась температура, равная 40-50°С.
Скорость добавления на каждой стадии определялась согласно кубическому уравнению, приведенному выше, величины этой скорости приведены в таблице 1.
После завершения добавления H2SO4 суспензию охлаждали до 20-25°С при перемешивании в течение, по меньшей мере, 1 ч. Суспензию перемешивали при 20-25°С в течение, по меньшей мере, 1 ч. Бисульфат отфильтровывали до блеска и маточную жидкость возвращали в цикл, что необходимо для осуществления полной передачи. Осадок на фильтре промывали ацетоном (5-10 мл/г свободного основания; 1200 мл ацетона). Бисульфат высушивали при NMT 55°С под вакуумом до достижения LOD<1% с получением кристаллического продукта.
Кристаллический продукт анализировали методами PXRD, DSC и TGA и получали спектры SSNMR, было установлено, что получены (несольватированные) кристаллы Формы А бисульфата, указанного в заголовке (см. Фиг.1-5).
Большинство водородов были опущены, включены только водороды на N9 и в кислоте.
Анизотропно очищенные атомы приведены в виде изотропного эквивалентного параметра замещения, определенного как (4/3)* [а2*В(1,1)+b2*B(2,2)+с2*В(3,3)+ab(cos gamma)*B(1,2)x+ac(cos beta)*B(1,3)+bc(cos alpha)*B(2,3)].
Форма А характеризовалась термограммой дифференциальной сканирующей калориметрии, на которой имелась эндотерма обычно в интервале от примерно 165,6°С до примерно 200,9°С, как показано на Фиг.3.
Форма А характеризовалась также кривой термогравиметрического анализа, свидетельствующей о незначительной потере веса до примерно 100-150°С. Кристаллы, полученные кубической кристаллизацией, когда H2SO4 добавляется с возрастающей скоростью в соответствии с кубическим уравнением, описанным выше, были больше и более четко определенными, характеризовались более узким интервалом размеров частиц и меньшим количеством мелких частиц, чем кристаллы, полученные при постоянной скорости добавления кислоты при кристаллизации.
Осадок на фильтре, полученный по методу кубической кристаллизации, был прессуемым в меньшей степени, чем осадок, полученный при постоянной скорости добавления, что способствовало эффективному обезвоживанию осадка и промывке, а также получению гомогенного продукта.
Пример 2.
Конфигурация С бисульфата атазанавира.
Метод А.
Кристаллы Формы А бисульфата атазанавира (полученные, как описано в Примере 1) (25,33 г) суспендировали в 200 мл воды и перемешивали смесь механически с получением толстого геля, который подвергали сушке.
Высушенную смесь разминали шпателем, получая Конфигурацию С. Рентгеновская порошковая дифрактограмма Конфигурации С показана на Фиг.6.
Метод В.
Кристаллы Формы А бисульфата атазанавира подвергали мокрому гранулированию, используя достаточное количество воды (около 40% вес./вес.), в подходящем смесителе-грануляторе. Влажную массу высушивали в печке. Полученный продукт просеивали через подходящее сито. Рентгеновская дифрактограмма полученного продукта соответствует Конфигурации С, как показано на Фиг.6.
Конфигурация С характеризовалась термограммой дифференциальной сканирующей калориметрии, показанной на Фиг.7, имеющей эндотермы обычно в интервале от примерно 76,7 до примерно 96,6°С и от примерно 156,8 до примерно 165,9°С.
Конфигурация С характеризовалась также кривой термогравиметрического анализа, свидетельствующей о потере веса, равной примерно 2,4% при примерно 125°С и равной примерно 4,4% при примерно 190°С, как показано на Фиг.8.
Пример 3.
Форма Е3 (триэтанольный сольват) бисульфата атазанавира.
Свободное основание атазанавира (полученное, как описано в Примере 1, раздел С) (3,0 г, 4,26 ммол) суспендировали в сухом этаноле с крепостью 200 (20,25 мл, 6,75 мл/г свободного основания) в трехгорлой круглодонной колбе объемом 100 мл, снабженной механической мешалкой, датчиком температуры и воронкой, уравновешивающей давление.
В суспензию свободного основания атазанавира при 20-25°С добавляли концентрированную H2SO4 (0,25 мл, 0,46 г, 4,69 ммол, 1,1 экв.). Полученный раствор (KF 0,2 до 1,0% воды) отфильтровывали до блеска (бумага Whatman #1), фильтр промывали 2,25 мл абсолютного этанола и промывной раствор добавляли к отфильтрованному раствору. Раствор нагревали до 37°С и добавляли зародыши, 10 мг, аморфного бисульфата атазанавира, полученные из кристаллов Формы Е3 (выдержкой кристаллов Формы Е3 при комнатной температуре), и перемешивали смесь в течение 15 мин. Через 1 ч добавляли гексан (380 мл, 8,25 мл/г свободного основания). Полученную кристаллизующуюся смесь перемешивали 8 ч при 15-25°С. Кристаллический бисульфат атазанавира отфильтровывали на воронке Бюхнера. Остаток на фильтре промывали 184 мл (4 мл/г свободного основания) смеси 1:1 этанол:гептан. Затем остаток промывали 46 мл (1 мл/г свободного основания) гептана. Полученный продукт сушили под вакуумом при температуре 40-50°С до достижения LOD=0,97%. Выход продукта составил 47,7 г (0,0594 мол, 74,3 мол.%), продукт представлял собой Форму Е3 бисульфата атазанавира (триэтанольный сольват) с ЖХВР HI=100,0 (см. Фиг.9 и 10).
Большинство водородов были опущены; включены только водороды на N9 и в кислоте.
Анизотропно очищенные атомы приведены в виде изотропного эквивалентного параметра замещения, определенного как (4/3)* [а2*В(1,1)+b2*B(2,2)+с2*В(3,3)+ab(cos gamma)*B(1,2)x+ac(cos beta)*B(1,3)+bc(cos alpha)*B(2,3)].
Форма Е3 характеризовалась термограммой дифференциальной сканирующей калориметрии с эндотермой обычно в интервале от примерно 89,4 до примерно 96,6°С, как показано на Фиг.11.
Форма Е3 характеризовалась также кривой термогравиметрического анализа с потерей веса, равной примерно 14,7% при температуре около 150°С, как показано на Фиг.11.
Пример 4.
Капсулы Конфигурации С бисульфата атазанавира получали из следующих композиций, как описано ниже.
Исходный состав при получении гранул бисульфата атазанавира получали следующим образом, при этом образовывалась Конфигурация С.
Форму А бисульфата атазанавира, гидрат лактозы и часть кросповидона (3 вес.% от общего количества кросповидона) смешивали в планетарном смесителе. Полученную смесь подвергали мокрому гранулированию вместе с очищенной водой для превращения Формы А в Конфигурацию С. Мокрый гранулят сушили в тарельчатой сушилке и измельчали в молотковой мельнице. Оставшийся кросповидон добавляли к измельченному грануляту и смесь перемешивали в смесителе PKV. Добавляли стеарат магния и перемешивали смесь до получения практически однородного продукта.
Полученным гранулятом заполняли капсулы 50 мг, 100 мг и 200 мг, содержащие бисульфат атазанавира.
Пример 5.
Порошок Формы А бисульфата атазанавира для орального введения получали из нижеследующей композиции.
Форму А бисульфата атазанавира смешивали с аспартамом, вкусовой добавкой Orange vanilla и сахарозой в подходящем смесителе. Смесь измельчали в молотковой мельнице, затем снова перемешивали до получения однородной смеси. Продукт помещали в бутылочки из полиэтилена высокой плотности.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВАЯ ПОЛИМОРФНАЯ ФОРМА КАЛЬЦИМИМЕТИЧЕСКОГО СОЕДИНЕНИЯ | 2011 |
|
RU2599789C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2007 |
|
RU2446156C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ТИОТРОПИЙБРОМИДА | 2006 |
|
RU2417224C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО 7Н-ПИРРОЛО[2,3-D] ПИРИМИДИНА И ЕГО ИНТЕРМЕДИАТА | 2016 |
|
RU2755618C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С НАТРИЕВОЙ СОЛЬЮ ТЕЛМИСАРТАНА | 2004 |
|
RU2372918C2 |
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИСУЛЬФАТА ИНГИБИТОРА JAK И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2015 |
|
RU2704795C2 |
| МАЛЕАТ 3-[2-(ДИМЕТИЛАМИНО)МЕТИЛ-(ЦИКЛОГЕКС-1-ИЛ)]ФЕНОЛА И ЕГО КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2006 |
|
RU2434845C2 |
| Способ получения полиморфа гидрохлорида 2-[4-(метиламинометил)фенил]-5-фтор-бензофуран-7-карбоксамида | 2018 |
|
RU2783418C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ГИДРОКСИ-4-МЕТИЛТИОБУТАНОВОЙ КИСЛОТЫ | 2000 |
|
RU2213729C2 |

Изобретение относится к способу получения ингибитора ВИЧ-протеазы сульфата атазанавира в виде кристаллов Формы А, который включает взаимодействие раствора свободного основания атазанавира в органическом растворителе, в котором сульфат атазанавира практически не растворяется, при температуре от 35°С до 55°С с первой порцией концентрированной серной кислоты в количестве, достаточном для реакции с менее, чем примерно 15 вес.% свободного основания атазанавира, добавление зародышей кристаллов Формы А сульфата атазанавира к реакционной смеси в качестве кристаллов сульфата атазанавира, добавление дополнительного количества концентрированной серной кислоты в несколько стадий, где кислота добавляется со все возрастающей скоростью, с образованием кристаллов сульфата атазанавира и сушку сульфата атазанавира с образованием кристаллов Формы А. Также предложен способ получения сульфата атазанавира в виде кристаллов Формы С. 6 н. и 14 з.п. ф-лы, 11 ил., 6 табл.
1. Способ получения сульфата атазанавира в виде кристаллов Формы А, который включает взаимодействие раствора свободного основания атазанавира в органическом растворителе, в котором сульфат атазанавира практически не растворяется, при температуре от 35 до 55°С с первой порцией концентрированной серной кислоты в количестве, достаточном для реакции с менее чем примерно 15 вес.% свободного основания атазанавира, добавление зародышей кристаллов Формы А сульфата атазанавира к реакционной смеси в качестве кристаллов сульфата атазанавира, добавление дополнительного количества концентрированной серной кислоты в несколько стадий, где кислота добавляется со все возрастающей скоростью, с образованием кристаллов сульфата атазанавира и сушку сульфата атазанавира с образованием кристаллов Формы А.
2. Способ по п.1, отличающийся тем, что раствор свободного основания атазанавира вначале реагирует с примерно 5-15% используемой серной кислоты в расчете на все количество серной кислоты.
3. Способ по п.1, отличающийся тем, что раствор свободного основания атазанавира вначале реагирует с примерно 8-12% используемой серной кислоты в расчете на все количество серной кислоты.
4. Способ по п.1, отличающийся тем, что в реакционную смесь свободного основания атазанавира и серной кислоты вводят от примерно 0,1 до примерно 80 вес.% кристаллов Формы А в расчете на вес свободного основания атазанавира.
5. Способ по п.1, отличающийся тем, что реакционную смесь, содержащую зародыши кристаллов, нагревают при температуре в пределах от примерно 35 до 55°С.
6. Способ по п.1, отличающийся тем, что органический растворитель для свободного основания атазанавира представляет собой ацетон, смесь ацетона с N-метилпирролидоном, этанол или смесь этанола с ацетоном.
7. Способ получения сульфата атазанавира Формы С, включающий выдержку кристаллов Формы А сульфата атазанавира в условиях высокой влажности, равной, по меньшей мере, примерно 95%, в течение, по меньшей мере, 24 ч.
8. Способ получения сульфата атазанавира Формы С, включающий:
а) взаимодействие раствора свободного основания атазанавира в органическом растворителе, в котором сульфат атазанавира практически не растворяется, при температуре от 35 до 55°С с первой порцией концентрированной серной кислоты в количестве, достаточном для реакции с менее чем примерно 15 вес.% свободного основания атазанавира, добавление зародышей кристаллов Формы А сульфата атазанавира к реакционной смеси в качестве кристаллов сульфата атазанавира, добавление дополнительного количества концентрированной серной кислоты в несколько стадий, где кислота добавляется со все возрастающей скоростью, с образованием кристаллов сульфата атазанавира и сушку сульфата атазанавира с образованием кристаллов Формы А;
б) суспендирование кристаллов сульфата атазанавира Формы А в воде и сушку суспензии с образованием Формы С.
9. Способ получения сульфата атазанавира Формы С, включающий:
а) взаимодействие раствора свободного основания атазанавира в органическом растворителе, в котором сульфат атазанавира практически не растворяется, при температуре от 35 до 55°С с первой порцией концентрированной серной кислоты в количестве, достаточном для реакции с менее чем примерно 15 вес.% свободного основания атазанавира, добавление зародышей кристаллов Формы А сульфата атазанавира к реакционной смеси в качестве кристаллов сульфата атазанавира, добавление дополнительного количества концентрированной серной кислоты в несколько стадий, где кислота добавляется со все возрастающей скоростью, с образованием кристаллов сульфата атазанавира и сушку сульфата атазанавира с образованием кристаллов Формы А;
б) выдержку кристаллов Формы А сульфата атазанавира в условиях относительной влажности, величина которой больше 95% в течение, по меньшей мере, 24 ч, с образованием Формы С.
10. Способ получения сульфата атазанавира Формы С, включающий:
а) взаимодействие раствора свободного основания атазанавира в органическом растворителе, в котором сульфат атазанавира практически не растворяется, при температуре от 35 до 55°С с первой порцией концентрированной серной кислоты в количестве, достаточном для реакции с менее чем примерно 15 вес.% свободного основания атазанавира, добавление зародышей кристаллов Формы А сульфата атазанавира к реакционной смеси в качестве кристаллов сульфата атазанавира, добавление дополнительного количества концентрированной серной кислоты в несколько стадий, где кислота добавляется со все возрастающей скоростью, с образованием кристаллов сульфата атазанавира и сушку сульфата атазанавира с образованием кристаллов Формы А;
б) мокрое гранулирование сульфата атазанавира и сушку мокрого гранулята с образованием Формы С.
11. Способ по одному из пп.1 или 8-10, отличающийся тем, что серную кислоту добавляют с увеличивающейся скоростью согласно следующему уравнению:

где Vвремя = объем серной кислоты, добавляемый во время прохождения кристаллизации,
Vвесь = общий объем кислоты, составляющий 90% от загрузки,
время = время прохождения кристаллизации,
времявсе = общее время кристаллизации.
12. Способ получения сульфата атазанавира:
в виде кристаллов Формы А, который включает получение триаминной соли формулы:
взаимодействие триаминной соли без ее выделения с активным эфиром кислоты формулы:
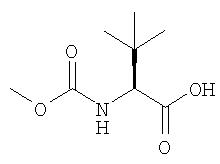
и основанием в присутствии органического растворителя с получением раствора свободного основания атазанавира:
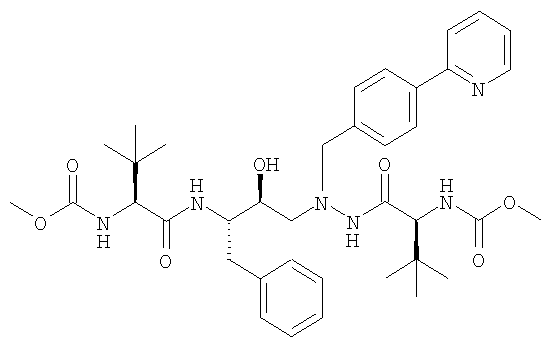
и превращение свободного основания в соответствующую сульфатную соль, при этом серную кислоту добавляют с увеличивающейся скоростью согласно следующему уравнению:
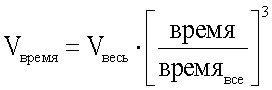
где Vвремя = объем серной кислоты, добавляемый во время прохождения кристаллизации,
Vвесь = общий объем кислоты, составляющий 90% от загрузки,
время = время прохождения кристаллизации,
времявсе = общее время кристаллизации.
13. Способ по п.12, отличающийся тем, что триаминная соль является гидрохлоридной солью:
14. Способ по п.12, отличающийся тем, что активный эфир кислоты имеет формулу:

15. Способ по п.12, отличающийся тем, что основание представляет собой гидроокись щелочного металла, гидроокись щелочно-земельного металла, карбонат щелочного металла, карбонат щелочно-земельного металла, фосфат щелочного металла, фосфат щелочно-земельного металла или органическое основание.
16. Способ по п.12, отличающийся тем, что основание представляет собой NaOH, KOH, Mg(OH)2, K2HPO4, MgCO3, Na2CO3, K2CO3, триэтиламин, диизопропилэтиламин или N-метилморфолин и органический растворитель является метиленхлоридом, этилацетатом, дихлорэтаном, тетрагидрофураном, ацетонитрилом или N,N-диметилформамидом.
17. Способ по п.12, отличающийся тем, что триаминная соль и активный эфир реагируют при температуре в пределах от примерно 30 до примерно 40°С.
18. Способ по п.17, отличающийся тем, что триаминная соль и активный эфир реагируют в присутствии K2HPO4 в качестве основания и метиленхлорида в качестве растворителя.
19. Способ по п.12, отличающийся тем, что он включает стадию введения зародышей кристаллов сульфата атазанавира в смесь свободного основания, ацетона и N-метилпирролидона.
20. Способ получения сульфата атазанавира в виде кристаллов формы А:
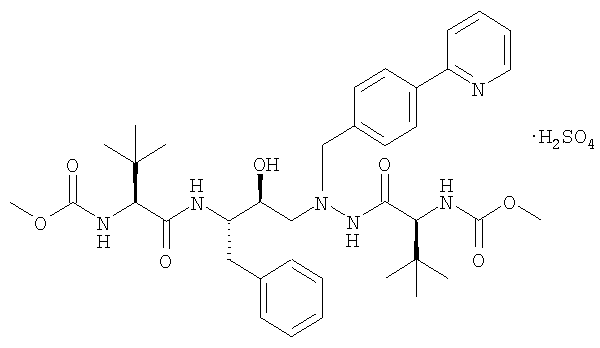
который включает получение гидрохлорида триамина формулы:

взаимодействие гидрохлорида триамина с активным эфиром формулы:
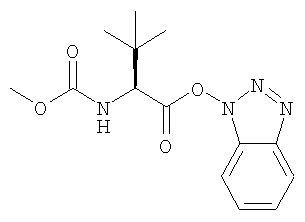
и K2HPO4 в присутствии метиленхлорида с образованием раствора свободного основания формулы:
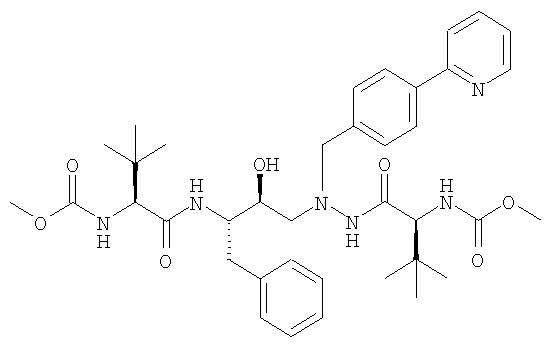
в метиленхлориде и превращение свободного основания в соответствующую сульфатную соль путем кубической кристаллизации, причем серную кислоту добавляют с увеличивающейся скоростью согласно следующему уравнению:

где Vвремя = объем серной кислоты, добавляемый во время прохождения кристаллизации,
Vвесь = общий объем кислоты, составляющий 90% от загрузки,
время = время прохождения кристаллизации,
времявсе = общее время кристаллизации.
| МОНОСУЛЬФАТ АЗАПЕПТИДНОГО ПРОИЗВОДНОГО И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ВИЧ-ПРОТЕАЗУ | 1998 |
|
RU2186070C2 |
| US 5849911 А, 15.12.1998 | |||

